chapter 9 ft-ir, ft-raman, ab initio and dft ......fundamental bands in the experimental ft-ir and...
TRANSCRIPT

CHAPTER – 9
FT-IR, FT-RAMAN, AB INITIO AND DFT STRUCTURAL,
VIBRATIONAL FREQUENCY AND HOMO-LUMO ANALYSIS
OF 1-NAPHTHALENACETIC ACID METHYL ESTER
9.1. INTRODUCTION
1-Naphthaleneacetic acid, commonly abbreviated as NAA, is an organic
compound with carboxylmethyl group (CH2CO2H) linked to the "1-position" of
naphthalene. Aromatic acids are very important in biochemical as well as in
biological fields. The derivatives of NAA are used in anti-inflammatory drugs [1] and
used as an intermediate for the synthesis of pharmaceuticals, photochemical, dyes etc.
It is a plant hormone and used as an ingredient in many commercial plant rooting
horticultural products.
9.2. LITERATURE SURVEY
Various studies have been conducted on the vibrational spectra, electron
exchange, structural stability, lattice vibrations, anti resonance of naphthalene using
normal co-ordinate analysis and resonance Raman spectral analysis by several
workers. Furthermore, the vibrational analysis of naphthoic acid, 2-naphthoic acid,
bromo naphthoic acid, 1-hydroxynaphthalene, 1-napthyl acetic acid and naphthalene
acetic acid have been extensively studied and analyzed by quantum mechanical
approach [2-12].
Lippincott et al [2] investigated the vibrational spectra of naphthalene and
naphthalene-d-8 molecules. IR and Raman spectra of the samples with high
deuterium content were taken and normal co-ordinate analysis was used for the
assignment of the frequencies.
Freeman et al [3] reported the normal coordinate analysis of the planar
vibrations of naphthalene. The 33 planar vibration frequencies of naphthalene have
been calculated using a simple quadratic valence force field with 24 independent
constants which initially derived from benzene molecule. Systematic attempts have
been made for the agreement with certain observed frequencies.

An experimental and theoretical study of vibrational spectra and electron
vibration interactions of the naphthalene radical anion of h8 and d8 were carried out by
Torii et al [4]. The vibrational force field and the IR intensities were calculated with
CASSCF and B3LYP methods. This study reported that C-C stretch occurred in one
ring while shrink in the other below 2000 cm-1
due to the changes in the electronic
structure induced by this vibration.
Szeghalmi et al [5] analysed 1-hydroxy-2- acetonapthone (HAN) using
quantum chemical calculations and Resonance Raman (RR) spectroscopy. A time
dependent approach for the analysis of the resonance Raman spectroscopy was
applied to determine the Franck-Condon parameters. RR spectra of HAN were
recorded in cyclohexane solution and about 15 vibrational modes were found to be
resonantly enhanced. DFT calculations at BPW91 and B3LYP levels of theory have
been carried out with 6-31G(d) and 6-311+G(d) split valence basis sets with
polarization and diffuse functions for the optimization. The quantum chemical
calculations for the ground and the first excited states have been performed at the
CASSCF level of theory using 6-31G(d) and 6-31G(d,p) basis sets. The computations
led to planar molecular structures of Cs symmetry for both ground and first excited
states. The excited state structural parameters significantly differed from the ground
state. The analysis of the molecular orbitals and the NPA calculations showed that
there was an increase of electron density on the carbonyl group in the excited state.
Alka Srivastava et al. [6] analysed the Raman and FTIR spectra of
naphthalene and its cation using HF/6-311++G** and B3LYP/6-311++G** level.
The structural parameters of naphthalene and its cation were investigated with DFT
calculations and reported that ionization causes small change in the optimized
geometry and vibrational frequencies but produced significant change in the intensity
of neutral naphthalene. A significant reduction of intensity in C-H stretching
vibrations and the intensity of C-C stretching vibrations along with C-H in-plane
bending vibrations have been observed after ionization.
Krishnakumar et al.[7] investigated the structures and vibrational frequencies
of 2-naphthoic acid and 6-bromo-2-naphthoic acid molecules using density functional
theory calculations with B3LYP/6-311+G* method. The FTIR and FT-Raman
spectra of these molecules were recorded in the regions 4000-400 cm-1
and 3500-

100cm-1
respectively. The presence of carbonyl group and bromine atom in the
molecules were discussed elaborately. The various modes of vibrations were assigned
based on TED calculations obtained from normal coordinate analysis.
The FTIR and FT-Raman spectra of 1-naphthyl acetic acid molecule by DFT
method with B3LYP/6-311+G** basis set was analyzed by Krishnakumar et al.[8].
The Cartesian representation of the theoretical force constants have been computed by
assuming Cs point group symmetry. Scaling of the force field was performed
according to SQM procedure using selective scaling in the natural internal coordinate
representation. Transformations of the force field and the subsequent normal
coordinate analysis using the least squares refinement of the scaling factors,
calculation of Total Energy Distribution (TED) and IR and Raman intensities were
calculated. Pure Lorentzian band shapes were used with bandwith of 10cm-1
to plot
the simulated IR and Raman spectra. The presence of OH, CH2, C-O, and C=O
vibrations were discussed in detail.
Chandra et al. [9] discussed the naphthoic acid molecule by DFT methods
using FTIR gas phase spectrum and FTIR and FT-Raman spectra in solid phase.
Quantum mechanical calculations were used to find out energies, geometrical
structure, infrared intensities and Raman scattering activities. This study reported
that the naphthalene ring was distorted by yielding ring angles smaller and larger
than the normal value of 120o due to the presence of carboxylic acid group on the
ring. Besides, the dimer parameters of the molecule by B3LYP/6-31G(d,p) method
were utilized to explain the intermolecular hydrogen bonding. Furthermore, the
polarizabilities, total dipole moment and Mullikan atomic charges of naphthoic acid
have been discussed ornately.
Ravikumar et al.[10] analyzed the FT Raman and IR spectra of the
biologically active molecule, 1-naphthalene acetamide (NA). The equilibrium
geometry, bonding features and harmonic vibrational wavenumbers of NA have been
calculated with the help of B3LYP density functional theory (DFT) method. The
assignments of the vibrational spectra have been carried out with the help of normal
coordinate analysis (NCA) following the scaled quantum mechanical force field
methodology (SQMFF). The downshifting of NH2 stretching wavenumber indicates
the formation of intermolecular N–H O hydrogen bonding. The NBO analysis

confirmed the occurrence of strong intermolecular hydrogen bonding in the
molecule.
Kavitha et al [11] investigated naphthalene acetic acid molecule on the basis
of its anharmonic vibrational frequencies, molecular structure, NBO, HOMO and
LUMO energies. The optimized geometrical parameters were compared with the
experimental X-ray data. The Anharmonic frequencies of the molecule were analysed
by DFT level of theory utilizing 6-311+G(d,p) basis set. Optimized geometry showed
that the molecular structure was not plannar due to the consequence of steric repulsion
with hydrogen atom and oxygen atom. The calculated dihedral angles showed that the
naphthalene ring was coplanar and in addition, the ring was distorted due to the
deviation of angles smaller and larger than the normal value of 120o. Stability of the
molecule arising from hyperconjugative interactions and charge delocalization has
been analyzed using natural bond orbital (NBO) analysis. The results showed that
charge in electron density (ED) in the * and * anti-bonding orbitals and E2
energies which confirmed the occurrence of ICT (Intermolecular Charge Transfer)
within the molecule. The calculated HOMO and LUMO energies implies that the
charge transfer occurs between ring and acid group. Moreover, these orbital
significantly overlap in their position for naphthalene acetic acid.
Govindarajan et al.[12] have been analysed the FT-IR and FT-Raman spectra
of 1-methoxy naphthalene using DFT method with B3LYP/3-21G, B3LYP/6-31G(d),
B3LYP/6-31G(d,) and B3LYP/6-311++G(d,p) basis sets by assuming Cs point group
symmetry. The FT-IR and FT-Raman spectra were taken in the range of 4000-400cm-
1 and 3500-100cm
-1 respectively. The optimized structural parameters were calculated
with the above said basis sets and the values are compared with the experimental
values of naphthoic acid. But these theoretical optimized parameters were slightly
overestimated with crystallographic literature values. The complete vibrational
assignments of wavenumbers are made on the basis of Potential Energy Distribution
(PED). The electronic properties such as excitation energies and wavelengths were
performed by time-dependent DFT (TD-DFT) approach. The frontier molecular
orbital energies have been calculated with B3LYP/6-311+G(d,p) level and the results
showed that charge transfer occurs within the molecule.

To the best of my knowledge, neither quantum chemical calculations nor the
vibrational spectra of 1-naphthaleneacetic acid methyl ester (abbreviated as
1-NAAME, is known as methyl 1-naphthylacetate) have been reported up to now.
This inadequacy observed in the literature encouraged to do this theoretical and
experimental vibrational spectroscopic research to give a correct assignment of the
fundamental bands in the experimental FT-IR and FT-Raman spectra on the basis of
the calculated total energy distribution (TED). Therefore, the present study aims to
give a complete description of the molecular geometry, molecular vibrations and
HOMO-LUMO energies of 1-NAAME. Besides, Frontier Molecular Orbitals (FMO)
and thermodynamic properties were performed. On the basis of the thermodynamic
properties of the title compound at different temperatures have been calculated,
revealing the correlations between standard heat capacities (C) standard entropies
(S), and standard enthalpy changes (H) and temperatures.
9.3. COMPUTATIONAL METHODS
The primary task for the computational work was to determine the
optimized geometry of the compound. Since the experimental geometries of free
1NAAME were not available, the spatial coordinate positions of Naphthalene acetic
acid, as obtained from an X-ray structural analysis [13] were used as the initial
coordinates for the theoretical calculations. To determine conformational features of
the molecule, the selected degree of torsional freedom, Г(C11-C18-C21-O23), was
varied from 00 to 360
0 in every 10
0 and the molecular energy profile was obtained
with the B3LYP/6-311G(d,p) method. The molecular structure optimization of the
title compound and corresponding vibrational harmonic frequencies were calculated
using HF and the DFT with hybrid Beckee-3-Lee-Yag-Parr (B3LYP) combined with
6-31G(d,p), 6-311G(d,p) basis sets using GAUSSIAN 03 program package without
any constraint on the geometry. Geometries have been first optimized with full
relaxation on the potential energy surfaces at HF/6-31G(d,p) and 6-311G(d,p) basis
sets. The geometry was then re-optimized at B3LYP level using 6-31G(d,p) and
6-311G(d,p) basis sets. The stability of the optimized geometries was confirmed by
wavenumber calculations, which gave positive values for all the obtained
wavenumbers. TED calculations, which show the relative contributions of the
redundant internal coordinates to each normal vibrational mode of the molecule and

thus enable us numerically to describe the character of each mode, were carried out
by the Scaled Quantum Mechanical (SQM) method [14] using PQS program [15] in
which the output files created at the end of the wavenumber calculations. The
optimized geometrical parameters, true rotational constants, fundamental vibrational
frequencies, IR and Raman intensity, Raman activity, electronic polarizability, atomic
charges, dipole moment, and other thermo dynamical parameters were calculated
using the Gaussian 03 package [16]. By combining the results of the GAUSSVIEW
[17] program with symmetry considerations, vibrational frequency assignments were
made with a high degree of accuracy.
The electronic properties, such as HOMO-LUMO energies, absorption
wavelengths and oscillator strengths were calculated using B3LYP method of the time
dependent TD-DFT, basing on the optimized structure in solvent (DMSO and
chloroform) and gas phase. The changes in the thermodynamic functions such as the
heat capacity, entropy, and enthalpy were investigated for the different temperatures
from the vibrational frequency calculations of the title molecule.
9.4. RESULTS AND DISCUSSION
9.4.1. Prediction of Raman Intensities
The Raman activities (SRa) calculated with the Gaussian 03 program were
converted to relative Raman intensities (IRa) using the following relationship derived
from the intensity theory of Raman scattering [18-19].
…… 9.1
Where, νo is the laser exciting wavenumber in cm−1
(in this work, the excitation
wavenumber is used as νo = 9398.5 cm−1
, which corresponds to the wavelength of
1064 nm of a Nd : YAG laser), νi the vibrational wavenumber of the ith
normal mode
(in cm−1
) and Si is the Raman scattering activity of the normal mode νi, f (is a constant
equal to 10−12
) is a suitably chosen common normalization factor for all peak
intensities. h, k, c and T are Planck constant, Boltzmann constant, speed of light and
temperature in Kelvin, respectively.

9.4.2. Molecular Geometry and Potential energy surface scan
The molecular structure along with numbering of atoms of 1-NAAME is
obtained from Gaussian 03 and GAUSSVIEW programs and is as shown in the
Fig 9.1. The global minimum energy obtained by HF and DFT structure optimization
using 6-31G(d,p) and 6-311G(d,p) basis sets for the title molecule as -649.052 a.u.,
-649.176 a.u, -653.098 a.u. and -653.245 a.u. respectively. The most optimized
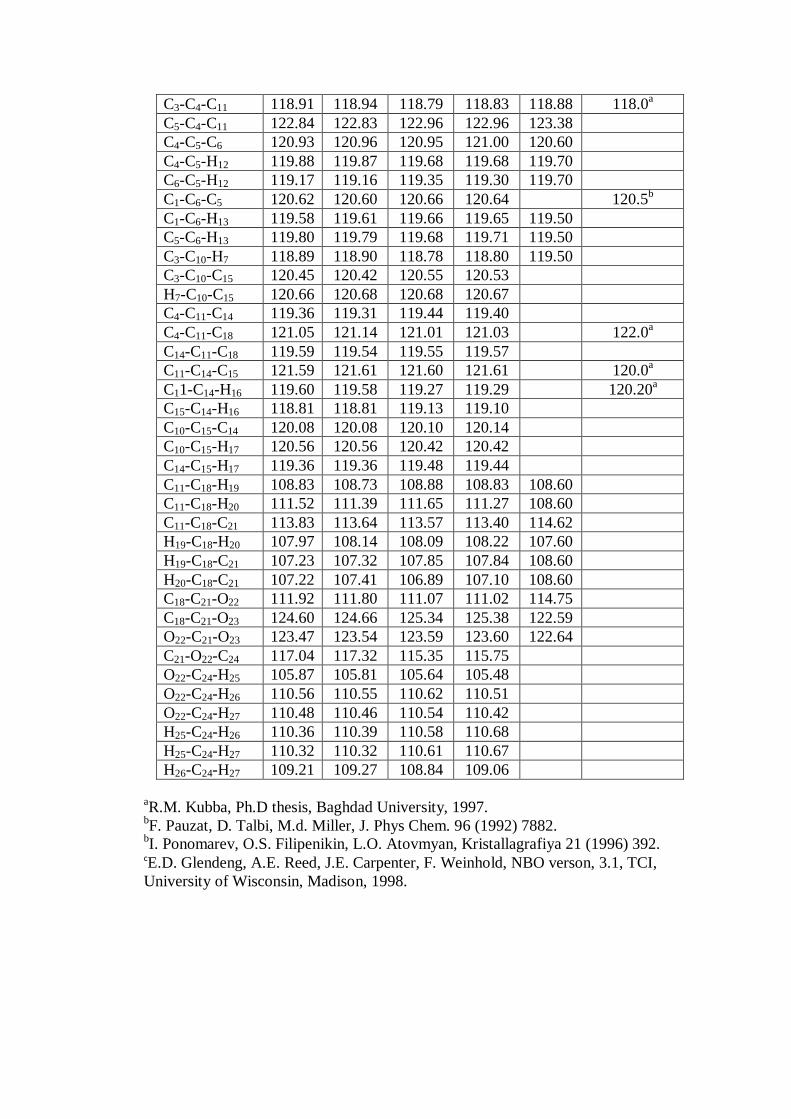
structural parameters (bond length, bond angle and dihedral angle) calculated by HF,
DFT/B3LYP with 6-31G(d,p) and 6-311G(d,p) basis sets are compared with the XRD
experimental data and represented in Table 9.1 in accordance with the atom
numbering scheme given in Fig. 9.1.
The crystal structure of 1-Naphthaleneacetic acid using intensity data
estimated from Weissenberg films [20] and the structure of 1-Naphthaleneacetic acid
was investigated with improved precision with respect to geometric parameters [21] .
Basically, the C-C bonds in naphthalene ring are not of the same length such
that the bonds C11–C14, C15–C10, C3–C4 and C2–C1 are about 1.36 Å, whereas the other
C-C bonds are of about 1.42 Å . From the theoretical values, one can find that most
of the optimized bond lengths are larger than the experimental values, because the
theoretical calculations refer to isolated molecules in the gaseous phase and the
experimental results are for molecules in the solid state. The C-H bond lengths
obtained from the experimental value [13] are ranging from 0.93 to 0.97 Å while of
theoretical values it ranges between 1.07 to 1.09 Å. This larger deviation of C-H bond
lengths may be due to the low scattering factors of hydrogen atoms in X-ray
diffraction experiments which are not included in the theoretical calculations. This
overestimation is also verified in our calculation as represented in Table 9.1.
The C18-C21, i.e. Cacetic-Ccarboxylic acid, bond length calculated by
B3LYP/6-311G(d,p) is 1.5182 Å which is very close to the experimental value. In the
earlier literature [22], the bond lengths of C=O and C-O were 1.266 Å and 1.305 Å,
respectively. In this study, the corresponding bond lengths are 1.220 Å and 1.2759 Å
[13], which are calculated as 1.2063 Å and 1.3469 by B3LYP/6-311G(d,p). The
C4-C11-C14 is 119.4o and C4-C5-C6 is 120.6
o which changed from the normal value of
120o shows that the naphthalene ring is distorted due to the substitution.

Hence, by comparing the calculated values of bond lengths with experimental
data [13], C-C, C-H, C=O and C-O varies between the ranges (0.01 Å – 0.035 Å),
(0.12 Å -0.16 Å), 0.071 Å, 0.014 Å respectively at B3LYP/6311G(d,p). The bond
lengths reported in the literatures [33-35] are much closer to the theoretical
values.The graphical representation of bond lengths between different atoms are as
shown in fig. 9.4.
In order to describe conformational flexibility of the title molecule, the energy
profile as a function of C-C-C-O torsion angle was achieved with
B3LYP/6-311G(d,p) method and shown in Fig. 9.2. In connection with the CH3
orientations of the oxygen atom of the carboxylic acid group, 1NAAME may have
two possible structures as shown in Fig.9.3. It is clear from Fig. 9.2, that there are
two local minima (two conformers) observed at 800, which is C2 conformer, and 260
0
that is C1 conformer, (-653.2437622690 Hartree for 800, -653.2446478280 Hartree
for 2600) for T (C-C-C-O). The difference between two conformers is 0.55570
kcal/mol. According to Fig.9.3 and obtained energy values, of the two forms, the
stable isomer is C1 since it possess lower energy when compared with C2 form and
the most stable conformer is for 2600 torsion angle.
9.4.3. Vibrational Assignments
1-NAAME molecule has 27 atoms with 75 normal modes of vibrations and
considered under C1 point group symmetry. The detailed analysis of fundamental
modes of vibration with FT-IR and FT-Raman experimental frequencies, unscaled
and scaled vibrational frequencies, and Total Energy distribution (TED) of
1-NAAME using HF and DFT methods with different basis sets basis sets are
reported in the Table 9.2. All vibrations are active both in IR and Raman. The IR and
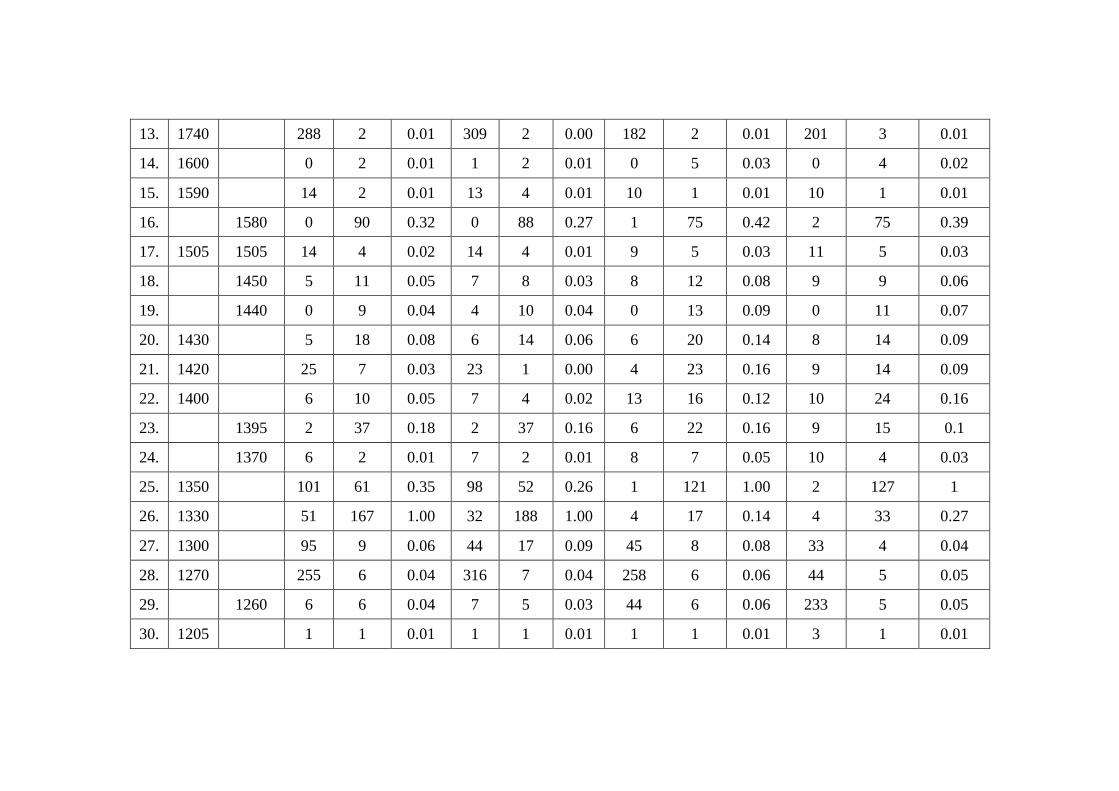
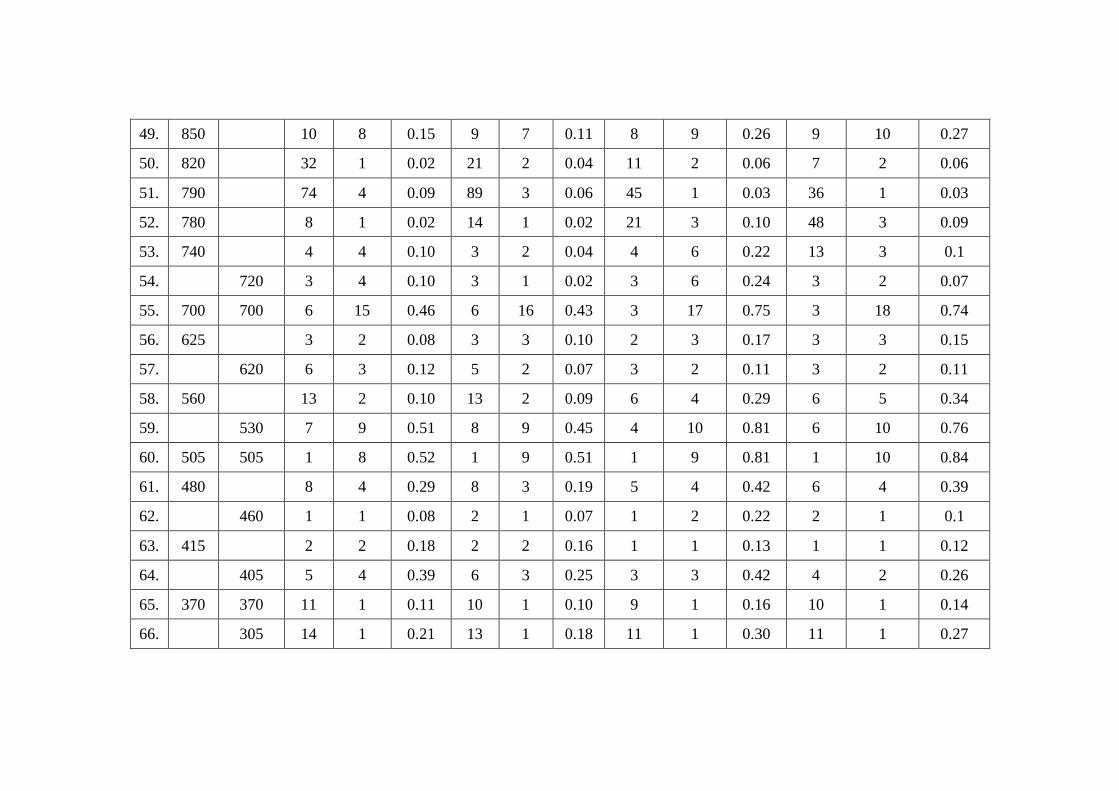
Raman intensity with different methods are depicted in table 9.3. The experimental
FTIR and FT-Raman spectra were shown in Figs 9.5 and 9.6 respectively. The TED
for each normal mode among the symmetry coordinates of the molecule was
calculated. A complete assignment of the fundamentals was proposed based on the
calculated TED values, infrared and Raman intensities. Calculated Raman and IR
intensities helped to distinguish and more precisely assign those fundamentals which
are close in frequency. The calculated harmonic force constants and wavenumbers are
usually higher than the corresponding experimental quantities because of the

combination of electron correlation effects and basis set deficiencies. The observed
slight disagreement between theory and experiment could be a consequence of the
anharmonicity and the general tendency of the quantum mechanical methods to
overestimate the force constants at the exact equilibrium geometry. Nevertheless, after
applying a uniform scaling factor, the theoretical calculation reproduces the
experimental data well. Therefore, in order to improve the calculated values in
agreement with the experimental values, it is necessary to scale down the calculated
harmonic frequencies.
9.4.2.1. C-H Vibrations
The heteroatomic structure shows the presence of C-H stretching vibrations in
the region 3100–3000 cm-1
and these vibrations are not found to be affected by the
nature and position of the substituent [26-27]. This is the characteristic region for the
identification of C-H stretching vibrations. Two benzene rings are fused together in
1-NAAME molecule, it has seven adjacent C-H moieties. The expected seven C-H
stretching vibrations corresponding to stretching modes of C1-H8, C2-H9, C5-H12,
C6-H13, C10-H7, C14-H16 and C15-H17 units. Most of the aromatic compounds have
nearly four infrared peaks in the region 3080-3010 cm-1
due to ring C-H stretching
bonds [28-29]. Accordingly, in the title compound, the symmetric stretching
vibrations are assigned to 3060 cm-1
of FTIR and 3100 cm-1
(vw), 3070 cm-1
of
FT-Raman respectively. The peaks at 3050, 3005 cm-1 of FTIR and the peaks at
3020, 2990 cm-1
FT-Raman are assigned to asymmetric stretching vibrations. The
corresponding calculated vibrational frequencies (mode nos. 1-7) by
B3LYP/6-311G(d,p) method fall within the recorded spectral range. However, one of
the asymmetric stretching vibration observed at 2990 cm-1
is comparatively lesser
than the expected range by 10 cm-1
is due to steric effect of acetic acid methyl ester in
the naphthalene ring. As expected, all the stretching modes are pure stretching
modes as is evident from TED column in Table 9.2; they almost contribute around
100%.
The C-H in plane bending vibrations usually occurs in the region 1430-990
cm-1
and is very useful for characterization purposes [30]. The bands due to C-H in-
plane ring vibration are coupled vibrations with C-C stretching vibration and are
observed in the region 1000-1300 cm-1
. In this study, the FT-IR peaks at 1205, 1195,

1160, 1140, 1070, 1045 and 1010 cm-1
(mode nos. 30,31,33,34,37,38,40) are assigned
to C-H in-plane-bending vibrations as reported in the Table 9.2 and figures 9.5 - 9.6.
The aromatic and hetroatomic compounds display strong out-of phase C-H
bending and ring bending absorption bands below 900 cm-1
that can be frequently be
correlated with the substitution pattern [31-32]. In the present study, the peaks at
1000, 960, 940, 900, 820, 790 and 780 cm-1
in FT-IR confirm the C-H out of plane
bending vibrations. The peaks 1000, 960 and 940 cm-1
are shifted above from the
expected range due to the vibrations of more groups in the molecule. However, the
frequencies assigned for C-H out-of plane vibrations shows good agreement with
theoretical anharmonic wavenumber values at 969-800 cm-1
by B3LY/6311G(d,p)
method. The TED also show mixed contribution of approximately 80% and 40% for
these vibrations (mode nos.42-44 and 50-52). By comparing the experimental and
calculated frequencies by HF and B3LYP, it is noticed that the difference produced by
B3LYP method is less and it makes better coincidence with the experimental
frequencies.
9.4.2.2. C-C and Ring Vibrations
The ring stretching vibrations are very much important in the spectrum of
naphthalene derivatives and are highly characteristic of the aromatic ring itself.
However, empirical assignments of vibrational modes for peaks in the fingerprint
region are not easy. The earlier literatures [33-35] pointed out that the bands between
1430-1650 cm-1
were due to C=C stretching modes. Naphthalene ring stretching
vibrations are expected in the region 1620-1390 cm-1
[11]. Naphthalene ring
vibrations are found to make a major contribution in the IR and Raman spectra
[36-37]. As predicted, the C=C aromatic stretching vibrations are noted here at 1600
(FTIR), 1590 (FTIR), 1580 (FT-R) and 1505 (both). These vibrations are in
agreement with the scaled theoretical assignments given by DFT.
The expected range of C-C vibration is between 1300- 1400 cm-1
. These C-C
stretching vibrations are coupled with skeletal ring and in-plane bending vibrations.
Here, it is assigned at 1440, 1370,1350, 1330 and 1270 cm-1
. There are deviation of
frequencies from the expected range is due to the presence of other vibrations such as
(C-O), CH2 and CH3 vibrations as shown in tables 9.2 – 9.5.

9.4.2.3. Methyl group vibrations
For the assignments of CH3 group frequencies, nine fundamentals can be
associated to each CH3 group [31]. The C-H stretching in CH3 occurs at lower
frequencies than those of aromatic ring (3100-3000 cm-1
). Moreover, the asymmetric
stretch is usually at higher wavenumber than the symmetric stretch. In this present
work, the CH3 symmetric stretching frequency is assigned at 2840 cm-1
, whereas CH3
asymmetric frequencies are assigned at 2950 cm-1
(FT-IR) and 2960 cm-1
(FT-
Raman). The deformation of CH3 group is usually observed in the range 1450-1400
cm-1
for methyl substituted aromatic rings [34, 38]. Accordingly, in 1-NAAME, the
peaks at 1420, 1400 cm-1
in FT-IR and 1450, 1395 cm-1
in FT-Raman are assigned to
CH3 in-plane bending deformation vibrations while the peaks at 1170 cm-1
(FT-IR)
and 1130 cm-1
(FT-Raman) are assigned to CH3 in-plane rocking modes. In this
study, the CH3 out-of-plane bending vibrations are mixed vibrations as shown at TED
in Table 9.2.
9.4.2.4. Methylene vibrations
For the assignments of CH2 group frequencies, basically six fundamentals can
be associated to each CH2 group namely CH2 symmetric stretch; CH2 asymmetric
stretch; CH2 scissoring and CH2 rocking which belongs to in-plane vibrations and two
out-of plane vibrations, viz., CH2 wagging and CH2 twisting modes, which are
expected to be depolarized for out-of plane vibrations. It is expected that, the
asymmetric CH2 stretching vibration generally observed in the region 3000-2900
cm-1
, while the CH2 symmetric stretch will appear between 2900 and 2800 cm-1
. In
accordance with the above statement, here, the asymmetric and symmetric stretching
vibrations were observed at 2930 cm-1
and 2850 cm−1
in FT-Raman respectively.
It is expected that the scissoring band in the spectra of hydrocarbons occurs
nearly at 1465 cm−1
while methylene twisting and wagging vibrations are observed in
the region 1350–1150 cm−1
which are weaker than those resulting from methylene
scissoring. Since the bending modes involving hydrogen atom attached to the central
carbon falls into 1450-875 cm-1
range, there is extensive vibrational coupling of these
modes with CH2 deformations particularly with the CH2 twist. It is notable that both
CH2 scissoring and CH2 rocking modes were sensitive to the molecular confirmations

[11]. A series of bands in this region, arising from the methylene group, is
characteristic of the spectra of solid samples of long-chain acids. According to the
above said references, in this work, the peaks at 1430 cm-1
with TED of 72% and
1300 cm-1
with TED of 83% are assigned to CH2 scissoring and CH2 wagging
vibrations while the peak at 930 cm-1
in both spectra is assigned to CH2 rocking which
is coupled with C-C stretching. Nevertheless, the scissoring vibration noted here, has
been shifted to a lower frequency and increased in intensity because of its proximity
to the carbonyl group. Also, the presence of asymmetrical vibration of CH3 overlaps
the scissoring vibration of the methylene group. All the vibrations of CH2 computed
by B3LYP method agrees well with the experimental observations.
9.4.2.5. C=O and C-O vibrations
The C=O stretch of carboxylic acids is identical to the C=O stretch in ketones,
which is expected in the region 1740–1660 cm−1
[39]. In literature [11], very strong
band at 1661 cm−1
in IR and 1648 cm−1
in Raman for NAA was assigned to C=O
stretching vibrations. The C=O bond formed by Pπ-Pπ between C and O, internal
hydrogen bonding reduces the frequencies of the C=O stretching absorption to a
greater degree than does intermolecular H bonding because of the different electro-
negativities of C and O, the bonding are not equally distributed between the two
atoms. In this present study, a very strong band observed in FT-IR spectrum at 1740
cm−1
(mode no. 13) is assigned to C=O stretching vibrations which the B3LYP
predicted value (1738 cm−1
) show a small deviation of about ca. 2 cm-1
with FT-IR
data and the TED value of 88% as reported in Table 9.2.
The C-O stretching vibrations of esters actually consists of two asymmetric
coupled vibrations ie., C-C(=O)-O and O-C-C, the former is more important. The
assignment of the carbonyl band to an ester should be confirmed by observation of a
strong band in the C-O stretching region, 1300-1100 cm-1
. The C-O stretch
correlations are less reliable than the C=O stretch correlations. Here, in this molecule,
the C-O stretching vibration is assigned to 1260 cm-1
in FT-IR which has the TED
value of 57%. The C-O stretching related to O-C-C is assigned at 1005 cm-1
which
has the TED value of 41% .Most of the C-O vibrations are mixed vibrations as shown
in the TED values in Table 9.2. The peaks at 850 cm-1
and 625 cm-1
are assigned to
C-O of C-O-CH3 (mode nos.:49,56).

9.4.2.6. Coupled vibrational assignments
Since the molecule has 75 vibrations and has lot of coupled vibrations below
1000 cm-1
, difficulty arrived to assign all the vibrations exactly. However, the effort
has been taken in this chapter to assign some coupled vibrations with the help of
Gaussview [17] programme.
C-(CH2-C(=O)-O-CH3), C-(CH2-C(=O)-O-CH3), C-(CH2-C(=O)-O-
CH3) vibrations are coupled vibrations and noted at 1030, 890 and 700 cm-1
respectively. The remainders of the observed and calculated frequencies are
accounted in Table 9.2 with TED values.
9.4.3. Interpretation on Atomic charges
Mulliken atomic charge calculation has an important act in the application of
quantum chemical calculation to molecular system because of atomic charges effect
molecular polarizability, dipole moment, electronic structure and more a lot of
properties of molecular systems. The total atomic charges of 1NAAME are obtained
by Mulliken population analysis with HF and B3LYP methods and are listed in Table
9.4. and graph is given in Fig. 9.10. The negative values on C24 atom of CH3 group
leads to a redistribution of electron density. On the other hand, atomic charges of
hydrogen atom in the CH3 group are also almost identical. The C21 atom connected
with highly electro negative oxygen atoms making that particular carbon more
electron deficient. So that it gets more positive and acidic. The negative charges
mainly located on atoms O21 and O22 will interact with the positive part of the
receptor. On the contrary, C21 is the most positively charged part, which can interact
with the negatively charged part of the receptor easily. Moreover, due to inductive
effect, the charges on C18 and C11 decrease as the distance increases.
9.4.4. Thermodynamic properties
Several calculated thermodynamical parameters such as the Zero-Point
Vibration Energies (ZPVE), the entropy, S, the molar capacity, C, at constant volume,
rotational constants, rotational temperature and dipole moment have been presented in
Table 9.5. The variations in the ZPVEs seem to be insignificant. The total energies are
found to decrease with the increase of the basis set dimension. The changes in the

total entropy of 1NAAME at room temperature at different basis sets are only
marginal.
On the basis of vibrational analysis at B3LYP/6-311G(d,p) level, the standard
statistical thermodynamic functions: standard heat capacity standard entropy
, and standard enthalpy changes for the title compounds were obtained
from the theoretical harmonic frequencies and listed in Table 9.6.
From Table 9.8, it can be observed that these thermodynamic functions are
increasing with temperature ranging from 100 to 700 K due to the fact that the
molecular vibrational intensities increases with temperature [40]. The correlation
equations between heat capacities, entropies, enthalpy changes and temperatures were
fitted by quadratic formulas and the corresponding fitting factors (R2) for these
thermodynamic properties are 0.9985, 1.0000 and 0.9999, respectively. The
corresponding fitting equations are as follows and the correlation graphics of those
shows in Fig 9.7 to Fig 9.9.
)9985.0R(T10x1815.6T1852.01118.0C 2250m,p
…… 9.2
)0000.1R(T10x5714.3T1951.05526.59S 2250m
…… 9.3
)9999.0R(T10x9731.6T0093.02674.0H 2250m
…… 9.4
All the thermodynamic data supply helpful information for the further study
on the 1NAAME. They can be used to compute the other thermodynamic energies
according to the relationships of thermodynamic functions and estimate directions of
chemical reactions according to the second law of thermodynamics in thermochemical
field. It should be kept in mind that all thermodynamic calculations were done in gas
phase and they could not be used in solution.
9.4.5. HOMO–LUMO analysis
The total energy, energy gap and dipole moment have an effect on the stability
of a molecule. The optimization was done in order to investigate the energetic
behavior and dipole moment of title compound in gas phase and solvent. The total

energy, dipole moment and frontier molecular orbital energies have been calculated
with B3LYP/6-311G(d,p) level. Results obtained from solvent and gas phase are
listed in Table 9.7.
The energy values of HOMO are computed as -6.16125, -6.10356 and
-5.99989 eV and LUMO as -1.48711, -1.43323 and -1.33745 eV, and the energy gap
values are 4.67414, 4.67033 and 4.66244 eV in DMSO, chloroform and gas phase for
1NAAME molecule, respectively. Lower value in the HOMO and LUMO energy gap
explains the eventual charge transfer interactions taking place within the molecule.
Surfaces for the frontier orbitals were drawn to understand the bonding scheme of
present compound. The four important molecular orbitals (MO) for title molecule: the
second highest and highest occupied MOs and the lowest and the second lowest
unoccupied MOs which were denoted as HOMO−1, HOMO, LUMO and LUMO+1,
respectively. These MOs for gas phase are outlined in Fig. 9.10. The positive phase is
red and the negative one is green. According to Fig 9.10, the HOMO of 1NAAME
submits a charge density localized at C=C bonds on the ring expect of CH3 group, but
LUMO is characterized by a charge distribution at C-H bonds on the ring.
Dipole moment shows the molecular charge distribution and is given as a vector
in three dimensions. Therefore, it can be used as a descriptor to depict the charge
movement across the molecule. Direction of the dipole moment vector in a molecule
depends on the centres of positive and negative charges. Dipole moments are strictly
determined for neutral molecules. From the table 9.9, it is clear that in going from the
gas phase to the solvent phase, the dipole moment value increases.
9.4.6. UV–VIS spectra analysis
Molecules allow strong –* and –* transition in the UV–vis region with
high extinction coefficients. Ultraviolet spectra analyses of 1NAAME have been
researched by theoretical calculation. In order to understand electronic transitions of
compound, TD-DFT calculations on electronic absorption spectra in gas phase and
solvent (DMSO and Chloroform) were performed. The calculated frontier orbital
energies, absorption wavelengths (λ), oscillator strengths ( f ) and excitation energies
(E) for gas phase and solvent (DMSO and Chloroform) are illustrated in Table 9.8.
The major contributions of the transitions were designated with the aid of Swizard

program [41]. The visible absorption maxima of this molecule from calculations of
the molecular orbital geometry show that correspond to the electron transition
between frontier orbitals such as translation from HOMO to LUMO. As can be seen
from the Table 9.8, the calculated absorption maxima values have been found to be
291.04, 280.48, 237.04 nm for gas phase, 292.68, 279.95, 237.17 nm for DMSO
solution and 293.07, 280.21, 237.32 nm for chloroform solution at
DFT/B3LYP/6-311G(d,p) method. As can be seen, all calculations performed are
very close. In view of calculated absorption spectra, the maximum absorption
wavelength corresponds to the electronic transition from HOMO to LUMO with 94%
contribution. This transition (HL) is predicted as -* transition. The other
wavelength, excitation energies, oscillator strength, calculated counterparts with
major contributions and assignments can be seen in Table 9.8.
9.5. CONCLUSION
The FT-IR and FT-Raman spectra were recorded and the detailed vibrational
assignments using HF and DFT methods with 6-31G(d,p) and 6-311G(d,p) basis sets
were presented for 1-Naphthaleneacetic acid methyl ester, for the first time. The
difference between the corresponding wavenumbers (observed and calculated) is very
small for most of fundamentals. Therefore, the results presented in this work for 1-
NAAME indicate that this level of theory is reliable for the prediction of both infrared
and Raman spectra of the title compound. The equilibrium geometries of 1-NAAME
have been determined and compared with X-ray crystal data. Furthermore, theoretical
calculations give the thermodynamic properties (heat capacity, entropy and enthalpy)
for the compound. It can be observed that these thermodynamic functions are
increasing with temperature ranging from 100 to 700 K due to the fact that the
molecular vibrational intensities increase with temperature. The optimization has been
done in order to investigate the energetic behavior and dipole moment of title
compound in the gas phase and solvent. UV-VIS spectral analyses of 1NAAME have
been analyzed by theoretical calculation. In order to understand electronic transitions
of compound, TD-DFT calculations on electronic absorption spectra in gas phase and
solvent (DMSO and Chloroform) were performed. The observation done regarding
this molecule is as follows:

From the theoretical values, one can find that most of the optimized bond
lengths are larger than the experimental values, because the theoretical
calculations refer to isolated molecules in the gaseous phase and the
experimental results are for molecules in the solid state.
The C-H bond lengths obtained from the experimental value are ranging from
0.93 to 0.97 Å while of theoretical values it ranges between 1.07 to 1.09 Å.
This larger deviation of C-H bond lengths may be due to the low scattering
factors of hydrogen atoms in X-ray diffraction experiments which are not
included in the theoretical calculations.
The C4-C11-C14 is 119.4o and C4-C5-C6 is 120.6
o which changed from the
normal value of 120o shows that the naphthalene ring is distorted due to the
substitution.
In connection with the CH3 orientations of the oxygen atom of the carboxylic
acid group, 1-Naphthaleneacetic acid methyl ester have two possible structures
(C1 and C2 conformers. It is clear that there are two local minima (two
conformers) observed at 800
(C2 conformer) and 2600 (C1 conformer). The
corresponding energies are -653.2437622690 Hartree for 800,
-653.2446478280 Hartree for 2600 for T (C-C-C-O). The difference between
two conformers is 0.55570 kcal/mol. According to obtained energy values, of
the two forms, the stable isomer is C1 since it possess lower energy when
compared with C2 form and the most stable conformer is for 2600 torsion
angle.
The occurrence of lesser frequency from the expected range (ie., 2990 cm-1
for
C-H Stretching) are due to the presence of functional groups in the first
position of naphthalene ring.
In methyl esters of long chain acids, there should be a 3 band pattern with
bonds near 1250 cm-1
, 1205 cm-1
and 1175 cm-1
and the 1175 cm-1
band is the
strongest band. This statement is confirmed in this work by the occurrence of
three bands at 1260 cm-1
, 1205 cm-1
and 1170 cm-1

In view with the study in this chapter, the assignment suggested for CH2
bending modes, assignments follow in decreasing wave number such that CH2
deformation > CH2 waging > CH2 twisting > CH2 rocking. The scissoring
vibration has been shifted to a lower frequency from the expected range and
increased in intensity because of its proximity to the carbonyl group and the
overlapping of asymmetric vibration of CH3.
The HOMO of 1NAAME submits a charge density localized at C=C bonds on
the ring expect of CH3 group, but LUMO is characterized by a charge
distribution at C-H bonds on the ring.
In view of calculated absorption spectra, the maximum absorption wavelength
corresponds to the electronic transition from HOMO to LUMO with 94%
contribution. This transition (HL) is predicted as -* transition. Hence, the
results of this study will help researchers to analyze and synthesis of new
materials.

Fig. 9.1. Molecular Structure and Numbering of atoms of
1-Naphthaneacetic Acid Methyl Ester

Fig. 9.2. PES Scan for the selected torsional angle T (C-C-C-O) of freedom
Fig. 9.3. Theoretically optimized two possible geometric structures of
1-Naphthaleneactic Acid Methyl Ester molecule
Tota
l E
ner
gy (
Hart
ree)
Scan Coordinate
C1 Conformer
-653.2446478280 Hartree
C2 Conformer
-653.2437622690 Hartree
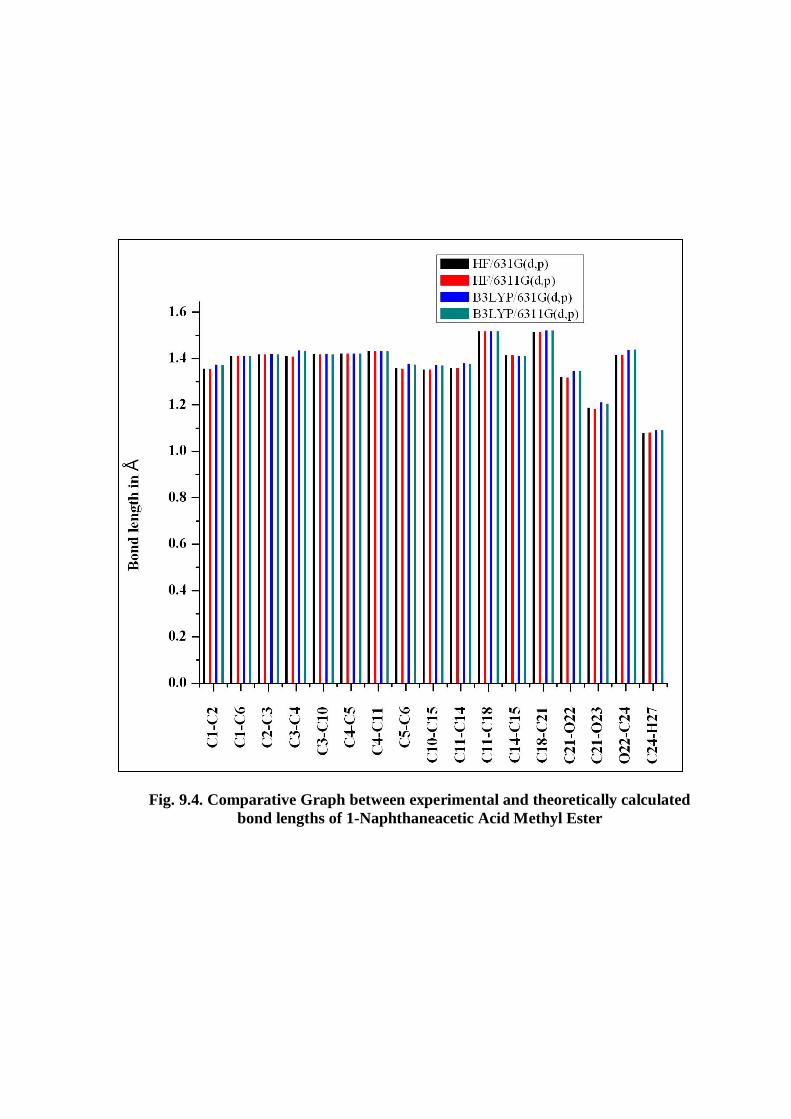
Fig. 9.4. Comparative Graph between experimental and theoretically calculated
bond lengths of 1-Naphthaneacetic Acid Methyl Ester

Fig. 9.5. Experimental FTIR spectrum of 1-Naphthaleneacetic acid methyl ester
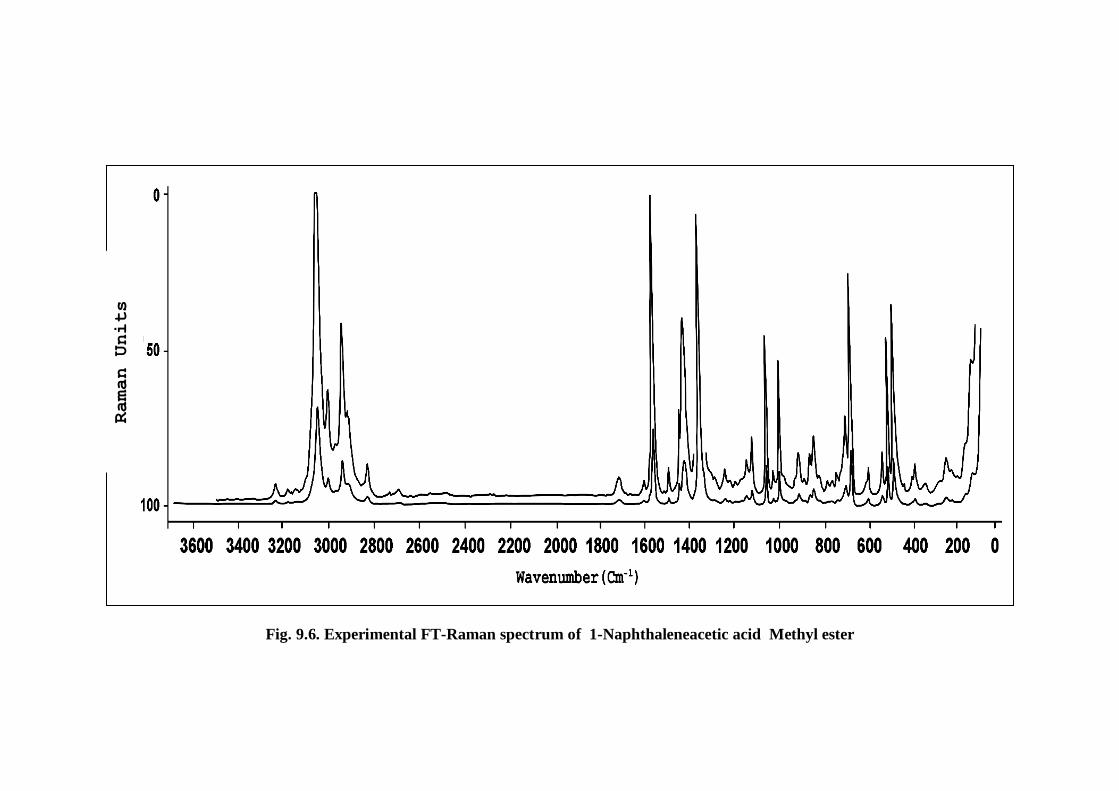
Fig. 9.6. Experimental FT-Raman spectrum of 1-Naphthaleneacetic acid Methyl ester
Raman Units
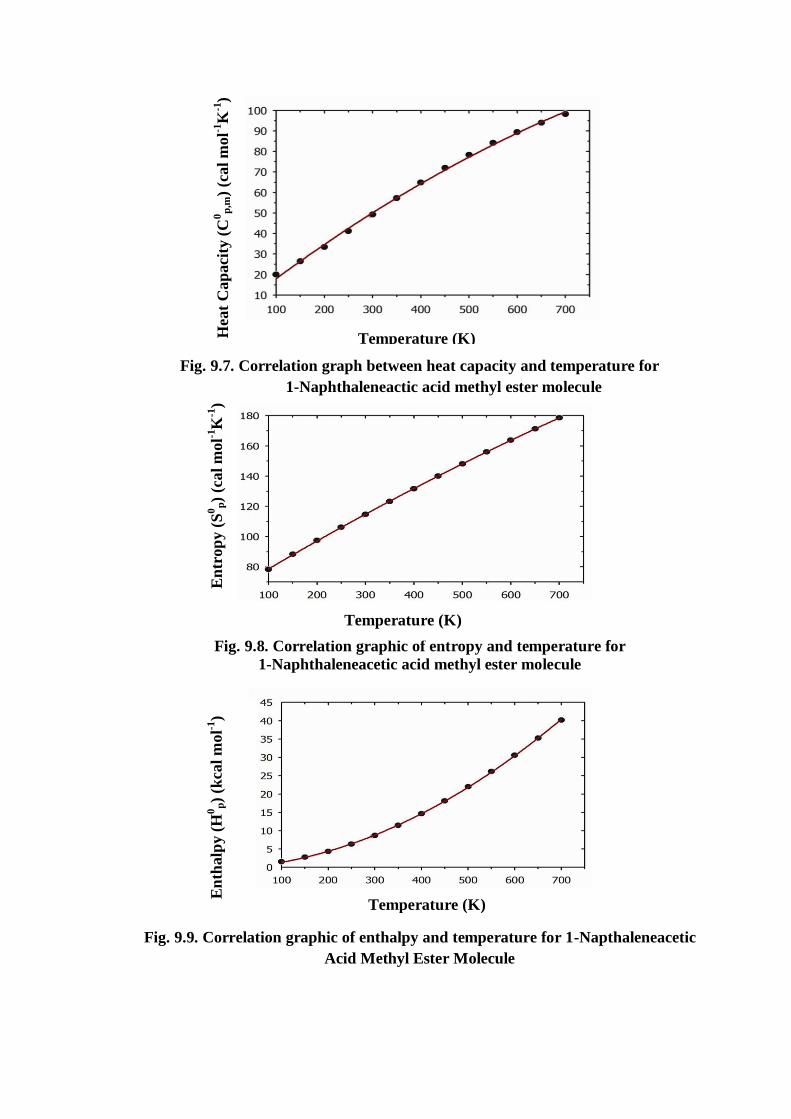
Fig. 9.7. Correlation graph between heat capacity and temperature for
1-Naphthaleneactic acid methyl ester molecule
Fig. 9.8. Correlation graphic of entropy and temperature for
1-Naphthaleneacetic acid methyl ester molecule
Fig. 9.9. Correlation graphic of enthalpy and temperature for 1-Napthaleneacetic
Acid Methyl Ester Molecule
En
trop
y (
S0
p)
(cal
mol-1
K-1
)
Temperature (K)
Hea
t C
ap
aci
ty (
C0
p,m
) (c
al
mol-1
K-1
)
Temperature (K)
En
thalp
y (
H0
p)
(kca
l m
ol-1
)
Temperature (K)

Fig. 9.10. Comparative graph for mullikan charges on carbon atoms of
1-Napthaleneacetic Acid Methyl Ester with HF and DFT for different basis sets

Fig. 9.11. The atomic orbital compositions of the frontier molecular orbital for
1-Napthaleneacetic acid Methyl ester molecule

Table 9.1
Optimized Geometrical Parameters (Bond lengths and Bond Angles) of
1-Naphthaleneacetic acid Methyl Ester
Parameters
HF B3LYP Experimental data
6-31
G(d,p)
6-311
G(d,p)
6-31
G(d,p)
6-311
G(d,p) XRD
c
Bond lengths in Å
C1-C2 1.3578 1.3568 1.3757 1.3728 1.3410
C1-C6 1.4130 1.4129 1.4141 1.4123 1.3860 1.41b
C1-H8 1.0756 1.0753 1.0861 1.0842 0.9300
C2-C3 1.4191 1.4188 1.4205 1.4191 1.4060
C2-H9 1.0763 1.0759 1.0870 1.0851 0.9300
C3-C4 1.4117 1.4103 1.4360 1.4336 1.4220 1.462a,1.421
b
C3-C10 1.4203 1.4197 1.4204 1.4188 1.4050
C4-C5 1.4228 1.4224 1.4235 1.4220 1.4130 1.422b
C4-C11 1.4335 1.4329 1.4341 1.4324 1.4160 1.449a
C5-C6 1.3596 1.3587 1.3777 1.3748 1.3600 1.377b
C5-H12 1.0731 1.0726 1.0842 1.0823 0.9300 1.095b
C6-H13 1.0756 1.0753 1.0860 1.0842 0.9300 1.098b
C10-H7 1.0762 1.0758 1.0868 1.0850 0.9300
C10-C15 1.3547 1.3537 1.3738 1.3710 1.3360
C11-C14 1.3607 1.3597 1.3805 1.3778 1.3650 1.380a
C11-C18 1.5181 1.5180 1.5195 1.5182 1.5000
C14-C15 1.4147 1.4146 1.4140 1.4121 1.3940 1.430a
C14-H16 1.0758 1.0754 1.0865 1.0846 0.9300 1.105a
C15-H17 1.0755 1.0753 1.0859 1.0842 0.9300
C18-H19 1.0837 1.0830 1.0938 1.0918 0.9700
C18-H20 1.0816 1.0812 1.0925 1.0906 0.9700
C18-C21 1.5145 1.5147 1.5230 1.5218 1.5000
C21-O22 1.3220 1.3198 1.3491 1.3469 1.2759
C21-O23 1.1897 1.1842 1.2128 1.2063 1.2200
O22-C24 1.4173 1.4169 1.4380 1.4396
C24-H25 1.0792 1.0791 1.0896 1.0876
C24-H26 1.0812 1.0817 1.0928 1.0911
C24-H27 1.0810 1.0816 1.0925 1.0909
Bond angle in degrees
C2-C1-C6 119.87 119.96 119.91 119.93 120.30
C2-C1-H8 120.38 120.38 120.24 120.27 119.80
C6-C1-H8 119.75 119.76 119.85 119.80 119.80
C1-C2-C3 120.93 120.93 121.02 121.03 121.20
C1-C2-H9 120.43 120.43 120.48 120.44 119.40
C3-C2-H9 118.65 118.63 118.50 118.53 119.40
C2-C3-C4 119.39 119.42 119.20 119.19 119.08
C2-C3-C10 121.00 120.95 121.29 121.33 121.88
C4-C3-C10 119.61 119.63 119.51 119.48 119.03
C3-C4-C5 118.25 118.23 118.25 118.21 117.74 119.0b
C3-C4-C11 118.91 118.94 118.79 118.83 118.88 118.0a
C5-C4-C11 122.84 122.83 122.96 122.96 123.38
C4-C5-C6 120.93 120.96 120.95 121.00 120.60
C4-C5-H12 119.88 119.87 119.68 119.68 119.70
C6-C5-H12 119.17 119.16 119.35 119.30 119.70
C1-C6-C5 120.62 120.60 120.66 120.64
120.5b
C1-C6-H13 119.58 119.61 119.66 119.65 119.50
C5-C6-H13 119.80 119.79 119.68 119.71 119.50
C3-C10-H7 118.89 118.90 118.78 118.80 119.50
C3-C10-C15 120.45 120.42 120.55 120.53
H7-C10-C15 120.66 120.68 120.68 120.67
C4-C11-C14 119.36 119.31 119.44 119.40
C4-C11-C18 121.05 121.14 121.01 121.03
122.0a
C14-C11-C18 119.59 119.54 119.55 119.57
C11-C14-C15 121.59 121.61 121.60 121.61
120.0a
C11-C14-H16 119.60 119.58 119.27 119.29
120.20a
C15-C14-H16 118.81 118.81 119.13 119.10
C10-C15-C14 120.08 120.08 120.10 120.14
C10-C15-H17 120.56 120.56 120.42 120.42
C14-C15-H17 119.36 119.36 119.48 119.44
C11-C18-H19 108.83 108.73 108.88 108.83 108.60
C11-C18-H20 111.52 111.39 111.65 111.27 108.60
C11-C18-C21 113.83 113.64 113.57 113.40 114.62
H19-C18-H20 107.97 108.14 108.09 108.22 107.60
H19-C18-C21 107.23 107.32 107.85 107.84 108.60
H20-C18-C21 107.22 107.41 106.89 107.10 108.60
C18-C21-O22 111.92 111.80 111.07 111.02 114.75
C18-C21-O23 124.60 124.66 125.34 125.38 122.59
O22-C21-O23 123.47 123.54 123.59 123.60 122.64
C21-O22-C24 117.04 117.32 115.35 115.75
O22-C24-H25 105.87 105.81 105.64 105.48
O22-C24-H26 110.56 110.55 110.62 110.51
O22-C24-H27 110.48 110.46 110.54 110.42
H25-C24-H26 110.36 110.39 110.58 110.68
H25-C24-H27 110.32 110.32 110.61 110.67
H26-C24-H27 109.21 109.27 108.84 109.06
aR.M. Kubba, Ph.D thesis, Baghdad University, 1997.
bF. Pauzat, D. Talbi, M.d. Miller, J. Phys Chem. 96 (1992) 7882.
bI. Ponomarev, O.S. Filipenikin, L.O. Atovmyan, Kristallagrafiya 21 (1996) 392.
cE.D. Glendeng, A.E. Reed, J.E. Carpenter, F. Weinhold, NBO verson, 3.1, TCI,
University of Wisconsin, Madison, 1998.
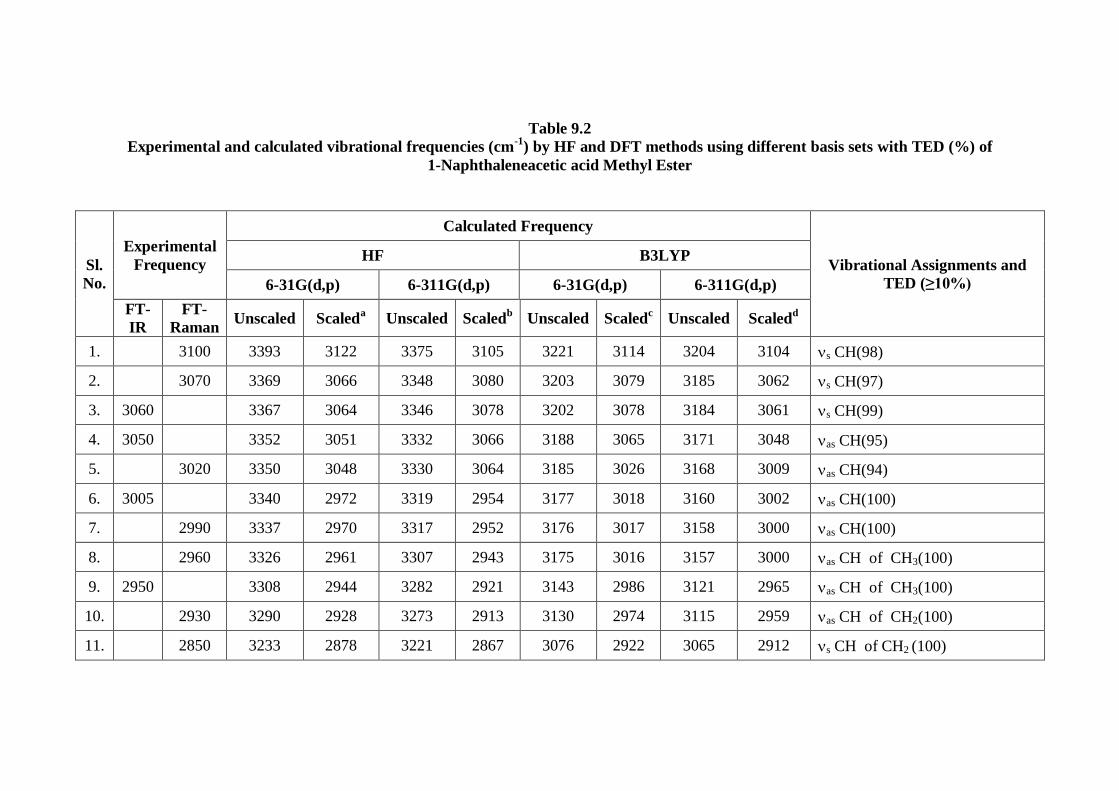
Table 9.2
Experimental and calculated vibrational frequencies (cm-1
) by HF and DFT methods using different basis sets with TED (%) of
1-Naphthaleneacetic acid Methyl Ester
Sl.
No.
Experimental
Frequency
Calculated Frequency
Vibrational Assignments and
TED (≥10%)
HF B3LYP
6-31G(d,p) 6-311G(d,p) 6-31G(d,p) 6-311G(d,p)
FT-
IR
FT-
Raman Unscaled Scaled
a Unscaled Scaled
b Unscaled Scaled
c Unscaled Scaled
d
1.
3100 3393 3122 3375 3105 3221 3114 3204 3104 s CH(98)
2.
3070 3369 3066 3348 3080 3203 3079 3185 3062 s CH(97)
3. 3060
3367 3064 3346 3078 3202 3078 3184 3061 s CH(99)
4. 3050
3352 3051 3332 3066 3188 3065 3171 3048 as CH(95)
5.
3020 3350 3048 3330 3064 3185 3026 3168 3009 as CH(94)
6. 3005
3340 2972 3319 2954 3177 3018 3160 3002 as CH(100)
7.
2990 3337 2970 3317 2952 3176 3017 3158 3000 as CH(100)
8.
2960 3326 2961 3307 2943 3175 3016 3157 3000 as CH of CH3(100)
9. 2950
3308 2944 3282 2921 3143 2986 3121 2965 as CH of CH3(100)
10.
2930 3290 2928 3273 2913 3130 2974 3115 2959 as CH of CH2(100)
11.
2850 3233 2878 3221 2867 3076 2922 3065 2912 s CH of CH2 (100)

12. 2840
3225 2870 3206 2853 3065 2912 3049 2897 s CH of CH3(100)
13. 1740
2002 1782 1982 1764 1817 1757 1798 1738 C=O(88)
14. 1600
1838 1635 1823 1623 1679 1623 1665 1610 C=C(75)
15. 1590
1806 1608 1791 1594 1655 1600 1641 1586 C=C(71)
16.
1580 1788 1591 1775 1580 1632 1578 1618 1564 C=C(67)
17. 1505 1505 1682 1497 1669 1486 1562 1510 1549 1498 C=C (45)
18.
1450 1633 1453 1625 1446 1509 1459 1499 1449 def CH2 of CH3(95)
19.
1440 1625 1446 1614 1436 1506 1456 1496 1446 C-C(72)
20. 1430
1624 1446 1612 1435 1496 1446 1487 1438 CH2(72)
21. 1420
1621 1442 1612 1434 1493 1443 1483 1434 def CH3(100)
22. 1400
1616 1438 1609 1432 1484 1435 1474 1425 def CH3(51)
23.
1395 1602 1425 1590 1415 1480 1431 1471 1422 def CH3(58)
24.
1370 1545 1375 1535 1366 1437 1389 1427 1380 C-C (74)
25. 1350
1499 1334 1489 1325 1410 1363 1396 1350 C-C (72)
26. 1330
1477 1314 1465 1304 1397 1351 1382 1336 C-C (61)
27. 1300
1459 1299 1448 1289 1331 1287 1326 1282 CH2(83)
28. 1270
1440 1281 1428 1271 1304 1261 1294 1251 C-C(18)
29.
1260 1391 1238 1383 1231 1296 1253 1288 1245 C-O of (O-C=O) (57)

30. 1205
1363 1213 1356 1207 1266 1224 1262 1220 CH(38)
31. 1195
1332 1186 1329 1183 1246 1205 1237 1196 CH(31)
32. 1170
1313 1169 1305 1162 1212 1172 1208 1168 CH3(90)
33. 1160
1295 1153 1289 1147 1198 1158 1193 1153 CH(60)
34. 1140
1291 1149 1287 1145 1193 1153 1188 1149 CH (54)
35.
1130 1286 1144 1278 1137 1179 1140 1173 1134 C
36.
1080 1270 1131 1262 1124 1177 1138 1170 1131 t CH2 (14)
37. 1070
1219 1085 1209 1076 1174 1135 1166 1127 CH(48)
38. 1045
1168 1040 1162 1035 1105 1068 1099 1063 CH(22)
39.
1030 1157 1030 1148 1022 1071 1035 1066 1031
C-(CH2-C=O-O-CH3)
{ CC(27)+CCH(14)+ C-O(13)
+ CCC(11)}
40. 1010
1134 1009 1126 1003 1054 1019 1046 1011 CH
41.
1005 1127 1003 1116 993 1036 1002 1029 995 C-O of C-O-CH3(41)
42. 1000
1112 989 1104 983 999 966 1002 969 CH(85)
43. 960
1095 975 1091 971 980 947 984 951 CH(78)
44. 940
1079 960 1068 950 966 934 971 939 CH(86)
45. 930 930 1041 926 1038 924 953 921 952 920 def C
46. 900
1034 920 1029 916 922 891 922 891 CH(78)

47. 890
982 874 977 869 894 864 894 864 C-(CH2-C=O-O-CH3)
48. 860
964 858 960 854 884 855 882 853 CCCH(37)
49. 850
945 841 944 840 862 833 858 830 C-O of C-O-CH3 (43)
50. 820
903 804 900 801 824 797 824 797 CH(35)
51. 790
885 788 883 786 808 781 806 779 CH(47)
52. 780
857 763 855 761 800 773 800 773 CH(36)
53. 740
850 756 849 756 779 753 781 755 CCCC(20) + CCCH(16)
54.
720 828 737 823 732 752 727 747 722 CCCH(82)
55. 700 700 768 684 764 680 717 693 715 691 C-(CH2-C=O-O-CH3)
56. 625
701 624 701 624 647 626 650 628 C-O of C-O-CH3 (26)
57.
620 688 612 686 611 638 617 639 618 CCCC(24) + COO(11)
+ CC(10)
58. 560
620 552 619 551 573 554 572 553 CCCC (26)
59.
530 587 522 585 521 548 530 546 528 CCCC(22) + CC(16)
60. 505 505 555 494 553 492 521 504 521 504 Ring(61) + CC(17)
61. 480
527 469 525 467 488 472 488 472 Ring(30)
62.
460 518 461 517 460 480 464 478 462 Ring(52)
63. 415
476 424 477 424 446 431 448 433 CCC(47) + CCO(16)
64.
405 462 411 461 410 425 411 424 410 CCCC(43)

65. 370 370 433 385 435 387 406 393 407 393 CCO(47)+ CCC(15)
66.
305 325 289 325 290 301 291 302 292 CO-CH3(47) + CCC(15)+
CCO(11)
67. 270 270 301 268 299 266 276 267 275 266 CCCC(45)
68. 240
251 224 248 221 230 222 228 220 COC(28)+ CCC(20)
+ CCCC(13)
69.
180 194 173 193 172 182 176 180 174 COCC(26) + CCCC(18)
+ CCC(14)
70. 170
186 165 186 165 175 169 174 168 CCCC(36) + COCC(13)
71.
140 163 145 163 145 135 131 132 128 CCCC(38) + CH3(23)
72.
138 123 136 121 120 116 109 105 CH3(88)
73.
70 62 71 63 65 63 65 63 COCC(33)+CCCC(24)
+CCC(23) +CCCH(10)
74.
25 22 64 57 56 54 55 53 CCCC(39) + CCCO(27)
+ CCCH(17)
75.
25 23 26 23 20 19 19 18 (CH3-CO2-C)(96)
a Scale factor of 0.92, 0.91, 0.89 for calculated wavenumbers;
b Scale factor of 0.92, 0.91, 0.89 for calculated wavenumbers;
c Scale factor of 0.9668, 0.9614, 0.95 for calculated wavenumbers;
d Scale factor of 0.9668, 0.9614, 0.95 for calculated wavenumbers
: stretching; s: symmetric stretching, as: asymmetric stretching, in-plane-bending; : out-of-plane bending; scissoring,
: rocking; wagging: twisting; : torsion; d: deformation. IIR-IR Intensity; SRa-Raman activity; IRa-Raman Intensity

Table 9.3
Theoretical (HF and B3LYP) Infra Red and Raman intensity of 1-Naphthaleneacetic acid Methyl Ester
with different basis sets
Sl.
No.
Experimental
frequency
HF B3LYP
6-31G(d,p) 6-311G(d,p) 6-31G(d,p) 6-311G(d,p)
FT -
IR
FT-
Raman IIR SRa IRa IIR SRa IRa IIR SRa IRa IIR SRa IRa
1.
3100 6 95 0.04 5 88 0.03 6 115 0.07 4 112 0.06
2.
3070 20 380 0.15 19 398 0.14 13 415 0.25 14 460 0.26
3. 3060
54 41 0.02 48 44 0.02 40 38 0.02 39 42 0.02
4. 3050
34 119 0.05 34 125 0.05 21 117 0.07 22 130 0.08
5.
3020 7 97 0.04 4 95 0.03 16 101 0.06 14 108 0.06
6. 3005
1 39 0.02 1 36 0.01 0 62 0.04 0 60 0.04
7.
2990 1 34 0.01 1 40 0.01 15 64 0.04 1 33 0.02
8.
2960 24 63 0.03 25 64 0.02 1 25 0.02 15 69 0.04
9. 2950
30 47 0.02 33 46 0.02 21 49 0.03 22 52 0.03
10.
2930 8 50 0.02 9 48 0.02 5 48 0.03 5 48 0.03
11.
2850 15 81 0.04 16 92 0.04 13 90 0.06 14 108 0.07
12. 2840
41 103 0.05 42 112 0.05 33 113 0.08 33 127 0.09
13. 1740
288 2 0.01 309 2 0.00 182 2 0.01 201 3 0.01
14. 1600
0 2 0.01 1 2 0.01 0 5 0.03 0 4 0.02
15. 1590
14 2 0.01 13 4 0.01 10 1 0.01 10 1 0.01
16.
1580 0 90 0.32 0 88 0.27 1 75 0.42 2 75 0.39
17. 1505 1505 14 4 0.02 14 4 0.01 9 5 0.03 11 5 0.03
18.
1450 5 11 0.05 7 8 0.03 8 12 0.08 9 9 0.06
19.
1440 0 9 0.04 4 10 0.04 0 13 0.09 0 11 0.07
20. 1430
5 18 0.08 6 14 0.06 6 20 0.14 8 14 0.09
21. 1420
25 7 0.03 23 1 0.00 4 23 0.16 9 14 0.09
22. 1400
6 10 0.05 7 4 0.02 13 16 0.12 10 24 0.16
23.
1395 2 37 0.18 2 37 0.16 6 22 0.16 9 15 0.1
24.
1370 6 2 0.01 7 2 0.01 8 7 0.05 10 4 0.03
25. 1350
101 61 0.35 98 52 0.26 1 121 1.00 2 127 1
26. 1330
51 167 1.00 32 188 1.00 4 17 0.14 4 33 0.27
27. 1300
95 9 0.06 44 17 0.09 45 8 0.08 33 4 0.04
28. 1270
255 6 0.04 316 7 0.04 258 6 0.06 44 5 0.05
29.
1260 6 6 0.04 7 5 0.03 44 6 0.06 233 5 0.05
30. 1205
1 1 0.01 1 1 0.01 1 1 0.01 3 1 0.01

31. 1195
5 2 0.02 5 2 0.01 14 2 0.02 20 1 0.01
32. 1170
26 4 0.03 35 4 0.03 4 3 0.04 5 2 0.02
33. 1160
47 11 0.09 16 5 0.04 18 3 0.04 19 2 0.02
34. 1140
4 5 0.04 45 11 0.08 3 2 0.03 5 1 0.01
35.
1130 5 8 0.07 6 7 0.05 1 5 0.07 2 3 0.04
36.
1080 3 10 0.09 4 8 0.06 72 12 0.16 28 9 0.11
37. 1070
9 3 0.03 11 3 0.03 12 7 0.09 89 8 0.1
38. 1045
31 4 0.04 31 5 0.05 4 12 0.19 5 14 0.2
39.
1030 15 7 0.08 14 9 0.09 19 2 0.03 18 3 0.05
40. 1010
10 2 0.02 10 3 0.03 12 10 0.17 9 12 0.2
41.
1005 0 2 0.02 0 1 0.01 18 2 0.04 28 2 0.03
42. 1000
3 1 0.01 2 1 0.01 1 0 0.00 1 0 0
43. 960
1 1 0.01 1 2 0.02 2 0 0.00 2 0 0
44. 940
3 4 0.05 4 4 0.05 2 1 0.02 1 0 0
45. 930 930 1 3 0.04 1 3 0.04 0 2 0.04 0 2 0.04
46. 900
1 4 0.06 1 2 0.03 0 4 0.10 0 1 0.02
47. 890
1 1 0.02 2 0 0.00 1 5 0.13 1 4 0.1
48. 860
3 6 0.11 4 7 0.11 2 4 0.11 3 4 0.1
49. 850
10 8 0.15 9 7 0.11 8 9 0.26 9 10 0.27
50. 820
32 1 0.02 21 2 0.04 11 2 0.06 7 2 0.06
51. 790
74 4 0.09 89 3 0.06 45 1 0.03 36 1 0.03
52. 780
8 1 0.02 14 1 0.02 21 3 0.10 48 3 0.09
53. 740
4 4 0.10 3 2 0.04 4 6 0.22 13 3 0.1
54.
720 3 4 0.10 3 1 0.02 3 6 0.24 3 2 0.07
55. 700 700 6 15 0.46 6 16 0.43 3 17 0.75 3 18 0.74
56. 625
3 2 0.08 3 3 0.10 2 3 0.17 3 3 0.15
57.
620 6 3 0.12 5 2 0.07 3 2 0.11 3 2 0.11
58. 560
13 2 0.10 13 2 0.09 6 4 0.29 6 5 0.34
59.
530 7 9 0.51 8 9 0.45 4 10 0.81 6 10 0.76
60. 505 505 1 8 0.52 1 9 0.51 1 9 0.81 1 10 0.84
61. 480
8 4 0.29 8 3 0.19 5 4 0.42 6 4 0.39
62.
460 1 1 0.08 2 1 0.07 1 2 0.22 2 1 0.1
63. 415
2 2 0.18 2 2 0.16 1 1 0.13 1 1 0.12
64.
405 5 4 0.39 6 3 0.25 3 3 0.42 4 2 0.26
65. 370 370 11 1 0.11 10 1 0.10 9 1 0.16 10 1 0.14
66.
305 14 1 0.21 13 1 0.18 11 1 0.30 11 1 0.27

67. 270 270 3 2 0.01 4 2 0.01 3 2 0.01 4 2 0.01
68. 240
1 1 0.00 1 1 0.00 1 1 0.01 1 1 0.01
69.
180 2 0 0.00 3 0 0.00 3 0 0.00 4 0 0
70. 170
3 0 0.00 4 0 0.00 3 0 0.00 3 0 0
71.
140 1 1 0.01 1 1 0.01 0 3 0.03 0 2 0.02
72.
1 2 0.02 1 2 0.01 1 0 0.00 1 0 0
73.
1 3 0.05 1 3 0.04 0 4 0.09 1 4 0.08
74.
1 5 0.24 1 4 0.06 1 6 0.15 1 4 0.1
75.
2 4 0.19 2 3 0.12 1 4 0.29 1 4 0.28
IIR – Infra Red Intensity; SRa – Raman Activity; IRa – Raman Intensity
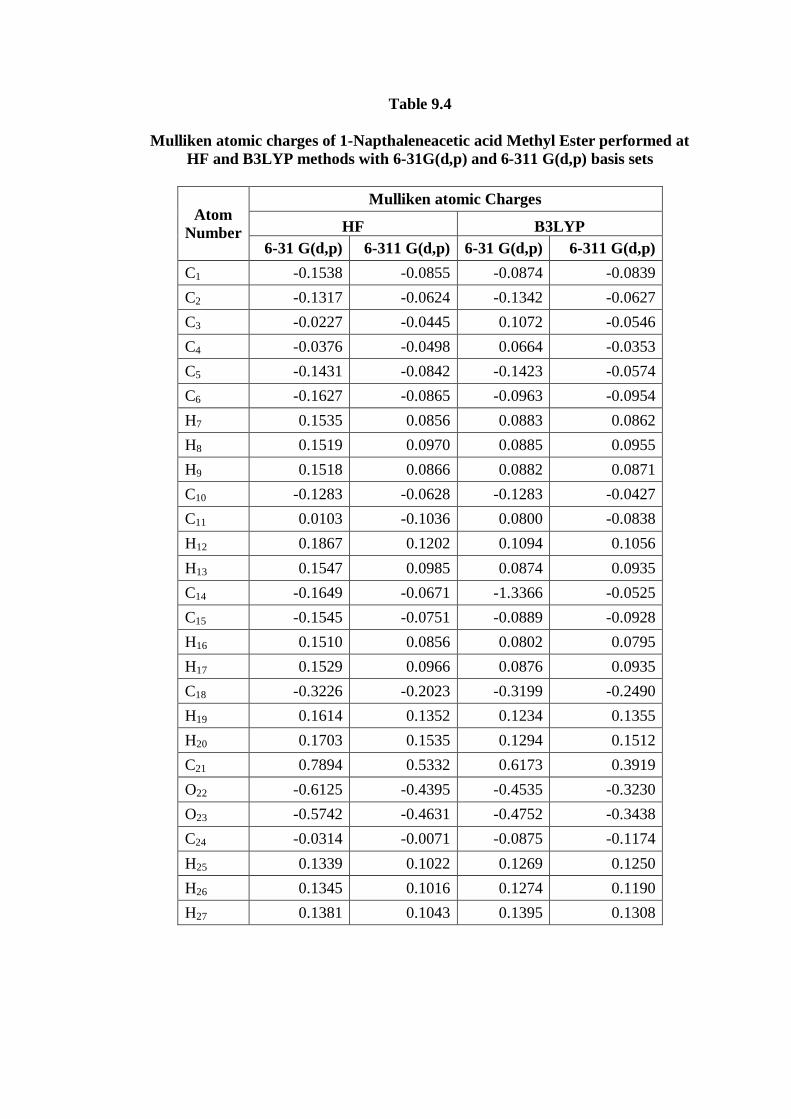
Table 9.4
Mulliken atomic charges of 1-Napthaleneacetic acid Methyl Ester performed at
HF and B3LYP methods with 6-31G(d,p) and 6-311 G(d,p) basis sets
Atom
Number
Mulliken atomic Charges
HF B3LYP
6-31 G(d,p) 6-311 G(d,p) 6-31 G(d,p) 6-311 G(d,p)
C1 -0.1538 -0.0855 -0.0874 -0.0839
C2 -0.1317 -0.0624 -0.1342 -0.0627
C3 -0.0227 -0.0445 0.1072 -0.0546
C4 -0.0376 -0.0498 0.0664 -0.0353
C5 -0.1431 -0.0842 -0.1423 -0.0574
C6 -0.1627 -0.0865 -0.0963 -0.0954
H7 0.1535 0.0856 0.0883 0.0862
H8 0.1519 0.0970 0.0885 0.0955
H9 0.1518 0.0866 0.0882 0.0871
C10 -0.1283 -0.0628 -0.1283 -0.0427
C11 0.0103 -0.1036 0.0800 -0.0838
H12 0.1867 0.1202 0.1094 0.1056
H13 0.1547 0.0985 0.0874 0.0935
C14 -0.1649 -0.0671 -1.3366 -0.0525
C15 -0.1545 -0.0751 -0.0889 -0.0928
H16 0.1510 0.0856 0.0802 0.0795
H17 0.1529 0.0966 0.0876 0.0935
C18 -0.3226 -0.2023 -0.3199 -0.2490
H19 0.1614 0.1352 0.1234 0.1355
H20 0.1703 0.1535 0.1294 0.1512
C21 0.7894 0.5332 0.6173 0.3919
O22 -0.6125 -0.4395 -0.4535 -0.3230
O23 -0.5742 -0.4631 -0.4752 -0.3438
C24 -0.0314 -0.0071 -0.0875 -0.1174
H25 0.1339 0.1022 0.1269 0.1250
H26 0.1345 0.1016 0.1274 0.1190
H27 0.1381 0.1043 0.1395 0.1308
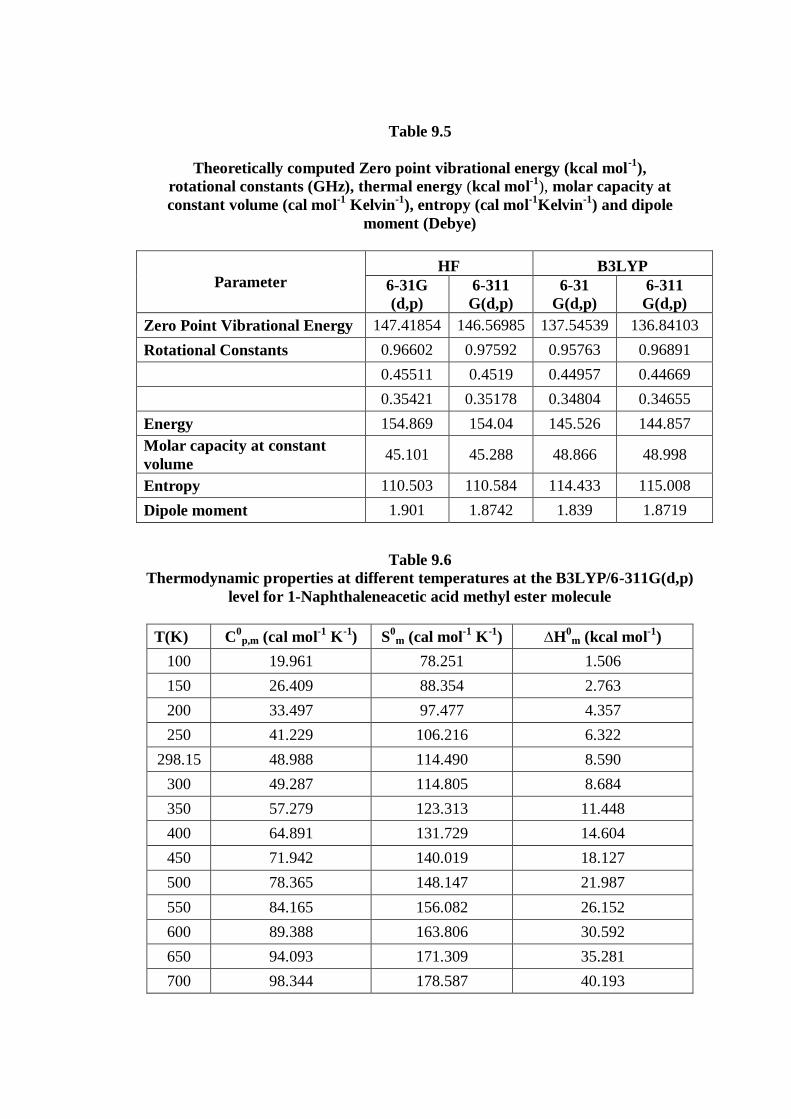
Table 9.5
Theoretically computed Zero point vibrational energy (kcal mol-1
),
rotational constants (GHz), thermal energy (kcal mol-1
), molar capacity at
constant volume (cal mol-1
Kelvin-1
), entropy (cal mol-1
Kelvin-1
) and dipole
moment (Debye)
Parameter HF B3LYP
6-31G
(d,p)
6-311
G(d,p)
6-31
G(d,p)
6-311
G(d,p)
Zero Point Vibrational Energy 147.41854 146.56985 137.54539 136.84103
Rotational Constants 0.96602 0.97592 0.95763 0.96891
0.45511 0.4519 0.44957 0.44669
0.35421 0.35178 0.34804 0.34655
Energy 154.869 154.04 145.526 144.857
Molar capacity at constant
volume 45.101 45.288 48.866 48.998
Entropy 110.503 110.584 114.433 115.008
Dipole moment 1.901 1.8742 1.839 1.8719
Table 9.6 Thermodynamic properties at different temperatures at the B3LYP/6-311G(d,p)
level for 1-Naphthaleneacetic acid methyl ester molecule
T(K) C0
p,m (cal mol-1
K-1
) S0
m (cal mol-1
K-1
) ∆H0
m (kcal mol-1
)
100 19.961 78.251 1.506
150 26.409 88.354 2.763
200 33.497 97.477 4.357
250 41.229 106.216 6.322
298.15 48.988 114.490 8.590
300 49.287 114.805 8.684
350 57.279 123.313 11.448
400 64.891 131.729 14.604
450 71.942 140.019 18.127
500 78.365 148.147 21.987
550 84.165 156.082 26.152
600 89.388 163.806 30.592
650 94.093 171.309 35.281
700 98.344 178.587 40.193
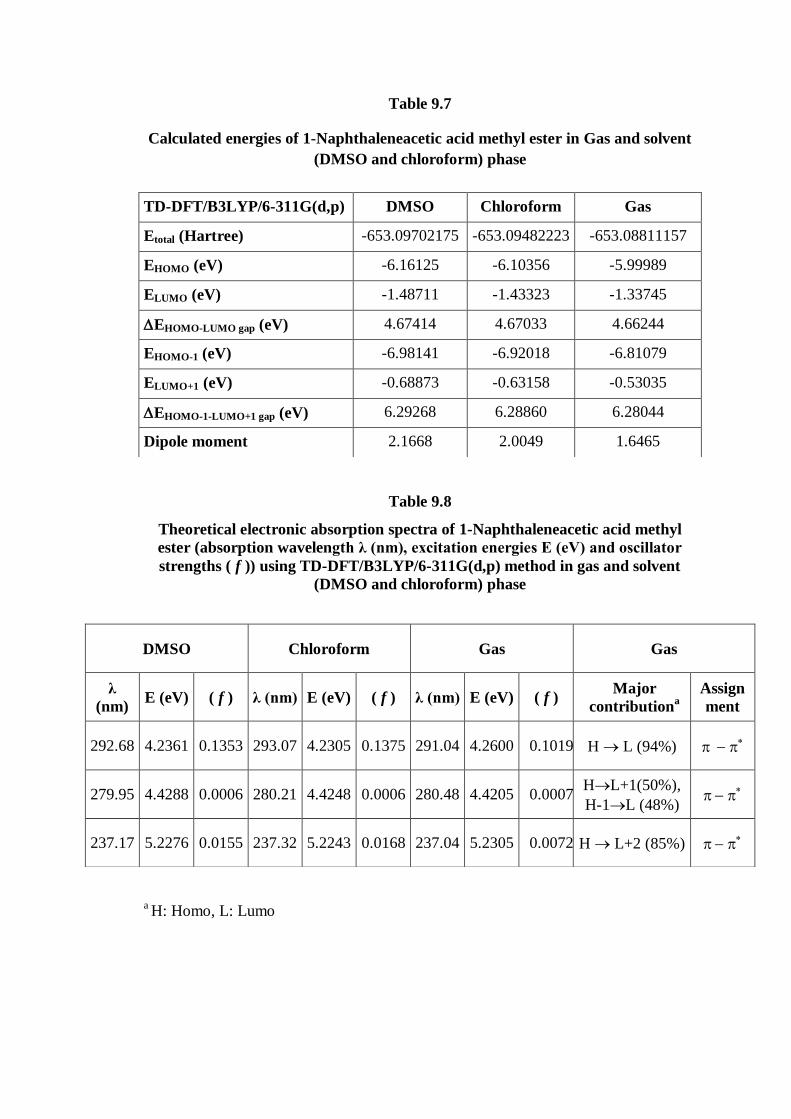
Table 9.7
Calculated energies of 1-Naphthaleneacetic acid methyl ester in Gas and solvent
(DMSO and chloroform) phase
Table 9.8
Theoretical electronic absorption spectra of 1-Naphthaleneacetic acid methyl
ester (absorption wavelength λ (nm), excitation energies E (eV) and oscillator
strengths ( f )) using TD-DFT/B3LYP/6-311G(d,p) method in gas and solvent
(DMSO and chloroform) phase
a H: Homo, L: Lumo
TD-DFT/B3LYP/6-311G(d,p) DMSO Chloroform Gas
Etotal (Hartree) -653.09702175 -653.09482223 -653.08811157
EHOMO (eV) -6.16125 -6.10356 -5.99989
ELUMO (eV) -1.48711 -1.43323 -1.33745
EHOMO-LUMO gap (eV) 4.67414 4.67033 4.66244
EHOMO-1 (eV) -6.98141 -6.92018 -6.81079
ELUMO+1 (eV) -0.68873 -0.63158 -0.53035
EHOMO-1-LUMO+1 gap (eV) 6.29268 6.28860 6.28044
Dipole moment 2.1668 2.0049 1.6465
DMSO Chloroform Gas Gas
λ
(nm) E (eV) ( f ) λ (nm) E (eV) ( f ) λ (nm) E (eV) ( f )
Major
contributiona
Assign
ment
292.68 4.2361 0.1353 293.07 4.2305 0.1375 291.04 4.2600 0.1019 H L (94%)
279.95 4.4288 0.0006 280.21 4.4248 0.0006 280.48 4.4205 0.0007 HL+1(50%),
H-1L (48%)
237.17 5.2276 0.0155 237.32 5.2243 0.0168 237.04 5.2305 0.0072 H L+2 (85%)