bioinformatics bio-informatics management of the biological information using computer technology....
TRANSCRIPT

Bioinformatics Bio-Informatics
Management of the biological information using computer technology
BCH463 BioinformaticsMd Ashrafuzzaman DScKnown as Dr AshrafEmail mashrafuzzamanksuedusa Emergency contact cell 0564174931Office 2B10 Bldg 5 KSU
Biological informationsHuge
What kind of info(structure and mechanism)
bull Discovered aspects related to biologybull Literature search using various routesbull Data bank exploration from different international sourcesbull Biological network databull Biological structure databull Data that will help understand the working mechanisms of
biological systemsbull etc
Searching Data
bull Why searching
bull How to search
bull Where to search
bull What is usually done with searched data
bull Who should be a Bioinformatician
A case studybull Bioinformatic-driven search for metabolic biomarkers in diseasebull httpwwwjclinbioinformaticscomcontent112bull The search and validation of novel disease biomarkers requires the complementary power of professional study planning and
execution modern profiling technologies and related bioinformatics tools for data analysis and interpretation Biomarkers have considerable impact on the care of patients and are urgently needed for advancing diagnostics prognostics and treatment of disease This survey article highlights emerging bioinformatics methods for biomarker discovery in clinical metabolomics focusing on the problem of data preprocessing and consolidation the data-driven search verification prioritization and biological interpretation of putative metabolic candidate biomarkers in disease In particular data mining tools suitable for the application to omic data gathered from most frequently-used type of experimental designs such as case-control or longitudinal biomarker cohort studies are reviewed and case examples of selected discovery steps are delineated in more detail This review demonstrates that clinical bioinformatics has evolved into an essential element of biomarker discovery translating new innovations and successes in profiling technologies and bioinformatics to clinical application
Data sequencing-GeneBank
What is GeneBank
GenBankreg is the National Institute of Health (NIH) genetic sequence database an annotated collection of all publicly available DNA sequences
As of 2008 there are approximately
100 billion bases in
100 million sequences
Consider the growth rate
Started in 1982 with 680338 base pairs in 606 sequences
GenBank is part of the International Nucleotide Sequence Database Collaboration which comprises the DNA DataBank of Japan (DDBJ) the European Molecular Biology Laboratory (EMBL) and GenBank at National Center for Biotechnology Information (NCBI) These three organizations exchange data on a daily basis
How GeneBank worksSubmissions to GenBankbull Many journals require submission of sequence information to a database prior to
publication so that an accession number may appear in the paper Sequin NCBIs stand-alone submission software for MAC PC and UNIX platforms is available When using Sequin the output files for direct submission should be sent to GenBank by electronic mail
Updating or Revising a Sequencebull Revisions or updates to GenBank entries can be made at any time and can be accepted
as BankIt or Sequin files or as the text of an e-mail message
Access to GenBankbull GenBank is available for searching at NCBI via several methodsbull The GenBank database is designed to provide and encourage access within the scientific
community to the most up to date and comprehensive DNA sequence information Therefore NCBI places no restrictions on the use or distribution of the GenBank data However some submitters may claim patent copyright or other intellectual property rights in all or a portion of the data they have submitted
New Developmentsbull NCBI is continuously developing new tools and enhancing existing ones to improve both
submission and access to GenBank The easiest way to keep abreast of these and other developments is to check the Whats New section of the NCBI Web page and to read the NCBI News which is also available by free subscription
Various bases of Bioinformaticsbull Count Bases at the Fraunhofer IGB GermanyThis system basically consists of modules that cover sequence analysis (Count
Bases ndash Next-Gen Sequence Assistant) statistics as well as visualization (Count Bases Viewer)
In a single run 106ndash109 DNA fragments with an average sequence length of 30ndash800 bases are simultaneously sequenced This results in huge amounts of data that require a storage volume of up to 10ndash100 gigabyte
Sources Genome and proteomic data bases
Major rersearch areasSequence analysis
Genome annotation
Literature
Analysis of gene expression regulation
Analysis of protein expression
Mutations in cancer
Etc
Organisms in GeneBank
bull 260000 different speciesbull 1000 new species being added per month
bull Human (Homo sapiens)
11551000 entries with 13149000000 basesbull Mouse (Mus musculus)
7256000 entries with 8361230000 bases
are top two species
GeneBank FormatGenBank format (GenBank Flat File Format) consists of an annotation
section and a sequence section
Annotation sectionThe start of the annotation section is marked by a line beginning with the word LOCUS
The only rule now applied in assigning a locus name is that it must be unique
Sequence sectionThe start of sequence section is marked by a line beginning with the word ORIGIN and the end of the section is marked by a line with only ldquo
GeneBank Flat File FormatLOCUS AF068625 200 bp mRNA linear ROD 06-DEC-1999
DEFINITION Mus musculus DNA cytosine-5 methyltransferase 3A (Dnmt3a) mRNA complete cds
ACCESSION AF068625 REGION 1200
VERSION AF0686252 GI6449467
KEYWORDS
SOURCE Mus musculus (house mouse)
ORGANISM Mus musculus Eukaryota Metazoa Chordata Craniata Vertebrata Euteleostomi Mammalia Eutheria Euarchontoglires Glires Rodentia Sciurognathi Muroidea Muridae Murinae Mus
REFERENCE1 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REFERENCE2 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REMARK Sequence update by submitter
COMMENT On Nov 18 1999 this sequence version replaced gi3327977
FEATURES LocationQualifiers
source 1200 organism=Mus musculus mol_type=mRNA db_xref=taxon10090 chromosome=12 map=40 cM
gene 1gt200 gene=Dnmt3a
ORIGIN 1 gaattccggc ctgctgccgg gccgcccgac ccgccgggcc acacggcaga gccgcctgaa 61 gcccagcgct gaggctgcac ttttccgagg gcttgacatc agggtctatg tttaagtctt 121 agctcttgct tacaaagacc acggcaattc cttctctgaa gccctcgcag ccccacagcg 181 ccctcgcagc cccagcctgc
GenBank sequence formatItrsquos a rich format for storing sequences and associated annotations It shares a feature
table vocabulary and format with the EMBL and DDJB formats
bull LOCUS CAA89576 109 aa linear PLN 11-AUG-1997 bull DEFINITION CYC1 [Saccharomyces cerevisiae] bull ACCESSION CAA89576 bull VERSION CAA895761 GI1015707 bull DBSOURCE embl locus SCYJR048W accession Z495481 bull KEYWORDS 5-10 or as many as neededbull SOURCE Saccharomyces cerevisiae (bakers yeast) bull ORGANISM Saccharomyces cerevisiae Eukaryota Fungi Ascomycota
Saccharomycotina Saccharomycetes Saccharomycetales Saccharomycetaceae Saccharomyces
bull REFERENCE1 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull REFERENCE2 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull FEATURES LocationQualifiers bull source 1109 organism=Saccharomyces cerevisiae db_xref=taxon4932
chromosome=X Protein 1109 name=CYC1 bull CDS 1109 gene=CYC1 coded_by=Z4954819541283 note=ORF YJR048w
db_xref=GOAP00044 db_xref=SGDS0003809 db_xref=UniProtKBSwiss-ProtP00044 bull ORIGIN 1 mtefkagsak kgatlfktrc lqchtvekgg phkvgpnlhg ifgrhsgqae gysytdanik 61 knvlwdennm
seyltnpkky ipgtkmafgg lkkekdrndl itylkkace
Online Mendelian Inheritance in Man (OMIM) Databasebull OMIM (since 1960s) catalogues all the known diseases with a genetic component
and tries to link them to the relevant genes in human genome
bull In 2004 there were 15000 records
bull One can request to download the mim2genetxt file from OMIM here httpwwwomimorgdownloads
bull The OMIM codebull Every disease and gene is assigned a six digit number of which the first number classifies
the method of inheritancebull If the initial digit is 1 the trait is deemed autosomal dominant if 2 autosomal recessive if 3
X-linked Wherever a trait defined in this dictionary has a MIM number the number from the 12th edition of MIM is given in square brackets with or without an asterisk (asterisks indicate that the mode of inheritance is known a number symbol () before an entry number means that the phenotype can be caused by mutation in any of two or more genes) as appropriate eg Pelizaeus-Merzbacher disease [MIM 312080] is an X-linked recessive disorder
bull For further studies visit httpwwwomimorg
OMIMExample httpwwwomimorgentry189911
189911 TRANSFER RNA GLYCINE 1 TRNAG1
Alternative titles symbols TRANSFER RNA GLYCINE-CCC-1 TRG1
Cytogenetic location Chr16 Genomic coordinates (GRCh37) 160 - 90354753 (from NCBI)
TEXT
Mapping McBride et al (1989) assigned a glycine tRNA(CCC) gene (TRG1) to human chromosome 1 (1pter-p34) on the basis of Southern analysis of a panel of hybrid cell DNAs They also assigned a cloned DNA fragment encompassing a glycine tRNA gene (tRNA-GCC) and pseudogene to human chromosome 16 by the same method
Evolution There are about 1300 tRNA genes in the haploid human genome (Hatlen and Attardi 1971) encoding 60 to 90 tRNA isoacceptors (Lin and Agris 1980) The studies by McBride et al (1989) as well as studies by others (see eg 180620 189930 189920 180640 189880) indicated that tRNA genes and pseudogenes are dispersed on at least 7 human chromosomes and suggested that these sequences would probably be found on most if not all human chromosomes McBride et al (1989) described short 8-12 nucleotide direct terminal repeats flanking many of the dispersed tRNA genes This finding combined with the dispersion of tRNA genes suggests that many of these genes may have arisen by an RNA-mediated retroposition mechanism There may have been selection for reiteration of genes encoding isoaccepting tRNAs since a single mutation in a single-copy tRNA gene could be devastating Moreover even a mutation in the anticodon of a single tRNA gene might not be crucial if competition was provided by the normal wildtype tRNA isoacceptor produced by multiple copies of the normal tRNA gene still present in the genome Dispersion of multiple copies of each tRNA gene could provide diversity of 5-prime-flanking sequences which are known to modulate the expression of some human tRNA genes Tissue-specific or differentiation-specific expression of tRNA isoacceptors might be provided for by this mechanism The recombination and unequal crossingover that can occur with tandem tRNA sequences can result in homogenization of the sequences with disastrous consequences
Nucleotide Databasebull NUCLEOTIDE DATABASESbull NCBIs sequence databases accept genome data from sequencing projects from around
the world and serve as the cornerstone of bioinformatics researchbull GenBankbull An annotated collection of all publicly available nucleotide and amino acid sequencesbull EST databasebull A collection of expressed sequence tags or short single-pass sequence reads from mRNA
(cDNA)bull GSS databasebull A database of genome survey sequences or short single-pass genomic sequencesbull HomoloGenebull A gene homology tool that compares nucleotide sequences between pairs of organisms in order
to identify putative orthologsbull HTG databasebull A collection of high-throughput genome sequences from large-scale genome sequencing
centers including unfinished and finished sequencesbull SNPs databasebull A central repository for both single-base nucleotide substitutions and short deletion and
insertion polymorphisms
Nucleotide Database
bull RefSeqbull A database of non-redundant reference sequences standards including genomic
DNA contigs mRNAs and proteins for known genes Multiple collaborations both within NCBI and with external groups support our data-gathering efforts
bull STS databasebull A database of sequence tagged sites or short sequences that are operationally
unique in the genomebull UniSTSbull A unified non-redundant view of sequence tagged sites (STSs)bull UniGenebull A collection of ESTs and full-length mRNA sequences organized into clusters each
representing a unique known or putative human gene annotated with mapping and expression information and cross-references to other sources
UniGene computationally identifies transcripts from the same locus analyzes expression by tissue age and health status and reports related proteins (protEST) and clone resources
Single Nucleotide Polymorphism (SNP) database
What it isThe SNP Database (also known as dbSNP) is an archive for genetic variation within and across different species developed and hosted by NCBI in collaboration with the National Human Genome Research Institute (NHGRI)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species related to biodiversity genetic variation and adaptation
-The dbSNP accepts apparently neutral polymorphisms polymorphisms corresponding to known phenotypes and regions of no variation
-It was created in September 1998 to supplement GenBank (NCBIrsquos nucleic acid and protein sequences)
Goal
Its goal is to act as a single database that contains all identified genetic variation which can be used to investigate a wide variety of genetically based natural phenomenon Specifically access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping population genetics investigations into evolutionary relationships as well as being able to quickly and easily quantify the amount of variation at a given site of interest
Application
Applied research genetic engineering drug discovery etc
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

Biological informationsHuge
What kind of info(structure and mechanism)
bull Discovered aspects related to biologybull Literature search using various routesbull Data bank exploration from different international sourcesbull Biological network databull Biological structure databull Data that will help understand the working mechanisms of
biological systemsbull etc
Searching Data
bull Why searching
bull How to search
bull Where to search
bull What is usually done with searched data
bull Who should be a Bioinformatician
A case studybull Bioinformatic-driven search for metabolic biomarkers in diseasebull httpwwwjclinbioinformaticscomcontent112bull The search and validation of novel disease biomarkers requires the complementary power of professional study planning and
execution modern profiling technologies and related bioinformatics tools for data analysis and interpretation Biomarkers have considerable impact on the care of patients and are urgently needed for advancing diagnostics prognostics and treatment of disease This survey article highlights emerging bioinformatics methods for biomarker discovery in clinical metabolomics focusing on the problem of data preprocessing and consolidation the data-driven search verification prioritization and biological interpretation of putative metabolic candidate biomarkers in disease In particular data mining tools suitable for the application to omic data gathered from most frequently-used type of experimental designs such as case-control or longitudinal biomarker cohort studies are reviewed and case examples of selected discovery steps are delineated in more detail This review demonstrates that clinical bioinformatics has evolved into an essential element of biomarker discovery translating new innovations and successes in profiling technologies and bioinformatics to clinical application
Data sequencing-GeneBank
What is GeneBank
GenBankreg is the National Institute of Health (NIH) genetic sequence database an annotated collection of all publicly available DNA sequences
As of 2008 there are approximately
100 billion bases in
100 million sequences
Consider the growth rate
Started in 1982 with 680338 base pairs in 606 sequences
GenBank is part of the International Nucleotide Sequence Database Collaboration which comprises the DNA DataBank of Japan (DDBJ) the European Molecular Biology Laboratory (EMBL) and GenBank at National Center for Biotechnology Information (NCBI) These three organizations exchange data on a daily basis
How GeneBank worksSubmissions to GenBankbull Many journals require submission of sequence information to a database prior to
publication so that an accession number may appear in the paper Sequin NCBIs stand-alone submission software for MAC PC and UNIX platforms is available When using Sequin the output files for direct submission should be sent to GenBank by electronic mail
Updating or Revising a Sequencebull Revisions or updates to GenBank entries can be made at any time and can be accepted
as BankIt or Sequin files or as the text of an e-mail message
Access to GenBankbull GenBank is available for searching at NCBI via several methodsbull The GenBank database is designed to provide and encourage access within the scientific
community to the most up to date and comprehensive DNA sequence information Therefore NCBI places no restrictions on the use or distribution of the GenBank data However some submitters may claim patent copyright or other intellectual property rights in all or a portion of the data they have submitted
New Developmentsbull NCBI is continuously developing new tools and enhancing existing ones to improve both
submission and access to GenBank The easiest way to keep abreast of these and other developments is to check the Whats New section of the NCBI Web page and to read the NCBI News which is also available by free subscription
Various bases of Bioinformaticsbull Count Bases at the Fraunhofer IGB GermanyThis system basically consists of modules that cover sequence analysis (Count
Bases ndash Next-Gen Sequence Assistant) statistics as well as visualization (Count Bases Viewer)
In a single run 106ndash109 DNA fragments with an average sequence length of 30ndash800 bases are simultaneously sequenced This results in huge amounts of data that require a storage volume of up to 10ndash100 gigabyte
Sources Genome and proteomic data bases
Major rersearch areasSequence analysis
Genome annotation
Literature
Analysis of gene expression regulation
Analysis of protein expression
Mutations in cancer
Etc
Organisms in GeneBank
bull 260000 different speciesbull 1000 new species being added per month
bull Human (Homo sapiens)
11551000 entries with 13149000000 basesbull Mouse (Mus musculus)
7256000 entries with 8361230000 bases
are top two species
GeneBank FormatGenBank format (GenBank Flat File Format) consists of an annotation
section and a sequence section
Annotation sectionThe start of the annotation section is marked by a line beginning with the word LOCUS
The only rule now applied in assigning a locus name is that it must be unique
Sequence sectionThe start of sequence section is marked by a line beginning with the word ORIGIN and the end of the section is marked by a line with only ldquo
GeneBank Flat File FormatLOCUS AF068625 200 bp mRNA linear ROD 06-DEC-1999
DEFINITION Mus musculus DNA cytosine-5 methyltransferase 3A (Dnmt3a) mRNA complete cds
ACCESSION AF068625 REGION 1200
VERSION AF0686252 GI6449467
KEYWORDS
SOURCE Mus musculus (house mouse)
ORGANISM Mus musculus Eukaryota Metazoa Chordata Craniata Vertebrata Euteleostomi Mammalia Eutheria Euarchontoglires Glires Rodentia Sciurognathi Muroidea Muridae Murinae Mus
REFERENCE1 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REFERENCE2 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REMARK Sequence update by submitter
COMMENT On Nov 18 1999 this sequence version replaced gi3327977
FEATURES LocationQualifiers
source 1200 organism=Mus musculus mol_type=mRNA db_xref=taxon10090 chromosome=12 map=40 cM
gene 1gt200 gene=Dnmt3a
ORIGIN 1 gaattccggc ctgctgccgg gccgcccgac ccgccgggcc acacggcaga gccgcctgaa 61 gcccagcgct gaggctgcac ttttccgagg gcttgacatc agggtctatg tttaagtctt 121 agctcttgct tacaaagacc acggcaattc cttctctgaa gccctcgcag ccccacagcg 181 ccctcgcagc cccagcctgc
GenBank sequence formatItrsquos a rich format for storing sequences and associated annotations It shares a feature
table vocabulary and format with the EMBL and DDJB formats
bull LOCUS CAA89576 109 aa linear PLN 11-AUG-1997 bull DEFINITION CYC1 [Saccharomyces cerevisiae] bull ACCESSION CAA89576 bull VERSION CAA895761 GI1015707 bull DBSOURCE embl locus SCYJR048W accession Z495481 bull KEYWORDS 5-10 or as many as neededbull SOURCE Saccharomyces cerevisiae (bakers yeast) bull ORGANISM Saccharomyces cerevisiae Eukaryota Fungi Ascomycota
Saccharomycotina Saccharomycetes Saccharomycetales Saccharomycetaceae Saccharomyces
bull REFERENCE1 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull REFERENCE2 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull FEATURES LocationQualifiers bull source 1109 organism=Saccharomyces cerevisiae db_xref=taxon4932
chromosome=X Protein 1109 name=CYC1 bull CDS 1109 gene=CYC1 coded_by=Z4954819541283 note=ORF YJR048w
db_xref=GOAP00044 db_xref=SGDS0003809 db_xref=UniProtKBSwiss-ProtP00044 bull ORIGIN 1 mtefkagsak kgatlfktrc lqchtvekgg phkvgpnlhg ifgrhsgqae gysytdanik 61 knvlwdennm
seyltnpkky ipgtkmafgg lkkekdrndl itylkkace
Online Mendelian Inheritance in Man (OMIM) Databasebull OMIM (since 1960s) catalogues all the known diseases with a genetic component
and tries to link them to the relevant genes in human genome
bull In 2004 there were 15000 records
bull One can request to download the mim2genetxt file from OMIM here httpwwwomimorgdownloads
bull The OMIM codebull Every disease and gene is assigned a six digit number of which the first number classifies
the method of inheritancebull If the initial digit is 1 the trait is deemed autosomal dominant if 2 autosomal recessive if 3
X-linked Wherever a trait defined in this dictionary has a MIM number the number from the 12th edition of MIM is given in square brackets with or without an asterisk (asterisks indicate that the mode of inheritance is known a number symbol () before an entry number means that the phenotype can be caused by mutation in any of two or more genes) as appropriate eg Pelizaeus-Merzbacher disease [MIM 312080] is an X-linked recessive disorder
bull For further studies visit httpwwwomimorg
OMIMExample httpwwwomimorgentry189911
189911 TRANSFER RNA GLYCINE 1 TRNAG1
Alternative titles symbols TRANSFER RNA GLYCINE-CCC-1 TRG1
Cytogenetic location Chr16 Genomic coordinates (GRCh37) 160 - 90354753 (from NCBI)
TEXT
Mapping McBride et al (1989) assigned a glycine tRNA(CCC) gene (TRG1) to human chromosome 1 (1pter-p34) on the basis of Southern analysis of a panel of hybrid cell DNAs They also assigned a cloned DNA fragment encompassing a glycine tRNA gene (tRNA-GCC) and pseudogene to human chromosome 16 by the same method
Evolution There are about 1300 tRNA genes in the haploid human genome (Hatlen and Attardi 1971) encoding 60 to 90 tRNA isoacceptors (Lin and Agris 1980) The studies by McBride et al (1989) as well as studies by others (see eg 180620 189930 189920 180640 189880) indicated that tRNA genes and pseudogenes are dispersed on at least 7 human chromosomes and suggested that these sequences would probably be found on most if not all human chromosomes McBride et al (1989) described short 8-12 nucleotide direct terminal repeats flanking many of the dispersed tRNA genes This finding combined with the dispersion of tRNA genes suggests that many of these genes may have arisen by an RNA-mediated retroposition mechanism There may have been selection for reiteration of genes encoding isoaccepting tRNAs since a single mutation in a single-copy tRNA gene could be devastating Moreover even a mutation in the anticodon of a single tRNA gene might not be crucial if competition was provided by the normal wildtype tRNA isoacceptor produced by multiple copies of the normal tRNA gene still present in the genome Dispersion of multiple copies of each tRNA gene could provide diversity of 5-prime-flanking sequences which are known to modulate the expression of some human tRNA genes Tissue-specific or differentiation-specific expression of tRNA isoacceptors might be provided for by this mechanism The recombination and unequal crossingover that can occur with tandem tRNA sequences can result in homogenization of the sequences with disastrous consequences
Nucleotide Databasebull NUCLEOTIDE DATABASESbull NCBIs sequence databases accept genome data from sequencing projects from around
the world and serve as the cornerstone of bioinformatics researchbull GenBankbull An annotated collection of all publicly available nucleotide and amino acid sequencesbull EST databasebull A collection of expressed sequence tags or short single-pass sequence reads from mRNA
(cDNA)bull GSS databasebull A database of genome survey sequences or short single-pass genomic sequencesbull HomoloGenebull A gene homology tool that compares nucleotide sequences between pairs of organisms in order
to identify putative orthologsbull HTG databasebull A collection of high-throughput genome sequences from large-scale genome sequencing
centers including unfinished and finished sequencesbull SNPs databasebull A central repository for both single-base nucleotide substitutions and short deletion and
insertion polymorphisms
Nucleotide Database
bull RefSeqbull A database of non-redundant reference sequences standards including genomic
DNA contigs mRNAs and proteins for known genes Multiple collaborations both within NCBI and with external groups support our data-gathering efforts
bull STS databasebull A database of sequence tagged sites or short sequences that are operationally
unique in the genomebull UniSTSbull A unified non-redundant view of sequence tagged sites (STSs)bull UniGenebull A collection of ESTs and full-length mRNA sequences organized into clusters each
representing a unique known or putative human gene annotated with mapping and expression information and cross-references to other sources
UniGene computationally identifies transcripts from the same locus analyzes expression by tissue age and health status and reports related proteins (protEST) and clone resources
Single Nucleotide Polymorphism (SNP) database
What it isThe SNP Database (also known as dbSNP) is an archive for genetic variation within and across different species developed and hosted by NCBI in collaboration with the National Human Genome Research Institute (NHGRI)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species related to biodiversity genetic variation and adaptation
-The dbSNP accepts apparently neutral polymorphisms polymorphisms corresponding to known phenotypes and regions of no variation
-It was created in September 1998 to supplement GenBank (NCBIrsquos nucleic acid and protein sequences)
Goal
Its goal is to act as a single database that contains all identified genetic variation which can be used to investigate a wide variety of genetically based natural phenomenon Specifically access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping population genetics investigations into evolutionary relationships as well as being able to quickly and easily quantify the amount of variation at a given site of interest
Application
Applied research genetic engineering drug discovery etc
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

Searching Data
bull Why searching
bull How to search
bull Where to search
bull What is usually done with searched data
bull Who should be a Bioinformatician
A case studybull Bioinformatic-driven search for metabolic biomarkers in diseasebull httpwwwjclinbioinformaticscomcontent112bull The search and validation of novel disease biomarkers requires the complementary power of professional study planning and
execution modern profiling technologies and related bioinformatics tools for data analysis and interpretation Biomarkers have considerable impact on the care of patients and are urgently needed for advancing diagnostics prognostics and treatment of disease This survey article highlights emerging bioinformatics methods for biomarker discovery in clinical metabolomics focusing on the problem of data preprocessing and consolidation the data-driven search verification prioritization and biological interpretation of putative metabolic candidate biomarkers in disease In particular data mining tools suitable for the application to omic data gathered from most frequently-used type of experimental designs such as case-control or longitudinal biomarker cohort studies are reviewed and case examples of selected discovery steps are delineated in more detail This review demonstrates that clinical bioinformatics has evolved into an essential element of biomarker discovery translating new innovations and successes in profiling technologies and bioinformatics to clinical application
Data sequencing-GeneBank
What is GeneBank
GenBankreg is the National Institute of Health (NIH) genetic sequence database an annotated collection of all publicly available DNA sequences
As of 2008 there are approximately
100 billion bases in
100 million sequences
Consider the growth rate
Started in 1982 with 680338 base pairs in 606 sequences
GenBank is part of the International Nucleotide Sequence Database Collaboration which comprises the DNA DataBank of Japan (DDBJ) the European Molecular Biology Laboratory (EMBL) and GenBank at National Center for Biotechnology Information (NCBI) These three organizations exchange data on a daily basis
How GeneBank worksSubmissions to GenBankbull Many journals require submission of sequence information to a database prior to
publication so that an accession number may appear in the paper Sequin NCBIs stand-alone submission software for MAC PC and UNIX platforms is available When using Sequin the output files for direct submission should be sent to GenBank by electronic mail
Updating or Revising a Sequencebull Revisions or updates to GenBank entries can be made at any time and can be accepted
as BankIt or Sequin files or as the text of an e-mail message
Access to GenBankbull GenBank is available for searching at NCBI via several methodsbull The GenBank database is designed to provide and encourage access within the scientific
community to the most up to date and comprehensive DNA sequence information Therefore NCBI places no restrictions on the use or distribution of the GenBank data However some submitters may claim patent copyright or other intellectual property rights in all or a portion of the data they have submitted
New Developmentsbull NCBI is continuously developing new tools and enhancing existing ones to improve both
submission and access to GenBank The easiest way to keep abreast of these and other developments is to check the Whats New section of the NCBI Web page and to read the NCBI News which is also available by free subscription
Various bases of Bioinformaticsbull Count Bases at the Fraunhofer IGB GermanyThis system basically consists of modules that cover sequence analysis (Count
Bases ndash Next-Gen Sequence Assistant) statistics as well as visualization (Count Bases Viewer)
In a single run 106ndash109 DNA fragments with an average sequence length of 30ndash800 bases are simultaneously sequenced This results in huge amounts of data that require a storage volume of up to 10ndash100 gigabyte
Sources Genome and proteomic data bases
Major rersearch areasSequence analysis
Genome annotation
Literature
Analysis of gene expression regulation
Analysis of protein expression
Mutations in cancer
Etc
Organisms in GeneBank
bull 260000 different speciesbull 1000 new species being added per month
bull Human (Homo sapiens)
11551000 entries with 13149000000 basesbull Mouse (Mus musculus)
7256000 entries with 8361230000 bases
are top two species
GeneBank FormatGenBank format (GenBank Flat File Format) consists of an annotation
section and a sequence section
Annotation sectionThe start of the annotation section is marked by a line beginning with the word LOCUS
The only rule now applied in assigning a locus name is that it must be unique
Sequence sectionThe start of sequence section is marked by a line beginning with the word ORIGIN and the end of the section is marked by a line with only ldquo
GeneBank Flat File FormatLOCUS AF068625 200 bp mRNA linear ROD 06-DEC-1999
DEFINITION Mus musculus DNA cytosine-5 methyltransferase 3A (Dnmt3a) mRNA complete cds
ACCESSION AF068625 REGION 1200
VERSION AF0686252 GI6449467
KEYWORDS
SOURCE Mus musculus (house mouse)
ORGANISM Mus musculus Eukaryota Metazoa Chordata Craniata Vertebrata Euteleostomi Mammalia Eutheria Euarchontoglires Glires Rodentia Sciurognathi Muroidea Muridae Murinae Mus
REFERENCE1 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REFERENCE2 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REMARK Sequence update by submitter
COMMENT On Nov 18 1999 this sequence version replaced gi3327977
FEATURES LocationQualifiers
source 1200 organism=Mus musculus mol_type=mRNA db_xref=taxon10090 chromosome=12 map=40 cM
gene 1gt200 gene=Dnmt3a
ORIGIN 1 gaattccggc ctgctgccgg gccgcccgac ccgccgggcc acacggcaga gccgcctgaa 61 gcccagcgct gaggctgcac ttttccgagg gcttgacatc agggtctatg tttaagtctt 121 agctcttgct tacaaagacc acggcaattc cttctctgaa gccctcgcag ccccacagcg 181 ccctcgcagc cccagcctgc
GenBank sequence formatItrsquos a rich format for storing sequences and associated annotations It shares a feature
table vocabulary and format with the EMBL and DDJB formats
bull LOCUS CAA89576 109 aa linear PLN 11-AUG-1997 bull DEFINITION CYC1 [Saccharomyces cerevisiae] bull ACCESSION CAA89576 bull VERSION CAA895761 GI1015707 bull DBSOURCE embl locus SCYJR048W accession Z495481 bull KEYWORDS 5-10 or as many as neededbull SOURCE Saccharomyces cerevisiae (bakers yeast) bull ORGANISM Saccharomyces cerevisiae Eukaryota Fungi Ascomycota
Saccharomycotina Saccharomycetes Saccharomycetales Saccharomycetaceae Saccharomyces
bull REFERENCE1 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull REFERENCE2 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull FEATURES LocationQualifiers bull source 1109 organism=Saccharomyces cerevisiae db_xref=taxon4932
chromosome=X Protein 1109 name=CYC1 bull CDS 1109 gene=CYC1 coded_by=Z4954819541283 note=ORF YJR048w
db_xref=GOAP00044 db_xref=SGDS0003809 db_xref=UniProtKBSwiss-ProtP00044 bull ORIGIN 1 mtefkagsak kgatlfktrc lqchtvekgg phkvgpnlhg ifgrhsgqae gysytdanik 61 knvlwdennm
seyltnpkky ipgtkmafgg lkkekdrndl itylkkace
Online Mendelian Inheritance in Man (OMIM) Databasebull OMIM (since 1960s) catalogues all the known diseases with a genetic component
and tries to link them to the relevant genes in human genome
bull In 2004 there were 15000 records
bull One can request to download the mim2genetxt file from OMIM here httpwwwomimorgdownloads
bull The OMIM codebull Every disease and gene is assigned a six digit number of which the first number classifies
the method of inheritancebull If the initial digit is 1 the trait is deemed autosomal dominant if 2 autosomal recessive if 3
X-linked Wherever a trait defined in this dictionary has a MIM number the number from the 12th edition of MIM is given in square brackets with or without an asterisk (asterisks indicate that the mode of inheritance is known a number symbol () before an entry number means that the phenotype can be caused by mutation in any of two or more genes) as appropriate eg Pelizaeus-Merzbacher disease [MIM 312080] is an X-linked recessive disorder
bull For further studies visit httpwwwomimorg
OMIMExample httpwwwomimorgentry189911
189911 TRANSFER RNA GLYCINE 1 TRNAG1
Alternative titles symbols TRANSFER RNA GLYCINE-CCC-1 TRG1
Cytogenetic location Chr16 Genomic coordinates (GRCh37) 160 - 90354753 (from NCBI)
TEXT
Mapping McBride et al (1989) assigned a glycine tRNA(CCC) gene (TRG1) to human chromosome 1 (1pter-p34) on the basis of Southern analysis of a panel of hybrid cell DNAs They also assigned a cloned DNA fragment encompassing a glycine tRNA gene (tRNA-GCC) and pseudogene to human chromosome 16 by the same method
Evolution There are about 1300 tRNA genes in the haploid human genome (Hatlen and Attardi 1971) encoding 60 to 90 tRNA isoacceptors (Lin and Agris 1980) The studies by McBride et al (1989) as well as studies by others (see eg 180620 189930 189920 180640 189880) indicated that tRNA genes and pseudogenes are dispersed on at least 7 human chromosomes and suggested that these sequences would probably be found on most if not all human chromosomes McBride et al (1989) described short 8-12 nucleotide direct terminal repeats flanking many of the dispersed tRNA genes This finding combined with the dispersion of tRNA genes suggests that many of these genes may have arisen by an RNA-mediated retroposition mechanism There may have been selection for reiteration of genes encoding isoaccepting tRNAs since a single mutation in a single-copy tRNA gene could be devastating Moreover even a mutation in the anticodon of a single tRNA gene might not be crucial if competition was provided by the normal wildtype tRNA isoacceptor produced by multiple copies of the normal tRNA gene still present in the genome Dispersion of multiple copies of each tRNA gene could provide diversity of 5-prime-flanking sequences which are known to modulate the expression of some human tRNA genes Tissue-specific or differentiation-specific expression of tRNA isoacceptors might be provided for by this mechanism The recombination and unequal crossingover that can occur with tandem tRNA sequences can result in homogenization of the sequences with disastrous consequences
Nucleotide Databasebull NUCLEOTIDE DATABASESbull NCBIs sequence databases accept genome data from sequencing projects from around
the world and serve as the cornerstone of bioinformatics researchbull GenBankbull An annotated collection of all publicly available nucleotide and amino acid sequencesbull EST databasebull A collection of expressed sequence tags or short single-pass sequence reads from mRNA
(cDNA)bull GSS databasebull A database of genome survey sequences or short single-pass genomic sequencesbull HomoloGenebull A gene homology tool that compares nucleotide sequences between pairs of organisms in order
to identify putative orthologsbull HTG databasebull A collection of high-throughput genome sequences from large-scale genome sequencing
centers including unfinished and finished sequencesbull SNPs databasebull A central repository for both single-base nucleotide substitutions and short deletion and
insertion polymorphisms
Nucleotide Database
bull RefSeqbull A database of non-redundant reference sequences standards including genomic
DNA contigs mRNAs and proteins for known genes Multiple collaborations both within NCBI and with external groups support our data-gathering efforts
bull STS databasebull A database of sequence tagged sites or short sequences that are operationally
unique in the genomebull UniSTSbull A unified non-redundant view of sequence tagged sites (STSs)bull UniGenebull A collection of ESTs and full-length mRNA sequences organized into clusters each
representing a unique known or putative human gene annotated with mapping and expression information and cross-references to other sources
UniGene computationally identifies transcripts from the same locus analyzes expression by tissue age and health status and reports related proteins (protEST) and clone resources
Single Nucleotide Polymorphism (SNP) database
What it isThe SNP Database (also known as dbSNP) is an archive for genetic variation within and across different species developed and hosted by NCBI in collaboration with the National Human Genome Research Institute (NHGRI)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species related to biodiversity genetic variation and adaptation
-The dbSNP accepts apparently neutral polymorphisms polymorphisms corresponding to known phenotypes and regions of no variation
-It was created in September 1998 to supplement GenBank (NCBIrsquos nucleic acid and protein sequences)
Goal
Its goal is to act as a single database that contains all identified genetic variation which can be used to investigate a wide variety of genetically based natural phenomenon Specifically access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping population genetics investigations into evolutionary relationships as well as being able to quickly and easily quantify the amount of variation at a given site of interest
Application
Applied research genetic engineering drug discovery etc
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-
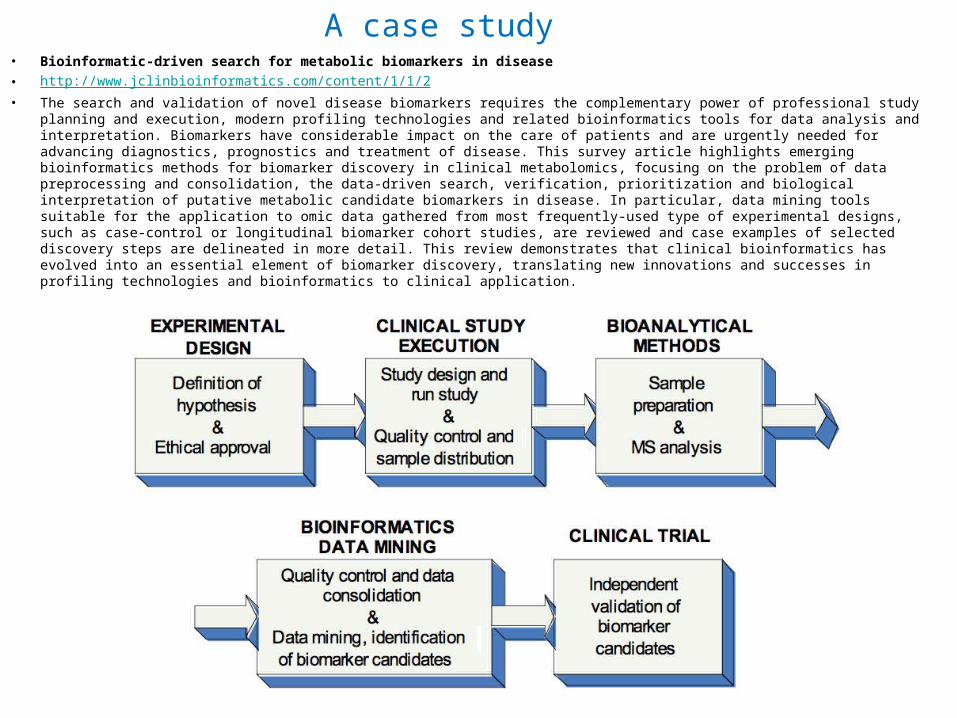
A case studybull Bioinformatic-driven search for metabolic biomarkers in diseasebull httpwwwjclinbioinformaticscomcontent112bull The search and validation of novel disease biomarkers requires the complementary power of professional study planning and
execution modern profiling technologies and related bioinformatics tools for data analysis and interpretation Biomarkers have considerable impact on the care of patients and are urgently needed for advancing diagnostics prognostics and treatment of disease This survey article highlights emerging bioinformatics methods for biomarker discovery in clinical metabolomics focusing on the problem of data preprocessing and consolidation the data-driven search verification prioritization and biological interpretation of putative metabolic candidate biomarkers in disease In particular data mining tools suitable for the application to omic data gathered from most frequently-used type of experimental designs such as case-control or longitudinal biomarker cohort studies are reviewed and case examples of selected discovery steps are delineated in more detail This review demonstrates that clinical bioinformatics has evolved into an essential element of biomarker discovery translating new innovations and successes in profiling technologies and bioinformatics to clinical application
Data sequencing-GeneBank
What is GeneBank
GenBankreg is the National Institute of Health (NIH) genetic sequence database an annotated collection of all publicly available DNA sequences
As of 2008 there are approximately
100 billion bases in
100 million sequences
Consider the growth rate
Started in 1982 with 680338 base pairs in 606 sequences
GenBank is part of the International Nucleotide Sequence Database Collaboration which comprises the DNA DataBank of Japan (DDBJ) the European Molecular Biology Laboratory (EMBL) and GenBank at National Center for Biotechnology Information (NCBI) These three organizations exchange data on a daily basis
How GeneBank worksSubmissions to GenBankbull Many journals require submission of sequence information to a database prior to
publication so that an accession number may appear in the paper Sequin NCBIs stand-alone submission software for MAC PC and UNIX platforms is available When using Sequin the output files for direct submission should be sent to GenBank by electronic mail
Updating or Revising a Sequencebull Revisions or updates to GenBank entries can be made at any time and can be accepted
as BankIt or Sequin files or as the text of an e-mail message
Access to GenBankbull GenBank is available for searching at NCBI via several methodsbull The GenBank database is designed to provide and encourage access within the scientific
community to the most up to date and comprehensive DNA sequence information Therefore NCBI places no restrictions on the use or distribution of the GenBank data However some submitters may claim patent copyright or other intellectual property rights in all or a portion of the data they have submitted
New Developmentsbull NCBI is continuously developing new tools and enhancing existing ones to improve both
submission and access to GenBank The easiest way to keep abreast of these and other developments is to check the Whats New section of the NCBI Web page and to read the NCBI News which is also available by free subscription
Various bases of Bioinformaticsbull Count Bases at the Fraunhofer IGB GermanyThis system basically consists of modules that cover sequence analysis (Count
Bases ndash Next-Gen Sequence Assistant) statistics as well as visualization (Count Bases Viewer)
In a single run 106ndash109 DNA fragments with an average sequence length of 30ndash800 bases are simultaneously sequenced This results in huge amounts of data that require a storage volume of up to 10ndash100 gigabyte
Sources Genome and proteomic data bases
Major rersearch areasSequence analysis
Genome annotation
Literature
Analysis of gene expression regulation
Analysis of protein expression
Mutations in cancer
Etc
Organisms in GeneBank
bull 260000 different speciesbull 1000 new species being added per month
bull Human (Homo sapiens)
11551000 entries with 13149000000 basesbull Mouse (Mus musculus)
7256000 entries with 8361230000 bases
are top two species
GeneBank FormatGenBank format (GenBank Flat File Format) consists of an annotation
section and a sequence section
Annotation sectionThe start of the annotation section is marked by a line beginning with the word LOCUS
The only rule now applied in assigning a locus name is that it must be unique
Sequence sectionThe start of sequence section is marked by a line beginning with the word ORIGIN and the end of the section is marked by a line with only ldquo
GeneBank Flat File FormatLOCUS AF068625 200 bp mRNA linear ROD 06-DEC-1999
DEFINITION Mus musculus DNA cytosine-5 methyltransferase 3A (Dnmt3a) mRNA complete cds
ACCESSION AF068625 REGION 1200
VERSION AF0686252 GI6449467
KEYWORDS
SOURCE Mus musculus (house mouse)
ORGANISM Mus musculus Eukaryota Metazoa Chordata Craniata Vertebrata Euteleostomi Mammalia Eutheria Euarchontoglires Glires Rodentia Sciurognathi Muroidea Muridae Murinae Mus
REFERENCE1 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REFERENCE2 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REMARK Sequence update by submitter
COMMENT On Nov 18 1999 this sequence version replaced gi3327977
FEATURES LocationQualifiers
source 1200 organism=Mus musculus mol_type=mRNA db_xref=taxon10090 chromosome=12 map=40 cM
gene 1gt200 gene=Dnmt3a
ORIGIN 1 gaattccggc ctgctgccgg gccgcccgac ccgccgggcc acacggcaga gccgcctgaa 61 gcccagcgct gaggctgcac ttttccgagg gcttgacatc agggtctatg tttaagtctt 121 agctcttgct tacaaagacc acggcaattc cttctctgaa gccctcgcag ccccacagcg 181 ccctcgcagc cccagcctgc
GenBank sequence formatItrsquos a rich format for storing sequences and associated annotations It shares a feature
table vocabulary and format with the EMBL and DDJB formats
bull LOCUS CAA89576 109 aa linear PLN 11-AUG-1997 bull DEFINITION CYC1 [Saccharomyces cerevisiae] bull ACCESSION CAA89576 bull VERSION CAA895761 GI1015707 bull DBSOURCE embl locus SCYJR048W accession Z495481 bull KEYWORDS 5-10 or as many as neededbull SOURCE Saccharomyces cerevisiae (bakers yeast) bull ORGANISM Saccharomyces cerevisiae Eukaryota Fungi Ascomycota
Saccharomycotina Saccharomycetes Saccharomycetales Saccharomycetaceae Saccharomyces
bull REFERENCE1 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull REFERENCE2 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull FEATURES LocationQualifiers bull source 1109 organism=Saccharomyces cerevisiae db_xref=taxon4932
chromosome=X Protein 1109 name=CYC1 bull CDS 1109 gene=CYC1 coded_by=Z4954819541283 note=ORF YJR048w
db_xref=GOAP00044 db_xref=SGDS0003809 db_xref=UniProtKBSwiss-ProtP00044 bull ORIGIN 1 mtefkagsak kgatlfktrc lqchtvekgg phkvgpnlhg ifgrhsgqae gysytdanik 61 knvlwdennm
seyltnpkky ipgtkmafgg lkkekdrndl itylkkace
Online Mendelian Inheritance in Man (OMIM) Databasebull OMIM (since 1960s) catalogues all the known diseases with a genetic component
and tries to link them to the relevant genes in human genome
bull In 2004 there were 15000 records
bull One can request to download the mim2genetxt file from OMIM here httpwwwomimorgdownloads
bull The OMIM codebull Every disease and gene is assigned a six digit number of which the first number classifies
the method of inheritancebull If the initial digit is 1 the trait is deemed autosomal dominant if 2 autosomal recessive if 3
X-linked Wherever a trait defined in this dictionary has a MIM number the number from the 12th edition of MIM is given in square brackets with or without an asterisk (asterisks indicate that the mode of inheritance is known a number symbol () before an entry number means that the phenotype can be caused by mutation in any of two or more genes) as appropriate eg Pelizaeus-Merzbacher disease [MIM 312080] is an X-linked recessive disorder
bull For further studies visit httpwwwomimorg
OMIMExample httpwwwomimorgentry189911
189911 TRANSFER RNA GLYCINE 1 TRNAG1
Alternative titles symbols TRANSFER RNA GLYCINE-CCC-1 TRG1
Cytogenetic location Chr16 Genomic coordinates (GRCh37) 160 - 90354753 (from NCBI)
TEXT
Mapping McBride et al (1989) assigned a glycine tRNA(CCC) gene (TRG1) to human chromosome 1 (1pter-p34) on the basis of Southern analysis of a panel of hybrid cell DNAs They also assigned a cloned DNA fragment encompassing a glycine tRNA gene (tRNA-GCC) and pseudogene to human chromosome 16 by the same method
Evolution There are about 1300 tRNA genes in the haploid human genome (Hatlen and Attardi 1971) encoding 60 to 90 tRNA isoacceptors (Lin and Agris 1980) The studies by McBride et al (1989) as well as studies by others (see eg 180620 189930 189920 180640 189880) indicated that tRNA genes and pseudogenes are dispersed on at least 7 human chromosomes and suggested that these sequences would probably be found on most if not all human chromosomes McBride et al (1989) described short 8-12 nucleotide direct terminal repeats flanking many of the dispersed tRNA genes This finding combined with the dispersion of tRNA genes suggests that many of these genes may have arisen by an RNA-mediated retroposition mechanism There may have been selection for reiteration of genes encoding isoaccepting tRNAs since a single mutation in a single-copy tRNA gene could be devastating Moreover even a mutation in the anticodon of a single tRNA gene might not be crucial if competition was provided by the normal wildtype tRNA isoacceptor produced by multiple copies of the normal tRNA gene still present in the genome Dispersion of multiple copies of each tRNA gene could provide diversity of 5-prime-flanking sequences which are known to modulate the expression of some human tRNA genes Tissue-specific or differentiation-specific expression of tRNA isoacceptors might be provided for by this mechanism The recombination and unequal crossingover that can occur with tandem tRNA sequences can result in homogenization of the sequences with disastrous consequences
Nucleotide Databasebull NUCLEOTIDE DATABASESbull NCBIs sequence databases accept genome data from sequencing projects from around
the world and serve as the cornerstone of bioinformatics researchbull GenBankbull An annotated collection of all publicly available nucleotide and amino acid sequencesbull EST databasebull A collection of expressed sequence tags or short single-pass sequence reads from mRNA
(cDNA)bull GSS databasebull A database of genome survey sequences or short single-pass genomic sequencesbull HomoloGenebull A gene homology tool that compares nucleotide sequences between pairs of organisms in order
to identify putative orthologsbull HTG databasebull A collection of high-throughput genome sequences from large-scale genome sequencing
centers including unfinished and finished sequencesbull SNPs databasebull A central repository for both single-base nucleotide substitutions and short deletion and
insertion polymorphisms
Nucleotide Database
bull RefSeqbull A database of non-redundant reference sequences standards including genomic
DNA contigs mRNAs and proteins for known genes Multiple collaborations both within NCBI and with external groups support our data-gathering efforts
bull STS databasebull A database of sequence tagged sites or short sequences that are operationally
unique in the genomebull UniSTSbull A unified non-redundant view of sequence tagged sites (STSs)bull UniGenebull A collection of ESTs and full-length mRNA sequences organized into clusters each
representing a unique known or putative human gene annotated with mapping and expression information and cross-references to other sources
UniGene computationally identifies transcripts from the same locus analyzes expression by tissue age and health status and reports related proteins (protEST) and clone resources
Single Nucleotide Polymorphism (SNP) database
What it isThe SNP Database (also known as dbSNP) is an archive for genetic variation within and across different species developed and hosted by NCBI in collaboration with the National Human Genome Research Institute (NHGRI)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species related to biodiversity genetic variation and adaptation
-The dbSNP accepts apparently neutral polymorphisms polymorphisms corresponding to known phenotypes and regions of no variation
-It was created in September 1998 to supplement GenBank (NCBIrsquos nucleic acid and protein sequences)
Goal
Its goal is to act as a single database that contains all identified genetic variation which can be used to investigate a wide variety of genetically based natural phenomenon Specifically access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping population genetics investigations into evolutionary relationships as well as being able to quickly and easily quantify the amount of variation at a given site of interest
Application
Applied research genetic engineering drug discovery etc
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-
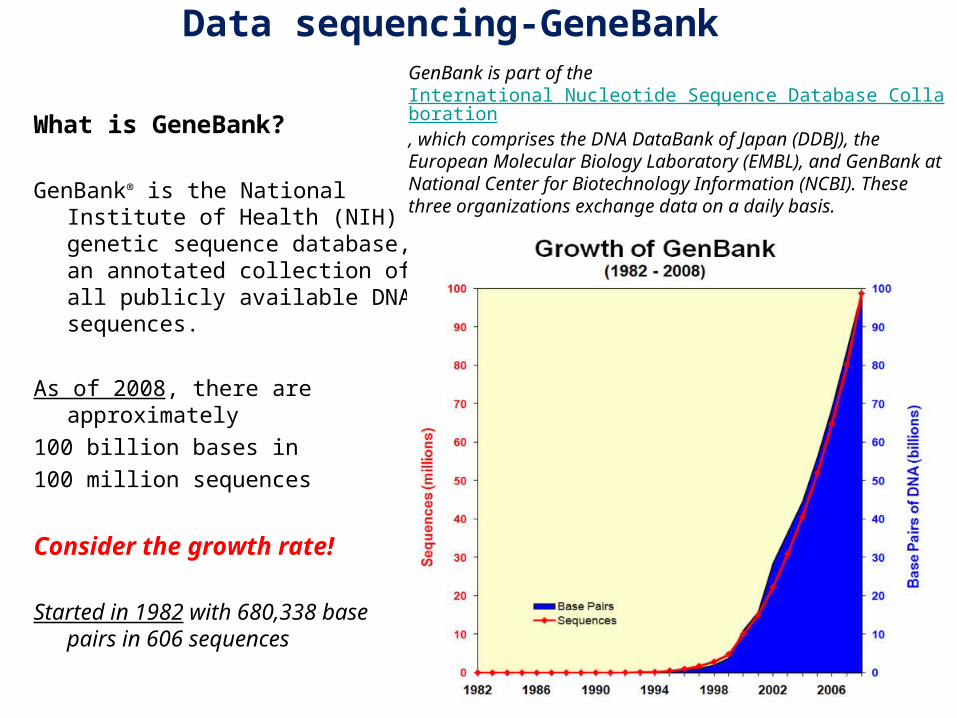
Data sequencing-GeneBank
What is GeneBank
GenBankreg is the National Institute of Health (NIH) genetic sequence database an annotated collection of all publicly available DNA sequences
As of 2008 there are approximately
100 billion bases in
100 million sequences
Consider the growth rate
Started in 1982 with 680338 base pairs in 606 sequences
GenBank is part of the International Nucleotide Sequence Database Collaboration which comprises the DNA DataBank of Japan (DDBJ) the European Molecular Biology Laboratory (EMBL) and GenBank at National Center for Biotechnology Information (NCBI) These three organizations exchange data on a daily basis
How GeneBank worksSubmissions to GenBankbull Many journals require submission of sequence information to a database prior to
publication so that an accession number may appear in the paper Sequin NCBIs stand-alone submission software for MAC PC and UNIX platforms is available When using Sequin the output files for direct submission should be sent to GenBank by electronic mail
Updating or Revising a Sequencebull Revisions or updates to GenBank entries can be made at any time and can be accepted
as BankIt or Sequin files or as the text of an e-mail message
Access to GenBankbull GenBank is available for searching at NCBI via several methodsbull The GenBank database is designed to provide and encourage access within the scientific
community to the most up to date and comprehensive DNA sequence information Therefore NCBI places no restrictions on the use or distribution of the GenBank data However some submitters may claim patent copyright or other intellectual property rights in all or a portion of the data they have submitted
New Developmentsbull NCBI is continuously developing new tools and enhancing existing ones to improve both
submission and access to GenBank The easiest way to keep abreast of these and other developments is to check the Whats New section of the NCBI Web page and to read the NCBI News which is also available by free subscription
Various bases of Bioinformaticsbull Count Bases at the Fraunhofer IGB GermanyThis system basically consists of modules that cover sequence analysis (Count
Bases ndash Next-Gen Sequence Assistant) statistics as well as visualization (Count Bases Viewer)
In a single run 106ndash109 DNA fragments with an average sequence length of 30ndash800 bases are simultaneously sequenced This results in huge amounts of data that require a storage volume of up to 10ndash100 gigabyte
Sources Genome and proteomic data bases
Major rersearch areasSequence analysis
Genome annotation
Literature
Analysis of gene expression regulation
Analysis of protein expression
Mutations in cancer
Etc
Organisms in GeneBank
bull 260000 different speciesbull 1000 new species being added per month
bull Human (Homo sapiens)
11551000 entries with 13149000000 basesbull Mouse (Mus musculus)
7256000 entries with 8361230000 bases
are top two species
GeneBank FormatGenBank format (GenBank Flat File Format) consists of an annotation
section and a sequence section
Annotation sectionThe start of the annotation section is marked by a line beginning with the word LOCUS
The only rule now applied in assigning a locus name is that it must be unique
Sequence sectionThe start of sequence section is marked by a line beginning with the word ORIGIN and the end of the section is marked by a line with only ldquo
GeneBank Flat File FormatLOCUS AF068625 200 bp mRNA linear ROD 06-DEC-1999
DEFINITION Mus musculus DNA cytosine-5 methyltransferase 3A (Dnmt3a) mRNA complete cds
ACCESSION AF068625 REGION 1200
VERSION AF0686252 GI6449467
KEYWORDS
SOURCE Mus musculus (house mouse)
ORGANISM Mus musculus Eukaryota Metazoa Chordata Craniata Vertebrata Euteleostomi Mammalia Eutheria Euarchontoglires Glires Rodentia Sciurognathi Muroidea Muridae Murinae Mus
REFERENCE1 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REFERENCE2 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REMARK Sequence update by submitter
COMMENT On Nov 18 1999 this sequence version replaced gi3327977
FEATURES LocationQualifiers
source 1200 organism=Mus musculus mol_type=mRNA db_xref=taxon10090 chromosome=12 map=40 cM
gene 1gt200 gene=Dnmt3a
ORIGIN 1 gaattccggc ctgctgccgg gccgcccgac ccgccgggcc acacggcaga gccgcctgaa 61 gcccagcgct gaggctgcac ttttccgagg gcttgacatc agggtctatg tttaagtctt 121 agctcttgct tacaaagacc acggcaattc cttctctgaa gccctcgcag ccccacagcg 181 ccctcgcagc cccagcctgc
GenBank sequence formatItrsquos a rich format for storing sequences and associated annotations It shares a feature
table vocabulary and format with the EMBL and DDJB formats
bull LOCUS CAA89576 109 aa linear PLN 11-AUG-1997 bull DEFINITION CYC1 [Saccharomyces cerevisiae] bull ACCESSION CAA89576 bull VERSION CAA895761 GI1015707 bull DBSOURCE embl locus SCYJR048W accession Z495481 bull KEYWORDS 5-10 or as many as neededbull SOURCE Saccharomyces cerevisiae (bakers yeast) bull ORGANISM Saccharomyces cerevisiae Eukaryota Fungi Ascomycota
Saccharomycotina Saccharomycetes Saccharomycetales Saccharomycetaceae Saccharomyces
bull REFERENCE1 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull REFERENCE2 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull FEATURES LocationQualifiers bull source 1109 organism=Saccharomyces cerevisiae db_xref=taxon4932
chromosome=X Protein 1109 name=CYC1 bull CDS 1109 gene=CYC1 coded_by=Z4954819541283 note=ORF YJR048w
db_xref=GOAP00044 db_xref=SGDS0003809 db_xref=UniProtKBSwiss-ProtP00044 bull ORIGIN 1 mtefkagsak kgatlfktrc lqchtvekgg phkvgpnlhg ifgrhsgqae gysytdanik 61 knvlwdennm
seyltnpkky ipgtkmafgg lkkekdrndl itylkkace
Online Mendelian Inheritance in Man (OMIM) Databasebull OMIM (since 1960s) catalogues all the known diseases with a genetic component
and tries to link them to the relevant genes in human genome
bull In 2004 there were 15000 records
bull One can request to download the mim2genetxt file from OMIM here httpwwwomimorgdownloads
bull The OMIM codebull Every disease and gene is assigned a six digit number of which the first number classifies
the method of inheritancebull If the initial digit is 1 the trait is deemed autosomal dominant if 2 autosomal recessive if 3
X-linked Wherever a trait defined in this dictionary has a MIM number the number from the 12th edition of MIM is given in square brackets with or without an asterisk (asterisks indicate that the mode of inheritance is known a number symbol () before an entry number means that the phenotype can be caused by mutation in any of two or more genes) as appropriate eg Pelizaeus-Merzbacher disease [MIM 312080] is an X-linked recessive disorder
bull For further studies visit httpwwwomimorg
OMIMExample httpwwwomimorgentry189911
189911 TRANSFER RNA GLYCINE 1 TRNAG1
Alternative titles symbols TRANSFER RNA GLYCINE-CCC-1 TRG1
Cytogenetic location Chr16 Genomic coordinates (GRCh37) 160 - 90354753 (from NCBI)
TEXT
Mapping McBride et al (1989) assigned a glycine tRNA(CCC) gene (TRG1) to human chromosome 1 (1pter-p34) on the basis of Southern analysis of a panel of hybrid cell DNAs They also assigned a cloned DNA fragment encompassing a glycine tRNA gene (tRNA-GCC) and pseudogene to human chromosome 16 by the same method
Evolution There are about 1300 tRNA genes in the haploid human genome (Hatlen and Attardi 1971) encoding 60 to 90 tRNA isoacceptors (Lin and Agris 1980) The studies by McBride et al (1989) as well as studies by others (see eg 180620 189930 189920 180640 189880) indicated that tRNA genes and pseudogenes are dispersed on at least 7 human chromosomes and suggested that these sequences would probably be found on most if not all human chromosomes McBride et al (1989) described short 8-12 nucleotide direct terminal repeats flanking many of the dispersed tRNA genes This finding combined with the dispersion of tRNA genes suggests that many of these genes may have arisen by an RNA-mediated retroposition mechanism There may have been selection for reiteration of genes encoding isoaccepting tRNAs since a single mutation in a single-copy tRNA gene could be devastating Moreover even a mutation in the anticodon of a single tRNA gene might not be crucial if competition was provided by the normal wildtype tRNA isoacceptor produced by multiple copies of the normal tRNA gene still present in the genome Dispersion of multiple copies of each tRNA gene could provide diversity of 5-prime-flanking sequences which are known to modulate the expression of some human tRNA genes Tissue-specific or differentiation-specific expression of tRNA isoacceptors might be provided for by this mechanism The recombination and unequal crossingover that can occur with tandem tRNA sequences can result in homogenization of the sequences with disastrous consequences
Nucleotide Databasebull NUCLEOTIDE DATABASESbull NCBIs sequence databases accept genome data from sequencing projects from around
the world and serve as the cornerstone of bioinformatics researchbull GenBankbull An annotated collection of all publicly available nucleotide and amino acid sequencesbull EST databasebull A collection of expressed sequence tags or short single-pass sequence reads from mRNA
(cDNA)bull GSS databasebull A database of genome survey sequences or short single-pass genomic sequencesbull HomoloGenebull A gene homology tool that compares nucleotide sequences between pairs of organisms in order
to identify putative orthologsbull HTG databasebull A collection of high-throughput genome sequences from large-scale genome sequencing
centers including unfinished and finished sequencesbull SNPs databasebull A central repository for both single-base nucleotide substitutions and short deletion and
insertion polymorphisms
Nucleotide Database
bull RefSeqbull A database of non-redundant reference sequences standards including genomic
DNA contigs mRNAs and proteins for known genes Multiple collaborations both within NCBI and with external groups support our data-gathering efforts
bull STS databasebull A database of sequence tagged sites or short sequences that are operationally
unique in the genomebull UniSTSbull A unified non-redundant view of sequence tagged sites (STSs)bull UniGenebull A collection of ESTs and full-length mRNA sequences organized into clusters each
representing a unique known or putative human gene annotated with mapping and expression information and cross-references to other sources
UniGene computationally identifies transcripts from the same locus analyzes expression by tissue age and health status and reports related proteins (protEST) and clone resources
Single Nucleotide Polymorphism (SNP) database
What it isThe SNP Database (also known as dbSNP) is an archive for genetic variation within and across different species developed and hosted by NCBI in collaboration with the National Human Genome Research Institute (NHGRI)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species related to biodiversity genetic variation and adaptation
-The dbSNP accepts apparently neutral polymorphisms polymorphisms corresponding to known phenotypes and regions of no variation
-It was created in September 1998 to supplement GenBank (NCBIrsquos nucleic acid and protein sequences)
Goal
Its goal is to act as a single database that contains all identified genetic variation which can be used to investigate a wide variety of genetically based natural phenomenon Specifically access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping population genetics investigations into evolutionary relationships as well as being able to quickly and easily quantify the amount of variation at a given site of interest
Application
Applied research genetic engineering drug discovery etc
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

How GeneBank worksSubmissions to GenBankbull Many journals require submission of sequence information to a database prior to
publication so that an accession number may appear in the paper Sequin NCBIs stand-alone submission software for MAC PC and UNIX platforms is available When using Sequin the output files for direct submission should be sent to GenBank by electronic mail
Updating or Revising a Sequencebull Revisions or updates to GenBank entries can be made at any time and can be accepted
as BankIt or Sequin files or as the text of an e-mail message
Access to GenBankbull GenBank is available for searching at NCBI via several methodsbull The GenBank database is designed to provide and encourage access within the scientific
community to the most up to date and comprehensive DNA sequence information Therefore NCBI places no restrictions on the use or distribution of the GenBank data However some submitters may claim patent copyright or other intellectual property rights in all or a portion of the data they have submitted
New Developmentsbull NCBI is continuously developing new tools and enhancing existing ones to improve both
submission and access to GenBank The easiest way to keep abreast of these and other developments is to check the Whats New section of the NCBI Web page and to read the NCBI News which is also available by free subscription
Various bases of Bioinformaticsbull Count Bases at the Fraunhofer IGB GermanyThis system basically consists of modules that cover sequence analysis (Count
Bases ndash Next-Gen Sequence Assistant) statistics as well as visualization (Count Bases Viewer)
In a single run 106ndash109 DNA fragments with an average sequence length of 30ndash800 bases are simultaneously sequenced This results in huge amounts of data that require a storage volume of up to 10ndash100 gigabyte
Sources Genome and proteomic data bases
Major rersearch areasSequence analysis
Genome annotation
Literature
Analysis of gene expression regulation
Analysis of protein expression
Mutations in cancer
Etc
Organisms in GeneBank
bull 260000 different speciesbull 1000 new species being added per month
bull Human (Homo sapiens)
11551000 entries with 13149000000 basesbull Mouse (Mus musculus)
7256000 entries with 8361230000 bases
are top two species
GeneBank FormatGenBank format (GenBank Flat File Format) consists of an annotation
section and a sequence section
Annotation sectionThe start of the annotation section is marked by a line beginning with the word LOCUS
The only rule now applied in assigning a locus name is that it must be unique
Sequence sectionThe start of sequence section is marked by a line beginning with the word ORIGIN and the end of the section is marked by a line with only ldquo
GeneBank Flat File FormatLOCUS AF068625 200 bp mRNA linear ROD 06-DEC-1999
DEFINITION Mus musculus DNA cytosine-5 methyltransferase 3A (Dnmt3a) mRNA complete cds
ACCESSION AF068625 REGION 1200
VERSION AF0686252 GI6449467
KEYWORDS
SOURCE Mus musculus (house mouse)
ORGANISM Mus musculus Eukaryota Metazoa Chordata Craniata Vertebrata Euteleostomi Mammalia Eutheria Euarchontoglires Glires Rodentia Sciurognathi Muroidea Muridae Murinae Mus
REFERENCE1 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REFERENCE2 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REMARK Sequence update by submitter
COMMENT On Nov 18 1999 this sequence version replaced gi3327977
FEATURES LocationQualifiers
source 1200 organism=Mus musculus mol_type=mRNA db_xref=taxon10090 chromosome=12 map=40 cM
gene 1gt200 gene=Dnmt3a
ORIGIN 1 gaattccggc ctgctgccgg gccgcccgac ccgccgggcc acacggcaga gccgcctgaa 61 gcccagcgct gaggctgcac ttttccgagg gcttgacatc agggtctatg tttaagtctt 121 agctcttgct tacaaagacc acggcaattc cttctctgaa gccctcgcag ccccacagcg 181 ccctcgcagc cccagcctgc
GenBank sequence formatItrsquos a rich format for storing sequences and associated annotations It shares a feature
table vocabulary and format with the EMBL and DDJB formats
bull LOCUS CAA89576 109 aa linear PLN 11-AUG-1997 bull DEFINITION CYC1 [Saccharomyces cerevisiae] bull ACCESSION CAA89576 bull VERSION CAA895761 GI1015707 bull DBSOURCE embl locus SCYJR048W accession Z495481 bull KEYWORDS 5-10 or as many as neededbull SOURCE Saccharomyces cerevisiae (bakers yeast) bull ORGANISM Saccharomyces cerevisiae Eukaryota Fungi Ascomycota
Saccharomycotina Saccharomycetes Saccharomycetales Saccharomycetaceae Saccharomyces
bull REFERENCE1 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull REFERENCE2 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull FEATURES LocationQualifiers bull source 1109 organism=Saccharomyces cerevisiae db_xref=taxon4932
chromosome=X Protein 1109 name=CYC1 bull CDS 1109 gene=CYC1 coded_by=Z4954819541283 note=ORF YJR048w
db_xref=GOAP00044 db_xref=SGDS0003809 db_xref=UniProtKBSwiss-ProtP00044 bull ORIGIN 1 mtefkagsak kgatlfktrc lqchtvekgg phkvgpnlhg ifgrhsgqae gysytdanik 61 knvlwdennm
seyltnpkky ipgtkmafgg lkkekdrndl itylkkace
Online Mendelian Inheritance in Man (OMIM) Databasebull OMIM (since 1960s) catalogues all the known diseases with a genetic component
and tries to link them to the relevant genes in human genome
bull In 2004 there were 15000 records
bull One can request to download the mim2genetxt file from OMIM here httpwwwomimorgdownloads
bull The OMIM codebull Every disease and gene is assigned a six digit number of which the first number classifies
the method of inheritancebull If the initial digit is 1 the trait is deemed autosomal dominant if 2 autosomal recessive if 3
X-linked Wherever a trait defined in this dictionary has a MIM number the number from the 12th edition of MIM is given in square brackets with or without an asterisk (asterisks indicate that the mode of inheritance is known a number symbol () before an entry number means that the phenotype can be caused by mutation in any of two or more genes) as appropriate eg Pelizaeus-Merzbacher disease [MIM 312080] is an X-linked recessive disorder
bull For further studies visit httpwwwomimorg
OMIMExample httpwwwomimorgentry189911
189911 TRANSFER RNA GLYCINE 1 TRNAG1
Alternative titles symbols TRANSFER RNA GLYCINE-CCC-1 TRG1
Cytogenetic location Chr16 Genomic coordinates (GRCh37) 160 - 90354753 (from NCBI)
TEXT
Mapping McBride et al (1989) assigned a glycine tRNA(CCC) gene (TRG1) to human chromosome 1 (1pter-p34) on the basis of Southern analysis of a panel of hybrid cell DNAs They also assigned a cloned DNA fragment encompassing a glycine tRNA gene (tRNA-GCC) and pseudogene to human chromosome 16 by the same method
Evolution There are about 1300 tRNA genes in the haploid human genome (Hatlen and Attardi 1971) encoding 60 to 90 tRNA isoacceptors (Lin and Agris 1980) The studies by McBride et al (1989) as well as studies by others (see eg 180620 189930 189920 180640 189880) indicated that tRNA genes and pseudogenes are dispersed on at least 7 human chromosomes and suggested that these sequences would probably be found on most if not all human chromosomes McBride et al (1989) described short 8-12 nucleotide direct terminal repeats flanking many of the dispersed tRNA genes This finding combined with the dispersion of tRNA genes suggests that many of these genes may have arisen by an RNA-mediated retroposition mechanism There may have been selection for reiteration of genes encoding isoaccepting tRNAs since a single mutation in a single-copy tRNA gene could be devastating Moreover even a mutation in the anticodon of a single tRNA gene might not be crucial if competition was provided by the normal wildtype tRNA isoacceptor produced by multiple copies of the normal tRNA gene still present in the genome Dispersion of multiple copies of each tRNA gene could provide diversity of 5-prime-flanking sequences which are known to modulate the expression of some human tRNA genes Tissue-specific or differentiation-specific expression of tRNA isoacceptors might be provided for by this mechanism The recombination and unequal crossingover that can occur with tandem tRNA sequences can result in homogenization of the sequences with disastrous consequences
Nucleotide Databasebull NUCLEOTIDE DATABASESbull NCBIs sequence databases accept genome data from sequencing projects from around
the world and serve as the cornerstone of bioinformatics researchbull GenBankbull An annotated collection of all publicly available nucleotide and amino acid sequencesbull EST databasebull A collection of expressed sequence tags or short single-pass sequence reads from mRNA
(cDNA)bull GSS databasebull A database of genome survey sequences or short single-pass genomic sequencesbull HomoloGenebull A gene homology tool that compares nucleotide sequences between pairs of organisms in order
to identify putative orthologsbull HTG databasebull A collection of high-throughput genome sequences from large-scale genome sequencing
centers including unfinished and finished sequencesbull SNPs databasebull A central repository for both single-base nucleotide substitutions and short deletion and
insertion polymorphisms
Nucleotide Database
bull RefSeqbull A database of non-redundant reference sequences standards including genomic
DNA contigs mRNAs and proteins for known genes Multiple collaborations both within NCBI and with external groups support our data-gathering efforts
bull STS databasebull A database of sequence tagged sites or short sequences that are operationally
unique in the genomebull UniSTSbull A unified non-redundant view of sequence tagged sites (STSs)bull UniGenebull A collection of ESTs and full-length mRNA sequences organized into clusters each
representing a unique known or putative human gene annotated with mapping and expression information and cross-references to other sources
UniGene computationally identifies transcripts from the same locus analyzes expression by tissue age and health status and reports related proteins (protEST) and clone resources
Single Nucleotide Polymorphism (SNP) database
What it isThe SNP Database (also known as dbSNP) is an archive for genetic variation within and across different species developed and hosted by NCBI in collaboration with the National Human Genome Research Institute (NHGRI)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species related to biodiversity genetic variation and adaptation
-The dbSNP accepts apparently neutral polymorphisms polymorphisms corresponding to known phenotypes and regions of no variation
-It was created in September 1998 to supplement GenBank (NCBIrsquos nucleic acid and protein sequences)
Goal
Its goal is to act as a single database that contains all identified genetic variation which can be used to investigate a wide variety of genetically based natural phenomenon Specifically access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping population genetics investigations into evolutionary relationships as well as being able to quickly and easily quantify the amount of variation at a given site of interest
Application
Applied research genetic engineering drug discovery etc
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

Various bases of Bioinformaticsbull Count Bases at the Fraunhofer IGB GermanyThis system basically consists of modules that cover sequence analysis (Count
Bases ndash Next-Gen Sequence Assistant) statistics as well as visualization (Count Bases Viewer)
In a single run 106ndash109 DNA fragments with an average sequence length of 30ndash800 bases are simultaneously sequenced This results in huge amounts of data that require a storage volume of up to 10ndash100 gigabyte
Sources Genome and proteomic data bases
Major rersearch areasSequence analysis
Genome annotation
Literature
Analysis of gene expression regulation
Analysis of protein expression
Mutations in cancer
Etc
Organisms in GeneBank
bull 260000 different speciesbull 1000 new species being added per month
bull Human (Homo sapiens)
11551000 entries with 13149000000 basesbull Mouse (Mus musculus)
7256000 entries with 8361230000 bases
are top two species
GeneBank FormatGenBank format (GenBank Flat File Format) consists of an annotation
section and a sequence section
Annotation sectionThe start of the annotation section is marked by a line beginning with the word LOCUS
The only rule now applied in assigning a locus name is that it must be unique
Sequence sectionThe start of sequence section is marked by a line beginning with the word ORIGIN and the end of the section is marked by a line with only ldquo
GeneBank Flat File FormatLOCUS AF068625 200 bp mRNA linear ROD 06-DEC-1999
DEFINITION Mus musculus DNA cytosine-5 methyltransferase 3A (Dnmt3a) mRNA complete cds
ACCESSION AF068625 REGION 1200
VERSION AF0686252 GI6449467
KEYWORDS
SOURCE Mus musculus (house mouse)
ORGANISM Mus musculus Eukaryota Metazoa Chordata Craniata Vertebrata Euteleostomi Mammalia Eutheria Euarchontoglires Glires Rodentia Sciurognathi Muroidea Muridae Murinae Mus
REFERENCE1 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REFERENCE2 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REMARK Sequence update by submitter
COMMENT On Nov 18 1999 this sequence version replaced gi3327977
FEATURES LocationQualifiers
source 1200 organism=Mus musculus mol_type=mRNA db_xref=taxon10090 chromosome=12 map=40 cM
gene 1gt200 gene=Dnmt3a
ORIGIN 1 gaattccggc ctgctgccgg gccgcccgac ccgccgggcc acacggcaga gccgcctgaa 61 gcccagcgct gaggctgcac ttttccgagg gcttgacatc agggtctatg tttaagtctt 121 agctcttgct tacaaagacc acggcaattc cttctctgaa gccctcgcag ccccacagcg 181 ccctcgcagc cccagcctgc
GenBank sequence formatItrsquos a rich format for storing sequences and associated annotations It shares a feature
table vocabulary and format with the EMBL and DDJB formats
bull LOCUS CAA89576 109 aa linear PLN 11-AUG-1997 bull DEFINITION CYC1 [Saccharomyces cerevisiae] bull ACCESSION CAA89576 bull VERSION CAA895761 GI1015707 bull DBSOURCE embl locus SCYJR048W accession Z495481 bull KEYWORDS 5-10 or as many as neededbull SOURCE Saccharomyces cerevisiae (bakers yeast) bull ORGANISM Saccharomyces cerevisiae Eukaryota Fungi Ascomycota
Saccharomycotina Saccharomycetes Saccharomycetales Saccharomycetaceae Saccharomyces
bull REFERENCE1 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull REFERENCE2 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull FEATURES LocationQualifiers bull source 1109 organism=Saccharomyces cerevisiae db_xref=taxon4932
chromosome=X Protein 1109 name=CYC1 bull CDS 1109 gene=CYC1 coded_by=Z4954819541283 note=ORF YJR048w
db_xref=GOAP00044 db_xref=SGDS0003809 db_xref=UniProtKBSwiss-ProtP00044 bull ORIGIN 1 mtefkagsak kgatlfktrc lqchtvekgg phkvgpnlhg ifgrhsgqae gysytdanik 61 knvlwdennm
seyltnpkky ipgtkmafgg lkkekdrndl itylkkace
Online Mendelian Inheritance in Man (OMIM) Databasebull OMIM (since 1960s) catalogues all the known diseases with a genetic component
and tries to link them to the relevant genes in human genome
bull In 2004 there were 15000 records
bull One can request to download the mim2genetxt file from OMIM here httpwwwomimorgdownloads
bull The OMIM codebull Every disease and gene is assigned a six digit number of which the first number classifies
the method of inheritancebull If the initial digit is 1 the trait is deemed autosomal dominant if 2 autosomal recessive if 3
X-linked Wherever a trait defined in this dictionary has a MIM number the number from the 12th edition of MIM is given in square brackets with or without an asterisk (asterisks indicate that the mode of inheritance is known a number symbol () before an entry number means that the phenotype can be caused by mutation in any of two or more genes) as appropriate eg Pelizaeus-Merzbacher disease [MIM 312080] is an X-linked recessive disorder
bull For further studies visit httpwwwomimorg
OMIMExample httpwwwomimorgentry189911
189911 TRANSFER RNA GLYCINE 1 TRNAG1
Alternative titles symbols TRANSFER RNA GLYCINE-CCC-1 TRG1
Cytogenetic location Chr16 Genomic coordinates (GRCh37) 160 - 90354753 (from NCBI)
TEXT
Mapping McBride et al (1989) assigned a glycine tRNA(CCC) gene (TRG1) to human chromosome 1 (1pter-p34) on the basis of Southern analysis of a panel of hybrid cell DNAs They also assigned a cloned DNA fragment encompassing a glycine tRNA gene (tRNA-GCC) and pseudogene to human chromosome 16 by the same method
Evolution There are about 1300 tRNA genes in the haploid human genome (Hatlen and Attardi 1971) encoding 60 to 90 tRNA isoacceptors (Lin and Agris 1980) The studies by McBride et al (1989) as well as studies by others (see eg 180620 189930 189920 180640 189880) indicated that tRNA genes and pseudogenes are dispersed on at least 7 human chromosomes and suggested that these sequences would probably be found on most if not all human chromosomes McBride et al (1989) described short 8-12 nucleotide direct terminal repeats flanking many of the dispersed tRNA genes This finding combined with the dispersion of tRNA genes suggests that many of these genes may have arisen by an RNA-mediated retroposition mechanism There may have been selection for reiteration of genes encoding isoaccepting tRNAs since a single mutation in a single-copy tRNA gene could be devastating Moreover even a mutation in the anticodon of a single tRNA gene might not be crucial if competition was provided by the normal wildtype tRNA isoacceptor produced by multiple copies of the normal tRNA gene still present in the genome Dispersion of multiple copies of each tRNA gene could provide diversity of 5-prime-flanking sequences which are known to modulate the expression of some human tRNA genes Tissue-specific or differentiation-specific expression of tRNA isoacceptors might be provided for by this mechanism The recombination and unequal crossingover that can occur with tandem tRNA sequences can result in homogenization of the sequences with disastrous consequences
Nucleotide Databasebull NUCLEOTIDE DATABASESbull NCBIs sequence databases accept genome data from sequencing projects from around
the world and serve as the cornerstone of bioinformatics researchbull GenBankbull An annotated collection of all publicly available nucleotide and amino acid sequencesbull EST databasebull A collection of expressed sequence tags or short single-pass sequence reads from mRNA
(cDNA)bull GSS databasebull A database of genome survey sequences or short single-pass genomic sequencesbull HomoloGenebull A gene homology tool that compares nucleotide sequences between pairs of organisms in order
to identify putative orthologsbull HTG databasebull A collection of high-throughput genome sequences from large-scale genome sequencing
centers including unfinished and finished sequencesbull SNPs databasebull A central repository for both single-base nucleotide substitutions and short deletion and
insertion polymorphisms
Nucleotide Database
bull RefSeqbull A database of non-redundant reference sequences standards including genomic
DNA contigs mRNAs and proteins for known genes Multiple collaborations both within NCBI and with external groups support our data-gathering efforts
bull STS databasebull A database of sequence tagged sites or short sequences that are operationally
unique in the genomebull UniSTSbull A unified non-redundant view of sequence tagged sites (STSs)bull UniGenebull A collection of ESTs and full-length mRNA sequences organized into clusters each
representing a unique known or putative human gene annotated with mapping and expression information and cross-references to other sources
UniGene computationally identifies transcripts from the same locus analyzes expression by tissue age and health status and reports related proteins (protEST) and clone resources
Single Nucleotide Polymorphism (SNP) database
What it isThe SNP Database (also known as dbSNP) is an archive for genetic variation within and across different species developed and hosted by NCBI in collaboration with the National Human Genome Research Institute (NHGRI)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species related to biodiversity genetic variation and adaptation
-The dbSNP accepts apparently neutral polymorphisms polymorphisms corresponding to known phenotypes and regions of no variation
-It was created in September 1998 to supplement GenBank (NCBIrsquos nucleic acid and protein sequences)
Goal
Its goal is to act as a single database that contains all identified genetic variation which can be used to investigate a wide variety of genetically based natural phenomenon Specifically access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping population genetics investigations into evolutionary relationships as well as being able to quickly and easily quantify the amount of variation at a given site of interest
Application
Applied research genetic engineering drug discovery etc
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

Organisms in GeneBank
bull 260000 different speciesbull 1000 new species being added per month
bull Human (Homo sapiens)
11551000 entries with 13149000000 basesbull Mouse (Mus musculus)
7256000 entries with 8361230000 bases
are top two species
GeneBank FormatGenBank format (GenBank Flat File Format) consists of an annotation
section and a sequence section
Annotation sectionThe start of the annotation section is marked by a line beginning with the word LOCUS
The only rule now applied in assigning a locus name is that it must be unique
Sequence sectionThe start of sequence section is marked by a line beginning with the word ORIGIN and the end of the section is marked by a line with only ldquo
GeneBank Flat File FormatLOCUS AF068625 200 bp mRNA linear ROD 06-DEC-1999
DEFINITION Mus musculus DNA cytosine-5 methyltransferase 3A (Dnmt3a) mRNA complete cds
ACCESSION AF068625 REGION 1200
VERSION AF0686252 GI6449467
KEYWORDS
SOURCE Mus musculus (house mouse)
ORGANISM Mus musculus Eukaryota Metazoa Chordata Craniata Vertebrata Euteleostomi Mammalia Eutheria Euarchontoglires Glires Rodentia Sciurognathi Muroidea Muridae Murinae Mus
REFERENCE1 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REFERENCE2 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REMARK Sequence update by submitter
COMMENT On Nov 18 1999 this sequence version replaced gi3327977
FEATURES LocationQualifiers
source 1200 organism=Mus musculus mol_type=mRNA db_xref=taxon10090 chromosome=12 map=40 cM
gene 1gt200 gene=Dnmt3a
ORIGIN 1 gaattccggc ctgctgccgg gccgcccgac ccgccgggcc acacggcaga gccgcctgaa 61 gcccagcgct gaggctgcac ttttccgagg gcttgacatc agggtctatg tttaagtctt 121 agctcttgct tacaaagacc acggcaattc cttctctgaa gccctcgcag ccccacagcg 181 ccctcgcagc cccagcctgc
GenBank sequence formatItrsquos a rich format for storing sequences and associated annotations It shares a feature
table vocabulary and format with the EMBL and DDJB formats
bull LOCUS CAA89576 109 aa linear PLN 11-AUG-1997 bull DEFINITION CYC1 [Saccharomyces cerevisiae] bull ACCESSION CAA89576 bull VERSION CAA895761 GI1015707 bull DBSOURCE embl locus SCYJR048W accession Z495481 bull KEYWORDS 5-10 or as many as neededbull SOURCE Saccharomyces cerevisiae (bakers yeast) bull ORGANISM Saccharomyces cerevisiae Eukaryota Fungi Ascomycota
Saccharomycotina Saccharomycetes Saccharomycetales Saccharomycetaceae Saccharomyces
bull REFERENCE1 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull REFERENCE2 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull FEATURES LocationQualifiers bull source 1109 organism=Saccharomyces cerevisiae db_xref=taxon4932
chromosome=X Protein 1109 name=CYC1 bull CDS 1109 gene=CYC1 coded_by=Z4954819541283 note=ORF YJR048w
db_xref=GOAP00044 db_xref=SGDS0003809 db_xref=UniProtKBSwiss-ProtP00044 bull ORIGIN 1 mtefkagsak kgatlfktrc lqchtvekgg phkvgpnlhg ifgrhsgqae gysytdanik 61 knvlwdennm
seyltnpkky ipgtkmafgg lkkekdrndl itylkkace
Online Mendelian Inheritance in Man (OMIM) Databasebull OMIM (since 1960s) catalogues all the known diseases with a genetic component
and tries to link them to the relevant genes in human genome
bull In 2004 there were 15000 records
bull One can request to download the mim2genetxt file from OMIM here httpwwwomimorgdownloads
bull The OMIM codebull Every disease and gene is assigned a six digit number of which the first number classifies
the method of inheritancebull If the initial digit is 1 the trait is deemed autosomal dominant if 2 autosomal recessive if 3
X-linked Wherever a trait defined in this dictionary has a MIM number the number from the 12th edition of MIM is given in square brackets with or without an asterisk (asterisks indicate that the mode of inheritance is known a number symbol () before an entry number means that the phenotype can be caused by mutation in any of two or more genes) as appropriate eg Pelizaeus-Merzbacher disease [MIM 312080] is an X-linked recessive disorder
bull For further studies visit httpwwwomimorg
OMIMExample httpwwwomimorgentry189911
189911 TRANSFER RNA GLYCINE 1 TRNAG1
Alternative titles symbols TRANSFER RNA GLYCINE-CCC-1 TRG1
Cytogenetic location Chr16 Genomic coordinates (GRCh37) 160 - 90354753 (from NCBI)
TEXT
Mapping McBride et al (1989) assigned a glycine tRNA(CCC) gene (TRG1) to human chromosome 1 (1pter-p34) on the basis of Southern analysis of a panel of hybrid cell DNAs They also assigned a cloned DNA fragment encompassing a glycine tRNA gene (tRNA-GCC) and pseudogene to human chromosome 16 by the same method
Evolution There are about 1300 tRNA genes in the haploid human genome (Hatlen and Attardi 1971) encoding 60 to 90 tRNA isoacceptors (Lin and Agris 1980) The studies by McBride et al (1989) as well as studies by others (see eg 180620 189930 189920 180640 189880) indicated that tRNA genes and pseudogenes are dispersed on at least 7 human chromosomes and suggested that these sequences would probably be found on most if not all human chromosomes McBride et al (1989) described short 8-12 nucleotide direct terminal repeats flanking many of the dispersed tRNA genes This finding combined with the dispersion of tRNA genes suggests that many of these genes may have arisen by an RNA-mediated retroposition mechanism There may have been selection for reiteration of genes encoding isoaccepting tRNAs since a single mutation in a single-copy tRNA gene could be devastating Moreover even a mutation in the anticodon of a single tRNA gene might not be crucial if competition was provided by the normal wildtype tRNA isoacceptor produced by multiple copies of the normal tRNA gene still present in the genome Dispersion of multiple copies of each tRNA gene could provide diversity of 5-prime-flanking sequences which are known to modulate the expression of some human tRNA genes Tissue-specific or differentiation-specific expression of tRNA isoacceptors might be provided for by this mechanism The recombination and unequal crossingover that can occur with tandem tRNA sequences can result in homogenization of the sequences with disastrous consequences
Nucleotide Databasebull NUCLEOTIDE DATABASESbull NCBIs sequence databases accept genome data from sequencing projects from around
the world and serve as the cornerstone of bioinformatics researchbull GenBankbull An annotated collection of all publicly available nucleotide and amino acid sequencesbull EST databasebull A collection of expressed sequence tags or short single-pass sequence reads from mRNA
(cDNA)bull GSS databasebull A database of genome survey sequences or short single-pass genomic sequencesbull HomoloGenebull A gene homology tool that compares nucleotide sequences between pairs of organisms in order
to identify putative orthologsbull HTG databasebull A collection of high-throughput genome sequences from large-scale genome sequencing
centers including unfinished and finished sequencesbull SNPs databasebull A central repository for both single-base nucleotide substitutions and short deletion and
insertion polymorphisms
Nucleotide Database
bull RefSeqbull A database of non-redundant reference sequences standards including genomic
DNA contigs mRNAs and proteins for known genes Multiple collaborations both within NCBI and with external groups support our data-gathering efforts
bull STS databasebull A database of sequence tagged sites or short sequences that are operationally
unique in the genomebull UniSTSbull A unified non-redundant view of sequence tagged sites (STSs)bull UniGenebull A collection of ESTs and full-length mRNA sequences organized into clusters each
representing a unique known or putative human gene annotated with mapping and expression information and cross-references to other sources
UniGene computationally identifies transcripts from the same locus analyzes expression by tissue age and health status and reports related proteins (protEST) and clone resources
Single Nucleotide Polymorphism (SNP) database
What it isThe SNP Database (also known as dbSNP) is an archive for genetic variation within and across different species developed and hosted by NCBI in collaboration with the National Human Genome Research Institute (NHGRI)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species related to biodiversity genetic variation and adaptation
-The dbSNP accepts apparently neutral polymorphisms polymorphisms corresponding to known phenotypes and regions of no variation
-It was created in September 1998 to supplement GenBank (NCBIrsquos nucleic acid and protein sequences)
Goal
Its goal is to act as a single database that contains all identified genetic variation which can be used to investigate a wide variety of genetically based natural phenomenon Specifically access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping population genetics investigations into evolutionary relationships as well as being able to quickly and easily quantify the amount of variation at a given site of interest
Application
Applied research genetic engineering drug discovery etc
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

GeneBank FormatGenBank format (GenBank Flat File Format) consists of an annotation
section and a sequence section
Annotation sectionThe start of the annotation section is marked by a line beginning with the word LOCUS
The only rule now applied in assigning a locus name is that it must be unique
Sequence sectionThe start of sequence section is marked by a line beginning with the word ORIGIN and the end of the section is marked by a line with only ldquo
GeneBank Flat File FormatLOCUS AF068625 200 bp mRNA linear ROD 06-DEC-1999
DEFINITION Mus musculus DNA cytosine-5 methyltransferase 3A (Dnmt3a) mRNA complete cds
ACCESSION AF068625 REGION 1200
VERSION AF0686252 GI6449467
KEYWORDS
SOURCE Mus musculus (house mouse)
ORGANISM Mus musculus Eukaryota Metazoa Chordata Craniata Vertebrata Euteleostomi Mammalia Eutheria Euarchontoglires Glires Rodentia Sciurognathi Muroidea Muridae Murinae Mus
REFERENCE1 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REFERENCE2 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REMARK Sequence update by submitter
COMMENT On Nov 18 1999 this sequence version replaced gi3327977
FEATURES LocationQualifiers
source 1200 organism=Mus musculus mol_type=mRNA db_xref=taxon10090 chromosome=12 map=40 cM
gene 1gt200 gene=Dnmt3a
ORIGIN 1 gaattccggc ctgctgccgg gccgcccgac ccgccgggcc acacggcaga gccgcctgaa 61 gcccagcgct gaggctgcac ttttccgagg gcttgacatc agggtctatg tttaagtctt 121 agctcttgct tacaaagacc acggcaattc cttctctgaa gccctcgcag ccccacagcg 181 ccctcgcagc cccagcctgc
GenBank sequence formatItrsquos a rich format for storing sequences and associated annotations It shares a feature
table vocabulary and format with the EMBL and DDJB formats
bull LOCUS CAA89576 109 aa linear PLN 11-AUG-1997 bull DEFINITION CYC1 [Saccharomyces cerevisiae] bull ACCESSION CAA89576 bull VERSION CAA895761 GI1015707 bull DBSOURCE embl locus SCYJR048W accession Z495481 bull KEYWORDS 5-10 or as many as neededbull SOURCE Saccharomyces cerevisiae (bakers yeast) bull ORGANISM Saccharomyces cerevisiae Eukaryota Fungi Ascomycota
Saccharomycotina Saccharomycetes Saccharomycetales Saccharomycetaceae Saccharomyces
bull REFERENCE1 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull REFERENCE2 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull FEATURES LocationQualifiers bull source 1109 organism=Saccharomyces cerevisiae db_xref=taxon4932
chromosome=X Protein 1109 name=CYC1 bull CDS 1109 gene=CYC1 coded_by=Z4954819541283 note=ORF YJR048w
db_xref=GOAP00044 db_xref=SGDS0003809 db_xref=UniProtKBSwiss-ProtP00044 bull ORIGIN 1 mtefkagsak kgatlfktrc lqchtvekgg phkvgpnlhg ifgrhsgqae gysytdanik 61 knvlwdennm
seyltnpkky ipgtkmafgg lkkekdrndl itylkkace
Online Mendelian Inheritance in Man (OMIM) Databasebull OMIM (since 1960s) catalogues all the known diseases with a genetic component
and tries to link them to the relevant genes in human genome
bull In 2004 there were 15000 records
bull One can request to download the mim2genetxt file from OMIM here httpwwwomimorgdownloads
bull The OMIM codebull Every disease and gene is assigned a six digit number of which the first number classifies
the method of inheritancebull If the initial digit is 1 the trait is deemed autosomal dominant if 2 autosomal recessive if 3
X-linked Wherever a trait defined in this dictionary has a MIM number the number from the 12th edition of MIM is given in square brackets with or without an asterisk (asterisks indicate that the mode of inheritance is known a number symbol () before an entry number means that the phenotype can be caused by mutation in any of two or more genes) as appropriate eg Pelizaeus-Merzbacher disease [MIM 312080] is an X-linked recessive disorder
bull For further studies visit httpwwwomimorg
OMIMExample httpwwwomimorgentry189911
189911 TRANSFER RNA GLYCINE 1 TRNAG1
Alternative titles symbols TRANSFER RNA GLYCINE-CCC-1 TRG1
Cytogenetic location Chr16 Genomic coordinates (GRCh37) 160 - 90354753 (from NCBI)
TEXT
Mapping McBride et al (1989) assigned a glycine tRNA(CCC) gene (TRG1) to human chromosome 1 (1pter-p34) on the basis of Southern analysis of a panel of hybrid cell DNAs They also assigned a cloned DNA fragment encompassing a glycine tRNA gene (tRNA-GCC) and pseudogene to human chromosome 16 by the same method
Evolution There are about 1300 tRNA genes in the haploid human genome (Hatlen and Attardi 1971) encoding 60 to 90 tRNA isoacceptors (Lin and Agris 1980) The studies by McBride et al (1989) as well as studies by others (see eg 180620 189930 189920 180640 189880) indicated that tRNA genes and pseudogenes are dispersed on at least 7 human chromosomes and suggested that these sequences would probably be found on most if not all human chromosomes McBride et al (1989) described short 8-12 nucleotide direct terminal repeats flanking many of the dispersed tRNA genes This finding combined with the dispersion of tRNA genes suggests that many of these genes may have arisen by an RNA-mediated retroposition mechanism There may have been selection for reiteration of genes encoding isoaccepting tRNAs since a single mutation in a single-copy tRNA gene could be devastating Moreover even a mutation in the anticodon of a single tRNA gene might not be crucial if competition was provided by the normal wildtype tRNA isoacceptor produced by multiple copies of the normal tRNA gene still present in the genome Dispersion of multiple copies of each tRNA gene could provide diversity of 5-prime-flanking sequences which are known to modulate the expression of some human tRNA genes Tissue-specific or differentiation-specific expression of tRNA isoacceptors might be provided for by this mechanism The recombination and unequal crossingover that can occur with tandem tRNA sequences can result in homogenization of the sequences with disastrous consequences
Nucleotide Databasebull NUCLEOTIDE DATABASESbull NCBIs sequence databases accept genome data from sequencing projects from around
the world and serve as the cornerstone of bioinformatics researchbull GenBankbull An annotated collection of all publicly available nucleotide and amino acid sequencesbull EST databasebull A collection of expressed sequence tags or short single-pass sequence reads from mRNA
(cDNA)bull GSS databasebull A database of genome survey sequences or short single-pass genomic sequencesbull HomoloGenebull A gene homology tool that compares nucleotide sequences between pairs of organisms in order
to identify putative orthologsbull HTG databasebull A collection of high-throughput genome sequences from large-scale genome sequencing
centers including unfinished and finished sequencesbull SNPs databasebull A central repository for both single-base nucleotide substitutions and short deletion and
insertion polymorphisms
Nucleotide Database
bull RefSeqbull A database of non-redundant reference sequences standards including genomic
DNA contigs mRNAs and proteins for known genes Multiple collaborations both within NCBI and with external groups support our data-gathering efforts
bull STS databasebull A database of sequence tagged sites or short sequences that are operationally
unique in the genomebull UniSTSbull A unified non-redundant view of sequence tagged sites (STSs)bull UniGenebull A collection of ESTs and full-length mRNA sequences organized into clusters each
representing a unique known or putative human gene annotated with mapping and expression information and cross-references to other sources
UniGene computationally identifies transcripts from the same locus analyzes expression by tissue age and health status and reports related proteins (protEST) and clone resources
Single Nucleotide Polymorphism (SNP) database
What it isThe SNP Database (also known as dbSNP) is an archive for genetic variation within and across different species developed and hosted by NCBI in collaboration with the National Human Genome Research Institute (NHGRI)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species related to biodiversity genetic variation and adaptation
-The dbSNP accepts apparently neutral polymorphisms polymorphisms corresponding to known phenotypes and regions of no variation
-It was created in September 1998 to supplement GenBank (NCBIrsquos nucleic acid and protein sequences)
Goal
Its goal is to act as a single database that contains all identified genetic variation which can be used to investigate a wide variety of genetically based natural phenomenon Specifically access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping population genetics investigations into evolutionary relationships as well as being able to quickly and easily quantify the amount of variation at a given site of interest
Application
Applied research genetic engineering drug discovery etc
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-
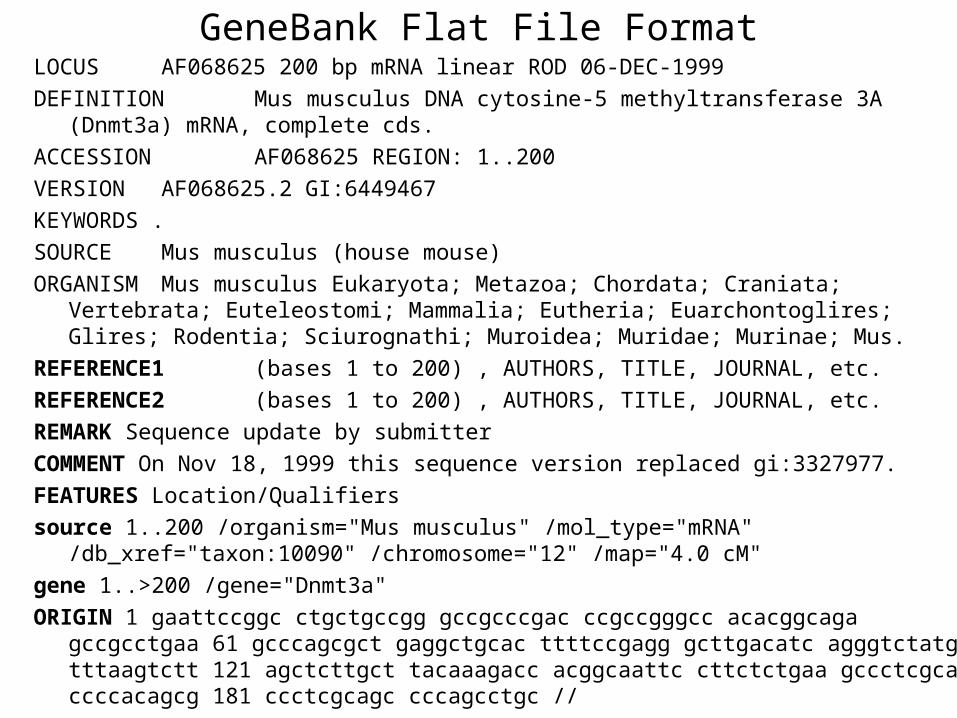
GeneBank Flat File FormatLOCUS AF068625 200 bp mRNA linear ROD 06-DEC-1999
DEFINITION Mus musculus DNA cytosine-5 methyltransferase 3A (Dnmt3a) mRNA complete cds
ACCESSION AF068625 REGION 1200
VERSION AF0686252 GI6449467
KEYWORDS
SOURCE Mus musculus (house mouse)
ORGANISM Mus musculus Eukaryota Metazoa Chordata Craniata Vertebrata Euteleostomi Mammalia Eutheria Euarchontoglires Glires Rodentia Sciurognathi Muroidea Muridae Murinae Mus
REFERENCE1 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REFERENCE2 (bases 1 to 200) AUTHORS TITLE JOURNAL etc
REMARK Sequence update by submitter
COMMENT On Nov 18 1999 this sequence version replaced gi3327977
FEATURES LocationQualifiers
source 1200 organism=Mus musculus mol_type=mRNA db_xref=taxon10090 chromosome=12 map=40 cM
gene 1gt200 gene=Dnmt3a
ORIGIN 1 gaattccggc ctgctgccgg gccgcccgac ccgccgggcc acacggcaga gccgcctgaa 61 gcccagcgct gaggctgcac ttttccgagg gcttgacatc agggtctatg tttaagtctt 121 agctcttgct tacaaagacc acggcaattc cttctctgaa gccctcgcag ccccacagcg 181 ccctcgcagc cccagcctgc
GenBank sequence formatItrsquos a rich format for storing sequences and associated annotations It shares a feature
table vocabulary and format with the EMBL and DDJB formats
bull LOCUS CAA89576 109 aa linear PLN 11-AUG-1997 bull DEFINITION CYC1 [Saccharomyces cerevisiae] bull ACCESSION CAA89576 bull VERSION CAA895761 GI1015707 bull DBSOURCE embl locus SCYJR048W accession Z495481 bull KEYWORDS 5-10 or as many as neededbull SOURCE Saccharomyces cerevisiae (bakers yeast) bull ORGANISM Saccharomyces cerevisiae Eukaryota Fungi Ascomycota
Saccharomycotina Saccharomycetes Saccharomycetales Saccharomycetaceae Saccharomyces
bull REFERENCE1 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull REFERENCE2 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull FEATURES LocationQualifiers bull source 1109 organism=Saccharomyces cerevisiae db_xref=taxon4932
chromosome=X Protein 1109 name=CYC1 bull CDS 1109 gene=CYC1 coded_by=Z4954819541283 note=ORF YJR048w
db_xref=GOAP00044 db_xref=SGDS0003809 db_xref=UniProtKBSwiss-ProtP00044 bull ORIGIN 1 mtefkagsak kgatlfktrc lqchtvekgg phkvgpnlhg ifgrhsgqae gysytdanik 61 knvlwdennm
seyltnpkky ipgtkmafgg lkkekdrndl itylkkace
Online Mendelian Inheritance in Man (OMIM) Databasebull OMIM (since 1960s) catalogues all the known diseases with a genetic component
and tries to link them to the relevant genes in human genome
bull In 2004 there were 15000 records
bull One can request to download the mim2genetxt file from OMIM here httpwwwomimorgdownloads
bull The OMIM codebull Every disease and gene is assigned a six digit number of which the first number classifies
the method of inheritancebull If the initial digit is 1 the trait is deemed autosomal dominant if 2 autosomal recessive if 3
X-linked Wherever a trait defined in this dictionary has a MIM number the number from the 12th edition of MIM is given in square brackets with or without an asterisk (asterisks indicate that the mode of inheritance is known a number symbol () before an entry number means that the phenotype can be caused by mutation in any of two or more genes) as appropriate eg Pelizaeus-Merzbacher disease [MIM 312080] is an X-linked recessive disorder
bull For further studies visit httpwwwomimorg
OMIMExample httpwwwomimorgentry189911
189911 TRANSFER RNA GLYCINE 1 TRNAG1
Alternative titles symbols TRANSFER RNA GLYCINE-CCC-1 TRG1
Cytogenetic location Chr16 Genomic coordinates (GRCh37) 160 - 90354753 (from NCBI)
TEXT
Mapping McBride et al (1989) assigned a glycine tRNA(CCC) gene (TRG1) to human chromosome 1 (1pter-p34) on the basis of Southern analysis of a panel of hybrid cell DNAs They also assigned a cloned DNA fragment encompassing a glycine tRNA gene (tRNA-GCC) and pseudogene to human chromosome 16 by the same method
Evolution There are about 1300 tRNA genes in the haploid human genome (Hatlen and Attardi 1971) encoding 60 to 90 tRNA isoacceptors (Lin and Agris 1980) The studies by McBride et al (1989) as well as studies by others (see eg 180620 189930 189920 180640 189880) indicated that tRNA genes and pseudogenes are dispersed on at least 7 human chromosomes and suggested that these sequences would probably be found on most if not all human chromosomes McBride et al (1989) described short 8-12 nucleotide direct terminal repeats flanking many of the dispersed tRNA genes This finding combined with the dispersion of tRNA genes suggests that many of these genes may have arisen by an RNA-mediated retroposition mechanism There may have been selection for reiteration of genes encoding isoaccepting tRNAs since a single mutation in a single-copy tRNA gene could be devastating Moreover even a mutation in the anticodon of a single tRNA gene might not be crucial if competition was provided by the normal wildtype tRNA isoacceptor produced by multiple copies of the normal tRNA gene still present in the genome Dispersion of multiple copies of each tRNA gene could provide diversity of 5-prime-flanking sequences which are known to modulate the expression of some human tRNA genes Tissue-specific or differentiation-specific expression of tRNA isoacceptors might be provided for by this mechanism The recombination and unequal crossingover that can occur with tandem tRNA sequences can result in homogenization of the sequences with disastrous consequences
Nucleotide Databasebull NUCLEOTIDE DATABASESbull NCBIs sequence databases accept genome data from sequencing projects from around
the world and serve as the cornerstone of bioinformatics researchbull GenBankbull An annotated collection of all publicly available nucleotide and amino acid sequencesbull EST databasebull A collection of expressed sequence tags or short single-pass sequence reads from mRNA
(cDNA)bull GSS databasebull A database of genome survey sequences or short single-pass genomic sequencesbull HomoloGenebull A gene homology tool that compares nucleotide sequences between pairs of organisms in order
to identify putative orthologsbull HTG databasebull A collection of high-throughput genome sequences from large-scale genome sequencing
centers including unfinished and finished sequencesbull SNPs databasebull A central repository for both single-base nucleotide substitutions and short deletion and
insertion polymorphisms
Nucleotide Database
bull RefSeqbull A database of non-redundant reference sequences standards including genomic
DNA contigs mRNAs and proteins for known genes Multiple collaborations both within NCBI and with external groups support our data-gathering efforts
bull STS databasebull A database of sequence tagged sites or short sequences that are operationally
unique in the genomebull UniSTSbull A unified non-redundant view of sequence tagged sites (STSs)bull UniGenebull A collection of ESTs and full-length mRNA sequences organized into clusters each
representing a unique known or putative human gene annotated with mapping and expression information and cross-references to other sources
UniGene computationally identifies transcripts from the same locus analyzes expression by tissue age and health status and reports related proteins (protEST) and clone resources
Single Nucleotide Polymorphism (SNP) database
What it isThe SNP Database (also known as dbSNP) is an archive for genetic variation within and across different species developed and hosted by NCBI in collaboration with the National Human Genome Research Institute (NHGRI)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species related to biodiversity genetic variation and adaptation
-The dbSNP accepts apparently neutral polymorphisms polymorphisms corresponding to known phenotypes and regions of no variation
-It was created in September 1998 to supplement GenBank (NCBIrsquos nucleic acid and protein sequences)
Goal
Its goal is to act as a single database that contains all identified genetic variation which can be used to investigate a wide variety of genetically based natural phenomenon Specifically access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping population genetics investigations into evolutionary relationships as well as being able to quickly and easily quantify the amount of variation at a given site of interest
Application
Applied research genetic engineering drug discovery etc
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

GenBank sequence formatItrsquos a rich format for storing sequences and associated annotations It shares a feature
table vocabulary and format with the EMBL and DDJB formats
bull LOCUS CAA89576 109 aa linear PLN 11-AUG-1997 bull DEFINITION CYC1 [Saccharomyces cerevisiae] bull ACCESSION CAA89576 bull VERSION CAA895761 GI1015707 bull DBSOURCE embl locus SCYJR048W accession Z495481 bull KEYWORDS 5-10 or as many as neededbull SOURCE Saccharomyces cerevisiae (bakers yeast) bull ORGANISM Saccharomyces cerevisiae Eukaryota Fungi Ascomycota
Saccharomycotina Saccharomycetes Saccharomycetales Saccharomycetaceae Saccharomyces
bull REFERENCE1 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull REFERENCE2 (residues 1 to 109) AUTHORS TITLE JOURNAL etcbull FEATURES LocationQualifiers bull source 1109 organism=Saccharomyces cerevisiae db_xref=taxon4932
chromosome=X Protein 1109 name=CYC1 bull CDS 1109 gene=CYC1 coded_by=Z4954819541283 note=ORF YJR048w
db_xref=GOAP00044 db_xref=SGDS0003809 db_xref=UniProtKBSwiss-ProtP00044 bull ORIGIN 1 mtefkagsak kgatlfktrc lqchtvekgg phkvgpnlhg ifgrhsgqae gysytdanik 61 knvlwdennm
seyltnpkky ipgtkmafgg lkkekdrndl itylkkace
Online Mendelian Inheritance in Man (OMIM) Databasebull OMIM (since 1960s) catalogues all the known diseases with a genetic component
and tries to link them to the relevant genes in human genome
bull In 2004 there were 15000 records
bull One can request to download the mim2genetxt file from OMIM here httpwwwomimorgdownloads
bull The OMIM codebull Every disease and gene is assigned a six digit number of which the first number classifies
the method of inheritancebull If the initial digit is 1 the trait is deemed autosomal dominant if 2 autosomal recessive if 3
X-linked Wherever a trait defined in this dictionary has a MIM number the number from the 12th edition of MIM is given in square brackets with or without an asterisk (asterisks indicate that the mode of inheritance is known a number symbol () before an entry number means that the phenotype can be caused by mutation in any of two or more genes) as appropriate eg Pelizaeus-Merzbacher disease [MIM 312080] is an X-linked recessive disorder
bull For further studies visit httpwwwomimorg
OMIMExample httpwwwomimorgentry189911
189911 TRANSFER RNA GLYCINE 1 TRNAG1
Alternative titles symbols TRANSFER RNA GLYCINE-CCC-1 TRG1
Cytogenetic location Chr16 Genomic coordinates (GRCh37) 160 - 90354753 (from NCBI)
TEXT
Mapping McBride et al (1989) assigned a glycine tRNA(CCC) gene (TRG1) to human chromosome 1 (1pter-p34) on the basis of Southern analysis of a panel of hybrid cell DNAs They also assigned a cloned DNA fragment encompassing a glycine tRNA gene (tRNA-GCC) and pseudogene to human chromosome 16 by the same method
Evolution There are about 1300 tRNA genes in the haploid human genome (Hatlen and Attardi 1971) encoding 60 to 90 tRNA isoacceptors (Lin and Agris 1980) The studies by McBride et al (1989) as well as studies by others (see eg 180620 189930 189920 180640 189880) indicated that tRNA genes and pseudogenes are dispersed on at least 7 human chromosomes and suggested that these sequences would probably be found on most if not all human chromosomes McBride et al (1989) described short 8-12 nucleotide direct terminal repeats flanking many of the dispersed tRNA genes This finding combined with the dispersion of tRNA genes suggests that many of these genes may have arisen by an RNA-mediated retroposition mechanism There may have been selection for reiteration of genes encoding isoaccepting tRNAs since a single mutation in a single-copy tRNA gene could be devastating Moreover even a mutation in the anticodon of a single tRNA gene might not be crucial if competition was provided by the normal wildtype tRNA isoacceptor produced by multiple copies of the normal tRNA gene still present in the genome Dispersion of multiple copies of each tRNA gene could provide diversity of 5-prime-flanking sequences which are known to modulate the expression of some human tRNA genes Tissue-specific or differentiation-specific expression of tRNA isoacceptors might be provided for by this mechanism The recombination and unequal crossingover that can occur with tandem tRNA sequences can result in homogenization of the sequences with disastrous consequences
Nucleotide Databasebull NUCLEOTIDE DATABASESbull NCBIs sequence databases accept genome data from sequencing projects from around
the world and serve as the cornerstone of bioinformatics researchbull GenBankbull An annotated collection of all publicly available nucleotide and amino acid sequencesbull EST databasebull A collection of expressed sequence tags or short single-pass sequence reads from mRNA
(cDNA)bull GSS databasebull A database of genome survey sequences or short single-pass genomic sequencesbull HomoloGenebull A gene homology tool that compares nucleotide sequences between pairs of organisms in order
to identify putative orthologsbull HTG databasebull A collection of high-throughput genome sequences from large-scale genome sequencing
centers including unfinished and finished sequencesbull SNPs databasebull A central repository for both single-base nucleotide substitutions and short deletion and
insertion polymorphisms
Nucleotide Database
bull RefSeqbull A database of non-redundant reference sequences standards including genomic
DNA contigs mRNAs and proteins for known genes Multiple collaborations both within NCBI and with external groups support our data-gathering efforts
bull STS databasebull A database of sequence tagged sites or short sequences that are operationally
unique in the genomebull UniSTSbull A unified non-redundant view of sequence tagged sites (STSs)bull UniGenebull A collection of ESTs and full-length mRNA sequences organized into clusters each
representing a unique known or putative human gene annotated with mapping and expression information and cross-references to other sources
UniGene computationally identifies transcripts from the same locus analyzes expression by tissue age and health status and reports related proteins (protEST) and clone resources
Single Nucleotide Polymorphism (SNP) database
What it isThe SNP Database (also known as dbSNP) is an archive for genetic variation within and across different species developed and hosted by NCBI in collaboration with the National Human Genome Research Institute (NHGRI)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species related to biodiversity genetic variation and adaptation
-The dbSNP accepts apparently neutral polymorphisms polymorphisms corresponding to known phenotypes and regions of no variation
-It was created in September 1998 to supplement GenBank (NCBIrsquos nucleic acid and protein sequences)
Goal
Its goal is to act as a single database that contains all identified genetic variation which can be used to investigate a wide variety of genetically based natural phenomenon Specifically access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping population genetics investigations into evolutionary relationships as well as being able to quickly and easily quantify the amount of variation at a given site of interest
Application
Applied research genetic engineering drug discovery etc
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

Online Mendelian Inheritance in Man (OMIM) Databasebull OMIM (since 1960s) catalogues all the known diseases with a genetic component
and tries to link them to the relevant genes in human genome
bull In 2004 there were 15000 records
bull One can request to download the mim2genetxt file from OMIM here httpwwwomimorgdownloads
bull The OMIM codebull Every disease and gene is assigned a six digit number of which the first number classifies
the method of inheritancebull If the initial digit is 1 the trait is deemed autosomal dominant if 2 autosomal recessive if 3
X-linked Wherever a trait defined in this dictionary has a MIM number the number from the 12th edition of MIM is given in square brackets with or without an asterisk (asterisks indicate that the mode of inheritance is known a number symbol () before an entry number means that the phenotype can be caused by mutation in any of two or more genes) as appropriate eg Pelizaeus-Merzbacher disease [MIM 312080] is an X-linked recessive disorder
bull For further studies visit httpwwwomimorg
OMIMExample httpwwwomimorgentry189911
189911 TRANSFER RNA GLYCINE 1 TRNAG1
Alternative titles symbols TRANSFER RNA GLYCINE-CCC-1 TRG1
Cytogenetic location Chr16 Genomic coordinates (GRCh37) 160 - 90354753 (from NCBI)
TEXT
Mapping McBride et al (1989) assigned a glycine tRNA(CCC) gene (TRG1) to human chromosome 1 (1pter-p34) on the basis of Southern analysis of a panel of hybrid cell DNAs They also assigned a cloned DNA fragment encompassing a glycine tRNA gene (tRNA-GCC) and pseudogene to human chromosome 16 by the same method
Evolution There are about 1300 tRNA genes in the haploid human genome (Hatlen and Attardi 1971) encoding 60 to 90 tRNA isoacceptors (Lin and Agris 1980) The studies by McBride et al (1989) as well as studies by others (see eg 180620 189930 189920 180640 189880) indicated that tRNA genes and pseudogenes are dispersed on at least 7 human chromosomes and suggested that these sequences would probably be found on most if not all human chromosomes McBride et al (1989) described short 8-12 nucleotide direct terminal repeats flanking many of the dispersed tRNA genes This finding combined with the dispersion of tRNA genes suggests that many of these genes may have arisen by an RNA-mediated retroposition mechanism There may have been selection for reiteration of genes encoding isoaccepting tRNAs since a single mutation in a single-copy tRNA gene could be devastating Moreover even a mutation in the anticodon of a single tRNA gene might not be crucial if competition was provided by the normal wildtype tRNA isoacceptor produced by multiple copies of the normal tRNA gene still present in the genome Dispersion of multiple copies of each tRNA gene could provide diversity of 5-prime-flanking sequences which are known to modulate the expression of some human tRNA genes Tissue-specific or differentiation-specific expression of tRNA isoacceptors might be provided for by this mechanism The recombination and unequal crossingover that can occur with tandem tRNA sequences can result in homogenization of the sequences with disastrous consequences
Nucleotide Databasebull NUCLEOTIDE DATABASESbull NCBIs sequence databases accept genome data from sequencing projects from around
the world and serve as the cornerstone of bioinformatics researchbull GenBankbull An annotated collection of all publicly available nucleotide and amino acid sequencesbull EST databasebull A collection of expressed sequence tags or short single-pass sequence reads from mRNA
(cDNA)bull GSS databasebull A database of genome survey sequences or short single-pass genomic sequencesbull HomoloGenebull A gene homology tool that compares nucleotide sequences between pairs of organisms in order
to identify putative orthologsbull HTG databasebull A collection of high-throughput genome sequences from large-scale genome sequencing
centers including unfinished and finished sequencesbull SNPs databasebull A central repository for both single-base nucleotide substitutions and short deletion and
insertion polymorphisms
Nucleotide Database
bull RefSeqbull A database of non-redundant reference sequences standards including genomic
DNA contigs mRNAs and proteins for known genes Multiple collaborations both within NCBI and with external groups support our data-gathering efforts
bull STS databasebull A database of sequence tagged sites or short sequences that are operationally
unique in the genomebull UniSTSbull A unified non-redundant view of sequence tagged sites (STSs)bull UniGenebull A collection of ESTs and full-length mRNA sequences organized into clusters each
representing a unique known or putative human gene annotated with mapping and expression information and cross-references to other sources
UniGene computationally identifies transcripts from the same locus analyzes expression by tissue age and health status and reports related proteins (protEST) and clone resources
Single Nucleotide Polymorphism (SNP) database
What it isThe SNP Database (also known as dbSNP) is an archive for genetic variation within and across different species developed and hosted by NCBI in collaboration with the National Human Genome Research Institute (NHGRI)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species related to biodiversity genetic variation and adaptation
-The dbSNP accepts apparently neutral polymorphisms polymorphisms corresponding to known phenotypes and regions of no variation
-It was created in September 1998 to supplement GenBank (NCBIrsquos nucleic acid and protein sequences)
Goal
Its goal is to act as a single database that contains all identified genetic variation which can be used to investigate a wide variety of genetically based natural phenomenon Specifically access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping population genetics investigations into evolutionary relationships as well as being able to quickly and easily quantify the amount of variation at a given site of interest
Application
Applied research genetic engineering drug discovery etc
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

OMIMExample httpwwwomimorgentry189911
189911 TRANSFER RNA GLYCINE 1 TRNAG1
Alternative titles symbols TRANSFER RNA GLYCINE-CCC-1 TRG1
Cytogenetic location Chr16 Genomic coordinates (GRCh37) 160 - 90354753 (from NCBI)
TEXT
Mapping McBride et al (1989) assigned a glycine tRNA(CCC) gene (TRG1) to human chromosome 1 (1pter-p34) on the basis of Southern analysis of a panel of hybrid cell DNAs They also assigned a cloned DNA fragment encompassing a glycine tRNA gene (tRNA-GCC) and pseudogene to human chromosome 16 by the same method
Evolution There are about 1300 tRNA genes in the haploid human genome (Hatlen and Attardi 1971) encoding 60 to 90 tRNA isoacceptors (Lin and Agris 1980) The studies by McBride et al (1989) as well as studies by others (see eg 180620 189930 189920 180640 189880) indicated that tRNA genes and pseudogenes are dispersed on at least 7 human chromosomes and suggested that these sequences would probably be found on most if not all human chromosomes McBride et al (1989) described short 8-12 nucleotide direct terminal repeats flanking many of the dispersed tRNA genes This finding combined with the dispersion of tRNA genes suggests that many of these genes may have arisen by an RNA-mediated retroposition mechanism There may have been selection for reiteration of genes encoding isoaccepting tRNAs since a single mutation in a single-copy tRNA gene could be devastating Moreover even a mutation in the anticodon of a single tRNA gene might not be crucial if competition was provided by the normal wildtype tRNA isoacceptor produced by multiple copies of the normal tRNA gene still present in the genome Dispersion of multiple copies of each tRNA gene could provide diversity of 5-prime-flanking sequences which are known to modulate the expression of some human tRNA genes Tissue-specific or differentiation-specific expression of tRNA isoacceptors might be provided for by this mechanism The recombination and unequal crossingover that can occur with tandem tRNA sequences can result in homogenization of the sequences with disastrous consequences
Nucleotide Databasebull NUCLEOTIDE DATABASESbull NCBIs sequence databases accept genome data from sequencing projects from around
the world and serve as the cornerstone of bioinformatics researchbull GenBankbull An annotated collection of all publicly available nucleotide and amino acid sequencesbull EST databasebull A collection of expressed sequence tags or short single-pass sequence reads from mRNA
(cDNA)bull GSS databasebull A database of genome survey sequences or short single-pass genomic sequencesbull HomoloGenebull A gene homology tool that compares nucleotide sequences between pairs of organisms in order
to identify putative orthologsbull HTG databasebull A collection of high-throughput genome sequences from large-scale genome sequencing
centers including unfinished and finished sequencesbull SNPs databasebull A central repository for both single-base nucleotide substitutions and short deletion and
insertion polymorphisms
Nucleotide Database
bull RefSeqbull A database of non-redundant reference sequences standards including genomic
DNA contigs mRNAs and proteins for known genes Multiple collaborations both within NCBI and with external groups support our data-gathering efforts
bull STS databasebull A database of sequence tagged sites or short sequences that are operationally
unique in the genomebull UniSTSbull A unified non-redundant view of sequence tagged sites (STSs)bull UniGenebull A collection of ESTs and full-length mRNA sequences organized into clusters each
representing a unique known or putative human gene annotated with mapping and expression information and cross-references to other sources
UniGene computationally identifies transcripts from the same locus analyzes expression by tissue age and health status and reports related proteins (protEST) and clone resources
Single Nucleotide Polymorphism (SNP) database
What it isThe SNP Database (also known as dbSNP) is an archive for genetic variation within and across different species developed and hosted by NCBI in collaboration with the National Human Genome Research Institute (NHGRI)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species related to biodiversity genetic variation and adaptation
-The dbSNP accepts apparently neutral polymorphisms polymorphisms corresponding to known phenotypes and regions of no variation
-It was created in September 1998 to supplement GenBank (NCBIrsquos nucleic acid and protein sequences)
Goal
Its goal is to act as a single database that contains all identified genetic variation which can be used to investigate a wide variety of genetically based natural phenomenon Specifically access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping population genetics investigations into evolutionary relationships as well as being able to quickly and easily quantify the amount of variation at a given site of interest
Application
Applied research genetic engineering drug discovery etc
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

Nucleotide Databasebull NUCLEOTIDE DATABASESbull NCBIs sequence databases accept genome data from sequencing projects from around
the world and serve as the cornerstone of bioinformatics researchbull GenBankbull An annotated collection of all publicly available nucleotide and amino acid sequencesbull EST databasebull A collection of expressed sequence tags or short single-pass sequence reads from mRNA
(cDNA)bull GSS databasebull A database of genome survey sequences or short single-pass genomic sequencesbull HomoloGenebull A gene homology tool that compares nucleotide sequences between pairs of organisms in order
to identify putative orthologsbull HTG databasebull A collection of high-throughput genome sequences from large-scale genome sequencing
centers including unfinished and finished sequencesbull SNPs databasebull A central repository for both single-base nucleotide substitutions and short deletion and
insertion polymorphisms
Nucleotide Database
bull RefSeqbull A database of non-redundant reference sequences standards including genomic
DNA contigs mRNAs and proteins for known genes Multiple collaborations both within NCBI and with external groups support our data-gathering efforts
bull STS databasebull A database of sequence tagged sites or short sequences that are operationally
unique in the genomebull UniSTSbull A unified non-redundant view of sequence tagged sites (STSs)bull UniGenebull A collection of ESTs and full-length mRNA sequences organized into clusters each
representing a unique known or putative human gene annotated with mapping and expression information and cross-references to other sources
UniGene computationally identifies transcripts from the same locus analyzes expression by tissue age and health status and reports related proteins (protEST) and clone resources
Single Nucleotide Polymorphism (SNP) database
What it isThe SNP Database (also known as dbSNP) is an archive for genetic variation within and across different species developed and hosted by NCBI in collaboration with the National Human Genome Research Institute (NHGRI)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species related to biodiversity genetic variation and adaptation
-The dbSNP accepts apparently neutral polymorphisms polymorphisms corresponding to known phenotypes and regions of no variation
-It was created in September 1998 to supplement GenBank (NCBIrsquos nucleic acid and protein sequences)
Goal
Its goal is to act as a single database that contains all identified genetic variation which can be used to investigate a wide variety of genetically based natural phenomenon Specifically access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping population genetics investigations into evolutionary relationships as well as being able to quickly and easily quantify the amount of variation at a given site of interest
Application
Applied research genetic engineering drug discovery etc
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

Nucleotide Database
bull RefSeqbull A database of non-redundant reference sequences standards including genomic
DNA contigs mRNAs and proteins for known genes Multiple collaborations both within NCBI and with external groups support our data-gathering efforts
bull STS databasebull A database of sequence tagged sites or short sequences that are operationally
unique in the genomebull UniSTSbull A unified non-redundant view of sequence tagged sites (STSs)bull UniGenebull A collection of ESTs and full-length mRNA sequences organized into clusters each
representing a unique known or putative human gene annotated with mapping and expression information and cross-references to other sources
UniGene computationally identifies transcripts from the same locus analyzes expression by tissue age and health status and reports related proteins (protEST) and clone resources
Single Nucleotide Polymorphism (SNP) database
What it isThe SNP Database (also known as dbSNP) is an archive for genetic variation within and across different species developed and hosted by NCBI in collaboration with the National Human Genome Research Institute (NHGRI)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species related to biodiversity genetic variation and adaptation
-The dbSNP accepts apparently neutral polymorphisms polymorphisms corresponding to known phenotypes and regions of no variation
-It was created in September 1998 to supplement GenBank (NCBIrsquos nucleic acid and protein sequences)
Goal
Its goal is to act as a single database that contains all identified genetic variation which can be used to investigate a wide variety of genetically based natural phenomenon Specifically access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping population genetics investigations into evolutionary relationships as well as being able to quickly and easily quantify the amount of variation at a given site of interest
Application
Applied research genetic engineering drug discovery etc
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

Single Nucleotide Polymorphism (SNP) database
What it isThe SNP Database (also known as dbSNP) is an archive for genetic variation within and across different species developed and hosted by NCBI in collaboration with the National Human Genome Research Institute (NHGRI)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species related to biodiversity genetic variation and adaptation
-The dbSNP accepts apparently neutral polymorphisms polymorphisms corresponding to known phenotypes and regions of no variation
-It was created in September 1998 to supplement GenBank (NCBIrsquos nucleic acid and protein sequences)
Goal
Its goal is to act as a single database that contains all identified genetic variation which can be used to investigate a wide variety of genetically based natural phenomenon Specifically access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping population genetics investigations into evolutionary relationships as well as being able to quickly and easily quantify the amount of variation at a given site of interest
Application
Applied research genetic engineering drug discovery etc
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-
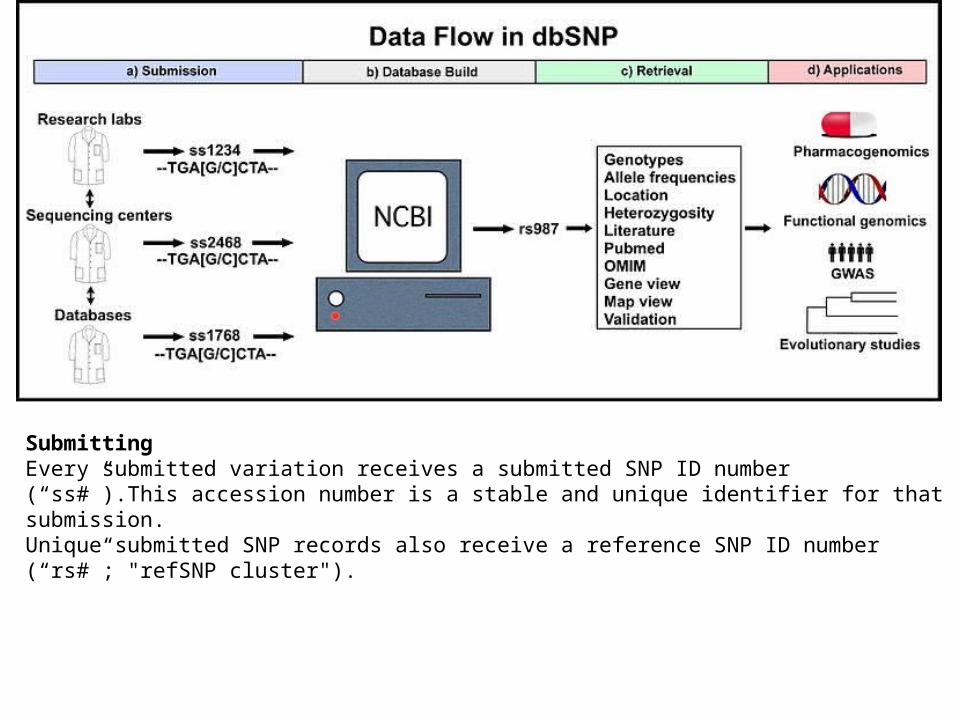
SubmittingEvery submitted variation receives a submitted SNP ID number (ldquossrdquo)This accession number is a stable and unique identifier for that submission Unique submitted SNP records also receive a reference SNP ID number (ldquorsrdquo refSNP cluster)
Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

Section Types for Submissions to dbSNPContact
TYPE CONT
HANDLEEGREEN
NAME Eric Green
EMAIL egreenwugenmailwustledu
LAB Biophysics laboratory
INST King Saud University
ADDR PO Box 2455 Riyadh 11451 Kingdom of Saudi Arabia
Publication section
TYPE PUB
HANDLE EGREEN
MEDUID Medline unique identifier Not obligatory
TITLE Human chromosome 7 STS
AUTHORS AshrafuzzamanM
YEAR 2012
STATUS 1 (unpublished) 2 (submitted) 3 (in press) 4 (published)
Population class
TYPEPOPULATION
HANDLEWHOEVER
IDYOUR_POP
POP_CLASS EUROPE
POPULATION ContinentEurope
Nation Some Nation
Phenotype You name it
How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

How to Submit
Element Explanation
Flanking DNA (region of DNA that is not transcribed to RNA region of DNA adjacent to 5rsquo end of the gene)
Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall
AllelesAlleles must be defined using A G C or T nomenclature IUPAC nomenclature will only be accepted in flanking regions See httpwwwncbinlmnihgovsitesentrezdb=snp
MethodA description of how the variation was detected (eg DNA sequencing) or how the allele frequencies were calculated A table of method classes is provided
PopulationA description of the initial group from which the variation was found or from which the allele frequency was calculated A table of population classes is provided
Sample sizeThe number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies
Population-specific allele frequency The allele frequency of the surveyed population
Population-specific genotype frequency The genotype frequency of the surveyed population
Population-specific heterozygosityThe proportion of individuals who are heterozygous for the variation
Individual genotypes The genotype of individuals from the study
Validation informationThe validation status lists the categories of evidence supporting the variation
To submit variations to dbSNP one must first acquire a submitter handle which identifies the laboratory responsible for the submission Next the author is required to complete a submission file containing the relevant information and data Submitted records must contain the ten essential pieces of information listed in the following tableOther information required for submissions includes contact information publication information (title journal authors year) molecule type (genomic DNA cDNA mitochondrial DNA chloroplast DNA) and organismA sample submission sheet can be found at (httpwwwncbinlmnihgovSNPget_htmlcgiwhichHtml=how_to_submitSECTION_TYPES)
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-
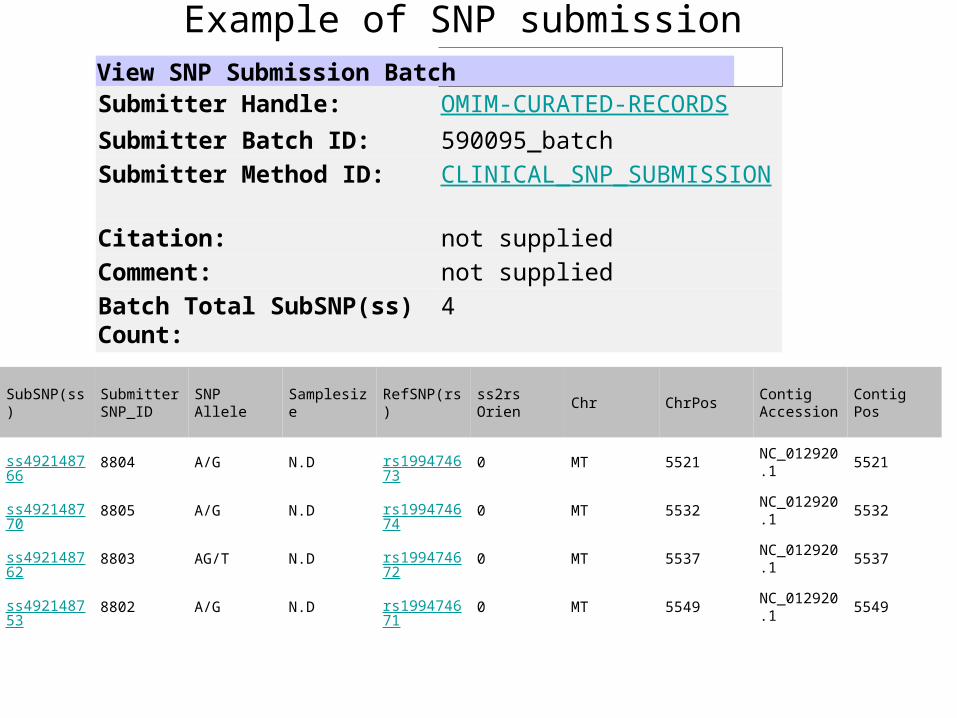
Example of SNP submissionView SNP Submission BatchSubmitter Handle OMIM-CURATED-RECORDS
Submitter Batch ID 590095_batch
Submitter Method ID CLINICAL_SNP_SUBMISSION
Citation not supplied
Comment not supplied
Batch Total SubSNP(ss) Count
4
SubSNP(ss)SubmitterSNP_ID
SNPAllele
Samplesize RefSNP(rs)ss2rsOrien
Chr ChrPosContigAccession
ContigPos
ss492148766
8804 AG ND rs199474673
0 MT 5521NC_0129201
5521
ss492148770
8805 AG ND rs199474674
0 MT 5532NC_0129201
5532
ss492148762
8803 AGT ND rs199474672
0 MT 5537NC_0129201
5537
ss492148753
8802 AG ND rs199474671
0 MT 5549NC_0129201
5549
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-
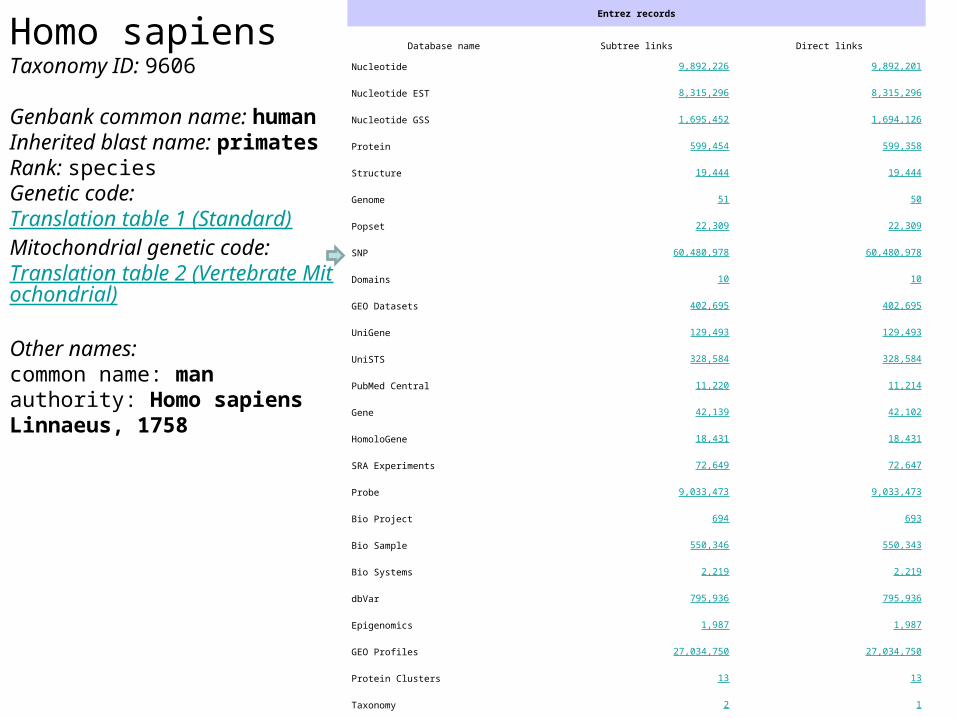
Homo sapiensTaxonomy ID 9606
Genbank common name humanInherited blast name primatesRank speciesGenetic code Translation table 1 (Standard)Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial)
Other namescommon name man authority Homo sapiens Linnaeus 1758
Entrez records
Database name Subtree links Direct links
Nucleotide 9892226 9892201
Nucleotide EST 8315296 8315296
Nucleotide GSS 1695452 1694126
Protein 599454 599358
Structure 19444 19444
Genome 51 50
Popset 22309 22309
SNP 60480978 60480978
Domains 10 10
GEO Datasets 402695 402695
UniGene 129493 129493
UniSTS 328584 328584
PubMed Central 11220 11214
Gene 42139 42102
HomoloGene 18431 18431
SRA Experiments 72649 72647
Probe 9033473 9033473
Bio Project 694 693
Bio Sample 550346 550343
Bio Systems 2219 2219
dbVar 795936 795936
Epigenomics 1987 1987
GEO Profiles 27034750 27034750
Protein Clusters 13 13
Taxonomy 2 1
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-
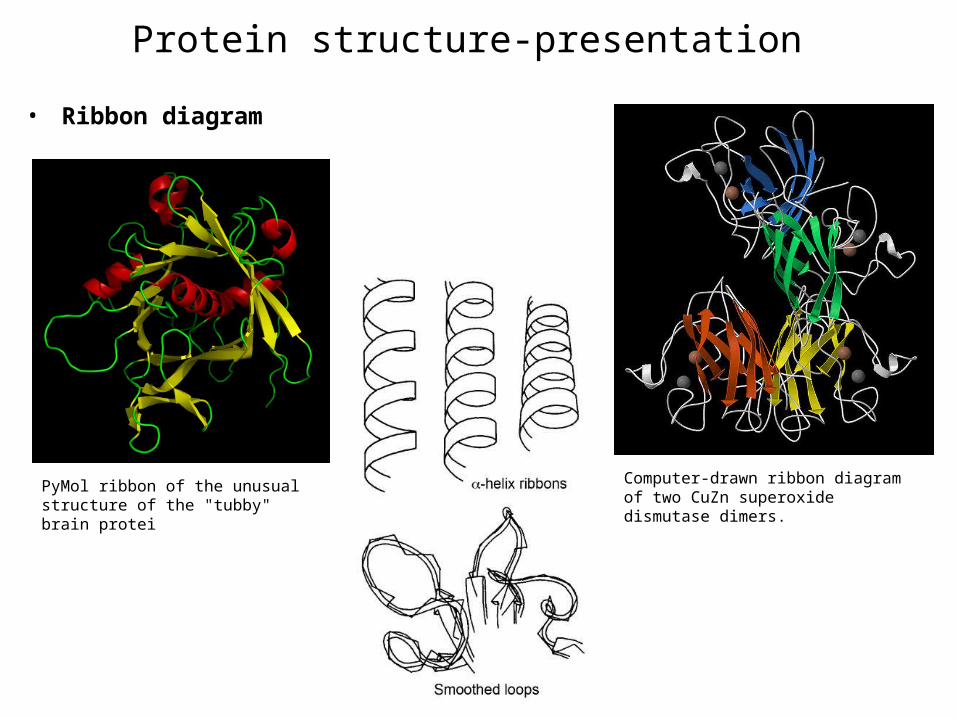
Protein structure-presentation
bull Ribbon diagram
Computer-drawn ribbon diagram of two CuZn superoxide dismutase dimers
PyMol ribbon of the unusual structure of the tubby brain protei
Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

Hollow 11 ndash Illustration software for Proteins
HOLLOW facilitates the production of surface images of proteins Hollow generates fake atoms that identifies voids pockets channels and depressions in a protein structure specified in the PDB format
channel surfaces (and electrostatic surfaces)
interior pathway surfaces
ligand-binding surfaces
Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

Softwares help addressing protein functionsMolecular dynamics (MD)
(mimicking the structureconformations)
PurposeTo understand statistical nature of conformationsMD requires the following parametersbull i Dimension parameters related to the state of the platform-initial conditionsbull ii Dimensions of the participating atomsbull iii Structure of the individual molecules or sections of the whole structure bull iv Physical properties like charges on the atoms
MD allows to locate agentsatoms involved in a structure by providing the following
bull i coordinates (in most cases time dependent)bull ii Projection
MD results can importantly be converted into energeticsbull i interactions between participating agentsatomsbull ii Interactions with the background
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-
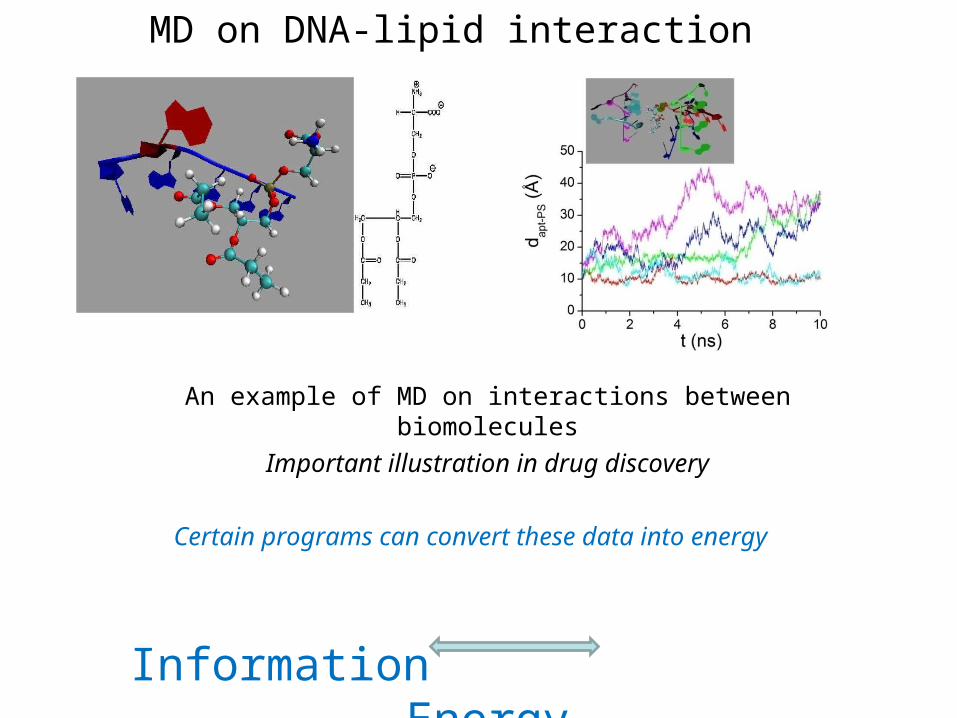
MD on DNA-lipid interaction
An example of MD on interactions between biomolecules
Important illustration in drug discovery
Certain programs can convert these data into energy
Information Energy
Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

Swiss Prot Database
bull UniProtKBSwiss-Prot
bull UniProtKBSwiss-Prot is the manually annotated and reviewed section of the UniProt Knowledgebase (UniProtKB)It is a high quality annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
bull Since 2002 it is maintained by the UniProt consortium and is accessible via the UniProt website httpwwwuniprotorg
bull Deals with
interactions protein modelling proteomics protein structure amp function and genome analysis amp annotation etc
UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

UniProtKBbull The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of
functional information on proteins with accurate consistent and rich annotation
The UniProt Knowledgebase consists of two sections
a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis
and a section with computationally analyzed records that await full manual annotation
For the sake of continuity and name recognition the two sections are referred to as UniProtKBSwiss-Prot (reviewed manually annotated) and UniProtKBTrEMBL (unreviewed automatically annotated) respectively
bull Why is UniProtKB composed of 2 sections UniProtKBSwiss-Prot and UniProtKBTrEMBLbull Where do the protein sequences come frombull About 85 of the protein sequences provided by UniProtKB are derived from the translation of
the coding sequences (CDS) which have been submitted to the public nucleic acid databases the EMBL-BankGenBankDDBJ databases (INSDC) All these sequences as well as the related data submitted by the authors are automatically integrated into UniProtKBTrEMBL
bull Where do the UniProtKB protein sequences come frombull Does UniProtKB contain all protein sequencesbull What are the differences between UniProtKBSwiss-Prot and UniProtKBTrEMBLbull UniProtKBTrEMBL (unreviewed) contains protein sequences associated with computationally
generated annotation and large-scale functional characterization UniProtKBSwiss-Prot (reviewed) is a high quality manually annotated and non-redundant protein sequence database which brings together experimental results computed features and scientific conclusions
PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

PCR-Polymerase Chain Reactionbull Polymerase Chain Reaction
bull Polymerase chain reaction (PCR) enables researchers to produce millions of copies of a specific DNA sequence in approximately two hours This automated process bypasses the need to use bacteria for amplifying DNA
bull PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude generating thousands to millions of copies of a particular DNA sequence
bull Developed in 1983 by Kary Mullis[1] PCR is now a common and often indispensable technique used in medical and biological research labs for a variety of applications [2][3] These include DNA cloning for sequencing DNA-based phylogeny or functional analysis of genes the diagnosis of hereditary diseases the identification of genetic fingerprints (used in forensic sciences and paternity testing) and the detection and diagnosis of infectious diseases In 1993 Mullis was awarded the Nobel Prize in Chemistry along with Michael Smith for his work on PCR[4]
bull The method relies on thermal cycling consisting of cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication of the DNA Primers (short DNA fragments) containing sequences complementary to the target region along with a DNA polymerase (after which the method is named) are key components to enable selective and repeated amplification As PCR progresses the DNA generated is itself used as a template for replication setting in motion a chain reaction in which the DNA template is exponentially amplified PCR can be extensively modified to perform a wide array of genetic manipulations
bull httpwwwyoutubecomDNALearningCenter
Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

Fast A and BLASTbull FASTA suite of programs to perform sequence searching of the EBI protein
databases using local or global similarity
bull In bioinformatics Basic Local Alignment Search Tool or BLAST is an algorithm for comparing primary biological sequence information such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and identify library sequences that resemble the query sequence above a certain threshold Different types of BLASTs are available according to the query sequences For example following the discovery of a previously unknown gene in the mouse a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence The BLAST program was designed by Stephen Altschul Warren Gish Webb Miller Eugene Myers and David J Lipman at the NIH and was published in the Journal of Molecular Biology in 1990
Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-

Phylogenetic tree tutorialAll life on Earth is united by evolutionary history we are all evolutionary cousins mdash twigs on the tree
of life Phylogenetic systematics is the formal name for the field within biology that reconstructs evolutionary history and studies the patterns of relationships among organisms Unfortunately history is not something we can see It has only happened once and only leaves behind clues as to what happened Systematists use these clues to try to reconstruct evolutionary history
See the attached tutorial pdf file provided
Evolutionary trees depict clades A clade is a group of organisms that includes an ancestor and all descendants of that ancestor You can think of a clade as a branch on the tree of life
A phylogeny or evolutionary tree represents the evolutionary relationships among a set of organisms or groups of organisms called taxa (singular taxon) The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants Two descendents that split from the same node are called sister groups In the tree below species A amp B are sister groups mdash they are each others closest relatives Many phylogenies also include an outgroup mdash a taxon outside the group of interest All the members of the group of interest are more closely related to each other than they are to the outgroup Hence the outgroup stems from the base of the tree An outgroup can give you a sense of where on the bigger tree of life the main group of organisms falls It is also useful when constructing evolutionary trees
- Bioinformatics Bio-Informatics
- Biological informations Huge What kind of info (structure and mechanism)
- Searching Data
- A case study
- Data sequencing-GeneBank
- How GeneBank works
- Various bases of Bioinformatics
- Organisms in GeneBank
- GeneBank Format
- GeneBank Flat File Format
- GenBank sequence format Itrsquos a rich format for storing sequences and associated annotations It shares a feature table vocabulary and format with the EMBL and DDJB formats
- Online Mendelian Inheritance in Man (OMIM) Database
- OMIM Example httpwwwomimorgentry189911
- Nucleotide Database
- Slide 15
- Single Nucleotide Polymorphism (SNP) database
- PowerPoint Presentation
- Section Types for Submissions to dbSNP
- How to Submit
- Example of SNP submission
- Homo sapiens Taxonomy ID 9606 Genbank common name human Inherited blast name primates Rank species Genetic code Translation table 1 (Standard) Mitochondrial genetic code Translation table 2 (Vertebrate Mitochondrial) Other names common name man authority Homo sapiens Linnaeus 1758
- Protein structure-presentation
- Hollow 11 ndash Illustration software for Proteins
- Softwares help addressing protein functions Molecular dynamics (MD) (mimicking the structureconformations)
- MD on DNA-lipid interaction
- Swiss Prot Database
- UniProtKB
- PCR-Polymerase Chain Reaction
- Fast A and BLAST
- Phylogenetic tree tutorial
-