biochemical characterization of the active anti …aac.asm.org/content/60/8/4659.full.pdfated viral...
TRANSCRIPT

Biochemical Characterization of the Active Anti-Hepatitis C VirusMetabolites of 2,6-Diaminopurine Ribonucleoside Prodrug Comparedto Sofosbuvir and BMS-986094
Maryam Ehteshami,a Sijia Tao,a Tugba Ozturk,a Longhu Zhou,a Jong Hyun Cho,a Hongwang Zhang,a Sheida Amiralaei,a
Jadd R. Shelton,a Xiao Lu,a Ahmed Khalil,a Robert A. Domaoal,a Richard A. Stanton,a Justin E. Suesserman,a Biing Lin,b Sam S. Lee,b
Franck Amblard,a Tony Whitaker,b Steven J. Coats,b Raymond F. Schinazia
Center for AIDS Research, Laboratory of Biochemical Pharmacology, Department of Pediatrics, Emory University School of Medicine, Atlanta, Georgia, USAa; CocrystalPharma, Inc., Tucker, Georgia, USAb
Ribonucleoside analog inhibitors (rNAI) target the hepatitis C virus (HCV) RNA-dependent RNA polymerase nonstructuralprotein 5B (NS5B) and cause RNA chain termination. Here, we expand our studies on �-D-2=-C-methyl-2,6-diaminopurine-ribo-nucleotide (DAPN) phosphoramidate prodrug 1 (PD1) as a novel investigational inhibitor of HCV. DAPN-PD1 is metabolizedintracellularly into two distinct bioactive nucleoside triphosphate (TP) analogs. The first metabolite, 2=-C-methyl-GTP, is a well-characterized inhibitor of NS5B polymerase, whereas the second metabolite, 2=-C-methyl-DAPN-TP, behaves as an adenosinebase analog. In vitro assays suggest that both metabolites are inhibitors of NS5B-mediated RNA polymerization. Additional fac-tors, such as rNAI-TP incorporation efficiencies, intracellular rNAI-TP levels, and competition with natural ribonucleotides,were examined in order to further characterize the potential role of each nucleotide metabolite in vivo. Finally, we found thatalthough both 2=-C-methyl-GTP and 2=-C-methyl-DAPN-TP were weak substrates for human mitochondrial RNA (mtRNA)polymerase (POLRMT) in vitro, DAPN-PD1 did not cause off-target inhibition of mtRNA transcription in Huh-7 cells. In con-trast, administration of BMS-986094, which also generates 2=-C-methyl-GTP and previously has been associated with toxicity inhumans, caused detectable inhibition of mtRNA transcription. Metabolism of BMS-986094 in Huh-7 cells leads to 87-fold higherlevels of intracellular 2=-C-methyl-GTP than DAPN-PD1. Collectively, our data characterize DAPN-PD1 as a novel and potentantiviral agent that combines the delivery of two active metabolites.
Chronic infection with hepatitis C virus (HCV) represents asignificant disease burden on global health. It is estimated that
3% of the human population carries a chronic HCV infection,which translates to roughly 170 million infections worldwide and3.2 million infections in the United States (1). Although up to 25%of acutely infected individuals clear the virus spontaneously,chronic HCV infection develops in the remaining 75% of thoseinfected and can lead to the development of liver cirrhosis andhepatocellular carcinoma (2–4).
HCV contains a 9.6-kb positive-strand RNA genome thatcodes for three structural and seven nonstructural proteins. Thenonstructural protein 5B (NS5B) is the viral RNA-dependentRNA polymerase and is responsible for genome replication. Be-cause of the high mutation rate of this enzyme, as well as the highvirus replication rate, HCV propagation results in the generationof thousands of viral quasispecies (5, 6). Therefore, drug combi-nation therapy is required in order to overcome development ofresistance and to achieve successful viral clearance. HCV treat-ment comprised of ribavirin and pegylated interferon alpha, withor without protease inhibitors, can achieve sustained virologicresponses in 40 to 80% of treated patients (7–9). However, cura-tive outcomes historically have been suboptimal due to host poly-morphisms, viral genotypic variability, and onset of adverseevents.
In pursuit of new direct-acting antiviral agents with improvedtherapeutic outcome, several different virus replication steps,such as entry, replication complex formation, and NS5B-medi-ated viral RNA synthesis, are currently being investigated (re-viewed in references 10–14). Members of a major class of anti-
NS5B compounds, termed ribonucleoside analog inhibitors(rNAI), block HCV RNA replication inside the host cell cytoplasmthrough viral RNA chain termination. In December 2013, sofos-buvir became the first anti-HCV rNAI phosphoramidate prodrugto receive FDA approval (http://www.fda.gov/forpatients/illness/hepatitisbc/ucm377920.htm). Once inside the cell, host kinasesmust first phosphorylate rNAIs into the active ribonucleosidetriphosphate (rNTP) form. Therefore, sufficient intracellular lev-els of triphosphorylated ribonucleoside analog (rNAI-TP) metab-olites are critical for antiviral activity (14–16). The presence of amember of the phosphoramidate prodrug group on rNAI, such assofosbuvir, allows the first, often rate-limiting, phosphorylationstep to be bypassed, leading to increased intracellular rNAI-TPgeneration.
Despite the high mutation rate of HCV, clinical emergence ofresistance toward sofosbuvir has rarely been observed. Although
Received 9 February 2016 Returned for modification 13 March 2016Accepted 12 May 2016
Accepted manuscript posted online 23 May 2016
Citation Ehteshami M, Tao S, Ozturk T, Zhou L, Cho JH, Zhang H, Amiralaei S,Shelton JR, Lu X, Khalil A, Domaoal RA, Stanton RA, Suesserman JE, Lin B, Lee SS,Amblard F, Whitaker T, Coats SJ, Schinazi RF. 2016. Biochemical characterization ofthe active anti-hepatitis C virus metabolites of 2,6-diaminopurine ribonucleosideprodrug compared to sofosbuvir and BMS-986094. Antimicrob AgentsChemother 60:4659 – 4669. doi:10.1128/AAC.00318-16.
Address correspondence to Raymond F. Schinazi, [email protected].
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
crossmark
August 2016 Volume 60 Number 8 aac.asm.org 4659Antimicrobial Agents and Chemotherapy
on May 25, 2018 by guest
http://aac.asm.org/
Dow
nloaded from

several resistance-associated substitutions and baseline polymor-phisms recently have been associated with treatment, their phe-notypic impact on conferring resistance to rNAI remains to bedetermined (17–19). For example, the infrequent emergence ofresistance-conferring mutation S282T (20–22) is probably due tothe unfit nature of the mutated virus (23). Sofosbuvir, which gen-erates 2=-C-methyl-2=-F-UTP as its active metabolite in vivo (Fig.1A), has demonstrated an exceedingly favorable safety profile inhumans (24–26). Conversely, clinical development of many otheranti-HCV rNAI has been terminated due to issues with toxicity(reviewed in reference 14). A recent important example pertainsto phase II clinical trials with BMS-986094 (formerly known asINX-189), a nucleoside prodrug that is metabolized to generate2=-C-methyl-GTP in vivo (Fig. 1B) (27, 28). Despite the high po-tency of this compound, clinical trials were halted after reports ofsevere adverse events and one death (29, 30). Although the exactcause of toxicity for BMS-986094 remains elusive (31), it is pos-tulated that off-target inhibition of host cell nucleic acid synthesisat least in part accounts for the observed deleterious effects. Inagreement with this hypothesis, recent studies have shown thathuman mtRNA polymerase (POLRMT) can indeed incorporateribonucleoside 5=-triphosphate analogs in vitro. This is in turncorrelated with inhibition of mtRNA transcription and interfer-
ence with mitochondrial function in cell culture (32, 33). Thus,mechanisms associated with ribonucleotide analog toxicity are ofsignificant interest as new rNAI are being developed for the treat-ment of infections with RNA viruses.
While sofosbuvir has paved the way as the gold standard inHCV therapy, development of novel rNAI will be warranted forseveral reasons. Patients with genotype (GT) 3a infection treatedwith sofosbuvir-containing regimens generally attain lower sus-tained virologic response rates than other genotypes (34, 35). Ad-ditionally, the high cost of direct-acting antiviral agents will limitaccess to treatment globally, especially in developing countries (3,36). Finally, considering the diversity of the chronically infectedpopulation, it is conceivable that niche populations require alter-native therapies. In our search for innovative, potent, and safeanti-HCV therapeutic agents, we have recently described thechemical synthesis of �-D-2=-C-Me-2,6-diaminopurine ribonu-cleoside (DAPN) phosphoramidate prodrugs (PD) as novel in-hibitors of HCV (37). We showed that cellular administration ofDAPN-PD led to the generation of two chemically distinct bioac-tive ribonucleoside triphosphate analogs, namely, 2=-C-methyl-DAPN-TP and 2=-C-methyl-GTP (Fig. 1C). In this study, weexamined the biochemical properties of each of the nucleoside5=-triphosphate metabolites of prototype compound DAPN-PD1
FIG 1 Intracellular metabolism of anti-HCV phosphoramidate prodrugs. (A) Administration of sofosbuvir leads to intracellular generation of 2=-C-methyl-2=-F-UTP. (B) BMS-986094 prodrug group cleavage and removal of methyl group at position 6 of the guanine base leads to the generation of 2=-C-methyl-GTP.(C) Prodrug groups from �-D-2=-C-Me-2,6-diaminopurine ribonucleoside phosphoramidate prodrug 1 (DAPN-PD1) are cleaved, and 2=-C-methyl-DAPN-TPis generated after intracellular phosphorylation of the parent ribonucleoside. Parent 2=-C-methyl-DAPN monophosphate also can be a substrate for cellularadenosine deaminases that remove the amino group at position 6 of the purine ring. This leads to the generation of 2=-C-methyl-GTP.
Ehteshami et al.
4660 aac.asm.org August 2016 Volume 60 Number 8Antimicrobial Agents and Chemotherapy
on May 25, 2018 by guest
http://aac.asm.org/
Dow
nloaded from

(compound 14c [37]) and showed both metabolites were inhibi-tors of viral RNA polymerization. We also showed that althoughboth active metabolites generated by DAPN-PD1 can be weaksubstrates for POLRMT in vitro, DAPN-PD1 did not inhibitmtRNA transcription. This is in contrast to BMS-986094, whichshowed significant untoward effects on the synthesis of mtRNAcompared with DAPN-PD1. The relative abundance of intracel-lular 2=-C-methyl-GTP levels produced in off-target cells and dif-ferences in inhibition of mitochondrial transcription likely ac-count for the distinct safety profiles of the two prodrugs. Workpresented here characterizes DAPN-PD1 as a potent and differen-tiated antiviral agent that combines the delivery of two inhibitorymetabolites with distinct incorporation profiles.
MATERIALS AND METHODSPurified NS5B enzymes for GT 5a and 6a were provided by CocrystalPharma, Inc. Expression plasmids for the GT 1b NS5B wild type (WT) andS282T mutant were a gift from Matthias Götte (University of Alberta).Plasmid for the NS5B S96T mutant was generated by QuikChange site-directed mutagenesis. GpG primer (Trilink) and other RNA oligomersubstrates (IDT) were 32P-radiolabeled as previously described (38) using[�-32P]ATP (PerkinElmer Life Sciences). Reactions with T4 polynucle-otide kinase (Fermentas) were allowed to proceed for 1 h at 37°C.
Chemical synthesis of rNAI-TP metabolites. Nucleoside analogtriphosphates were prepared as previously described (39–41). Briefly,high-performance liquid chromatography (HPLC) purification was per-formed using a Dionex NucleoPac PA-200 (9 by 250 mm) column, elutingwith a gradient of 0 to 100% of 0.5 M triethylammonium bicarbonate(TEAB) over 25 min. The appropriate fractions were collected, solventswere evaporated by lyophilization, and the products were freeze-driedwith deionized water several times to remove excess buffer. Nucleosideanalog triphosphates were isolated as trimethylammonium salts with�95% purity as measured by liquid chromatography-tandem mass spec-trometry (LC-MS/MS).
Expression and purification of HCV NS5B. NS5B purification wasperformed essentially as previously described (42). Briefly, HCV GT 1bNS5B (accession number AJ238799.1), in which the 21 amino acids at theC terminus were replaced by a His tag, was inserted into the expressionvector pET-21b (Novagen). WT, S282T, or S96T mutant proteins wereexpressed in 3 liters of transformed BL21(DE3) cells grown to an opticaldensity (OD) of 0.6. After induction with 1 mM isopropyl-�-D-thiogalac-topyranoside (IPTG), cultures were shaken for 16 h at 25°C. Protein waspurified using a Talon Ni2� column (Clontech) and eluted with increas-ing concentrations of imidazole. Following additional purification with aDEAE anion exchange column, samples were eluted from the cation ex-change SP column using increasing concentrations of NaCl. Purified pro-tein was stored in 10 mM HEPES buffer, pH 7.5, with 50% glycerol, 1 mMdithiothreitol (DTT), and 600 mM NaCl. GT 2a JFH1 (accession numberAB114136), GT 3a (accession number EF523597), and GT 4a (accessionnumber EF523599) were purified using the same protocol.
In vitro NS5B-mediated RNA polymerization inhibition assay. C-terminal His-tagged NS5B�21 enzyme was incubated at 30°C with a syn-thetic 20mer RNA template (T20; 5=-AACCGUAUCCAAAACAGUCC-3=) and 1 �M 32P-radiolabeled GpG primer in a buffer containing 40 mMTris, pH 7.5, 6 mM NaCl, and 2 mM MgCl2. Reactions were initiated withthe addition of 10 �M nucleotide triphosphate (NTP) mix, 1 �M com-peting NTP, and various concentrations of inhibitor. Reactions were al-lowed to proceed for 90 min and subsequently stopped with the additionof 10 mM EDTA and formamide. Samples were visualized on 20% dena-turing polyacrylamide gel and quantified using Quantity One software.Fifty percent inhibitory concentrations (IC50s) were calculated using Ka-leidaGraph software.
Rapid single-ribonucleotide incorporation assay. NS5B�21 (3 �M)was incubated with 1 �M RNA template (T20G10; 5=-AAUGUAUAAGC
AUUAUAUCC-3=) (43) and 1 �M 32P-radiolabeled GpG primer. ATPand UTP (20 �M) were added to the mix, and the elongation complex wasallowed to form for 1 h at 30°C and paused at position �10. Variousamounts of GTP or GTP analog subsequently were added, and synthesisof �11 RNA product was measured over time. A similar experimentalsetup was used when assessing ATP or ATP analog incorporation wherethe RNA template (T20A16; 5=-AACCUGAGAAGGAGAAAGCC-3=)(38) was preincubated with 20 �M CTP and UTP. Reactions were stoppedwith the addition of 10 mM EDTA and formamide. Samples were subse-quently denatured at 95°C for 3 min and resolved in a 20% denaturingpolyacrylamide gel. A single-exponential equation, y � ymax(1 exp-(kobs[S])), was used to obtain rates of incorporation (kobs) in Kaleida-Graph software, where S represents substrate concentration and y repre-sents amount of product formed. Obtained rates were fitted to a nonlinearregression using the hyperbolic equation y � (kpol[S])/(Kd,app� [S]) inorder to obtain Kd,app and kpol values. Statistical significance (unpaired ttest) was calculated using GraphPad Prism software.
In vitro IC50 assay with host DNA polymerases. Increasing concen-trations of 2=-C-methyl-DAPN-TP or 2=-C-methyl-GTP (up to 200 �M)were incubated with recombinant human DNA polymerase (catalog num-ber 1075; CHIMERx), recombinant human polymerase � (catalog number1077; CHIMERx), or recombinant human polymerase � (catalog number1076; CHIMERx). Each enzyme was incubated with a DNA/DNA hybrid,and DNA synthesis was performed essentially according to the manufac-turer’s protocol. Briefly, nucleotide incorporation was allowed to proceedfor 5 min at 37°C for human DNA polymerase and � and for 200 min at37°C for human DNA polymerase �.
In vitro POLRMT assay. In vitro RNA synthesis assays with POLRMT(Indigo Biosciences) were performed as previously described (32). Briefly,32P-radiolabeled RNA primer (5=-UUUUGCCGCGCC) was hybridizedto a 3 M excess of the appropriate DNA template (5=-GGGAATGCANGGCGCGGC, where position N was replaced by A, T, or C). POLRMT (125nM) was incubated with 500 nM 5=-radiolabeled RNA/DNA hybrid, 10mM MgCl2, and 100 �M corresponding NTP or rNAI-TP. Incorporationwas allowed to proceed for 2 h at 30°C, and reactions were stopped by theaddition of 10 mM EDTA and formamide. Samples were visualized on20% denaturing polyacrylamide gel. Data were analyzed by normalizingthe product fraction for each rNAI-TP to that of the corresponding nat-ural NTP.
Measurement of intracellular rNAI-TP levels. Huh-7 cells were ex-posed to medium containing 50 �M the nucleotide prodrugs for 4 h at37°C. Cells were washed with 3� PBS to remove extracellular com-pounds. Intracellular prodrugs and metabolites were extracted from 1 �106 cells using 1 ml 70% ice-cold methanol (containing 20 nM internalstandard ddATP). Samples were dried and resuspended in HPLC duringmobile phase before analysis by LC-MS/MS (44). The calibration curveswere generated from standards of parent nucleotides, prodrugs, 2=-C-methyl-DAPN-TP, and 2=-C-methyl-GTP. Similar protocols were re-peated for primary human hepatocytes incubated with 50 �M nucleotideprodrugs for 24 h at 37°C.
Mitochondrial transcription inhibition assay. Huh-7 cells weremaintained in growth medium containing advanced Dulbecco’s modifiedEagle medium (DMEM) (Gibco, Life Sciences) supplemented with 10%fetal bovine serum (FBS), 100 U/ml penicillin, 2 nM L-glutamine, and 5mM HEPES. Huh-7 cells were cultured in a 24-well plate at a density of0.5 � 106 cells. After attachment of the cells, medium with 50 ng/mlethidium bromide (EtBr) was added to each well and placed at 37°C in a5% CO2 incubator (32). Following EtBr treatment for 24 h, medium wasremoved and cells were washed with PBS two times. Fresh medium con-taining 10 �M different ribonucleoside analog prodrugs was added toeach well in duplicates. The plate was placed at 37°C in a 5% CO2 incuba-tor for 48 h with medium/compound replenishment every 24 h. At 48 h,cells were washed with PBS and harvested to perform reverse transcrip-tion-PCR (RT-PCR). Total RNA was isolated using an RNeasy minikit(Qiagen), and RNA samples were treated with DNase RQ1 (Promega).
Inhibition of HCV Polymerase by DAPN-PD1 Metabolites
August 2016 Volume 60 Number 8 aac.asm.org 4661Antimicrobial Agents and Chemotherapy
on May 25, 2018 by guest
http://aac.asm.org/
Dow
nloaded from

Extracted RNA was converted to cDNA using SuperScript-VILO (Invit-rogen). RT-PCR was performed using a LightCycler 480 probe master(Roche) and 900 nM forward and reverse primers for mitochondrial ND1transcript (forward, 5=-AACCTCTCCACCCTTATCACAA-3=; reverse,5=-TCATATTATGGCCAAGGGTCA-3=) and nuclear glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcript (forward, 5=-TCCACTGGCGTCTTCACC-3=; reverse, 5=-GGCAGAGATGATGACCCTTTT-3=).Amplicons were detected using Roche FastStart universal probe masternumbers 45 and 60 for ND1 and GAPDH, respectively. RT-PCR wasconducted at 95°C for 10 min, followed by 95°C for 15 s and 60°C for 1min for 40 cycles. The threshold cycle value (CT) of each amplicon wasnormalized to the internal control (GAPDH). Data were analyzed by the2��CT relative quantification method (45). Statistical significance (un-paired t test) was calculated using GraphPad Prism software.
RESULTSDAPN-PD1 generates two nucleoside 5=-triphosphate metabo-lites that are active against HCV NS5B polymerase. We have pre-viously described the chemical synthesis of DAPN-PD1 as a novelribonucleoside analog with anti-HCV activity (median effectiveconcentration [EC50] of 0.7 �M in HCV GT 1b replicon) (37).The intracellular metabolism of 2,6-diaminopurine prodrug re-sulted in the generation of two nucleoside metabolites, namely,2=-C-methyl-GTP and 2=-C-methyl-DAPN-TP, in both primaryhuman hepatocytes and Huh-7 cells (37). Upon prodrug groupcleavage, 2=-C-methyl-DAPN monophosphate was subjected tointracellular phosphorylation and 2=-C-methyl-DAPN-TP wasgenerated (Fig. 1C). In parallel, 2=-C-methyl-DAPN nucleosidesalso can be a substrate for intracellular deamination at position 6of the purine ring, which in turn leads to the generation of 2=-C-methyl-GTP (Fig. 1C). Prior studies have described an inhibitoryrole for 2=-C-methyl-GTP in both HCV replicon-based and cell-free NS5B polymerase assays (20, 28, 46). However, 2=-C-methyl-DAPN-TP has not previously been examined as an inhibitor ofHCV NS5B-mediated RNA polymerization. In order to assesschain termination, we chemically synthesized 2=-C-methyl-DAPN-TP and performed cell-free RNA polymerization assayswith purified NS5B�21 enzyme. 2=-C-methyl-DAPN-TP inhib-ited RNA synthesis at an IC50 of 3.4 � 1.1 �M for HCV GT 1b(Table 1), while the IC50 for 2=-C-methyl-GTP was 5.6 � 1.6 �M.Similar results were obtained for NS5B enzymes from HCV GT 2a,3a, 4a, 5a, and 6a. The IC50s obtained for each metabolite ofDAPN-PD1 were comparable to that of 2=-C-methyl-2=-F-UTP,the active metabolite of sofosbuvir (Table 1).
Incorporation profiles for active nucleoside 5=-triphosphatemetabolites of DAPN-PD1. In vitro nucleotide incorporation as-
says preformed on NS5B/RNA elongation complexes (describedin Materials and Methods) were conducted in order to confirmthe incorporation of 2=-C-methyl-DAPN-TP as an A analog. TheRNA templates used allowed for ATP or GTP analog incorpora-tion at position �16 or �11, respectively (Fig. 2A and C). Ourbiochemical data confirmed that as expected, 2=-C-methyl-DAPN-TP behaves as an ATP analog, while 2=-C-methyl-GTPmaintained a GTP analog incorporation profile. No incorpora-tion was observed with up to 250 �M 2=-C-methyl-DAPN-TPwhen cytidine was present in the RNA template at position �11,indicating that 2=-C-methyl-DAPN-TP is not a GTP analog(Fig. 2B and D). Overall, these data suggest that DAPN-PD1 can
FIG 2 NS5B elongation complex single-nucleotide extension. (A) Radiola-beled GpG primer is extended to position �15 with the addition of CTP andUTP. Incorporation of ATP or ATP analog subsequently is measured at posi-tion �16. (B) Nucleoside triphosphate incorporation was resolved on a dena-turing polyacrylamide gel where incubation with ATP (7.5 �M) or 2=-C-meth-yl-DAPN-TP (75 �M) results in extension by one position over time. (C)Radiolabeled GpG primer is extended to position �9 with the addition of ATPand UTP. Incorporation of GTP or GTP analog subsequently was measured atposition �10. (D) Nucleoside 5=-triphosphate incorporation was resolved ona denaturing polyacrylamide gel where GTP (2.2 �M) and 2=-C-methyl-GTP(2.2 �M), but not 2=-C-methyl-DAPN-TP (250 �M), show extension by oneposition over time. Assays were repeated at various NTP concentrations in orderto obtain Kd,app and kpol values for each inhibitor (summarized in Table 2).
TABLE 1 In vitro inhibition of NS5B-mediated RNA synthesis bynucleoside 5=-triphosphate analogs
NS5B GT
IC50a (�M)
2=-C-Methyl-DAPN-TP
2=-C-Methyl-GTP
2=-C-Methyl-2=-F-UTP
1b 3.4 � 1.1 5.6 � 1.6 2.9 � 0.62a 3.6 � 2.1 5.1 � 1.2 3.0 � 0.93a 1.6 � 0.1 3.6 � 0.8 7.2 � 2.14a 1.8 � 1.4 7.4 � 4.2 12.8 � 10.75a 4.7 � 3.5 3.5 � 1.0 ND b
6a 5.1 � 5.1 2.1 � 0.4 NDa In vitro IC50s are the averages from two to four replicates � standard deviations.b ND, not determined.
Ehteshami et al.
4662 aac.asm.org August 2016 Volume 60 Number 8Antimicrobial Agents and Chemotherapy
on May 25, 2018 by guest
http://aac.asm.org/
Dow
nloaded from

deliver two rNAI-TP metabolites with distinct incorporation pro-files.
Kinetics of incorporation for rNAI-TPs generated by DAPN-PD1. Rapid nucleotide incorporation by NS5B in the elongationcomplex was assessed in order to determine the apparent nucleo-tide dissociation equilibrium constant (Kd,app) and maximal in-corporation rate (kpol). Increasing concentrations of nucleosidetriphosphate analogs were incubated with the appropriate NS5B/RNA elongation complex, and incorporation was measured over ashort time period (Fig. 2). The amount of product formed wasfitted to a single-turnover nucleotide incorporation plot whereproductively bound NS5B/RNA complexes could be extended byone nucleotide (Fig. 3). Kd,app and kpol values are summarized inTable 2, with incorporation efficiency reported as a kpol/Kd,app
ratio. 2=-C-methyl-GTP appears to have over 13-fold lower cata-lytic efficiency than GTP substrate, while 2=-C-methyl-DAPN-TPappears to be more severely compromised with regard to bothbinding affinity and incorporation rate (over 900-fold) (Table 2).Although earlier results suggest that both 2=-C-methyl-DAPN-TPand 2=-C-methyl-GTP are inhibitors of NS5B-mediated RNA syn-thesis, the kinetic parameters measured here suggest that 2=-C-methyl-GTP is more readily incorporated by NS5B polymerase(see Discussion).
In addition to enzymatic measurements of incorporation, cy-toplasmic rNAI-TP levels are another important factor to takeinto consideration when estimating in vivo incorporation rates ofnucleoside analogs. The rate constant for incorporation (keff, NS5B
[per second]) values can be calculated through experimentallydetermined kinetic parameters and measurement of intracellularrNAI-TP levels according to the equation Keff, NS5B (per second) �(kpol � [NTP])/(Kd,app � [NTP]) (Table 3) (32). Intracellular me-tabolism of DAPN-PD1 was monitored in primary human hepa-tocyte cells, which represent the primary host cell environmentwhere inhibition of HCV viral replication occurs. By taking intoaccount intracellular levels of natural NTPs and rNAI-TPs, wewere able to determine keff, NS5B values for each nucleoside species.Based on these variables, we estimated that fold differences be-tween rate constants for incorporation of 2=-C-methyl-DAPN-TPto be 43-fold lower than those for ATP. A difference of 8-foldwas observed for 2=-C-methyl-GTP compared to GTP (Table 3).
Resistance profiles of 2=-C-methyl-DAPN-TP and 2=-C-methyl-GTP metabolites. In order to assess resistance to activerNAI-TP metabolites of DAPN-PD1, cell-free IC50 assays wereperformed with purified NS5B�21 containing a mutation eitherat position 96 or 282, each of which has been associated with drugresistance. The presence of S96T mutation did not affect suscep-tibility to either 2=-C-methyl-DAPN-TP or 2=-C-methyl-GTP(IC50 of 2.5 �M and 8.8 �M, respectively). However, compared toWT enzyme, addition of S282T mutation resulted in IC50s ofabove 50 �M, indicating at least 15-fold and 9-fold resistance to2=-C-methyl-DAPN-TP and 2=-C-methyl-GTP, respectively (Ta-ble 4). Similar trends were observed for 2=-C-methyl-2=-F-UTP.
Incorporation of 2=-C-methyl-DAPN-TP by human hostDNA polymerases. In order to examine potential off-target inhi-bition of host cellular polymerases by 2=-C-methyl-DAPN-TP, in-creasing amounts of the rNAI-TP were incubated with humanDNA polymerases , �, and �. No inhibition of DNA synthesiswas observed for up to 200 �M 2=-C-methyl-DAPN-TP or 2=-C-methyl-GTP (Table 5), suggesting these nucleoside analogtriphosphates are not substrates for host DNA polymerases. The
active metabolite of sofosbuvir, 2=-C-methyl-2=-F-UTP, did notshow any inhibitory effect up to 100 �M (Table 5).
Incorporation of 2=-C-methyl-DAPN-TP by human host mito-chondrial RNA polymerase POLRMT. We next examined whethernucleoside 5=-triphosphate metabolites of DAPN-PD1 were sub-
FIG 3 Kinetic parameters for nucleoside 5=-triphosphate incorporation byNS5B polymerase. Single representative plots are shown for kinetic parametersreported in Table 2. Incorporation of increasing amounts of rNTP coincu-bated with preformed NS5B/RNA elongation complexes was monitored overtime. The amount of product formed was quantified and fitted to a singleexponential equation, y � ymax(1 exp(kobs[S]), in order to obtain rates ofincorporation. Obtained kobs(S 1) values were plotted as a function of NTPconcentration and fitted to a nonlinear regression using the hyperbolic equa-tion y � (kpol[S])/(Kd,app� [S]). kpol and Kd,app values are indicated on eachplot for ATP (A), 2=-C-methyl-DAPN-TP (B), GTP (C), and 2=-C-methyl-GTP (D).
Inhibition of HCV Polymerase by DAPN-PD1 Metabolites
August 2016 Volume 60 Number 8 aac.asm.org 4663Antimicrobial Agents and Chemotherapy
on May 25, 2018 by guest
http://aac.asm.org/
Dow
nloaded from
strates for host mitochondrial RNA polymerase (POLRMT). Whenpurified POLRMT was incubated with annealed 32P-radiolabeledRNA/DNA hybrids containing an A, C, or T on the DNA template atposition 9 (Fig. 4A), we found that the addition of 100 �M 2=-C-methyl-DAPN-TP resulted in 34% incorporation when normal-ized to incorporation of natural ATP substrate (Fig. 4B). Consis-tent with previous findings (32), 39% incorporation was observedfor 2=-C-methyl-GTP normalized to natural GTP substrate (Fig.4B), while the active metabolite of sofosbuvir, 2=-C-methyl-2=-F-UTP, was not found to be a substrate for POLRMT when tested upto 100 �M.
Effect of DAPN-PD1 on mitochondrial transcription inHuh-7 cells. A phenotypic cell-based assay that measures the im-pact of DAPN-PD1 on mtRNA transcription was performed.Considering the observed clinical safety of sofosbuvir, and con-versely the clinical toxicity of BMS-986094, these compoundswere used as negative and positive controls, respectively. WhenHuh-7 cells were coincubated with EtBr (Fig. 5A), which selec-tively reduces mtRNA synthesis, we observed a 2-fold reduction inmtRNA transcript levels. mtRNA replenishment subsequentlywas observed after EtBr removal (Fig. 5B). Interestingly, we foundthat cells previously treated with EtBr exhibited increased mito-chondrial transcription as normalized to nuclear GAPDH (thusaccounting for increased cell growth). This suggests that cellsovercompensate for EtBr-induced mtRNA suppression by selec-tively upregulating mtRNA synthesis. Conversely, treatment with10 �M BMS-986094 (which generates 2=-C-methyl-GTP intracel-lularly) was sufficient to fully inhibit mtRNA replenishment afterEtBr removal (Fig. 5B). Treatment with DAPN-PD1 (which gen-erates 2=-C-methyl-GTP and 2=-C-methyl-DAPN-TP intracellu-larly) did not impede mtRNA replenishment. Treatment with thecomparator sofosbuvir did not inhibit mtRNA replenishment.Our data suggest that although both DAPN-PD1 and BMS-986094 generate rNAI-TPs that can be substrates for POLRMT,only BMS-986094 demonstrates a deleterious effect with regard toreplenishment of mitochondrial transcription. The phenotypicimpact of the observed rNAI-mediated mtRNA suppression onmitochondrial function is not yet fully understood.
Intracellular concentrations of nucleoside 5=-triphosphatemetabolites. In order to address the underlying cause of selectivemtRNA depletion with BMS-986094 but not DAPN-PD1, we ex-amined the intracellular metabolism of each inhibitor. Huh-7 cellswere incubated with 50 �M either BMS-986094 or DAPN-PD1for 4 h at 37°C, and intracellular 2=-C-methyl-GTP levels were
measured using quantitative LC-MS/MS. We found that metabo-lism of BMS-986094 generated 87-fold higher intracellular 2=-C-methyl-GTP levels than DAPN-PD1 (680 pmol/106 cells and 7.8pmol/106 cells, respectively) (Table 6). Taking into account pre-viously reported kinetic parameters of incorporation of 2=-C-methyl-GTP by POLRMT (32), we calculated the rate constant forthe incorporation value keff, POLRMT (also known as the mitovirscore [32]) for this inhibitor. When the intracellular 2=-C-methyl-GTP levels generated by BMS-986094 or DAPN-PD1 were com-pared, mitovir scores of 0.04 s1 and 0.0007 s1, respectively, wereobtained (Table 6). The mitovir score for 2=-C-methyl-GTP gen-erated by BMS-986094 was determined to be 57-fold higher thanthe mitovir score for 2=-C-methyl-GTP generated by DAPN-PD1.
DISCUSSION
�-D-2=-C-Methyl-2,6-diaminopurine ribonucleoside is a newphosphoramidate prodrug with selective antiviral activity againstHCV in vitro. This prodrug can produce two nonobligate chain-terminator rNAI-TPs. While 2=-C-methyl-GTP behaves as a GTPanalog, we observed that 2=-C-methyl-DAPN-TP is an ATP ana-log. This is consistent with previous reports that modified 2,6-diaminopurine nucleosides form base pairs opposite thymidine oruridine residues (47, 48). We found that both 2=-C-methyl-DAPN-TP and 2=-C-methyl-GTP inhibited NS5B-mediated RNAsynthesis in cell-free assays against all NS5B genotypes tested (Ta-ble 1). These findings suggest that both nucleoside 5=-triphos-phate metabolites have the potential to contribute to anti-HCVinhibition in vivo. In order to address this hypothesis, we took intoconsideration the kinetic parameters of nucleotide incorporationby NS5B, as well as intracellular rNAI-TP levels for each activemetabolite. Rapid single-nucleotide incorporation assays demon-strated that incorporation efficiency was reduced for both rNAI-TPs, albeit to different degrees. The incorporation efficiency of2=-C-methyl-GTP was reduced by 13-fold compared to that ofGTP, largely due to changes in the binding affinity (P � 0.0005).On the other hand, 2=-C-methyl-DAPN-TP was 900-fold less ef-ficiently incorporated than ATP. This effect is largely attributed to
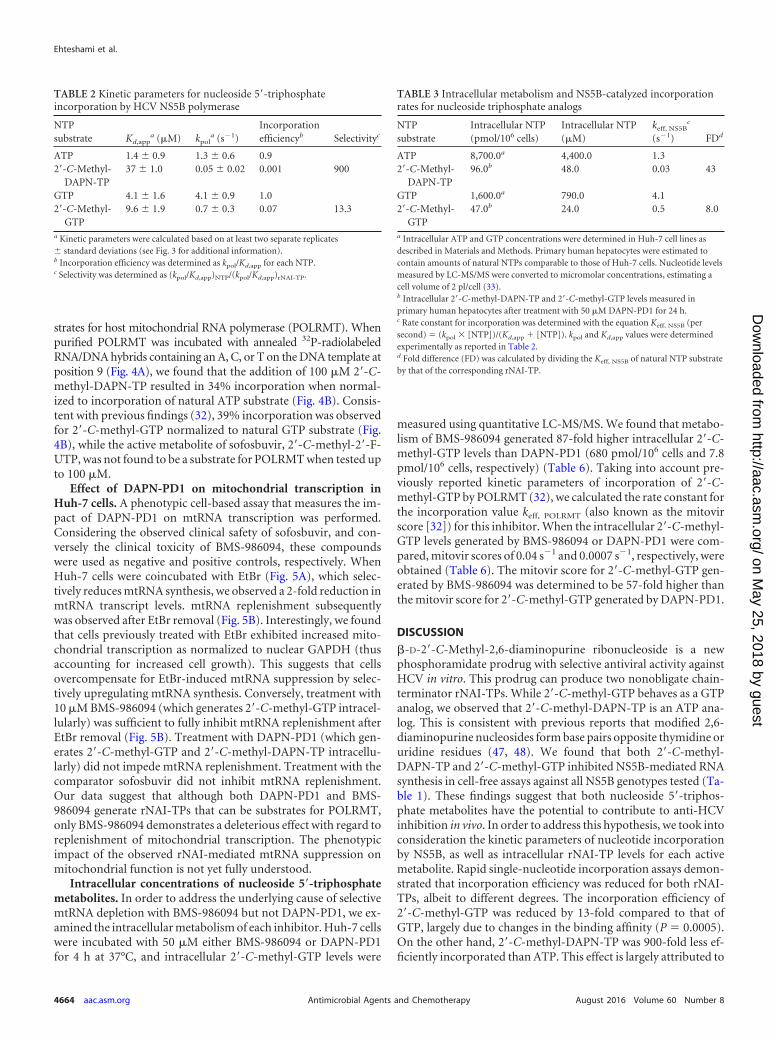
TABLE 3 Intracellular metabolism and NS5B-catalyzed incorporationrates for nucleoside triphosphate analogs
NTPsubstrate
Intracellular NTP(pmol/106 cells)
Intracellular NTP(�M)
keff, NS5Bc
(s1) FDd
ATP 8,700.0a 4,400.0 1.32=-C-Methyl-
DAPN-TP96.0b 48.0 0.03 43
GTP 1,600.0a 790.0 4.12=-C-Methyl-
GTP47.0b 24.0 0.5 8.0
a Intracellular ATP and GTP concentrations were determined in Huh-7 cell lines asdescribed in Materials and Methods. Primary human hepatocytes were estimated tocontain amounts of natural NTPs comparable to those of Huh-7 cells. Nucleotide levelsmeasured by LC-MS/MS were converted to micromolar concentrations, estimating acell volume of 2 pl/cell (33).b Intracellular 2=-C-methyl-DAPN-TP and 2=-C-methyl-GTP levels measured inprimary human hepatocytes after treatment with 50 �M DAPN-PD1 for 24 h.c Rate constant for incorporation was determined with the equation Keff, NS5B (persecond) � (kpol � [NTP])/(Kd,app � [NTP]). kpol and Kd,app values were determinedexperimentally as reported in Table 2.d Fold difference (FD) was calculated by dividing the Keff, NS5B of natural NTP substrateby that of the corresponding rNAI-TP.
TABLE 2 Kinetic parameters for nucleoside 5=-triphosphateincorporation by HCV NS5B polymerase
NTPsubstrate Kd,app
a (�M) kpola (s1)
Incorporationefficiencyb Selectivityc
ATP 1.4 � 0.9 1.3 � 0.6 0.92=-C-Methyl-
DAPN-TP37 � 1.0 0.05 � 0.02 0.001 900
GTP 4.1 � 1.6 4.1 � 0.9 1.02=-C-Methyl-
GTP9.6 � 1.9 0.7 � 0.3 0.07 13.3
a Kinetic parameters were calculated based on at least two separate replicates� standard deviations (see Fig. 3 for additional information).b Incorporation efficiency was determined as kpol/Kd,app for each NTP.c Selectivity was determined as (kpol/Kd,app)NTP/(kpol/Kd,app)rNAI-TP.
Ehteshami et al.
4664 aac.asm.org August 2016 Volume 60 Number 8Antimicrobial Agents and Chemotherapy
on May 25, 2018 by guest
http://aac.asm.org/
Dow
nloaded from

reductions in the rate of polymerization (kpol) of this inhibitor(Table 2).
The incorporation rate constants (keff, NS5B) for rNAI-TP me-tabolites of DAPN-PD1 were calculated based on their intracellu-lar metabolism in primary human hepatocytes. We estimated a43-fold difference in keff, NS5B values between 2=-C-methyl-DAPN-TP and ATP, while the difference in keff, NS5B values is8-fold for 2=-C-methyl-GTP and GTP (Table 3). It is worth notingthat the 9.6-kb genome of HCV GT 1b is comprised of 58% G·Ccontent, indicating that slightly more opportunities exist for GTPor CTP analog incorporation. Conversely, the 3=-untranslatedpoly(U/UC) region of the viral genome (81% U·A content) canrepresent a hot spot for chain termination by ATP analogs, such as2=-C-methyl-DAPN-TP. Overall, calculations described abovesuggest that under the cellular conditions tested, 2=-C-methyl-GTP has a higher probability for incorporation by NS5B enzyme.At the same time, considering that a single incorporation event issufficient to abrogate virus replication, the contribution of 2=-C-methyl-DAPN-TP to chain termination of viral RNA synthesis invivo should not be ruled out.
Cell culture selection of resistance-conferring mutations suchas S282T or S96T has been associated with rNAIs containing a2=-C-methyl or 4=-azido group, respectively, on the ribose ring(15, 23, 49, 50). Here, we report a low level of resistance conferredby recombinant S282T mutant NS5B enzyme when each rNAI-TPwas tested separately. However, we have previously reported thatthe GT 1b replicon harboring the S282T mutation does not showresistance to DAPN-PD1 (37). We hypothesize that the combineddelivery of both metabolites in cell culture can overcome resis-tance development. Additional studies are under way to addressthe distinct resistance profiles seen in cell-based and cell-free sys-tems.
In addition to antiviral activity, a thorough examination of thesafety profile of rNAI is critical for preclinical development of
investigational compounds. As highlighted recently, phase II clin-ical trials with BMS-986094 were halted after reports of severetoxicity and one death (51). Because 2=-C-methyl-GTP is an in-tracellular metabolite common to both BMS-986094 and DAPNphosphoramidate prodrugs, we previously reported head-to-headcomparative assays wherein the cytotoxicity of each prodrug wasmonitored in various cell lines. We were unable to detect cytotox-icity with DAPN phosphoramidate prodrugs in Vero, Huh-7,HepG2, CEM, peripheral blood mononuclear cells (PBMC), orPC3 cell lines up to 100 �M. Similarly, no bone marrow toxicity orany changes in lactic acid production were observed with DAPNprodrug treatment. On the other hand, BMS-986094 treatmentled to mitochondrial DNA toxicity, increased lactic acid produc-tion, and Huh-7 cell death (37). In this study, we aimed to lookmore closely at correlates of cytotoxicity with BMS-986094 treat-ment in order to better understand the lack of cytotoxicity ob-
TABLE 4 Effect of resistance-conferring mutations on nucleoside analog potency in cell-free assays
NS5B
2=-C-Methyl-DAPN-TP 2=-C-Methyl-GTP 2=-C-Methyl-2=-F-UTP
IC50a (�M) FDb IC50 (�M) FD IC50 (�M) FD
GT 1b WT 3.4 � 1.1 1 5.6 � 1.6 1 2.9 � 0.6 1GT 1b S96T 2.5 � 0.6 0.7 8.8 � 2.9 1.6 3.5 � 0.9 1.2GT 1b S282T �50 �15 �50 �9 �50 �19a In vitro IC50s were determined with cell-free assays using purified recombinant NS5B�21. Each value is the average from two to three replicates (� standard deviations).b FD, fold difference.
TABLE 5 In vitro inhibition of host DNA polymerase-mediated DNAsynthesis
Inhibitor
IC50 (�M)
DNA Pol DNA Pol � DNA Pol �
2=-C-Methyl-DAPN-TP �200 �200 �2002=-C-Methyl-GTP �200 �200 �2002=-C-Methyl-2=-F-UTP �100 �100 �1003=-dTTPa NDb 17.7 41.7Aphidicolinc 5.4 ND NDa 3=-dTTP was used as a positive control for inhibition of DNA synthesis by host DNApolymerase (Pol) � and �.b ND, not determined.c Aphidicolin was used as a positive control for inhibition of DNA synthesis by hostDNA polymerase .
FIG 4 Ribonucleoside 5=-triphosphate analog incorporation by POLRMT.(A) Schematic representation of RNA/DNA primer/template substrates usedfor incorporation of each nucleoside triphosphate. (B) Nucleotide incorpora-tion by POLRMT was allowed to proceed for 2 h in the presence of 100 �Meach NTP or rNAI-TP. The percentage of RNA product at n � 1 for eachrNAI-TP was normalized to that of natural NTP substrates. Error bars repre-sent standard deviation (SD) values from three separate experiments.
Inhibition of HCV Polymerase by DAPN-PD1 Metabolites
August 2016 Volume 60 Number 8 aac.asm.org 4665Antimicrobial Agents and Chemotherapy
on May 25, 2018 by guest
http://aac.asm.org/
Dow
nloaded from

served with DAPN phosphoramidate prodrugs. It was recentlyreported that human mitochondrial RNA polymerase (POLRMT)incorporates 2=-C-methyl-GTP as well as a number of other anti-HCV ribonucleoside analogs (32, 33). Here, we show that 2=-C-methyl-DAPN-TP is also a substrate for POLRMT (Fig. 4), sug-gesting that similar to BMS-986094, metabolites of DAPN-PD1also have the potential to interfere with mitochondrial transcrip-tion. However, the biochemical data alone do not fully explain thedistinct cell-based toxicity profiles of BMS-986094 and DAPNphosphoramidate prodrugs. When changes in mtRNA levels weremeasured after treatment with inhibitors, we observed that treat-ment with BMS-986094 prevented RNA replenishment whiletreatment with either DAPN-PD1 or sofosbuvir did not have aneffect in this regard (Fig. 5). These data were consistent with the
observation that BMS-986094, but not DAPN-PD1, causes mito-chondrial toxicity in Huh-7 cells in a 14-day assay (37).
In order to address the underlying cause of selective mtRNAinhibition by BMS-986094, we monitored the intracellular metab-olism of each prodrug in Huh-7 cells. BMS-986094 prodrug gen-erated 87 times higher 2=-C-methyl-GTP levels than DAPN-PD1 (Table 6). This finding is consistent with the low-nanomolarmedian effective concentration (EC50) value reported for BMS-986094 (28). As proposed previously (32), the data support thehypothesis that inhibition of mtRNA transcription by rNAIs isdependent not only on POLRMT substrate specificity but alsoon intracellular concentrations of rNAI-TP generated by eachprodrug.
As a caveat, it is worth noting that all rNAI-TP measurementsreported in this study are cytoplasmic, and the calculated mitovirscore values are based on the previously reported assumption thatintramitochondrial rNAI-TP levels correlate with rNAI-TP levelsdetected in the cytoplasm (32). Therefore, the obtained mitovirscores may change with more accurate measurements of intrami-tochondrial rNAI-TP levels. It is also possible that prodrug groupchoice may differently affect entry or accumulation of rNAI-TPsin the mitochondria, partially accounting for the distinct inhibi-tory profile of mtRNA transcription observed with DAPN-PD1and BMS-986094 treatment. For example, BMS-986094 may beparticularly well suited for targeting the mitochondria. Con-versely, the DAPN prodrug group may modulate mitochondrialentry, or the mitochondria may not have the necessary adenosinedeaminases for 2,6-diaminopurine-to-guanine base conversion.Further studies are under way to examine the dynamics ofrNAI-TP generation inside the mitochondria relative to prodrugchoice.
Although we and others (32) have reported on a correlationbetween cytotoxicity and mtRNA suppression, it is worth high-
FIG 5 Effect of ribonucleotide prodrugs on mitochondrial RNA transcription. (A) Huh-7 cells were incubated with ethidium bromide (EtBr) for 24 h. EtBr wassubsequently removed by washing the cells with PBS buffer in the absence or presence of 10 �M each prodrug. RT-PCR was employed to measure mtRNAtranscript after 48 h of replenishment. (B) Representative plot of mtRNA transcript levels after treatment with EtBr and nucleoside prodrugs as normalized tono-EtBr treatment controls (EtBr). “�EtBr” sample represents mtRNA levels at 24 h (light gray bars) after EtBr treatment. “No inhibitor” sample representsthe amount of mtRNA replenishment at 48 h (dark gray bars) after EtBr removal in the absence of inhibitors. All mtRNA measurements are normalized to nuclearGAPDH RNA, which also serves as an internal control for cell growth. Error bars represent SD values from two to four separate replicates. The differencesbetween the EtBr control sample, �EtBr sample, and no inhibitor control were significant (P � 0.01). The differences between BMS-986094-treated samplescompared to either DAPN-PD1-treated or sofosbuvir-treated samples were significant (P � 0.05). Differences between no inhibitor, DAPN-PD1-treatedsamples, and sofosbuvir-treated samples were not statistically significant. An unpaired t test (GraphPad Prism software) was used to determine significance forall samples.
TABLE 6 Generation of 2=-C-methyl-GTP from DAPN-PD1 and BMS-986094 in Huh-7 cells
Prodrug2=-C-Methyl-GTPa
(pmol/106 cells)
2=-C-Methyl-GTPb (�M)
keff, POLRMTc
(s1) Selectivityd
DAPN-PD1 7.8 � 0.2 3.9 0.0007BMS-986094 680 � 40 340 0.04 57.0a rNAI prodrug (50 �M) was incubated with Huh-7 cells for 4 h at 37°C. Intracellular2=-C-methyl-GTP levels generated from each prodrug were determined using LC-MS/MS. Each value is the average from two separate replicates (� standard deviations).b Amount of nucleoside 5=-triphosphate metabolites was converted from picomoles permillion cells to molarity using an estimated cellular volume of 2 pl per cell (33).c Rate constant for incorporation was determined with the equation Keff, POLRMT (persecond) � (kpol � [NTP])/(Kd,app � [NTP]). Keff, POLRMT was calculated based onreported kinetic properties of 2=-C-methyl-GTP incorporation by POLRMT enzyme(33).d Selectivity was determined as (Keff, POLRMT)2=-C-methyl-GTP from DAPN-PD1/(Keff, POLRMT) 2=-Cmethyl-GTP from BMS-986094.
Ehteshami et al.
4666 aac.asm.org August 2016 Volume 60 Number 8Antimicrobial Agents and Chemotherapy
on May 25, 2018 by guest
http://aac.asm.org/
Dow
nloaded from

lighting that little is known about the phenotypic effects of rNAI-mediated mtRNA inhibition on mitochondrial protein produc-tion and function. Mitochondrial damage caused by inhibition ofDNA polymerase � by anti-HIV 3=-deoxynucleoside inhibitorshas been shown to gradually accumulate over time (52). It is notclear whether inhibition of mtRNA transcription with ribonucle-oside analog inhibitors would show similar kinetics with regard tocytotoxicity or whether deleterious effects would appear in a moreimmediate fashion. As more antiviral rNAIs are developed againstRNA viruses, a better understanding of the dynamics of transcrip-tion-mediated mitochondrial toxicity will be warranted. Further-more, it is conceivable that cells that require higher ATP con-sumption are more sensitive to changes in mtRNA transcriptlevels. Considering that cardiotoxicity was observed in BMS-986094 phase II clinical trials (31), high 2=-C-methyl-GTP mayaccumulate in cardiac tissue in a somewhat selective matter. Thisaccumulation may in turn have more immediate consequencesbecause of increased energy requirements for cardiac tissue. Inagreement with this hypothesis, we have observed that the metab-olism of BMS-986094 in cardiomyocytes leads to high intracellu-lar 2=-C-methyl-GTP accumulation (S. Tao, personal communi-cation).
Finally, we cannot exclude the possibility that other non-rNAI-TP components of ribonucleoside prodrugs contribute tocytotoxicity. Indeed, the intracellular metabolism of BMS-986094leads to the generation of potentially hazardous by-products, suchas 1-naphtol, neopentanol, and methanol. To address this issue,we synthesized each of the aforementioned metabolites separatelyand tested their effect on cytotoxicity in VERO, CEM, PBM, andHuh-7 cells. We did not observe toxicity with up to 100 �M eachmetabolite in any of the cell lines tested (data not shown). Thesedata suggest that the observed toxicity involves the antiviral mol-ecule as a whole. Importantly, it is worth noting that cellularmetabolism of the prodrug moiety of DAPN-PD1 results in thegeneration of dihydrocoumarin metabolites, a nontoxic foodadditive that has been in use for human consumption for over40 years (53, 54).
In conclusion, this study describes the biochemical propertiesof nucleoside metabolites of DAPN-PD1. Our cell-based (37) andcell-free assays demonstrate that the potency and resistance pro-files of DAPN-PD1 metabolites are comparable to that of thesofosbuvir metabolite 2=-C-methyl-2=-F-UTP. Assessment ofDAPN-PD1 and sofosbuvir metabolism also shows that compa-rable levels of intracellular rNAI-TP are achieved with each prod-rug in primary human hepatocytes (S. Tao, personal communica-tion). Direct comparisons between DAPN-PD1, BMS-986094,and sofosbuvir highlight the importance of prodrug group choicein maintaining antiviral activity while minimizing off-target inhi-bition of host polymerases. These findings have important impli-cations for addressing cytotoxicity with ribonucleoside analogs indevelopment. Finally, DAPN-PD1 can deliver intracellularly tworibonucleotide analog chain terminators with distinct incorpora-tion profiles, making it an attractive prodrug that needs to befurther preclinically developed.
ACKNOWLEDGMENTS
Emmanuela Anandarajah and Dana Ditje are acknowledged for technicalassistance with regard to NS5B protein purification. Emily Hammond isacknowledged for experimental assistance. Joseph Hollenbaugh and JudyMathew are acknowledged for careful reading of the manuscript.
S.S.L. and R.F.S. are founders and shareholders of Cocrystal Pharma,Inc. Emory received no funding from Cocrystal Pharma, Inc., to performthis work and vice versa.
FUNDING INFORMATIONThis work, including the efforts of Raymond F. Schinazi, was funded byCFAR. This work, including the efforts of Maryam Ehteshami, was fundedby NCRTP. This work, including the efforts of Raymond F. Schinazi, wasfunded by HHS | NIH | National Institute of Allergy and Infectious Dis-eases (NIAID) (5P30-AI-50409). This work, including the efforts ofMaryam Ehteshami, was funded by American Liver Foundation (ALF).
REFERENCES1. Hutin HKM, Dore GJ, Perz JF, Armstrong GL, Dusheiko G. 2004.
Global burden of disease (GBD) for hepatitis C. J Clin Pharmacol 44:20 –29. http://dx.doi.org/10.1177/0091270003258669.
2. Rein DB, Wittenborn JS, Weinbaum CM, Sabin M, Smith BD, LesesneSB. 2011. Forecasting the morbidity and mortality associated with preva-lent cases of pre-cirrhotic chronic hepatitis C in the United States. DigLiver Dis 43:66 –72. http://dx.doi.org/10.1016/j.dld.2010.05.006.
3. Hagan LM, Schinazi RF. 2013. Best strategies for global HCV eradication.Liver Int 33(Suppl 1):S68 –S79.
4. Wong JB, McQuillan GM, McHutchison JG, Poynard T. 2000. Estimat-ing future hepatitis C morbidity, mortality, and costs in the United States.Am J Public Health 90:1562–1569. http://dx.doi.org/10.2105/AJPH.90.10.1562.
5. Ogata N, Alter HJ, Miller RH, Purcell RH. 1991. Nucleotide sequenceand mutation rate of the H strain of hepatitis C virus. Proc Natl Acad SciU S A 88:3392–3396. http://dx.doi.org/10.1073/pnas.88.8.3392.
6. Cuevas JM, Gonzalez-Candelas F, Moya A, Sanjuan R. 2009. Effect ofribavirin on the mutation rate and spectrum of hepatitis C virus in vivo. JVirol 83:5760 –5764. http://dx.doi.org/10.1128/JVI.00201-09.
7. Schaefer EA, Chung RT. 2011. The impact of human gene polymor-phisms on HCV infection and disease outcome. Semin Liver Dis 31:375–386. http://dx.doi.org/10.1055/s-0031-1297926.
8. Zeuzem S, Berg T, Moeller B, Hinrichsen H, Mauss S, Wedemeyer H,Sarrazin C, Hueppe D, Zehnter E, Manns MP. 2009. Expert opinion onthe treatment of patients with chronic hepatitis C. J Viral Hepat 16:75–90.http://dx.doi.org/10.1111/j.1365-2893.2008.01012.x.
9. Zoulim F, Liang TJ, Gerbes AL, Aghemo A, Deuffic-Burban S, Dush-eiko G, Fried MW, Pol S, Rockstroh JK, Terrault NA, Wiktor S. 2015.Hepatitis C virus treatment in the real world: optimising treatment andaccess to therapies. Gut 64:1824 –1833. http://dx.doi.org/10.1136/gutjnl-2015-310421.
10. Bartenschlager R, Lohmann V, Penin F. 2013. The molecular and struc-tural basis of advanced antiviral therapy for hepatitis C virus infection. NatRev Microbiol 11:482– 496. http://dx.doi.org/10.1038/nrmicro3046.
11. Scheel TK, Rice CM. 2013. Understanding the hepatitis C virus life cyclepaves the way for highly effective therapies. Nat Med 19:837– 849. http://dx.doi.org/10.1038/nm.3248.
12. Wendt A, Adhoute X, Castellani P, Oules V, Ansaldi C, Benali S,Bourliere M. 2014. Chronic hepatitis C: future treatment. Clin Pharmacol6:1–17.
13. Kohler JJ, Nettles JH, Amblard F, Hurwitz SJ, Bassit L, Stanton RA,Ehteshami M, Schinazi RF. 2014. Approaches to hepatitis C treatmentand cure using NS5A inhibitors. Infect Drug Resist 7:41–56.
14. Coats SJ, Garnier-Amblard EC, Amblard F, Ehteshami M, Amiralaei S,Zhang H, Zhou L, Boucle SR, Lu X, Bondada L, Shelton JR, Li H, LiuP, Li C, Cho JH, Chavre SN, Zhou S, Mathew J, Schinazi RF. 2014.Chutes and ladders in hepatitis C nucleoside drug development. AntiviralRes 102:119 –147. http://dx.doi.org/10.1016/j.antiviral.2013.11.008.
15. Sofia MJ. 2013. Nucleotide prodrugs for the treatment of HCV infection.Adv Pharmacol 67:39 –73. http://dx.doi.org/10.1016/B978-0-12-405880-4.00002-0.
16. Schinazi RF, Shi J, Whitaker T. 2015. Sofosbuvir (Sovaldi): the first-in-class HCV NS5B nucleotide polymerase inhibitor, p 61– 80. Innovativedrug synthesis. John Wiley & Sons, Inc, Hoboken, NJ.
17. Donaldson EF, Harrington PR, O’Rear JJ, Naeger LK. 2015. Clinicalevidence and bioinformatics characterization of potential hepatitis C virusresistance pathways for Sofosbuvir. Hepatology 61:56 – 65. http://dx.doi.org/10.1002/hep.27375.
Inhibition of HCV Polymerase by DAPN-PD1 Metabolites
August 2016 Volume 60 Number 8 aac.asm.org 4667Antimicrobial Agents and Chemotherapy
on May 25, 2018 by guest
http://aac.asm.org/
Dow
nloaded from

18. Tong X, Le Pogam S, Li L, Haines K, Piso K, Baronas V, Yan JM, So SS,Klumpp K, Najera I. 2014. In vivo emergence of a novel mutant L159F/L320F in the NS5B polymerase confers low-level resistance to the HCVpolymerase inhibitors mericitabine and sofosbuvir. J Infect Dis 209:668 –675. http://dx.doi.org/10.1093/infdis/jit562.
19. Svarovskaia ES, Dvory-Sobol H, Parkin N, Hebner C, Gontcharova V,Martin R, Ouyang W, Han B, Xu S, Ku K, Chiu S, Gane E, Jacobson IM,Nelson DR, Lawitz E, Wyles DL, Bekele N, Brainard D, Symonds WT,McHutchison JG, Miller MD, Mo H. 2014. Infrequent development ofresistance in genotype 1-6 hepatitis C virus-infected subjects treated withsofosbuvir in phase 2 and 3 clinical trials. Clin Infect Dis 59:1666 –1674.http://dx.doi.org/10.1093/cid/ciu697.
20. Migliaccio G, Tomassini JE, Carroll SS, Tomei L, Altamura S, Bhat B,Bartholomew L, Bosserman MR, Ceccacci A, Colwell LF, Cortese R, DeFrancesco R, Eldrup AB, Getty KL, Hou XS, LaFemina RL, LudmererSW, MacCoss M, McMasters DR, Stahlhut MW, Olsen DB, Hazuda DJ,Flores OA. 2003. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replica-tion in vitro. J Biol Chem 278:49164 – 49170. http://dx.doi.org/10.1074/jbc.M305041200.
21. Olsen DB, Eldrup AB, Bartholomew L, Bhat B, Bosserman MR, Cec-cacci A, Colwell LF, Fay JF, Flores OA, Getty KL, Grobler JA, LaFeminaRL, Markel EJ, Migliaccio G, Prhavc M, Stahlhut MW, Tomassini JE,MacCoss M, Hazuda DJ, Carroll SS. 2004. A 7-deaza-adenosine analog isa potent and selective inhibitor of hepatitis C virus replication with excel-lent pharmacokinetic properties. Antimicrob Agents Chemother 48:3944 –3953. http://dx.doi.org/10.1128/AAC.48.10.3944-3953.2004.
22. Le Pogam S, Jiang WR, Leveque V, Rajyaguru S, Ma H, Kang H, JiangS, Singer M, Ali S, Klumpp K, Smith D, Symons J, Cammack N, NajeraI. 2006. In vitro selected Con1 subgenomic replicons resistant to 2=-C-methyl-cytidine or to R1479 show lack of cross resistance. Virology 351:349 –359. http://dx.doi.org/10.1016/j.virol.2006.03.045.
23. Ludmerer SW, Graham DJ, Boots E, Murray EM, Simcoe A, Markel EJ,Grobler JA, Flores OA, Olsen DB, Hazuda DJ, LaFemina RL. 2005.Replication fitness and NS5B drug sensitivity of diverse hepatitis C virusisolates characterized by using a transient replication assay. AntimicrobAgents Chemother 49:2059 –2069. http://dx.doi.org/10.1128/AAC.49.5.2059-2069.2005.
24. Asselah T. 2014. Sofosbuvir for the treatment of hepatitis C virus. ExpertOpin Pharmacother 15:121–130. http://dx.doi.org/10.1517/14656566.2014.857656.
25. Jacobson IM, Gordon SC, Kowdley KV, Yoshida EM, Rodriguez-TorresM, Sulkowski MS, Shiffman ML, Lawitz E, Everson G, Bennett M, SchiffE, Al-Assi MT, Subramanian GM, An D, Lin M, McNally J, Brainard D,Symonds WT, McHutchison JG, Patel K, Feld J, Pianko S, Nelson DR.2013. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treat-ment options. N Engl J Med 368:1867–1877. http://dx.doi.org/10.1056/NEJMoa1214854.
26. Lawitz E, Mangia A, Wyles D, Rodriguez-Torres M, Hassanein T,Gordon SC, Schultz M, Davis MN, Kayali Z, Reddy KR, Jacobson IM,Kowdley KV, Nyberg L, Subramanian GM, Hyland RH, Arterburn S,Jiang D, McNally J, Brainard D, Symonds WT, McHutchison JG,Sheikh AM, Younossi Z, Gane EJ. 2013. Sofosbuvir for previously un-treated chronic hepatitis C infection. N Engl J Med 368:1878 –1887. http://dx.doi.org/10.1056/NEJMoa1214853.
27. McGuigan C, Madela K, Aljarah M, Gilles A, Brancale A, Zonta N,Chamberlain S, Vernachio J, Hutchins J, Hall A, Ames B, Gorovits E,Ganguly B, Kolykhalov A, Wang J, Muhammad J, Patti JM, Henson G.2010. Design, synthesis and evaluation of a novel double pro-drug: INX-08189. A new clinical candidate for hepatitis C virus. Bioorg Med ChemLett 20:4850 – 4854.
28. Vernachio JH, Bleiman B, Bryant KD, Chamberlain S, Hunley D,Hutchins J, Ames B, Gorovits E, Ganguly B, Hall A, Kolykhalov A, LiuY, Muhammad J, Raja N, Walters CR, Wang J, Williams K, Patti JM,Henson G, Madela K, Aljarah M, Gilles A, McGuigan C. 2011. INX-08189, a phosphoramidate prodrug of 6-O-methyl-2=-C-methyl guanos-ine, is a potent inhibitor of hepatitis C virus replication with excellentpharmacokinetic and pharmacodynamic properties. Antimicrob AgentsChemother 55:1843–1851. http://dx.doi.org/10.1128/AAC.01335-10.
29. Sheridan C. 2012. Calamitous HCV trial casts shadow over nucleosidedrugs. Nat Biotechnol 30:1015–1016. http://dx.doi.org/10.1038/nbt1112-1015.
30. Pollack A, Gale J. 2 August 2012. BMS suspends study of nucleotide
BMS094 formerly INX189. http://www.natap.org/2012/HCV/080312_01.htm.
31. Ahmad T, Yin P, Saffitz J, Pockros PJ, Lalezari J, Shiffman M, FreilichB, Zamparo J, Brown K, Dimitrova D, Kumar M, Manion D, Heath-Chiozzi M, Wolf R, Hughes E, Muir AJ, Hernandez AF. 2014. Cardiacdysfunction associated with a nucleotide polymerase inhibitor for treat-ment of hepatitis C. Hepatology 62:409 – 416.
32. Arnold JJ, Sharma SD, Feng JY, Ray AS, Smidansky ED, Kireeva ML,Cho A, Perry J, Vela JE, Park Y, Xu Y, Tian Y, Babusis D, Barauskus O,Peterson BR, Gnatt A, Kashlev M, Zhong W, Cameron CE. 2012.Sensitivity of mitochondrial transcription and resistance of RNA poly-merase II dependent nuclear transcription to antiviral ribonucleosides.PLoS Pathog 8:e1003030. http://dx.doi.org/10.1371/journal.ppat.1003030.
33. Feng JY, Xu Y, Barauskas O, Perry JK, Ahmadyar S, Stepan G, Yu H,Babusis D, Park Y, McCutcheon K, Perron M, Schultz BE, Sakowicz R,Ray AS. 2015. Role of the mitochondrial RNA polymerase in the toxicityof nucleotide inhibitors of the hepatitis C virus. Antimicrob Agents Che-mother 60:806 – 817.
34. Sofia MJ. 2014. Beyond sofosbuvir: what opportunity exists for a betternucleoside/nucleotide to treat hepatitis C? Antiviral Res 107:119 –124.http://dx.doi.org/10.1016/j.antiviral.2014.04.008.
35. McQuaid T, Savini C, Seyedkazemi S. 2015. Sofosbuvir, a significantparadigm change in HCV treatment. J Clin Transl Hepatol 3:27–35. http://dx.doi.org/10.14218/JCTH.2014.00041.
36. Hagan LM, Wolpe PR, Schinazi RF. 2013. Treatment as prevention andcure towards global eradication of hepatitis C virus. Trends Microbiol21:625– 633. http://dx.doi.org/10.1016/j.tim.2013.09.008.
37. Zhou L, Zhang HW, Tao S, Bassit L, Whitaker T, McBrayer TR,Ehteshami M, Amiralaei S, Pradere U, Cho JH, Amblard F, BobeckD, Detorio M, Coats SJ, Schinazi RF. 2015. �-D-2=-C-methyl-2,6-diaminopurine ribonucleoside phosphoramidates are potent and se-lective inhibitors of hepatitis C virus (HCV) and are bioconvertedintracellularly to bioactive 2,6-diaminopurine and guanosine 5=-triphosphate forms. J Med Chem 58:3445–3458. http://dx.doi.org/10.1021/jm501874e.
38. Powdrill MH, Tchesnokov EP, Kozak RA, Russell RS, Martin R,Svarovskaia ES, Mo H, Kouyos RD, Gotte M. 2011. Contribution of amutational bias in hepatitis C virus replication to the genetic barrier in thedevelopment of drug resistance. Proc Natl Acad Sci U S A 108:20509 –20513. http://dx.doi.org/10.1073/pnas.1105797108.
39. Ludwig W, Follmann H. 1978. The specificity of ribonucleoside triphos-phate reductase. Multiple induced activity changes and implications fordeoxyribonucleotide formation. Eur J Biochem 82:393– 403.
40. Ludwig J, Eckstein F. 1989. Rapid and efficient synthesis of nucleoside5=-0-(1-thiotriphosphates), 5=-triphosphates and 2=,3=-cyclophosphoro-thioates using 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one. J OrgChem 54:631– 635. http://dx.doi.org/10.1021/jo00264a024.
41. Kovács T, Otvös L. 1988. Simple synthesis of 5-vinyl- and 5-ethynyl-2=-deoxyuridine-5=-triphosphates. Tetrahedron Lett 29:4525– 4528. http://dx.doi.org/10.1016/S0040-4039(00)80537-7.
42. Powdrill MH, Deval J, Narjes F, De Francesco R, Gotte M. 2010.Mechanism of hepatitis C virus RNA polymerase inhibition with dihy-droxypyrimidines. Antimicrob Agents Chemother 54:977–983. http://dx.doi.org/10.1128/AAC.01216-09.
43. Jin Z, Leveque V, Ma H, Johnson KA, Klumpp K. 2012. Assembly,purification, and pre-steady-state kinetic analysis of active RNA-dependent RNA polymerase elongation complex. J Biol Chem 287:10674 –10683. http://dx.doi.org/10.1074/jbc.M111.325530.
44. Zhang HW, Detorio M, Herman BD, Solomon S, Bassit L, Nettles JH,Obikhod A, Tao SJ, Mellors JW, Sluis-Cremer N, Coats SJ, Schinazi RF.2011. Synthesis, antiviral activity, cytotoxicity and cellular pharmacologyof l-3=-azido-2=,3=-dideoxypurine nucleosides. Eur J Med Chem 46:3832–3844. http://dx.doi.org/10.1016/j.ejmech.2011.05.051.
45. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression datausing real-time quantitative PCR and the 2(-delta delta C(T)) method.Methods 25:402– 408. http://dx.doi.org/10.1006/meth.2001.1262.
46. Eldrup AB, Allerson CR, Bennett CF, Bera S, Bhat B, Bhat N,Bosserman MR, Brooks J, Burlein C, Carroll SS, Cook PD, Getty KL,MacCoss M, McMasters DR, Olsen DB, Prakash TP, Prhavc M, SongQ, Tomassini JE, Xia J. 2004. Structure-activity relationship of purineribonucleosides for inhibition of hepatitis C virus RNA-dependent
Ehteshami et al.
4668 aac.asm.org August 2016 Volume 60 Number 8Antimicrobial Agents and Chemotherapy
on May 25, 2018 by guest
http://aac.asm.org/
Dow
nloaded from

RNA polymerase. J Med Chem 47:2283–2295. http://dx.doi.org/10.1021/jm030424e.
47. Cheong C, Tinoco I, Jr, Chollet A. 1988. Thermodynamic studies of basepairing involving 2,6-diaminopurine. Nucleic Acids Res 16:5115–5122.http://dx.doi.org/10.1093/nar/16.11.5115.
48. Strobel SA, Cech TR, Usman N, Beigelman L. 1994. The 2,6-diaminopurine riboside.5-methylisocytidine wobble base pair: an isoen-ergetic substitution for the study of GU pairs in RNA. Biochemistry 33:13824 –13835. http://dx.doi.org/10.1021/bi00250a037.
49. Le Pogam S, Kang H, Harris SF, Leveque V, Giannetti AM, Ali S, JiangWR, Rajyaguru S, Tavares G, Oshiro C, Hendricks T, Klumpp K, SymonsJ, Browner MF, Cammack N, Najera I. 2006. Selection and characterizationof replicon variants dually resistant to thumb- and palm-binding nonnucleo-side polymerase inhibitors of the hepatitis C virus. J Virol 80:6146–6154. http://dx.doi.org/10.1128/JVI.02628-05.
50. Klumpp K, Leveque V, Le Pogam S, Ma H, Jiang WR, Kang H,
Granycome C, Singer M, Laxton C, Hang JQ, Sarma K, Smith DB,Heindl D, Hobbs CJ, Merrett JH, Symons J, Cammack N, Martin JA,Devos R, Najera I. 2006. The novel nucleoside analog R1479 (4=-azidocytidine) is a potent inhibitor of NS5B-dependent RNA synthesisand hepatitis C virus replication in cell culture. J Biol Chem 281:3793–3799. http://dx.doi.org/10.1074/jbc.M510195200.
51. Pockros P. 2013. Lessons learned from failed clinical trials in hepatitis Cdrug development. HepDART, Big Island, HI.
52. Kohler JJ, Lewis W. 2007. A brief overview of mechanisms of mitochon-drial toxicity from NRTIs. Environ Mol Mutagen 48:166 –172. http://dx.doi.org/10.1002/em.20223.
53. Rastogi SC, Johansen JD, Menne T. 1996. Natural ingredients basedcosmetics. Content of selected fragrance sensitizers. Contact Dermat 34:423– 426.
54. Fenaroli G. 2010. Fenaroli’s handbook of flavor ingredients, 6th ed. CRCPress, Boca Raton, FL.
Inhibition of HCV Polymerase by DAPN-PD1 Metabolites
August 2016 Volume 60 Number 8 aac.asm.org 4669Antimicrobial Agents and Chemotherapy
on May 25, 2018 by guest
http://aac.asm.org/
Dow
nloaded from