acidosis and alkalosis - mcgraw-hill education - … · metabolic acidosis ... from the...
TRANSCRIPT

Thomas D. DuBose, Jr.
NORMAL ACID–BASE HOMEOSTASIS
Systemic arterial pH is maintained between 7.35 and7.45 by extracellular and intracellular chemical bufferingtogether with respiratory and renal regulatory mecha-nisms. The control of arterial CO2 tension (PaCO2
) bythe central nervous system (CNS) and respiratory sys-tem and the control of the plasma bicarbonate by thekidneys stabilize the arterial pH by excretion or reten-tion of acid or alkali. The metabolic and respiratorycomponents that regulate systemic pH are described bythe Henderson-Hasselbalch equation:
pH = 6.1 + log
Under most circumstances, CO2 production and excre-tion are matched, and the usual steady-state PaCO2
is main-tained at 40 mmHg. Underexcretion of CO2 produceshypercapnia, and overexcretion causes hypocapnia. Never-theless, production and excretion are again matched at anew steady-state PaCO2
. Therefore, the PaCO2is regulated
primarily by neural respiratory factors (Chap. 22) and isnot subject to regulation by the rate of CO2 production.Hypercapnia is usually the result of hypoventilation ratherthan of increased CO2 production. Increases or decreasesin PaCO2
represent derangements of neural respiratorycontrol or are caused by compensatory changes inresponse to a primary alteration in the plasma [HCO3
–].
ACIDOSIS AND ALKALOSIS
The kidneys regulate plasma HCO3− through three
main processes: (1) “reabsorption” of filtered HCO3–,
(2) formation of titratable acid, and (3) excretion ofNH4
+ in the urine. The kidney filters ~4000 mmol ofHCO3
– per day.To reabsorb the filtered load of HCO3−,
the renal tubules must therefore secrete 4000 mmol ofhydrogen ions. Between 80 and 90% of HCO3
− is reab-sorbed in the proximal tubule.The distal nephron reab-sorbs the remainder and secretes protons, as generatedfrom metabolism, to defend systemic pH. Although thisquantity of protons, 40–60 mmol/d, is small, it must besecreted to prevent chronic positive H+ balance andmetabolic acidosis. This quantity of secreted protons isrepresented in the urine as titratable acid and NH4
+.Metabolic acidosis in the face of normal renal functionincreases NH4
+ production and excretion. NH4+ pro-
duction and excretion are impaired in chronic renal fail-ure, hyperkalemia, and renal tubular acidosis.
In sum, these regulatory responses, including chemicalbuffering, the regulation of PaCO2
by the respiratory system,and the regulation of HCO3
− by the kidneys, act in concertto maintain a systemic arterial pH between 7.35 and 7.45.
DIAGNOSIS OF GENERAL TYPES OF DISTURBANCES
The most common clinical disturbances are simpleacid–base disorders (i.e., metabolic acidosis or alkalosis
CHAPTER 40CHAPTER 40
410
� Normal Acid–Base Homeostasis . . . . . . . . . . . . . . . . . . . . . .410� Diagnosis of General Types of Disturbances . . . . . . . . . . . . .410
Simple Acid–Base Disorders . . . . . . . . . . . . . . . . . . . . . . . . .411Mixed Acid–Base Disorders . . . . . . . . . . . . . . . . . . . . . . . . . .412
� Metabolic Acidosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .413High-Anion-Gap Acidoses . . . . . . . . . . . . . . . . . . . . . . . . . . .414Hyperchloremic (Nongap) Metabolic Acidoses . . . . . . . . . . . .417
� Metabolic Alkalosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .418Pathogenesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .418
Differential Diagnosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .418Metabolic Alkalosis Associated with Extracellular Fluid Volume Contraction, K+ Depletion, and Secondary Hyperreninemic Hyperaldosteronism . . . . . . . . . . . . . . . . . .419
Metabolic Alkalosis Associated with Extracellular Fluid Volume Expansion, Hypertension, and Hyperaldosteronism . . . . . . .420
� Respiratory Acidosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .421� Respiratory Alkalosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .422� Further Readings . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .423
HCO3–
PaCO2× 0.0301
Loscalzo_Pulmonary-Ch40_p410-p423.qxd 2/11/10 4:04 PM Page 410
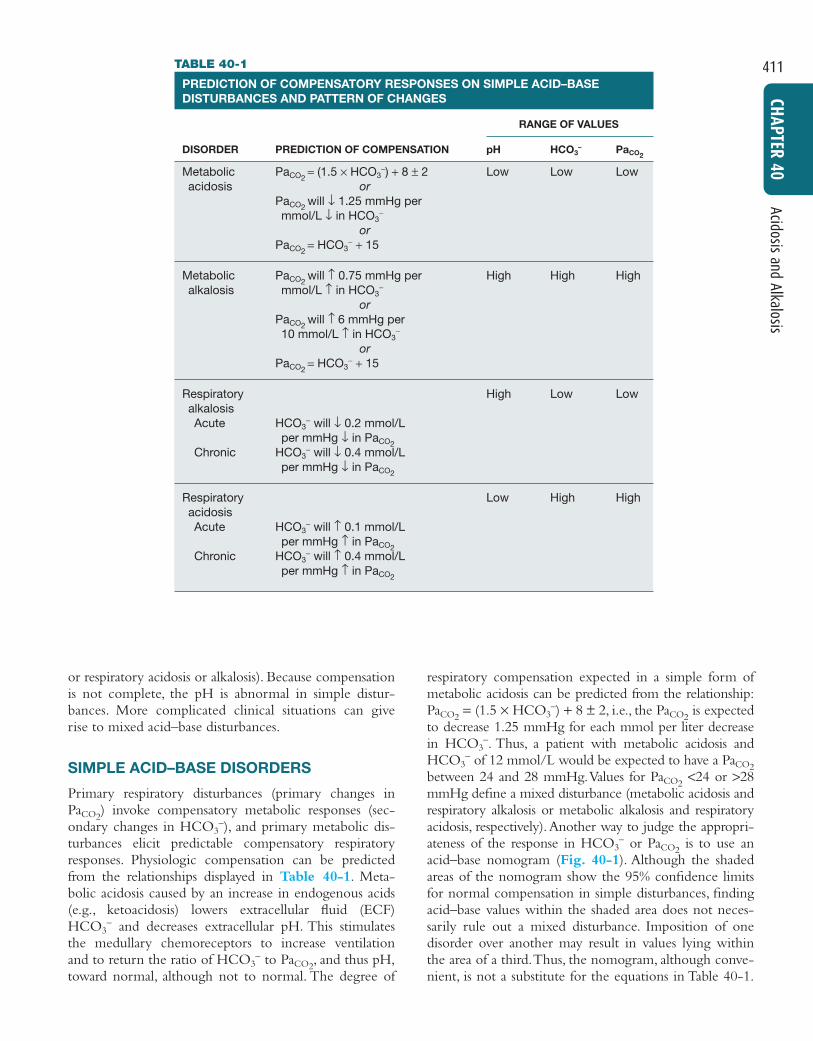
or respiratory acidosis or alkalosis). Because compensationis not complete, the pH is abnormal in simple distur-bances. More complicated clinical situations can giverise to mixed acid–base disturbances.
SIMPLE ACID–BASE DISORDERS
Primary respiratory disturbances (primary changes inPaCO2
) invoke compensatory metabolic responses (sec-ondary changes in HCO3
−), and primary metabolic dis-turbances elicit predictable compensatory respiratoryresponses. Physiologic compensation can be predictedfrom the relationships displayed in Table 40-1. Meta-bolic acidosis caused by an increase in endogenous acids(e.g., ketoacidosis) lowers extracellular fluid (ECF)HCO3
− and decreases extracellular pH. This stimulatesthe medullary chemoreceptors to increase ventilationand to return the ratio of HCO3
− to PaCO2, and thus pH,
toward normal, although not to normal. The degree of
Acidosis and Alkalosis
411
CHAPTER 40
respiratory compensation expected in a simple form ofmetabolic acidosis can be predicted from the relationship:PaCO2
= (1.5 × HCO3−) + 8 ± 2, i.e., the PaCO2
is expectedto decrease 1.25 mmHg for each mmol per liter decreasein HCO3
−. Thus, a patient with metabolic acidosis andHCO3
− of 12 mmol/L would be expected to have a PaCO2between 24 and 28 mmHg.Values for PaCO2
<24 or >28mmHg define a mixed disturbance (metabolic acidosis andrespiratory alkalosis or metabolic alkalosis and respiratoryacidosis, respectively).Another way to judge the appropri-ateness of the response in HCO3
− or PaCO2is to use an
acid–base nomogram (Fig. 40-1). Although the shadedareas of the nomogram show the 95% confidence limitsfor normal compensation in simple disturbances, findingacid–base values within the shaded area does not neces-sarily rule out a mixed disturbance. Imposition of onedisorder over another may result in values lying withinthe area of a third.Thus, the nomogram, although conve-nient, is not a substitute for the equations in Table 40-1.
TABLE 40-1
PREDICTION OF COMPENSATORY RESPONSES ON SIMPLE ACID–BASEDISTURBANCES AND PATTERN OF CHANGES
RANGE OF VALUES
DISORDER PREDICTION OF COMPENSATION pH HCO3– PaCO2
Metabolic PaCO2= (1.5 × HCO3
−) + 8 ± 2 Low Low Lowacidosis or
PaCO2will ↓ 1.25 mmHg per
mmol/L ↓ in HCO3−
orPaCO2
= HCO3− + 15
Metabolic PaCO2will ↑ 0.75 mmHg per High High High
alkalosis mmol/L ↑ in HCO3−
orPaCO2
will ↑ 6 mmHg per 10 mmol/L ↑ in HCO3
−
orPaCO2
= HCO3− + 15
Respiratory High Low LowalkalosisAcute HCO3
− will ↓ 0.2 mmol/L per mmHg ↓ in PaCO2
Chronic HCO3− will ↓ 0.4 mmol/L
per mmHg ↓ in PaCO2
Respiratory Low High HighacidosisAcute HCO3
− will ↑ 0.1 mmol/L per mmHg ↑ in PaCO2
Chronic HCO3− will ↑ 0.4 mmol/L
per mmHg ↑ in PaCO2
Loscalzo_Pulmonary-Ch40_p410-p423.qxd 2/11/10 4:04 PM Page 411

MIXED ACID–BASE DISORDERS
Mixed acid–base disorders—defined as independentlycoexisting disorders, not merely compensatory responses—are often seen in patients in critical care units and can leadto dangerous extremes of pH (Table 40-2). A patientwith diabetic ketoacidosis (DKA; metabolic acidosis)may develop an independent respiratory problem lead-ing to respiratory acidosis or alkalosis. Patients withunderlying pulmonary disease may not respond to meta-bolic acidosis with an appropriate ventilatory responsebecause of insufficient respiratory reserve. Such imposi-tion of respiratory acidosis on metabolic acidosis canlead to severe acidemia and a poor outcome. Whenmetabolic acidosis and metabolic alkalosis coexist in thesame patient, the pH may be normal or near normal.When the pH is normal, an elevated anion gap (AG; seebelow) denotes the presence of a metabolic acidosis. Adiscrepancy in the ΔAG (prevailing minus normal AG)and the ΔHCO3
− (normal minus prevailing HCO3− )
indicates the presence of a mixed high-gap acidosis—metabolic alkalosis (see example below). A diabeticpatient with ketoacidosis may have renal dysfunctionresulting in simultaneous metabolic acidosis. Patientswho have ingested an overdose of drug combinationssuch as sedatives and salicylates may have mixed distur-bances as a result of the acid–base response to the indi-vidual drugs (metabolic acidosis mixed with respiratoryacidosis or respiratory alkalosis, respectively). Even morecomplex are triple acid–base disturbances. For example,
412
Disorders Complicating Critical Illnesses and Their M
anagement
SECTION V
patients with metabolic acidosis caused by alcoholicketoacidosis (AKA) may develop metabolic alkalosisfrom vomiting and superimposed respiratory alkalosisbecause of the hyperventilation of hepatic dysfunctionor alcohol withdrawal.
FIGURE 40-1Acid–base nomogram. Shown are the 90% confidence lim-its (range of values) of the normal respiratory and metaboliccompensations for primary acid–base disturbances. (FromDuBose, used with permission.)
Arterial blood [H+] (nmol/L)
100 90 80 70 60 50 40 30 2060
56
52
48
44
40
36
32
28
24
20
16
12
8
4
07.0 7.1 7.2 7.3 7.4 7.5 7.6 7.7 7.8
Arterial blood, pH
120 100 90 80 70 60 50 40110
35
30
25
20
15
10
Normal
PCO2(mmHg)
Acuterespiratoryalkalosis
Acuterespiratoryacidosis
Chronicrespiratoryalkalosis
Chronicrespiratoryacidosis
Metabolicacidosis
Metabolicalkalosis
Art
eria
l pla
sma
[HC
O3]
(m
mol
/L)
–TABLE 40-2
EXAMPLES OF MIXED ACID–BASE DISORDERS
Mixed Metabolic and Respiratory
Metabolic acidosis—respiratory alkalosisKey: High- or normal-AG metabolic acidosis; prevailingPaCO2
below predicted value (Table 40-1)Example: Na+, 140; K+, 4.0; Cl−, 106; HCO3
−, 14; AG, 20;PaCO2
, 24; pH, 7.39 (lactic acidosis, sepsis in the ICU)
Metabolic acidosis—respiratory acidosisKey: High- or normal-AG metabolic acidosis; prevailing PaCO2
above predicted value (Table 40-1)Example: Na+, 140; K+, 4.0; Cl−, 102; HCO3
−, 18; AG, 20; PaCO2
, 38; pH, 7.30 (severe pneumonia, pulmonary edema)
Metabolic alkalosis—respiratory alkalosisKey: PaCO2
does not increase as predicted; pH higher than expected
Example: Na+, 140; K+, 4.0; Cl−, 91; HCO3−, 33; AG, 16;
PaCO2, 38; pH, 7.55 (liver disease and diuretics)
Metabolic alkalosis—respiratory acidosisKey: PaCO2
higher than predicted; pH normalExample: Na+, 140; K+, 3.5; Cl−, 88; HCO3
−, 42; AG, 10; PaCO2
, 67; pH, 7.42 (COPD on diuretics)
Mixed Metabolic Disorders
Metabolic acidosis—metabolic alkalosisKey: Only detectable with high-AG acidosis; ΔAG >>ΔHCO3
–
Example: Na+, 140; K+, 3.0; Cl−, 95; HCO3−, 25; AG, 20;
PaCO2, 40; pH, 7.42 (uremia with vomiting)
Metabolic acidosis—metabolic acidosisKey: Mixed high-AG–normal-AG acidosis; ΔHCO3
−
accounted for by combined change in ΔAG and ΔCl−
Example: Na+, 135; K+, 3.0; Cl−, 110; HCO3−, 10; AG, 15;
PaCO2, 25; pH, 7.20 (diarrhea and lactic acidosis,
toluene toxicity, treatment of diabetic ketoacidosis)
Note: AG, anion gap; COPD, chronic obstructive pulmonary disease;ICU, intensive care unit.
Approach to the Patient:ACID–BASE DISORDERS
A stepwise approach to the diagnosis of acid–basedisorders follows (Table 40-3). Care should be takenwhen measuring blood gases to obtain the arterialblood sample without using excessive heparin. Bloodfor electrolytes and arterial blood gases should be
Loscalzo_Pulmonary-Ch40_p410-p423.qxd 2/11/10 4:04 PM Page 412

Acidosis and Alkalosis
413
CHAPTER 40
drawn simultaneously before therapy because anincrease in HCO3
− occurs with metabolic alkalosisand respiratory acidosis. Conversely, a decrease inHCO3
− occurs in metabolic acidosis and respiratoryalkalosis. In the determination of arterial blood gasesby the clinical laboratory, both pH and PaCO2
aremeasured, and the HCO3
− is calculated from theHenderson-Hasselbalch equation. This calculatedvalue should be compared with the measured HCO3
−
(total CO2) on the electrolyte panel.These two valuesshould agree within 2 mmol/L. If they do not, thevalues may not have been drawn simultaneously, alaboratory error may be present, or an error couldhave been made in calculating the HCO3
−.After veri-fying the blood acid–base values, one can then iden-tify the precise acid–base disorder.
CALCULATE THE ANION GAP All evaluationsof acid–base disorders should include a simple calcu-lation of the AG; it represents those unmeasuredanions in plasma (normally 10–12 mmol/L) and iscalculated as follows: AG = Na+ − (Cl− + HCO3
−).The unmeasured anions include anionic proteins,phosphate, sulfate, and organic anions. When acidanions, such as acetoacetate and lactate, accumulate inECF, the AG increases, causing a high-AG acidosis.Anincrease in the AG is most often caused by an increasein unmeasured anions and less commonly is caused bya decrease in unmeasured cations (calcium, magnesium,potassium). In addition, the AG may increase with anincrease in anionic albumin because of either increasedalbumin concentration or alkalosis, which alters albu-min charge.A decrease in the AG can be caused by (1)an increase in unmeasured cations; (2) the addition tothe blood of abnormal cations, such as lithium (lithiumintoxication) or cationic immunoglobulins (plasma celldyscrasias); (3) a reduction in the major plasma anionalbumin concentration (nephrotic syndrome); (4) adecrease in the effective anionic charge on albuminby acidosis; or (5) hyperviscosity and severe hyperlipi-
TABLE 40-3
STEPS IN ACID–BASE DIAGNOSIS
1. Obtain arterial blood gas (ABG) and electrolytes simultaneously.
2. Compare HCO3− on ABG and electrolytes to verify
accuracy.3. Calculate anion gap (AG).4. Know four causes of high-AG acidosis (ketoacidosis,
lactic acid acidosis, renal failure, and toxins).5. Know two causes of hyperchloremic or nongap acidosis
(bicarbonate loss from GI tract, renal tubular acidosis).6. Estimate compensatory response (Table 40-1).7. Compare ΔAG and ΔHCO3
−.8. Compare change in Cl− with change in Na+.
demia, which can lead to an underestimation ofsodium and chloride concentrations. A decrease inserum albumin by 1 g/dL from the normal value(4.5 g/dL) decreases the AG by 2.5 meq/L. It isimportant to know the common causes of high-AGacidosis (Table 40-3).
In the face of a normal serum albumin, a high AGis usually caused by non–chloride-containing acidsthat contain inorganic (phosphate, sulfate), organic(ketoacids, lactate, uremic organic anions), exogenous(salicylate or ingested toxins with organic acid pro-duction), or unidentified anions.The high AG is sig-nificant even if an additional acid–base disorder issuperimposed to modify the HCO3
− independently.Simultaneous metabolic acidosis of the high-AG vari-ety plus either chronic respiratory acidosis or meta-bolic alkalosis represents such a situation in whichHCO3
− may be normal or even high (Table 40-2).Compare the change in HCO3
− (ΔHCO3−) and the
change in the AG (ΔAG).Similarly, normal values for HCO3
−, PaCO2, and pH
do not ensure the absence of an acid–base disturbance.For instance, an alcoholic who has been vomiting maydevelop a metabolic alkalosis with a pH of 7.55, PaCO2of 48 mmHg, HCO3
− of 40 mmol/L, Na+ of 135, Cl−
of 80, and K+ of 2.8. If such a patient were then todevelop a superimposed AKA with a β-hydroxybu-tyrate concentration of 15 mM, arterial pH woulddecrease to 7.40, HCO3
− to 25 mmol/L, and PaCO2to
40 mmHg.Although these blood gas levels are normal,the AG is elevated at 30 mmol/L, indicating a mixedmetabolic alkalosis and metabolic acidosis. A mixtureof high-gap acidosis and metabolic alkalosis is recog-nized easily by comparing the differences (Δ values) inthe normal to prevailing patient values. In this exam-ple, the ΔHCO3
– is 0 (25 − 25 mmol/L) but the ΔAGis 20 (30 − 10 mmol/L). Therefore, 20 mmol/L isunaccounted for in the Δ/Δ value (ΔAG to ΔHCO3
−).
METABOLIC ACIDOSIS
Metabolic acidosis can occur because of an increase inendogenous acid production (e.g., lactate and ketoacids),loss of bicarbonate (as in diarrhea), or accumulation ofendogenous acids (as in renal failure). Metabolic acidosishas profound effects on the respiratory, cardiac, and ner-vous systems. The decrease in blood pH is accompaniedby a characteristic increase in ventilation, especially thetidal volume (Kussmaul respiration). Intrinsic cardiac con-tractility may be depressed, but inotropic function can benormal because of catecholamine release. Both peripheralarterial vasodilation and central venoconstriction can bepresent; the decrease in central and pulmonary vascular
Loscalzo_Pulmonary-Ch40_p410-p423.qxd 2/11/10 4:04 PM Page 413

414
Disorders Complicating Critical Illnesses and Their M
anagement
SECTION V
compliance predisposes to pulmonary edema with evenminimal volume overload. CNS function is depressed,with headache, lethargy, stupor, and, in some cases, evencoma. Glucose intolerance may also occur.
The two major categories of clinical metabolic acido-sis are high-AG and normal-AG, or hyperchloremic aci-dosis (Table 40-3 and Table 40-4).
HIGH-ANION-GAP ACIDOSESTABLE 40-4
CAUSES OF HIGH-ANION-GAP METABOLICACIDOSIS
Lactic acidosis ToxinsKetoacidosis Ethylene glycol
Diabetic MethanolAlcoholic SalicylatesStarvation Propylene glycol
Pyroglutamic acidRenal failure (acute and chronic)
Treatment:METABOLIC ACIDOSIS
Treatment of metabolic acidosis with alkali should bereserved for severe acidemia except when the patienthas no “potential HCO3
−” in plasma. Potential HCO3− can
be estimated from the increment (Δ) in the AG (ΔAG =patient’s AG − 10). It must be determined if the acidanion in plasma is metabolizable (i.e., β-hydroxybu-tyrate, acetoacetate, and lactate) or nonmetabolizable(anions that accumulate in chronic renal failure andafter toxin ingestion). The latter requires return of renalfunction to replenish the HCO3
− deficit, a slow and oftenunpredictable process. Consequently, patients with anormal AG acidosis (hyperchloremic acidosis), a slightlyelevated AG (mixed hyperchloremic and AG acidosis), oran AG attributable to a nonmetabolizable anion in theface of renal failure should receive alkali therapy, eitherPO (NaHCO3 or Shohl’s solution) or IV (NaHCO3), in anamount necessary to slowly increase the plasma HCO3
−
into the 20–22 mmol/L range.Controversy exists, however, regarding the use of
alkali in patients with a pure AG acidosis owing toaccumulation of a metabolizable organic acid anion(ketoacidosis or lactic acidosis). In general, severeacidosis (pH < 7.20) warrants the IV administration of50–100 meq of NaHCO3 over 30–45 min, during the ini-tial 1–2 h of therapy. Provision of such modest quanti-ties of alkali in this situation seems to provide an addedmeasure of safety, but it is essential to monitor plasmaelectrolytes during the course of therapy because the K+
may decline as pH increases. The goal is to increase theHCO3
− to 10 meq/L and the pH to 7.15, not to increasethese values to normal.
Approach to the Patient:HIGH-ANION-GAP ACIDOSES
The four principal causes of a high-AG acidosis are lac-tic acidosis, ketoacidosis, ingested toxins, and acute andchronic renal failure (see Table 40-4).The initial screen-ing to differentiate the high-AG acidoses shouldinclude (1) a probe of the history for evidence of drugand toxin ingestion and measurement of arterial bloodgas to detect coexistent respiratory alkalosis (salicylates);(2) determination of whether diabetes mellitus is pre-sent (DKA); (3) a search for evidence of alcoholism orincreased levels of β-hydroxybutyrate (AKA); (4) obser-vation for clinical signs of uremia and determination ofthe blood urea nitrogen (BUN) and creatinine (uremicacidosis); (5) inspection of the urine for oxalate crystals(ethylene glycol); and (6) recognition of the numerousclinical settings in which lactate levels may be increased(hypotension, shock, cardiac failure, leukemia, cancer,drug or toxin ingestion).
Lactic Acidosis
An increase in plasma L-lactate may be secondary to poortissue perfusion (type A)—circulatory insufficiency (shock,cardiac failure), severe anemia, mitochondrial enzymedefects, and inhibitors (carbon monoxide, cyanide)—or toaerobic disorders (type B)—malignancies, nucleoside ana-logue reverse transcriptase inhibitors in HIV, diabetes mel-litus, renal or hepatic failure, thiamine deficiency, severeinfections (cholera, malaria), seizures, or drugs or toxins(biguanides, ethanol, methanol, propylene glycol, isoniazid,and fructose). Propylene glycol may be used as a vehiclefor IV medications, including lorazepam, and toxicity hasbeen reported in several settings. Unrecognized bowelischemia or infarction in a patient with severe atheroscle-rosis or cardiac decompensation receiving vasopressors is acommon cause of lactic acidosis. Pyroglutamic acidemiahas been reported in critically ill patients receiving aceta-minophen, which is associated with depletion of glu-tathione. D-Lactic acid acidosis, which may be associatedwith jejunoileal bypass, short bowel syndrome, or intestinalobstruction, is caused by formation of D-lactate by gutbacteria.
Approach to the Patient:LACTIC ACID ACIDOSIS
The underlying condition that disrupts lactate metabo-lism must first be corrected, and tissue perfusion mustbe restored when inadequate. Vasoconstrictors shouldbe avoided, if possible, because they may worsen tissueperfusion. Alkali therapy is generally advocated for
Loscalzo_Pulmonary-Ch40_p410-p423.qxd 2/11/10 4:04 PM Page 414

Ketoacidosis
Diabetic KetoacidosisThis condition is caused by increased fatty acid metabo-lism and the accumulation of ketoacids (acetoacetateand β-hydroxybutyrate). DKA usually occurs in patientswith insulin-dependent diabetes mellitus in associationwith cessation of insulin or an intercurrent illness, suchas an infection, gastroenteritis, pancreatitis, or myocardialinfarction, which increases insulin requirements tem-porarily and acutely. The accumulation of ketoacidsaccounts for the increment in the AG and is accompa-nied most often by hyperglycemia [glucose >17 mmol/Lor (>300 mg/dL)]. The relationship between the ΔAGand ΔHCO3
− is ~1:1 in DKA but may decrease in well-hydrated patients with preservation of renal function.Ketoacid excretion in the urine reduces the AG in thissituation. It should be noted that because insulin pre-vents production of ketones, bicarbonate therapy israrely needed except with extreme acidemia (pH <7.1)and then in only limited amounts. Patients with DKAare typically volume depleted and require fluid resuscita-tion with isotonic saline.Volume overexpansion is com-mon, however, after IV fluid administration, and con-tributes to the development of hyperchloremic acidosisduring treatment of DKA because volume expansionincreases urinary ketoacid anion excretion (loss of poten-tial bicarbonate).
Alcoholic KetoacidosisChronic alcoholics can develop ketoacidosis when alco-hol consumption is abruptly curtailed and nutrition ispoor. AKA is usually associated with binge drinking,vomiting, abdominal pain, starvation, and volume deple-tion. The glucose concentration is variable, and acidosis
may be severe because of elevated ketones, predomi-nantly β-hydroxybutyrate. Hypoperfusion may enhancelactic acid production, chronic respiratory alkalosis mayaccompany liver disease, and metabolic alkalosis canresult from vomiting (refer to the relationship betweenΔAG and ΔHCO3
−).Thus, mixed acid–base disorders arecommon in patients with AKA. As the circulation isrestored by administration of isotonic saline, the prefer-ential accumulation of β-hydroxybutyrate is then shiftedto acetoacetate. This explains the common clinicalobservation of an increasingly positive nitroprussidereaction as the patient improves.The nitroprusside ketonereaction (Acetest) can detect acetoacetic acid but not β-hydroxybutyrate, so the degree of ketosis and ketonuriacan not only change with therapy but can also be underes-timated initially. Patients with AKA usually present withrelatively normal renal function, as opposed to those withDKA, in whom renal function is often compromisedbecause of volume depletion (osmotic diuresis) or diabeticnephropathy. AKA patients with normal renal functionmay excrete relatively large quantities of ketoacids in theurine, therefore, and may have a relatively normal AG anda discrepancy in the ΔAG/ΔHCO3
− relationship.Typically,insulin levels are low, and concentrations of triglyceride,cortisol, glucagon, and growth hormone are increased.
Acidosis and Alkalosis
415
CHAPTER 40
acute, severe acidemia (pH <7.15) to improve cardiacfunction and lactate utilization. However, NaHCO3
therapy may paradoxically depress cardiac performanceand exacerbate acidosis by enhancing lactate production(HCO3
− stimulates phosphofructokinase).Although theuse of alkali in moderate lactic acidosis is controversial,it is generally agreed that attempts to return the pH orHCO3
− to normal by administration of exogenousNaHCO3 are deleterious. A reasonable approach is toinfuse sufficient NaHCO3 to increase the arterial pH tono more than 7.2 over 30–40 min.
NaHCO3 therapy can cause fluid overload andhypertension because the amount required can bemassive when accumulation of lactic acid is relentless.Fluid administration is poorly tolerated because ofcentral venoconstriction, especially in patients witholiguria.When the underlying cause of the lactic aci-dosis can be remedied, blood lactate will be convertedto HCO3
− and may result in an overshoot alkalosis.
Treatment:ALCOHOLIC KETOACIDOSIS
ECF deficits almost always accompany AKA and shouldbe repleted by IV administration of saline and glucose(5% dextrose in 0.9% NaCl). Hypophosphatemia,hypokalemia, and hypomagnesemia may coexist andshould be corrected. Hypophosphatemia usually emerges12–24 h after admission; may be exacerbated by glucoseinfusion; and, if severe, may induce rhabdomyolysis.Upper gastrointestinal hemorrhage, pancreatitis, andpneumonia may accompany this disorder.
Drug- and Toxin-Induced Acidosis
SalicylatesSalicylate intoxication in adults usually causes respiratoryalkalosis or a mixture of high-AG metabolic acidosis andrespiratory alkalosis. Only a portion of the AG is caused bysalicylates. Lactic acid production is also often increased.
Treatment:SALICYLATE-INDUCED ACIDOSIS
Vigorous gastric lavage with isotonic saline (not NaHCO3)should be initiated immediately followed by administra-tion of activated charcoal per nasogastric tube. In aci-dotic patients, to facilitate removal of salicylate, IV NaHCO3
is administered in amounts adequate to alkalinize the
Loscalzo_Pulmonary-Ch40_p410-p423.qxd 2/11/10 4:04 PM Page 415

AlcoholsUnder most physiologic conditions, sodium, urea, andglucose generate the osmotic pressure of blood. Plasmaosmolality is calculated according to the following expres-sion: Posm = 2Na+ + Glu + BUN (all in mmol/L), or usingconventional laboratory values in which glucose andBUN are expressed in milligrams per deciliter: Posm =2Na+ + Glu/18 + BUN/2.8. The calculated and deter-mined osmolality should agree within 10–15 mmol/kgH2O. When the measured osmolality exceeds the calcu-lated osmolality by >15–20 mmol/kg H2O, one of twocircumstances prevails. Either the serum sodium is spuri-ously low, as with hyperlipidemia or hyperproteinemia(pseudohyponatremia), or osmolytes other than sodiumsalts, glucose, or urea have accumulated in plasma. Exam-ples include mannitol, radiocontrast media, isopropylalcohol, ethylene glycol, propylene glycol, ethanol,methanol, and acetone. In this situation, the differencebetween the calculated osmolality and the measuredosmolality (osmolar gap) is proportional to the concentra-tion of the unmeasured solute.With an appropriate clini-cal history and index of suspicion, identification of anosmolar gap is helpful in identifying the presence ofpoison-associated AG acidosis. Three alcohols may causefatal intoxications: ethylene glycol, methanol, and iso-propyl alcohol.All cause an elevated osmolal gap, but onlythe first two cause high-AG acidosis.
Ethylene GlycolIngestion of ethylene glycol (commonly used inantifreeze) leads to a metabolic acidosis and severe damageto the CNS, heart, lungs, and kidneys. The increased AGand osmolar gap are attributable to ethylene glycol and itsmetabolites, oxalic acid, glycolic acid, and other organicacids. Lactic acid production increases secondary to inhibi-tion of the tricarboxylic acid cycle and altered intracellularredox state. Diagnosis is facilitated by recognizing oxalate
crystals in the urine, the presence of an osmolar gap inserum, and a high-AG acidosis. If antifreeze containing afluorescent dye is ingested, a Wood’s lamp applied to theurine may be revealing.Treatment should not be delayedwhile awaiting measurement of ethylene glycol levels inthis setting.
416
Disorders Complicating Critical Illnesses and Their M
anagement
SECTION V
urine and to maintain urine output (urine pH >7.5).Although this form of therapy is straightforward in aci-dotic patients, a coexisting respiratory alkalosis maymake this approach hazardous. Alkalemic patientsshould not receive NaHCO3
−. Acetazolamide may beadministered in the face of alkalemia, when an alkalinediuresis cannot be achieved, or to ameliorate volumeoverload associated with NaHCO3
− administration, butthis drug can cause systemic metabolic acidosis if HCO3
−
is not replaced. Hypokalemia should be anticipated withan alkaline diuresis and should be treated promptly andaggressively. Glucose-containing fluids should be admin-istered because of the danger of hypoglycemia. Exces-sive insensible fluid losses may cause severe volumedepletion and hypernatremia. If renal failure preventsrapid clearance of salicylate, hemodialysis can be per-formed against a bicarbonate dialysate.
Treatment:ETHYLENE GLYCOL–INDUCED ACIDOSIS
Treatment should include prompt institution of a saline orosmotic diuresis, thiamine and pyridoxine supplements,fomepizole or ethanol, and hemodialysis. IV administrationof the alcohol dehydrogenase inhibitor fomepizole (4-methylpyrazole; 7 mg/kg as a loading dose) or ethanolIV to achieve a level of 22 mmol/L (100 mg/dL) serves tolessen toxicity because they compete with ethylene glycolfor metabolism by alcohol dehydrogenase. Fomepizole,although expensive, offers the advantages of a predictabledecline in ethylene glycol levels without excessive obtun-dation during ethyl alcohol infusion.
MethanolThe ingestion of methanol (wood alcohol) causes meta-bolic acidosis, and its metabolites formaldehyde andformic acid cause severe optic nerve and CNS damage.Lactic acid, ketoacids, and other unidentified organic acidsmay contribute to the acidosis. Because of its low molec-ular weight (32 Da), an osmolar gap is usually present.
Treatment:METHANOL-INDUCED ACIDOSIS
This is similar to that for ethylene glycol intoxication,including general supportive measures, fomepizole orethanol administration, and hemodialysis.
Isopropyl AlcoholIngested isopropanol is absorbed rapidly and may be fatalwhen as little as 150 mL of rubbing alcohol, solvent, orde-icer is consumed. A plasma level >400 mg/dL is lifethreatening. Isopropyl alcohol differs from ethylene glycoland methanol in that the parent compound, not themetabolites, causes toxicity, and acidosis is not presentbecause acetone is rapidly excreted.
Treatment:ISOPROPYL ALCOHOL TOXICITY
Isopropanol alcohol toxicity is treated by watchful wait-ing and supportive therapy; IV fluids, pressors, ventila-tory support if needed, and occasionally hemodialysisfor prolonged coma or levels >400 mg/dL.
Loscalzo_Pulmonary-Ch40_p410-p423.qxd 2/11/10 4:04 PM Page 416

Renal Failure
(See also Chap. 37) The hyperchloremic acidosis ofmoderate renal insufficiency is eventually converted tothe high-AG acidosis of advanced renal failure. Poor fil-tration and reabsorption of organic anions contributeto the pathogenesis. As renal disease progresses, thenumber of functioning nephrons eventually becomesinsufficient to keep pace with net acid production.Uremic acidosis is characterized, therefore, by a reducedrate of NH4
+ production and excretion, primarilybecause of decreased renal mass. HCO3
− rarely decreasesto <15 mmol/L, and the AG is rarely >20 mmol/L.Theacid retained in chronic renal disease is buffered byalkaline salts from bone. Despite significant retention ofacid (�20 mmol/d), the serum HCO3
− does notdecrease further, indicating participation of buffers out-side the extracellular compartment. Chronic metabolicacidosis results in significant loss of bone mass becauseof reduction in bone calcium carbonate. Chronic acido-sis also increases urinary calcium excretion proportionalto cumulative acid retention.
Acidosis and Alkalosis
417
CHAPTER 40
Treatment:RENAL FAILURE
Because of the association of renal failure acidosiswith muscle catabolism and bone disease, both ure-mic acidosis and the hyperchloremic acidosis of renalfailure require oral alkali replacement to maintain theHCO3
− between 20 and 24 mmol/L. This can be accom-plished with relatively modest amounts of alkali(1.0–1.5 mmol/kg body weight per day). Sodium cit-rate (Shohl’s solution) or NaHCO3 tablets (650-mgtablets contain 7.8 meq) are equally effective alkaliniz-ing salts. Citrate enhances the absorption of alu-minum from the gastrointestinal tract and shouldnever be given together with aluminum-containingantacids because of the risk of aluminum intoxication.When hyperkalemia is present, furosemide (60–80 mg/d)should be added.
HYPERCHLOREMIC (NONGAP) METABOLIC ACIDOSES
Alkali can be lost from the gastrointestinal tract indiarrhea or from the kidneys [renal tubular acidosis(RTA)]. In these disorders (Table 40-5), reciprocalchanges in Cl− and HCO3
− result in a normal AG. Inpure hyperchloremic acidosis, therefore, the increase inCl− above the normal value approximates the decreasein HCO3
−.The absence of such a relationship suggestsa mixed disturbance.
TABLE 40-5
CAUSES OF NON–ANION-GAP ACIDOSIS
I. Gastrointestinal bicarbonate lossA. DiarrheaB. External pancreatic or small bowel drainageC. Ureterosigmoidostomy, jejunal loop, ileal loopD. Drugs
1. Calcium chloride (acidifying agent)2. Magnesium sulfate (diarrhea)3. Cholestyramine (bile acid diarrhea)
II. Renal acidosisA. Hypokalemia
1. Proximal RTA (type 2)2. Distal (classic) RTA (type 1)
B. Hyperkalemia1. Generalized distal nephron dysfunction
(type 4 RTA)a. Mineralocorticoid deficiencyb. Mineralocorticoid resistance (autosomal
dominant PHA I)c. Voltage defect (autosomal dominant
PHA I and PHA II)d. Tubulointerstitial disease
III. Drug-induced hyperkalemia (with renal insufficiency)A. Potassium-sparing diuretics (amiloride,
triamterene, spironolactone)B. TrimethoprimC. PentamidineD. ACE inhibitors and ARBsE. NSAIDsF. Cyclosporine and tacrolimus
IV. OtherA. Acid loads (ammonium chloride, hyperalimentation)B. Loss of potential bicarbonate: ketosis with ketone
excretionC. Expansion acidosis (rapid saline administration)D. HippurateE. Cation exchange resins
Note: ACE, angiotensin-converting enzyme; ARB, angiotensin receptorblocker; NSAID, nonsteroidal anti-inflammatory drug; PHA, pseudohy-poaldosteronism; RTA, renal tubular acidosis.
Approach to the Patient:HYPERCHLOREMIC METABOLIC ACIDOSES
In diarrhea, stools contain a higher HCO3− and
decomposed HCO3− than plasma so that metabolic
acidosis develops along with volume depletion.Instead of an acid urine pH (as anticipated with sys-temic acidosis), urine pH is usually ∼6 because meta-bolic acidosis and hypokalemia increase renal synthesisand excretion of NH4
+, thus providing a urinarybuffer that increases urine pH. Metabolic acidosiscaused by gastrointestinal losses with a high urine pHcan be differentiated from RTA because urinaryNH4
+ excretion is typically low in RTA and highwith diarrhea. Urinary NH4
+ levels can be estimated
Loscalzo_Pulmonary-Ch40_p410-p423.qxd 2/11/10 4:04 PM Page 417

METABOLIC ALKALOSIS
Metabolic alkalosis is manifested by an elevated arterialpH, an increase in the serum HCO3
−, and an increase inPaCO2
as a result of compensatory alveolar hypoventilation(Table 40-1). It is often accompanied by hypochloremiaand hypokalemia.The arterial pH establishes the diagnosisbecause it is increased in metabolic alkalosis and decreasedor normal in respiratory acidosis. Metabolic alkalosis fre-quently occurs in association with other disorders such asrespiratory acidosis or alkalosis or metabolic acidosis.
PATHOGENESIS
Metabolic alkalosis occurs as a result of net gain ofHCO3
− or loss of nonvolatile acid (usually HCl by vom-iting) from the ECF. Because it is unusual for alkali to beadded to the body, the disorder involves a generativestage, in which the loss of acid usually causes alkalosis,and a maintenance stage, in which the kidneys fail tocompensate by excreting HCO3
−.Under normal circumstances, the kidneys have an
impressive capacity to excrete HCO3−. Continuation of
metabolic alkalosis represents a failure of the kidneys toeliminate HCO3
− in the usual manner. For HCO3− to
be added to the ECF, it must be administered exoge-nously or synthesized endogenously, partly or entirely bythe kidneys.The kidneys will retain, rather than excrete,the excess alkali and maintain the alkalosis if (1) volumedeficiency, chloride deficiency, and K+ deficiency existin combination with a reduced GFR, which augmentsdistal tubule H+ secretion, or (2) hypokalemia existsbecause of autonomous hyperaldosteronism. In the firstexample, alkalosis is corrected by administration ofNaCl and KCl; in the latter, it is necessary to repair thealkalosis by pharmacologic or surgical intervention, notwith saline administration.
DIFFERENTIAL DIAGNOSIS
To establish the cause of metabolic alkalosis (Table 40-6),it is necessary to assess the status of the ECF volume(ECFV), the recumbent and upright blood pressure,the serum K+, and the renin–aldosterone system. Forexample, the presence of chronic hypertension andchronic hypokalemia in an alkalotic patient suggests
418
Disorders Complicating Critical Illnesses and Their M
anagement
SECTION V
by calculating the urine AG (UAG): UAG = [Na+ +K+]u − [Cl−]u.When [Cl−]u >[Na+ + K+], the urine gapis negative by definition.This indicates that the urineammonium level is appropriately increased, suggestingan extrarenal cause of the acidosis. Conversely, whenthe urine UAG is positive, the urine ammonium levelis low, suggesting a renal cause of the acidosis.
Loss of functioning renal parenchyma by progressiverenal disease leads to hyperchloremic acidosis whenthe glomerular filtration rate (GFR) is between 20 and50 mL/min and to uremic acidosis with a high AGwhen the GFR decreases to <20 mL/min. Such a pro-gression occurs commonly with tubulointerstitialforms of renal disease, but hyperchloremic metabolicacidosis can persist with advanced glomerular disease.In advanced renal failure, ammoniagenesis is reducedin proportion to the loss of functional renal mass, andammonium accumulation and trapping in the outermedullary collecting tubule may also be impaired.Because of adaptive increases in K+ secretion by thecollecting duct and colon, the acidosis of chronic renalinsufficiency is typically normokalemic.
Proximal RTA (type 2 RTA) is most often causedby generalized proximal tubular dysfunction mani-fested by glycosuria, generalized aminoaciduria, andphosphaturia (Fanconi syndrome). With a lowplasma HCO3
−, the urine pH is acidic (pH <5.5).The fractional excretion of HCO3
− may exceed10–15% when the serum HCO3
− >20 mmol/L.Because HCO3
− is not reabsorbed normally in theproximal tubule, therapy with NaHCO3 willenhance renal potassium wasting and hypokalemia.
The typical findings in acquired or inheritedforms of classic distal RTA (type 1 RTA) includehypokalemia, hyperchloremic acidosis, low urinaryNH4
+ excretion (positive UAG, low urine [NH4+]),
and inappropriately high urine pH (pH >5.5). Suchpatients are unable to acidify the urine below a pHof 5.5. Most patients have hypocitraturia and hyper-calciuria, so nephrolithiasis, nephrocalcinosis, andbone disease are common. In generalized distalnephron dysfunction (type 4 RTA), hyperkalemia isdisproportionate to the reduction in GFR becauseof coexisting dysfunction of potassium and acidsecretion. Urinary ammonium excretion is invariablydepressed, and renal function may be compromised,for example, because of diabetic nephropathy, amy-loidosis, or tubulointerstitial disease.
Hyporeninemic hypoaldosteronism typically causeshyperchloremic metabolic acidosis, most commonlyin older adults with diabetes mellitus or tubulointer-stitial disease and renal insufficiency. Patients usuallyhave mild to moderate renal insufficiency (GRF,20–50 mL/min) and acidosis, with elevation in serumK+ (5.2−6.0 mmol/L), concurrent hypertension, and
congestive heart failure. Both the metabolic acidosisand the hyperkalemia are out of proportion toimpairment in GFR. Nonsteroidal anti-inflammatorydrugs, trimethoprim, pentamidine, and angiotensin-converting enzyme inhibitors can also cause hyper-kalemia with hyperchloremic metabolic acidosis inpatients with renal insufficiency (Table 40-5).
Loscalzo_Pulmonary-Ch40_p410-p423.qxd 2/11/10 4:04 PM Page 418

either mineralocorticoid excess or that the hypertensivepatient is receiving diuretics. Low plasma renin activityand normal urine Na+ and Cl– in a patient who is nottaking diuretics indicate a primary mineralocorticoidexcess syndrome. The combination of hypokalemia andalkalosis in a normotensive, nonedematous patient may
be attributable to Bartter’s or Gitelman’s syndrome,magnesium deficiency, vomiting, exogenous alkali, ordiuretic ingestion. Determination of urine electrolytes(especially the urine Cl−) and screening of the urine fordiuretics may be helpful. If the urine is alkaline, with anelevated Na+ and K+ but low Cl−, the diagnosis is usuallyeither vomiting (overt or surreptitious) or alkali inges-tion. If the urine is relatively acid and has low concen-trations of Na+, K+, and Cl−, the most likely possibilitiesare prior vomiting, the posthypercapnic state, or priordiuretic ingestion. If, on the other hand, the urine sodium,potassium, and chloride concentrations are not depressed,magnesium deficiency, Bartter’s or Gitelman’s syndrome,or current diuretic ingestion should be considered. Bart-ter’s syndrome is distinguished from Gitelman’s syndromebecause of hypocalciuria and hypomagnesemia in the lat-ter disorder.The genetic and molecular basis of these twodisorders has been elucidated recently.
Alkali Administration
Chronic administration of alkali to individuals with nor-mal renal function rarely, if ever, causes alkalosis. However,in patients with coexistent hemodynamic disturbances,alkalosis can develop because the normal capacity toexcrete HCO3
− may be exceeded or there may beenhanced reabsorption of HCO3
−. Such patients includethose who receive HCO3
− (PO or IV), acetate loads (par-enteral hyperalimentation solutions), citrate loads (transfu-sions), or antacids plus cation-exchange resins (aluminumhydroxide and sodium polystyrene sulfonate).
METABOLIC ALKALOSIS ASSOCIATED WITH EXTRACELLULAR FLUID VOLUMECONTRACTION, K++ DEPLETION, AND SECONDARY HYPERRENINEMICHYPERALDOSTERONISM
Gastrointestinal Origin
Gastrointestinal loss of H+ from vomiting or gastric aspi-ration results in retention of HCO3
−. The loss of fluidand NaCl in vomitus or nasogastric suction results incontraction of the ECFV and an increase in the secre-tion of renin and aldosterone. Volume contractionthrough a reduction in GFR results in an enhancedcapacity of the renal tubule to reabsorb HCO3
−. Duringactive vomiting, however, the filtered load of bicarbon-ate is acutely increased to the point that the reabsorptivecapacity of the proximal tubule for HCO3
− is exceeded.The excess NaHCO3 issuing out of the proximal tubulereaches the distal tubule, where H+ secretion is enhancedby an aldosterone and the delivery of the poorly reab-sorbed anion, HCO3
−. Correction of the contractedECFV with NaCl and repair of K+ deficits corrects theacid–base disorder, and chloride deficiency.
Acidosis and Alkalosis
419
CHAPTER 40TABLE 40-6
CAUSES OF METABOLIC ALKALOSIS
I. Exogenous HCO3− loads
A. Acute alkali administrationB. Milk-alkali syndrome
II. Effective ECFV contraction, normotension, K+ deficiency, and secondary hyperreninemic hyperaldosteronismA. Gastrointestinal origin
1. Vomiting2. Gastric aspiration3. Congenital chloridorrhea4. Villous adenoma
B. Renal origin1. Diuretics2. Posthypercapnic state3. Hypercalcemia or hypoparathyroidism4. Recovery from lactic acidosis or ketoacidosis5. Nonreabsorbable anions, including penicillin,
carbenicillin6. Mg2+ deficiency7. K+ depletion8. Bartter’s syndrome (loss of function mutations
in TALH)9. Gitelman’s syndrome (loss of function mutation
in Na+-Cl− cotransporter in DCT)III. ECFV expansion, hypertension, K+ deficiency, and
mineralocorticoid excessA. High renin
1. Renal artery stenosis2. Accelerated hypertension3. Renin-secreting tumor4. Estrogen therapy
B. Low renin1. Primary aldosteronism
a. Adenomab. Hyperplasiac. Carcinoma
2. Adrenal enzyme defectsa. 11 β-Hydroxylase deficiencyb. 17 α-Hydroxylase deficiency
3. Cushing’s syndrome or disease4. Other
a. Licoriceb. Carbenoxolonec. Chewer’s tobacco
IV. Gain-of-function mutation of renal sodium channelwith ECFV expansion, hypertension, K+ deficiency, andhyporeninemic-hypoaldosteronismA. Liddle’s syndrome
Note: DCT, distal convoluted tubule; ECFV, extracellular fluid vol-ume; TALH, thick ascending limb of the loop of Henle.
Loscalzo_Pulmonary-Ch40_p410-p423.qxd 2/11/10 4:04 PM Page 419

Renal Origin
DiureticsDrugs that induce chloruresis, such as thiazides and loopdiuretics (furosemide, bumetanide, torsemide, andethacrynic acid), acutely diminish the ECFV withoutaltering the total body bicarbonate content. The serumHCO3
− increases because the reduced ECFV “contracts”the HCO3
− in the plasma (contraction alkalosis). Thechronic administration of diuretics tends to generate analkalosis by increasing distal salt delivery, so that K+ andH+ secretion are stimulated.The alkalosis is maintained bypersistence of the contraction of the ECFV, secondaryhyperaldosteronism, K+ deficiency, and the direct effect ofthe diuretic (as long as diuretic administration continues).Repair of the alkalosis is achieved by providing isotonicsaline to correct the ECFV deficit.
Nonreabsorbable Anions and MagnesiumDeficiencyAdministration of large quantities of nonreabsorbableanions, such as penicillin or carbenicillin, can enhance dis-tal acidification and K+ secretion by increasing thetransepithelial potential difference (lumen negative). Mg2+
deficiency results in hypokalemic alkalosis by enhancingdistal acidification through stimulation of renin and hencealdosterone secretion.
Potassium DepletionChronic K+ depletion may cause metabolic alkalosis byincreasing urinary acid excretion. Both NH4
+ productionand absorption are enhanced and HCO3
− reabsorption isstimulated. Chronic K+ deficiency upregulates the renalH+, K+-ATPase to increase K+ absorption at the expenseof enhanced H+ secretion.Alkalosis associated with severeK+ depletion is resistant to salt administration, but repairof the K+ deficiency corrects the alkalosis.
After Treatment of Lactic Acidosis or KetoacidosisWhen an underlying stimulus for the generation of lacticacid or ketoacid is removed rapidly, as with repair of circu-latory insufficiency or with insulin therapy, the lactate orketones are metabolized to yield an equivalent amount ofHCO3
−. Other sources of new HCO3− are additive with
the original amount generated by organic anion metabo-lism to create a surfeit of HCO3
–. Such sources include(1) new HCO3
− added to the blood by the kidneys as aresult of enhanced acid excretion during the preexistingperiod of acidosis and (2) alkali therapy during the treat-ment phase of the acidosis. Acidosis-induced contractionof the ECFV and K+ deficiency act to sustain the alkalosis.
PosthypercapniaProlonged CO2 retention with chronic respiratory acido-sis enhances renal HCO3
− absorption and the generationof new HCO3
− (increased net acid excretion). If the
PaCO2is returned to normal, metabolic alkalosis results
from the persistently elevated HCO3−. Alkalosis develops
if the elevated PaCO2is abruptly returned toward normal
by a change in mechanically controlled ventilation. Asso-ciated ECFV contraction does not allow complete repairof the alkalosis by correction of the PaCO2
alone, and alka-losis persists until Cl− supplementation is provided.
METABOLIC ALKALOSIS ASSOCIATED WITH EXTRACELLULAR FLUID VOLUMEEXPANSION, HYPERTENSION, AND HYPERALDOSTERONISM
Increased aldosterone levels may be the result ofautonomous primary adrenal overproduction or of sec-ondary aldosterone release caused by renal overproduc-tion of renin. Mineralocorticoid excess increases net acidexcretion and may result in metabolic alkalosis, whichmay be worsened by associated K+ deficiency. ECFVexpansion from salt retention causes hypertension. Thekaliuresis persists because of mineralocorticoid excessand distal Na+ absorption, causing enhanced K+ excre-tion, continued K+ depletion with polydipsia, inabilityto concentrate the urine, and polyuria.
Liddle’s syndrome results from increased activity ofthe collecting duct Na+ channel (ENaC) and is a rareinherited disorder associated with hypertension causedby volume expansion manifested as hypokalemic alkalo-sis and normal aldosterone levels.
Symptoms
With metabolic alkalosis, changes in central and periph-eral nervous system function are similar to those ofhypocalcemia; symptoms include mental confusion,obtundation, and a predisposition to seizures, paresthesia,muscular cramping, tetany, aggravation of arrhythmias,and hypoxemia in chronic obstructive pulmonary disease.Related electrolyte abnormalities include hypokalemiaand hypophosphatemia.
420
Disorders Complicating Critical Illnesses and Their M
anagement
SECTION VTreatment:METABOLIC ALKALOSIS
Treatment is primarily directed at correcting the under-lying stimulus for HCO3
– generation. If primary aldos-teronism, renal artery stenosis, or Cushing’s syndrome ispresent, correction of the underlying cause will reversethe alkalosis. H+ loss by the stomach or kidneys can bemitigated by the use of proton pump inhibitors or thediscontinuation of diuretics. The second aspect of treat-ment is to remove the factors that sustain the inappro-priate increase in HCO3
− reabsorption, such as ECFV con-traction or K+ deficiency. Although K+ deficits should berepaired, NaCl therapy is usually sufficient to reverse the
Loscalzo_Pulmonary-Ch40_p410-p423.qxd 2/11/10 4:04 PM Page 420

RESPIRATORY ACIDOSIS
Respiratory acidosis can be caused by severe pulmonarydisease, respiratory muscle fatigue, or abnormalities inventilatory control and is recognized by an increase inPaCO2
and decrease in pH (Table 40-7). In acute respi-ratory acidosis, there is an immediate compensatory ele-vation (because of cellular buffering mechanisms) inHCO3
−, which increases 1 mmol/L for every 10-mmHgincrease in PaCO2
. In chronic respiratory acidosis (>24 h),renal adaptation increases the HCO3
− by 4 mmol/L for
every 10-mmHg increase in PaCO2. The serum HCO3
−
usually does not increase above 38 mmol/L.The clinical features vary according to the severity
and duration of the respiratory acidosis, the underlyingdisease, and whether accompanying hypoxemia is pre-sent. A rapid increase in PaCO2
may cause anxiety, dysp-nea, confusion, psychosis, and hallucinations and mayprogress to coma. Lesser degrees of dysfunction inchronic hypercapnia include sleep disturbances; loss ofmemory; daytime somnolence; personality changes;impairment of coordination; and motor disturbancessuch as tremor, myoclonic jerks, and asterixis. Headachesand other signs that mimic increased intracranial pres-sure, such as papilledema, abnormal reflexes, and focalmuscle weakness, are caused by vasoconstriction sec-ondary to loss of the vasodilator effects of CO2.
Depression of the respiratory center by a variety ofdrugs, injury, or disease can produce respiratory acidosis.This may occur acutely with general anesthetics, sedatives,and head trauma or chronically with sedatives, alcohol,intracranial tumors, and the syndromes of sleep-disorderedbreathing, including the primary alveolar and obesity-hypoventilation syndromes (Chaps. 22 and 23).Abnormal-ities or disease in the motor neurons, neuromuscular junc-tion, and skeletal muscle can cause hypoventilation via
Acidosis and Alkalosis
421
CHAPTER 40
alkalosis if ECFV contraction is present, as indicated by alow urine Cl−.
If associated conditions preclude infusion of saline,renal HCO3
– loss can be accelerated by administration ofacetazolamide, a carbonic anhydrase inhibitor, which isusually effective in patients with adequate renal func-tion but can worsen K+ losses. Dilute hydrochloric acid(0.1 N HCl) is also effective but can cause hemolysis andmust be delivered centrally and slowly. Hemodialysisagainst a dialysate low in HCO3
− and high in Cl− can beeffective when renal function is impaired.
TABLE 40-7
RESPIRATORY ACID–BASE DISORDERS
I. AlkalosisA. Central nervous system stimulation
1. Pain2. Anxiety, psychosis3. Fever4. Cerebrovascular accident5. Meningitis, encephalitis6. Tumor7. Trauma
B. Hypoxemia or tissue hypoxia1. High altitude, ↓ PaCO22. Pneumonia, pulmonary edema3. Aspiration4. Severe anemia
C. Drugs or hormones1. Pregnancy, progesterone2. Salicylates3. Cardiac failure
D. Stimulation of chest receptors1. Hemothorax2. Flail chest3. Cardiac failure4. Pulmonary embolism
E. Miscellaneous1. Septicemia2. Hepatic failure3. Mechanical hyperventilation4. Heat exposure5. Recovery from metabolic acidosis
II. AcidosisA. Central
1. Drugs (anesthetics, morphine, sedatives)
2. Stroke3. Infection
B. Airway1. Obstruction2. Asthma
C. Parenchyma1. Emphysema2. Pneumoconiosis3. Bronchitis4. Adult respiratory
distress syndrome5. Barotrauma
D. Neuromuscular1. Poliomyelitis2. Kyphoscoliosis3. Myasthenia4. Muscular dystrophies
E. Miscellaneous1. Obesity2. Hypoventilation3. Permissive hypercapnia
Loscalzo_Pulmonary-Ch40_p410-p423.qxd 2/11/10 4:04 PM Page 421

respiratory muscle fatigue. Mechanical ventilation, whennot properly adjusted and supervised, may result in respira-tory acidosis, particularly if CO2 production suddenlyincreases (because of fever, agitation, sepsis, or overfeeding)or alveolar ventilation decreases because of worsening pul-monary function. High levels of positive end-expiratorypressure in the presence of reduced cardiac output maycause hypercapnia as a result of large increases in alveolardead space (Chap. 5). Permissive hypercapnia is being usedwith increasing frequency because of studies suggestinglower mortality rates than with conventional mechanicalventilation, especially with severe CNS or heart disease.The potential beneficial effects of permissive hypercapniamay be mitigated by correction of the acidemia by admin-istration of NaHCO3.
Acute hypercapnia occurs after sudden occlusion ofthe upper airway or generalized bronchospasm as insevere asthma, anaphylaxis, inhalational burn, or toxininjury. Chronic hypercapnia and respiratory acidosis occurin end-stage obstructive lung disease. Restrictive disordersinvolving both the chest wall and the lungs can cause res-piratory acidosis because the high metabolic cost of respi-ration causes ventilatory muscle fatigue. Advanced stagesof intrapulmonary and extrapulmonary restrictive defectspresent as chronic respiratory acidosis.
The diagnosis of respiratory acidosis requires, bydefinition, the measurement of PaCO2
and arterial pH.A detailed history and physical examination oftenindicate the cause. Pulmonary function studies (Chap. 5),including spirometry, diffusion capacity for carbonmonoxide, lung volumes, and arterial PaCO2
and O2
saturation, usually make it possible to determine if res-piratory acidosis is secondary to lung disease. Theworkup for nonpulmonary causes should include adetailed drug history; measurement of hematocrit; andassessment of upper airway, chest wall, pleura, andneuromuscular function.
RESPIRATORY ALKALOSIS
Alveolar hyperventilation decreases PaCO2and increases the
HCO3−/PaCO2
ratio, thus increasing pH (see Table 40-7).Nonbicarbonate cellular buffers respond by consumingHCO3
−. Hypocapnia develops when a sufficiently strongventilatory stimulus causes CO2 output in the lungs toexceed its metabolic production by tissues. Plasma pH andHCO3
− appear to vary proportionately with PaCO2over a
range from 40–15 mmHg.The relationship between arter-ial H+ concentration and PaCO2
is ∼0.7 mmol/L permmHg (or 0.01 pH unit/mmHg), and that for plasmaHCO3
− is 0.2 mmol/L per mmHg. Hypocapnia sustainedfor >2–6 h is further compensated by a decrease in renalammonium and titratable acid excretion and a reductionin filtered HCO3
− reabsorption. Full renal adaptation torespiratory alkalosis may take several days and requires nor-mal volume status and renal function.The kidneys appearto respond directly to the lowered PaCO2
rather than toalkalosis per se. In chronic respiratory alkalosis, a 1-mmHgdecrease in PaCO2
causes a 0.4- to 0.5-mmol/L decrease inHCO3
− and a 0.3-mmol/L decrease (or 0.003-mmol/Lincrease in pH) in H+.
The effects of respiratory alkalosis vary according toduration and severity but are primarily those of the under-lying disease. Reduced cerebral blood flow as a conse-quence of a rapid decline in PaCO2
may cause dizziness,mental confusion, and seizures, even in the absence ofhypoxemia.The cardiovascular effects of acute hypocapniain conscious humans are generally minimal, but in anes-thetized or mechanically ventilated patients, cardiac outputand blood pressure may decrease because of the depressanteffects of anesthesia and positive-pressure ventilation onheart rate, systemic resistance, and venous return. Cardiacarrhythmias may occur in patients with heart disease as aresult of changes in oxygen unloading by blood from aleft shift in the hemoglobin-oxygen dissociation curve(Bohr effect). Acute respiratory alkalosis causes intracel-lular shifts of Na+, K+, and PO4
− and reduces free Ca2+
by increasing the protein-bound fraction. Hypocapnia-induced hypokalemia is usually minor.
422
Disorders Complicating Critical Illnesses and Their M
anagement
SECTION V
Treatment:RESPIRATORY ACIDOSIS
The management of respiratory acidosis depends on itsseverity and rate of onset. Acute respiratory acidosiscan be life threatening, and measures to reverse theunderlying cause should be undertaken simultaneouslywith restoration of adequate alveolar ventilation. Thismay necessitate tracheal intubation and assistedmechanical ventilation. Oxygen administration shouldbe titrated carefully in patients with severe obstructivepulmonary disease and chronic CO2 retention who arebreathing spontaneously (Chap. 18). When oxygen isused injudiciously, these patients may experience pro-gression of the respiratory acidosis. Aggressive andrapid correction of hypercapnia should be avoided
because the decreasing PaCO2may provoke the same
complications noted with acute respiratory alkalosis(i.e., cardiac arrhythmias, reduced cerebral perfusion,and seizures). The PaCO2
should be lowered gradually inchronic respiratory acidosis, aiming to restore the PaCO2to baseline levels and to provide sufficient Cl− and K+ toenhance the renal excretion of HCO3
−.Chronic respiratory acidosis is frequently difficult to
correct, but measures aimed at improving lung function(Chap. 18) can help some patients and forestall furtherdeterioration in most.
Loscalzo_Pulmonary-Ch40_p410-p423.qxd 2/11/10 4:04 PM Page 422

Chronic respiratory alkalosis is the most commonacid–base disturbance in critically ill patients and, whensevere, portends a poor prognosis. Many cardiopul-monary disorders manifest respiratory alkalosis in theirearly to intermediate stages, and the finding of normo-capnia and hypoxemia in a patient with hyperventilationmay herald the onset of rapid respiratory failure andshould prompt an assessment to determine if the patientis becoming fatigued. Respiratory alkalosis is commonduring mechanical ventilation.
The hyperventilation syndrome may be disabling.Paresthesia, circumoral numbness, chest wall tightness orpain, dizziness, inability to take an adequate breath, and(rarely) tetany may themselves be sufficiently stressful toperpetuate the disorder. Arterial blood gas analysisdemonstrates an acute or chronic respiratory alkalosis,often with hypocapnia in the range of 15–30 mmHgand no hypoxemia. CNS diseases or injury can pro-duce several patterns of hyperventilation and sustainedPaCO2
levels of 20–30 mmHg. Hyperthyroidism, highcaloric loads, and exercise raise the basal metabolicrate, but ventilation usually increases in proportion sothat arterial blood gases are unchanged and respiratoryalkalosis does not develop. Salicylates are the mostcommon cause of drug-induced respiratory alkalosis asa result of direct stimulation of the medullary chemore-ceptor. The methylxanthines, theophylline, and amino-phylline stimulate ventilation and increase the ventilatoryresponse to CO2. Progesterone increases ventilation andlowers arterial PaCO2
by as much as 5–10 mmHg.There-fore, chronic respiratory alkalosis is a common feature ofpregnancy. Respiratory alkalosis is also prominent in indi-viduals with liver failure, and the severity correlates withthe degree of hepatic insufficiency. Respiratory alkalosis isoften an early finding in gram-negative septicemia beforefever, hypoxemia, or hypotension develops.
The diagnosis of respiratory alkalosis depends onmeasurement of arterial pH and PaCO2
.The plasma K+ isoften reduced and the Cl− increased. In the acute phase,respiratory alkalosis is not associated with increased renalHCO3
− excretion, but within hours, net acid excretionis reduced. In general, the HCO3
– concentrationdecreases by 2.0 mmol/L for each 10-mmHg decrease inPaCO2
. Chronic hypocapnia reduces the serum HCO3−
FURTHER READINGS
DUBOSE TD JR: Acid-base disorders, in Brenner and Rector’s The Kidney,8th ed, BM Brenner (ed). Philadelphia, Saunders, 2007, pp 505–546
———, ALPERN RJ: Renal tubular acidosis, in The Metabolic andMolecular Bases of Inherited Disease, 8th ed, CR Scriver et al (eds).New York, McGraw-Hill, 2001, pp 4983–5021
GALLA JH: Metabolic alkalosis, in Acid-Base and Electrolyte Disorders—A Companion to Brenner and Rector’s The Kidney, TD DuBose, LLHamm (eds). Philadelphia, Saunders, 2002, pp 109–128
LASKI ME, WESSON DE: Lactic acidosis, in Acid-Base and ElectrolyteDisorders—A Companion to Brenner and Rector’s The Kidney, TDDuBose, LL Hamm (eds). Philadelphia, Saunders, 2002, pp 83–107
MADIAS NE: Respiratory alkalosis, in Acid-Base and Electrolyte Disor-ders—A Companion to Brenner and Rector’s The Kidney,TD DuBose,LL Hamm (eds). Philadelphia, Saunders, 2002, pp 147–164
WESSON DE et al: Clinical syndromes of metabolic alkalosis, in TheKidney: Physiology and Pathophysiology, 3rd ed, DW Seldin, GGiebisch (eds). Philadelphia, Lippincott Williams and Wilkins,2000, pp 2055–2072
Acidosis and Alkalosis
423
CHAPTER 40
Treatment:RESPIRATORY ALKALOSIS
The management of respiratory alkalosis is directedtoward alleviation of the underlying disorder. If respira-tory alkalosis complicates ventilator management,changes in dead space, tidal volume, and frequencycan minimize the hypocapnia. Patients with the hyper-ventilation syndrome may benefit from reassurance,rebreathing from a paper bag during symptomaticattacks, and attention to underlying psychologicalstress. Antidepressants and sedatives are not recom-mended. β-Adrenergic blockers may ameliorate periph-eral manifestations of the hyperadrenergic state.
by 4.0 mmol/L for each 10-mmHg decrease in PaCO2.
It is unusual to observe a plasma HCO3− <12 mmol/L as
a result of a pure respiratory alkalosis.When a diagnosis of respiratory alkalosis is made, its
cause should be investigated.The diagnosis of hyperven-tilation syndrome is made by exclusion. In difficultcases, it may be important to rule out other conditionssuch as pulmonary embolism, coronary artery disease,and hyperthyroidism.
Loscalzo_Pulmonary-Ch40_p410-p423.qxd 2/11/10 4:04 PM Page 423