a supramolecular porous organic cage platform promotes
TRANSCRIPT

doi.org/10.26434/chemrxiv.14213528.v1
A Supramolecular Porous Organic Cage Platform PromotesElectrochemical Hydrogen Evolution from Water Catalyzed by CobaltPorphyrinsPeter T. Smith, Bahiru Punja Benke, Lun An, Younghoon Kim, Kimoon Kim, Christopher Chang
Submitted date: 13/03/2021 • Posted date: 16/03/2021Licence: CC BY-NC-ND 4.0Citation information: Smith, Peter T.; Benke, Bahiru Punja; An, Lun; Kim, Younghoon; Kim, Kimoon; Chang,Christopher (2021): A Supramolecular Porous Organic Cage Platform Promotes Electrochemical HydrogenEvolution from Water Catalyzed by Cobalt Porphyrins. ChemRxiv. Preprint.https://doi.org/10.26434/chemrxiv.14213528.v1
We report a supramolecular porous organic cage platform composed of cobalt porphyrins for catalyzing theelectrochemical hydrogen evolution reaction (HER) from water at neutral pH. Owing to its permanent porosity,the supramolecular structure yields a catalyst film with a 5-fold increase in the number of electrochemicallyactive cobalt atoms and an improvement in Tafel slope from 170 mV/decade to 119 mV/decade compared toa planar cobalt porphyrin analog, reaching activities over 19,000 turnovers for HER over a 24-hour period with100% Faradaic efficiency.
File list (2)
download fileview on ChemRxivBox_HER_ChemRxiv_Manuscript.pdf (1.20 MiB)
download fileview on ChemRxivBox_HER_Supporting_Information_031321.pdf (496.07 KiB)

1
A Supramolecular Porous Organic Cage Platform Promotes Electrochemical Hydrogen Evolution from Water Catalyzed by Cobalt Porphyrins Peter T. Smith,[a] Bahiru Punja Benke,[b] Lun An,[a] Younghoon Kim,[b, c] Kimoon Kim,*[b, c] and Christopher J. Chang*[a, d]
[a] Department of Chemistry, University of California, Berkeley, Chemical Sciences Division, Lawrence Berkeley
National Laboratory, Berkeley, CA 94720-1460 (USA), [email protected] [b] Center for Self-assembly and Complexity (CSC), Institute for Basic Science (IBS), Pohang 37673 (Republic of
Korea), [email protected] [c] Department of Chemistry, Pohang University of Science and Technology, Pohang 37673 (Republic of Korea) [d] Department of Molecular and Cell Biology, University of California, Berkeley, Berkeley, CA 94720-1460 (USA) We dedicate this paper to Prof. Jean-Michel Savéant.
Abstract: We report a supramolecular porous organic cage platform composed of cobalt porphyrins for catalyzing the electrochemical hydrogen evolution reaction (HER) from water at neutral pH. Owing to its permanent porosity, the supramolecular structure yields a catalyst film with a 5-fold increase in the number of electrochemically active cobalt atoms and an improvement in Tafel slope from 170 mV/decade to 119 mV/decade compared to a planar cobalt porphyrin analog, reaching activities over 19,000 turnovers for HER over a 24-hour period with 100% Faradaic efficiency.
The generation of high-purity hydrogen gas from renewable sources for use as a fuel or synthetic feedstock is an attractive alternative over conventional energy-intensive and environmentally harmful processes including natural gas reforming.[1] In this context, water is an abundant and sustainable source of protons that can be reduced to form hydrogen, and the development of catalysts for the electrochemical hydrogen evolution reaction (HER) using earth-abundant metals has been the subject of intense research.[2] In particular, electrochemical HER using a variety of cobalt complexes has been thoroughly investigated,[3] and cobalt porphyrins specifically have been shown to be efficient hydrogen evolving catalysts in homogeneous organic or aqueous conditions,[4] as catalyst films,[5] and even within artificial metalloenzymes.[6] More broadly, porphyrin-based catalysts have proven to be privileged scaffolds for many small-molecule activation reactions owing to facile synthetic tuning of the porphyrin electronics and the
associated outer-coordination sphere.[7] In materials platforms for energy catalysis, a common strategy for tuning catalytic activity is altering the nanostructure of the electrode material in an effort to elucidate how different shapes, sizes, or assemblies can influence the active sites.[8] At even smaller length scales, the supramolecular architecture of electrochemical molecular catalysts at the electrode can enable tuning of catalytic activity in confined spaces,[9] with extended framework materials being tailored matrices using a similar approach.[10] As part of a major thrust in our research program in unifying design strategies from molecular, materials, and biological catalysts for sustainable electrosynthesis and photosynthesis,[11] we report here an investigation into the effects of embedding cobalt porphyrin electrocatalysts for HER within a supramolecular porous organic cage.
Based on recent work from our laboratories using porous supramolecular architectures to enhance the electrochemical activity and selectivity of embedded

2
molecular catalysts for CO2 reduction reaction (CO2RR) and O2 reduction reaction (ORR) processes,[9i, 9j] we reasoned that electrochemical HER would likewise benefit from permanent porosity to more efficiently expose molecular active sites. Indeed, we have focused on exploring porous organic cages as catalyst platforms owing to the ability to rationally design and synthesize supramolecules built from robust covalent linkages that feature permanent porosity.[12] These architectures can provide both chemical and electrochemical stability,[13] effective gas adsorption and separation properties,[14] and promote gas solubility in the liquid phase.[15] We have previously demonstrated the utility of various derivatives of the porphyrin box platform as synthetic ion channels,[16] building blocks for metal-organic frameworks,[17] materials for photoinduced charge separation,[18] and catalysts for electrochemical CO2 or O2 reduction.[9i, 9j] These collective efforts establish porphyrin-based porous organic cages as a versatile and modular system where metal and strut substitutions can enable a broad range of applications.[19]
To start, the free-base porphyrin box was first obtained by imine condensation between six tetra(formylphenyl)porphyrins and eight triamine linkers bearing solubilizing C8H17 alkyl groups (Scheme 1). Successful and complete metalation of all six porphyrin moieties in the porphyrin box using CoCl2 to obtain Co-PB-1(8) was confirmed using MALDI mass spectrometry and UV/Vis spectroscopy (Figure S1-S2). Gas sorption studies of Co-PB-1(8) show permanent porosity and indicate a Brunauer–Emmett–Teller surface area of 519 m2/g (Figure S3).
In order to utilize the porous organic cage system with an aqueous electrolyte, the supramolecular Co-PB-1(8) catalyst was heterogenized by drop-casting onto a glassy carbon working electrode that had first been coated with carbon nanotubes to enhance conductivity. Linear sweep voltammetry (LSV) of this heterogeneous Co-PB-1(8) electrode in 1 M pH 7 phosphate buffer shows a large cathodic current enhancement when scanning beyond –0.45 V vs. the reversible hydrogen electrode (RHE), suggesting catalytic proton reduction to
hydrogen (Figure 1). For comparison, we investigated Co-TPP as an electrochemical HER catalyst under the same conditions since it is the base molecular unit of the supramolecular Co-PB-1(8) architecture. Six molar equivalents of Co-TPP were deposited onto the electrode in order to normalize to the number of Co centers in the Co-PB-1(8) and Co-TPP heterogeneous catalyst films. Accordingly, LSV of this Co-TPP electrode shows a much weaker current enhancement with an onset potential for HER shifted slightly more negative compared to Co-PB-1(8). This enhanced catalytic response is observed despite the supramolecular catalyst possessing electron-withdrawing imine groups that may reduce the nucleophilicity of the metal center and thus hinder catalysis, as observed in reported scaling relationships.[20]
Figure 1. LSV traces for supramolecular porous cage catalyst Co-PB-1(8) (blue), its planar molecular analog Co-TPP (red), and electrode background (black) obtained in 1 M pH 7 phosphate buffer saturated with N2.
Notably, Tafel analyses plotted in Figure 2 indicate that the supramolecular Co-PB-1(8) catalyst displays improved kinetics compared to the molecular Co-TPP congener. Co-PB-1(8) exhibits a Tafel slope of 119 mV/dec, which is in agreement with the theory that a single electron transfer to a proton from water is rate-determining.[21] In contrast, Co-TPP has a larger Tafel
Scheme 1. (A) Synthetic scheme for the preparation of the porous organic cage Co-PB-1(8). (i) Trifluoroacetic acid (cat.), CHCl3, 55 ºC, 12 h; (ii) CoCl2, 2,6-lutidine, THF, 70 ºC, 12 h. (B) Comparison of a model of the porous organic cage catalyst Co-PB-1(8) (left) and its monomeric analog Co-TPP (right). Alkyl chains and hydrogen atoms are omitted in the molecular model for clarity.

3
slope of 170 mV/dec. We attribute this observed difference to inferior electron transfer throughout the Co-TPP catalyst film and/or to a lower local concentration of water within the film relative to the Co-PB-1(8) system. We speculate that the structural porosity of Co-PB-1(8) facilitates the delivery of both electrons and protons to the active Co centers. Indeed, heterogeneous molecular electrochemical catalysts commonly suffer from large Tafel slopes, indicating that the initial electron transfer is greatly hindered and/or that substrate adsorption or product desorption steps are slow.[22] As such, embedding the catalyst into a supramolecular architecture may accelerate this rate-limiting step by facilitating charge, electrolyte, and substrate transport.
Figure 2. Tafel plot for Co-PB-1(8) and Co-TPP catalysts. Each catalyst electrode was prepared by depositing 13.8 nmol/cm2 of Co centers (2.3 nmol/cm2 of hexa-cobalt Co-PB-1(8) and 13.8 nmol/cm2 of mono-cobalt Co-TPP).
We next analyzed the electrochemically active surface areas of the Co-PB-1(8) and Co-TPP heterogeneous electrodes by measuring the scan rate dependence of the reversible CoIII/CoII redox couple observed at 0.76 V vs. RHE for both Co-PB-1(8) and Co-TPP in 1 M pH 7 phosphate buffer (Figure 3A, B). Both catalyst films were prepared by depositing 13.8 nmol/cm2
of Co centers (2.3 nmol/cm2 of Co-PB-1(8) and 13.8 nmol/cm2 of Co-TPP). Variation of the cathodic and anodic peak currents with the scan rate was linear for both catalysts, (Figure 3C, D) indicating catalyst immobilization and reversible electron transfer.[23] From these data, we calculated that the Co-PB-1(8) electrode had 1.0 ± 0.1 nmol/cm2 of electrochemically active Co centers compared to 0.2 ± 0.05 nmol/cm2 in the Co-TPP electrode. These values correspond to 1.4% of Co centers being electroactive in the Co-TPP film and 7.2% of the Co centers are electroactive in the Co-PB-1(8) film, resulting in a 5-fold enhancement in electrochemically active surface area observed for the supramolecular Co-PB-1(8) catalyst over the molecular Co-TPP control, reinforcing that the permanent porosity derived from supramolecular incorporation of the Co-porphyrin unit can contribute to making the active sites more accessible in the film. Indeed, aggregation of planar macrocyclic catalysts has been shown to greatly hinder electrocatalytic activity.[24]
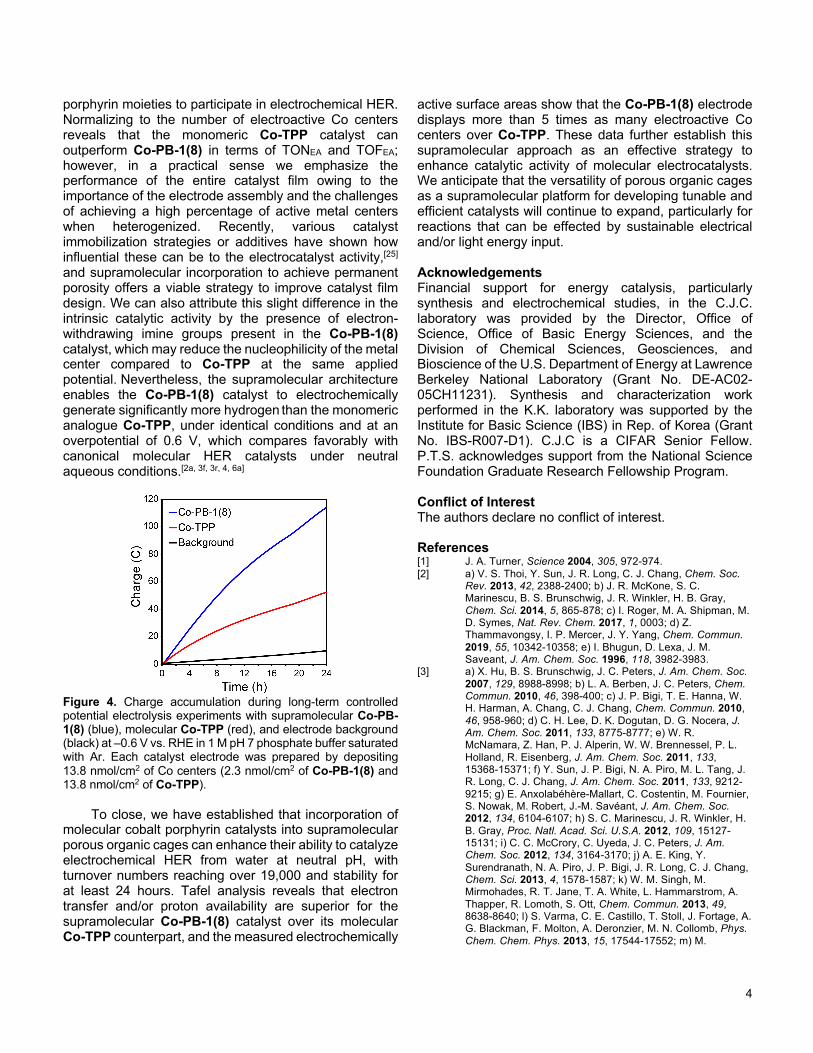
With these data in hand, we performed controlled potential electrolysis experiments with both Co-PB-1(8) and Co-TPP heterogeneous electrodes as HER electrocatalysts (13.8 nmol/cm2 Co deposited for each) in 1 M pH 7 phosphate buffer. A potential of –0.6 V vs. RHE was applied for 24 h, and Co-PB-1(8) and Co-TPP consumed 114 C and 52 C of charge, respectively (Figure 4). Analyzing the headspace after each experiment by gas chromatography revealed that both catalysts gave ca. 100% Faradaic efficiency for HER, with Co-PB-1(8) producing more than twice the amount of H2 compared to Co-TPP (14.0 mL vs. 6.6 mL of hydrogen). For Co-PB-1(8), these results correspond to a turnover number (TON) of 19,030 [TON per electroactive Co (TONEA) = 262,660] and an average turnover frequency (TOF) of 0.22 s–1 [TOF per electroactive Co (TOFEA) = 3.04 s–1]. For Co-TPP, these data give a TON of 8,710 (TONEA = 601,090) and an average TOF of 0.10 s–1 (TOFEA = 6.96 s–1). The data support the notion that the supramolecular structure allows for more of the deposited catalytic cobalt
Figure 3. Scan rate dependence from 0.025 to 0.2 V/s of the CoIII/CoII redox couple for supramolecular Co-PB-1(8) (A) and molecular Co-TPP (B) catalyst films in 1 M pH 7 phosphate buffer saturated with N2. Each catalyst electrode was prepared by depositing 13.8 nmol/cm2 of Co centers (2.3 nmol/cm2 of Co-PB-1(8) and 13.8 nmol/cm2 of Co-TPP). The corresponding cathodic and anodic peak currents are plotted against the scan rate in (C) for Co-PB-1(8) and in (D) for Co-TPP.
4
porphyrin moieties to participate in electrochemical HER. Normalizing to the number of electroactive Co centers reveals that the monomeric Co-TPP catalyst can outperform Co-PB-1(8) in terms of TONEA and TOFEA; however, in a practical sense we emphasize the performance of the entire catalyst film owing to the importance of the electrode assembly and the challenges of achieving a high percentage of active metal centers when heterogenized. Recently, various catalyst immobilization strategies or additives have shown how influential these can be to the electrocatalyst activity,[25] and supramolecular incorporation to achieve permanent porosity offers a viable strategy to improve catalyst film design. We can also attribute this slight difference in the intrinsic catalytic activity by the presence of electron-withdrawing imine groups present in the Co-PB-1(8) catalyst, which may reduce the nucleophilicity of the metal center compared to Co-TPP at the same applied potential. Nevertheless, the supramolecular architecture enables the Co-PB-1(8) catalyst to electrochemically generate significantly more hydrogen than the monomeric analogue Co-TPP, under identical conditions and at an overpotential of 0.6 V, which compares favorably with canonical molecular HER catalysts under neutral aqueous conditions.[2a, 3f, 3r, 4, 6a]
Figure 4. Charge accumulation during long-term controlled potential electrolysis experiments with supramolecular Co-PB-1(8) (blue), molecular Co-TPP (red), and electrode background (black) at –0.6 V vs. RHE in 1 M pH 7 phosphate buffer saturated with Ar. Each catalyst electrode was prepared by depositing 13.8 nmol/cm2 of Co centers (2.3 nmol/cm2 of Co-PB-1(8) and 13.8 nmol/cm2 of Co-TPP).
To close, we have established that incorporation of molecular cobalt porphyrin catalysts into supramolecular porous organic cages can enhance their ability to catalyze electrochemical HER from water at neutral pH, with turnover numbers reaching over 19,000 and stability for at least 24 hours. Tafel analysis reveals that electron transfer and/or proton availability are superior for the supramolecular Co-PB-1(8) catalyst over its molecular Co-TPP counterpart, and the measured electrochemically
active surface areas show that the Co-PB-1(8) electrode displays more than 5 times as many electroactive Co centers over Co-TPP. These data further establish this supramolecular approach as an effective strategy to enhance catalytic activity of molecular electrocatalysts. We anticipate that the versatility of porous organic cages as a supramolecular platform for developing tunable and efficient catalysts will continue to expand, particularly for reactions that can be effected by sustainable electrical and/or light energy input. Acknowledgements Financial support for energy catalysis, particularly synthesis and electrochemical studies, in the C.J.C. laboratory was provided by the Director, Office of Science, Office of Basic Energy Sciences, and the Division of Chemical Sciences, Geosciences, and Bioscience of the U.S. Department of Energy at Lawrence Berkeley National Laboratory (Grant No. DE-AC02-05CH11231). Synthesis and characterization work performed in the K.K. laboratory was supported by the Institute for Basic Science (IBS) in Rep. of Korea (Grant No. IBS-R007-D1). C.J.C is a CIFAR Senior Fellow. P.T.S. acknowledges support from the National Science Foundation Graduate Research Fellowship Program. Conflict of Interest The authors declare no conflict of interest. References [1] J. A. Turner, Science 2004, 305, 972-974. [2] a) V. S. Thoi, Y. Sun, J. R. Long, C. J. Chang, Chem. Soc.
Rev. 2013, 42, 2388-2400; b) J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler, H. B. Gray, Chem. Sci. 2014, 5, 865-878; c) I. Roger, M. A. Shipman, M. D. Symes, Nat. Rev. Chem. 2017, 1, 0003; d) Z. Thammavongsy, I. P. Mercer, J. Y. Yang, Chem. Commun. 2019, 55, 10342-10358; e) I. Bhugun, D. Lexa, J. M. Saveant, J. Am. Chem. Soc. 1996, 118, 3982-3983.
[3] a) X. Hu, B. S. Brunschwig, J. C. Peters, J. Am. Chem. Soc. 2007, 129, 8988-8998; b) L. A. Berben, J. C. Peters, Chem. Commun. 2010, 46, 398-400; c) J. P. Bigi, T. E. Hanna, W. H. Harman, A. Chang, C. J. Chang, Chem. Commun. 2010, 46, 958-960; d) C. H. Lee, D. K. Dogutan, D. G. Nocera, J. Am. Chem. Soc. 2011, 133, 8775-8777; e) W. R. McNamara, Z. Han, P. J. Alperin, W. W. Brennessel, P. L. Holland, R. Eisenberg, J. Am. Chem. Soc. 2011, 133, 15368-15371; f) Y. Sun, J. P. Bigi, N. A. Piro, M. L. Tang, J. R. Long, C. J. Chang, J. Am. Chem. Soc. 2011, 133, 9212-9215; g) E. Anxolabéhère-Mallart, C. Costentin, M. Fournier, S. Nowak, M. Robert, J.-M. Savéant, J. Am. Chem. Soc. 2012, 134, 6104-6107; h) S. C. Marinescu, J. R. Winkler, H. B. Gray, Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 15127-15131; i) C. C. McCrory, C. Uyeda, J. C. Peters, J. Am. Chem. Soc. 2012, 134, 3164-3170; j) A. E. King, Y. Surendranath, N. A. Piro, J. P. Bigi, J. R. Long, C. J. Chang, Chem. Sci. 2013, 4, 1578-1587; k) W. M. Singh, M. Mirmohades, R. T. Jane, T. A. White, L. Hammarstrom, A. Thapper, R. Lomoth, S. Ott, Chem. Commun. 2013, 49, 8638-8640; l) S. Varma, C. E. Castillo, T. Stoll, J. Fortage, A. G. Blackman, F. Molton, A. Deronzier, M. N. Collomb, Phys. Chem. Chem. Phys. 2013, 15, 17544-17552; m) M.

5
Vennampalli, G. Liang, L. Katta, C. E. Webster, X. Zhao, Inorg. Chem. 2014, 53, 10094-10100; n) D. Basu, S. Mazumder, X. Shi, H. Baydoun, J. Niklas, O. Poluektov, H. B. Schlegel, C. N. Verani, Angew. Chem. Int. Ed. 2015, 54, 2105-2110; o) J. W. Jurss, R. S. Khnayzer, J. A. Panetier, K. A. El Roz, E. M. Nichols, M. Head-Gordon, J. R. Long, F. N. Castellano, C. J. Chang, Chem. Sci. 2015, 6, 4954-4972; p) A. Lewandowska-Andralojc, T. Baine, X. Zhao, J. T. Muckerman, E. Fujita, D. E. Polyansky, Inorg. Chem. 2015, 54, 4310-4321; q) J. Willkomm, N. M. Muresan, E. Reisner, Chem. Sci. 2015, 6, 2727-2736; r) B. Kandemir, L. Kubie, Y. Guo, B. Sheldon, K. L. Bren, Inorg. Chem. 2016, 55, 1355-1357; s) A. G. Maher, G. Passard, D. K. Dogutan, R. L. Halbach, B. L. Anderson, C. J. Gagliardi, M. Taniguchi, J. S. Lindsey, D. G. Nocera, ACS Catal. 2017, 7, 3597-3606; t) S. Khandelwal, A. Zamader, V. Nagayach, D. Dolui, A. Q. Mir, A. Dutta, ACS Catal. 2019, 9, 2334-2344; u) L. Kohler, J. Niklas, R. C. Johnson, M. Zeller, O. G. Poluektov, K. L. Mulfort, Inorg. Chem. 2019, 58, 1697-1709; v) J.-W. Wang, K. Yamauchi, H.-H. Huang, J.-K. Sun, Z.-M. Luo, D.-C. Zhong, T.-B. Lu, K. Sakai, Angew. Chem. Int. Ed. 2019, 58, 10923-10927; w) J. L. Dempsey, B. S. Brunschwig, J. R. Winkler, H. B. Gray, Acc. Chem. Res. 2009, 42, 1995-2004; x) V. Artero, M. Chavarot-Kerlidou, M. Fontecave, Angew. Chem. Int. Ed. 2011, 50, 7238-7266; y) N. Kaeffer, M. Chavarot-Kerlidou, V. Artero, Acc. Chem. Res. 2015, 48, 1286-1295; z) D. Z. Zee, T. Chantarojsiri, J. R. Long, C. J. Chang, Acc. Chem. Res. 2015, 48, 2027-2036.
[4] a) R. M. Kellett, T. G. Spiro, Inorg. Chem. 2002, 24, 2373-2377; b) B. B. Beyene, S. B. Mane, C. H. Hung, Chem. Commun. 2015, 51, 15067-15070.
[5] a) R. M. Kellett, T. G. Spiro, Inorg. Chem. 2002, 24, 2378-2382; b) T. Abe, F. Taguchi, H. Imaya, F. Zhao, J. Zhang, M. Kaneko, Polym. Adv. Technol. 1998, 9, 559-562; c) C. Canales, F. Varas-Concha, T. E. Mallouk, G. Ramirez, Appl. Catal. B Environ. 2016, 188, 169-176; d) A. M. Beiler, D. Khusnutdinova, B. L. Wadsworth, G. F. Moore, Inorg. Chem. 2017, 56, 12178-12185.
[6] a) J. G. Kleingardner, B. Kandemir, K. L. Bren, J. Am. Chem. Soc. 2014, 136, 4-7; b) D. J. Sommer, M. D. Vaughn, G. Ghirlanda, Chem. Commun. 2014, 50, 15852-15855.
[7] a) C. Y. Yeh, C. J. Chang, D. G. Nocera, J. Am. Chem. Soc. 2001, 123, 1513-1514; b) C. J. Chang, L. L. Chng, D. G. Nocera, J. Am. Chem. Soc. 2003, 125, 1866-1876; c) S. Samanta, K. Sengupta, K. Mittra, S. Bandyopadhyay, A. Dey, Chem. Commun. 2012, 48, 7631-7633; d) C. T. Carver, B. D. Matson, J. M. Mayer, J. Am. Chem. Soc. 2012, 134, 5444-5447; e) D. J. Graham, D. G. Nocera, Organometallics 2014, 33, 4994-5001; f) I. Azcarate, C. Costentin, M. Robert, J. M. Saveant, J. Am. Chem. Soc. 2016, 138, 16639-16644; g) A. Rana, B. Mondal, P. Sen, S. Dey, A. Dey, Inorg. Chem. 2017, 56, 1783-1793; h) E. M. Nichols, J. S. Derrick, S. K. Nistanaki, P. T. Smith, C. J. Chang, Chem. Sci. 2018, 9, 2952-2960; i) R. Zhang, J. J. Warren, J. Am. Chem. Soc. 2020, 142, 13426-13434; j) C. G. Margarit, C. Schnedermann, N. G. Asimow, D. G. Nocera, Organometallics 2018, 38, 1219-1223; k) S. Sinha, M. Ghosh, J. J. Warren, ACS Catal. 2019, 9, 2685-2691; l) W. Zhang, W. Lai, R. Cao, Chem. Rev. 2017, 117, 3717-3797; m) B. B. Beyene, C. H. Hung, Coord. Chem. Rev. 2020, 410, 213234; n) S. Amanullah, P. Saha, A. Nayek, M. E. Ahmed, A. Dey, Chem. Soc. Rev. 2021.
[8] a) T. F. Jaramillo, K. P. Jorgensen, J. Bonde, J. H. Nielsen, S. Horch, I. Chorkendorff, Science 2007, 317, 100-102; b) C. Wang, H. Daimon, T. Onodera, T. Koda, S. Sun, Angew. Chem. Int. Ed. 2008, 47, 3588-3591; c) C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang, V. R. Stamenkovic, Science 2014, 343, 1339-1343; d) M. S. Faber, S. Jin,
Energy Environ. Sci. 2014, 7, 3519-3542; e) E. J. Popczun, C. W. Roske, C. G. Read, J. C. Crompton, J. M. McEnaney, J. F. Callejas, N. S. Lewis, R. E. Schaak, J. Mater. Chem. A 2015, 3, 5420-5425; f) H. Mistry, A. S. Varela, S. Kuhl, P. Strasser, B. Roldan Cuenya, Nat. Rev. Mater. 2016, 1, 16009; g) Y. P. Zhu, T. Y. Ma, M. Jaroniec, S. Z. Qiao, Angew. Chem. Int. Ed. 2017, 56, 1324-1328.
[9] a) A. M. Kluwer, R. Kapre, F. Hartl, M. Lutz, A. L. Spek, A. M. Brouwer, P. W. van Leeuwen, J. N. Reek, Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 10460-10465; b) C. W. Machan, J. Yin, S. A. Chabolla, M. K. Gilson, C. P. Kubiak, J. Am. Chem. Soc. 2016, 138, 8184-8193; c) M. Schulze, V. Kunz, P. D. Frischmann, F. Wurthner, Nat. Chem. 2016, 8, 576-583; d) S. Donck, J. Fize, E. Gravel, E. Doris, V. Artero, Chem. Commun. 2016, 52, 11783-11786; e) M. Gong, Z. Cao, W. Liu, E. M. Nichols, P. T. Smith, J. S. Derrick, Y. S. Liu, J. Liu, X. Wen, C. J. Chang, ACS Cent. Sci. 2017, 3, 1032-1040; f) A. N. Oldacre, A. E. Friedman, T. R. Cook, J. Am. Chem. Soc. 2017, 139, 1424-1427; g) S. S. Nurttila, R. Becker, J. Hessels, S. Woutersen, J. N. H. Reek, Chem. Eur. J. 2018, 24, 16395-16406; h) A. N. Oldacre, M. R. Crawley, A. E. Friedman, T. R. Cook, Chem. Eur. J. 2018, 24, 10984-10987; i) P. T. Smith, B. P. Benke, Z. Cao, Y. Kim, E. M. Nichols, K. Kim, C. J. Chang, Angew. Chem. Int. Ed. 2018, 57, 9684-9688; j) P. T. Smith, Y. Kim, B. P. Benke, K. Kim, C. J. Chang, Angew. Chem. Int. Ed. 2020, 59, 4902-4907.
[10] a) S. Pullen, H. Fei, A. Orthaber, S. M. Cohen, S. Ott, J. Am. Chem. Soc. 2013, 135, 16997-17003; b) A. J. Clough, J. W. Yoo, M. H. Mecklenburg, S. C. Marinescu, J. Am. Chem. Soc. 2015, 137, 118-121; c) S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi, C. J. Chang, Science 2015, 349, 1208-1213; d) R. Dong, M. Pfeffermann, H. Liang, Z. Zheng, X. Zhu, J. Zhang, X. Feng, Angew. Chem. Int. Ed. 2015, 54, 12058-12063; e) I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha, J. T. Hupp, ACS Catal. 2015, 5, 6302-6309; f) E. M. Miner, T. Fukushima, D. Sheberla, L. Sun, Y. Surendranath, M. Dinca, Nat. Commun. 2016, 7, 10942; g) C. S. Diercks, S. Lin, N. Kornienko, E. A. Kapustin, E. M. Nichols, C. Zhu, Y. Zhao, C. J. Chang, O. M. Yaghi, J. Am. Chem. Soc. 2018, 140, 1116-1122.
[11] a) P. T. Smith, E. M. Nichols, Z. Cao, C. J. Chang, Acc. Chem. Res. 2020, 53, 575-587; b) A. H. Proppe, Y. G. C. Li, A. Aspuru-Guzik, C. P. Berlinguette, C. J. Chang, R. Cogdell, A. G. Doyle, J. Flick, N. M. Gabor, R. van Grondelle, S. Hammes-Schiffer, S. A. Jaffer, S. O. Kelley, M. Leclerc, K. Leo, T. E. Mallouk, P. Narang, G. S. Schlau-Cohen, G. D. Scholes, A. Vojvodic, V. W. W. Yam, J. Y. Yang, E. H. Sargent, Nat. Rev. Mater. 2020, 5, 828-846.
[12] a) N. M. Rue, J. L. Sun, R. Warmuth, Isr. J. Chem. 2011, 51, 743-768; b) T. Hasell, A. I. Cooper, Nat. Rev. Mater. 2016, 1, 16053.
[13] a) T. Hasell, M. Schmidtmann, C. A. Stone, M. W. Smith, A. I. Cooper, Chem. Commun. 2012, 48, 4689-4691; b) S. Hong, M. R. Rohman, J. Jia, Y. Kim, D. Moon, Y. Kim, Y. H. Ko, E. Lee, K. Kim, Angew. Chem. Int. Ed. 2015, 54, 13241-13244; c) X. Y. Hu, W. S. Zhang, F. Rominger, I. Wacker, R. R. Schroder, M. Mastalerz, Chem. Commun. 2017, 53, 8616-8619.
[14] a) Y. Jin, B. A. Voss, R. D. Noble, W. Zhang, Angew. Chem. Int. Ed. 2010, 49, 6348-6351; b) T. Mitra, K. E. Jelfs, M. Schmidtmann, A. Ahmed, S. Y. Chong, D. J. Adams, A. I. Cooper, Nat. Chem. 2013, 5, 276-281.
[15] R. L. Greenaway, D. Holden, E. G. B. Eden, A. Stephenson, C. W. Yong, M. J. Bennison, T. Hasell, M. E. Briggs, S. L. James, A. I. Cooper, Chem. Sci. 2017, 8, 2640-2651.
[16] B. P. Benke, P. Aich, Y. Kim, K. L. Kim, M. R. Rohman, S. Hong, I. C. Hwang, E. H. Lee, J. H. Roh, K. Kim, J. Am. Chem. Soc. 2017, 139, 7432-7435.

6
[17] Y. Kim, J. Koo, I. C. Hwang, R. D. Mukhopadhyay, S. Hong, J. Yoo, A. A. Dar, I. Kim, D. Moon, T. J. Shin, Y. H. Ko, K. Kim, J. Am. Chem. Soc. 2018, 140, 14547-14551.
[18] X. Yu, B. Wang, Y. Kim, J. Park, S. Ghosh, B. Dhara, R. D. Mukhopadhyay, J. Koo, I. Kim, S. Kim, I. C. Hwang, S. Seki, D. M. Guldi, M. H. Baik, K. Kim, J. Am. Chem. Soc. 2020, 142, 12596-12601.
[19] R. D. Mukhopadhyay, Y. Kim, J. Koo, K. Kim, Acc. Chem. Res. 2018, 51, 2730-2738.
[20] a) C. Costentin, J. M. Saveant, J. Am. Chem. Soc. 2018, 140, 16669-16675; b) Y. H. Wang, M. L. Pegis, J. M. Mayer, S. S. Stahl, J. Am. Chem. Soc. 2017, 139, 16458-16461.
[21] T. Shinagawa, A. T. Garcia-Esparza, K. Takanabe, Sci. Rep. 2015, 5, 13801.
[22] N. Morlanés, K. Takanabe, V. Rodionov, ACS Catal. 2016, 6, 3092-3095.
[23] C. M. Hanna, C. D. Sanborn, S. Ardo, J. Y. Yang, ACS Appl. Mater. Interfaces 2018, 10, 13211-13217.
[24] M. H. Zhu, R. Q. Ye, K. Jin, N. Lazouski, K. Manthiram, ACS Energy Lett. 2018, 3, 1381-1386.
[25] a) G. Q. Zhao, K. Rui, S. X. Dou, W. P. Sun, Adv. Funct. Mater. 2018, 28, 1803291; b) A. Wagner, C. D. Sahm, E. Reisner, Nat. Catal. 2020, 3, 775-786.

download fileview on ChemRxivBox_HER_ChemRxiv_Manuscript.pdf (1.20 MiB)

S1
Table of Contents
Experimental details S2 Synthetic details for Co-PB-1(8) S4 Figure S1. UV/Vis spectra S5 Figure S2. MALDI-MS spectrum S6 Figure S3. CO2 (195 K) sorption isotherms for Co-PB-1(8) S6 Figure S4. IR spectra S7 References S8

S2
General Methods. All reagents and solvents were purchased from commercial sources and used without further purification unless specified. Hydrogen (99.999%), ethylene (99.9%), argon (99.999%) and nitrogen (99.998%) were purchased from Praxair. Multi-walled carbon nanotubes (MWCNTs) were purchased from Sigma-Aldrich and were washed with 10% HCl followed by deionized water until the filtrate was neutral and then dried in vacuo before use. 2,6-Lutidine was purchased from Sigma-Aldrich and dried over AlCl3 then purified by distillation. Co(II) tetraphenylporphyrin and CoCl2 were purchased from Strem Chemicals and used as received. Chelex chelating resin, sodium phosphate monobasic, and sodium phosphate dibasic were purchased from Sigma-Aldrich and used as received. The free-base porphyrin box was synthesized and characterized as previously described.1 THF and CH3CN were dried using a JC Meyer solvent purification system. Deionized water was obtained from a Millipore Autopure system. UV/Vis spectra were collected using an Agilent Cary 60 spectrophotometer at room temperature. Matrix-assisted laser desorption ionization (MALDI) mass spectrometry spectra were obtained using an Applied Biosystems Voyager DE Pro at the QB3 Mass Spectrometry Facility at the University of California, Berkeley. FT-IR spectra were collected using a Bruker ALPHA-P spectrometer. Gas Sorption Measurements. Gas sorption isotherms of Co-PB-1(8) were measured with an Autosorp-iQ volumetric adsorption analyzer. Typically, a sample of as-synthesized material (~30 mg) was loaded and, prior to the measurements, residual solvents were evacuated by heating to 100 ºC under high vacuum (10-2 Pa) overnight. Sorption isotherms were measured for CO2 using a dry-ice/acetone dewar (for 195 K). Cyclic Voltammetry. Cyclic voltammetry experiments were performed using either a Bioanalytical Systems, Inc. (BASi) Epsilon potentiostat or a CH Instruments, Inc. 600E potentiostat. Aqueous voltammetry studies were performed with a three-electrode cell equipped with a glassy carbon disk (3.0 mm diameter) working electrode (BASi), a platinum wire counter electrode, and a Ag/AgCl reference electrode. The electrolyte used was 1 M aq. potassium phosphate buffer with pH 7 and sparged with Ar. The electrolyte was pre-treated with Chelex resin overnight to remove any trace metal ion impurities.2 No iR compensation was applied. Preparative Scale Electrolysis. Controlled potential electrolyses were performed using either a Bioanalytical Systems, Inc. (BASi) Epsilon potentiostat or a CH Instruments, Inc. 600E potentiostat. A custom-made three-piece glass electrochemical cell was used (Adams & Chittenden Scientific Glass), including a working compartment body, a working compartment lid, and a counter compartment body. A cation exchange membrane (Nafion 117) separates the working electrode and Ag/AgCl (sat. KCl) reference electrode from the graphite rod counter electrode. This experimental setup is a smaller version of what we have described elsewhere.3-6 The cathode compartment holds 50 mL of electrolyte and the anode compartment holds 12 mL of electrolyte. The electrolyte used was 1 M aq. potassium phosphate buffer with pH 7 and sparged with Ar for 20 min immediately before electrolysis. The electrolyte was pre-treated with Chelex resin overnight to remove any trace metal ion impurities.2 Once assembled, an internal standard of 0.5 mL ethylene was injected into the headspace for GC analysis. Electrolysis was then performed at a constant potential for a specified amount of time. No iR compensation was applied. After this period, the headspace was immediately analyzed by GC, and the amount of gas generated was

S3
determined on the basis of the internal standard and from calibration curves plotted from injecting known amounts of H2 and ethylene into the GC. Gas production was measured on an SRI gas chromatography instrument (model #8610C) equipped with a Haysep D column (1/8” × 6’) and a 13X Mol Sieve column (1/8” × 6’). Hydrogen and ethylene were quantified using a thermal conductivity detector (TCD). Preparation of Working Electrodes. For cyclic voltammetry experiments, a glassy carbon (GC) disk electrode (0.071 cm2) was thoroughly polished with 0.5 µm alumina powder, rinsed with isopropanol, and dried in air. To increase conductivity, a dispersion of MWCNTs in iPrOH (1 mg/mL) was sonicated for 20 min, and then 10 µL was slowly deposited onto the 3 mm diameter GC electrode and dried in air. Then, either 14.2 µL of a Co-PB-1(8) solution in THF (0.1 mg/mL) or 6.6 µL of a Co-TPP solution in THF (0.1 mg/mL) was deposited on top of the MWCNT/GC electrode and allowed to dry in air. This electrode was then thoroughly rinsed with iPrOH, H2O, and 1 M pH 7 phosphate buffer, and then dried in air again. This preparation results in 0.14 mg/cm2 of MWCNTs and either 0.02 mg/cm2 (2.31 nmol/cm2) of Co-PB-1(8) or 0.01 mg/cm2 (13.84 nmol/cm2) of Co-TPP. For controlled potential electrolyses, a glassy carbon plate electrode (2.25 cm2) was thoroughly polished with 0.5 µm alumina powder, rinsed with isopropanol, and dried in air. A dispersion of MWCNTs in iPrOH (2 mg/mL) was sonicated for 20 min, and then 190 µL was slowly deposited onto the GC plate electrode and thoroughly dried in air. Then, either 450 µL of a Co-PB-1(8) solution in THF (0.1 mg/mL) or 209 µL of a Co-TPP solution in THF (0.1 mg/mL) was deposited on top of the MWCNT/GC electrode and allowed to dry in air. This electrode was then thoroughly rinsed with iPrOH, H2O, and 1 M pH 7 phosphate buffer, and then dried in air again. This preparation results in 0.17 mg/cm2 of MWCNTs and either 0.02 mg/cm2 (2.31 nmol/cm2) of Co-PB-1(8) or 0.01 mg/cm2 (13.84 nmol/cm2) of Co-TPP. Determination of Electroactive Metal Centers. The scan rate dependence was measured from 0.025-0.2 V/s of the CoIII/CoII redox couple for Co-PB-1(8) and Co-TPP catalyst films in 1 M pH 7 phosphate buffer saturated with N2. Each catalyst electrode was prepared by depositing 13.8 nmol/cm2 of Co centers (2.3 nmol/cm2 of Co-PB-1(8) and 13.8 nmol/cm2 of Co-TPP). The number of electroactive Co centers in the catalyst films (G, mol/cm2) was determined from these plots using the following equation:
𝑖! = 𝑛"𝐹𝑆Γ𝐹𝑣4𝑅𝑇
where ip is the peak current of the CoIII/CoII redox couple, n is the number of electrons, F is Faraday’s constant, S is the geometrical surface area of the electrode, v is the scan rate, R is the gas constant, and T is the temperature. The number of electrons, n, is taken to be 1 for the single electron transfer. This treats each cobalt porphyrin moiety in Co-PB-1(8) or Co-TPP as independent of one another. Cathodic Current Anodic Current Co-PB-1(8) y = 65.84x + 0.75; R2 = 0.998 y = –75.18x – 0.74; R2 = 0.996 Co-TPP y = 14.57x + 0.18; R2 = 0.997 y = –16.68x – 0.21; R2 = 0.994

S4
Synthesis of Co-PB-1(8). Under a dinitrogen atmosphere, free-base porphyrin box (25 mg, 2.9 µmol) and CoCl2 (40 mg, 0.31 mmol, 106 equiv.) were dissolved in anhydrous THF (50 mL). Subsequently, 2,6-lutidine (10 µL, 0.083 mmol, 29 equiv.) was injected, and the reaction mixture was heated at 70 ºC for 12 h. After cooling to room temperature, solvent was removed under reduced pressure. Then, acetonitrile (25 mL) was added, and after sonication for 30 s to suspend the product, the mixture was transferred to a conical tube and centrifuged. The supernatant was then decanted off. This washing cycle with acetonitrile was repeated six times. Co-PB-1(8) was obtained as a dark red-purple powder (95% yield). MALDI-MS (matrix: DCTB) m/z calcd. for C552H624Co6N48O24 [M]+: 8667.52, found: 8667.48. Elemental analysis calc. for C552H624Co6N48O24(C2H6O)2: C, 76.22; H, 7.24; N, 7.72. Found: C, 75.97; H, 7.28; N, 7.58. UV/Vis (CH2Cl2) labs 418, 531 nm.
N
NH N
HN
O
H
H O
H
O
HO
O
O O
(CH2)7CH3
(CH2)7CH3
H3C(H2C)7
H2N
NH2
NH2
N
NH N
HN
N
H
H N
H
N
HN
O
O
O
N
N
H3C(H2C)7
H3C(H2C)7(CH2)7CH3
6
8
N
N N
N
N
H
H N
H
N
HN
O
O
O
N
N
H3C(H2C)7
H3C(H2C)7(CH2)7CH3
Co(i) (ii)

S5
Figure S1. UV/Vis spectra of (A) free-base porphyrin box (B) Co-PB-1(8), and (C) Co-TPP.
0.0
0.2
0.4
0.6
0.8
1.0
300 350 400 450 500 550 600 650 700
No
rma
lize
d A
bso
rban
ce (
AU
)
Wavelength (nm)
0.0
0.2
0.4
0.6
0.8
1.0
300 350 400 450 500 550 600 650 700
No
rma
lize
d A
bso
rban
ce (
AU
)
Wavelength (nm)
A
B
C
0.0
0.2
0.4
0.6
0.8
1.0
300 350 400 450 500 550 600 650 700
No
rma
lize
d A
bso
rban
ce (
AU
)
Wavelength (nm)

S6
Figure S2. MALDI-MS spectrum of Co-PB-1(8) (DCTB matrix).
Figure S3. CO2 sorption isotherms for Co-PB-1(8) at 195 K.
0
50
100
150
200
250
300
350
4000 6000 8000 10000 12000
Inte
nsity
Mass (m/z)
[M+] 8667.48
0
50
100
150
200
250
300
0.0 0.2 0.4 0.6 0.8 1.0
V Ads
(cm
3 /g)
Relative Pressure (P/P0)
Adsorption
Desorption

S7
Figure S4. IR spectra of (A) free-base porphyrin box, (B) Co-PB-1(8), and (C) Co-TPP.
A
B
C
40080012001600200024002800320036004000
Tra
nsm
itta
nce
Wavenumbers (cm–1)
40080012001600200024002800320036004000
Tra
nsm
itta
nce
Wavenumbers (cm–1)
40080012001600200024002800320036004000
Tra
nsm
itta
nce
Wavenumbers (cm–1)

S8
References: 1. B. P. Benke, P. Aich, Y. Kim, K. L. Kim, M. R. Rohman, S. Hong, I. C. Hwang, E. H.
Lee, J. H. Roh and K. Kim, J. Am. Chem. Soc., 2017, 139, 7432-7435. 2. A. Wuttig and Y. Surendranath, ACS Catal., 2015, 5, 4479-4484. 3. S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D.
Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208-1213. 4. Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong, J. A.
Reimer, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 8120-8125. 5. E. M. Nichols, J. J. Gallagher, C. Liu, Y. Su, J. Resasco, Y. Yu, Y. Sun, P. Yang, M. C.
Chang and C. J. Chang, Proc. Natl. Acad. Sci. U.S.A., 2015, 112, 11461-11466. 6. Z. Cao, J. S. Derrick, J. Xu, R. Gao, M. Gong, E. M. Nichols, P. T. Smith, X. Liu, X.
Wen, C. Coperet and C. J. Chang, Angew. Chem. Int. Ed., 2018, 57, 4981-4985.

download fileview on ChemRxivBox_HER_Supporting_Information_031321.pdf (496.07 KiB)