a new partitioning scheme for molecular interacting systems within a multiconfigurational or...
TRANSCRIPT

— —< <
A New Partitioning Scheme forMolecular Interacting SystemsWithin a Multiconfigurationalor MonoconfigurationalHartree]Fock Formalism
´ ˜ANDRE MELO, MARIA JOAO RAMOSCEQUPrDepartamento de Quımica, Faculdade de Ciencias, Universidade do Porto,´ ˆRua do Campo Alegre, 687, 4150 Porto, Portugal
Received 20 January 1998; revised 10 September 1998; accepted 20 October 1998
ABSTRACT: A new method, based on the spatial decomposition of the reduced-density and pair-density matrices and the indistinguishable integrals formalism, isintroduced to partition the molecular and stabilization energies into meaningfulfragments. These are defined as entirely flexible variable-size entities, for example,atoms, group of atoms, ions, and interacting monomers. This new partitioning scheme isespecially appropriated to study systems in which a directly bonded group-transferprocess occurs. In these cases, the stabilization energies are partitioned into anintrafragment component, associated with the difference of intrinsic affinity to thetransferred group between the involved fragments, and an interfragment component,associated with the difference of the magnitude of the interaction between the fragmentsin the initial and final binding complexes. This method was applied to the study of thearginine]carboxylate interactions, allowing us to have insight into what really happens in
Ž .this system. Two zwitterionic and neutral binding complexes can be considered. Themain effects accountable for the preferential stabilizations of the binding complexes aredetermined to be basis-set independent. The zwitterionic complex is favored by theinterfragment component, while the neutral complex is favored by the larger intrinsicproton affinity of the acetate relatively to the methylguanidium. Q 1999 John Wiley &Sons, Inc. Int J Quant Chem 72: 157]176, 1999
Key words: partitioning scheme; molecular and stabilization energies; reduced densitymatrices; indistinguishable integrals; arginine]carboxylate interactions
Correspondence to: M. J. Ramos.
( )International Journal of Quantum Chemistry, Vol. 72, 157]176 1999Q 1999 John Wiley & Sons, Inc. CCC 0020-7608 / 99 / 030157-20

MELO AND RAMOS
Introduction
he physical interpretation of phenomena suchT as chemical bonding, molecular associationprocesses, and proton-transfer reactions requires apartitioning of the energy into components andrormeaningful molecular fragments associated withthe different types of interactions involved. Therehave been a series of proposals for the above-mentioned partitioning of the energy which havebeen described in the literature. They will be brieflyreferred to next.
One methodology consists in a perturbationalexpansion of the stabilization energy of a super-molecular system as a function of electrostatic,polarization, exchange, and dispersion energy
w xterms 1]4 . An alternative method, within theHartree]Fock variational approach, was suggested
w xby Kitaura and Morokuma 5 . According to it,certain blocks of the Fock matrix for a supermolec-ular system, constructed using the molecular or-bitals of its fragments, are set to zero. The result-ing matrix is diagonalized iteratively to obtain thestabilization energy components, that is, electro-static, polarization, exchange, charge transfer, andcoupling. The perturbational and variational Mo-rokuma decomposition method allows a very use-ful interpretation of the interactions in terms of theelectronic properties of the isolated fragments.However, it presents some problems in describingcertain phenomena such as the internal fragmentsgeometry relaxation or the exchange of atoms be-tween fragments, and these can be very importantto obtain a meaningful physical interpretation ofthe supermolecular system formation from its iso-lated pieces.
A third approach to the problem of energy par-titioning is based on the fact that the energy of amolecular system can be completely determinedby the reduced-density and pair-density matrices.If a Hartree]Fock approximation is used, the den-sity and pair-density populations can be parti-tioned in terms associated with the atomic basisfunctions. This decomposition procedure can beextended also, within a supermolecule approach,to the stabilization energy of the system. In thiscontext, several partition schemes have been intro-duced to decompose molecular energy quantities
w xin atomic components 6]10 .
In his study of the physical nature of the chemi-w xcal bond, Ruedenberg 6 devised a method to
decompose the stabilization energy, combiningŽspatial components intratomic, interatomic, and
. Žinterbonds and components promotional, quasi-classical, electrostatic, sharing-penetration, inter-
.ference, and charge transfer associated with aphysical partition of the interactions involving thereduced-density and pair-density matrices.
Another molecular energy decomposition meth-odology, using the one and two-electron integraland the reduced-density matrices, was proposed
w xby Clementi 7 . Within this formalism, intra-Ž .atomic and bicentric and multicentric interatomic
components were considered and introduced.w xKollmar 8 proposed a method to partition the
molecular energy using the one-electron integral,the reduced density, and the Fock matrices. Withinhis scheme, only monocentric intraatomic and bi-centric interatomic effective terms arise.
w xBofill et al. 9 suggested a formalism in whichthe molecular energy is partitioned using the one-and two-electron integral and the reduced pair-density matrices. This allows the molecular struc-ture and energies to be analyzed using only thepair-density populations.
w xMelo and Gomes 10 performed a decomposi-tion of the orbital energies in its components, fol-lowing its nature and location in terms of atomiccenters. According to its nature, the orbital ener-gies are partitioned in kinetic and potential com-ponents; with respect to location, the orbital ener-gies are partitioned in monocentric and bicentriccomponents. This energy decomposition was per-formed within a semiempirical formalism.
Here, we propose a general partition method,within a monoconfigurational or multiconfigura-tional Hartree]Fock formalism, which follows theapproach based on the general knowledge that theenergy of a molecular system can be completelydetermined by the reduced-density and pair-density matrices. According to this scheme, themolecular and stabilization energies are parti-tioned into meaningful fragments. This allows fora useful and flexible interpretation of the interac-tion within a particular system. The philosophy ofthe present method is similar to the Clementischeme to partition the molecular energy into
w xatomic components 7 . Therefore, compared to thew x w x w xRuedenberg 6 , Kollmar 8 , and Bofill 9 schemes,
a more rigorous criterion is adopted to deal with
VOL. 72, NO. 3158

PARTITIONING FOR MOLECULAR INTERACTING SYSTEMS
the inherent arbitrariness associated with the over-lap density and monoelectronic energy spatial de-compositions. However, the present method ismuch more general than Clementi’s, presentingadditionally the following advantages:
Ž .i The partitioning is applied not only to themolecular energy, but also to the stabiliza-tion energy of the system.
Ž .ii The fragments, in which the molecular sys-tem is subdivided, are absolutety general
Žentities atoms, group of atoms, ions, inter-.acting monomers, etc. .
Ž .iii The model is flexibly defined, accommo-dating several different important effectssuch as polarization, Coulombic interac-tion, overlap, electronic charge transfer, in-ternal fragment geometry relaxation, anddirectly bonded group transfer.
Ž .iv The formalism is expressed in terms of thedensity and pair-density spatial partition-ing. This provides a meaningful physicalinterpretation of fragment interactions ac-cording to the nature of chemical bondsestablished among them.
Ž .v The components of density, pair density,energy, and stabilization energy are calcu-lated using only the indistinguishable inte-grals, which reduces significantly the com-putational time and the storage disk spacenecessary to implement the partitioningscheme.
Ž .vi In the study of molecular systems where adirectly bonded group-transfer process oc-curs, the present scheme allows a decom-position of the relative stabilization energyto be made into meaningful intra- and in-terfragment components. The first compo-nent is associated with the difference ofintrinsic affinity, to the transferred group,between the involved fragments, while thesecond component is associated with thedifference of magnitude of the interaction,between the fragments, in the final andinitial binding complexes. This allows for apowerful interpretation methodology whichprovides a better understanding of the maineffects accountable for the preferential sta-bilization of the relevant stationary minimain the respective potential-energy surface.
Methodology
GENERAL
The total energy of a molecular system, within aŽ .multiconfigurational or monoconfigurationalHartree]Fock formalism, is given by
Ž0. Ž1. Ž2. Ž .E s E q E q E . 1
Ž . Ž0.In Eq. 1 , E is the nuclear repulsion energydefined as
N Xy1 Z ZX YŽ0. Ž .E s , 2Ý Ý« rX YXs1 Ys1
where « is the dielectric constant of the environ-ment; N, the total number of atoms of the system;Z and Z the charges of the nuclei X and Y,X Yrespectively; and r , the distance between themX YŽall the physical quantities in this article are ex-
. Ž1.pressed in au , E is the monoelectronic energygiven by
m mŽ1. Ž .E s P h , 3Ý Ý p q p q
ps1 qs1
where m is the total number of atomic basis func-tions; P , an element of the reduced-density ma-p qtrix; and h , the monoelectronic integral associ-p q
ated with the atomic basis functions x and x ,p q
ˆ² Ž . < < Ž .: Ž .h s x 1 h x 1 , 4p q p 1 q
ˆwith h given by1
N1 ZX2ˆ Ž .h s y = y . 5Ý1 12 « r1 XXs1
Ž . Ž2.Still, in Eq. 1 , E is the bielectronic energydefined as
m m m m1Ž2. Ž . Ž .E s D pq ¬ rs , 6Ý Ý Ý Ý p qr s2 ps1 qs1 rs1 ss1
where D is an element of the reduced pair-p qr sŽ .density matrix and pq ¬ rs is the bielectronic inte
gral associated with the atomic basis functions x ,px , x , and x :q r s
Ž . ² Ž . Ž . < Ž . Ž .: Ž .pq ¬ rs s x 1 x 2 ¬ 1r« r x 1 x 2 . 7p r 12 q s
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 159

MELO AND RAMOS
The reduced-density and pair-density matrices areassociated with the following conservation rela-
w xtions 11 :
m m
Ž .P S s n , 8Ý Ý p q p qps1 qs1
and
m m m m Ž .1 n n y 1Ž .D S S s , 9Ý Ý Ý Ý p qr s p q r s2 2ps1 qs1 rs1 ss1
where n is the total number of electrons in thesystem, and S , the overlap integral between thep qatomic basis functions x and x :p q
² Ž . Ž .: Ž .S s x 1 ¬ x 1 . 10p q p q
The latter conservation equations can be inter-w Ž .xpreted also as density Eq. 8 and pair-density
w Ž .xEq. 9 population analysis in the discrete atomicbasis set space.
w Ž .xThe density populations Eq. 8 and monoelec-w Ž .xtronic energy Eq. 3 are calculated by adding the
Ž .contributions over all ordered pairs p, q of atomicŽbasis functions. However, some of them indis-
.tinguishable pairs give the same contribution tothe density populations and the monoelectronic
w xenergy 12 . Therefore, it is possible to select theŽpairs whose contributions differ by symmetry dis-
. Ž . Ž .tinguishable pairs and express Eqs. 3 and 8 asa function of them:
py1m mŽ1. Ž .E s P h q 2 P h 11Ý Ý Ýp p p p p q p q
ps1 ps1 qs1
and
py1m m
Ž .P q 2 P s n. 12Ý Ý Ýp p p qps1 ps1 qs1
This methodology is extended to the pair-densityw Ž .xpopulations Eq. 9 and the bielectronic energy
w Ž .xEq. 6 . These quantities are usually expressed byadding the contributions over all ordered quartetsŽ .p, q, r, s . However, it is also possible to definethe concept of indistinguishable quartets as thosewhich give the same contributions to the pair-
w Ž .xdensity populations Eq. 9 and the bielectronicw x Ženergy 13 . For example, the quartet p, q, r, s
.with p / q / r / s is associated with a set of
eight indistinguishable quartets:
Ž . Ž . Ž . Ž .p , q , r , s ; p , q , s, r ; q , p , r , s ; q , p , s, r ;Ž . Ž . Ž . Ž .r , s, p , q ; r , s, q , p ; s, r , p , q ; s, r , q , p .
Ž .13
Ž . Ž .Accordingly, in this work, Eqs. 6 and 9 areexpressed as a function of the indistinguishablequartets yielding
p pm lim1Ž2. Ž . Ž .E s f pq ¬ rs , 14Ý Ý Ý Ý p qr s2 ps1 qs1 rs1 ss1
and
p pm lim Ž .1 n n y 1Ž .f S S s . 15Ý Ý Ý Ý p qr s p q r s2 2ps1 qs1 rs1 ss1
In the latter equations, lim and f are defined asp qr s
Ž .lim s q if p s r ; lim s r if p / r 16
and
Ž .f s n D , 17p qr s p qr s p qr s
where n is the number of indistinguishablep qr sintegrals associated with the set of indicesŽ .p, q, r, s . The coefficients n for the differentp qr s
types of quartets are presented in Table I.
DECOMPOSITION METHOD
Here, we propose a general decompositionmethod according to which the molecular energy
Žis partitioned into k meaningful fragments atoms,
TABLE ICoefficients n for the different typespqrsof quartets.
Types of quartets( )p, q, r , s Examples npqrs
( )p = q n r / s; p, p, r , s p / r / s 4( )pqrs / 0 p, p, p, s p / s( )p = q n r = s; p, p, r , r p / r 2
pqrs / 0( )p = q n r = s; p, p, p, p 1
pqrs = 0( )p / q n r / s; p, q, r , s p / q / r / s 8
pqrs / 0( )p / q n r / s; p, q, p, q p / q 4
pqrs = 0
( ) ( )pqrs = 0 « p = r n q = s k p = s n q = r . pqrs / 0 «( ) ( )p / r k q / s n p / s k q / r .
VOL. 72, NO. 3160

PARTITIONING FOR MOLECULAR INTERACTING SYSTEMS
.group of atoms, ions, interacting monomers, etc .This decomposition method, within a Hartree]
ŽFock formalism multiconfigurational or mono-.configurational , is thus based on the following
methodology:
Ž .a The density populations and monoelectronicenergies are expressed in terms of contribu-
w Ž .tions of distinguishable pairs Eqs. 11 andŽ .x12 .
Ž .b The pair-density populations and bielec-tronic energies are expressed in terms ofcontributions of distinguishable quartetsw Ž . Ž .xEqs. 14 and 15 .
Ž . Ž .The procedure used in a and b reduces sig-nificantly the computation time necessary to calcu-
Ž .late the monoelectronic integrals h and bielec-prŽ .tronic integrals pq ¬ rs and the space needed to
store them.
Ž .c The density and pair-density populations ofthe molecular system are partitioned interms associated with its meaningful frag-ments.
Ž .d The monoelectronic energy decomposition isobtained by substituting the density parti-
Ž .tioning into Eq. 11 .Ž .e The bielectronic energy decomposition is ob-
tained by substituting the pair-density parti-Ž .tioning into Eq. 14 .
Ž .f This decomposition energy methodology isextended to the stabilization energy, withina supermolecule approach.
Ž .g If the fragments involved in the molecularsystem share electrons, some type of arbi-trariness is always inherent to this type of
w xdecomposition 6 . This is originated by thelack of a rigorous criterion in the spatialdecomposition of the density and pair-density populations associated with theoverlap between two fragments. Accordingto the Mulliken populations analysis methodw x14 , used in Ruedenberg’s decomposition
w xscheme 6 , the overlap density populationsare equally divided by the two fragmentsinvolved. In this work, this approximation isnot used and the overlap density is consid-ered as having a genuine interfragmentnature.
Ž . w xh A frequently used criterion 8, 9 is to asso-Ž .ciate the monoelectronic energies P hp q p q
with the same spatial region to which thecorresponding elements of the reduced-
Ž .density matrix P belong. However, it hasp q
to be considered also that integrals h con-p qtain a nuclear]electronic component whichrepresents the interactions of all the nuclei
Ž . Ž . w xwith the overlap density P x 1 x 1 7 .p q p qTherefore, these energies are partitioned as
kA Ž .P h s P T q P V , 18Ýp q p q p q p q p q p q
As1
with
1 2² Ž . < < Ž .: Ž .T s y x 1 = x 1 , 19p q p 1 q2
and
MA ZXA ² Ž . < < Ž .: Ž .V s y x 1 x 1 . 20Ýp q p q« r1 XXsNA
Ž .In Eq. 18 , P T is the kinetic energy ofp q p q
Ž . Ž . Athe overlap density P x 1 x 1 and P Vp q p q p q p qis the attraction energy between the nucleiof fragment A and the same overlap den-
Ž .sity. In Eq. 20 , N and M are the first andA Athe last nuclei of fragment A, respectively.
The kinetic and nuclear]electronic com-ponents of the monoelectronic energies canhave a different spatial location according tothe atomic basis functions and nuclei in-volved. This information is presented inTable II.
Ž .i In some cases, the formation of the molecu-lar system from its isolated fragments caninvolve the exchange of atoms between themŽ .e.g., proton transfer . To describe this typeof occurrence, some flexibility has been in-troduced in the model to allow fragment-sizevariations during the mentioned process.This is dealt with next.
SPATIAL ENERGIES, DENSITY, ANDPAIR-DENSITY POPULATIONS
When a molecular system is formed from itsfragments, the following states can be considered:
Ž .isolated fragments initial state ` , nonoverlappingŽ .fragments binding state no , and covalent binding
Ž .state ov . The first two states are noncovalentŽ .nrcov and, consequently, the density and pair-density populations associated with the interfrag-ment overlap vanish. In this context, there is no
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 161

MELO AND RAMOS
TABLE IISpatial location of the kinetic and nuclear]electronic components of the monoelectronic energiesassociated with x and x .p q
Spatial location of the atomic basis Spatial location of the kinetic Spatial location of the nuclear]A( ) ( )functions x and x component P T electronic component P Vp q pq pq pq pq
x g A and x g A Intrafragment A Intrafragment Ap qx g B and x g B Intrafragment B Interfragment A]Bp qx g A and x g B Interfragment A]B Interfragment A]Bp qx g B and x g C Interfragment B]C Interfragment A]B]Cp q
ambiguity in the spatial partitioning, neither in thedensity and pair density populations nor in thecorrespondent monoelectronic and bielectronic en-
Ž .ergies. In the latter state, approximations g andŽ .h are used to deal with the arbitrariness inherentto the partition of the overlapping quantities.
In this section, we present the general formal-ism which can be applied to any type of state.However, in a noncovalent state, several compo-nents of the energy, stabilization energy, density,and pair-density populations, could vanish. Therigorous definition of the states as well as thespecific details of the decomposition scheme arepresented in the Appendix.
Ž .In a general state G , the density populationsw GŽ .xcan be partitioned in intrafragment P A and
w GŽ .xinterfragment P AB components:
k k Ay1G GŽ . Ž . Ž .P A q P AB s n. 21Ý Ý Ý
As1 As1 Bs1
The pair-density populations can then be ex-Žpanded as a sum of intrafragment and bicentric,
.tricentric, and tetracentric interfragment compo-nents:
k k Ay1G GŽ . Ž .D A q D ABÝ Ý Ý
As1 As1 Bs1
k Ay1 By1G Ž .q D ABCÝ Ý Ý
As1 Bs1 Cs1
k Ay1 By1 Cy1 Ž .n n y 1G Ž .q D ABCD s .Ý Ý Ý Ý 2As1 Bs1 Cs1 Ds1
Ž .22
The total energy of the system can be partitionedaccording to the equation
k k Ay1G G GŽ . Ž .E s E A q E ABÝ Ý Ý
As1 As1 Bs1
k Ay1 By1G Ž .q E ABCÝ Ý Ý
As1 Bs1 Cs1
k Ay1 By1 Cy1G Ž . Ž .q E ABCD 23Ý Ý Ý Ý
As1 Bs1 Cs1 Ds1
Within a supermolecule approach, the stabilizationŽ G .energy D E of the molecular system can be de-
fined as
G G ` Ž .D E s E y E 24
and can be partitioned according to the equation
k k Ay1G G GŽ . Ž .D E s D E A q E ABÝ Ý Ý
As1 As1 Bs1
k Ay1 By1G Ž .q E ABCÝ Ý Ý
As1 Bs1 Cs1
k Ay1 By1 Cy1G Ž . Ž .q E ABCD , 25Ý Ý Ý Ý
As1 Bs1 Cs1 Ds1
where
G Ž . G Ž . `Ž . Ž .D E A s E A y E A . 26
Ž . GŽ .In Eq. 25 , D E A is the energy variation of afragment A between the initial state and the pre-
GŽ . GŽ . GŽ .sent state. E AB , E ABC , and E ABCD rep-resent the interactions between fragments A and
Ž .B, within the trimer ABC and within the te-Ž .tramer ABCD , in the state G.
VOL. 72, NO. 3162

PARTITIONING FOR MOLECULAR INTERACTING SYSTEMS
Applications
There has been a great deal of interest focusedon determining the interactions between pairs or
w xcollections of molecules 15]31 . In particular, sta-bilization reactions involving the transfer of di-rectly bonded groups have been subjected to ex-
w xtensive studies by both experimental 32]41 andw xtheoretical procedures 42]70 .
In a molecular system, where a reaction occursinvolving the transfer of only one directly bondedgroup, two regions can be considered: a reactiveregion including the two fragments involved inthe transfer process and an environmental regionnot directly involved in this process.
In vacuo, the environmental region does notexist and a transfer of the group G between frag-ments A and B can be expressed by the followingchemical equation:
G group transferAssociation 6 6w xA y G q B A y G : BIsolated Binding
fragments complex 1Ž .Initial state ` Ž .1 BC1
Dissociation 6w xA : G y B A q G y B .Isolated fragmentsBinding complex 2
Ž .Final state `Ž .BC 22
Ž .27
Selecting the initial state as a reference, the stabi-lization energy of the system can be calculated as
G G `1 Ž .D E s E y E , 28
with
Ž .G s BC , BC , or ` , 291 2 2
and partitioned as
G G G Ž .D E s D E q D E . 30intra inter
Ž . GIn Eq. 30 , D E is the intrafragment componentintraof the stabilization energy and represents the in-trinsic stability of the monomers of state G inrelation to the monomers of the initial state,
G G Ž . G Ž . Ž .D E s D E A q D E B , 31intra
with
G Ž . G Ž . `1 Ž . Ž .D E A s E A y E A 32
and
G Ž . G Ž . `1 Ž . Ž .D E B s E B y E B . 33
It should be noted that group G is included infragment A for the initial state and binding com-plex 1 and in fragment B for the other two states.D EG is the interfragment component of the sta-interbilization energy and represents the interactionbetween fragments A and B in state G:
G G Ž . Ž .D E s E AB . 34inter
This component is obviously null in the final state.The binding complexes are of special interest in
the study of this type of systems. Their relativeŽ BC1ª BC2 .stabilization energy DD E can be calcu-
lated as
DD E BC1ª BC2 s D E BC2 y D E BC1 s E BC2 y E BC1
Ž .35
and can be partitioned as
DD E BC1ª BC2 s DD E BC1ª BC2 q DD E BC1ª BC2 .intra inter
Ž .36
Ž . BC1ª BC2In Eq. 36 , DD E is the interfragment com-intra
ponent of DD E BC1ª BC2 calculated as
BC1ª BC2 Ž BC2 Ž . BC2 Ž ..DD E s E A q E Bintra
Ž BC1 Ž . BC1 Ž .. Ž .y E A q E B 37
and DD E BC1ª BC2 is the interfragment componentinter
of DD E BC1ª BC2 given by
BC1ª BC2 BC2 Ž . BC1 Ž . Ž .DD E s E AB y E AB . 38inter
The interfragment component represents the dif-ference of the energy of interaction between pairsŽ . Ž .A]G : B and A : G]B . Usually, this componentshould be essentially electrostatic and, conse-quently, should favor the charge separation in themonomers.
The intrafragment component represents the en-Ž .ergy difference between monomers A, G]B and
Ž .A]G, B . This component evaluates the differenceof intrinsic affinity to group G between fragmentsA and B. If DD E BC1ª BC2 - 0, fragment B has aintralarger intrinsic affinity to group G than has frag-ment A. If DD E BC1ª BC2 ) 0, the opposite situa-intration occurs.
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 163

MELO AND RAMOS
ŽIn the presence of an environment protein,.water, etc. , two additional effects should be
considered:
Dielectric Effect
The magnitude of the Coulombic interactionsŽ .depends on the value of the dielectric constant «
of the environment. In hydrophobic environmentsŽ . Ž .« close to 1 and in vacuo « s 1 , the magnitudeof these interactions is similar. In hydrophilic envi-
Ž .ronments « c 1 , this magnitude is significantlyreduced.
Fragment Effect
Some parts of the environment could have di-Žrect interaction with the reactive region e.g., the
first hydration shell in an aqueous environment orneighbor amino acid residues and buried waters in
.a proteic environment . In this case, these partsshould be included in the environment fragmentand, therefore, two additional components of the
Ž G .stabilization D E and relative stabilizationŽ BC1ª BC2 .DD E energies should be considered: acomponent of interaction between the environ-ment and the reactive regions and a componentassociated with the environment region reorgani-zation. In this context, the hydrophilic environ-ments preferentially stabilize charged structures,while the hydrophobic environments preferentiallystabilize neutral structures.
Proton transfer is one of the most importantreactions of this type and has a key role in manybiological mechanisms such as enzymatic reactions
w xand drug]receptor interactions 71]73 . Usually, areaction of this type can be expressed by a chemi-
Ž . w xcal equation similar to Eq. 27 74 :
Association Proton transfer6 6w xA y H q B A y H : BIsolated Binding
fragments complex 1Ž .Initial state ` Ž .1 BC1
Dissociation 6w xA : H y B A q H y BIsolated fragmentsBinding complex 2
Ž .Final state `Ž .BC 22
Ž .39
and is associated with a characteristic double-w x Žwell minima potential energy surface 74, 75 see
.Fig. 1 . In this context, the stabilization energy
FIGURE 1. Characteristic double-well minimapotential-energy surface for proton transfer betweenfragments A and B.
partitioning analysis presented in this section isalso valid for proton-transfer reactions.
The properties of functional proteins depend ontheir three-dimensional structures, which, in turn,depend on the charge of amino acid side chains.Proton-transfer reactions between acidic and basicside chains can produce conformational changes in
w xproteins and influence their functions 76, 77 . Theinteractions between acid amino acids, namely,
Ž . Ž .aspartic ASP and glutamic GLU acids, and argi-Ž .nine ARG are among the most important occur-
ring between charged side chains in proteinsw x78, 79 .
The interactions ARG]ASP and ARG]GLU oc-cur in two different binding complexes: zwitteri-onic and neutral. These complexes can be wellsimulated using appropriate molecular models
Ž qsuch as methylguanidinium]acetate MGH :y. Ž .Ac and methylguanidine]acetic acid MG : HAc ,
Ž .respectively see Fig. 2 using these molecularmodels the siderchain ARG]GLU and ARG]ASPinteractions can be expressed by a chemical equa-
VOL. 72, NO. 3164

PARTITIONING FOR MOLECULAR INTERACTING SYSTEMS
( )FIGURE 2. Schematic representation of A( )methylguanidinium]acetate and B
methylguanidine]acetic acid. In both systems, theintermolecular parameters a, h, and d are also shown.
Ž .tion similar to Eq. 39 :
transferAssociation Protonq y q y6 6w xMGH q Ac MGH : Ac
Isolated Zwitterionicfragments complex
Ž .Initial state ` Ž .zwitt.1
Dissociation 6w xMG : HAc MG q HAc .Isolated fragmentsNeutral complex
Ž .Final state `Ž .neut. 2
Ž .40
The strength of these interactions has been inter-w xpreted both by experimentalists 79]85 and theo-
w xreticians 85]89 on the basis of a large Coulombicinteraction occurring to the zwitterionic complex.
w xHowever, some recent theoretical works 90]92strongly suggest that, in some environments theneutral complex should be more stable. Two con-
Ž . Ž .formational minima, trans H and trans CH , have3w xbeen identified 79, 84]89, 91 for both zwitterionic
and neutral complexes. These conformations arefavored because they are the only ones where it ispossible to establish two hydrogen bonds of the
Ž .type N——H——O——C see Fig. 3 .In this work, the proton-transfer potential en-
Ž . Ž .ergy surfaces, for both trans H and trans CH3conformational minima, were determined at the
UU w x6-31G ab initio level 93 . The transition statesfor a proton-transfer reaction between zwitterionicand neutral complexes were located for both po-tential-energy surfaces.
The effects accountable for the preferentialstabilization of the two mentioned complexes inarginine]carboxylate interactions are not yetcompletely understood. To provide an appropriatemolecular interpretation of the nature of this inter-action, the stabilization energy partitioning schemepresented here was applied to relevant minima of
FIGURE 3. Schematic representation of conformational( ) ( )minima trans H and trans CH . In both conformations,3
the reaction coordinate for proton-transfer reaction[ ( )]d N—H is also shown.
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 165

MELO AND RAMOS
proton-transfer potential-energy surfaces. It hasbeen observed that these surfaces are basis-set-
w xdependent 71, 72, 74]76, 94]96 . Therefore, toevaluate the influence of the relative stability ofthe mentioned minima, this decomposition proce-dure was also performed using a series of differentbasis sets.
Computational Details
The following methodology was used to con-struct the 6-31GUU potential-energy surfaces asso-ciated with the proton transfer between the bind-
w q yx w xing complexes MGH : Ac and MG : HAc :Initially, the energy for the isolated fragments
Ž q y .MG, MGH , Ac , and HAc was calculated fol-lowed by geometry optimizations of binding com-
Ž . Ž .plexes performed for both trans H and trans CH3conformational minima. In these calculations, theheavy atoms of both fragments were constrainedto the same plane, because this is the region wheremost of the arginine]carboxylate interactions seem
Žto occur. All the remainder intra- and inter- a, h,. Ž .and d fragment parameters see Figs. 2 and 3
were optimized.The proton-transfer potential-energy surfaces,
for both conformational minima, were then deter-mined as a function of the reaction coordinateŽ . Ž .d N—H see Fig. 3 . For each point, d wasN—H
fixed and the remainder intra- and interfragmentparameters were optimized. The planarity of theheavy atoms of both fragments were conserved in
Ž .these calculations. The increments in d N—H˚ wwere 0.1 A far from the transition state from
˚ ˚Ž . xd N—H s 1.3 to 1.7 A and 0.01 A in the regionw Ž .of the transition state from d N—H s 1.1 to
˚ x1.3 A . The transition states were located for bothproton-transfer potential-energy surfaces by verify-ing if they had only one negative eigenvalue of theforce constant matrix corresponding to one imagi-nary vibration.
The following methodology was used in thestabilization energy partitioning calculations: The
w q yx w xbinding complexes MGH : Ac and MG : HAcŽwere completely optimized without planarity re-
. UUstrictions within a 6-31G ab initio level, and thepartitioning scheme presented in the third sectionwas applied to these relevant stationary minima ofthe system.
The same procedure was repeated using 11 ad-Ž w x U w xditional basis sets 6-31G 97, 98 , 6-31G 93 ,
w x U w x UU w x w x6-311G 99 , 6-311G 99 , 6-311G 99 , D95 100 ,U w x UU w x w xD95 100 , D95 100 , 6-31 q G 101]104 ,
U w x UU w x6-31 q G 101]104 and 6-31 q G 101]104 .All the calculations were done with the GAUSS-
w xIAN 92 package 105 on an IBM Risc 6000 workstation.
Results
ŽThe energies of the initial reference state iso-.lated zwitterionic fragments , obtained with differ-
ent basis sets, are presented in Table III. The 6-31GUU proton-transfer potential-energy surfaces,
Ž . Ž .for trans H and trans CH conformational min-3ima, are presented in Figures 4 and 5 and in TablesIV and V, respectively. The analysis of these re-sults enable us to conclude that the potential-
TABLE III( )Energies au of zwitterionic monomers and initial reference state.
+ y `1( ) ( )Basis set E MGH E Ac E
6-31G y243.464298741 y227.122764392 y470.587063133U6-31G y243.555028314 y227.225068500 y470.780096814UU6-31G y243.580556480 y227.229867500 y470.810423980
6-311G y243.517451894 y227.185340948 y470.702792842U6-311G y243.609552668 y227.284624010 y470.894176678UU6-311G y243.630240589 y227.289288754 y470.919529343
D95 y243.500812148 y227.174532158 y470.675344306UD95 y243.594929074 y227.283498897 y470.878427971UUD95 y243.620911527 y227.288878742 y470.909790269
6-31 + G y243.467791270 y227.145434981 y470.613226251U6-31 + G y243.558737174 y227.249509399 y470.808246573UU6-31 + G y243.584012624 y227.254207637 y470.838220261
VOL. 72, NO. 3166

PARTITIONING FOR MOLECULAR INTERACTING SYSTEMS
FIGURE 4. Proton-transfer potential-energy surface,UU ( )obtained at the 6-31G ab initio level, for trans H
conformational minimum.
energy barrier for proton transfer is very smallw Ž . Ž .6.94 and 4.97 kJrmol, for trans H and trans CH ,3
x w xrespectively . Zheng and Ornstein 92 determinedthe proton-transfer energy barriers in the guani-dinium]formate model system, using different en-vironments and ab initio-approximated levels. In
Žvacuo, their values are significantly larger from.14.6 to 33.1 kJrmol than those which have been
obtained in this work. The main reason for thisdisagreement is, probably, that these authors used
Ž .a symmetric model h s 0 in their calculations. Inthe present work, due to the possibility of the
Ž .horizontal displacements h / 0 during the opti-mization procedure, the proton-transfer potential-energy surface is much more flexible and realistic.Consequently, the associated barriers are smaller.
In the binding complexes’ global optimizations,no significant deviations from planarity were ob-served. This indicates that the molecular modelused in the proton-transfer potential-energy sur-faces is appropriate.
The energy-partitioning scheme was appliedhere to the stabilization energy, rather than to themolecular energy, as the former is a much moreconvenient way of understanding the process ofproton transfer. The results of this exercise, ob-tained with the 12 different basis sets, are pre-
FIGURE 5. Proton-transfer potential-energy surface,UU ( )obtained at the 6-31G ab initio level, for trans CH3
conformational minimum.
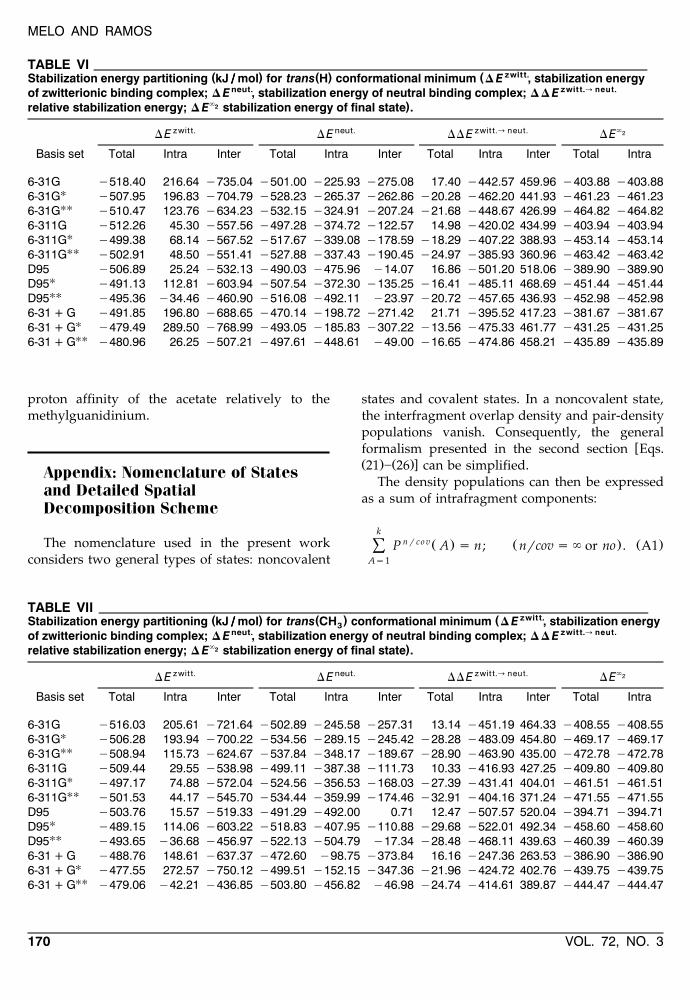
sented in Tables VI and VII. The last two columnsŽ `2 .of each table D E show that the energy-partition
process is being properly carried out, in the finalstate with the fragments at infinite distances, sincethe contribution to the stabilization energy of thesystem should arise exclusively from the intrafrag-ment components, as is indeed the case.
From the observation of Tables VI and VII, it isnoticeable that the poorer basis sets favor the zwit-terionic complex; however, the inclusion of polar-ization functions consistently reverse this result,favoring the neutral complex. This behavior ex-plains the positive sign of the total DD Ezwitt.ª neut.
quantity readily becoming more negative as thenumber of polarization functions used increases ineach type of basis set.
In the zwitterionic complex, and for bothŽ . Ž .trans H and trans CH conformational minima,3
the interfragment component is consistently nega-tive, whereas the intrafragment component is al-ways positive with few exceptions. It is interestingto note that the improvement in the basis setsleads to a much less negative interfragment com-ponent and a much less positive intrafragment oneŽwith the exception of the 6-311G basis set inwhich the inclusion of polarization functions does
.not seem to affect the results much . This differ-
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 167

MELO AND RAMOS
TABLE IVUU ( )Stabilization energies D E, obtained at the 6-31G ab initio level, versus reaction coordinate d N—H for the
( )proton-transfer potential-energy surface of the trans H conformational minimum, and the optimizedinterfragment parameters a , d, and h.
˚ ˚ ˚( ) ( ) ( ) ( ) ( ) ( )d N—H A DE kJ / mol a degrees d A h A
( )1.042 zwitt. y510.461 58.896 3.877 y0.0711.100 y507.713 62.527 3.863 0.0401.110 y507.441 64.895 3.856 0.1691.120 y506.756 65.026 3.853 0.1911.130 y506.089 66.264 3.850 0.2111.140 y505.471 66.906 3.846 0.2341.150 y504.915 67.545 3.843 0.2491.160 y504.440 68.086 3.841 0.2671.170 y504.054 68.676 3.838 0.2841.180 y503.769 69.247 3.836 0.3001.190 y503.591 69.795 3.834 0.3151.200 y503.522 70.311 3.833 0.330
( )1.201 TS y503.521 70.376 3.833 0.3331.210 y503.560 70.791. 3.832 0.3461.220 y503.706 71.230 3.832 0.3541.230 y503.952 71.607 3.831 0.3671.240 y504.294 71.987 3.831 0.3761.250 y504.724 72.352 3.832 0.3821.260 y505.234 72.664 3.833 0.3921.270 y505.812 72.991 3.835 0.4041.280 y506.455 73.312 3.836 0.4151.290 y507.156 73.481 3.839 0.4141.300 y507.901 73.680 3.841 0.4181.400 y516.443 74.684 3.878 0.4381.500 y524.062 74.536 3.929 0.4331.600 y529.140 73.660 3.987 0.4071.700 y531.686 74.469 4.048 0.373
( )1.776 neut. y532.213 71.391 4.096 0.339
ence in both energy components, and in withineach basis set, is of the same order of magnitude.However, and on the whole the stabilization of thezwitterionic complex is associated with the inter-fragment component which contributes with amuch larger effect consistent with the also largerelectrostatic interfragment interaction occurring inthis complex relatively to the neutral complex.
Ž .In the neutral complex, and for both trans HŽ .and trans CH conformational minima, the inter-3
fragment component to the stabilization energy isalso negative, just as it was in the previous ana-lyzed case of the zwitterionic complex; however,here, this component takes on a very different rolewith values much inferior than it did previously,contributing sometimes very little to the total valueof the energy. Therefore, on the whole, the stabi-lization of the neutral complex is associated with
the intrafragment component of the energy. Thispoints to the fact that the acetate has a largerintrinsic proton affinity than has the methylguani-dinium.
w xRecent theoretical works 90]92 indicate thathydrophilic environments stabilize the zwitterioniccomplex relatively to the neutral one while hy-drophobic environments have the opposite effect.
w xIt was also demonstrated 92 that the potential-energy barriers are significantly decreased withthe increasing of environment hydrophilicity.
Conclusions
In this work, a new general energy-decomposi-tion scheme is introduced based on the indistin-
VOL. 72, NO. 3168

PARTITIONING FOR MOLECULAR INTERACTING SYSTEMS
TABLE VUU ( )Stabilization energies D E, obtained at the 6-31G ab initio level, versus reaction coordinate d N—H for the
( )proton-transfer potential-energy surface of the trans CH conformational minimum, and the optimized3interfragment parameters a , d, and h.
˚ ˚ ˚( ) ( ) ( ) ( ) ( ) ( )d N—H A DE kJ / mol a degrees d A h A
( )1.042 zwitt. y508.945 178.920 3.873 y0.0461.100 y506.778 y178.212 3.860 0.0671.110 y506.131 y177.682 3.857 0.0891.120 y506.084 y175.651 3.849 0.2201.130 y505.555 y175.093 3.846 0.2401.140 y505.084 y174.645 3.843 0.2601.150 y504.676 y174.184 3.839 0.2771.160 y504.352 y173.690 3.837 0.3001.170 y504.125 y173.284 3.834 0.3151.180 y503.997 y172.892 3.832 0.330
( )1.186 TS y503.978 y172.588 3.831 0.3421.190 y503.986 y172.470 3.831 0.3461.200 y504.081 y172.112 3.829 0.3611.210 y504.285 y171.777 3.828 0.3731.220 y504.595 y171.472 3.827 0.3841.230 y505.000 y171.231 3.828 0.3961.240 y505.508 y170.955 3.828 0.4041.250 y506.094 y170.789 3.828 0.4141.260 y506.762 y170.553 3.830 0.4211.270 y507.496 y170.352 3.831 0.4271.280 y508.290 y170.218 3.833 0.4331.290 y509.130 y170.024 3.835 0.4391.300 y510.021 y169.979 3.838 0.4421.400 y519.765 y169.674 3.876 0.4631.500 y528.261 y170.315 3.928 0.4581.600 y533.986 y171.404 3.987 0.4401.700 y537.024 y172.926 4.050 0.397
( )1.798 neut. y537.890 y174.608 4.112 0.349
guishable integrals formalism and in the spatialpartitioning of reduced-density and pair-densitymatrices. According to this method, the molecularand stabilization energies are partitioned intomeaningful fragments. This partitioning schemehas a flexible form, allowing the exchange of atomsbetween fragments, and is especially appropriateto study systems in which a directly bondedgroup-transfer process occurs.
Proton transfer is one of the most importantreactions of this type and has a key role in theside-chain interactions between arginine and car-
Ž .boxylate aspartate and glutamate amino acidresidues. Here, methylguanidium]acetate andmethylguanidine]acetic acid were used to simu-late the zwitterionic and neutral complexes of
arginine]carboxylate interactions. Potential-energysurfaces, calculated at the 6-31GUU level for both
Ž . Ž .trans H and trans CH conformational minima3
indicate that the proton-transfer barriers are verysmall. The partitioning scheme introduced in thiswork was used, with 12 different basis sets, toprovide a better understanding of the main factorsaccountable for the relative stabilities of the sta-tionary minima on the potential-energy surface.The best basis sets favor the neutral complex,while the poorer basis sets favor the zwitterioniccomplex. Despite this disagreement, all the calcu-lations indicate that the zwitterionic complex isfavored by the interfragment electrostatic compo-nent while the preferential stabilization of the neu-tral complex is favored with the larger intrinsic
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 169
MELO AND RAMOS
TABLE VI( ) ( ) ( zwit t.Stabilization energy partitioning kJ ///// mol for trans H conformational minimum D E , stabilization energy
neut. zwit t.ª neut.of zwitterionic binding complex; D E , stabilization energy of neutral binding complex; D D E`2 )relative stabilization energy; D E stabilization energy of final state .
zwit t. neut. zwit t.ª neut. `2DE DE DDE DE
Basis set Total Intra Inter Total Intra Inter Total Intra Inter Total Intra
6-31G y518.40 216.64 y735.04 y501.00 y225.93 y275.08 17.40 y442.57 459.96 y403.88 y403.88U6-31G y507.95 196.83 y704.79 y528.23 y265.37 y262.86 y20.28 y462.20 441.93 y461.23 y461.23UU6-31G y510.47 123.76 y634.23 y532.15 y324.91 y207.24 y21.68 y448.67 426.99 y464.82 y464.82
6-311G y512.26 45.30 y557.56 y497.28 y374.72 y122.57 14.98 y420.02 434.99 y403.94 y403.94U6-311G y499.38 68.14 y567.52 y517.67 y339.08 y178.59 y18.29 y407.22 388.93 y453.14 y453.14UU6-311G y502.91 48.50 y551.41 y527.88 y337.43 y190.45 y24.97 y385.93 360.96 y463.42 y463.42
D95 y506.89 25.24 y532.13 y490.03 y475.96 y14.07 16.86 y501.20 518.06 y389.90 y389.90UD95 y491.13 112.81 y603.94 y507.54 y372.30 y135.25 y16.41 y485.11 468.69 y451.44 y451.44UUD95 y495.36 y34.46 y460.90 y516.08 y492.11 y23.97 y20.72 y457.65 436.93 y452.98 y452.98
6-31 + G y491.85 196.80 y688.65 y470.14 y198.72 y271.42 21.71 y395.52 417.23 y381.67 y381.67U6-31 + G y479.49 289.50 y768.99 y493.05 y185.83 y307.22 y13.56 y475.33 461.77 y431.25 y431.25UU6-31 + G y480.96 26.25 y507.21 y497.61 y448.61 y49.00 y16.65 y474.86 458.21 y435.89 y435.89
proton affinity of the acetate relatively to themethylguanidinium.
Appendix: Nomenclature of Statesand Detailed SpatialDecomposition Scheme
The nomenclature used in the present workconsiders two general types of states: noncovalent
states and covalent states. In a noncovalent state,the interfragment overlap density and pair-densitypopulations vanish. Consequently, the general
wformalism presented in the second section Eqs.Ž . Ž .x21 ] 26 can be simplified.
The density populations can then be expressedas a sum of intrafragment components:
kn r co v Ž . Ž . Ž .P A s n; nrcov s ` or no . A1Ý
As1
TABLE VII( ) ( ) ( zwit t.Stabilization energy partitioning kJ ///// mol for trans CH conformational minimum D E , stabilization energy3
neut. zwit t.ª neut.of zwitterionic binding complex; D E , stabilization energy of neutral binding complex; D D E`2 )relative stabilization energy; D E stabilization energy of final state .
zwit t. neut. zwit t.ª neut. `2DE DE DDE DE
Basis set Total Intra Inter Total Intra Inter Total Intra Inter Total Intra
6-31G y516.03 205.61 y721.64 y502.89 y245.58 y257.31 13.14 y451.19 464.33 y408.55 y408.55U6-31G y506.28 193.94 y700.22 y534.56 y289.15 y245.42 y28.28 y483.09 454.80 y469.17 y469.17UU6-31G y508.94 115.73 y624.67 y537.84 y348.17 y189.67 y28.90 y463.90 435.00 y472.78 y472.78
6-311G y509.44 29.55 y538.98 y499.11 y387.38 y111.73 10.33 y416.93 427.25 y409.80 y409.80U6-311G y497.17 74.88 y572.04 y524.56 y356.53 y168.03 y27.39 y431.41 404.01 y461.51 y461.51UU6-311G y501.53 44.17 y545.70 y534.44 y359.99 y174.46 y32.91 y404.16 371.24 y471.55 y471.55
D95 y503.76 15.57 y519.33 y491.29 y492.00 0.71 12.47 y507.57 520.04 y394.71 y394.71UD95 y489.15 114.06 y603.22 y518.83 y407.95 y110.88 y29.68 y522.01 492.34 y458.60 y458.60UUD95 y493.65 y36.68 y456.97 y522.13 y504.79 y17.34 y28.48 y468.11 439.63 y460.39 y460.39
6-31 + G y488.76 148.61 y637.37 y472.60 y98.75 y373.84 16.16 y247.36 263.53 y386.90 y386.90U6-31 + G y477.55 272.57 y750.12 y499.51 y152.15 y347.36 y21.96 y424.72 402.76 y439.75 y439.75UU6-31 + G y479.06 y42.21 y436.85 y503.80 y456.82 y46.98 y24.74 y414.61 389.87 y444.47 y444.47
VOL. 72, NO. 3170

PARTITIONING FOR MOLECULAR INTERACTING SYSTEMS
Ž . n r co vŽ .In Eq. A1 , p A is the density population offragment A in a noncovalent state, defined as
m` m` py1A An r co v n r co v n r co vŽ .P A s P q 2 P SÝ Ý Ýp p p q p q
` ` `psn psn qsnA A A
` Ž . Ž .s q ; nrcov s ` or no , A2A
in which P n r co v are the elements of the reducedp q
density matrix, x ` and x ` are the first and then mA A
last atomic basis functions of fragment A, and q`A
is its electronic charge in the initial state.In a state of this type, the pair-density popula-
tion associated with fragment A, which representsthe number of electron pairs where both partnersbelong to this fragment, is given by
m` p p limA1n r co v n r co vŽ .D A s f S SÝ Ý Ý Ý p qr s p q r s2 ` ` ` `psn qsn rsn ssnA A A A
` Ž ` .q q y 1A A Ž .s ; nrcov s ` or no .2
Ž .A3
Considering that the interfragment overlap densitypopulations vanish, the only allowed interfrag-
Ž .ment A]B pairs are those in which one partnerbelongs to fragment A and the other belongs tofragment B:
m` m`py1 ry1A B1n r co v n r co vŽ .D A s f S SÝ Ý Ý Ý p qr s p q r s2 ` ` ` `psn qsn rsn ssnA A A A
` ` Ž . Ž .s q q ; nrcov s ` or no . A4A B
Ž . Ž . n r co vIn Eqs. A3 and A4 , the coefficients f havep qr sthe same physical meaning previously definedw Ž . xEq. 17 and Table I and are associated with anoncovalent state.
Because the high-order terms vanish, the pair-density populations can be only expanded as asum of intra- and interfragment components:
k k Ay1n r co v n r co vŽ . Ž .D A q D ABÝ Ý Ý
As1 As1 Bs1
Ž .n n y 1Ž . Ž .s ; nrcov s ` or no . A5
2
The main difference between the two states of thistype is that, while in the initial state the fragmentsare at infinite distances, in a nonoverlapping frag-ment binding state, the fragments can interact witheach other. Consequently, while in the first state
the energy of the system is expressed as a sum ofintrafragment components,
k` `Ž . Ž .E s E A , A6Ý
As1
in the second state, bicentric components associ-ated with the Coulombic interactions between thefragments has also to be considered:
k k Ay1no no noŽ . Ž . Ž .E s E A q E AB . A7Ý Ý Ý
As1 As1 Bs1
Ž . w n oŽ .xIn Eq. A7 , a interfragment component E ABincludes the attraction energies between the elec-tronic density of fragment A and the nuclei offragment B, the attraction energies between theelectronic density of fragment B and the nuclei offragment A, the repulsion energies between theelectronic densities of fragments A and B and therepulsion energies between the nuclei of the twofragments A and B:
n o Ž .E ABM` M`
A B Z ZX Ys Ý Ý no« r` ` X YXsN YsNA B
m` m`A A
n o B n o Aq P V q P VÝ Ýp p p p p p p p` `psn psnA B
m` m`py1 py1A Bno B n o Aq 2 P V q P VÝ Ý Ý Ýp q p q p q p qž /` ` ` `psn qsn psn qsnA A B B
m` m`py1 ry1A B1no Ž .q f pq ¬ rs .Ý Ý Ý Ý p qr s2 ` ` ` `psn qsn rsn ssnA A B B
Ž .A8
In both noncovalent states, the monocentric com-w n r co vŽ .xponents E A can be defined by a common
equation:
n r co v Ž .E AM` m`Xy1A AZ ZX Y n r co v As q P hÝ Ý Ý p p p pn r co v« r` ` `X YXsN YsN psnA A A
m` py1An r co v Aq 2 P hÝ Ý p q p q
` `psn qsnA A
m` p p limA1n r co v Ž .q f pq ¬ rs ;Ý Ý Ý Ý p qr s2 ` ` ` `psn qsn rsn ssnA A A A
Ž . Ž .nrcov s ` or no . A9
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 171

MELO AND RAMOS
Ž . Ž . ` `In Eqs. A8 and A9 , N and M are the first andA Athe last nuclei of fragment A in the initial stateand h A is defined asp q
A A Ž .h s T q V . A10p q p q p q
However, in a nonoverlapping binding state, theinterfragment interactions originate polarizationeffects which preserve the fragment’s populationsbut alter their distributions over the atomic basis
Ž n ofunctions. Therefore, the matrix elements P andp qno .f usually differ from the correspondent ele-p qr s
Ž ` ` .ments P and f in the initial state. Geometryp q p qr s
Ž no ` .relaxations could also occur r / r , X, Y g AX Y X Ywithin each fragment.
Within a supermolecule approach, the stabiliza-Ž n o.tion energy D E of the molecular system in a
nonoverlapping binding state is definedŽ .by Eq. 24 , with G s no, and can be partitioned
only in monocentric and bicentric components:
k k Ay1no no noŽ . Ž .D E s D E A q E AB .Ý Ý Ý
As1 As1 Bs1
Ž .A11
w Ž .xAs in total energy Eq. A7 , the high-order com-ponents vanish due to the null interfragment den-
Ž . n oŽ .sity populations. In Eq. A11 , D E A is theenergy variation of a fragment A between theinitial state and the nonoverlapping fragmentbinding state, due to polarization and internal ge-ometry relaxation effects which arise from theCoulombic interactions with the other fragments.
The formalism presented in the second sectionis fully applied to covalent states. In this context,the density, pair-density, energy, and stabilizationenergy decomposition can be obtained directly
Ž . Ž .from Eqs. 21 to 26 , changing the generic super-script G by the more specific one ov.
In addition to the Coulombic interaction andpolarization effects, observed in the nonoverlap-ping binding state, the most important alterationoccurring in a covalent state is that the overlapinterfragment density and pair-density popula-tions become nonnull. This can induce electroniccharge transfer and an exchange of atoms, ions, orgroup of atoms between the fragments.
When some atoms are exchanged between afragment A and other fragments, their nuclei dif-fer in a covalent state and in the other states. In acovalent state, the concept of fragment density
population has been associated with some kindŽ .of arbitrariness. Using approximations g , only the
w o vŽ .xnet density population P A is associatedwith fragment A. Consequently, the density popu-lations of this fragment should usually differ froma covalent state to the other states, due to overlapand electronic charge transfer with other frag-ments.
The density populations can now be partitionedw o vŽ .xin intrafragment P A and interfragment
w o vŽ .xP AB components:
mov mov py1A Ao v o v o vŽ .P AB s P q 2 P SÝ Ý Ýp p p q p q
ov ov ovpsn psn qsnA A A
o v Ž . Ž .s q A , A12mov mov
A Bo v o v o vŽ . Ž . Ž .P AB s 2 P S s q AB . A13Ý Ý p p p q
ov ovpsn qsnA B
In the latter equations, P o v are the elements of thep qreduced-density matrix, and x o v and x o v are then mA A
first and the last atomic basis functions of frag-o vŽ .ment A in the covalent state. Additionally, q A
is the net electronic charge of fragment A ando vŽ .q AB is the overlap electronic charge between
fragments A and B.As the interfragment overlap density popula-
tions are non null, there are pair-density popula-tions where one or both partners are associatedwith the overlap density between fragments. Ad-ditionally, high-order pair-density terms, asso-ciated with trimers and tetrameters, could alsooccur.
The intrafragment components are given by
mov p p limA1o v o vŽ .D A s f S S ,Ý Ý Ý Ý p qr s p q r s2 ov ov ov ovpsn qsn rsn ssnA A A A
Ž .A14
the bicentric interfragment components are calcu-lated as
ov ovm mpy1 ry1A B1o v o vŽ .D AB s f S SÝ Ý Ý Ý p qr s p q r s2 ov ov ov ovpsn qsn rsn ssnA A B B
mov mov movpy1A A Bo vq f S SÝ Ý Ý Ý p qr s p q r s
ov ov ov ovpsn qsn rsn ssnA A A B
mov mov mov ry1A B Bo vq f S SÝ Ý Ý Ý p qr s p q r s
ov ov ov ovpsn qsn rsn ssnA B B B
VOL. 72, NO. 3172

PARTITIONING FOR MOLECULAR INTERACTING SYSTEMS
ov ov ov ovm m m mA B A Bo vq f S S ,Ý Ý Ý Ý p qr s p q r s
ov ov ov ovpsn qsn rsn ssnA B A B
Ž .A15
the tricentric interfragment components are givenby
ov ov ovm m mpy1A B C1o v o vŽ .D ABC s f S SÝ Ý Ý Ý p qr s p q r s2 ov ov ov ovpsn qsn rsn ssnA A B C
mov mov movpy1B A Co vq f S SÝ Ý Ý Ý p qr s p q r s
ov ov ov ovpsn qsn rsn ssnB B A C
mov mov movpy1C A Bo vq f S SÝ Ý Ý Ý p qr s p q r s
ov ov ov ovpsn qsn rsn ssnC C A B
mov mov mov movA B B C
o vq f S SÝ Ý Ý Ý p qr s p q r sov ov ov ovpsn qsn rsn ssnA B B C
mov mov mov movA C C B
o vq f S SÝ Ý Ý Ý p qr s p q r sov ov ov ovpsn qsn rsn ssnA C C B
ov ov ov ovm m m mB A A Co vq f S S ,Ý Ý Ý Ý p qr s p q r s
ov ov ov ovpsn qsn rsn ssnB A A C
Ž .A16
and the tetracentric interfragment components areobtained as
o v Ž .D ABCDov ov ov ovm m m mA B C D1
o vŽs f S SÝ Ý Ý Ý p qr s p q r s2 ov ov ov ovpsn qsn rsn ssnA B C D
o v o v . Ž .qf S S q f S S . A17pr qs pr qs p sqr p s qr
Several new energy terms arise from the non-null interfragment density populations:
B Monoelectronic kinetic energies associatedwith the overlap density of two fragments A
w o vŽ .xand B included in E AB .B Monoelectronic attraction energies between
the overlap density of two fragments A andwB and the nuclei of both fragments included
o vŽ .xin E AB .B Monoelectronic attraction energies between
the overlap density of two fragments A and
wB and the nuclei of a third fragment C in-o vŽ .xcluded in E ABC .
B Bielectronic energies associated with electronpairs where a partner belongs to a fragmentA and the other belongs to the overlap den-
wsity between fragments A and B includedo vŽ .xin E AB .
B Bielectronic energies associated with electronpairs where both partners belong to the over-lap density between fragments A and Bw o vŽ .xincluded in E AB .
B Bielectronic energies associated with electronpairs where a partner belongs to a fragmentA and the other belongs to the overlap den-
wsity between fragments B and C includedo vŽ .xin E ABC .
B Bielectronic energies associated with electronpairs where a partner belongs to the overlapdensity between fragments A and B and theother belongs to the overlap density between
w o vŽ .xfragments A and C included in E ABC .B Bielectronic energies associated with electron
pairs where a partner belongs to the overlapdensity between fragments A and B andthe other belongs to the overlap density be-
wtween fragments C and D included ino vŽ .xE ABCD .
In this context, the intrafragment componentsare given by
M ov movXy1A AZ ZX Yo v o v AŽ .E A s q P hÝ Ý Ý p p p po v« rov ov ovX YXsN YsN psnA A A
mov py1Ao v Aq 2 P hÝ Ý p q p q
ov ovpsn qsnA A
mov p p limA1o v Ž .q f pq ¬ rs .Ý Ý Ý Ý p qr s2 ov ov ov ovpsn qsn rsn ssnA A A A
Ž .A18
The bicentric interfragment components are calcu-lated as
M ov M ovA B Z ZX Yo v Ž .E AB s Ý Ý o v« rov ov X YXsN YsNA B
mov movA B
o v B o v Aq P V q P VÝ Ýp p p p p p p pov ovpsn psnA B
ovm py1Ao v Bq 2 P VÝ Ý p q p q
ov ovpsn qsnA A
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 173

MELO AND RAMOS
mov py1Bo v Aq P VÝ Ý p q p q
ov ovpsn qsnB B
ov ovm mA Bo v A Bq P h q hŽ .Ý Ý p q p q p q
ov ovpsn qsnA B
ov ovm mpy1 ry1A B1o v Ž .q f pq ¬ rsÝ Ý Ý Ý p qr s2 ov ov ov ovpsn qsn rsn ssnA A B B
mov mov movpy1A A Bo v Ž .q f pq ¬ rsÝ Ý Ý Ý p qr s
ov ov ov ovpsn qsn rsn ssnA A A B
mov mov mov ry1A B Bo v Ž .q f pq ¬ rsÝ Ý Ý Ý p qr s
ov ov ov ovpsn qsn rsn ssnA B B B
ov ov ov ovm m m mA B A Bo v Ž .q f pq ¬ rs .Ý Ý Ý Ý p qr s
ov ov ov ovpsn qsn rsn ssnA B A B
Ž .A19
The tricentric interfragment components are givenby
o v Ž .E ABCov ov ov ovm m m mA B B C
o v C o v As 2 P V q P VÝ Ý Ý Ýp q p q p q p qov ov ov ovpsn qsn psn qsnA B B C
ov ovm mA Co v Bq P VÝ Ý p q p q
ov ovpsn qsnA C
ov ov ovm m mpy1A B C1o v Ž .q f pq ¬ rsÝ Ý Ý Ý p qr s2 ov ov ov ovpsn qsn rsn ssnA A B C
mov mov movpy1B A Co v Ž .q f pq ¬ rsÝ Ý Ý Ý p qr s
ov ov ov ovpsn qsn rsn ssnB B A C
mov mov movpy1C A Bo v Ž .q f pq ¬ rsÝ Ý Ý Ý p qr s
ov ov ov ovpsn qsn rsn ssnC C A B
mov mov mov movA B B C
o v Ž .q f pq ¬ rsÝ Ý Ý Ý p qr sov ov ov ovpsn qsn rsn ssnA B B C
mov mov mov movA C C B
o v Ž .q f pq ¬ rsÝ Ý Ý Ý p qr sov ov ov ovpsn qsn rsn ssnA C C B
ov ov ov ovm m m mB A A Co v Ž . Ž .q f pq ¬ rs . A20Ý Ý Ý Ý p qr s
ov ov ov ovpsn qsn rsn ssnB A A C
The tetracentric interfragment components are ob-tained as
o v Ž .E ABCDov ov ov ovm m m mA B C D1
o v Ž .s f pq ¬ rs�Ý Ý Ý Ý p qr s2 ov ov ov ovpsn qsn rsn ssnA B C D
o v o vŽ . Ž . Ž .qf pr ¬ qs q f ps ¬ qr . A214pr qs p sqr
Within a supermolecule approach, the stabilizationŽ o v.energy D E of the molecular system, in this
Ž .state, can be calculated by Eq. 24 and can beŽ .partitioned by Eq. 25 , with G s ov.
o vŽ .In this context, D E A is the energy variationof a fragment A between the initial state and thecovalent state due to polarization, internal geome-try relaxation, electronic charge transfer, and atomexchange effects which arise from the Coulombicinteractions and overlap with the other fragments.
References
1. Murrell, J. M.; Randic, M.; Williams, D. R. Proc R SocŽ .Lond A 1965, 284, 566.
2. Fukui, K.; Fujimoto, H. Bull Chem Soc Jpn 1968, 41, 1989.3. Fujimoto, H.; Kato, S.; Yamabe, S.; Fukui, K. J Chem Phys
1974, 60, 572.
4. Daudey, J. P.; Claverie, P.; Malrieu, J. P. Int J QuantumChem 1974, 8, 1.
5. Kitaura, K.; Morokuma, K. Int J Quantum Chem 1976, 10,325.
6. Ruedenberg, K. Rev Mod Phys 1962, 34, 326.7. Clementi, E. J Chem Phys 1967, 46, 3842.
Ž .8. Kollmar, H. Theor Chim Acta Berl 1978, 50, 235.
9. Bofill, J. M.; Povill, A.; Rubio, J. Chem Phys Lett 1994, 222,51.
10. Melo, A.; Gomes, J. A. N. F. Int J Quantum Chem 1993, 46,651.
11. McWeeny, R.; Sutcliffe, B. T. Methods of Molecular Quan-tum Mechanics; Academic: New York, 1969.
12. Daudel, R.; Leroy, G.; Peeters, D.; Sana, M. QuantumChemistry; Wiley: New York, 1983.
13. Cook, D. B. Ab initio Valence Calculations in Chemistry;Butterworths: London, 1974.
14. Mulliken, R. S. J Chem Phys 1955, 23, 1833.
15. van der Avoird, A.; Wolmer, P. E. S.; Mulder, F.; Berns,R. M. Top Curr Chem 1980, 93, 1.
16. Molecular Interactions, Ratajczak, H.; Orville-Thomas,W. J. Eds.; Wiley, Chichester, 1982; Vols. 1]3.
17. Hydrogen Bonds, Schuster, P. Ed.; Springer: Berlin, 1984;Vol. 120.
VOL. 72, NO. 3174

PARTITIONING FOR MOLECULAR INTERACTING SYSTEMS
18. Hobza, P.; Zahradnık, R. Intermolecular Complexes, the´Role of Van der Waals Systems in Physical Chemistry andin the Biodisciplines; Elsevier: Amsterdam, 1988.
19. Tang, T.-H.; Hu, W.-J.; Yan, D.-Y.; Cui, Y.-P. J Mol StructŽ .Theochem 1990, 207, 319.
20. Kubicki, M.; Codding, P. W. Can J Chem 1993, 71, 68.
21. Aida, M. Bull Chem Soc Jpn 1993, 66, 3423.
22. Hobza, P.; Selzle, H. L.; Schlag, E. W. J Am Chem Soc1994, 116, 3500.
23. Bures, M.; Berus, J. Collect Czech Chem Commun 1994, 59,1251.
24. Magalhaes, A.; Maigret, B.; Hotflack, J.; Gomes, J. A. N. F.;˜Scheraga, H. A. J Protein Chem 1994, 13, 195.
Ž .25. Sapse, A. M.; Kabir, S.; Snyder, G. J Mol Struct Theochem1995, 339, 227.
26. Pirard, B.; Baudoux, G.; Durant, F. Acta Crystallogr B1995, 51, 103.
27. Tang, T.-H., Ciu, Y.-P. Can J Chem 1996, 74, 1162.
28. Malliavin, M.-J.; Coudray, J Chem Phys 1997, 106, 2323.
29. Yu, M.; Dolg, M. Chem Phys Lett 1997, 273, 229.
30. Cordeiro, J. M. M. Int J Quantum Chem 1997, 65, 709.
31. McKee, M. L.; Worley, S. D. J Phys Chem A 1997, 101,5600.
32. Brown, D. A.; Glass, W. K.; Mageswaran, R.; Girmay, B.Magn Reson Chem 1988, 26, 970.
33. Rabinowitz, M. R.; Sutherland, J. W.; Patterson, P. M.;Klemm, R. B. J Phys Chem 1991, 95, 674.
34. Heberger, K.; Walbiner, M.; Fisher, H. Angew Chem Int´Ed Engl 1992, 31, 635.
35. Kondo, N.; Fueno, H.; Fujimoto, H.; Makino, M.; Nakaoka,H.; Aaoki, I.; Uemura, S. J Org Chem 1994, 59, 5254.
36. Shimoni, L.; Glusker, J. P.; Bock, C. W. J Phys Chem 1996,100, 2957.
37. Fulle, D.; Hamann, H. F.; Hipler, H.; Trol, J. J Chem Phys1996, 105, 983.
38. Kandel, S. A.; Rakitzis, T. P.; Levon, T.; Zare, R. N. J ChemPhys 1996, 105, 7550.
39. Griffin, J. B.; Armentrout, P. B. J Chem Phys 1997, 107,5345.
40. Richardz, T.; Gale, J.; Klose, G. Chem Phys Lett 1997, 271,79.
41. More, M. B.; Ray, D.; Armentrout, P. B. J Phys Chem A1997, 101, 7007.
42. Arnaud, R. New J Chem 1991, 15, 615.
43. Schroder, S.; Buckley, N.; Oppenheimer, N. J.; Kollman,¨P. A. J Am Chem Soc 1992, 114, 8232.
44. Arnaud, R.; Postlethwaite, H.; Barone, V. J Phys Chem1994, 98, 5913.
45. Lee, I.; Kim, C. K.; Chung, D. S.; Lee, B.-S. J Org Chem1994, 59, 4490.
46. Jursic, B. S.; Zdravkovski, Z. J Org Chem 1994, 59, 3015.
47. McAllister, M. A.; Tidwell, T. T. J Org Chem 1994, 59,4506.
48. Buttar, D.; Hirst, D. M. J Chem Soc Faraday Trans 1994, 90,1811.
49. Dobbs, K. D.; Dixon, D. A. J Phys Chem 1994, 98, 5290.
50. Andres, J.; Moliner, V.; Krechl, J.; Silla, E. J Phys Chem´1994, 98, 3664.
51. Sola, M.; Andres, J. L.; Duran, M.; Lledos, A.; Bertran, J.` ´ ´Ž .Theor Chim Acta Berl 1995, 91, 333.
52. Ruiz-Lopez, M. F.; Rinaldi, D. J Chem Phys 1995, 103,´9249.
53. Muguruma, C.; Koga, N.; Kitaura, K.; Morokuma, K.J Chem Phys 1995, 103, 9274.
54. Sakai, S. J Phys Chem 1995, 99, 5883.
55. Jurcic, B. S. Chem Phys Lett 1995, 244, 263.
Ž56. Bouyacoub, A.; Jean, Y.; Volatron, F. J Mol Struct Theo-.chem 1996, 371, 51.
57. Assfeld, X.; Garapon, J.; Rinaldi, D., Ruiz-Lopez, M. F.;´Ž .Rivail, J. L. J Mol Struct Theochem 1996, 371, 107.
58. Tunon, I.; Tortonda, F. R.; Pascual-Ahuir, J. L.; Silla, E.˜´Ž .J Mol Struct Theochem 1996, 371, 117.
59. Sumathi, R.; Engels, B.; Peyerimhoff, S. D. J Chem Phys1996, 105, 8117.
60. Cui, Q.; Musaev, D. G.; Svenson, M.; Morokuma, K. J PhysChem 1996, 100, 10936.
61. Lee, W. T.; Masel, R. I. J Phys Chem 1996, 100, 10945.
62. Peng, T.; Zang, D. H.; Zang, J. Z. H.; Schinke, R. ChemPhys Lett 1996, 248, 37.
63. Sinclair, P. E.; Catlow, C. R. A. J Chem Soc Faraday Trans1996, 92, 2099.
64. Thompson, W. H.; Miller, W. H. J Chem Phys 1997, 106,142.
65. Takayanagi, T.; Schatz, G. C. J Chem Phys 1997, 106, 3227.
66. Corchado, J. C.; Espinosa-Garcia, J. J Chem Phys 1997, 106,4013.
67. Sengupta, D.; Mguyen, M. T. J Chem Phys 1997, 106, 9703.
68. Ramachandra, R.; Semekowitsch, J.; Wyatt, R. E. ChemPhys Lett 1997, 270, 387.
69. Espinosa-Garcia, J.; Corchado, J. C. J Phys Chem A 1997,101, 7336.
70. Jemmis, E.; Giju, K. T.; Leszczynski, J. J Phys Chem A1997, 101, 7389.
71. Dijkman, J. P.; Osman, R.; Weinstein, M. Int J QuantumChem Quantum Biol Symp 1987, 14, 211.
Ž .72. Hadzi, D. J Mol Struct Theochem 1988, 177, 1.
73. Dijkman, J. P.; Osman, R.; Weinstein, M. Int J QuantumChem 1989, 35, 241.
74. Scheiner, S. Acc Chem Res 1985, 18, 174.
75. Pardo, L.; Mazurek, A. P.; Osman, R. Int J Quantum Chem1990, 37, 701.
76. Cassaday, C. J.; Carr, S. R.; Zhang, K.; Chung-Philiphs, A.J Org Chem 1995, 60, 1704.
77. Scharnagl, C.; Hettenkofer, J.; Fisher, S. F. Int J QuantumChem Quantum Biol Symp 1994, 21, 33.
78. Barlow, D. J.; Thornton, J. M. J Mol Biol 1983, 168, 867.
79. Singh, J.; Thornton, J. M.; Snarey, M.; Campbell, S. F. FEBSLett 1987, 224, 161.
80. Salunke, D. M.; Vijayan, M. Int J Peptide Protein Res 1981,18, 348.
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 175

MELO AND RAMOS
81. Eggleston, D. S.; Hodgson, D. J. Int J Peptide Protein Res1985, 25, 242.
82. Gorbitz, C. H. Acta Crystallogr B 1989, 45, 390.¨83. Ippolito, J. A.; Alexander, R. S.; Christianson, D. W. J Mol
Biol 1990, 215, 457.84. Chakrabarti, P. Int J Peptide Protein Res 1994, 43, 284.85. Mitchel, J. B. O.; Thornton, J. M.; Singh, J. J Mol Biol 1992,
226, 91.86. Sapse, A. M.; Russell, C. S. J Quantum Chem 1984, 26, 291.
Ž .87. Sapse, A. M.; Russell, C. S. J Mol Struct Theochem 1986,137, 43.
88. Deerfield, D. W.; Nicholas, H. B., Jr.; Hiskey, R. G.; Peder-sen, L. G. Proteins Struct Fun Genet 1989, 6, 168.
89. Mitchell, J. B. O.; Nandi, C. L.; Thornton, J. M.; Price, S. L.;Singh, J.; Snarey, M. J Chem Soc Faraday Trans 1993, 89,2619.
90. Thery, V.; Rinaldi, D.; Rivail, J. L.; Maigret, B.; Ferenczy,´G. G. J Comput Chem 1994, 15, 269.
91. Melo, A.; Ramos, M. J. Chem Phys Lett 1995, 245, 498.92. Zheng, Y.-J.; Ornstein, R. L. J Am Chem Soc 1996, 118,
11237.Ž .93. Hariharan, P. C.; Pople, J. A. Theor Chim Acta Berl 1973,
28, 213.94. Hrouda, V., Florian, J.; Polasek, M.; Hobza, P. J Phys´ ´
Chem 1994, 98, 4742.
Ž .95. Scheiner, S. J Mol Struct Theochem 1994, 307, 65.96. Sobolewsky, A. L.; Adamowicz, L. Chem Phys Lett 1995,
234, 94.97. Ditchfield, R.; Hehre, W. J.; Pople, J. A. J Chem Phys 1971,
54, 724.98. Hehre, W. J.; Ditchfield, R.; Pople, J. A. J Chem Phys 1972,
56, 2257.99. Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. J Chem
Phys 1980, 72, 650.100. Dunning, T. H.; Hay, P. J. Modern Theoretical Chemistry;
Plenum: New York, 1976.Ž .101. Hariharen, P. C.; Pople, J. A. Theor Chim Acata Berl
1973, 28, 213.102. Hehre, W. J.; Ditchfield, R.; Pople, J. A. J Chem Phys 1972,
56, 2257.103. Clark, T.; Chandrasekhar, J.; Spitznagel, G. W.; Schleyer,
P. v. R. J Comput Chem 1983, 4, 294.104. Frisch, M. J.; Pople, J. A.; Binkley, J. S. J Chem Phys 1984,
80, 3265.105. Frisch, M. J.; Trucks, G. W.; Head-Gordon, M.; Gill,
P. M. W.; Wong, M. W. Foresman, J. B.; Johnson, B. G.;Schlegel, H. B.; Robb, M. A.; Replogle, E. S.; Gomperts, R.;Andres, J. L.; Raghavachari, K.; Binkley, J. S.; Gonzalez, C.;Martin, R. L.; Fox, D. J.; Defrees, D. J.; Baker, J.; Stewart,J. J. P.; Pople, J. A. Gaussian 92, Revision A; Gaussian:Pittsburgh, PA, 1992.
VOL. 72, NO. 3176