02 assaigs clínics, disseny, fonts de financiació ... · de trabajo escritos, de forma que los...
TRANSCRIPT

“Assaigs clínics. Disseny, fonts de financiació, responsabilitats del promotor i
del investigador”Magí Farré
Grupo de Investigación Clínica en Farmacología Humana y Neurociencias. Programa de Neurociencias, IMIM
26-04-2012

Contenido
• Investigación vs asistencia• Ensayo clínico• Investigación clínica independiente• Regulación legal investigación clínica• Investigador y sus responsabilidades• Promotor y sus responsabilidades• Obligaciones legales investigación con
medicamentos y productos sanitarios • Obligaciones legales investigación sin
medicamentos

Investigación vs. asistencia• ¿Qué es investigación y que es asistencia
(práctica clínica)?– La práctica clínica o asistencia se refiere a las
actividades encaminadas a diagnosticar, prevenir, o tratar una enfermedad en un paciente concreto o en un grupo de pacientes con el objetivo de cubrir las necesidades y beneficiar al individuo o colectivo y debe basarse en la mejor evidencia disponible para ofrecer el máximo beneficio y el mínimo riesgo.

Investigación vs. asistencia• ¿Qué es investigación y que es asistencia
(práctica clínica)?– La investigación clínica incluye estrategias
dirigidas a testar hipótesis que permitan alcanzar conclusiones útiles para mejorar el cuidado médico y la salud pública.

Investigación vs. asistencia• ¿Qué es investigación y que es asistencia
(práctica clínica)?– Práctica clínica = beneficio del paciente– Investigación = es posible que los participantes
no obtengan de forma directa un beneficio ya que los resultados definitivos se conocerán al final de la investigación y serán entonces aplicables a futuros pacientes.

Investigación vs. asistencia• Investigación terapéutica y no terapéutica
– Investigación terapéutica• Se investiga algún procedimiento que puede tener
efecto terapéutico en la enfermedad del paciente• p.e. ensayo clínico de un nuevo medicamento para
el tratamiento de la depresión
– Investigación no terapéutica• Se investiga algún procedimiento que no tiene
efecto terapéutico en la enfermedad del paciente• p.e. ensayo clínico de un nuevo medicamento para
estudiar su efecto sobre la función pulmonar (espirometría)

Investigación vs. asistencia• Investigación en humanos
– Pacientes (sujetos enfermos)• Terapéuticos• No terapéuticos• Uso de placebo• Equilibrio clínico (clinical equipoise)• Beneficencia vs no maleficencia
– Voluntarios sanos (sujetos no enfermos)• No terapéuticos• Modelos experimentales de enfermedad• Maleficencia vs beneficencia

Ensayo clínico
• Puede tener un significado distinto según se hable en términos epidemiológico o en investigación con medicamentos
• En epidemiología: es un estudio experimental en que la asignación a cada grupo se realiza de forma aleatoria
• En la investigación con medicamentos se denomina ensayo clínico a cualquier estudio para evaluar los efectos o la farmacocinética de un medicamento. No significa que sea un estudio experimental, aleatorio y controlado
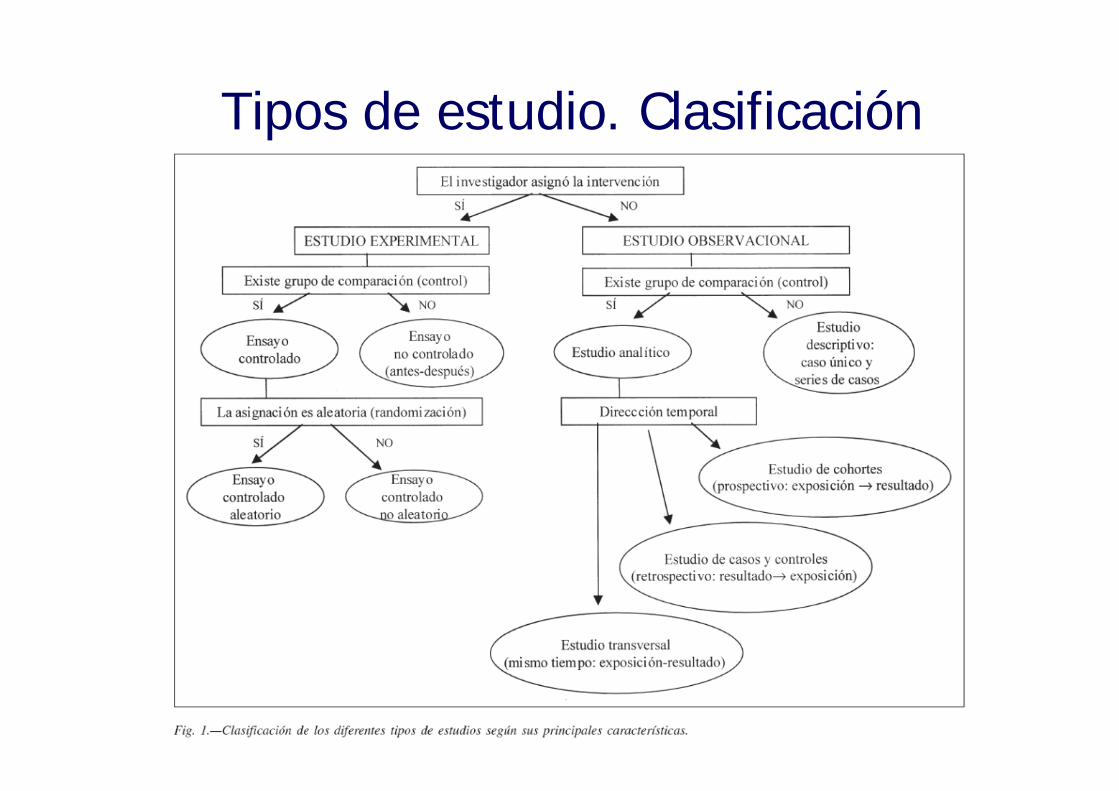
Tipos de estudio. Clasificación

Definición ensayo clínico• Ensayo clínico con medicamentos desde el punto de
vista legal en España: Directiva 2001/20/CE y Ley deacompañamiento 31 Diciembre de 2002 (modifica LeyMedicamento 25/1990) y Real Decreto 223/2004 deensayos clínicos con medicamentos:– “ensayo clínico: toda investigación efectuada en
seres humanos, con el fin de determinar o confirmarlos efectos clínicos, farmacológicos, y/o demásefectos farmacodinámicos, y/o de detectar lasreacciones adversas, y/o estudiar la absorción,distribución, metabolismo y eliminación de uno ovarios medicamentos en investigación con el fin dedeterminar su inocuidad y/o su eficacia”.

Definiciones• Investigación clínica independiente
• Promotor no pertenece a la industria farmacéutica-sanitaria o a organizaciones de investigación bajo contrato(CRO)
• Promotor puede ser:• Investigador individual o asociación de investigadores• Centros sanitarios o de investigación• Universidades o centros asociados• Sociedades científicas• Otras figuras mixtas
• El promotor debe tener entidad jurídica• No puede ser un médico de un servicio clínico• Obligatorio que sea el Centro Sanitario, Fundación, …

Definiciones• Investigación clínica independiente
• Fondos económicos pueden proceder de:• Ayudas a investigación competitiva (nacional: FIS, Ministerio de
Sanidad, Ministerio de Educación; internacional: europea o de otros países u organizaciones)
• Fundaciones (privadas o públicas, incluso relacionadas con industria farmacéutica)
• Programas especiales de promoción y apoyo de la investigación clínica independiente• Ministerio de Sanidad• FIS y CAIBER• Programas específicos de algunas industrias farmacéuticas
(fundaciones, interés por sus medicamentos)• Casi siempre el presupuesto es insuficiente• Industria farmacéutica
• Protocolos de ensayos clínicos de sociedades científicas (Fase IV)• Aportan apoyo económico parcial• Casi nunca subvencionan el gasto del medicamento (Fase IV)

Investigación clínica independiente• Si se trata de un ensayo clínico con
medicamentos o productos sanitarios las obligaciones legales son idénticas a las de cualquier promotor
• Debe asegurarse la calidad en cualquier tipo de investigación que sea ensayo clínico, sea independiente o promovida por industria farmacéutica-sanitaria
• La Directiva de ensayos clínicos europea está pensada para desarrollar medicamentos• Estudios no terapéuticos pueden ser excepciones si no
se va a desarrollar un medicamento. Se debe garantizar calidad.

Investigación clínica independiente• Cualquier ensayo clínico puede ser objeto de
inspección por la AEMPS en colaboración con las autoridades autonómicas
• Las inspecciones en principio no difieren dependiendo del tipo de promotor
• Debe por tanto asegurarse la calidad

Regulación legal investigación• La investigación clínica está regulada
legalmente en España :– Investigación en general (excepto medicamentos y
productos sanitarios)• Ley 14/2007, de 3 de julio, de Investigación
biomédica.– Medicamentos y productos sanitarios
• Ley 29/2006, de 26 de julio, de garantías y usoracional de los medicamentos y productossanitarios, RD 223/2004 de ensayos clínicos yOrden SAS/3470/2009 de estudiosposautorización
• Corresponde a legislación europea

Autoregulación investigación• La investigación clínica está autoregulada por los
propios científicos:– Códigos deontológicos
• P. e. Col.legi Oficial Metges Barcelona
– Manual de ética médica• P.e. Asociación médica Mundial
– Códigos de Buenas Prácticas científicas• P.e. PRBB

Guía de BPC (ICH). InvestigadorInvestigador
Según RD 223/2004:Médico o persona que ejerce una profesión reconocida para llevar a cabo investigaciones en razón de su formación científica y de su experiencia en la atención sanitaria requerida.El investigador dirige y se responsabiliza de la realización práctica del ensayo clínico en un centro, y firma junto con el promotor la solicitud, corresponsabilizándose con él.

Guía de BPC (ICH). InvestigadorSon responsabilidades del investigador:
a) Estar de acuerdo y firmar junto con el promotor el protocolo del ensayo.b) Conocer a fondo las propiedades de los medicamentos en investigación.c) Garantizar que el consentimiento informado se recoge de conformidad a lo establecido en este Real Decreto.d) Recoger, registrar y notificar los datos de forma correcta y garantizar su veracidad.

Guía de BPC (ICH). InvestigadorSon responsabilidades del investigador:
e) Notificar inmediatamente los acontecimientos adversos graves o inesperados al promotor.f) Garantizar que todas las personas implicadas respetarán la confidencialidad de cualquier información acerca de los sujetos del ensayo, así como la protección de sus datos de carácter personal.g) Informar regularmente al Comité Ético de Investigación Clínica de la marcha del ensayo.h) Corresponsabilizarse con el promotor de la elaboración del informe final del ensayo, dando su acuerdo con su firma.

Guía de BPC (ICH). Promotor
PromotorPromotor (RD 223/2004):
Individuo, empresa, institución u organización responsable del inicio, gestión y/o financiación de un ensayo clínico. El promotor o su representante legal habrá de estar establecido en uno de los Estados miembros de la Unión Europea.Corresponde al promotor firmar las solicitudes de dictamen y autorización dirigidas al Comité Ético de Investigación Clínica y a la Agencia Española de Medicamentos y Productos Sanitarios.

Guía de BPC (ICH). PromotorSon responsabilidades del promotor:a) Establecer y mantener un sistema de garantías y
control de calidad, con procedimientos normalizados de trabajo escritos, de forma que los ensayos sean realizados y los datos generados, documentados y comunicados de acuerdo con el protocolo, las normas de buena práctica clínica y lo dispuesto en este real decreto.
b) Firmar, junto con el investigador que corresponda, el protocolo y cualquiera de sus modificaciones.
c) Seleccionar al investigador más adecuado según su cualificación y medios disponibles, y asegurarse de que éste llevará a cabo el estudio tal como está especificado en el protocolo.

Guía de BPC (ICH). Promotor
d) Proporcionar la información básica y clínica disponible del producto en investigación y actualizarla a lo largo del ensayo.
e) Solicitar el dictamen del Comité Ético de Investigación Clínica y la autorización de la Agencia Española de Medicamentos y Productos Sanitarios, así como suministrarles la información y recabar las autorizaciones que procedan, sin perjuicio de la comunicación a las comunidades autónomas, en caso de modificación o violación del protocolo o interrupción del ensayo, y las razones para ello.

Guía de BPC (ICH). Promotor
f) Suministrar de forma gratuita los medicamentos en investigación, garantizar que se han cumplido las normas de correcta fabricación y que las muestras están adecuadamente envasadas y etiquetadas. También es responsable de la conservación de muestras y sus protocolos de fabricación y control, del registro de las muestras entregadas y de asegurarse que en el centro donde se realiza el ensayo existirá un procedimiento correcto de manejo, conservación y uso de dichas muestras. Excepcionalmente, se podrán acordar con el centro otras vías de suministro

Guía de BPC (ICH). Promotorg) Designar el monitor que vigilará la marcha del
ensayo.h) Comunicar a las autoridades sanitarias, a los
investigadores y a los Comités Éticos de Investigación Clínica involucrados en el ensayo las sospechas de reacciones adversas graves e inesperadas de conformidad con lo establecido en los artículos 43 a 46.
i) Proporcionar al investigador y al CEIC, de forma inmediata, cualquier información de importancia a la que tenga acceso durante el ensayo.
j) Proporcionar compensación económica a los sujetos en caso de lesión o muerte relacionadas con el ensayo.

Guía de BPC (ICH). Promotork) Proporcionar al investigador cobertura legal y
económica en estos casos excepto cuando la lesión sea consecuencia de negligencia o mala práctica del investigador.
l) Acordar con el investigador las obligaciones en cuanto al tratamiento de datos, elaboración de informes y publicación de resultados. En cualquier caso, el promotores responsable de elaborar los informes finales parciales del ensayo y comunicarlos a quien corresponda.
El promotor dispondrá de un punto de contacto, donde los sujetos del ensayo puedan obtener mayor información sobre éste, que podrá delegar en el investigador.

Obligaciones legales investigación en humanos
• Ensayos clínicos con medicamentos y productos sanitarios– Protocolo y consentimiento informado– Aprobación por CEIC– Aprobación por Agencia Española de Medicamentos y
Productos Sanitarios• Estudios posautorización
– Protocolo y consentimiento informado– Aprobación por CEIC– Según el tipo aprobación por Agencia Española de
Medicamentos y Productos Sanitarios y/o CCAA• Investigación sin medicamentos o productos sanitarios
– Protocolo y consentimiento informado– Aprobación por CEIC

Obligaciones legales EECC medicamentos I
– Redacción protocolo según RD 223/2004– Consentimiento informado participantes– Seguro de responsabilidad civil (no terapéutico,
nuevos fármacos, condiciones distintas utilización aprobada)
– Respeto Declaración Helsinki– Respeto normas de buena práctica clínica– Confidencialidad (Ley orgánica protección de
datos carácter personal 15/1999, LOPD)– Comunicación urgente de efectos indeseables
graves e inesperados

Obligaciones legales EECC medicamentos II
– Presentación a Comité Ético de Investigación Clínica (CEIC) para aprobación, si aprobación, remitir a AEMPS)
– Presentación Agencia Española Medicamento (AEMPS-MSC) para evaluación (si aprobación, puede iniciarse el ensayo)
– Registro (Eudra-CT clinicaltrialsregister.eu ; clintrials.gov)
– Inicio - final: informe de progreso anual, informe final

Obligaciones legales Estudios Posautorización I
• Estudio posautorización: (Real Decreto 1344/2007, de 11 de octubre): – Cualquier estudio clínico o epidemiológico
realizado durante la comercialización de un medicamento según las condiciones autorizadas en su ficha técnica, o bien en condiciones normales de uso, en el que el medicamento o los medicamentos de interés son el factor de exposición fundamental investigado. Este estudio podrá adoptar la forma de un ensayo clínico o un estudio observacional.

Obligaciones legales Estudios Posautorización II• EPA-LA: Estudios posautorización cuya realización tiene lugar a
instancia de las autoridades reguladoras y ligada a la autorización de comercialización (AEMPS o Agencia Europea de Medicamentos). Se incluyen en esta vía administrativa tanto los estudios ligados a la autorización como los estudios de seguridad a requerimiento de las autoridades sanitarias y los incluidos en los planes de gestión de riesgos.
• EPA-AS: Estudio posautorización promovido por las Administraciones Sanitarias (AS) o financiado con fondos públicos.
• EPA-SP: Estudio posautorización de seguimiento prospectivo que no corresponde a ninguna de las dos categorías anteriores.
• EPA-OD: Estudios posautorización con diseño diferente al de seguimiento prospectivo, por ejemplo, estudios transversales o retrospectivos.
• No-EPA: Estudios en los que el factor de exposición fundamental investigado no es un medicamento; por ejemplo estudios de incidencia o de prevalencia de enfermedades, etc.

Obligaciones legales Estudios Posautorización III

Obligaciones legales proyectos de investigación (no EECC medicamentos)– Redacción protocolo– Consentimiento informado participantes– Seguro de responsabilidad civil – Respeto Declaración Helsinki– Confidencialidad (Ley orgánica protección de
datos carácter personal 15/1999, LOPD)– Presentación a Comité Ético de Investigación
Clínica (CEIC)(si aprobación se pude iniciar le estudio)
– Inicio - final: informe de progreso y final

“Gracias por su atención”
Magí FarréGrupo de Investigación Clínica en Farmacología
Humana y NeurocienciasPrograma de Neurociencias, IMIM
26-04-2012