what do genome sequences reveal? non-random nucleotide distribution a dynamic nature genes,...
TRANSCRIPT

What do genome sequences reveal?
• Non-random nucleotide distribution
• A dynamic nature
• Genes, metabolic pathways and potential

Randomness in genome sequences
• How long do you make primers to ensure specificity?
• A random probability would predict ~1000 EcoRI restriction sites in the E. coli genome.– Later in the semester you can write a Perl
program to validate or refute this prediction

Examples of nucleotide skew
• CpG islands
• Purine loaded RNA
• GC Skew
• Codon usage preference
• Transposons and repeats

CpG islands
• CpG refers to the dinucleotide, while C-G to the base pair
• In the human genome, the C of CpG is typically methylated, and there is a high chance of this methyl C mutating to a T (transition)
• As a consequence, CpG are rarer in this genome than one would expect

CpG islands
• However, for biologically important reasons methylation is suppressed in short stretches, such as around promoters
• As a result, CpG islands have been used to define start sites for putative genes

Purine-loaded RNA
• When transcription is to the right of the promoter, the “top”, mRNA template strand rends to be purine-rich. When transcription is to the left of the promoter, the top, mRNA strand tends to be pyrimidine rich.
• Selective pressure for purine loading mRNA, thought to prevent unwanted RNA-RNA interactions, affects codon usage
• Szybalski’s rule
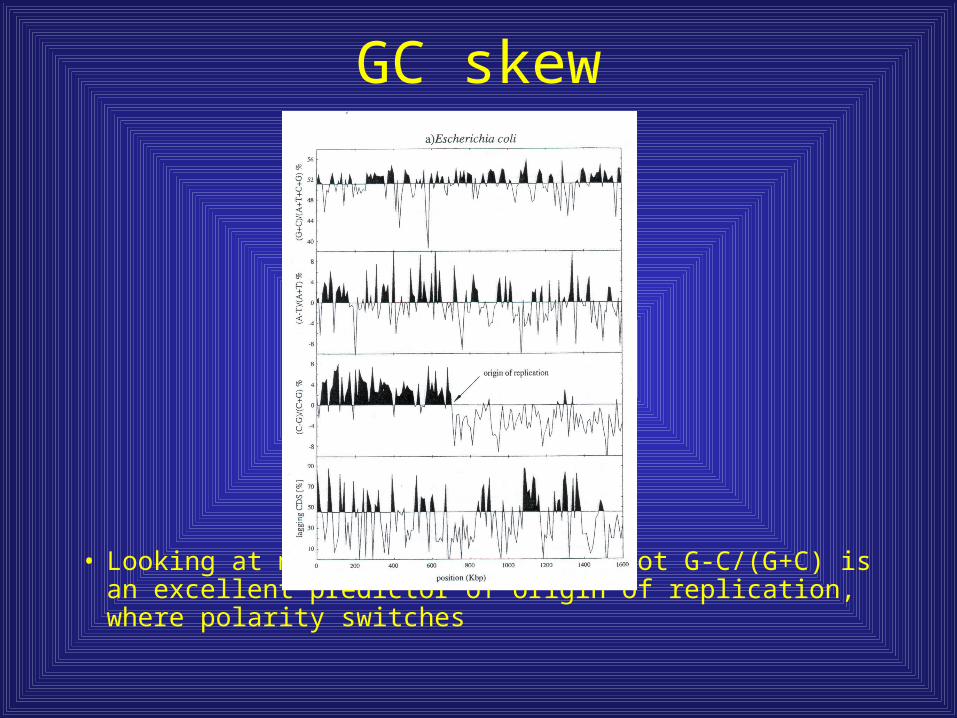
GC skew
• Looking at microbial genomes, a plot G-C/(G+C) is an excellent predictor of origin of replication, where polarity switches
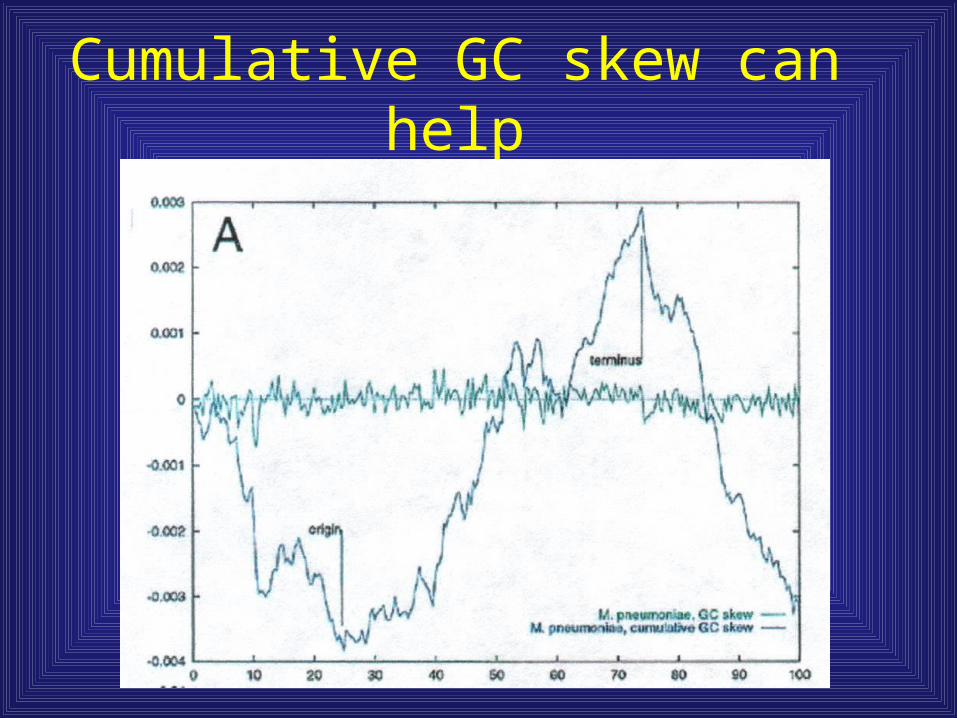
Cumulative GC skew can help

Why is there GC skew?
• Consequence of asymmetry in replication or repair• Leading vs. lagging strand synthesis• Transcription-repair coupling
– Removes most frequent types of DNA damge (deaminated cytosines and pyrimidine dimers, both of which lead to base substitution
– This repair would occur on template strand (not coding), which then should become pyrimidine rich
– Also, the template strand is significantly protected against DNA damage during transcription, while the coding strand is exposed. This should help increase the purine load of the coding strand.
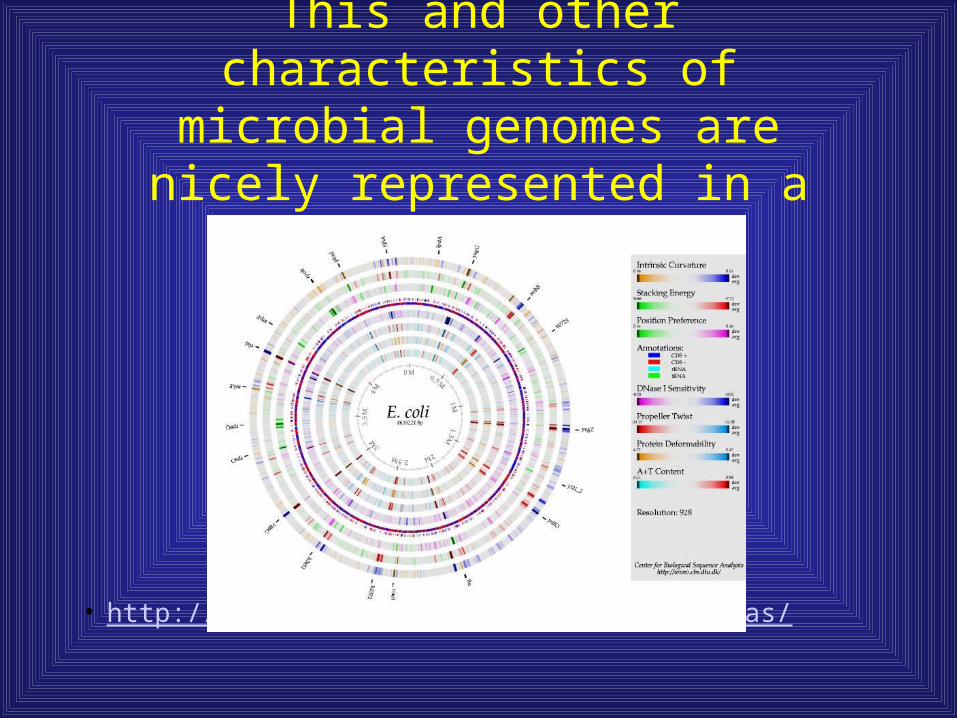
This and other characteristics of microbial genomes are nicely represented in a genome atlas.
• http://www.cbs.dtu.dk/services/GenomeAtlas/

Codon usage
• Factors shaping codon usage are complex
– Suggested at one time that the third codon position was neutral – therefore all codons coding for the same amino acid should be equally frequent. – not true
– Codon preference has been thought to be a result of translation efficiency, ie. selection against GC-rich anti-codons/codons due to increase energy in binding, slowing down translation of highly expressed genes (observed in E. coli; highly expressed genes show one profile, low expressed genes another).
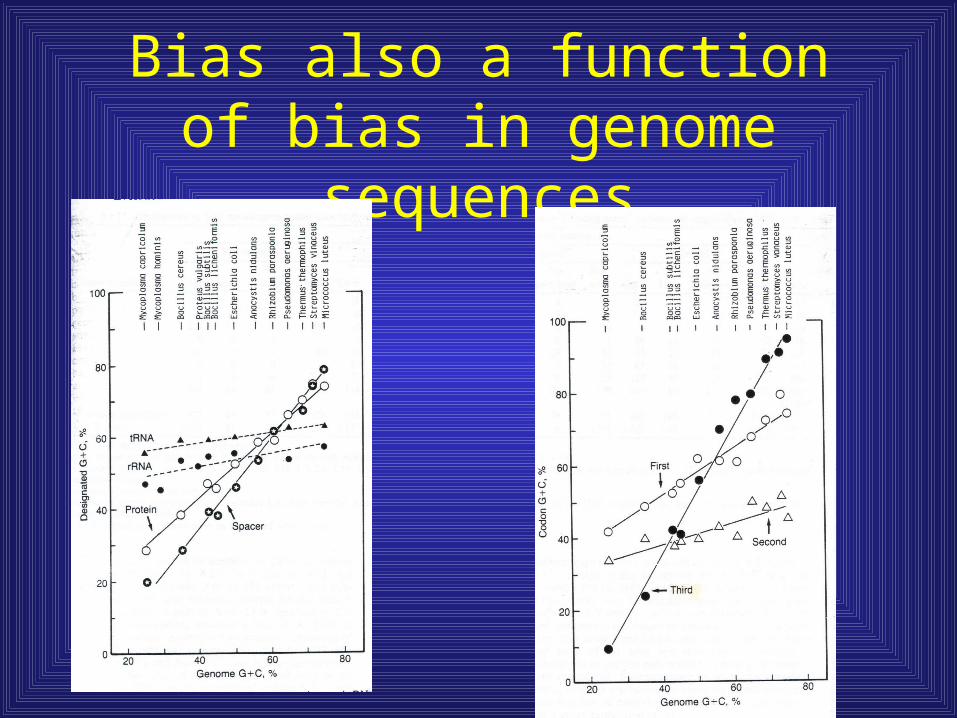
Bias also a function of bias in genome sequences

Math to approximate codon nucleotide composition
• GC1st = 0.615 X GC genome + 26.9
• GC2nd = 0.27 X GC genome + 26.7
• GC3rd = 1.692 x GC genome – 32.3
• Violation of these relationships offers support for lateral gene transfer events
• What about amelioration? – acetate kinase story

The genome as a plastic, dynamic, evolving molecule
• Pervasiveness of repeat sequences– Transposons– Pseudogenes– Simple repeats
• Gene duplication, paralogs, and expansion of protein families
• Synteny• Deletions

Transposable elements found in the human genome
• SINEs – short interspersed elements
• LINEs – long interspersed elements
• LTR retrotransposons
• DNA transposons

AluI is a SINE example
• SINEs comprise ~13% of genome• Alu – 282 consensus containing AluI restriction
site concentrated around genes, selected for?• SINEs are transcribed under stress conditions,
resulting in RNAs that hybridize to a particular protein kinase that normally inhibits translation. Thus, SINEs enable cells to make more proteins under stress.

A mechanism for SINE proliferation – reverse transcriptase
• Reverse transcriptase is consider one of the most influential macromolecules in genome evolution
• First discovered in retroviruses
• Repetitive sequences are thought to be generated from one or few “master” genes
• For instance, the small cytoplasmic BC1 RNA appears responsible for a subclass of ID repetitive elements in rodents

Reverse transcriptase generate retrogenes
• mRNA is reversed transcribed into cDNA, and integrated into the genome
• Hallmarks of this process include:– Short direct repeats at their flanks– A-rich regions at their 3’ end– Lack of introns compared to their founder gene
• Often the retrogene is left without a promoter or other regulatory elements

Lots of retrogenes…• Inactivity and lack of selective pressure on retrogenes
leads to acquisition of nonsense mutations and indels, generating pseudogenes
• Numerous genes have spawned retrogenes, generally ranging in numbers from 1 to ten, but occasionally greater
• The single intron-containing source gene for glyceraldehyde 3 phosphate dehydrogenase has yielded more than 300 intronless retrogenes in rats and mice

Lots more retrogenes
• Small RNA’s such as small nuclear RNAs, SRP RNA, or tRNA’s also produce retrogenes
• Alu RNA’s are derived from SRP RNA, can reach over 105 copies in genome
• Apparently, reverse transcriptase has a higher affinity for certain tRNA, or small RNA structures

LINEs make up 21% of the human genome
• Along with SINEs considered the “litter” of the genomes of eukaryotes
• Several hundred thousand long interspersed DNA elements up to 7 kb in length without long terminal repeats (LTR’s)

Retrotransposons and DNA transposons
• Comprise ~8%, and ~3% of human genome, respectively.
• Retrotransposons move through an RNA intermediate, DNA transposons do not
• DNA transposons are characterized by terminal inverted repeats of 10-500 bp recognized by a transposase which cuts and pastes the sequence
• RepBase – www.girinst.org

Human genome repeat summary• Repeats make up ~45% of genome• Evolutionary biologists use repeat sequences to calibrate a
“molecular clock”• The X chromosome has the highest [] of transposable
elements, with a 525 kb segment being 89% transposon. Contrasted by the hox locus which is only 2%
• Y chromosome contains the most LINE transposons, and looks the “youngest” due to most recent insertions
• ~50 human genes appear to be derived from transposons (exaptations)

Vertebrate comparison - Fugu
• Only 2.7% of genome is interspersed repeats• ~40 families of transposons, with <5% sequence
divergence reflecting young age and potential “current” activity (contrasts 6 families of low divergence levels in humans)
• Analysis of Fugu repeat sequences suggests that frequency of deletion relative to point mutations is higher than in humans

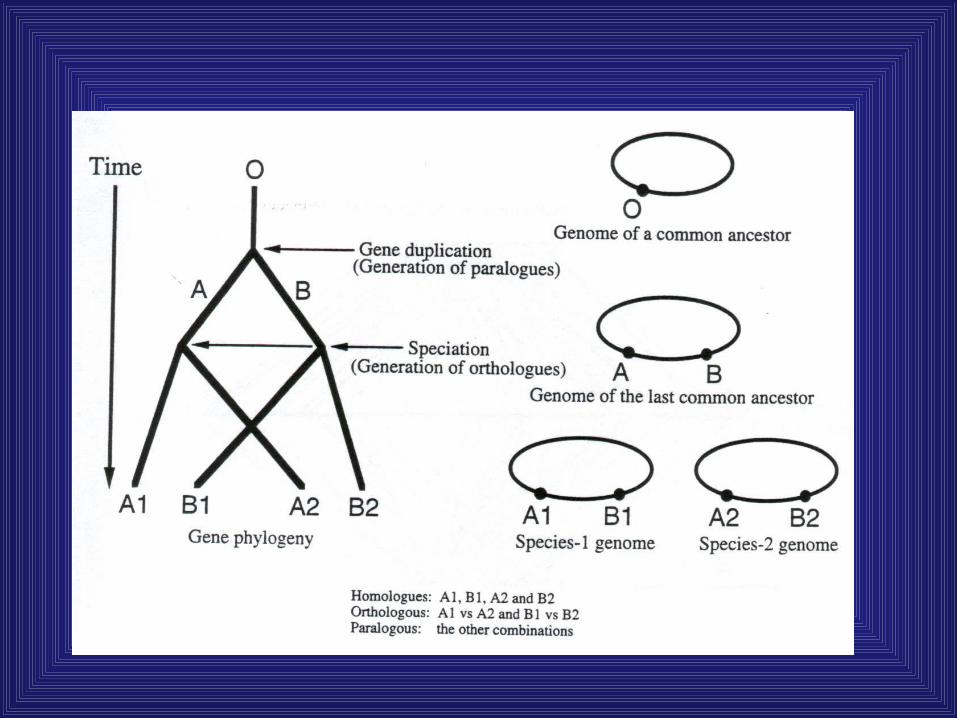
Gene duplication is a central mechanism for evolution
• Gene duplications result in paralogs, genes arising from a common ancestral gene within one species
• Orthologs are genes in two organisms evolved from a common ancestral gene in another species
• Homology refers to common evolutionary origin, not sequence identity
• An example or two:

A visit to the M. acetivorans database
• http://www-genome.wi.mit.edu/annotation/microbes/methanosarcina/
• Gene families were generated based on cut-off values of identity and similarity in this genome
• Take note of putative transcriptional regulatory proteins

Isocitrate dehydrogenase is ubiquitous
• Go to the OMIM database at NCBI and search for IDH, click on IDH3A and read summary to understand function of this protein
• What sort of diversity is found in genes encoding these proteins, orthologs and paralogs?

Clusters of Orthologous Genes (COGS)
• www.ncbi.nlm.nih.gov/COG• Scroll down and on the right side under the
heading of Functional categories click on Letter C – Energy production and conversion
• Do a Ctrl-F function for isocitrate and then click on COG0538 next to isocitrate dehydrogenase
• You will find a table of 29 proteins, and links to various information about this COG

COG eye candy• At the bottom you will see a dendogram
• Click on the dendogram to bring up figure with a circle and colored diamonds. Select “centered” and click on the word “Rotate”
• Notice the two major parts, indicating there are two isoforms (distinct genes encoding similar proteins with similar roles but no assumptions implied about their evolution)
• As you read in OMIM, IDH is split into two major groups
Currently, COG is limited
• Can use Swiss-Prot
• http://www.ebi.ac.uk/swissprot/
• I’ve had difficulties with the www.expasy.ch

Loci with common gene order share synteny
• With the completion of so many genomes, now comparative genomics allows genome vs. genome comparisons
• Often closely related species share extensive identity in gene order – synteny
• Such comparisons have been insightful for many reasons -
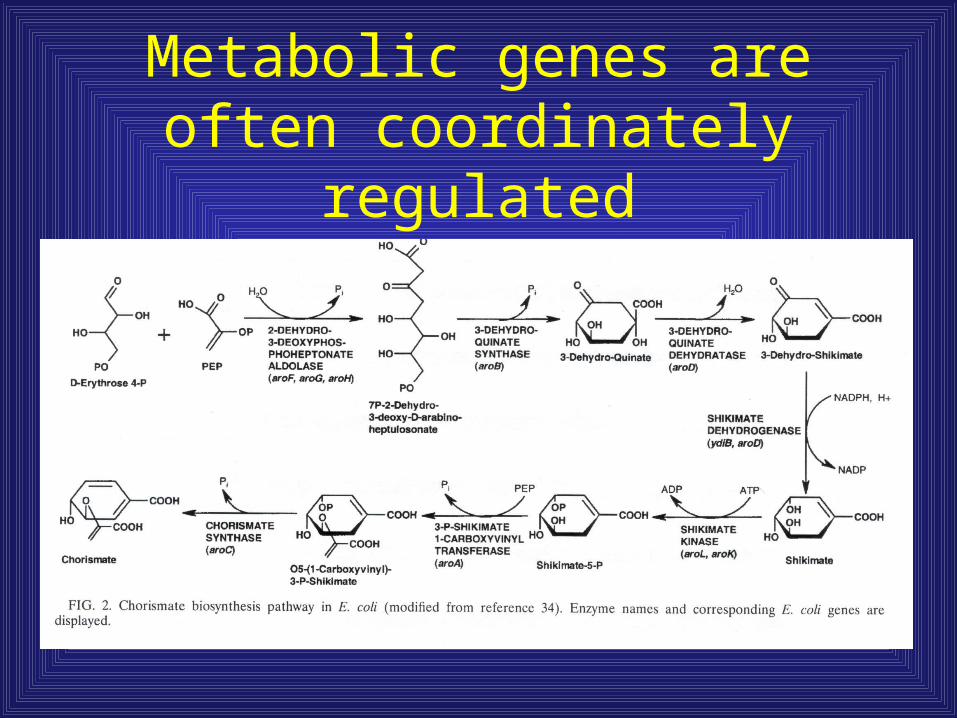
Metabolic genes are often coordinately regulated
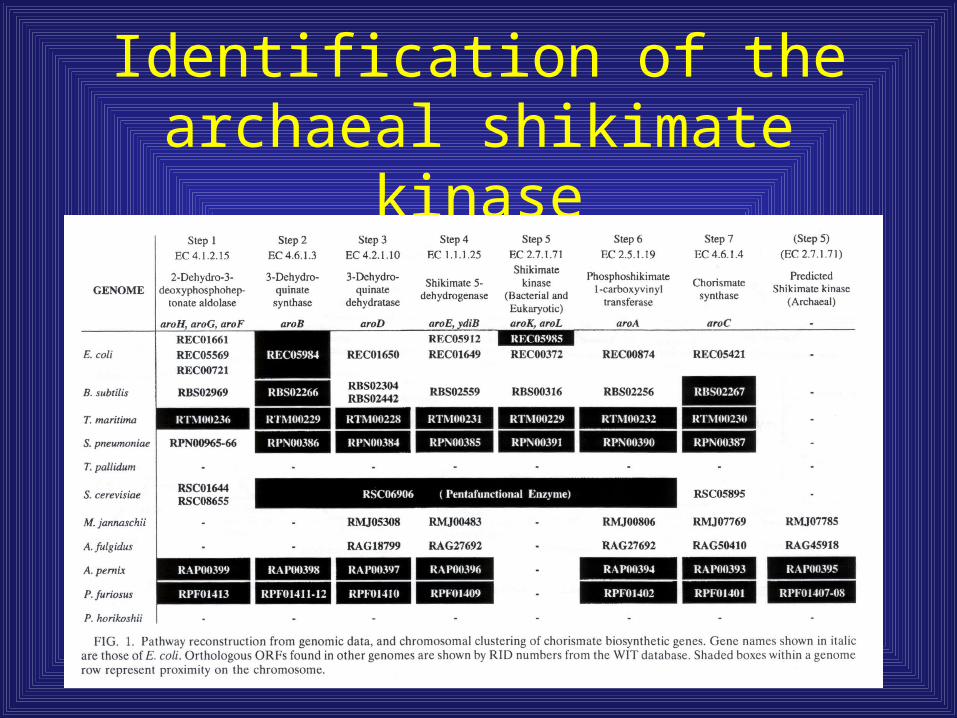
Identification of the archaeal shikimate kinase

Nitrogenous waste and genome deletions
• Animals produce nitrogenous waste• Aphids are exceptions to this rule – how?• Contain bacteriocytes housing Buchnera• The Buchnera genome is ~650,000 bp• A close look at this genome reveals only
583 predicted genes, 500 with known biological roles, 79 hypotheticals found in other genomes, 4 unique to Buchnera

Buchnera and E. coli
• Most of the genes found in Buchnera were very similar to those found in E. coli, indicating that Buchnera was a close relative of modern E. coli, but lost 75% of it’s genome – as a result, Buchnera has become a aphid symbiont
• Amino acid biosynthetic symbiosis – Buchnera and aphids exhibit reciprocal genetic capacity for amino acid biosynthesis

Not a totally free ride
• Buchnera has maintained it’s ability to carry out ATP synthesis; keeping ATP synthase and electron transport so that it is not a weight on the aphids energy generation
• In this way, Buchnera resembles a mitochondrion

An interesting perspective on deletions
• Prader-Willi Syndrome (PWS) is caused by a mega-based deletion in the region of 15q11-q13
• Angelman Syndrome is caused by loss of the same section of genome
• Two different conditions are caused by loss of the same section of our genome, yet symptoms differ!

Inheritance has a role• Whether you have AS or PWS depends on which
parent gave you the mutated chromosome, but not sex-linked
• If from father – PWS; Mother – AS• PWS causes weakened muscles, fail to suckle, will not
grow very tall, short hands, short feet, obesity, and mental retardation.
• AS problem with balance and motor skills, appear hyperactive, overly happy, and more severely retarded

How? - Imprinting
• Mammals mark a small set of genes during gametogenesis so that only the paternal or maternal copy will be transcribed, and the other allele at the same locus will be silent
• >20 mammalian genes known to be imprinted
• Why imprinting?

The dichotomy of sex and gene expression
• Male wants offspring to extract maximum nutrients from mother to promote survival
• Female needs to save some for herself (Save the brain!!)
• Paternally expressed genes should promote growth, maternally expressed genes should reduce growth as long as Mom is sole source of nutrients

Nice hypothesis, where’s the data?
• One paternally expressed gene, insulin-like growth factor 2 (IGF2) promotes growth of embryo. Only the paternal allele is expressed in embryo.
• The IGF2 receptor is only expressed by the maternally inherited chromosome, allowing the mother to maintain control of this cascade
• Many more examples…

What is the mechanism for imprinting?
• DNA methylation; Dnmt-1, a specific methyltransferase adds a methyl group to many but not all cytosines
• Methylation of promoter regions may block transcription of genes
• Similarly, may cause an enhancer to stimulate expression of another gene
• Methylation may inactivate chromatin insulator
• Investigators interested in the methylome

Functional genomics approach to imprinting
• Hypomethylation of genome is generally associated with senescence and cell death
• Using tissue culture, a group led by Laurie Jackson-Grusby examined how the genome would respond to a Dnmt1 deletion
• Looking at ~6000 genes, 10% showed altered expression levels, most induced, few repressed.
• Affected imprinted genes, cell cycle control, etc. genes are available online
• Bottomline – the genome has five bases G, C, A, T, and methyl-C.

Genome annotation tools
• Blast, Psi-BLAST• Transmembrane domain prediction
– http://www.bio.davidson.edu/courses/compbio/slr/kyte-doolittle-background.htm
– www.expasy.ch
• Hmmer - http://pfam.wustl.edu/• Prosite – amino acid signatures in protein
families, and more…

Scoring for hydrophobicity
• Segments of 18-22 amino acids with an average polarity value greater than 1.6 (KD) can be predicted with a rather high probability as TM segments when averaging over 19 residues
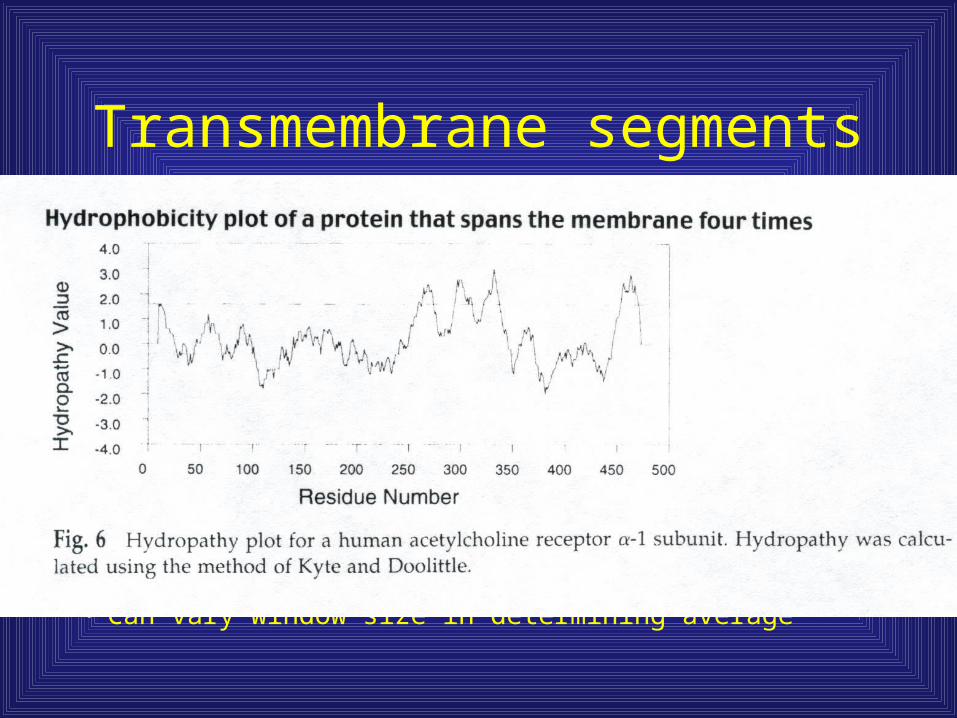
Transmembrane segments
• Can vary window size in determining average

100 pts.
• What two motifs do transmembrane proteins exhibit?

Amphipathic helices and such
• Look for periodicity of hydrophobic moment-helix that has one face buried in protein core, otherexposed to solvent. (n, n + 3
n + 4, n + 7) -sheet (n, n +2, n + 4,n + 8, etc., polar at n + 1n + 3, n + 5, etc.)
LDSALRTLEAYL

Protein secondary structure (RNA next week)
• Current methods using single sequence (ie. info like proline is a good helix initiator) give ~60% accuracy for alpha, beta, and coil
• Current methods based on multiple sequence alignments and neural nets ~70% accurate

Secondary structure predictors
• www.pbil.ibcp.fr
• www.bmm.icnet.uk/~prof/
• www. Embl-heidelberg.de/predictprotein/predictprotein.html
• Last one is PredictProtein

PredictProtein
• Generates alignments looking for similar sequences via blastp
• A multiple sequence alignment is generated by a weighted dynamic programming method
• PROSITE motifs are retrieved from PROSITE• Protein is compared to ProDom (domain database)• Prediction of protein structure in 1D. The
multiple alignment is used for profile-based neural network predictions

Validation
• Expected helix, sheet, coil, overall accuracy is >72% for water-soluble globular proteins
• >95% for transmembrane vs. non-transmembrane

Other secondary structure predictors
• www.embl-heidelberg.de/argos/predator/predator_info.html
• http://Globin.bio.warwick.ac.uk/psipred
• http://Circinus.ebi.ac.uk:8888