wes ino80d circg sm r3 - circulation: genomic and...
TRANSCRIPT

1
SUPPLEMENTAL MATERIAL

2
Library preparation, whole exome capture and sequencing
Paired-‐end indexed libraries were prepared following the manufacturer’s (Agilent
Technologies, Santa Clara, CA) protocol. Briefly, target DNA (3ηg in 120 ul TE buffer) was
fragmented using a Covaris E210 sonicator (Covaris Inc, Woburn, Mass.,) using a duty cycle of 10%,
intensity 5, cycles 200, time 360 seconds, resulting in double-‐stranded DNA fragments with blunt or
sticky ends with a fragment size between 150-‐200 bp. The ends were repaired and phosphorylated
using Klenow, T4 polymerase, and T4 polynucleotide kinase, after which “A” base is added to the 3’
ends of double-‐stranded DNA using Klenow exo-‐ (3’ to 5’ exo minus). Paired end Index DNA
adaptors (Agilent Technologies, Santa Clara, CA) with a single “T” base overhang at the 3’ end were
ligated and resulting constructs were purified using AMPure SPRI beads from Agencourt. The
adapter-‐modified DNA fragments were enriched by 4 cycles of PCR using InPE 1.0 forward and
SureSelect Pre-‐Capture Indexing reverse (Agilent Technologies, Santa Clara, Ca) primers. The
concentration and size distribution of the libraries were determined on an Agilent Bioanalyzer DNA
1000 chip (Agilent Technologies, Santa Clara, Ca).
Exome capture was carried out using the protocol for Agilent’s SureSelect Human All Exon
50MB kit (Agilent Technologies, Santa Clara, Ca). This kit encompasses coding exons annotated by
the GENCODE project (www.sanger.ac.uk/gencode/) as well as consensus coding sequence (CCDS,
www.ncbi.nlm.nih.gov/CCDS/) and RefSeq (www.ncbi.nlm.nih.gov/refseq/) databases and
incorporates exomic regions and non-‐coding RNAs from miRBase (v.13) and Rfam databases to
provide a capture size of approximately 50 Mb. 500 ng of the prepped library was incubated for 24
hours at 65 °C with whole exon biotinylated RNA capture baits supplied in the kit. The captured
DNA:RNA hybrids were recovered using Dynabeads MyOne Streptavidin T1 from Dynal (Invitrogen,
Carlsbad, CA). DNA was eluted from the beads and purified using Ampure XP beads from Agencourt
(Beckman Coulter, Brea, CA). The purified capture products were then amplified using the
SureSelect Post-‐Capture Indexing forward and Index PCR reverse primers (Agilent) for 12 cycles.
Libraries were validated and quantified on the Agilent Bioanalyzer (Agilent Technologies, Santa
Clara, Ca).
For individuals L6-‐L9, libraries were loaded onto paired end flow cells at concentrations of
4-‐5 pM to generate cluster densities of 300,000-‐500,000/mm2 following Illumina’s standard
protocol using the Illumina cBot and HiSeq Paired end cluster kit version 1 (Illumina, San Diego,
CA). The flow cells were sequenced as 101 X 2 paired end reads on an Illumina HiSeq 2000 using

3
TruSeq SBS sequencing kit version 1 and HiSeq data collection version 1.1.37.0 software. Base-‐
calling was performed using Illumina’s RTA version 1.7.45.0.
For individual L10, libraries were loaded onto paired end flow cells at concentrations of 7.5
pM to generate cluster densities of 500,000-‐600,000/mm2 following Illumina’s standard protocol
using the Illumina cBot and HiSeq Paired end cluster kit version 3. The flow cells were sequenced as
101 X 2 paired end reads on an Illumina HiSeq 2000 using TruSeq SBS sequencing kit version 3 and
HiSeq data collection version 1.4.8 software. Base calling was performed using Illumina’s RTA
version 1.12.4.2.
Genotype calling and variant filtration
The technical challenge of the modern era of genomic medicine and personalized exome
analytics is in the effective use of combination of tools to find software-‐agnostic, highly concordant,
high-‐quality genetic variants underlying complex, familial diseases[1]. To address this challenge, we
used two computational genomic data analysis pipelines and two complementary variant filtering
methods. Both pipelines included analyses modules for quality control, sequence alignment (two
different aligners: BWA and Novoaligner), base quality score recalibration, and variant calling and
complimentary variant filtering methods.
Illumina fastq files were converted to Sanger fastq files using the MAQ software
(http://maq.sourceforge.net/). We used the FASTX-‐toolkit
(http://hannonlab.cshl.edu/fastx_toolkit/) for preprocessing short-‐read fastq files. The
preprocessing steps included clipping sequencing primers/adapter sequences, trimming sequences
based on the quality scores, and filtering artifacts and low quality sequences. We used FastQC
(http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/) to perform QC on raw and QC filtered
sequence data. We aligned sequence-‐reads with the reference genome GRCh37/hg19 from the
1000 Genomes project, using the Burrows-‐Wheeler Aligner (BWA) software. We used a Perl script
(cmpfastq.pl [http://compbio.brc.iop.kcl.ac.uk/software/cmpfastq.php]) to identify paired and un-‐
paired reads. BWA software was then used to align single-‐end and paired-‐end data separately. The
generated SAM files were merged using PICARD (http://picard.sourceforge.net/index.shtml) to
generate sorted BAM files. The BAM files were indexed using SAMtools
(http://samtools.sourceforge.net/).
We used the Genome Analysis Toolkit[2] (GATK v1.0.5777,
http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit) for post-‐
alignment processing of BAM files, including local realignment around insertions or deletions

4
(indels), removal of duplicates, and base quality score recalibration. Due to alignment artifacts or
false positive SNPs, sequence aligners are unable to perfect map reads containing indels. Multiple
steps were employed in the realignment process, i.e., determining (small) suspicious intervals
which are likely in need of realignment, running the realigner over those intervals, and fixing the
mate pairs of realigned reads. The realigned, fixed, and sorted BAM files were generated for each
sample. Duplicate reads were located and removed using PICARD tools (MarkDuplicates). Finally,
we corrected for variation in quality with machine cycle and sequence context by analyzing the co-‐
variation among several features of the base (i.e., reported quality score, the position within the
read, the preceding and current nucleotide observed by the sequencing machine, probability of
mismatching the reference genome, and known SNPs taken into account). The recalibrated quality
scores are more accurate.[2]
Analysis of depth of coverage in the final BAM files indicated that approximately 80% of the
exomic regions were present >8 times in the five patients (-‐-‐minMappingQuality=10 and -‐-‐
minBaseQuality=20). We then used GATK ‘UnifiedGenotyper’ for multiple-‐sample calling to
generate raw variants. GATK applies a Bayesian algorithm for variant discovery and genotyping
that simultaneously estimates the probability that two alleles A (the reference allele), and B (the
alternative allele), are segregating in a sample of N individuals and the likelihoods for each of the
AA, AB and BB genotypes for each of individual samples.[2] If the genotype for this individual could
not be assigned based on the genotype likelihood model, an unknown genotype ‘N’ was assigned.
To generate analysis-‐ready variants, the GATK ‘VariantFiltration’ was used to annotate
suspicious calls from variant calling format (VCF) files based on their failing given filters. Raw SNP
calls were filtered using empirically derived cut-‐offs for the following GATK filter expressions: –
filterExpression “QUAL<30.0 || QD<5.0 || HRun>5 || SB>−0.10 || SNPcluster || InDel” –filterName
“StandardFilters” –filterExpression “DP<8” –filterName “LOW_DEPTH” -‐filterExpression “MQ0 >= 4
&& ((MQ0 / (1.0 * DP)) > 0.1)” –filterName “Hard2Validate”, where DP–sequencing depth at the
SNP position; QD–QUAL/DP ratio at the SNP position; HRun–maximal length of the homopolymer
run; SB–strand bias at the SNP position; SNPcluster– 3 SNPs with 10 bp of each other; InDel–SNP
calls around the raw InDels calls; and MQ0–the number of mapping-‐quality zero reads at the
position. The resulting VCF file was annotated using SeattleSeq Annotation Server and filtered
based on variant quality and localization of variants overlapping the ROH regions.
In the confirmatory pipeline, the fastq files were processed using GenomeGPS pipeline v2.0.
Briefly, the Illumina paired end reads were aligned to the hg19 reference genome using Novoalign

5
(http://novocraft.com) followed by the sorting and marking of duplicate reads using Picard. Local
realignment of INDELs and base quality score recalibration were then performed using the Genome
Analysis Toolkit (GATK). Single nucleotide variants (SNVs) and insertions/deletions (INDELs) were
called across all of the samples simultaneously using GATK's UnifiedGenotyper with variant quality
score recalibration. Variants were annotated using a custom annotation workflow and filtered
using VAAST v1.04 and knowledge-‐based gene lists relevant to the phenotype observed.
Genotype calling and variant filtration
Results from the primary pipeline based were filtered using annotation database following
strict criteria and localization of variants in ROH regions shared by the family (Table S2).
Results from the confirmatory pipeline were filtered using two different methods. The first
variant filtering method focused on annotation driven filtering followed by presence of variants
based on localization of a variant on ROH region shared by the pedigree. The second filtering
method followed a knowledge-‐based approach to look for rare variants in the known genes
associated with leading phenotypes followed by a probabilistic disease variant identification using
VAAST. The analysis was carried out to identify high-‐confidence variants in the affected individuals
using a pipeline that use a different short-‐read aligner. Given the fact that two siblings were
affected, we ruled out the possibility that the mutation arose from a de novo mutation as such a
possibility is extremely low in likelihood.. We also ruled out the possibility of uniparental disomy
(UPD) or a single copy deletion occurring in both siblings, as this is also extremely unlikely.
(i) Method 1: Single-‐ homozygous alternative mutation at a position where both parents are
heterozygous
Variants were processed using VAAST v1.04 (3) configured to fit a recessive mode of
inheritance in a trio-‐mode assessment. Variant calls from the two affected siblings were intersected
and input as the “proband”. The resulting statistically significant candidate variants were filtered to
exclude any findings positive in the unaffected sibling. The program was configured with the
following options:
m lrt -‐o output_trio_pnt_i_02_21_2013 -‐pnt c -‐-‐mp1 8 -‐-‐less_ram -‐-‐fast_gp -‐-‐gp 1e10 -‐-‐
significance 2.4e-‐6 -‐-‐codon_bias -‐iht r -‐-‐locus_heterogeneity n –trio
The VAAST program was configured to run in a recessive mode with no locus heterogeneity. Given
the limitations of the current version of the software, we ran it with the affected siblings’ genotype

6
data intersected and compared against the parents. A post-‐analysis filter was then applied to
remove potential candidates that were found in the unaffected daughter.
(ii) Method 2: Compound heterozygous variant where each parent carries only one variant
Variant calls were filtered for positions that were homozygous alternative in the affected
siblings, heterozygous in the parents, and either heterozygous or homozygous reference in the
unaffected sibling. Results were then filtered to select non-‐synonymous mutations with an MAF <
0.1 in the ESP6500 datasets and all HapMap and 1k Genome population datasets. Results from the
two methods were combined and manually reviewed for relevance to the disease state using
GLAD4U (http://bioinfo.vanderbilt.edu/glad4u/). Two gene lists were created, one with eight
targets using the specific term “aortic hypoplasia-‐atherosclerosis syndrome” and a second list that
generated 196 targets using the generic term “atherosclerosis”.
Initial results were processed to find Method (i) variants. Results were filtered to restrict
the unaffected sibling to being either heterozygous or homozygous reference. Additionally, variants
were be filtered for non-‐synonymous changes. Variant list were initially filtered on the basis of
MAF (< 0.1) are as follows: 595 variants (both affected siblings were homozygous alternative and
both parents were heterozygous). Of these, there were 387 variants (unaffected sister was either
homozygous reference or heterozygous). Of these, 68 variants encoded non-‐synonymous changes.
Further results from variant filtering using MAF were compared against the candidate gene lists
and a common variant in APOB (chr2:21250914 G>A; A618V) was observed. 4/68 variants that
had population frequencies < 0.1 in all population datasets queried. This includes two deleterious
and damaging variants in INO80D (Chr2:206869724 T>A) and NPIPA5 (Chr16: 15463612 G>A),
and two tolerated and benign variants in SLC35B1 (Chr17:47783663 C>T) and SALL3
(Chr18:76754549 T>C).
Sanger sequencing
To validate the presence of the mutation we used Sanger sequencing. The FASTA sequence
of two genes of interest (that satisfied strict filtering of discovery and validation pipelines, VAAST
analyses and ROH filtering results) were obtained using NCBI nucleotide search to design the
primers http://www.ncbi.nlm.nih.gov/nuccore. This FASTA sequence was used as a query to
search in NCBI Primer BLAST http://www.ncbi.nlm.nih.gov/tools/primer-‐
blast/index.cgi?LINK_LOC=BlastHome. Primers pairs were selected based on primer length (18-‐30

7
bp), GC content, theoretical melting temperature (Tm = 59-‐60°C), and product size. BLAST was used
to check the specificity of primers. PAGE-‐purified oligos (Integrated DNA Technologies, IA, USA)
were used for real-‐time PCR. Primers used for resequencing mutation sites in INO80D and
TMPRS11E is provided (Table S4). Sanger sequencing for loci mapping was performed using Big
Dye terminator chemistries on ABI 3730xl (Life technologies; Carlsbad, CA) sequencer.
Sequence, structure and functional annotation of INO8OD
Conserved domains and motifs in INO80D:
We performed a comparative sequence analysis of wild-‐type isoforms and derived sequence
with Ser818Cys mutation. Two wild-‐type isoform sequences and mutated sequences were used to
assess the secondary structure, solvent accessibility and distribution of LCRs. Secondary structure
and solvent accessibility were predicted from the sequence using SABBLE [3], and LCRs were
characterized using SEG program integrated in SMART database[4].
We also characterized conserved functional domains in INO80D using sequence based
protein domain searches: protein sequences (Q53TQ3-‐1 and Q53TQ3-‐2) were scanned against
Pfam database. INO80D encodes two copies of zf-‐C3Hc3H domains in both isoforms. Seven low-‐
complexity regions (LCRs) were found in the sequence of Q53TQ3-‐1 and nine different segments of
(LCR) were found on Q53TQ3-‐2 using SEG [5] program integrated in SMART[4, 6]. LCRs are tandem
sequence repeats in the protein universe and were often excluded in the past prior to detailed
sequence analysis. For example, sequence search algorithms like BLAST mask off sequence with
low compositional complexity. Recent studies on LCR function suggest that protein sequences with
LCRs have several important functional roles. LCRs are common in protein sequence space and
observed in diverse proteins. Proteins with LCRs have the higher number of first-‐degree interaction
partners when compared to proteins without LCRs (Wilcoxon-‐Mann-‐Whitney test; P<0.05). LCRs in
proteins were preferentially positioned in sequence extremities and relative position of functions
could have an impact on the function (Kolmogorov-‐Smirnov test; P = 7.6 x 10-‐6). Gene Ontology
based analysis indicated that LCRs encoded in different regions of proteins may also mediate
different biological roles. From a structural perspective, LCRs do not adopt a definite 3D structure
but could exist as a solvent exposed disordered coils [5, 7]. Scanning the INO80D protein sequence
using Pfam database indicated that presence of two copies of zf-‐C3Hc3H domains in the protein.
The E-‐values of the associations were 4.3e-‐12 and 1e-‐16 respectively. According to Pfam

8
annotations, zf-‐C3Hc3H is considered as a potential DNA binding domain and may found in
chromatin remodeling proteins and helicases (http://pfam.sanger.ac.uk/family/zf-‐C3Hc3H).
Protein structure modeling and fold prediction of INO80D:
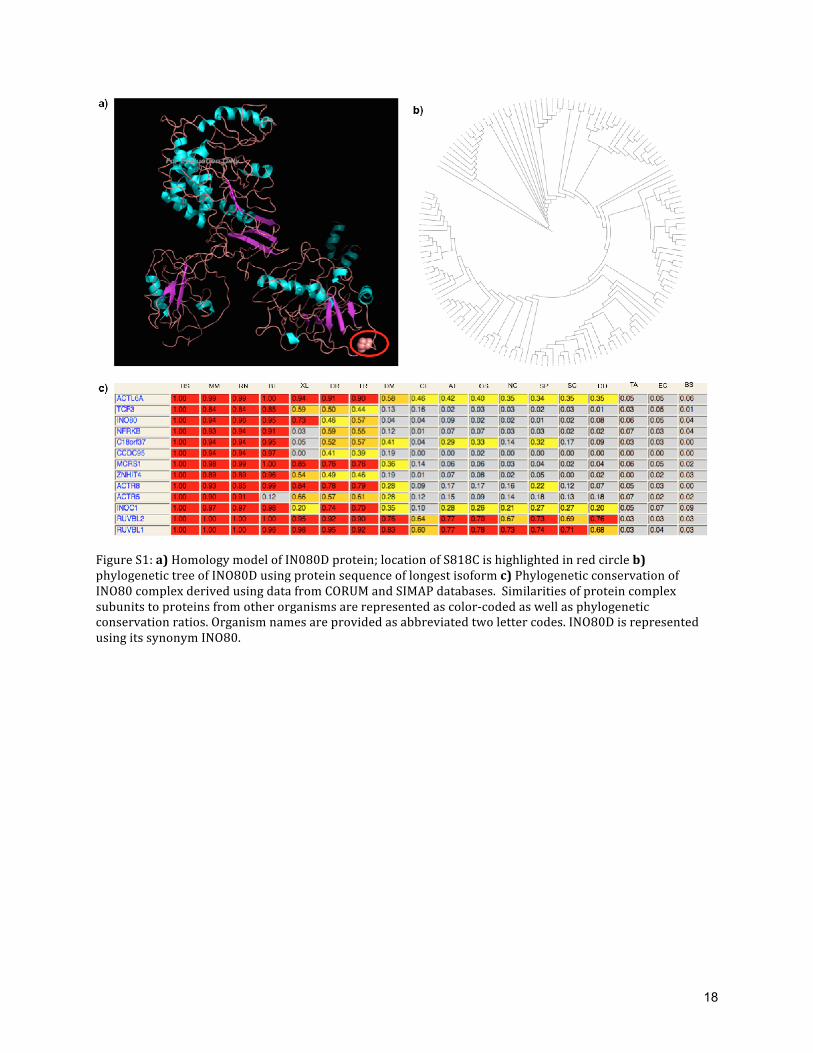
For structural characterization of INO80D, we performed a sequence-‐structure template
search using ModBase and ModWeb. No single structural homologs were found with sequence
similarity above the twilight zone (>30%)[8]. A remote homolog (16%) of INO80D sequence was
found in PDB identifier 2VZ9, A chain (structure of mammalian fatty acid synthase) [9]. This could
be a further pointer that IN080D may encode a novel fold or a fold similar to fatty acid synthase
(Figure: S1(a)). As 2VZ9 is not yet incorporated in the database of Structural Classification of
Proteins (SCOP) [10], hence an objective fold recognition approach was not possible to detect
additional structural relationships.
Interactome of INO80D:
To understand the functional context of INO80D from a network perspective, first-‐degree
interactome of INO80D was obtained from IntAct, a database of experimentally characterized
protein-‐protein interactions and visualized using Cytoscape (Figure 3(d)). 20 interactions
originated from spoke-‐expanded co-‐complexes were reported in IntAct. This indicates that INO80D
gene product is involved in multiple protein-‐protein interactions, a hallmark feature of proteins
with LCRs in addition to an important subunit of the INO80 complex.
Phylogenetic analysis of IN080D:
INO80D is a component of human INO80 complex, which has multiple functions including
chromatin remodeling. INO80D is a non-‐conserved subunit in human, yeast and drosophila. Exact
evolutionary lineage of INO80D is unclear. To understand functional role from homologs of
INO80D, we performed a detailed phylogenetic analysis. Sequence of the longest isoform was used
for homology search using PSI-‐BLAST, and a phylogenetic tree was constructed using Phylip v3.6
(http://evolution.genetics.washington.edu/phylip.html)and visualized using iTOL [11]. PSI-‐BLAST
search [12] (E-‐value: 0.05) was performed against non-‐redundant database (nr) with sequences
from GenBank CDS translations, PDB, SwissProt, PIR and PRF. From first iteration, 146 sequences
were obtained. 146 sequences were aligned using Clustal-‐Omega [13]. Bootstrapping of the output
from Clustal-‐Omega was performed using seqboot (1000 iterations). ‘protdist’ program was used to
derive the pairwise distance between 146 sequences. Phylogenetic trees were derived from

9
‘protdist’ output using ‘neighbor’ program (Neighbor-‐joining tree method). Consensus trees with
bootstrap values were derived from ‘neighbor’ output using ‘consense’ program. Nodes of INO80D
phylogenetic tree co-‐clustered with the query sequence (Q53TQ3-‐2) indicate that INO80D is
conserved exclusively in higher eukaryotes and the functions of the co-‐clustered proteins are
largely unknown (Figure S1(b)). This indicates that INO80D is a metazoan specific protein, and it
may have a recent evolutionary history.
MicroRNAs targeting INO80D:
MicroRNA (miRNA) molecules have established role in the regulating genes involved in
cardiovascular and aging phenotypes [14-‐17] via translational repression pathways [18]. To
understand whether any known miRNAs implicated in cardiovascular or aging phenotypes, we
compiled literature reports and miRNA expression data. To perform this analysis we retrieved all
miRNAs targeting the UTR region of INO80D. A list of putative miRNAs that could target INO80D
was identified by TargetScan search [19-‐21] using a library of regulatory targets of mammalian and
vertebrate miRNAs. A list of 27 miRNAs was retrieved, and clinical phenotypes associated with
these miRNAs were obtained from Human MiRNA & Disease Database (HMDD). We noted that
several miRNAs implicated in cardiovascular and aging phenotypes target INO80D (See
Supplementary Table S6) suggesting a regulatory perturbation of INO80D in the setting of various
disease phenotypes.
Disease or quantitative traits associated with INO80 complex subunits:
We compiled results from published genome-‐wide association studies to understand the
genetic role of different subunits of INO80 complex. Subunits of INO80 complex and their
phylogenetic similarity compiled from protein databases are provided (Figure S1(c)). Published
GWAS reports suggest that that subunits of INO80 complex were associated with phenotypes like
extreme obesity, heart rate and capecitabine sensitivity (Table S7).

10
Table S1. Application, results and inferences from various tools employed for functional analysis of Ser818Cys mutation on INO80D Application Tool Result Inference Prediction of conserved domains and motifs
SMART No conserved domains predicted, LCRs are predicted
INO80D encodes multiple LCRs and the mutation site is part of LCR-‐7
Prediction of conserved domains and motifs
Pfam Encodes 2 copies of Potential DNA-‐binding domain “zf-‐C3Hc3H”
Presence of zf-‐C3Hc3H indicates its functional role in mediating protein-‐DNA binding and related functional mechanisms
Prediction of unassigned region
PURE No distant domain association predicted
No known domains could be assigned to INO80D using PURE
Homology modeling (template search)
ModWeb/ModBase Template search identified remote homolog (16%) A chain of structure of mammalian fatty acid synthase, PDB ID: 2VZ9 with no SCOP classification
IN080D may encode a novel fold or a fold similar to fatty acid synthase
Homology modeling ModWeb/ModBase No single structural homologs were found with sequence similarity above the twilight zone (>30%).
Homology model derived using low-‐similarity (< 30%) templates are not ideal for structure analysis
Phylogenetic analysis PSI-‐BLAST, Phylip v3.6, iTOL
A phylogenetic tree was derived using protein homologs. (Figure S1)
Tree depicts that INO80D is conserved exclusively in higher eukaryotes and the functions of the co-‐clustered proteins are largely unknown
Protein-‐protein interaction analysis
IntAct First-‐degree interactome of INO80D was obtained
20 interactions originating from spoke expanded co-‐complexes were reported in IntAct indicating that INO80D is involved in multiple protein-‐protein

11
interactions, a feature of proteins with LCRs
Secondary structure and solvent accessibility
SABBLE Secondary structure and solvent accessibility predicted using the sequence of INO80D isoforms
Comparative analysis of wild-‐type and mutant sequence revealed changes in solvent accessibility due to mutation

12
Table S2: Homozygous regions identified using Runs of Homozygosity (ROH) routine in PLINK
!"#$ %&'($ %&')$ %*+#*$ ,-.$ /0$ &%&'$&1234#$
56$74-48$
!5.9-7$
:589*95-8$
!" #$%!&%'()*" #$&)+)!)," !(&-.(+-(%%" !()-)%)-,,," %!%,/,+" !!," %%" !'-!%&"
;! #8<)=>?@?! #8(=)@?(=($ (A;B@;(B=AC$ (A@B>;;BA>@$ ))>AD;=$ )@<$ (C$ A;BA<C$
?$ #8CA;)@);$ #8CA<<)(($ @(B>;=BC(@$ @)BA<CB@(;$ ()C(DC$ (;;$ E$ E$
(>$ #8())<@;(@$ #8)ACA)A>$ ;=B=A?B>>C$ ;CB>)@B<(=$ ()C)D<($ (;A$ ($ =A@$
F5*+G$ $ $ $ $ $ $ A($ ?(BC>A$
13
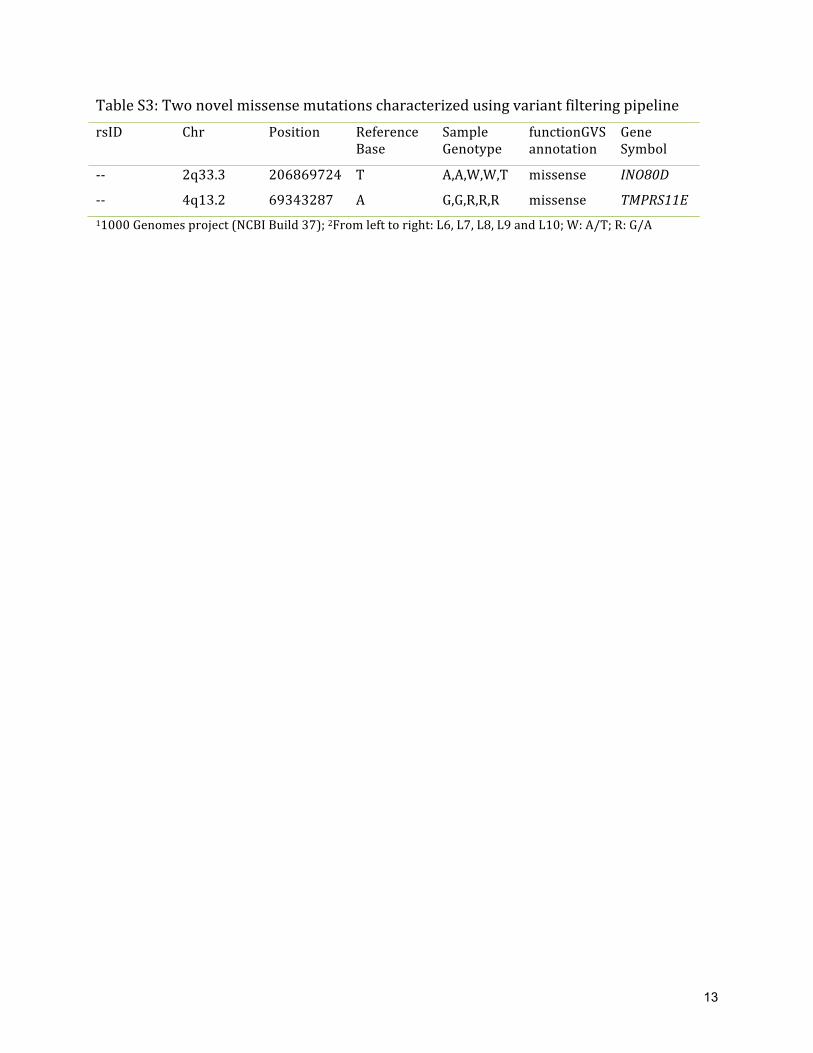
Table S3: Two novel missense mutations characterized using variant filtering pipeline rsID Chr Position Reference
Base Sample Genotype
functionGVS annotation
Gene Symbol
-‐-‐ 2q33.3 206869724 T A,A,W,W,T missense INO80D
-‐-‐ 4q13.2 69343287 A G,G,R,R,R missense TMPRS11E 11000 Genomes project (NCBI Build 37); 2From left to right: L6, L7, L8, L9 and L10; W: A/T; R: G/A

14
Table S4: Forward and reverse primers used for amplifying genomic regions around INO80D and TMPRSS11E primers Primer ID Sequence Theoretical
Tm (⁰C) Tm (⁰C)
GC Content(%)
INO80D-‐FP CACGCCTCCAAGTGGCACCTC 60.1 63.1 66.6
INO80D-‐RP ACCCACCTACACCCCTGGCA 57.8 63.5 65
TMPRSS11E-‐FP ACCTGGTCAGAACCCTGAGCCTT 58.7 62.3 56.5
TMPRSS11E-‐RP TGAGTTCCTCTTCCGATGCTCACC 59 60.5 54.1
FP=Forward Primer; RP=Reverse Primer

15
Table S5: Syndromes associated with mutations in genes involved in chromatin remodeling
Syndrome Clinical features Genes Chromatin remodeling complex or gene
References
ATRX-‐syndrome (α-‐thalassemia X-‐linked mental retardation) and α-‐thalassemia myelodysplasia syndrome
Distinctive craniofacial features, genital anomalies, severe developmental delays, hypotonia, intellectual disability, and mild-‐to-‐moderate anemia secondary to alpha-‐thalassemia
ATRX SWI/SNF family
[22]
CHARGE syndrome
Coloboma, heart defect, atresia choanae, retarded growth and development, genital hypoplasia, ear anomalies/deafness
CHD7 CHD family
[23]
Williams-‐Beuren syndrome
Diverse phenotypes including supravalvular aortic stenosis, peripheral pulmonary arterial stenosis, dysmorphism, mental and growth deficiency, aberrant vitamin D metabolism, and hypercalcemia
Deletion of 28 genes in 7q11.23 including a chromatin remodeling complex associated gene
BAZ1B [24-‐26]
COFS (cerebro-‐oculo-‐facio-‐skeletal syndrome) and Cockayne syndrome B (CSB)
UV sensitivity, cataracts, growth failure, and neurological degeneration
ERCC6 and ERCC8
Mutations in chromatin remodelers or proteins interacting with components of remodeling complexes
[27, 28]
Werner's Syndrome
Cataract, premature arteriosclerosis, subcutaneous calcification, diabetes mellitus, wizened and prematurely aged facies, scleroderma-‐like skin changes, especially in the extremities. Chromosomal deletion or instabilities were also reported.
WRN WRN is a member of the RecQ Helicase family
http://www. pathology. washington.edu/ research/werner/ database/

16
Table S6: Clinical phenotypes implicated to microRNAs targeting INO80D
miRNA* PCT Score^ Observed phenotypes Related phenotypes has-‐miR-‐30abcdef/30abe-‐5p > 0.99 Vascular calcification [29]
Periodontitis [30] Periodontal disease [31]
Left ventricular hypertrophy [32]
hsa-‐miR-‐384-‐5p >0.99 _ Myocardial ischemia [33] hsa-‐miR-‐125a-‐5p/125b-‐5p 0.99 _ Heart failure [34]
Hypertrophic cardiomyopathy [35] Myocardial ischemia [36]
hsa-‐miR-‐351 0.99 NR NR hsa-‐miR-‐670 0.99 NR NR hsa-‐miiR-‐4319 0.99 NR NR has-‐miR-‐181abcd 0.99 Coronary artery disease
[37]; Periodontitis [30] Heart failure [34] Hypertrophic cardiomyopathy [38]
hsa-‐miR-‐4262 0.99 NR NR hsa-‐miR-‐128/128ab 0.94 _ _ has-‐miR-‐101/101ab 0.93 _ _ hsa-‐miR-‐27abc/27a-‐p 0.93 Vascular disease [39] Heart failure [34]
Hypertrophic cardiomyopathy [35] Ischemic heart disease [39] Cardiomyopathies [40]
hsa-‐miR-‐199ab-‐5p 0.92 _ Hypertrophic cardiomyopathy [38] Cardiomegaly, expression cardiac myocytes [41] Heart failure [42] Aging [43]
hsa-‐let-‐7 0.91 Cataract [44] Heart failure [34] Cardiac hypertrophy [45]
hsa-‐miR-‐98 0.91 _ Heart failure [34] Cardiac hypertrophy [45] Myocardial infarction [46]
hsa-‐miR-‐4458 0.91 NR NR hsa-‐miR-‐4500 0.91 NR NR hsa-‐miR-‐124/124ab/506 0.89 _ _ hsa-‐miR-‐103a 0.87 Hypertension [47] _ hsa-‐miR-‐107/107ab 0.87 _ Heart failure [34, 48] hsa-‐miR-‐19ab 0.83 Vascular disease [49] Age-‐related heart failure [50] hsa-‐miR-‐17/17-‐5p 0.83 Vascular disease [49]
Periodontitis [51] Heart failure [34]
hsa-‐20ab/20b-‐5p 0.83 Vascular disease [49] Hypertension [52]
_
hsa-‐miR-‐93 0.83 _ _ hsa-‐miR-‐106ab 0.83 Periodontal disease [31] Heart failure [34]
Myocardial infarction [53] hsa-‐miR-‐427 0.83 NR NR hsa-‐miR-‐518a-‐3p 0.83 _ _ hsa-‐miR-‐519d 0.83 _ _
* Preferentially conserved targeting (PCT) score is a Bayesian estimate of the probability that an miRNA binding site in the upstream of INO80D is conserved due to selective maintenance of miRNA targeting rather than by chance or any other reason not pertinent to miRNA targeting, allowing for uncertainty in the S/B ratio * miRNAs targeting INO80D identified using TargetScan 6.2 (human); high confidence miRNAs with PCT <= 0.8 considered for phenotype mapping NR: No phenotype data reported in Human MiRNA & Disease Database -‐: No reported phenotype implications in Human MiRNA & Disease Database

17
Table S7: Published genomics associations of INO80 complex subunits from genome-‐wide
association studies
Gene * Synonyms^ SNP Disease / Trait P-‐value OR/β Genes1; Context
Ref
ACTL6A BAF53, BAF53A,
INO80K
rs7612445 Heart rate 2.00E-‐14 0.36 GNB4-‐ACTL6A; Intergenic
[54]
ACTR5 ARP5 -‐ -‐ -‐ -‐ -‐ -‐
ACTR8 INO80N -‐ -‐ -‐ -‐ -‐ -‐
INO80C C18orf37 rs7603514 Capecitabine sensitivity
3.00E-‐06 NR INO80C-‐MIR3975; Intergenic
[55]
INO80E CCDC95 -‐ -‐ -‐ -‐ -‐ -‐
INO80D INO80 rs7603514 Obesity (extreme) 8.00E-‐06 1.36 NRP2-‐INO80D; Intergenic
[56]
INOC1 INOC1, KIAA1259 -‐ -‐ -‐ -‐ -‐ -‐
MCRS1 INO80Q, MSP58 -‐ -‐ -‐ -‐ -‐ -‐
NFRKB INO80G -‐ -‐ -‐ -‐ -‐ -‐
RUVBL1 INO80H, NMP238, TIP49, TIP49A
-‐ -‐ -‐ -‐ -‐ -‐
RUVBL2 INO80J, TIP48, TIP49B, CGI-‐46
-‐ -‐ -‐ -‐ -‐ -‐
TCF3 E2A, ITF1 -‐ -‐ -‐ -‐ -‐ -‐
ZNHIT4 HMGA1L4, PAPA1, ZNHIT4
-‐ -‐ -‐ -‐ -‐ -‐
* Complex subunits, gene names were obtained from CORUM database (http://mips.helmholtz-‐muenchen.de/genre/proj/corum/complexdetails.html?id=302) ^ Synonyms were compiled from HUGO Gene Nomenclature Committee (HGNC) portal (http://www.genenames.org/) and GeneCards (http://www.genecards.org/) 1 Reported/mapped genes from GWAS Catalog (http://www.genome.gov/gwastudies/)
18
Figure S1: a) Homology model of IN080D protein; location of S818C is highlighted in red circle b) phylogenetic tree of INO80D using protein sequence of longest isoform c) Phylogenetic conservation of INO80 complex derived using data from CORUM and SIMAP databases. Similarities of protein complex subunits to proteins from other organisms are represented as color-‐coded as well as phylogenetic conservation ratios. Organism names are provided as abbreviated two letter codes. INO80D is represented using its synonym INO80.

19
References:
1. O'Rawe J, Jiang T, Sun G, Wu Y, Wang W, Hu J, et al. Low concordance of multiple variant-‐calling pipelines: practical implications for exome and genome sequencing. Genome Med. 2013; 5(3):28.
2. Caliskan M, Chong JX, Uricchio L, Anderson R, Chen P, Sougnez C, et al. Exome sequencing reveals a novel mutation for autosomal recessive non-‐syndromic mental retardation in the TECR gene on chromosome 19p13. Hum Mol Genet. 2011; 20(7):1285-‐1289.
3. Adamczak R, Porollo A, Meller J. Combining prediction of secondary structure and solvent accessibility in proteins. Proteins. 2005; 59(3):467-‐475.
4. Letunic I, Doerks T, Bork P. SMART 6: recent updates and new developments. Nucleic Acids Res. 2009;37(Database issue):D229-‐232.
5. Wootton JC. Non-‐globular domains in protein sequences: automated segmentation using complexity measures. Comput Chem. 1994; 18(3):269-‐285.
6. Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Aca Sci. U S A 1998; 95(11):5857-‐5864.
7. Coletta A, Pinney JW, Solis DY, Marsh J, Pettifer SR, Attwood TK. Low-‐complexity regions within protein sequences have position-‐dependent roles. BMC Syst Biol. 2010; 4:43.
8. May AC, Johnson MS, Rufino SD, Wako H, Zhu ZY, Sowdhamini R, et al. The recognition of protein structure and function from sequence: adding value to genome data. Philos Trans R Soc Lond B Biol Sci. 1994; 344(1310):373-‐381.
9. Maier T, Leibundgut M, Ban N. The crystal structure of a mammalian fatty acid synthase. Science 2008, 321(5894);1315-‐1322.
10. Andreeva A, Howorth D, Chandonia JM, Brenner SE, Hubbard TJ, Chothia C, et al. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res. 2008; 36(Database issue):D419-‐425.
11. Letunic I, Bork P: Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 2007; 23(1):127-‐128.
12. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-‐BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25(17):3389-‐3402.
13. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, et al. Fast, scalable generation of high-‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011; 7:539.
14. Lu M, Zhang Q, Deng M, Miao J, Guo Y, Gao W, et al. An analysis of human microRNA and disease associations. PLoS One 2008; 3(10):e3420.
15. Small EM, Frost RJ, Olson EN. MicroRNAs add a new dimension to cardiovascular disease. Circulation 2010; 121(8):1022-‐1032.
16. Smith-‐Vikos T, Slack FJ. MicroRNAs and their roles in aging. J Cell Sci. 2012; 125(Pt 1):7-‐17.
17. Stather PW, Sylvius N, Wild JB, Choke E, Sayers RD, Bown MJ. Differential MicroRNA Expression Profiles in Peripheral Arterial Disease. Circ Cardiovasc Genet. 2013.

20
18. He L, Hannon GJ. MicroRNAs. small RNAs with a big role in gene regulation. Nat Rev Genet. 2004; 5(7):522-‐531.
19. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005; 120(1):15-‐20.
20. Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009; 19(1):92-‐105.
21. Grimson A, Farh KK, Johnston WK, Garrett-‐Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol cell. 2007; 27(1):91-‐105.
22. Xue Y, Gibbons R, Yan Z, Yang D, McDowell TL, Sechi S, et al. The ATRX syndrome protein forms a chromatin-‐remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci. U S A. 2003; 100(19):10635-‐10640.
23. Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004; 36(9):955-‐957.
24. Lu X, Meng X, Morris CA, Keating MT. A novel human gene, WSTF, is deleted in Williams syndrome. Genomics. 1998; 54(2):241-‐249.
25. Meng X, Lu X, Li Z, Green ED, Massa H, Trask BJ, et al. Complete physical map of the common deletion region in Williams syndrome and identification and characterization of three novel genes. Hum Genet. 1998; 103(5):590-‐599.
26. Meng X, Lu X, Morris CA, Keating MT. A novel human gene FKBP6 is deleted in Williams syndrome. Genomics. 1998; 52(2):130-‐137.
27. Woudstra EC, Gilbert C, Fellows J, Jansen L, Brouwer J, Erdjument-‐Bromage H, et al. A Rad26-‐Def1 complex coordinates repair and RNA pol II proteolysis in response to DNA damage. Nature. 2002; 415(6874):929-‐933.
28. Citterio E, Van Den Boom V, Schnitzler G, Kanaar R, Bonte E, Kingston RE, Hoeijmakers JH, et al. ATP-‐dependent chromatin remodeling by the Cockayne syndrome B DNA repair-‐transcription-‐coupling factor. Mol Cell Biol. 2000; 20(20):7643-‐7653.
29. Balderman JA, Lee HY, Mahoney CE, Handy DE, White K, Annis S, et al. Bone morphogenetic protein-‐2 decreases microRNA-‐30b and microRNA-‐30c to promote vascular smooth muscle cell calcification. J Am Heart Assoc. 2012; 1(6):e003905.
30. Lee YH, Na HS, Jeong SY, Jeong SH, Park HR, Chung J. Comparison of inflammatory microRNA expression in healthy and periodontitis tissues. Biocell. 2011; 35(2):43-‐49.
31. Perri R, Nares S, Zhang S, Barros SP, Offenbacher S. MicroRNA modulation in obesity and periodontitis. J Dent Res. 2012; 91(1):33-‐38.
32. Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, et al. miR-‐133 and miR-‐30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. 2009; 104(2):170-‐178, 176p following 178.
33. Bao Y, Lin C, Ren J, Liu J. MicroRNA-‐384-‐5p regulates ischemia-‐induced cardioprotection by targeting phosphatidylinositol-‐4,5-‐bisphosphate 3-‐kinase, catalytic subunit delta (PI3K p110delta). Apoptosis. 2013; 18(3):260-‐270.

21
34. Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, et al: MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007; 116(3):258-‐267.
35. Busk PK, Cirera S. MicroRNA profiling in early hypertrophic growth of the left ventricle in rats. Biochem Biophys Res Commun. 2010; 396(4):989-‐993.
36. Ren D, Wang X, Ha T, Liu L, Kalbfleisch J, Gao X, et al. SR-‐A deficiency reduces myocardial ischemia/reperfusion injury; involvement of increased microRNA-‐125b expression in macrophages. Biochim Biophys Acta. 2013; 1832(2):336-‐346.
37. Hulsmans M, Sinnaeve P, Van der Schueren B, Mathieu C, Janssens S, Holvoet P. Decreased miR-‐181a expression in monocytes of obese patients is associated with the occurrence of metabolic syndrome and coronary artery disease. J Clin Endocrinol Metab. 2012; 97(7):E1213-‐1218.
38. van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, et al. A signature pattern of stress-‐responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. 2006; 103(48):18255-‐18260.
39. Bang C, Fiedler J, Thum T. Cardiovascular importance of the microRNA-‐23/27/24 family. Microcirculation. 2012; 19(3):208-‐214.
40. Yeh CH, Chen TP, Wang YC, Lin YM, Fang SW. MicroRNA-‐27a regulates cardiomyocytic apoptosis during cardioplegia-‐induced cardiac arrest by targeting interleukin 10-‐related pathways. Shock. 2012; 38(6):607-‐614.
41. Song XW, Li Q, Lin L, Wang XC, Li DF, Wang GK, et al. MicroRNAs are dynamically regulated in hypertrophic hearts, and miR-‐199a is essential for the maintenance of cell size in cardiomyocytes. J Cell Physiol. 2010; 225(2):437-‐443.
42. Haghikia A, Missol-‐Kolka E, Tsikas D, Venturini L, Brundiers S, Castoldi M, et al. Signal transducer and activator of transcription 3-‐mediated regulation of miR-‐199a-‐5p links cardiomyocyte and endothelial cell function in the heart: a key role for ubiquitin-‐conjugating enzymes. Eur Heart J. 2011; 32(10):1287-‐1297.
43. Ukai T, Sato M, Akutsu H, Umezawa A, Mochida J. MicroRNA-‐199a-‐3p, microRNA-‐193b, and microRNA-‐320c are correlated to aging and regulate human cartilage metabolism. J Orthop Res. 2012; 30(12):1915-‐1922.
44. Peng CH, Liu JH, Woung LC, Lin TJ, Chiou SH, Tseng PC, et al. MicroRNAs and cataracts: correlation among let-‐7 expression, age and the severity of lens opacity. Br J Ophthalmol. 2012; 96(5):747-‐751.
45. Yang Y, Ago T, Zhai P, Abdellatif M, Sadoshima J. Thioredoxin 1 negatively regulates angiotensin II-‐induced cardiac hypertrophy through upregulation of miR-‐98/let-‐7. Circ Res. 2011; 108(3):305-‐313.
46. Zhu W, Yang L, Shan H, Zhang Y, Zhou R, Su Z, et al. MicroRNA expression analysis: clinical advantage of propranolol reveals key microRNAs in myocardial infarction. PLoS One. 2011; 6(2):e14736.
47. Wu WH, Hu CP, Chen XP, Zhang WF, Li XW, Xiong XM, et al. MicroRNA-‐130a mediates proliferation of vascular smooth muscle cells in hypertension. Am J Hypertens. 2011; 24(10):1087-‐1093.
48. Voellenkle C, van Rooij J, Cappuzzello C, Greco S, Arcelli D, Di Vito L, et al. MicroRNA signatures in peripheral blood mononuclear cells of chronic heart failure patients. Physiol Genomics. 2010; 42(3):420-‐426.

22
49. Parmacek MS: MicroRNA-‐modulated targeting of vascular smooth muscle cells. J Clin Invest. 2009; 119(9):2526-‐2528.
50. van Almen GC, Verhesen W, van Leeuwen RE, van de Vrie M, Eurlings C, Schellings MW, et al. MicroRNA-‐18 and microRNA-‐19 regulate CTGF and TSP-‐1 expression in age-‐related heart failure. Aging Cell. 2011; 10(5):769-‐779.
51. Liu Y, Liu W, Hu C, Xue Z, Wang G, Ding B, et al. MiR-‐17 modulates osteogenic differentiation through a coherent feed-‐forward loop in mesenchymal stem cells isolated from periodontal ligaments of patients with periodontitis. Stem Cells. 2011; 29(11):1804-‐1816.
52. Brock M, Samillan VJ, Trenkmann M, Schwarzwald C, Ulrich S, Gay RE, et al. AntagomiR directed against miR-‐20a restores functional BMPR2 signalling and prevents vascular remodelling in hypoxia-‐induced pulmonary hypertension. Eur Heart J. 2012.
53. Liu Z, Yang D, Xie P, Ren G, Sun G, Zeng X, et al. MiR-‐106b and MiR-‐15b modulate apoptosis and angiogenesis in myocardial infarction. Cell Physiol Biochem. 2012; 29(5-‐6):851-‐862.
54. den Hoed M, Eijgelsheim M, Esko T, Brundel BJ, Peal DS, Evans DM, et al. Identification of heart rate-‐associated loci and their effects on cardiac conduction and rhythm disorders. Nat Genet. 2013; 45(6):621-‐631.
55. O'Donnell PH, Stark AL, Gamazon ER, Wheeler HE, McIlwee BE, Gorsic L, et al. Identification of novel germline polymorphisms governing capecitabine sensitivity. Cancer. 2012; 118(16):4063-‐4073.
56. Cotsapas C, Speliotes EK, Hatoum IJ, Greenawalt DM, Dobrin R, Lum PY, et al. Common body mass index-‐associated variants confer risk of extreme obesity. Hum Mol Genet. 2009; 18(18):3502-‐3507.