welcome to the ucla – igert 2005 summer bioinformatics workshop igert – sponsored bioinformatics...
TRANSCRIPT

Welcome to the UCLA – IGERT 2005 Summer Bioinformatics
Workshop
IGERT – Sponsored Bioinformatics Workshop SeriesMichael Janis and Max Kopelevich, Ph.D.
Dept. of Chemistry & Biochemistry, UCLA

Who this workshop is for
• Biologists: –Integrating bioinformatics methods into their existing projects
• Bioinformatics students: –Preparing for and augmenting existing bioinformatics coursework
• Computational researchers: –Identifying and understanding common bioinformatics language classes and data structures

Why this workshop?
• Current graduate curriculum: – Assumes substantial knowledge base upon entry into
bioinformatics program
• Researchers: – No general overview of computational research
environments • some aimed at tools (suites), others deep in theory base

Why learn bioinformatics?
• The 1 gene, 1 protein research model ignores the deluge of inferential biological data at our disposal
• This information must be combined for any thorough study of biological phenomena
• Assembly of qualitative (sequence, etc.) and quantitative (measured, inferential) data is beyond the capacity of a human to interpret in a systematic fashion
• Computational methods must be addressed both to interpret and to share research data in a meaningful way – Yet Another Tool in a biologist’s arsenal

What is Bioinformatics?
• A tool in a molecular biologist’s arsenal such as PCR, gels, or microarrays; or:
• A support machine for handling and storing large data sets; or:
• A discipline in its’ own right, enabled by a critical mass of biological data and mirroring the evolution of a number of scientific sub-disciplines

An argument for learning: as a tool for molecular biologists?
• 1 gene, 1 protein becomes an interactive series of pathways and protein interactions
• A pathway allows novel interaction discovery and ontological groupings of data
• Moving from identification of a gene that has biological function to elucidation of a pathway from a given biological function (just for example)

An argument for learning: as support for data management?
• Completion of the human genome working draft in June 2000 resulted in an absolute requirement for a logical, hierarchical way to store, modify, and retrieve data
• Cross-referencing information is necessary to incorporate disparate and sometimes conflicting information
• Combination of data and necessary algorithm development to find signal among the noise of the genome

An argument for learning: as an entirely new discipline?
• Evolutionary biology through homology – (COG for example)
• Gene expression and structure prediction – (alternative splicing for example)
• Protein and ncRNA modeling• Genome mapping
– (statistical genetics and disease correlations)
• Pathway prediction• Towards an end: Systems Biology

What has bioinformatics allowed us to do?
• Evolutionary Biology – Clusters of Orthologous Genes (COG database)
http://www.ncbi.nlm.nih.gov/COG/
• Structure and alignments – PFAM/RFAM http://www.sanger.ac.uk/Software/Rfam/
• Expression inferences – clustering to find interactions (for example through SMD:
http://genome-www5.stanford.edu/
• Genome mapping – (for example the UCSC Genome Browser: http://genome.ucsc.edu/cgi-
bin/hgGateway

What do we need to create our own genomic inferences?
• That’s the problem… The data is encoded…
• AGCTAGCGACTAGCGATTATA… – Now predict what this sequence codes for, how it is
transcriptionally regulated, in what pathway does it function …
• The human genome is 3 billion bases. – Clearly we need a new set of tools
• Problem: no human being could ever look at all this data, so the patterns must be discovered computationally

The magic wand analogy
• The sum of all human knowledge can be encrypted and stored by marking a single scratch on a metal rod. It all depends upon RESOLUTION and LANGUAGE.
• We can apply this analogy to our analytical process:– High RESOLUTION is required to decode the genome– A robust LANGUAGE is required to achieve that RESOLUTION– Computational biology seeks to describe that LANGUAGE

How do discover meaningful biological patterns?
• Need to be able to pick “needles from haystacks”• Need to be able to assemble needles• Need to orient and classify needles• And so on…• This requires a strong, flexible environment• The Shell, PERL, R, PostGreSQL/MySQL – integration,
data exploration• Statistical development environments – making
inferences from the data• Databases and resources – combining the data

How our workshop will help you (and how it won’t?)
• We cover common, portable, powerful resources – but require suitable activation energy!
• Integration for a project model is key
• Thus, easily understood but limited GUI based suites are not covered
• Likewise theory of algorithm development is covered in other courses and will be ignored (for the time being!)

How this workshop is structured
• Two lectures a week for six weeks (fast!)
• Lots of reading material (need to keep pace)
• Hands – on practical experience with biological data and programming tasks
• Peer interaction

What you’ll need for this workshop
• Access to either the online version of the text through O’Reilly Media (linked on the website, $20 for two months) or through purchase of the print bound version (approx $130, we’ll put in an order during the second week)
• Access to a computer. Any computer will do.
• Motivation to learn, and to ask questions
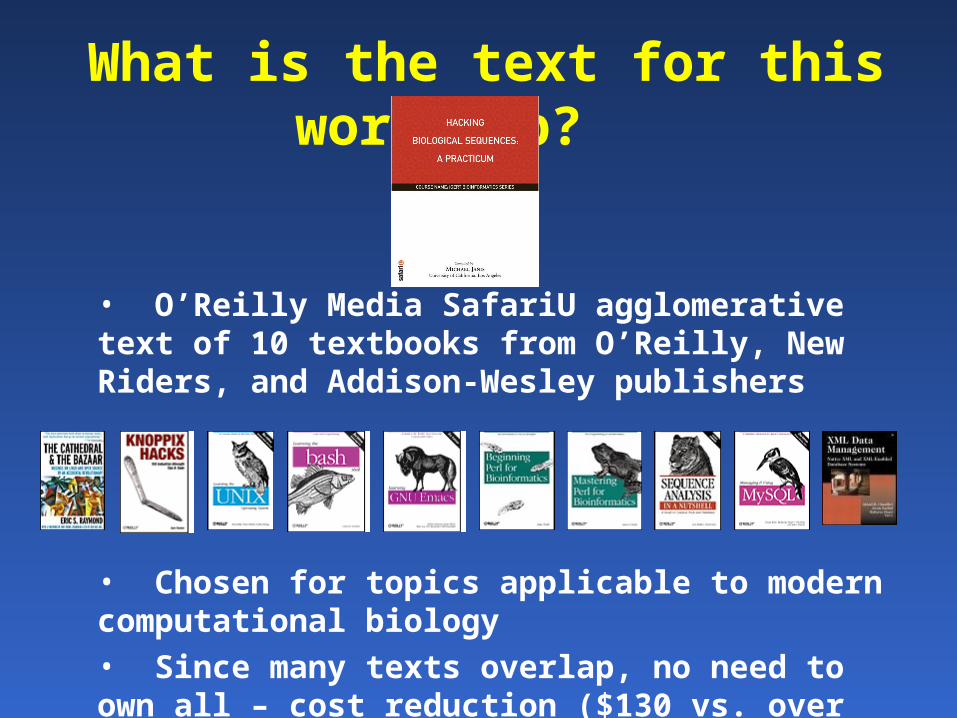
What is the text for this workshop?
• O’Reilly Media SafariU agglomerative text of 10 textbooks from O’Reilly, New Riders, and Addison-Wesley publishers
• Chosen for topics applicable to modern computational biology
• Since many texts overlap, no need to own all – cost reduction ($130 vs. over $500)

Class information
• Office: 4054 Young Hall
• http://www.bol.ucla.edu/~mjanis/biohackers2005.html• http://lists.ucla.edu/cgibin/mailman/listinfo/bioinformatics
• IRC: #uclaBioinformatics, freenode server (mako)
• T / R 2-4 pm 1054 Young Hall, Aug 11- Sept. 16 2005

Class information
BRIEF SUBJECT MATTER OUTLINE (SUBJECT TO CHANGE!)
08/11 Unix: Introduction to Unix; Bioknoppix08/16 Unix basics, editors, remote logins08/18 Shell programming08/23 Regular expressions; biological data formats08/25 Perl: data and control structures08/30 Modular programming09/01 Object oriented perl09/06 Biological classes; Bioperl09/08 NCBI tools; BLAST09/13 Database design; perl DBI09/15 Net programming; XML; CGI09/16 R; Bioconductor

We used to call it “biohackers”…
• What is a hacker?
• One with technical adpetness who delights in solving problems and overcoming limits…
• Hackers can be found in all walks of life – music, art, science
• Hackers create things – the internet, Unix, NCBI, the Human Genome project
• Not what the media would call a “hacker”…

The hacker attitude*
• Hackers believe the world is full of fascinating problems waiting to be solved. • Nobody should ever have to solve the same problem twice. • Boredom and drudgery are evil.• Freedom is good• Attitude is no substitute for competence
* From “The Cathedral and the Bazaar”

Why would I want to become a “hacker” or a “biohacker” or
whatever?
• Spend less time brutally munging your data. Spend more time gaining biological insight. Publish faster.
• Begin to assemble complex and disparate data in ways not achievable by humans. Think systematically about your data. Think systemically about your research.
• Smugly travel easily through electronic medium. Automate your life
• Be cool. Get chicks! (or whomever you want)…

It worked for “neo”…

In reality, it worked for someone else too …

How does one become a hacker?
• Learn to program. The more languages, the better. (Don’t think in terms of languages… Think in terms of PROGRAMMING)
• Get an extensible, modifiable OS and learn to use it
• Learn how to access and use information
• Practice, practice, practice. This workshop can only show you the door. You have to walk through it.

So what is biohacking?
• Inferential analysis of disparate biological data
• Discovering and creating new ways of looking at biological phenomenon
• Creation of novel tools
• Decoding the nature of life (BHAG)

So show me the tool to find the gene… I’ll be in the lab…
• What if your research looks at biology from a novel angle? What if there are no tools? Where to start?
• We need a modular framework. Something that will scale nicely so that we can incorporate more data later. Some set of tools that integrate well, each doing one job and doing it well. We need COMBINATORIC TOOLSETS!

A tale of two philosophies
• Windows works extremely well at solving one problem at a time, if it falls within defined parameters… It does not scale well.
• This monolithic approach is good for small scale biology but not for complex biological analysis

We need a construct where we can call upon modular tools to
assemble our own analysis…
I need bioinformatics tools… Lots of bioinformatics tools…

The construct – UNIX (the other philosophy)
• UNIX is an operating system that provides powerful modular utilities that function together to create computer environments that can solve a wide variety of tasks. • Unix Operating System
–created 1960s–provides a multi-user, multitasking system
•It’s a server by design•Networks extremely well due to ports – X11 is an example
–command line based •Bioinformatics relies heavily upon UNIX – and a fundamentally different philosophy:•modular by its’ very nature, UNIX is scaleable, robust, and … FREE!
• But more than anything else, Unix has THE SHELL

The Unix Shell
• The shell is the unix user environment• The shell is started automatically when you log in• Everything is controlled from a shell• The shell interprets commands
– It’s also a programming language itself!
• http://www.looking-glass.org/shell.html– http://www.strath.ac.uk/CC/Courses/IntroToUnix/node46.html
• More than one shell can be open at a time

Commands to the system• Specific commands used to perform functions in
the shell– Ex: changing directories, copying files, mv files,
sorting, cutting, pasting, etc…– Many of these can be done in XL, but…
• What if you need a subsequent command? ChiSquare analysis for example?
• What if you need to do this to 10 files? 100? 10000?
• Each command is itself a program and take command line arguments– The Shell is the “glue” that allows us to
combinatorically daisy-chain our tools together to do a job
• The Shell allows us to batch these commands into a pipeline or framework for analysis

The Shell controls input / output
STDERRCommandOr
Program
STDIN
STDOUT

Input / Output II
STDERRCommandOr
Program
STDIN
STDOUT-Keyboard-File-Previous STDOUT
-Terminal/ monitor-File-Next STDIN

Input / Output III
Command 1
Command 2Pipe
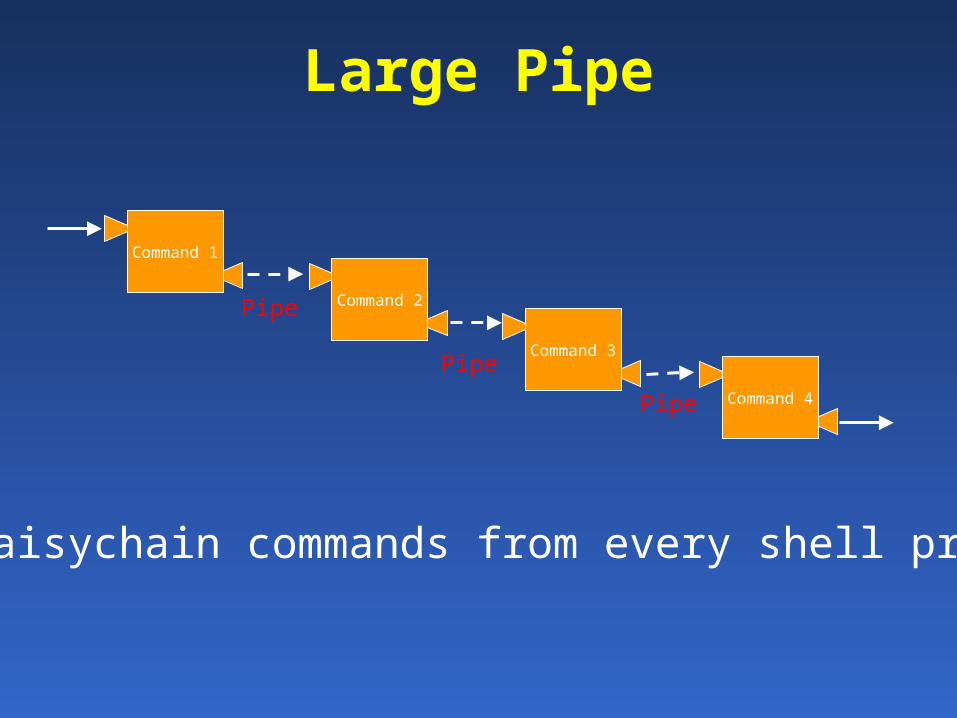
Large Pipe
Command 3
Command 4Pipe
Command 1
Command 2Pipe
Pipe
Can daisychain commands from every shell program

UNIX then… Linux? BSD? OS X? (over 360 distributions according to linux.org)

How can I get *nix to use?
• Linux is open source – Available for a variety of platforms
• BSD is likewise open source– Forms the basis of Mac OS X, and many MSWin utilities
• There are live distributions as well – Example: Bioknoppix - http://bioknoppix.hpcf.upr.edu/
• Bioknoppix is a customized distribution of Knoppix Linux Live CD. With this distribution you just boot from the CD and you have a fully functional Linux OS distribution with open source bioinformatics (sic) applications.
• Beside using some RAM, Bioknoppix doesn't touch the host computer.

What about administrating the system? I heard it’s hard!

Package managers
• Most modern distributions have a package manager– Handles installation– Handles dependencies– Handles conflicts– Packages are written in such a way as to ensure that they do not
interfere with other parts of the OS install
• With BioKnoppix, you won’t really need one• With Debian, there is aptitude, and synaptic• There are a wealth of bioinformatics tools packaged for
use with package managers• We’ll be exploring these as we go along…

For our Apple brethren…

There is fink
• Mac OS X forms a natural basis for a end user bioinformatics analysis station
• It’s UNIX underneath!• The GUI is advanced• It’s easy to administrate• http://fink.sourceforge.net/• Finks installs into its own protected directory /sw• Easy to remove if things go south…

What is fink?
• Fink is a distribution of Unix Open Source software for Mac OS X and Darwin. – It brings a wide range of free command-line and
graphical software developed for Linux and similar operating systems to your Mac.
• Installation is accomplished via a tarball. – After that, it’s simple command line- interaction
(based upon the debian model, which I’ll show in class)
• http://fink.sourceforge.net/doc/users-guide/install.php?phpLang=en

For our MS brethren…
Welcome to Earth
A subsidiary of Microsoft

There is cygwin
• Shell utilities written as windows executables• Creates its own protected / filesystem• Problems with networking• No real package manager beyond the shell utils• Can be problematic to install packages with deep
*nix library dependencies• Created / maintained by RedHat developers• http://www.cygwin.com/

But mainly have remote access to a real *nix machine

Configure Putty

SSH Option

We’ll be using…
• BioKnoppix Beta 0.2.1 (but the Knoppix part is 3.3…)• This distribution is included in your folder• Debian stable 3.1 (“Sarge”) install – which I’ll do in class
– will be our main server. We’ll call it “subi” (subi.chem.ucla.edu) for SUmmer BIonformatics.
• The text has details about the use of knoppix
• Let’s take a look …

The shell, files, directories, commands, and editors

File System
visualize the Unix file system as an upside down tree. At the very top of the tree is the root directory, named "/".

Common filesystem locations/ Root directory of the file system.
/bin/User utilities fundamental to both single-
user and multi-user environments.
/boot/Programs and configuration files used
during operating system bootstrap.
/boot/defaults/Default bootstrapping configuration files;
see loader.conf(5).
/dev/ Device nodes; see intro(4).
/etc/ System configuration files and scripts.
/etc/defaults/Default system configuration files; see
rc(8).
/etc/mail/Configuration files for mail transport
agents such as sendmail(8).
/etc/namedb/ named configuration files; see named(8).
/etc/periodic/Scripts that are run daily, weekly, and
monthly, via cron(8); see periodic(8).
/etc/ppp/ ppp configuration files; see ppp(8).
/mnt/Empty directory commonly used by system
administrators as a temporary mount point.
/proc/Process file system; see procfs(5),
mount_procfs(8).
/rescue/Statically linked programs for emergency
recovery; see rescue(8).
/root/ Home directory for the root account.
/sbin/
System programs and administration utilities fundamental to both single-user and multi-user environments.
/stand/Programs used in a standalone
environment.
/tmp/
Temporary files, usually a mfs(8) memory-based file system (the contents of /tmp are usually NOT preserved across a system reboot).
/usr/The majority of user utilities and applications.
/usr/bin/Common utilities, programming tools, and applications.
/usr/include/ Standard C include files.
/usr/lib/ Archive libraries.
/usr/libdata/ Miscellaneous utility data files.
/usr/libexec/System daemons & system utilities (executed by other programs).
/usr/local/
Local executables, libraries, etc. Also used as the default destination for the FreeBSD ports framework. Within /usr/local, the general layout sketched out by hier(7) for /usr should be used. Exceptions are the man directory, which is directly under /usr/local rather than under /usr/local/share, and the ports documentation is in share/doc/port.
/usr/obj/Architecture-specific target tree produced by building the /usr/src tree.
/usr/ports The FreeBSD Ports Collection (optional).
/usr/sbin/System daemons & system utilities (executed by users).
/usr/share/ Architecture-independent files.
/usr/src/ BSD and/or local source files.
/usr/X11R6/X11R6 distribution executables, libraries, etc (optional).
/var/Multi-purpose log, temporary, transient, and spool files.
/var/log/ Miscellaneous system log files.
/var/mail/ User mailbox files.
/var/spool/Miscellaneous printer and mail system spooling directories.
/var/tmp/Temporary files that are kept between system reboots.
/var/yp NIS maps.

Types Files
• ordinary files– text files – data files – command text files (shell scripts), – executable files (binaries);
• directories; • links special device files (physical hardware)• In unix, everything is a file… and every file is a
device… including the display – which is a port …

Directories
All directories are subdirectories of the root directory
Every directory can contain files or subdirectories
Every directory can contain a device. Which is a file. Which really means that from anyplace in the filestructure hierarchy, you can pipe IO to any other place (device… err, file…)

Relevant Terminology
• Path
• File permission
• Home directory
• Current working directory
• Account
• Login Name / Username
• Password
• Root

Logging Into Machine
Red Hat Linux release 9
Kernel 2.4.18-14 on an i686
localhost login:

The Unix Shell
• http://www.looking-glass.org/shell.html• http://www.strath.ac.uk/CC/Courses/IntroToUnix/node46.html
• When you type commands you are communicating with the shell
• The shell is started automatically when you log in• Everything is controlled from a shell• More than one shell can be open at a time

Commands
• Specific commands used to perform functions in the shell
• Ex: changing directories, copying files, mv files,etc…
• Each command is itself a program and take command line arguments
• For help on a specific command type:man command

ls
• pwd “returns current working dir path”
• ls “view contents of directory”
• ls “lists all files (except hidden files)”
• ls –A “all files INCLUDING hidden files”
• ls -lt “lists all files in descending time
• ls –1 “what does this do?”
• man ls
• ls *.CEL wildcards can be used

cd
• cd “change directory”• cd “changes to user’s home directory”
– ‘home directory’ –top of users dir tree
• cd some_dir “moves you to some_dir”• cd ~ “moves you to your home directory”• cd ../.. “moves you two directories up”• Cd – “returns you to previous working dir”• mkdir – creates a new directory with default permissions.

more / less
• More “view contents of a file”– Use return or space to move through file– Use b to go backwards– ASCII only -> dos2unix command
• Less “view contents of a file”– Use page up and page down to scrolol– Does not buffer whole file
man more “less is more!!”man less

grep
• Search contents of a file or directory of files– “Get Regex” – uses regular expressions we learned
– File wildcards can be used like with ls
• grep 1sq ~/DATA/*.CEL -> array type used

rm
• rm “remove a file or directory”• rm –R “remove all subdirectories”• rm –f “dangerous, forces, no prompt”• man rm
• HOW DO YOU UNDELETE???

UNDELETE
• THERE IS NO UNDELETE• (no trash, no recycle bin to open / browse)• Be careful what you do
– But, that’s why there are permissions!!!
– Also a reason for *nix stability

An Example ls –l output: Unix Permissions
Permissions listings
File permissions prevent users from deleting or tampering others files

chmod
• chmod “change file permissions”• chmod 777 “change permissions to 777”• chmod –R 775 “change recursively”• man chmod

Permissions Explained
• --- --- ---• you group world
• --- READ WRITE EXECUTE
• rwx rwx rwx• Binary 421 421 421 = 777• rw- r– r-- 6 44

cp
• cp “copy from x to y”
• cp /home/igert/bin/a /home/igert
• cp –R directory /folder/new_dir
• man cp
• cd /home/igert/temp
• cp a ..
• cp /home/igert/temp/a ~

Process Control, I/O Redirection & Pipes

Overview
• Unix provides powerful features for:– Managing your jobs (process control)
– Redirecting standard input and standard output
– Reading and writing files
– This is the ‘power of the shell’

Input / Output
STDERRCommandOr
Program
STDIN
STDOUT

Input / Output II
STDERRCommandOr
Program
STDIN
STDOUT-Keyboard-File-Previous STDOUT
-Terminal/ monitor-File-Next STDIN
Input / Output III
Command 1
Command 2Pipe
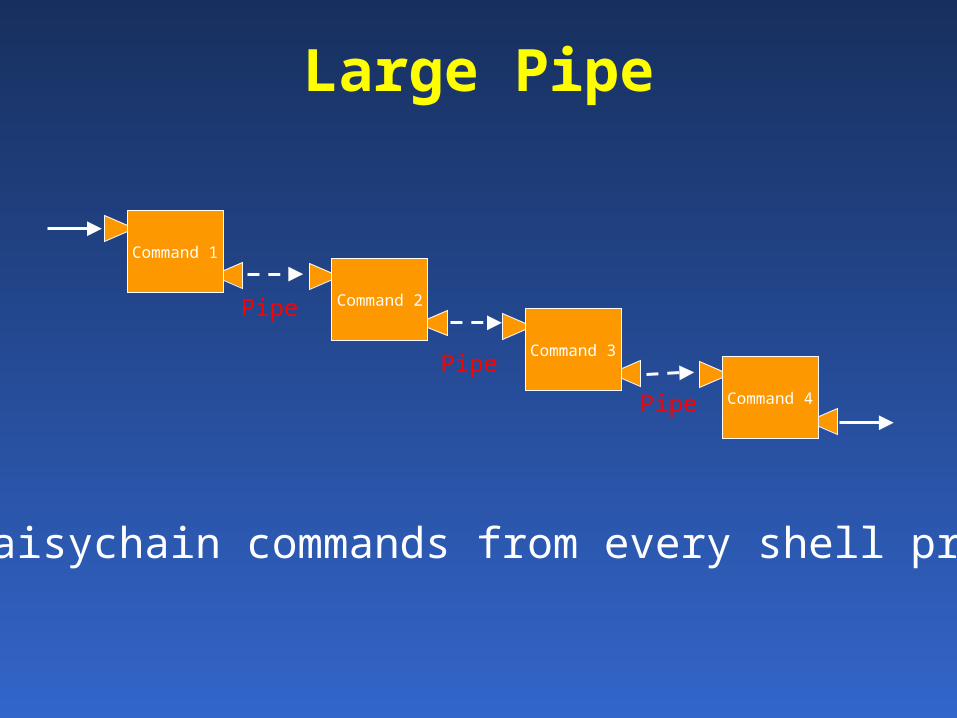
Large Pipe
Command 3
Command 4Pipe
Command 1
Command 2Pipe
Pipe
Can daisychain commands from every shell program

Example
CommandOr
Program
STDIN
STDOUT
echo “Hello Genome Hacker!”

Example (details)
ECHO
“Hello Genome Hacker!”
Hello Genome Hacker!
echo “Hello Genome Hacker!”

What can we pipe to?
• Many useful shell commands exist – use apropos to find
• awk, sed, perl –e, wc, diff, cut, paste, sort, uniq… just to name a few
• What do these do? Try man ‘command’ (as: man paste)

AWK is your pre-perl friend
• Use to print a subset of fields• Default field delimiter is “ “ (white space)• Useful for grabbing a subset of fields• Useful for rearranging fields
field1 filed2 field3 field4 . . .
$1 $2 $3 $4 . . . .

Using AWK
| awk –F” “ ‘{print $1}’
| awk –F” “ ‘{print $1” “$2}’
| awk –F” “ ‘{print $1”\t”$2}’
\t = TAB
\n = newline
pipe

Example of a pipe
Command 1(ECHO)
Command 2(AWK}Pipe
echo “Hello Genome Hacker!” | awk ‘{print $3”\t”$2”\t”$1}’
“Hello Genome Hacker!”
Hacker! Genome Hello

Example: redirecting STDOUT
CommandOr
Program
STDIN
STDOUT
echo “Hello Genome Hacker!” > output_file
more output_file
OUTPUT_FILE
“redirection operator”

Overwrite versus Append
• > OVERWRITE – delete and replace
• >> APPEND – add to end of existing file

Example: microarray data tracking
• grep 1sq ~/DATA/*.CEL (gives array info)• grep 1sq ~/DATA/*.CEL | awk ‘{print $12}’ gives
array type only• grep 1sq ~/DATA/*.CEL | awk ‘{print $12}’ >
arrayTypes.txt (store results in file)• ls ~/DATA/*.DAT | wc (gives a count)

Process Control
• Each specific job / command is called a process• Each process runs in a shell
– BEFORE: prompt available
– DURING: prompt NOT available
– AFTER: prompt available

Two Ways to monitor Processes
• “top”– Lists all jobs
– Uses a table format
– Dynamically changes
• “ps”– man ps
– static content
– Command options

Top output

Background / Foreground
• Commands running in foreground prevent prompt from being used until command completes
• Commands can also run in BACKGROUND• “Backgrounded” commands DO NOT AFFECT
the prompt

Two Ways to Background jobs
• “&”– Running a command with
“&” automacically sends it to the background
– Backgrounded commands return the prompt
• “bg”– Once a command is run
from the prompt
– Stop the command
– Then background it• Starts the command again
• Returns the prompt for use

Creating Our Own Commands
• Use programming language to create the new command• We will use perl• We’ll start USING OUR ENVIRONMENT (*NIX) NEXT
WEEK!

Homework Set 1
• What is the purpose of a pipe?• What does the pipe do?• What symbol is used for pipe?• How are commands put in background?• Why would you want to background?• What is the top command?• Why is it useful?• Submit one question about the *NIX environment to the
bioinformatics mailing list• ANSWER one question from the bioinformatics mailing
list!
(estimated time: 10 – 15 minutes)

Homework Problem 2
• Create a two command pipe• What does it do• What are the inputs and outputs for each command?• Can you extend it to three commands?• How about four?• (command hints: echo, grep, awk, wc)• Redirect output to a file• Run it a second time, and redirect (append) this output to
previous output
(estimated time: 15 to 30 minutes)