volume 29 number 7 biopharm - ubmimages2.advanstar.com/pixelmags/biopharm/pdf/2016-07.pdf · volume...
TRANSCRIPT

The Science & Business of Biopharmaceuticals
BioPharmINTERNATIONAL
Bio
Ph
arm
Intern
atio
nal
JULY 2
016
Sin
gle
-Use
Syste
ms I C
lean
roo
m S
tan
dard
s I Raw
Mate
rials
Vo
lum
e 2
9 N
um
ber 7
July 2016
Volume 29 Number 7
MANAGING BIOMANUFACTURING
CAPACITY EXPECTATIONS
QUALITY
MICROBIOLOGICAL
TESTING: TIME IS
OF THE ESSENCE
PEER-REVIEWED
BIOPROCESSING TECHNOLOGY
TRENDS OF RNA-BASED
THERAPEUTICS AND VACCINES
ANALYTICAL TESTING
FORCED DEGRADATION
STUDIES FOR
BIOPHARMACEUTICALS
www.biopharminternational.com

HARVEST
YOUR
DISCOVERYThe Biological Process Development
Facility has the expertise and flexibility to transfer your discovery to large-scale GMP manufacturing. Let our decades of experience and
comprehensive strategies help realize your product’s potential.
• Master and working cell banks
• Upstream and downstream process development
• Microbial manufacture of API and other biologics
• Analytical method development and qualification
• Quality control and stability testing services
• Regulatory Support
Learn more at • bpdf.unl.edu • [email protected] • 402.472.1983
ES808007_BP0716_CV2_FP.pgs 07.05.2016 19:40 ADV blackyellowmagentacyan

INTERNATIONAL
BioPharmThe Science & Business of Biopharmaceuticals
EDITORIAL
Editorial Director Rita Peters [email protected]
Senior Editor Agnes Shanley [email protected]
Managing Editor Susan Haigney [email protected]
Science Editor Randi Hernandez [email protected]
Science Editor Adeline Siew, PhD [email protected]
Community Manager Caroline Hroncich [email protected]
Art Director Dan Ward [email protected]
Contributing Editors Jill Wechsler, Jim Miller, Eric Langer, Anurag Rathore, Jerold Martin, Simon Chalk, and Cynthia A. Challener, PhD
Correspondent Sean Milmo (Europe, [email protected])
ADVERTISING
Publisher Mike Tracey [email protected]
National Sales Manager Steve Hermer [email protected]
East Coast Sales Manager Scott Vail [email protected]
European Sales Manager Linda Hewitt [email protected]
C.A.S.T Data and List Information Ronda Hughes [email protected]
Reprints 877-652-5295 ext. 121/ [email protected] Outside US, UK, direct dial: 281-419-5725. Ext. 121
PRODUCTION
Production Manager Jesse Singer [email protected]
AUDIENCE DEVELOPMENT
Audience Development Rochelle Ballou [email protected]
© 2016 UBM. All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical including by photocopy, recording, or information storage and retrieval without permission in writing from the publisher. Authorization to photocopy items for internal/educational or personal use, or the internal /educational or personal use of specific clients is granted by UBM for libraries and other users registered with the Copyright Clearance Center, 222 Rosewood Dr. Danvers, MA 01923, 978-750-8400 fax 978-646-8700 or visit http://www.copyright.com online. For uses beyond those listed above, please direct your written request to Permission Dept. fax 440-756-5255 or email: [email protected].
UBM Americas provides certain customer contact data (such as customers’ names, addresses, phone numbers, and e-mail addresses) to third parties who wish to promote relevant products, services, and other opportunities that may be of interest to you. If you do not want UBM Americas to make your contact information available to third parties for marketing purposes, simply call toll-free 866-529-2922 between the hours of 7:30 a.m. and 5 p.m. CST and a customer service representative will assist you in removing your name from UBM Life Sciences’ lists. Outside the U.S., please phone 218-740-6477.
BioPharm International does not verify any claims or other information appearing in any of the advertisements contained in the publication, and cannot take responsibility for any losses or other damages incurred by readers in reliance of such content.
BioPharm International welcomes unsol ic i ted ar t ic les, manuscr ipts, photographs, illustrations, and other materials but cannot be held responsible for their safekeeping or return.
To subscribe, call toll-free 888-527-7008. Outside the U.S. call 218-740-6477.
EDITORIAL ADVISORY BOARDBioPharm International’s Editorial Advisory Board comprises distinguished
specialists involved in the biologic manufacture of therapeutic drugs,
diagnostics, and vaccines. Members serve as a sounding board for the
editors and advise them on biotechnology trends, identify potential
authors, and review manuscripts submitted for publication.
K. A. Ajit-Simh President, Shiba Associates
Rory Budihandojo Director, Quality and EHS Audit
Boehringer-Ingelheim
Edward G. Calamai Managing Partner
Pharmaceutical Manufacturing
and Compliance Associates, LLC
Suggy S. Chrai President and CEO
The Chrai Associates
Leonard J. Goren Global Leader, Human Identity
Division, GE Healthcare
Uwe Gottschalk Vice-President,
Chief Technology Officer,
Pharma/Biotech
Lonza AG
Fiona M. Greer Global Director,
BioPharma Services Development
SGS Life Science Services
Rajesh K. Gupta Vaccinnologist and Microbiologist
Jean F. Huxsoll Senior Director, Quality
Product Supply Biotech
Bayer Healthcare Pharmaceuticals
Denny Kraichely Associate Director
Johnson & Johnson
Stephan O. Krause Director of QA Technology
AstraZeneca Biologics
Steven S. Kuwahara Principal Consultant
GXP BioTechnology LLC
Eric S. Langer President and Managing Partner
BioPlan Associates, Inc.
Howard L. Levine President
BioProcess Technology Consultants
Herb Lutz Principal Consulting Engineer
Merck Millipore
Jerold Martin Independent Consultant
Hans-Peter Meyer Lecturer, University of Applied Sciences
and Arts Western Switzerland,
Institute of Life Technologies.
K. John Morrow President, Newport Biotech
David Radspinner Global Head of Sales—Bioproduction
Thermo Fisher Scientific
Tom Ransohoff Vice-President and Senior Consultant
BioProcess Technology Consultants
Anurag Rathore Biotech CMC Consultant
Faculty Member, Indian Institute of
Technology
Susan J. Schniepp Fellow
Regulatory Compliance Associates, Inc.
Tim Schofield Senior Fellow
MedImmune LLC
Paula Shadle Principal Consultant,
Shadle Consulting
Alexander F. Sito President,
BioValidation
Michiel E. Ultee Principal
Ulteemit BioConsulting
Thomas J. Vanden Boom VP, Biosimilars Pharmaceutical Sciences
Pfizer
Krish Venkat Managing Partner
Anven Research
Steven Walfish Principal Scientific Liaison
USP
Gary Walsh Professor
Department of Chemical and
Environmental Sciences and Materials
and Surface Science Institute
University of Limerick, Ireland

4 BioPharm International www.biopharminternational.com July 2016
Contents
BioPharmINTERNATIONAL
BioPharm International integrates the science and business of
biopharmaceutical research, development, and manufacturing. We provide practical,
peer-reviewed technical solutions to enable biopharmaceutical professionals
to perform their jobs more effectively.
COLUMNS AND DEPARTMENTS
BioPharm International ISSN 1542-166X (print); ISSN 1939-1862 (digital) is published monthly by UBM Life Sciences 131 W. First Street, Duluth, MN 55802-2065. Subscription rates: $76 for one year in the United States and Possessions; $103 for one year in Canada and Mexico; all other countries $146 for one year. Single copies (prepaid only): $8 in the United States; $10 all other countries. Back issues, if available: $21 in the United States, $26 all other countries. Add $6.75 per order for shipping and handling. Periodicals postage paid at Duluth, MN 55806, and additional mailing offices. Postmaster Please send address changes to BioPharm International, PO Box 6128, Duluth, MN 55806-6128, USA. PUBLICATIONS MAIL AGREEMENT NO. 40612608, Return Undeliverable Canadian Addresses to: IMEX Global Solutions, P. O. Box 25542, London, ON N6C 6B2, CANADA. Canadian GST number: R-124213133RT001. Printed in U.S.A.
BioPharm InternationalJTTFMFDUJWFMZBCTUSBDUFEPSJOEFYFEJOrBiological Sciences Database (Cambridge Scientifi c Abstracts)rBiotechnology and Bioengineering Database (Cambridge Scientifi c Abstracts)rBiotechnology Citation Index (ISI/Thomson Scientifi c)rChemical Abstracts (CAS) rŞScience Citation Index Expanded (ISI/Thomson Scientifi c)rWeb of Science (ISI/Thomson Scientifi c)
The Science & Business of Biopharmaceuticals
BioPharmINTERNATIONAL
July 2016
Volume 29 Number 7
MANAGING BIOMANUFACTURING
CAPACITY EXPECTATIONS
QUALITY
MICROBIOLOGICAL
TESTING: TIME IS
OF THE ESSENCE
PEER-REVIEWED
BIOPROCESSING TECHNOLOGY
TRENDS OF RNA-BASED
THERAPEUTICS AND VACCINES
ANALYTICAL TESTING
FORCED DEGRADATION
STUDIES FOR
BIOPHARMACEUTICALS
www.biopharminternational.com
Cover: JurgaR/Getty Images
6 From the Editor
CPhI Pharma Awards seek nominations for excellence in biopharma.Rita Peters
8 US Regulatory Beat
Agency guidance and industry standards aim to reduce lapses and improve quality operations. Jill Wechsler
12 Perspectives on Outsourcing
CDMOs need to be aware that unfavorable public markets put emerging bio/pharma R&D spending at risk in 2017. Jim Miller
44 Troubleshooting
The author provides a review of the concepts of design and qualification that apply to single-use systems.Jerold M. Martin
49 New Technology Showcase
49 Product Spotlight
49 Ad Index
50 Biologics News Pipeline
CAPACITY
Managing Biomanufacturing
Capacity Expectations
Randi HernandezCapacity for complex
therapeutics is becoming
increasingly difficult to predict. 14
QUALITY
Microbiological Testing:
Time is of the Essence
Cynthia A. ChallenerPressures to accelerate
current and next-gen therapies
are challenging traditional
microbiological testing methods. 20
ANALYTICAL TESTING
Forced Degradation Studies
for Biopharmaceuticals
Anette Skammelsen SchmidtThe author addresses critical issues to consider
prior to performing forced degradation studies
and provides best practice recommendations
for these types of studies. 24
PEER-REVIEWED
Bioprocessing Technology
Trends of RNA-Based
Therapeutics and Vaccines
Claire Scanlan, Priyabrata Pattnaik, Ruta Waghmare, Elina Gousseinov, Mikhail Kozlov, Aaron Hammons, Ling Bei, Youssef Benchek, and Karim PiraniThis article reviews the current dynamics
in the RNA therapeutics/vaccines market. 30
CLEANROOM STANDARDS
Revised ISO Cleanroom
Standards Improve Air
Cleanliness Classification
Jennifer MarkarianRevised versions of ISO 14644 Parts
1 and 2 introduce changes to sampling
procedures and monitoring plans for
cleanrooms and clean zones. 38
PACKAGING TRENDS
Raw Materials
Packaging Innovations
for Biopharmaceutical
Manufacturing
Nandu DeorkarRecent trends in raw materials packaging
may impact manufacturing, quality,
and cost of biopharmaceuticals. 40
Volume 29 Number 7 July 2016
FEATURES

It’s time to make a change.
© 2016 BD. BD, the BD Logo and BD OneFeed are trademarks of Becton, Dickinson and Company. 23-18517-00
BD™ CHO CD Medium and Feed Kit
BD7 Loveton CircleSparks, MD 21152bdbiosciences.com/advbio
RIGHT NUTRIENTS?
Think your cells are getting the
BD CHO CD Medium and Feed Kit, and the BD OneFeed supplement are prototypes in the BD Preview Series. Larger quantities are available through BD Custom Products Services.
Specifically designed for CHO cells, BD™ CHO CD Medium and Feed Kit, rich in carbohydrates, amino acids, and vitamins, gives you another option when it comes to supplying your cells with the nourishment they need to thrive.
Supporting the direct adaptation of CHO cells, BD’s newest chemically defined, animal-free medium can be used in both batch and fed-batch culture, while the versatile BD OneFeed™ medium supplement is a chemically defined, animal-free, protein-free feed shown to enhance protein production when used in a variety of commercially available media.
To order a BD Preview Pak of the BD CHO CD Medium and Feed Kit or the BD OneFeed supplement, please call 877.362.2700 or go to bdbiosciences.com/go/cho-cd.

6 BioPharm International www.biopharminternational.com July 2016
From the Editor
CPhI Pharma
Awards seek
nominations
for excellence
in biopharma
development and
manufacturing.
Recognizing Biopharma Industry Excellence
Each day, the editors of BioPharm International receive press releases promot-
ing potential: announcements of new products, services, methods, and
processes destined to solve a problem, cure a disease, save money, and—in
some cases of extreme hyperbole—change the world.
As natural cynics, the editors question the validity of these projections of
great success. We also appreciate the true successes, as recognized by indepen-
dent evaluations by industry experts. The CPhI Pharma Awards are one such
program to recognize excellence in biopharma development and manufacturing.
The awards program recognizes companies and individuals helping to accel-
erate the development of biopharmaceuticals through the introduction of
innovations, new technologies, and strategies that support drug development,
manufacturing, and distribution. The program is organized by CPhI Worldwide,
a global tradeshow for the pharma industry, scheduled for Oct. 4–6, 2016 in
Barcelona, Spain. CPhI and BioPharm International are UBM plc brands.
The 2016 awards are organized into 12 categories including:
t Excellence in Pharma: Bioprocessing. Technologies, products, processes, and
services for the manufacture of biologic drugs.
t Excellence in Pharma: Contract Services & Outsourcing. Contracted services
and processes for the bio/pharmaceutical industry including research,
development, formulation, manufacturing, analysis, and consulting.
t Excellence in Pharma: Regulatory Procedures and Compliance. Technologies,
products, processes, and services designed to ensure that bio/pharma com-
panies comply with standards, rules, and guidances established by regula-
tory and compendial authorities.
t Excellence in Pharma: Supply Chain, Logistics, & Distribution. Technologies,
products, processes, and services for ensuring the safe handling and track-
ing of drug substances, raw materials, and drug products.
t Excellence in Pharma: Analysis, Testing, and Quality Control. Technologies, prod-
ucts, processes, and services for the analysis and testing of drug substances, raw
materials, and drug products in a laboratory or production-line setting.
t Excellence in Pharma: Packaging. Technologies, products, processes, and ser-
vices related to primary and secondary packaging.
t Excellence in Pharma: Drug Delivery Devices. Technologies, products, pro-
cesses, and services related to the delivery of drug products to patients.
t Excellence in Pharma: Corporate Social Responsibility. Innovation in improv-
ing transparency and public outreach.
t Excellence in Pharma: CEO of the Year. The chief executive officer of an innova-
tor or generic-drug company is eligible for nomination. Attributes to be con-
sidered include financial performance, product performance, leadership skills,
management capability, marketing, acquisitions, and corporate strategy.
t Excellence in Pharma: API Development. Technologies, products, processes,
and services for the development and manufacture of APIs.
t Excellence in Pharma: Formulation and Excipients. Technologies, products,
processes, and services related to the formulation of drug products.
t Excellence in Pharma: Manufac turing Technology and Equipment.
Technologies, products, processes, and services for the manufacture of
solid, semi-solid, parenteral, inhalation, or other dosage drugs.
Applications for the 2016 CPhI Pharma Awards must be submitted by Aug. 14,
2016 via the awards website at awards.cphi.com. Finalists will be announced
in September 2016. The awards will be presented on Oct. 4, 2016 during the
Pharma Awards Gala at CPhI Worldwide. X
Rita Peters is the
editorial director of
BioPharm International.

Accelerate your bioprocess journey6SHHGDQGHFLHQF\DUHFUXFLDODVSHFWVRIELRPDQXIDFWXULQJ7KHULJKWVXSSOLHUFDQFRQWULEXWHWR\RXUVXFFHVV'LVFRYHU KRZRXUSLRQHHULQJWHFKQRORJLHVDJLOHVHUYLFHVDQGDELOLW\WRGHVLJQDQGFRQVWUXFWFRPSOHWHIDFLOLWLHVLPSURYHVVSHHG WRPDUNHW
(QJDJHZLWK*(WRDFFHVVLQGXVWU\H[SHUWLVHDQG LQVLJKWVWRDFFHOHUDWH\RXUELRSURFHVVMRXUQH\
gelifesciences.com/bioprocess
*(DQG*(PRQRJUDPDUHWUDGHPDUNVRI*HQHUDO(OHFWULF&RPSDQ\*HQHUDO(OHFWULF&RPSDQ\)LUVWSXEOLVKHG$SU*(+HDOWKFDUH%LR6FLHQFHV$%%M|UNJDWDQ8SSVDOD6ZHGHQ
$$

8 BioPharm International www.biopharminternational.com July 2016
Regulatory Beat
Vis
ion
so
fAm
eri
ca
/Jo
e S
oh
m/G
ett
y I
ma
ge
s
FDA has long emphasized the importance
of reliable and accurate data in ensuring
drug safety, quality, and purity, but cur-
rent good manufacturing practice (CGMP) vio-
lations involving data integrity failings seem to
be on the rise, especially at overseas bio/phar-
maceutical operations. Current FDA policies for
ensuring that manufacturers maintain accurate
records and submit complete information stem
from its Application Integrity Policy, which
was established in the wake of the generic-drug
scandal of the 1980s. And while most data
integrity citations tend to involve sloppy prac-
tices and inadvertent violations, as opposed to
outright fraud, FDA officials are taking stronger
action to emphasize the importance of main-
taining secure systems for collecting and retain-
ing records to document the production of
quality drugs and biologics.
To highlight its concerns about the rise in
serious data and recordkeeping lapses in the
United States and abroad, FDA issued in April
2016 a long-awaited draft guidance (1). The
guidance uses a Q&A format to outline key
strategies for ensuring that manufacturing data
are reliable and accurate, and that companies
establish risk-based approaches for prevent-
ing and detecting problems in docu-
menting processes and tests and in
retaining records. FDA emphasizes
the need to ensure that all records are
complete and that effective systems
are in place for retaining and tracking
information and for preventing the
deterioration and loss of stored data.
The draft guidance defines key
terms, such as “backup” and “audit
trail,” and discusses methods for
restr ict ing unauthorized access
to computer IT systems. There’s
an explanation for why “testing
into compliance” is an inappropriate way to
achieve desired test results, a practice that is
cited increasingly in inspection reports. At the
same time, FDA seeks to avoid adding onerous
requirements or to complicate efficient drug
production. The regulators are all too aware
that a hard slap on a large producer could lead
to production delays that create drug short-
ages or reduce competition that helps maintain
lower drug prices.
INSPECTIONS FIND PROBLEMS FDA notes in the guidance that it is observing
an increased number of violations involving
data integrity in CGMP inspections, including
instances of poor records, inadequate written
procedures, and deficient systems for ensuring
effective production processes and controls at
manufacturing facilities all over the world. FDA
inspections cite a range of serious deficiencies
in how employees handle important records
and documents. There are reports of records
found in trash bins, data that do not match test
results, data manipulation, sample retesting to
achieve desired results, and deletion of undesir-
able results. These violations have led to warn-
ing letters, import alerts, and consent decrees,
particularly at facilities in India and China.
A scathing letter was sent in April to
Mumba i -based A PI producer Pol id r ug
Laboratories, following an in-depth inspection
in March 2015 (2). FDA cited the firm for failing
to record or investigate quality-related customer
complaints and for production deviations and
inadequate controls on computerized systems
to prevent unauthorized access or changes to
manufacturing data. FDA banned all imports
from this site in September 2015, as has Health
Canada and other regulatory authorities (3, 4).
Another warning letter to contract manufac-
turer Sri Krishna Pharmaceuticals highlights
FDA and Manufacturers Intensify Concerns about Data IntegrityAgency guidance and industry standards aim to reduce lapses and improve quality operations.
Jill Wechsler is BioPharm
International’s Washington editor,
Chevy Chase, MD, 301.656.4634,

July 2016 www.biopharminternational.com BioPharm International 9
Regulatory Beat
data integrity violations involv-
ing incomplete laboratory records,
inappropriate controls on com-
puter systems, a lack of written
procedures, plus a failure to fol-
low those procedures that are in
place (5). The letter cites multiple
situations where the firm deleted
non-conforming test results and
repeated tests to gain desired
results. FDA wants Sri Krishna to
conduct a comprehensive inves-
tigation into the extent of record
inaccuracies and to develop a
global corrective action and pre-
vention plan.
Of 28 warning letters issued by
FDA’s Center for Drug Evaluation
and Research (CDER) from January
2015 to May 2016, 21 cite data
integrity issues, reported Thomas
Cosgrove, acting director of CDER’s
Office of Compliance, at the ISPE/
FDA/PQRI Quality Manufacturing
conference in Bethesda, MD, in
June 2016. He noted that FDA is
seeing fewer problems at “top
tier” pharma companies, but more
violations in China and other
foreign countries. FDA and indus-
try experts further discussed key
components of FDA data integrity
requirements and industry best
practices at a special data integrity
workshop held in conjunction with
the quality conference.
Dealing with such problems
can carry high legal costs, noted
attorney Neil DiSpirito of Ballard
Spahr LLP at the May 2016 annual
meeting of the Food and Drug Law
Institute (FDLI) in Washington,
DC. Consequently, the importance
of fixing data problems is draw-
ing more attention in executive
offices and prompting more corpo-
rate initiatives to prevent and fix
data problems. The International
S oc ie t y for Pha r maceut ic a l
Engineering (ISPE) is developing
a white paper to make a strong
business case for investing in sys-
tems able to ensure that all data
are appropriately recorded and
reviewed and that appropriate con-
trols can detect any problems. The
impact of data breaches and com-
pliance problems are now “hitting
the bottom line” at pharma com-
panies, pointed out Frances Lipp,
president of Lachman Consultant
Services, at the FDLI conference.
Situations involving falsified data,
she noted, can lead to delays in
product launches, recalls, and
major overhauls of information
systems.
PDA OFFERS GUIDELINESThe Parenteral Drug Association
(PDA) has formed a task force to
address the “spectrum of issues”
related to the complexities man-
ufacturers face in ensuring the
integrity of processes generating
key production and regulatory
information, reported PDA presi-
dent and CEO Richard Johnson
at the FDLI conference. A lack of
accountability in production sys-
tems has led to improper data
manipulation, adjustment of time
clocks, record backdating, exclu-
sion of adverse informat ion,
and trashing of original records,
Johnson observed.
To remedy these problems, the
PDA group has developed guide-
l ines to help manufacturers
develop internal codes of conduct
for ensuring data integrity. Such
policies can emphasize to employ-
ees, suppliers, and contractors the
importance of meeting require-
ments for ensuring the accuracy
of information and records. They
apply to organizations that con-
duct clinical trials and laboratory
tests and that contract to provide
services to bio/pharma companies,
as well as to manufacturers.
The PDA task force also is pre-
paring a points-to-consider docu-
ment on the fundamental concepts
for data integrity, as well as tech-
nical reports on ensuring data
accuracy in laboratory systems.
The aim is to assist manufactur-
ers in establishing mechanisms for
detecting and remediating non-
compliance situations, to encour-
age harmonized standards for
ensuring data integrity in different
regions, and ultimately to restore
confidence of regulators and the
public in quality production sys-
tems, Johnson said. PDA will dis-
cuss its guidelines for company
codes and other related initiatives
at a workshop on data integrity
in September in Washington, DC
(in conjunction with its annual
FDA/PDA regulatory conference)
and at similar workshops in Berlin,
Germany, and San Diego.
In the old days of paper records,
it was relatively easy to destroy or
replace production files. Today’s
computerized systems require
audit trails for every operation,
which make discrepancies easier
to detect—by both manufacturers
and by FDA investigators. Thus it is
important for biopharma compa-
nies to ensure that all contractors
and suppliers—for IT, manufactur-
ing, and clinical trials—understand
and follow the rules, and report
quickly when problems emerge.
REFERENCES 1. FDA, Data Integrity and Compliance With
CGMP Guidance for Industry, Draft
Guidance (CDER, CBER, CVM, April
2016), www.fda.gov/downloads/
Drugs/GuidanceCompliance
RegulatoryInformation/Guidances/
UCM495891.pdf.
2. FDA, Warning Letter to Polydrug
Laboratories Pvt. Ltd. Corporate Office,
April 14, 2016, www.fda.gov/ICECI/
EnforcementActions/
WarningLetters/2016/ucm496623.
htm.
3. FDA, Import Alert 66–40, FDA.gov,
www.accessdata.fda.gov/cms_ia/
importalert_189.html
4. “FDA Bans Drugs From India’s Polydrug
Labs, Citing GMP Issues, FDANews.
com, www.fdanews.com/
articles/173104-fda-bans-drugs-from-
indias-polydrug-labs-citing-gmp-
issues?v=preview
5. FDA, Warning Letter to Sri Krishna
Pharmaceuticals Ltd.–Unit II, April 1,
2016, www.fda.gov/ICECI/Enforcement
Actions/WarningLetters/2016/
ucm495535.htm.

10 BioPharm International July 2016
AdvertorialProduct & Service Innovations
With a proven track record of providing quality
testing services for the largest pharmaceutical and
biopharmaceutical companies in the world, Eurofins
Lancaster Laboratories is a global leader in bio/
pharmaceutical laboratory services providing,
comprehensive, innovative, and timely solutions to
streamline all of your CMC testing requirements.
As a member of Eurofins BioPharma Product
Testing Group—the largest network of harmonized
bio/pharmaceutical GMP product testing laboratories
worldwide—Eurofins Lancaster Laboratories provides
comprehensive laboratory services to support all
functional areas of bio/pharmaceutical production.
FacilitiesWith a global capacity of more than 625,000 square feet,
our network of GMP laboratories operates under the
same strict quality procedures, LIMS, and centralized
billing system across 22 locations worldwide to make
working with any of our global operations seamless.
In addition to these laboratory locations, we also have
teams of scientists placed at more than 60 client facilities
throughout the US and Europe through our Professional
Scientific Services® (PSS) insourcing program. We
also provide secure 24-hour data access from all of our
laboratories via LabAccess.com.
Markets ServicedWe provide complete CMC Testing Services to support
virtual and large bio/pharmaceutical companies and
CMOs, including testing of all starting materials, process
intermediates, drug substances, finished products and
manufacturing support through our broad technical
expertise in Biochemistry, Molecular & Cell Biology,
Virology, Chemistry, and Microbiology.
Comprehensive Testing Servicesr.FUIPEFTUBCMJTINFOU JODMVEJOHNFUIPEEFWFMPQNFOU
feasibility, optimization, cGMP qualification, and
validation, as well as verification of compendial methods
r$PNQSFIFOTJWFTUBCJMJUZBOESFMFBTFQSPHSBNTGPSDMJOJDBM
and marketed products
r$PNQMFUFCJPDIFNJDBMBOEDIFNJDBMDIBSBDUFSJ[BUJPOBOE
microbial identification
r3BXNBUFSJBMTBOEFYDJQJFOUUFTUJOHUSP/NF, EP, JP)
r1SPEVDUJPOBOEOPOQSPEVDUJPODFMMCBOLJOH JODMVEJOH
full characterization
r-PUSFMFBTFVOQSPDFTTFECVMLUFTUJOH
r1SPDFTTGBDJMJUJFTWBMJEBUJPO JODMVEJOHWJSBMDMFBSBODF
residual impurities testing, extractables & leachables,
water testing, environmental monitoring, disinfectant
efficacy and on-site sample collection
r$POTVMUJOHQSPUPDPMXSJUJOH
Major Product InnovationsWe offer the flexibility to manage your testing programs
more efficiently through your choice of three unique
service models, including standard Fee for Service, as well
as our award-winning Professional Scientific Services®
(PSS) insourcing solutions and Full-Time-Equivalent
(FTE) service models.
Our (PSS) service model places our people at the
client’s site and is dedicated to running and managing
laboratory services while eliminating headcount,
co-employment, and project-management worries.
Currently providing services in over 13 countries to
more than 60 client sites throughout the US and Europe,
our PSS insourcing program also solves the challenges
associated with the EU Temporary Agency’s Workers
Directive 2008/104.
Our FTE service Model offers a dedicated team to a
single customer within our laboratory. Providing expertise
with technical and productivity enhancements, FTE offers
integration & customization to meet specific testing needs
with the ability to track the performance of the team.
Eurofins Lancaster Laboratories, Inc.
Eurofins Lancaster Laboratories, Inc.2425 New Holland Pike
Lancaster, PA 17601
Phone: 717.656.2300
www.EurofinsLancasterLabs.com

Eliminate the ups and downs of continually replacing
and retraining temps by retaining our scientists at your
site. Hired, trained and managed by us, our award-
winning Professional Scientific Services® (PSS):
t Eliminates headcount, co-employment and
project management worries
tAvoids Temp turnover rate with managed insourcing
tCosts you less than your own full-time employees
t Delivers a 50-year history of regulatory compliant
technical expertise in your lab
t Holds numerous client awards as the top
insourcing service provider for the past 10 years
Choose the PSS Insourcing solution that enables
us to keep staff grounded.
Tired of Your
Temps Bouncing?
Partner and prosper with our award-winning PSS.
www.EurofinsLancasterLabs.com
12 BioPharm International www.biopharminternational.com July 2016
Perspectives on Outsourcing
Do
n F
arr
all/G
ett
y I
ma
ge
s
All
fig
ure
s a
re c
ou
rte
sy o
f th
e a
uth
or.
Contract research organizations (CROs)
and contract development and manufac-
turing organizations (CDMOs) may not
be feeling it yet, but the downturn in external
financing for early-stage bio/pharma companies
is real. The impact on CDMOs and CROs will be
delayed, but there is no doubt that service pro-
viders will be feeling it in coming months.
Two recent articles in the financial press
underscore what has been happening. An item
on Bloomberg.com chronicled the challenges
early-stage public companies are facing as they
go out for further funding beyond their initial
public offerings (IPOs) (1). The article high-
lights the plight of two bio/pharma companies,
Aldeyra Therapeutics and Ovascience, whose
stock prices took big hits when they floated
secondary public offerings of their stock on
May 26th. The shares of Ovascience went down
30% on the day of the offering while those of
Aldeyra fell 10%.
The Bloomberg article noted that Aldeyra and
Ovascience’s experiences are reflective of what
has been happening to many bio/pharma com-
panies trying to tap public markets.
According to the article, the num-
ber of secondary offerings from
bio/pharma companies is down
40% in 2016, with 64 offerings
vs. 106 in 2015; but the amount
raised is down 70% from $9 bil-
lion to $2.6 billion. So it’s not just
the decline in the number of offer-
ings that is hurting early-stage bio/
pharma; they are also raising fewer
dollars per offering.
The market has also not been
friendly to young bio/pharma com-
panies trying to tap public markets
for the first time. There have been
half the number of IPOs, 8 in 2016
vs. 17 in 2015, but the amount raised, just $483
million by Bloomberg’s count, is down 75%.
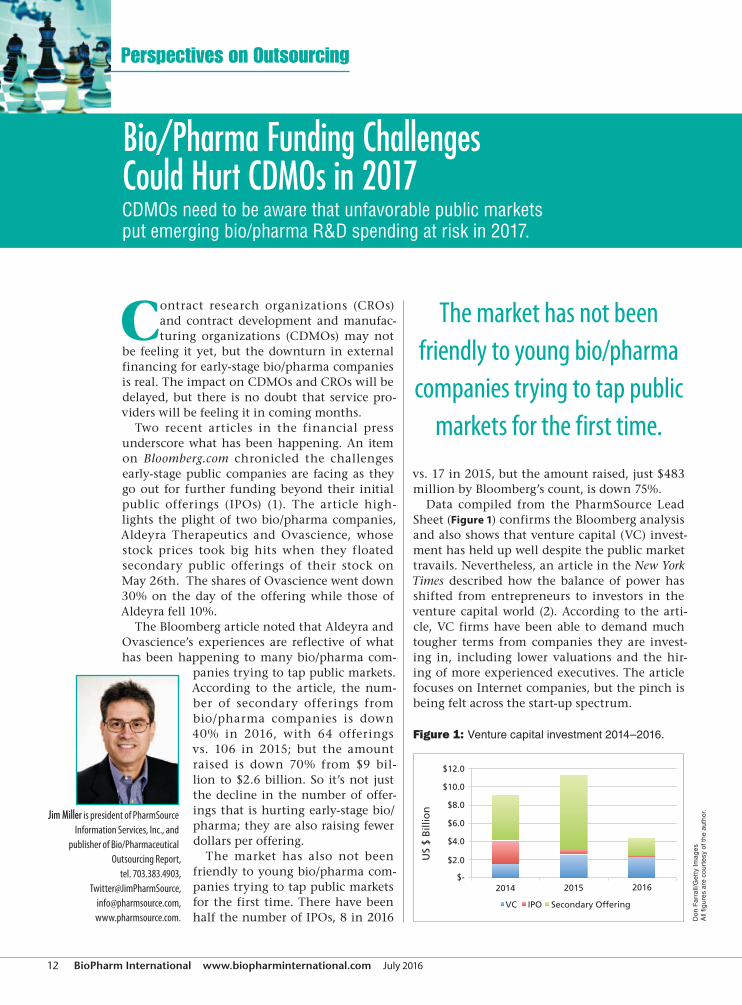
Data compiled from the PharmSource Lead
Sheet (Figure 1) confirms the Bloomberg analysis
and also shows that venture capital (VC) invest-
ment has held up well despite the public market
travails. Nevertheless, an article in the New York
Times described how the balance of power has
shifted from entrepreneurs to investors in the
venture capital world (2). According to the arti-
cle, VC firms have been able to demand much
tougher terms from companies they are invest-
ing in, including lower valuations and the hir-
ing of more experienced executives. The article
focuses on Internet companies, but the pinch is
being felt across the start-up spectrum.
Bio/Pharma Funding Challenges Could Hurt CDMOs in 2017CDMOs need to be aware that unfavorable public markets put emerging bio/pharma R&D spending at risk in 2017.
Jim Miller is president of PharmSource
Information Services, Inc., and
publisher of Bio/Pharmaceutical
Outsourcing Report,
tel. 703.383.4903,
Twitter@JimPharmSource,
www.pharmsource.com.
Figure 1: Venture capital investment 2014–2016.
VC
2014
$12.0
$10.0
$8.0
$6.0
$4.0
$2.0
$-
2015 2016
IPO Secondary Offering
US $
Billio
n
The market has not been
friendly to young bio/pharma
companies trying to tap public
markets for the first time.

July 2016 www.biopharminternational.com BioPharm International 13
Perspectives on Outsourcing
IMPACT ON EARLY-STAGE COMPANIESIt’s not surprising that CROs and
CDMOs may not be feeling the
impact of the funding down-
turn quite yet. After the 2008
global f inancial crisis, it took
two years for investigational new
drug (IND) filings and Phase I
clinical trial starts to reflect the
funding decline (Figure 2). That’s
because early-stage companies
focused their remaining cash on
getting their lead candidates into
the clinic as quickly as possible
in hopes of demonstrating proof
of concept (POC). POC is typi-
cally the prerequisite for licens-
ing deals and other partnering
arrangements from larger bio/
pharma as well as funding from
public sources. Funding from
partnering arrangements is prob-
ably the most secure funding
source because large bio/pharma
companies now depend on in-
licensed and acquired candidates
for at least half of the products
they ultimately take to commer-
cial markets.
Ea rly- stage companies t r y-
ing to get into the clinic are an
important source of business
for CDMOs because most are
dependent on service providers
to manufacture APIs and formu-
lated dose forms. Those compa-
nies represent the majority of
customers at most CDMOs, but
they typically have just a small
number of new drug candidates,
so CDMOs need to constantly
replenish their customer port-
folios to thrive. That replenish-
ment is highly dependent on a
rebound in public bio/pharma
equity markets: venture capital
might get candidates through
discovery and into preclinical,
but emerging bio/pharma compa-
nies need the larger tranches of
public funding to sustain a clini-
cal development program.
IMPACT ON CROS AND CDMOSThe most recent downturn in
external financing is barely a year
old, so CROs and CDMOs aren’t yet
feeling the pinch. Emerging bio/
pharma companies are using the
funds they have to get their can-
didates into the clinic, as in past
funding cycles, which is good for
CDMO business in the near term.
But without public markets, the
industry could see a sharp drop in
IND filings, and in the demand for
CDMO services, as it did in 2010.
One way for CDMOs and CROs
to prepare themselves for the
worst case is to focus on resur-
recting their business develop-
ment skills. After several years of
just answering unsolicited exter-
nal inquiries, they will soon have
to be prospecting for new busi-
ness and selling the customer on
why they should use them rather
than a competitor. As they were in
the last downturn, new business
development skills will be a key to
sustained success.
REFERENCES 1. M. Nilsen, “Biotech’s Vicious Second-
ary Cycle,” Bloomberg.com, May 26,
2016, www.bloomberg.com/gadf ly/ar-
ticles/2016-05-26/biotech-secondary-
offerings-get-punished
2. K. Benner, “Start-Ups Once Showered
With Cash Now Have to Work for It,”
New York Times, May 20, 2016, www.
nytimes.com/2016/05/21/technology/
start-ups-once-showered-with-cash-
now-have-to-work-for-it.html?rref=col
lection%2Ftimestopic%2FVenture%20
Capital&action=click&contentCollec
tion=timestopics®ion=stream&m
odule=stream_unit&version=latest&
contentPlacement=5&pgtype=collect
ion&_r=0
Figure 2: Investigational new drug filings 2009–2015.
900
800
700
600
500
400
300
200
100
0
2009 2010 2011 2012 2013 2014 2015
Nu
mb
er o
f I
ND
Fil
ing
s
Early-stage companies
trying to get into
the clinic are an
important source of
business for CDMOs.

14 BioPharm International www.biopharminternational.com July 2016
Jurg
aR
/Gett
y Im
ag
es
Demand for any given new prod-
uct is typically only known
after significant investments
have already been made.
Because executives commonly plan capac-
ity requirements based on launch fore-
casts, there are many factors that can lead
to miscalculations of capacity, making
it challenging to know what capacity to
build into a facility. According to a new
survey by BioPlan Associates, more than
half of respondents (60%) expect facility
constraints to create biopharmaceutical pro-
duction capacity constraints by 2021 (1).
BioPlan found that the development of
more efficient single-use products, better
downstream purification technologies, the
introduction of continuous downstream
operations, and increased modularization
of production systems were identified as
the top things the industry must do to
avoid further capacity restrictions at bio-
manufacturing plants (Figure 1). Analytical
testing concerns and adequate hiring of
qualified personnel to manage the facili-
ties are among the other top issues that are
expected to create capacity limitations in
the future (Figure 2).
FLEXIBLE SOLUTIONSEven if demand is accurately predicted,
changes to a development plan can also
occur that require facility changes, says
Christian Wyss, attorney at Vischer AG,
who specializes in drafting and negotiat-
ing contracts for clients in the life sciences.
These developments can arise because an
opportunity presents itself to improve a
drug or add more indications—or, scien-
tific issues may have to be addressed that
Managing Biomanufacturing Capacity Expectations
Randi Hernandez
Capacity for complex therapeutics is becoming increasingly
difficult to predict.
Capacity

Biosimilar Development in CHO, NS0 & Sp2/0
with enhanced PQA assessment
ďnjĞŶĂŽīĞƌƐŽƌŝŐŝŶĂƚŽƌďŝŽƐŝŵŝůĂƌĐĞůůůŝŶĞĚĞǀĞůŽƉŵĞŶƚƉƌŽŐƌĂŵŵĞƐǁŝƚŚďĞƐƉŽŬĞ
quality assessment tailored to individual projects.
ŶƟƚŽƉĞWŽůLJdŚĞƌŝĐƐdZ^ĂŶĚWĂĐŝĮĐ'DWĂƌĞŶŽǁƚƌĂĚŝŶŐĂƐďnjĞŶĂ
Enhanced PQA assessment
ŽŶƟŶƵŽƵƐĂƐƐĞƐƐŵĞŶƚŽĨƉƌŽĚƵĐƚƋƵĂůŝƚLJ
ĂƩƌŝďƵƚĞƐ;WY!ĚƵƌŝŶŐĐĞůůůŝŶĞĚĞǀĞůŽƉŵĞŶƚ
helps ensure biosimilars retains the desired
ƉƌŽĚƵĐƚĐŚĂƌĂĐƚĞƌŝƐƟĐƐĂŶĚĨƵŶĐƟŽŶ"ĂǀŽŝĚŝŶŐ
ƌĞĚĞǀĞůŽƉŵĞŶƚĐŽƐƚƐĂŶĚĚĞůĂLJƐƚŽƚŚĞŵĂƌŬĞƚ#
PQA assessment includes:
• WƌŽĚƵĐƚŝŶƚĞŐƌŝƚLJ
• WƌŽĚƵĐƚĂĐƟǀŝƚLJ
• WƌŽĚƵĐƚĂŐŐƌĞŐĂƟŽŶ
• WƌŽĚƵĐƚŐůLJĐĂŶƉƌŽĮůŝŶŐ
• YƚĞƐƟŶŐ
ĞůŝǀĞƌŝŶŐŽŶĞdžƉĞĐƚĂƟŽŶƐ
ĸĐŝĞŶƚƉƌŽĚƵĐƟŽŶŝƐŬĞLJƚŽďŝŽƐŝŵŝůĂƌĚĞǀĞůŽƉŵĞŶƚ
ĂŶĚďnjĞŶĂŚĂƐĚĞǀĞůŽƉĞĚƚĞĐŚŶŽůŽŐŝĞƐƚŽĞŶƐƵƌĞ
ŬĞLJĐĞůůůŝŶĞĚĞǀĞůŽƉŵĞŶƚƌĞƋƵŝƌĞŵĞŶƚƐĐĂŶďĞŵĞƚ"
ŝŶĐůƵĚŝŶŐ*
• ,ŝŐŚĞdžƉƌĞƐƐŝŽŶůĞǀĞůƐŽĨLJŽƵƌĂŶƟďŽĚŝĞƐŽƌƉƌŽƚĞŝŶƐ
• &ƌĞĞŽĨĂŶŝŵĂůĚĞƌŝǀĞĚƉƌŽĚƵĐƚƐƚŚƌŽƵŐŚŽƵƚƚŚĞ
ƉƌŽĐĞƐƐŝŶĂĐŚĞŵŝĐĂůůLJĚĞĮŶĞĚŵĞĚŝƵŵ
• DĂŶĂŐĞŵĞŶƚŽĨƚƌĂŶƐĨĞƌƚŽĂ'DWĨĂĐŝůŝƚLJĨŽƌĂ
ƐŵŽŽƚŚƚƌĂŶƐŝƟŽŶƚŽƐĐĂůĞ-ƵƉ
• No downstream milestones or royalty payments
Start your project today. Visit www.abzena.com

16 BioPharm International www.biopharminternational.com July 2016
AL
L F
IGU
RE
S A
RE
CO
UR
TE
SY
OF
BIO
PL
AN
AS
SO
CIA
TE
S.
were not planned. Wyss notes that
there could also be problems with a
technology transfer. “Either the man-
ufacturing process was not as robust
as the sponsor thought it was, or the
tech transfer failed to successfully
convey all subtleties to the contract
manufacturing organization [CMO].”
To alter technical capacity, a facil-
ity has to have “solution-oriented
professionals that are willing and
able to find room for flexibility in
a highly regulated environment,”
says Wyss. The change can become
more complicated if there is a change
in product type, which may even
require a completely different facil-
ity, says Tom Ransohoff, vice-pres-
ident and principal consultant at
BioProcess Technology Consultants.
It is more difficult to respond to shift-
ing demands if process equipment
and clean utility systems are hard
piped into the infrastructure, says
Parrish Galliher, CTO for upstream
at GE Healthcare’s Life Sciences busi-
ness. Multiple closed-off cleanroom
sections in facilities, numerous heat-
ing/ventilation/air conditioning
zones, and low ceilings (which limit
types and scales of new equipment)
can also serve as barriers to rapid
capacity expansion, Galliher states.
Forecasting long-term demand
during the transition from clinical
to commercial is challenging, says
Ransohoff. He adds that to meet
uncertain or changing demands,
one strategy is to “number up,” or
use multiple single-use bioreac-
tors to achieve a range of upstream
scales. Galliher concurs that adding
extra operating shifts to an existing
facility helps rapidly expand capac-
ity, as well as overlapping or “stag-
gering” of batches to meet need.
According to a report compiled by
Patheon, ORC International, and
PharmSource, demand and capac-
ity forecast inaccuracies have
prompted biopharmaceutical com-
panies to embrace the use of out-
sourcing with more fervor than
ever before (2).
Capacity
Figure 1: The top 10 areas to address to avoid capacity constraints, according to
a survey of biomanufacturers.
Figure 2: The top 10 factors creating future capacity constraints, as identified by
a survey of biomanufacturers in 2016.
Avoiding capacity constraints“If this industry is to avoid significant capacity constraints, the
most important areas to beaddressed are:”
46.0%
38.8%
37.4%
36.0%
33.8%
30.9%
29.5%
28.1%
27.3%
26.6%
Develop more cost-effective disposable, single-use products
Source: Figure adapted from the Thirteenth Annual Report and Survey of BiopharmaceuticalManufacturing Capacity and Production, BioPlan Associates Inc., 2016. Used with permission.
Develop better-performing disposable, single-useproducts
Develop better continuous bioprocessing - downstream technologies
Develop better downstream purificationtechnologies
Develop more ‘modularized’ production systems
Standardize international regulatory processes
Streamline FDA regulatory process
Optimize cell culture systems to increaseupstream performance
Fund more research to maximize productionefficiencies
Optimize systems to improve downstreampurification performance
Which factors are likely to create biopharmaceuticalproduction capacity constraints at your facility
in 5 years (by 2021)? (n=140)
Facility constraints
Analytical testing and drug product release
Inability to hire new, experienced technical and productionstaff
Inability to retain experienced technical and production staff
Physical capacity of downstream purification equipment
Inability to hire new, experienced scientific staff
Inability to retain experienced scientific staff
Costs associated with downstream purification
Physical capacity of fermentation/bioreactor equipment
Inability for me to optimize my overall system, given mycurrent technology and resources
60.0%
37.1%
30.7%
29.3%
27.1%
25.0%
23.6%
20.7%
20.0%
17.9%
Source: Figure adapted from the Thirteenth Annual Report and Survey of BiopharmaceuticalManufacturing Capacity and Production, BioPlan Associates Inc., 2016. Used with permission.

July 2016 www.biopharminternational.com BioPharm International 17
FORECASTING CAPACITY NEEDSA multitude of unforeseen circum-
stances can skew capacity fore-
casts. Some of these could include
reports of a serious adverse event,
slow enrollment in clinical trials, sale
of a parent company that is devel-
oping the drug, an unusually suc-
cessful marketing strategy, provider
motives and incentives, final cost to
the patient, willingness for a payer or
pharmacy benefit manager to reim-
burse a drug, a change in raw mate-
rial availability, availability of new
therapeutic alternatives, or new regu-
latory legislation.
In a Nature Reviews Drug Discovery
study from 2013, investigators
concluded that more than 60% of
companies miss their demand fore-
casts by at least 40% (3). A signifi-
cant number of companies were
also overly optimistic by more than
160% of the actual peak revenues
that a product could pull in. Even
up to six years post-launch, fore-
casts were still found to be off the
mark by percentages as high as 45%.
The researchers found that demand
for oncology drugs was most com-
monly underestimated, most likely
because of the additional indica-
tions for which these drugs earned
approval by FDA after initial launch.
This demand underestimation is an
important finding considering the
large number of biologic, immune-
oncology therapeutics (with various
proposed indications) that are cur-
rently in the pipeline. The authors
found that analyst forecasts for
generic therapies were also mark-
edly off-target (3). These findings
could have implications for future
demand calculations for biologics, as
well as biosimilars with numerous
market competitors—especially if the
Centers for Medicare and Medicaid
Service’s proposal to use reference
pricing for all groups of therapeu-
tically equivalent drugs under
Medicare Part B (even for biosimilars
that are not interchangeable) comes
into effect.
A 2007 article in Pharmaceutical
Executive estimated that a launch
delay costs an average of $15 mil-
lion per drug per day (4). This num-
ber changes, however, depending on
the market demand of the drug in
question. “The general rule is that
a biologic will generate, on average,
$300 million per year. So, each day
delayed is a loss of $1 million,” esti-
mates Galliher. “I have seen much
larger numbers in print for blockbust-
ers,” he adds.
Including post-approval R&D
costs, as well as costs associated with
unsuccessful projects, the estimate
for the average out-of-pocket cost to
develop a new compound was found
to be $2870 million (in 2013 dollars),
according to an analysis by DiMasi
et al. that appeared in the May
2016 issue of the Journal of Health
Economics (5). Even though there
have been slight methodological
differences in DiMasi et al.’s studies
since 2003—when the authors began
looking at the cost of bringing a drug
to market—this cost of development
has still increased substantially since
2003. Additionally, said the authors
of the study, “clinical success rates
are substantially lower for the stud-
ies focused on more recent periods”
(5). Thus, because failure rates have
increased and the cost of developing
a drug has also increased so mark-
edly, it is increasingly difficult to
accurately predict the demand for a
drug—as well as that drug’s associ-
ated capacity requirements.
Indeed, many industry experts
agree that predicting capacity will
become even more problematic for
pharmaceutical manufacturers in the
future because of market access issues.
In Europe, because physicians seem
to be more accepting of biosimilars,
market penetration forecasts may be
a bit more clear—but in the United
States, physician acceptance and pre-
scribing practices (as well as the inter-
changeability status of a biosimilar)
may make launch and capacity pre-
dictions increasingly challenging.
HYBRID CAPACITY VS. OTHER MODELSWhile it seems like a number of phar-
maceutical companies still rely on a
largely in-house approach to manag-
ing capacity, most large firms have
been open to the concept of using
outside CMOs to meet short-term
requirements. Small firms often use
a completely outsourced model to
meet capacity. The percentage of pro-
duction that is outsourced at each
biomanufacturing firm depends
largely on what type of product is
being manufactured. According
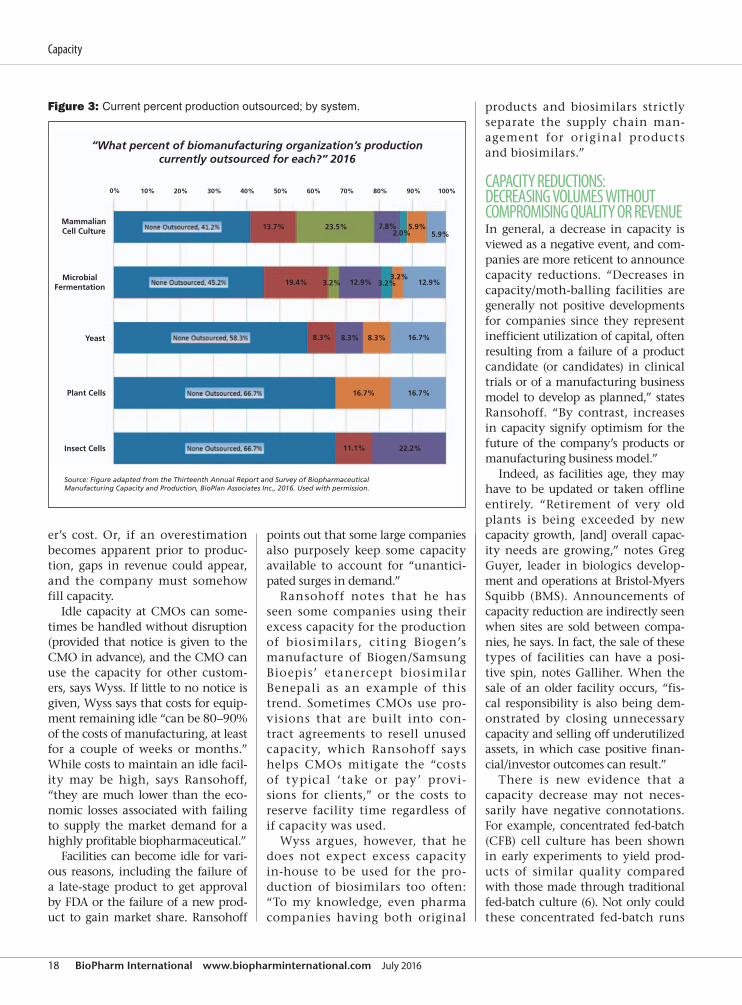
to numbers from the 2016 BioPlan
report (1), approximately 59% of
respondents used at least some out-
sourced capacity for mammalian cell
culture, 55% used outsourcing for
microbial fermentation, 42% used
outsourcing for production in yeast,
33% outsourced for production in
plant cells, and 33% outsourced
capacity for production of therapies
in insect cells (see Figure 3).
Companies such as Amgen,
Bristol-Myers Squibb, and Roche use
hybrid approaches for the production
of their medications. Wyss estimates
that almost all biotech companies
that have several products on the
market use a mixed approach to
manufacturing, but most companies
keep the number of CMOs that they
work with to a minimum. An excep-
tion would be a small biopharma
company with few products, says
Wyss. “Drug development companies
with no product on the market or
one-product companies often rely on
CMOs only and do not use in-house
manufacturing. When the date for
market launch is set, these companies
will often look for additional CMOs
to back up their supply chain.”
EXCESS CAPACITYAs mentioned, it is common to over-
estimate or underestimate demand
for a drug. Overestimating can lead
to the manufacture of too much
product, which would then have
to be disposed at the manufactur-
Capacity
18 BioPharm International www.biopharminternational.com July 2016
Capacity
er’s cost. Or, if an overestimation
becomes apparent prior to produc-
tion, gaps in revenue could appear,
and the company must somehow
fill capacity.
Idle capacity at CMOs can some-
times be handled without disruption
(provided that notice is given to the
CMO in advance), and the CMO can
use the capacity for other custom-
ers, says Wyss. If little to no notice is
given, Wyss says that costs for equip-
ment remaining idle “can be 80–90%
of the costs of manufacturing, at least
for a couple of weeks or months.”
While costs to maintain an idle facil-
ity may be high, says Ransohoff,
“they are much lower than the eco-
nomic losses associated with failing
to supply the market demand for a
highly profitable biopharmaceutical.”
Facilities can become idle for vari-
ous reasons, including the failure of
a late-stage product to get approval
by FDA or the failure of a new prod-
uct to gain market share. Ransohoff
points out that some large companies
also purposely keep some capacity
available to account for “unantici-
pated surges in demand.”
Ransohoff notes that he has
seen some companies using their
excess capacity for the production
of biosimilars, citing Biogen’s
manufacture of Biogen/Samsung
Bioepis’ etanercept biosimilar
Benepali as an example of this
trend. Sometimes CMOs use pro-
visions that are built into con-
tract agreements to resell unused
capacity, which Ransohoff says
helps CMOs mitigate the “costs
of typical ‘take or pay’ provi-
sions for clients,” or the costs to
reserve facility time regardless of
if capacity was used.
Wyss argues, however, that he
does not expect excess capacity
in-house to be used for the pro-
duction of biosimilars too often:
“To my knowledge, even pharma
companies having both original
products and biosimilars strictly
separate the supply chain man-
agement for original products
and biosimilars.”
CAPACITY REDUCTIONS: DECREASING VOLUMES WITHOUT COMPROMISING QUALITY OR REVENUEIn general, a decrease in capacity is
viewed as a negative event, and com-
panies are more reticent to announce
capacity reductions. “Decreases in
capacity/moth-balling facilities are
generally not positive developments
for companies since they represent
inefficient utilization of capital, often
resulting from a failure of a product
candidate (or candidates) in clinical
trials or of a manufacturing business
model to develop as planned,” states
Ransohoff. “By contrast, increases
in capacity signify optimism for the
future of the company’s products or
manufacturing business model.”
Indeed, as facilities age, they may
have to be updated or taken offline
entirely. “Retirement of very old
plants is being exceeded by new
capacity growth, [and] overall capac-
ity needs are growing,” notes Greg
Guyer, leader in biologics develop-
ment and operations at Bristol-Myers
Squibb (BMS). Announcements of
capacity reduction are indirectly seen
when sites are sold between compa-
nies, he says. In fact, the sale of these
types of facilities can have a posi-
tive spin, notes Galliher. When the
sale of an older facility occurs, “fis-
cal responsibility is also being dem-
onstrated by closing unnecessary
capacity and selling off underutilized
assets, in which case positive finan-
cial/investor outcomes can result.”
There is new evidence that a
capacity decrease may not neces-
sarily have negative connotations.
For example, concentrated fed-batch
(CFB) cell culture has been shown
in early experiments to yield prod-
ucts of similar quality compared
with those made through traditional
fed-batch culture (6). Not only could
these concentrated fed-batch runs
Figure 3: Current percent production outsourced; by system.
“What percent of biomanufacturing organization’s productioncurrently outsourced for each?” 2016
MammalianCell Culture
MicrobialFermentation
Yeast
Plant Cells
Insect Cells 11.1% 22.2%
16.7%
16.7%8.3%8.3%8.3%
12.9%3.2%
5.9%5.9%
2.0%7.8%23.5%13.7%
0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100%
3.2%3.2% 12.9%19.4%
16.7%
Source: Figure adapted from the Thirteenth Annual Report and Survey of BiopharmaceuticalManufacturing Capacity and Production, BioPlan Associates Inc., 2016. Used with permission.

July 2016 www.biopharminternational.com BioPharm International 19
be manufactured at lower volumet-
ric capacities (meaning smaller facili-
ties could accommodate volumes
typically seen at larger facilities), the
resulting products were also shown
to enhance cell-line charge hetero-
geneity, proving that concentrated
fed-batch could be associated with
“both process and product quality
benefits” (6).
Despite these benefits, concen-
trated fed-batch used more perfusion
and feed media, required numerous
filters, and also overloaded down-
stream processes, causing filter
fouling in some cases. The yields
obtained in the Yang study (6) were
not sufficient enough to suggest
totally replacing larger facilities, but
the technique has potential for some
slow-growing cell lines. While it may
not be economically feasible for a leg-
acy system to be converted to CFB,
Yang et al. wrote that new compa-
nies seeking flexibility in capacity
operations might want to consider
trying CFB to meet their production
needs. The authors concluded, “The
key to unlocking the cost and capac-
ity savings of concentrated fed-batch
is increasing the specific productivity
of the process through cell line and
process development.”
WORKING WITH CMOSThe use of CMOs can be helpful
when there are fluctuating capac-
ity and demand conditions, but
sometimes a biopharma company
may be wary of the CMO model. A
biopharma company may initially
choose an “in-house” approach
because it fears the leverage a CMO
can gain over its business. “The
leverage stems from the fact that
the CMO/contract development
and manufacturing organization
[CDMO] has all manufacturing
knowledge, and there is always a
substantial risk that technology
transfer will not be successful imme-
diately,” states Wyss. Thus, he says,
detailed technology transfer plans
are crucial.
If a CMO/CDMO has its own pro-
prietary manufacturing platform, a
biopharma company cannot easily
transfer the process back in-house
or to another CMO, Wyss points
out. “If the contract manufacturing
agreement does not give the sponsor
a license to this technology solely
for the continued production of this
specific biologic drug, the manu-
facturing process will have to be
partially re-designed, which is prac-
tically not feasible from a time and
cost perspective.”
Intellectual property
barriers to capacity outsourcing
Manufacturers typically choose an
in-house model to keep better con-
trol of their supply chain, handle
development risks, manage speed
of development and launch, and for
tax purposes, says Ransohoff—but
another important reason to keep
production in house is to ensure pro-
tection of a company’s intellectual
property (IP).
There seems to be mixed responses
about whether or not the protection
of IP is a significant problem when
working with CMOs. Guyer says
when BMS outsources, it establishes
clear contract provisions to protect its
IP, and if it cannot reach agreeable IP
terms with CMO partners, it simply
does not work with that CMO. He
says BMS rarely finds IP to be a bar-
rier to successful relationships with
outsourcing partners. Conversely,
GE’s Galliher sees IP as a “major
issue” for CMOs when joint owner-
ship of technology platforms and/
or inventions exist. “The customer
usually wants to retain its rights to
the drug and cell line and how it is
made, especially if proprietary tech-
niques are used. The CMO wants the
business freedom to use the process
techniques for other customers and
its own cell line if it is providing it.”
Although there many be signif-
icant advantages in terms of labor
costs when using foreign outsourc-
ing operations, specifically, concerns
about keeping IP secure often prevail,
says Wyss, and as a result, pharma
companies rarely outsource to coun-
tries with perceived weak patent pro-
tection or in areas where national
laws provide for mandatory licenses
to local generic drug manufacturers.
“It seems that originator companies
have been able to solve all quality
related issues when manufacturing
in those countries, but are still reluc-
tant to expose themselves to these
legal risks before patent expiry.” Wyss
tells this publication that intellec-
tual property issues come into play
most when a manufacturer is decid-
ing between different CMOs in vari-
ous parts of the country. He adds,
“many countries have regulations
requiring that at least a part of the
manufacturing of the drugs sold is
accomplished within that country,
either directly by relevant legislation
(such as in Russia), or indirectly by
making this a requirement to obtain
research funding or collaborate with
public academic institutions (e.g.,
the standard Cooperative Research
and Development Agreement in the
United States requires manufactur-
ing in the US).” On the other hand,
Galliher mentions that it is also rela-
tively common for some companies
to choose a foreign CMO to handle
capacity specifically to avoid local
IP legislation.
REFERENCES 1. BioPlan Associates, Inc., Thirteenth
Annual Report and Survey of
Biopharmaceutical Manufacturing
Capacity and Production, April 2016.
2. ORC International, Patheon, and
PharmSource, “Impact of Incorrect
Forecasts on New Product Launches,”
Industry Report, 2016.
3. M. Cha, B. Rifai, and P. Sarraf, Nat. Rev.
Drug Disc. 12, pp. 737–738 (2013).
4. T. Noffke, “Successful Product
Manager’s Handbook”, a supplement
to Pharmaceutical Executive (March
2007), www.pharmexec.com/no-time-
delay, accessed April 30, 2016.
5. J.A. DiMasi, H.G. Grabowski,
and R.W. Hansen, J. Health Econ.
47, pp. 20–33 (2016).
6. W.C. Yang et al., J. Biotechnol.
217, pp. 1–11 (2016).
Capacity

20 BioPharm International www.biopharminternational.com July 2016
Sto
ckb
yte
/Gett
y Im
ag
es
Effective microbiological test-
ing during biopharmaceutical
drug development and manu-
facturing is crucial for ensur-
ing sterility, determining antimicrobial
effectiveness, detecting microbial con-
tamination or bioburden levels, ana-
lyzing endotoxins, and implementing
environmental monitoring programs.
Growing pressures to increase produc-
tivity, flexibility, and cost-effectiveness
and the unique properties of many
next-generation therapies are challeng-
ing today’s microbiologists.
CONVENTIONAL PROTOCOLS NO LONGER SUITABLE FOR TRADITIONAL BIOLOGICSMicrobiology testing of pharmaceutical
products is performed with a view to
detection, enumeration, and identifica-
tion of microbial contaminants, accord-
ing to Marian McKee, senior director of
BioReliance operational development
services at MilliporeSigma. Traditional
microbiological methods for sterility
testing take 14 days from inoculation to
detection and conclusive results, while
culture-based methods for mycoplasma
take 28 days. “The time to obtain results
for these traditional microbiological test
methods is lengthy,” McKee notes.
For release of traditional biologics that
are produced in large lots with longer
stability profiles, the turnaround time on
conventional microbiological tests, which
are sensitive and robust, is not a concern
with respect to the ability to obtain reli-
able results. The faster release of bulk drug
substances and batches during in-pro-
cess testing, however, is desirable. “More
rapid alternatives to traditional methods
are needed to speed the manufacturing
Microbiological Testing: Time is of the Essence
Cynthia A. Challener
Pressures to accelerate
current and next-gen
therapies are challenging
traditional microbiological testing methods.
Cynthia A. Challener, PhD
is a contributing editor to
BioPharm International.
Quality: Microbiological Testing

July 2016 BioPharm International 21
Product & Service InnovationsAdvertorial
Company DescriptionBio-Rad Laboratories is a leading provider of innovative tools
to the life science and clinical diagnostics markets, where the
company’s products are used for scientific discovery, drug
development, and biopharmaceutical production.
Bio-Rad’s Life Science Group has long served the
bioprocessing industry by supplying advanced purification and
process technologies. Bio-Rad provides a full line of scalable
process chromatography resins to meet your purification needs
with worldwide access and personalized service and support.
Markets ServedThe Bio-Rad Laboratories Life Sciences Group is
ISO 9001:2008 registered and focuses on meeting the
needs of global biopharmaceutical companies and contract
manufacturing organizations.
Products and ServicesBio-Rad products are used in the purification processes of
many FDA and EMA approved biological therapeutics. Bio-
Rad products are designed to deliver the scalability, lot-to-lot
reproducibility, and security of supply needed in today’s
demanding commercial downstream processes.
r /VWJB™ resins—a family of next generation ion-exchange
and mixed-mode products built on an industry proven
rigid polymer base matrix. They offer superior flow
properties and low nonspecific binding while delivering
IJHIDBQBDJUZBOEVOJRVFTFMFDUJWJUZ/VWJB)34JTB
high-resolution cation exchanger capable of separating
very challenging high molecular weight impurities.
/VWJBSFTJOTCSJOHUPHFUIFSBVOJRVFTFUPGQSPQFSUJFT
specifically designed to meet the demands of current and
GVUVSFEPXOTUSFBNQSPDFTTFT/VWJBSFTJOTBSFGMFYJCMF
and robust with a large operational window, making
them effective in capture and/or polish steps.
r6/0TQIFSF™ S & Q resins. Based on a single-step
QPMZNFSJ[BUJPOQSPDFTT 6/0TQIFSFSFTJOTEFMJWFSTIJHI
binding capacities at high flow rates and low backpressures.
r $)5DFSBNJDIZESPYZBQBUJUF"NJYFENPEFNFEJBUIBU
significantly reduces a full range of impurities in a single
polishing step that is easy to develop for a wide range of
proteins. This industry-proven media is easy to pack and
selected for late-stage and commercially approved processes.
r 6/0TQIFSF™461S" BOBGGJOJUZDISPNBUPHSBQIZSFTJO
S1SPUFJO"CVJMUPOBSJHJEQPMZBDSZMBNJEFNBUSJY
designed for fast-flow and high recovery of monoclonal
antibodies.
r .BDSP1SFQ®SFTJOT1PMZNFSJDNFUIBDSZMBUFTVQQPSU
available with strong or weak ion exchange functionalities.
These rigid
macroporous resins
provide excellent
dynamic binding
capacity, resolution,
and throughput
at high flow rates
for the purification
of biomolecules.
.BDSP1SFQSFTJOT
are an excellent
choice for process-
scale applications
such as blood
fractionation
purification.
t$IFMFY® resins.
$IFMBUJOHSFTJOT
that bind
polyvalent cations with high selectivity, remove metal ions
from samples and buffers.
r"(® resins. Extensively processed to remove both organic
and inorganic impurities, AG resins are used for the
separation of low molecular weight molecules such as
inorganic ions, organic acids, and carbohydrates.
r #JP#FBET™ SM-2 resin—nonpolar polystyrene-based resin
for hydrophobic interaction chromatography,
Bio-Beads are used extensively for the removal of nonpolar
detergents from biological preparations for manufacturing
at both laboratory- and process-scale.
r 7JTJUCJPSBEDPNSFTJOMJCSBSZUPSFRVFTUBSFTJOTBNQMF
and Process Chromatography Resin Selection Guide.
Technical Services:
The Bio-Rad Laboratories Life Sciences Group’s technical
support offerings include technical telephone and on-line
support, regulatory support files, and process-scale
applications development services.
Bio-Rad Laboratories
Bio-Rad Laboratories2000 Alfred Nobel Drive
Hercules, CA 94547
Phone: 510.741.1000
Toll free: 1.800.424.6723
www.bio-rad.com/process
Email: [email protected]

22 BioPharm International www.biopharminternational.com July 2016
process while ensuring process and
product safety,” says McKee.
Protein-based biologic drugs can
also present real technical challenges
with respect to reading assay results.
“Occasionally the therapeutic mol-
ecule can have toxic effects on the
detector cells that are used to detect
viruses in in-vitro assays that comply
with ICH Q5A (1) and related regu-
latory guidelines,” observes Archie
Lovatt, scientific director of biosafety
for SGS. He does add, however, that
this issue can usually be overcome
by dilution of the sample. Results of
the tests performed on diluted sam-
ples can also be supported by viral
clearance data generated during pro-
cess development.
For biopharmaceutical manufactur-
ers including contract manufacturers
that produce many different prod-
ucts, the time and resources involved
in performing required microbial
testing is a challenge, says Aaron
Ortiz, QC manager of microbiology
at GSK Biologics’ GMS Rockville site.
“Qualifying the bioburden method
for each step of each process is
extremely time consuming. In addi-
tion, the many different matrices of
the different in-process samples lead
to challenges with recovery of differ-
ent organisms depending on the pro-
cess step,” he explains.
NEXT-GEN THERAPIES HAVE UNIQUE NEEDSThe lengthy time to obtain results
for traditional microbiological test
methods, while not a technical issue
for more stable proteins, antibodies,
and other older types of biologics,
is typically unacceptable for newer
cell-based therapeutics. “Some next-
generation products have short shelf
lives and require novel technologies
to detect microbes that are more
rapid than traditional methods,”
says Lovatt. Cellular- and gene-ther-
apy products, which not only have
short shelf lives, but also nontradi-
tional lot sizes, are driving the need
for rapid microbiological methods
(RMMs) to reduce testing times and
sample volumes, agrees McKee.
In addition, for cellular thera-
pies that involve the modification
of patient cells followed by their
injection back into the patient,
the process is typically completed
within just a few days. As a result,
Lovatt points out that the use of
contract research organizations/
third-party testing laboratories may
not be practical. “Microbiological
testing for these therapies may
need to be performed at the pro-
duction site in order to avoid addi-
tional delays due to the need for
the shipment of samples,” he says.
Ortiz also notes that developing
and implementing appropriate tech-
nologies for the detection of organ-
isms in many of the new types of
biologic drug substances and drug
products classified as next-genera-
tion therapies is also a challenge.
VACCINES DRIVE TEST DEVELOPMENTThe need for a rapid response to a
possible influenza pandemic in the
first decade of the 2000s spurred
the development of RMMs, accord-
ing to McKee. Alternative strategies
for in-process release of vaccine
batches were needed to accelerate
production of flu vaccines, with
the 14-day sterility test seen as a
prime target for decreasing the
turnaround time.
The high cell content in many
vaccine batches can also interfere
with the ability to visually read
traditional test results. “Alternative
test methods based on detection of
metabolites or luminescence have
been developed that can overcome
this obstacle.” McKee notes.
Viral vaccines in particular can
cause challenges due to the need
to generate neutralizing antisera to
neutralize the cytopathic effect of
the vaccines on the cells that are
used to detect viruses/mycoplasma,
such as MRC-5 and Vero, etc. “The
generation of the antisera can sig-
nificantly extend the time required
for the testing process and, con-
sequently, manufacturing of viral
vaccines,” says Lovatt.
Quality: Microbiological Testing
US and EU Regulators Seek to Reduce Inspections
FDA is assessing an information exchange with the European Medicines Agency
(EMA) that would identify facilities with strong records of compliance with good
manufacturing practices based on inspections by competent local inspectorates,
potentially reducing the number of facility inspections.
Although FDA has been receiving inspection reports from EMA for years, current
FDA policy prevents sharing of trade secret information that appears in field inspection
reports, explained Dara Corrigan, associate commissioner for global regulatory policy
at FDA at the ISPE/FDA/PQRI conference in Bethesda, MD, in June 2016. Other US
government agencies share sensitive, classified information with European Union (EU)
authorities, offering a precedent for FDA to act similarly. Legislation enacted in 2012
permits FDA to share confidential information in situations where the agency can
certify that the other country can keep this information secret.
FDA has been negotiating for three years to devise a system for mutual reliance
on inspection reports by local regulators found to meet acceptable standards. The
situation is complicated due to each of the 28 EU member states conducting its
own pharma facility inspections following different practices and standards.
Currently, regulatory officials from FDA, EMA, and EU member states are auditing
inspection programs by other authorities, with the goal of building confidence in the
capabilities of the other inspectorates and their inspection findings.

July 2016 www.biopharminternational.com BioPharm International 23
Influenza vaccines, for example,
must be manufactured in a very
limited time due to their seasonal
nature, and it is not possible to gen-
erate antisera quickly enough. As a
result, microbiological testing using
rapid nucleic acid methods is allowed
in the pharmacopeias, while poly-
merase chain reaction (PCR) methods
for mycobacteria detection, which
typically take one week, can be used
as alternatives to traditional culture-
based methods, which take up to 56
days, according to Lovatt. Nucleic
acid testing is also an effective alter-
native for microbial testing of viral
vaccines for which the viral drug sub-
stance is difficult to neutralize.
REGULATORY FLEXIBILITY CAN BE BENEFICIALSignificant changes to regulatory
requirements for microbial testing
have, in fact, been crucial for acceler-
ating the adoption of more advanced
methods. As mentioned previously,
the pharmacopeias have added chap-
ters to address the needs of next-gen-
eration therapies for RMMs that can
assure product safety. McKee points
to the European Pharmacopoeia (Ph.
Eur.) chapters 2.6.7 Mycoplasmas and
2.6.27 Microbiological Control of
Cellular Products as two important
examples. Ph.Eur. 2.6.27 outlines an
alternative to traditional compendial
sterility testing. “The 2.6.27 method
is suitable for qualification and vali-
dation of rapid methods for sterility
testing and incorporates conditions
and control organisms that yield a
more sensitive and broader range of
detection for cellular products,” she
explains. Ph.Eur. 2.6.7 Mycoplasmas,
meanwhile, includes specific guid-
ance for validation of nucleic acid
amplification techniques (NAT) that
may be used as alternatives to the
28-day culture and indicator cell
methods for detection of adventi-
tious mycoplasmas.
In addition, FDA has made
changes to its microbial testing
requirements that have had a posi-
tive impact on the development
of more rapid, advanced microbio-
logical methods (2). Amendments
to the Sterility Test for Biological
Products rule (21 Code of Federal
Regulations 610.12), which estab-
lishes FDA’s microbiological testing
requirements for biological products,
have been particularly important,
according to McKee. “The changes
to the rule provide manufacturers
of biologic products greater flexibil-
ity. Biopharmaceutical manufactur-
ers are in fact encouraged to use the
most appropriate and state-of-the-art
test methods to assure the safety of
biologic products,” she explains.
In addition, McKee notes that the
changes to the rule promote innova-
tion in the development of sterility
testing and are thus paving the way
for the use of novel methods such
as adenosine triphosphate (ATP) bio-
luminescence, chemiluminescence,
and even non-growth based RMMs.
“Not only was the use of a mandated
method for sterility testing removed,
the change in the rule allows greater
flexibility in sampling, putting the
onus of sample size on the manu-
facturer; the sample must be appro-
priate to the material tested both in
volume and representation of the lot
size,” she says.
There are some changes to regula-
tions that have created challenges,
however. Ortiz points to the rela-
tively recent low endotoxin recovery
(LER) phenomenon. For biologics,
FDA is requesting studies to deter-
mine if drug substances or drug
products demonstrate LER when
performing endotoxin analyses (3).
Due to the recent discovery of this
phenomenon and the various formu-
lations of drug substances and drug
products, there have been difficulties
in developing proper studies for LER.
RAPID SOLUTIONS: AUTOMATED VS. ALTERNATIVEThe development of new test meth-
ods has been pursued using two
fairly different strategies. Some
methods are still culture-based, but
with automation to help accelerate
the process. Others involve com-
pletely new testing technologies.
“Some automated RMMs use
a culture phase coupled with an
automated end-point to reduce the
overall testing time, but are still
in keeping with the traditional
tests described in the compendia.
Alternative methods, on the other
hand, are different from traditional
methods and often do not incorpo-
rate a culture phase,” McKee says.
The results for alternative methods
are also often reported in units other
than colony forming units (CFUs),
which is the typical format for cul-
ture-based assays. The results of these
tests, therefore, are not directly com-
parable with those obtained using
traditional or automated growth-
based methods, according to McKee.
“The prevailing concern is that
these alternative methods require
extensive validation studies to dem-
onstrate equivalency to traditional
microbiological methods. The need
for such extensive method vali-
dation in addition to the capital
expenditures necessary to develop
and implement these new technolo-
gies poses a hurdle for most bio-
pharmaceutical manufacturers that
wish to employ rapid microbial
methods,” McKee concludes. The
strong value proposition that RMMs
offer, however, extends to the devel-
opment of assays targeting other
adventitious contaminants includ-
ing viruses, mycoplasmas, residual
DNA, and residual proteins. Assays
that can be performed near the bio-
reactor location and in real time are
definitely the assays for the future.
REFERENCES 1. International Council for Harmonisation,
Q5A (R1) Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin (September 1999).
2. FDA, “FDA issues final rule on sterility testing of biological products,” Press Release, May 3, 2012.
3. K. Williams, BioPharm International, 28 (7) 28-33 (2015).
Quality: Microbiological Testing

24 BioPharm International www.biopharminternational.com July 2016
Photo
by M
asakazu M
ats
um
oto
/Gett
y Im
ag
es
Forced degradation studies are
performed by means of various
stressing agents such as pH,
temperature, light, chemical
agents (e.g., oxidizing, deamidating
agents, etc.), and mechanical stress to
speed up the chemical degradation,
physical degradation, or instability
of a molecule. Currently, there are no
industry guidelines available defin-
ing how to perform forced degrada-
tion studies for biopharmaceuticals.
The guidelines only provide useful
definitions, general comments, and
a rough concept about degradation
studies (1–5). Strict guidelines with
specific ranges or exact conditions
for forced degradation studies are not
necessarily possible, as every mole-
cule is different, and certain freedoms
for selecting stress conditions for bio-
pharmaceuticals are inherent (4, 5).
Hence, conditions should be carefully
selected on a case-by-case basis (3).
Regulatory guidance documents
specify the following expectations on
forced degradation:
t 5IFNBOVGBDUVSFSTIPVMEQSPQPTFB
stability-indicating profile that pro-
vides assurance that changes in the
identity, purity, and potency of the
product can be detected (2, 3).
t 3FTVMUT G SPN GPSDFE EFHSBEBUJPO
studies will form an integral part of
the information provided to regula-
tory authorities (4, 5).
Furthermore, studies exposing the
biopharmaceuticals to stress condi-
Forced Degradation Studies for Biopharmaceuticals
Anette Skammelsen
Schmidt
The author addresses
critical issues to consider prior to
performing forced degradation studies and
provides best practice
recommendations for these types
of studies.
Anette Skammelsen Schmidt, PhD,
is senior research scientist,
API analytical development, at
Novo Nordisk A/S, Denmark.
Analytical Testing

July 2016 BioPharm International 25
Product & Service InnovationsAdvertorial
Company DescriptionTosoh Bioscience LLC is a major supplier of chromatography
products worldwide, particularly to the pharmaceutical,
biotechnology, and chemical industries. The company is a
division of Tosoh Corporation, a global chemical company
with headquarters and manufacturing facilities in Japan. We
provide bulk chromatographic resins for the purification
of biopharmaceutical drugs in commercial manufacturing
processes, and we manufacture analytical HPLC columns.
Tosoh’s portfolio of more than 500 TSKgel® and
TOYOPEARL® products encompasses all common modes of
liquid chromatography. CaPure-HA™, a hydroxyapatite resin
for biomolecule purification, is a unique resin from Tosoh in
that it is both the ligand and base bead.
Facilitiesr 5PTPI$PSQPSBUJPO#JPTDJFODF%JWJTJPOTFSWFT+BQBO
r 5PTPI#JPTDJFODF--$TFSWFT/PSUIBOE4PVUI
America
r 5PTPI#JPTDJFODF(NC)TFSWFT&VSPQF .JEEMF&BTU
and Africa
r 5PTPI#JPTDJFODF4IBOHIBJ$P -UETFSWFT$IJOB
r 5PTPI"TJB15&4JOHBQPSFTFSWFT"TJB1BDJGJDBOE
India
Servicesr 0OTJUFQBDLJOHBTTJTUBODFGPSQSPDFTTTDBMFDPMVNOT
r .FUIPETEFWFMPQNFOUBTTJTUBODF
r 3FHVMBUPSZFYQFSUJTF
r 3FEVOEBOUWBMJEBUFENBOVGBDUVSJOHMJOFTGPSQSPDFTT
media products
r $VTUPNBOBMZUJDBM TFNJQSFQ BOEQSFQTDBMF)1-$
columns
r 1SPGFTTJPOBMUFDIOJDBMTFSWJDFTGPSBMMPGPVSQSPEVDUMJOFT
Major Product Innovationsr $B1VSF)" IZESPYZBQBUJUFNFEJBGPSUIFQVSJGJDBUJPO
of biomolecules
r 50:01&"3-/)'BOJPOFYDIBOHFSFTJOGPSUIF
purification of proteins in elevated salt conditions
r 50:01&"3-"'S1SPUFJO")$'IJHIDBQBDJUZ
resin for the capture and purification of monoclonal
antibodies
r 50:01&"3-(JHB$BQ® high capacity/high resolution
low elution volume ion exchange resins for protein
purifications:
50:01&"3-(JHB$BQ44
50:01&"3-(JHB$BQ24
50:01&"3-(JHB$BQ$.4
r 54,HFM18UZQFIJHISFTPMVUJPOSFTJOT
54,HFM4VQFS218GPSPMJHPOVDMFPUJEF
purification
54,HFM4118GPSTNBMMFSQSPUFJOBOE
peptide purification
54,HFM4118GPSJOTVMJOQVSJGJDBUJPO
r 50:01&"3-#VUZM 1IFOZM BOE11(
resins for mAb purification
r 50:01&"3-)FYZM$SFTJOGPSGMPXUISPVHI
polishing applications
Markets Servedr E. Coli and mammalian cell expressed biologics such
as monoclonal antibodies, cytokines, growth factors,
insulin, blood factors, plasma, and other large and small
proteins and peptides
r 0UIFSNBSLFUTTFSWFEJODMVEFPMJHPOVDMFPUJEFT %/"
3/" BOEQFHZMBUFEQSPUFJOT
Tosoh Bioscience LLC
Tosoh Bioscience LLC3604 Horizon Drive, Suite 100
King of Prussia, PA
Phone: 484.805.1219
Fax: 610.272.3028
www.tosohbioscience.com
Email: [email protected]

26 BioPharm International www.biopharminternational.com July 2016
AL
L F
IGU
RE
S A
RE
CO
UR
TE
SY
OF
TH
E A
UT
HO
R
tions may be useful in determin-
ing whether accidental exposures
to conditions other than those
proposed (e.g., during transpor-
tation) generate changes in the
molecule. Stress studies are also
useful for evaluating which spe-
cific test parameters may be the
best indicators of product sta-
bility and should be monitored
under proposed storage condi-
tions (3).
THE PURPOSE OF FORCED DEGRADATIONForced deg radat ion st udy i s
defined as an intentional break-
dow n of a molec u le to a n
appropriate extent by means of
various stressing agents (includ-
ing mechanical stress) to speed
up the chemical and physical
degradation and instability of a
biopharmaceutical. A forced deg-
radation study can give a range of
information regarding the likely
degradation products of a specific
biological drug. This informa-
tion can be useful for many pur-
poses, and can help to establish
the degradation pathways and the
intrinsic stability of the molecule.
Challenging the analytical proce-
dures helps validate the method’s
stability-indicating power (4, 5).
Prior to performing a forced
degradation study, the goal of the
study needs to be defined. Several
purposes might be addressed
in one study. When relevant, a
forced degradation study can be
performed at different develop-
ment stages. Figure 1 shows exam-
ples of the various reasons that
forced degradation studies are per-
formed.
Degradation products for bio-
pharmaceuticals may be either
product-related substances or
product-related impurit ies, as
some degradation products may
retain biological activity (1–3). An
example of this is illustrated in
Figure 2 and describes a situation
in which oxidation is not associ-
ated with a decrease in activity.
DEGRADATION PATHWAYS OF BIOPHARMACEUTICALSBiopharmaceuticals can usually
degrade in many different path-
ways following different kinet-
ics. The extent of stress needs
to provide a measurable change
and confirm the most relevant
degradation pathways. Too much
stress, however, might form sec-
ondary degradation products not
seen in formal stability studies,
and the level of stress might not
ref lect actual potential stress-
ors. An extent of degradation of
approximately 5–20% is assumed
to be suitable for most purposes
and for most analytical meth-
ods. An adequate level of stress
should be carefully selected on a
case-by-case basis (3).
The selection of the degradation
pathways to be investigated dur-
ing forced degradation should be
based on known and anticipated
degradation pathways—as well as
prior knowledge from similar mol-
ecules, if such knowledge exists.
The degradation pathways are typ-
ically either physical (e.g., aggrega-
tion) or chemical (e.g., oxidation)
in nature.
Aggregation can be noncovalent
in nature, such as an association of
monomers that are dissociable at
the right conditions (e.g., solvent,
temperature). These noncovalent
aggregates are mainly formed by
denaturation and unfolding of the
molecule, or by an interaction with
interfaces such as liquid-air, liquid-
solid, or even liquid-liquid. These
associations are typically a result of
mechanical stress such as shaking,
stirring, rotation, pumping; freeze-
thaw cycles; heating; or exposure
to acidic pH.
Analytical Testing
Figure 1: The purpose of forced degradation studies (FDS).
Figure 2: Determined oxidized forms (A) and activity (B) of drug substance (DS)
and drug product (DP) after exposure to 0.003% H2O2 at 22 °C for 0, 2, 4, 6, and
24 hours.
Identify likely
degradation
productsSupport process
development
Information to
process robustness
studies
Validation of
analytical methods,
including stability
indicating power
Impurity / variant
characterization Support
formulation
development
Support formal
stability studies
Provide samples for
analytical development
FDS
Establish and
understand
degradation
pathways
Determine
intrinsic stability
of molecule
60
50
40
30
20
10
0
Oxid
ized
fo
rm
s (
%)
0 10 20 30
Time (hours)
0 10 20 30
Time (hours)
A B
DS
DP 2 mg
DP 1 mg
DS
DP 2 mg
DP 1 mg
Activ
ity

July 2016 www.biopharminternational.com BioPharm International 27
Aggregation can also be cova-
lent in nature, such as chemical
bonding between the molecules,
and is non-dissociable during buf-
fer change. These chemical bonds
are often formed by rearranged
disulfide bridges or other altered
intramolecular bridges. They are
typically a result of reactions of the
amino acid residues with trace met-
als (copper or iron) or an incom-
plete reduction of the protein.
Side chains of methionine,
cysteine, histidine, tryptophan,
or tyrosine residues are suscep-
tible to oxidation, where methio-
nine is the most reactive residue.
Oxidation can alter the physico-
chemical properties of a protein,
such as folding and subunit asso-
ciations. The oxidation is mainly
due to exposure to atmospheric
O2 under condit ions of l ight,
heat, moisture, agitation, or to
exposure to oxidizing agents.
Deamidation is a hydrolytic con-
version of asparagine or gluta-
mine to a free carboxylic acid
residue and is typically due to
changes in pH, ionic strength,
temperature, and humidity in
the case of lyophilized proteins.
The overall effect of a chemical
modification of a single amino
acid residue depends on its posi-
tion in the protein and on the
specific role the residue has in
the functionality and active site
of the protein.
Photolysis by exposure to light
involves a free radical mecha-
nism that affects many functional
groups (e.g., carbonyl groups). The
free radicals can result in oxida-
tion, aggregation, or peptide bond
cleavage. The photolysis is due
to exposure to photo-irradiation,
which is typically in the form of
ultraviolet irradiation.
Hydrolysis (f ragmentat ion)
is a cleavage of peptide bonds
between amino acid residues
releasing smaller peptide chains.
The peptide bonds of Asp-Pro
and Asp-Gly are the most suscep-
tible to hydrolysis. Hydrolysis is
mainly a result from exposure to
acidic or alkaline pH.
Disu l f ide br idge exchange
might cause incorrect paired
disulfide bridges, which affects
the tertiary structure of a protein.
Such incorrect disulfide bridges
might be a result of partial cleav-
ing and reformation of disulfide
bonds as a result from denatur-
ing/reducing conditions (expo-
sure to reagents such as GnHCl,
u rea, and 1,4 -Dith iothre itol
[DTT]) and oxidation of cysteine
residues such as oxidation by Cu
(II) or Fe (II) ions.
Severa l biopharmaceut ica ls
contain ligands or conjugates.
Such bound moieties (e.g., acyla-
tion and conjugation) might be
lost due to chemical or physical
stress on the molecule.
SELECTION OF MATERIALS FOR A FORCED DEGRADATION STUDYWhen performing forced degra-
dation studies, it is important
to use a single batch of material.
Forced degradation studies usu-
ally require a large amount of
material. However, the material
could be non-GMP, a test batch,
or even an out-of-specification
batch (if such is available), as long
as the choice of batch is justified.
A l l re levant sample t y pes
should be included in the forced
degradation study. Drug prod-
uct at both high- and low-dose
levels can be included for drug
product-specific methods. If the
molecule is modif ied (e.g., by
acylation, glycosylation, or con-
jugation), the inclusion of the
intermediate is highly recom-
mended to aid understanding of
Analytical Testing
Table I: Examples of selected analytical methods for evaluation of degradation
pathways.
PathwayRP–HPLC
SE–HPLC
IE–HPLC
Peptide mapping
SDS–PAGE
Activity*
Aggregation - xxx - x x x
Fragmentation/clips
xx x x x xxx x
Oxidation xxx - - xxx - x
Deamidation x - xxx xx x x
Disulfide bridge exchange
x x - x - x
*Effect depends on location and extent of change in molecule
- indicates no effect, x indicates small effect, xx indicates significant effect, xxx indicates severe effect
RP–HPLC: reversed-phase high-performance liquid chromatography
SE–HPLC: size-exclusion high-performance liquid chromatography
IE–HPLC: ion-exchange high-performance liquid chromatography
SDS–PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis
Due to the complexity
of biopharmaceuticals,
there is no single
stability-indicating
method that can
profile all its stability
characteristics.

28 BioPharm International www.biopharminternational.com July 2016
Analytical Testing
the changes seen in the underly-
ing structure of the molecule.
Solution/buffer blanks and con-
trols (excipients) are included for
evaluation of peak profile regard-
ing occurrence of new peaks
as a result of stress conditions.
Always include reference samples
in each experiment.
SELECTION OF ANALYTICAL METHODS FOR FORCED DEGRADATION STUDIESDue to the complexity of bio-
pharmaceuticals, there is no sin-
gle stability-indicating method
that can profile all its stability
characteristics (2, 3). The nature
of biopharmaceuticals will dic-
tate which test methods to use.
In general, methods that are
used in stability studies should
be included in forced degrada-
tion studies, as well as methods
that determine identity, purity,
content, and methods for moni-
toring impurities. The methods
should provide reliable data—as
measured by a satisfactory selec-
tivity between the main peak
and impur it ies—an adequate
intermediate precision, and be
able to detect the change i f/
when it occurs. Examples of ana-
lytical methods to evaluate deg-
radation pathways are shown in
Table I.
Examples of common methods
to employ for analysis of biophar-
maceuticals during forced degra-
dation are appearance (i.e., color,
clarity, particulate matter); activ-
ity measurement; sodium dodecyl
sulfate polyacrylamide gel electro-
phoresis (SDS–PAGE); microchip
gel electrophoresis; size-exclusion
high-performance liquid chro-
matography (SE–HPLC) (e.g., for
protein content and aggregates);
reversed-phase high-performance
liquid chromatography (RP–HPLC)
(e.g., for purity and specific impu-
rities); isoelectric focusing (IEF)/
imaged capillary isoelectric focus-
ing (iCE)/ion-exchange HPLC (IE–
HPLC) (e.g., for deamidated forms);
peptide mapping; and physico-
chemical analysis (e.g., differen-
tial scanning calorimetry [DSC],
circular dichroism [CD], and fluo-
rescence). Additional analysis can
be employed based on the results
obtained by the initially selected
analytical methods.
SUITABLE CONDITIONS FOR FORCED DEGRADATION STUDIESAll molecules can be degraded
by some chemical or physical
means. Figure 3 shows examples
of common st ress condit ions
known to induce different deg-
radation pathways for biophar-
maceuticals. The conditions used
in forced degradation have to be
harsher than conditions used in
accelerated studies. If the condi-
tions result in no change, longer
exposure time is recommended,
rather than the use of a more
ext reme temperat u re. W hen
selecting the relevant stress con-
dit ions, the fol lowing points
must be considered:
t8FSFBMMEFHSBEBUJPOQBUIXBZT
covered?
Figure 3: Examples of common stress conditions, including light for forced
degradation studies.
DTT: 1,4-Dithiothereitol; ICH: International Council for Harmonization
Oxidation
8 e.g., 0.003% H2O
2 overnight 25 ºC, or
atmospheric O2
8 pH
8 e.g., overnight at 5 ºC and 25 ºC
8 Elevated temperature
8 e.g., 1-2 weeks or even up to 4 weeks
8 Depends on normal storage temperature
(and accelerated studies)
8 Normal storage (e.g., 25 ºC), then
stressed study (e.g., 50 ºC to 60 ºC)
8 Mechanical stress
8 e.g., rotation, shaking, agitation,
freeze-thaw
8 Freeze-thaw (e.g., 5-10-15 cycles)
8 Reduction
8 e.g., 0.01 M DTT overnight 25 ºC
Light
8 Exposure - ICH Q1B conditions
8 ≥ 1.2 million lux hours and ≥ 200 W
hours per m2 (25 ºC); ICH Confirmatory
conditions
8 Dark control (25 ºC); wrapped in
aluminium foil and placed next to
exposed sample
8 Exposure - stressed conditions
8 ≥ 2.4 million lux hours and ≥ 400 W
hours per m2 (25 ºC); stress conditions
8 Dark control (25 ºC); wrapped in
aluminium foil and placed next to
exposed sample
8 Depending on the biologics, even
harsher conditions can be employed
Figure 4: (A) Determined high-molecular weight proteins (HMWP) and (B) total
protein content after storage at elevated temperature (5 °C, 25 °C, and 37 °C)
at one and two weeks. The chromatogram (C) from the high-performance liquid
chromatography illustrates the increasing HMWP peak at high temperature.
45
40
35
30
25
20
15
10
5
0
1.2
1.0
0.8
0.6
0.4
0.2
0.0
8.00 10.00 12.00 14.00 16.00 18.00 20.00
A
C
B
T=0 (as is)
1 week
2 weeks
T=0 (as is)
1 week
2 weeks
5 25 37
Temperature (ºC)
5 25 37
Temperature (ºC)
HMWP
Mo
no
mer
Retention time (minutes)
37ºC 2 weeks
25ºC 2 weeks
5ºC 2 weeks
T = 0 (as it)
HM
WP
(%
)
Ab
sorb
an
ce U
nit
s
12
10
8
6
4
2
0
Co
nte
nt
(mg
.ml)

July 2016 www.biopharminternational.com BioPharm International 29
t )PXNBOZ UJNF QPJOUT XFSF
used?
t )PXNBOZ FYUSB TBNQMFT GPS
new methods and character-
ization) were used?
The total protein content should
be measured for all samples (as
shown in Figure 4) to evaluate
the presence of insoluble aggre-
gates. As the determined total
protein content is constant under
the applied conditions, insoluble
aggregates are not formed under
these conditions. Conditions of
high temperature and long peri-
ods of time, however, lead to high
amount of high-molecular weight
proteins (HMWP). Reference
samples have to be placed next
to forced degradation samples in
order to evaluate the cause for an
observed effect. All samples from a
specific study need to be analyzed
in the same analytical series to
exclude the effect of possible ana-
lytical variation.
FORCED DEGRADATION DURING THE DEVELOPMENT PHASESForced degradation studies can be
performed in early development
or late development depending on
the purpose of the study and the
amount of material available. The
health authorities expect forced
degradation studies to be carried
out during development Phase
III at the latest, but no guide or
specific requirements exist about
when to perform forced degrada-
tion studies.
A forced degradation study will
provide knowledge about the deg-
radation pathways of the molecule.
By performing such studies early
in development, this knowledge
about the molecule will be avail-
able for optimal process and for-
mulation development.
The degraded samples can aid
the development of stability-indi-
cating analytical methods by dem-
onstrating if the current methods
are sufficient to evaluate stabil-
ity (e.g., use oxidized samples to
develop method for determination
of oxidized forms) and by identify-
ing which test parameters are the
best indicators of stability.
Degraded samples are also use-
ful during analytical validation,
as they can be spiked in valida-
tion samples. However, a limited
amount of material is usually
available at the early stage of
development and the analytical
package might be incomplete.
During development, the process
steps and the formulation might
change. Additionally, the analyti-
cal methods might change due to
further optimization of the ana-
lytical conditions. Hence, forced
degradation studies most likely
need to be repeated or extended
at a later stage of development. In
conclusion, a limited forced degra-
dation study should be performed
as early as possible during develop-
ment, and a more comprehensive
forced degradation study during
Phase III should be performed.
GENERAL EVALUATION OF FORCED DEGRADATION STUDIESResults f rom forced degrada-
t ion st ud ies shou ld be pre -
sented graphically and should
include compare plots for chro-
matographic methods. A result
matrix is an excellent way to
show results, as such a matrix
will be able to indicate which
forced degradation conditions
resulted in changes for which
degradation pathway. Statistical
and kinetic tools should be used
for evaluation of data when pos-
sible to aid the understanding
of the degradation kinetics. A
forced degradation study reveals
the most important degrada-
tion pathways. Such pathways
can, for example, be indicators
of aggregation or the formation
of specific impurities that could
cause concern. The forced deg-
radat ion study a lso indicates
which analytical methods are
most concerning and whether
these methods are able to detect
the change that occurs. In sum-
mary, degradation pathway stud-
ies can help investigators predict
whether an analytical package
is suff icient for a molecule or
whether a need exists for the
development of other analytical
methods.
REFERENCES 1. ICH, Q6A, Specifications: New
Chemical Drug Substances and
Products, Step 4 version (1999).
2. ICH, Q6B, Specifications Test
Procedures and Acceptance Criteria
for Biotechnological/Biological
Products, Step 4 version (1999).
3. ICH, Q5C, Stability Testing of
Biotechnological/Biological
Products, Step 4 version (1995).
4. ICH, Q1A(R2), Stability Testing
of New Drug Substances and
Products, Step 4 version (2003).
5. EMEA, Guideline On Stability Testing:
Stability Testing of Existing Active
Substances and Related Finished
Products (London, October 2003).
Analytical Testing
Forced degradation studies can be performed
in early development or late development
depending on the purpose of the study and the
amount of material available.

30 BioPharm International www.biopharminternational.com July 2016
This article reviews the current
dynamics in the RNA thera-
peutics/vaccines market as well
as differences between small-
interfering RNA (siRNA), RNA interfer-
ence (RNAi), microRNA (miRNA), and
messenger RNA (mRNA). In addition,
the authors outline the general produc-
tion processes for these platforms, the
challenges encountered during process
development and production, and the
strategies to overcome them.
MARKET OVERVIEW RNA-based therapeutics target the treat-
ment of diseases such as diabetes, cancer,
tuberculosis, and some cardiovascular
conditions. There is currently a great
deal of money being put into this rela-
tively new class of therapeutics and vac-
cines, which is projected to grow 12%
in 2016 and reach $1.2 billion by 2020
(1). The 2015 research and develop-
ment (R&D) biotech pipeline is shown
in Figure 1. There are more than 700
nucleic acid-based therapeutics (DNA
and RNA) in the pipeline and more than
60% of the nucleic acid-based therapeu-
tic pipeline is in preclinical development.
It is interesting to note that 35% of such
pipeline is focused on oncology (2, 3).
Several companies (approximately 160)
and many academic institutes (approxi-
mately 65) are developing RNA-based
therapeutics. Table I provides a non-
comprehensive list of a few (4). Two
companies have marketed R NA-
based therapies: NeXstar and Ionis
Pharmaceuticals. There are 12 mRNA
vacc ines in development, seven
of which are being developed by
Curevac (Germany). Based on current
outlook, the RNA therapeutics market
seems more promising than the mar-
ket for DNA therapeutics.
From a partnership perspective, Ionis
Pharmaceuticals has entered into a global
collaboration with Janssen Biotech, Inc.
to discover and develop antisense drugs
to treat autoimmune disorders of the gas-
trointestinal tract (5), and Merck & Co.
(MSD) has bet $100 million on Moderna’s
mRNA technology (6). Moderna also has
previously announced collaborations
ABSTRACTIn 2014, the monoclonal antibodies market had the highest growth rate (19%)
for the number of new molecules in the pipeline. DNA and RNA therapeutics were not far behind, achieving 12% year-over-year growth. Industry analytics data
suggest that the RNA-based therapeutics market will reach $1.2 billion by 2020.
Bioprocessing Technology Trends of
RNA-Based Therapeutics and Vaccines
Elina Gousseinov, Mikhail Kozlov,Claire Scanlan, Aaron Hammons, Ling Bei, Youssef Benchek, Karim Pirani, and Ruta
Waghmare are all from MilliporeSigma.
Priyabrata Pattnaik works in the
Biomanufacturing Sciences and Training
Centre at Merck Pte Ltd., Singapore.
PEER-REVIEWED
Article submitted: Nov. 9, 2015.
Article accepted: April 5, 2016.
LA
GU
NA
DE
SIG
N/G
ett
y I
ma
ge
sPeer-Reviewed
Claire Scanlan, Priyabrata Pattnaik, Ruta Waghmare, Elina Gousseinov,
Mikhail Kozlov, Aaron Hammons, Ling Bei, Youssef Benchek, and Karim Pirani

July 2016 BioPharm International 31
Product & Service InnovationsAdvertorial
Our CompanyAs a premier contract discovery, development, and
manufacturing organization, WuXi Biologics (a wholly-
owned subsidiary of WuXi AppTec Inc.) provides
our worldwide clients with the necessary expertise,
quality, and capacities to develop biologic drugs from
concept to commercialization. Through the open-access
technology platforms we provide in our Shanghai,
Wuxi and Suzhou facilities and along with our WuXi
AppTec affiliates, we provide the world with the ONE
true single-source approach that saves our clients critical
time and money.
FacilitiesOccupying more than 750,000 sq. ft. of lab and
manufacturing space, our network of facilities operates
under global regulatory standards and provides
unparalleled capacities. This network is comprised of
three main facilities: 1.) Shanghai facility—supports all
pilot-scale projects, including antibody discovery, process
development and preclinical studies; 2.) Wuxi facility—
offers cGMP manufacturing of DS and DP using fully
disposable bioreactors; 3.) Suzhou facility—supports
toxicology studies and biosafety testing. All the facilities
in the network are located within 1-2 hours’ drive from
each other and provide a simplified supply chain for our
clients worldwide.
Markets ServedWe support biologics drug developers of all sizes in all
major markets, from top 20 pharmaceutical companies to
virtual biotech companies by providing integrated CMC
package (from DNA to IND filing) as well as standalone
services to meet the customers’ outsourcing needs at
different stages of development.
Services/InnovationsIn addition to full CMC development capabilities, we also
provide research material generation services that utilize a
variety of technologies to produce high-quality antibodies,
recombinant proteins, and cell lines for early-stage
drug screening and lead selection, assay development,
structure analysis, and other research purposes. Our team
has extensive experience with various types of proteins,
including monoclonal antibodies, bi-specific antibodies,
kinases, proteases, cell surface receptors, cytokines,
metabolic enzymes, nuclear receptors, DNA binding
proteins, and viral envelope proteins.
Research Grade Protein
Expression and Purification
r &YQSFTTJPOTZTUFNTE. coli, insect (baculovirus) and
mammalian Cells (HEK 293, CHO-S, and CHO-K1)
r 1VSJGJDBUJPOTZTUFNT"GGJOJUZ JPOFYDIBOHF )*$ 4&$
and endotoxin removal columns
r 1SPUFJODIBSBDUFSJ[BUJPOT4%41"(&*&' QSPUFJO
determination, UV spectrophotometry, western blot,
gel filtration, mass spectrometry, isothermal titration,
DBMPSJNFUSZ CJPDIFNJDBM BOEFOEPUPYJOUFTUTBTTBZT
Assay Cell Line Generation
r 1BSFOUBMDFMMMJOFT)&,#B'$$)0
r 7FDUPSTZTUFNTQD%/"Q%JTQMBZ™; pLEX;
Q-&/55BHTDPVMEJODMVEF7 DNZD 'MBH
r 5SBOTGFDUJPO-JQJENFEJBUFE&MFDUSJDUSBOTGFDUJPO
Virus-mediated
r 4DSFFOJOHNFUIPET'"$4.JSSPSCBMM®
WuXi Biologics, a wholly owned subsidiary of WuXi AppTec
WuXi Biologics, a wholly owned
subsidiary of WuXi AppTec288 Fute Zhang Road Waigaoqiao
Free Trade Zone, Shanghai, China 200131
www.wuxibiologics.com

32 BioPharm International www.biopharminternational.com July 2016
with Alexion, AstraZeneca, and the Defense
Advanced Research Projects Agency (DARPA)
totalling $450 million. Moderna has raised $625
million in equity funding (7, 8).
RNA interference (RNAi) and RNA anti-
sense technologies appear to be dominating
the market. RNAi is a gene-silencing technol-
ogy in which RNA molecules inhibit gene
expression by targeting and destroying spe-
cific mRNA molecules. RNA antisense tech-
nology involves synthesizing an RNA strand
that binds to a specific mRNA or to a splic-
ing site on a pre-mRNA molecule to prevent
translation. The major challenges associated
with the commercialization of these RNA-
based therapies are toxicity and drug delivery.
RNA-BASED THERAPEUTICSWith the advent of RNA-based therapeu-
tics and their potential in treating a variety
of chronic diseases, it is important to note
the number of enabled technologies used to
exploit the RNA mechanism/pathway, some
of which are discussed in the following.
RNAi
RNAi technologies work by “silencing” or
turning off a gene through the use of its own
DNA sequence (Figure 2). The process is initi-
ated by double-stranded RNA (dsRNA) that
expresses either as a small or short hairpin
RNA (shRNA) or as a microRNA (miRNA) tran-
script. Using this silencing mechanism, RNAi
is commonly used to gain a better under-
standing of gene function, which can then
be used to generate additional targeted thera-
peutics (9). Small interfering RNA (siRNA) and
miRNA are the core elements of RNAi tech-
nology based therapeutics.
siRNA
RNAi utilizes a “dicer” enzyme to cut dsRNA
into 21 oligonucleotide segments, called
siRNA. These siRNAs can then bind to a
specific family of proteins called Argonaute
proteins, of which there are two classes: Ago
and Piwi. Ago proteins bind to siRNAs or
miRNAs, while Piwi proteins bind to Piwi-
interacting RNA (piRNA) and are used to
silence mobile genetic elements. The siRNA,
miRNA, or piRNA complex bound to the
Argonaute protein is called the RNA-induced
silencing complex (RISC). Once bound to the
Argonaute protein, one strand of the dsRNA
is removed and the remaining strand binds
to and directs the degradation of the comple-
mentary RNA target sequence, which then
leads to the loss of protein expression (10). AL
L F
IGU
RE
S A
RE
CO
UR
TE
SY
OF
TH
E A
UT
HO
RS
Peer-Reviewed
Figure 1: R&D biotech pipeline expansion.
9765
2209
727
2331
427
1104
15631488
976
295
1952
377
660
733
2009
8490
20152014
487
917
Monoclonal antibody
Bioengineered vaccine
Cell therapy
DNA & RNA therapeutics
Gene therapy
Antibody-drug conjugates
Other biotechnology product
Recombinant product
Figure 2: An insight into the RNAi pathway. Small hairpin RNA
(shRNA) is a class of double-stranded RNA (dsRNA). The dsRNA is
cleaved or degraded by a “dicer” enzyme into oligonucleotide segments
called small interfering RNA (siRNA), which then enter a cell to form
the RNA-inducing silencing complex (RISC). The siRNA strands then
separate or unwind to form the activated RISC complex, which can then
target messenger RNA (mRNA), bind to it, and cleave it.
Dicer
dsRNA cleavage
Target mRNA
Unwound siRNA
Target mRNA cleavage
RISC Complex
RISC Assembly
siRNA
or shRNA
dsRNA

July 2016 www.biopharminternational.com BioPharm International 33
It has been reported that synthetic siRNA
is able to knock down targets in various dis-
eases in vivo, including hepatitis B, human
papilloma virus, ovarian cancer, bone cancer,
hypercholesterolemia, and liver cirrhosis. Only
a few molecules of siRNA per cell are required
to produce effective gene silencing (11). siR-
NAs are most commonly delivered into cells
using microinjection or a transfection agent.
Many companies now offer siRNA-delivering
reagents to simplify this process (12).
miRNA
miRNA do not code for proteins, as they
belong to specific class of non-coding RNAs.
miRNA are 19–25 nucleotides in length and
are encoded within introns (i.e., the portions
of the gene sequence that are not expressed in
the protein) (13). miRNA acts as a guide strand
for the RISC complex to its mRNA target in
vertebrates. Approximately 30% of genes in the
human genome are regulated by miRNA (14).
Though siRNA silencing requires exact
match between target and small interfering
RNA, miRNA are non-specific and can exert
action through imperfect base pairing. In
addition, miRNA triggers translation inhibi-
tion (i.e., prevents the RNA from synthesiz-
ing protein from amino acids), while siRNA
triggers mRNA degradation.
mRNA
mRNA, which codes for protein, is an essen-
tial component of the central dogma of life
(DNAmRNAprotein). mRNA is tran-
scribed from a DNA template. mRNA takes
the genetic code from DNA to the ribosome
where the mRNA is translated to protein.
There has been a significant increase in
mRNA-based therapies in large part due to
the many advantages that mRNA has over
DNA in relation to gene expression and
transfer. While RNAi and antisense RNA
technologies are used primarily for gene
Peer-Reviewed
Table I: Biopharmaceutical companies developing RNA-based therapeutics and vaccines.
siRNA=small interfering RNA, miRNA=microRNA, and mRNA=messenger RNA.
siRNA miRNA mRNA
Kyowa Hakko Kirin Andes Biotechnologies CureVac
Silence Therapeutics Mirna Therapeutics Biontech RNA Pharmaceuticals
Debiopharm miRagen Therapeutics Boehringer Ingelheim
Marina Biotech Marina Biotech Johnson & Johnson
Ipsen Moderna Therapeutics Ludwig Institute for Cancer Research
Alnylam Pharmaceuticals Alnylam Pharmaceuticals BioNTech
Sanofi Pasteur Sanofi Pasteur Sanofi Pasteur
Tekmira Pharmaceuticals Tekmira Pharmaceuticals
NanoCarrier Regulus Therapeutics
Dicerna Pharmaceuticals Biogen Idec
BioCancell Therapeutics GlaxoSmithKline
Samyang Group AstraZeneca
Silenseed Ionis Pharmaceuticals
siRNAsense Les Laboratoires Servier
Reference Biolabs Celsion
Avena Therapeutics Rosetta Genomics
Lipella Pharmaceuticals Santaris Pharma
Arrowhead Research Shire
InteRNA Technologies
Alexion Pharmaceuticals
t2cure
Rigontec
Microlin Bio

34 BioPharm International www.biopharminternational.com July 2016
silencing, mRNA technologies are often used
in vaccines or gene therapy (15). In both
cases, after injection into the human body,
mRNA is translated to protein, which can
ultimately replace a missing protein (ther-
apeutic) or induce an immune response
(preventive approach). The production of
synthetic mRNA for therapeutic use is rela-
tively straightforward, and the challenges
associated with its stability and delivery have
been tackled through scientific advances in
recent years (16).
RNA-BASED VACCINESConceptually, mRNA-based vaccines are a
simple approach to inducing an immuno-
logical response by delivering the coding
genetic element as a translation-ready mol-
ecule. Upon direct vaccination with mRNA
molecules, dendritic cells (antigen-present-
ing cells) take-up, process, and encode the
target antigen, which in turn induces an
immune response. Typically, mRNA vaccines
are produced by in-vitro synthesis through
an enzymatic process. Such a synthetic pro-
cess can be tightly controlled, resulting in
a quality and predictable product profile.
mRNA can be easily tailored to offer a spe-
cific immunogenic profile and pharmacoki-
netics (17). mRNA’s stability and antigenic
properties can be easily manipulated by
changing codon or modifying base pairs.
Ongoing clinical trials show that mRNA
can be delivered as naked mRNA; immobi-
lized on particles or in liposome nanopar-
ticle; or transfected in dendritic cells in vitro
resulting in a discernible immune response
and protective efficacy. mRNA can also act
as an adjuvant and mRNA also has been
explored to stimulate the innate immune
system through toll-like receptors (18). RNA-
based vaccines are comparatively simple
to produce and can be developed, manu-
factured, and administered in a short time
period, therefore, they are suitable for pan-
demic situations. Thermostability of mRNA
vaccines can also significantly contribute to
their low cost, as they do not require cold-
chain distribution.
MANUFACTURING RNA-BASED BIOPHARMACEUTICALSAs more experimental RNA drugs move
through the clinic and into large-scale tri-
als, the demand for efficient and cost-effec-
tive manufacturing strategies will grow (19).
RNA-based biopharmaceuticals are inher-
ently susceptible to endonucleases, so spe-
cial handling is required for production
and purification. Degradation of product
during manufacturing adds heterogeneity
and chemical instability to the product.
Therefore, the manufacturing and purifi-
cation methods used in RNA-based thera-
peutics differ from that of DNA and other
proteins (20).
mRNA purification (post-chemical syn-
thesis) includes concentration precipitation,
extraction, and chromatographic methods
(including high-performance liquid chroma-
tography) (19). The purpose of the upstream
concentration and diafiltration step is to
concentrate (if lower titer) and change the
buffer to the necessary pH and conductiv-
ity for the first chromatography step. The
objective of the final concentration and dia-
filtration step is to de-salt and achieve the
necessary final concentration prior to ster-
ile filtration. A 5-kD membrane cut-off is
generally used for concentration and diafil-
tration in mRNA processes. Because siRNA
are smaller than mRNA, a 1-kD membrane
cut-off is used for adequate retention of the
siRNA product (21).
CHROMATOGRAPHIC PURIFICATION STEPSSince the breakthrough discoveries of cat-
alytic RNAs in the early 1980s and RNA
interference in the late 1990s, more than
50 RNA or RNA-derived therapeutics have
reached clinical testing. In RNA purifica-
tion, despite the different techniques such
as arginine-affinity, ion-pairing reversed-
phase, or pellicular anion exchange, the
traditional ion-exchange (IEX) media—
especially anion exchange (AEX)—remains
the most popular technique used in both
pure RNA and RNA packaged for delivery
(22, 23, 24, 25).
Sm and Sm-like proteins, which can form
heteromeric complexes or bind to vari-
ous RNAs, were proven to contain ancient
RNA-binding motifs (Sm domain) with
oligo(U) specificity (26). Fractogel TMAE
(MilliporeSigma), a strong anion-exchange
resin, was used for the purification of small
nuclear ribonucleoproteins (snRNP). The
snRNP molecule was eluted with Tris/HCl
Peer-Reviewed

July 2016 www.biopharminternational.com BioPharm International 35
and 300 mM NaCl. Ribonucleoprotein and
uncoupled RNA were separated from free pro-
tein, and the sample was immediately used
for negatively stained electron microscopy.
Both AEX and reversed-phase (RP) tech-
nologies are widely used in the RNA puri-
fication process. Quarternary amine (Q)
and Dimethylaminoethyl (DMAE) chemis-
try are among the choices for AEX (27, 28,
29, 30). One study proved that a few AEX
resins can be used for RNA purification
with optimized experimental conditions to
achieve high dynamic binding capacity. In
this study, among the 18 AEX medias that
were screened, only four resins—Q Sepharose
FF (GE Healthcare), POROS 50HQ (Applied
Biosystems), Q Ceramic HyperD F (Pall), and
Fractogel DEAE (MilliporeSigma)—showed
baseline separations of RNA and plasmid
DNA (31). After optimized loading and elut-
ing conditions, Fractogel DEAE had a wider
range of operation, higher dynamic binding
capacity, and complete separation of RNA
in the breakthrough from plasmid in the
elute. The high recovery, robustness, and
reproducibility also met the requirement for
large-scale manufacturing. These binding
and elution conditions can be utilized as a
starting point for optimal experimental con-
ditions in RNA purification.
Overall, many biochromatography res-
ins are suitable for RNA purification similar
to use in other biomolecule separations. In
many cases, the Fractogel resins showcased
superior capacity and efficiency, largely due
to the “tentacular” structure whereby func-
tional groups are located at the end of long
arms grafted to the bead surface, which cir-
cumvent the steric hindrance caused by large
biomolecules (32).
FORMULATION AND DELIVERYThe most challenging aspect of RNA-based
therapeutics is its delivery to target cells.
Several methods have been explored and
tested in clinical trials. Some of the most
promising approaches are explained in the
following passages.
Polymer conjugation/
chemical modification
Native RNA and RNA-based therapies are
vulnerable to degradation from the many
r ibonucleases found within the cel l .
Chemical modification is one method for
hardening the RNA against such enzymatic
attacks. Modifications to the molecule can
also increase its target affinity, decrease its
undesired immunogenicity, and improve its
overall efficacy. Hardening strategies include
modifications to the backbone, sugar, or base
of the RNA molecule.
Conjugation of the RNA therapeutic is
a strategy that is increasingly being used
for improved delivery and uptake. Alnylam
Pharmaceuticals has adopted a method of
conjugating an amino sugar derivative of
galactose, N-Acetylgalactosamine (GalNac)
to improve the delivery of siRNA therapies
to the liver. The GalNac-conjugated siRNA
is taken up by asialoglycoprotein receptors
in the liver resulting in a fivefold increase in
efficacy versus the parent molecule (33).
Arrowhead Research is developing a com-
peting conjugation strategy. Arrowhead’s
delivery technology, termed Dynamic
Polyconjugates (DPCs), is a siRNA bound to
an endosomolytic polymer backbone via a
disulfide bond. The endosomolytic polymer
enables the quick and efficient release of the
siRNA from the endosome. Arrowhead’s most
recent strategy includes attaching cholesterol
to the siRNA and GalNac to the endosomo-
lytic polymer, ensuring they are both deliv-
ered to the hepatocytes. The co-injection
therapy was shown to increase the efficacy
of siRNA-cholesterol 500-fold with a 90%
knockdown (34).
Encapsulation
The dominant and most-studied strategy for
the delivery of RNA-based therapeutics is
lipid-based delivery systems. One successful
platform is the use of stable nucleic acid lipid
particles (SNALPs), which are lipid particles
formed from a fusogenic lipid, cationic lipid,
and PEG-lipid mixture. The SNALP deliv-
ery system has been developed and champi-
oned by Tekmira Pharma; the company now
refers to it as LNP technology. According to
Tekmira, the LNP “encapsulates siRNAs (also
mRNA) with high efficiency in uniform lipid
nanoparticles that are effective in delivering
RNAi therapeutics to disease sites in numer-
ous preclinical models” (35).
Another promising lipid delivery tech-
nology is the proprietary Smarticles deliv-
ery platform developed by Novosom and
Peer-Reviewed

36 BioPharm International www.biopharminternational.com July 2016
now owned by Marina Biotech. Similar to
SNALPs, the Smarticles technology can
change their surface charge to facilitate both
stability and endosomal release. Smarticles
are capable of encapsulating both single-
and double-stranded nucleic acid therapies.
Smarticles are comprised of cationic, anionic,
and neutral lipids. The negatively charged
Smarticles avoid the often seen toxic effects
of positively charged lipids at physiologi-
cal pH but convert to a positive charge in
the acidic environment of the endosome,
facilitating its release. Other interesting
encapsulation techniques involve PLGA
nanoparticles (36, 37).
CONCLUSIONRNA-based therapeutics are a relatively
new class of therapies that has bright pros-
pects in the treatment and prevention of
difficult-to-treat chronic and rare diseases.
RNAi work by interfering with the tran-
scription process, and thereby inhibit pro-
tein translation. Though such a therapeutic
approach is highly selective and targeted,
special care is required during the produc-
tion of these therapies and vaccines because
of their susceptibility to ubiquitous RNAse-
induced degradation. Large-scale manufac-
turing of new class of therapeutics would
require bioprocessing components, chemi-
cals, and tools free from RNAse. Technology
and tool providers need to consider making
such products available to enable large-scale
production of RNA-based therapeutics. The
surmounting challenges related to poten-
tial toxicity and drug delivery need to be
addressed before such products can be com-
mercialized. However, new technologies are
emerging to overcome some of these chal-
lenges, and the future of RNA-based thera-
peutics is very promising.
REFERENCES 1. Allied Market Research, “RNA Therapeutics
Market is Expected to Reach $1.2 Billion,
Globally, by 2020,” Press Release, www.
prnewswire.com/news-releases/rna-therapeutics-
market-is-expected-to-reach-12-billion-globally-by-
2020---allied-market-research-274471461.html,
accessed May 24, 2016.
2. Personal communication, Donia Slimani, EMD
Millipore (now MilliporeSigma).
3. EvaluatePharma, World Preview 2015,
Outlook to 2020 (8th Edition, June 2015).
www.evaluategroup.com/public/reports/
EvaluatePharma-World-Preview-2015.aspx,
accessed May 24, 2016.
4. E. Gousseinov et al., Genetic Engineering &
Biotechnology News (Sept. 15, 2015), www.
genengnews.com/insight-and-intelligence/rna-
based-therapeutics-and-vaccines/77900520/,
accessed May 24, 2016.
5. Ionis Pharmaceuticals (n.d.), www.ionispharma.
com/, accessed May 24, 2016.
6. D. Garde, “Merck Bets $100M on Moderna and
its Pioneering RNA Tech,” www.fiercebiotech.com/
partnering/merck-bets-100m-on-moderna-and-its-
pioneering-rna-tech, accessed May 24, 2016.
7. Moderna Messenger Therapeutics, “Our
Core ‘Expression’ Platform: Messenger RNA
Therapeutics,” www.modernatx.com/mrna-
expression-platform, accessed May 24, 2016.
8. B. Fidler, “With Massive Venture Round, Moderna
Has $450M Reasons to Stay Private,” www.
xconomy.com/boston/2015/01/05/with-massive-
venture-round-moderna-has-450m-reasons-to-
stay-private/2/, accessed May 24, 2016.
9. UMass Medical School, “How RNAi Works,” www.
umassmed.edu/rti/biology/how-rnai-works,
accessed May 24, 2016.
10. J. Höck and G. Meister, Genome Biol. 9 (2):210.
doi:10.1186/gb-2008-9-2-210. Feb. 26, 2008).
11. Gene Link, “What is RNAi and siRNA?”, www.
genelink.com/sirna/RNAiwhatis.asp, accessed
May 24, 2016.
12. M. Gujrati and Z.R. Lu, “Targeted Delivery of
Therapeutic siRNA,” in Gene Therapy of Cancer:
Translational Approaches from Preclinical Studies to
Clinical Implementation, E.C. Lattime and S.L. Gerson,
Eds. (Academic Press, 3rd ed., 2013), pp. 47–65.
13. Sigma-Aldrich, “miRNA (microRNA) Introduction,”
www.sigmaaldrich.com/life-science/functional-
genomics-and-rnai/mirna/learning-center/mirna-
introduction.html, accessed May 24, 2016.
14. Qiagen, “MicroRNA–Why Study It and How,” www.
sabiosciences.com/pathwaymagazine/pathways7/
microrna.php, accessed May 24, 2016.
15. R. Scott McIvor, Mol. Therapy 19 (5), pp. 822–
823 (2011).
16. U. Sahin, K. Karikó, Ö Türeci, Nat. Rev. Drug
Discov. 13 (10), pp. 759 –780 (October 2014).
17. T. Kramps and L. Probst, Wiley Interdisp. Rev.
RNA 4 (6), pp. 737–749 (July 25, 2013).
18. S. Pascolo, Handb. Exp. Pharmacol. 183, pp.
221–235 (2008).
Peer-Reviewed
Contin. on page 48

ON-DEMAND WEBCAST Aired June 21, 2016
Register for free at www.biopharminternational.com/bp/development
Functional Comparability Studies for
BiosimilarDevelopment
EVENT OVERVIEW:
Regulatory authorities require applicants for biosimilar
therapies to demonstrate that the proposed product is
biosimilar to the reference product using analytical and
other studies. Comparative analytical testing that evaluates
factors including structure and function can be used to
make decisions about the scope of subsequent studies and
could result in a shortened clinical development process.
This webcast will review key factors in developing,
optimizing and implementing comprehensive func-
tional comparability studies to provide data needed to
advance the development process. Experts will discuss
the regulatory requirements and guidelines for functional
comparability studies and the types of studies that should
be conducted; review approaches to understand multiple
mechanisms of action and assay development; and the
qualification of assays used in these studies.
Key learning objectives
Review the regulatory requirements for comparability studies
Learn which functional comparability studies should be conducted
Understand the role of reference products for comparability studies
Learn about development & qualification of assays for comparability studies
For questions contact Kristen Moore at [email protected]
Who should attend
Laboratory managers
Product development scientists
Process development leaders
Quality control professionals
Sponsored by
Presenters
Hoss A Dowlat, PhD
Vice President,
Regulatory Affairs, Global
Strategy
PharmaBio Consulting
Abhi Saharia, PhD
Director, Cell-based
Assays and Biologics
DiscoverX
Nicolas Fourrier, PhD
Director Biomarker
and Biopharmaceutical
Testing
SGS Life Sciences
Moderator
Rita Peters
Editorial Director
BioPharm International
Presented by

38 BioPharm International www.biopharminternational.com July 2016
The International Organization
for Standard izat ion ( ISO)
published the long-awaited
revisions to its standards for
classification and monitoring of air
cleanliness in cleanrooms on Dec. 15,
2015. ISO 14644-1:2015 “Cleanrooms
and associated controlled environments
Part 1: Classification of air cleanliness
by particle concentration” (1) replaces
ISO 14644-1:1999, and ISO 14644-
2:2015 “Cleanrooms and associated con-
trolled environments Part 2: Monitoring
to provide evidence of cleanroom per-
formance related to air cleanliness by
particle concentration” (2) replaces ISO
14644-2:2000.
The 2015 editions are the result of a
systematic review and include changes
made in response to requests by users and
experts in the cleanroom community. In
particular, the requests for reviewing Part 1
were related to “the basis for the number of
sampling locations and, most importantly,
the whole statistical basis of classification
of cleanliness using the Student T-test for
one to nine sampling locations,” notes
Gordon Farquharson, convenor of the ISO
TC209 working group 1, which performed
the review and revisions. Because Part 2 is
closely aligned with Part 1, the committee
reviewed both parts together.
CLASSIFICATION BY PARTICLE CONCENTRATIONThe addition of “by particle concen-
tration” to the title of the standard is
a long-overdue clarif ication, com-
ments Karen Ginsbury, CEO at PCI
Pharmaceutical Consulting. “For years,
I have heard cleanroom contractors and
practitioners alike wrongly describe ISO
14644-1 and 2 as cleanroom ‘validation’
or ‘qualification’ standards.” She notes
that the standards only address airborne
particles, not other factors crucial to
cleanroom qualification, such as smoke
tests to determine airflow patterns.
The introduction of Part 1 explains,
“This part of ISO 14644 specifies classes
of air cleanliness in terms of the num-
ber of particles expressed as a concentra-
tion in air volume. It also specifies the
standard method of testing to deter-
mine cleanliness class, including selec-
tion of sampling locations” (1).
SAMPLING CHANGESThe primary changes to Part 1 involve the
number of samples and the selection of
sampling locations. “The number of sam-
ples will increase from what was required
previously,” explains Marsha Stabler
Hardiman, senior consultant at ValSource.
“The minimum number of samples is now
determined from a lookup table (instead
of an equation), and that number is set to
be statistically significant.”
According to ISO, the new method
for selecting the sites and number of
sampling collections uses a more con-
sistent statistical approach based “where
samples are drawn randomly without
replacement from a finite population.
The new approach allows each location
to be treated independently with at least
a 95% level of confidence that at least
90% of the cleanroom or clean zone
areas will comply with the maximum
particle concentration limit for the target
class of air cleanliness. No assumptions
are made regarding the distribution of
the actual particle counts over the area
of the cleanroom or clean zone; while in
ISO 14644-1:1999 an underlying assump-
tion was that the particle counts follow
the same normal distribution across the
room” (1). The sampling locations are
to be chosen representatively, meaning
that “features such as cleanroom or clean
Revised ISO Cleanroom Standards Improve Air Cleanliness Classification
Jennifer Markarian
Revised versions of ISO 14644
Parts 1 and 2 introduce changes to
sampling procedures
and monitoring plans for
cleanrooms and clean zones.
Cleanroom Standards

July 2016 www.biopharminternational.com BioPharm International 39
zone layout, equipment disposition,
and airflow systems should be con-
sidered when selecting sampling
locations” (1).
This representative selection of
sample locations is a big change from
the previous random selection, says
Hardiman. “A company now has to
have a rationale and justification for
sample site location to ensure that
the sample locations selected are rep-
resentative of the characteristics of
that section. Companies will have to
look at the new number of sample
locations and then determine where
the representative sample locations
will be collected. If contracting out
the classification activities, you
should make sure that your contrac-
tor is now using your new, represen-
tative sample locations.”
CLASSIFICATION OF MACRO AND NANO-PARTICLESThe revision makes a change
regarding large (≥ 5 μm) particles,
which are required to be measured
for some classifications in the EU
Annex 1 GMP guidelines (3) and
others. “The experts working on
the revision of ISO 14644-1 were
of the opinion that particles ≥ 5
μm diameter should not be used
to classify ISO class 5 and cleaner
environments because of the
uncertainty associated with par-
ticle collection efficiency and
accuracy of counting low con-
centrations,” says Farquharson.
“In order that the European
Union (EU), the Pharmaceutical
Inspec t ion Convent ion and
Ph a r m a c e ut i c a l I n s p e c t io n
Co-operat ion Scheme, World
Health Organization, and Chinese
GMPs are not left without a clas-
sification tool for their Grades A (at
rest and operational) and B (at rest),
ISO 14644-1:2015 provides a mech-
anism of extrapolating the macro-
particle descriptor for class limits of
20 and 29 particles ≥ 5 μm.”
The new document does not
address nano-scale particles, which
were formerly defined as ultrafine
particles in ISO 14644-1:1999, but
will address these under a new
Part 12, notes Farquharson, who
explains, “These particles are mea-
sured using a different particle
counter, and industries such as
semi-conductor monitor for con-
centration of these very small air-
borne particles at critical control
points. These particles are not gen-
erally of interest to the pharmaceu-
tical and life sciences industries.”
MONITORING AND TESTINGISO 14644-2:2015 now requires
monitoring to provide evidence of
cleanroom performance, explains
Fa rqu ha r son. T he s t a nda rd
addresses airborne particle concen-
tration, airflow, and device pres-
sure difference. New topics include
monitoring of critical parameters
and setting action and alert alarms.
The revised standard now allows
companies to use risk management
to set their periodic classification
testing schedules, notes Hardiman.
“In the past, the retesting was pre-
scribed in a table and the timing was
based on the ISO class of the clean-
room or clean zone. Now, compa-
nies can put more emphasis on the
day-to-day data that they generate in
their own facilities to help determine
the appropriate testing and frequen-
cies needed for continued cleanroom
compliance. If a company is generat-
ing great data and the risk to contin-
ued cleanroom compliance is low,
then they can set a longer periodic
classification frequency,” explains
Hardiman. She notes that risk-based
sample site selection is crucial for
environmental monitoring. “The key
is understanding your unique prod-
ucts and processes and selecting sites
that best address the risks that your
products and processes present. It is
important to be able to identify all of
the potential contamination sources
in each cleanroom and to select
environmental monitoring sample
locations in close proximity to these
sources. It is also very important to
understand the people, material, and
waste flows,” concludes Hardiman.
Pharmaceutical cleanrooms typi-
cally already have monitoring plans,
which are required by Annex 1 of
the EU GMPs, says Ginsbury (3). She
notes that users should, however,
check with their cleanroom contrac-
tors to determine whether a contrac-
tor is qualified and familiar with
the revisions, because the regulators
reference the ISO standards and they
must be followed, says Ginsbury. “I
would recommend having the dis-
cussion now so that, by 2017, your
contractors and in-house staff are
fully up to speed and following the
new standard,” she notes. “Even if
you use contractors, the responsi-
bility for review and approval of
their results and compliance with
regulatory standards lies with you.
ISO may not require 5 μm particles
and may let you use risk assessment
to determine frequency of classifi-
cation. However, EU Annex 1 still
requires measuring 5 μm particles
as part of classification, and current
industry practice (EU [3] and FDA
[4]) is to perform cleanroom classi-
fication twice a year for aseptic core.
We haven’t heard the regulators
on this one but I wouldn’t rush to
reduce that frequency based on risk
assessment,” cautions Ginsbury.
REFERENCES 1. ISO, ISO 14644-1:2015, Cleanrooms
and associated controlled
environments – Part 1: Classification
of air cleanliness by particle
concentration (Geneva, 2015).
2. ISO, ISO 14644-2:2015, Cleanrooms
and associated controlled
environments – Part 2: Monitoring
to provide evidence of cleanroom
performance related to air cleanliness by
particle concentration (Geneva, 2015).
3. EC, EudraLex Volume 4: Good
manufacturing practice Guidelines,
“Annex 1, Manufacture of Sterile
Medicinal Products,” (Brussels, 2008).
4. FDA, Guidance for Industry:
Sterile Drug Products Produced
by Aseptic Processing—Current
Good Manufacturing Practice
(Rockville, MD, 2004).
Cleanroom Standards

40 BioPharm International www.biopharminternational.com July 2016
Biopharmaceutical manufactur-
ing is a complex process involv-
ing many unit operations,
precise amounts of materials, and
numerous variables. These elements must
be addressed properly to ensure consistent
product quality and drug product yield.
While a strong focus on quality, safety,
and drug efficacy is absolutely essential,
biopharmaceutical manufacturers are
also committed to finding ways to reduce
costs and improve manufacturing efficien-
cies. One production step with significant
costs (and potential risk to quality) is the
buffer and cell-culture materials prepara-
tion process: it is labor-intensive, requires
investment in storage and environmental
resources, and involves repeated quality
assurance (QA) testing as bulk materials
are subdivided for individual process runs.
New innovations in raw materials
packaging technologies can directly
impact this process—streamlining oper-
ations, mitigating risks, and contribut-
ing to operational excellence (OpEx). In
this article, the author examines new
methods of raw materials packaging
and how they may lead to manufac-
turing efficiencies, elimination of raw
material yield losses, reduction in QA
testing time and costs, and lower costs
in weigh and dispense production.
TRADITIONAL RAW MATERIAL DELIVERY METHODSUpstream biopharmaceutical processes
consume various raw materials, includ-
ing cell-culture media, carbohydrates,
amino acids, and buffers, which are typ-
ically supplied in powder form. The bio-
reactors and medium preparation tanks
that use these materials often oper-
ate around the clock. This operation
includes both large-scale reactors with
10,000 L capacity that run con-
t i nu o u s l y f o r a ny w h e r e f r o m
15–35 days, to newer generation single-
use technologies, with multiple 2000-L
bioreactors operating in overlapping
sequences to achieve similar or greater
productivity.
In general, two kinds of packaging
systems are in use today: Traditional
steel 100 kg drums with one or two plas-
tic liners, and smaller cardboard boxes
with plastic liners holding 50 kg. Both
bulk-packaging systems are part of stan-
dard practices that most raw materials
suppliers have established for their sup-
ply chain systems. The end user (i.e.,
the biopharmaceutical producer) typi-
cally orders, receives, and stores enough
salts, buffers, and other cell-culture
powdered materials to last several weeks
or months. The material consumption
rates can be substantial: a typical buf-
fer media preparation/usage cycle may
consume between 150 and 400 kg of
dry products per run, and uses multiple
raw materials per cycle. These materials
are then subdivided by the biopharma-
ceutical producer and used in smaller
amounts depending on the processes
they are running. In essence, this bulk
material packaging and delivery meth-
odology satisfies the raw material sup-
plier’s operational requirements, without
full consideration of how that material is
used by the biopharmaceutical producer.
OPERATIONAL INEFFICIENCIES AND RISKSFollowing the more traditional method
described above, the biopharmaceuti-
cal manufacturer must follow multiple
processing steps to properly manage and
use these bulk raw materials.
t #VML NBUFSJBMT BSF SFDFJWFE BOE
inventoried in storage areas that
must have the appropriate temper-
Raw Materials Packaging Innovations for Biopharmaceutical Manufacturing
Nandu Deorkar
Recent trends in raw
materials packaging
may impact manufacturing,
quality, and cost of
biopharma-ceuticals.
Nandu Deorkar, PhD, MBA, is
vice-president of R&D for Avantor
Performance Materials. He has
more than 25 years of experience in
materials technology research and
development, and has worked on
various aspects of chemical/polymer
R&D, drug development, formulation,
drug delivery technologies, process
development, and technology transfer.
Packaging Trends

July 2016 www.biopharminternational.com BioPharm International 41
ature and humidity controls
to maintain the mater ia l’s
integrity.
t5IF DPOUBJOFSTPVUFSQBDLBHJOH
is cleaned and sanitized. A sam-
ple is taken to independently
confirm via lab analysis that the
product’s quality, purity, and
characterization match what was
ordered. Once this is confirmed
(and analysis can take multi-
ple days), the bulk material is
cleared for use in the producer’s
dispensing operation.
t 0ODF DMFBSFE UIF DPOUFOUT PG
the drum are subdivided by
hand according to manufactur-
ing requirements. For example,
if 45 kg are initially required,
that quantity is removed and the
remainder is put back into stor-
age. For a 2000-L bioreactor, a
manufacturer may need to sub-
divide a 100 kg drum of material
between two and five times.
t 5IJT TVCEJWJTJPO TUFQ JT UJNF
consuming and risks cross-con-
taminating the remaining bulk
material. Some biopharmaceu-
tical producers conduct multi-
ple lab analyses of these drums,
each time a new quantity of
material is removed, to confirm
that no issues have occurred.
Much of this activity precedes,
and is not directly related to, the
value-added process of protein
manufacturing. It is time and
effort spent on materials manage-
ment and warehousing activities,
particularly the sub-dividing step,
to supply the bioreactors with the
precise amount of material needed
for production.
COMPLICATIONS DUE TO MATERIAL CAKING/CLUMPINGVarious raw materials, such as salts,
buffers, amino acids, and carbohy-
drates, have an intrinsic propensity
to form clumps or cake due to their
crystal structure and surface mois-
ture content. The presence of avail-
able surface moisture catalyzes the
process of caking when free mois-
ture migrates onto the surface of
the crystals and dissolves a small
portion, forming a salt bridge. This
caking, or clumping (Figure 1), is
a common problem with packag-
ing, storing, and sub-dividing pow-
dered materials in bulk containers.
Changes in ambient temperature
or humidity are the principal fac-
tors driving this process; the num-
ber of temperature-change cycles
will increase the strength of the
cake. Severe cases of caking can
result in complete solidification of
the entire package. Caked materi-
als must be completely broken up
in order to measure out the precise
amounts needed for bioreactor pro-
cesses. This is a time-consuming
manual activity with an open con-
tainer, which is at risk for cross-
contamination and absorption of
additional moisture, potentially
extending the problem. This prac-
tice also creates a potential safety
risk, as operators work to manu-
ally break up clumps while the
container is open, and can lead to
material loss.
MATCHING PACKAGING SIZE TO PROCESS NEEDSOver the past few years, pharma-
ceutical and biopharmaceutical
manufacturers have worked with
bulk material suppliers to modify
packaging approaches. The goal
was to enhance operational excel-
lence and reduce wasted time
and impact on process and prod-
uct quality. The initial focus was
on bulk material container sizes,
which typically held many more
materials than were needed, and
forced on-site storage and subdi-
viding steps. While continuing
to offer the standard 50 kg and
100 kg containers, suppliers began
to provide smaller, more manage-
able container sizes, such as 12 kg
and 25 kg.
This approach reduced the
length of time containers were
kept in storage and the number of
times a container was subdivided.
Subdividing was still required, how-
ever, because the exact amounts
of sugars, buffers, salts, and other
powdered bulk materials varied
greatly by manufacturer, based on
the specific process, protein, and
bioreactor equipment being used.
Subdividing also adds a measure of
uncertainty to the amount of raw
materials dispensed in a particu-
lar process step. Biopharmaceutical
production uses extremely tight
control on process parameters to
protect the final drug’s safety and
quality, and finding ways to strictly
control the amount of raw mate-
rial that goes into bioreactors can
enhance operational excellence and
process yield.
NEW PACKAGING FOR PRE-WEIGHED, FREE-FLOWING MATERIALSInnovative chemicals suppliers
have begun offering single-use,
pre-weighed product bags that pro-
vide biopharmaceutical produc-
ers with an easy-to-use method
for dispensing salts, buffers, and
other cell-culture materials directly
into their media or buffer prepara-
tion tanks, in the exact amounts
they specify for a given process.
The packaging options that are
now used essentially complete the
evolution from 50 kg and 100 kg
bulk product drums, to individual
direct dispense bags. The packag-
ing is constructed of transparent
polymers, the same materials that
have already been used to line the
traditional bulk drums; therefore,
biopharmaceutical manufactur-
ers do not have to re-validate the
material as safe for use.
The packaging’s design offers
biopharmaceutical manufactur-
ers more choices with regard to
packaging size. Manufacturers can
order the materials in a wide range
of smaller, precise quantities (e.g.,
from 250 g to 100 kg). The size,
shape, sealing, and seams of these
Packaging Trends

42 BioPharm International www.biopharminternational.com July 2016
AL
L F
IGU
RE
S A
RE
CO
UR
TE
SY
OF
TH
E A
UT
HO
R
bags are designed so that when they
are inverted, they dispense virtually
all the pre-weighed material into
the bioreactor. An important con-
sideration here is that pre-weighed
amounts should be within a 1%
tolerance of the amount of mate-
rial required. This is crucial, espe-
cially when the direct dispense bags
are used in single-use bioreactors.
If a biopharmaceutical manufac-
turer has determined that an exact
amount of glucose or sodium citrate
is needed to achieve maximum
yield on a process, the ability of
the packaging to freely deliver that
amount to very tight tolerances
must be assured.
To help ensure a free-flowing
dispensing system, the bags also
incorporate design features to
eliminate clumping, including
outer and inner layers with spe-
cial desiccant materials installed
between the two layers (Figure 2).
The inner layer uses a vapor per-
meable material already in use in
other container-lining applica-
tions. Any moisture that develops
within the bag passes through this
material and is controlled by the
desiccant, to maintain the cor-
rect moisture levels and reduce
clumping to an absolute minimum
(Figure 3).
DIRECT DISPENSE BAGS SIMPLIFY SAMPLING AND TESTINGTraditional large-volume bulk
container packaging also necessi-
tates the time-consuming process
of sampling and testing to verify
the material properties of a newly
delivered drum of product. Direct
dispense bag systems use trans-
parent polymers that are compat-
ible with non-destructive identity
testing tools, such as contact-free
Raman spectroscopy. With Raman
testing, there is no need to open
the bag and take a physical sample
to verify the product, the closed
bag can be scanned and verified
upon delivery, saving multiple
testing steps. The packaging is also
tamper-evident to ensure validity
and supply chain security.
In addition to near Raman test-
ing, some suppliers also will pro-
vide a tailgate sample along with
the bags. Biopharmaceutical pro-
ducers that are required to conduct
full analyses of all materials used in
their processes don’t need to open
the bag to obtain a product sample.
The tailgate sample process must be
validated to ensure that the material
in the tailgate sample is equivalent
to the materials packed in the bag.
DIRECT DISPENSE SYSTEMS: TIME AND COST SAVINGSExpanding the use of these direct
dispense systems can help advance
operational excellence initiatives
and reduce costs within the bio-
pharmaceutical industry’s supply
chain. By adopting flexible manu-
facturing technology, such as the
direct dispense systems, biophar-
maceutical manufacturers may
be able to reduce their operating
costs in certain areas by up to 40%.
There are multiple savings associ-
ated with the use of these systems:
t -BCPS&MJNJOBUFT UIF UJNF BOE
cost of personnel who need to
weigh, subdivide, and dispense
materials from bulk containers.
The savings can be significant.
Depending on the process, each
1000 L run of a cell-culture reac-
tor may require approximately
1000 to 2000 kg of more than
50 raw materials for the produc-
tion process. The media/buf-
fer preparation activities could
require 30–50 labor hours for
dispensing and adding the mate-
rials to the reactors.
t 'BDJMJUJFT 6TF PG EJSFDU EJT-
pense systems can eliminate the
need for dedicated raw material
preparation areas, drum storage
and handling equipment, and
environmental (temperature
and humidity) control equip-
ment. In some cases, these sys-
tems can reduce f loor space
needs by 40–70%.
t 5FTU JOH WB M JEBU JOH 6TF PG
Raman testing and tailgate sam-
ples greatly simplifies the testing/
validating step. Products do not
have to be re-validated each time
material is sub-divided from a
bulk container. In addition, the
primary packaging material typ-
Packaging Trends
Figure 1. A common problem with packaging, storing and sub-dividing powdered
materials in bulk containers is material caking or clumping.

July 2016 www.biopharminternational.com BioPharm International 43
Packaging Trends
ically used for these bags is the
same as standard drum liners, so
contact material does not have to
be re-validated.
t 2VBMJUZ1SFXFJHIFEEJSFDUEJT-
pense systems eliminate the need
to clean the weighing and dis-
pensing area for another opera-
tion, saving time and eliminating
risk of cross-contamination and
employee exposure.
t .BUFSJBMTUBCJMJUZBOEFGGJDJFOUVTF
Anti-clumping packaging design
improves raw material yields by
avoiding material non-confor-
mities and inaccurate ingredi-
ent measurements from clumped
materials.
t 4BGFUZ "OUJDMVNQJOH QBDLBH-
ing leads to sound environmen-
tal, health, and safety practices,
as employees no longer need to
engage in the potentially unsafe
practice of breaking up clumps
that can form in large drums.
t 3BX NBUFSJBMT TBWJOHT 1SF
weighed amounts in direct dis-
pense bags that match specific
biopharmaceut ica l process
requirements eliminate the need
to buy and store material in bulk,
reducing overages, out-of-date
materials, and disposal costs.
ADVANCES IN BULK MATERIAL PACKAGING FOCUS ON IMPROVING OVERALL OPEXBy evolving raw material packag-
ing and delivery options to align
more closely with the operational
requirements of biopharmaceutical
producers, raw materials suppli-
ers are helping to eliminate inef-
ficiencies and drive down costs
within the overall supply chain
and production environment. The
success of these bag-based, pre-
weighed, free-flowing direct dis-
pense systems is also encouraging
the development of streamlined
systems for other types of materi-
als beyond bulk powders. Some
raw-materials suppliers are inves-
tigating new packaging methods
to deliver ready-made liquid solu-
tions to customers, which would
eliminate the biopharmaceutical
manufacturing step of taking solid
materials and creating solutions.
While the transportation and
storage considerations for liquid
solutions are more complex than
solids, there are opportunities to
apply innovations to raw-materi-
als packaging designs to improve
the efficiency, productivity, safety,
and quality of biopharmaceutical
manufacturing.
Figure 2. To help ensure a free-flowing dispensing system and eliminate
clumping, some bags have outer and inner layers with special desiccant materials
installed between the two layers. The inner layer uses a vapor permeable material;
any moisture that develops within the bag passes through this material and is
controlled by the desiccant, to maintain the correct moisture levels.
INNER TYVEK®
LAYER
4” TC FERRULE
BLANKING CAP
4” TC CLAMP
4” TC CASKET
(EPDM)
2-OFF DESI-PAK
ACTIVATED CLAY
DESICCANT
(4 UNITS)
DESICCANT PACKS
FITTED BETWEEN
TYVEK® LAYER AND
OUTER PE LAYER
Figure 3. With new developments in raw material packaging, even after four
weeks at 40 °C and 90% relative humidity, the material in this 10 kg bag remains
free-flowing.

44 BioPharm International www.biopharminternational.com July 2016
Sve
ta D
em
ido
ff/G
ett
y I
ma
ge
s
Troubleshooting
Single-use technologies (SUT) have made sig-
nificant inroads in biopharmaceutical and
vaccine manufacturing. Greater adoption in
the years to come can be anticipated, given their
broad use in pre-clinical and clinical manufactur-
ing and expanding use for approved therapeutics.
With increasing regulatory oversight of SUT pro-
cesses, it’s worthwhile to review basic concepts of
design and qualification that apply to single-use
components and systems (SUS).
EQUIPMENT DESIGN REGULATIONS AND GUIDANCEWhile drug and vaccine manufacturers are sub-
ject to regulatory review and inspection of how
equipment is used, that is not the case for the
manufacturers of process equipment, including
SUT components and systems, even when sold for
use under good manufacturing practice (GMP).
Suitability of design and qualification for use must
be determined by the therapeutic manufacturer.
The US Code of Federal Regulations (21 CFR 211.63),
Section 211.63 states, “Equipment used in the
manufacture, processing, packing, or holding of a
drug product shall be of appropriate design, ade-
quate size, and suitably located to facilitate opera-
tions for its intended use and for its cleaning and
maintenance” (1). While this is the responsibility
of the user, equipment manufacturers need to
understand and design to user requirements for
what may be considered appropriate,
adequate, or suitable.
US 21 CFR does provide some
information for process equipment
designers: Section 211.65, paragraph
(a) states, “Equipment shall be con-
structed so that surfaces that contact
components, in-process materials, or
drug products shall not be reactive,
additive, or absorptive so as to alter
the safety, identity, strength, qual-
ity, or purity of the drug product
beyond the official or other established require-
ments” (1). While not specifying those require-
ments, this requirement highlights the need for
SUT equipment designers to consider operational
performance criteria, as well as potential chemical
interactions and equipment-derived impurities or
particulate contaminants that may be introduced
into applicable processes and potentially impact
intermediates or final dosages.
Much of SUT use occurs in therapeutic pro-
tein API production. International Council
on Harmonization (ICH) Q7, European
Medicines Agency (EMA) Q7, and FDA Q7A
Good Manufacturing Practice Guidance for Active
Pharmaceutical Ingredients incorporate essen-
tially the same design requirement statement:
“Equipment should be constructed so that surfaces
that contact raw materials, intermediates, or APIs
do not alter the quality of the intermediates and
APIs beyond the official or other established speci-
fications” (2–4).
Fortunately, established SUT manufacturers
have significant experience working with drug and
vaccine companies and have learned to adopt SUT
equipment designs to meet user needs in GMP-
regulated processes. Adoption of established good
engineering practices for design and deployment
of SUTs is crucial and requires close collaboration
between users, component designers and system
engineers having experience in polymer materials,
device manufacturing, human interface engineer-
ing, and other related practices (5).
SUT DESIGN CONSIDERATIONSDesign considerations for SUT manufacturers can
be divided into six categories:
t 1IZTJDPDIFNJDP QSPQFSUJFT DPWFS TFMFDUJPO
and performance of the materials of construc-
tion and finished component under antici-
pated use conditions, with reasonable safety
factors. These properties can include pressure
requirements (e.g., operating, burst, creep, pres-
Design and Qualification of Single-Use Systems The author provides a review of the concepts of design and qualification that apply to single-use systems.
Jerold M. Martin is an independent
consultant and Chairman Emeritus,
BPSA, [email protected].

July 2016 www.biopharminternational.com BioPharm International 45
TVSFESPQ NBYNJO UFNQFSBUVSF
requirements (e.g., operating,
thermal sterilization, melt, cryo-
genic, impact of temperature on
pressure performance), mechani-
cal properties (e.g., flexibility,
rigidity, and tensile strength),
optical properties (e.g., clarity or
opacity), cleanliness (e.g., surface
or embedded particles), biocom-
patibility, material oxidation and
radiation stability, gas barrier
properties, polymer additives,
transmissible spongiform enceph-
alopathy-bovine spongiform
encephalopathy (TSE–BSE) status,
and other raw material supplier
data, etc.
t 'MVJE DPOUBDU QSPQFSUJFT UIBU
should be considered in product
design include chemical compat-
ibility (e.g., polymer solubility,
swelling, embrittlement, etc.),
chemical reactivity, extractables
under appropriate solvents and
extraction conditions, particle
TIFEEJOH BOEQSPUFJOCJOEJOH
adsorption to contact surfaces
(along with any other formulation
ingredients).
t 'PSN GJU BOE GVODUJPO SFMBUFT
to the “fitness for purpose” of
the physical design of the
component(s) and assemblies.
It includes handling and other
ergonomic properties, accessibil-
ity for installation and removal,
microbial barrier and physical
and fitment (leak) integrity prop-
erties, sterilizability, drainability,
incorporation into an automated
platform as the disposable fluid
path, etc.
t .BOVGBDUVSBCJMJUZ FOTVSFT UIBU
the SUT production equipment
and environment are suitable to
effectively produce the compo-
nent or system as designed and
maintain quality specifications
with a high degree of assurance.
Manufacturability should also
include operator training and in-
process and final product testing
(as needed) for quality factors such
as integrity (i.e., retention prop-
erties of filters, leak absence of
containers, connector and hose fit-
ments, microbial barrier, etc.) bio-
burden, endotoxins, and particle
contamination or generation.
t 1BDLBHJOHBOETIJQNFOUDPOUBJO-
ers must be designed to effectively
protect the SUT component or
assembly during transport from
the manufacturing or assembly
site, as well as ease and security of
unpacking and installation at the
user site.
t %PDVNFOUBUJPO TIPVME BMTPCF
considered as part of product
design, to include support data
for performance claims, design
and production validation data
reports, operation guides, and
other information that may be
requested by users or from regu-
latory authorities.
QUALIFICATIONQualification is the action of proving
that any equipment works correctly
(as designed) and can be expected
to perform as intended. For the user,
qualification includes confirming
that the equipment is the right tool
for the job. While the term valida-
tion is sometimes applied to incor-
porate the concept of qualification,
validation means verifying (and doc-
umenting) that the equipment con-
sistently functions within a specified
range of operations to produce an
intended result. Qualification studies
are, therefore, done with representa-
tive samples prior to use. Validation
is conducted on process equipment
(or scaled-down models) under actual
use conditions.
While the word qualification is not
specifically mentioned in 21 CFR 211,
the interpretation of these regula-
tions by both industry and regulatory
agencies has introduced terms such
as design qualification (DQ) (i.e., is
the design suitable for user require-
ments?), installation qualification
(IQ) (i.e., is the equipment installed
properly?), operational qualification
(OQ) (i.e., does it operate according
to the manufacturer’s specifications?),
and performance qualification (PQ)
(i.e., does it consistently perform
to meet the user’s requirements?).
Additional terms that combine these
concepts include factory acceptance
qualification (FAQ) or test (FAT) (e.g.,
DQ, IQ, OQ) and site acceptance
qualification (SAQ) or test (SAT) (e.g.,
IQ, OQ, PQ).
The first step of qualification
should be DQ. For SUT, this occurs
in two phases: confirmation by the
equipment supplier that the equip-
ment meets its’ design and opera-
tion criteria, and evaluation by the
user that the component design and
performance is suitable for use in the
intended application. To the degree
that the equipment supplier can
anticipate the user’s requirements,
some portion of the manufacturer’s
qualification data can also serve as
the user’s evaluation and be incor-
porated into the user’s documenta-
tion to be presented to regulatory
authorities. This is of course provided
UIBU UIFNBOVGBDUVSFSTVQQMJFSQSP-
cedures and data are suitable for GMP
use, the user review of the data is
documented, and a user audit is con-
ducted to confirm data validity.
Conformance of the SUT design
with the manufacturer’s claims
and user’s process requirements
should be demonstrated and docu-
mented. According to European
Union GMP Annex 15, Section
3, “Qualification activities should
consider all stages from initial
development of the user require-
ments specification (URS) through
to the end of use of the equip-
ment, facility, utility or system.”
Once the URS is developed, “The
next element in the qualification
of equipment, facilities, utilities,
or systems is DQ where the com-
pliance of the design with GMP
should be demonstrated and doc-
umented. The requirements of
Troubleshooting
Contin. on page 48

Product & Service Innovations Advertorial
Company DescriptionDistek, Inc., headquartered in North Brunswick, NJ, is
a leading manufacturer of laboratory testing instruments
for the pharmaceutical and biotechnology industry, as well
as an experienced provider of validation and qualification
services.
Distek’s robust product portfolio includes water
bath and bathless dissolution, dissolution media heating,
degassing, dispensing and disposal, in-situ fiber optic UV,
bathless tablet disintegration, content uniformity, digital
video monitoring, programmable automated sampling,
and now a single-use bioreactor system for mammalian cell
culture applications.
Founded in 1976, Distek has an excellent reputation
for innovation, product reliability, and customer
support, and we believe that these, as well as continual
improvement, are essential to our current and future
growth and crucial to ongoing success.
For technological advancements that offer reliable
and consistent performance, the pharmaceutical industry
trusts Distek.
Markets ServedDistek’s core market includes brand name and generic
pharmaceutical drug manufacturers, CROs, CMOs,
Nutraceuticals, government agencies, and universities.
To ensure consistent quality and to meet the international
standards of our world market, Distek has been ISO
certified since 2002.
Services & InnovationsDistek does not simply design ‘me too’ products. Distek
instruments are considered to be highly innovative, which
is exemplified in the product portfolio. Distek engineers
technically modern and user-friendly instruments that
have provided many innovative features to customers,
worldwide. Distek holds several instrumentation and
accessory patents and has been ISO certified since 2002.
Distek’s robust product portfolio includes water
bath and bathless dissolution, dissolution media heating,
degassing, dispensing and disposal, in-situ fiber optic UV,
bathless tablet disintegration, content uniformity, digital
video monitoring, programmable automated sampling,
and now a single-use bioreactor system for mammalian cell
culture applications.
Major Product InnovationsYou don’t need to make a large capital investment to
convert your existing benchtop glass bioreactor to a single-
use bioreactor. Simply remove your existing headplate
and place the preassembled and irradiated Distek BIOne
System into the glass vessel. The “patent pending”
bioreactor liner will mold to and mimic your existing
glass system allowing you to continue using your existing
cabinet, probes, motor, heating jacket and recipes. Since
the liner conforms to the dimensions and aspect ratio
of your existing vessel, there is no need to change your
process. All materials are USP Class VI, animal derivative
free and utilized in existing single-use products.
Distek, Inc.
Distek, Inc.121 North Center Drive
North Brunswick, NJ 08902
Phone: 732.422.7585
Fax: 732.422.7310
www.distekinc.com
Email: [email protected]
46 BioPharm International July 2016

• CONVERT Your Existing BIOREACTOR to a SINGLE-USE BIOREACTOR in Seconds
• Available in a 2L & 5L WORKING VOLUME
• SINGLE-USE HEADPLATE with Multiple Sensor Ports
• Materials are USP CLASS VI & ANIMAL DERIVATIVE FREE
• ‘Patent Pending’ SINGLE-USE BIOREACTOR Liner & Headplate
• INCREASED Throughput
• BIOne is COMPATIBLE with Most Manufacturer’s GLASS VESSELS Including:
– Applikon
– Sartorius
– Broadley-James
– Eppendorf
– Finesse
– Infors & more...
VISIT Bioprocessing
Summit
#501
BIONE SINGLE-USE BIOREACTOR SYSTEMNEW!
Proudly Assembled in the USA
distekinc.com
• MAANN
• ‘‘PPUSUSSHe
• ININ
• BIBIIMVEVEE
–
–
–
–
–
SINGLE-USELINER
SINGLE-USEHEADPLATE
GLASSVESSEL

48 BioPharm International www.biopharminternational.com July 2016
Troubleshooting
the user requirements specifica-
tion should be verified during the
design qualification” (6).
SUPPLIER QUALITY SYSTEM AND QUALIFICATION PRACTICESSUT and SUS are unique from tra-
ditional stainless equipment in that
the user depends on the supplier’s
quality system. Review of the suppli-
er’s quality system should be part of
supplier qualification. Furthermore,
users cannot normally perform
incoming testing prior to using SUSs
intended for implementation in pro-
duction, so are also more dependent
on suppliers’ product qualification
practices.
Supplier qualification of SUT
products can be summarized in
product qualification and process
validation reports suitable for user
documentation and submittal to
regulatory authorities. Applicable
test methods for pre-use qualifica-
tion are detailed in Bio-Process
Systems Alliance (BPSA’s) Quality Test
Reference Matrices document, along
with supplemental BPSA guides
with specific recommendations on
qualification of extractables, steril-
ization, and particulates testing (7).
Additional tests for SUS qualification
have been described by users (8–12).
The United States Pharmacopeial
Convention (USP) has published a
draft standard on extractables test-
ing for plastic process equipment
(13). A BPSA guide on SUS leak test-
ing is also under development. In
addition, BPSA provides an indus-
try consensus-generated Quality
Agreement Template that can be
used to document accountabili-
ties (7), and audits should be used
to review these under the supplier’s
quality management system.
Also important for SUS users is
to qualify the SUT equipment sup-
plier’s reliability and delivery his-
tory. Potential supplier performance
can be assessed by the experience
of integrators and users who have
previously worked with them.
Attendance at industry meetings,
participation on scientific asso-
ciation SUS committees, or trade
associations such as BPSA pro-
vide important access to suppli-
ers, integrators, and users to aid in
EFUFSNJOJOH BQQSPQSJBUF TVQQMJFS
integrator qualification practices.
REFERENCES 1. FDA, US Code of Federal Regulations (21
CFR 211.63), Section 211.63, Current
Good Manufacturing Practice for Finished
Pharmaceuticals (FDA, April 2014).
2. ICH, Q7 Good Manufacturing Practice
Guide forActive Pharmaceutical Ingredients
(ICH, November 2000).
3. EMA, Note for Guidance on Good
Manufacturing Practice for Active
Pharmaceutical Ingredients (CPMP/
ICH/4106/00), November 2000.
4. FDA, Guidance for Industry, Q7A Good
Manufacturing Practice Guidance for Active
Pharmaceutical Ingredients, August 2001
5. M. Botterill and B. Rawlings, BioProcess
Int. (December 2008).
6. EMA, EU Guidelines for Good
Manufacturing Practice for Medicinal
Products for Human and Veterinary Use,
Annex 15: Qualification and Validation,
February 2014.
7. BPSA, Component Quality Test Matrices,
Gamma Irradiation and Sterilization,
Extractables and Leachables, Particulates,
Quality Agreement Template, www.
bpsalliance.org
8. PDA, Technical Report No. 66,Application
of Single-Use Systems in Pharmaceutical
Manufacturing (PDA, 2014).
9. D. M. Stephenson, J. Val. Technol.
(February 2003).
10. M. A. Petrich, Amer. Pharm. Rev. 16 (7)
(November/December 2013).
11. W. Ding, BioPharm Int. 28 (9) (September
2015) pp. 32–39.
12. D. Riedman and J. Martin, BioProcess Int.
9 (S2) (2011) pp. 28-35.
13. USP, <661.3> (draft): Plastic Systems
Used for Manufacturing Pharmaceutical
Products, USP—Pharmacopoeal Forum
42(3) (USP, March 2016).
19. R. Martins, J.A. Queiroz, and F. Souza, J. Chrom. A 1355,
(August 2014).
20. T. Schlake et al., RNA Biol. 9 (11), pp. 1319–1330 (Nov. 1, 2012).
21. Personal communication with BSN team, EMD Millipore (now
MilliporeSigma).
22. R. Martins, J.A. Queiroz, and F. Sousa, Anal. Bioanal. Chem.
405 (27), pp. 8849–8858 (November 2013).
23. P.M. Swiderski et al., Anal. Biochem. 216, pp. 83–88 (1994).
24. J.R. Thayer et al., Chrom. Today, (March 2011), www.
chromatographytoday.com/article_read/985/, accessed
May 24, 2016.
25. F. Wincott et al., Nucl. Acids. Res. 23 (14), pp. 2677 –2684 (1995).
26. T. Achsel, H. Stark, and R. Luhrmann, Proc. Natl. Acad. Sci.
U.S.A. 98 (7), pp. 3685–3689 (March 27, 2001).
27. C. Miller et al., Mol. Syst. Biol. 7 (458), doi: 10.1038/
msb.2010.112 (Jan. 4, 2011).
28. L.E. Easton et al., RNA 16 (3), pp. 647–653 (2010).
29. W.J. Issa et al., “Ion Exchange Purification of mRNA,” US
patent WO2014144767 A1, Sept. 18, 2014.
30. W.H. Pan et al., Mol. Ther. 9 (4), pp. 596–606 (April 2004).
31. A. Eon-Duval et al., J. Chrom. B Analyt. Technol. Biomed. Life
Sci. 804 (2), pp. 327–335 (2004).
32. I. Theodossiou, M. SØndergaard, and O.R. Thomas,
Bioseparation 10 (1–3), pp. 31–44 (2001).
33. J.K. Nair et al., J. Am. Chem. Soc. 136 (49), pp. 16958–
16961 (Dec. 10, 2014).
34. S.C. Wong et al., Nucleic Acid Ther. 22 (6), pp. 380–390
(December 2012).
35. Arbutus Biopharma, “Tekmira Presents Recent Advances in
mRNA Delivery at Scientific Symposium,” Press Release, Feb.
25, 2014, http://investor.tekmirapharm.com/releasedetail.
cfm?ReleaseID=827952, accessed May 24, 2016.
36. P. Pantazis et al., “Preparation of siRNA-Encapsulated PLGA
Nanoparticles for Sustained Release of siRNA and Evaluation of
Encapsulation Efficiency,” in Nanoparticles in Biology and Medicine:
Methods and Protocols, Methods in Molecular Biology, M. Soloviei,
Ed. (Springer Science+Business Media, 2012), pp. 311–319.
37. M. Chen et al., ACS Nano 6 (6), pp. 4835–4844 (2012).
Peer-Reviewed—Contin. from page 36
Contin. from page 45

July 2016 www.biopharminternational.com BioPharm International 49
Pa
ge h
ea
de
r im
ag
e: A
rth
ur
S. A
ub
ry/G
ett
y I
ma
ge
s
CMC BIOLOGICS, RIGHT. ON TIME.CMC Biologics is a global contract process
development and manufacturer of
biopharmaceuticals. We offer our clients a
full suite of contract services from DNA to
API and leverage our technical expertise to
transform these services into solutions that meet our clients’ near- and long-
term objectives. With cGMP-compliant facilities in both the United States
and Europe and a team of more than 500 dedicated employees, every project
is performed with our absolute commitment to quality manufacturing
and customer satisfaction. CMC Biologics Labs, www.cmcbio.com
LABORATORY SERVICESAs a member of Eurofins’ BioPharma
Product Testing Group—the
largest network of harmonized
bio/pharmaceutical GMP product
testing laboratories worldwide—
Eurofins Lancaster Laboratories supports all functional areas of bio/
pharmaceutical manufacturing, including method development,
microbiology, process validation, and quality control throughout
all stages of the drug development process. Eurofins Lancaster
Labs, tel. 717.656.2300, www.EurofinsLancasterLabs.com
IMPROVING AGGREGATE REMOVAL FROM MAB FEED USING HIGH RESOLUTION CATION EXCHANGE CHROMATOGRAPHYNuvia™ HR-S media is a cation exchanger
(CEX) designed for high resolution of closely related product impurities such as
aggregates. It delivers excellent resolution with a final aggregate content of
<0.3% and a high recovery of >80% from a heterogeneous feed containing mAb
aggregates and monomer. Furthermore, aggregate content and recovery in the
eluate were shown to be a function of the target conductivity measured at the
end of collection. Bio-Rad Laboratories, www.bio-rad.com/resin-library
NEW INTEGRATED BIOLOGICS SOLUTION CENTERWuXi has started construction of a state-of-
the-art, integrated R&D and manufacturing
center at the company’s headquarters (see
press release). This 250,000 sq. ft. facility
will be operational in 2017 and can accommodate 800 scientists. The center will
provide comprehensive concept to clinic biologics discovery, development, and GMP
manufacturing services on one consolidated campus. This facility utilizes WuXi’s
single-source technology platforms. WuXi Biologics (A wholly owned subsidiary
of WuXi AppTec), [email protected], www.wuxibiologics.com/news-events.
1.8
1.6
1.4
1.2
1
0.8
0.6
0.4
0.2
0
0 10 20 30 40 50 60 70 80 90 100
Nuvia HR-S
Media 1
Ag
greg
ate, %
Monomer recovery, %
New Technology Showcase
Modular Bioreactor Simplifies Process Development
The ambr 250 mini benchtop bioreactor system from Sartorius Stedim Biotech can be used for parallel fermentation and cell culture. The system combines a unique single-use bioreactor vessel and expandable system design for process development. The
new ambr 250 modular system consists of a workstation with two, four, six, or eight single-use bioreactors, with a working volume range of 100–250 mL. These mini bioreactors, based on the same stirred tank bioreactors in the high throughput system, contain impellers suitable for fermentation or cell culture, and scale up to larger bioreactors. They are also fully integrated with liquid reservoirs and syringe pumps, allowing rapid experimental set up and turn around, thus significantly increasing lab efficiency. Once installed, the bioreactor has all the required process services for parameter control, including pH, DO, temperature, and agitation. Feeds can also be delivered from the reagent reservoirs via syringe pumps into the bioreactor. One control unit is capable of controlling up to eight bioreactor stations independently using a touch screen user interface.
Sartorius Stedim Biotech
www.sartorius.com
Ad Index
Company Page
BD BIOSCIENCES 5
BIO RAD LABORATORIES Cover Tip, 21
DISTEK INC 46–47
EUROFINS LANCASTER LABORATORIES 10–11
GE HEALTHCARE 7
POLYTHERICS LTD 15
SGS LIFE SCIENCE SERVICES 37
TOSOH BIOSCIENCE 25, 51
UNIVERSITY OF NEBRASKA-LINCOLN 2
WUXI APP TEC 31, 52
PRODUCT SPOTLIGHT

50 BioPharm International www.biopharminternational.com July 2016
BIOLOGICS NEWS PIPELINE
IN THE PIPELINE
Using a New CRISPR Effector to Edit RNA
A naturally occurring CRISPR (clustered regularly
interspaced short palindromic repeats) system that spe-
cifically can be used to modify the RNA of an organ-
ism is the newest development in the technology’s
evolution. A new study, published in Science, identifies
C2c2, a bacterial protein that can be used as a tool
to cleave single-stranded RNA sequences at desired
locations. As an RNA-guided RNAse, C2c2 can be har-
nessed to defend against viral intruders and turn off
gene expression of certain conditions, according to
researchers.
Typically, gene silencing is a gene expression manip-
ulation performed by small interfering RNA (siRNA),
but the study researchers attest that the C2c2 complex
is even more efficient when it comes to editing RNA.
Specifically, C2c2 can be used to add to or delete infor-
mation from existing RNA sequences, and can also be
used to tag RNA to learn more information about the
function of certain sequences. In addition, C2c2 can
be programmed to knock out certain messenger RNA
(mRNA), disrupting the DNA-->mRNA-->protein tran-
scription process. C2c2 requires only a single RNA to
function, and is “genetically encodable,” the researchers
said in a Broad Institute/Harvard press release accompa-
nying the study. As an RNA-targeting immune system
mechanism, they say C2c2 is a promising tool for future
RNA manipulation.
The authors concluded that the CRISPR-C2c2 complex
is probably not the only system that can be programmed
to alter RNA sequence, and other, patentable editing tools
may be on the horizon. They wrote, “It is likely that other,
broadly analogous Class 2 RNA-targeting immune systems
exist, and further characterization of the diverse members
of Class 2 systems will provide a deeper understanding of
bacterial immunity and provide a rich starting point for
the development of programmable molecular tools for in
vivo RNA manipulation.”
Although the research on CRISPR-C2c2 was conducted
by various teams at MIT, the Broad Institute, Harvard
University, the National Institutes of Health, Rutgers
University-New Brunswick, and the Skolkovo Institute
of Science and Technology, the Broad Institute’s Feng
Zhang is at the helm of the research and serves as one of
the paper’s senior authors. Zhang is notorious for being
one of the founders of the CRISPR-Cas9 complex and its
applications for editing DNA, however, there is currently
an ongoing legal battle between the Broad Institute and
the University of California (involving work by researcher
Jennifer Doudna and by French researcher Emmanuelle
Charpentier) for the patent rights to the technology.
Study: HIV-1 Neutralizing Antibodies
in Infants May Impact Vaccine Development
A new article in Cell that studied HIV-neutralizing antibod-
ies in an infant may have important implications in the
development of an HIV vaccine. The novel study exam-
ined broadly neutralizing antibodies (bnAbs) in a Nairobi
infant prior to receiving antiretrovirals and approximately
one-year post infection. Researchers isolated and character-
ized infant HIV-1 neutralizing antibodies in order to fur-
ther understand their impact on the HIV virus.
Multiple studies have been done on HIV-1 bnAbs in
adults, but little is known about infant bnAbs contributing
to broad plasma responses. While a subset of adults with
HIV-1 develop bnAbs, these antibodies exhibit high levels
of somatic hypermutation (SHM) and only neutralize the
disease after years of affinity maturation. By comparison,
infant’s bnAbs develop broad responses early, and “HIV-1-
specific neutralization breadth can develop without pro-
longed affinity maturation and extensive SHM.” Therefore,
infants may have the ability to neutralize the disease more
quickly than adults.
“Most studies of adults have looked at responses many
years after infection and these have suggested that broad
and potent HIV neutralizing antibodies take years to
develop and that they require extensive hypermutation to
be effective,” study author Julie Overbaugh, PhD, member
of the Human Biology Division at the Fred Hutchinson
Cancer Research Center, told BioPharm International. “This
is the first example of broadly neutralizing antibodies
detected this early in infection. These infant antibodies
have much less hypermutation and yet, they are nonethe-
less broad and potent.”
Overbaugh notes the infant studied had a polyclonal
response to the virus, meaning, the infant’s antibodies
targeted multiple epitopes, making it more difficult for the
virus to survive. This presents an interesting comparison
to studies done on adult antibodies, which target only one
dominant epitope, Overbaugh says.
The challenge is now finding ways to harness these
unique attributes of infant bnAbs for practical applications
such as vaccine development. The goal, researchers say,
is to better understand infant bnAbs that develop early
in natural infection in order to develop vaccines that can
elicit neutralizing antibodies more quickly.
“Infants mount a more rapid potent antibody
response to HIV than adults—understanding how they
do this is important, both in terms of how they do it
more rapidly and whether there are fundamental dif-
ferences in the pathway,” said Overbaugh. “Adults take
a long time to develop these responses [naturally], lon-
ger than is possible to imagine for a vaccine regimen.”

www.tosohbioscience.com
Tosoh Bioscience and TOYOPEARL are registered trademarks of Tosoh Corporation.
Every mAb is unique.
Your Protein A should be as well.
TOSOH BIOSCIENCE LLC • Customer service: 866-527-3587 • Technical service: 800-366-4875, option #3
TOYOPEARL® AF-rProtein A HC-650FHigh Capacity Protein A Resin for Monoclonal Antibody Purifi cation
0
10
20
30
40
50
60
70
80
2 3.5 5
Residence time (minutes)
DBC
for I
gG (g
/L)
1 g/L
5 g/L
10 g/LResin: TOYOPEARL AF-rProtein A HC-650FColumn size: 5 mm ID × 5 cmMobile phase: 0.02 mol/L sodium phosphate, 0.15 mol/L NaCl, pH 7.4Residence time: 2, 3.5, 5 minDetection: UV @ 280 nm (10% breakthrough)Sample: human IgG @ 1, 5, 10 g/L in mobile phase

The world’s ONE true single-source platform
from concept to commercialization.
Since 2014
The Dawn of a New CDMO Paradigm
How does a single source save time and reduce risk?(Scan QR code below to find out.)
[email protected] OR www.wuxibiologics.com
4785
40IND-enabling global CMC
projects completed 99percent of the CMC and
preclinical work required for
an ADC drug’s global IND filing
stable cell lines developed
for GMP Manufacturing
cell-based assays developed
including 15 qualified for GMP