volume 17 number 16 april 28, 2011 · world journal of gastroenterology volume 17 number 16 april...
TRANSCRIPT

World Journal of Gastroenterology
Volume 17 Number 16April 28, 2011
ISSN 1007-9327 CN 14-1219/R Local Post Offices Code No. 82-261
Published by Baishideng Publishing Group Co., Limited,Room 1701, 17/F, Henan Building,
No. 90 Jaffe Road, Wanchai, Hong Kong, ChinaFax: +852-3115-8812
Telephone: +852-5804-2046E-mail: [email protected]
http://www.wjgnet.com
World Journal of GastroenterologyWorld J Gastroenterol 2011 April 28; 17(16): 2063-2160
ISSN 1007-9327 (print)ISSN 2219-2840 (online)
www.wjgnet.com
World Journal of G
astroenterology ww
w.w
jgnet.com Volum
e 17 Num
ber 16 Apr 28 2011
I S S N 1 0 0 7 - 9 3 2 7
9 7 7 1 0 07 9 3 2 0 45
1 6

The World Journal of Gastroenterology Editorial Board consists of 1144 members, representing a team of worldwide experts in gastroenterology and hepatology. They are from 60 countries, including Albania (1), Argentina (8), Australia (29), Austria (14), Belgium (12), Brazil (10), Brunei Darussalam (1), Bulgaria (2), Canada (20), Chile (3), China (69), Colombia (1), Croatia (2), Cuba (1), Czech (4), Denmark (8), Ecuador (1), Egypt (2), Estonia (2), Finland (8), France (24), Germany (75), Greece (14), Hungary (10), India (26), Iran (6), Ireland (7), Israel (12), Italy (101), Japan (112), Jordan (1), Kuwait (1), Lebanon (3), Lithuania (2), Malaysia (1), Mexico (10), Moldova (1), Netherlands (29), New Zealand (2), Norway (11), Pakistan (2), Poland (11), Portugal (4), Romania (3), Russia (1), Saudi Arabia (3), Serbia (3), Singapore (10), South Africa (2), South Korea (32), Spain (38), Sweden (18), Switzerland (11), Thailand (1), Trinidad and Tobago (1), Turkey (24), United Arab Emirates (2), United Kingdom (82), United States (249), and Uruguay (1).
Editorial Board2010-2013
HONORARY EDITORS-IN-CHIEFJames L Boyer, New HavenKe-Ji Chen, BeijingMartin H Floch, New HavenEmmet B Keeffe, Palo AltoGeng-Tao Liu, BeijingLein-Ray Mo, TainanEamonn M Quigley, CorkRafiq A Sheikh, SacramentoNicholas J Talley, RochesterMing-Lung Yu, Kaohsiung
PRESIDENT AND EDITOR-IN-CHIEFLian-Sheng Ma, Beijing
ACADEMIC EDITOR-IN-CHIEFTauseef Ali, Oklahoma CityMauro Bortolotti, BolognaTarkan Karakan, AnkaraWeekitt Kittisupamongkol, BangkokAnastasios Koulaouzidis, EdinburghBo-Rong Pan, Xi’anSylvia LF Pender, SouthamptonMax S Petrov, AucklandGeorge Y Wu, Farmington
STRATEGY ASSOCIATE EDITORS-IN-CHIEFPeter Draganov, FloridaHugh J Freeman, VancouverMaria C Gutiérrez-Ruiz, MexicoKazuhiro Hanazaki, KochiAkio Inui, KagoshimaKalpesh Jani, BarodaJavier S Martin, Punta del Este
Natalia A Osna, OmahaWei Tang, TokyoAlan BR Thomson, EdmontonHarry HX Xia, HanoverJesus K Yamamoto-Furusho, MexicoYoshio Yamaoka, Houston
ASSOCIATE EDITORS-IN-CHIEFYou-Yong Lu, BeijingJohn M Luk, SingaporeHiroshi Shimada, Yokohama
GUEST EDITORIAL BOARD MEMBERSChien-Jen Chen, TaipeiYang-Yuan Chen, ChanghuaJen-Hwey Chiu, TaipeiSeng-Kee Chuah, KaohsiungWan-Long Chuang, KaohsiunMing-Chih Hou, TaipeiKevin Cheng-Wen Hsiao, TaipeiPo-Shiuan Hsieh, TaipeiTsung-Hui Hu, KaohsiungWen-Hsin Huang, TaichungChao-Hung Hung, KaohsiungI-Rue Lai, TaipeiTeng-Yu Lee, TaichungChing Chung Lin, TaipeiHui-Kang Liu, TaipeiHon-Yi Shi, KaohsiungChih-Chi Wang, KaohsiungJin-Town Wang, TaipeiCheng-Shyong Wu, Chia-YiJaw-Ching Wu, TaipeiJiunn-Jong Wu, TainanMing-Shiang Wu, Taipei
Ta-Sen Yeh, TaoyuanHsu-Heng Yen, ChanghuaMing-Whei Yu, Taipei
MEMBERS OF THE EDITORIAL BOARD
Albania
Bashkim Resuli, Tirana
Argentina
Julio H Carri, CórdobaEduardo de Santibañes, Buenos AiresBernardo Frider, Buenos AiresCarlos J Pirola, Buenos AiresBernabe Matias Quesada, Buenos AiresSilvia Sookoian, Buenos AiresAdriana M Torres, RosarioMaria Ines Vaccaro, Buenos Aires
Australia
Leon Anton Adams, NedlandsRichard Anderson, VictoriaMinoti V Apte, New South WalesAndrew V Biankin, SydneyFilip Braet, SydneyChristopher Christophi, MelbournePhilip G Dinning, KoagarahGuy D Eslick, SydneyMichael A Fink, Melbourne
January 7, 2011IWJG|www.wjgnet.com

Robert JL Fraser, Daw ParkJacob George, WestmeadMark D Gorrell, SydneyAlexander G Heriot, MelbourneMichael Horowitz, AdelaideJohn E Kellow, SydneyWilliam Kemp, MelbourneFinlay A Macrae, VictoriaDaniel Markovich, BrisbaneVance Matthews, MelbournePhillip S Oates, PerthShan Rajendra, TasmaniaRajvinder Singh, Elizabeth ValeRoss C Smith, SydneyKevin J Spring, BrisbaneNathan Subramaniam, BrisbanePhil Sutton, MelbourneCuong D Tran, North AdelaideDebbie Trinder, FremantleDavid Ian Watson, Bedford Park
Austria
Herwig R Cerwenka, GrazAshraf Dahaba, GrazPeter Ferenci, ViennaValentin Fuhrmann, ViennaAlfred Gangl, ViennaAlexander M Hirschl, WienKurt Lenz, LinzDietmar Öfner, SalzburgMarkus Peck-Radosavljevic, ViennaMarkus Raderer, ViennaStefan Riss, ViennaGeorg Roth, ViennaMichael Trauner, GrazThomas Wild, Kapellerfeld
Belgium
Rudi Beyaert, GentBenedicte Y De Winter, AntwerpInge I Depoortere, LeuvenOlivier Detry, LiègePhilip Meuleman, GhentMarc Peeters, De PintelaanFreddy Penninckx, LeuvenJean-Yves L Reginster, LiègeMark De Ridder, BrusselsEtienne M Sokal, BrusselsKristin Verbeke, LeuvenEddie Wisse, Keerbergen
Brazil
José LF Caboclo, São José do Rio PretoRoberto J Carvalho-Filho, São PauloJaime Natan Eisig, São PauloAndre Castro Lyra, SalvadorMarcelo Lima Ribeiro, Braganca Paulista Joao Batista Teixeira Rocha, Santa MariaHeitor Rosa, GoianiaDamiao C Moraes Santos, Rio de JaneiroAna Cristina Simões e Silva, Belo HorizonteEduardo Garcia Vilela, Belo Horizonte
Brunei Darussalam
Vui Heng Chong, Bandar Seri Begawan
Bulgaria
Zahariy Krastev, SofiaMihaela Petrova, Sofia
Canada
Alain Bitton, MontrealMichael F Byrne, VancouverKris Chadee, CalgaryWangxue Chen, OttawaRam Prakash Galwa, OttawaPhilip H Gordon, MontrealWaliul Khan, OntarioQiang Liu, SaskatoonJohn K Marshall, OntarioAndrew L Mason, AlbertaKostas Pantopoulos, QuebecNathalie Perreault, SherbrookeBaljinder Singh Salh, VancouverEldon Shaffer, CalgaryMartin Storr, CalgaryPingchang Yang, HamiltonEric M Yoshida, VancouverClaudia Zwingmann, Montreal
Chile
Marcelo A Beltran, La SerenaXabier De Aretxabala, SantiagoSilvana Zanlungo, Santiago
China
Hui-Jie Bian, Xi’anSan-Jun Cai, ShanghaiGuang-Wen Cao, ShanghaiXiao-Ping Chen, WuhanChi-Hin Cho, Hong KongZong-Jie Cui, Beijing Jing-Yuan Fang, ShanghaiDe-Liang Fu, ShanghaiZe-Guang Han, ShanghaiChun-Yi Hao, BeijingMing-Liang He, Hong KongChing-Lung Lai, Hong KongSimon Law, Hong KongYuk-Tong Lee, Hong KongEn-Min Li, ShantouFei Li, BeijingYu-Yuan Li, GuangzhouZhao-Shen Li, ShanghaiXing-Hua Lu, BeijingYi-Min Mao, ShanghaiQin Su, BeijingPaul Kwong-Hang Tam, Hong KongYuk Him Tam, Hong KongRen-Xiang Tan, NanjingWei-Dong Tong, ChongqingEric WC Tse, Hong Kong
Fu-Sheng Wang, BeijingXiang-Dong Wang, ShanghaiNathalie Wong, Hong KongJustin CY Wu, Hong KongWen-Rong Xu, ZhenjiangAn-Gang Yang, Xi’an Wei-Cheng You, BeijingChun-Qing Zhang, JinanJian-Zhong Zhang, Beijing Xiao-Peng Zhang, BeijingXuan Zhang, Beijing
Colombia
Germán Campuzano-Maya, Medellín
Croatia
Tamara Cacev, ZagrebMarko Duvnjak, Zagreb
Cuba
Damian C Rodriguez, Havana
Czech
Jan Bures, Hradec KraloveMilan Jirsa, PrahaMarcela Kopacova, Hradec KralovePavel Trunečka, Prague
Denmark
Leif Percival Andersen, CopenhagenAsbjørn M Drewes, AalborgMorten Frisch, CopenhagenJan Mollenhauer, OdenseMorten Hylander Møller, HolteSøren Rafaelsen, VejleJorgen Rask-Madsen, SkodsborgPeer Wille-Jørgensen, Copenhagen
Ecuador
Fernando E Sempértegui, Quito
Egypt
Zeinab Nabil Ahmed, CairoHussein M Atta, El-Minia
Estonia
Riina Salupere, TartuTamara Vorobjova, Tartu
Finland
Saila Kauhanen, Turku
January 7, 2011IIWJG|www.wjgnet.com

Thomas Kietzmann, OuluKaija-Leena Kolho, HelsinkiJukka-Pekka Mecklin, JyvaskylaMinna Nyström, HelsinkiPauli Antero Puolakkainen, TurkuJuhani Sand, TampereLea Veijola, Helsinki
France
Claire Bonithon-Kopp, DijonLionel Bueno, ToulouseSabine Colnot, ParisCatherine Daniel, Lille CedexAlexis Desmoulière, LimogesThabut Dominique, ParisFrancoise L Fabiani, AngersJean-Luc Faucheron, GrenobleJean Paul Galmiche, Nantes cedexBoris Guiu, DijonPaul Hofman, NiceLaurent Huwart, ParisJuan Iovanna, MarseilleAbdel-Majid Khatib, ParisPhilippe Lehours, BordeauxFlavio Maina, MarseillePatrick Marcellin, ParisRene Gerolami Santandera, MarseilleAnnie Schmid-Alliana, Nice cedexAlain L Servin, Châtenay-MalabryStephane Supiot, NantesBaumert F Thomas, StrasbourgJean-Jacques Tuech, RouenFrank Zerbib, Bordeaux Cedex
Germany
Erwin Biecker, SiegburgHubert Blum, Freiburg Thomas Bock, TuebingenDean Bogoevski, HamburgElfriede Bollschweiler, KölnJürgen Borlak, HannoverChrista Buechler, RegensburgJürgen Büning, LübeckElke Cario, EssenBruno Christ, Halle/SaaleChristoph F Dietrich, Bad Mergentheim Ulrich R Fölsch, Kiel Nikolaus Gassler, AachenMarkus Gerhard, MunichDieter Glebe, GiessenRalph Graeser, FreiburgAxel M Gressner, AachenNils Habbe, MarburgThilo Hackert, HeidelbergWolfgang Hagmann, HeidelbergDirk Haller, FreisingPhilip D Hard, GiessenClaus Hellerbrand, RegensburgKlaus R Herrlinger, StuttgartEberhard Hildt, BerlinAndrea Hille, GoettingenJoerg C Hoffmann, BerlinPhilipe N Khalil, MunichAndrej Khandoga, MunichJorg Kleeff, MunichIngmar Königsrainer, TübingenPeter Konturek, Erlangen
Stefan Kubicka, HannoverJoachim Labenz, SiegenMichael Linnebacher, RostockJutta Elisabeth Lüttges, RiegelsbergPeter Malfertheiner, MagdeburgOliver Mann, HamburgPeter N Meier, HannoverSabine Mihm, GöttingenKlaus Mönkemüller, BottropJonas Mudter, ErlangenSebastian Mueller, HeidelbergRobert Obermaier, FreiburgMatthias Ocker, ErlangenStephan Johannes Ott, KielGustav Paumgartner, MunichChristoph Reichel, Bad Brückenau Markus Reiser, BochumSteffen Rickes, MagdeburgElke Roeb, GiessenChristian Rust, MunichHans Scherubl, BerlinMartin K Schilling, HomburgJoerg F Schlaak, EssenRene Schmidt, FreiburgAndreas G Schreyer, RegensburgKarsten Schulmann, BochumHenning Schulze-Bergkamen, MainzManfred V Singer, MannheimJens Standop, BonnJurgen M Stein, Frankfurt Ulrike S Stein, BerlinWolfgang R Stremmel, Heidelberg Harald F Teutsch, Ulm Hans L Tillmann, LeipzigChristian Trautwein, AachenJoerg Trojan, FrankfurtArndt Vogel, HannoverSiegfried Wagner, DeggendorfFrank Ulrich Weiss, GreifswaldFritz von Weizsäcker, BerlinThomas Wex, MagdeburgStefan Wirth, WuppertalMarty Zdichavsky, Tübingen
Greece
Helen Christopoulou-Aletra, ThessalonikiT Choli-Papadopoulou, ThessalonikiTsianos Epameinondas, IoanninaIoannis Kanellos, ThessalonikiElias A Kouroumalis, Heraklion Ioannis E Koutroubakis, HeraklionMichael Koutsilieris, AthensAndreas Larentzakis, AthensEmanuel K Manesis, AthensSpilios Manolakopoulos, AthensKonstantinos Mimidis, AlexandroupolisGeorge Papatheodoridis, AthensSpiros Sgouros, Athens Evangelos Tsiambas, Ag Paraskevi Attiki
Hungary
György M Buzás, BudapestLászló Czakó, SzegedGyula Farkas, SzegedPeter Hegyi, SzegedPeter L Lakatos, Budapest
Yvette Mándi, SzegedZoltan Rakonczay, SzegedFerenc Sipos, BudapestZsuzsa Szondy, DebrecenGabor Veres, Budapest
India
Philip Abraham, MumbaiVineet Ahuja, New DelhiGiriraj Ratan Chandak, HyderabadDevinder Kumar Dhawan, ChandigarhRadha K Dhiman, Chandigarh Pankaj Garg, PanchkulaPramod Kumar Garg, New DelhiDebidas Ghosh, MidnporeUday C Ghoshal, LucknowBhupendra Kumar Jain, DelhiAshok Kumar, LucknowBikash Medhi, ChandigarhSri P Misra, Allahabad Gopal Nath, VaranasiSamiran Nundy, New DelhiJagannath Palepu, MumbaiVandana Panda, MumbaiBenjamin Perakath, Tamil NaduRamesh Roop Rai, JaipurNageshwar D Reddy, HyderabadBarjesh Chander Sharma, New DelhiVirendra Singh, ChandigarhRupjyoti Talukdar, GuwahatiRakesh Kumar Tandon, New DelhiJai Dev Wig, Chandigarh
Iran
Mohammad Abdollahi, TehranPeyman Adibi, IsfahanSeyed-Moayed Alavian, TehranSeyed Mohsen Dehghani, ShirazReza Malekzadeh, TehranAlireza Mani, Tehran
Ireland
Billy Bourke, DublinTed Dinan, CorkCatherine Greene, DublinRoss McManus, DublinAnthony P Moran, GalwayMarion Rowland, Dublin
Israel
Simon Bar-Meir, HashomerAlexander Becker, AfulaAbraham R Eliakim, Haifa Sigal Fishman, Tel AvivBoris Kirshtein, Beer ShevaEli Magen, AshdodMenachem Moshkowitz, Tel-AvivAssy Nimer, SafedShmuel Odes, Beer ShevaMark Pines, Bet DaganRon Shaoul, HaifaAmi D Sperber, Beer-Sheva
January 7, 2011IIIWJG|www.wjgnet.com

Italy
Donato F Altomare, BariPiero Amodio, PadovaAngelo Andriulli, San Giovanni RotondoPaolo Angeli, PadovaBruno Annibale, RomePaolo Aurello, RomeSalvatore Auricchio, NaplesAntonio Basoli, RomeClaudio Bassi, VeronaGabrio Bassotti, Perugia Mauro Bernardi, BolognaAlberto Biondi, RomeLuigi Bonavina, Milano Guglielmo Borgia, NaplesRoberto Berni Canani, NaplesMaria Gabriella Caruso, BariFausto Catena, BolognaGiuseppe Chiarioni, ValeggioMichele Cicala, RomeDario Conte, Milano Francesco Costa, PisaAntonio Craxì, PalermoSalvatore Cucchiara, RomeGiuseppe Currò, MessinaMario M D’Elios, FlorenceMirko D’Onofrio, VeronaSilvio Danese, MilanoRoberto de Franchis, MilanoPaola De Nardi, MilanGiovanni D De Palma, NaplesGiuliana Decorti, TriesteGianlorenzo Dionigi, VareseMassimo Falconi, VeronaSilvia Fargion, MilanGiammarco Fava, AnconaFrancesco Feo, SassariAlessandra Ferlini, FerraraAlessandro Ferrero, TorinoMirella Fraquelli, MilanLuca Frulloni, VeronaGiovanni B Gaeta, NapoliAntonio Gasbarrini, RomeEdoardo G Giannini, Genoa Alessandro Granito, BolognaFabio Grizzi, MilanSalvatore Gruttadauria, PalermoPietro Invernizzi, MilanAchille Iolascon, NaplesAngelo A Izzo, NaplesEzio Laconi, CagliariGiovanni Latella, L’AquilaMassimo Levrero, RomeFrancesco Luzza, CatanzaroLucia Malaguarnera, CataniaFrancesco Manguso, NapoliPier Mannuccio Mannucci, MilanGiancarlo Mansueto, VeronaGiulio Marchesini, Bologna Mara Massimi, CoppitoGiovanni Milito, RomeGiuseppe Montalto, Palermo Giovanni Monteleone, RomeLuca Morelli, TrentoGiovanni Musso, TorinoMario Nano, TorinoGerardo Nardone, NapoliRiccardo Nascimbeni, BresciaValerio Nobili, RomeFabio Pace, MilanNadia Peparini, Rome
Marcello Persico, NaplesMario Pescatori, RomeRaffaele Pezzilli, Bologna Alberto Piperno, MonzaAnna C Piscaglia, RomePiero Portincasa, Bari Michele Reni, MilanVittorio Ricci, PaviaOliviero Riggio, RomeMario Rizzetto, TorinoBallarin Roberto, ModenaGerardo Rosati, PotenzaFranco Roviello, SienaCesare Ruffolo, TrevisoMassimo Rugge, PadovaMarco Scarpa, PadovaC armelo Scarpignato, ParmaGiuseppe Sica, RomeMarco Silano, RomePierpaolo Sileri, RomeVincenzo Stanghellini, BolognaFiorucci Stefano, PerugiaGiovanni Tarantino, NaplesAlberto Tommasini, TriesteGuido Torzilli, Rozzano MilanCesare Tosetti, Porretta TermeAntonello Trecca, RomeVincenzo Villanacci, BresciaLucia Ricci Vitiani, RomeMarco Vivarelli, Bologna
Japan
Kyoichi Adachi, Izumo Yasushi Adachi, SapporoTakafumi Ando, Nagoya Akira Andoh, OtsuMasahiro Arai, Tokyo Hitoshi Asakura, TokyoKazuo Chijiiwa, MiyazakiYuichiro Eguchi, SagaItaru Endo, YokohamaMunechika Enjoji, FukuokaYasuhiro Fujino, AkashiMitsuhiro Fujishiro, TokyoKouhei Fukushima, SendaiMasanori Hatakeyama, TokyoKeiji Hirata, KitakyushuToru Hiyama, HigashihiroshimaMasahiro Iizuka, Akita Susumu Ikehara, OsakaKenichi Ikejima, Bunkyo-kuYutaka Inagaki, KanagawaHiromi Ishibashi, Nagasaki Shunji Ishihara, Izumo Toru Ishikawa, Niigata Toshiyuki Ishiwata, Tokyo Hajime Isomoto, NagasakiYoshiaki Iwasaki, OkayamaSatoru Kakizaki, GunmaTerumi Kamisawa, TokyoMototsugu Kato, Sapporo Naoya Kato, TokyoTakumi Kawaguchi, KurumeYohei Kida, KainanShogo Kikuchi, AichiTsuneo Kitamura, Chiba Takashi Kobayashi, TokyoYasuhiro Koga, IseharaTakashi Kojima, SapporoNorihiro Kokudo, TokyoMasatoshi Kudo, OsakaShin Maeda, Tokyo
Satoshi Mamori, HyogoAtsushi Masamune, SendaiYasushi Matsuzaki, Tsukuba Kenji Miki, TokyoToshihiro Mitaka, SapporoHiroto Miwa, Hyogo Kotaro Miyake, TokushimaManabu Morimoto, YokohamaYoshiharu Motoo, Kanazawa Yoshiaki Murakami, HiroshimaYoshiki Murakami, KyotoKunihiko Murase, Tusima Akihito Nagahara, TokyoYuji Naito, Kyoto Atsushi Nakajima, YokohamaHisato Nakajima, Tokyo Hiroki Nakamura, Yamaguchi Shotaro Nakamura, FukuokaAkimasa Nakao, NagogyaShuhei Nishiguchi, HyogoMikio Nishioka, Niihama Keiji Ogura, TokyoSusumu Ohmada, Maebashi Hirohide Ohnishi, AkitaKenji Okajima, NagoyaKazuichi Okazaki, OsakaMorikazu Onji, EhimeSatoshi Osawa, Hamamatsu Hidetsugu Saito, TokyoYutaka Saito, TokyoNaoaki Sakata, SendaiYasushi Sano, ChibaTokihiko Sawada, TochigiTomohiko Shimatan, HiroshimaYukihiro Shimizu, KyotoShinji Shimoda, FukuokaYoshio Shirai, Niigata Masayuki Sho, NaraShoichiro Sumi, KyotoHidekazu Suzuki, TokyoMasahiro Tajika, NagoyaYoshihisa Takahashi, TokyoToshinari Takamura, KanazawaHiroaki Takeuchi, KochiYoshitaka Takuma, OkayamaAkihiro Tamori, OsakaAtsushi Tanaka, TokyoShinji Tanaka, Hiroshima Satoshi Tanno, HokkaidoShinji Togo, YokohamaHitoshi Tsuda, TokyoHiroyuki Uehara, OsakaMasahito Uemura, KashiharaYoshiyuki Ueno, SendaiMitsuyoshi Urashima, TokyoTakuya Watanabe, NiigataSatoshi Yamagiwa, NiigataTaketo Yamaguchi, ChibaMitsunori Yamakawa, YamagataTakayuki Yamamoto, Yokkaichi Yutaka Yata, MaebashiHiroshi Yoshida, Tokyo Norimasa Yoshida, Kyoto Yuichi Yoshida, OsakaKentaro Yoshika, ToyoakeHitoshi Yoshiji, NaraKatsutoshi Yoshizato, HigashihiroshimaTomoharu Yoshizumi, Fukuoka
Jordan
Ismail Matalka, Irbid
January 7, 2011IVWJG|www.wjgnet.com

Kuwait
Islam Khan, Safat
Lebanon
Bassam N Abboud, BeirutAla I Sharara, BeirutRita Slim, Beirut
Lithuania
Giedrius Barauskas, KaunasLimas Kupcinskas, Kaunas
Malaysia
Andrew Seng Boon Chua, Ipoh
Mexico
Richard A Awad, MexicoAldo Torre Delgadillo, MexicoDiego Garcia-Compean, MonterreyPaulino M Hernández Magro, CelayaMiguel Angel Mercado, Distrito FederalArturo Panduro, JaliscoOmar Vergara-Fernandez, TlalpanSaúl Villa-Trevio, Mexico
Moldova
Igor Mishin, Kishinev
Netherlands
Ulrich Beuers, AmsterdamLee Bouwman, LeidenAlbert J Bredenoord, NieuwegeinLodewijk AA Brosens, UtrechtJ Bart A Crusius, AmsterdamWouter de Herder, RotterdamPieter JF de Jonge, RotterdamRobert J de Knegt, RotterdamWendy W Johanna de Leng, UtrechtAnnemarie de Vries, RotterdamJames CH Hardwick, LeidenFrank Hoentjen, HaarlemMisha Luyer, SittardJeroen Maljaars, MaastrichtGerrit A Meijer, AmsterdamServaas Morré, AmsterdamChris JJ Mulder, Amsterdam John Plukker, Groningen Albert Frederik Pull ter Gunne, TilburgPaul E Sijens, GroningenBW Marcel Spanier, ArnhemShiri Sverdlov, MaastrichtMaarten Tushuizen, AmsterdamJantine van Baal, HeidelberglaanAstrid van der Velde, The HagueKarel van Erpecum, Utrecht Loes van Keimpema, Nijmegen
Robert Christiaan Verdonk, GroningenErwin G Zoetendal, Wageningen
New Zealand
Andrew S Day, Christchurch
Norway
Olav Dalgard, OsloTrond Peder Flaten, TrondheimReidar Fossmark, TrondheimRasmus Goll, TromsoOle Høie, ArendalAsle W Medhus, OsloEspen Melum, OsloTrine Olsen, TromsoEyvind J Paulssen, TromsoJon Arne Søreide, StavangerKjetil Soreide, Stavanger
Pakistan
Shahab Abid, KarachiSyed MW Jafri, Karachi
Poland
Marek Bebenek, WroclawTomasz Brzozowski, Cracow Halina Cichoż-Lach, LublinAndrzej Dabrowski, BialystokHanna Gregorek, WarsawMarek Hartleb, KatowiceBeata Jolanta Jablońska, KatowiceStanislaw J Konturek, KrakowJan Kulig, KrakowDariusz M Lebensztejn, BialystokJulian Swierczynski, Gdansk
Portugal
Raquel Almeida, PortoAna Isabel Lopes, Lisboa CodexRicardo Marcos, PortoGuida Portela-Gomes, Estoril
Romania
Dan L Dumitrascu, ClujAdrian Saftoiu, CraiovaAndrada Seicean, Cluj-Napoca
Russia
Vasiliy I Reshetnyak, Moscow
Saudi Arabia
Ibrahim A Al Mofleh, RiyadhAbdul-Wahed Meshikhes, QatifFaisal Sanai, Riyadh
Serbia
Tamara M Alempijevic, BelgradeDusan M Jovanovic, Sremska KamenicaZoran Krivokapic, Belgrade
Singapore
Madhav Bhatia, SingaporeKong Weng Eu, SingaporeBrian Kim Poh Goh, SingaporeKhek-Yu Ho, Singapore Kok Sun Ho, SingaporeFock Kwong Ming, SingaporeLondon Lucien Ooi, SingaporeNagarajan Perumal, SingaporeFrancis Seow-Choen, Singapore
South Africa
Rosemary Joyce Burnett, PretoriaMichael Kew, Cape Town
South Korea
Sang Hoon Ahn, SeoulSung-Gil Chi, SeoulMyung-Gyu Choi, SeoulHoon Jai Chun, SeoulYeun-Jun Chung, SeoulYoung-Hwa Chung, SeoulKim Donghee, SeoulKi-Baik Hahm, IncheonSun Pyo Hong, Geonggi-doSeong Gyu Hwang, SeongnamHong Joo Kim, SeoulJae J Kim, SeoulJin-Hong Kim, Suwon Nayoung Kim, Seongnam-siSang Geon Kim, SeoulSeon Hahn Kim, SeoulSung Kim, SeoulWon Ho Kim, SeoulJeong Min Lee, SeoulKyu Taek Lee, Seoul Sang Kil Lee, SeoulSang Yeoup Lee, Gyeongsangnam-doYong Chan Lee, SeoulEun-Yi Moon, SeoulHyoung-Chul Oh, SeoulSeung Woon Paik, SeoulJoong-Won Park, GoyangJi Kon Ryu, SeoulSi Young Song, SeoulMarie Yeo, Suwon Byung Chul Yoo, SeoulDae-Yeul Yu, Daejeon
Spain
Maria-Angeles Aller, MadridRaul J Andrade, MálagaLuis Aparisi, ValenciaGloria González Aseguinolaza, NavarraMatias A Avila, Pamplona
January 7, 2011VWJG|www.wjgnet.com

Fernando Azpiroz, Barcelona Ramon Bataller, BarcelonaBelén Beltrán, ValenciaAdolfo Benages, ValenciaJosep M Bordas, Barcelona Lisardo Boscá, MadridLuis Bujanda, San SebastiánJuli Busquets, BarcelonaMatilde Bustos, PamplonaJosé Julián calvo Andrés, SalamancaAndres Cardenas, BarcelonaAntoni Castells, Barcelona Fernando J Corrales, PamplonaJ E Domínguez-Muñoz, Santiago de CompostelaJuan Carlos Laguna Egea, BarcelonaIsabel Fabregat, BarcelonaAntoni Farré, BarcelonaVicente Felipo, ValenciaLaureano Fernández-Cruz, BarcelonaLuis Grande, BarcelonaAngel Lanas, Zaragoza Juan-Ramón Larrubia, GuadalajaraMaría IT López, JaénJuan Macías, SevilleJavier Martin, GranadaJosé Manuel Martin-Villa, MadridJulio Mayol, MadridMireia Miquel, SabadellAlbert Parés, BarcelonaJesús M Prieto, Pamplona Pedro L Majano Rodriguez, MadridJoan Roselló-Catafau, BarcelonaEva Vaquero, Barcelona
Sweden
Lars Erik Agréus, StockholmMats Andersson, StockholmRoland Andersson, LundMauro D’Amato, HuddingeEvangelos Kalaitzakis, GothenburgGreger Lindberg, Stockholm Annika Lindblom, StockholmSara Lindén, GöteborgHanns-Ulrich Marschall, StockholmPär Erik Myrelid, LinköpingÅke Nilsson, LundHelena Nordenstedt, StockholmKjell Öberg, UppsalaLars A Pahlman, UppsalaStefan G Pierzynowski, LundSara Regnér, MalmöBobby Tingstedt, LundZongli Zheng, Stockholm
Switzerland
Pascal Bucher, GenevaMichelangelo Foti, GenevaJean L Frossard, GenevaAndreas Geier, ZürichPascal Gervaz, GenevaGerd A Kullak-Ublick, ZürichFabrizio Montecucco, GenevaPaul M Schneider, ZürichFelix Stickel, BerneBruno Stieger, ZürichInti Zlobec, Basel
Trinidad and Tobago
Shivananda Nayak, Mount Hope
Turkey
Sinan Akay, TekirdagMetin Basaranoglu, IstanbulYusuf Bayraktar, AnkaraA Mithat Bozdayi, AnkaraHayrullah Derici, BalıkesirEren Ersoy, AnkaraMukaddes Esrefoglu, MalatyaCan Goen, KutahyaSelin Kapan, IstanbulAydin Karabacakoglu, KonyaCuneyt Kayaalp, MalatyaKemal Kismet, AnkaraSeyfettin Köklü, AnkaraMehmet Refik Mas, Etlik-AnkaraOsman C Ozdogan, IstanbulBülent Salman, AnkaraOrhan Sezgin, MersinIlker Tasci, AnkaraMüge Tecder-Ünal, AnkaraAhmet Tekin, MersinMesut Tez, AnkaraEkmel Tezel, AnkaraÖzlem Yilmaz, Izmir
United Arab Emirates
Fikri M Abu-Zidan, Al-AinSherif M Karam, Al-Ain
United Kingdom
Simon Afford, BirminghamNavneet K Ahluwalia, StockportMohamed H Ahmed, SouthamptonBasil Ammori, SalfordLesley A Anderson, BelfastChin Wee Ang, LiverpoolYeng S Ang, WiganAnthony TR Axon, Leeds Kathleen B Bamford, LondonJim D Bell, LondonJohn Beynon, SwanseaChris Briggs, SheffieldGeoffrey Burnstock, LondonAlastair D Burt, NewcastleJeff Butterworth, ShrewsburyJeremy FL Cobbold, LondonJean E Crabtree, LeedsTatjana Crnogorac-Jurcevic, LondonWilliam Dickey, LondonderrySunil Dolwani, Cardiff Emad M El-Omar, AberdeenA M El-Tawil, BirminghamCharles B Ferguson, BelfastAndrew Fowell, SouthamptonPiers Gatenby, LondonDaniel R Gaya, EdinburghAnil George, LondonRob Glynne-Jones, NorthwoodJason CB Goh, BirminghamGianpiero Gravante, Leicester
Brian Green, BelfastWilliam Greenhalf, Liverpool Indra N Guha, NottinghamStefan G Hübscher, BirminghamRobin Hughes, LondonPali Hungin, StocktonNawfal Hussein, NottinghamClement W Imrie, GlasgowJanusz AZ Jankowski, Oxford Sharad Karandikar, BirminghamPeter Karayiannis, LondonShahid A Khan, LondonPatricia F Lalor, BirminghamJohn S Leeds, SheffieldIan Lindsey, OxfordHong-Xiang Liu, Cambridge Dileep N Lobo, NottinghamGraham MacKay, GlasgowMark Edward McAlindon, SheffieldAnne McCune, BristolDonald Campbell McMillan, GlasgowGiorgina Mieli-Vergani, London Jamie Murphy, LondonGuy Fairbairn Nash, PooleJames Neuberger, Birmingham Patrick O’Dwyer, GlasgowChristos Paraskeva, BristolRichard Parker, North StaffordshireThamara Perera, BirminghamKondragunta Rajendra Prasad, LeedsD Mark Pritchard, LiverpoolAlberto Quaglia, LondonAkhilesh B Reddy, CambridgeKevin Robertson, GlasgowSanchoy Sarkar, LiverpoolJohn B Schofield, KentMarco Senzolo, PadovaVenkatesh Shanmugam, DerbyPaul Sharp, LondonChew Thean Soon, ManchesterAravind Suppiah, East YorkshireNoriko Suzuki, MiddlesexSimon D Taylor-Robinson, London Frank I Tovey, LondonA McCulloch Veitch, WolverhamptonVamsi R Velchuru, LowestoftSumita Verma, BrightonCatherine Walter, CheltenhamJulian RF Walters, LondonRoger Williams, London
United States
Kareem M Abu-Elmagd, PittsburghSami R Achem, FloridaGolo Ahlenstiel, BethesdaBhupinder S Anand, HoustonM Ananthanarayanan, New YorkBalamurugan N Appakalal, MinneapolisDimitrios V Avgerinos, New YorkShashi Bala, WorcesterAnthony J Bauer, PittsburghKevin E Behrns, GainesvilleRoberto Bergamaschi, New York Henry J Binder, New HavenEdmund J Bini, New YorkWojciech Blonski, PhiladelphiaMark Bloomston, ColumbusEdward L Bradley III, SarasotaCarla W Brady, Durham
January 7, 2011VIWJG|www.wjgnet.com

David A Brenner, San DiegoAdeel A Butt, PittsburghShi-Ying Cai, New HavenJustin MM Cates, NashvilleEugene P Ceppa, DurhamJianyuan Chai, Long BeachRonald S Chamberlain, LivingstonFei Chen, MorgantownXian-Ming Chen, Omaha Ramsey Chi-man Cheung, Palo AltoDenesh Chitkara, East BrunswickClifford S Cho, MadisonParimal Chowdhury, ArkansasJohn David Christein, BirminghamThomas Clancy, BostonAna J Coito, Los AngelesRicardo Alberto Cruciani, New YorkJoseph J Cullen, Iowa CityMark J Czaja, New YorkMariana D Dabeva, BronxJessica A Davila, HoustonConor P Delaney, ClevelandLaurie DeLeve, Los AngelesAnthony J Demetris, PittsburghSharon DeMorrow, TempleBijan Eghtesad, ClevelandYoram Elitsur, HuntingtonMohamad A Eloubeidi, AlabamaWael El-Rifai, NashvilleSukru H Emre, New HavenGiamila Fantuzzi, ChicagoAshkan Farhadi, Irvine Ronnie Fass, TucsonMartín E Fernández-Zapico, RochesterAlessandro Fichera, ChicagoJosef E Fischer, BostonPiero Marco Fisichella, Maywood Fritz Francois, New YorkGlenn T Furuta, AuroraT Clark Gamblin, Pittsburgh Henning Gerke, Iowa CityJean-Francois Geschwind, BaltimoreR Mark Ghobrial, TexasJohn F Gibbs, BuffaloShannon S Glaser, TempleAjay Goel, DallasJon C Gould, MadisonEileen F Grady, San FranciscoJames H Grendell, New YorkJohn R Grider, RichmondAnna S Gukovskaya, Los Angeles Chakshu Gupta, St. JosephGrigoriy E Gurvits, New YorkHai-Yong Han, PhoenixYuan-Ping Han, Los AngelesImran Hassan, SpringfieldCharles P Heise, MadisonLisa J Herrinton, OaklandOscar Joe Hines, Los AngelesSamuel B Ho, San DiegoSteven Hochwald, GainesvilleRichard Hu, Los AngelesEric S Hungness, ChicagoJamal A Ibdah, ColumbiaAtif Iqbal, Omaha Hartmut Jaeschke, TucsonDonald M Jensen, ChicagoRobert Jensen, BethesdaLeonard R Johnson, MemphisAndreas M Kaiser, Los AngelesJingXuan Kang, CharlestownJohn Y Kao, MichiganRandeep Singh Kashyap, New YorkRashmi Kaul, Tulsa
Jonathan D Kaunitz, Los AngelesStephen M Kavic, BaltimoreAli Keshavarzian, ChicagoAmir Maqbul Khan, MarshallKusum K Kharbanda, OmahaChang Kim, West LafayetteDean Y Kim, DetroitMiran Kim, ProvidenceBurton I Korelitz, New York Josh Korzenik, BostonRichard A Kozarek, Seattle Alyssa M Krasinskas, PittsburghShiu-Ming Kuo, Buffalo Michelle Lai, BostonMichael Leitman, New YorkDong-Hui Li, HoustonMing Li, New Orleans Zhiping Li, BaltimoreGary R Lichtenstein, Philadelphia Chen Liu, GainesvilleZhang-Xu Liu, Los AngelesCraig D Logsdon, HoustonKaye M Reid Lombardo, RochesterMichael R Lucey, MadisonKirk Ludwig, WisconsinJames D Luketich, Pittsburgh Patrick M Lynch, HoustonJohn S Macdonald, New YorkWillis C Maddrey, DallasMercedes Susan Mandell, AuroraChristopher Mantyh, DurhamWendy M Mars, PittsburghJohn Marshall, ColumbiaRobert CG Martin, LouisvilleLaura E Matarese, PittsburghCraig J McClain, LouisvilleLynne V McFarland, WashingtonDavid J McGee, ShreveportValentina Medici, SacramentoStephan Menne, New YorkDidier Merlin, AtlantaGeorge Michalopoulos, PittsburghJames M Millis, ChicagoPramod K Mistry, New HavenEmiko Mizoguchi, BostonHuanbiao Mo, DentonRobert C Moesinger, OgdenSmruti R Mohanty, ChicagoJohn Morton, StanfordPeter L Moses, BurlingtonSandeep Mukherjee, OmahaMillion Mulugeta, Los AngelesMichel M Murr, TampaPete Muscarella, ColumbusEce A Mutlu, ChicagoMasaki Nagaya, BostonLaura E Nagy, ClevelandAejaz Nasir, TampaUdayakumar Navaneethan, CincinnatiStephen JD O’Keefe, PittsburghRobert D Odze, BostonGiuseppe Orlando, Winston SalemPal Pacher, RockvilleGeorgios Papachristou, PittsburghJong Park, TampaWilliam R Parker, DurhamMansour A Parsi, ClevelandMarco Giuseppe Patti, ChicagoZhiheng Pei, New York CS Pitchumoni, New Brunswiuc Parviz M Pour, OmahaXiaofa Qin, NewarkFlorencia Georgina Que, RochesterMassimo Raimondo, Jacksonville
Raymund R Razonable, MinnesotaKevin Michael Reavis, OrangeRobert V Rege, DallasDouglas K Rex, IndianapolisVictor E Reyes, Galveston Basil Rigas, New YorkRichard A Rippe, Chapel HillAlexander S Rosemurgy, TampaPhilip Rosenthal, San FranciscoRaul J Rosenthal, WestonJoel H Rubenstein, Ann ArborShawn D Safford, NorfolkRabih M Salloum, RochesterBruce E Sands, BostonTor C Savidge, GalvestonMichael L Schilsky, New HavenBeat Schnüriger, CaliforniaRobert E Schoen, PittsburghMatthew James Schuchert, PittsburghEkihiro Seki, La JollaLe Shen, ChicagoPerry Shen, Winston-SalemStuart Sherman, Indianapolis Mitchell L Shiffman, RichmondShivendra Shukla, ColumbiaBronislaw L Slomiany, NewarkScott Steele, Fort LewisBranko Stefanovic, TallahasseeLygia Stewart, San FranciscoLuca Stocchi, ClevelandDaniel S Straus, RiversideRobert Todd Striker, MadisonJonathan Strosberg, TampaChristina Surawicz, SeattlePatricia Sylla, BostonWing-Kin Syn, DurhamYvette Taché, Los AngelesKazuaki Takabe, RichmondKam-Meng Tchou-Wong, New York Klaus Thaler, ColumbiaCharles Thomas, OregonNatalie J Torok, SacramentoGeorge Triadafilopoulos, Stanford Chung-Jyi Tsai, LexingtonThérèse Tuohy, Salt Lake CityAndrew Ukleja, FloridaSanthi Swaroop Vege, RochesterAaron Vinik, NorfolkDinesh Vyas, WashingtonArnold Wald, WisconsinScott A Waldman, PhiladelphiaJack R Wands, ProvidenceJiping Wang, BostonIrving Waxman, ChicagoWilfred M Weinstein, Los AngelesSteven D Wexner, Weston John W Wiley, Ann ArborJackie Wood, OhioJian Wu, SacramentoWen Xie, PittsburghGuang-Yin Xu, GalvestonFang Yan, NashvilleRadha Krishna Yellapu, New YorkAnthony T Yeung, PhiladelphiaZobair M Younossi, VirginiaLiqing Yu, Winston-SalemRun Yu, Los AngelesRuben Zamora, Pittsburgh Michael E Zenilman, New YorkMark A Zern, SacramentoLin Zhang, PittsburghMartin D Zielinski, RochesterMichael A Zimmerman, Colorado
January 7, 2011VIIWJG|www.wjgnet.com

S
2063 Targetingthecellcycleinesophagealadenocarcinoma:Anadjunctto
anticancertreatment
Dibb M, Ang YS
2070 Optimizingmanagementinautoimmunehepatitiswithliverfailureatinitial
presentation
Potts JR, Verma S
2076 Apracticalapproachtothediagnosisofautoimmunepancreatitis
Frulloni L, Amodio A, Katsotourchi AM, Vantini I
2080 Endoscopicultrasonographyfindingsinautoimmunepancreatitis
Buscarini E, De Lisi S, Arcidiacono PG, Petrone MC, Fuini A, Conigliaro R, Manfredi G,
Manta R, Reggio D, De Angelis C
2086 Effectsofα-mangostinonapoptosisinductionofhumancoloncancer
Watanapokasin R, Jarinthanan F, Nakamura Y, Sawasjirakij N, Jaratrungtawee A,
Suksamrarn S
2096 Chemometricsofdifferentiallyexpressedproteinsfromcolorectalcancer
patients
Yeoh LC, Dharmaraj S, Gooi BH, Singh M, Gam LH
2104 Dietarytreatmentofcoliccausedbyexcessgasininfants:Biochemical
evidence
Infante D, Segarra O, Luyer BL
2109 Levelsofmatrixmetalloproteinase-1andtissueinhibitorsof
metalloproteinase-1ingastriccancer
Kemik O, Kemik AS, Sümer A, Dulger AC, Adas M, Begenik H, Hasirci I, Yilmaz O,
Purisa S, Kisli E, Tuzun S, Kotan C
Contents
EDITORIAL
Weekly Volume 17 Number 16 April 28, 2011
� April 28, 2011|Volume 17|�ssue 16|WJG|www.wjgnet.com
ORIGINAL ARTICLE
BRIEF ARTICLE
TOPIC HIGHLIGHT

ContentsWorld Journal of Gastroenterology
Volume 17 Number 16 April 28, 2011
2113 SunitinibforTaiwanesepatientswithgastrointestinalstromaltumorafter
imatinibtreatmentfailureorintolerance
Chen YY, Yeh CN, Cheng CT, Chen TW, Rau KM, Jan YY, Chen MF
2120 MELDscorecanpredictearlymortalityinpatientswithrebleedingafterband
ligationforvaricealbleeding
Chen WT, Lin CY, Sheen IS, Huang CW, Lin TN, Lin CJ, Jeng WJ, Huang CH, Ho YP,
Chiu CT
2126 StudyonchronicpancreatitisandpancreaticcancerusingMRSandpancreatic
juicesamples
Wang J, Ma C, Liao Z, Tian B, Lu JP
2131 Ku80 geneG-1401Tpromoterpolymorphismandriskofgastriccancer
Li JQ, Chen J, Liu NN, Yang L, Zeng Y, Wang B, Wang XR
2137 Effectsofpenehyclidinehydrochlorideonratintestinalbarrierfunctionduring
cardiopulmonarybypass
Sun YJ, Cao HJ, Jin Q, Diao YG, Zhang TZ
2143 p53 genetherapyincombinationwithtranscatheterarterial
chemoembolizationforHCC:One-yearfollow-up
Guan YS, Liu Y, He Q, Li X, Yang L, Hu Y, La Z
2150 Celiacdiseaseandmicroscopiccolitis:Areportof4cases
Barta Z, Zold E, Nagy A, Zeher M, Csipo I
2155 Pureredcellaplasiacausedbypegylatedinterferon-α-2aplusribavirininthe
treatmentofchronichepatitisC
Chang CS, Yan SL, Lin HY, Yu FL, Tsai CY
2159 Enucleationforgastrointestinalstromaltumorsattheesophagogastric
junction:Isthisanadequatesolution?
Peparini N, Carbotta G, Chirletti P
�� April 28, 2011|Volume 17|�ssue 16|WJG|www.wjgnet.com
CASE REPORT
BRIEF ARTICLE
LETTERS TO THE EDITOR

ContentsWorld Journal of Gastroenterology
Volume 17 Number 16 April 28, 2011
FLYLEAF
APPENDIX
EDITORS FOR THIS ISSUE
Responsible Assistant Editor: Xiao-Fang Liu Responsible Science Editor: Hong SunResponsible Electronic Editor: Wen-Hua Ma Proofing Editorial Office Director: Jian-Xia ChengProofing Editor-in-Chief: Lian-Sheng Ma
NAMEOFJOURNALWorld Journal of Gastroenterology
LAUNCHDATEOctober 1, 1995
RESPONSIBLEINSTITUTIONDepartment of Science and Technology of Shanxi Province
SPONSORTaiyuan Research and Treatment Center for Digestive Diseases, 77 Shuangta Xijie, Taiyuan 030001, Shanxi Province, China
EDITINGEditorial Board of World Journal of Gastroenterology Room 903, Building D, Ocean International Center, No. 62 Dongsihuan Zhonglu, Chaoyang District, Beijing 100025, ChinaTelephone: +86-10-5908-0039Fax: +86-10-8538-1893E-mail: [email protected]://www.wjgnet.com
PUBLISHINGBaishideng Publishing Group Co., LimitedRoom 1701, 17/F, Henan Building, No.90 Jaffe Road, Wanchai, Hong Kong, ChinaFax: +852-3115-8812Telephone: +852-5804-2046E-mail: [email protected]://www.wjgnet.com
SUBSCRIPTIONBeijing Baishideng BioMed Scientific Co., Ltd. Room 903, Building D, Ocean International Center, No. 62 Dongsihuan Zhonglu, Chaoyang District, Beijing 100025, ChinaTelephone: +86-10-8538-1892Fax: +86-10-8538-1893E-mail: [email protected]://www.wjgnet.com
PRINTSUBSCRIPTIONRMB 245 Yuan for each issue, RMB 11760 Yuan for one year.
PUBLICATIONDATEApril 28, 2011
ISSNANDEISSNISSN 1007-9327 (print)ISSN 2219-2840 (online)
HONORARYEDITORS-IN-CHIEFJames L Boyer, New HavenKe-Ji Chen, BeijingMartin H Floch, New Haven Geng-Tao Liu, BeijingEmmet B Keeffe, Palo AltoLein-Ray Mo, TainanEamonn M Quigley, CorkRafiq A Sheikh, SacramentoNicholas J Talley, RochesterMing-Lung Yu, Kaohsiung
PRESIDENTANDEDITOR-IN-CHIEFLian-Sheng Ma, Beijing
ACADEMICEDITOR-IN-CHIEFTauseef Ali, OklahomaMauro Bortolotti, BolognaTarkan Karakan, AnkaraWeekitt Kittisupamongkol, BangkokAnastasios Koulaouzidis, EdinburghGerd A Kullak-Ublick, ZürichBo-Rong Pan, Xi’anSylvia LF Pender, Southampton Max S Petrov, AucklandGeorge Y Wu, Farmington
STRATEGYASSOCIATEEDITORS-IN-CHIEFPeter Draganov, FloridaHugh J Freeman, VancouverMaria Concepción Gutiérrez-Ruiz, MéxicoKazuhiro Hanazaki, KochiAkio Inui, Kagoshima
Kalpesh Jani, BarodaJavier S Martin, Punta del EsteNatalia A Osna, OmahaWei Tang, TokyoAlan BR Thomson, EdmontonHarry HX Xia, Hanover
ASSOCIATEEDITORS-IN-CHIEFYou-Yong Lu, BeijingJohn M Luk, PokfulamHiroshi Shimada, Yokohama
EDITORIALOFFICEJian-Xia Cheng, DirectorWorld Journal of GastroenterologyRoom 903, Building D, Ocean International Center, No. 62 Dongsihuan Zhonglu, Chaoyang District, Beijing 100025, ChinaTelephone: +86-10-5908-0039Fax: +86-10-8538-1893E-mail: [email protected]://www.wjgnet.com
COPYRIGHT© 2011 Baishideng. Articles published by this Open-Access journal are distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and repro-duction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license.
SPECIALSTATEMENTAll articles published in this journal represent the viewpoints of the authors except where indicated otherwise.
INSTRUCTIONSTOAUTHORSFull instructions are available online at http://www.wjgnet.com/1007-9327/g_info_20100315215714.htm.
ONLINESUBMISSIONhttp://www.wjgnet.com/1007-9327office
ABOUT COVER
ACKNOWLEDGMENTS I AcknowledgmentstoreviewersofWorldJournalofGastroenterology
I Meetings
I-VI Instructionstoauthors
WatanapokasinR,JarinthananF,NakamuraY,SawasjirakijN,JaratrungtaweeA,SuksamrarnS.Effectsofα-mangostinonapoptosisinductionofhumancoloncancer.WorldJGastroenterol 2011;17(16):2086-2095http://www.wjgnet.com/1007-9327/full/v17/i16/2086.htm
World Journal of Gastroenterology (World J Gastroenterol, WJG, print ISSN 1007-9327, DOI: 10.3748) is a weekly, open-access, peer-reviewed journal supported by an editorial board of 1144 experts in gastroenterology and hepatology from 60 countries.
The major task of WJG is to report rapidly the most recent results in basic and clinical research on esophageal, gastrointestinal, liver, pancreas and biliary tract diseases, Helicobacter pylori, endoscopy and gastrointestinal surgery, including: gastroesophageal reflux disease, gastrointestinal bleeding, infection and tumors; gastric and duodenal disorders; intestinal inflammation, microflora and immunity; celiac disease, dyspepsia and nutrition; viral hepatitis, portal hypertension, liver fibrosis, liver cirrhosis, liver transplantation, and metabolic liver disease; molecular and cell biology; geriatric and pediatric gastroenterology; diagnosis and screening, imaging and advanced technology.
I-VII EditorialBoard
��� April 28, 2011|Volume 17|�ssue 16|WJG|www.wjgnet.com
AIM AND SCOPE

EDITORIAL
Targeting the cell cycle in esophageal adenocarcinoma: An adjunct to anticancer treatment
Martyn Dibb, Yeng S Ang
Martyn Dibb, Department of Gastroenterology, Royal Albert Ed-ward Infirmary, Wigan Lane, Wigan WN1 2NN, United KingdomYeng S Ang, School of Translational Medicine, Faculty of Medical and Human Sciences, The University of Manchester, Manchester M13 9PL, United KingdomAuthor contributions: Literature review and manuscript by Dibb M, concepts and corrections by Ang YS.Supported by UK National Institute of Health Research/Can-cer Research Network and Research and Development Depart-ment of Wrightington Wigan and Leigh NHS Foundation Trust (to Ang YS); Wrightington Wigan and Leigh NHS Foundation Trust Cancer Therapy Fund (to Dibb M)Correspondence to: Yeng S Ang, MD, FRCP, FRCPI, FEBG, Consultant Gastroenterologist/Honorary Senior Lecturer, Royal Albert Edward Infirmary, Wigan Lane, Wigan WN1 2NN, United Kingdom. [email protected]: +44-1942-773119 Fax: +44-1942-822340Received: December 2, 2010 Revised: January 11, 2011Accepted: January 18, 2011Published online: April 28, 2011
AbstractEsophageal adenocarcinoma is a major cause of cancer death in men in the developed world. Continuing poor outcomes with conventional therapies that predomi-nantly target apoptosis pathways have lead to increas-ing interest in treatments that target the cell cycle. A large international effort has led to the development of a large number of inhibitors, which target cell cycle kinases, including cyclin-dependent kinases, Aurora ki-nases and polo-like kinase. Initial phase Ⅰ/Ⅱ trials in solid tumors have often demonstrated only modest clini-cal benefits of monotherapy. This may relate in part to a failure to identify the patient populations that will gain the most clinical benefit. Newer compounds lacking the side effect profile of first-generation compounds may show utility as adjunctive treatments targeted to an in-dividual’s predicted response to treatment.
© 2011 Baishideng. All rights reserved.
Key words: Esophageal adenocarcinoma; Cell cycle; Cyclin-dependent kinase; Aurora kinases; Polo-like kinase
Peer reviewer: Luis Grande, Professor, Department of Surgery, Hospital del Mar, Passeig Marítim 25-29, Barcelona 08003, Spain
Dibb M, Ang YS. Targeting the cell cycle in esophageal adenocarcinoma: An adjunct to anticancer treatment. World J Gastroenterol 2011; 17(16): 2063-2069 Available from: URL: http://www.wjgnet.com/1007-9327/full/v17/i16/2063.htm DOI: http://dx.doi.org/10.3748/wjg.v17.i16.2063
INTRODUCTIONEsophageal cancer is a major cause of cancer death world-wide[1]. It was the fourth most common cause of death from cancer in men in the United Kingdom between 2004 and 2006[2]. Although in the developed world the incidence and mortality of cancer in general has decreased with advances in diagnosis and treatment, the incidence and mortality of esophageal carcinoma have increased[1].
Esophageal cancer carries a poor prognosis with a 5-year survival rate of < 10%[3]. This probably reflects the fact that the majority of esophageal cancers pres-ent late with symptoms after invasion of the muscularis propria and lymph node metastasis have occurred[4]. Ex-tensive disease means that few patients are suitable for definitive surgical therapy[4,5]. Poor outcomes from con-ventional therapies including surgery and radiochemo-therapy have led to increasing interest in understanding the molecular mechanisms that underpin the develop-ment of esophageal cancer. This may assist in develop-ing new diagnostic techniques and identifying potential therapeutic targets.
The mechanism by which cells reproduce has fasci-nated biologists since Virchow’s 1855 observation that cells could only arise from pre-existing cells. By the
2063
World J Gastroenterol 2011 April 28; 17(16): 2063-2069 ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2063
April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

Dibb M et al . Esophageal adenocarcinoma and cell cycle
early 20th century, pathologists had recorded extensive descriptions of the cytological events of cell division, including division of the nucleus and partitioning of the cytoplasm to the formation of two daughter cells[6]. It has become increasingly clear since those early de-scriptions of the normal cell cycle that disorders in this process can lead to disease. It was not however until the 1970s, that molecular biology allowed a deeper under-standing of the cell cycle and its role in health, disease and cancer development. The past three decades, in particular, have seen major advances in our understand-ing of the genetic and molecular mechanisms by which cells reproduce and how this process is regulated and controlled. It has also been aptly described that cell cycle deregulation, in the form of growth self-sufficiency and insensitivity to growth inhibitory signals, have be-come fundamental hallmarks of cancer development[7-9]. Targeting these pathways in cancer development for diagnostic and therapeutic use has become increasingly important. We assess in this review the potential for tar-geting the cell cycle to treat esophageal adenocarcinoma.
HALLMARKS OF CANCERIt is clear that cellular reproduction is carefully con-trolled and regulated to prevent uncontrolled prolifera-tion of cells[10]. A number of alterations in cell physiol-ogy are required to lead to carcinogenesis[7]. First, a cell must become able to move from its dormant inactive state (known as quiescence) to enter the cell cycle with-out stimulation from external growth factors. Second, the cell must lose response to growth-inhibitory signals. Cells must evade senescence and programmed cell death to gain limitless replicative potential. Finally, it must be able to develop and maintain an adequate blood supply (angiogenesis), which allows the cancer cell to invade and metastasize throughout the organism[11].
Many genes responsible for the carcinogenesis have been identified. Broadly, they fall into two categories: oncogenes and tumor suppressor genes. Oncogenes are created by mutations in genes that cause them to be-come constitutively active, whereas in tumor suppressor genes, mutations reduce or inactive the gene product[12]. Oncogenes and tumor suppressor genes increase tumor cell number by stimulation of cell division or prevention of cell death.
CELL CYCLEEmbyronic cells can undergo DNA replication and nuclear division at rapid rates. A full cycle of embyronic cell division can last just 30 min[13]. Division of adult stem cells requires more complex control (Figure 1). Gaps or pauses are inserted between the phases of nuclear division (M phase) and DNA synthesis (S phase). These gaps are known as G1 (between M and S phases) and G2 (between S and M phases).
Events in the cell cycle happen in a temporally orga-nized sequence, with later events depending on success-ful completion of earlier events[14].
Control of the cell cycle is driven by the cyclin-depen-dent kinases (CDKs), a family of serine/threonine kinas-es. Cells cannot enter S phase, without CDK activation. In order to become catalytically active, CDKs need to bind to a cyclin subunit that acts as an activator. CDKs can also be modulated by inhibitors such as CDK inhibitor 1A (p21CIP1), CDK inhibitor 1B (p27KIP1) or CDK inhibitor 2B (p15INK4B)[10]. It has previously been thought that mam-malian cells require the sequential activation of a number of the CDKs to complete the cell cycle successfully[15]. Recent evidence from mouse models has suggested that CDK1 alone is sufficient to complete the cell cycle, al-though other CDKs are required for normal development and cell type specialization[16]. Cell cycle defects can con-tribute to esophageal cancer development in a number of different ways (Figure 2).
Mitosis itself contains a series of phases that lead to chromosome separation and cell division. Mitosis is a vital step in the cell cycle, which involves carefully regu-lated interactions between multiple proteins. Abnormali-ties throughout the cell cycle can lead to genomic insta-bility through unrestrained proliferation or defects in the transmission of genetic information to daughter cells. A number of established chemotherapy agents, including
2064 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Figure 1 Cell cycle.
Figure 2 Compounds targeting the cell cycle.
PLK1 AURORA A
• • •
• •
Legend
KinaseActive replicationGap phaseCyclin/CD complexMain actionCheckpoint
G2/M checkpointCell division
DNA synthesis
CDK1
Cylin
B
CDK4/6
Cylin D
G1/S checkpoint
Legend
DrugActive replicationGap phaseCell cycle targetInhibition
Bl-2536
Danusertib
AZD1152
Flavopiridol
CDK1
Cylin
B
CDK4/6
Cylin D
BUB-1MPS-1
Aurora B
Polo like
Auro
ra A
kinase 1

the vinca alkaloids and the taxanes work by targeting the mitotic phase of the cell cycle.
CELL CYCLE CHECKPOINTSCells need mechanisms that prevent progression of the cell cycle if there is significant genomic damage, until the damage is repaired or the cell undergoes apoptosis. These have become known as cell cycle checkpoints. There are two major checkpoints: the G1/S checkpoint and the G2/M checkpoint. Checkpoint kinases ATM and ATR mediate these checkpoints, through effector kinases such as CHK1 and CHK2, by preventing activa-tion of CDKs and progression through the cell cycle[17]. Double-stranded DNA breaks activate preferentially ATM, whereas UV light activates ATR kinase. Defects in this DNA damage response can contribute to cancer formation by allowing tumor cell survival despite ge-nome instability and enhanced mutation rates[18,19]. The DNA damage response is commonly activated in early neoplastic lesions[20,21].
G1/S checkpointThe G1/S checkpoint occurs towards the end of the G1 phase, prior to entry into G2. During G1, the cell remains responsive to external mitogenic and anti-mitogenic stimuli. These can either cause the cell to become quies-cent (entering the GO phase) or allow re-entry to the cell cycle. This decision is controlled by the pocket protein RB. Immediately after mitosis, RB is dephosphorylated by protein phosphatase type 1. Whilst in this dephosphory-lated state, RB binds to a group of transcription factors called E2Fs and inhibits their activity. During G1, RB is hypophosphorylated by the complex of CDK4 and cyclin D. CDK2 and cyclin E complexes then act to hyperphos-phorylate RB, which causes dissociation from E2Fs. Free E2Fs trigger increased transcription of CDK2 and cyclin E, which creates a positive feedback loop that drives the cell into DNA synthesis (S phase). CDC25 phosphatases act to regulate CDK and cyclin complexes by removing inhibitory phosphate groups thereby promoting cell cycle progression[13]. In genomic damage, CHK2 activates the p53 pathway, which stimulates production of p21CIP1 as well as phosphorylation of CDC25A. This prevents acti-vation of the CDK/cyclin complexes[13].
G2/M CheckpointThe G2/M checkpoint acts as a final check to prevent mitosis occurring if the genome is damaged. A complex of cyclin B and CDK1 regulates this transition. Through-out G2, the inhibitory kinases CHK1, WEE1 and MYT1 phosphorylate CDK1, which prevents its activation and progression to mitosis. Polo-like kinase 1 (PLK1) protein levels begin to accumulate during S phase and G2/M phases, having been relatively low during G1[22,23]. PLK1 transcription is most abundant in cells that are in G2/M phase[24]. In the absence of DNA damage, PLK1 is phosphorylated by Aurora A at its phosphorylation site
at T210[25]. Phosphorylated PLK1 then activates CDC25 phosphatases that remove inhibitory phosphates from the ATP-binding site located at Thr14 and Tyr15 in human CDK1. This causes the activation of the CDK1/cyclin B complex and drives the cell into mitosis[26]. PLK1 also increases phosphorylation-dependent cyclin B import to the nucleus[27]. PLK1 phosphorylates WEE1 and MYT1, which leads to ubiquitination and degradation of WEE1 and inhibition of MYT1[28,29]. PLK1 is then inactivated and degraded during anaphase by ubiquitin-dependent degradation mediated by the anaphase promoting com-plex[30]. Cell cultures show severely impaired growth when PLK1 is either overexpressed or functionally depleted[31,32].
CELL CYCLE AS A TARGET FOR CANCER THERAPEUTICSMany oncogenes and tumor suppressors have downstream effects on cellular functions involving cell cycle entry and exit. Healthy or normal cells have the ability to stop at pre-determined checkpoints in the cell cycle in the presence of damage or unfavorable conditions. Cancer cells develop mechanisms that eliminate these checkpoints, which leads to uncontrolled proliferation. One example of this is the INK4 family member p16. This occurs as a result of epi-genetic silencing by DNA hypermethylation at the p16 promoter, which leads to reduced transcription and loss of gene expression. p16 is a CDK inhibitor and loss of p16 function leads to unrestrained cellular proliferation. This has been demonstrated to occur with a number of different tu-mors[33]. Abnormalities of p16 function have been described in Barrett’s esophagus and esophageal adenocarcinoma[34]. DNA hypermethylation of the p16 promoter has also been shown to be a strong predictor of the progression to high-grade dysplasia and esophageal adenocarcinoma[35].
CDK inhibitorsAbnormal expression of CDKs and their partner cyclins has been noted in esophageal cancer[36-39]. Polymorphisms of CCND1, which encodes cyclin D1 has been shown to be associated with an increased risk of esophageal adeno-carcinoma[40]. CCND1 amplification and nuclear staining of cyclin D1 have been shown to correlate negatively with survival[41,42]. Abnormal activity of the CDK/cyclin com-plexes in esophageal adenocarcinoma has been shown to be a marker of acquired chemoradioresistance[42,43]. The observation that inhibition of CDKs leads to cell cycle arrest and apoptosis has lead to the development of CDK inhibitors as antitumor drugs. There are a number of drugs that target these pathways. The pioneer compound for this group is flavopiridol, a semi-synthetic inhibitory flavonoid of CDKs. Flavopiridol prevents the phos-phorylation and activation of CDK1, CDK2, CDK4 and CDK6, which leads to reduced expression of cyclin D1, cell cycle arrest, and induction of apoptosis[44].
In vitro, it has been demonstrated that even at nanomo-lar doses, flavopiridol can enhance the antitumor activity
2065 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Dibb M et al . Esophageal adenocarcinoma and cell cycle

of cytotoxic drugs by increasing apoptosis[45]. Phase Ⅰ and Ⅱ studies have been undertaken with various combina-tions of chemotherapeutic agents with variable results. Most promising is the combination with irinotecan and cisplatin. A phase I trial of relapsed gastric and esophageal cancer patients showed that eight out 14 patients achieved a partial response[46]. Further clinical studies are awaited.
Aurora kinases inhibitorsThe Aurora kinase family is an important family of ser-ine/threonine kinases that are evolutionarily conserved and act as mitotic regulators throughout the cell cycle. There are three mammalian aurora kinases, Aurora A, Aurora B, and Aurora C, which have differing roles throughout mitosis[47]. Aurora A is required for centro-some maturation and spindle formation, in addition to its role at the G2/M checkpoint described above. Aurora B is required for chromosome segregation and cytokinesis. Small molecule inhibitors of Aurora B lead to premature mitotic exit without successful chromosome separation. Continued inhibition of Aurora B results in large mul-tiploid cells that eventually undergo apoptosis[48]. This potentially has the advantage that Aurora B inhibitors could be combined with other agents that act during other phases of the cell cycle. Aurora C is abundant in the testes. Its global functions are unclear, however, it has recently been shown to have some overlap with the func-tions of Aurora B during mitosis[49]. Aurora kinases have been shown to be overexpressed in a number of different tumors. Aurora A has been shown to be overexpressed in Barrett’s esophagus and esophageal adenocarcinoma[50,51]. Cell line models suggest that Aurora A overexpression protects developing esophageal adenocarcinoma cells against drug-induced apoptosis[51]. In other forms of cancer, Aurora A expression has been shown to correlate with chromosomal instability[52]. A number of Aurora ki-nase inhibitors are undergoing phase Ⅰ and Ⅱ evaluation. Danusertib, a pan-Aurora kinase inhibitor has undergone phase I testing in patients with advanced solid tumors. Forty-six percent of patients treated with danuserib had stable disease following treatment and a number of pro-longed objective responses were noted[53,54]. The major dose limiting effect of these drugs is neutropenia.
PLK1 inhibitorsPLKs form a group of prominent mitotic kinases. They were first described in mutants that failed to undergo a normal mitosis in Drosophilia melanogaster (polo)[55,56]. They are highly conserved from yeast to humans. There are four members of the polo family in mammals (PLK1-4)[57,58]. They are involved in multiple functions throughout the cell cycle in mitosis and meiosis. PLK1 is the best charac-terized of the four known PLKs[58].
PLK1 is a candidate for development as a therapeu-tic target because it contains two functionally relevant sites: a C-terminal regulatory region containing two polo box domains (PBDs) and an N-terminal catalytic kinase domain[59]. The highly conserved PBD has been identi-
fied as a phosphopeptide-binding motif[60]. The polo box motif is only observed in the PLK family and contains a characteristic sequence. Drugs that target the PBD are specific to the human family of PLKs.
PLK1 is overexpressed in a broad range of primary gastrointestinal tumors, including gastric, colorectal and pancreatic carcinoma[61-63]. In contrast, one study has noted downregulation of PLK1 within tumor cells[64]. There is now increasing evidence that PLK1 expres-sion levels have prognostic significance within different cancers, including esophageal cancer[63]. Two separate re-ports of PLK1 overexpression in esophageal carcinoma primarily relate to squamous cell carcinoma (SCC) in the far east[63,65]. Given the high impact of environmen-tal factors (e.g. aflatoxin) on SCC development in these populations, it is unclear whether the findings can be di-rectly applied to western populations. There are no data on PLK1 expression in adenocarcinoma patients. Some reports of other cancers have suggested that PLK1 ex-pression is a reliable marker of metastasis[66]. PLK1 has also been used in the context of larger arrays of genes as a prognostic marker to predict metastasis in breast cancers[67]. Current cancer staging systems and histologi-cal assessments often fail to predict individual outcomes reliably but correlation of PLK1 protein and mRNA expression levels with clinical stage has the potential to improve clinical decision making in a number of differ-ent tumors[68].
The unique PLD of PLK1 also makes it a good can-didate for the development of alternative cancer thera-pies. Initial efforts have focused on specific phosphoro-thioate antisense oligonucleotides that are able to block protein translation[69]. Use of siRNAs, which cause de-pletion of PLK1, has also been considered. Whilst there are drawbacks of siRNAs, including off-target effects and nuclease sensitivity, these hold promise in cancers such as bladder cancer in which they can act locally[70]. There are now a number of small molecule inhibitors of PLK1, which act either in an ATP-competitive or non-ATP-competitive manner[68]. The multiple actions of PLK1 throughout the cell cycle mean that these new agents need to be carefully assessed for specificity and side effects. In particular, it is possible that anti-PLK1 agents have similar toxicity to other microtubule inhibi-tors. PLK1 inhibitors are now in early clinical testing (phase Ⅰ and Ⅱ). Early clinical experience suggests that neutropenia and thrombocytopenia are dose-related ef-fects, although neuropathy has not been seen[71].
MPS1 inhibitorsCell cycle translational research has focused on the de-velopment of inhibitors of the major kinases discussed above. There are additional mitotic kinases that may have relevance for inhibiting tumor growth. Inhibitors of MPS1, a kinetochore-associated kinase that is involved in the spindle assembly checkpoint, have been shown to ar-rest tumor cell proliferation in vitro[72,73]. This appears to be mediated at least in part by impaired Aurora B func-
2066 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Dibb M et al . Esophageal adenocarcinoma and cell cycle

tion at centromeres, which leads to impaired alignment of chromosomes[74]. Detailed information on MPS1 in esophageal cancer is lacking, however, MPS1 inhibition has been demonstrated as a chemotherapy sensitization strategy in vitro[75].
CONCLUSIONEstablished esophageal carcinoma chemotherapy regimes are relatively blunt tools that predominantly target apop-tosis pathways and are often associated with significant side effects. This has led to a large international effort to develop targeted therapy.
Current therapies that target the cell cycle have largely disappointed with relatively modest effects seen in phase Ⅰ/Ⅱ trials (Table 1). This may be in part related to failure to identify the patient populations that will gain the most clinical benefit. Few treatments are targeted towards spe-cific pathways or personalized to the individual tumor pro-teome or genomic signature.
Efforts are now being made to assess gene expression profiles from histological specimens from solid tumors such as breast cancer in an attempt to predict response to che-motherapy[76]. Initial steps in this direction have been taken by the UK Oesophageal Cancer Clinical and Molecular Stratification (OCCAMS) Study Group, which has demon-strated a four-gene signature associated with poor prognosis in esophageal adenocarcinoma, as well as a larger group of genes associated with lymph node metastasis[77]. Efforts have also been made to identify Barrett’s esophagus patients who are likely to progress to adenocarcinoma, however, little work has been undertaken on response to chemotherapy in the esophagus[78]. Careful studies are needed in esophageal adenocarcinoma to define patient populations that are likely to respond well to treatment with both established and novel chemotherapy regimes. Optimizing individual chemotherapy regimens for patients will assume greater significance as health economies demand most clinical benefit from limited resources. In this setting of personalized targeted therapy, new cell cycle treatments may hold promise as carefully se-lected adjuncts to existing chemotherapy regimes.
Patients with esophageal adenocarcinoma unfortu-nately often still present late with a large burden of dis-
ease. Given the large number of cells involved it is likely that some tumor cells will abrogate the inhibited path-ways and escape from chemotherapy-induced apoptosis. Targeted cell cycle therapy in esophageal cancer presents an alternate strategy as cell cycle inhibitors affect mul-tiple essential pathways involved in replication and DNA damage repair. They may provide a useful adjunct in pa-tients with late presenting esophageal tumors who have failed standard chemotherapy regimens.
REFERENCES1 Umar SB, Fleischer DE. Esophageal cancer: epidemiology,
pathogenesis and prevention. Nat Clin Pract Gastroenterol Hepatol 2008; 5: 517-526
2 National Statistics Online - Product - Cancer Registration Statistics England. Available from: URL: http://www.sta-tistics.gov.uk/StatBase/Product.asp?vlnk=7720
3 Coleman MP, Gatta G, Verdecchia A, Estève J, Sant M, Storm H, Allemani C, Ciccolallo L, Santaquilani M, Berrino F. EU-ROCARE-3 summary: cancer survival in Europe at the end of the 20th century. Ann Oncol 2003; 14 Suppl 5: v128-v149
4 Bird-Lieberman EL, Fitzgerald RC. Early diagnosis of oe-sophageal cancer. Br J Cancer 2009; 101: 1-67
5 Perlmutter AE, Karimi KM, McFadden DW, Nigam A. Man-agement of esophageal carcinoma. W V Med J 2005; 101: 60-63
6 Nurse P, Masui Y, Hartwell L. Understanding the cell cycle. Nat Med 1998; 4: 1103-1106
7 Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57-70
8 Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Mar-kowitz SD, Kinzler KW, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature 1998; 392: 300-303
9 Lane DP. Cancer. p53, guardian of the genome. Nature 1992; 358: 15-16
10 Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009; 9: 153-166
11 Morales CP, Souza RF, Spechler SJ. Hallmarks of cancer pro-gression in Barrett’s oesophagus. Lancet 2002; 360: 1587-1589
12 Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004; 10: 789-799
13 Massagué J. G1 cell-cycle control and cancer. Nature 2004; 432: 298-306
14 Nurse P. A long twentieth century of the cell cycle and be-yond. Cell 2000; 100: 71-78
15 Satyanarayana A, Kaldis P. Mammalian cell-cycle regula-tion: several Cdks, numerous cyclins and diverse compen-satory mechanisms. Oncogene 2009; 28: 2925-2939
16 Santamaría D, Barrière C, Cerqueira A, Hunt S, Tardy C,
2067 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Dibb M et al . Esophageal adenocarcinoma and cell cycle
Table 1 Compounds targeting the cell cycle under active development
Inhibitor Main target Sponsor Clinical trials
BI2536 PLK1 (partial inhibition of PLK2/3) Boehringer Ingelheim Phase Ⅱ pancreatic cancerDanusertib(Formerly PHA-739358)
Pan-aurora kinase inhibitor Pfizer Italia Phase Ⅱ advanced solid tumors
MLN8237 Aurora a inhibitor Millennium Phase Ⅰ/Ⅱ advanced solid tumoursBI6267 PLK1 inhibitor Boehringer Ingelheim Phase Ⅱ ovarian cancer/phase Ⅰ advanced solid tumorsP276-00 Small molecule cyclin inhibitor Piramal Life Sciences Phase Ⅰ advanced malignancy/phase Ⅱ head and neck
malignancyNMS-1286937 PLK1 selective inhibitor Nerviano Medical Sciences Phase Ⅰ advanced solid tumoursP1446A-05 CDK selective inhibitor Piramal Life Sciences Phase Ⅰ advanced malignancySCH727965 CDK inhibitor Schering–Plough Phase Ⅰ advanced malignancySeliciclib (Roscovitine) CDK inhibitor Cyclacel Pharmaceuticals Phase Ⅰ advanced malignancy
CDK: Cyclin-dependent kinase; PLK1: Polo-like kinase 1.

Newton K, Cáceres JF, Dubus P, Malumbres M, Barbacid M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007; 448: 811-815
17 Takaki T, Trenz K, Costanzo V, Petronczki M. Polo-like kinase 1 reaches beyond mitosis--cytokinesis, DNA damage response, and development. Curr Opin Cell Biol 2008; 20: 650-660
18 Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature 2009; 458: 719-724
19 Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-in-duced DNA damage model for cancer development. Science 2008; 319: 1352-1355
20 Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Ørntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005; 434: 864-870
21 Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr, Kastrina-kis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Na-ture 2005; 434: 907-913
22 Golsteyn RM, Mundt KE, Fry AM, Nigg EA. Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. J Cell Biol 1995; 129: 1617-1628
23 Hamanaka R, Smith MR, O’Connor PM, Maloid S, Mihalic K, Spivak JL, Longo DL, Ferris DK. Polo-like kinase is a cell cycle-regulated kinase activated during mitosis. J Biol Chem 1995; 270: 21086-21091
24 Uchiumi T, Longo DL, Ferris DK. Cell cycle regulation of the human polo-like kinase (PLK) promoter. J Biol Chem 1997; 272: 9166-9174
25 Macůrek L, Lindqvist A, Lim D, Lampson MA, Klomp-maker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008; 455: 119-123
26 Pelengaris S, Khan M. The Molecular biology of Cancer. 2006
27 Toyoshima-Morimoto F, Taniguchi E, Shinya N, Iwamatsu A, Nishida E. Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature 2001; 410: 215-220
28 Nakajima H, Toyoshima-Morimoto F, Taniguchi E, Nishida E. Identification of a consensus motif for Plk (Polo-like ki-nase) phosphorylation reveals Myt1 as a Plk1 substrate. J Biol Chem 2003; 278: 25277-25280
29 Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, Osada H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci USA 2004; 101: 4419-4424
30 Tsvetkov L. Polo-like kinases and Chk2 at the interface of DNA damage checkpoint pathways and mitotic regulation. IUBMB Life 2004; 56: 449-456
31 Mundt KE, Golsteyn RM, Lane HA, Nigg EA. On the regu-lation and function of human polo-like kinase 1 (PLK1): effects of overexpression on cell cycle progression. Biochem Biophys Res Commun 1997; 239: 377-385
32 Smith MR, Wilson ML, Hamanaka R, Chase D, Kung H, Longo DL, Ferris DK. Malignant transformation of mamma-lian cells initiated by constitutive expression of the polo-like kinase. Biochem Biophys Res Commun 1997; 234: 397-405
33 Wajed SA, Laird PW, DeMeester TR. DNA methylation: an alternative pathway to cancer. Ann Surg 2001; 234: 10-20
34 Barrett MT, Sanchez CA, Galipeau PC, Neshat K, Emond M, Reid BJ. Allelic loss of 9p21 and mutation of the CDKN2/p16 gene develop as early lesions during neoplastic progres-sion in Barrett’s esophagus. Oncogene 1996; 13: 1867-1873
35 Wang JS, Guo M, Montgomery EA, Thompson RE, Cosby H, Hicks L, Wang S, Herman JG, Canto MI. DNA promoter hypermethylation of p16 and APC predicts neoplastic pro-
gression in Barrett’s esophagus. Am J Gastroenterol 2009; 104: 2153-2160
36 Kawakubo H, Ozawa S, Ando N, Kitagawa Y, Mukai M, Ueda M, Kitajima M. Alterations of p53, cyclin D1 and pRB expression in the carcinogenesis of esophageal squamous cell carcinoma. Oncol Rep 2005; 14: 1453-1459
37 Arber N, Gammon MD, Hibshoosh H, Britton JA, Zhang Y, Schonberg JB, Roterdam H, Fabian I, Holt PR, Weinstein IB. Overexpression of cyclin D1 occurs in both squamous carci-nomas and adenocarcinomas of the esophagus and in adeno-carcinomas of the stomach. Hum Pathol 1999; 30: 1087-1092
38 Hirai T, Kuwahara M, Yoshida K, Osaki A, Toge T. The prog-nostic significance of p53, p21 (Waf1/Cip1), and cyclin D1 protein expression in esophageal cancer patients. Anticancer Res 1999; 19: 4587-4591
39 Morgan RJ, Newcomb PV, Hardwick RH, Alderson D. Amplification of cyclin D1 and MDM-2 in oesophageal carci-noma. Eur J Surg Oncol 1999; 25: 364-367
40 Casson AG, Zheng Z, Evans SC, Geldenhuys L, van Zanten SV, Veugelers PJ, Porter GA, Guernsey DL. Cyclin D1 polymorphism (G870A) and risk for esophageal adenocarci-noma. Cancer 2005; 104: 730-739
41 Miller CT, Moy JR, Lin L, Schipper M, Normolle D, Brenner DE, Iannettoni MD, Orringer MB, Beer DG. Gene amplifi-cation in esophageal adenocarcinomas and Barrett’s with high-grade dysplasia. Clin Cancer Res 2003; 9: 4819-4825
42 Bani-Hani K, Martin IG, Hardie LJ, Mapstone N, Briggs JA, Forman D, Wild CP. Prospective study of cyclin D1 overex-pression in Barrett’s esophagus: association with increased risk of adenocarcinoma. J Natl Cancer Inst 2000; 92: 1316-1321
43 Milas L, Akimoto T, Hunter NR, Mason KA, Buchmiller L, Yamakawa M, Muramatsu H, Ang KK. Relationship be-tween cyclin D1 expression and poor radioresponse of mu-rine carcinomas. Int J Radiat Oncol Biol Phys 2002; 52: 514-521
44 Syrigos KN, Zalonis A, Kotteas E, Saif MW. Targeted ther-apy for oesophageal cancer: an overview. Cancer Metastasis Rev 2008; 27: 273-288
45 Motwani M, Rizzo C, Sirotnak F, She Y, Schwartz GK. Flavo-piridol enhances the effect of docetaxel in vitro and in vivo in human gastric cancer cells. Mol Cancer Ther 2003; 2: 549-555
46 Shah MA, Kortmansky J, Motwani M, Drobnjak M, Gonen M, Yi S, Weyerbacher A, Cordon-Cardo C, Lefkowitz R, Brenner B, O’Reilly E, Saltz L, Tong W, Kelsen DP, Schwartz GK. A phase I clinical trial of the sequential combination of irinotecan fol-lowed by flavopiridol. Clin Cancer Res 2005; 11: 3836-3845
47 Kimura M, Uchida C, Takano Y, Kitagawa M, Okano Y. Cell cycle-dependent regulation of the human aurora B pro-moter. Biochem Biophys Res Commun 2004; 316: 930-936
48 Gizatullin F, Yao Y, Kung V, Harding MW, Loda M, Sha-piro GI. The Aurora kinase inhibitor VX-680 induces en-doreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint func-tion. Cancer Res 2006; 66: 7668-7677
49 Slattery SD, Mancini MA, Brinkley BR, Hall RM. Aurora-C kinase supports mitotic progression in the absence of Auro-ra-B. Cell Cycle 2009; 8: 2984-2994
50 Agnese V, Cabibi D, Calcara D, Terrasi M, Pantuso G, Fio-rentino E, Intrivici C, Colucci G, Aragona F, Gebbia N, Bazan V, Russo A. Aurora-A overexpression as an early marker of reflux-related columnar mucosa and Barrett’s oesophagus. Ann Oncol 2007; 18 Suppl 6: vi110-vi115
51 Dar AA, Zaika A, Piazuelo MB, Correa P, Koyama T, Belkhiri A, Washington K, Castells A, Pera M, El-Rifai W. Frequent overexpression of Aurora Kinase A in upper gas-trointestinal adenocarcinomas correlates with potent anti-apoptotic functions. Cancer 2008; 112: 1688-1698
52 Miyoshi Y, Iwao K, Egawa C, Noguchi S. Association of centrosomal kinase STK15/BTAK mRNA expression with chromosomal instability in human breast cancers. Int J Can-cer 2001; 92: 370-373
53 Cohen RB, Jones SF, Aggarwal C, von Mehren M, Cheng
2068 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Dibb M et al . Esophageal adenocarcinoma and cell cycle

J, Spigel DR, Greco FA, Mariani M, Rocchetti M, Ceruti R, Comis S, Laffranchi B, Moll J, Burris HA. A phase I dose-escalation study of danusertib (PHA-739358) administered as a 24-hour infusion with and without granulocyte colony-stimulating factor in a 14-day cycle in patients with ad-vanced solid tumors. Clin Cancer Res 2009; 15: 6694-6701
54 Steeghs N, Eskens FA, Gelderblom H, Verweij J, Nortier JW, Ouwerkerk J, van Noort C, Mariani M, Spinelli R, Carpinelli P, Laffranchi B, de Jonge MJ. Phase I pharmaco-kinetic and pharmacodynamic study of the aurora kinase inhibitor danusertib in patients with advanced or metastatic solid tumors. J Clin Oncol 2009; 27: 5094-5101
55 Sunkel CE, Glover DM. polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J Cell Sci 1988; 89 (Pt 1): 25-38
56 Llamazares S, Moreira A, Tavares A, Girdham C, Spruce BA, Gonzalez C, Karess RE, Glover DM, Sunkel CE. polo encodes a protein kinase homolog required for mitosis in Drosophila. Genes Dev 1991; 5: 2153-2165
57 Nigg EA. Polo-like kinases: positive regulators of cell divi-sion from start to finish. Curr Opin Cell Biol 1998; 10: 776-783
58 Martin BT, Strebhardt K. Polo-like kinase 1: target and reg-ulator of transcriptional control. Cell Cycle 2006; 5: 2881-2885
59 Leung GC, Hudson JW, Kozarova A, Davidson A, Den-nis JW, Sicheri F. The Sak polo-box comprises a structural domain sufficient for mitotic subcellular localization. Nat Struct Biol 2002; 9: 719-724
60 Elia AE, Cantley LC, Yaffe MB. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic sub-strates. Science 2003; 299: 1228-1231
61 Weichert W, Kristiansen G, Schmidt M, Gekeler V, Noske A, Niesporek S, Dietel M, Denkert C. Polo-like kinase 1 expres-sion is a prognostic factor in human colon cancer. World J Gastroenterol 2005; 11: 5644-5650
62 Macmillan JC, Hudson JW, Bull S, Dennis JW, Swallow CJ. Comparative expression of the mitotic regulators SAK and PLK in colorectal cancer. Ann Surg Oncol 2001; 8: 729-740
63 Tokumitsu Y, Mori M, Tanaka S, Akazawa K, Nakano S, Niho Y. Prognostic significance of polo-like kinase expres-sion in esophageal carcinoma. Int J Oncol 1999; 15: 687-692
64 Simizu S, Osada H. Mutations in the Plk gene lead to insta-bility of Plk protein in human tumour cell lines. Nat Cell Biol 2000; 2: 852-854
65 Feng YB, Lin DC, Shi ZZ, Wang XC, Shen XM, Zhang Y, Du XL, Luo ML, Xu X, Han YL, Cai Y, Zhang ZQ, Zhan QM, Wang MR. Overexpression of PLK1 is associated with poor survival by inhibiting apoptosis via enhancement of sur-vivin level in esophageal squamous cell carcinoma. Int J Cancer 2009; 124: 578-588
66 Kneisel L, Strebhardt K, Bernd A, Wolter M, Binder A, Kaufmann R. Expression of polo-like kinase (PLK1) in thin
melanomas: a novel marker of metastatic disease. J Cutan Pathol 2002; 29: 354-358
67 Ahr A, Karn T, Solbach C, Seiter T, Strebhardt K, Holtrich U, Kaufmann M. Identification of high risk breast-cancer pa-tients by gene expression profiling. Lancet 2002; 359: 131-132
68 Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer 2006; 6: 321-330
69 Spänkuch B, Steinhauser I, Wartlick H, Kurunci-Csacsko E, Strebhardt KI, Langer K. Downregulation of Plk1 expression by receptor-mediated uptake of antisense oligonucleotide-loaded nanoparticles. Neoplasia 2008; 10: 223-234
70 Nogawa M, Yuasa T, Kimura S, Tanaka M, Kuroda J, Sato K, Yokota A, Segawa H, Toda Y, Kageyama S, Yoshiki T, Okada Y, Maekawa T. Intravesical administration of small interfering RNA targeting PLK-1 successfully prevents the growth of bladder cancer. J Clin Invest 2005; 115: 978-985
71 Jackson JR, Patrick DR, Dar MM, Huang PS. Targeted anti-mitotic therapies: can we improve on tubulin agents? Nat Rev Cancer 2007; 7: 107-117
72 Dorer RK, Zhong S, Tallarico JA, Wong WH, Mitchison TJ, Murray AW. A small-molecule inhibitor of Mps1 blocks the spindle-checkpoint response to a lack of tension on mitotic chromosomes. Curr Biol 2005; 15: 1070-1076
73 Schmidt M, Budirahardja Y, Klompmaker R, Medema RH. Ablation of the spindle assembly checkpoint by a com-pound targeting Mps1. EMBO Rep 2005; 6: 866-872
74 Jelluma N, Brenkman AB, van den Broek NJ, Cruijsen CW, van Osch MH, Lens SM, Medema RH, Kops GJ. Mps1 phos-phorylates Borealin to control Aurora B activity and chro-mosome alignment. Cell 2008; 132: 233-246
75 Janssen A, Kops GJ, Medema RH. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc Natl Acad Sci USA 2009; 106: 19108-19113
76 Gianni L, Zambetti M, Clark K, Baker J, Cronin M, Wu J, Mariani G, Rodriguez J, Carcangiu M, Watson D, Valagussa P, Rouzier R, Symmans WF, Ross JS, Hortobagyi GN, Pusz-tai L, Shak S. Gene expression profiles in paraffin-embed-ded core biopsy tissue predict response to chemotherapy in women with locally advanced breast cancer. J Clin Oncol 2005; 23: 7265-7277
77 Peters CJ, Rees JR, Hardwick RH, Hardwick JS, Vowler SL, Ong CA, Zhang C, Save V, O’Donovan M, Rassl D, Alder-son D, Caldas C, Fitzgerald RC. A 4-gene signature predicts survival of patients with resected adenocarcinoma of the esophagus, junction, and gastric cardia. Gastroenterology 2010; 139: 1995-2004.e15
78 Lao-Sirieix P, Boussioutas A, Kadri SR, O’Donovan M, Debi-ram I, Das M, Harihar L, Fitzgerald RC. Non-endoscopic screening biomarkers for Barrett’s oesophagus: from micro-array analysis to the clinic. Gut 2009; 58: 1451-1459
S- Editor Tian L L- Editor Kerr C E- Editor Ma WH
2069 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Dibb M et al . Esophageal adenocarcinoma and cell cycle

EDITORIAL
Optimizing management in autoimmune hepatitis with liver failure at initial presentation
Jonathan R Potts, Sumita Verma
2070
World J Gastroenterol 2011 April 28; 17(16): 2070-2075 ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2070
April 28|Volume 17|Issue 16|WJG|www.wjgnet.com
Jonathan R Potts, Sumita Verma, Department of Medicine, Brighton and Sussex Medical School, Brighton, BN1 9PX, United KingdomAuthor contributions: Verma S conceived the idea; Potts JR performed the literature search; Verma S wrote the initial draft; Potts JR and Verma S contributed equally to writing the final draft of the manuscript.Correspondence to: Dr. Sumita Verma, Senior Lecturer Medicine, Honorary Consultant Hepatologist, Department of medcine Brighton and Sussex Medical School, Falmer, Depart-ment of medcine Brighton, BN1 9PX, United Kingdom. [email protected]: +44-1273-877890 Fax: +44-1273-877576Received: October 12, 2010 Revised: November 4, 2010Accepted: November 11, 2010Published online: April 28, 2011
AbstractAutoimmune hepatitis (AIH) is a disease of unknown etiology, its hallmark being ongoing hepatic inflamma-tion. By its very nature, it is a chronic condition, al-though increasingly, we are becoming aware of patients with acute presentations, some of whom may have liver failure. There are very limited published data on patients with AIH with liver failure at initial diagnosis, which consist mostly of small retrospective studies. As a consequence, the clinical features and optimal man-agement of this cohort remain poorly defined. A subset of patients with AIH who present with liver failure do respond to corticosteroids, but for the vast majority, an urgent liver transplantation may offer the only hope of long-term survival. At present, there is uncertainty on how best to stratify such a cohort into responders and non- responders to corticosteroids as soon as possible after hospitalization, thus optimizing their management. This editorial attempts to answer some of the unre-solved issues relating to management of patients with AIH with liver failure at initial presentation. However, it must be emphasized that, at present, this editorial is based mostly on small retrospective studies, and it is an
understatement that multicenter prospective studies are urgently needed to address this important clinical issue.
© 2011 Baishideng. All rights reserved. Key words: Autoimmune hepatitis; Liver failure; Liver transplantation; Corticosteroids
Peer reviewer: Atsushi Tanaka, MD, PhD, Associate Professor, Department of Medicine, Teikyo University School of Medicine, 2-11-1, Kaga, Itabashi-ku, Tokyo 173-8605, Japan
Potts JR, Verma S. Optimizing management in autoimmune hepatitis with liver failure at initial presentation. World J Gas-troenterol 2011; 17(16): 2070-2075 Available from: URL: http://www.wjgnet.com/1007-9327/full/v17/i16/2070.htm DOI: http://dx.doi.org/16.3748/wjg.v17.i16.2070
INTRODUCTIONAutoimmune hepatitis (AIH) is a disease that is character-ized by chronic hepatic inflammation, presence of autoan-tibodies [antinuclear antibody (ANA), anti-smooth muscle antibody (SMA), and liver kidney microsomal (LKM) anti-body], female preponderance and elevated serum gamma-globulins, especially IgG[1]. Earlier studies have established the beneficial effects of corticosteroids in AIH and up to 80% of patients can now achieve remission with im-munosuppressants[2,3]. At accession, 10%-20% of patients with AIH can be negative for the conventional autoanti-bodies[4], although their outcomes, especially response to immunosuppression, are no different from those that are autoantibody-positive[5].
AIH can have protean manifestations, with the majority of patients presenting with subclinical or chronic disease. However, in > 25%, the disease may present acutely with jaundice, a subset of whom may have fulminant or sub-acute liver failure (LF)[6-8]. Fulminant hepatic failure (FHF) is a devastating clinical condition that occurs in patients

Potts JR et al . Autoimmune hepatitis and liver failure
with no prior history of liver disease, and is characterized by development of hepatic encephalopathy and coagulopa-thy within 8 wk after onset of jaundice[9]. In contrast, those with subacute LF present with encephalopathy at 8-26 wk after onset of symptoms[10]. In a survey in the United States carried out between 1998 and 2008, the major eti-ologies of FHF in 1147 patients were acetaminophen over-dose (46%), followed by indeterminate causes (14%), drug-induced (11%), hepatitis B virus (7%), other causes (7%), AIH (5%), ischemic hepatitis (4%), hepatitis A virus (3%) and Wilson’s disease (2%)[11]. Similar data were reported from Europe where 2%-5% of patients with FHF have AIH as the underlying etiology[12,13]. Unfortunately, neither the International Autoimmune Hepatitis Group (IAIHG) criteria[14] nor the simplified diagnostic criteria for diagnosis of AIH[15] have been extensively validated in patients with LF; largely because of the small number of cases encoun-tered. Thus, diagnosis of AIH and LF remains clinical and is supported by positive autoantibodies, negative viral serology, absence of alcohol excess and culprit drugs, and compatible liver biopsy. This has been corroborated by an earlier study in which 28 patients with FHF were clinically diagnosed with AIH, but after application of the IAIHG criteria and simplified scoring systems only 50% and 46%, respectively, fulfilled the criteria, with the concordance of the two scoring systems being only 46%[16].
Immunoparesis is commonly seen in critically ill pa-tients with LF in whom both autoantibodies and/or el-evated IgG concentrations may be absent[17]. In addition, because of the severity of the hepatic insult (massive/sub-massive necrosis), histological evaluation may be difficult or impossible[16]. Although challenging, AIH can still be diagnosed in such a scenario by excluding other liver dis-eases, and by testing for other autoantibodies [perinuclear antineutrophil cytoplasmic antibodies (pANCA), and an-tibodies to soluble liver antigen (SLA)][18,19]. Furthermore, if the patient is HLA B8, DR3 or DR4 positive, has a con-current immunological disorder, and responds to cortico-steroid therapy, this further lends credence to the diagnosis of AIH[4]. Nonetheless, the decision to initiate corticoste-roids in patients who do not fulfill conventional diagnostic criteria for AIH must be made on an individual basis, and remains the prerogative of the treating hepatologist.
AIH AND LFThere is a paucity of published data on patients with AIH with LF at initial diagnosis; consisting mostly of anecdotal case reports or small case series[20,21]. Thus the clinical char-acteristics, response to immunosuppression, and outcomes with/without liver transplantation (LT) of this cohort re-main poorly described. Much of the controversy hinges on a critical management issue, namely should such patients be given a trial of corticosteroids, be priority listed for LT, or both. If corticosteroids are indeed initiated, how and at what time point do we define failure of medical treatment? This editorial attempts to address some of these controver-sies with the aim to develop strategies that could optimize
management of patients with AIH that present with LF. We therefore searched the medical literature (PubMed)
to collect published data on AIH with initial presentation with LF. Only studies providing data on type and duration of immunosuppressive therapy and outcomes were includ-ed. Case reports/small case series, and studies in which authors reported acute AIH in the absence of LF were excluded. We identified five studies that met our inclusion criteria and these included a total of 85 patients with AIH and LF[7,22-25] (Table 1). In three of the five studies[7,23,24], patients were diagnosed with AIH according to IAIHG criteria, although information regarding probable or defi-nite AIH was only available in two[7,24]. In the remaining two studies[22,25], the diagnosis of AIH was based on the presence of autoantibodies, elevated IgG levels, exclusion of Wilson’s disease, negative viral serology, absence of cul-prit drugs, and compatible liver histology (1). The patients were very heterogeneous as regards ethnicity, presence/ab-sence of cirrhosis, and inclusion of acute and subacute LF. It is well known that these factors have a prognostic value in patients with AIH and in those with LF[7,26-28]. In addi-tion, all the studies were retrospective, and one has only been published in an abstract form[22]. Nonetheless, these five studies do provide valuable information about the natural history of AIH with LF at initial presentation.
In these five studies, the prevalence of LF at initial presentation in patients with AIH varied from 8.7% to 19.8%[7,23]. In all but one patient this was the first presenta-tion of their disease. The majority (> 75%) were women in the third to the sixth decade with type 1 AIH. Almost all patients had either encephalopathy at admission and/or had significant coagulopathy (Table 1). IgG levels were available in two studies[24,25], and 74% had levels in excess of 1800 mg/dL.
OUTCOMES IN PATIENTS WITH AIH AND LFTable 2 shows treatment data and outcomes in these five above studies. Of the total of 85 patients, 69 (89.2%) re-ceived immunosuppression, mostly corticosteroids (Table 2). For the majority of the patients, there was no rationale provided for initiation or withholding corticosteroids, and the decision appeared to have been made on an ad hoc basis. The remission rates with immunosuppression varied from 8.3% to 50% (average: 33.3%, 23/69) (Table 2). Overall, 43.5% (37/85) either underwent or were listed for LT and 32.9% (28/85) died. These outcomes are certainly poorer than those reported in patients with chronic AIH (remission with corticosteroids ~80%[2,3], need for LT 1.4%-8.4% and mortality 1.8%-4.9%[27, 29]), and makes for dismal reading.
The variability in remission rates with corticosteroid therapy in these five studies is most certainly a reflection of the heterogeneous patient population. Unsurprisingly, the lowest remission rates were seen in the study of Ichai et al[25], which had the sickest patients, as reflected by their high admission MELD scores. However, those patients with AIH and LF that did respond to corticosteroid
2071 April 28|Volume 17|Issue 16|WJG|www.wjgnet.com

therapy survived, obviating the need for a subsequent LT. Unfortunately, among the non-responders to corticoste-roids in these five studies (n = 46), death was the inevitable outcome in the absence of LT (Table 2). The duration of steroid therapy prior to death was highly variable (3-95 d). Clearly, in some, the illness was so fulminant that death occurred rapidly after hospitalization, thereby precluding LT, and in others, there were active contraindications to transplantation, such as sepsis (Table 2). Nevertheless, in these five studies, there were a subset of patients with AIH and LF in whom death may have been preventable had LT been more aggressively pursued. It is conceivable that initi-ation of steroids provided a false sense of security, thereby delaying transplant evaluation.
One could argue that the low remission rates to cor-ticosteroids in this cohort were partly related to delay in initiating therapy. However, where available, the data do not support this conclusion, as corticosteroids were initi-ated promptly, especially in the sicker patients. In our study, subsequent non-responders to corticosteroids were com-menced on therapy within 2.6 ± 1.8 d of admission, com-pared to 6.4 ± 5.5 d in those who eventually responded to
corticosteroids[7]. It is more likely that non-responders to corticosteroids had aggressive disease at the time of diag-nosis with a critical degree of liver cell death already having occurred prior to the introduction of medical treatment[24]. This hypothesis is supported by the study of Ichai et al[25], in which all patients had massive/sub-massive liver necro-sis (median MELD score at admission: 37), with only 8.3% responding to corticosteroids and > 80% needing LT.
OPTIMIZING MANAGEMENT IN PA-TIENTS WITH AIH AND LFAssessing patients with LF for LT is a complex process. The most widely used criteria for prioritizing patients for LT are the King’s College criteria[30]. However, neither the King’s College criteria[29] nor the more recently developed MELD score[31] have been validated in patients with AIH and LF. This is most likely due to the fact that the preva-lence of AIH in patients with LF being evaluated for LT is low (0%-5%)[12,13,32]. As is evident from the published data[7,22-25], there certainly are a subset of patients with AIH and LF who will respond to corticosteroids. Inappropri-
2072 April 28|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 1 Clinical characteristics of patients with autoimmune hepatitis with liver failure at initial presentation
Villamil et al [22]1 (n = 28)
Kessler et al [23]
(n = 10) Miyake et al [24]
(n = 11)Ichai et al [25] (n = 16)
Verma et al [7] (n = 20)
Study design Retrospective Retrospective Retrospective Retrospective RetrospectiveAge (yr)2 41 40 ± 15.9 53 (16-75) 36 ± 13.1 41.3 ± 14.2Definition of LF NA NA PT < 40% and HE
≥ grade 2HE within 12 wk of
jaundiceAny grade HE and/or
INR > 2Symptoms duration2 NA 3.2 wk 24 (16-52) d NA 2.1 ± 2.5 mo3
Female NA 8 (80%) 11 (100%) 14/16 (87.5%) 15 (75%)Ethnicity or country of origin South American 80% White Japanese French 70% blackDefinite/probable AIH (IAIHG4 criteria)
NA NA5 3(36%)/8 (64%) NA 9(45%)/11(55%)
LC/LKM6 positiveANA/SMA7 positive
6 (21.4) 1 (10%) 3 (18.7%) NA 22 (78.5%) 7 (70%) NA 11 (68.7%) 20 (100%)
Bilirubin2 (mg/dL) 3988 16.97 ± 9.83 20.6 (5.9-31) 425 (278-850)8 19.3 ± 10.3
AST or ALT2 NA 1179 ± 1127.17 220 (59-1094) 678 (60-2867) 1147.1 ± 711.4INR2 or PT 30% 49.3 ± 66.9 29% (6%-38%) 5.36 (1.7-12.2) 2.7 ± 1.4HE9 at onset 28 (100%) 8 (80%) 11 (100%) 10 (62.5%) 19 (95%)Cirrhosis None 2/10 (20%) NA None 8/20 (40%)MELD2 NA NA NA 37 (24-47) 28 ± 7.41Sub-massive or massive necrosis (SMN, MN)
19/23 (82.6%) 5/10 (50%) NA 16/16 (100%) 15 needed LT and/or
died
12/19 (63.1%), 10 needed LT and or
died 17 needed LT and/or
diedImmunosuppressant regimen used Prednisone 60 mg/d Corticosteroids
(Dose NA) and other10
Prednisolone Prednisone 1 mg/kg per day and other10
Corticosteroids11 40-60 mg/d and
steroid pulse 20-1250 mg/d
Poor prognostic criteria 1: PT < 20%; 2: Grade 4 HE; 3: SMN at diagno-sis; 4: 20% increase in PT at day 3 of steroids
NA 1: High bilirubin at onset; 2: Worsening bilirubin during days 8-15 of steroid therapy
NA 1: Absence of cirrhosis; 2: MELD > 28; 3: Worsening trend in bilirubin and INR after 3.7 ± 0.6 d of steroid therapy
Septic events NA NA NA 7 (43.7%), of whom 6 had received steroids
2 (10%), of whom 1 received steroids
1Published only in abstract form; 2Data presented as mean ± SD or median (range); 3Duration from first symptom (and not necessarily jaundice/hepatic en-cephalopathy) to hospitalization; 4IAIHG: International Autoimmune Hepatitis Group; 5Met IAIHG criteria, data on probable or definite disease unavailable; 6LKM/LC: Liver kidney microsomal antibody/liver cytosol antibody; 7ANA/SMA: antinuclear antibody/anti-smooth muscle antibody; 8Values in µmol/L; 9HE: Hepatic encephalopathy; 10Additional immunosuppression was used in nine patients in the study of Kessler et al (azathioprine, tacrolimus, mycopheno-late mofetil, 6-mercaptopurine, cyclosporine) and in one patient in the study of Ichai et al (azathioprine and cyclosporine); 11Included prednisone, hydrocorti-sone and methylprednisone, (converted to equivalent doses of prednisone); LT:Liver transplantation; PT: Prothrombin time; AIH: Autoimmune hepatitis.
Potts JR et al . Autoimmune hepatitis and liver failure

Table 2 Outcomes of patients with autoimmune hepatitis and initial presentation with liver failure
ate transplantation in such patients would mean subjecting them to unnecessary surgery (and its attendant complica-tions) and lifelong immunosuppression. In addition, it would deprive another more suitable recipient from receiv-ing the graft[33]. On the other hand, denying LT to a pa-tient with AIH and LF who is unlikely to respond to cor-ticosteroids means condemning them to a certain death, which is unacceptable, especially since post-transplant survival for AIH is excellent [estimated 5-year survival probability after first LT is 0.73 (95% CI: 0.67-0.77)][34].
The contentious issue thus is how best to stratify pa-tients with AIH and LF into likely responders and non-responders to corticosteroids as soon as possible after hospitalization; hence optimizing their management. In our study[7], all responders to corticosteroid therapy had a MELD score ≤ 28 at admission. This is also supported by Ichai et al[25], who showed that the only patient to respond to corticosteroids had a MELD score of 24, and none with an initial MELD score > 28 responded to corticosteroids. Furthermore, in our study, responders to corticosteroids were more likely to have either an improvement or stabiliza-tion in bilirubin and INR within 3.7 ± 0.6 d of initiation of corticosteroid therapy, whereas non-responders tended to have a trend for higher bilirubin and INR[7]. Villamil et al[22] also observed that a 20% increase in prothrombin time (PT) at day 3 of corticosteroid therapy to be a predictor of poor outcome, along with PT < 20% , grade 4 encephalopathy, and LKM antibody/liver cytosol (LC) antibody positivity at diagnosis. Histological evidence of sub-massive/massive necrosis is also invariably associated with need for LT and/or death (Table 1). Surprisingly, in our study, the presence of cirrhosis was more likely was associated with response to corticosteroids[7]. Although the impact of cirrhosis on the natural history of AIH remains controversial[27,28,35,36], it is likely that this group has long-standing indolent disease that progresses to cirrhosis, with LF representing an acute relapse of AIH[37]. This is in contrast with the study of Ichai et al[25], in which absence of significant hepatic fibrosis in all the patients indicated a de novo fulminant disease process.
CORTICOSTEROIDS AND INFECTIONSWhether steroids increase the risk of septic complications in patients with severe liver disease is subject to an ongo-ing debate. The issue becomes even more contentious in the presence of LF because in itself that has been associ-ated with an increased risk of bacterial and fungal infec-tions[25,38,39]. In fact, earlier studies have shown that up to 35% of patients with LF can develop bacteremia in the pre-transplant period[39]. This increased propensity for sep-sis is further aggravated in the post-transplant setting due to use of immunosuppression. Therefore, not surprisingly, sepsis with or without multiorgan failure, accounts for almost one-third of all deaths in patients undergoing LT for LF, and is the most common cause of mortality in this cohort[40]. In the study of Ichai et al[25] (which had the sick-est cohort of patients with a median MELD score of 37 at admission), 42.3% developed a septic event, and this prev-alence is not higher than that reported previously[39]. It is however noteworthy that in Ichai et al’s study septic events were more likely to occur in those initiated (6/12) versus those not initiated (1/4) on corticosteroids[25]. It is unclear whether patients received prophylactic antibiotics in this study. Reich et al[41] also have reported an increased trend for wound infection in corticosteroid-treated patients with AIH undergoing LT (30.7% vs 5.2%). In a recent publica-tion that analyzed data from the European Transplant Registry, in comparison with transplantation for primary biliary cirrhosis and alcoholic cirrhosis, the probability of infectious complications limiting patient survival was sig-nificantly increased after transplantation for AIH. This was especially relevant to patients aged > 50 years and within the first 3 mo of transplantation[34]. Unfortunately, data on disease severity and use of pre-transplant immunosuppres-sion and prophylactic antibiotics were not available in that study. On the other hand, others have reported corticoste-roids not to be associated with increased risk of infections in patients with severe AIH[42]. These discordant results most likely reflect the heterogeneous patient groups (in-
2073 April 28|Volume 17|Issue 16|WJG|www.wjgnet.com
Study Villamil et al [22] (n = 28)
Kessler et al [23] (n = 10)
Miyake et al [24] (n = 11)
Ichai et al [25] (n = 16)
Verma et al [7] (n = 20)
Treated with IS1 25 10 8 12 14Responders to steroids 9 (36%) (alive) 4 (40%) (alive) 2 (25%) (alive) 1 (8.3%) (alive) 7 (50%) (alive)Non responders 16 6 6 11 7LT 11 (2 Died) 3 1 10 (1 Died) 1 (Died)Listed for LT - 1 - - 1 (Died)Died without LT 5 2 5 14 52
Not treated with IS1 3 - 33 4 6Spontaneous survival - - 3 - -LT 1 - - 3 5 (1 Died)Listed for LT - - - - -Died 2 - - 1 1Overall underwent LT or listed for LT 12/28 (42.8%) 4/10 (40%) 1/11 (9%) 13/16 (81.2%) 7/20 (35%)Overall mortality 9/28 (32.1%) 2/10 (20%) 5/11 (45.4%) 3/16 (18.7%) 9/20 (45%)
1IS: Immunosuppression; 2Four died while being evaluated for liver transplantation, in 1 sepsis precluded liver transplantation evaluation; 3Treated with plasmapheresis and or stronger neo-minophagen; 4Not evaluated for LT due to sepsis; LT: Liver transplantation. Additional outcome data obtained by per-sonal communication with authors.
Potts JR et al . Autoimmune hepatitis and liver failure

cluding the whole spectrum from chronic disease to FHF), use of varying immunosuppressive regimens, and incon-sistent use of prophylactic antibiotics. Nonetheless, Ichai et al[25] caution against injudicious use of corticosteroids in patients with AIH and LF, and on the contrary, emphasize the need for expedited LT evaluation in such a cohort. Furthermore, it lends credence to the argument for the use of prophylactic antibiotics and antifungal agents, because such a strategy has been shown to reduce the risk of infec-tions in the pre-transplant setting[43].
THE FUTUREProspective multicenter studies are clearly needed to ad-dress this complex and important clinical issue. In future, testing for additional autoantibodies and HLA typing might also help risk-stratify patients. For example, presence of antibodies to SLA have been associated with DRB1 *0301, and such patients have aggressive disease and are more likely to require LT and/or die[44,45].
CONCLUSIONThe diagnosis and management of patients with AIH with AF at initial diagnosis can be challenging. Although there are only limited published data available, mostly in the form of small retrospective studies, up to 8.7%-19.8% of patients with AIH may have this form of presentation. On the whole, about one-third can respond to corticosteroids and have a good outcome, although for the vast major-ity, LT may offer the only hope of long-term survival. A MELD score at admission of ≤ 28, more severe hepatic fibrosis, absence of sub-massive/massive necrosis, and early (within 4 d) improvement or stabilization in biliru-bin and INR, identify those who are likely to respond to corticosteroid therapy, and thus survive without the need for LT. If clinical and biochemical improvement does not occur within the first few days, then continuation of corti-costeroids may be a futile exercise, as it would be unlikely to change the clinical outcome, and on the contrary, may result in adverse events, especially sepsis. Nonetheless, if a decision is made to continue therapy with corticosteroids it is imperative that LT be actively pursued concomitantly. Furthermore, it may not be unreasonable to consider prophylactic antimicrobial and antifungal agents in such high-risk patients. It must however be emphasized that, at present, these recommendations are based on small retrospective studies. This underlines the urgent need for prospective multicenter studies to address this important clinical issue.
ACKNOWLEDGEMENTSSV is grateful to Dr. Villamil, Dr. Kessler and Dr. Mi-yake for providing additional data upon request.
REFERENCES 1 Czaja AJ, Freese DK. Diagnosis and treatment of autoim-
mune hepatitis. Hepatology 2002; 36: 479-4972 Murray-Lyon IM, Stern RB, Williams R. Controlled trial
of prednisone and azathioprine in active chronic hepatitis. Lancet 1973; 1: 735-737
3 Soloway RD, Summerskill WH, Baggenstoss AH, Geall MG, Gitnićk GL, Elveback IR, Schoenfield LJ. Clinical, bio-chemical, and histological remission of severe chronic active liver disease: a controlled study of treatments and early prognosis. Gastroenterology 1972; 63: 820-833
4 McFarlane IG. Autoimmune hepatitis: diagnostic criteria, subclassifications, and clinical features. Clin Liver Dis 2002; 6: 605-621
5 Czaja AJ, Hay JE, Rakela J. Clinical features and prognostic implications of severe corticosteroid-treated cryptogenic chronic active hepatitis. Mayo Clin Proc 1990; 65: 23-30
6 Ferrari R, Pappas G, Agostinelli D, Muratori P, Muratori L, Lenzi M, Verucchi G, Cassani F, Chiodo F, Bianchi FB. Type 1 autoimmune hepatitis: patterns of clinical presentation and differential diagnosis of the ‘acute’ type. QJM 2004; 97: 407-412
7 Verma S, Torbenson M, Thuluvath PJ. The impact of ethnic-ity on the natural history of autoimmune hepatitis. Hepatol-ogy 2007; 46: 1828-1835
8 Verma S, Maheshwari A, Thuluvath P. Liver failure as initial presentation of autoimmune hepatitis: clinical char-acteristics, predictors of response to steroid therapy, and outcomes. Hepatology 2009; 49: 1396-1397
9 Trey C, Davidson LS. The management of fulminant hepatic failure. In: Popper H, Schaffner F, editors. Progress in liver disease. New York: Grune and Stratton, 1970: 282-298
10 Gimson AE, O’Grady J, Ede RJ, Portmann B, Williams R. Late onset hepatic failure: clinical, serological and histologi-cal features. Hepatology 1986; 6: 288-294
11 Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: Summary of a workshop. Hepatology 2008; 47: 1401-1415
12 Escorsell A, Mas A, de la Mata M. Acute liver failure in Spain: analysis of 267 cases. Liver Transpl 2007; 13: 1389-1395
13 Brandsaeter B, Höckerstedt K, Friman S, Ericzon BG, Kirkeg-aard P, Isoniemi H, Olausson M, Broome U, Schmidt L, Foss A, Bjøro K. Fulminant hepatic failure: outcome after listing for highly urgent liver transplantation-12 years experience in the nordic countries. Liver Transpl 2002; 8: 1055-1062
14 Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, Chapman RW, Cooksley WG, Czaja AJ, Des-met VJ, Donaldson PT, Eddleston AL, Fainboim L, Heath-cote J, Homberg JC, Hoofnagle JH, Kakumu S, Krawitt EL, Mackay IR, MacSween RN, Maddrey WC, Manns MP, McFarlane IG, Meyer zum Büschenfelde KH, Zeniya M. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol 1999; 31: 929-938
15 Hennes EM, Zeniya M, Czaja AJ, Parés A, Dalekos GN, Krawitt EL, Bittencourt PL, Porta G, Boberg KM, Hofer H, Bianchi FB, Shibata M, Schramm C, Eisenmann de Torres B, Galle PR, McFarlane I, Dienes HP, Lohse AW. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatol-ogy 2008; 48: 169-176
16 Yeoman AD, Westbrook RH, Al-Chalabi T, Carey I, Heaton ND, Portmann BC, Heneghan MA. Diagnostic value and utility of the simplified International Autoimmune Hepatitis Group (IAIHG) criteria in acute and chronic liver disease. Hepatology 2009; 50: 538-545
17 Gregorio GV, Portmann B, Reid F, Donaldson PT, Doherty DG, McCartney M, Mowat AP, Vergani D, Mieli-Vergani G. Autoimmune hepatitis in childhood: a 20-year experience. Hepatology 1997; 25: 541-547
18 Mulder AH, Horst G, Haagsma EB, Limburg PC, Kleibeu-ker JH, Kallenberg CG. Prevalence and characterization of neutrophil cytoplasmic antibodies in autoimmune liver dis-eases. Hepatology 1993; 17: 411-447
2074 April 28|Volume 17|Issue 16|WJG|www.wjgnet.com
Potts JR et al . Autoimmune hepatitis and liver failure

19 Han S, Tredger M, Gregorio GV, Mieli-Vergani G, Vergani D. Anti-liver cytosolic antigen type 1 (LC1) antibodies in child-hood autoimmune liver disease. Hepatology 1995; 21: 58-62
20 Viruet EJ, Torres EA. Steroid therapy in fulminant hepatic failure secondary to autoimmune hepatitis. P R Health Sci J 1998; 17: 297-300
21 Herzog D, Rasquin-Weber AM, Debray D, Alvarez F. Sub-fulminant hepatic failure in autoimmune hepatitis type 1: an unusual form of presentation. J Hepatol 1997; 27: 578-582
22 Villamil AG, Casciato P, Eduardo M, Bustos D, Giunta D, Ciardullo M, et al. Fulminant autoimmune hepatitis type 1: Clinical presentation, outcome and prognostic factors. (Ab-stract 480) AJT 2005; 5 (Suppl 11): 278
23 Kessler WR, Cummings OW, Eckert G, Chalasani N, Lu-meng L, Kwo PY. Fulminant hepatic failure as the initial presentation of acute autoimmune hepatitis. Clin Gastroen-terol Hepatol 2004; 2: 625-631
24 Miyake Y, Iwasaki Y, Terada R, Onishi T, Okamoto R, Sakai N, Sakaguchi K, Shiratori Y. Clinical characteristics of fulminant-type autoimmune hepatitis: an analysis of eleven cases. Aliment Pharmacol Ther 2006; 23: 1347-1353
25 Ichai P, Duclos-Vallée JC, Guettier C, Hamida SB, Antonini T, Delvart V, Saliba F, Azoulay D, Castaing D, Samuel D. Usefulness of corticosteroids for the treatment of severe and fulminant forms of autoimmune hepatitis. Liver Transpl 2007; 13: 996-1003
26 O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342: 273-275
27 Verma S, Gunuwan B, Mendler M, Govindrajan S, Redeker A. Factors predicting relapse and poor outcome in type I au-toimmune hepatitis: role of cirrhosis development, patterns of transaminases during remission and plasma cell activity in the liver biopsy. Am J Gastroenterol 2004; 99: 1510-1516
28 Roberts SK, Therneau TM, Czaja AJ. Prognosis of histologi-cal cirrhosis in type 1 autoimmune hepatitis. Gastroenterol-ogy 1996; 110: 848-857
29 Montano-Loza AJ, Carpenter HA, Czaja AJ. Features associ-ated with treatment failure in type 1 autoimmune hepatitis and predictive value of the model of end-stage liver disease. Hepatology 2007; 46: 1138-1145
30 O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastro-enterology 1989; 97: 439-445
31 Kamath PS, Wiesner RH, Malinchoc M, Kremers W, Ther-neau TM, Kosberg CL, D’Amico G, Dickson ER, Kim WR. A model to predict survival in patients with end-stage liver disease. Hepatology 2001; 33: 464-470
32 Gow PJ, Jones RM, Dobson JL, Angus PW. Etiology and out-come of fulminant hepatic failure managed at an Australian liver transplant unit. J Gastroenterol Hepatol 2004; 19: 154-159
33 Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376: 190-201
34 Schramm C, Bubenheim M, Adam R, Karam V, Buckels J, O’Grady JG, Jamieson N, Pollard S, Neuhaus P, Manns MM, Porte R, Castaing D, Paul A, Traynor O, Garden J, Friman S, Ericzon BG, Fischer L, Vitko S, Krawczyk M, Metselaar HJ, Foss A, Kilic M, Rolles K, Burra P, Rogiers X, Lohse AW. Primary liver transplantation for autoimmune hepatitis: a comparative analysis of the European Liver Transplant Reg-istry. Liver Transpl 2010; 16: 461-469
35 Verma S, Redeker A. In type 1 autoimmune hepatitis, is cir-rhosis at presentation or follow-up associated with a poorer outcome? Hepatology 2005; 42: 1237; author reply 1237-1238
36 Feld JJ, Dinh H, Arenovich T, Marcus VA, Wanless IR, Heathcote EJ. Autoimmune hepatitis: effect of symptoms and cirrhosis on natural history and outcome. Hepatology 2005; 42: 53-62
37 Nikias GA, Batts KP, Czaja AJ. The nature and prognostic implications of autoimmune hepatitis with an acute presen-tation. J Hepatol 1994; 21: 866-871
38 Rolando N, Harvey F, Brahm J, Philpott-Howard J, Alexan-der G, Casewell M, Fagan E, Williams R. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12: 1-9
39 Karvellas CJ, Pink F, McPhail M, Cross T, Auzinger G, Bernal W, Sizer E, Kutsogiannis DJ, Eltringham I, Wendon JA. Predictors of bacteraemia and mortality in patients with acute liver failure. Intensive Care Med 2009; 35: 1390-1396
40 Kolber MA, Hill P. Vincristine potentiates cytochalasin B-induced DNA fragmentation in vitro. Cancer Chemother Pharmacol 1992; 30: 286-290
41 Reich DJ, Fiel I, Guarrera JV, Emre S, Guy SR, Schwartz ME, Miller CM, Sheiner PA. Liver transplantation for auto-immune hepatitis. Hepatology 2000; 32: 693-700
42 Ahmed M, Mutimer D, Hathaway M, Hubscher S, McMas-ter P, Elias E. Liver transplantation for autoimmune hepati-tis: a 12-year experience. Transplant Proc 1997; 29: 496
43 Rolando N, Wade JJ, Stangou A, Gimson AE, Wendon J, Philpott-Howard J, Casewell MW, Williams R. Prospective study comparing the efficacy of prophylactic parenteral an-timicrobials, with or without enteral decontamination, in pa-tients with acute liver failure. Liver Transpl Surg 1996; 2: 8-13
44 Czaja AJ, Donaldson PT, Lohse AW. Antibodies to soluble liver antigen/liver pancreas and HLA risk factors for type 1 autoimmune hepatitis. Am J Gastroenterol 2002; 97: 413-4139
45 Ma Y, Okamoto M, Thomas MG, Bogdanos DP, Lopes AR, Portmann B, Underhill J, Dürr R, Mieli-Vergani G, Vergani D. Antibodies to conformational epitopes of soluble liver antigen define a severe form of autoimmune liver disease. Hepatology 2002; 35: 658-664
S- Editor Tian L L- Editor Kerr C E- Editor Ma WH
2075 April 28|Volume 17|Issue 16|WJG|www.wjgnet.com
Potts JR et al . Autoimmune hepatitis and liver failure

TOPIC HIGHLIGHT
A practical approach to the diagnosis of autoimmune pancreatitis
Luca Frulloni, Antonio Amodio, Anna Maria Katsotourchi, Italo Vantini
Luca Frulloni, Antonio Amodio, Anna Maria Katsotourchi, Italo Vantini, Department of Medicine, University of Verona, 37134 Verona, ItalyLuca Frulloni, Professor, Cattedra di Gastroenterologia, Poli-clinico GB Rossi, P.le LA Scuro, 10, 37134 Verona, ItalyAuthor contributions: Frulloni L and Amodio A wrote the paper; Katsotourchi AM searched the literature for papers on autoimmune pancreatitis up to December 2009 and for papers on the frequency of benign lesions in resected pancreatic masses; Vantini I revised the paper.Correspondence to: Luca Frulloni, Professor, Cattedra di Gas-troenterologia, Policlinico GB Rossi, P.le LA Scuro, 10, 37134 Verona, Italy. [email protected]: +39-45-8124191 Fax: +39-45-8027495Received: September 21, 2010 Revised: January 29, 2011Accepted: February 5, 2011Published online: April 28, 2011
AbstractAutoimmune pancreatitis is a disease characterized by specific pathological features, different from those of other forms of pancreatitis, that responds dramatically to steroid therapy. The pancreatic parenchyma may be diffusely or focally involved with the possibility of a low-density mass being present at imaging, mimicking pancreatic cancer. Clinically, the most relevant problems lie in the diagnosis of autoimmune pancreatitis and in distinguishing autoimmune pancreatitis from pancreatic cancer. Since in the presence of a pancreatic mass the probability of tumour is much higher than that of pancre-atitis, the physician should be aware that in focal autoim-mune pancreatitis the first step before using steroids is to exclude pancreatic adenocarcinoma. In this review, we briefly analyse the strategies to be followed for a correct diagnosis of autoimmune pancreatitis.
© 2011 Baishideng. All rights reserved.
Key words: Autoimmune diseases; Pancreatitis; Thera-py; Diagnosis
Peer reviewers: Yoshiharu Motoo, MD, PhD, FACP, FACG, Professor and Chairman, Department of Medical Oncology, Kanazawa Medical University, 1-1 Daigaku, Uchinada, Ishi-kawa 920-0293, Japan; Richard Hu, MD, MSc, Division of Gastroenterology, Department of Medicine, Olive view-UCLA Medical Center, 14445 Olive View Drive, Los Angeles, CA 91342, United States
Frulloni L, Amodio A, Katsotourchi AM, Vantini I. A practical approach to the diagnosis of autoimmune pancreatitis. World J Gastroenterol 2011; 17(16): 2076-2079 Available from: URL: http://www.wjgnet.com/1007-9327/full/v17/i16/2076.htm DOI: http://dx.doi.org/10.3748/wjg.v17.i16.2076
INTRODUCTIONAutoimmune pancreatitis (AIP) is now a well defined en-tity among the inflammatory diseases of the pancreas[1-3]. The number of studies in literature has constantly in-creased since the first one published in 1995 by Yoshida et al[4] (Figure 1). Despite the fact that in the first paper from Japan the disease was described as diffusely involv-ing the pancreatic gland[5-8], later publications pointed out that the pancreas may also be focally involved by the autoimmune process[3,9-12]. Therefore, some authors have classified AIP as focal or diffuse[3]. Focal AIP is charac-terized by a segmental involvement of the parenchyma with the possibility of a low-density mass being present at imaging.
Clinically, the focal form, particularly in the presence of a low-density pancreatic mass, requires a more care-ful patient evaluation, since it may be easily confused with pancreatic cancer. Several series indicate that in 5%-21% of resected pancreatic masses suspected of being cancer, the final diagnosis excluded malignancy (Table 1)[13-20]. Since AIP responds dramatically to steroid treatment[1], a correct diagnosis of the disease is important to avoid surgery. On the other hand, in the presence of a resectable pancreatic mass, the probability of cancer is very high (> 90%). A
2076
World J Gastroenterol 2011 April 28; 17(16): 2076-2079 ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2076
April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Luca Frulloni, MD, PhD, Professor, Series Editor

Table 1 Frequency of benign lesions in patients who undergo pancreatico-duodenectomy in the presence of a pancreatic mass suspected of being pancreatic adenocarcinoma
Frulloni L et al . A practical approach to the diagnosis of autoimmune pancreatitis
misdiagnosis of AIP implies 2-3 week’s steroid treatment and a one month delay in surgery, with the consequent risk of not operating because of the progression of the malig-nancy with the onset of metastasis or of vascular involve-ment. A correct and quick diagnosis of AIP is therefore an important goal in clinical practice, particularly in focal AIP.
AIP diagnosis may be attained through well established diagnostic criteria. There is agreement on the use of four main criteria based on histological findings, radiological fea-tures, other organ involvement and clinical and instrumental response to steroid therapy. HISORt criteria introduced by Chari et al[21] in 2006 and based on surgical specimens of op-erated AIP patients can be considered standard criteria for the diagnosis of AIP. Serum IgG4[22-24] and positive IgG4+ plasma cells in pancreatic surgical specimens or pancreatic biopsies may also support the diagnosis of AIP[25-29].
There is agreement on the use of these diagnostic cri-teria (pathology, imaging, presence of other organ involve-ment, response to steroids), but not on the strategy to be followed in making the diagnosis.
THE STRATEGIES IN THE DIAGNOSIS OF AIPThree main strategies, from Japan, the USA and Italy, have been suggested. The clinical approach to the disease by these strategies is different.
In the USA distinguishing the different pathological subtypes of AIP[21,30,31] is considered prominent for the di-agnosis. In the USA and in Europe, AIP may be classified as type 1 (or Lympho-Plasmacytic Sclerosing Pancreatitis-LPSP) and type 2 (or Idiopathic Duct-centric Chronic Pancreatitis- IDCP)[31-34]. Since the clinical evolution of these forms seems to be different, some authors have suggested obtaining the diagnosis of AIP subtypes from EUS-guided biopsy[31,35].
The main pathological and serological features in type 1 AIP are[31,36]: (1) Prevalence of storiform fibrosis, with obstructive phlebitis; (2) high levels of serum IgG4; (3) presence of IgG4+ plasma cells in the involved pancreatic tissue; and (4) absence of granulocytic epithelial lesions (GEL), that are the expression of an aggression against epithelial ductal cells, with rupture and destruction of duc-tal structures. The pathological characteristics in type 2 AIP are on the contrary[31,36]: (1) prevalence of inflammation; (2) presence of GEL; and (3) absence of serum IgG4 and of IgG4+ plasmavcells in the inflamed pancreatic tissue.
The clinical aspects and the evolution are different in type 1 and 2 AIP[31,36,37]. In type 1 AIP (LPSP), there is a prevalence of males, patients are older, other organs may be involved (more commonly salivary glands, biliary tract, kidney, lung, retroperitoneum) and the relapse of the dis-ease is more frequent after steroid treatment. In type 2 (IDCP), male/female ratio is about 1, patients are younger, the colon only may be involved (ulcerative colitis) and relapse after steroids is infrequent. Both forms respond quickly to steroid treatment[31,36-38].
The diagnostic approach is therefore aimed at diagnosing
AIP subtypes, mainly through pancreatic core biopsy, and this appears to have a good sensitivity and specificity[28,29,39].
In Japan, only type 1 AIP (LPSP) is considered an auto-immune disorder and an IgG4-mediated systemic disorder associated with pancreatic lesions[40]. Only in a few cases has type 2 AIP (IDCP) been described in Japan and it is not considered an autoimmune disease, despite its quick re-sponse to steroids just as type 1 AIP. Instrumentally, in the majority of cases the disease diffusely involves the pancreas. Several diagnostic algorithms have been suggested in Japan and Korea[8,41-44]. A comprehensive diagnosis should be based on pancreatic imaging (including ERCP), serological tests (IgG4, total IgG, non organ specific autoantibodies, antibodies to carbonic anhydrase type Ⅰ and Ⅱ, antibodies to lactoferrin) and pathological findings. The presence of extrapancreatic lesions may suggest the possibility of AIP.
THE ITALIAN STRATEGY: A CLINICAL APPROACH TO THE DISEASEThe Italian proposal for the diagnosis of AIP, which is dif-ferent from that suggested in Japan and the USA, is based on the instrumental distinction between focal and diffuse forms of the disease[2,3].
2077 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
120
100
80
60
40
20
0
No.
of
publ
ishe
d pa
pers
Figure 1 Increased number of published papers on autoimmune pancre-atitis obtained by searching in Pubmed up to 2009 (search terms: Autoim-mune pancreatitis, limit: Field title).
1995
1996
1997
1998
1999
2000
2001
2002
2003
2004
2005
2006
2007
2008
2009
Authors Yr No. of pts Frequency of benign lesions
n %
Smith et al[13] 1994 603 29 5Barens et al[14] 1996 510 108 21van Gulik et al[15] 1997 220 14 6Abraham et al[16] 2003 442 47 10.4Weber et al[17] 2003 1287 159 12Kennedy et al[18] 2006 162 21 12.9De La Fuente et al[19] 2010 494 37 7.4Hurtuk et al[20] 2010 461 35 8All studies - 4179 450 10.8

A wide range of symptoms are reported by patients at the clinical onset of the disease. Jaundice, abdominal pain, usually mild, symptoms secondary to pancreatic exocrine and endocrine insufficiency (weight loss, diabetes), and persistent elevation of serum levels of pancreatic enzymes may be observed in AIP patients. In a few cases AIP is discovered incidentally by US or other imaging techniques performed without an indication for a pancreatic disorder.
On the basis of imaging, these patients can be divided in those with focal involvement of the pancreas and those with diffuse enlargement of the pancreatic gland[12]. In the case of focal AIP, particularly in the presence of a low-density pancreatic mass, the clinical challenge is to exclude pancreatic cancer and correctly diagnose AIP. Therefore, focal and diffuse types AIP should be strictly separated, since the problem of differential diagnosis with pancreatic cancer involves only focal AIP.
Diffuse AIP may be confused with acute pancreatitis. The clinical picture of diffuse AIP, however, differs from those observed in acute pancreatitis. In AIP, pain, if pres-ent, is mild, no risk factors for pancreatitis (biliary lithiasis, alcohol) are present, a persistent increase in serum pan-creatic enzymes may be observed, jaundice is caused by enlargement of the pancreas without the presence of a mass, with a stricture on the intrapancreatic tract of the common bile duct. Since pancreatic necrosis has never been described in AIP, the differential diagnosis should be with oedematous pancreatitis. This can be achieved through imaging, since the radiologic features of AIP are different from those observed in acute oedematous pan-creatitis. Hypodensity of the pancreas in arterial phase and the absence of a peripancreatic strand appear to differen-tiate AIP from acute oedematous pancreatitis, where the pancreatic gland shows normal perfusion and the peripan-creatic strand is a common radiological picture (personal unpublished data). We do not suggest pancreatic biopsy in diffuse AIP. The diagnosis may be definitely made after treatment with steroids, which produces complete disap-pearance of the pancreatic changes.
In the diffuse form associated with jaundice secondary to a common bile duct stricture, a diagnosis of cholangio-carcinoma should be considered and, if necessary, ruled out before steroid therapy through ERCP with biliary bi-opsies and/or intraductal biliary ultrasonography.
In the focal form, particularly in the presence of a low-density pancreatic mass at imaging, the first diagnostic goal is to exclude pancreatic cancer, even if the presence of clinical (young age, other organ involvement), radiological (perfusion of the pancreatic mass suggestive of inflamma-tion, no or mild dilation of the main pancreatic duct) and serological (high level of IgG4, presence of autoantibod-ies, low serum levels of Ca 19-9) findings are suggestive of AIP. Therefore, pancreatic biopsy is mandatory, preferably EUS-guided, first of all to exclude neoplasia and possibly to confirm the diagnosis of AIP.
If pancreatic biopsy confirms the diagnosis of AIP, a 3 wk steroid treatment is indicated. The diagnosis of AIP is final in the presence of a significant clinical and radio-logical response. Since significant improvement/resolution
of jaundice is an indication of response to steroid therapy, biliary stenting is not recommended, unless serum biliru-bin levels are very high.
If pancreatic biopsy is only suggestive of AIP or non diagnostic, a careful evaluation of HISORt criteria is nec-essary to decide whether the patient should be treated with steroids or undergo resective surgery. The decision is actu-ally a challenge and should be made in experienced centres only, because it requires expert clinicians, radiologists, pathologists and surgeons. After a complete or significant response to steroid therapy, a definitive diagnosis of AIP may be made.
CONCLUSIONThe diagnosis of AIP still remains difficult. The diagnostic algorithm is different in the diffuse and focal forms of the disease, particularly in the presence of a low-density pancre-atic mass at imaging. Biopsy or fine needle aspiration cytol-ogy is mandatory in the presence of a low-density pancre-atic mass. In some cases, only a full or significant response to steroids allows a final diagnosis of AIP to be made. Agreement among experienced clinicians, radiologists, pa-thologists and surgeons is needed to adopt the response to steroid therapy as a diagnostic criterion in patients where the diagnosis cannot be made through pancreatic biopsy.
REFERENCES1 Finkelberg DL, Sahani D, Deshpande V, Brugge WR. Au-
toimmune pancreatitis. N Engl J Med 2006; 355: 2670-26762 Buscarini E, Frulloni L, De Lisi S, Falconi M, Testoni PA,
Zambelli A. Autoimmune pancreatitis: a challenging dia-gnostic puzzle for clinicians. Dig Liver Dis 2010; 42: 92-98
3 Frulloni L, Scattolini C, Falconi M, Zamboni G, Capelli P, Manfredi R, Graziani R, D’Onofrio M, Katsotourchi AM, Amodio A, Benini L, Vantini I. Autoimmune pancreatitis: differences between the focal and diffuse forms in 87 patien-ts. Am J Gastroenterol 2009; 104: 2288-2294
4 Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pan-creatitis. Dig Dis Sci 1995; 40: 1561-1568
5 Okazaki K, Chiba T. Autoimmune related pancreatitis. Gut 2002; 51: 1-4
6 Okazaki K. Autoimmune pancreatitis is increasing in Japan. Gastroenterology 2003; 125: 1557-1558
7 Nishimori I, Tamakoshi A, Otsuki M. Prevalence of autoim-mune pancreatitis in Japan from a nationwide survey in 2002. J Gastroenterol 2007; 42 Suppl 18: 6-8
8 Otsuki M, Chung JB, Okazaki K, Kim MH, Kamisawa T, Kawa S, Park SW, Shimosegawa T, Lee K, Ito T, Nishimori I, Notohara K, Naruse S, Ko SB, Kihara Y. Asian diagnostic crite-ria for autoimmune pancreatitis: consensus of the Japan-Korea Symposium on Autoimmune Pancreatitis. J Gastroenterol 2008; 43: 403-408
9 Taniguchi T, Seko S, Azuma K, Tamegai M, Nishida O, Inoue F, Okamoto M, Mizumoto T, Kobayashi H. Autoim-mune pancreatitis detected as a mass in the tail of the pan-creas. J Gastroenterol Hepatol 2000; 15: 461-464
10 Wakabayashi T, Kawaura Y, Satomura Y, Watanabe H, Mo-too Y, Okai T, Sawabu N. Clinical and imaging features of autoimmune pancreatitis with focal pancreatic swelling or mass formation: comparison with so-called tumor-forming pancreatitis and pancreatic carcinoma. Am J Gastroenterol 2003;
2078 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Frulloni L et al . A practical approach to the diagnosis of autoimmune pancreatitis

98: 2679-268711 Kajiwara M, Gotohda N, Konishi M, Nakagohri T, Takahashi
S, Kojima M, Kinoshita T. Incidence of the focal type of au-toimmune pancreatitis in chronic pancreatitis suspected to be pancreatic carcinoma: experience of a single tertiary cancer center. Scand J Gastroenterol 2008; 43: 110-116
12 Manfredi R, Graziani R, Cicero C, Frulloni L, Carbognin G, Mantovani W, Mucelli RP. Autoimmune pancreatitis: CT patterns and their changes after steroid treatment. Radiology 2008; 247: 435-443
13 Smith CD, Behrns KE, van Heerden JA, Sarr MG. Radical pancreatoduodenectomy for misdiagnosed pancreatic mass. Br J Surg 1994; 81: 585-589
14 Barens SA, Lillemoe KD, Kaufman HS, Sauter PK, Yeo CJ, Talamini MA, Pitt HA, Cameron JL. Pancreaticoduode-nectomy for benign disease. Am J Surg 1996; 171: 131-134; discussion 134-135
15 van Gulik TM, Reeders JW, Bosma A, Moojen TM, Smits NJ, Allema JH, Rauws EA, Offerhaus GJ, Obertop H, Gou-ma DJ. Incidence and clinical findings of benign, inflamma-tory disease in patients resected for presumed pancreatic head cancer. Gastrointest Endosc 1997; 46: 417-423
16 Abraham SC, Wilentz RE, Yeo CJ, Sohn TA, Cameron JL, Boitnott JK, Hruban RH. Pancreaticoduodenectomy (Whip-ple resections) in patients without malignancy: are they all ‘chronic pancreatitis’? Am J Surg Pathol 2003; 27: 110-120
17 Weber SM, Cubukcu-Dimopulo O, Palesty JA, Suriawinata A, Klimstra D, Brennan MF, Conlon K. Lymphoplasmacytic sclerosing pancreatitis: inflammatory mimic of pancreatic car-cinoma. J Gastrointest Surg 2003; 7: 129-137; discussion 137-139
18 Kennedy T, Preczewski L, Stocker SJ, Rao SM, Parsons WG, Wayne JD, Bell RH, Talamonti MS. Incidence of benign in-flammatory disease in patients undergoing Whipple proce-dure for clinically suspected carcinoma: a single-institution experience. Am J Surg 2006; 191: 437-441
19 de la Fuente SG, Ceppa EP, Reddy SK, Clary BM, Tyler DS, Pappas TN. Incidence of benign disease in patients that un-derwent resection for presumed pancreatic cancer diagno-sed by endoscopic ultrasonography (EUS) and fine-needle aspiration (FNA). J Gastrointest Surg 2010; 14: 1139-1142
20 Hurtuk MG, Shoup M, Oshima K, Yong S, Aranha GV. Pan-creaticoduodenectomies in patients without periampullary neoplasms: lesions that masquerade as cancer. Am J Surg 2010; 199: 372-376; discussion 376
21 Chari ST, Smyrk TC, Levy MJ, Topazian MD, Takahashi N, Zhang L, Clain JE, Pearson RK, Petersen BT, Vege SS, Farnell MB. Diagnosis of autoimmune pancreatitis: the Mayo Clinic experience. Clin Gastroenterol Hepatol 2006; 4: 1010-1016; quiz 934
22 Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Aka-matsu T, Fukushima M, Nikaido T, Nakayama K, Usuda N, Kiyosawa K. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001; 344: 732-738
23 Ghazale A, Chari ST, Smyrk TC, Levy MJ, Topazian MD, Takahashi N, Clain JE, Pearson RK, Pelaez-Luna M, Petersen BT, Vege SS, Farnell MB. Value of serum IgG4 in the diagno-sis of autoimmune pancreatitis and in distinguishing it from pancreatic cancer. Am J Gastroenterol 2007; 102: 1646-1653
24 Morselli-Labate AM, Pezzilli R. Usefulness of serum IgG4 in the diagnosis and follow up of autoimmune pancreatitis: A systematic literature review and meta-analysis. J Gastroen-terol Hepatol 2009; 24: 15-36
25 Zhang L, Notohara K, Levy MJ, Chari ST, Smyrk TC. IgG4-positive plasma cell infiltration in the diagnosis of autoim-mune pancreatitis. Mod Pathol 2007; 20: 23-28
26 Adsay NV, Basturk O, Thirabanjasak D. Diagnostic features and differential diagnosis of autoimmune pancreatitis. Semin Diagn Pathol 2005; 22: 309-317
27 Kojima M, Sipos B, Klapper W, Frahm O, Knuth HC, Yana-
gisawa A, Zamboni G, Morohoshi T, Klöppel G. Autoimmu-ne pancreatitis: frequency, IgG4 expression, and clonality of T and B cells. Am J Surg Pathol 2007; 31: 521-528
28 Detlefsen S, Mohr Drewes A, Vyberg M, Klöppel G. Dia-gnosis of autoimmune pancreatitis by core needle biopsy: application of six microscopic criteria. Virchows Arch 2009; 454: 531-539
29 Mizuno N, Bhatia V, Hosoda W, Sawaki A, Hoki N, Hara K, Takagi T, Ko SB, Yatabe Y, Goto H, Yamao K. Histological diagnosis of autoimmune pancreatitis using EUS-guided trucut biopsy: a comparison study with EUS-FNA. J Ga-stroenterol 2009; 44: 742-750
30 Levy MJ. Endoscopic ultrasound-guided trucut biopsy of the pancreas: prospects and problems. Pancreatology 2007; 7: 163-166
31 Sah RP, Chari ST, Pannala R, Sugumar A, Clain JE, Levy MJ, Pearson RK, Smyrk TC, Petersen BT, Topazian MD, Takahashi N, Farnell MB, Vege SS. Differences in clinical profile and relapse rate of type 1 versus type 2 autoimmune pancreatitis. Gastroenterology 2010; 139: 140-148; quiz e12-e13
32 Klöppel G, Detlefsen S, Chari ST, Longnecker DS, Zam-boni G. Autoimmune pancreatitis: the clinicopathological characteristics of the subtype with granulocytic epithelial lesions. J Gastroenterol 2010; 45: 787-793
33 Chari ST, Kloeppel G, Zhang L, Notohara K, Lerch MM, Shimosegawa T. Histopathologic and clinical subtypes of autoimmune pancreatitis: the Honolulu consensus docu-ment. Pancreas 2010; 39: 549-554
34 Chari ST, Longnecker DS, Klöppel G. The diagnosis of au-toimmune pancreatitis: a Western perspective. Pancreas 2009; 38: 846-848
35 Sah RP, Pannala R, Chari ST, Sugumar A, Clain JE, Levy MJ, Pearson RK, Smyrk TC, Petersen BT, Topazian MD, Takahashi N, Vege SS. Prevalence, diagnosis, and profile of autoimmune pancreatitis presenting with features of acute or chronic pan-creatitis. Clin Gastroenterol Hepatol 2010; 8: 91-96
36 Sugumar A, Klöppel G, Chari ST. Autoimmune pancreatitis: pathologic subtypes and their implications for its diagnosis. Am J Gastroenterol 2009; 104: 2308-2310; quiz 2311
37 Maire F, Le Baleur Y, Rebours V, Vullierme MP, Couvelard A, Voitot H, Sauvanet A, Hentic O, Lévy P, Ruszniewski P, Ham-mel P. Outcome of patients with type 1 or 2 autoimmune pan-creatitis. Am J Gastroenterol 2011; 106: 151-156
38 Sugumar A, Chari ST. Diagnosis and treatment of autoimmu-ne pancreatitis. Curr Opin Gastroenterol 2010; 26: 513-518
39 Hirano K, Fukushima N, Tada M, Isayama H, Mizuno S, Yamamoto K, Yashima Y, Yagioka H, Sasaki T, Kogure H, Nakai Y, Sasahira N, Tsujino T, Kawabe T, Fukayama M, Omata M. Diagnostic utility of biopsy specimens for autoim-mune pancreatitis. J Gastroenterol 2009; 44: 765-773
40 Okazaki K, Kawa S, Kamisawa T, Shimosegawa T, Tanaka M. Japanese consensus guidelines for management of autoim-mune pancreatitis: I. Concept and diagnosis of autoimmune pancreatitis. J Gastroenterol 2010; 45: 249-265
41 Choi EK, Kim MH, Kim JC, Han J, Seo DW, Lee SS, Lee SK. The Japanese diagnostic criteria for autoimmune chronic pan-creatitis: is it completely satisfactory? Pancreas 2006; 33: 13-19
42 Kamisawa T, Chung JB, Irie H, Nishin T, Ueki T, Takase M, Kawa S, Nishimori I, Okazaki K, Kim MH, Otsuki M. Japan-Korea symposium on autoimmune pancreatitis (KOKURA 2007). Pancreas 2007; 35: 281-284
43 Kamisawa T, Okazaki K, Kawa S. Diagnostic criteria for au-toimmune pancreatitis in Japan. World J Gastroenterol 2008; 14: 4992-4994
44 Okazaki K, Kawa S, Kamisawa T, Ito T, Inui K, Irie H, Irisawa A, Kubo K, Notohara K, Hasebe O, Fujinaga Y, Ohara H, Ta-naka S, Nishino T, Nishimori I, Nishiyama T, Suda K, Shirato-ri K, Shimosegawa T, Tanaka M. Japanese clinical guidelines for autoimmune pancreatitis. Pancreas 2009; 38: 849-866
S- Editor Tian L L- Editor O’Neill M E- Editor Ma WH
2079 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Frulloni L et al . A practical approach to the diagnosis of autoimmune pancreatitis

Endoscopic ultrasonography findings in autoimmune pancreatitis
Elisabetta Buscarini, Stefania De Lisi, Paolo Giorgio Arcidiacono, Maria Chiara Petrone, Arnaldo Fuini, Rita Conigliaro, Guido Manfredi, Raffaele Manta, Dario Reggio, Claudio De Angelis
Elisabetta Buscarini, Guido Manfredi, Department of Gastro-enterology, Maggiore Hospital, Crema 26013, ItalyStefania De Lisi, Department of Gastroenterology, Di.Bi.M.I.S., University of Palermo, Palermo 90121, ItalyPaolo Giorgio Arcidiacono, Maria Chiara Petrone, Department of Gastroenterology, I.R.C.C.S San Raffaele, Università Vita e Salute San Raffaele, Milan 20122, ItalyArnaldo Fuini, Department of Gastroenterology and Digestive Endoscopy, Ospedale Civile Maggiore, Verona 37142, ItalyRita Conigliaro, Guido Manfredi, Raffaele Manta, Department of Gastroenterology and Digestive Endoscopy, New Civil Hospi-tal S. Agostino Estense, Modena 41126, ItalyDario Reggio, Chirurgia Trapianti di Fegato, San Giovanni Bat-tista Hospital, Torino 10156, ItalyClaudio De Angelis, Department of Gastroenterology, San Giovanni Battista Hospital, Torino 10156, Italy Author contributions: All authors contributed to conception and design, drafting the article or revising it, and to final approval.Correspondence to: Elisabetta Buscarini, MD, Department of Gastroenterology, Maggiore Hospital, Largo Dossena 2, Crema 26013, Italy. [email protected]: +39-373-280278 Fax: +39-373-280654Received: September 21, 2010 Revised: November 16, 2010Accepted: November 23, 2010Published online: April 28, 2011
AbstractEndoscopic ultrasonography is an established diagnostic tool for pancreatic masses and chronic pancreatitis. In recent years there has been a growing interest in the worldwide medical community in autoimmune pancre-atitis (AIP), a form of chronic pancreatitis caused by an autoimmune process. This paper reviews the cur-rent available literature about the endoscopic ultraso-nographic findings of AIP and the role of this imaging technique in the management of this protean disease.
© 2011 Baishideng. All rights reserved.
Key words: Pancreatitis; Autoimmune; Endoscopic ul-trasound; IgG4 cholangitis
Peer reviewer: Dr. Jeremy FL Cobbold, PhD, Clinical Lecturer in Hepatology, Department of Hepatology and Gastroenterology, Liver Unit, Imperial College London, St Mary’s Hospital, 10th Floor, QEQM building, Praed Street, London, W2 1NY, United Kingdom
Buscarini E, De Lisi S, Arcidiacono PG, Petrone MC, Fuini A, Conigliaro R, Manfredi G, Manta R, Reggio D, De Angelis C. Endoscopic ultrasonography findings in autoimmune pancreatitis. World J Gastroenterol 2011; 17(16): 2080-2085 Available from: URL: http://www.wjgnet.com/1007-9327/full/v17/i16/2080.htm DOI: http://dx.doi.org/10.3748/wjg.v17.i16.2080
INTRODUCTIONEndoscopic ultrasonography (EUS) is superior to stan-dard imaging techniques in detecting pancreatic cancer or masses and in the assessment of early parenchymal changes in chronic pancreatitis[1,2]; however, its role in the diagnosis of autoimmune pancreatitis (AIP) has yet to be standardized, even though its high accuracy together with its safety make it a promising tool in the management of this disease.
To date, no consensus about the diagnosis of AIP has been reached[3]. Several criteria have recently been pro-posed, reflecting the different clinical entities that AIP can adopt worldwide[4-9]. The diffuse form of AIP has typically been included in the first set of criteria[4], and in 2008 only, a focal pancreatic enlargement evident upon imaging was classified as a form of AIP.
Obstructive jaundice is the most common presenta-tion of AIP and, together with biochemical and imaging features, can mimic neoplastic conditions[10,11]. Because
TOPIC HIGHLIGHT
World J Gastroenterol 2011 April 28; 17(16): 2080-2085ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2080
2080 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Luca Frulloni, MD, PhD, Professor, Series Editor

Buscarini E et al . EUS and autoimmune pancreatitis
AIP is a benign disease, a definitive diagnosis without the need for surgery is desirable.
AIP can present with extrapancreatic lesions, the most frequent being IgG4-related sclerosing cholangitis (IgG4-SC), followed by hilar lymphadenopathy[12].
EUS can display both of these conditions in addition to parenchymal, ductal and vascular lesions. Moreover, this technique offers the advantage over other diagnos-tic tools of allowing clinicians to perform biopsies to achieve a definitive diagnosis[13-16].
In this article, we describe the EUS findings of AIP (Table 1) by reviewing the currently available literature in this field.
PANCREATIC FINDINGSThe predominant finding in the diffuse form of AIP is a diffuse pancreatic enlargement with altered echotexture (Figure 1)[14,15]. A recent retrospective study proposed to differentiate early from advanced stage AIP according to EUS findings[15]. In 19 patients with AIP, the presence of parenchymal lobularity and a hyperechoic pancreatic duct margin were significantly correlated with early stage AIP[17]. Other EUS findings that are indicative of AIP, such as reduced echogenicity, hyperechoic foci and hyper-echoic strands (Figure 1), are found in both AIP stages[17]. Should these results be confirmed in prospective studies, EUS would acquire an essential role in the identification of early stage AIP, which is characterized by a prompt response to steroid therapy. Stones and cysts similar to those described in chronic alcoholic pancreatitis can oc-cur in the late stage of AIP[18].
The Sahai criteria for chronic pancreatitis were found
to be inadequate to evaluate parenchymal and ductal changes in AIP[19]. According to the scoring system, a se-ries of 25 patients with AIP were classified as normal or displaying mild disease[16].
In the focal form of AIP a solitary (Figure 2), irregu-lar hypoechoic mass, generally located in the head of the pancreas, is observed[13-15]. In addition, upstream dilata-tion of the main pancreatic duct could be observed[13,17]. In this setting, the overlap with EUS findings of pancre-atic cancer is remarkable, and EUS-elastography (Figure 1) can provide further information about pancreatic lesions. In a case-control study of five patients with AIP, EUS-elastography showed a typical and homogeneous stiff-ness pattern of the focal lesions and of the surrounding parenchyma that is different from that observed in ductal adenocarcinoma[20].
COMMON BILE DUCT FINDINGSThe common bile duct is the most frequent extrapancre-atic organ involved in AIP and was found to affect 58% of patients in a Japanese survey[12]. Biliary strictures can mimic both sclerosing cholangitis and biliary cancer. EUS allows visualization of the entire common bile duct and enables identification of the cause of a biliary stricture. In patients with either diffuse or focal AIP, EUS can show dilatation of the common bile duct and thickening of its wall better than other diagnostic techniques[3,13-16,21]. The typical EUS feature of the common bile duct is a homogeneous, regular thickening of the bile duct wall, called “sandwich-pattern”, which is characterized by an echopoor intermediate layer and hyperechoic outer and inner layers, has been described as a EUS feature of the
2081 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 1 Endoscopic ultrasonography features of autoimmune pancreatitis
Pancreas Extrahepatic bile duct Gallbladder Lymph nodes1 Peripancreatic vessels
Diffuse AIP Focal AIP
EUS Gland volume: increased
Gland volume: focal enlargement/s
Caliber: dilated Wall: diffuse and uniform thickening, “sandwich-pattern”1
Volume: enlarged (also substantially)
Loss of interface between pancreas and portal or mesenteric veins1
Echotexture: echopoor, with echogenic interlobular septa
Echotexture: echopoor, with echogenic interlobular septa
Wall: diffuse and uniform thickening, “sandwich-pattern”1
Echotexture: echopoor
Gland border: thickened
Sites: liver hylum, peripancreatic, celiac
Wirsung: narrowed Wirsung: narrowed within the lesion, dilated upstream to the lesion
Wirsung wall: thickened1
Wirsung wall: thickened1
IDUS Wall: diffuse and uniform thickening, “sandwich-pattern”; differential diagnosis with cholangiocarcinoma1
1Indicates the features which are detected only or substantially better by endoscopic ultrasonography (EUS) and not seen in conventional cross-sectional imaging. AIP: Autoimmune pancreatitis; IDUS: Intraductal ultrasonography.

2082 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
SV
D1
Soft
Hard
c
DC
BA
Figure 1 Diffuse form of autoimmune pancreatitis. A: Endoscopic ultrasonography (EUS) linear scanning shows a diffuse pancreatic enlargement (arrowheads) with echopoor echotexture, and with loss of interface with splenic vein (arrows); B: Parenchymal lobularity and hyperechoic strands (arrows) are visible in the enlarged gland; C: Pancreatic duct calliper is 1.8 mm; D: EUS-elastography demonstrates the diffuse pancreatic stiffness (arrowheads).
c
w
DC
BA
Figure 2 Focal form of autoimmune pancreatitis. A: Endoscopic ultrasonography (EUS) shows a focal lesion (arrows) of pancreatic head which is echopoor with hyperechoic strands; B: A EUS-guided fine needle aspiration is performed (arrow) for tissue characterization; C: Another case of focal autoimmune pancreatitis (AIP) with echopoor lesion of pancreatic head (between callipers) and marked echopoor thickening of the choledochal wall (arrow); D: In this case of focal AIP EUS shows a echopoor lesion (arrows) of pancreatic head, with upstream dilatation of both common bile duct (c) and pancreatic duct (w); notice the thickened choledochal wall.
Buscarini E et al . EUS and autoimmune pancreatitis

common bile duct; the cause of the biliary stricture is the thickened wall itself rather than extrinsic pancreatic com-pression (Figure 3)[3,13].
A further application of EUS is intraductal ultraso-nography (IDUS), which can be performed during endo-scopic retrograde cholangiography for the characteriza-tion of biliary stenosis. Naitoh et al[22] recently evaluated IDUS findings in 23 patients with IgG4-SC. They found that a circular, symmetric wall thickness, smooth inner and outer margins and a homogeneous intermediate layer in the stricture were significantly more common in AIP than in cholangiocarcinoma. The wall thickness in IgG4-SC in regions of non-stricture on the cholangiogram was significantly greater than that in cholangiocarcinoma and therefore a bile duct wall thickness exceeding 0.8 mm in regions of non-stricture on the cholangiogram was
highly suggestive of IgG4-SC. Contrast enhancement of conventional EUS and IDUS showed an inflammatory pattern of the bile duct wall, with a long-lasting enhance-ment starting in the early phase instead of the poor en-hancement found in bile duct cancer (Figure 3)[21].
PERIPANCREATIC FINDINGSHilar lymphadenopathy is one of the most frequently de-scribed extrapancreatic lesions[12]. Other sites of enlarged lymph nodes are the peripancreatic and celiac regions. EUS nodal features that accurately predict nodal metasta-sis have been previously identified in patients with esoph-ageal cancer[23]; they include size (> 1 cm in diameter on the short axis), hypoechoic appearance, round shape, and smooth border. However, these conventional EUS cri-
2083 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
c
c
c
g
c
DC
BA
E
Figure 3 Biliary and peripancreatic findings in autoimmune pancreatitis. Autoimmune pancreatitis presenting with jaundice: A: Endoscopic ultrasonography (EUS) shows a dilated common bile duct (c) upstream to a distal funnel-shaped stenosis; EUS demonstrates the diffuse thickening of the biliary wall (between arrows) with “sandwich-pattern”, either of common bile duct or of cystic duct (arrowheads). This thickening is equally visible both in the dilated region of the common bile duct; B: In the distal strictured tract (arrows); C: After contrast administration (Sonovue, Bracco) the biliary wall shows an early and persistent enhancement (arrowheads); D: EUS shows the same thickening of the gallbladder (g) wall (arrowheads); E: Enlarged lymph nodes to the hepatic hylum (arrows).
Buscarini E et al . EUS and autoimmune pancreatitis

teria have proven inaccurate for staging non-esophageal cancers, including those that are biliopancreatic[24,25]. EUS can detect single or multiple enlarged lymph nodes in patients with AIP (Figure 3), reflecting the underlying inflammatory process, which can involve extra-pancreatic organs[14,15].
Hoki et al[16] reported a significant difference in detec-tion of lymphadenopathy by EUS imaging over CT (72% vs 8%) in patients with AIP. Moreover, in the same series, a trend toward a higher prevalence of lymphadenopathy in AIP compared to pancreatic cancer was reported.
In the absence of specific nodal features indicating malignancy, the differential diagnosis with biliopancreatic neoplasms is arduous but can be achieved by evaluating the broad spectrum of clinical and imaging data of AIP patients.
EUS criteria for vascular invasion of pancreatic can-cer have been established[26,27].
In a series of 14 patients with AIP, EUS suspected invasion of the portal or mesenteric veins in 21% of patients compared to 14% on CT. No pancreatic cancer developed during the follow-up of these patients. Such EUS features, easily mistaken for malignancy, are due to the inflammatory process of AIP, which can involve me-dium and large-sized vessels (Figure 1)[15].
Peripancreatic fluid collections are less common and not specific for AIP.
INTERVENTIONAL EUS IN AIPIn recent years, the possibility of guiding tissue sampling with either fine needle aspiration (EUS-FNA) (Figure 2) or Tru-cut biopsy (EUS-TCB) has increased the diagnos-tic potential of EUS by the acquisition of cytological and histological specimens from gastrointestinal lesions[28-30].
In the setting of AIP, EUS-FNA can be employed to yield specimens of pancreatic lesions, the common bile duct wall or lymph nodes[13,15]. Although a cytologic pat-tern specific for AIP has not been identified, high cellular-ity of stromal fragments with lymphoplasmacytic infiltrate has emerged as a discriminating feature in a retrospective series of 16 patients with an AIP diagnosis confirmed by histology of the respective specimens. Indeed, 56% of AIP patients presented such a feature vs 19% of patients with pancreatic carcinoma, and none of the chronic pan-creatitis controls exhibited this feature[31]. Immunohisto-chemical staining can show IgG4-positive plasma cells which are a useful marker for the tissue diagnosis of AIP.
EUS-FNA can fail in diagnosis because of the small size of the specimens that do not have preserved tissue architecture. Moreover, sampling error due to the patchy distribution of AIP can occur. EUS-TCB can overcome these limitations by acquiring large samples fit for histo-logical examination[30-32].
A recent study compared EUS-FNA and EUS-TCB performed in 14 patients for the diagnosis of AIP. EUS-TCB showed higher sensitivity (100%) and specificity (100%) compared to EUS-FNA (36% and 33%, respec-
tively). Both procedures were found to be safe, with no complications[33].
However, the diagnostic accuracy of EUS-FNA for pancreatic cancer has been reported to range between 60% and 90%[34-36], and the shortcomings of EUS-TCB due to technical difficulties of the sampling of lesions in the pancreatic head should also be considered.
Hence, when AIP is suspected, a sequential sampling strategy has been proposed based on using EUS-FNA first, which is followed by EUS-TCB when cytologic ex-amination is inconclusive[33].
In cases of inconclusive cytology, an additional aid for AIP diagnosis could come from molecular analysis of EUS-FNA samples, which has shown high accuracy in the differential diagnosis between AIP and pancreatic cancer[37].
CONCLUSIONAIP represents 20%-25% of benign diagnoses undergo-ing resection for presumed malignancy[38,39]. Thus, a defin-itive diagnosis based on safe and reliable methods should be obtained. In this setting, EUS could play an important role in diagnosis, identifying typical features of AIP and distinguishing it from biliopancreatic neoplasms. The higher sensitivity over standard imaging for pancreatic, biliary and nodal lesions should make it a cornerstone in the process of diagnosing AIP. If validated in large prospective series, newly available techniques such as EUS-elastography and CE-EUS could add useful infor-mation about focal lesions without resorting to invasive procedures. Finally, EUS-FNA or EUS-TCB can provide pathological specimens, as required by some diagnostic criteria.
REFERENCES1 Chang DK, Nguyen NQ, Merrett ND, Dixson H, Leong RW,
Biankin AV. Role of endoscopic ultrasound in pancreatic cancer. Expert Rev Gastroenterol Hepatol 2009; 3: 293-303
2 Kahl S, Glasbrenner B, Leodolter A, Pross M, Schulz HU, Malfertheiner P. EUS in the diagnosis of early chronic pan-creatitis: a prospective follow-up study. Gastrointest Endosc 2002; 55: 507-511
3 Buscarini E, Frulloni L, De Lisi S, Falconi M, Testoni PA, Zambelli A. Autoimmune pancreatitis: a challenging diag-nostic puzzle for clinicians. Dig Liver Dis 2010; 42: 92-98
4 Japan Pancreas Society. Diagnostic criteria for autoimmune pancreatitis. J Jpn Pancreas Soc 2002; 17: 585-587
5 Okazaki K, Kawa S, Kamisawa T, Naruse S, Tanaka S, Nishi-mori I, Ohara H, Ito T, Kiriyama S, Inui K, Shimosegawa T, Koizumi M, Suda K, Shiratori K, Yamaguchi K, Yamaguchi T, Sugiyama M, Otsuki M. Clinical diagnostic criteria of auto-immune pancreatitis: revised proposal. J Gastroenterol 2006; 41: 626-631
6 Kim MH, Kwon S. Diagnostic criteria for autoimmune chronic pancreatitis. J Gastroenterol 2007; 42 Suppl 18: 42-49
7 Chari ST, Smyrk TC, Levy MJ, Topazian MD, Takahashi N, Zhang L, Clain JE, Pearson RK, Petersen BT, Vege SS, Farnell MB. Diagnosis of autoimmune pancreatitis: the Mayo Clinic experience. Clin Gastroenterol Hepatol 2006; 4: 1010-1016; quiz 934
8 Otsuki M, Chung JB, Okazaki K, Kim MH, Kamisawa T,
2084 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Buscarini E et al . EUS and autoimmune pancreatitis

Kawa S, Park SW, Shimosegawa T, Lee K, Ito T, Nishimori I, Notohara K, Naruse S, Ko SB, Kihara Y. Asian diagnostic criteria for autoimmune pancreatitis: consensus of the Japan-Korea Symposium on Autoimmune Pancreatitis. J Gastroen-terol 2008; 43: 403-408
9 Frulloni L, Gabbrielli A, Pezzilli R, Zerbi A, Cavestro GM, Marotta F, Falconi M, Gaia E, Uomo G, Maringhini A, Mu-tignani M, Maisonneuve P, Di Carlo V, Cavallini G. Chronic pancreatitis: report from a multicenter Italian survey (Pan-CroInfAISP) on 893 patients. Dig Liver Dis 2009; 41: 311-317
10 Kim KP, Kim MH, Song MH, Lee SS, Seo DW, Lee SK. Au-toimmune chronic pancreatitis. Am J Gastroenterol 2004; 99: 1605-1616
11 Kamisawa T, Egawa N, Nakajima H, Tsuruta K, Okamoto A, Kamata N. Clinical difficulties in the differentiation of autoimmune pancreatitis and pancreatic carcinoma. Am J Gastroenterol 2003; 98: 2694-2699
12 Kamisawa T, Okamoto A. Autoimmune pancreatitis: pro-posal of IgG4-related sclerosing disease. J Gastroenterol 2006; 41: 613-625
13 De Lisi S, Buscarini E, Arcidiacono PG, Petrone M, Menozzi F, Testoni PA, Zambelli A. Endoscopic ultrasonography find-ings in autoimmune pancreatitis: be aware of the ambiguous features and look for the pivotal ones. JOP 2010; 11: 78-84
14 Sahani DV, Kalva SP, Farrell J, Maher MM, Saini S, Mueller PR, Lauwers GY, Fernandez CD, Warshaw AL, Simeone JF. Autoimmune pancreatitis: imaging features. Radiology 2004; 233: 345-352
15 Farrell JJ, Garber J, Sahani D, Brugge WR. EUS findings in patients with autoimmune pancreatitis. Gastrointest Endosc 2004; 60: 927-936
16 Hoki N, Mizuno N, Sawaki A, Tajika M, Takayama R, Shi-mizu Y, Bhatia V, Yamao K. Diagnosis of autoimmune pan-creatitis using endoscopic ultrasonography. J Gastroenterol 2009; 44: 154-159
17 Kubota K, Kato S, Akiyama T, Fujita K, Yoneda M, Taka-hashi H, Ogawa M, Inamori M, Abe Y, Kirikoshi H, Ko-bayashi N, Saito S, Hisatomi K, Matsuhashi N, Nakajima A. A proposal for differentiation between early- and advanced-stage autoimmune pancreatitis by endoscopic ultrasonogra-phy. Dig Endosc 2009; 21: 162-169
18 Takayama M, Hamano H, Ochi Y, Saegusa H, Komatsu K, Muraki T, Arakura N, Imai Y, Hasebe O, Kawa S. Recurrent attacks of autoimmune pancreatitis result in pancreatic stone formation. Am J Gastroenterol 2004; 99: 932-937
19 Sahai AV, Zimmerman M, Aabakken L, Tarnasky PR, Cun-ningham JT, van Velse A, Hawes RH, Hoffman BJ. Prospec-tive assessment of the ability of endoscopic ultrasound to diagnose, exclude, or establish the severity of chronic pan-creatitis found by endoscopic retrograde cholangiopancrea-tography. Gastrointest Endosc 1998; 48: 18-25
20 Dietrich CF, Hirche TO, Ott M, Ignee A. Real-time tissue elastography in the diagnosis of autoimmune pancreatitis. Endoscopy 2009; 41: 718-720
21 Hyodo N, Hyodo T. Ultrasonographic evaluation in patients with autoimmune-related pancreatitis. J Gastroenterol 2003; 38: 1155-1161
22 Naitoh I, Nakazawa T, Ohara H, Ando T, Hayashi K, Tanaka H, Okumura F, Takahashi S, Joh T. Endoscopic transpapil-lary intraductal ultrasonography and biopsy in the diagnosis of IgG4-related sclerosing cholangitis. J Gastroenterol 2009; 44: 1147-1155
23 Catalano MF, Sivak MV Jr, Rice T, Gragg LA, Van Dam J. Endosonographic features predictive of lymph node metas-tasis. Gastrointest Endosc 1994; 40: 442-446
24 Bhutani MS, Hawes RH, Hoffman BJ. A comparison of the accuracy of echo features during endoscopic ultrasound (EUS) and EUS-guided fine-needle aspiration for diagnosis of
malignant lymph node invasion. Gastrointest Endosc 1997; 45: 474-479
25 Gleeson FC, Rajan E, Levy MJ, Clain JE, Topazian MD, Hare-wood GC, Papachristou GI, Takahashi N, Rosen CB, Gores GJ. EUS-guided FNA of regional lymph nodes in patients with unresectable hilar cholangiocarcinoma. Gastrointest En-dosc 2008; 67: 438-443
26 Rösch T, Dittler HJ, Strobel K, Meining A, Schusdziarra V, Lorenz R, Allescher HD, Kassem AM, Gerhardt P, Siewert JR, Höfler H, Classen M. Endoscopic ultrasound criteria for vascular invasion in the staging of cancer of the head of the pancreas: a blind reevaluation of videotapes. Gastrointest En-dosc 2000; 52: 469-477
27 Snady H, Bruckner H, Siegel J, Cooperman A, Neff R, Kiefer L. Endoscopic ultrasonographic criteria of vascular invasion by potentially resectable pancreatic tumors. Gastrointest En-dosc 1994; 40: 326-333
28 Vilmann P, Jacobsen GK, Henriksen FW, Hancke S. Endo-scopic ultrasonography with guided fine needle aspiration biopsy in pancreatic disease. Gastrointest Endosc 1992; 38: 172-173
29 Levy MJ, Reddy RP, Wiersema MJ, Smyrk TC, Clain JE, Harewood GC, Pearson RK, Rajan E, Topazian MD, Yusuf TE, Chari ST, Petersen BT. EUS-guided trucut biopsy in es-tablishing autoimmune pancreatitis as the cause of obstruc-tive jaundice. Gastrointest Endosc 2005; 61: 467-472
30 Wiersema MJ, Levy MJ, Harewood GC, Vazquez-Sequeiros E, Jondal ML, Wiersema LM. Initial experience with EUS-guided trucut needle biopsies of perigastric organs. Gastroin-test Endosc 2002; 56: 275-278
31 Deshpande V, Mino-Kenudson M, Brugge WR, Pitman MB, Fernandez-del Castillo C, Warshaw AL, Lauwers GY. En-doscopic ultrasound guided fine needle aspiration biopsy of autoimmune pancreatitis: diagnostic criteria and pitfalls. Am J Surg Pathol 2005; 29: 1464-1471
32 Zamboni G, Lüttges J, Capelli P, Frulloni L, Cavallini G, Pederzoli P, Leins A, Longnecker D, Klöppel G. Histopatho-logical features of diagnostic and clinical relevance in auto-immune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch 2004; 445: 552-563
33 Mizuno N, Bhatia V, Hosoda W, Sawaki A, Hoki N, Hara K, Takagi T, Ko SB, Yatabe Y, Goto H, Yamao K. Histological di-agnosis of autoimmune pancreatitis using EUS-guided trucut biopsy: a comparison study with EUS-FNA. J Gastroenterol 2009; 44: 742-750
34 Suits J, Frazee R, Erickson RA. Endoscopic ultrasound and fine needle aspiration for the evaluation of pancreatic mass-es. Arch Surg 1999; 134: 639-642; discussion 642-643
35 Chang KJ, Nguyen P, Erickson RA, Durbin TE, Katz KD. The clinical utility of endoscopic ultrasound-guided fine-needle aspiration in the diagnosis and staging of pancreatic carci-noma. Gastrointest Endosc 1997; 45: 387-393
36 Wiersema MJ, Vilmann P, Giovannini M, Chang KJ, Wi-ersema LM. Endosonography-guided fine-needle aspiration biopsy: diagnostic accuracy and complication assessment. Gastroenterology 1997; 112: 1087-1095
37 Khalid A, Nodit L, Zahid M, Bauer K, Brody D, Finkelstein SD, McGrath KM. Endoscopic ultrasound fine needle as-pirate DNA analysis to differentiate malignant and benign pancreatic masses. Am J Gastroenterol 2006; 101: 2493-2500
38 Abraham SC, Wilentz RE, Yeo CJ, Sohn TA, Cameron JL, Boitnott JK, Hruban RH. Pancreaticoduodenectomy (Whip-ple resections) in patients without malignancy: are they all 'chronic pancreatitis'? Am J Surg Pathol 2003; 27: 110-120
39 Weber SM, Cubukcu-Dimopulo O, Palesty JA, Suriawinata A, Klimstra D, Brennan MF, Conlon K. Lymphoplasmacytic sclerosing pancreatitis: inflammatory mimic of pancreatic car-cinoma. J Gastrointest Surg 2003; 7: 129-137; discussion 137-139
S- Editor Tian L L- Editor O’Neill M E- Editor Zheng XM
2085 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Buscarini E et al . EUS and autoimmune pancreatitis

Effects of α-mangostin on apoptosis induction of human colon cancer
Ramida Watanapokasin, Faongchat Jarinthanan, Yukio Nakamura, Nitisak Sawasjirakij, Amornmart Jaratrungtawee, Sunit Suksamrarn
Ramida Watanapokasin, Department of Biochemistry, Fac-ulty of Medicine, Srinakharinwirot University, Bangkok 10110, ThailandFaongchat Jarinthanan, Faculty of Medical Technology, Rang-sit University, Pratumthani 12130, ThailandYukio Nakamura, Clinical Research Center, Murayama Medical Center, Gakuen 2-37-1, Tokyo 208-0011, JapanNitisak Sawasjirakij, Department of Research, North Bangkok University, Bangkok 10220, ThailandAmornmart Jaratrungtawee, Sunit Suksamrarn, Department of Chemistry, Faculty of Science, Srinakharinwirot University, Bangkok 10110, ThailandAuthor contributions: Watanapokasin R conceived, initiated the project and designed the research, provided project guidance and supervision and wrote the paper; Jarinthanan F performed the experiments; Nakamura Y and Sawasjirakij N revised the manu-script and analyzed the data; Jaratrungtawee A and Suksamrarn S participated in purifying of the α-mangostin. Supported by The Thailand Research Fund, Grant No. RMU 4980043Correspondence to: Ramida Watanapokasin, PhD, Associ-ate Professor and Head, Department of Biochemistry, Faculty of Medicine, Srinakharinwirot University, Sukhumvit 23, Bang-kok 10110, Thailand. [email protected]: +66-2-6495369 Fax: +66-2-6495834Received: November 11, 2010 Revised: January 10, 2011Accepted: January 17, 2011Published online: April 28, 2011
AbstractAIM: To investigate the effect of α-mangostin on the growth and apoptosis induction of human colon cancer cells.
METHODS: The three colorectal adenocarcinoma cell lines tested (COLO 205, MIP-101 and SW 620) were treated with α-mangostin to determine the effect on cell proliferation by MTT assay, cell morphology, chro-matin condensation, cell cycle analysis, DNA fragmen-tation, phosphatidylserine exposure and changing of
mitochondrial membrane potential. The molecular mechanisms of α-mangostin mediated apoptosis were further investigated by Western blotting analysis in-cluding activation of caspase cascade, cytochrome c release, Bax, Bid, p53 and Bcl-2 modifying factor.
RESULTS: The highest inhibitory effect of α-mangostin on cell proliferation of COLO 205, MIP-101 and SW 620 were 9.74 ± 0.85 μg/mL, 11.35 ± 1.12 μg/mL and 19.6 ± 1.53 μg/mL, respectively. Further study showed that α-mangostin induced apoptotic cell death in COLO 205 cells as indicated by membrane blebbing, chromatin condensation, DNA fragmentation, cell cycle analysis, sub-G1 peak (P < 0.05) and phosphatidylserine expo-sure. The executioner caspase, caspase-3, the initiator caspase, caspase-8, and caspase-9 were expressed upon treatment with α-mangostin. Further studies of apoptotic proteins were determined by Western blotting analysis showing increased mitochondrial cytochrome c release, Bax, p53 and Bmf as well as reduced mito-chondrial membrane potential (P < 0.05). In addition, up-regulation of tBid and Fas were evident upon treat-ment with α-mangostin (P < 0.01).
CONCLUSION: α-Mangostin may be effective as an anti-cancer agent that induced apoptotic cell death in COLO 205 via a link between extrinsic and intrinsic pathways.
© 2011 Baishideng. All rights reserved.
Key words: α-mangostin; Apoptosis; Caspases; Colon cancer; Mitochondria
Peer reviewer: Masahiro Iizuka, MD, PhD, Director, Akita Health Care Center, Akita Red Cross Hospital, 3-4-23, Nakadori, Akita, 010-0001, Japan
Watanapokasin R, Jarinthanan F, Nakamura Y, Sawasjirakij N, Jaratrungtawee A, Suksamrarn S. Effects of α-mangostin on apoptosis induction of human colon cancer. World J Gastroenterol
ORIGINAL ARTICLE
World J Gastroenterol 2011 April 28; 17(16): 2086-2095ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2086
2086 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

Watanapokasin R et al . Colon cancer apoptosis with mangostin
2011; 17(16): 2086-2095 Available from: URL: http://www.wjgnet.com/1007-9327/full/v17/i16/2086.htm DOI: http://dx.doi.org/10.3748/wjg.v17.i16.2086
INTRODUCTIONSearching for new biologically active compounds, novel chemotherapeutic agents derived from active phytochem-icals, could be used to improve the anti-carcinogenicity of standard drug treatment. A variety of tropical plants have useful biological activities and some offer potential thera-peutic applications. Mangosteen (Garcinia mangostana L.) in the Clusiaceae family has been used in Southeast Asia as traditional medicine for treatment of wounds, skin infection, diarrhea and chronic ulcer[1]. Phytochemical studies showed that the fruit hull of mangosteen is rich in a variety of oxygenated and prenylated xanthones[2,3] which possess different biological properties, such as anti-mycobacterial[4], anti-fungal[5], anti-oxidant[6-8], cyto-toxicity[9-12] and anti-inflammatory activities[13]. However, the underlying molecular mechanisms of α-mangostin in COLO 205 cells are not yet reported.
Apoptosis plays a vital role in controlling cell number in many physiological and developmental stages, tissue homeostasis, and regulation of immune system[14], while insufficient apoptosis is an integral part of cancer devel-opment[15]. Mammalian cells have two major apoptotic pathways. One pathway (extrinsic pathway) is triggered when ligands [Fas/CD95, tumor necrosis factor (TNF)-α] bind to receptors on cell surface leading to the activation of caspase-8 and -3[16], respectively. The other involves mitochondrial (intrinsic) pathway induced by anti-cancer drugs, prostaglandin, etc. resulting in disruption of mito-chondrial membrane and release of various pro-apoptotic factors[17-19]. The pro-apoptotic and anti-apoptotic mem-bers of B-cell CLL/lymphoma 2 (Bcl-2) family regulate the release of cytochrome c, a mitochondrial protein that can activate caspases.
Fas/CD95 belongs to the TNF superfamily and is the prototype of death receptor that initiates an apop-totic cascade[20]. FasL, the tumor necrosis factor-related cytokine, is a ligand of Fas and synthesized as a type Ⅱ membrane protein[21]. Upon FasL binding, activated death receptors engage the Fas associated death domain (FADD)[22], which in turn recruits caspase-8 and forming the death inducing signaling complex (DISC). The DISC then activates caspase-8 through a proximity-inducing dimerization mechanism. In type Ⅰ cells, caspase-8 di-rectly activates caspase-3, -6 and -7, leading to cell death. In contrast, in type Ⅱ cells the small amount of active caspase-8 generated at the DISC is not sufficient to in-duce cell death, therefore the mitochondria-dependent apoptosis pathway is needed[23]. As such, the pro-apoptot-ic signal has to be amplified via cleavage of the BH3-only protein Bid, Bax/Bak-assisted release of cytochrome c from the mitochondria, an activation of caspase-9 and subsequently caspase-3[24].
The objective of the present study was to purify the α-mangostin from the fruit hull of Garcinia mangostana L. and explore its effect on apoptosis induction and mecha-nisms involved in COLO 205 cells.
MATERIALS AND METHODSα-mangostin preparationMangosteen fruit (G. mangostana) was collected from Kombang District, Chantaburi Province, Thailand in April, 2007. A voucher specimen (Ms Porntip Wongnapa No. 002) was deposited at the Faculty of Science, Ram-khamhaeng University. The dried and pulverized fruit hull of G. mangostana (0.5 kg) was thoroughly extracted with ethyl acetate (EtOAc) at 50℃. The combined extract af-ter filtration was concentrated under reduced pressure to yield the extract as a yellowish solid (285 g). A portion of the extract was subjected to repeated column chromatog-raphy over silica gel using a gradient of hexane/acetone which yielded the pure major compound, α-mangostin, including other minor xanthones. Purity of α-mangostin exceeded 98% as determined by LC analysis and its spec-troscopic data (NMR and MS) was consistent with the reported values[12].
Cell lines and culture conditionsThree human colorectal cancer cell lines were used: COLO 205 (colorectal adenocarcinoma), MIP-101 (colorectal car-cinoma) and SW620 (colorectal adenocarcinoma). COLO 205 and SW620 were obtained from the American Type Culture Collection (Manassas, VA). MIP-101 was a gener-ous gift from Peter Thomas, Boston University School of Medicine, Boston, MA. COLO 205 and MIP-101 were maintained in the RPMI 1640 medium (Invitrogen), supplemented with 10% fetal calf serum (Invitrogen). SW620 were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen), supplemented with 10% fetal calf serum. All cell lines were maintained in culture at 37°C in an atmosphere of 5% CO2.
Cell proliferation and cell viability assaysThe cytotoxic activity of α-mangostin was determined by cell proliferation analysis using MTT assays as previ-ously described[25]. Briefly, cells were cultured in 96-well plates at a density of 1 × 104/well in complete medium. Then the cells were treated with varying concentrations of α-mangostin and incubated at 37℃ for 24 h. The final DMSO concentration in each well was 0.05%, at which concentration no appreciable effect on cell proliferation was seen. Then, 100 μL of 5.0 mg/mL MTT in culture me-dia was added to each well and incubated at 37℃ for 2 h. The metabolic product of MTT, formazan, in each well was dissolved in DMSO, and the absorbance was deter-mined at 595 nm. Effect of α-mangostin on the viability of COLO 205 cells was analyzed by using a trypan blue exclusion method. Briefly, cells were cultured in 96-well plates at cell density of 1 × 104/well at 37℃ for 24 h. α-mangostin was then added to culture wells at 0, 10, 20,
2087 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

30 and 40 μg/mL or vehicle (in DMSO), incubated at 37℃ then collected periodically (0, 3, 6, 9 and 12 h). The number of viable cells was determined with hemocytom-eters under a light microscope. Cell viability was expressed as a percentage of the number of viable cells to that of the control, to which no α-mangostin was applied.
Apoptosis assayCharacterization of the anticancer activity of α-mangostin in COLO 205 cells was further conducted. Based on the preliminary experiments, 20 μg/mL of α-mangostin was used to study cell and nuclear morphology, cytochrome c release, mitochondrial transmembrane potential, expres-sion of pro-apoptotic proteins, Fas and truncated-Bid (t-Bid). However, a selective range of test concentrations, ranging from 10 to 30 μg/mL was used to study DNA fragmentation and for cell cycle analysis and annexin V-FITC assays.
Microscopic analysis of cell and nuclear morphologyCOLO 205 cells were cultured in 24-well culture plates at the initial number of 2 × 104/well in the presence of 20 μg/mL α-mangostin for 3, 6, 9 and 12 h. As controls, cells were cultured in the same fashion in the absence of α-mangostin. The cells were examined under a phase-contrast inverted microscope (model CKX31/CKX41, Olympus) for cell morphology.
The nuclear morphology was analyzed by treatment of COLO 205 cells with 20 μg/mL α-mangostin for 3, 6, 9 and 12 h. Control cells were grown in the same manner in the absence of α-mangostin. Cells were trypsinized and fixed with methanol. Then, cell nuclei were stained by treatment with 1 μg/mL Hoechst 33342 (Sigma) at 37℃ for 15 min in the dark. Stained cells were examined under a fluorescence inverted microscope (model BX50, Olympus).
Analysis of DNA fragmentationCOLO 205 cells were treated with varying concentrations, 10, 20 and 30 μg/mL, of α-mangostin for 12 h and then lysed in 500 μL of lysis solution, consisting of 5 mmol/L Tris-Cl (pH 8.0), 0.5% Triton X-100, and 20 mmol/L ethylenediaminetetraacetic acid (EDTA). The cells were then treated with RNase A (0.5 mg/mL) for 1 h at 37℃. DNA fractions were prepared using phenol-chloroform-isoamyl alcohol (25:24:1) and electrophoresed on 1.8% agarose gels. Approximately 20 μg of DNA was loaded in each well and the agarose gels were run at 50 V for 2 h in Tris-borate/EDTA electrophoresis buffer. DNA was stained with ethidium bromide and visualized under a UV light trans-illuminator and photographed.
Flow cytometer analysis The cell cycle analysis, annexin V binding and mitochon-drial transmembrane potential were investigated by flow cytometric analysis (FACScan, Becton Dickinson). The proper filters and optimal setting of the instrument were chosen, the histograms generated by FACS were analyzed by Cell Quest™ software (Becton Dickinson).
Cell cycle analysis using flow cytometer COLO 205 cells were cultured in 6-well culture plates at 4 × 106 cells/well and treated with 0, 10, 20 and 30 μg/mL of α-mangostin for 3 h. The cells were then har-vested, washed with PBS and resuspended in 200 μL PBS and fixed in 800 μL of ice-cold 70% ethanol at -20℃, overnight. The cells were stained with 1 mL of 50 μg/mL propidium iodide solution (containing 0.1% Triton X-100, and 0.1% sodium citrate) for 30 min at 37℃. The samples were then analyzed by a flow cytometer (FACScan, Bec-ton Dickinson). Excitation was done at 488 nm, and emis-sion filter at 600 nm. Histograms generated by FACS were analyzed by Cell Quest™ software (Becton Dickinson) to determine the percentage of cells in each phase.
Annexin V-FITC assayPercentage of α-mangostin-treated cells undergoing ap-optosis was determined using an annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit (BD) Bioscience, San Jose, CA). COLO 205 cells at 1 × 105 cells/mL were treated with 0, 10, 20 and 30 μg/mL of α-mangostin for 3 h and resuspended in 100 μL of annex-in V binding buffer (10 mmol/L HEPES, 150 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 1.8 mmol/L CaCl2), then incubated with 5 μL of 1 μg/mL FITC-conjugated annexin V and 1 μg/mL propidium iodide for 15 min at room temperature prior to analysis on a FACScan, Becton Dickinson.
Analysis of mitochondrial transmembrane potential COLO 205 cells at 1 × 106 cells/mL were treated with 20 μg/mL of α-mangostin for 3 h, and then incubated with 10 μg/mL JC-1 (5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazolcarbocyanine iodide) at 37℃ for 10 min in darkness. Stained cells were washed with PBS, followed by FACS analysis. The mitochondrial function was assessed as JC-1 green (uncoupled mitochondria) or red (contact mitochondria).
Western blotting analysis of cytochrome cCOLO 205 cells were cultured in 6-well culture plates at 4 × 106 cells/well and then incubated in the absence or presence of 20 μg/mL α-mangostin for 3, 6, and 9 h. Cell lysates was prepared as previously described[25]. Briefly, cell suspensions were sonicated for 10 s and the cell lysates was centrifuged at 4℃ at 10 000 × g for 30 min. The super-natants (cytosol fractions) were subjected to SDS-PAGE using 12% polyacrylamide gels, and transferred onto Im-mobilon P membrane and subjected to immuno-detection of cytochrome c using a mouse monoclonal antibody against human cytochrome c (7H8, mouse monoclonal Ig G 2b, Santa Cruz, Biotechnology) with goat anti-mouse IgG conjugates to horse radish peroxidase (Cell Signal-ing Technology) and detected using an ECL Plus Western Blotting Detection System (Amersham Biosciences).
Western blotting analysis of caspases, Bid, p53, Bax, Bmf and FasCOLO 205 cells were treated with 20 μg/mL α-mangostin
2088 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Watanapokasin R et al . Colon cancer apoptosis with mangostin
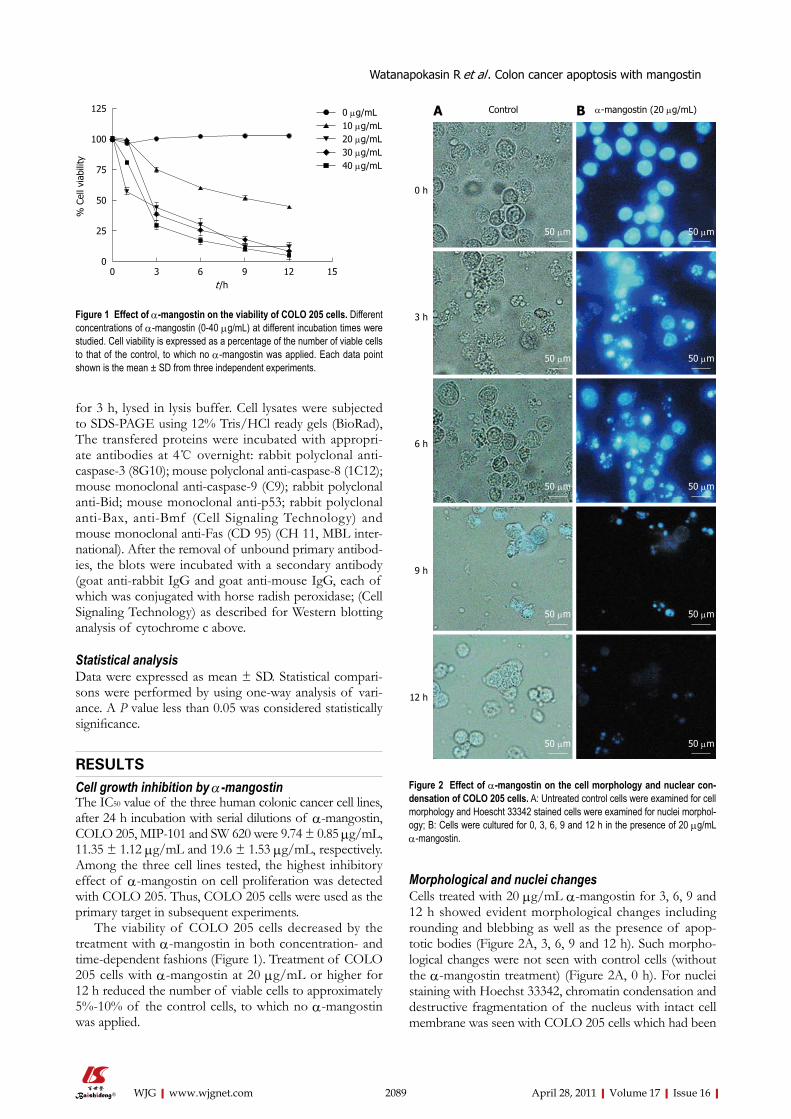
for 3 h, lysed in lysis buffer. Cell lysates were subjected to SDS-PAGE using 12% Tris/HCl ready gels (BioRad), The transfered proteins were incubated with appropri-ate antibodies at 4℃ overnight: rabbit polyclonal anti-caspase-3 (8G10); mouse polyclonal anti-caspase-8 (1C12); mouse monoclonal anti-caspase-9 (C9); rabbit polyclonal anti-Bid; mouse monoclonal anti-p53; rabbit polyclonal anti-Bax, anti-Bmf (Cell Signaling Technology) and mouse monoclonal anti-Fas (CD 95) (CH 11, MBL inter-national). After the removal of unbound primary antibod-ies, the blots were incubated with a secondary antibody (goat anti-rabbit IgG and goat anti-mouse IgG, each of which was conjugated with horse radish peroxidase; (Cell Signaling Technology) as described for Western blotting analysis of cytochrome c above.
Statistical analysisData were expressed as mean ± SD. Statistical compari-sons were performed by using one-way analysis of vari-ance. A P value less than 0.05 was considered statistically significance.
RESULTS Cell growth inhibition by α-mangostin The IC50 value of the three human colonic cancer cell lines, after 24 h incubation with serial dilutions of α-mangostin, COLO 205, MIP-101 and SW 620 were 9.74 ± 0.85 μg/mL, 11.35 ± 1.12 μg/mL and 19.6 ± 1.53 μg/mL, respectively. Among the three cell lines tested, the highest inhibitory effect of α-mangostin on cell proliferation was detected with COLO 205. Thus, COLO 205 cells were used as the primary target in subsequent experiments.
The viability of COLO 205 cells decreased by the treatment with α-mangostin in both concentration- and time-dependent fashions (Figure 1). Treatment of COLO 205 cells with α-mangostin at 20 μg/mL or higher for 12 h reduced the number of viable cells to approximately 5%-10% of the control cells, to which no α-mangostin was applied.
Morphological and nuclei changes Cells treated with 20 μg/mL α-mangostin for 3, 6, 9 and 12 h showed evident morphological changes including rounding and blebbing as well as the presence of apop-totic bodies (Figure 2A, 3, 6, 9 and 12 h). Such morpho-logical changes were not seen with control cells (without the α-mangostin treatment) (Figure 2A, 0 h). For nuclei staining with Hoechst 33342, chromatin condensation and destructive fragmentation of the nucleus with intact cell membrane was seen with COLO 205 cells which had been
2089 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
125
100
75
50
25
0
% C
ell v
iabi
lity
0 μg/mL10 μg/mL20 μg/mL30 μg/mL40 μg/mL
0 3 6 9 12 15 t /h
Figure 1 Effect of α-mangostin on the viability of COLO 205 cells. Different concentrations of α-mangostin (0-40 μg/mL) at different incubation times were studied. Cell viability is expressed as a percentage of the number of viable cells to that of the control, to which no α-mangostin was applied. Each data point shown is the mean ± SD from three independent experiments.
0 h
3 h
6 h
9 h
12 h
Control α-mangostin (20 μg/mL)BA
Figure 2 Effect of α-mangostin on the cell morphology and nuclear con-densation of COLO 205 cells. A: Untreated control cells were examined for cell morphology and Hoescht 33342 stained cells were examined for nuclei morphol-ogy; B: Cells were cultured for 0, 3, 6, 9 and 12 h in the presence of 20 μg/mL α-mangostin.
50 μm 50 μm
50 μm50 μm
50 μm 50 μm
50 μm50 μm
50 μm 50 μm
Watanapokasin R et al . Colon cancer apoptosis with mangostin

treated with 20 μg/mL of α-mangostin for 3, 6, 9 and 12 h (Figure 2B, 3, 6, 9 and 12 h), indicated early apopto-sis while the nuclei of control cells without α-mangostin treatment showed normal morphology (Figure 2B, 0 h).
α-mangostin mediated apoptotic cell death The early apoptosis was detected upon treatment of COLO 205 cells with 0, 10, 20 and 30 μg/mL of α-mangostin for 3 h using Annexin V-FITC assay. The results indicated significant increases in apoptotic populations in COLO 205 cells approximately 0.50% ± 0.02%, 4.00% ± 0.25%, 10.00% ± 0.50% and 13.00% ± 0.30%, (P < 0.05) re-spectively (Figure 3). Exposure of COLO 205 cells to increasing concentrations (0, 10, 20 and 30 μg/mL) of α-mangostin for 3 h resulted in increased percentage of cells arrested in sub G-1 phase (apoptotic cell death) of 1.03% ± 0.15%, 7.00% ± 0.20%, 51.00% ± 1.53% and 80.00% ± 2.08% (P < 0.05), respectively (Figure 4). It is evident that the formation of apoptotic cells at sub-G1 phase was directly proportional to the increased concen-tration of α-mangostin.
Fragmentation of chromosomal DNA Fragmentation of genomic DNA was apparent by the presence of DNA ladders when COLO 205 cells were treated with 10, 20 and 30 μg/mL α-mangostin for 12 h (Figure 5). The characteristic ladder pattern of discon-tinuous DNA fragments was observed only in the treated cells, whereas the untreated control showed no DNA fragmentation.
Activation of caspases upon α-mangostin treatmentThe activation of caspases-3, -8 and -9 was detected (Figure 6). The amount of the pro-enzyme form of cas-pase-3 (pro-caspase-3, 35 kDa), pro-caspase-8 (57 kDa) and pro-caspase-9) (47 kDa) decreased with increasing concentration of α-mangostin (10, 20, 30 and 40 μg/mL).
Accordingly, cleaved activated forms of caspase-3 (19 and 20 kDa), caspase-8 (41 and 43 kDa) and caspase-9 (35 and 37 kDa) (P < 0.01) became apparent upon treat-ment of α-mangostin at 20 μg/mL or higher. Activated caspase-3 was apparent upon treatment with 20 μg/mL α-mangostin but not at higher concentrations (30 and 40 μg/mL α-mangostin) due to cell death at higher con-centrations, as the effector caspase-3 is the late event of the pathways. On the other hand, caspase-8 and -9 were also detected implying the consecutive activation of cas-pases of the intrinsic pathway. In addition, induction of apoptosis by α-mangostin was accompanied by increased phospho-p53, pro-apoptotic Bax and Bmf (P < 0.01) (Figure 7). The release of cytochrome c from mitochondria to cytosol was evident upon treatment of COLO 205 cells with 20 μg/mL α-mangostin for 3 h and 6 h (Figure 8). The non-cytosolic fraction (pellet) of the treated cells showed no cytochrome c expression (data not shown). At 9 h of α-mangostin treatment, cytochrome c was reduced due to increasing cell death. No appreciable amount of cytochrome c was detected in the cytosol fraction of con-trol COLO 205 cells, to which no α-mangostin was ap-plied (Figure 8). This implied that α-mangostin mediated apoptosis is accompanied by mitochondrial dysfunction, which could be further strengthened by mitochondrial membrane depolarization detected by a carbocyanine fluorescence dye, JC-1, upon treatment with α-mangostin. The results indicated the increased percentage of cells with depolarized mitochondrial membrane potential (red to green) approximately 82% after α-mangostin treatment for 3 h (Figure 9). These results further confirmed that α-mangostin is an efficient inducer of apoptosis that both extrinsic and intrinsic pathway may be involved. Thus, we further conducted the Western blotting analysis of Bid, t-Bid, the linker between extrinsic and intrinsic pathway, and Fas receptor and found that they were up-regulated upon α-mangostin treatment (P < 0.01) (Figure 7).
2090 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
105
104
103
102
FL-2
H
102 103 104 105
FL-1H
2% 10 μg/mL
4%Negative
105
104
103
102
FL-2
H
102 103 104 105
FL-1H
1% 0 μg/mL
0.5%Negative
105
104
103
102
FL-2
H
102 103 104 105
FL-1H
10% 30 μg/mL
13%Negative
105
104
103
102
FL-2
H
102 103 104 105
FL-1H
8% 20 μg/mL
10%Negative
15
10
5
0
Apop
totic
cel
l (%
)
a
a
a
a
0 10 20 30 α-mangostin (μg/mL)
Figure 3 Fluorescent-activated cell sorter analysis of COLO 205 cells stained with Annexin V-FITC. Cells were treated with 10, 20 and 30 μg/mL α-mangostin for 3 h. α-mangostin induced early apoptosis in a concentration dependent manner. The values are expressed as mean ± SD; aP < 0.05.
Watanapokasin R et al . Colon cancer apoptosis with mangostin

DISCUSSIONThe evident goal of medical research is to be able to ma-nipulate the machinery of cell death. Regulation of apop-tosis might also lead to new possibilities for cancer ther-
apy[26]. In the present study, we showed that α-mangostin treatment of human colon COLO 205 induced cytotoxic effects in a dose and time dependent fashion. We then investigated the apoptotic effects of α-mangostin in COLO 205 cells to advance our knowledge of its biologi-cal functions and also health advantages. The membrane shrinkage, chromatin condensation and fragmentation were detected. As cancer growth is associated with the loss of cell cycle checkpoints, which regulate the DNA integrity and ensure that the genes are co-ordinately ex-pressed[27]. The sub-G1 fraction and phosphatidylserine translocation is an indication of apoptosis cell death that naturally occurs in cells and is beneficial for cancer therapy[28]. Therefore, to characterize apoptotic cells upon treatment of COLO 205 cells with α-mangostin, a bi-parametric cytofluorimetric analysis was performed using PI and annexin V-FITC, which stained DNA and phos-phatidylserine residues, respectively. In the early apoptotic process, a phosphatidylserine residue became exposed on the cell surface by flipping from the inner to outer leaflet of the cytoplasmic membrane[29,30]. Our results demon-strated the increased sub-G1 population and numbers of early apoptotic cells upon treatment of COLO 205 cells with 20 μg/mL α-mangostin for 3 h as compared to untreated control. The Bcl-2 family of proteins regulates apoptosis and it has been shown that the gene prod-ucts of Bcl-2 and Bax play important roles in apoptotic cell death[14]. The Bcl-2 family comprises of both pro-apoptotic and anti-apoptotic proteins that elicit opposite effects on mitochondria. Anti-apoptotic members include Bcl-2, Bcl-xL, Bcl-W, Mcl-1, whereas pro-apoptotic mem-bers are Bid, Bax, Bakm, Bmf and others. Several path-ways involve p53-mediated apoptosis, and one of these is the Bcl-2 and Bax proteins. The Bax protein is a p53 target and known to promote cytochrome c release from mitochondria which in turn activates caspase-3. Regula-tion of Bax/Bcl-2 and caspases activity becomes impor-
2091 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Figure 4 Flow analysis of cell cycle. Representative plots of PI staining of COLO 205 cells that were treated with 0 (control), 10, 20 and 30 μg/mL α-mangostin for 3 h. The values are expressed as mean ± SD; aP < 0.05.
100
75
50
25
0
Apop
totic
cel
ls (
%)
a
a
aa
0 10 20 30 α-mangostin (μg/mL)
800700600500400300200100
0
Coun
ts
Sub-G1G0/G1
SG2/M
Sub-G1: 80%
G0/G1: 14%
S: 5%
G2/M: 1%
50 100 150 200 250
PI-A (× 1000)
30 μg/mL
800700600500400300200100
0
Coun
ts
Sub-G1G0/G1
SG2/M
Sub-G1: 52%
G0/G1: 36%
S: 9%
G2/M: 3%
50 100 150 200 250
PI-A (× 1000)
20 μg/mL
800700600500400300200100
0
Coun
ts
Sub-G1G0/G1
SG2/M
Sub-G1: 7%
G0/G1: 55%
S: 23%
G2/M: 15%
50 100 150 200 250
PI-A (× 1000)
10 μg/mL
800700600500400300200100
0
Coun
ts
Sub-G1G0/G1
SG2/M
Sub-G1: 1%
G0/G1: 55%
S: 29%
G2/M: 15%
50 100 150 200 250
PI-A (× 1000)
0 μg/mL
α-mangostin (3 h)
200 kb120 kb100 kb50 kb
40 kb
30 kb
20 kb
6 kb
M 0 10 20 30 (μg/mL)
α-mangostin (12 h)
Figure 5 Analysis of DNA integrity in COLO 205 cells upon treatment with α-mangostin. COLO 205 cells were treated with varying concentrations of α-mangostin (10, 20 and 30 μg/mL) for 12 h. Cells were lysed, followed by phenol-chloroform extraction. DNA fractions were then electrophoresed on 1.8% agarose gels. DNA was stained with ethidium bromide and visualized under a UV light trans-illuminator.
Watanapokasin R et al . Colon cancer apoptosis with mangostin

2092 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
20 cl
eave
d-for
m
1.00
0.75
0.50
0.25
0.00
Rela
tive
quan
tity
bb b
20 pr
oform
10 pr
oform
0 pro
form 30 40
α-mangostin (μg/mL)
Caspase-3/actin
30 pr
oform
1.00
0.75
0.50
0.25
0.00
Rela
tive
quan
tity
bb b
20 pr
oform
10 pr
oform
0 pro
form
Cleav
ed-fo
rm
40 cl
eave
d-for
m
α-mangostin (μg/mL)
Caspase-8/actin
b
30 pr
oform
1.00
0.75
0.50
0.25
0.00
Rela
tive
quan
tity
20 pr
oform
10 pr
oform
0 pro
form
40 pr
oform
40 cl
eave
d-for
m
α-mangostin (μg/mL)
Caspase-9/actin
b
b
b
30 cl
eave
d-for
m
20 cl
eave
d-for
m
0 10 20 30 40 (μg/mL)
α-mangostin (3 h)
Pro-caspase-3
P20P19
Pro-caspase-8
P43P41
Pro-caspase-9P37P35
Actin (42 kDa)
Figure 6 Effects of α-mangostin on the activation of caspase-3, -8 and -9 in COLO 205 cells. Cells were treated with 10, 20, 30 and 40 μg/mL α-mangostin for 3 h. Cell lysates were separated by SDS-PAGE using 12% polyacrylamide gels. Proteins were subjected to immuno-detection of caspases-3, -8, and -9 using appropriate anti-caspase antibodies at 4℃. The expression of cleaved-caspase-3, -8 and -9 were detected. The density of each band was determined, equal protein loading was verified by β-actin staining. The values are expressed as mean ± SD; bP < 0.01.
Bax20 kDa
Bmf18 kDa
p5353 kDa
Phospho-p53 (Ser15)53 kDa
FAS clone CH1143 kDa36 kDa
Bid22 kDa15 kDa Cleaved-Bid
42 kDa Actin
0 20 (μg/mL)
Time (3 h)
p-p5
3
Rela
tive
quan
tity
of p
rote
ins
(pro
tein
s/ac
tin)
Bmf
Bax
Cont
rol
Bid t-Bid
b
b
FAS
p53
25
20
15
10
5
0
b b
Figure 7 Effects of α-mangostin on the activation of Bax, Bmf, p53, Fas and Bid. Cells were treated with 20 μg/mL α-mangostin for 3 h. Cell lysates were separated by SDS-PAGE using 12% polyacrylamide gels. Proteins were subjected to immuno-detection of Bax, Bmf, p53, p-p53, Fas, Bid and t-Bid using appropriate antibodies. The density of each band was determined, equal protein loading was verified by β-actin staining. The values are expressed as mean ± SD; bP < 0.01.
Watanapokasin R et al . Colon cancer apoptosis with mangostin

2093 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
0 μg/mL α-mangostin
50 μm
20 μg/mL α-mangostin
50 μm
300
260
200
160
100
60
0
Freq
of
even
ts
102 103 104 105
FITC-A
8% 82%
20 μg/mL α-mangostin
360
300
260
200
160
100
60
0
Freq
of
even
ts
102 103 104 105
FITC-A
97% 3%
0 μg/mL α-mangostin
Figure 9 Measurements of mitochondrial membrane depolarization in COLO 205 cells. Cells were treated with 0 (control) and 20 μg/mL α-mangostin for 3h and then incubated with JC-1 (10 μg/mL in PBS) at 37℃ for 10 min. Stained cells were subjected to FACS analysis. The mitochondrial function was assessed as JC-1 green. The values are expressed as mean ± SD; aP < 0.05.
100
75
50
25
0
% J
C-1
posi
tive
cell a
Control 20 μg/mL α-mangostin
a
0 3 6 9 h
α-mangostin (20 μg/mL)
Cytochrome c (15 kDa)
Actin (42 kDa)
Figure 8 Release of cytochrome c from the mitochondria in COLO 205 cells. Upon treatment with 20 μg/mL α-mangostin for 3, 6 and 9 h, cytosol fractions were prepared from these cells and separated by SDS-PAGE using 12% polyacrylamide gels. Proteins were subjected to immuno-detection of cytochrome c using a mouse monoclonal antibody against human cytochrome c. The density of each band was determined, equal protein loading was verified by β-actin staining. The values are expressed as mean ± SD; aP < 0.05.
1.5
1.0
0.5
0.0Rela
tive
quan
tity
of c
ytoc
hrom
e c
a a
a
0 3 6 9 t /h
Watanapokasin R et al . Colon cancer apoptosis with mangostin

2094 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
DYm loss
O
HO OH
OHO
OMitochondria
Cell membrane
FasL
Fas
p53
Bid t-Bid p53 BmfBax
Procaspase 8
Caspase 8
Cytochrome c
Procaspase 9
Caspase 9
Procaspase 3
Caspase 3Apoptosis
Figure 10 A proposed diagram for α-mangostin-induced apoptosis in COLO 205 cells. Upon α-mangostin treatment, extrinsic pathway was activated, procas-pase-8 was cleaved to caspase-8 which then further activated the cleavage of Bid to t-Bid. The t-Bid then translocates to mitochondria resulting in the activa-tion of mitochondrial apoptotic pathway.
tant targets for cancer intervention. Caspase-3 being the major executioner caspase[31], thus we examined whether activated caspase-3, -8 and -9 is involved in apoptotic induction and found evident expression of activated cas-pase-3, -8 and -9.
Under the influence of α-mangostin treatment in COLO 205 cells, a cell death pathway both via death receptor pathway and mitochondrial pathway may be involved, as caspase-8 and -9 were expressed. We fur-ther demonstrated the loss of mitochondrial membrane potential, release of cytochrome c into the cytosol and DNA fragmentation. Our results are in agreement with a study showing that apoptosis was associated with a loss of mitochondrial membrane potential, which may correspond to the opening of an outer membrane pore, leading to cytochrome c release from mitochondria into the cytosol. The released cytochrome c later triggered the cleavage and activation of caspases and onset of apop-tosis[32]. The expression of Fas and caspase-8 implies the death receptor pathway, while regulation of mitochon-drial membrane permeability by upregulation of Bax and p-Bad triggering the release of cytochrome c from mi-tochondria to cytosol. Our further investigation showed the expression of Fas, Bid, t-Bid, p-53, phospho-p53 and the proteins in Bcl-2 family (Bax and Bmf). Expression of t-Bid is the important linkage between death receptor and mitochondrial pathway. When Bid is cleaved into t-Bid by the activated caspase-8 and translocated towards the mitochondria to activate Bax and Bak, then changing the mitochondrial membrane permeability and the release of cytochrome c[33] which form a complex with apoptotic enzyme activators and caspase-9. It activates and starts a caspase cascade reaction, which further activates the downstream caspase-3 and other caspase family members for apoptosis induction. We also noticed that α-mangostin
up-regulated the expression of Bax in COLO 205 cells at protein level, suggesting that Bcl-2 family protein regulate α-mangostin mediated apoptotic cell death.
Taken together our results evaluating the molecular mechanism that α-mangostin induced apoptosis cell death in COLO 205 cells may occur via caspase-8 depen-dent cleavage of Bid to tBid providing a link between ex-trinsic and intrinsic pathways (Figure 10). This could be a promising chemotherapeutic agent and may also serve as a model to develop and design new derivatives which may be more potent.
COMMENTSBackgroundCancer causes significant morbidity and mortality and is a major public health problem worldwide. Globally, colorectal cancer is one of the most common types of cancer in both men and women. Phytochemical studies showed that a variety of tropical plants have useful biological activities and some offer poten-tial therapeutic applications. However, the molecular mechanisms underlying α-mangostin induced apoptosis in human colorectal adenocarcinoma have not yet been fully understood. Research frontiersApoptosis plays a vital role in controlling cell number in many physiological and developmental stages, tissue homeostasis, and regulation of the immune system, while insufficient apoptosis is an integral part of cancer development. The main function of apoptosis is to dispose of a cell without causing damage or stress to neighbouring cells. Thus, the anti-cancer drug that induces apop-totic cell death would be more suitable for use in patients and should be further developed. Therefore, the effect of α-mangostin on the growth and apoptosis induction of human colon cancer cells was investigated in this study.Innovations and breakthroughsThe results showed that α-mangostin induced apoptotic cell death in COLO 205 cells indicating by membrane blebbing, chromatin condensation, DNA fragmentation, cell cycle analysis, sub-G1 peak, and phosphatidylserine ex-posure. The expression of caspase-3, caspase-8 and caspase-9, cytochrome c release, Bax, p53 and Bcl-2 modifying factor (Bmf) as well as reduced mitochondrial membrane potential were demonstrated. In addition, upon treat-ment with α-mangostin, up-regulation of tBid and Fas were evident. Therefore, α-mangostin may be effective as an anti-cancer agent that induces apoptotic cell death in COLO 205 via a link between extrinsic and intrinsic pathways. Applicationsα-mangostin could be used as a future promising anti-cancer agent for the treatment of colorectal adenocarcinoma cells. TerminologyThe fruit hull of mangosteen (Garcinia mangostana L.) in the Clusiaceae fam-ily is rich in a variety of oxygenated and prenylated xanthones which possess different biological properties, such as anti-mycobacterial, anti-fungal, anti-oxidant, cytotoxicity and anti-inflammatory activities.Peer reviewIn this study the authors examined the effect of α-mangostin on the growth and apoptosis induction of human colon cancer cells. They showed that α-mangostin induced apoptotic cell death in COLO 205 cells, activated caspase-3, -8, and -9, increased the mitochondrial cytochrome c release, Bax, p53 and Bmf, reduced mitochondrial membrane potential, and up-regulated tBid and Fas. From these results, the authors suggested that α-mangostin may be effective as an anti-cancer agent via a link between extrinsic and intrinsic pathways. It is interesting that the authors clearly showed the mechanism through which α-mangostin induced apoptosis in colon cancer cells.
REFERENCES1 Mahabusarakam W, Wiriyachitra P, Taylor WC. Chemi-
cal constituents of garcinia mangostana. J Nat Prod 1987; 50: 474-478
Watanapokasin R et al . Colon cancer apoptosis with mangostin
COMMENTS

2095 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
2 Peres V, Nagem TJ. Trioxygenated naturally occurring xan-thones. Phytochemistry 1997; 44: 191-214
3 Peres V, Nagem TJ, de Oliveira FF. Tetraoxygenated natu-rally occurring xanthones. Phytochemistry 2000; 55: 683-710
4 Suksamrarn S, Suwannapoch N, Phakhodee W, Thanuhiran-lert J, Ratananukul P, Chimnoi N, Suksamrarn A. Antimy-cobacterial activity of prenylated xanthones from the fruits of Garcinia mangostana. Chem Pharm Bull (Tokyo) 2003; 51: 857-859
5 Gopalakrishnan G, Banumathi B, Suresh G. Evaluation of the antifungal activity of natural xanthones from Garcinia mangostana and their synthetic derivatives. J Nat Prod 1997; 60: 519-524
6 Williams P, Ongsakul M, Proudfoot J, Croft K, Beilin L. Mangostin inhibits the oxidative modification of human low density lipoprotein. Free Radic Res 1995; 23: 175-184
7 Jung HA, Su BN, Keller WJ, Mehta RG, Kinghorn AD. Anti-oxidant xanthones from the pericarp of Garcinia mangostana (Mangosteen). J Agric Food Chem 2006; 54: 2077-2082
8 Mahabusarakam W, Proudfoot J, Taylor W, Croft K. Inhibi-tion of lipoprotein oxidation by prenylated xanthones de-rived from mangostin. Free Radic Res 2000; 33: 643-659
9 Ho CK, Huang YL, Chen CC. Garcinone E, a xanthone de-rivative, has potent cytotoxic effect against hepatocellular carcinoma cell lines. Planta Med 2002; 68: 975-979
10 Matsumoto K, Akao Y, Ohguchi K, Ito T, Tanaka T, Iinuma M, Nozawa Y. Xanthones induce cell-cycle arrest and apoptosis in human colon cancer DLD-1 cells. Bioorg Med Chem 2005; 13: 6064-6069
11 Nakagawa Y, Iinuma M, Naoe T, Nozawa Y, Akao Y. Char-acterized mechanism of alpha-mangostin-induced cell death: caspase-independent apoptosis with release of endonucle-ase-G from mitochondria and increased miR-143 expression in human colorectal cancer DLD-1 cells. Bioorg Med Chem 2007; 15: 5620-5628
12 Suksamrarn S, Komutiban O, Ratananukul P, Chimnoi N, Lartpornmatulee N, Suksamrarn A. Cytotoxic prenylated xanthones from the young fruit of Garcinia mangostana. Chem Pharm Bull (Tokyo) 2006; 54: 301-305
13 Chen CN, Hsieh FJ, Cheng YM, Chang KJ, Lee PH. Expres-sion of inducible nitric oxide synthase and cyclooxygenase-2 in angiogenesis and clinical outcome of human gastric cancer. J Surg Oncol 2006; 94: 226-233
14 Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell 1997; 88: 347-354
15 Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science 1995; 267: 1456-1462
16 Leist M, Jäättelä M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol 2001; 2: 589-598
17 Rudner J, Lepple-Wienhues A, Budach W, Berschauer J, Fried-rich B, Wesselborg S, Schulze-Osthoff K, Belka C. Wild-type, mitochondrial and ER-restricted Bcl-2 inhibit DNA damage-induced apoptosis but do not affect death receptor-induced apoptosis. J Cell Sci 2001; 114: 4161-4172
18 Nencioni A, Lauber K, Grünebach F, Van Parijs L, Denzlinger C, Wesselborg S, Brossart P. Cyclopentenone prostaglandins induce lymphocyte apoptosis by activating the mitochondrial apoptosis pathway independent of external death receptor signaling. J Immunol 2003; 171: 5148-5156
19 Castedo M, Ferri K, Roumier T, Métivier D, Zamzami N, Kroemer G. Quantitation of mitochondrial alterations associ-ated with apoptosis. J Immunol Methods 2002; 265: 39-47
20 Salvesen GS, Dixit VM. Caspase activation: the induced-proximity model. Proc Natl Acad Sci USA 1999; 96: 10964-10967
21 Tanaka M, Suda T, Haze K, Nakamura N, Sato K, Kimura F, Motoyoshi K, Mizuki M, Tagawa S, Ohga S, Hatake K, Drum-mond AH, Nagata S. Fas ligand in human serum. Nat Med 1996; 2: 317-322
22 Siegel RM, Frederiksen JK, Zacharias DA, Chan FK, John-son M, Lynch D, Tsien RY, Lenardo MJ. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science 2000; 288: 2354-2357
23 Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME. Two CD95 (APO-1/Fas) signaling pathways. EMBO J 1998; 17: 1675-1687
24 Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998; 94: 491-501
25 Watanapokasin R, Jarinthanan F, Jerusalmi A, Suksamrarn S, Nakamura Y, Sukseree S, Uthaisang-Tanethpongtamb W, Ratananukul P, Sano T. Potential of xanthones from tropical fruit mangosteen as anti-cancer agents: caspase-dependent apoptosis induction in vitro and in mice. Appl Biochem Bio-technol 2010; 162: 1080-1094
26 Tamatani T, Azuma M, Motegi K, Takamaru N, Kawashima Y, Bando T. Cepharanthin-enhanced radiosensitivity through the inhibition of radiation-induced nuclear factor-kappaB activity in human oral squamous cell carcinoma cells. Int J Oncol 2007; 31: 761-768
27 Nojima H. Cell cycle checkpoints, chromosome stability and the progression of cancer. Hum Cell 1997; 10: 221-230
28 Lopéz L, Villavicencio MA, Albores A, Martínez M, de la Garza J, Meléndez-Zajgla J, Maldonado V. Cupressus lusitan-ica (Cupressaceae) leaf extract induces apoptosis in cancer cells. J Ethnopharmacol 2002; 80: 115-120
29 Diaz C, Schroit AJ. Role of translocases in the generation of phosphatidylserine asymmetry. J Membr Biol 1996; 151: 1-9
30 Earnshaw WC. Nuclear changes in apoptosis. Curr Opin Cell Biol 1995; 7: 337-343
31 Cohen GM. Caspases: the executioners of apoptosis. Biochem J 1997; 326 (Pt 1): 1-16
32 Kantrow SP, Piantadosi CA. Release of cytochrome c from liver mitochondria during permeability transition. Biochem Biophys Res Commun 1997; 232: 669-671
33 Terrones O, Antonsson B, Yamaguchi H, Wang HG, Liu J, Lee RM, Herrmann A, Basañez G. Lipidic pore formation by the concerted action of proapoptotic BAX and tBID. J Biol Chem 2004; 279: 30081-30091
S- Editor Tian L L- Editor O’Neill M E- Editor Zheng XM
Watanapokasin R et al . Colon cancer apoptosis with mangostin

Chemometrics of differentially expressed proteins from colorectal cancer patients
Lay-Chin Yeoh, Saravanan Dharmaraj, Boon-Hui Gooi, Manjit Singh, Lay-Harn Gam
Lay-Chin Yeoh, Lay-Harn Gam, School of Pharmaceutical Sci-ences, Universiti Sains Malaysia, Penang, 11800, MalaysiaSaravanan Dharmaraj, Centre for Drug Research, Universiti Sains Malaysia, Penang, 11800, MalaysiaBoon-Hui Gooi, Manjit Singh, Department of Surgery, Penang General Hospital, Penang, 10990, MalaysiaAuthor contributions: Gam LH conceived the design of the study and edited the manuscript; Yeoh LC carried out the experi-mental work and manuscript writing; Dharmaraj S carried out the statistical analyses; Gooi BH and Singh M provided the colorec-tal cancer specimens and patient information.Supported by Research Universiti Grant, Grant No. 1001/PFAR MASI/815007Correspondence to: Lay-Harn Gam, PhD, School of Phar-maceutical Sciences, Universiti Sains Malaysia, Penang, 11800, Malaysia. [email protected]: +60-4-6533888 Fax: +60-4-6570017Received: August 13, 2010 Revised: September 18, 2010Accepted: September 25, 2010Published online: April 28, 2011
AbstractAIM: To evaluate the usefulness of differentially ex-pressed proteins from colorectal cancer (CRC) tissues for differentiating cancer and normal tissues.
METHODS: A Proteomic approach was used to iden-tify the differentially expressed proteins between CRC and normal tissues. The proteins were extracted using Tris buffer and thiourea lysis buffer (TLB) for extraction of aqueous soluble and membrane-associated proteins, respectively. Chemometrics, namely principal compo-nent analysis (PCA) and linear discriminant analysis (LDA), were used to assess the usefulness of these proteins for identifying the cancerous state of tissues.
RESULTS: Differentially expressed proteins identified were 37 aqueous soluble proteins in Tris extracts and 24 membrane-associated proteins in TLB extracts. Based on the protein spots intensity on 2D-gel images, PCA
by applying an eigenvalue > 1 was successfully used to reduce the number of principal components (PCs) into 12 and seven PCs for Tris and TLB extracts, respectively, and subsequently six PCs, respectively from both the extracts were used for LDA. The LDA classification for Tris extract showed 82.7% of original samples were cor-rectly classified, whereas 82.7% were correctly classified for the cross-validated samples. The LDA for TLB extract showed that 78.8% of original samples and 71.2% of the cross-validated samples were correctly classified.
CONCLUSION: The classification of CRC tissues by PCA and LDA provided a promising distinction between normal and cancer types. These methods can pos-sibly be used for identification of potential biomarkers among the differentially expressed proteins identified.
© 2011 Baishideng. All rights reserved.
Key words: Colorectal cancer; Proteomics; Marker pro-tein; Principal component analysis; Linear discriminant analysis
Peer reviewer: Ki-Baik Hahm, MD, PhD, Professor, Gachon Graduate School of Medicine, Department of Gastroenterology, Lee Gil Ya Cancer and Diabetes Institute, Lab of Translational Medicine, 7-45 Songdo-dong, Yeonsu-gu, Incheon, 406-840, South Korea
Yeoh LC, Dharmaraj S, Gooi BH, Singh M, Gam LH. Chemomet-rics of differentially expressed proteins from colorectal cancer pa-tients. World J Gastroenterol 2011; 17(16): 2096-2103 Available from: URL: http://www.wjgnet.com/1007-9327/full/v17/i16/2096.htm DOI: http://dx.doi.org/10.3748/wjg.v17.i16.2096
INTRODUCTIONProteomic research has made great achievements in bio-marker discovery, especially when incorporated with high-
ORIGINAL ARTICLE
World J Gastroenterol 2011 April 28; 17(16): 2096-2103ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2096
2096 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

Yeoh LC et al . Chemometrics of colorectal cancer biomarkers
throughput analytical tools and technology, for example 2D-PAGE and LC-MS/MS[1]. Two-dimensional gel elec-trophoresis is a fundamental tool for protein analysis to detect alterations in protein expression between control and disease states of cells, which can lead to the discov-ery of various biomarkers that contribute to pathogenesis or carcinogenesis[2]. Biomarkers can be used to discrimi-nate variables for subsequent classification of normal and diseased groups[3]. The complexity of variables generated by mass spectra, microarray and immunohistochemistry often requires advanced statistical techniques or chemo-metrics to evaluate their clinical value.
Multivariate analyses including the dimension reduc-tion method known as principal component analysis (PCA), and classification methods such as linear discrimi-nant analysis (LDA) are often employed in proteomic studies. PCA reduces the number of variables for further data analysis and interpretation while identifying the vari-ables that retain most of the data variance[4]. A principal component (PC) is defined as a new variable to explain the maximum amount of variance in the original data and corresponds to a linear combination of the original variables. PCs are presented orthogonally to each other, which provides a more effective representation of the data than the original variables[2]. LDA is a multivariate technique to classify observations into groups or catego-ries. LDA forms new variables from the original data and identifies the variables that provide the best discrimina-tion between the groups[5].
Djidja et al[6] have used a novel approach that combines matrix-assisted laser desorption ionization-ion mobility separation-mass spectrometry (MALDI-IMS-MS) and PCA-discriminant analysis (PCA-DA) to generate tumor classification models based on pancreatic cancer protein patterns. Furthermore, Kamath et al[7] have used PCA-based k-nearest neighbor analysis to classify normal and cancerous autofluorescence spectra of colonic mucosal tissues. Zwielly et al[8] have investigated the use of Fourier transform infrared microscopy for colon cancer diagnosis. Their model uses PCA to define spectral changes among normal and cancerous human biopsied colon tissues. Ragazzi et al[9] have reported the use of multivariate tech-niques on plasma proteins to diagnose colorectal cancer (CRC). The plasma protein profile generated by MALDI-MS is analyzed by PCA and LDA to discriminate ionic species from normal subjects and CRC patients.
In this study, we carried out the comparison of 2-D images of cancerous and normal colorectal tissues. The differentially expressed proteins from Tris and thiourea lysis buffer (TLB) extractions were respectively tested on a PCA-LDA model to find out the possibility of using protein expression to classify the disease and non-disease tissues of CRC.
MATERIALS AND METHODSTissue specimen collectionMatching pairs of normal colonic mucosa and cancerous colonic tissue (located 10 cm from each other) from 26
CRC patients were collected after surgery at the Penang General Hospital, Penang, Malaysia. The study was ap-proved by the Human Ethical Committee of Universiti Sains Malaysia. Informed written consent was received from all patients before the study was conducted. Prior to surgery, the patients did not receive preoperative neoad-juvant chemotherapy and radiotherapy. The tissues were confirmed as cancerous and normal, respectively, by the hospital’s pathologist. The cancerous tissues were classi-fied using the TNM system. Surgically removed samples were stored at -80℃ until use.
Protein analysisThe method of protein analysis was as described in Yeoh et al[10]. Frozen tissue (250 mg) was rinsed in distilled water to remove cell debris and excess blood. The tissues were homogenized in ice-cold Tris buffer (0.5 g tissue/mL buf-fer) [40 mmol/L Tris and 1 × Protease Inhibitor Cocktail (Sigma, St Louis, MO, USA)] and centrifuged at 12 000 rpm for 15 min at 18℃. The supernatant was recovered and labeled as Tris extract. The pellet was subjected to further extraction using TLB (1 g tissue/1 mL buffer) [8 mol/L urea, 2 mol/L thiourea, 4% (w/v) CHAPS, 0.4% (w/v) carrier ampholytes and 50 mmol/L dithiothreitol] and centrifuged at 12 000 rpm for 15 min at 18℃. The su-pernatant was recovered and labeled as TLB extract. The extracts were subjected to 2D gel separation on 11 cm ReadyStrip™ IPG strip (linear pH 4-7, Bio-Rad, USA) followed by separation on 10% (w/v) PAGE at a constant voltage of 200 V. The gels were stained with Coomassie Blue. The images obtained were analyzed by PDQuest version 7.3 (Bio-Rad). Comparison of the protein ex-pression levels was carried out between cancerous and normal tissues. Differentially expressed proteins were defined as proteins with a spot intensity that was 1.5-fold higher or lower in cancerous tissues when compared to that in the corresponding normal tissues. A differentially expressed protein was defined as upregulated when it was found at greater intensity in cancerous tissue than in the corresponding normal tissue. The downregulated pro-teins were detected at greater intensity in normal tissues than in the corresponding CRC cancerous tissues.
Protein identificationThe differentially expressed proteins were excised from the gel and subjected to in-gel digestion using trypsin and the tryptic peptides were analyzed by LC/MS/MS using an electrospray ionization ion trap mass analyzer (Agilent Technologies, Santa Clara, CA, USA). The MS/MS data were subjected to the MASCOT protein database search engine for protein identification. The identities of a few proteins (dependent on the availability of antibodies) were further confirmed using western blotting.
Statistical analysisThe differential expression of the proteins was tested by the paired Student’s t test that is included in PDQuest, to determine their statistical significance (P < 0.05). For
2097 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

PCA and LDA, the protein spot intensities were exported out from PDQuest and imported into SPSS version 15.0 (Chicago, IL, USA) to perform multivariate analyses. Pro-tein spot intensities were used as variables.
RESULTSThe tissues specimens from each patient were collected in pairs of cancerous and normal tissues. Table 1 shows the details of the tissues used in the analysis. The tissues were subjected to a sequential extraction method to ex-tract aqueous soluble proteins and membrane-associated proteins in two different fractions using Tris and TLB, re-spectively. Tables 2 and 3 show the 37 and 24 differential-ly expressed proteins identified in Tris and TLB extracts, respectively. The average fold change indicates the degree of differentiation in expression levels of the protein in cancerous tissues compared to normal tissues in all the patients tested, where a positive sign indicates a greater expression level in cancerous tissues, whereas a negative sign indicates a greater expression level in normal tis-sues. The MOWSE score refers to the score values given by the MASCOT search. Tables 4 and 5 show the mean intensity of spots and SD, and percentage coefficient of variation (%CV) of spot intensity of differentially ex-
pressed proteins in all patients for Tris and TLB extracts, respectively. An example of the differentially expressed protein, as represented by different intensities of protein spots between normal and cancerous tissues for glutathi-one S-transferase P (GST-P), is shown in Figure 1; the bar chart was plotted according to the intensity of the respec-tive protein spots. GST-P was detected as upregulated in cancerous tissues.
Data analysisThe significance of the expression levels of the differ-entially expressed proteins in both Tris and TLB extracts was analyzed by Student’s t test. After univariate analysis was performed, the normalized intensities of 37 dif-ferentially expressed protein spots in Tris extracts were subjected to PCA. The PCA reduced the original data to 12 PCs based on an eigenvalue of > 1, and these 12 PCs contributed 76.43% of the total data variance of the Tris extract data. Figure 2 shows the 3D PC plot with the x- y- and z-axes representing the first, second and third PC number. The variables that had the highest loadings were those that contributed most to the differentiation of the disease state. Figure 3 shows the scree plot of Tris ex-tracts. Six PCs were chosen and these components con-tributed 53.97% of the total variance of the Tris extract
2098 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 1 Clinicopathological features of 26 colorectal cancer patients involved in study
Patient No. Age (yr) Race Sex pTNM Stage Degree of differentiation Tumor location
1 62 Malay Male pT3N1Mx ⅢB MD Sigmoid colon2 79 Malay Male pT2NoM0 Ⅰ MD Descending colon3 74 Malay Male pT3N0M0 ⅡA MD Ascending colon4 - Malay Male pT3N2Mx ⅢC MD Rectum5 37 Malay Male pT3N0M0 ⅡA MD Transverse colon6 58 Malay Female pT3N0Mx ⅡA MD Recto-sigmoid7 59 Malay Female pT4N2Mx ⅢC MD Ileocecal8 69 Malay Male pT3N0Mx ⅡA MD Sigmoid colon9 63 Malay Female pT3N0Mx ⅡA MD Recto-sigmoid10 84 Chinese Female pT4N0M0 ⅡB MD Rectum11 58 Chinese Male pT3N0Mx ⅡA MD Recto-sigmoid
MD: Moderately differentiated adenocarcinoma.
Normal
Cancer
30 000
25 000
20 000
15 000
10 000
5000
0
Prot
ein
inte
nsity
Normal Cancer
Figure 1 Comparison of protein spot intensity between normal and colorectal cancer tissues for glutathione S-transferase P.
Yeoh LC et al . Chemometrics of colorectal cancer biomarkers

data. Table 6 shows the LDA results for Tris extract pro-teins, where 22 out of 26 original normal tissues, and 21
out of 26 original cancer tissues were correctly classified. In cross-validated samples, 22 out of 26 normal tissues and 21 out of 26 cancer tissues were correctly classified.
2099 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 2 List of proteins found in 2D gel of Tris extracts
Spot No.
Protein name Swissprot No.1
MOWSE score2
MW (Da) pI Sequence coverage (%)
GRAVY Average fold change3
1 Proteasome subunit β type 6 P28072 134 25573 4.80 16 0.034 -2.9672 14-3-3 protein ζ P63104 336 35567 6.97 40 -0.744 11.6593 Tropomyosin α-3C-like protein A6NL28 127 27407 4.71 31 -0.992 44.1834 Rho GDP-dissociation inhibitor 1 P52565 167 23120 5.03 29 -0.700 -7.6075 14-3-3 protein ζ P63104 282 27919 4.73 16 -0.621 4.1276 Tubulin β-2C chain P68371 524 50304 4.83 40 -0.362 -52.1847 Cathepsin B P07858 74 22981 5.20 18 -0.433 33.1498 Rho GDP-dissociation inhibitor 2 P52566 48 22901 5.10 18 -0.799 -10.6259 SEC13 homolog P55735 78 36040 5.22 9 -0.372 6.87310 Hsc70-interacting protein P50502 164 28464 8.92 21 -0.653 20.95911 Apolipoprotein A-I P02647 143 30777 5.56 26 -0.717 -4.47812 Proteasome subunit α type 3 P25788 201 15958 6.82 41 0.008 4.24913 Actin, cytoplasmic 2 P63261 105 26169 5.65 14 -0.156 28.60114 60 kDa heat shock protein P10809 151 61348 5.70 14 -0.074 131.21915 Peroxiredoxin-2 P32119 283 21935 5.67 42 -0.210 1.25016 Guanine nucleotide binding protein subunit β 2 P62879 112 37954 5.60 11 -0.183 -14.44217 F-actin-capping protein subunit β P47756 259 34187 6.02 37 -0.574 33.55418 GST-P P09211 730 23442 5.44 60 -0.131 4.83419 Haptoglobin-related protein P00739 49 39529 6.42 3 -0.308 56.20920 Cathepsin Z Q9UBR2 100 27787 5.48 15 -0.545 -60.76621 F-actin-capping protein subunit β P47756 245 21280 7.93 34 -0.540 13.27822 Actin-related protein 3 P61158 148 47704 5.61 27 -0.271 15.88123 Abhydrolase domain-containing protein 14B Q96IU4 200 25429 6.82 26 -0.023 0.76524 Nucleoside diphosphate kinase A P15531 87 19873 5.42 36 -0.075 73.12025 L-lactate dehydrogenase B chain P07195 228 36928 5.71 14 0.056 3.51326 Fibrinogen β chain P02675 151 56624 8.54 22 -0.758 41.32927 Leukocyte elastase inhibitor P30740 170 42857 5.90 11 -0.249 10.45828 PDI A3 P30101 674 57202 5.98 35 -0.506 7.57929 Gelsolin P06396 238 86103 5.90 20 -0.415 -11.91730 Heat shock 27 kDa protein P04792 256 22840 5.98 47 -0.567 -1.50831 DJ-1 protein Q99497 122 20079 6.33 54 0.004 4.98132 Fibrinogen β chain P02675 75 56624 8.54 22 -0.758 -72.72233 Selenium-binding protein 1 Q13228 502 52971 5.93 21 -0.254 -26.54434 Selenium-binding protein 1 Q13228 592 52938 5.93 30 -0.254 27.40335 Selenium-binding protein 1 Q13228 979 52938 5.93 37 -0.254 -1.88736 Leukotriene A-4 hydrolase P09960 215 69792 5.80 22 -0.259 29.75937 Proteasome subunit α type 6 P60900 71 20988 8.57 39 -0.247 0.768
1Protein accession number at SwissProt at http://www.expasy.org/uniprot; 2MOWSE score from MASCOT protein database search at http://www.matrixscience.com, where score > 41 is statistically significant (P < 0.05); 3Average ratio of the spot intensity in normal mucosa over tumor tissue (negative variation or decrease) or tumor tissue over normal tissue (positive variation or increase). GST-P: Glutathione S-transferase P; PDI: Protein disulfide isomerase.
4.0
3.0
2.0
1.0
0.0
-1.0
-2.0
PC2
3.0 2.0 1.0 0.0 -1.0 -2.0 -3.0
-3.0
-2.0
-1.0
0.0
1.0
2.0
3.0 PC3PC1
Type
CancerNormal
Figure 2 Principal component plot of Tris proteins.
7
6
5
4
3
2
1
0
Eige
nval
ue
1 5 9 13 17 21 25 29 33 37 Principal component
Scree plot
Figure 3 Scree plot showing principal components and their eigenvalues in Tris extracts.
Yeoh LC et al . Chemometrics of colorectal cancer biomarkers

Both original and cross-validation samples had an average 82.7% correct classification.
Figure 4 shows the 3D view of the PCs plot for the TLB extract. PCA reduced the original data of the TLB extract to seven PCs based on an eigenvalue one of > 1, and the seven PCs accounted for 72.46% of the total data variance. The 3D view indicates that tissues can be grouped according to CRC disease state. Figure 5 shows the scree plot of the TLB extracts. Six PCs were cho-sen based on the slope of scree plot, which contributed
67.61% of the total data variance of TLB extracts. Table 7 shows the LDA results of TLB extracts, where 22 out of 26 original normal tissues, and 19 out of 26 original can-cerous tissues were correctly classified. In cross-validated samples, 21 out of 26 normal tissues and 16 out of 26 cancerous tissues were correctly classified. The average percentages of correct classification for original and cross-validation samples were 78.8% and 71.2%, respectively.
DISCUSSIONThe expression levels of the differentially expressed pro-tein between colorectal cancerous and normal tissues were
2100 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 3 List of proteins found in 2D gel in thiourea lysis buffer extracts
Spot No.
Protein name SwissProt No.1
MOWSE score2
MW (Da) pI Sequence coverage (%)
GRAVY Average fold change3
1 Tropomyosin α-4 chain P67936 139 28506 4.67 33 -1.033 -51.1512 Putative tropomyosin α-3-chain-like protein A6NL28 53 27407 4.71 25 -0.992 4.9223 GC1q-R, mitochondrial Q07021 123 31768 4.74 20 -0.461 -3.3334 Calreticulin P27797 73 47092 4.30 11 -1.191 1.3945 Prohibitin P35232 421 29890 5.57 41 0.024 0.0326 Heat shock 70 kDa protein P11021 775 72488 5.07 42 -0.487 -32.9407 Tubulin β-2C chain P68371 299 48142 4.70 25 -0.347 -9.0608 PDI P07237 266 57510 4.82 42 -0.450 -1.5159 ATP synthase subunit β, mitochondrial P06576 1096 56559 5.26 43 0.018 -15.66110 ATP synthase D chain O75947 117 18406 5.22 32 -0.569 -5.12911 Chloride intracellular channel protein 1 O00299 299 27123 5.09 30 -0.293 20.28812 Tubulin α-1 chain Q71U36 61 50800 4.94 6 -0.229 -30.29113 Apolipoprotein A-I P02647 129 28078 5.27 37 -0.840 78.13514 Actin, cytoplasmic 2 P63261 52 42009 5.31 4 -0.205 -26.71615 Actin, aortic smooth muscle P62736 261 42154 5.23 21 -0.233 46.18116 Stomatin-like protein 2 Q9UJZ1 151 38644 6.88 28 -0.161 -29.70917 60 kDa heat shock protein, mitochondrial P10809 451 61386 5.70 28 -0.074 14.02318 Triosephosphate isomerase P60174 167 26828 6.51 24 -0.126 16.75719 Annexin A5 P08758 195 35994 4.94 39 -0.330 -2.01920 Cytochrome b-c1 complex subunit 1, mitochondrial P31930 96 53342 5.94 18 -0.141 13.15121 Annexin A3 P12429 140 36396 5.63 22 -0.430 31.24422 Annexin A4 P09525 165 35983 5.85 33 -0.447 11.89023 α-enolase P06733 143 47385 6.99 12 -0.226 85.96024 Lamin-A/C P02545 198 65192 6.40 25 -0.947 -3.378
1Protein accession number as SwissProt at http://www.expasy.org/uniprot; 2MOWSE score from MASCOT protein database search at http://www.matrixscience.com, where score > 41 is statistically significant (P < 0.05); 3Average ratio of the spot intensity in normal mucosa over tumor tissue (negative variation or decrease) or tumor tissue over normal tissue (positive variation or increase). PDI: Protein disulfide isomerase.
4.0
3.0
2.0
1.0
0.0
-1.0
-2.0
PC2
3.0 2.0 1.0 0.0 -1.0 -2.0 -3.0
PC3
-3.0
-2.0
-1.0
0.0
1.0
2.0
3.0
PC1
Type
CancerNormal
Figure 4 Principal component plot of thiourea lysis buffer proteins.
Eige
nval
ue
1 3 5 7 9 12 15 18 21 24 Principal component
Scree plot
Figure 5 Scree plot showing principal components and their eigenvalues in thiourea lysis buffer extracts.
5
4
3
2
1
0
Yeoh LC et al . Chemometrics of colorectal cancer biomarkers
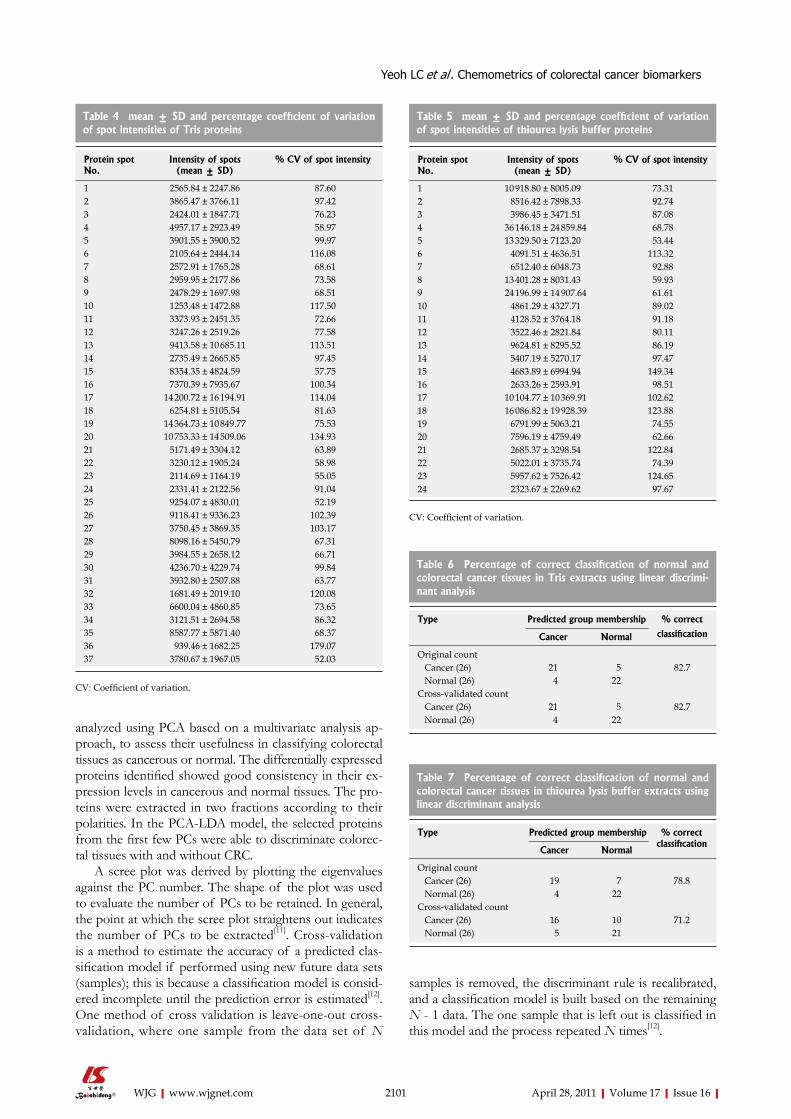
analyzed using PCA based on a multivariate analysis ap-proach, to assess their usefulness in classifying colorectal tissues as cancerous or normal. The differentially expressed proteins identified showed good consistency in their ex-pression levels in cancerous and normal tissues. The pro-teins were extracted in two fractions according to their polarities. In the PCA-LDA model, the selected proteins from the first few PCs were able to discriminate colorec-tal tissues with and without CRC.
A scree plot was derived by plotting the eigenvalues against the PC number. The shape of the plot was used to evaluate the number of PCs to be retained. In general, the point at which the scree plot straightens out indicates the number of PCs to be extracted[11]. Cross-validation is a method to estimate the accuracy of a predicted clas-sification model if performed using new future data sets (samples); this is because a classification model is consid-ered incomplete until the prediction error is estimated[12]. One method of cross validation is leave-one-out cross-validation, where one sample from the data set of N
samples is removed, the discriminant rule is recalibrated, and a classification model is built based on the remaining N - 1 data. The one sample that is left out is classified in this model and the process repeated N times[12].
2101 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 4 mean ± SD and percentage coefficient of variation of spot intensities of Tris proteins
Protein spot No.
Intensity of spots (mean ± SD)
% CV of spot intensity
1 2565.84 ± 2247.86 87.602 3865.47 ± 3766.11 97.423 2424.01 ± 1847.71 76.234 4957.17 ± 2923.49 58.975 3901.55 ± 3900.52 99.976 2105.64 ± 2444.14 116.087 2572.91 ± 1765.28 68.618 2959.95 ± 2177.86 73.589 2478.29 ± 1697.98 68.5110 1253.48 ± 1472.88 117.5011 3373.93 ± 2451.35 72.6612 3247.26 ± 2519.26 77.5813 9413.58 ± 10 685.11 113.5114 2735.49 ± 2665.85 97.4515 8354.35 ± 4824.59 57.7516 7370.39 ± 7935.67 100.3417 14 200.72 ± 16 194.91 114.0418 6254.81 ± 5105.54 81.6319 14 364.73 ± 10 849.77 75.5320 10 753.33 ± 14 509.06 134.9321 5171.49 ± 3304.12 63.8922 3230.12 ± 1905.24 58.9823 2114.69 ± 1164.19 55.0524 2331.41 ± 2122.56 91.0425 9254.07 ± 4830.01 52.1926 9118.41 ± 9336.23 102.3927 3750.45 ± 3869.35 103.1728 8098.16 ± 5450.79 67.3129 3984.55 ± 2658.12 66.7130 4236.70 ± 4229.74 99.8431 3932.80 ± 2507.88 63.7732 1681.49 ± 2019.10 120.0833 6600.04 ± 4860.85 73.6534 3121.51 ± 2694.58 86.3235 8587.77 ± 5871.40 68.3736 939.46 ± 1682.25 179.0737 3780.67 ± 1967.05 52.03
CV: Coefficient of variation.
Table 5 mean ± SD and percentage coefficient of variation of spot intensities of thiourea lysis buffer proteins
Protein spot No.
Intensity of spots (mean ± SD)
% CV of spot intensity
1 10 918.80 ± 8005.09 73.312 8516.42 ± 7898.33 92.743 3986.45 ± 3471.51 87.084 36 146.18 ± 24 859.84 68.785 13 329.50 ± 7123.20 53.446 4091.51 ± 4636.51 113.327 6512.40 ± 6048.73 92.888 13 401.28 ± 8031.43 59.939 24 196.99 ± 14 907.64 61.6110 4861.29 ± 4327.71 89.0211 4128.52 ± 3764.18 91.1812 3522.46 ± 2821.84 80.1113 9624.81 ± 8295.52 86.1914 5407.19 ± 5270.17 97.4715 4683.89 ± 6994.94 149.3416 2633.26 ± 2593.91 98.5117 10 104.77 ± 10 369.91 102.6218 16 086.82 ± 19 928.39 123.8819 6791.99 ± 5063.21 74.5520 7596.19 ± 4759.49 62.6621 2685.37 ± 3298.54 122.8422 5022.01 ± 3735.74 74.3923 5957.62 ± 7526.42 124.6524 2323.67 ± 2269.62 97.67
CV: Coefficient of variation.
Table 6 Percentage of correct classification of normal and colorectal cancer tissues in Tris extracts using linear discrimi-nant analysis
Type Predicted group membership % correct
classification Cancer Normal
Original count Cancer (26) 21 5 82.7 Normal (26) 4 22Cross-validated count Cancer (26) 21 5 82.7 Normal (26) 4 22
Table 7 Percentage of correct classification of normal and colorectal cancer tissues in thiourea lysis buffer extracts using linear discriminant analysis
Type Predicted group membership % correct classification
Cancer Normal
Original count Cancer (26) 19 7 78.8 Normal (26) 4 22Cross-validated count Cancer (26) 16 10 71.2 Normal (26) 5 21
Yeoh LC et al . Chemometrics of colorectal cancer biomarkers

2102 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
PCA and LDA results from Tris extract indicated that six out of 37 proteins were reliable to determine the tis-sues with CRC. The proteins comprised five upregulated proteins, namely GST-P, tropomyosin α-3C-like protein, F-actin capping protein subunit β, selenium binding pro-tein 1 and DJ-1 protein, and one downregulated protein, namely, proteasome subunit β type 6. DJ-1 protein and GST-P contributed the most to the first PC based on the weight of their loadings. This was followed by the tropomyosin α-3C-like protein and proteasome subunit β type 6 that contributed to the second PC, while F-actin capping protein subunit β and selenium binding protein 1 contributed to the third PC. The initial PCA reduced the original data and therefore enabled LDA to be carried out because LDA is sensitive to the number of variables. In LDA, the six PCs chosen were shown to be capable of predicting whether the tissues were with or without CRC. Two-way validation by using original and cross-validation analyses was applied to validate the state of the tissues, where the cancerous and normal tissues were classified correctly at 82.7% for both original and cross-validation samples.
Two proteins that contributed most to PC1 in Tris ex-tract were DJ-1 and GST. DJ-1 is a putative oncoprotein that is able to transform cells with H-Ras[13]. Overexpres-sion of DJ-1 activates protein kinase B, which subsequent-ly increases cell survival. Furthermore, increased DJ-1 expression also activates Nrf2 (nuclear factor erythroid 2-related factor), which in turn increases expression of antioxidant enzymes that confer a survival advantage to tumor cells[14]. Upregulation of DJ-1 protein in esopha-geal squamous cell carcinoma is correlated with lymph node metastasis[15]. Although there is no reported role of DJ-1 in CRC, its upregulation in CRC is undeniable, and we have shown that its expression can be used to dis-criminate between CRC cancerous and normal tissues.
GST catalyzes the conjugation of reduced glutathione to electrophiles[16]. GST functions to remove peroxides from endogenous compounds such as lipids and DNA[17]. Overexpression of GST-P1 in CRC may be involved in cell proliferation, differentiation and apoptosis[18]. GST-P1 is overexpressed in liver cancer cells[19].
In TLB extract, six of the 24 differentially expressed proteins identified were found to be useful in discrimi-nating CRC cancerous from normal tissues. These pro-teins were protein disulfide isomerase (PDI), comple-ment component 1 Q subcomponent-binding protein (GC1q-R), chloride intracellular channel protein 1, trios-ephosphate isomerase, annexin A5 and actin cytoplasmic 2. All the proteins were downregulated in TLB extracts, except chloride intracellular channel protein 1 and trios-ephosphate isomerase. PDI and GC1q-R contributed the most to the first PC based on the weight of their loadings. This was followed by the chloride intracellular channel protein 1 and triosephosphate isomerase that contributed the most to the second PC, while annexin A5 and actin cytoplasmic 2 contributed most to the third PC. In LDA, the six PCs that explained 67.61% of the total variance were able to distinguish CRC cancerous from
normal tissues. The leave-one-out cross-validation ob-tained 71.2% correct classification of normal and cancer-ous tissues. The value for original grouped samples was higher with 78.8% correct classification.
Two proteins that contributed most to PC1 in TLB extracts were PDI and GC1q-R. PDI catalyzes the for-mation and breakage of disulfide bonds between two cysteine residues[20]. PDI regulates cell transformation and intracellular and extracellular redox activities via its reductase activity[21]. PDI regulates STAT3 signaling and proliferation, which is thought to induce malignancy[22]. PDI is upregulated in CRC cell lines and its upregulation is correlated with cancer cell differentiation[23,24].
GC1q-R is a cell surface glycoprotein, which binds to the globular heads of C1q molecules[25]. C1q molecules bind to a variety of cells such as B cells, monocytes, mac-rophages, endothelial and smooth muscle cells[26]. C1q elicits responses such as phagocytosis in monocytes and activation of tumor cytotoxicity of macrophages[27,28]. GC1q-R is overexpressed in colon cancer cells and may be involved in tumor metastasis. However, PDI and GC1q-R were downregulated when using average fold change to determine their expression levels.
Proteins are the expression components that regulate cell activity. Differential expression of proteins is ex-pected upon transformation of normal cells to cancerous cells. These differentially expressed proteins are useful in diagnosis and prognosis of the disease. In the pres-ent study, the specimens used in the analysis comprised tissues from female and male patients who were diag-nosed with various stages, grades and locations of CRC. Regardless of the sex of the patients and pathological specification of the tissues, we showed that the differ-entially expressed protein identified from 2D protein profiles of cancerous and normal tissues could be used to separate and classify normal and cancerous tissues by combining PCA and LDA. The data reduction tech-nique of PCA was sufficient to provide a classification of tissues according to CRC disease state. These statisti-cal models simplify the data management through the reduced dimensionality of protein spots from the 2D gel images. Therefore, multivariate analysis of differentially expressed proteins identified from cancerous and normal tissues may be used as a tool for diagnosis and prognosis of CRC disease state.
ACKNOWLEDGMENTSWe want to express our appreciation to Universiti Sains Malaysia for provided the grant under RU funding to conduct this project. We would also like to thank Na-tional Institute of Pharmaceutical and Neutraceutical for their generosity in allowing us to conduct the LC/MS/MS experiments.
COMMENTSBackgroundColorectal cancer (CRC) is one of the leading causes of death worldwide. Dif-
Yeoh LC et al . Chemometrics of colorectal cancer biomarkers
COMMENTS

2103 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
ferentially expressed proteins between cancerous and normal colonic tissues were identified using 2D gel separation followed by LC/MS/MS analysis. The protein spot intensities of the 2D gel images were analyzed using principal com-ponent analysis (PCA) and linear discriminant analysis (LDA) for their possible use in classification of disease state.Research frontiersMultivariate analyses, including the dimension reduction method known as PCA and classification methods such as LDA, are used in cancer proteomic studies to identify the protein variables that provide the best discrimination between the cancerous and normal tissues.Innovations and breakthroughsThe authors used sequential protein extraction to extract aqueous soluble and membrane-associated proteins from colorectal tissues. Differentially expressed proteins were analyzed using a combination of PCA and LDA to determine their usability in differentiating normal and cancerous colonic tissues. Using this method, the authors successfully classified the tissues according to their respective types. DJ-1 protein and glutathione S transferase P1 of the aqueous soluble proteins, protein disulfide isomerase and complement component 1 Q subcomponent-binding protein of the membrane-associated proteins gave the best classification of the tissues.ApplicationsThe identified biomarkers may be used for the diagnosis and prognosis of CRC.TerminologyChemometrics is defined as the information aspects of complex biological and chemical systems. Chemometrics utilize mathematical, statistical or formal logic-based methods to extract chemical information, which in this case, is for biomarker discovery.Peer reviewThis study investigated the use of PCA and LDA of differential protein expres-sion between normal and cancerous tissues for classification of disease state. The method gave good classification of cancerous and normal colonic tissues.
REFERENCES1 Cowan ML, Vera J. Proteomics: advances in biomarker dis-
covery. Expert Rev Proteomics 2008; 5: 21-232 Rodríguez-Piñeiro AM, Rodríguez-Berrocal FJ, Páez de la
Cadena M. Improvements in the search for potential biomark-ers by proteomics: application of principal component and discriminant analyses for two-dimensional maps evaluation. J Chromatogr B Analyt Technol Biomed Life Sci 2007; 849: 251-260
3 Hilario M, Kalousis A. Approaches to dimensionality reduc-tion in proteomic biomarker studies. Brief Bioinform 2008; 9: 102-118
4 Karson MJ. Multivariate statistical methods: An introduction. Iowa: Iowa State University Press, 1982: 159, 191
5 Giri NC. Multivariate statistical analysis. New York: Marcel Dekker, 1996: 293-294
6 Djidja MC, Claude E, Snel MF, Francese S, Scriven P, Caro-lan V, Clench MR. Novel molecular tumour classification using MALDI-mass spectrometry imaging of tissue micro-array. Anal Bioanal Chem 2010; 397: 587-601
7 Kamath SD, Mahato KK. Principal component analysis (PCA)-based k-nearest neighbor (k-NN) analysis of colonic mucosal tissue fluorescence spectra. Photomed Laser Surg 2009; 27: 659-668
8 Zwielly A, Mordechai S, Sinielnikov I, Salman A, Bogomolny E, Argov S. Advanced statistical techniques applied to com-prehensive FTIR spectra on human colonic tissues. Med Phys 2010; 37: 1047-1055
9 Ragazzi E, Pucciarelli S, Seraglia R, Molin L, Agostini M, Lise M, Traldi P, Nitti D. Multivariate analysis approach to the plasma protein profile of patients with advanced colorectal cancer. J Mass Spectrom 2006; 41: 1546-1553
10 Yeoh LC, Loh CK, Gooi BH, Singh M, Gam LH. Hydrophobic protein in colorectal cancer in relation to tumor stages and grades. World J Gastroenterol 2010; 16: 2754-2763
11 McGarigal K, Cushman S, Stafford S. Ordination: Principal component analysis. In: McGarigal, editor. Multivariate sta-tistics for wildlife and ecology research. New York: Springer-Verlag, 2000: 41-42
12 Dziuda DM. Biomarker discovery and classification. In: Dziuda, editor. Data mining for genomics and proteomics: Analysis of gene and protein expression data. Hoboken: John Wiley and Sons, 2010: 110-112
13 Nagakubo D, Taira T, Kitaura H, Ikeda M, Tamai K, Iguchi-Ariga SM, Ariga H. DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem Biophys Res Commun 1997; 231: 509-513
14 Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP. DJ-1, a cancer- and Parkinson's disease-associated protein, stabi-lizes the antioxidant transcriptional master regulator Nrf2. Proc Natl Acad Sci USA 2006; 103: 15091-15096
15 Yuen HF, Chan YP, Law S, Srivastava G, El-Tanani M, Mak TW, Chan KW. DJ-1 could predict worse prognosis in esoph-ageal squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev 2008; 17: 3593-3602
16 Mannervik B, Danielson UH. Glutathione transferases--structure and catalytic activity. CRC Crit Rev Biochem 1988; 23: 283-337
17 Park HJ, Lee KS, Choo SH, Kong KH. Functional studies of cysteine residues in human glutathione S-transferase P1-1 by site-directed mutagenesis. Bull Korean Chem Soc 2001; 22: 77-83
18 Lo HW, Antoun GR, Ali-Osman F. The human glutathione S-transferase P1 protein is phosphorylated and its metabolic function enhanced by the Ser/Thr protein kinases, cAMP-de-pendent protein kinase and protein kinase C, in glioblastoma cells. Cancer Res 2004; 64: 9131-9138
19 Tsuchida S, Sato K. Glutathione transferases and cancer. Crit Rev Biochem Mol Biol 1992; 27: 337-384
20 Wilkinson B, Gilbert HF. Protein disulfide isomerase. Bio-chim Biophys Acta 2004; 1699: 35-44
21 Hirano N, Shibasaki F, Sakai R, Tanaka T, Nishida J, Yazaki Y, Takenawa T, Hirai H. Molecular cloning of the human glucose-regulated protein ERp57/GRP58, a thiol-dependent reductase. Identification of its secretory form and inducible expression by the oncogenic transformation. Eur J Biochem 1995; 234: 336-342
22 Coe H, Jung J, Groenendyk J, Prins D, Michalak M. ERp57 modulates STAT3 signaling from the lumen of the endoplas-mic reticulum. J Biol Chem 2010; 285: 6725-6738
23 Katayama M, Nakano H, Ishiuchi A, Wu W, Oshima R, Sakurai J, Nishikawa H, Yamaguchi S, Otsubo T. Protein pattern difference in the colon cancer cell lines examined by two-dimensional differential in-gel electrophoresis and mass spectrometry. Surg Today 2006; 36: 1085-1093
24 Stierum R, Gaspari M, Dommels Y, Ouatas T, Pluk H, Jes-persen S, Vogels J, Verhoeckx K, Groten J, van Ommen B. Proteome analysis reveals novel proteins associated with pro-liferation and differentiation of the colorectal cancer cell line Caco-2. Biochim Biophys Acta 2003; 1650: 73-91
25 Ghebrehiwet B, Lim BL, Peerschke EI, Willis AC, Reid KB. Isolation, cDNA cloning, and overexpression of a 33-kD cell surface glycoprotein that binds to the globular "heads" of C1q. J Exp Med 1994; 179: 1809-1821
26 Ghebrehiwet B. Functions associated with the C1q receptor. Behring Inst Mitt 1989; 204-215
27 Bobak DA, Frank MM, Tenner AJ. C1q acts synergistically with phorbol dibutyrate to activate CR1-mediated phagocy-tosis by human mononuclear phagocytes. Eur J Immunol 1988; 18: 2001-2007
28 Leu RW, Zhou AQ, Shannon BJ, Herriott MJ. Inhibitors of C1q biosynthesis suppress activation of murine macrophages for both antibody-independent and antibody-dependent tu-mor cytotoxicity. J Immunol 1990; 144: 2281-2286
S- Editor Sun H L- Editor Kerr C E- Editor Zheng XM
Yeoh LC et al . Chemometrics of colorectal cancer biomarkers

Dietary treatment of colic caused by excess gas in infants: Biochemical evidence
Dámaso Infante, Oscar Segarra, Bernard Le Luyer
Dámaso Infante, Oscar Segarra, Unit of Gastroenterology, Hepatology and Nutrition, Children’s Hospital Vall d’Hebron, Autonomous University, Barcelona 08035, SpainBernard Le Luyer, Medical Director UP International I.C.C Route de Pré-Bois, CH-1215, Geneva, SwitzerlandAuthor contributions: Infante D designed, performed the re-search, analyzed the data and wrote the paper; Segarra O per-formed the research; Luyer BL revised the data and wrote the paper.Supported by United Pharmaceuticals SAS, 55 Avenue Hoche, 75008 Paris, FranceCorrespondence to: Dámaso Infante, MD, Chief, Unit of Gastroenterology, Hepatology and Nutrition, Children’s Hospi-tal Vall d’Hebron, Autonomous University, Pg Vall d´Hebro nº 119-129, Barcelona 08035, Spain. [email protected]: +34-93-4893000 Fax: +34-93-4174892Received: June 22, 2010 Revised: September 9, 2010Accepted: September 16, 2010Published online: April 28, 2011
AbstractAIM: To evaluate the impact of feeding colicky infants with an adapted formula on the hydrogen breath test and clinical symptoms.
METHODS: Hydrogen expiration was measured by SC MicroLyzer gas chromatography at inclusion and 15 d after treatment with an adapted low-lactose formula in 20 colicky infants.
RESULTS: All babies were symptomatic: 85% with excess gas, 75% with abnormal feeding pattern, and 85% with excessive crying. The hydrogen breath test at inclusion was abnormal: 35 ± 3.1 ppm. After 15 d feeding with an adapted low-lactose formula, crying and flatulence decreased in 85% of patients (P < 0.001). For infants in whom no decrease of gas was reported, crying was still reduced (P < 0.01). Moreover, the feed-ing pattern was improved in 50% of infants when it was initially considered as abnormal. Finally, the hydrogen
breath test decreased significantly (10 ± 2.5 ppm, P < 0.01).
CONCLUSION: This study showed an association be-tween clinical improvement and evidence of decreased levels of hydrogen when the infants were fed with a specially designed, low-lactose formula.
© 2011 Baishideng. All rights reserved.
Key words: Infants; Colic; Lactose; Hydrogen breath test
Peer reviewers: Guang-Yin Xu, MD, PhD, Assistant Professor, Division of Gastroenterology, Department of Internal Medicine, University of Texas Medical Branch, Galveston, TX 77555-0655, United States; Loes van Keimpema, MSc, PhD, Deparment of Gastroenterology and Hepatology, Radboud University Ni-jmegen Medical Center, PO Box 9101, 6500 HB, Nijmegen, The Netherlands
Infante D, Segarra O, Luyer BL. Dietary treatment of colic caused by excess gas in infants: Biochemical evidence. World J Gastroenterol 2011; 17(16): 2104-2108 Available from: URL: http://www.wjgnet.com/1007-9327/full/v17/i16/2104.htm DOI: http://dx.doi.org/10.3748/wjg.v17.i16.2104
INTRODUCTIONInfant colic continues to be one of the most disconcert-ing issues in pediatric medicine. Wessel in 1954 estab-lished the famous “rule of three” criteria: “a symptomatic disorder characterized by paroxysms of fussing, agitation or crying, lasting more than 3 h a day and occurring more than 3 d a week for at least 3 wk”[1]. These criteria are now outdated and as there is no clear definition for the condition, studies on its causes, prevalence and treatment inevitably include a heterogeneous group of infants with different problems[2-4]. The definition of “excessive in-fant crying syndrome”[5] is preferred, although the word
BRIEF ARTICLE
World J Gastroenterol 2011 April 28; 17(16): 2104-2108ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2104
2104 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

Infante D et al . Dietary treatment of functional colic
“colic” is still used and can be defined as an acronym standing for “Cause Obscure Lengthy Infant Crying”. It is characterized by paroxysms of excessive and inconsol-able crying. The infant might present with a tense abdo-men, flex the leg to the abdomen, and appear flushed. Symptoms typically start around the second week of life, peak around 3-6 wk and resolve by 3 mo[4,6]. This term now includes digestive disorders such as constipation, gastroesophageal reflux, allergy to cow milk proteins, and excess intestinal gas due to malabsorption of lactose, and its prevalence has been established[6,7].
These disorders, although not serious from a medical point of view, can be very distressing for the baby and his/her family, and can be associated with symptoms of depression in the mother in the first months after birth[8]. For most of these disorders, some dietary solutions have been developed in compliance with the international expert group coordinated by ESPGHAN (European Society for Paediatric Gastroenterology, Hepatology and Nutrition)[9].
The objective of this study was to provide clinical and biochemical evidence of the efficacy of an adapted formula in colic caused by excessive gas, due to physi-ological hypolactasia, which led to excessive infant crying syndrome.
MATERIALS AND METHODSWe included formula-fed infants who were referred to their pediatrician and/or the Unit of Gastroenterology, Hepatol-ogy and Nutrition, Children’s Hospital, Vall d´Hebron, Bar-celona, Spain because of excessive crying reported by their parents. Infants with vomiting/regurgitation, constipation or cutaneous rash were excluded. When the parents reported “rumbling tummies” excessive flatus, and frothy stools, we considered it as suggestive of carbohydrate malabsorption. Other symptoms were taken into account such as feeding difficulties (e.g. crying during meals) and excessive crying time per 24 h. The hydrogen breath test was performed in case of suspected excess intestinal gas, or infant crying for > 3 h/d to diagnose possible reduced lactose absorption.
Twenty consecutive infants with positive hydrogen breath test were included in this study. All infants were fed with an adapted formula, from various brands having a lactose content of 7 g/100 mL equivalent to 10.4 g of lac-tose/100 kcal (caloric density of formulas 67 kcal/100 mL).
All of them were eutrophic, Caucasian, healthy full term infants whose growth and development had been normal since birth. Infants were 3.7 wk old on average (range: 1.7-6 wk). They were switched to an adapted for-mula, Novalac AC (United Pharmaceuticals SA, Paris, France) during the intervention period (Table 1).
Duration of crying, intestinal bloating and behavior during feeding (such as interruption of the meal due to crying) were evaluated through a questionnaire at baseline and during a second consultation 15 d later. A second hy-drogen breath test was also performed during the second visit in these 20 infants.
The persons legally responsible for the children were invited to give their informed consent for participation in the study. The study was approved by the local ethics committee.
Method of hydrogen breath testBreath samples were collected before the start of feeding, as well as at 90, 120 and 180 min after the beginning of the meal. The feeding schedule was not modified. Samples were taken using face masks with a two-way valve and a pot system. Breath samples were collected in duplicate. The samples were injected in an SC MicroLyzer gas chro-matograph (Quinton Instrument Company, USA) for simultaneous detection of hydrogen, CO2 and methane[10]. This model had an internal gas chromatographic column through which the sample was flushed. Material in the column retarded components which might have interfered with the measurement, and hydrogen thus appeared by itself at the detector and was accurately measured. The gas was inserted in a SvRite-10 cartridge before being analyzed. Prior calibration was performed with a standard gas that contained 102 ppm hydrogen, 23 ppm methane, and 5% CO2. The results were expressed as parts per mil-lion (ppm). The result was considered to be positive when there was an increase > 20 ppm; the normal level for methane being < 10 ppm applying a correction factor for CO2. The hydrogen breath data were analyzed as a nested factorial design by analysis of variance. For each infant, the maximum hydrogen value was defined as being the highest mean of the hydrogen breath test, because the ac-tual time from the beginning of the feeding did not have a consistent impact on the value.
Statistical analysisHydrogen values were expressed as mean ± SE. The val-ues of expired hydrogen were compared using Student’s t test. A χ2 test was used for categorical variables when both groups were compared. P < 0.05 was considered significant. The statistical analysis was performed using SPSS version 18.0 (SPSS Inc., Chicago, IL, USA).
RESULTSTable 2 shows the clinical evolution (data reported by
2105 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 1 Composition of Novalac AC®
Average composition for 100 mL
Energy (kcal) 65.7Proteins (g) 1.4Carbohydrates (g) 7.5Lactose (g) 3.0Maltodextrin (g) 4.5Fat (g) 3.3C18:2 (mg) 610.0C18:3 (mg) 59.8Calcium (mg) 50.7Phosphorus (mg) 31.2

relatives) of infants who were included because of cry-ing secondary to excess gas. The duration of crying was reduced in all infants regardless of the initial duration, and 85% cried for < 1 h/d. Moreover, of the 85% of infants reported with excessive gas at inclusion, only 25% still had excessive gas at the end of the study period (P < 0.001). Four out of the five infants who were described by their parents as having excessive gas were crying for < 1 h/d. The proportion of infants for whom feeding was described as abnormal decreased from 75% at inclusion to 30% after 2 wk feeding with Novalac AC (P < 0.01). The level of hydrogen expired (biochemical evidence) de-creased from 35 ± 3.1 ppm at inclusion to 10 ± 2.5 ppm (P < 0.01) after 2 wk feeding with Novalac AC. The methane excretion was unchanged (Figure 1).
DISCUSSIONMany authors tend to include digestive disorders in the etiology of “excessive infant crying syndrome” (COLIC), and recommend dietary treatment of this functional disorder[6,7]. Therefore, this study focused on transient lactose intolerance in infants with colic[11,12] and we do not discuss other etiologies such as food hypersensitivity, which has been reviewed by Hill et al[13].
As a result of the failure to break down all the lactose ingested, part of it enters the large bowel where it be-comes a substrate for lactobacilli and bifidobacteria. This reaction (fermentation) produces hydrogen and other sub-stances. Therefore, subsequent increases in breath hydro-gen are accepted as an indirect sign of hypolactasia[6,11,12,14].
Miller et al[12] have shown that the expired hydrogen in 65 infants with colic was significantly higher than in control subjects (29 ppm vs 11 ppm, P < 0.001). Sixty-two percent of children with colic had expired hydrogen > 20 ppm, but 38% of control subjects also had abnormal levels of expired hydrogen. Similar results have been shown by Moore et al[14], with 80% of colicky infants having positive expired hydrogen vs 36% in normal infants. The assign-ment of infants to the colicky or control groups accord-ing to the mothers’ perception of the duration of crying might not distinguish clearly colicky or non-colicky in-fants. This might explain the dissociation between clinical and hydrogen values reported by some studies[12-14]. More-over, among infants with positive expired hydrogen, indi-vidual response to abdominal distension might vary, and this is why some infants cry more than others. Therefore, the issue of personal susceptibility to stimuli and a lengthy crying response varies considerably between infants.
Our baseline data are close to those reported by Miller et al[12]. In this pilot study, we assessed the efficacy of the studied formula only in children younger than 6 wk who had a positive expired hydrogen test and clearly defined symptoms.
Mature breast milk contains 7 g lactose per 100 mL (10.2 g/100 kcal), as do several standard infants formulas. In the first few weeks, infants present a physiological or functional lactase deficiency that limits the amount of lactose that they can digest[15]. Twenty-seven point five percent of neonates present a positive hydrogen breath test (i.e. > 20 ppm) after lactose ingestion, regardless of sex or gestational age, with only weight playing a signifi-cant role: hydrogen expired is more important in infants with a birth weight < 2.5 kg than in those > 2.5 kg[16]. One study[17] has found that infants who weigh < 1.8 kg, fed with a low-lactose formula (< 5% lactose) ingested more calories, finished their bottles sooner, presented less milk residue in the stomach, and required feeding more often and with less interruptions than those fed with a formula with normal lactose content (7 g/100 mL). The study of Douwes et al[18] has indicated that abnormal ex-pired hydrogen is more frequent in breast-fed infants or those fed with a 7.5% lactose formula, than in infants fed with a 1% lactose formula.
Furthermore, the concentration of hydrogen in the breath of healthy infants increases from birth to reach its highest level in the second month of life, and declines to low concentrations by 3-4 mo of age. This pattern is par-allel to the evolution of crying in infants with COLIC[4]. Children selected in our study were almost as old as those in the clinical studies of Barr et al[4] or Moore et al[14] (3.7 wk vs 28.4 d and 2.6 wk, respectively) and younger than those studied by Miller et al[12] (median age: 8 wk).
Fermentation of the disaccharide generates osmotic active substances such as lactic acid, short chain fatty ac-ids, and hydrogen and/or methane. Methane production in lactose malabsorbers is normal, and without signifi-cance[10].
Colic can result directly from the hyper-peristaltic
2106 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 2 Clinical evaluation of associated symptoms in infants with crying secondary to excess gas n (%)
Inclusion After 15 d χ2 P
Excess gas 17 (85) 5 (25) 14.56 < 0.001Abnormal feeding 15 (75) 6 (30) 8.22 < 0.01Duration of crying < 1 h/d 85% 1-3 h/d 13 (65) 3 (15) 10.41 < 0.001 > 3 h/d 7 (35) 0 (0) 8.48 < 0.01
40
30
20
10
0
Gas
(pp
m)
HydrogenMethane
D0 D15
SE 0.2
SE 3.1
SE 2.5
SE 0.1
Figure 1 Evolution of breath hydrogen and methane before (D0) and after 15 d (D15) of consumption of low-lactose formula. Values are expressed as mean ± SE. Only the change in expired hydrogen was significant (P < 0.01).
Infante D et al . Dietary treatment of functional colic

stimulus of the fluid load imposed by the osmotic action of unabsorbed lactose in the small intestine, and gas or pharmacologically active metabolites might be respon-sible for the symptomatic signs. In many susceptible in-fants, this excess of gas is responsible for triggering colic.
Several studies have investigated lengthy crying and have related it to excess intestinal gas[14,19,20]. The rapid production of hydrogen in the lower bowel distends the colon, which causes different symptoms. This model of colic implies that symptoms could be relieved by reducing the lactose content of the infant feed. Four clinical trials that have used artificial lactase have been published[19,21-23]. When lactase is added to a formula 30 min before it is fed, 30% of the lactose is not hydrolyzed. In a double-blind, placebo-controlled crossover study, 10 colicky infants were fed breast milk and cows’ milk formulas, untreated and treated with lactase[19]. This study showed no evidence that low-lactose milk reduced the severity and amount of cry-ing[19]. In another double-blind, placebo-controlled cross-over trial in 12 infants, no effects on duration of crying and fussing were demonstrated[21]. In a third study with the same methodology, the lactase-treated formula reduced crying time by 1.14 h/d[22]. Yet another double-blind placebo-controlled crossover study was performed on 53 infants with colic who were treated with placebo or lactase added to their formula 4 h before they were fed. Data on 46 infants were available for crying time analysis and hy-drogen breath tests were available in 34. Only 32 infants complied with treatment. In these infants, crying time and median expired hydrogen were significantly lower in the active group than in the placebo group[23]. The results of our study were in agreement with Kanabar’s study that also included an expired hydrogen measurement.
Differences in hydrogen breath excretion between colicky and control infants might also be associated with factors other than the amount or rate of delivery of lac-tose to the colon. The microbiota, the colonic bacterial metabolic pathways, the partial pressure of hydrogen in the colon, the buffering capacity of the colon, gut perfu-sion, and incomplete monosaccharide absorption might all play a part. Therefore, the volume of gas released by a fecal sample reflects the end result of a complex interac-tion of several factors.
This pathophysiological mechanism explains the clinical and biochemical response of these infants to an adapted, low-lactose diet[22,23]. However, when calcium absorption is enhanced by the presence of lactose[24], the formula has a lactose content (3 g/100 mL) that provides a daily amount close to absorption capacity in young in-fants of 4.5 g/kg per day)[25].
Even in the absence of a placebo control group, we believe that the clinical improvement observed in our study was related to dietary management. This clinical improvement appeared earlier (after 15 d feeding with the test formula only) than the usual resolution of colic. Moreover this improvement was endorsed by a decrease in expired hydrogen.
In conclusion this non-randomized, non-placebo-con-
trolled pilot study demonstrates that the use of an adapted infant formula with a low lactose concentration leads to clinical improvement and a decrease in expired hydrogen in colicky infants. Thus, infants with colic might benefit from a switch from standard formula to this specific adapted formula. Larger randomized clinical trials on the efficacy of this formula are needed.
COMMENTSBackgroundInfant colic is still one of the most disconcerting issues in pediatric medicine. This term now includes digestive disorders such as constipation, gastroesopha-geal reflux, allergy to cow milk proteins, and excess intestinal gas due to lac-tose malabsorption.Research frontiersIn infants with colic, the possibility of functional lactase deficiency has led to clinical trials with lactase supplementation of infant formulas. These studies have given conflicting results.Innovations and breakthroughsThis article reports the clinical and biological efficiency of an adapted low lac-tose formula in infants with colic and positive hydrogen expiration.ApplicationsIn colicky infants with excess abdominal gas, a diet with an adapted low lactose formula can be tried for several days. If the results are positive, the diet can be continued for several months.Peer reviewThe experiments were well designed and the paper is well written. However, there is one major concern about the data analysis.
REFERENCES1 Wessel MA, Cobb JC, Jackson EB, Harris GS Jr, Detwiler AC.
Paroxysmal fussing in infancy, sometimes called colic. Pediat-rics 1954; 14: 421-435
2 St James-Roberts I. What is distinct about infants' "colic" cries? Arch Dis Child 1999; 80: 56-61; discussion 62
3 Lucassen PL, Assendelft WJ, van Eijk JT, Gubbels JW, Dou-wes AC, van Geldrop WJ. Systematic review of the occur-rence of infantile colic in the community. Arch Dis Child 2001; 84: 398-403
4 Barr RG, Rotman A, Yaremko J, Leduc D, Francoeur TE. The crying of infants with colic: a controlled empirical descrip-tion. Pediatrics 1992; 90: 14-21
5 Poole SR. The infant with acute, unexplained, excessive cry-ing. Pediatrics 1991; 88: 450-455
6 Savino F. Focus on infantile colic. Acta Paediatr 2007; 96: 1259-1264
7 Infante Pina D, Badia Llach X, Ariño-Armengol B, Villegas Iglesias V. Prevalence and dietetic management of mild gas-trointestinal disorders in milk-fed infants. World J Gastroen-terol 2008; 14: 248-254
8 Vik T, Grote V, Escribano J, Socha J, Verduci E, Fritsch M, Carlier C, von Kries R, Koletzko B. Infantile colic, prolonged crying and maternal postnatal depression. Acta Paediatr 2009; 98: 1344-1348
9 Koletzko B, Baker S, Cleghorn G, Neto UF, Gopalan S, Hernell O, Hock QS, Jirapinyo P, Lonnerdal B, Pencharz P, Pzyrembel H, Ramirez-Mayans J, Shamir R, Turck D, Yamashiro Y, Zong-Yi D. Global standard for the composi-tion of infant formula: recommendations of an ESPGHAN coordinated international expert group. J Pediatr Gastroenterol Nutr 2005; 41: 584-599
10 Tormo R, Bertaccini A, Conde M, Infante D, Cura I. Methane and hydrogen exhalation in normal children and in lactose malabsorption. Early Hum Dev 2001; 65 Suppl: S165-S172
2107 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Infante D et al . Dietary treatment of functional colic
COMMENTS

11 Hyams JS, Geertsma MA, Etienne NL, Treem WR. Colonic hydrogen production in infants with colic. J Pediatr 1989; 115: 592-594
12 Miller JJ, McVeagh P, Fleet GH, Petocz P, Brand JC. Breath hydrogen excretion in infants with colic. Arch Dis Child 1989; 64: 725-729
13 Hill DJ, Hosking CS. Infantile colic and food hypersensitiv-ity. J Pediatr Gastroenterol Nutr 2000; 30 Suppl: S67-S76
14 Moore DJ, Robb TA, Davidson GP. Breath hydrogen re-sponse to milk containing lactose in colicky and noncolicky infants. J Pediatr 1988; 113: 979-984
15 Barr RG, Hanley J, Patterson DK, Wooldridge J. Breath hy-drogen excretion in normal newborn infants in response to usual feeding patterns: evidence for "functional lactase insuf-ficiency" beyond the first month of life. J Pediatr 1984; 104: 527-533
16 Laforgia N, Benedetti G, Altavilla T, Baldassarre ME, Grassi A, Bonsante F, Mautone A. [Significance of lactose breath test in the newborn]. Minerva Pediatr 1995; 47: 433-436
17 Griffin MP, Hansen JW. Can the elimination of lactose from formula improve feeding tolerance in premature infants? J Pediatr 1999; 135: 587-592
18 Douwes AC, Oosterkamp RF, Fernandes J, Los T, Jongbloed AA. Sugar malabsorption in healthy neonates estimated by breath hydrogen. Arch Dis Child 1980; 55: 512-515
19 Ståhlberg MR, Savilahti E. Infantile colic and feeding. Arch Dis Child 1986; 61: 1232-1233
20 Barr RG, Wooldridge J, Hanley J. Effects of formula change on intestinal hydrogen production and crying and fussing behavior. J Dev Behav Pediatr 1991; 12: 248-253
21 Miller JJ, McVeagh P, Fleet GH, Petocz P, Brand JC. Effect of yeast lactase enzyme on "colic" in infants fed human milk. J Pediatr 1990; 117: 261-263
22 Kearney PJ, Malone AJ, Hayes T, Cole M, Hyland M. A trial of lactase in the management of infant colic. J Hum Nutr Diet 1998; 11: 281-285
23 Kanabar D, Randhawa M, Clayton P. Improvement of symp-toms in infant colic following reduction of lactose load with lactase. J Hum Nutr Diet 2001; 14: 359-363
24 Abrams SA, Griffin IJ, Davila PM. Calcium and zinc absorp-tion from lactose-containing and lactose-free infant formulas. Am J Clin Nutr 2002; 76: 442-446
25 Fomon S. Carbohydrate. In: Craven L, editor. Nutrition of Normal Infants. St. Louis: Mosby, 1993: 178-180
S- Editor Shi ZF L- Editor Kerr C E- Editor Zheng XM
2108 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Infante D et al . Dietary treatment of functional colic

Levels of matrix metalloproteinase-1 and tissue inhibitors of metalloproteinase-1 in gastric cancer
Ozgur Kemik, Ahu Sarbay Kemik, Aziz Sümer, Ahmet Cumhur Dulger, Mine Adas, Huseyin Begenik, Ismail Hasirci, Ozkan Yilmaz, Sevim Purisa, Erol Kisli, Sefa Tuzun, Cetin Kotan
Ozgur Kemik, Aziz Sümer, Ismail Hasirci, Ozkan Yilmaz, Erol Kisli, Cetin Kotan, Department of General Surgery, Yuzuncu Yıl University Medical Faculty, Van, 6500, TurkeyAhu Sarbay Kemik, Department of Biochemistry, Cerrahpasa Medical Faculty, University of Istanbul, Istanbul, 3400, TurkeyAhmet Cumhur Dulger, Department of Gastroenterology, Medi-cal Faculty, University of Yüzüncü Yıl, Van, 6500, TurkeyMine Adas, Department of Endocrinology, Okmeydani Educa-tion and Research Hospital, Istanbul, 3400, TurkeyHuseyin Begenik, Department of Internal Medicine, Medical Faculty, University of Yüzüncü Yıl, Van, 6500, TurkeySevim Purisa, Department of Biostatistics, Istanbul Medical Faculty, University of Istanbul, Istanbul, 3400, TurkeySefa Tuzun, II. General Surgery, Haseki Education and Research Hospital, Istanbul, 3400, TurkeyAuthor contributions: Kemik O, Kemik AS and Sümer A de-signed the study and wrote the paper; Kemik AS performed the biochemical evaluation, and collected and analyzed the data; Purisa S performed the statistical analyzis; Adas M, Begenik H, Yilmaz O, Hasirci I, Dulger AC, Kisli E, Tuzun S and Kotan C contributed to the discussion. Correspondence to: Ozgur Kemik, MD, Assistant Professor, Department of General Surgery, Yuzuncu Yıl University Medical Faculty, Van, 6500, Turkey. [email protected]: +90-432-2251024 Fax: +90-432-2164705Received: August 10, 2010 Revised: January 18, 2011Accepted: January 25, 2011Published online: April 28, 2011
AbstractAIM: To evaluate the levels of preoperative serum ma-trix metalloproteinase-1 (MMP-1) and tissue inhibitor of metalloproteinase-1 (TIMP-1) in gastric cancer.
METHODS: One hundred gastric cancer patients who underwent gastrectomy were enrolled in this study. The serum concentrations of MMP-1 and TIMP-1 in these patients and in fifty healthy controls were determined
using an enzyme-linked immunosorbent assay.
RESULTS: Higher serum MMP-1 and TIMP-1 levels were observed in patients than in controls (P < 0.001). Serum MMP-1 and TIMP-1 levels were positively associ-ated with morphological appearance, tumor size, depth of wall invasion, lymph node metastasis, liver metasta-sis, perineural invasion, and pathological stage. They were not significantly associated with age, gender, tu-mor location, or histological type.
CONCLUSION: Increased MMP-1 and TIMP-1 were associated with gastric cancer. Although these mark-ers are not good markers for diagnosis, these markers show in advanced gastric cancer.
© 2011 Baishideng. All rights reserved.
Key words: Gastric cancer; Matrix metalloproteinase-1; Tissue matrix metalloproteinase-1
Peer reviewer: Peter JK Kuppen, PhD, Associate Professor, De-partment of Surgery, Leiden University Medical Center, 2300 RC Leiden, The Netherlands
Kemik O, Kemik AS, Sümer A, Dulger AC, Adas M, Begenik H, Hasirci I, Yilmaz O, Purisa S, Kisli E, Tuzun S, Kotan C. Levels of matrix metalloproteinase-1 and tissue inhibitors of metal-loproteinase-1 in gastric cancer. World J Gastroenterol 2011; 17(16): 2109-2112 Available from: URL: http://www.wjgnet.com/1007-9327/full/v17/i16/2109.htm DOI: http://dx.doi.org/10.3748/wjg.v17.i16.2109
INTRODUCTIONMatrix metalloproteinases (MMPs) are a family of zinc-dependent neutral endopeptidases that play a significant role in the degradation of all matrix partitions, which
BRIEF ARTICLE
World J Gastroenterol 2011 April 28; 17(16): 2109-2112ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2109
2109 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

Kemik O et al . MMP-1 and TIMP-1 in gastric cancer
are crucial for malignant tumor growth, invasion, and metastasis[1,2]. MMPs are inhibited by tissue inhibitors of metalloproteinase (TIMPs), which are secreted proteins. TIMPs bind to enzymatically active MMPs at a 1:1 molar stoichiometry, thus inhibiting proteolysis[3]. The role of TIMPs in the imbalance of the extracellular matrix is sig-nificant and may inhibit or stimulate tumorigenesis[4].
MMP-1 is also known as collagenase (EC 3.4.23.7)[5]. Saffarian et al[6] showed that activated MMP-1 acts by processing on the collagen fibril. The biological implica-tions of MMP-1 acting as a molecular retainer, tied to the cell surface, prompted recent mechanisms for its status in tissue remodeling and cell-matrix interaction to be pro-posed. MMP-1 in the stromal tumor microenvironment can change the behavior of cancer cells to promote cell migration and invasion[7].
TIMP-1 is a 28.5 kDa glycoprotein that has been stud-ied in many human malignancies, including gastric can-cer[8]. TIMP-1 mRNA expression is increased in gastric, esophageal, and pancreatic cancer[9-11]. TIMP-1 is present in human peripheral blood and body fluids[12]. MMP-1 and TIMP-1 levels have been studied in plasma or serum of patients with cumulative malignancies[13,14].
Our study was carried out to analyze serum MMP-1 and TIMP-1 levels in gastric cancer patients and to inves-tigate their clinicopathological correlations.
MATERIALS AND METHODSA total of 100 patients who underwent gastrectomy with gastric cancer between December 2007 and April 2010 were enrolled. Their median age was 58.5 years (range, 34-78 years), and the ratio of men/women was 47/53. There were 50 healthy volunteer controls without family history of cancer, whose average age was 56 years (range, 48-65 years) (22 men, 28 women). Peripheral venous blood of patients and controls was taken before gastrec-tomy and stored at 4℃. Blood from controls was taken on the day of a physical examination. The blood samples were centrifuged 1000 rpm, in 15 min, at 20℃ to separate the serum, which was stored at -70℃ until analysis. The mean storage time of all samples was 2 mo (45-80 d).
Resected tumor specimens were studied pathologi-cally according to the criteria of the UICC’s pTNM clas-sification[15]. Information recorded included age, gender, tumor location, tumor size, wall invasion, resection mar-gin, histological type, lymph node metastasis, vascular invasion, lymphatic invasion, and perineural invasion. The histological features were classified into two types: (1) intestinal or differentiated type, consisting of papil-lary and/or tubular adenocarcinomas; and (2) diffuse or undifferentiated type, consisting of poorly differentiated, signet-ring cells, and/or mucinous adenocarcinomas.
Enzyme-linked immunosorbent assay (ELISA) for se-rum MMP-1 and TIMP-1 was performed using an ELISA kit (R&D System, USA) following the manufacturer’s instructions.
As appropriate, the Mann-Whitney U test or Fisher’s exact test was used for group comparisons. Correlations
between parameters were tested by Spearman’s correla-tion coefficient. A P < 0.05 was considered statistically significant.
RESULTSSerum MMP-1 and TIMP-1 levels in gastric cancer patients and controls are shown in Table 1 and Figure 1A and B. The serum levels of MMP-1 and TIMP-1 in gastric can-cer patients were significantly higher than in the control group (P < 0.0001). Clinicopathological variables are shown in Table 2. Serum MMP-1 and TIMP-1 levels were positively associated with the depth of wall invasion (P < 0.01), lymph node metastasis (P < 0.001), and lymphatic invasion (P < 0.001). The serum levels of MMP-1 and TIMP-1 were closely associated with distant metastasis (P < 0.001). In particular, higher MMP-1 and TIMP-1 levels were significantly associated with positive lymphovascu-lar invasion (P < 0.001), tumor size ≥ 4 cm (P < 0.001), positive lymph node metastasis (P < 0.001), T stage
2110 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 1 Serum matrix metalloproteinase-1 and tissue inhibi-tor of metalloproteinase-1 levels in patients and controls
Variables Controls (n = 50) Patients (n = 100) P
Age (yr) 56 (48-65) 58 (47-64)Gender female (%) 37 40MMP-1 (ng/mL) 256 (109-342) 785 (457-900) < 0.0001TIMP-1 (ng/mL) 220 (198-267) 725 (417-1134) < 0.0001
MMP-1: Matrix metalloproteinase-1; TIMP-1: Tissue inhibitor of metallo-proteinase-1.
1600
1400
1200
1000
800
600
400
200
0
MM
P-1
(ng/
mL)
PatientsControls
1800
1600
1400
1200
1000
800
600
400
200
0
TIM
P-1
(ng/
mL)
PatientsControls
B
A
Figure 1 Serum matrix metalloproteinase-1 (A) and tissue inhibitor of me-talloproteinase-1 (B) levels of controls and patients. MMP-1: Matrix metal-loproteinase-1; TIMP-1: Tissue inhibitor of metalloproteinase-1.

(T3-T4) (P < 0.001), or TNM stage (Ⅲ and Ⅳ) (P < 0.001). MMP-1 and TIMP-1 levels were not significantly associ-ated with negative lymphovascular invasion, tumor size < 4 cm, negative lymph node metastasis, T stage (T0-T2), and TNM stage (Ⅰ and Ⅱ). Overall, they were associated with pathological stage (P < 0.001). Serum MMP-1 and TIMP-1 levels were not associated with age (P = 0.237), gender (P = 0.281), tumor location (P < 0.142), histologi-cal type (P = 0.103), vascular invasion (P = 0.247), or peri-toneal seeding (P = 0.271).
Higher serum MMP-1 and TIMP-1 levels were cor-related with gastric cancer (P < 0.001, r = 0.77). Figure 1A shows that MMP-1 levels in patients with gastric cancer were significantly higher than in control groups. Figure 1B shows that TIMP-1 levels in patients with gastric cancer were significantly higher than in control groups.
DISCUSSIONIn our study, we investigated MMP-1 and TIMP-1 levels in gastric cancer patients and compared them with a con-trol group. We also investigated their associations with clinicopathological features.
Matrix metalloproteinases are involved in many nor-mal biological processes (e.g. embryonic development, blastocyst implantation, organ morphogenesis, nerve growth, ovulation, cervical dilatation, postpartum uterine involution, endometrial cycling, hair follicle cycling, bone remodeling, wound healing, angiogenesis, and apoptosis) and pathological processes (e.g. arthritis, cancer, cardio-vascular disease, nephritis, neurological disease, break-down of the blood brain barrier, periodontal disease, skin ulceration, corneal ulceration, liver fibrosis, emphysema, and fibrotic lung disease). Although the main function of matrix metalloproteinases is elevation of ECM during tissue resorption and progression of many diseases, it is
obvious that matrix metalloproteinases also alter the bio-logical functions of ECM molecules by definite proteoly-sis. MMP-1 and TIMP-1 are thought out to be involved in dissemination of cancer cells by dissolving the ECM, but they are also important in creating an environment that supports the initiation and growth of primary and metastatic tumors. These effects may be associated with proteolytic release of growth factors and/or modification of cellular environments[16].
The most important finding in our study was the as-sociation between high MMP-1 and TIMP-1 levels in gastric cancer patients. In addition, high MMP-1 and TIMP-1 levels were significantly associated with certain clinicopathological variables. High MMP-1 expression has been associated with hematogenous metastasis[17,18], rising depth of invasion, and metastasis in colorectal can-cer[18,19]. Our study also suggested that MMP-1 levels are associated with depth of invasion and metastasis.
Patients with colorectal cancer, ovary, lung, and liver diseases have increased TIMP-1 levels compared to control groups[14,20-22]. Wang et al[23] suggested that serum TIMP-1 levels were higher in gastric cancer patients than con-trol groups and were associated with clinicopathological variables. However, they suggested that serum TIMP-1 levels were associated with depth of wall invasion, distant metastasis, peritoneal seeding, lymphatic invasion, lymph node metastasis, and perineural invasion. However, we did not find that serum TIMP-1 levels were associated with peritoneal seeding and perineural invasion.
MMP-1 is associated with the primal pace of inva-sion and angiogenesis in gastric cancer, which may make it a useful marker for prognosis. TIMP-1 is more simply released into the blood[24]; therefore, the sensitivity of the assay is higher than that for MMP-1.
High blood levels of MMP-1 and TIMP-1 are associ-ated with poor prognosis of malignancies. Thus, they might useful as markers for malignant potential (i.e. tu-mor growth and/or differentiation) for cancer. Notably, serum TIMP-1 levels have been established as an inde-pendent factor in gastric cancer[23].
Some metalloproteinases have been shown to degrade over time when measured in stored blood samples. How-ever, we do not think that such protein decay is a signifi-cant factor when proteins are stored for 2 mo. This as-sumption is supported by the work of Papazoglou et al[25], Kardeşler et al[26] and Karapanagiotidis et al[27].
MMP-1 and TMP-1 can be considered as ‘traditional’ and conventional serum biomarkers; many studies have measured both of these proteins as serum biomarkers[28].
This study demonstrated that high serum MMP-1 and TIMP-1 levels in gastric cancer patients are signifi-cantly associated with disease progression. Their levels are important markers of tumor progression or advanced tumor stages.
COMMENTSBackgroundThe incidence of gastric cancer is rising worldwide. Collagenases may play a role
2111 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 2 Clinicopathological variables of serum matrix metal-loproteinase-1 and tissue inhibitor of metalloproteinase-1 in patients
Variables MMP-1 TIMP-1 P
Lymphovascular invasion Negative 543 (500-678) 489 (450-573) Positive 801 (768-845) 642 (567-703) < 0.001Tumor size (cm) < 4 478 (460-501) 429 (425-479) ≥ 4 675 (509-725) 671 (532-690) < 0.001Lymph node metastasis Negative 563 (503-650) 642 (598-709) Positive 742 (657-799) 756 (570-876) < 0.001T stage T0-2 521 (498-599) 598 (564-783) T3-4 674 (578-783) 749 (570-794) < 0.001TNM stage Ⅰ 469 (458-502) 476 (423-512) Ⅱ 534 (467-563) 521 (478-589) Ⅲ 714 (546-857) 753 (512-699) < 0.001 Ⅳ 765 (699-900) 975 (812-1134) < 0.001
MMP-1: Matrix metalloproteinase-1; TIMP-1: Tissue inhibitor of metallo-proteinase-1.
Kemik O et al . MMP-1 and TIMP-1 in gastric cancer
COMMENTS

in degradation of the cell matrix, possibly leading to growth of malignant tumors, lymph node metastasis, increased depth of invasion and other metastases.Research frontiersMatrix metalloproteinase-1 (MMP-1) and tissue inhibitor of matrix metallopro-teinase-1 (TIMP-1) change the environment of cancer cells to promote cell migration and invasion. Changes caused by these endopeptidases have a role in the progression of the gastric cancer.Innovations and breakthroughsHigh blood levels of MMP-1 and TIMP-1 are associated with poor prognosis of malignancies, making them potentially useful biomarkers for the malignant potential (i.e. tumor growth and/or differentiation) of cancer. These effects may be associated with proteolytic release of growth factors and/or modification of tumor cells.ApplicationsThe date generated in this paper might be used to explain the development of gastric cancer, to prevent metastasis, and to aid early diagnosis. TerminologyMMP-1 and TIMP-1 zinc-dependent neutral endopeptidases. The role of MMP-1 and TIMP-1 in the imbalance of the extracellular matrix is significant and may inhibit or stimulate tumorigenesis. These effects have been demonstrated, and these molecules may represent useful markers of tumorigenesis.Peer reviewIt is a nice study, with interesting results.
REFERENCES1 Zucker S, Vacirca J. Role of matrix metalloproteinases (MMPs)
in colorectal cancer. Cancer Metastasis Rev 2004; 23: 101-1172 Ala-aho R, Kähäri VM. Collagenases in cancer. Biochimie
2005; 87: 273-2863 Matrisian LM. Metalloproteinases and their inhibitors in ma-
trix remodeling. Trends Genet 1990; 6: 121-1254 Jiang Y, Goldberg ID, Shi YE. Complex roles of tissue in-
hibitors of metalloproteinases in cancer. Oncogene 2002; 21: 2245-2252
5 Nagase H, Barrett AJ, Woessner JF Jr. Nomenclature and glossary of the matrix metalloproteinases. Matrix Suppl 1992; 1: 421-424
6 Saffarian S, Collier IE, Marmer BL, Elson EL, Goldberg G. Interstitial collagenase is a Brownian ratchet driven by prote-olysis of collagen. Science 2004; 306: 108-111
7 Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005; 120: 303-313
8 Curran S, Murray GI. Matrix metalloproteinases: molecular aspects of their roles in tumour invasion and metastasis. Eur J Cancer 2000; 36: 1621-1630
9 Nomura H, Fujimoto N, Seiki M, Mai M, Okada Y. Enhanced production of matrix metalloproteinases and activation of matrix metalloproteinase 2 (gelatinase A) in human gastric carcinomas. Int J Cancer 1996; 69: 9-16
10 Mori M, Mimori K, Sadanaga N, Inoue H, Tanaka Y, Mafune K, Ueo H, Barnard GF. Prognostic impact of tissue inhibitor of matrix metalloproteinase-1 in esophageal carcinoma. Int J Cancer 2000; 88: 575-578
11 Gress TM, Müller-Pillasch F, Lerch MM, Friess H, Büchler M, Adler G. Expression and in-situ localization of genes coding for extracellular matrix proteins and extracellular matrix de-grading proteases in pancreatic cancer. Int J Cancer 1995; 62: 407-413
12 Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim Biophys Acta 2000; 1477: 267-283
13 Baker T, Tickle S, Wasan H, Docherty A, Isenberg D, Wax-man J. Serum metalloproteinases and their inhibitors: mark-ers for malignant potential. Br J Cancer 1994; 70: 506-512
14 Oberg A, Höyhtyä M, Tavelin B, Stenling R, Lindmark G. Limited value of preoperative serum analyses of matrix metal-loproteinases (MMP-2, MMP-9) and tissue inhibitors of matrix metalloproteinases (TIMP-1, TIMP-2) in colorectal cancer. An-ticancer Res 2000; 20: 1085-1091
15 Sobin LH, Wittekand CH, editors. TNM Classification of Ma-lignant Tumors. 6th ed. New York: Wiley-Liss, 2002
16 Nagase H, Woessner JF Jr. Matrix metalloproteinases. J Biol Chem 1999; 274: 21491-21494
17 Sunami E, Tsuno N, Osada T, Saito S, Kitayama J, Tomo-zawa S, Tsuruo T, Shibata Y, Muto T, Nagawa H. MMP-1 is a prognostic marker for hematogenous metastasis of colorectal cancer. Oncologist 2000; 5: 108-114
18 Hilska M, Roberts PJ, Collan YU, Laine VJ, Kössi J, Hirsimäki P, Rahkonen O, Laato M. Prognostic significance of matrix metalloproteinases-1, -2, -7 and -13 and tissue inhibitors of metalloproteinases-1, -2, -3 and -4 in colorectal cancer. Int J Cancer 2007; 121: 714-723
19 Shiozawa J, Ito M, Nakayama T, Nakashima M, Kohno S, Sekine I. Expression of matrix metalloproteinase-1 in human colorectal carcinoma. Mod Pathol 2000; 13: 925-933
20 Manenti L, Paganoni P, Floriani I, Landoni F, Torri V, Buda A, Taraboletti G, Labianca R, Belotti D, Giavazzi R. Expression levels of vascular endothelial growth factor, matrix metallo-proteinases 2 and 9 and tissue inhibitor of metalloproteinases 1 and 2 in the plasma of patients with ovarian carcinoma. Eur J Cancer 2003; 39: 1948-1956
21 Ylisirniö S, Höyhtyä M, Mäkitaro R, Pääakkö P, Risteli J, Kinnula VL, Turpeenniemi-Hujanen T, Jukkola A. Elevated serum levels of type I collagen degradation marker ICTP and tissue inhibitor of metalloproteinase (TIMP) 1 are associated with poor prognosis in lung cancer. Clin Cancer Res 2001; 7: 1633-1637
22 Muzzillo DA, Imoto M, Fukuda Y, Koyama Y, Saga S, Nagai Y, Hayakawa T. Clinical evaluation of serum tissue inhibitor of metalloproteinases-1 levels in patients with liver diseases. J Gastroenterol Hepatol 1993; 8: 437-441
23 Wang CS, Wu TL, Tsao KC, Sun CF. Serum TIMP-1 in gastric cancer patients: a potential prognostic biomarker. Ann Clin Lab Sci 2006; 36: 23-30
24 Brennan FM, Browne KA, Green PA, Jaspar JM, Maini RN, Feldmann M. Reduction of serum matrix metalloproteinase 1 and matrix metalloproteinase 3 in rheumatoid arthritis patients following anti-tumour necrosis factor-alpha (cA2) therapy. Br J Rheumatol 1997; 36: 643-650
25 Papazoglou D, Papatheodorou K, Papanas N, Papadopoulos T, Gioka T, Kabouromiti G, Kotsiou S, Maltezos E. Matrix metalloproteinase-1 and tissue inhibitor of metalloprotein-ases-1 levels in severely obese patients: what is the effect of weight loss? Exp Clin Endocrinol Diabetes 2010; 118: 730-734
26 Kardeşler L, Biyikoğlu B, Cetinkalp S, Pitkala M, Sorsa T, Buduneli N. Crevicular fluid matrix metalloproteinase-8, -13, and TIMP-1 levels in type 2 diabetics. Oral Dis 2010; 16: 476-81
27 Karapanagiotidis GT, Antonitsis P, Charokopos N, Foroulis CN, Anastasiadis K, Rouska E, Argiriadou H, Rammos K, Pa-pakonstantinou C. Serum levels of matrix metalloproteinases -1,-2,-3 and -9 in thoracic aortic diseases and acute myocar-dial ischemia. J Cardiothorac Surg 2009; 4: 59
28 Sutnar A, Pesta M, Liska V, Treska V, Skalicky T, Kormunda S, Topolcan O, Cerny R, Holubec L Jr. Clinical relevance of the expression of mRNA of MMP-7, MMP-9, TIMP-1, TIMP-2 and CEA tissue samples from colorectal liver metastases. Tu-mour Biol 2007; 28: 247-252
S- Editor Tian L L- Editor Stewart GJ E- Editor Zheng XM
2112 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Kemik O et al . MMP-1 and TIMP-1 in gastric cancer

Sunitinib for Taiwanese patients with gastrointestinal stromal tumor after imatinib treatment failure or intolerance
Yen-Yang Chen, Chun-Nan Yeh, Chi-Tung Cheng, Tsung-Wen Chen, Kun-Ming Rau, Yi-Yin Jan, Miin-Fu Chen
Yen-Yang Chen, Kun-Ming Rau, Division of Hematology-On-cology, Department of Internal Medicine, Chang Gung Memorial Hospital, Kaohsiung Medical Center, Taoyuan 333, Taiwan, ChinaChun-Nan Yeh, Chi-Tung Cheng, Tsung-Wen Chen, Yi-Yin Jan, Miin-Fu Chen, GIST team, Department of Surgery, Chang Gung Memorial Hospital and University, Taoyuan 333, Taiwan, ChinaAuthor contributions: Chen YY helped collect the data and wrote the manuscript; Yeh CN was in charge of this project and revised the manuscript; Cheng CT, Chen TW, Rau KM, Jan YY and Chen MF helped to review this paper.Supported by Chang Gung Medical Research Program 380711 Grant to Dr. Yeh CNCorrespondence to: Chun-Nan Yeh, MD, GIST team, Depart-ment of Surgery, Chang Gung Memorial Hospital and University, 5, Fu-Hsing Street, Kwei-Shan, Taoyuan 333, Taiwan, China. [email protected]: +886-3-3281200 Fax: +886-3-3285818Received: November 8, 2010 Revised: January 3, 2011Accepted: January 10, 2011Published online: April 28, 2011
AbstractAIM: To report preliminary results of the efficacy and safety of sunitinib in the management of Taiwanese gastrointestinal stromal tumors (GIST) patients facing imatinib mesylate (IM) intolerance or failure.
METHODS: Between 2001 and May 2010, 199 Taiwan-ese patients with metastatic GIST were treated at Chang Gung Memorial Hospital. Among them, 23 (11.6%) pa-tients receiving sunitinib were investigated.
RESULTS: Sixteen male and 7 female patients with a median age of 59 years (range: 24-83 years) received sunitinib. Twenty-two GIST patients changed to suni-tinib because of IM failure and 1 because of intoler-ance. The median duration of sunitinib administration was 6.0 mo (range: 2-29 mo). The clinical benefit was 65.2% [2 complete response (CR), 4 partial response
(PR), and 9 stationary disease (SD); 15/23]. In 12 pa-tients harboring mutations of the kit gene at exon 11, the clinical benefit rate (CR, PR, and SD) was 75.0% and 6 patients with tumors containing kit exon 9 muta-tions had a clinical benefit of 50.0% (not significant, P = 0.344). The progression free survival (PFS) and overall survival (OS) did not differ between patients whose GISTs had wild type, KIT exon 9, or KIT exon 11 mutations. Hand-foot syndrome was the most common cause of grade Ⅲ adverse effect (26.1%), followed by anemia (17.4%), and neutropenia (13.0%). During the median 7.5-mo follow-up after sunitinib use, the medi-an PFS and OS of these 23 GIST patients after sunitinib treatment were 8.4 and 14.1 mo, respectively.
CONCLUSION: Sunitinib appears to be an effective treatment for Taiwanese with IM-resistant/intolerant GISTs and induced a sustained clinical benefit in more than 50% of Taiwanese advanced GIST patients.
© 2011 Baishideng. All rights reserved.
Key words: Suintinib; Gastrointestinal stromal tumors; Imaitinib; Failure or intolerance
Peer reviewer: I-Rue Lai, Assistant professor, Department of Anatomy and Cell Biology, Medical College, National Taiwan University, 7, Chun-San S. Rd, Taipei 106, Taiwan, China
Chen YY, Yeh CN, Cheng CT, Chen TW, Rau KM, Jan YY, Chen MF. Sunitinib for Taiwanese patients with gastrointestinal stro-mal tumor after imatinib treatment failure or intolerance. World J Gastroenterol 2011; 17(16): 2113-2119 Available from: URL: http://www.wjgnet.com/1007-9327/full/v17/i16/2113.htm DOI: http://dx.doi.org/10.3748/wjg.v17.i16.2113
INTRODUCTIONGastrointestinal stromal tumors (GISTs) primarily arise from mesenchymal tissue in the gastrointestinal (GI) tract
BRIEF ARTICLE
World J Gastroenterol 2011 April 28; 17(16): 2113-2119ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2113
2113 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

Chen YY et al . Sunitinib for GIST after imatinib failure
and abdomen. Although GISTs are rare, representing only an estimated 0.1% to 3% of all GI tract tumors[1] GISTs account for the most common mesenchymal malignancy of the GI tract with unknown incidence[2]. GISTs appear to be related to the interstitial cells of Cajal[3] and express the cell surface transmembrane re-ceptor KIT, which has tyrosine kinase activity. Gain-of-function mutations of KIT are frequent in GISTs and result in constitutive activation of KIT signaling and lead to uncontrolled cell proliferation and resistance to apoptosis[4,5]. The KIT tyrosine kinase inhibitor imatinib mesylate (IM) has shown a promising clinical result for an advanced GIST patient[6], and several trials have shown a promising effect of this target therapy[6,7]. Our previous study showed that IM significantly affected survival in GIST patients[8,9].
Surgical resection remains the mainstay therapy for GIST, but recurrence is common. The 5-year survival rates for GIST after complete resection range from 40% to 65%[6,10-13]. Unresectable or metastatic GIST is a fatal disease that resists conventional chemotherapy. IM selec-tively inhibits certain protein tyrosine kinases: intracellular ABL kinase, chimeric BCR-ABL fusion oncoprotein of chronic myeloid leukemia, the transmembrane receptor KIT, and platelet-derived growth factor receptors (PDG-FR)[14-17]. IM induced a sustained objective response in more than 50% of patients with advanced GISTs in the West and in Taiwan[8,9]. However, progression of GIST eventually develops and emerges as a challenge.
Sunitinib is an oral multi-targeted tyrosine kinase in-hibitor with activity against KIT and PDGFRs, as well as vascular endothelial growth factor receptors (VEGFRs), glial cell line-derived neurotrophic factor receptor (rear-ranged during transfection; RET), colony-stimulating fac-tor 1 receptor (CSF-1R), and FMS- like tyrosine kinase-3 receptor (FLT3)[18-23]. Sunitinib received multi-national approval for the treatment of GIST after failure of IM because of resistance or intolerance based on the results of an international, randomized, double-blind, placebo-controlled phase Ⅲ trial[24]. The clinical safety and ef-ficacy of both IM and sunitinib in GIST have primarily been established in Western patients living in the USA or Europe and have not been thoroughly studied in Asian patients. Fifty-six centers in 11 countries participated in the phase Ⅲ trial of sunitinib in GIST, but only 15 of the 312 patients were of Asian descent (10 and 5 in the suni-tinib and placebo groups, respectively)[25]. Therefore, we report our preliminary results to clarify the efficacy and safety of sunitinib in management of Taiwanese GIST patients facing IM intolerance or failure.
MATERIALS AND METHODSBetween 2001 and May 2010, 199 patients who had his-tologically confirmed, recurrent, unresectable, or meta-static GIST that expressed CD117 or CD34 and were treated at the Department of Medical Oncology, and Surgery, Chang Gung Memorial Hospital were retro-
spectively reviewed. Failure of prior IM therapy, as dem-onstrated by disease progression [based on Response Evaluation Criteria in Solid Tumors (RECIST)][26] or dis-continuation of IM due to toxicity was the inclusion cri-teria in this study. Additional eligibility criteria included an Eastern Cooperative Oncology Group (ECOG) per-formance status of 0 or 1 and adequate cardiac, hepatic, renal, coagulation, and hematologic function. Key exclu-sion criteria included lack of recovery from the acute toxic effects of previous anticancer therapy or imatinib treatment, discontinuation of imatinib therapy within 2 wk or of any other approved or investigational drug for GIST within 4 wk before starting sunitinib treatment, clinically significant cardiovascular events or disease in the previous 12 mo, diabetes mellitus with clinical evi-dence of peripheral vascular disease or diabetic ulcers, or a diagnosis of any second malignancy within the previous 5 years. Patients could have previously received chemotherapeutic regimens (the last chemotherapy treat-ment must have been at least 4 wk before study entry) and undergone radiotherapy, or surgery, or both. The study was approved by the local institutional review board of Chang Gung Memorial Hospital, and written informed consent for drug administration and the analy-sis of tumor-associated genetic alteration was obtained independently from each patient.
Study design and follow-up studyA retrospective study was conducted to evaluate the effect of sunitinib in inducing objective response in Taiwanese GIST patients. Patients were administered 50 mg (4 wk on and 2 wk off; for clinical trial) or 37.5 mg continu-ously of sunitinib in 12.5 mg capsules taken orally daily with food. Patients had regular physical examinations and evaluations of performance status, body weight, complete blood count, and serum chemistry. The administration of each dose and any adverse events were recorded for each patient. Standard computed tomography (CT) was per-formed on each patient every 3 mo for the first 3 years and every 6 mo for the following 2 years to assess patient response. Measurement of efficacy was based on objec-tive tumor assessments made using RECIST with a minor modification to allow use of standard radiographic pro-tocols for spiral CT. Time to response (TTR) was defined as the interval for better drug response during sunitinib treatment. Time to progression (TTP) was defined as the interval for worse drug response during sunitinib treat-ment. Progression free survival (PFS) was defined as no progression after sunitinib use. Overall survival (OS) was defined as survival after administration of sunitinib and death was the endpoint of the study. Response rate, PFS, OS, TTR, duration of response, and TTP were recorded. Safety and tolerability were assessed by analysis of adverse events, physical examinations, vital signs, ECOG perfor-mance status, and laboratory abnormality assessments (for example, complete blood count with differential count, serum electrolyte measurements, and electrocardiogram). Cardiac function was assessed at screening, at day 28 of
2114 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

all treatment cycles, and treatment end with 12-lead elec-trocardiogram and multigated acquisition scans. Toxic effects were recorded in accordance with the National Cancer Institute Common Toxicity Criteria[27].
Analysis of KIT and PDGFRA mutationsSections were prepared from formalin-fixed, paraffin-embedded pretreatment specimens trimmed to enrich tumor cells. Polymerase chain reaction amplification of genomic DNA for KIT and PDGFRA was performed and amplification was analyzed for mutations as previ-ously described[28].
Statistical analysisAll data are presented as percentages of patients or means with standard deviation. Pearson χ2 test and Fisher exact test were used for nominal variables. Survival rate was cal-culated and plots constructed by the Kaplan-Meier method and compared between groups with a log-rank test. All statistical analyses were performed using the SPSS com-puter software package (Version 10.0, Chicago, IL, USA). A P-value < 0.05 was considered statistically significant.
RESULTSClinical featuresTable 1 summarizes the demographic features of 23 GIST patients receiving sunitinib. There were 16 male and 7 female patients with a median age of 59 years (range from 24 to 83 years). The stomach was the most com-mon site for GISTs treated with sunitinib (8/23; 35%), followed by the jejunum (5/23; 22%), the ileum (5/23; 22%), and the rectum (3/23; 13%) (Table 1).
Treatment and outcomes before and after use of sunitinibIn Taiwan, sunitinib has been approved for treatment of metastatic GIST patients facing IM intolerance or failure since February 2009. Before 2009, sunitinib was administered to selected patients with unresectable or metastatic (advanced) GISTs facing IM failure or intoler-ance because they were enrolled in clinical trials. Sunitinib (12.5-50 mg/d) was given to 23 patients and all 23 pa-tients were followed after administration of sunitinib at regular intervals until death or until the time of this man-uscript writing. The median follow-up time after sunitinib was 7.5 mo, range: 1.2-58.0 mo. Overall, 2 patients (8.7%) had a complete response (CR), 4 (17.4%) had a partial re-sponse (PR), 9 had stationary disease (SD) (39.1 %), and 8 had progressive disease (PD) (34.8%). A clinical benefit was observed in 65.2% of GIST patients. Among the 23 patients, the median TTR for 2 patients with CR was 3.73 mo and was 3.67 mo for 4 PR patients. The median TTP was 2.37 mo and the median survival is still un-known in the 8 PD patients (Table 2). During the median 7.5 mo follow-up after sunitinib use, the median PFS and OS of these 23 GIST patients after sunitinib treatment was 8.4 and 14.1 mo, respectively (Figures 1 and 2).
Spectrum of mutations in 23 advanced GIST patients Tumor specimens suitable for genetic analysis were avail-able from 21 (84.4%) of the 23 GIST patients with IM failure or intolerance. Overall, 18 (85.7%) of the 21 ex-amined GISTs had activated mutations of KIT exon 9 and 11. Six of 21 (28.6%) GISTs had exon 9 mutation, 12 (57.1%) had exon 11 mutation, and 1 (4.8%) had no mutation of KIT. One PDGFRA exon 18 mutation was found. One patient had a concurrent deletion mutation in exon 11 and a missense mutation in exon 13; however, the exon 13 mutation was followed by the deletion mutation in exon 11. This patient developed acquired resistance and expired from disease progression. All 6 GISTs had KIT exon 9 mutation and displayed in-frame duplication of nu-cleotides, resulting in insertion of alanine (A) and tyrosine (Y) at codons 502 and 503. The KIT exon 11 mutations in the 12 GIST patients included insertion and deletion mu-tations, deletion mutations, and missense mutations.
2115 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 1 Demographic and genetic data of 23 Taiwanese gastrointestinal stromal tumor patients with imatinib failure or intolerance treated with sunitinib n (%)
Sunitinib (n = 23)
Age (median/range, yr) 59.0/24-83Gender (male:female) 16:7Location Stomach 8 (26.6) Duodenum 1 (12.5) Jejunum 5 (23.4) Ileum 5 (14.1) Mesentery 1 (18.8) Rectum 3 (4.7)Tumor recurrence Liver 15 Peritoneum 6 Local recurrence 2Genetic spectrum 21 (84.4) Exon 11 12 Deletion mutation Deletion and insertion mutation Missense mutation Exon 9 (insertion mutation) 6 Exon 13 1 No mutation (wild type) 1 PDGFRA (exon 18) 1 Median duration of sunitinib use (mo) 6
PDGFRA: Platelet derived growth factor α.
Table 2 Antitumor response of 23 Taiwanese with advanced gastrointestinal stromal tumor treated with sunitinib
n (%) Sunitinib duration (median, mo)
TTR/TTP (median, mo)
OS (median, mo)
CR 2 (8.7) 9.85 3.73/NA NAPR 4 (17.4) 12.3 3.67/12.71 NASD 9 (39.1) 11.9 1.87/13.53 14.03PD 8 (34.8) 3.63 2.37 NA
CR: Complete response; PR: Partial response; SD: Stationary disease; PD: Progression of disease; TTR: Time to response; TTP: Time to progression; OS: Overall survival.
Chen YY et al . Sunitinib for GIST after imatinib failure

Treatment and outcomes after use of sunitinib in terms of mutation statusIn 12 patients with GISTs harboring KIT exon 11 muta-tions, the clinical benefit rate was 75% (2 CR, 2PR, and 5 PR) and 3 of 6 patients with tumors containing a KIT exon 9 mutation had a clinical benefit of 50% (1 PR and 2 SD) (not significant, P = 0.344) (Table 3). The median PFS and OS for the 12 GIST patients who had KIT exon 11 mutations after sunitinib use was 8.8 mo and still not reached, respectively. The median PFS and OS for the 6
patients with tumors containing a KIT exon 9 mutation were 3.7 and 13.5 mo, respectively. The twelve GIST pa-tients who had KIT exon 11 mutations had similar PFS and OS to that of 6 patients with tumors containing a KIT exon 9 mutation (Figures 3 and 4).
Adverse events in 23 advanced GIST patients receiving sunitinibHand-foot syndrome was the most common cause of grade Ⅲ adverse effects (26.1%), followed by anemia (17.4%), and neutropenia (13.0%). None of 11 patients had hypothyroidism after use of sunitinib (Table 4).
DISCUSSIONWe had shown that IM significantly prolongs the post-recurrence and OS of Taiwanese patients with advanced GISTs[8,9]. However, approximately 50% of GIST pa-tients eventually develop progression in 24 mo after IM treatment and emerge as a challenge[7]. This study con-firmed the positive effect of sunitinib on improving PFS
2116 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
1.0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0.0
Cum
ulat
ive
surv
ival
rat
e
0 2 4 6 8 10 12 14 16 18 20 t /mo
Use of sunitinib (n = 23)
Figure 1 Progression free survival of 23 Taiwanese with metastatic gas-trointestinal stromal tumor treated with sunitinib after imatinib failure or intolerance.
Median/mean: 8.4/8.37 mo
1.0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0.0
Cum
ulat
ive
surv
ival
rat
e
0 6 12 18 24 30 t /mo
Use of sunitinib (n = 23)
Figure 2 Overall survival of 23 Taiwanese with metastatic gastrointestinal stromal tumor treated with sunitinib after imatinib failure or intolerance.
Median/mean: 14.1/19.3 mo
Table 3 Correlation between antitumor response and muta-tion status of 21 Taiwanese with advanced gastrointestinal stromal tumor treated with sunitinib
CR PR SD PD P CR + PR + SD
PD P
Exon 9 (n = 6) 0 1 2 3 0.6101 3 3 0.3441
Exon 11 (n = 12) 2 2 5 3 9 3Exon 13 (n = 1) 0 0 0 1No mutation (n = 1)
0 0 1 1
PDGFRA (n = 1) 0 1 0 0
1Exon 9 vs exon 11. CR: Complete response; PR: Partial response; SD: Stationary disease; PD: Progression of disease; PDGFRA: Platelet derived growth factor α.
1.0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0.0
Cum
ulat
ive
surv
ival
rat
e
0 2 4 6 8 10 12 14 16 18 20 t /mo
Figure 3 Progression free survival of 18 Taiwanese with metastatic gas-trointestinal stromal tumor treated with sunitinib after imatinib failure or intolerance (exon 9 vs exon 11). Median/mean (mo): 3.7/5.6 (exon 9), 8.8/10.2 (exon 11); 95% CI: 0-7.9/2.5-8.6 (exon 9), 2.24-14.4/6.5-14 (exon 11). Log-rank test, P = 0.221.
Exon 9 (n = 6)Exon 11 (n = 12)
1.0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0.0
Cum
ulat
ive
surv
ival
rat
e
Figure 4 Overall survival of 18 Taiwanese with metastatic gastrointestinal stromal tumor treated with sunitinib after imatinib failure or intolerance (exon 9 vs exon 11). Median/mean (mo): 13.5/13.7 (exon 9), Not achieved/22.6 (exon 11); 95% CI: 0.9-26.1/9.8-17.7 (exon 9), NA/16.1-29.1 (exon 11). Log-rank test, P = 0.473.
Exon 9 (n = 6)Exon 11 (n = 12)
0 6 12 18 24 30 t /mo
Chen YY et al . Sunitinib for GIST after imatinib failure

and OS of advanced GIST patients facing IM failure or intolerance. This study reported a median PFS and OS for 23 advanced GIST patients of 8.4 and 14.1 mo, respectively, after sunitinib administration for a median period of 6.0 mo.
Sunitinib induced a sustained clinical benefit in more than 50% of Taiwanese patients with advanced GISTs (15/23; 65.2%)[29] in our study, which was better than Henrich’s report. A CR induced by tyrosine kinase inhibi-tors on GIST patients has been sporadically reported. The US S0033 phase Ⅲ study revealed that the CR rate was 3% for 751 metastatic or unresectable GIST patients re-ceiving 400 or 800 mg IM daily[30]. In the EORTC 62005 phase Ⅲ study, the CR rate was 4.76% for 923 metastatic or unresectable patients receiving 400 or 800 mg imatinib daily[31]. In contrast to the 2 previous studies, the CR rate in this study was 8.7% (2/23) and the median TTR for 2 patients that had a CR was 3.73 mo. The high incidence of CR in this study, even for patients using the second line tyrosine kinase, is because 1 of these 2 patients un-derwent surgery to achieve complete tumor removal. The limited experience on CR after sunitinib treatment for advanced or metastatic GIST patients facing IM failure or intolerance may still not justify the use of surgery as an adjunct method for target therapy in selected patients.
Regarding the relationship between response rate and kinase mutation, KIT exon 11 and exon 9 mutations predict a favorable response to IM[32]. Henrich reported that the clinical activity of sunitinib after IM failure is
significantly impacted by both primary and secondary mutations in the predominant pathogenic kinases, which has implications on optimal treatment of patients with GIST. Heinrich reported that both the clinical benefit and the objective response rates with sunitinib were higher in patients with primary KIT exon 9 mutations than with exon 11 mutations. Similarly, PFS and OS were significantly longer in patients with primary KIT exon 9 mutations or a wild-type genotype than in those with KIT exon 11 mutations[29]. A possible explanation is that the potency of sunitinib against wild-type and exon 9 mutant KIT was superior to that of imatinib in vitro, whereas both drugs exhibited similar potency against KIT exon 11 mutant kinases. These results suggest that the greater clinical benefit seen in sunitinib-treated pa-tients with exon 9 mutant or wild-type imatinib-resistant GISTs may be related to the greater potency of sunitinib against these kinases[29]. In contrast to Heinrich’s study, the clinical benefit, PFS, and OS did not differ between the groups of patients whose GISTs had KIT exon 9 or exon 11 mutation. Although the KIT oncoproteins encoded by exon 9 and exon 11 mutants were unequally sensitive to sunitinib in vitro[29], the limited case number and racial difference might partly explain the similar clini-cal response rate of sunitinib in terms of KIT exon mu-tations in Taiwanese GIST patients.
Sunitinib was reasonably well tolerated in our study and the most common treatment-related adverse events were fatigue, diarrhea, skin discoloration, and nausea. Treatment-related adverse events of any severity grade were reported in 83% of sunitinib-treated patients, and serious treatment-related adverse events were reported in 20% of patients[24]. In contrast to western GIST pa-tients, hand-foot syndrome was the most common cause of grade Ⅲ adverse events in our study. The reason for this discrepant incidence of hand-foot syndrome is still unknown and needs to be fully clarified. Racial differ-ences in drug metabolism or pharmacokinetics are pos-sible reasons for this observation[33]. However, Lee et al[34] reported a higher frequency of hand-foot syndrome in Asian patients at Asian sites compared to Asian patients at non-Asian sites and in non-Asian patients in more than 4000 renal cell carcinoma patients receiving sunitinib. A lower frequency of some GI-related adverse events (AEs) in Asian patients at non-Asian sites compared to frequen-cies in Asian patients at Asian sites and in non-Asian patients has been observed. Recent evidence suggest that heterogeneity in toxicity and efficacy among patients re-ceiving anti-VEGF therapy can be partially explained by genomic variability, including single-nucleotide polymor-phisms, providing a possible explanation for the differ-ences in AE frequencies between Asians and non-Asians in this analysis[34].
Sunitinib-induced hypothyroidism was reported as a side effect in 12% of GIST patients. No hypothyroidism was noted in our series and primary hypothyroidism is not a common complication of therapeutic drugs. Drugs known to affect thyroid function are lithium, thioamides,
2117 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 4 Adverse events and selected laboratory abnormalities
Variable Sunitinib (n = 23)
Grade 1 Grade 2 Grade 3 Grade 4
Adverse event Anorexia 5 1 0 0 Diarrhea 6 8 0 0 Constipation 1 2 0 0 Fatigue 2 2 0 0 Nausea 0 0 0 0 Mucositis/stomatitis 4 0 0 0 Vomiting 1 0 0 0 Hypertension 4 4 1 0 Hand-foot syndrome 1 2 6 0 Rash 1 4 0 0 Skin discoloration 5 0 0 0 Fever 2 0 1 0Laboratory abnormalities Leukopenia 4 6 1 0 Neutropenia 2 4 3 0 Febrile neutropenia 0 0 1 0 Anemia 8 6 4 0 Elevated creatinine 4 4 0 0 Thrombocytopenia 6 7 0 0 AST 7 1 0 0 ALT 4 0 0 0 Total bilirubin 3 1 1 0 GFR 3 2 0 0 Hypothyroidism 0 0 0 0
AST: Aspartate aminotransferase; ALT: Alanine aminotransferase; GFR: Glomerular filtration rate.
Chen YY et al . Sunitinib for GIST after imatinib failure

amiodarone, and cytokines such as interferon and inter-leukin-2. The molecular mechanisms of sunitinib-induced hypothyroidism are currently unknown but one possible mechanism by which sunitinib direct affects the thyroid is through the inhibition of VEGFR and/or PDGFR. Re-cent studies in a mouse model have shown that VEGFR inhibition can induce capillary regression in various organs, including the thyroid. Moreover, the vasculature of the thyroid showed the greatest regression of all organs[35,36].
In conclusion, sunitinib appears to be a safe and ef-fective treatment for Taiwanese patients with imatinib-resistant/intolerant GIST. Sunitinib induced a sustained clinical benefit in more than 50% of Taiwanese advanced GIST patients, even those facing imatinib failure or in-tolerance, with a median 8.4 mo PFS. ORR, PFS, and OS did not differ between patients whose GISTs had wild type KIT, KIT exon 9 mutation, or KIT exon 11 muta-tion. However, hand-foot syndrome accounted for the most common cause of grade Ⅲ adverse event.
ACKNOWLEDGMENTSWe would like to thank Novartis (Taiwan) Co., Ltd. for financial support of genetic analysis.
COMMENTSBackgroundThe clinical safety and efficacy of both imatinib mesylate (IM) and sunitinib in gastrointestinal stromal tumors (GIST) have primarily been established in West-ern patients living in the USA or Europe and have not been thoroughly studied in Asian patients. Fifty-six centers in 11 countries participated in the phase Ⅲ trial of sunitinib in GIST, but only 15 of the 312 patients were of Asian descent (10 and 5 in the sunitinib and placebo groups, respectively). Research frontiersTo clarify the efficacy and safety of sunitinib in management of Taiwanese GIST patients facing IM intolerance or failure. The response of this second line target therapy also correlates with genetic status of the tumor.Innovations and breakthroughsSunitinib appears to be a safe and effective treatment for Taiwanese patients with imatinib-resistant/intolerant GIST. Sunitinib induced a sustained clinical benefit in more than 50% of Taiwanese advanced GIST patients, even those facing imatinib failure or intolerance, with a median 8.4 mo progression free survival (PFS). ORR, PFS, and overall survival did not differ between patients whose GISTs had wild type KIT, KIT exon 9 mutation, or KIT exon 11 mutation. However, hand-foot syndrome accounted for the most common cause of grade Ⅲ adverse event.ApplicationsThe preliminary report helps to clarify the efficacy and safety of sunitinib in management of Taiwanese GIST patients facing IM intolerance or failure.Peer reviewThis is a review of therapeutic effects of sunitinib on 22 Taiwanese patients with metastatic GISTs after IM failure. Their data showed that sunitinib was helpful in 15 of the 23 patients, and the clinical benefits of sunitinib did not differ in pa-tients with either primary KIT exon 9 or exon 11 mutation. Although the finding of this study is not new for sunitinib has been shown effective for IM failure, it is an interesting report showing authors’ experience in using sunitinib in Taiwan-ese patients.
REFERENCES1 Lewis JJ, Brennan MF. Soft tissue sarcomas. Curr Probl Surg
1996; 33: 817-872
2 Rossi CR, Mocellin S, Mencarelli R, Foletto M, Pilati P, Nitti D, Lise M. Gastrointestinal stromal tumors: from a surgical to a molecular approach. Int J Cancer 2003; 107: 171-176
3 DeMatteo RP, Lewis JJ, Leung D, Mudan SS, Woodruff JM, Brennan MF. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg 2000; 231: 51-58
4 Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastro-intestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol 1998; 152: 1259-1269
5 Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Mu-hammad Tunio G, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998; 279: 577-580
6 Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tu-mors. N Engl J Med 2002; 347: 472-480
7 Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, Issels R, van Oosterom A, Hogendoorn PC, Van Glabbeke M, Bertulli R, Judson I. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet 2004; 364: 1127-1134
8 Yeh CN, Chen TW, Wu TJ, Hsueh S, Jan YY. Treatment of patients with advanced gastrointestinal stromal tumor of small bowel: implications of imatinib mesylate. World J Gas-troenterol 2006; 12: 3760-3765
9 Yeh CN, Chen TW, Lee HL, Liu YY, Chao TC, Hwang TL, Jan YY, Chen MF. Kinase mutations and imatinib mesylate response for 64 Taiwanese with advanced GIST: preliminary experience from Chang Gung Memorial Hospital. Ann Surg Oncol 2007; 14: 1123-1128
10 Akwari OE, Dozois RR, Weiland LH, Beahrs OH. Leio-myosarcoma of the small and large bowel. Cancer 1978; 42: 1375-1384
11 Shiu MH, Farr GH, Papachristou DN, Hajdu SI. Myosarco-mas of the stomach: natural history, prognostic factors and management. Cancer 1982; 49: 177-187
12 McGrath PC, Neifeld JP, Lawrence W Jr, Kay S, Horsley JS 3rd, Parker GA. Gastrointestinal sarcomas. Analysis of prog-nostic factors. Ann Surg 1987; 206: 706-710
13 Ng EH, Pollock RE, Munsell MF, Atkinson EN, Romsdahl MM. Prognostic factors influencing survival in gastrointesti-nal leiomyosarcomas. Implications for surgical management and staging. Ann Surg 1992; 215: 68-77
14 Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 1996; 2: 561-566
15 Buchdunger E, Cioffi CL, Law N, Stover D, Ohno-Jones S, Druker BJ, Lydon NB. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther 2000; 295: 139-145
16 Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zi-gler AJ. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 2000; 96: 925-932
17 Wang WL, Healy ME, Sattler M, Verma S, Lin J, Maulik G, Stiles CD, Griffin JD, Johnson BE, Salgia R. Growth inhibi-tion and modulation of kinase pathways of small cell lung cancer cell lines by the novel tyrosine kinase inhibitor STI 571. Oncogene 2000; 19: 3521-3528
18 Osusky KL, Hallahan DE, Fu A, Ye F, Shyr Y, Geng L. The receptor tyrosine kinase inhibitor SU11248 impedes endo-
2118 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Chen YY et al . Sunitinib for GIST after imatinib failure
COMMENTS

2119 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
thelial cell migration, tubule formation, and blood vessel for-mation in vivo, but has little effect on existing tumor vessels. Angiogenesis 2004; 7: 225-233
19 Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther 2003; 2: 471-478
20 Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, Miller T, Shirazian S, McMahon G, Cherrington JM. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic re-lationship. Clin Cancer Res 2003; 9: 327-337
21 Murray LJ, Abrams TJ, Long KR, Ngai TJ, Olson LM, Hong W, Keast PK, Brassard JA, O'Farrell AM, Cherrington JM, Pryer NK. SU11248 inhibits tumor growth and CSF-1R-dependent osteolysis in an experimental breast cancer bone metastasis model. Clin Exp Metastasis 2003; 20: 757-766
22 O'Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, Wong LM, Hong W, Lee LB, Town A, Smolich BD, Manning WC, Murray LJ, Heinrich MC, Cherrington JM. SU11248 is a novel FLT3 tyrosine kinase inhibitor with po-tent activity in vitro and in vivo. Blood 2003; 101: 3597-3605
23 Schueneman AJ, Himmelfarb E, Geng L, Tan J, Donnelly E, Mendel D, McMahon G, Hallahan DE. SU11248 maintenance therapy prevents tumor regrowth after fractionated irradia-tion of murine tumor models. Cancer Res 2003; 63: 4009-4016
24 Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour af-ter failure of imatinib: a randomised controlled trial. Lancet 2006; 368: 1329-1338
25 Shirao K, Nishida T, Doi T, Komatsu Y, Muro K, Li Y, Ueda E, Ohtsu A. Phase I/II study of sunitinib malate in Japanese patients with gastrointestinal stromal tumor after failure of prior treatment with imatinib mesylate. Invest New Drugs 2010; 28: 866-875
26 Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European Organi-zation for Research and Treatment of Cancer, National Can-cer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000; 92: 205-216
27 Cancer Therapy Evaluation Program. Common toxicity criteria (CTC) version 2.0. Bethesda, MD: National Cancer Institute, April 30, 1999. Available from: URL: http://ctep.
cancer.gov/protocolDevelopment/electronic_applications/docs/ctcv20_4-30-992.pdf
28 Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den Abbeele AD, Druker BJ, Kiese B, Eisenberg B, Roberts PJ, Singer S, Fletcher CD, Silberman S, Dimitrijevic S, Fletcher JA. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003; 21: 4342-4349
29 Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, Town A, McKinley A, Ou WB, Fletcher JA, Fletcher CD, Huang X, Cohen DP, Baum CM, Demetri GD. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 2008; 26: 5352-5359
30 Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, Raymond AK, Bramwell VH, Baker LH, Maki RG, Tanaka M, Hecht JR, Heinrich MC, Fletcher CD, Crowley JJ, Borden EC. Phase III randomized, intergroup tri-al assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol 2008; 26: 626-632
31 Zalcberg JR, Verweij J, Casali PG, Le Cesne A, Reichardt P, Blay JY, Schlemmer M, Van Glabbeke M, Brown M, Judson IR. Outcome of patients with advanced gastro-intestinal stromal tumours crossing over to a daily imatinib dose of 800 mg after progression on 400 mg. Eur J Cancer 2005; 41: 1751-1757
32 Miettinen M, Sarlomo-Rikala M, Lasota J. Gastrointestinal stromal tumors: recent advances in understanding of their biology. Hum Pathol 1999; 30: 1213-1220
33 Anderson R, Jatoi A, Robert C, Wood LS, Keating KN, Lacouture ME. Search for evidence-based approaches for the prevention and palliation of hand-foot skin reaction (HFSR) caused by the multikinase inhibitors (MKIs). Oncologist 2009; 14: 291-302
34 Lee S, Chung HC, Mainwaring P, Ng C, Chang JWC, Kwong P. An Asian subpopulation analysis of the safety and efficacy of sunitinib in metastatic renal cell carcinoma. Eur J Cancer Suppl 2009; 7: 428
35 Baffert F, Le T, Sennino B, Thurston G, Kuo CJ, Hu-Lowe D, McDonald DM. Cellular changes in normal blood capillaries undergoing regression after inhibition of VEGF signaling. Am J Physiol Heart Circ Physiol 2006; 290: H547-H559
36 Kamba T, Tam BY, Hashizume H, Haskell A, Sennino B, Mancuso MR, Norberg SM, O'Brien SM, Davis RB, Gowen LC, Anderson KD, Thurston G, Joho S, Springer ML, Kuo CJ, McDonald DM. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physi-ol Heart Circ Physiol 2006; 290: H560-H576
S- Editor Tian L L- Editor O’Neill M E- Editor Zheng XM
Chen YY et al . Sunitinib for GIST after imatinib failure

BRIEF ARTICLE
MELD score can predict early mortality in patients with rebleeding after band ligation for variceal bleeding
Wei-Ting Chen, Chun-Yen Lin, I-shyan Sheen, Chang-Wen Huang, Tsung-Nan Lin, Chun-Jung Lin, Wen-Juei Jeng, Chien-Hao Huang, Yu-Pin Ho, Cheng-Tang Chiu
Wei-Ting Chen, Chun-Yen Lin, I-Shyan Sheen, Chang-Wen Huang, Tsung-Nan Lin, Chun-Jung Lin, Wen-Juei Jeng, Chien-Hao Huang, Yu-Pin Ho, Cheng-Tang Chiu, Department of Hepato-Gastroenterology, Linkou Medical Center, Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Taoyuan 333, Taipei, Taiwan, ChinaAuthor Contributions: Ho YP and Chiu CT designed the research; Chen WT, Jeng WJ and Huang CH performed data collection; Chen WT, Lin CY and Ho YP analyzed and interpreted the data; Chen WT, Ho YP and Lin CJ wrote the manuscript; Lin CY and Sheen IS contributed critical revision of the manuscript for important intellectual content; Huang CW contributed statistical analysis; Lin CJ provided material support; the final version of the manuscript was approved by all authors.Correspondence to: Yu-Pin Ho, MD, Assistant Professor, Department of Hepato-Gastroenterology, Linkou Medical Center, Chang Gung Memorial Hospital and Chang Gung University College of Medicine, 5 Fu-Shin Street, Kweishan, Taoyuan 333, Taipei, Taiwan, China. [email protected]: +886-3-3281200 Fax: +886-3-3272236Received: October 7, 2010 Revised: December 1, 2010 Accepted: December 8, 2010Published online: April 28, 2011
AbstractAIM: To investigate the outcomes, as well as risk factors for 6-wk mortality, in patients with early rebleeding after endoscopic variceal band ligation (EVL) for esophageal variceal hemorrhage (EVH).
METHODS: Among 817 EVL procedures performed for EVH between January 2007 and December 2008, 128 patients with early rebleeding, defined as rebleeding within 6 wk after EVL, were enrolled for analysis.
RESULT: The rate of early rebleeding after EVL for acute EVH was 15.6% (128/817). The 5-d, 6-wk, 3-mo, and 6-mo mortality rates were 7.8%, 38.3%, 55.5%, and 58.6%, respectively, in these early rebleeding patients. The use of beta-blockers, occurrence of hypovolemic
shock, and higher model for end-stage liver disease (MELD) score at the time of rebleeding were independent predictors for 6-wk mortality. A cut-off value of 21.5 for the MELD score was found with an area under ROC curve of 0.862 (P < 0.001). The sensitivity, specificity, positive predictive value, and negative predictive value were 77.6%, 81%, 71.7%, and 85.3%, respectively. As for the 6-mo survival rate, patients with a MELD score ≥ 21.5 had a significantly lower survival rate than patients with a MELD score < 21.5 (P < 0.001).
CONCLUSION: This study demonstrated that the MELD score is an easy and powerful predictor for 6-wk mortality and outcomes of patients with early rebleeding after EVL for EVH.
© 2011 Baishideng. All rights reserved.
Key words: Model for end-stage liver disease score; Es-ophageal variceal hemorrhage; Rebleeding; Cirrhosis; Mortality
Peer reviewer: Igor Mishin, MD, PhD, First Department of Surgery “N.Anestiadi” and Laboratory of Hepato-Pancreato-Biliary Surgery, Medical University "N.Testemitsanu", National Center of Emergency Medicine, Str. T Chorba 1, 2001 Kishinev, Republic of Moldova
Chen WT, Lin CY, Sheen IS, Huang CW, Lin TN, Lin CJ, Jeng WJ, Huang CH, Ho YP, Chiu CT. MELD score can pre-dict early mortality in patients with rebleeding after band ligation for variceal bleeding. World J Gastroenterol 2011; 17(16): 2120-2125 Available from: URL: http://www.wjgnet.com/1007-9327/full/v17/i16/2120.htm DOI: http://dx.doi.org/10.3748/wjg.v17.i16.2120
INTRODUCTIONEsophageal variceal hemorrhage (EVH) is a serious com
2120
World J Gastroenterol 2011 April 28; 17(16): 2120-2125 ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2120
April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

Chen WT et al . Predictors for early mortality with rebleeding
plication of liver cirrhosis and causes 70% of all upper gastrointestinal bleeding episodes in patients with portal hypertension[1]. According to the Baveno Consensus Workshop in portal hypertension, endoscopic variceal band ligation (EVL) therapy is recommended for acute EVH, although endoscopic sclerotherapy may be used if ligation is technically difficult[2]. According to the natural course of EVH, the risk of a recurrent episode of EVH increases after the first EVH but becomes similar to nonbleeding esophageal varices (EV) after 6 wk[3]. Therefore, rebleeding within 6 wk after the first EVH is coined as early rebleeding. Secondary prophylaxis could reduce the early rebleeding rate to 20%[1]. Several factors have been identified as predictors of mortality after EVH, including early rebleeding, bacterial infection[4], hepatic venous pressure gradient (HVPG) > 20 mmHg measured shortly after admission[5], active bleeding at initial endoscopy, severity of initial bleeding, hematocrit level, AST levels, presence of portal vein thrombosis or of hepatocellular carcinoma (HCC), alcoholic liver disease, serum bilirubin and albumin levels, ChildTurcottePugh (CTP) score[1], and Model for Endstage Liver Disease (MELD) score[68]. Among these predictors, early rebleeding is the most important one[3,9]. However, little information is known about the risk factors for mortality in the group of patients with early rebleeding. Thus, the goal of this retrospective study was to investigate the predictive factors for mortality in patients with early rebleeding.
MATERIALS AND METHODSA total of 817 consecutive EVL procedures for esophageal variceal bleeding were recorded and evaluated in a 3500bed tertiary referral medical center between January 2007 and December 2008. All of the patients with early rebleeding, defined as rebleeding between one day and 6 wk after ligation, were enrolled. The patients without endoscopic confirmation of rebleeding focus were excluded. The appropriately convened Institutional Review Board approved this study. Finally, 128 cirrhotic patients (15.6%) with early rebleeding were enrolled in our study. Among these patients, 49 patients who died within 6wk after rebleeding were classified as the mortality group. The remaining 79 patients who survived more than 6 wk were classified as the survival group. The clinical characteristics and laboratory data of the patients in these 2 groups were collected for comparison. Vasoactive drug therapy (terlipressin, somatostatin, or octreotide) was routinely administered before diagnostic endoscopic examination and was continued for at least 3 d according to national insurance guidelines of Taiwan for variceal hemorrhage. Prophylactic antibiotic treatment with intravenous ceftriaxone and nonselective betablockers were prescribed for some, depending on the patients’ clinical condition, contraindication, adverse effect with tolerability, and physician’s preference. Diagnosis of liver cirrhosis was based on a previous liver biopsy or compatible clinical, laboratory, and imaging findings. Hepatocellular carcinoma (HCC) was diagnosed by liver biopsy, fine needle aspiration cytology, or com
bined typical dynamic imaging appearance and elevated αfetoprotein (AFP). According to the tumor size, patients with HCC were divided into early (one nodule ≤ 5 cm or maximum three nodules, each < 3 cm) or advanced (one nodule > 5 cm or > 3 nodules). The diagnosis of infection was made by positive results of blood, sputum, urine, and ascites bacterial culture or elevated ascites fluid and absolute neutrophil count (ANC) ≥ 250 cells/μL. In addition, 6-wk mortality was defined as death occurring within 6 wk after rebleeding. The first EVL procedure applied to esophageal variceal bleeding during our study period was considered as the index EVL and index bleeding. The following definitions were used on the basis of the recommendations of the Baveno Consensus Workshop: (1) esophageal variceal bleeding: (a) visible oozing or spurting of blood from a esophageal varix, (b) white nipple sign or blood clot adherent to a varix, (c) presence of medium or large esophageal varices with no other potential bleeding lesion; (2) EVL ulcer bleeding: bleeding from esophageal ulcers after endoscopic EVL with one of the following: (a) active bleeding from the ulcer site, (b) adherent clot at the ulcer site, or (c) absence of other potential bleeding lesions; (3) bleeding duration: the acute bleeding episode was considered finished at the beginning of the first 24-h interval with no hematemesis, stable hemoglobin concentration without blood transfusions, and stable hemodynamic condition; (4) early rebleeding: recurrence of clinically significant hemorrhage (hematemesis/melena, aspiration of greater than 100 mL of fresh blood from nasogastric tube or > 3 g/dL decrease of Hb if no transfusion is given) within 6 wk after index bleeding episode was considered finished; (5) rebleeding 5-d failure: uncontrolled bleeding, death, or recurrent hemorrhage within 5 d since rebleeding; and (6) portal hypertensive gastropathy (PHG) bleeding: a macroscopic finding of a characteristic mosaic-like pattern of gastric mucosa with redpoint lesions, cherry red spots, and/or blackbrown spots (severe PHG) and the absence of other potential bleeding lesions.
Statistical analysisStatistical analysis was performed after proper tabulation of data. Continuous variables were expressed as mean with range, and categorical variables were expressed as count with percentage. Groups were compared using Student’s software ttest for continuous variables and χ2 test for categorical variables. Multivariate analysis was performed using logistic regression, and a receiver operating characteristic (ROC) curve was generated to assess the predictive accuracy of the variables. All of these values were considered statistically significant if the Pvalue was < 0.05. Cumulative survival estimates were calculated by using the KaplanMeier method. All statistical analyses were performed with SPSS statistical for Windows (Version 16; SPSS. Inc., Chicago, IL, USA).
RESULTSThe relevant characteristics of these 128 rebleeding pa
2121 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

tients are reported in Table 1. The mean age of the patients was 54 years old (range 1482); 83.6% were male and 16.4% were female. The etiologies of liver cirrhosis were virus (47.7%), alcohol (24.2%), or combined virus and alcohol (21.9%). The endoscopic findings at index bleeding were active EV bleeding (30.5%), blood in the esophageal or gastric lumen (38.3%), and clean esophagogastroduodenal lumen (31.2%). The average interval between index EVL and rebleeding was 14 ± 10.8 d. The average CTP scores at the time of index bleeding and rebleeding were 9.8 ± 2.3 and 10.1 ± 2.6, and the average MELD scores were 19.8 ± 8.9 and 21.4 ± 9.8, respectively.
The surveillance of rebleeding sites was carried out by upper GI endoscopy within 1 day for all enrolled patients; the rebleeding site, therapeutic methods, and outcomes are show in Table 2. The surveillance revealed 75 patients with residual EV bleeding (58.6%), 24 patients with EVLrelated esophageal ulcer bleeding (18.8%), 7 patients with gastric variceal bleeding (5.5%), 11 patients with peptic ulcer bleeding (8.6%), and 11 patients with PHGrelated bleeding (8.6%). The management methods for rebleeding included combined endoscopic and pharmacologic therapy (71.9%) and pharmacologic therapy only for EVL ulcers or PHG with mild oozing (27.3%). Only one patient was treated with surgical intervention (0.8%). In total, 24 pa
tients (18.8%) were associated with 5d failure at rebleeding of which 8 patients had uncontrolled bleeding and died within 5 d, 2 patients died of other causes within 5 d, and 14 patients had recurrent bleeding within 5 d. The rebleeding mortality rates at 5 d, 6 wk, 3 mo, and 6 mo were 7.8%, 38.3%, 55.5%, and 58.6%, respectively. The causes of death within 6 wk after rebleeding were sepsisinduced multiple organ failure (53.1%), upper gastrointestinal tract hemorrhage (36.7%), and liver failure (10.2%).
We then further divided the patients into two groups according to their mortality or survival during the 6wk period after rebleeding. The clinical characteristics of patients in the 6wk mortality group and the survival group are displayed and compared in Table 3. There were no significant differences between these two groups with regard to gender, age, etiology of cirrhosis, HCC, PVT, duration between rebleeding and index EVL, rebleeding focus and treatment methods, antibiotic use, serum platelet count, and sodium and potassium level at rebleeding. However, higher CTP score (11.9 vs 8.9), higher MELD score (28.9 vs 16.8), and hypovolemic shock during rebleeding (P < 0.001); higher serum total bilirubin, creatinine, and white cell count levels; lower serum albumin and hemoglobin levels; longer prothrombin time (INR) and active bleeding on endoscopy; and higher hepatic encephalopathy grade were markedly seen in the 6wk mortality group. Betablocker use after rebleeding was also significantly associated with 6wk mortality.
Furthermore, by multivariate logistic regression analysis, hypovolemic shock (OR = 9.25, 95% CI: 1.6850.93, P = 0.011), betablocker use after rebleeding (OR = 0.18, 95%
2122 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 2 Rebleeding focus, therapy and outcome of patients with rebleeding after endoscopic variceal band ligation for esophageal varices bleeding n (%)
Overall (n = 128)
Rebleeding focus EV bleeding 75 (58.6) Post-EVL ulcer bleeding 24 (18.8) GV bleeding 7 (5.5) Peptic ulcer bleeding 11 (8.6) PHG 11 (8.6)Therapy for rebleeding Endoscopic and pharmacologic therapy 92 (71.9) Pharmacologic therapy only 35 (27.3) Surgery 1 (0.8)Rebleeding 5-d failure 24 (18.8) Uncontrolled bleeding and died within 5 d 8 (6.3) Died of non-bleeding cause 2 (1.6) Recurrent bleeding within 5 d 14 (10.9)5-d mortality 10 (7.8)6-wk mortality 49 (38.3)6-wk mortality causes Sepsis-induced multi-organ failure 26 (53.1) GI bleeding 18 (36.7) Liver failure 5 (10.2)3-mo mortality 71 (55.5)6-mo mortality 75 (58.6)
EV: Esophageal varices; EVL: Endoscopic variceal band ligation; GV: Gas-tric varices; PHG: Portal hypertensive gastropathy; GI: Gastrointestinal.
Table 1 Characteristics of patients with rebleeding after en-doscopic variceal band ligation for esophageal varices bleeding (mean ± SD) n (%)
Variable Overall (n = 128)
Sex (male/female) 107 (83.6)/21 (16.4)Age (yr) 53.6 ± 13.9Etiology Virus 61 (47.7) Alcohol 31 (24.2) Virus + alcohol 28 (21.9) Others 8 (6.2)Index EGD finding Active EV bleeding 39 (30.5) Blood in lumen 49 (38.3) Clean 40 (31.2)Duration between rebleeding and Index EVL 14.0 (10.8) 1-5 d 33 (25.8) 6-42 d 95 (74.2)Index EVL CTP score 9.8 ± 2.3 CTP classification A 12 (9.40) B 44 (34.4) C 72 (56.2) MELD 19.8 ± 8.9Rebleeding CTP score 10.1 ± 2.6 CTP classification A 14 (10.9) B 36 (28.1) C 78 (60.9) MELD 21.4 ± 9.8
EVL: Endoscopic variceal band ligation; EV: Esophageal varices; CTP: Child-turcotte-pugh; MELD: Model for end-stage liver disease; EGD: Esophago-gastroduodenoscopy.
Chen WT et al . Predictors for early mortality with rebleeding

CI: 0.050.63, P = 0.007), and higher MELD score (OR = 1.17, 95% CI: 1.101.25, P < 0.001) at rebleeding were found to be independent factors for 6wk mortality in these patients and this is reported in Table 4. The ROC curve
was used for predicting 6wk mortality in cirrhotic patients with early rebleeding, and the area under ROC curve (AUROC) of the MELD score for predicting 6wk mortality was 0.862 (95% CI: 0.800.93, P < 0.001). An optimized
2123 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 3 Variables associated with 6-wk mortality in patients with rebleeding after endoscopic variceal band ligation for esophageal varices bleeding (mean ± SD) n (%)
Variable 6-wk mortality (n = 49, 38.3%) 6-wk survival (n = 79, 61.7%) P -value
Sex (male/female) 40 (81.6)/9 (18.8) 67 (84.8)/12 (17.6) 0.637Age (yr) 53.3 ± 13.3 53.7 ± 14.3 0.866Etiology 0.683 Virus 23 (46.9) 38 (48.1) Alcohol 11 (22.4) 20 (25.3) Virus + alcohol 13 (26.5) 15 (19.0) Others 2 (4.1) 6 (7.6)HCC 0.291 No or small 29 (59.2) 54 (68.4) Advanced 20 (40.8) 25 (31.6)PVT 0.786 Main trunk 12 (24.5) 18 (22.8) Branch 5 (10.2) 9 (11.4)Duration between rebleeding and index EVL 12.8 ± 9.0 14.7 ± 11.7 0.528 1-5 d 13 (26.5) 20 (25.3) 0.879 6-42 d 36 (73.5) 59 (74.7)Rebleeding laboratory findings CTP score 11.9 ± 1.9 8.9 ± 2.3 0.000 CTP classification A 1 (2.0) 13 (16.5) 0.017 B 3 (6.1) 33 (41.8) 0.000 C 45 (91.8) 33 (41.8) 0.000 MELD score 28.9 ± 9.0 16.8 ± 7.0 0.000 HE (grade) 1.3 ± 1.5 0.3 ± 0.8 0.000 Cr (μmol/L) 203.3 ± 159.1 114.9 ± 97.2 0.000 Na (mmol/L) 138.7 ± 9.6 134.6 ± 14.3 0.084 K (mmol/L) 4.2 ± 1.0 3.9 ± 0.9 0.171 Bil (μmol/L) 263.3 ± 236.0 90.6 ± 135.1 0.000 Alb (g/L) 25 ± 5 29 ± 6 0.000 Hb (g/L) 83 ± 18 90 ± 17 0.041 WBC (109/L) 12.5 ± 6.4 8.2 ± 4.7 0.000 Platelets (109/L) 102.4 ± 69.2 101.0 ± 72.3 0.915 INR (PT) 2.2 ± 0.9 1.5 (0.4) 0.000Rebleeding 5-d failure 18 (36.7) 6 (7.6) 0.000 Died within 5 d 10 0 0.000 Recurrent bleeding within 5 d 8 6 0.150Hypovolemic shock 21 (42.9) 3 (3.8) 0.000Rebleeding focus 0.500 EV bleeding 28 (57.1) 47 (59.5) Post-EVL ulcer bleeding 9 (18.4) 15 (19.0) GV bleeding 3 (6.1) 4 (5.1) Peptic ulcer bleeding 6 (12.2) 5 (6.3) PHG 3 (6.1) 8 (10.1)Rebleeding EGD finding 0.019 Active bleeding 20 (40.8) 30 (38.0) Blood in lumen 25 (51.0) 27 (34.2) Clean 4 (8.2) 22 (27.8)EGD Tx 0716 Endoscopic therapy + medical 36 (73.5) 56 (70.9) Medical therapy only 13 (26.5) 22 (27.8) Surgery 0 (0.0) 1 (1.3)Rebleeding infection 42 (85.7) 36 (45.6) 0.000Rebleeding antibiotic use 35 (71.4) 46 (58.2) 0.132Rebleeding Inderal use 8 (16.3) 37 (46.8) 0.000
The P-values were calculated by using Student’s t-test for continuous variables and χ2 test for categorical variables. EV: Esophageal varices; EVL: Endoscopic variceal band ligation; HCC: Hepatocellular carcinoma; PVT: Portal vein thrombosis; CTP: Child-turcotte-pugh; MELD: Model for end-stage liver disease; INR (PT): International normalised ratio of a patient's prothrombin time to a normal control sample; PHG: portal hypertensive gastropathy; EGD: Esophagogastro-duodenoscopy; GV: Gastric varices.
Chen WT et al . Predictors for early mortality with rebleeding

cutoff value of the MELD score is 21.5. As shown in Table 5, the MELD score had a good sensitivity of 78%, specificity of 81%, positive predictive value (PPV) of 72%, and negative predictive value (NPV) of 85% for predicting 6wk mortality. By the above analyses, a MELD score of ≥ 21.5 was subsequently chosen as the value for identifying patients with a high risk of death at 6 wk after rebleeding.
The Kaplan-Meier survival curves in patients classified according to a MELD score of < 21.5 and ≥ 21.5 revealed a significant difference as shown in Figure 1. The mortality rate was 14.7% in patients with MELD <21.5 and 71.7% in patients with MELD ≥ 21.5 at 6 wk (P < 0.001); 36% in patients with MELD < 21.5 and 83% in patients MELD ≥ 21.5 at 3 mo (P < 0.001); and 40% in patients with MELD < 21.5 and 84.9% in patients MELD ≥ 21.5 at 6 mo (P < 0.001), respectively.
DISCUSSIONOur study has revealed that potential rebleeding sources in cirrhosis cases after index EVL for EVH were esophageal varices (58.6%), esophageal ulcer (18.8%), peptic ulcer (8.6%), PHG (8.6%), and gastric varices (5.5%). The results were consistent with a previous study[1] reporting that residual esophageal varices were a major source of rebleeding. In addition, betablocker usage after rebleeding, hypovolemic shock, and higher MELD score at the time of rebleeding were independent predictors of 6wk mortality. In addition, we found that the 6wk mortality rate was 14.7% for patients with MELD scores less than 21.5 and
71.7% for patients with MELD scores more than 21.5. The mortality rate within 6 wk in our study was 38.3%,
which is higher than the mortality rate of patients after acute variceal bleeding[1,6]. This is probably because the patients with rebleeding after EVH were more advanced in disease severity than patients with initial acute EVH. This difference in severity could be reflected in the mean MELD score; the mean score was 21.4 in the rebleeding patients of our study group, higher than the group of acute variceal bleeding with a MELD score of 12 as previously reported[6].
The presence of HCC could influence both early rebleeding and mortality in patients with EVH, as reported previously[1,10]. However, in the present study, advanced HCC and portal vein thrombosis were not predictors of 6wk mortality, which is consistent with previous reports that advanced HCC is not an independent risk factor but MELD score is a good predictor for early mortality after EVH[11].
Another independent factor associated with 6wk mortality in our study was rebleeding related to hypovolemic shock. This observation was similar to previous studies that found that the severity of the hemorrhage was predictive of 6wk mortality in acute EVH of all cirrhotic patients[1,6]. The third independent factor associated with 6wk mortality was the use of beta-blockers, which reflected the general consensus that the use of betablockers for the secondary prevention of EVH could reduce mortality[12]. Overall, 49 patients (38.3%) died within 6 wk after early rebleeding;
2124 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 5 Sensitivity, specificity, positive predictive value, and negative predictive value for predicting 6-wk mortality in pa-tients with rebleeding after endoscopic variceal band ligation for esophageal varices bleeding n (%)
6-wk mortality Sensitivity Specificity PPV NPV
MELD score ≥ 21.5 77.55 81.01 71.7 85.33Hypovolemic shock (+) 42.86 96.20 87.5 26.92Beta-blocker use after rebleeding 83.67 46.84 49.4 82.22
PPV: Positive predictive value; NPV: Negative predictive value; MELD: Model for end-stage liver disease.
Rebleeding MELD < 21.5
Rebleeding MELD ≥ 21.5
0 30 60 90 120 150 180
Time since rebleeding (d)
1.0
0.8
0.6
0.4
0.2
0.0
Cum
sur
viva
l
Figure 1 Kaplan-Meier survival curves in patients classified according to model for end-stage liver disease score < 21.5 or ≥ 21.5 (P < 0.001). MELD: Model for end-stage liver disease.
Table 4 Logistic regression models for variables associated with 6-wk mortality in patients with rebleeding after endoscopic variceal band ligation for esophageal varices bleeding
Variables UV MVEstimate P -value Estimate OR 95% CI P -value
Rebleeding laboratory CTP score 0.61 < 0.001 MELD score 0.17 < 0.001 0.16 1.17 1.10-1.25 < 0.001 HE (grade) 0.76 < 0.001 Cr 0.61 0.001 Bil 0.09 < 0.001 Alb -1.18 0.001 Hb -0.23 0.045 WBC 0.15 < 0.001 INR (PT) 1.85 < 0.001Rebleeding 5-d failure
1.96 < 0.001
Hypovolemic shock
2.94 < 0.001 2.23 9.25 1.68-50.93 0.011
Rebleeding EGD finding
-1.47 0.011
Rebleeding infection
1.97 < 0.001
Rebleeding Inderal use
-1.51 0.001 -1.7 0.18 0.05-0.63 0.007
UV: Univariate analysis; MV: Multivariate analysis; CTP: Child-turcotte-pugh; MELD: Model for end-stage liver disease; HE: Hepatic encephalopa-thy; INR (PT): International normalised ratio of a patient's prothrombin time to a normal control sample; EGD: Esophagogastroduodenoscopy.
Chen WT et al . Predictors for early mortality with rebleeding

among of them, 10 died within 5 d, and 39 died from day 6 to day 42. In our study, the causes of death were sepsisrelated multiple organ failure (53.1%), GI bleedingrelated complications (36.7%), and liver failurerelated complications (10.2%); our results were similar to a recent study reporting that early mortality after cessation of initial EV bleeding is significantly associated with bacterial infection and rebleeding[13]. This finding provides evidence to support the AASLD guidelines for the treatment of acute variceal bleeding regarding the early use of pharmacological agents and emergent endoscopic procedure within 12 h[14]. Additionally, for reducing sepsisrelated multiorgan failure, prophylactic use of antibiotics for all patients with cirrhosis and GI hemorrhage should be encouraged[2,15]. Patients with a CTP classification of A respond well to current therapies with minimal risk of death and represented only 2% of the patients in the 6wk mortality group in our study. Whether current treatment recommendations should be applied to all patients should be further investigated[16].
In conclusion, this study examined the focuses of rebleeding and treatment outcomes in cirrhotic patients with early rebleeding after EVL for acute EVH. Specifically, the study revealed that hypovolemic shock and MELD scores ≥ 21.5 at the time of rebleeding are predictors for 6wk mortality in patients with early rebleeding after EVL for acute EVH. Also, betablocker use after rebleeding was associated with lower 6wk mortality.
COMMENTSBackgroundThe management of variceal bleeding remains a clinical challenge with high mortality. At the present time, available treatments have reduced the 6-wk re-bleeding rate to 20%. Early rebleeding is a strong predictor of death from vari-ceal bleeding. Endoscopic therapy increases control of bleeding and decreases the risk of rebleeding and mortality. Despite the fact that endoscopic variceal ligation (EVL) is recommended for acute esophageal variceal bleeding in recent practice guidelines, there has been relatively little research investigating the situation of early rebleeding after EVL for esophageal variceal bleeding.Research frontiersCurrently, treatment recommendations are applied to all patients with variceal bleeding. At present, only 40% of deaths are directly related to bleeding, while the majority are caused by infection-related multiple organ failure that is paralleled with the severity of liver cirrhosis. Patients with Child-Pugh classification A have good response to current therapy, with a minimal risk of mortality. However, treat-ment strategies might be different with different Child-Pugh classification.Innovations and breakthroughsThis study provides evidence that there are independent predictors for 6-wk mortality and rebleeding origin in cirrhotic patients with early rebleeding after therapeutic endoscopic band ligation of initial esophageal varices bleeding. Applications This article shows significantly less beta blocker use in the mortality group and recognizes this as an independent poor predictor. To prevent rebleeding with associated mortality, secondary prophylaxis with beta blockers should start as soon as possible from the day after stopping usage of vasoactive drugs. Furthermore, the authors demonstrate that Model for End-Stage Liver Disease (MELD) score is an easy and accurate predictor of 6-wk mortality of patients with early rebleeding after EVL for esophageal variceal bleeding. Accurate pre-dictive rules are provided for early recognition of high risk patients.TerminologyThe Model for End-Stage Liver Disease, or MELD, is a scoring system for assessing the severity of chronic liver disease. It was initially developed
to predict death within three months of surgery in patients who had under-gone a transjugular intrahepatic protosystemic shut procedure, and was subsequently found to be useful in determining prognosis and prioritizing for receipt of liver transplant instead of the older Child-Pugh score. MELD Score = [0.957 × In(Serum Cr) + 0.378 × In(Serum Bilirubin) + 1.120 × In(INR) + 0.643] × 10.Peer reviewThe manuscript is a well designed retrospective study with the aim to investigate the predictive factors for mortality in patients with early rebleeding.
REFERENCES1 D'Amico G, De Franchis R. Upper digestive bleeding in cir-
rhosis. Post-therapeutic outcome and prognostic indicators. Hepatology 2003; 38: 599-612
2 de Franchis R. Evolving consensus in portal hypertension. Report of the Baveno IV consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol 2005; 43: 167-176
3 Graham DY, Smith JL. The course of patients after variceal hemorrhage. Gastroenterology 1981; 80: 800-809
4 Goulis J, Armonis A, Patch D, Sabin C, Greenslade L, Bur-roughs AK. Bacterial infection is independently associated with failure to control bleeding in cirrhotic patients with gas-trointestinal hemorrhage. Hepatology 1998; 27: 1207-1212
5 Abraldes JG, Villanueva C, Bañares R, Aracil C, Catalina MV, Garci A-Pagán JC, Bosch J. Hepatic venous pressure gradient and prognosis in patients with acute variceal bleeding treated with pharmacologic and endoscopic therapy. J Hepatol 2008; 48: 229-236
6 Bambha K, Kim WR, Pedersen R, Bida JP, Kremers WK, Ka-math PS. Predictors of early re-bleeding and mortality after acute variceal haemorrhage in patients with cirrhosis. Gut 2008; 57: 814-820
7 Kamath PS, Wiesner RH, Malinchoc M, Kremers W, Therneau TM, Kosberg CL, D'Amico G, Dickson ER, Kim WR. A model to predict survival in patients with end-stage liver disease. Hepatology 2001; 33: 464-470
8 Kamath PS, Kim WR. The model for end-stage liver disease (MELD). Hepatology 2007; 45: 797-805
9 Bosch J, Abraldes JG, Berzigotti A, Garcia-Pagan JC. Portal hy-pertension and gastrointestinal bleeding. Semin Liver Dis 2008; 28: 3-25
10 Lo GH, Lai KH, Chang CF, Shen MT, Jeng JS, Huang RL, Hwu JH. Endoscopic injection sclerotherapy vs. endoscopic variceal ligation in arresting acute variceal bleeding for patients with ad-vanced hepatocellular carcinoma. J Hepatol 1994; 21: 1048-1052
11 Amitrano L, Guardascione MA, Bennato R, Manguso F, Balz-ano A. MELD score and hepatocellular carcinoma identify pa-tients at different risk of short-term mortality among cirrhotics bleeding from esophageal varices. J Hepatol 2005; 42: 820-825
12 D'Amico G, Pagliaro L, Bosch J. Pharmacological treatment of portal hypertension: an evidence-based approach. Semin Liver Dis 1999; 19: 475-505
13 Lee SW, Lee TY, Chang CS. Independent factors associated with recurrent bleeding in cirrhotic patients with esophageal variceal hemorrhage. Dig Dis Sci 2009; 54: 1128-1134
14 Garcia-Tsao G, Sanyal AJ, Grace ND, Carey W. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology 2007; 46: 922-938
15 Fernández J, Ruiz del Arbol L, Gómez C, Durandez R, Serra-dilla R, Guarner C, Planas R, Arroyo V, Navasa M. Norfloxacin vs ceftriaxone in the prophylaxis of infections in patients with advanced cirrhosis and hemorrhage. Gastroenterology 2006; 131: 1049-1056; quiz 1285
16 Garcia-Tsao G, Bosch J. Management of varices and variceal hemorrhage in cirrhosis. N Engl J Med 2010; 362: 823-832
S- Editor Tian L L- Editor Logan S E- Editor Ma WH
2125 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Chen WT et al . Predictors for early mortality with rebleeding
COMMENTS

BRIEF ARTICLE
Study on chronic pancreatitis and pancreatic cancer using MRS and pancreatic juice samples
Jian Wang, Chao Ma, Zhuan Liao, Bing Tian, Jian-Ping Lu
Jian Wang, Chao Ma, Bing Tian, Jian-Ping Lu, Department of Radiology, Changhai Hospital, The Second Military Medical University, Shanghai 200433, ChinaZhuan Liao, Department of Gastroenterology, Changhai Hospital, The Second Military Medical University, Shanghai 200433, ChinaAuthor contributions: Wang J and Ma C contributed equally to this work and performed the majority of experiments and data analysis; Liao Z provided the vital reagents and analytical tools; Tian B collected of pancreatic juice samples; Lu JP designed the study and wrote the manuscript.Supported by Grants from the National Natural Science Foundation of China, No. 30870709 and the Program of Shanghai Subject Chief Scientist, No. 08XD14002(A)Correspondence to: Jian-Ping Lu, MD, Department of Radiology, Changhai Hospital, The Second Military Medical University, 168 Changhai Road, Shanghai 200433, China. [email protected]: +862181873637 Fax: +862181873637Received: August 17, 2010 Revised: December 10, 2010 Accepted: December 17, 2010Published online: April 28, 2011
AbstractAIM: To investigate the markers of pancreatic diseases and provide basic data and experimental methods for the diagnosis of pancreatic diseases.
METHODS: There were 15 patients in the present study, among whom 10 had pancreatic cancer and 5, chronic pancreatitis. In all patients, pancreatic cancer or chronic pancreatitis was located on the head of the pancreas. Pathology data of all patients was confirmed by biopsy and surgery. Among the 10 patients with pa-ncreatic cancer, 3 people had a medical history of long-term alcohol consumption. Of 5 patients with chronic pancreatitis, 4 men suffered from alcoholic chronic pancreatitis. Pancreatic juice samples were obtained from patients by endoscopic retrograde cholangio-pancreatography. Magnetic resonance spectroscopyn was performed on an 11.7-T scanner (Bruker DRX-500) using Call-Purcell-Meiboom-Gill pulse sequences.
The parameters were as follows: spectral width, 15 KHz; time domain, 64 K; number of scans, 512; and acquisition time, 2.128 s.
RESULTS: The main component of pancreatic juice inc-luded leucine, iso-leucine, valine, lactate, alanine, acetate, aspartate, lysine, glycine, threonine, tyrosine, histidine, tryptophan, and phenylalanine. On performing 1D 1H and 2D total correlation spectroscopy, we found a triplet peak at the chemical shift of 1.19 ppm, which only ap-peared in the spectra of pancreatic juice obtained from patients with alcoholic chronic pancreatitis. This triplet peak was considered the resonance of the methyl of ethoxy group, which may be associated with the metab-olism of alcohol in the pancreas.
CONCLUSION: The triplet peak, at the chemical shift of 1.19 ppm is likely to be the characteristic metabolite of alcoholic chronic pancreatitis.
© 2011 Baishideng. All rights reserved.
Key words: Pancreatic juice; Pancreatic cancer; Chronic pancreatitis; Magnetic resonance spectroscopy; Magnetic resonance imaging
Peer reviewer: Vinay Kumar Kapoor, Professor, Department of Surgical Gastroenterology, Sanjay Gandhi PostGraduate Institute of Medical Sciences, Lucknow 226014, India
Wang J, Ma C, Liao Z, Tian B, Lu JP. Study on chronic pancreatitis and pancreatic cancer using MRS and pancreatic juice samples. World J Gastroenterol 2011; 17(16): 21262130 Available from: URL: http://www.wjgnet.com/10079327/full/v17/i16/2126.htm DOI: http://dx.doi.org/10.3748/wjg.v17.i16.2126
INTRODUCTIONPancreatic cancer accounts for about 2% of all cancer cases, but it has the worst prognosis of all cancers with a 5-year
2126
World J Gastroenterol 2011 April 28; 17(16): 2126-2130 ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2126
April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

Wang J et al . MRS for pancreatic diseases
survival rate of less than 3%[1,2]. Because of the deep-seated location of the pancreas and no apparent symptoms at the initial stages of pancreatic cancer, it is difficult to diagnose this disease in the early stages. Chronic pancreatitis is a kind of localized or diffuse inflammation, and is caused by many factors. One of the medical dilemmas is to distinguish pancreatic cancer from chronic pancreatitis with a mass in the head of the pancreas; both these diseases have similar clinical behavior and imaging features[3]. A puncture biopsy is usually preferred over an operation when diagnosing the disease, but this is an injurious procedure and may lead to some complications[4].
Magnetic resonance imaging (MRI) is the most com-mon procedure used in the diagnosis of pancreatic cancer. An MRI helps obtain images of the pancreas and its sur-rounding structures[5]. The deep-seated location of the pancreas and similar clinical manifestations of chronic pancreatitis and pancreatic cancer are the main barriers in differentiating between these two diseases even with ad-vanced MRI techniques. Magnetic resonance spectroscopy (MRS) has high sensitivity and resolution, allows in vitro testing of metabolites, and has been widely used in the field of metabolomics[6-12]. MRS will be the most potent tool to help differentiate between pancreatic cancer and chronic pancreatitis, and it will be the most effective tool for the early diagnosis of a pancreatic tumor.
Usually, in the process of cancerization, gene and metabolite abnormalities appear before tissue structure transformation. Detection of abnormalities in metabolites facilitates early diagnoses of tumors. Clinically, the serum marker CA19-9[13] and gene tumor markers such as the K-ras gene[14,15], p53 anti-oncogene, and p53 protein[16-19] are widely used as markers of pancreatic cancer; however, these markers are not sensitive, show low specificity, and are used as auxiliary tools[20]. Beger et al[10] had successfully used MRS and mass spectrum (MS) to analyze blood constituents of patients with pancreatic cancer and of healthy volunteers; they were able to make a good distinction between pancre-atic cancer and the control group by performing lipid pro-filing of the blood. Pancreatic juice is the exocrine of the pancreas, and is closely related with pancreatic tissues. We wanted to investigate whether it is possible to obtain some information to help differentiate between chronic pancre-atitis and pancreatic cancer by analyzing pancreatic juice samples. In the present study, we used MRS technology to analyze the pancreatic juice of patients with pancreatic can-cer or chronic pancreatitis with a mass in the head of the pancreas and tried to explore the markers of these diseases. We provided basic data and experimental methods for the study of pancreatopathy.
MATERIALS AND METHODSThe initial subject population comprised 35 patients with pancreatic cancer (24 men and 11 women; mean age, 67.2 years; age range, 47-85 years) recruited between January 2006 and June 2009. We selected 10 subjects (7 men and 3 women; mean age, 67.7 years; age range, 57-74 years) with surgically confirmed pancreatic cancer. The mean maxi-
mum lesion diameter was 26.1 mm (range, 11-51 mm), and all lesions were located in the head of the pancreas. We chose 5 patients (4 men and 1 woman; mean age, 58.3 years) with chronic pancreatitis, confirmed by in vivo bi-opsy, and lesions located at the head of the pancreas. The medical history of every patient was recorded in detail. All study protocols were approved by our Institutional Re-view Board, and informed consent was obtained from all patients before they were enrolled in this study.
Pancreatic juice samples were obtained from patients by endoscopic retrograde cholangio-pancreatography (ERCP) in frozen tubes and immediately placed in liquid nitrogen before storing the tubes in a -80℃ refrigerator for MRS experiments. All patients were diagnosed by bi-opsy analyses of pathology data. Both the tumor marker CA19-9 and the cancer gene maker p53 were detected in patients with pancreatic cancer, whereas these markers were not detected in patients with chronic pancreatitis.
MRS experimentsMRS experiments were performed on a Bruker DRX-500 spectrometer (1H frequency, 500.13 MHz; Bruker Bio-spin, Rheinstetten, Germany). Pancreatic juice samples were diluted with phosphate buffer in D2O and placed in sample tubes (diameter, 5 mm), which are used in MRS experiments. Spectra were acquired at 300.0 K using Call-Purcell-Meiboom-Gill (CPMG) pulse sequence[21,22] along with water presaturation during the relaxation delay of 2 s. The CPMG pulse sequence was applied as a T2 filter to suppress signals from molecules with short T2 values (such as macromolecules and lipids), using a total echo time (TE) time of 320 ms. The main parameters for the 1D 1H-MRS spectra were as follows: spectral width (SW), 15 KHz; time domain (TD), 64 K; number of scans (NS), 512; and acquisition time (AQ), 2.128 s. Spectral assignments were confirmed by 2D 1H-1H TOCSY[23] and J-resolved (JRES) along with the values obtained from the literature[24]. The main parameters used for TOCSY were as follows: TD (F1-dimensional), 512; TD, (F2-dimensional), 1 K; SW (F1 and F2-dimensional), 5 KHz; and NS, 32. The main param-eters used for JRES were as follows: TD (F1-dimensional), 256; TD (F2-dimensional), 8 K; SW (F1-dimensional), 78 Hz; SW (F2-dimensional), 8 KHz; and NS, 32. In both the 2D MRS experiments, the delay time was 2 s. The stability of the pancreatic juice samples was evaluated by repeat-ing a 1D MRS experiment after overall acquisition. No biochemical degradation of any of the pancreatic juice samples was observed.
RESULTS On combining 2D MRS experimental results (Figure 1) obtained in the present study with results from related lit-erature[25-27] we identified the main resonances in 1H MRS spectra of pancreatic juice. TOCSY is a useful 2D MRS technology, it can be used to distinguish the frequencies in the total spin system and improve the sensitivity of de-tecting small J couplings[28]. On the basis of TOCSY data, the resonances in 1D 1H MRS spectra (Figure 2) of some
2127 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

amino acids were identified. The components, including leucine (Leu), iso-leucine (Ileu), valine (Val), lactate (Lac), alanine (Ala), acetate (Ace), aspartate (Asp), lysine (Lys), glycine (Gly), threonine (Thr), tyrosine (Tyr), histidine (His), tryptophan (Trp) and phenylalanine (Phe), and the locations were also identified.
From the analysis of 1D 1H MRS spectra of all panc-reatic juice samples, it was easy to find a triplet peak at the chemical shift of 1.19 ppm (Figure 3), which only appeared in some spectra. Four of the pancreatic juice samples of patients with chronic pancreatitis showed a triplet peak at the chemical shift of 1.19 ppm on 1H MRS, whereas the spectra of the pancreatic juice of patients with pancreatic cancer did not show a peak at the chemical shift of 1.19 ppm. When subjected to 2D TOCSY, the spectra of pancreatic juice samples of patients with chronic pancreatitis only showed one correlation peak at the chemical shift of 1.19 ppm and 3.36 ppm (Figure 4). By chemical shift and J coupling constant, we found that the peak at the chemical shift of 1.19 ppm was the 1H peak of the methyl of ethoxy group (CH3CH2O-). The 1D 1H spectra of the 4 panc-reatic juice samples of patients with chronic pancreatitis showed a triplet peak at the chemical shift of 1.19 ppm; further, these patients had a history of drinking, which was found from the analysis of pathology data. The 1D 1H spectra of pancreatic juice obtained from the female patient with chronic pancreatitis did not show a triplet peak at the chemical shift of 1.19 ppm.
Among the 10 patients with pancreatic cancer, diag-nosed by postoperative analyses of pathology data, 3 peo-ple had a medical history of long-term alcohol consump-tion. Of 5 patients with chronic pancreatitis, 4 men had a medical history of long-term alcohol consumption, and they were typical patients with alcoholic chronic pancreati-tis. The female patient suffered from auto-immune chronic pancreatitis by analyses of the pathology data.
DISCUSSIONIn recent years, there has been much debate on the relati-on between chronic pancreatitis and pancreatic cancer. While some scholars[29] believe that both diseases have a close connection, others[30] disagree. The results of our study show that there is no apparent difference between the components of pancreatic juice obtained from patien-ts with chronic pancreatitis and pancreatic cancer, except that 1D 1H spectra of pancreatic juice obtained from the former group of patients who suffered from alcoholic chronic pancreatitis shows a triplet peak of the methyl of ethoxy group (CH3CH2O-) at the chemical shift of 1.19 ppm. This finding may be used to differentiate pancreatic can-cer from alcoholic chronic pancreatitis with a mass in the head of the pancreas.
In the present study of the 10 patients with pancre-atic cancer, 3 had a medical history of long-term alcohol consumption, but the 1D 1H spectra of their pancreatic juice did not show the triplet peak of the ethoxy group (CH3CH2O-); further, the pathology data of these patients did not show symptoms related to chronic pancreatitis. Hence, we can conclude that alcohol is not the main factor that causes pancreatic cancer. It is controversial whether
2128 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
His
Tyr
Trp
Phe
7.8 7.6 7.4 7.2 7.0
δH
His
Tyr
Trp
4.0 3.5 3.0 2.5 2.0 1.5 1.0
δH
Tyr ValAspLacThr
Ala + Lys Gly Lys
ThrAsp
Ace
Val + Leu + Ileu
Lac
Ala
Figure 2 The assignment of proton magnetic resonance spectroscopy spec-tra of pancreatic juice with chronic pancreatitis. Ace: Acetate; Ala: Alanine; Asp: Aspartate; Gly: Glycine; His: Histidine; Ileu: Iso-leucine; Lac: Lactate; Leu: Leucine; Lys: Lysine; Phe: Phenylalanine; Thr: Threonine; Tyr: Tyrosine; Trp: Tryptophan; Val: Valine.
Figure 1 Assignments of partial resonances in 1H spectra of pancreatic juice. A: 2D Total correlation spectroscopy (TOCSY) of human pancreatic juice; B: JRES spectra of human pancreatic juice. Tyr: Tyrosine; His: Histidine; Trp: Trypto-phan; Phe: Phenylalanine.
7.8 7.6 7.4 7.2 7.0 F2
δH
F1
7.0
7.2
7.4
7.6
7.8
δH
His
His
Tyr
Trp
Phe
A
F1
-0.05
0
0.05
0.10
δH
3.7 3.6 1.2 F2
δH
B
Wang J et al . MRS for pancreatic diseases

long-term alcohol consumption can cause pancreatic can-cer. Riediger et al[31] found that the association between al-cohol and pancreatic cancer was not apparent during epide-miological investigation, which is in accord with our results.
We applied MRS to study pancreatic juice obtained from patients with chronic pancreatitis and pancreatic cancer, and separated the various components of differ-ent amino acids in human pancreatic juice by 1D and 2D 1H spectra. Recently, many analyses on the components of pancreatic juice have focused on the aspect of pro-teomics. Our study in the field of metabolomics is auxil-iary to proteomics, and goes a step further in the study of pancreatopathy.
It is difficult to differentiate between pancreatic cancer and chronic pancreatitis with a mass in the head of the pancreas solely by MRI, because both these diseases have similar clinical behaviors and imaging features. There is no noninvasive method that can be successfully used to diagnose these diseases. Biopsy is frequently used to diag-nose pancreatic cancer or chronic pancreatitis with a mass in the head of the pancreas but has some disadvantages, e.g. false negative results, many complications (bleeding, seepage of bile and pancreatic juice), and risk of tumor metastasis. ERCP causes some injury, but it is better than biopsy, because it causes fewer complications. ERCP, when combined with MRS technology, helps obtain more information on metabolites, which cannot be obtained from biopsy or MRI, and is likely to be used to distinguish pancreatic cancer from alcoholic chronic pancreatitis with a mass in the head of pancreas on the basis of 1H MRS spectra of pancreatic juice.
There are some limitations to this study. Lack of con-trol groups and the content of metabolites in the pancre-atic juice being small meant we could not perform a quan-titative analysis, and only obtained some qualitative results. The excreta of patients with pancreatic cancer or chronic pancreatitis may not only have different components but also differ in quantity.
In conclusions, MRS is a powerful tool that can be applied to the study of pancreatic juice obtained from patients by ERCP, and does not cause injury. The triplet peak, which is at the chemical shift of 1.19 ppm in 1D 1H-MRS data of pancreatic juice obtained from the pa-tients with alcoholic chronic pancreatitis, was identified as resonance of the methyl of ethoxy group (CH3CH2O-). The ethoxy group may be associated with alcohol metabo-lism in the pancreas, and is likely to be used to distinguish pancreatic cancer from alcoholic chronic pancreatitis with a mass in the head of the pancreas. In view of small num-bers, further confirmation of the results in a larger num-ber of patients is required.
COMMENTSBackgroundPancreatic cancer is a malignant neoplasm of the pancreas, and it has a high death rate. One of the medical dilemmas is to distinguish pancreatic cancer from chronic pancreatitis with a mass in the head of the pancreas; both diseas-es have similar clinical behavior and imaging features. Exploring the markers to distinguish the diseases is very important to the therapies of patients in clinic.Research frontiersCA19-9, K-ras gene, p53 anti-oncogene, and p53 protein are widely used as markers of pancreatic cancer, but they are not sensitive and show low specific-ity. Searching characteristic markers of the diseases is still the goal of tireless pursuit. Magnetic resonance spectroscopy (MRS) has high sensitivity and
2129 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
δH
4.0 3.5 3.0 2.5 2.0 1.5 F2
δH
F1
1.5
2.0
2.5
3.0
3.5
4.0
ThrLys Lys Lys
Val
IleuLeu
Phe
Tyr
Val
AlaLac
A
4.0 3.5 3.0 2.5 2.0 1.5 F2
δH
F1
1.5
2.0
2.5
3.0
3.5
4.0
δH
B
Figure 4 2D total correlation spectroscopy spectra of pancreatic juice with chronic pancreatitis and pancreatic cancer. A: Total correlation spec-troscopy (TOCSY) spectra of pancreatic juice from patients with alcoholic chronic pancreatitis; B: TOCSY spectra of pancreatic juice from patients with pancreatic cancer. Ala: Alanine; Ileu: Iso-leucine; Lac: Lactate; Leu: Leucine; Lys: Lysine; Phe: Phenylalanine; Thr: Threonine; Tyr: Tyrosine; Val: Valine.
A
B
C
D
E
F
G
H
4.0 3.5 3.0 2.5 2.0 1.5
δH
Figure 3 1H magnetic resonance spectroscopy spectra of human pan-creatic juice with chronic pancreatitis and pancreatic cancer. A-E: 1D 1H MRS spectra of pancreatic juice of patients with chronic pancreatitis; F-H: 1D 1H magnet-ic resonance spectroscopy spectra of pancreatic juice of patients with pancreatic cancer, other 7 similar spectra of pancreatic juice of patients with pancreatic cancer are not shown.
Wang J et al . MRS for pancreatic diseases
COMMENTS

resolution, allows in vitro testing of metabolites, and has been widely used in the field of metabolomics. MRS will be the most potent tool to help differentiate between pancreatic cancer and chronic pancreatitis.Innovations and breakthroughsIt is difficult to differentiate between pancreatic cancer and chronic pancreatitis with a mass in the head of the pancreas solely by Magnetic Resonance Imaging (MRI). Biopsy is frequently used to diagnose the diseases but has many complications and risk of tumor metastasis. Endoscopic retrograde cholangio-pancreatography (ERCP) combined with MRS helps obtain more information on metabolites, which cannot be obtained from biopsy or MRI. We separated the various components of different amino acids in human pancreatic juice. The triplet peak which is at the chemical shift of 1.19 ppm in 1D 1H MRS spectra was identified as resonance of the methyl of ethoxy group (CH3CH2O-), and it may be the characteristic metabolites of the patients with alcoholic chronic pancreatitis.ApplicationsThis study provides basic data and experimental methods for the diagnosis of pancreatic diseases. The ethoxy group is likely to be used to distinguish pancreatic cancer from alcoholic chronic pancreatitis with a mass in the head of the pancreas.TerminologyChemical shift is a basic concept in MRS, the pick appearing in the 1H MRS spec-tra with different chemical shift means the nuclear of proton spins with different frequency. The same rotation frequency of nuclear of protons only shows one peak in the 1H MRS spectra. The triplet peak at the chemical shift of 1.19 ppm is caused by the interaction of nearby nuclear of protons. In fact, it is split by one peak.Peer reviewIn my opinion, the article is acceptable for publication.
REFERENCES1 Greenlee RT, Murray T, Bolden S, Wingo PA. Cancer statis-
tics, 2000. CA Cancer J Clin 2000; 50: 7-332 Warshaw AL, Fernández-del Castillo C. Pancreatic carcinoma.
N Engl J Med 1992; 326: 455-4653 Shimosegawa T, Kume K, Satoh K. Chronic pancreatitis and
pancreatic cancer: prediction and mechanism. Clin Gastroen-terol Hepatol 2009; 7: S23-S28
4 Al-Haddad M, Eloubeidi MA. Diagnostic and therapeutic ap-plications of endoscopic ultrasound-guided punctures. Dig Dis 2008; 26: 390-397
5 Bolog N, Constantinescu G, Oancea I, Beuran M, Albu R, Tanţă M, Nicolau E, Iordache F. Magnetic resonance imaging of bile and pancreatic ducts: a retrospective study. Rom J Gas-troenterol 2004; 13: 91-97
6 Parsons HM, Ludwig C, Viant MR. Line-shape analysis of J-resolved NMR spectra: application to metabolomics and quantification of intensity errors from signal processing and high signal congestion. Magn Reson Chem 2009; 47 Suppl 1: S86-S95
7 Beckwith-Hall BM, Thompson NA, Nicholson JK, Lindon JC, Holmes E. A metabonomic investigation of hepatotoxicity using diffusion-edited 1H NMR spectroscopy of blood serum. Analyst 2003; 128: 814-818
8 Tang H, Wang Y, Nicholson JK, Lindon JC. Use of relaxation-edited one-dimensional and two dimensional nuclear mag-netic resonance spectroscopy to improve detection of small metabolites in blood plasma. Anal Biochem 2004; 325: 260-272
9 Potts BC, Deese AJ, Stevens GJ, Reily MD, Robertson DG, The-iss J. NMR of biofluids and pattern recognition: assessing the impact of NMR parameters on the principal component analy-sis of urine from rat and mouse. J Pharm Biomed Anal 2001; 26: 463-476
10 Beger RD, Schnackenberg LK, Holland RD, Li DH, Dragan Y. Metabonomic models of human pancreatic cancer using 1D proton NMR spectra of lipids in plasma. Metabolomics 2006; 2: 125-134
11 Lenz EM, Bright J, Wilson ID, Morgan SR, Nash AF. A 1H NMR-based metabonomic study of urine and plasma samples
obtained from healthy human subjects. J Pharm Biomed Anal 2003; 33: 1103-1115
12 Duarte IF, Goodfellow BJ, Barros A, Jones JG, Barosa C, Diogo L, Garcia P, Gil AM. Metabolic characterisation of plasma in juveniles with glycogen storage disease type 1a (GSD1a) by high-resolution (1)H NMR spectroscopy. NMR Biomed 2007; 20: 401-412
13 Balzano G, Di Carlo V. Is CA 19-9 useful in the management of pancreatic cancer? Lancet Oncol 2008; 9: 89-91
14 Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988; 53: 549-554
15 Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res 1988; 16: 7773-7782
16 Algül H, Schmid RM. Pancreatic cancer: a plea for good and comprehensive morphological studies. Eur J Gastroenterol Hepatol 2008; 20: 713-715
17 Redston MS, Caldas C, Seymour AB, Hruban RH, da Costa L, Yeo CJ, Kern SE. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res 1994; 54: 3025-3033
18 Kirsch DG, Kastan MB. Tumor-suppressor p53: implications for tumor development and prognosis. J Clin Oncol 1998; 16: 3158-3168
19 Yokoyama M, Yamanaka Y, Friess H, Buchler M, Korc M. p53 expression in human pancreatic cancer correlates with enhanced biological aggressiveness. Anticancer Res 1994; 14: 2477-2483
20 Fry LC, Mönkemüller K, Malfertheiner P. Molecular markers of pancreatic cancer: development and clinical relevance. Lan-genbecks Arch Surg 2008; 393: 883-890
21 Carr HY, Purcell EM. Effects of Diffusion on Free Precession in Nuclear Magnetic Resonance Experiments. Phys. Rev 1954; 94: 630-638
22 Meiboom S, Gill D. Modified spin-echo method for measuring nuclear relaxation times. Rev Sci Instrum 1958; 29: 688-691
23 Polshakov VI, Frenkiel TA, Westley B, Chadwick M, May F, Carr MD, Feeney J. NMR-based structural studies of the pNR-2/pS2 single domain trefoil peptide. Similarities to por-cine spasmolytic peptide and evidence for a monomeric struc-ture. Eur J Biochem 1995; 233: 847-855
24 Gheysen K, Mihai C, Conrath K, Martins JC. Rapid identifica-tion of common hexapyranose monosaccharide units by a sim-ple TOCSY matching approach. Chemistry 2008; 14: 8869-8878
25 Govindaraju V, Young K, Maudsley AA. Proton NMR chemi-cal shifts and coupling constants for brain metabolites. NMR Biomed 2000; 13: 129-153
26 Waters NJ, Holmes E, Waterfield CJ, Farrant RD, Nicholson JK. NMR and pattern recognition studies on liver extracts and intact livers from rats treated with alpha-naphthylisothiocya-nate. Biochem Pharmacol 2002; 64: 67-77
27 Commodari F, Khiat A, Ibrahimi S, Brizius AR, Kalkstein N. Comparison of the phytoestrogen trans-resveratrol (3, 4', 5-tri-hydroxystilbene) structures from x-ray diffraction and solution NMR. Magn Reson Chem 2005; 43: 567-572
28 Mandelshtam VA. The multidimensional filter diagonaliza-tion method. J Magn Reson 2000; 144: 343-356
29 Beger HG, Rau BM, Gansauge F, Poch B. Duodenum-preserv-ing subtotal and total pancreatic head resections for inflamma-tory and cystic neoplastic lesions of the pancreas. J Gastrointest Surg 2008; 12: 1127-1132
30 Nair RJ, Lawler L, Miller MR. Chronic pancreatitis. Am Fam Physician 2007; 76: 1679-1688
31 Riediger H, Adam U, Fischer E, Keck T, Pfeffer F, Hopt UT, Makowiec F. Long-term outcome after resection for chronic pancreatitis in 224 patients. J Gastrointest Surg 2007; 11: 949-959; discussion 959-960
S- Editor Sun H L- Editor O’Neill M E- Editor Ma WH
2130 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Wang J et al . MRS for pancreatic diseases

Ku80 gene G-1401T promoter polymorphism and risk of gastric cancer
Jia-Qi Li, Jie Chen, Nan-Nan Liu, Li Yang, Ying Zeng, Bin Wang, Xue-Rong Wang
Jia-Qi Li, Jie Chen, Nan-Nan Liu, Ying Zeng, Bin Wang, Xue-Rong Wang, Key Laboratory of Reproductive Medicine, Department of Pharmacology, Nanjing Medical University, Nan-jing 210029, Jiangsu Province, ChinaLi Yang, Department of General Surgery, First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, Jiangsu Prov-ince, ChinaAuthor contributions: Yang L, Zeng Y, Wang B and Wang XR designed the research and enrolled the patients; Li JQ, Chen J and Liu NN performed the research; Li JQ analyzed the data; Li JQ wrote the paper. Supported by Grants from the National Natural Science Foun-dation of China, No. 30672486; the Natural Science Foundation of Jiangsu Province, No. BK2006525; “333 Project” and “Qinglan Project” Funds for the Young Academic Leader of Jiangsu Prov-ince to Wang BCorrespondence to: Xue-Rong Wang, PhD, Key Labora-tory of Reproductive Medicine, Department of Pharmacology, Nanjing Medical University, Nanjing 210029, Jiangsu Province, China. [email protected]: +86-25-86862884 Fax: +86-25-86862884Received: December 12, 2009 Revised: January 20, 2010Accepted: January 27, 2010Published online: April 28, 2011
AbstractAIM: To evaluate the possible relationship between the Ku80 gene polymorphism and the risk of gastric cancer in China.
METHODS: In this hospital-based case-control study of gastric cancer in Jiangsu Province, China, we investi-gated the association of the Ku80 G-1401T (rs828907) polymorphism with gastric cancer risk. A total of 241 patients with gastric cancer and 273 age- and sex-matched control subjects were genotyped and analyzed by polymerase chain reaction-restriction fragment length polymorphism.
RESULTS: The frequencies of genotypes GG, GT and
TT were 65.6%, 22.8% and 11.6% in gastric cancer cases, respectively, and 75.8%, 17.6% and 6.6% in controls, respectively. There were significant differ-ences between gastric cancer and control groups in the distribution of their genotypes (P = 0.03) and allelic frequencies (P = 0.002) in the Ku80 promoter G-1401T polymorphism.
CONCLUSION: The T allele of Ku80 G-1401T may be associated with the development of gastric cancer.
© 2011 Baishideng. All rights reserved.
Key words: Ku80; Gastric cancer; Polymorphism; Pro-moter; Carcinogenesis
Peer reviewer: Pete Muscarella, MD, Division of Gastrointesti-nal Surgery, The Ohio State University, N711 Doan Hall, 410 W. 10th Ave., Columbus, OH 43210, United States
Li JQ, Chen J, Liu NN, Yang L, Zeng Y, Wang B, Wang XR. Ku80 gene G-1401T promoter polymorphism and risk of gas-tric cancer. World J Gastroenterol 2011; 17(16): 2131-2136 Available from: URL: http://www.wjgnet.com/1007-9327/full/v17/i16/2131.htm DOI: http://dx.doi.org/10.3748/wjg.v17.i16.2131
INTRODUCTIONGastric cancer is one of the most frequent malignancies in many countries, accounting for 8.7% of all cancers and 10.4% of all cancer deaths in the year of 2000[1]. In China, gastric cancer remains the leading cause of cancer-related mortality among men and women[1,2]. It is estimated that about 39% of gastric cancer cases occur in Chinese population[1,2]. The environmental factors, diet, tobacco, alcohol and Helicobacter pylori infection are well-known causes of gastric cancer in China[3-5]. However, only a fraction of individuals exposed to these factors
BRIEF ARTICLE
World J Gastroenterol 2011 April 28; 17(16): 2131-2136ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2131
2131 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

Li JQ et al . Ku80 and gastric cancer
develop gastric cancer, suggesting that individual suscep-tibility to gastric cancer should be different. Currently, the genomic etiology of gastric cancer is of great interest but largely unknown.
DNA damage drives the formation and development of malignant tumors that ameliorate this damage, and its sequelae can be categorized as either gatekeeper or caretaker tumor suppressors, depending on their mode of action[6]. Nonhomologous end joining (NHEJ) repairs DNA double-strand breaks (DSBs) by joining ends with-out using a homologous template strand and has been described as a caretaker[7,8]. Many studies have shown that NHEJ is the predominant repair system in humans, which included the DNA ligase IV and its associated protein XRCC4, and the three components of the DNA-dependent protein kinase (DNA-PK) complex, Ku70, Ku80, and the catalytic subunit PKcs[9]. The Ku80 gene, also known as XRCC5, is an important and specific mem-ber of NHEJ. Ku70 and Ku80 form a heterodimer called Ku that is well known for its role in NHEJ pathway[10].
Ku acts as a regulator of transcription by interacting with the recombination signal binding protein Jκ and the nuclear factor (NF)-κB p50 homodimer to up-regulate p50 expression, which may regulate the proliferation of gastric cancer cells[11]. Gastric cancer cells with a low level of constitutive NF-κB had a lower expression level of Ku70 and Ku80, which was reflected in the lower nuclear levels of Ku proteins, than the wild-type cells and the cells transfected with control vector[12,13]. In addition, several studies reported that gastric cancer patients with a lower Ku80 expression level had a slightly prolonged survival after neoadjuvant chemotherapy[14-16].
Genetic polymorphisms in Ku80 genes influence DNA repair capacity and change predisposition of several can-cers, including colorectal[17], bladder[18] and oral cancers[19]. In addition, in these hospital-based case-control studies of other cancers, it was reported that the frequency of GT/TT type of the Ku80 gene at promoter G-1401T (rs828907) was significantly higher in cases than in con-trols[17-19]. Thus, we assumed that the specific polymor-phism of Ku80 gene may also contribute to gastric cancer. To test the hypothesis that the promoter G-1401T poly-morphism is associated with the risk of gastric cancer, we used polymerase chain reaction-restriction fragment length polymorphism (PCR-RELP) to genotype this poly-morphism in a hospital-based case-control study of 241 patients with gastric cancer and 273 age- and sex-matched cancer-free controls. The results of this research will lead to a better understanding of the role of SNPs in the Ku80 genes in gastric cancer carcinogenesis. Such knowledge may eventually lead to the development of better preven-tive measures for gastric cancer.
MATERIALS AND METHODSStudy populationThe case-control study consisted of 241 patients with gastric cancer and 273 cancer-free control subjects. The
gastric cancer patients were confirmed histologically. Ge-netically unrelated cancer-free individuals were recruited as controls who were selected by matching for age and gender during the same period. All subjects were Han Chinese from the eastern region of China and randomly selected from the Department of General Surgery of the First Affiliated Hospital of Nanjing Medical University between 2005 and 2009. All patients and control subjects voluntarily participated in the study, completed a self-administered questionnaire and donated 5 mL of blood samples. The questionnaire included questions on sex, age, residence, diabetes, hypertension and smoking status. Smoking was defined as ≥ 10 cigarettes per day. This re-search protocol was approved by the Institutional Review Board of Nanjing Medical University.
Genotyping analysisGenomic DNA was isolated from peripheral blood lym-phocytes using standard phenol-chloroform extraction, as previously described[20,21]. PCR-RELP assay was used to type the Ku80 G-1401T (rs828907) polymorphisms. In brief, the primers of the Ku80 G-1401T polymorphism were 5'-TAGCTGACAACCTCACAGAT-3' (forward) and 5'-ATTCAGAGGTGCTCATAGAG-3' (reverse)[19], which generated a 252-bp fragment. The PCR reaction was performed in a total volume of 20 μL containing 2 μL 10 × PCR buffer, 1.25 mmol/L MgCl2, 0.1 mmol/L dNTPs, 0.25 μmol/L each primer, 200 ng of genomic DNA and 1 U of Taq DNA polymerase (MBI Fermen-tas). The PCR was performed at 94℃ for 5 min and fol-lowed by 35 cycles of 30 s at 94℃, 30 s at 55℃ and 30 s at 72℃, with a final elongation at 72℃ for 10 min. The restriction enzyme BfaI (New England BioLabs) was used to distinguish the PCR product, and the genotypes were discriminated on 3% agarose gel and visualized by staining with 0.5 μg/mL ethidium bromide. The wild-type G-allele produced a single 252-bp fragment, and the polymorphic T-allele produced 2 fragments of 81-bp and 171-bp. Ap-proximately, 10%-15% of the samples were randomly selected for repeated assays, and the results were 100% concordant.
Statistical analysis Continuous variables are presented as mean ± SD and compared by unpaired Student’s t test. Continuous vari-ables departing from the normal distribution were pre-sented as median and interquartile range and analyzed by Mann-Whitney U-test. Discrete variables were repre-sented as frequencies and percentages and evaluated by the Pearson’s χ2 test. Pearson’s χ2 test was also used to compare the distribution of the Ku80 genotypes between cases and controls. The association between the Ku80 G-1401T polymorphism and the risk of gastric cancer was estimated by odds ratio (OR) and 95% CI using multivariate logistic regression. P < 0.05 was considered statistically significant. All statistical analyses were per-formed using SPSS version 13.0 for Windows (SPSS Inc., Chicago, IL, USA).
2132 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

RESULTSBaseline characteristicsThe frequency distributions of selected characteristics of the cases and controls are presented in Table 1. There was no significant difference between the cases and con-trols in sex (male: 75.1% vs 70.7%, P = 0.43) and age (57.9 ± 12.9 years vs 56.9 ± 14.1 years, P = 0.52), indicating that the matching for the subjects was successful. More smokers were found among gastric cancer cases com-pared with controls (25.3% vs 14.3%, P = 0.014). No significant differences were noted in residing in the rural area (46.1% vs 52.0%, P = 0.32), hypertension (8.7% vs 10.3%, P = 0.58) and diabetes (6.2% vs 8.1%, P = 0.51).
Genotype distributions and allele frequenciesTable 2 shows the distribution of the genotypic for the Ku80 G-1401T (rs828907) between gastric cancer pa-tients and controls. The genotypic frequencies in both gastric cancer and control groups were in agreement with those predicted by Hardy-Weinberg equilibrium (P = NS). The distribution of the Ku80 G-1401T genotypes (GG, GT and TT) was markedly different between cases (65.6%, 22.8%, and 11.6%) and controls (75.8%, 17.6%, and 6.6%, P = 0.03). A significantly different distribu-tion of the Ku80 G-1401T genotype was demonstrated among the cases and controls. As shown in Table 3, the frequency of T allele was significantly higher in gastric cancer patients than in control subjects (23.0% vs 15.4%, P = 0.002).
Stratified analyses for the variant Ku80 genotype in cases and controls The multivariate logistic regression analysis was further used to evaluate the association between the G-1401T polymorphism and gastric cancer stratified by risk factors including age, sex, smoking and residence under control (Table 4). Adjusted OR (for age, sex, smoking status,
residence, diabetes and hypertension) with 95% CI for mutant genotypes was all described. In statistical analy-ses stratified by the median age of controls (58 years), the increased risk associated with the GT/TT genotypes tended to be more evident in the younger subjects aged < 58 years (adjusted OR = 1.97, 95% CI: 1.05-2.90). How-ever, we did not note a statistically significant inverse as-sociation with gastric cancer risk in older subjects aged ≥ 58 years (adjusted OR = 1.31, 95% CI: 0.88-1.96). The adjusted OR for the GT/GT genotypes was 1.81 (95% CI: 1.28-2.52) in male subjects and 1.33 (95% CI: 0.81-2.24) in female subjects. We did not note a statisti-cally significant inverse association with gastric cancer risk in both non-smokers (adjusted OR = 1.48; 95% CI: 1.08-2.02) and smokers (adjusted OR = 2.52; 95% CI: 1.25-5.18). In urban subjects, there was significant evi-dence of an increased risk of gastric cancer in the vari-ant genotypes (adjusted OR = 1.88; 95% CI: 1.26-2.76), while the association was not statistically significant in rural subjects (adjusted OR = 1.48; 95% CI: 0.99-2.15).
DISCUSSIONIn this hospital-based, case-control study, we assessed the potential association between the Ku80 G-1401T poly-morphism and the presence of gastric cancer in Chinese population. To our best knowledge, this is the first study linking the Ku80 G-1401T polymorphism with gastric cancer risk. Our data showed that the Ku80 -1401 G to T variant was associated with the increased risk of gastric cancer.
Gastric cancer is a genetic disease developing from a multifactorial, multigenetic and multistage process[22,23]. It was widely accepted that both genetic and environmental factors may be involved in the etiology of gastric can-cer[24]. During the multistage carcinogenesis, Ku80 may be involved in multiple important cellular processes. To date, several studies have reported abnormal expression of Ku80 protein in various cancers[13,25-28]. Over-expression of Ku80 increased the capability of cancer acquired resistance to radiation and chemical drugs[29-31], while suppression of Ku80 expression decreased cellular proliferation, colony formation and inhibited tumorigenicity in a xenograft model[32]. As an important component of NHEJ, Ku80 and Ku70 form a heterodimer, which acts as a regulatory subunit of the DNA-dependent protein kinase complex DNA-PK by increasing the affinity of the catalytic sub-unit PRKDC to DNA[17]. The Ku80 gene plays an impor-tant and specific role in removing DSBs. Chang et al[18]
2133 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 1 Baseline characteristics of cases and controls n (%)
Characteristics Cases (n = 241) Controls (n = 273) P
Sex (male) 181 (75.1) 193 (70.7) 0.43Age (yr) 57.9 ± 12.9 56.9 ± 14.1 0.52Smoking 61 (25.3) 39 (14.3) 0.014Residence (rural) 111 (46.1) 142 (52.0) 0.32Hypertension 21 (8.7) 28 (10.3) 0.58Diabetes 15 (6.2) 22 (8.1) 0.51
Table 2 Genotype of Ku80 G-1401T polymorphism in cases and controls n (%)
Genotype Cases Control
GG 158 (65.6) 207 (75.8)GT 55 (22.8) 48 (17.6)TT 28 (11.6) 18 (6.6)
χ2 = 7.26, df = 2, P = 0.03.
Table 3 Allele distribution of Ku80 G-1401T polymorphism in cases and controls n (%)
Allele Cases Controls
G 371 (77.0) 462 (84.6)T 111 (23.0) 84 (15.4)
χ2 = 9.73, df = 1, P = 0.002.
Li JQ et al . Ku80 and gastric cancer

found evidence that the Ku80 G-1401T variant was associ-ated with increased risk of bladder cancer in a central Tai-wanese population. A recent study, involving 362 patients with colorectal cancer and 362 age- and gender-matched healthy controls, showed that the T allele Ku80 G-1401T conferred a significantly (P = 0.0069) increased risk of colorectal cancer[17]. These observations were consistent with the findings previously described by other investiga-tors from Asian populations[19].
To further investigate the association between the Ku80 promoter G-1401T polymorphism and the risk of gastric cancer, we conducted this hospital-based case-control study in a Chinese population which incorporated the information on exposure to smoking, residence and other potential confounding factors (age and sex) that were frequency matched between cases and controls and further adjusted in the analysis. In our study, a significant difference of the Ku80 G-1401T genotype distribution was found between gastric cancer cases and controls. The frequency of T allele was significantly higher in gastric cancer patients than in control subjects.
The precise mechanisms underlying the relationship between Ku80 polymorphism and stomach carcinogene-sis remain unclear. Although the Ku80 promoter G-1401T genetic variation does not directly lead to amino acid coding change, presumably, it is plausible that this SNP influences the expression level or stability of the Ku80 protein by the alternative spicing, intervention, modifica-tion, determination or involvement. It is similar to anoth-er important member of NHEJ, XRCC4. A few reports provided evidence that its SNPs located on the promoter region are significant in various cancers[33,34].
Our data also showed that the association between increased gastric cancer risk and the mutant genotypes (GT + TT) was more evident in younger subjects aged < 58 years than in older subjects. We also found an interac-tion between genotype and sex. The adjusted OR was 1.81 (95% CI: 1.28-2.52) for GT/TT genotype compared with GG genotype among male subjects. But the OR (adjusted OR = 1.33; 95% CI: 0.81-2.24) was not statis-tically significant among female subjects. Our findings
were inconsistent with previous observations by Yang et al[17] and Chang et al[18]. The reason for the different ob-servations remains unclear.
In addition, we did not note a statistically significant inverse association with gastric cancer risk in both non-smokers (adjusted OR = 1.48; 95% CI: 1.08-2.02) and smokers (adjusted OR = 2.52; 95% CI: 1.25-5.18). But Yang et al[17] reported that the GT and TT genotypes, in association with smoking, conferred an increased risk (adjusted OR = 2.537; 95% CI: 1.398-4.601) for colorec-tal cancer. Similarly, Chang et al[18] and Hsu et al[19] found a significantly decreased risk of bladder cancer (adjusted OR = 2.053; 95% CI: 1.232-3.419) and oral cancer in smokers with GT or TT genotypes[18,19]. The results are inconsistent with our findings. The reason for the differ-ent observations remains unclear. Several studies have reported that smoking is associated with free radical-induced DNA damage and strand breaks[26], and tobacco smoke contains some potential carcinogens including polycyclic aromatic hydrocarbons, tobacco nitro-amines, aromatic amines and BPDE, which form DNA bulky ad-ducts and DNA strand breaks[27,35].
The stratified analyses by residence revealed that the association was significant in variant genotypes in urban subjects (OR = 1.88; 95% CI: 1.26-2.76) but not in rural subjects (adjusted OR = 1.48; 95% CI: 0.99-2.15). The different results may be explained, at least in part, be-tween rural and urban subjects. Environmental factors, including air, soil, diet, occupation and lifestyle, may be responsible for the different observations between rural and urban subjects. It was plausible, considering the bet-ter environment in rural areas[36].
The potential limitations of the present study should be stressed. Firstly, in this hospital-based case-control study, we selected controls from individuals with a variety of nonmalignant diseases. These may cause the possibil-ity of selection bias and confound the results. Neverthe-less, the frequencies of Ku80 G-1401T polymorphism variant alleles were similar to those reported in the NCBI Website in the Asian population studies. T allele fre-quencies of Ku80 promoter G-1401T are 15.4% in our
2134 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 4 Stratification analyses of the association between Ku80 polymorphism and risk of gastric cancer n (%)
Variable Cases (n = 241) Controls (n = 273) Adjusted OR (95% CI)1 P
GG GT + TT GG GT + TT
Age (yr) (median) < 58 76 (62.3) 46 (37.7) 109 (76.8) 33 (23.2) 1.97 (1.05-2.90) 0.01 ≥ 58 82 (68.9) 37 (31.1) 98 (74.8) 33 (25.2) 1.31 (0.88-1.96) 0.3Sex Male 118 (65.2) 63 (34.8) 149 (77.2) 44 (22.8) 1.81 (1.28-2.52) 0.01 Female 40 (66.7) 20 (33.3) 58 (72.5) 22 (27.5) 1.33 (0.81-2.24) 0.46Smoking status Smokers 39 (63.9) 22 (36.1) 32 (82.1) 7 (17.9) 2.52 (1.25-5.18) 0.051 Non-smokers 119 (66.1) 61 (33.9) 175 (74.8) 59 (25.2) 1.48 (1.08-2.02) 0.054Residence Urban 85 (65.4) 45 (34.6) 102 (77.7) 29 (22.3) 1.88 (1.26-2.76) 0.025 Rural 73 (65.8) 38 (34.2) 105 (73.9) 37 (26.1) 1.48 (0.99-2.15) 0.16
1Adjusted for age, sex, smoking status, hypertension, diabetes and residence.
Li JQ et al . Ku80 and gastric cancer

control group and 17.4% for Asian population in NCBI. The genotype distribution of controls in our study met Hardy-Weinberg equilibrium conditions. Secondly, the sample size of the present study was relatively small, which may limit the statistical power. Finally, our study was conducted in Chinese population. Caution should be exercised when extrapolating the data to other ethnic groups.
In conclusion, we found a significant difference in the Ku80 G-1401T polymorphism distribution between the patients with gastric cancer and the control group. The T allele of the Ku80 G-1401T was found more frequently in patients with gastric cancer and it may be associated with an increased risk of gastric cancer, suggesting that the polymorphism of Ku80 G-1401T, involved in the gastric tract carcinogenesis, may be a useful marker for primary prevention and anticancer intervention. Further studies are needed to determine the exact nature of this relation-ship.
COMMENTSBackgroundThe Ku80 gene is an important and specific member of NHEJ. Genetic polymor-phisms in Ku80 genes (G-1401T) influence DNA repair capacity and change the predisposition of several cancers, including colorectal, bladder and oral cancer. Whether genetic variants are involved in the risk of gastric cancer in a Chinese population is unknown.Research frontiersIn this study, the frequency of the Ku80 G-1401T GT/TT genotypes was sig-nificantly higher in the gastric cancer patients than in control subjects. This is the first analysis of the association between genetic predisposition and gastric cancer risk in Chinese population.Innovations and breakthroughsThe Ku80 G-1401T polymorphisms may modulate the development of gastric cancer in a Chinese population.ApplicationsThe Ku80 G-1401T GT/TT genotypes can be used as biomarkers for selecting patients from the individuals at high risk for gastric cancer in China. Identifying such susceptibility polymorphisms may lead to the development of tests that al-low more focused follow-ups of high-risk groups.TerminologyThe Ku80 gene, also known as XRCC5, is an important and specific member of NHEJ. As an important component of NHEJ, Ku80 and Ku70 form a heterodi-mer, which acts as a regulatory subunit of the DNA-dependent protein kinase complex DNA-PK by increasing the affinity of the catalytic subunit PRKDC to DNA.Peer reviewThe quality of the work and the methodology are sound. The conclusions are appropriate, although it seems unlikely that these findings represent a major breakthrough.
REFERENCES1 Parkin DM. Global cancer statistics in the year 2000. Lancet
Oncol 2001; 2: 533-5432 Sun XD, Mu R, Zhou YS, Dai XD, Zhang SW, Huangfu XM,
Sun J, Li LD, Lu FZ, Qiao YL. [Analysis of mortality rate of stomach cancer and its trend in twenty years in China]. Zhon-ghua Zhongliu Zazhi 2004; 26: 4-9
3 Takezaki T, Gao CM, Ding JH, Liu TK, Li MS, Tajima K. Comparative study of lifestyles of residents in high and low risk areas for gastric cancer in Jiangsu Province, China; with special reference to allium vegetables. J Epidemiol 1999; 9:
297-3054 Galanis DJ, Lee J, Kolonel LN. The influence of cigarette
smoking, alcohol, and green tea consumption on the risk of carcinoma of the cardia and distal stomach in Shanghai, China. Cancer 1997; 79: 1840-1841
5 Wu AH, Crabtree JE, Bernstein L, Hawtin P, Cockburn M, Tseng CC, Forman D. Role of Helicobacter pylori CagA+ strains and risk of adenocarcinoma of the stomach and esophagus. Int J Cancer 2003; 103: 815-821
6 Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gate-keepers and caretakers. Nature 1997; 386: 761, 763
7 Roth DB, Gellert M. New guardians of the genome. Nature 2000; 404: 823-825
8 Burma S, Chen BP, Chen DJ. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst) 2006; 5: 1042-1048
9 Jackson SP. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002; 23: 687-696
10 Liang F, Romanienko PJ, Weaver DT, Jeggo PA, Jasin M. Chromosomal double-strand break repair in Ku80-deficient cells. Proc Natl Acad Sci USA 1996; 93: 8929-8933
11 Kim H. DNA repair Ku proteins in gastric cancer cells and pancreatic acinar cells. Amino Acids 2008; 34: 195-202
12 Lim JW, Kim H, Kim KH. The Ku antigen-recombination signal-binding protein Jkappa complex binds to the nuclear factor-kappaB p50 promoter and acts as a positive regulator of p50 expression in human gastric cancer cells. J Biol Chem 2004; 279: 231-237
13 Lim JW, Kim H, Kim KH. Expression of Ku70 and Ku80 mediated by NF-kappa B and cyclooxygenase-2 is related to proliferation of human gastric cancer cells. J Biol Chem 2002; 277: 46093-46100
14 Napieralski R, Ott K, Kremer M, Specht K, Vogelsang H, Becker K, Müller M, Lordick F, Fink U, Rüdiger Siewert J, Höfler H, Keller G. Combined GADD45A and thymidine phosphorylase expression levels predict response and sur-vival of neoadjuvant-treated gastric cancer patients. Clin Cancer Res 2005; 11: 3025-3031
15 Höfler H, Langer R, Ott K, Keller G. Prediction of response to neoadjuvant chemotherapy in carcinomas of the upper gas-trointestinal tract. Adv Exp Med Biol 2006; 587: 115-120
16 Höfler H, Langer R, Ott K, Keller G. Prediction of response to neoadjuvant chemotherapy in carcinomas of the upper gas-trointestinal tract. Recent Results Cancer Res 2007; 176: 33-36
17 Yang MD, Hsu YM, Kuo YS, Chen HS, Chang CL, Wu CN, Chang CH, Liao YM, Wang HC, Wang MF, Bau DT. Signifi-cant association of Ku80 single nucleotide polymorphisms with colorectal cancer susceptibility in Central Taiwan. Anti-cancer Res 2009; 29: 2239-2242
18 Chang CH, Chiu CF, Liang SY, Wu HC, Chang CL, Tsai CW, Wang HC, Lee HZ, Bau DT. Significant association of Ku80 single nucleotide polymorphisms with bladder cancer sus-ceptibility in Taiwan. Anticancer Res 2009; 29: 1275-1279
19 Hsu CF, Tseng HC, Chiu CF, Liang SY, Tsai CW, Tsai MH, Bau DT. Association between DNA double strand break gene Ku80 polymorphisms and oral cancer susceptibility. Oral On-col 2009; 45: 789-793
20 Zhu H, Yang L, Zhou B, Yu R, Tang N, Wang B. Myeloper-oxidase G-463A polymorphism and the risk of gastric cancer: a case-control study. Carcinogenesis 2006; 27: 2491-2496
21 Wang LS, Tang NP, Zhu HJ, Zhou B, Yang L, Wang B. Endothelin-converting enzyme-1b C-338A polymorphism is associated with the increased risk of coronary artery disease in Chinese population. Clin Chim Acta 2007; 384: 75-79
22 Correa P, Haenszel W, Cuello C, Tannenbaum S, Archer M. A model for gastric cancer epidemiology. Lancet 1975; 2: 58-60
23 Gao L, Nieters A, Brenner H. Meta-analysis: tumour inva-sion-related genetic polymorphisms and gastric cancer sus-ceptibility. Aliment Pharmacol Ther 2008; 28: 565-573
2135 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Li JQ et al . Ku80 and gastric cancer
COMMENTS

24 Crandall WV, Mackner LM. Infusion reactions to infliximab in children and adolescents: frequency, outcome and a pre-dictive model. Aliment Pharmacol Ther 2003; 17: 75-84
25 Tonotsuka N, Hosoi Y, Miyazaki S, Miyata G, Sugawara K, Mori T, Ouchi N, Satomi S, Matsumoto Y, Nakagawa K, Miyagawa K, Ono T. Heterogeneous expression of DNA-dependent protein kinase in esophageal cancer and normal epithelium. Int J Mol Med 2006; 18: 441-447
26 Korabiowska M, Voltmann J, Hönig JF, Bortkiewicz P, König F, Cordon-Cardo C, Jenckel F, Ambrosch P, Fischer G. Altered expression of DNA double-strand repair genes Ku70 and Ku80 in carcinomas of the oral cavity. Anticancer Res 2006; 26: 2101-2105
27 Hosoi Y, Matsumoto Y, Enomoto A, Morita A, Green J, Na-kagawa K, Naruse K, Suzuki N. Suramin sensitizing cells to ionizing radiation by inactivating DNA-dependent protein kinase. Radiat Res 2004; 162: 308-314
28 Chen TY, Chen JS, Su WC, Wu MS, Tsao CJ. Expression of DNA repair gene Ku80 in lymphoid neoplasm. Eur J Haema-tol 2005; 74: 481-488
29 Wilson CR, Davidson SE, Margison GP, Jackson SP, Hendry JH, West CM. Expression of Ku70 correlates with survival in carcinoma of the cervix. Br J Cancer 2000; 83: 1702-1706
30 Shintani S, Mihara M, Li C, Nakahara Y, Hino S, Nakashiro K, Hamakawa H. Up-regulation of DNA-dependent protein kinase correlates with radiation resistance in oral squamous cell carcinoma. Cancer Sci 2003; 94: 894-900
31 Chang HW, Kim SY, Yi SL, Son SH, Song do Y, Moon SY, Kim JH, Choi EK, Ahn SD, Shin SS, Lee KK, Lee SW. Ex-pression of Ku80 correlates with sensitivities to radiation in cancer cell lines of the head and neck. Oral Oncol 2006; 42: 979-986
32 Yang QS, Gu JL, Du LQ, Jia LL, Qin LL, Wang Y, Fan FY. ShRNA-mediated Ku80 gene silencing inhibits cell prolif-eration and sensitizes to gamma-radiation and mitomycin C-induced apoptosis in esophageal squamous cell carcinoma lines. J Radiat Res (Tokyo) 2008; 49: 399-407
33 Chiu CF, Wang CH, Wang CL, Lin CC, Hsu NY, Weng JR, Bau DT. A novel single nucleotide polymorphism in XRCC4 gene is associated with gastric cancer susceptibility in Tai-wan. Ann Surg Oncol 2008; 15: 514-518
34 Chiu CF, Tsai MH, Tseng HC, Wang CL, Wang CH, Wu CN, Lin CC, Bau DT. A novel single nucleotide polymorphism in XRCC4 gene is associated with oral cancer susceptibility in Taiwanese patients. Oral Oncol 2008; 44: 898-902
35 Leanderson P, Tagesson C. Cigarette smoke-induced DNA damage in cultured human lung cells: role of hydroxyl radi-cals and endonuclease activation. Chem Biol Interact 1992; 81: 197-208
36 Wu MS, Chen CJ, Lin JT. Host-environment interactions: their impact on progression from gastric inflammation to carcinogenesis and on development of new approaches to prevent and treat gastric cancer. Cancer Epidemiol Biomarkers Prev 2005; 14: 1878-1882
S- Editor Wang YR L- Editor Ma JY E- Editor Zheng XM
2136 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Li JQ et al . Ku80 and gastric cancer

Effects of penehyclidine hydrochloride on rat intestinal barrier function during cardiopulmonary bypass
Ying-Jie Sun, Hui-Juan Cao, Qiang Jin, Yu-Gang Diao, Tie-Zheng Zhang
Ying-Jie Sun, Hui-Juan Cao, Qiang Jin, Yu-Gang Diao, Tie-Zheng Zhang, Department of Anesthesiology, General Hospital of Shenyang Commend, Shenyang 110840, Liaoning Province, ChinaAuthor contributions: Sun YJ and Jin Q contributed equally to this work; Sun YJ, Jin Q and Zhang TZ designed the research; Sun YJ, Cao HJ and Jin Q performed the research; Diao YG con-tributed new reagents/analytic tools; Sun YJ, Cao HJ and Jin Q analyzed the data; Sun YJ, Cao HJ and Jin Q wrote the paper.Supported by A grant from the Doctor Priming Foundation of Liaoning Province, No. 20091099Correspondence to: Qiang Jin, PhD, Associate Chief Phy-sician, Department of Anesthesiology, General Hospital of Shenyang Commend, Shenyang 110840, Liaoning Province, China. [email protected]: +86-24-28851366 Fax: +86-24-28851368Received: October 21, 2010 Revised: December 20, 2010Accepted: December 27, 2010Published online: April 28, 2011
AbstractAIM: To test the ability of penehyclidine hydrochloride (PHC) to attenuate intestinal injury in a rat cardiopul-monary bypass (CPB) model.
METHODS: Male Sprague-Dawley rats were randomly divided into six groups (eight each): sham-operated con-trol; sham-operated low-dose PHC control (0.6 mg/kg); sham-operated high-dose PHC control (2.0 mg/kg); CPB vehicle control; CPB low-dose PHC (0.6 mg/kg); and CPB high-dose PHC (2.0 mg/kg). Blood samples were collected from the femoral artery 2 h after CPB for de-termination of plasma diamine oxidase (DAO), D-lactate and endotoxin levels. Spleen, liver, mesenteric lymph nodes and lung were removed for biochemical analyses. Intestinal tissue ultrastructure was examined by elec-tron microscopy.
RESULTS: In the sham-operated groups, high- and low-dose-PHC had no significant impact on the levels of DAO, D-lactate and endotoxin, or the incidence of
intestinal bacterial translocation (BT). Serum levels of DAO, D-lactate, endotoxin and the incidence of intestinal BT were significantly increased in the surgi-cal groups, compared with the sham-operated groups (0.543 ± 0.061, 5.697 ± 0.272, 14.75 ± 2.46, and 0/40 vs 1.038 ± 0.252, 9.377 ± 0.769, 60.37 ± 5.63, and 30/40, respectively, all P < 0.05). PHC alleviated the biochemical and histopathological changes in a dose-dependent manner. Serum levels of DAO, D-lactate, and endotoxin and the incidence of intestinal BT in the high-dose PHC group were significantly lower than in the low-dose PHC group (0.637 ± 0.064, 6.972 ± 0.349, 29.64 ± 5.49, and 14/40 vs 0.998 ± 0.062, 7.835 ± 0.330, 38.56 ± 4.28, and 6/40, respectively, all P < 0.05).
CONCLUSION: PHC protects the structure and func-tion of the intestinal mucosa from injury after CPB in rats.
© 2011 Baishideng. All rights reserved.
Key words: Penehyclidine hydrochloride; Intestinal mu-cosa injury; Cardiopulmonary bypass
Peer reviewer: Dr. Richard A Rippe, Department of Medicine, The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7038, United States
Sun YJ, Cao HJ, Jin Q, Diao YG, Zhang TZ. Effects of pene-hyclidine hydrochloride on rat intestinal barrier function dur-ing cardiopulmonary bypass. World J Gastroenterol 2011; 17(16): 2137-2142 Available from: URL: http://www.wjgnet.com/1007-9327/full/v17/i16/2137.htm DOI: http://dx.doi.org/10.3748/wjg.v17.i16.2137
INTRODUCTIONCardiopulmonary bypass (CPB) is essential during some cardiovascular surgical procedures; however, it can cause
BRIEF ARTICLE
World J Gastroenterol 2011 April 28; 17(16): 2137-2142ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2137
2137 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

Sun YJ et al . Intestinal protection by penehyclidine
peripheral hypoperfusion as a result of non-pulsatile flow, low blood pressure, hemodilution, and other non-physiological conditions. Furthermore, an increase in in-testinal permeability and bacterial translocation (BT) has been demonstrated, not only in animal models, but also in patients during CPB[1-3]. Perioperative gastrointestinal integrity is therefore now recognized as an important fac-tor determining the outcome of cardiac surgical proce-dures[4]. In general, changes in mucosal permeability and morphology during CPB reflect the degree of damage to intestinal mucosal barrier function.
Previous in vitro studies have demonstrated that tropane alkaloids can stabilize the cell membrane and prevent oxi-dative stress. The new anticholinergic drug, penehyclidine hydrochloride (PHC), has been evaluated for its protec-tive effects on the cardiovascular system[5-7]. Its selective blocking of M1, M3 and N receptors means that PHC has few M2 receptor-associated cardiovascular side ef-fects. It has been shown to reduce endotoxin-stimulated acute lung injury and to attenuate liver damage during CPB in a rat model[8-10].
Based on the potential roles of PHC as an antioxidant and a cell membrane stabilizer, we hypothesized that its administration might reverse CPB-associated intestinal damage. Zhan et al[11] have suggested that PHC concen-trations of 0.18-3.60 mg/kg were found to be safe, and we therefore tested this hypothesis in a rat CPB model, using high- (2.0 mg/kg) and low-dose (0.6 mg/kg) PHC.
MATERIALS AND METHODSAnimals and treatmentsForty-eight male Sprague-Dawley rats (weighing 300-450 g, 18-22 wk old) were randomly assigned to one of six groups (eight each): sham-operated control; sham-operat-ed control + low-dose PHC (0.6 mg/kg) (sham L-PHC); sham-operated control + high-dose PHC (2 mg/kg) (sham H-PHC); CPB + vehicle (control); CPB + low-dose PHC (0.6 mg/kg) (L-PHC); and CPB + high-dose PHC (2.0 mg/kg) (H-PHC). PHC (Lisite Pharmacology Co. Chendou, China, No. 080301) was dissolved in abso-lute ethanol and diluted in saline (final concentration of ethanol < 1.0%), and added to the priming solution for CPB. All animals received humane care in compliance with the Principles of Laboratory Animal Care. The ex-perimental protocol was approved by the local animal use and care committee at the General Hospital of Shenyang Commend, China.
Surgical procedure None of the sham-operated control groups underwent CPB. After PHC injection, the respiratory rate (RR), heart rate (HR), blood pressure (BP) and electrocardi-ography (ECG) were continually monitored. The rat CPB model was established as previously described, with some modifications[12]. In brief, rats were anesthetized by intraperitoneal administration of 10% chloral hydrate (0.3 mL/100 g body weight) to provide stable anesthesia,
while maintaining spontaneous ventilation during the en-tire operative procedure. All subsequent procedures were performed under aseptic conditions.
After surgical-level anesthesia was achieved, the left femoral artery was cannulated using a 22-gauge Teflon heparinized catheter. Arterial pressure was monitored and blood samples were collected for gas analysis, using a blood gas analyzer (GEM Premier 3000; Mallinckrodt, Lexington, MA, USA). Following administration of hepa-rin (250 U/kg), an 18-gauge catheter was inserted into the right jugular vein and advanced to the right atrium. A 22-gauge catheter was cannulated into the tail artery, to serve as an arterial infusion line for the CPB circuit. The mini-CPB circuit comprised a venous reservoir, a specially designed membrane oxygenator, a roller pump, and ster-ile tubing with inner diameters of 4 mm for the venous line and 1.6 mm for the arterial line. The CPB circuit was primed with a total volume of 15 mL of synthetic colloid solution. The perfusion flow rate was gradually adjusted to sustain a mean arterial pressure of 60-80 mmHg. The gas flow (95% O2, 5% CO2) was initiated at around 50- 75 mL/kg per minute and adjusted to maintain blood gas analysis parameters within the physiological range. When the flow rate reached 80-100 mL/kg per minute, it was maintained for 60 min. At the end of CPB, the flow rate was reduced stepwise to achieve hemodynamic stabiliza-tion. Throughout the experiment, central body tempera-ture was monitored with a rectal probe and kept at 37.5 ± 1.0℃ using a heat lamp placed above the animal and CPB equipment. The mean arterial pressure was maintained at 60-80 mmHg.
Specimen collectionRats from each group were sacrificed by decapitation, and arterial blood (2.0 mL) and terminal ileums were sampled at 2 h after CPB. Plasma was prepared by centrifugation at 3000 g for 5-10 min at 4℃ and stored at -70℃ for de-termination of serum diamine oxidase (DAO), D-lactate, and endotoxin. Intestinal (ileum) tissue samples were ob-tained for electron microscopy.
Intestinal permeabilityThe permeability of the intestinal mucosa was assayed by measuring D-lactate and DAO levels in plasma. Plas-ma D-lactate levels were measured by enzymatic spectro-photometric assay using a centrifugal analyzer at 30℃, as described previously[13]. Plasma DAO activities were also determined by enzymatic spectrophotometry, as de-scribed previously[14]. D-lactate, D-lactate dehydrogenase, NAD+, O-dianisidine, cadaverine dihydrochloride and DAO were purchased from Sigma Chemical Company (Milan, Italy).
Plasma endotoxin determinationThe endotoxin content of the plasma sample was assayed using the Limulus amebocyte lysate test using the Endo-chrome K test kit (CoaChrom, Vienna, Austria). In brief, heparinized plasma was diluted 1:10 in pyrogen-free water
2138 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

and kept heated at 75℃ for 10 min to remove non-specif-ic inhibitors. A quantitative chromogenic kinetic method was used, as specified by the manufacturer, using a Ther-mo microplate reader (Tecan Spectra, Salzburg, Austria). The method had a detection limit of 0.75 pg/mL at a 1:10 plasma dilution.
Bacteriological culturesA midline incision was made using a sterile technique. Mesenteric lymph nodes (MLNs), portal vein, and sam-ples from the liver, spleen, and lung were harvested and weighed prior to determination of bacterial growth. The samples were homogenized in test tubes containing 3 mL Brain Heart Infusion Broth (Difco, Detroit, IL, USA). The supernatant (0.2 mL) was cultured for growth of aerobic, microaerophilic and anaerobic bacteria. All me-dia for aerobic cultures were incubated at 37℃ for at least 3-5 d, while organ samples for anaerobic bacteria were cultured at 37℃ for 7 d. Enteric Gram-negative bacteria were identified using the API 20 system (BioMérieux SA, Marcy-l’Etoile, France) and Lactobacillus acidophilus by API 50 CH (Analytab Products Inc., Plainview, NY, USA). All other aerobic, microaerophilic and anaerobic microbes isolated were identified by standard procedures. The numbers of living bacteria were calculated and expressed as the numbers of living organisms per gram of organ tissue.
Transmission electron microscopyFor transmission electron microscopy, ileum tissues were removed immediately from anesthetized rats 2 h after CPB, and then fixed with 2% paraformaldehyde and 2.5% glutaraldehyde in PBS (pH 7.3) for 2 h at room tempera-ture (25℃). The tissues were washed with PBS, fixed
with 1% osmium tetroxide for 2 h, washed again, and then embedded in Araldite 6005. Tissue sections were cut with a Leica EM FCS (Vienna, Austria) ultramicro-tome. Tissue sections (1 μm) were initially stained with toluidine blue-Azur Ⅱ to select the region of interest for subsequent procedures. Thin sections (60-70 nm) were stained with uranyl acetate and lead citrate and examined and photographed using an H-7200 transmission electron microscope (80 kV; Oberkochen, Germany). Electron microscopy pictures were evaluated twice by two inde-pendent histologists with at least 10 years of experience, who were blinded to our study.
Statistical analysisAll experimental data were expressed as mean ± SD and analyzed using a SPSS for Windows v. 13.0 (Chicago, IL, USA). One-way ANOVA was used for comparisons among various treatment groups. Post-hoc comparisons were analyzed using least significant difference test or Dunnett’s T3 test. P < 0.05 was considered to be statisti-cally significant.
RESULTSPHC reverses the increase in intestinal mucosal permeability after CPBThere were no obvious changes in the RR, HR, BP or ECG at any time points (0, 30, 60, or 120 min) after an-esthesia in the sham-operated groups (Table 1). Hemody-namic changes in the vehicle control, L-PHC and H-PHC groups are shown in Table 1. As shown in Table 2, DAO and D-lactate levels increased significantly in vehicle-treated CPB rats, compared with the sham group (P < 0.05), which effect was largely reversed by treatment with
2139 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 1 Physiological data (mean ± SD, n = 8)
Groups Pre-CPB CPB 30 min CPB 60 min Post-CPB 2 h
MAP (mmHg) Controls 86.33 ± 16.82 84.26 ± 14.55 85.45 ± 10.36 83.63 ± 11.24CPB + vehicle 84.50 ± 7.05 62.14 ± 15.23a,c 67.08 ± 19.12a,c 72.18 ± 17.39CPB + L-PHC 85.45 ± 9.36 66.58 ± 11.26a,c 69.78 ± 14.05a,c 76.15 ± 13.65CPB + H-PHC 87.54 ± 10.43 65.73 ± 9.85a,c 70.85 ± 12.36a,c 77.84 ± 12.36
HR (beats/min) Controls 325 ± 34 320 ± 25 315 ± 20 324 ± 15CPB + vehicle 315 ± 30 286 ± 28 305 ± 42 310 ± 37CPB + L-PHC 305 ± 26 315 ± 34 302 ± 38 304 ± 27CPB + H-PH 310 ± 20 296 ± 25 301 ± 35 305 ± 30
PH Controls 7.40 ± 0.02 7.41 ± 0.03 7.39 ± 0.02 7.40 ± 0.02CPB + vehicle 7.41 ± 0.03 7.38 ± 0.05 7.35 ± 0.06 7.39 ± 0.02CPB + L-PHC 7.38 ± 0.04 7.43 ± 0.02 7.41 ± 0.03 7.36 ± 0.04CPB + H-PHC 7.42 ± 0.01 7.42 ± 0.03 7.38 ± 0.04 7.43 ± 0.02
BE (mmol/L) Controls -1.96 ± 0.45 -2.36 ± 0.75 -1.54 ± 0.85 -1.95 ± 0.54 CPB + vehicle -1.86 ± 0.35 -1.36 ± 0.26 -1.60 ± 0.84 -2.30 ± 1.50
CPB + L-PHC -1.75 ± 0.26 -1.55 ± 0.96 -1.95 ± 0.63 -2.04 ± 0.72CPB + H-PHC -1.85 ± 0.36 -2.05 ± 0.90 -1.74 ± 0.42 -2.45 ± 0.14
HCT (%) Controls 41.10 ± 1.85 40.20 ± 1.53 40.60 ± 1.46 39.80 ± 1.35CPB + vehicle 41.80 ± 3.73 26.45 ± 4.24a,c 21.54 ± 3.71a,c 27.82 ± 3.66a,c
CPB + L-PHC 42.20 ± 2.45 27.65 ± 3.65a,c 23.05 ± 5.30a,c 29.53 ± 5.45a,c
CPB + H-PHC 42.45 ± 2.50 28.76 ± 5.38a,c 22.46 ± 4.25a,c 28.75 ± 4.56a,c
Controls summarize the results from sham-operated rats treated with saline or penehyclidine hydrochloride. aP < 0.05 vs baseline; cP < 0.05 vs controls. CPB: Cardiopulmonary bypass; MAP: Mean arterial pressure; HCT: Hematocrit; BE: Buffer excess; HR: Heart rate.
Sun YJ et al . Intestinal protection by penehyclidine

PHC in a dose-dependent manner (P < 0.05). H-PHC had significantly greater effects on the values of DAO and D-lactate than L-PHC (Table 2, P < 0.05).
PHC prevents CPB-induced BT Bacteriological cultures from all sham-operated animals were negative. The incidence of Escherichia coli-positive cultures was significantly increased in the CPB-vehicle group, while pretreatment with PHC seemed to prevent systemic dissemination. The incidence of intestinal BT to the MLNs, spleen, liver, lung and blood was significantly higher in the CPB-vehicle group, compared with that in the sham groups (Table 3, P < 0.05), which was largely reversed by treatment with PHC in a dose-dependent manner (Table 3, P < 0.05). The plasma endotoxin level was increased in the CPB-vehicle group compared with the sham groups (Table 2, P < 0.05) and PHC decreased the plasma endotoxin levels in a dose-dependent manner (Table 2, P < 0.05).
PHC prevents CPB-induced damage to the intestinal mucosa ultrastructureTransmission electronic microscopy demonstrated nor-mal intestinal ultrastructure in the sham-operated group, including regularly aligned microvilli in the intestinal epithelium, integral mitochondria and rough endoplas-mic reticulum (RER) and distinct junction complexes
(Figure 1A). In the vehicle control group, the microvilli were reduced in number, and showed irregular lengths and arrangements. The mitochondria were swollen with cracked and vacuolated cristae. Some RER structures were destroyed, and the intercellular spaces between epithelial cells were widened. The structure of the tight junctions became shortened (Figure 1B). Mitochondrial swelling with damage to mitochondrial cristae and vacu-olar degeneration was present in the L-PHC group. The nuclear structure was incomplete (Figure 1C). In the H-PHC group, microvilli in the intestinal epithelium were regularly aligned, and the tight junction structure became tight. However, mild swelling of the mitochondria and RER were seen (Figure 1D). These results indicated dose-dependent effects of PHC on the reduction of cellular damage after CPB.
DISCUSSIONThe present study focused on intestinal barrier injury af-ter CPB and the potential of PHC as a therapeutic agent. We demonstrated that application of PHC during CPB preserved intestinal barrier function in a dose-dependent manner, and histological evidence is provided to support these biochemical results.
DAO reduces the concentration of polyamines re-
2140 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
DC
BA
Figure 1 Effect of cardiopulmonary bypass on the ileal epithelial cells. A: Control group (× 5800), regularly-aligned microvilli in the intestinal epithelium, intact mitochondria and rough endoplasmic reticulum (RER), and distinct junc-tional complexes were observed; B: Cardiopulmonary bypass group (× 7200), epithelial damage was demonstrated by swollen mitochondria and loss of cris-tae, and tight junctions were disrupted; C: Mitochondrial swelling with damage to mitochondrial cristae and vacuolar degeneration were present. The nuclear structure was incomplete; D: Microvilli in the intestinal epithelium were regularly aligned, and the structure of the tight junctions became tight. However, mild swelling of mitochondria and RER were observed.
Table 2 Plasma diamine oxidase, D-lactate and endotoxin levels (mean ± SD, n = 8)
DAO (U/L) D-lactate (mg/L)
Endotoxin (pg/mL)
Controls 0.543 ± 0.061 5.697 ± 0.272 14.75 ± 2.46CPB + vehicle 1.038 ± 0.252a 9.377 ± 0.769a 60.37 ± 5.63a
CPB + L-PHC 0.998 ± 0.062a,e 7.835 ± 0.330a,e 38.56 ± 4.28a,e
CPB + H-PHC 0.637 ± 0.064a,c 6.972 ± 0.349a,c 29.64 ± 5.49a,c
Controls summarize the results from sham-operated rats treated with saline or penehyclidine hydrochloride (PHC). Data are shown as medians, n = 8 rats for each group. CPB: Cardiopulmonary bypass; H-PHC: High-dose PHC; L-PHC: Low-dose PHC; DAO: Diamine oxidase. The level of significance was set at P < 0.05. aP < 0.05 vs baseline; cP < 0.05 vs CPB group; eP < 0.05 vs H-PHC group.
Table 3 Numbers of animals pretreated with vehicle or pene-hyclidine hydrochloride with positive bacteriological cultures from blood (portal vein), mesenteric lymph nodes, liver, spleen and lungs after induction of cardiopulmonary bypass
Portal vein MLN Liver Lungs Spleen Total
Controls 0 0 0 0 0 0/40CPB + vehicle 7 8 6 4 5 30/40a,c
CPB + L-PHC 3 4 3 2 2 14/30a,c
CPB + H-PHC 2 3 1 0 0 6/40a
Controls summarize the results from sham-operated rats treated with saline or penehyclidine hydrochloride (PHC). MLN: Mesenteric lymph node; CPB: Cardiopulmonary bypass; H-PHC: High-dose PHC; L-PHC: Low-dose PHC. aP < 0.05 vs controls; cP < 0.05 vs L-PHC group.
Sun YJ et al . Intestinal protection by penehyclidine

quired for cell proliferation. DAO is localized to the small intestine and placenta, which are both organs with rapid cell turnover rates. In humans, DAO activity is especially high in the upper portion of the small intestinal villi, and has therefore been used as an index of small intestinal mucosal mass and integrity. Serum DAO levels have been found to increase markedly when the small intestine is strangulated, and elevations are thus believed to reflect small intestine mucosal ischemia[15]. Tsunooka et al[4,16] have demonstrated simultaneous increases in serum DAO activity and peptidoglycan concentrations during clinical CPB, suggesting the occurrence of small intestinal muco-sal ischemia and BT.
D-Lactate is habitually tested for in the intensive care unit. Mammals only have one type of enzyme: L-lactate dehydrogenase. L-Lactate is a marker of cell hypoxemia, and its levels correlate with survival in patients with septic shock[17,18]. Microorganisms however, particularly bacteria, are equipped with D-lactate dehydrogenase and produce D-lactate during fermentation, and D-lactate therefore acts as a marker of bacterial infection. D-Lactate has also recently been proposed as a sensitive, specific and early marker of translocation in gut ischemia[19].
In the current study, all the rats survived the CPB procedures. DAO and D-lactate activities remained low in the sham-operated animals (based on previously reported normal DAO value of 0.46 ± 0.087 U/L, and D-lactate value of 5.245 ± 0.653 mg/L[13,14]). However, significant increases in DAO, D-lactate and endotoxin levels were found in the vehicle-treated CPB group. The incidence of intestinal BT to the MLNs, spleen, liver, lung and blood was also significantly higher in the vehicle-treated CPB group, compared with that in the sham groups. These results indicate the occurrence of severe intestinal bar-rier injury after CPB. Transmission electron microscopic examination of intestinal tissues further confirmed the intestinal barrier injury in this rat CPB model.
There is increasing interest in developing PHC as a novel therapeutic agent. PHC is a new anticholinergic drug derived from hyoscyamine, which has few M2 receptor-associated cardiovascular side effects, because of its selec-tive blocking of M1, M3 and N receptors. Recent clinical results have demonstrated that PHC has curative effects in soman poisoning and pulmonary dysfunction associ-ated with chronic obstructive pulmonary disease[6,9,20]. In addition to improving microcirculation, PHC can inhibit lipid peroxidation, attenuate the release of lysosomes, and depress microvascular permeability[9,20]. Moreover, it can significantly decrease brain nuclear factor (NF)-κB expres-sion in cerebral ischemia/reperfusion (I/R) injury. Fur-thermore, PHC can improve acute lung injury stimulated by endotoxin and attenuate liver damage during CPB in a rat model[11].
In the present study, PHC lowered DAO, D-lactate and endotoxin levels in PHC-treated rats in a dose-depen-dent manner, suggesting its potential clinical application. The incidences of intestinal BT to MLNs, spleen, liver, lung and blood were lower in PHC-treated CPB rats than
in untreated CPB ones, suggesting that the use of PHC resulted in an overall decrease in bacteria. In intestinal mu-cosal injury, PHC can efficiently inhibit NF-κB expression in intestinal mucosal I/R injury. More importantly, PHC can improve the microcirculation, inhibit lipid peroxida-tion, attenuate the release of lysosomes, and decrease microvascular permeability, leading to the inhibition of inflammation[21].
However, our research was subject to some limita-tions. PHC treatment was only performed prior to CPB; although this pretreatment was effective, this study did not establish the efficacy of PHC given after CPB. Future studies are required to assess the effects of postoperative treatment with PHC. Additional research is also needed to establish the optimal time of PHC administration.
In conclusion, the present study demonstrated that PHC protected rat intestine from morphological and functional mucosal injury after CPB. These results sug-gest that PHC could be clinically useful for the treatment of intestinal injury induced by CPB.
COMMENTSBackgroundAn increase in intestinal permeability and bacterial translocation has been demonstrated not only in animal models, but also in patients during cardiopul-monary bypass (CPB). Previous in vitro studies have demonstrated that tropane alkaloids could stabilize the cell membrane and prevent oxidative stress. The new anticholinergic drug penehyclidine hydrochloride (PHC) has been evalu-ated for its protective effects on the cardiovascular system.Research frontiersPHC can improve microcirculation, inhibit lipid peroxidation, attenuate the release of lysosomes, and decrease microvascular permeability, leading to the inhibition of inflammation. The effects of PHC on intestinal barrier function during CPB have not been unequivocally addressed. We hypothesized that the administration of PHC could reverse CPB-associated intestinal damage.Innovations and breakthroughsThe selectivity of PHC in blocking M1, M3 and N receptors means that it has few M2 receptor-associated cardiovascular side effects. It can reduce endotox-in-stimulated acute lung injury and attenuate liver damage during CPB in a rat model. The present study demonstrated that the application of PHC during CPB preserved intestinal barrier function in a dose-dependent manner, and provided histological findings to support these biochemical results.ApplicationsThe present study demonstrated the ability of PHC to protect rat intestine from morphological and functional mucosal injury after CPB. These results suggest that PHC could be clinically useful in the treatment of intestinal injury induced by CPB.TerminologyPHC is a new anticholinergic drug with antioxidant and cell membrane-stabiliz-ing activities.Peer reviewThe authors have demonstrated a protective role for PHC in preventing intes-tinal breakdown during CPB. The conclusion was reached that the administra-tion of PHC could prevent intestinal damage and its sequelae following CPB surgery. This manuscript describes an interesting and well-performed study with convincing results.
REFERENCES1 Aydin NB, Gercekoglu H, Aksu B, Ozkul V, Sener T, Kiygil I,
Turkoglu T, Cimen S, Babacan F, Demirtas M. Endotoxemia in coronary artery bypass surgery: a comparison of the off-
2141 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Sun YJ et al . Intestinal protection by penehyclidine
COMMENTS

pump technique and conventional cardiopulmonary bypass. J Thorac Cardiovasc Surg 2003; 125: 843-848
2 Riddington DW, Venkatesh B, Boivin CM, Bonser RS, Elliott TS, Marshall T, Mountford PJ, Bion JF. Intestinal permeabil-ity, gastric intramucosal pH, and systemic endotoxemia in patients undergoing cardiopulmonary bypass. JAMA 1996; 275: 1007-1012
3 Watarida S, Mori A, Onoe M, Tabata R, Shiraishi S, Sugita T, Nojima T, Nakajima Y, Matsuno S. A clinical study on the effects of pulsatile cardiopulmonary bypass on the blood en-dotoxin levels. J Thorac Cardiovasc Surg 1994; 108: 620-625
4 Tsunooka N, Hamada Y, Imagawa H, Nakamura Y, Shiozaki T, Suzuki H, Kikkawa H, Miyauchi K, Watanabe Y, Kawachi K. Ischemia of the intestinal mucosa during cardiopulmonary bypass. J Artif Organs 2003; 6: 149-151
5 Shoji T, Omasa M, Nakamura T, Yoshimura T, Yoshida H, Ikeyama K, Fukuse T, Wada H. Mild hypothermia amelio-rates lung ischemia reperfusion injury in an ex vivo rat lung model. Eur Surg Res 2005; 37: 348-353
6 Liang SW, Chen YM. [Effects of penehyclidine hydrochloride or atropine combined with neostigmine for antagonizing residual neuromuscular block on patient's hemodynamics]. Diyi Junyi Daxue Xuebao 2005; 25: 1581-1582
7 Han XY, Liu H, Liu CH, Wu B, Chen LF, Zhong BH, Liu KL. Synthesis of the optical isomers of a new anticholinergic drug, penehyclidine hydrochloride (8018). Bioorg Med Chem Lett 2005; 15: 1979-1982
8 Yin L, Li K, Lü L. [Clinical observation of penehyclidine hy-drochloride as the preanesthetic medication before operation for patients with cleft lip/palate]. Huaxi Kouqiang Yixue Zazhi 2008; 26: 413-415
9 Gong P, Zhang Y, Liu H, Zhao GK, Jiang H. [Effects of penehyclidine hydrochloride on the splanchnic perfusion of patients with septic shock]. Zhongguo Weizhongbing Jijiu Yixue 2008; 20: 183-186
10 Cai DS, Jin BB, Pei L, Jin Z. Protective effects of penehycli-dine hydrochloride on liver injury in a rat cardiopulmonary bypass model. Eur J Anaesthesiol 2010; 27: 824-828
11 Zhan J, Wang Y, Wang C, Li J, Zhang Z, Jia B. Protective ef-fects of penehyclidine hydrochloride on septic mice and its
mechanism. Shock 2007; 28: 727-73212 Gourlay T, Ballaux PK, Draper ER, Taylor KM. Early experi-
ence with a new technique and technology designed for the study of pulsatile cardiopulmonary bypass in the rat. Perfu-sion 2002; 17: 191-198
13 Fürst W, Schiesser A. Test for stereospecifity of an automated Dd-lactate assay based on selective removal of Ll-lactate. Anal Biochem 1999; 269: 214-215
14 Li JY, Lu Y, Hu S, Sun D, Yao YM. Preventive effect of glu-tamine on intestinal barrier dysfunction induced by severe trauma. World J Gastroenterol 2002; 8: 168-171
15 Bounous G, Echavé V, Vobecky SJ, Navert H, Wollin A. Acute necrosis of the intestinal mucosa with high serum lev-els of diamine oxidase. Dig Dis Sci 1984; 29: 872-874
16 Tsunooka N, Maeyama K, Hamada Y, Imagawa H, Takano S, Watanabe Y, Kawachi K. Bacterial translocation secondary to small intestinal mucosal ischemia during cardiopulmonary bypass. Measurement by diamine oxidase and peptidogly-can. Eur J Cardiothorac Surg 2004; 25: 275-280
17 Bakker J, Gris P, Coffernils M, Kahn RJ, Vincent JL. Serial blood lactate levels can predict the development of multiple organ failure following septic shock. Am J Surg 1996; 171: 221-226
18 Dell'Aglio DM, Perino LJ, Kazzi Z, Abramson J, Schwartz MD, Morgan BW. Acute metformin overdose: examining serum pH, lactate level, and metformin concentrations in survivors versus nonsurvivors: a systematic review of the literature. Ann Emerg Med 2009; 54: 818-823
19 Isbir CS, Ergen A, Tekeli A, Zeybek U, Gormus U, Arsan S. The effect of NQO1 polymorphism on the inflammatory re-sponse in cardiopulmonary bypass. Cell Biochem Funct 2008; 26: 534-538
20 Lei LR, Wang YL, Jia BH. [Protective effect of penehycli-dine hydrochloride on lung injury in mice with sepsis and its mechanism]. Zhongguo Weizhongbing Jijiu Yixue 2007; 19: 623-624
21 Shi H, Dong CM. [The effect of penehyclidine hydrochloride on the expression of inflammatory factor in rat with sepsis-associated lung injury]. Zhongguo Weizhongbing Jijiu Yixue 2009; 21: 685-687
S- Editor Sun H L- Editor Kerr C E- Editor Zheng XM
2142 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Sun YJ et al . Intestinal protection by penehyclidine

BRIEF ARTICLE
p53 gene therapy in combination with transcatheter arterial chemoembolization for HCC: One-year follow-up
Yong-Song Guan, Yuan Liu, Qing He, Xiao Li, Lin Yang, Ying Hu, Zi La
Yong-Song Guan, Yuan Liu, Qing He, Xiao Li, Lin Yang, Ying Hu, Zi La, Department of Oncology, West China Hospital, Sichuan University, Chengdu 610041, Sichuan Province, ChinaAuthor contributions: Guan YS supervised the entire process of design and execution of the study and writing of the manuscript, and corrected the paper; Liu Y drafted the paper and organized the figures and patient data; He Q, Li X, Yang L, Hu Y and La Z completed the follow-up of patients and data collection. Correspondence to: Yong-Song Guan, Professor, Depart-ment of Oncology West China Hospital, Sichuan University, 37 Guoxuexiang, Chengdu 610041, Sichuan Province, China. [email protected] Telephone: +86-28-85421008 Fax: +86-28-85538359Received: August 15, 2010 Revised: November 13, 2010Accepted: November 20, 2010Published online: April 28, 2011
AbstractAIM: To evaluate the efficacy and safety of combina-tion therapy with recombinant adenovirus p53 injection (rAdp53) and transcatheter hepatic arterial chemoem-bolization (TACE) for advanced hepatocellular carci-noma (HCC).
METHODS: A total of 82 patients with advanced HCC treated only with TACE served as control group. Anoth-er 68 patients with HCC treated with TACE in combina-tion with recombinant adenovirus-p53 injection served as p53 treatment group. Patients were followed up for 12 mo. Safety and therapeutic effects were evaluated according to the improvement in clinical symptoms, leukocyte count, Karnofsky and RECIST criteria. Sur-vival rate was calculated with Kaplan-Meier method.
RESULTS: The total effective rate was 58.3% for p53 treatment group, and 26.5% for control group (P < 0.05). The incidence of gastrointestinal symptoms was lower in p53 treatment group than in control group (P < 0.05). The 3-, 6- and 12-mo survival rates were significantly higher for p53 treatment group than for
control group (P < 0.01). The combination treatment was well tolerated with such adverse events as fever (51.5%, P = 0.006) and pain of muscles and joints (13.2%, P = 0.003), which were significantly higher than the chemotherapy. Except for these minor ad-verse effects, no severe vector-related complications were identified. With respect to the efficacy, patients in p53 treatment group had less gastrointerestinal symp-toms (P = 0.062), better improvement in tumor-related pain (P = 0.003), less downgrade of leukocyte counts (P = 0.003) and more upgrade of Karnofsky performance score (P = 0.029) than those in control group. The total effective rate (CR + PR) for p53 treatment group and control group was 58.3% and 26.5%, respectively, with distributions of different effect in two groups (P = 0.042). The survival rates were 89.71%, 76.13%, and 43.30% for p53 treatment group, and 68.15%, 36.98%, and 24.02% for control group, respectively, 3, 6 and 12 mo after treatment, suggesting that the survival rates are significantly higher for p53 treatment group than for control group (P = 0.0002).
CONCLUSION: The rAd-p53 gene therapy in combi-nation with TACE is a safe and effective treatment mo-dality for advanced HCC.
© 2011 Baishideng. All rights reserved.
Key words: Adenovirus p53 ; Clinical trial; Hepatocel-lular carcinoma; Transcatheter hepatic arterial chemo-embolization; p53 gene therapy
Peer reviewer: Dr. Jun Qin, Baylor College of Medicine, One Baylor Plaza, Houston 77030, United States
Guan YS, Liu Y, He Q, Li X, Yang L, Hu Y, La Z. p53 gene therapy in combination with transcatheter arterial chemoembo-lization for HCC: One-year follow-up. World J Gastroenterol 2011; 17(16): 2143-2149 Available from: URL: http://www.wjgnet.com/1007-9327/full/v17/i16/2143.htm DOI: http://dx.doi.org/10.3748/wjg.v17.i16.2143
2143
World J Gastroenterol 2011 April 28; 17(16): 2143-2149 ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2143
April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

Guan YS et al . p53 combing TACE for HCC
INTRODUCTIONGene therapy is a potentially new treatment modality for cancer patients and an engineered recombinant replication-defective adenovirus can express the tumor suppressor gene p53 (rAd-p53) with encouraging clinical responses[1-3]. rAd-p53 has been recently approved by the State Food and Drug Administration of China as the very first gene ther-apy product for head and neck squamous cell carcinoma (HNSCC)[4].
Hepatocellular carcinoma (HCC) is one of the major cancers in China with a poor prognosis due to its occult onset, rapid infiltrating growth and complicating liver cirrhosis. No effective treatment modality is available for it at present. Although transcatheter hepatic arterial chemoembolization (TACE) is currently one of the most popular treatment modalities for unresectable advanced HCC, the long-term survival rate of such patients re-mains low with a reported 5-year survival rate of 17%[5]. In this study, the safety and efficacy of rAd-p53 therapy in combination with TACE were examined in patients with advanced HCC.
MATERIALS AND METHODSrAd-p53rAd-p53 is a recombinant human serotype 5 adenovirus in which the E1 region is replaced by a human wild-type p53 expression cassette. The p53 gene is driven by a Rous sarcoma virus promoter with a bovine growth hormone poly (A) tail. The recombinant adenovirus is produced in human embryonic kidney 293 cells and manufactured by Shenzhen SiBionoGenTech Co. Ltd (Shenzhen, China) and marketed under the trade name of Gendince®. Before p53 gene therapy, a vial of rAd-p53 is taken out from a re-frigerator in which the temperature is about -20℃. When thawed, the solution, diluted with 1 mL NS, is sucked into a 5-mL syringe for intra-tumor injection.
Patients and trial designOne hundred and fifty patients (83 men and 67 women) with advanced HCC were enrolled in this study from March to July 2004. Patients with Child C disease[6], tu-mor thrombus in the main portal trunk, or extrahepatic metastasis were excluded. These exclusion criteria were implemented to ensure at least a 3-mo life span in the enrolled patients so as to have enough time to follow up. All patients did not receive local ethanol injection, microwave coagulation, systemic chemotherapy or radio-therapy before and after TACE or gene therapy. All tu-mors were diagnosed according to pathologic examina-tion, distinctive findings on computed tomography (CT), conventional angiography, magnetic resonance imaging (MRI), or serum tumor markers [alpha-fetoprotein (AFP) or ferritin]. The patients were divided into gene treat-ment group (n = 68) with a mean age of 43 years (range 20-72 years) and control group (n = 82) with a mean age of 45 years (range 18-75 years). No patient was classified as stage Ⅰ or Ⅱ while 91 patients were classified as stage
Ⅲ and 59 patients as stage Ⅳ according to the Interna-tional Union against Cancer TNM classification[7].
Patients who gave their informed consent to receive Ad-p53 gene therapy served as gene treatment group, while those not willing to receive gene therapy served as control group. Patients in gene treatment group underwent rAd-p53 gene therapy and TACE while those in control group received only TACE. Although this was a retrospec-tive nonrandomized study, no statistical difference was observed in baseline between the two groups. The charac-teristics of the two groups are illustrated in Table 1.
Procedure of rAd-p53 intra-tumor injection The patients in gene treatment group were placed in a su-pine, prone or lateral position on the CT scanning bed and asked to hold their breath after an inhalation. The slice for puncture was carefully determined, the puncture site on the surface of body as well as the needle-traveling depth and angle within the body were determined. The bed was moved to the slice and a marker for puncture was made on the body surface according to the laser beam emitted from the gantry. The bed was then moved out and the puncture site was sterilized. After local anesthesia, a 19-G needle was inserted into the puncture site according to the determined angle and depth as the operator asked the pa-tient to hold his or her breath after an inhalation. Finally, another scan was performed to make sure that the tip of the needle was within the tumor, and the rAd-p53 gene was injected into the tumor in a multi-point fashion. Usu-ally, this procedure is repeated according to the patient’s clinical condition and the interval between two procedures is about 1 wk. At each injection, 1-4 rAd-p53 injections are administered at a viral dose of 1-4 × 1012 VP (viral particles) according to the diameter of the lesion, and the intra-tumor injection usually lasts 1-2 min.
TACE TACE was performed through the femoral artery using the Seldinger technique with local anesthesia. Arteriog-raphy of the celiac trunk and superior mesenteric artery was performed to visualize the arterial vascularization of liver and evaluate portal vein patency. An angiographic
Table 1 Characteristics of enrolled patients with hepatocel-lular carcinoma
2144 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Characteristics Gene group (n = 68)
Control group (n = 82)
Statistic analysis
Age 43.5 (20 - 72) 45.7 (18 - 75) NSSex (M/F) 43/25 40/42 NSChild class A 41 43 NSChild class B 27 39 NSUICC TNM classification Stage Ⅰ and Ⅱ 0 0 NS Stage Ⅲ 31 (46.5%) 60 (73.2%) NS Stage Ⅳ 37 (54.4%) 22 (26.8%) NSSize of main tumors ≥ 5 cm 53 (77.9%) 61 (74.3%) NS < 5 cm 15 (22.1%) 21 (25.7%) NS
NS: No statistical difference.

Group Change degree (×109/L) n (%)
< 4.0 < 3.0 < 2.0
Gene group 12 (25.0) 4 (8.3) 2 (4.2) 18 (37.5)Control group 8 (13.3) 20 (33.3) 11 (18.3) 39 (65.0)
catheter was inserted into the right or left hepatic artery where the target tumor was located. TACE agents, in-volving embolic agent (Lipiodol) and anticancer drugs, were injected through the right or left hepatic artery. In both groups, the dose of Lipiodol, ranging 3-20 mL, was determined according to the tumor location, tumor size, number of tumors, and functional hepatic reserve. An-ticancer drugs used were 5-Fluorouracil (800-1000 mg) and vinorelbine (30-40 mg). TACE was repeated accord-ing to the patient’s clinical condition at a 1-mo interval.
Follow-up protocolClinical symptoms, leukocyte counts and Karnofsky in-dex evaluation were recorded before and after treatment. After treatment, CT scan or MRI was performed every three months with or without contrast enhancement to evaluate the features of Lipiodol deposit and the thera-peutic effect according to the response evaluation crite-ria for solid tumors[8]. If elevated tumor markers (AFP and ferritin), diminished Lipiodol, or enlarged lesions or new nodules were observed, the patients were readmit-ted for angiography and treatment. The starting point of survival analysis was regulated as the day of initial treat-ment. The Kaplan-Meier method was used to analyze the survival rates in the two groups.
Statistical analysis Statistical analysis was performed to assess the baseline, leukocyte counts, Karnofsky index, clinical symptoms and survival curve between the two groups using the SPSS 11.0. P < 0.05 was considered statistically significant.
RESULTSTwo hundred and fifty-one p53 intra-tumor injections were performed for 83 lesions in 68 patients of gene treat-ment group. Of the 68 patients, 9 received one injection, 13 received two injections, 15 received three injections, 20 received four injections, 7 received five injections, 3 received six injections and 1 received seven injections. One hundred and ninety-two 2 (mean 2.82 procedures) and 167 (mean 2.03 procedures) procedures of TACE were per-formed in gene treatment and control groups, respectively. Arterial portal vein shunt (AVS), arterial hepatic vein shunt (APS) or/and portal vein involvement, signs that meant a high invasion and a poor prognosis were found in 27.9% (19/68) patients of gene treatment group and 36.6% (30/82) patients of control group, respectively, during the TACE. Although the patients with tumor thrombus in the main portal trunk were excluded, some of them developed vascular invasion because of tumor progression after they were enrolled in this study. No difference was observed in the incidence of malignancy signs such as AVS, APS or portal vein involvement between the two groups.
SafetyThe clinical symptoms were carefully recorded after treatment (Table 2). Overall, rAd-p53 gene therapy in combination with TRCE was well tolerated. The most
frequent adverse event occurred in patients receiving rAd-p53 gene therapy in combination with TACE was the flu-like symptom associated with fever. Of the 68 patients in gene treatment group, 35 (51.5%) had a fever at 38-39.5℃, usually occurred 3-10 h after p53 intra-tumor injection and decreased after physic cooling, and 9 (13.2%) had pain of muscles or joints which often faded away (Table 2). No other severe gene therapy-associated complications were encountered in this study.
EfficacyThe clinical symptoms were carefully recorded after treat-ment (Table 2). The patients in gene treatment group had less gastrointerestinal symptoms such as nausea, vomiting, abdominal pain or belling than those in control group. The palliative rate of mass-associated pain one week after treatment was 44.1% (30/68) for patients in gene treat-ment group, higher than that for those in control group.
Before and one week after treatment, the number of leukocytes was calculated (Table 3). Statistical analysis showed that the number of leukocytes was smaller in gene treatment group than in control group (P = 0.003).
Karnofsky index was changed in gene treatment group one month after treatment (Table 4). Generally speaking, the patients in gene treatment group had a higher Karnof-sky index than those in control group (P = 0.029).
The therapeutic effect was evaluated following the response evaluation criteria for solid tumors after treat-ment. CR, PR, NC and PD in the two groups are listed in Table 5. The total effective rate (CR + PR) was 58.3% and 26.5% for the gene treatment group and control group, respectively (P < 0.05). Chi-square test showed that the distributions of therapeutic effect were statisti-cally different (P = 0.042, Figures 1 and 2)
The patients were followed up for 12 mo. The num-ber of withdrawal patients in gene treatment group and control group was 4 and 7, respectively. The survival rate was 89.71% (standard error 0.036), 76.13% (stan-dard error 0.052), and 43.30% (standard error 0.061),
2145 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Table 2 Clinical synptoms after treatments
Group Fever Gastrointestinal symptoms
Palliation of mass-associated
pain
Pain of muscles or
joint
Gene group 35 (51.5)a 20 (29.4)b 30 (44.1)c 9 (13.2)d
Control group 24 (29.3) 28 (34.1) 21 (25.6) 1 (1.2)
χ2 = 7.679, aP = 0.006; χ2 = 4.001, bP = 0.062; χ2 = 5.674, cP = 0.017; χ2 = 8.626, dP = 0.003.
Table 3 Changes in leukocytes before and after treatment
Rank sum tests (Wilcoxon text), T = -3.018, P = 0.003 < 0.05.
Guan YS et al . p53 combing TACE for HCC

respectively, for the patients in gene treatment group 3, 6, and 12 mo after treatment. The survival rate was 68.15% (standard error 0.051), 36.98% (standard error 0.054), and 24.02% (standard error 0.049), respectively, for those in control group 3, 6, and 12 mo after treatment. Log-rank test showed that the survival rate for the two groups was significantly different (P = 0.0002, Figure 3).
DISCUSSIONHepatocellular carcinoma (HCC) is a highly malignant tumor with a very high morbidity and mortality. Since TACE was introduced as a palliative treatment of unre-sectable HCC, it has become one of the most common
interventional therapies[9-12]. However, its therapeutic ef-fect is also limited due to the lack of appropriate and reli-able embolic agents, and the infiltrative or hypovascular nature, too large or small in size[13-15]. Another limitation of TACE is the need for repeated treatment, thus result-ing in deterioration of liver function[16]. So, lots of efforts have been made to explore other new therapies in order to achieve the better efficacy of multiple treatments. PEI or RFA gene therapy in combination with TACE may improve the survival rate of HCC patients and decrease the risk of liver failure[17-19]. In this study, p53 gene therapy in combination with TACE could overcome the downside of TACE and improve the prognosis of HCC patients.
The p53 tumor suppressor gene is a gene guardian and loss of p53 is responsible for the lack of apoptotic signals
2146 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Figure 1 Contrast computed tomography showing a nodule (3.5 cm in diameter) in the right upper liver lobe manifested as homogenous enhancement (A); computed tomography scan (b) demonstrating the course of fine needle biopsy under computed tomography guidance with the diagnosis of hepatocellular carcinoma confirmed (B); computed tomography follow-up (c) revealing lipiodol deposit in the mass and spleen infarction after spleen embolization (C) in a 52-year-old man with multiple hepatic nodules, liver cirrhosis, splenomegaly and elevated alpha-fetoprotein.
A B C
A B
Table 4 Changes in Karnofsky index before and after treat-ment
Group Upgrade > 20 points
Upgrade > 10 points
No changes
Downgrade > 10 points
Total upgrade [n (%)]
Gene group 14 28 18 8 42 (61.8)Control group 12 24 18 28 36 (43.9)
χ2 = 4.752, P = 0.029.
Table 5 Therapeutic effect evaluated following response evaluation criteria for solid tumors 2 mo after treatment
Group n CR PR NC PD Effective rate (CR + PR)
Gene group 68 0 46 15 7 67.60%Control group 82 0 42 27 13 51.20%
χ2 = 4.137, P = 0.042 < 0.05. CR: Complete response; PR: Partial response; NC: No change; PD: Progressed disease.
Figure 2 Contrast computed tomography scan showing a 15 cm × 11.5 cm hepatocellular carcinoma in the right liver lobe manifested as a heterogenous lower density, partial enhancement and well-differentiated contour (A) and computed tomography follow-up displaying the significant regression of a 8.5 cm x 6 cm lesion with compact lipiodol deposit in a 72-year-old man after 3 p53 gene injections and 4 courses of transcatheter hepatic arterial chemoembolization.
Guan YS et al . p53 combing TACE for HCC

in tumor cells and thus for their uncontrolled prolifera-tion and recurrence[20]. Many human tumors carry muta-tions in the p53 gene[21,22] and mutant or absent p53 gene is associated with the resistance to radiotherapy and apoptosis-inducing chemotherapy[23]. It has been shown that p53 gene therapy in combination with radiotherapy or chemotherapy can control local tumor, suggesting that it is superior to either radiotherapy or chemotherapy alone[24,25]. It was reported that the incidence of p53 mu-tation is 61% in HCC[22]. Chen et al[26] also reported that mutations in the p53 gene are frequently detectable in recurrent HCC and the interval between surgical resection and recurrence of HCC is significantly longer in patients with the wild-type p53 gene than in those with mutant p53 gene mutations, strongly suggesting that the mutant p53 gene plays a role in pathogenesis of HCC. Jeng et al[27] demonstrated that the biological behavior of the mu-tant p53 gene is strongly related to the invasiveness of HCC and may also influence the postoperative course of HCC. Many scholars suggest that immunopositivity of the mutant p53 gene plays a role in predicting the prog-nosis of patients with HCC after resection[27-29].
The rAd-p53 gene has been approved in China under the trade name of Gendicine for the treatment of head and neck squamous cell carcinoma (HNSCC). In one of the trials[3], 75% tumors experienced complete regression following 8 wk of therapy involving 1 injection per week, which was significantly higher than that in control group, and combined chemotherapy and radiotherapy improved the treatment efficacy of over 3-fold. Although its rec-ommended indications are limited in HNSCC accord-ing to the specification, good treatment efficacy can be achieved in HCC patients when rAd-p53 is used[30]. In the current study, Gendicine was used in treatment of HCC to evaluate its effect in order to provide some evidence for its off-table use in treatment of HCC.
As for the safety of rAd-p53 used in treatment of ad-vanced HCC, just fever at 38-39.5℃ was observed in our study, which was returned to normal after symptomatic treatment. In addition, some patients suffered from pain of muscles or joints and its cause is still controversial. However, no severe complications caused by Gendicine
were observed. Although these adverse events have been observed in clinical practice, they can be well tolerated by most patients with no severe physical and mental harm.
The patients receiving p53 gene therapy had less severe post embolization syndrome than others after TACE. Gastrointestinal symptoms, such as nausea, vomiting and abdominal pain or belling, were less frequently observed in gene treatment group than in control group. The de-creased number of leukocytes in gene treatment group was a pleasing phenomenon. However, its mechanism re-mains to be studied. The Karnofsky index was significantly higher, suggesting that the life quality of patients is largely improved in gene treatment group. It could be concluded that the rAd-p53 gene therapy could reduce the side effects of chemical drugs and Lipiodol embolization. Also, it was noticed that many patients in gene treatment group had a compact Lipiodol deposit manifested as a high homoge-nous density occupying the majority of tumor mass (Figures 1 and 2). Compact deposit means tumor necrosis. Further study is needed to observe whether p53 gene therapy is re-lated to the better deposit of Lipiodol in lesions.
Theoretically, in-vitro p53 protein can bring about specif-ic anti-tumor cells into effect in such ways as induction of apoptosis or necrosis, incentive of body immune response, regulation of cell cycle, etc. Two months after treatment, the distributions of therapeutic effect in the two groups were statistically different and the effective rate (CR + PR) was higher for p53 gene treatment group than for control group, suggesting that p53 gene therapy can enhance the efficacy of TACE, radiotherapy and chemotherapy.
Kaplan-Meier analysis showed that the survival rate was higher for gene treatment group than for control group. Because no other control study is available, the outcome of p53 gene therapy for such a large number of patients was not compared with that in other studies. The 1-year survival rate was lower in our study than in anoth-er study (67% vs 81%)[31], which may be attributed to the different baselines, in which our enrolled patients might have a larger lesion and a poorer liver function reserve.
Although it seems that the higher survival rate in gene treatment group may be attributed to the longer mean TACE time in patients of gene treatment group than in those of control group (2.82 vs 2.03), it was the clinical im-provement after p53 gene therapy that made the patients in gene treatment group have more chance to receive repeat-ed TACE. On the other hand, no difference was found in the incidence of malignancy DSA signs between the two groups. However, these signs appeared later with a lower incidence in gene treatment group than in control group, which is an interesting phenomena, and further study with a larger sample size is needed to confirm it.
Usually, the rAd-p53 gene begins to express p53 protein 3 h after intra-tumor injection, reaches its peak on day 3, and then gradually decreases according to the specification of Gendicine®. On day 5 after injection, the expression decreases to 30%. Because most of the che-motherapeutic drugs can affect DNA or RNA duplica-tion or expression, cell cycle or nucleic acid metabolism would likewise affect the expression of p53 gene in
2147 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Control group
Combo group
Control group-censored
Combo group-censored
Treatment measures1.2
1.0
0.8
0.6
0.4
0.2-2 0 2 4 6 8 10 12
Cum
sur
viva
l
Survival time-month
Figure 3 Survival curves for patients following treatment.
Guan YS et al . p53 combing TACE for HCC

tumor tissue. In this study, TACE was started 3-4 d after p53 injection when the p53 protein was highly expressed in tumor tissue, indicating that these anti-tumor drugs do not interfere with the expression of p53. However, the optimal interval remains to be further studied.
In conclusion, rAd-p53 gene therapy in combination with TACE is well tolerated and its anti-tumor efficacy is superior to that of TACE alone in terms of the survival rate and improved symptoms of HCC patients. Further clinical study with a large sample size is warranted to optimize the administration procedure and assess the impact of anti-p53 antibody on its therapeutic effect.
COMMENTSBackgroundHepatocellular carcinoma (HCC) is one of the major cancers in China with a poor prognosis due to its occult onset, rapid infiltrating growth and complicating liver cirrhosis. Although transcatheter arterial chemoembolization (TACE) has been used in treatment of HCC for years, its effect is often unsatisfactory. Research frontiersAmong the actively studied novel treatment modalities for HCC, the majority of experts hold that comprehensive or combination ones are most promising. In addition, gene therapy with p53 (rAd-p53) is a potentially new treatment modality for cancer.Innovations and breakthroughsTACE in combination of rAd-p53 injection has a synergistic effect on HCC and its strategy is gene addition. Tumor with mutant of the rAd-p53 gene is a better candidate for p53 therapy. However, this treatment is also effective in those with inactivated wild-type p53, a common condition in tumors. Injection of rAd-p53 can lead to apoptosis of tumor cells and TACE can result in necrosis of tumor tissue.Applications The results of this study demonstrate that TACE in combination with rAd-p53 with is well tolerated and its anti-tumor efficacy is superior to that of TACE alone with respect to the survival rate and improved symptoms. Further study with a large sample size would provide an alternative treatment modality for HCC. Terminologyp53 gene is a tumor suppressor gene which can prevent the formation of tu-mors. Mutations in p53 are found in most tumor types and contribute to complex molecular events leading to tumor formation. Recombinant adenovirus is one of the viral vectors which are commonly used to deliver genetic materials into cells. Gene therapy for diseases is to insert, alterate, or remove such materials in cells.Peer reviewThis is a well-designed study in which the authors analyzed the clinical effect of rAd-p53 injection and TACE on advanced HCC. The data show that the combination therapy is a safe and effective treatment modality for advanced HCC, and can significantly improve the survival rate of HCC patients.
REFERENCES1 Robson T, Hirst DG. Transcriptional Targeting in Cancer
Gene Therapy. J Biomed Biotechnol 2003; 2003: 110-1372 Zhang SW, Xiao SW, Liu CQ, Sun Y, Su X, Li DM, Xu G,
Cai Y, Zhu GY, Xu B, Lü YY. [Treatment of head and neck squamous cell carcinoma by recombinant adenovirus-p53 combined with radiotherapy: a phase II clinical trial of 42 cases]. Zhonghua Yixue Zazhi 2003; 83: 2023-2028
3 Chen CB, Pan JJ, Xu LY. [Recombinant adenovirus p53 agent injection combined with radiotherapy in treatment of nasopharyngeal carcinoma: a phase II clinical trial]. Zhong-hua Yixue Zazhi 2003; 83: 2033-2035
4 Cancer gene therapy is first to be approved. Available from: URL: http://www.lucifer.com/pipermail/ectropy-chat/at-tachments/20030019/ee07cea0/attachment.htm
5 Ueno K, Miyazono N, Inoue H, Nishida H, Kanetsuki I, Na-
kajo M. Transcatheter arterial chemoembolization therapy using iodized oil for patients with unresectable hepatocel-lular carcinoma: evaluation of three kinds of regimens and analysis of prognostic factors. Cancer 2000; 88: 1574-1581
6 Takayasu K, Arii S, Matsuo N, Yoshikawa M, Ryu M, Ta-kasaki K, Sato M, Yamanaka N, Shimamura Y, Ohto M. Comparison of CT findings with resected specimens after chemoembolization with iodized oil for hepatocellular car-cinoma. AJR Am J Roentgenol 2000; 175: 699-704
7 Sobin LH, Wittekind CH, editors. UICC TNM classification of malignant tumours. 5th ed. New York: John Wiley and Sons, 1997
8 Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European Orga-nization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Insti-tute of Canada. J Natl Cancer Inst 2000; 92: 205-216
9 Llovet JM, Real MI, Montaña X, Planas R, Coll S, Aponte J, Ayuso C, Sala M, Muchart J, Solà R, Rodés J, Bruix J. Arte-rial embolisation or chemoembolisation versus symptom-atic treatment in patients with unresectable hepatocellular carcinoma: a randomised controlled trial. Lancet 2002; 359: 1734-1739
10 Li L, Wu PH, Li JQ, Zhang WZ, Lin HG, Zhang YQ. Seg-mental transcatheter arterial embolization for primary hepa-tocellular carcinoma. World J Gastroenterol 1998; 4: 511-512
11 Mizoe A, Yamaguchi J, Azuma T, Fujioka H, Furui J, Kane-matsu T. Transcatheter arterial embolization for advanced hepatocellular carcinoma resulting in a curative resec-tion: report of two cases. Hepatogastroenterology 2000; 47: 1706-1710
12 Llovet JM, Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoemboliza-tion improves survival. Hepatology 2003; 37: 429-442
13 Qian J, Truebenbach J, Graepler F, Pereira P, Huppert P, Eul T, Wiemann G, Claussen C. Application of poly-lactide-co-glycolide-microspheres in the transarterial chemoemboliza-tion in an animal model of hepatocellular carcinoma. World J Gastroenterol 2003; 9: 94-98
14 Fan J, Ten GJ, He SC, Guo JH, Yang DP, Wang GY. Arterial chemoembolization for hepatocellular carcinoma. World J Gastroenterol 1998; 4: 33-37
15 Lin DY, Lin SM, Liaw YF. Non-surgical treatment of hepato-cellular carcinoma. J Gastroenterol Hepatol 1997; 12: S319-S328
16 Ahrar K, Gupta S. Hepatic artery embolization for hepa-tocellular carcinoma: technique, patient selection, and out-comes. Surg Oncol Clin N Am 2003; 12: 105-126
17 Qian J, Feng GS, Vogl T. Combined interventional therapies of hepatocellular carcinoma. World J Gastroenterol 2003; 9: 1885-1891
18 Chen XM, Luo PF, Lin HH, Zhou ZJ, Shao PJ, Fu L, Li WK. [Long-term result of combination of transcatheter arterial chemoembolization and percutaneous ethanol injection for treatment of hepatocellular carcinoma]. Ai Zheng 2004; 23: 829-832
19 Guo WJ, Yu EX, Liu LM, Li J, Chen Z, Lin JH, Meng ZQ, Feng Y. Comparison between chemoembolization combined with radiotherapy and chemoembolization alone for large hepatocellular carcinoma. World J Gastroenterol 2003; 9: 1697-1701
20 Kouraklis G. Progress in cancer gene therapy. Acta Oncol 1999; 38: 675-683
21 Friedmann T. Gene therapy of cancer through restoration of tumor-suppressor functions? Cancer 1992; 70: 1810-1817
22 Hsia CC, Nakashima Y, Thorgeirsson SS, Harris CC, Mine-mura M, Momosaki S, Wang NJ, Tabor E. Correlation of im-munohistochemical staining and mutations of p53 in human hepatocellular carcinoma. Oncol Rep 2000; 7: 353-356
23 Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE,
2148 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Guan YS et al . p53 combing TACE for HCC
COMMENTS

Fisher DE, Housman DE, Jacks T. p53 status and the efficacy of cancer therapy in vivo. Science 1994; 266: 807-810
24 Gjerset RA, Turla ST, Sobol RE, Scalise JJ, Mercola D, Collins H, Hopkins PJ. Use of wild-type p53 to achieve complete treatment sensitization of tumor cells expressing endogenous mutant p53. Mol Carcinog 1995; 14: 275-285
25 Nguyen DM, Spitz FR, Yen N, Cristiano RJ, Roth JA. Gene therapy for lung cancer: enhancement of tumor suppres-sion by a combination of sequential systemic cisplatin and adenovirus-mediated p53 gene transfer. J Thorac Cardiovasc Surg 1996; 112: 1372-1376; discussion 1376-1377
26 Chen GG, Merchant JL, Lai PB, Ho RL, Hu X, Okada M, Huang SF, Chui AK, Law DJ, Li YG, Lau WY, Li AK. Muta-tion of p53 in recurrent hepatocellular carcinoma and its association with the expression of ZBP-89. Am J Pathol 2003; 162: 1823-1829
27 Jeng KS, Sheen IS, Chen BF, Wu JY. Is the p53 gene muta-tion of prognostic value in hepatocellular carcinoma after
resection? Arch Surg 2000; 135: 1329-133328 Qin LX, Tang ZY, Ma ZC, Wu ZQ, Zhou XD, Ye QH, Ji Y,
Huang LW, Jia HL, Sun HC, Wang L. P53 immunohisto-chemical scoring: an independent prognostic marker for patients after hepatocellular carcinoma resection. World J Gastroenterol 2002; 8: 459-463
29 Sheen IS, Jeng KS, Wu JY. Is p53 gene mutation an indicati-or of the biological behaviors of recurrence of hepatocellular carcinoma? World J Gastroenterol 2003; 9: 1202-1207
30 Guan YS, Liu Y, Zhou XP, Li X, He Q, Sun L. p53 gene (Gendicine) and embolisation overcame recurrent hepato-cellular carcinoma. Gut 2005; 54: 1318-1319
31 Kamada K, Nakanishi T, Kitamoto M, Aikata H, Kawakami Y, Ito K, Asahara T, Kajiyama G. Long-term prognosis of patients undergoing transcatheter arterial chemoemboliza-tion for unresectable hepatocellular carcinoma: comparison of cisplatin lipiodol suspension and doxorubicin hydrochlo-ride emulsion. J Vasc Interv Radiol 2001; 12: 847-854
S- Editor Sun H L- Editor Wang XL E- Editor Ma WH
2149 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Guan YS et al . p53 combing TACE for HCC

CASE REPORT
Celiac disease and microscopic colitis: A report of 4 cases
Zsolt Barta, Eva Zold, Arpad Nagy, Margit Zeher, Istvan Csipo
Zsolt Barta, Eva Zold, Arpad Nagy, Margit Zeher, Istvan Csipo, 3rd Department of Medicine, Institute for Internal Medi-cine, Medical and Health Science Center, University of Debre-cen, 4032 Moricz Zs. krt. 22, Debrecen, HungaryAuthor contributions: All authors gave substantial contributions to acquisition, analysis and interpretation of data and participated in writing the paper; Barta Z gave final approval of the version to be published.Correspondence to: Zsolt Barta, MD, PhD, 3rd Depart-ment of Medicine, Institute for Internal Medicine, Medical and Health Science Center, University of Debrecen, 4032 Moricz Zs. krt. 22, Debrecen, Hungary. [email protected]: +36-52-255218 Fax: +36-52-255218Received: October 27, 2010 Revised: December 30, 2010Accepted: January 7, 2011 Published online: April 28, 2011
AbstractCeliac disease (CD) is an autoimmune disorder of the small intestine that occurs in genetically predisposed people at all ages. However, it can be associated also to other immunopathological disorders, and may be associated with abnormal histology in segments of the gut other than the small bowel including colonic inflammation. While guidelines for endoscopic investigation of the jejunum are well defined, no indication is defined for colonic investigation. We describe four cases of concurrent CD and microscopic colitis (MC) diagnosed at our department over a 10year period and analyzed the main features and outcomes of CD in this setting. The symptoms of these patients were improved initially by a glutenfree diet before the onset of MC symptoms. Two of the patients were siblings and had an atypical form of CD. The other two patients with CD and MC also presented with fibrosing alveolitis and were antiSaccharomyces cerevisiae antibody positive. The coexistence of immunemediated small bowel and colonic inflammatory and pulmonary diseases are not wellknown, and no systematic approach has been used to identify the lifelong patterns of these immunebased diseases. Patients can develop, or present with CD at any stage in life, which can coexist with other gastrointestinal diseases of (auto) immune origin. In addition, the fa
milial coexistence and prevalence of MC in patients with a prior diagnosis of CD are unclear. Clinicians managing celiac disease should be aware of these associations and understand when to consider colon investigation.
© 2011 Baishideng. All rights reserved.
Key words: Collagen colitis; Lymphocytic colitis; Celiac disease; Fibrosing alveolitis; Antisaccharomyces cerevisiae antibody
Peer reviewer: Dr. Alberto Tommasini, MD, Professor, Labo-ratory of Immunopathology, Institute for Maternal and Child Health, IRCCS Burlo Garofolo, Via dell’Istria 65/1, Trieste 34137, Italy
Barta Z, Zold E, Nagy A, Zeher M, Csipo I. Celiac disease and microscopic colitis: A report of 4 cases. World J Gastroenterol 2011; 17(16): 2150-2154 Available from: URL: http://www.wjgnet.com/1007-9327/full/v17/i16/2150.htm DOI: http://dx.doi.org/10.3748/wjg.v17.i16.2150
INTRODUCTIONCeliac disease (CD) is an immune-mediated disorder, an autoimmune enteropathy, triggered by the ingestion of gluten in genetically susceptible persons. The dis-ease primarily affects the gastrointestinal tract and is characterized by chronic inflammation of the small bowel mucosa that may result in atrophy of intestinal villi, ma-labsorption, and a variety of clinical manifestations. Of genetic factors, the strongest recognized association is with HLA-DQ2 and/or -DQ8: 95%-100% of the patients carry these molecules. Dietary glutens interact with these HLA molecules to activate an abnormal mucosal immune response and induce tissue damage. Most affected individuals experience remission after gluten is excluded from their diet.
The diagnosis of CD is established by serologic test-ing, biopsy evidence of villous atrophy, and improvement of symptoms on a gluten-free diet. Avoidance of gluten
2150
World J Gastroenterol 2011 April 28; 17(16): 2150-2154 ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2150
April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

Barta Z et al . CD and MC
exposure is crucial for CD patients to reduce the risk of complications so the follow-up serological assessment of treatment effectiveness should be added to be sure of a good compliance.
There are atypical forms of CD. For example, silent CD is found in individuals who are asymptomatic but have a positive serologic test and villous atrophy on biopsy, and latent CD is defined by a positive serology but no villous atrophy on biopsy. These individuals are asymptomatic, but later may develop symptoms and/or histological changes[1]. The late concordance in the appearance of CD in mono-zygotic twins also suggests that the disorder may remain in the latent stage for a long time[2,3]. Small bowel villous atrop-hy with crypt hyperplasia and recovery of the lesion on a gluten-free diet suggest that villous atrophy comprises only the end stage of the clinical course of the disease and that CD clearly develops gradually from mucosal inflammation to crypt hyperplasia and finally to overt villous atrophy.
A typical feature of CD, in addition to mucosal changes, is gluten-dependent serum IgA class autoantibodies against transglutaminase 2 (TG2). These serum autoantibodies, endomysial and TG2, are powerful tools in disclosing CD with overt villous atrophy. Furthermore, positive serum celiac autoantibodies can predict impending CD in many patients evincing normal small bowel mucosal villous ar-chitecture. Hence, patients having “false-positive” celiac autoantibodies in serum are in fact at risk of developing overt CD. Some patients with positive serum endomysial or tissue TG2 antibodies may still seroconvert negatively during follow-up. However, it is well recognized that se-rum celiac autoantibodies in some cases fluctuate before a patient eventually develops overt CD after a longer follow-up period. The reason for this still remains obscure.
Transglutaminases are a family of 8 currently known calcium-dependent enzymes that catalyze the cross-linking or deamidation of proteins and are involved in important biological processes such as wound healing, tissue repair, fi-brogenesis, apoptosis, inflammation, and cell-cycle control. Therefore, they play an important role in the pathomecha-nisms of autoimmune, inflammatory, and degenerative diseases, many of which affect the gastrointestinal system. Transglutaminase 2 is prominent, since it is central to the pathogenesis of CD, and modulates inflammation and fi-brosis in inflammatory bowel and chronic liver diseases[4]. Respiratory disease and subclinical pulmonary abnormali-ties are the recognized complications of both CD and inflammatory bowel disease (IBD) but the mechanisms of lung disease in CD differ from that in IBD and support the hypothesis of a common mucosal defect in lung and small intestine in CD that allow increased permeability[5].
Lymphocytic colitis (LC), together with collagenous colitis (CC), is included under the umbrella term “micro-scopic colitis” (MC), in which chronic gastrointestinal symptoms, including diarrhea, abdominal pain, fecal urgen-cy, incontinence, and nausea, are not associated with endo-scopic or radiological alterations. It is not known whether LC and CC are two different diseases or distinct manifesta-tions of the same clinical condition. Data on pathophysiol-ogy conflict and different hypotheses refer to genetic pre-
disposition, immune dysregulation, autoimmunity, bile acid malabsorption, infection, and drug effect. Familial occur-rence of MC has been identified in some families[6-13]. The central role of an altered immune system in MC pathogen-esis is supported by the association with several conditions in which an immune dysregulation is involved, such as CD, rheumatoid arthritis, and hypo- and hyperthyroidism. Up to now, it has not been clear whether CC (or LC) is a distinct entity or only an epiphenomenon of another disease that leads to thickening of the collagen layer. However, whether MC (both CC and LC) is an autoimmune disease has not been conclusively established[14]. Diagnosis of MC can be established only by colonic biopsies and subsequent histo-pathological examination, when an increase in inflamma-tory infiltration and/or a thickening of the collagen layer are found. A number of papers have documented an asso-ciation between CD and MC[15-18]. However, the prevalence of MC in patients with a prior diagnosis of CD is unclear, but it does feature prominently in several series of patients with CD who have persisting symptoms despite gluten exclusion. When continuing gluten ingestion, inadvertent or covert, has been excluded, colon investigation should be considered as part of the investigation of these patients. The link may be genetic, at least in part. Both types of MC are known to resolve spontaneously in a majority of cases. Data are limited regarding pharmacological therapies, but budesonide appears best documented as showing an effi-cacy against CC and MC[19].
We report here 4 cases with sequential development of CD and MC and discuss the possible connection of these co-existences.
CASE REPORTCase 1A 42-year-old female with a previous history of both cognitive and neurovegetative symptoms of depression, including depressed mood, anhedonia, feelings of wor-thlessness, low energy, troubled sleep, and poor concen-tration, was evaluated in the local medical center for com-plaints of watery diarrhea. She had longstanding lactose intolerance for which she was taking a lactose-free diet. As her mother had manifestations of CD, enteroscopy was performed. However, the first endoscopic and histological evaluation showed no duodenal mucosal alterations (Marsh 0). Six months later, endoscopic findings were persistent and duodenal biopsies were taken which were not diagnostic for CD, and biochemical laboratory tests were within normal ranges. She was then referred to our clinic and additional laboratory tests showed increased antibody titers against gliadin, endomysium, and tTG. Her psychiatric disease was controlled after treatment and then remained stable. She was given a gluten-free diet, which resolved her diarrhea and allowed her to regain her lost body weight. Five years later, the patient presented with watery diarrhea occurring 8-10 times daily and mild body weight loss. At the beginning, this condition was associated with urgency, nocturnal stools, abdominal cramping, nausea, mild body weight loss, and fatigue, and persisted
2151 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

despite strict adherence to the gluten- and lactose-free diet. With the loss of patience of the diet and of her symptoms she broke her diet and clinical signs remained. Small bo-wel biopsies at upper endoscopy demonstrated nothing (Marsh 0) but the gluten panel was unambiguously positive. Stool cultures and Clostridium difficile toxin assay were negative. After consultation with dietitians, the patient was maintained on a gluten- and lactose-free diet for six months but with mild improvement. Colonoscopy was performed later, and biopsies from her colon demonstrated LC. Her symptoms responded partially to mesalazine treatment over the subsequent two months, at which point her medical therapy was changed to budesonide (9 mg/d). After she was put on budesonide with a strict diet, both abdominal complaints and psychiatric problems resolved (Figure 1).
Case 2A 45-year-old woman with suspected irritable bowel syn-drome was admitted to the hospital. Her bowel movements increased from one to six or eight a day with watery stools. She did not note any mucus or blood in the stool and could not identify any alleviating or aggravating factors. She consumed a normal diet, including meat, wheat, and dairy. Over-the-counter anti-diarrheal medications did not relieve her symptoms. She had no fevers, chills, or night sweats, but body weight loss. Her medical hist-ory included major depression for 10 years, which was controlled after treatment and remained stable at admis-sion. Results of basic laboratory tests, including thyroid-stimulating hormone (TSH), complete blood count, blood chemistries, renal function, and liver function, were nor-mal. Colonoscopy showed normal mucosa as far as the cecum. Colonic biopsy revealed a mildly expanded lamina propria and intraepithelial lymphocytosis with significantly thickening of the subepithelial collagen table. This set of features was consistent with CC, a variant of MC. Her symptoms were eventually controlled after a 6-mo course of oral budesonide (9 mg) and ongoing intermittent use of loperamide (Imodium). Six years later, similar problems with body weight loss caused her to be hospitalized at our clinic. A detailed previous history unraveled the familial connection with Case 1 and her mother with known CD. Psychiatric disease was controlled, so control and further GI investigations were organized. The histopathology report of colonic biopsy showed aspecific inflammation without MC. Further laboratory investigation revealed that the entire celiac antibody panel was positive. Results of duodenal biopsy did not reveal typical lymphocyte infiltration, crypt hyperplasia, and villous atrophy but normal mucosal architecture, without significant intraepi-thelial lymphocytic infiltration (Marsh 0). The diagnosis was latent CD, as the patient had abnormal antibody blood tests for CD but normal small intestines. After she was put on a strict gluten-free diet, both abdominal complaints and psychiatric problems resolved (Figure 1).
Case 3A 56-year-old woman with a previous history of chronic (non-specific) colitis and fibrosing alveolitis was referred
to our hospital from an outside hospital because of contin-ued signs and symptoms of CD that persisted despite self-reported adherence to a gluten-free diet. The patient rep-orted abdominal pain, bowel distension, and body weight loss over the past few years. Diagnosis of CD was made 4 years ago, based on the small bowel biopsy results showing evidence of villous blunting with increased chronic infl-ammatory cells, positive laboratory tests, and typical gastrointestinal signs and symptoms with negative stool cultures and Clostridium difficile toxin assay. Repeated laboratory tests showed elevated antibodies against gliadin, endomysium, and tTG, and small bowel biopsy proved villous atrophy. The patient met with a nutritionist and implemented recommended dietary changes to eliminate gluten. Her symptoms temporarily improved with her bowel function returned to normal, but after a short time her symptoms recurred. Results of further tests excluded conditions known to complicate or coexist with CD, including bacterial overgrowth and lactose intolerance. Because of chronic watery stools, a colonoscopy was do-ne with random biopsies from the colon for histological investigations. Based on the typical picture of prominent intraepithelial lymphocytes but no thickened collagenous layer, the pathologist diagnosed her with LC. She was started on strict gluten-free diet and budesonide with success. Five years later, she was free of complaints of CD and LC.
Case 4A man at age 31, with a previous history of bronchial ast-hma, was investigated for abdominal pain, chronic watery diarrhea, and body weight loss with negative stool samples (both the cultures and Clostridium difficile toxin assay). Findings from an upper gastrointestinal endoscopy were normal, but distal duodenal biopsies showed subtotal villous atrophy, inflammatory infiltration of the lamina propria, and an increase in intraepithelial lymphocytes. Based on the histology and positive laboratory tests, CD was diagnosed and the patient was started on a gluten-free diet. Abdominal pain ceased but he did not gain body weight and diarrhea remained a problem. Compliance with a gluten-free diet was confirmed by the assessment of dietitians. Repeated biopsies of the duodenal mucosa showed mild improvement in villous atrophy but serology
2152 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Mother
Celiacdisease
Case 1Latent CD
and MCLC
Case 2Latent CD
and MCCC
Figure 1 Family of cases 1 and 2.
Barta Z et al . CD and MC

was negative. Four years later, a dietitian again confirmed adherence to a strict gluten-free diet and colonic biopsies showed no alteration. A barium follow-through showed mild jejunal and rather featureless ileal mucosa but no obstructive lesion of the small bowel, nothing abnormal was seen on an ultrasound scan of the abdomen. Because of bloody stools and in view of his worsening symptoms despite the gluten-free diet, repeated colonoscopy with random biopsies was done for histological investigations from ileal and colonic samples. Both proved a submucosal thickened collagen layer, thus the diagnosis of collagenous entero-colitis (with CD) was made. He was started on mesalazine and budesonide but without e’clat. The next step was methylprednisolone, initially 32 mg/d, and then the dose was decreased to 4 mg/d. This therapy was continued with corticosteroids for three months. Over the next year, his clinical condition improved, with resolution of his diarrhea and a body weight gain of 3 kg. Three years later, his symptoms recurred. Results of further tests
excluded conditions known to complicate or coexist with CD, including bacterial overgrowth and lactose intolerance. Repeated biopsies excluded collagenous entero-colitis, thus fibrosing alveolitis was diagnosed by the pulmonologist based on the lung function, laboratory and radio-imaging tests, chest X-ray, and high-resolution CT scanning (HRCT). Because the abdominal symptoms of the patient were refractory to treatment, he was treated again with budesonide and his clinical condition improved.
The diagnoses of CD, MC, and fibrosing alveolitis in all cases were made according to the formally accepted crite-ria. Two independent pathologists certified the diagnosis of MC by verifying the subsequent sections and completing the check with additional investigations (intraepithelial lym-phocytes, tenascin labeling of the collagen layer, mast cells, and other lamina propria cell components). Fibrosing al-veolitis was proved by HRCT and upon the ATS/ERS clin-ical criteria. The laboratory tests were performed. In brief, the HLA-DQ alleles were determined from whole blood samples by PCR with sequence-specific primers, traditional IgG and IgA AGA were detected by ELISA (α-gliatest IgG and IgA; Eurospital, Trieste, Italy), anti-tTG was measured
by ELISA using recombinant human tissue transglutamin-ase as an antigen (EutTG, Eurospital), IgG and IgA EmA were investigated by indirect immunofluorescence using human umbilical cord cryostat sections prepared in our laboratory as a substrate, and both serum IgG and IgA levels in anti-Saccharomyces cerevisiae antibody (ASCA) were evaluated (separately) according to the manufacturer’s protocol (ASCA IgG, ASCA IgA, QUANTA Lite, INOVA Diagnostics) (Table 1).
DISCUSSIONIs CD much ado about nothing? This report presents four cases of CD with MC. The symptoms of these patients were improved initially by a gluten-free diet before the onset of MC symptoms. Their history indicates and underlines that patients can develop, or present with CD at any stage in life and that CD can co-exist with other gastrointestinal diseases of an (auto-) immune origin. Patients with CD can fail to respond to the initial introduction of a gluten-free diet or have a recurrence of symptoms after initial improvement, despite maintaining gluten exclusion. The most feared causes of either scenario are complicating malignancy, notably enteropathy-associated T-cell lymp-homa, or refractory sprue. Other causes of persistent sy-mptoms with increased prevalence in CD include lactose intolerance, exocrine pancreatic insufficiency, bacterial overgrowth, and microscopic (lymphocytic or collagenous) colitis. Thus, in patients whose symptoms fail to respond or who later relapse, despite the exclusion of gluten from their diet, the possibility of additional pathology should be considered and colonoscopy should, therefore, be part of the follow-up in patients who present with chronic watery diarrhea, even if initial tests indicate only CD.
Relations between CD and ulcerative ileojejunitis, polymyositis, and fibrosing alveolitis have been previously described[20,21], and it is of interest that an auto-immune pathophysiology has been implicated in each of these con-ditions. An association has been suggested between CD and diffuse interstitial lung disease of the hypersensitivity pneumonitis type in several reports from Europe[22]. A case
2153 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Barta Z et al . CD and MC
Table 1 Summary of the cases
Case 1 Case 2 Case 3 Case 4
Sex/birth (yr) Female/1956 Female/1955 Female/1945 Male/1964Small bowel histology Normal small-bowel
mucosal structureNormal small-bowel
mucosal structurePartial villous atrophy (Marsh 3A)
according to the modified Marsh criteriaTotal villous atrophy (Marsh 3C)
according to the modified Marsh criteriaHLA-DQ2 Present Present Present PresentlgA TTG + + + +lgG/lgA EMA +/+ -/+ -/+ +/+lgG/lgA Gliadin +/+ -/+ -/+ +/+lgG/lgA ASCA -/- -/- +/+ +/+Celiac disease Latent Latent Manifest ManifestColon histology Lymphocytic colitis Collagenous colitis Lymphocytic colitis Collagenous colitisTherapy GFD and budesonide GFD and budesonide GFD and budesonide GFD and budesonideOther disease - - Fibrosing alveolitis Fibrosing alveolitis
Laboratory tests and histology of the small bowel before gluten-free diet, colon histology after gluten-free diet, therapy, and concomitant lung disease. TTG: Tissue transglutaminase; EMA: Endomysial antibody; ASCA: Anti-saccharomyces cerevisiae antibody; GFD: Gluten-free diet; HLA: Human leukocyte antigen.

of lymphocytic bronchoalveolitis and CD with improve-ment following a gluten free diet was also reported[23].
Our patients with manifestations of CD and MC presented with fibrosing alveolitis and were ASCA (anti-yeast antibodies to yeast antigens that are found in bread and other cereal derived products) positive (both IgG and IgA types). Previously, ASCA positivity was shown to be evident in up to 40%-60% of CD patients and 13%-15% of MC patients, but its implication is disputed[24]. A pos-sible connection between alveolitis and ASCA is also not known. Only one case of a Japanese patient was published: lung biopsy specimens showed alveolitis and serum-precipitating antibody gave a positive reaction for an extract from S. cerevisiae[25].
In conclusion, the co-existence of immune-mediated small bowel and colonic inflammatory diseases (i.e. CD and IBD) and pulmonary diseases is not well-known and no systematic approach has been used to identify the life-long patterns of these immune-based diseases[26]. Such information may be useful for both disease preven-tion and treatment approaches. Clinicians managing CD should be aware of these associations and when to con-sider colon investigation.
REFERENCES1 Dewar DH, Ciclitira PJ. Clinical features and diagnosis of ce-
liac disease. Gastroenterology 2005; 128: S19-S242 Kaukinen K, Mäki M, Partanen J, Sievänen H, Collin P. Ce-
liac disease without villous atrophy: revision of criteria called for. Dig Dis Sci 2001; 46: 879-887
3 Kaukinen K, Collin P, Mäki M. Latent coeliac disease or co-eliac disease beyond villous atrophy? Gut 2007; 56: 1339-1340
4 Elli L, Bergamini CM, Bardella MT, Schuppan D. Transgluta-minases in inflammation and fibrosis of the gastrointestinal tract and the liver. Dig Liver Dis 2009; 41: 541-550
5 Robertson DA, Taylor N, Sidhu H, Britten A, Smith CL, Holdstock G. Pulmonary permeability in coeliac disease and inflammatory bowel disease. Digestion 1989; 42: 98-103
6 Vernier G, Cocq P, Baron P, Paquet PY, Colombel JF. [Familial occurrence of collagenous colitis]. Gastroenterol Clin Biol 2005; 29: 474-476
7 Kong SC, Keogh S, Carter MJ, Lobo AJ, Sanders DS. Familial occurrence of microscopic colitis: an opportunity to study the relationship between microscopic colitis and coeliac disease? Scand J Gastroenterol 2002; 37: 1344-1345
8 Thomson A, Kaye G. Further report of familial occurrence of collagenous colitis. Scand J Gastroenterol 2002; 37: 1116
9 Freeman HJ. Familial occurrence of lymphocytic colitis. Can J Gastroenterol 2001; 15: 757-760
10 Järnerot G, Hertervig E, Grännö C, Thorhallsson E, Eriksson
S, Tysk C, Hansson I, Björknäs H, Bohr J, Olesen M, Willén R, Kagevi I, Danielsson A. Familial occurrence of microscopic colitis: a report on five families. Scand J Gastroenterol 2001; 36: 959-962
11 Abdo AA, Zetler PJ, Halparin LS. Familial microscopic coli-tis. Can J Gastroenterol 2001; 15: 341-343
12 Chutkan R, Sternthal M, Janowitz HD. A family with col-lagenous colitis, ulcerative colitis, and Crohn’s disease. Am J Gastroenterol 2000; 95: 3640-3641
13 van Tilburg AJ, Lam HG, Seldenrijk CA, Stel HV, Blok P, Dekker W, Meuwissen SG. Familial occurrence of collage-nous colitis. A report of two families. J Clin Gastroenterol 1990; 12: 279-285
14 Abdo AA, Urbanski SJ, Beck PL. Lymphocytic and collag-enous colitis: the emerging entity of microscopic colitis. An update on pathophysiology, diagnosis and management. Can J Gastroenterol 2003; 17: 425-432
15 Green PH, Yang J, Cheng J, Lee AR, Harper JW, Bhagat G. An association between microscopic colitis and celiac disease. Clin Gastroenterol Hepatol 2009; 7: 1210-1216
16 Matteoni CA, Goldblum JR, Wang N, Brzezinski A, Achkar E, Soffer EE. Celiac disease is highly prevalent in lymphocytic colitis. J Clin Gastroenterol 2001; 32: 225-227
17 Fine KD, Do K, Schulte K, Ogunji F, Guerra R, Osowski L, McCormack J. High prevalence of celiac sprue-like HLA-DQ genes and enteropathy in patients with the microscopic coli-tis syndrome. Am J Gastroenterol 2000; 95: 1974-1982
18 DuBois RN, Lazenby AJ, Yardley JH, Hendrix TR, Bayless TM, Giardiello FM. Lymphocytic enterocolitis in patients with ‘refractory sprue’. JAMA 1989; 262: 935-937
19 Rubio-Tapia A, Talley NJ, Gurudu SR, Wu TT, Murray JA. Gluten-free diet and steroid treatment are effective therapy for most patients with collagenous sprue. Clin Gastroenterol Hepatol 2010; 8: 344-349.e3
20 Coupe MO, Barnard ML, Stamp G, Hodgson HJ. Ulcerative ileojejunitis associated with pulmonary fibrosis and poly-myositis. Hepatogastroenterology 1988; 35: 144-146
21 Ben M’rad S, Dogui MH, Merai S, Djenayah F. [Respiratory manifestation of celiac disease]. Presse Med 1998; 27: 1384
22 Tarlo SM, Broder I, Prokipchuk EJ, Peress L, Mintz S. As-sociation between celiac disease and lung disease. Chest 1981; 80: 715-718
23 Brightling CE, Symon FA, Birring SS, Wardlaw AJ, Robinson R, Pavord ID. A case of cough, lymphocytic bronchoalveolitis and coeliac disease with improvement following a gluten free diet. Thorax 2002; 57: 91-92
24 Holstein A, Burmeister J, Plaschke A, Rosemeier D, Widjaja A, Egberts EH. Autoantibody profiles in microscopic colitis. J Gastroenterol Hepatol 2006; 21: 1016-1020
25 Yamamoto Y, Osanai S, Fujiuchi S, Akiba Y, Honda H, Na-kano H, Ohsaki Y, Kikuchi K. [Saccharomyces-induced hy-persensitivity pneumonitis in a dairy farmer: a case report]. Nihon Kokyuki Gakkai Zasshi 2002; 40: 484-488
26 Hemminki K, Li X, Sundquist J, Sundquist K. Subsequent autoimmune or related disease in asthma patients: clustering of diseases or medical care? Ann Epidemiol 2010; 20: 217-222
S- Editor Sun H L- Editor Wang XL E- Editor Ma WH
2154 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Barta Z et al . CD and MC

CASE REPORT
Pure red cell aplasia caused by pegylated interferon-α-2a plus ribavirin in the treatment of chronic hepatitis C
Cheng-Shyong Chang, Sheng-Lei Yan, Hsuan-Yu Lin, Fu-Lien Yu, Chien-Yu Tsai
Cheng-Shyong Chang, Hsuan-Yu Lin, Fu-Lien Yu, Chien-Yu Tsai, Division of Hematology and Oncology, Department of In-ternal Medicine, Changhua Christian Hospital, Changhua City 500, Taiwan, ChinaSheng-Lei Yan, Division of Gastroenterology and Hepatology, Department of Internal Medicine, Chang-Bing Show Chwan Memorial Hospital, Changuha County 505, Taiwan, ChinaSheng-Lei Yan, Department and Graduate Program of Bio-industrial Technology, Dayeh University, Changhua County 51591, Taiwan, ChinaFu-Lien Yu, Graduate Institute of Nursing, Chung Shan Medi-cal University, Taichung City 40201, Taiwan, ChinaAuthor contributions: Chang CS substantial contributions to conception and design; Yan SL draft the article and final ap-proval of the version; Lin HY revise it critically for important intellectual content; Yu FL and Tsai CY help acquisition and interpretation of data.Correspondence to: Sheng-Lei Yan, MD, Division of Gastro-enterology and Hepatology, Department of Internal Medicine, Chang-Bing Show Chwan Memorial Hospital, No 6, Lu-Gong Rd., Lugang Township, Changhua County 505, Taiwan, China. [email protected]: +886-4-7813888 Fax: +886-4-7812401Received: December 19, 2010 Revised: January 18, 2011Accepted: January 25, 2011Published online: April 28, 2011
AbstractPure red cell aplasia (PRCA) is a rare hematological dis-order which is characterized by severe anemia, reticulo-cytopenia and almost complete absence of erythroid pre-cursors in bone marrow. The pathophysiology of PRCA may be congenital or acquired. To our knowledge, there is only one case report in the English literature of PRCA after pegylated interferon combination therapy for chron-ic hepatitis C. We report a second case of PRCA after pegylated interferon combination treatment for chronic hepatitis C. The diagnosis of PRCA was confirmed by the typical findings of bone marrow biopsy. The possible eti-ologies of our case are also discussed in this paper.
Key words: Chronic hepatitis C; Pegylated interferon-α-2a; Pure red cell aplasia; Ribavirin
© 2011 Baishideng. All rights reserved.
Peer reviewer: William Dickey, Professor, Altnagelvin Hospital, Londonderry, BT47 6SB, Northern Ireland, United Kingdom
Chang CS, Yan SL, Lin HY, Yu FL, Tsai CY. Pure red cell apla-sia caused by pegylated interferon-α-2a plus ribavirin in the treatment of chronic hepatitis C. World J Gastroenterol 2011; 17(16): 2155-2158 Available from: URL: http://www.wjgnet.com/1007-9327/full/v17/i16/2155.htm DOI: http://dx.doi.org/10.3748/wjg.v17.i16.2155
INTRODUCTIONPure red cell aplasia (PRCA) is a rare hematological disor-der which is characterized by severe anemia, reticulocytope-nia and almost complete absence of erythroid precursors in bone marrow[1]. Patients typically present with symptoms of severe anemia in the absence of hemorrhagic phenom-ena. Common causes of PRCA include human parvovirus B19 infection, lymphoproliferative disorder, humoral or cel-lular immunity, production of erythropoietin-neutralizing antibody, and drugs such as ribavirin and standard inter-feron[1-5]; however, to our knowledge, there is only one case report in the English literature of PRCA after pegylated in-terferon combination therapy for chronic hepatitis C[6]. We report a second case of PRCA during combination therapy for chronic hepatitis C.
CASE REPORTA 69-year-old male was referred to our hospital for treat-ment of chronic hepatitis C. His past medical history was remarkable for megaloblastic anemia due to vitamin B12 deficiency. He received regular vitamin B12 injection ther-apy with a stable hemoglobin level of around 11 g/dL. At
2155
World J Gastroenterol 2011 April 28; 17(16): 2155-2158 ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2155
April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

Chang CS et al . PRCA caused by chronic HCV treatment
initial presentation, elevated aminotransferase levels and positive antibody to hepatitis C virus were detected. HCV RNA level was 85 000 IU/mL. The genotype was 2. The pretreatment hemoglobin level was 11.2 g/dL and the reticulocyte count was 1.8% (normal range 0.5-1.5%). Se-rum level of vitamin B12 was 288 pg/mL (normal range 272-1078 pg/mL). Serum level of folic acid was 11.0 ng/mL (normal range 5-26 ng/mL). Treatment with peginter-feron alfa-2a 180 ug weekly and ribavirin 800 mg daily was started. After four weeks of treatment, the ribavirin dose was reduced to 600 mg due to a prominent decrease in the hemoglobin level to 8.4 g/dL. HCV RNA level at the 4th wk was undetected. Three weeks after dose reduc-tion of ribavirin, the hemoglobin level continued to drop to 6.9 g/dL. The combination therapy was discontinued after 7 wk of treatment due to the patient’s intolerance of anemia (Figure 1). Serum levels of indirect bilirubin, lac-tate dehydrogenase and haptoglobin level remained nor-mal during combination treatment. Parvovirus serologies revealed positive immunoglobin (Ig) G but negative IgM antibodies, which were consistent with past exposure.
He received follow up at a previous institution after termination of combination treatment. However, his ane-mia persisted and became transfusion-dependent. Blood transfusion with packed red blood cells was administered every month (Figure 1). Three months after discontinua-tion of combination treatment, the hemoglobin dropped to 5.5 g/dL and the reticulocyte count was 0.1% (nor-mal range 0.5-1.5%). Levels of vitamin B12 and folic acid were within the normal range. Bone marrow biopsy revealed severe hypocellularity of 10% (Figure 2) with normal maturation of myeloid precursor cells (Figure 3). Erythroid precursor cells were markedly decreased with a ratio of myeloid to erythroid precursors of 10. The diagnosis of PRCA was made based on these histopatho-logical findings. Oral methylprednisolone 15 mg daily was administered. After four weeks of oral methylpredniso-lone therapy, his hemoglobin level increased to 9.2 g/dL and the reticulocyte count increased to 1.7% (Figure 1). He needed no further blood transfusions in the following months.
2156 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Figure 1 Hemoglobin level and reticulocyte count over time. RBC: Red blood cells.
2
1.8
1.6
1.4
1.2
1
0.8
0.6
0.4
0.2
0
Hem
oglo
bin
(g/d
L)
0 4 7 9 12 16 20 24 28 32
Weeks after combination treatment
12
10
8
6
4
2
0
Retic
uloc
yte
conm
t (%
)RBC transfusions
Termination of combination treatment MethylprednisoloneHemoglobin
Reticulocyte count
Figure 2 Photomicrograph of bone marrow biopsy showing overall hypo-cellularity of 10% (HE, x 40).
Figure 3 Photomicrograph of higher magnification showing normal matu-ration of myeloid precursors and absence of erythroid precursors in this field (HE, x 100).

DISCUSSIONCombination treatment of pegylated interferon α and ribavirin has become the standard therapy for patients with chronic hepatitis C infection[7]. Patients with chronic hepatitis C receiving combination treatment develop anemia because of ribavirin-induced hemolysis[8] and interferon-induced bone marrow suppression[9]. The ribavirin-induced anemia is dose-dependent and revers-ible[10]. Conversely, interferon directly suppresses the bone marrow synthesis of granulocytes, erythrocytes and mega-karyocytes[9]. PRCA is rarely encountered in chronic hepa-titis C patients receiving combination treatment. To our knowledge, there is only one previous case report in the English literature of PRCA after combination treatment in patients with chronic hepatitis C[6].
The pathophysiology of PRCA is heterogenous, which may be congenital or acquired[1]. Diamond-Black-fan anemia is a congenital form of PRCA with genetic defects affecting erythropoietic lineage. Acquired causes of PRCA include human parvovirus B19 infection, lym-phoproliferative disorder, humoral or cellular immunity, production of erythropoietin-neutralizing antibody, and drugs such as ribavirin and standard interferon[1-5]. Other reported causes of PRCA include hepatitis A infection[11], malignant thymoma[12], and systemic lupus erythemato-sus[13]. The diagnosis of PRCA is based on bone marrow findings such as severe hypocellularity, a markedly elevated myeloid: erythroid ratio, and severe decreased erythroid precursors[6,14]. With regard to the treatment of PRCA, corticosteroid therapy is considered the treatment of first choice, although relapse is not uncommon[15]. In the study of Clark et al[16], 80% of patients relapsed as the dosage of steroid was tapered during the first year after remission. In contrast, cyclosporine A is suggested when the long-term feasibility of maintenance is considered[15].
As to the etiology of PRCA in our case, several pos-sibilities should be considered. Firstly, although HCV infection itself has been associated with PRCA[17], the HCV RNA in our case was undetected at the 4th wk of combination treatment. Furthermore, parvovirus serolo-gies revealed positive IgG but negative IgM antibodies, which excluded parvovirus infection as the cause of PRCA in our case. Secondly, although ribavirin-induced PRCA has been reported previously[3], the anemia in our case might not have been caused solely by ribavirin since the anemia persisted for 3 mon after discontinuation of ribavirin. Drug-induced PRCA generally resolves within 1-2 wk after removal of the causative agent[18]. Thirdly, one might consider the possible association of underly-ing pernicious anemia with PRCA in this case. Pernicious anemia is generally considered an autoimmune disease resulting in deficiency of intrinsic factor and subsequent vitamin B12 deficiency[19]. Coexistence of pernicious ane-mia and PRCA in the same patient has been reported in earlier literature[20-22], suggesting that an immunological process is involved in the pathogenesis of PRCA in these cases. In our case, serum levels of vitamin B12 did not
alter significantly before and after combination treatment, thus excluding vitamin B12 deficiency as the cause of se-vere anemia after combination treatment. Therefore, it is reasonable to assume that pegylated interferon might have played a role in the pathogenesis of PRCA in our case. Several studies have also suggested that interferon may play a role in the development of acquired PRCA[4,5,23]. However, further investigations may be needed to deter-mine whether PRCA is caused by pegylated interferon alone or by combination treatment.
In conclusion, this case highlights the importance of considering PRCA when severe anemia associated with reticulocytopenia develops during the combination treat-ment of patients with chronic hepatitis C.
REFERENCES1 Fisch P, Handgretinger R, Schaefer HE. Pure red cell apla-
sia. Br J Haematol 2000; 111: 1010-10222 Schecter JM, Mears JG, Alobeid B, Gaglio PJ. Anti-erythro-
poietin antibody-mediated pure red cell aplasia in a living donor liver transplant recipient treated for hepatitis C virus. Liver Transpl 2007; 13: 1589-1592
3 Tanaka N, Ishida F, Tanaka E. Ribavirin-induced pure red-cell aplasia during treatment of chronic hepatitis C. N Engl J Med 2004; 350: 1264-1265
4 Tomita N, Motomura S, Ishigatsubo Y. Interferon-alpha-in-duced pure red cell aplasia following chronic myelogenous leukemia. Anticancer Drugs 2001; 12: 7-8
5 Hirri HM, Green PJ. Pure red cell aplasia in a patient with chronic granulocytic leukaemia treated with interferon-alpha. Clin Lab Haematol 2000; 22: 53-54
6 Miura Y, Kami M, Yotsuya R, Toda N, Komatsu T. Pure red-cell aplasia associated with pegylated interferon-alpha-2b plus ribavirin. Am J Hematol 2008; 83: 758-759
7 Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Gonçales FL Jr, Häussinger D, Diago M, Carosi G, Dhumeaux D, Craxi A, Lin A, Hoffman J, Yu J. Peginterfer-on alfa-2a plus ribavirin for chronic hepatitis C virus infec-tion. N Engl J Med 2002; 347: 975-982
8 Peck-Radosavljevic M, Wichlas M, Homoncik-Kraml M, Kreil A, Hofer H, Jessner W, Gangl A, Ferenci P. Rapid suppression of hematopoiesis by standard or pegylated interferon-alpha. Gastroenterology 2002; 123: 141-151
9 De Franceschi L, Fattovich G, Turrini F, Ayi K, Brugnara C, Manzato F, Noventa F, Stanzial AM, Solero P, Corrocher R. Hemolytic anemia induced by ribavirin therapy in patients with chronic hepatitis C virus infection: role of membrane oxidative damage. Hepatology 2000; 31: 997-1004
10 Bodenheimer HC Jr, Lindsay KL, Davis GL, Lewis JH, Thung SN, Seeff LB. Tolerance and efficacy of oral ribavirin treatment of chronic hepatitis C: a multicenter trial. Hepatol-ogy 1997; 26: 473-477
11 Tomida S, Matsuzaki Y, Nishi M, Ikegami T, Chiba T, Abei M, Tanaka N, Osuga T, Sato Y, Abe T. Severe acute hepatitis A associated with acute pure red cell aplasia. J Gastroenterol 1996; 31: 612-617
12 Handa SI, Schofield KP, Sivakumaran M, Short M, Pum-phrey RS. Pure red cell aplasiaassociated with malignant thymoma, myasthenia gravis, polyclonal large granular lymphocytosis and clonal thymic T cell expansion. J Clin Pathol 1994; 47: 676-679
13 Linardaki GD, Boki KA, Fertakis A, Tzioufas AG. Pure red cell aplasia as presentation of systemic lupus erythematosus: anti-bodies to erythropoietin. Scand J Rheumatol 1999; 28: 189-191
14 Arcasoy MO, Rockey DC, Heneghan MA. Pure red cell aplasia following pegylated interferon alpha treatment. Am
2157 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Chang CS et al . PRCA caused by chronic HCV treatment

J Med 2004; 117: 619-62015 Sawada K, Fujishima N, Hirokawa M. Acquired pure red cell
aplasia: updated review of treatment. Br J Haematol 2008; 142: 505-514
16 Clark DA, Dessypris EN, Krantz SB. Studies on pure red cell aplasia. XI. Results of immunosuppressive treatment of 37 patients. Blood 1984; 63: 277-286
17 Al-Awami Y, Sears DA, Carrum G, Udden MM, Alter BP, Conlon CL. Pure red cell aplasia associated with hepatitis C infection. Am J Med Sci 1997; 314: 113-117
18 Mamiya S, Itoh T, Miura AB. Acquired pure red cell aplasia in Japan. Eur J Haematol 1997; 59: 199-205
19 Lahner E, Annibale B. Pernicious anemia: new insights from a gastroenterological point of view. World J Gastroenterol 2009; 15: 5121-5128
20 Dan K, Ito T, Nomura T. Pure red cell aplasia following per-nicious anemia. Am J Hematol 1990; 33: 148-150
21 Robins-Browne RM, Green R, Katz J, Becker D. Thymoma, pure red cell aplasia, pernicious anaemia and candidiasis: a defect in immunohomeostasis. Br J Haematol 1977; 36: 5-13
22 Goldstein C, Pechet L. Chronic Erythrocytic Hypoplasia Following Pernicious Anemia. Blood 1965; 25: 31-36
23 Schattner A. The possible involvement of interferons in ac-quired pure red cell aplasia. Am J Hematol 1988; 27: 72-73
S- Editor Tian L L- Editor Webster JR E- Editor Ma WH
2158 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
Chang CS et al . PRCA caused by chronic HCV treatment

LETTERS TO THE EDITOR
Enucleation for gastrointestinal stromal tumors at the esophagogastric junction: Is this an adequate solution?
Nadia Peparini, Giovanni Carbotta, Piero Chirletti
Nadia Peparini, Giovanni Carbotta, Piero Chirletti, Depart-ment of General Surgery “Francesco Durante”, La Sapienza Uni-versity, viale del Policlinico 155, 00161 Rome, ItalyAuthor contributions: Peparini N and Chirletti P contrib-uted equally to this manuscript, conceived and drafted the manuscript, critically revised the manuscript and gave its final approval; Carbotta G contributed to the manuscript draft and gave its final approval.Correspondence to: Nadia Peparini, MD, PhD, Department of General Surgery “Francesco Durante” viale del Policlinico 155, 00161 Rome, Italy. [email protected] Telephone: +39-339-2203940 Fax: +39-6-49970385Received: October 16, 2010 Revised: December 17, 2010Accepted: December 24,2010Published online: April 28, 2011
AbstractThe authors discussed the proposal by Coccolini and colleagues to treat gastrointestinal stromal tumors (GISTs) at the esophagogastric junction with enucle-ation and, if indicated, adjuvant therapy, reducing the risks related to esophageal and gastroesophageal re-section. They concluded that, because the prognostic impact of a T1 high-mitotic rate on esophageal GIST is worse than that of a T1 high-mitotic rate on gastric GIST, enucleation may not be an adequate surgery for esophagogastric GISTs with a high mitotic rate in which the guarantee of negative resection margins and adjuvant therapies can be the only chance of survival.
© 2011 Baishideng. All rights reserved.
Key words: Gastrointestinal stromal tumor; Esophagogastric junction; Surgery; Resection; Enucleation
Peer reviewer: Alexander Becker, MD, Department of Surgery, Haemek Medical Center, Afula 18000, Israel
Peparini N, Carbotta G, Chirletti P. Enucleation for gastrointestinal stromal tumors at the esophagogastric junction: Is this an adequate solution? World J Gastroenterol 2011;
17(16): 2159-2160 Available from: URL: http://www.wjgnet.com/10079327/full/v17/i16/2159.htm DOI: http://dx.doi.org/10.3748/wjg.v17.i16.2159
TO THE EDITORWe read with great interest the article by Coccolini and colleagues on the treatment of gastrointestinal stromal tumors (GISTs) at the esophagogastric junction[1]. They stated the problems related to the choice of extended esophageal and gastroesophageal resection (i.e. a bet-ter guarantee of R0 resection but a higher prevalence of morbidity and mortality) or enucleation (i.e. a higher risk of microscopically positive margins but a better postop-erative outcome).
The impact of microscopically negative margins on long-term survival remains controversial and there is no evidence that extensive resections are related to a better survival rate. The authors suggested that, for GISTs at the esophagastric junction, enucleation and adjuvant therapies can be useful alternatives to avoid the high prevalence of morbidity and mortality associated with esophageal and es-ophagogastric resections. However, the 2009 edition of the TNM Classification of Malignant Tumors states that, in the absence of nodal metastasis, esophageal GISTs ≤ 2 cm (T1, i.e. tumors that may be treated with enucleation more fre-quently) are classified as stage Ⅰ in the case of a low mitotic rate but as stage ⅢA in the case of a high mitotic rate. This case is different from T1 gastric GISTs that are classified as stage Ⅰ or stage Ⅱ in the presence of a low or high mitotic rate, respectively[2]. In the case of a high mitotic rate, the prognostic impact of a T1 esophageal GIST is worse than that of a gastric GIST with an identical size. Prospective, multicenter evaluation of the different treatment strategies for esophagogastric GISTs is sorely needed. However, enu-cleation may not be an adequate surgery for esophagogas-tric GISTs with a high-mitotic rate in which the guarantee of negative resection margins and adjuvant therapies can be the only chance of survival.
2159
World J Gastroenterol 2011 April 28; 17(16): 2159-2160 ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]:10.3748/wjg.v17.i16.2159
April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

Peparini N et al . GISTs at esophagogastric junction
REFERENCES1 Coccolini F, Catena F, Ansaloni L, Lazzareschi D, Pinna AD.
Esophagogastric junction gastrointestinal stromal tumor: resec-
tion vs enucleation. World J Gastroenterol 2010; 16: 4374-43762 Sobin LH, Gospodarowicz MK, Wittekind Ch, editors.
TNM Classification of Malignant Tumors. Seventh edition 2009. Wiley-Blackwell, 2010
S- Editor Tian L L- Editor Wang XL E- Editor Ma WH
2160 April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

World J Gastroenterol 2011 April 28; 17(16): I ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]
ACKNOWLEDGMENTS
Acknowledgments to reviewers of World Journal of Gastroenterology
Many reviewers have contributed their expertise and time to the peer review, a critical process to ensure the quality of World Journal of Gastroenterology. The editors and authors of the articles submitted to the journal are grateful to the following reviewers for evaluating the articles (including those published in this issue and those rejected for this issue) during the last editing time period.
Luis Bujanda, PhD,Professor, Departament of Gastroenterology, CIBEREHD, University of Country Basque, Donostia Hospital, Paseo Dr. Beguiristain s/n, 20014 San Sebastián, Spain
Olivier Detry, Dr., Department of Abdominal Surgery and Transplantation, University of Liège, CHU Sart Tilman B35, B-4000 Liège, Belgium
Marek Hartleb, Professor, Department of Gastroenterology, Silesian Medical School, ul. Medyków 14, Katowice 40-752, Poland
Kok Sun Ho, Dr., Department of Colorectal Surgery, Singapore General Hospital, Outram Road, Singapore 169608, Singapore
Richard Hu, MD, MSc, Division of Gastroenterology, Department of Medicine, Olive view-UCLA Medical Center, 14445 Olive View Drive, Los Angeles, CA 91342, United States
Hartmut Jaeschke, Professor, Liver Research Institute, University of Arizona, College of Medicine, 1501 N Campbell Ave, Room 6309, Tucson, Arizona 85724, United States
Ioannis Kanellos, Professor, 4th Surgical Department, Aristotle University of Thessaloniki, Antheon 1, Panorama, Thessaloniki, 55236, Greece
Islam Khan, PhD, Professor, Departmenet of Biochemistry, Faculty of Medicine, Kuwait University, PO box 24923, Safat 13110, Kuwait
Jae J Kim, MD, PhD, Associate Professor, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, 50, Irwon-dong, Gangnam-gu, Seoul 135-710, South Korea
Won Ho Kim, MD, Professor, Department of Internal Medicine, Yonsei Uiversity College of Medicine, 134 Shinchon-dong Seodaemun-ku, Seoul 120-752, South Korea
Ashok Kumar, MD, Dr., Department of Surgical Gastroenterology, Sanjay Gandhi Post Graduate Institute of Medical Sciences, Lucknow, 226014, India
Rene Lambert, Professor, International Agency for Research on Cancer, 150 Cours Albert Thomas, Lyon 69372 cedex 8, France
Giulio Marchesini, Professor, Department of Internal Medicine and Gastroenterology, “Alma Mater Studiorum” University of Bologna, Polic-linico S. Orsola, Via Massarenti 9, Bologna 40138, Italy
Julio Mayol, MD, PhD, Department of Digestive surgery, Hospital Clinico San Carlos, MARTIN-LAGOS S/n, Madrid, 28040, Spain
C Bart Rountree, MD, Assistant Professor of Pediatrics and Phar-macology, Penn State College of Medicine, 500 University Drive, H085, Hershey, PA 17033, United States
Frank G Schaap, PhD, Tytgat Institute for Liver and Intestinal Research, Academic Medical Center, Meibergdreef 69-71, 1105 BK Amsterdam, The Netherlands
Richie Soong, Associate Professor, Centre for Life Sciences #02-15, National University of Singapore, 28 Medical Drive, Singapore 117456, Singapore
Gisela Sparmann, MD, Division of Gastroenterology, Department of Internal Medicine, University of Rostock, Ernst-Heydemann-Str. 6, Rostock D-18057, Germany
Yoshitaka Takuma, MD, PhD, Department of Gastroenterology, Kurashiki Central Hospital, 1-1-1 Miwa, Kurashiki, Okayama, 710-8602 Japan
Yasuhito Tanaka, MD, PhD, Professor, Department of Virology and Liver unit, Nagoya City University Graduate School of Medical Sciences, Kawasumi, Mizuho, Nagoya 467-8601, Japan
Kam-Meng Tchou-Wong, Assistant Professor, Departments of Enviro-nmental Medicine and Medicine, NYU School ofMedicine, 57 Old Forge Road, Tuxedo, New York, NY 10987, United States
Luca VC Valenti, MD, Internal Medicine, Università degli Studi di Milano, Fondazione IRCCS Ospedale Maggiore Policlinico, Padiglione Granelli, via F Sforza 35, Milano, 20122, Italy
Marco Vivarelli, MD, Assistant Professor, Department of Surgery and Transplantation, University of Bologna, S.Orsola Hospital, Bologna 40123, Italy
Dinesh Vyas, Dr., Department of Minimally and Endosopic Surgery, St John Mercy Hospital, 851 E Fifth Street, Washington, MO 63090, United States
Yutaka Yata, MD, PhD, Director, Department of Gastroenterology, Saiseikai Maebashi Hospital, 564-1 Kamishinden-machi, Maebashi-city, Gunma 371-0821, Japan
Huiping Zhou, PhD, Assistant Professor, Department of Microbiology and Immunology, School of Medicine, Virginia Commonwealth University, 1217 East Marshall Street, MSB#533, Richmond, VA 23298, United States
I April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

Events Calendar 2011January 14-15, 2011AGA Clinical Congress of Gastroenterology and Hepatology: Best Practices in 2011 Miami, FL 33101, United States
January 20-22, 2011Gastrointestinal Cancers Symposium 2011, San Francisco, CA 94143,United States
January 27-28, 2011Falk Workshop, Liver and Immunology, Medical University, Franz-Josef-Strauss-Allee 11, 93053 Regensburg, Germany
January 28-29, 20119. Gastro Forum München, Munich, Germany
February 4-5, 201113th Duesseldorf International Endoscopy Symposium, Duesseldorf, Germany
February 13-27, 2011Gastroenterology: New Zealand CME Cruise Conference, Sydney, NSW, Australia
February 17-20, 2011APASL 2011-The 21st Conference of the Asian Pacific Association for the Study of the Liver Bangkok, Thailand
February 22, 2011-March 04, 2011Canadian Digestive Diseases Week 2011, Vancouver, BC, Canada
February 24-26, 2011Inflammatory Bowel Diseases 2011-6th Congress of the European Crohn's and Colitis Organisation, Dublin, Ireland
February 24-26, 20112nd International Congress on Abdominal Obesity, Buenos Aires, Brazil
February 24-26, 2011International Colorectal Disease Symposium 2011, Hong Kong, China
February 26-March 1, 2011Canadian Digestive Diseases Week, Westin Bayshore, Vancouver, British Columbia, Canada
February 28-March 1, 2011Childhood & Adolescent Obesity:
A whole-system strategic approach, Abu Dhabi, United Arab Emirates
March 3-5, 201142nd Annual Topics in Internal Medicine, Gainesville, FL 32614, United States
March 7-11, 2011Infectious Diseases: Adult Issues in the Outpatient and Inpatient Settings, Sarasota, FL 34234, United States
March 14-17, 2011British Society of Gastroenterology Annual Meeting 2011, Birmingham, England, United Kingdom
March 17-19, 201141. Kongress der Deutschen Gesellschaft für Endoskopie und Bildgebende Verfahren e.V., Munich, Germany
March 17-20, 2011Mayo Clinic Gastroenterology & Hepatology 2011, Jacksonville, FL 34234, United States
March 18, 2011UC Davis Health Informatics: Change Management and Health Informatics, The Keys to Health Reform, Sacramento, CA 94143, United States
March 25-27, 2011MedicReS IC 2011 Good Medical Research, Istanbul, Turkey
March 26-27, 201126th Annual New Treatments in Chronic Liver Disease, San Diego, CA 94143, United States
April 6-7, 2011IBS-A Global Perspective, Pfister Hotel, 424 East Wisconsin Avenue, Milwaukee, WI 53202, United States
April 7-9, 2011International and Interdisciplinary Conference Excellence in Female Surgery, Florence, Italy
April 15-16, 2011Falk Symposium 177, Endoscopy Live Berlin 2011 Intestinal Disease Meeting, Stauffenbergstr. 26, 10785 Berlin, Germany
April 18-22, 2011Pediatric Emergency Medicine: Detection, Diagnosis and Developing
Treatment Plans, Sarasota, FL 34234, United States
April 20-23, 20119th International Gastric Cancer Congress, COEX, World Trade Center, Samseong-dong, Gangnam-gu, Seoul 135-731, South Korea
April 25-27, 2011The Second International Conference of the Saudi Society of Pediatric Gastroenterology, Hepatology & Nutrition, Riyadh, Saudi Arabia
April 25-29, 2011Neurology Updates for Primary Care, Sarasota, FL 34230-6947, United States
April 28-30, 20114th Central European Congress of Surgery, Budapest, Hungary
May 7-10, 2011Digestive Disease Week, Chicago, IL 60446, United States
May 12-13, 20112nd National Conference Clinical Advances in Cystic Fibrosis, London, England, United Kingdom
May 19-22, 20111st World Congress on Controversies in the Management of Viral Hepatitis (C-Hep), Palau de Congressos de Catalunya, Av. Diagonal, 661-671 Barcelona 08028, Spain
May 21-24, 201122nd European Society of Gastrointestinal and Abdominal Radiology Annual Meeting and Postgraduate Course, Venise, Italy
May 25-28, 20114th Congress of the Gastroenterology Association of Bosnia and Herzegovina with international participation, Hotel Holiday Inn, Sarajevo, Bosnia and Herzegovina
June 11-12, 2011The International Digestive Disease Forum 2011, Hong Kong, China
June 13-16, 2011Surgery and Disillusion XXIV SPIGC, II ESYS, Napoli, Italy
June 14-16, 2011International Scientific Conference on Probiotics and Prebiotics-IPC2011, Kosice, Slovakia
June 22-25, 2011ESMO Conference: 13th World Congress on Gastrointestinal Cancer, Barcelona, Spain
June 29-2, 2011XI Congreso Interamericano de Pediatria "Monterrey 2011", Monterrey, Mexico
September 2-3, 2011 Falk Symposium 178, Diverticular Disease, A Fresh Approach to a Neglected Disease, Gürzenich Cologne, Martinstr. 29-37, 50667 Cologne, Germany
September 10-11, 2011New Advances in Inflammatory Bowel Disease, La Jolla, CA 92093, United States
September 10-14, 2011ICE 2011-International Congress of Endoscopy, Los Angeles Convention Center, 1201 South Figueroa Street Los Angeles, CA 90015, United States
September 30-October 1, 2011Falk Symposium 179, Revisiting IBD Management: Dogmas to be Challenged, Sheraton Brussels Hotel, Place Rogier 3, 1210 Brussels, Belgium
October 19-29, 2011Cardiology & Gastroenterology | Tahiti 10 night CME Cruise, Papeete, French Polynesia
October 22-26, 201119th United European Gastroenterology Week, Stockholm, Sweden
October 28-November 2, 2011ACG Annual Scientific Meeting & Postgraduate Course, Washington, DC 20001, United States
November 11-12, 2011Falk Symposium 180, IBD 2011: Progress and Future for Lifelong Management, ANA Interconti Hotel, 1-12-33 Akasaka, Minato-ku, Tokyo 107-0052, Japan
December 1-4, 20112011 Advances in Inflammatory Bowel Diseases/Crohn's & Colitis Foundation's Clinical & Research Conference, Hollywood, FL 34234, United States
World J Gastroenterol 2011 April 28; 17(16): I ISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
Online Submissions: http://www.wjgnet.com/[email protected]
I April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
MEETINGS

GENERAL INFORMATIONWorld Journal of Gastroenterology (World J Gastroenterol, WJG, print ISSN 1007-9327, online ISSN 2219-2840, DOI: 10.3748) is a weekly, open-access (OA), peer-reviewed journal supported by an editorial board of 1144 experts in gastroenterology and hepatol-ogy from 60 countries.
The biggest advantage of the OA model is that it provides free, full-text articles in PDF and other formats for experts and the public without registration, which eliminates the obstacle that traditional journals possess and usually delays the speed of the propagation and communication of scientific research results. The open access model has been proven to be a true ap-proach that may achieve the ultimate goal of the journals, i.e. the maximization of the value to the readers, authors and society.
Maximization of personal benefitsThe role of academic journals is to exhibit the scientific levels of a country, a university, a center, a department, and even a scien-tist, and build an important bridge for communication between scientists and the public. As we all know, the significance of the publication of scientific articles lies not only in disseminating and communicating innovative scientific achievements and academic views, as well as promoting the application of scientific achieve-ments, but also in formally recognizing the “priority” and “copy-right” of innovative achievements published, as well as evaluating research performance and academic levels. So, to realize these desired attributes of WJG and create a well-recognized journal, the following four types of personal benefits should be maximized. The maximization of personal benefits refers to the pursuit of the maximum personal benefits in a well-considered optimal manner without violation of the laws, ethical rules and the benefits of oth-ers. (1) Maximization of the benefits of editorial board members: The primary task of editorial board members is to give a peer re-view of an unpublished scientific article via online office system to evaluate its innovativeness, scientific and practical values and deter-mine whether it should be published or not. During peer review, editorial board members can also obtain cutting-edge information in that field at first hand. As leaders in their field, they have prior-ity to be invited to write articles and publish commentary articles. We will put peer reviewers’ names and affiliations along with the article they reviewed in the journal to acknowledge their contribu-tion; (2) Maximization of the benefits of authors: Since WJG is an open-access journal, readers around the world can immediately download and read, free of charge, high-quality, peer-reviewed articles from WJG official website, thereby realizing the goals and significance of the communication between authors and peers as well as public reading; (3) Maximization of the benefits of readers: Readers can read or use, free of charge, high-quality peer-reviewed articles without any limits, and cite the arguments, viewpoints, concepts, theories, methods, results, conclusion or facts and data of pertinent literature so as to validate the innovativeness, scientific and practical values of their own research achievements, thus en-suring that their articles have novel arguments or viewpoints, solid
evidence and correct conclusion; and (4) Maximization of the ben-efits of employees: It is an iron law that a first-class journal is un-able to exist without first-class editors, and only first-class editors can create a first-class academic journal. We insist on strengthening our team cultivation and construction so that every employee, in an open, fair and transparent environment, could contribute their wisdom to edit and publish high-quality articles, thereby realiz-ing the maximization of the personal benefits of editorial board members, authors and readers, and yielding the greatest social and economic benefits.
Aims and scopeThe major task of WJG is to report rapidly the most recent re-sults in basic and clinical research on esophageal, gastrointestinal, liver, pancreas and biliary tract diseases, Helicobacter pylori, endos-copy and gastrointestinal surgery, including: gastroesophageal reflux disease, gastrointestinal bleeding, infection and tumors; gastric and duodenal disorders; intestinal inflammation, micro-flora and immunity; celiac disease, dyspepsia and nutrition; viral hepatitis, portal hypertension, liver fibrosis, liver cirrhosis, liver transplantation, and metabolic liver disease; molecular and cell biology; geriatric and pediatric gastroenterology; diagnosis and screening, imaging and advanced technology.
ColumnsThe columns in the issues of WJG will include: (1) Editorial: To introduce and comment on major advances and developments in the field; (2) Frontier: To review representative achievements, com-ment on the state of current research, and propose directions for future research; (3) Topic Highlight: This column consists of three formats, including (A) 10 invited review articles on a hot topic, (B) a commentary on common issues of this hot topic, and (C) a com-mentary on the 10 individual articles; (4) Observation: To update the development of old and new questions, highlight unsolved problems, and provide strategies on how to solve the questions; (5) Guidelines for Basic Research: To provide guidelines for basic research; (6) Guidelines for Clinical Practice: To provide guidelines for clinical diagnosis and treatment; (7) Review: To review systemi-cally progress and unresolved problems in the field, comment on the state of current research, and make suggestions for future work; (8) Original Article: To report innovative and original find-ings in gastroenterology; (9) Brief Article: To briefly report the novel and innovative findings in gastroenterology and hepatology; (10) Case Report: To report a rare or typical case; (11) Letters to the Editor: To discuss and make reply to the contributions published in WJG, or to introduce and comment on a controversial issue of general interest; (12) Book Reviews: To introduce and comment on quality monographs of gastroenterology and hepatology; and (13) Guidelines: To introduce consensuses and guidelines reached by international and national academic authorities worldwide on basic research and clinical practice gastroenterology and hepatology.
Name of journalWorld Journal of Gastroenterology
Online Submissions: http://www.wjgnet.com/[email protected]
World J Gastroenterol 2011 April 28; 17(16): I-VIISSN 1007-9327 (print) ISSN 2219-2840 (online)
© 2011 Baishideng. All rights reserved.
I April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com
INSTRUCTIONS TO AUTHORS

ISSN and EISSNISSN 1007-9327 (print)ISSN 2219-2840 (online)
Indexed and Abstracted inCurrent Contents®/Clinical Medicine, Science Citation Index Expanded (also known as SciSearch®), Journal Citation Reports®, Index Medicus, MEDLINE, PubMed, PubMed Central, Digital Object Identifer, and Directory of Open Access Journals. ISI, Thomson Reuters, 2009 Impact Factor: 2.092 (33/65 Gastroen-terology and Hepatology).
Published byBaishideng Publishing Group Co., Limited
SPECIAL STATEMENTAll articles published in this journal represent the viewpoints of the authors except where indicated otherwise.
Biostatistical editingStatisital review is performed after peer review. We invite an expert in Biomedical Statistics from to evaluate the statistical method used in the paper, including t-test (group or paired comparisons), chi-squared test, Ridit, probit, logit, regression (linear, curvilinear, or stepwise), correlation, analysis of variance, analysis of covariance, etc. The reviewing points include: (1) Statistical methods should be described when they are used to verify the results; (2) Whether the statistical techniques are suitable or correct; (3) Only homoge-neous data can be averaged. Standard deviations are preferred to standard errors. Give the number of observations and subjects (n). Losses in observations, such as drop-outs from the study should be reported; (4) Values such as ED50, LD50, IC50 should have their 95% confidence limits calculated and compared by weighted probit analysis (Bliss and Finney); and (5) The word ‘significantly’ should be replaced by its synonyms (if it indicates extent) or the P value (if it indicates statistical significance).
Conflict-of-interest statementIn the interests of transparency and to help reviewers assess any potential bias, WJG requires authors of all papers to declare any competing commercial, personal, political, intellectual, or religious interests in relation to the submitted work. Referees are also asked to indicate any potential conflict they might have reviewing a particular paper. Before submitting, authors are suggested to read “Uniform Requirements for Manuscripts Submitted to Biomedi-cal Journals: Ethical Considerations in the Conduct and Reporting of Research: Conflicts of Interest” from International Committee of Medical Journal Editors (ICMJE), which is available at: http://www.icmje.org/ethical_4conflicts.html.
Sample wording: [Name of individual] has received fees for serving as a speaker, a consultant and an advisory board member for [names of organizations], and has received research funding from [names of organization]. [Name of individual] is an employ-ee of [name of organization]. [Name of individual] owns stocks and shares in [name of organization]. [Name of individual] owns patent [patent identification and brief description].
Statement of informed consentManuscripts should contain a statement to the effect that all hu-man studies have been reviewed by the appropriate ethics com-mittee or it should be stated clearly in the text that all persons gave their informed consent prior to their inclusion in the study. Details that might disclose the identity of the subjects under
study should be omitted. Authors should also draw attention to the Code of Ethics of the World Medical Association (Declara-tion of Helsinki, 1964, as revised in 2004).
Statement of human and animal rightsWhen reporting the results from experiments, authors should follow the highest standards and the trial should conform to Good Clinical Practice (for example, US Food and Drug Admin-istration Good Clinical Practice in FDA-Regulated Clinical Trials; UK Medicines Research Council Guidelines for Good Clinical Practice in Clinical Trials) and/or the World Medical Association Declaration of Helsinki. Generally, we suggest authors follow the lead investigator’s national standard. If doubt exists whether the research was conducted in accordance with the above stan-dards, the authors must explain the rationale for their approach and demonstrate that the institutional review body explicitly ap-proved the doubtful aspects of the study.
Before submitting, authors should make their study ap-proved by the relevant research ethics committee or institutional review board. If human participants were involved, manuscripts must be accompanied by a statement that the experiments were undertaken with the understanding and appropriate informed consent of each. Any personal item or information will not be published without explicit consents from the involved patients. If experimental animals were used, the materials and methods (experimental procedures) section must clearly indicate that ap-propriate measures were taken to minimize pain or discomfort, and details of animal care should be provided.
SUBMISSION OF MANUSCRIPTSManuscripts should be typed in 1.5 line spacing and 12 pt. Book Antiqua with ample margins. Number all pages consecutively, and start each of the following sections on a new page: Title Page, Abstract, Introduction, Materials and Methods, Results, Discussion, Acknowledgements, References, Tables, Figures, and Figure Legends. Neither the editors nor the publisher are responsible for the opinions expressed by contributors. Manu-scripts formally accepted for publication become the permanent property of Baishideng Publishing Group Co., Limited, and may not be reproduced by any means, in whole or in part, without the written permission of both the authors and the publisher. We reserve the right to copy-edit and put onto our website accepted manuscripts. Authors should follow the relevant guidelines for the care and use of laboratory animals of their institution or national animal welfare committee. For the sake of transparency in regard to the performance and reporting of clinical trials, we endorse the policy of the ICMJE to refuse to publish papers on clinical trial results if the trial was not recorded in a publicly-accessible registry at its outset. The only register now available, to our knowledge, is http://www.clinicaltrials.gov sponsored by the United States National Library of Medicine and we encourage all potential contributors to register with it. However, in the case that other registers become available you will be duly notified. A letter of recommendation from each author’s organization should be provided with the contributed article to ensure the pri-vacy and secrecy of research is protected.
Authors should retain one copy of the text, tables, photo-graphs and illustrations because rejected manuscripts will not be returned to the author(s) and the editors will not be responsible for loss or damage to photographs and illustrations sustained dur-ing mailing.
Online submissionsManuscripts should be submitted through the Online Submission
Instructions to authors
II April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

System at: http://www.wjgnet.com/1007-9327office. Authors are highly recommended to consult the ONLINE INSTRUC-TIONS TO AUTHORS (http://www.wjgnet.com/1007-9327/g_info_20100315215714.htm) before attempting to submit on-line. For assistance, authors encountering problems with the On-line Submission System may send an email describing the prob-lem to [email protected], or by telephone: +86-10-5908-0039. If you submit your manuscript online, do not make a postal contri-bution. Repeated online submission for the same manuscript is strictly prohibited.
MANUSCRIPT PREPARATIONAll contributions should be written in English. All articles must be submitted using word-processing software. All submissions must be typed in 1.5 line spacing and 12 pt. Book Antiqua with ample margins. Style should conform to our house format. Required in-formation for each of the manuscript sections is as follows:
Title pageTitle: Title should be less than 12 words.
Running title: A short running title of less than 6 words should be provided.
Authorship: Authorship credit should be in accordance with the standard proposed by ICMJE, based on (1) substantial contribu-tions to conception and design, acquisition of data, or analysis and interpretation of data; (2) drafting the article or revising it critically for important intellectual content; and (3) final approval of the version to be published. Authors should meet conditions 1, 2, and 3.
Institution: Author names should be given first, then the com-plete name of institution, city, province and postcode. For ex-ample, Xu-Chen Zhang, Li-Xin Mei, Department of Pathology, Chengde Medical College, Chengde 067000, Hebei Province, China. One author may be represented from two institutions, for example, George Sgourakis, Department of General, Viscer-al, and Transplantation Surgery, Essen 45122, Germany; George Sgourakis, 2nd Surgical Department, Korgialenio-Benakio Red Cross Hospital, Athens 15451, Greece.
Author contributions: The format of this section should be: Author contributions: Wang CL and Liang L contributed equally to this work; Wang CL, Liang L, Fu JF, Zou CC, Hong F and Wu XM designed the research; Wang CL, Zou CC, Hong F and Wu XM performed the research; Xue JZ and Lu JR contributed new reagents/analytic tools; Wang CL, Liang L and Fu JF analyzed the data; and Wang CL, Liang L and Fu JF wrote the paper.
Supportive foundations: The complete name and number of supportive foundations should be provided, e.g. Supported by National Natural Science Foundation of China, No. 30224801
Correspondence to: Only one corresponding address should be provided. Author names should be given first, then author title, affiliation, the complete name of institution, city, postcode, prov-ince, country, and email. All the letters in the email should be in lower case. A space interval should be inserted between country name and email address. For example, Montgomery Bissell, MD, Professor of Medicine, Chief, Liver Center, Gastroenterology Division, University of California, Box 0538, San Francisco, CA 94143, United States. [email protected]
Telephone and fax: Telephone and fax should consist of +,
country number, district number and telephone or fax number, e.g. Telephone: +86-10-59080039 Fax: +86-10-85381893
Peer reviewers: All articles received are subject to peer review. Normally, three experts are invited for each article. Decision for acceptance is made only when at least two experts recommend an article for publication. Reviewers for accepted manuscripts are acknowledged in each manuscript, and reviewers of articles which were not accepted will be acknowledged at the end of each issue. To ensure the quality of the articles published in WJG, reviewers of accepted manuscripts will be announced by publish-ing the name, title/position and institution of the reviewer in the footnote accompanying the printed article. For example, review-ers: Professor Jing-Yuan Fang, Shanghai Institute of Digestive Disease, Shanghai, Affiliated Renji Hospital, Medical Faculty, Shanghai Jiaotong University, Shanghai, China; Professor Xin-Wei Han, Department of Radiology, The First Affiliated Hospital, Zhengzhou University, Zhengzhou, Henan Province, China; and Professor Anren Kuang, Department of Nuclear Medicine, Huaxi Hospital, Sichuan University, Chengdu, Sichuan Province, China.
AbstractThere are unstructured abstracts (no more than 256 words) and structured abstracts (no more than 480). The specific re-quirements for structured abstracts are as follows:
An informative, structured abstracts of no more than 480 words should accompany each manuscript. Abstracts for original contributions should be structured into the following sections. AIM (no more than 20 words): Only the purpose should be included. Please write the aim as the form of “To investigate/study/…”; MATERIALS AND METHODS (no more than 140 words); RESULTS (no more than 294 words): You should present P values where appropriate and must provide relevant data to illustrate how they were obtained, e.g. 6.92 ± 3.86 vs 3.61 ± 1.67, P < 0.001; CONCLUSION (no more than 26 words).
Key wordsPlease list 5-10 key words, selected mainly from Index Medicus, which reflect the content of the study.
TextFor articles of these sections, original articles and brief arti-cles, the main text should be structured into the following sec-tions: INTRODUCTION, MATERIALS AND METHODS, RESULTS and DISCUSSION, and should include appropri-ate Figures and Tables. Data should be presented in the main text or in Figures and Tables, but not in both. The main text format of these sections, editorial, topic highlight, case report, letters to the editors, can be found at: http://www.wjgnet.com/1007-9327/g_info_20100315215714.htm.
IllustrationsFigures should be numbered as 1, 2, 3, etc., and mentioned clear-ly in the main text. Provide a brief title for each figure on a sep-arate page. Detailed legends should not be provided under the figures. This part should be added into the text where the figures are applicable. Figures should be either Photoshop or Illustra-tor files (in tiff, eps, jpeg formats) at high-resolution. Examples can be found at: http://www.wjgnet.com/1007-9327/13/4520.pdf; http://www.wjgnet.com/1007-9327/13/4554.pdf; http://www.wjgnet.com/1007-9327/13/4891.pdf; http://www.wjgnet.com/1007-9327/13/4986.pdf; http://www.wjgnet.com/1007-9327/13/4498.pdf. Keeping all elements compiled is necessary in line-art image. Scale bars should be
Instructions to authors
III April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

used rather than magnification factors, with the length of the bar defined in the legend rather than on the bar itself. File names should identify the figure and panel. Avoid layer-ing type directly over shaded or textured areas. Please use uniform legends for the same subjects. For example: Figure 1 Pathological changes in atrophic gastritis after treatment. A:...; B:...; C:...; D:...; E:...; F:...; G: …etc. It is our principle to publish high resolution-figures for the printed and E-versions.
TablesThree-line tables should be numbered 1, 2, 3, etc., and men-tioned clearly in the main text. Provide a brief title for each table. Detailed legends should not be included under tables, but rather added into the text where applicable. The informa-tion should complement, but not duplicate the text. Use one horizontal line under the title, a second under column heads, and a third below the Table, above any footnotes. Vertical and italic lines should be omitted.
Notes in tables and illustrationsData that are not statistically significant should not be noted. aP < 0.05, bP < 0.01 should be noted (P > 0.05 should not be noted). If there are other series of P values, cP < 0.05 and dP < 0.01 are used. A third series of P values can be expressed as eP < 0.05 and fP < 0.01. Other notes in tables or under illustra-tions should be expressed as 1F, 2F, 3F; or sometimes as other symbols with a superscript (Arabic numerals) in the upper left corner. In a multi-curve illustration, each curve should be la-beled with ●, ○, ■, □, ▲, △, etc., in a certain sequence.
AcknowledgmentsBrief acknowledgments of persons who have made genuine contributions to the manuscript and who endorse the data and conclusions should be included. Authors are responsible for obtaining written permission to use any copyrighted text and/or illustrations.
REFERENCESCoding systemThe author should number the references in Arabic numerals ac-cording to the citation order in the text. Put reference numbers in square brackets in superscript at the end of citation content or after the cited author’s name. For citation content which is part of the narration, the coding number and square brackets should be typeset normally. For example, “Crohn’s disease (CD) is associated with increased intestinal permeability[1,2]”. If references are cited directly in the text, they should be put together within the text, for example, “From references[19,22-24], we know that...”.
When the authors write the references, please ensure that the order in text is the same as in the references section, and also ensure the spelling accuracy of the first author’s name. Do not list the same citation twice.
PMID and DOIPleased provide PubMed citation numbers to the reference list, e.g. PMID and DOI, which can be found at http://www.ncbi.nlm.nih.gov/sites/entrez?db=pubmed and http://www.cross-ref.org/SimpleTextQuery/, respectively. The numbers will be used in E-version of this journal.
Style for journal referencesAuthors: the name of the first author should be typed in bold-faced letters. The family name of all authors should be typed with the initial letter capitalized, followed by their abbreviated
first and middle initials. (For example, Lian-Sheng Ma is ab-breviated as Ma LS, Bo-Rong Pan as Pan BR). The title of the cited article and italicized journal title (journal title should be in its abbreviated form as shown in PubMed), publication date, volume number (in black), start page, and end page [PMID: 11819634 DOI: 10.3748/wjg.13.5396].
Style for book referencesAuthors: the name of the first author should be typed in bold-faced letters. The surname of all authors should be typed with the initial letter capitalized, followed by their abbreviated middle and first initials. (For example, Lian-Sheng Ma is abbreviated as Ma LS, Bo-Rong Pan as Pan BR) Book title. Publication number. Publica-tion place: Publication press, Year: start page and end page.
FormatJournalsEnglish journal article (list all authors and include the PMID where ap-
plicable)1 Jung EM, Clevert DA, Schreyer AG, Schmitt S, Rennert J,
Kubale R, Feuerbach S, Jung F. Evaluation of quantitative contrast harmonic imaging to assess malignancy of liver tumors: A prospective controlled two-center study. World J Gastroenterol 2007; 13: 6356-6364 [PMID: 18081224 DOI: 10.3748/wjg.13.6356]
Chinese journal article (list all authors and include the PMID where ap-plicable)
2 Lin GZ, Wang XZ, Wang P, Lin J, Yang FD. Immunolog-ic effect of Jianpi Yishen decoction in treatment of Pixu-diarrhoea. Shijie Huaren Xiaohua Zazhi 1999; 7: 285-287
In press3 Tian D, Araki H, Stahl E, Bergelson J, Kreitman M.
Signature of balancing selection in Arabidopsis. Proc Natl Acad Sci USA 2006; In press
Organization as author4 Diabetes Prevention Program Research Group. Hyper-
tension, insulin, and proinsulin in participants with impaired glucose tolerance. Hypertension 2002; 40: 679-686 [PMID: 12411462 PMCID:2516377 DOI:10.1161/01.HYP.00000 35706.28494.09]
Both personal authors and an organization as author 5 Vallancien G, Emberton M, Harving N, van Moorse-
laar RJ; Alf-One Study Group. Sexual dysfunction in 1, 274 European men suffering from lower urinary tract symptoms. J Urol 2003; 169: 2257-2261 [PMID: 12771764 DOI:10.1097/01.ju.0000067940.76090.73]
No author given6 21st century heart solution may have a sting in the tail. BMJ
2002; 325: 184 [PMID: 12142303 DOI:10.1136/bmj.325. 7357.184]
Volume with supplement7 Geraud G, Spierings EL, Keywood C. Tolerability and
safety of frovatriptan with short- and long-term use for treatment of migraine and in comparison with sumatrip-tan. Headache 2002; 42 Suppl 2: S93-99 [PMID: 12028325 DOI:10.1046/j.1526-4610.42.s2.7.x]
Issue with no volume8 Banit DM, Kaufer H, Hartford JM. Intraoperative frozen
section analysis in revision total joint arthroplasty. Clin Orthop Relat Res 2002; (401): 230-238 [PMID: 12151900 DOI:10.1097/00003086-200208000-00026]
No volume or issue9 Outreach: Bringing HIV-positive individuals into care.
HRSA Careaction 2002; 1-6 [PMID: 12154804]
Instructions to authors
IV April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

BooksPersonal author(s)10 Sherlock S, Dooley J. Diseases of the liver and billiary
system. 9th ed. Oxford: Blackwell Sci Pub, 1993: 258-296Chapter in a book (list all authors)11 Lam SK. Academic investigator’s perspectives of medical
treatment for peptic ulcer. In: Swabb EA, Azabo S. Ulcer disease: investigation and basis for therapy. New York: Marcel Dekker, 1991: 431-450
Author(s) and editor(s)12 Breedlove GK, Schorfheide AM. Adolescent pregnancy.
2nd ed. Wieczorek RR, editor. White Plains (NY): March of Dimes Education Services, 2001: 20-34
Conference proceedings13 Harnden P, Joffe JK, Jones WG, editors. Germ cell tu-
mours V. Proceedings of the 5th Germ cell tumours Con-ference; 2001 Sep 13-15; Leeds, UK. New York: Springer, 2002: 30-56
Conference paper14 Christensen S, Oppacher F. An analysis of Koza’s compu-
tational effort statistic for genetic programming. In: Foster JA, Lutton E, Miller J, Ryan C, Tettamanzi AG, editors. Ge-netic programming. EuroGP 2002: Proceedings of the 5th European Conference on Genetic Programming; 2002 Apr 3-5; Kinsdale, Ireland. Berlin: Springer, 2002: 182-191
Electronic journal (list all authors)15 Morse SS. Factors in the emergence of infectious dis-
eases. Emerg Infect Dis serial online, 1995-01-03, cited 1996-06-05; 1(1): 24 screens. Available from: URL: http://www.cdc.gov/ncidod/eid/index.htm
Patent (list all authors)16 Pagedas AC, inventor; Ancel Surgical R&D Inc., as-
signee. Flexible endoscopic grasping and cutting device and positioning tool assembly. United States patent US 20020103498. 2002 Aug 1
Statistical dataWrite as mean ± SD or mean ± SE.
Statistical expressionExpress t test as t (in italics), F test as F (in italics), chi square test as χ2 (in Greek), related coefficient as r (in italics), degree of free-dom as υ (in Greek), sample number as n (in italics), and probabil-ity as P (in italics).
UnitsUse SI units. For example: body mass, m (B) = 78 kg; blood pres-sure, p (B) = 16.2/12.3 kPa; incubation time, t (incubation) = 96 h, blood glucose concentration, c (glucose) 6.4 ± 2.1 mmol/L; blood CEA mass concentration, p (CEA) = 8.6 24.5 mg/L; CO2 volume fraction, 50 mL/L CO2, not 5% CO2; likewise for 40 g/L formal-dehyde, not 10% formalin; and mass fraction, 8 ng/g, etc. Arabic numerals such as 23, 243, 641 should be read 23 243 641.
The format for how to accurately write common units and quantums can be found at: http://www.wjgnet.com/1007-9327/g_info_20100315223018.htm.
AbbreviationsStandard abbreviations should be defined in the abstract and on first mention in the text. In general, terms should not be ab-breviated unless they are used repeatedly and the abbreviation is helpful to the reader. Permissible abbreviations are listed in Units, Symbols and Abbreviations: A Guide for Biological and Medical Editors and Authors (Ed. Baron DN, 1988) published
by The Royal Society of Medicine, London. Certain commonly used abbreviations, such as DNA, RNA, HIV, LD50, PCR, HBV, ECG, WBC, RBC, CT, ESR, CSF, IgG, ELISA, PBS, ATP, EDTA, mAb, can be used directly without further explanation.
ItalicsQuantities: t time or temperature, c concentration, A area, l length, m mass, V volume.Genotypes: gyrA, arg 1, c myc, c fos, etc.Restriction enzymes: EcoRI, HindI, BamHI, Kbo I, Kpn I, etc.Biology: H. pylori, E coli, etc.
Examples for paper writingEditorial: http://www.wjgnet.com/1007-9327/g_info_20100315 220036.htm
Frontier: http://www.wjgnet.com/1007-9327/g_info_20100315 220305.htm
Topic highlight: http://www.wjgnet.com/1007-9327/g_info_20 100315220601.htm
Observation: http://www.wjgnet.com/1007-9327/g_info_201003 12232427.htm
Guidelines for basic research: http://www.wjgnet.com/1007-93 27/g_info_20100315220730.htm
Guidelines for clinical practice: http://www.wjgnet.com/1007- 9327/g_info_20100315221301.htm
Review: http://www.wjgnet.com/1007-9327/g_info_20100315 221554.htm
Original articles: http://www.wjgnet.com/1007-9327/g_info_20 100315221814.htm
Brief articles: http://www.wjgnet.com/1007-9327/g_info_2010 0312231400.htm
Case report: http://www.wjgnet.com/1007-9327/g_info_2010 0315221946.htm
Letters to the editor: http://www.wjgnet.com/1007-9327/g_info_ 20100315222254.htm
Book reviews: http://www.wjgnet.com/1007-9327/g_info_2010 0312231947.htm
Guidelines: http://www.wjgnet.com/1007-9327/g_info_2010 0312232134.htm
RESUBMISSION OF THE REVISED MANUSCRIPTSPlease revise your article according to the revision policies of WJG. The revised version includes manuscript and high-reso-lution image figures. The author should re-submit the revised manuscript online, along with printed high-resolution color or black and white photos; Copyright transfer letter, and responses to the reviewers, and science news are sent to us via email.
Editorial Office World Journal of GastroenterologyEditorial Department: Room 903, Building D,Ocean International Center,No. 62 Dongsihuan Zhonglu,
Instructions to authors
V April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com

Chaoyang District, Beijing 100025, ChinaE-mail: [email protected]://www.wjgnet.comTelephone: +86-10-5908-0039Fax: +86-10-85381893
Language evaluation The language of a manuscript will be graded before it is sent for revision. (1) Grade A: priority publishing; (2) Grade B: minor language polishing; (3) Grade C: a great deal of language polish-ing needed; and (4) Grade D: rejected. Revised articles should reach Grade A or B.
Copyright assignment formPlease download a Copyright assignment form from http://www.wjgnet.com/1007-9327/g_info_20100315222818.htm.
Responses to reviewersPlease revise your article according to the comments/sugges-tions provided by the reviewers. The format for responses to the reviewers’ comments can be found at: http://www.wjgnet.com/1007-9327/g_info_20100315222607.htm.
Proof of financial supportFor paper supported by a foundation, authors should provide a copy of the document and serial number of the foundation.
Links to documents related to the manuscript WJG will be initiating a platform to promote dynamic interac-
tions between the editors, peer reviewers, readers and authors. After a manuscript is published online, links to the PDF version of the submitted manuscript, the peer-reviewers’ report and the revised manuscript will be put on-line. Readers can make com-ments on the peer reviewer’s report, authors’ responses to peer reviewers, and the revised manuscript. We hope that authors will benefit from this feedback and be able to revise the manuscript accordingly in a timely manner.
Science news releasesAuthors of accepted manuscripts are suggested to write a science news item to promote their articles. The news will be released rap-idly at EurekAlert/AAAS (http://www.eurekalert.org). The title for news items should be less than 90 characters; the summary should be less than 75 words; and main body less than 500 words. Science news items should be lawful, ethical, and strictly based on your original content with an attractive title and interesting pictures.
Publication feeWJG is an international, peer-reviewed, Open-Access, online jour-nal. Articles published by this journal are distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. Authors of accepted articles must pay a publication fee. The related standards are as follows. Publication fee: 1300 USD per article. Editorial, topic highlights, book reviews and letters to the editor are published free of charge.
Instructions to authors
VI April 28, 2011|Volume 17|Issue 16|WJG|www.wjgnet.com