vol.271,no.23,issueofjune7,pp.13834–13843,1996 he ... · pdf...
TRANSCRIPT

Phagocytosed Live Listeria monocytogenes InfluencesRab5-regulated in Vitro Phagosome-Endosome Fusion*
(Received for publication, June 30, 1995, and in revised form, March 20, 1996)
Carmen Alvarez-Dominguez‡§, Alejandro M. Barbieri‡, Walter Beron‡¶i,Angela Wandinger-Ness**, and Philip D. Stahl‡ ‡‡
From the ‡Department of Cell Biology and Physiology, Washington University School of Medicine,St. Louis, Missouri 63110, ¶Instituto de Histologia y Embriologia, CONICET, Facultad de Ciencias Medicas,Universidad Nacional de Cuyo, Mendoza, Argentina, and the **Department of Biochemistry, Molecular Biologyand Cell Biology, Northwestern University, Evanston, Illinois 60208
Survival or destruction of a pathogen following phag-ocytosis depends, in part, on fusion events between thephagosome and the endosomal or lysosomal compart-ments. Here we use an in vitro assay to show that pha-gosome-endosome fusion is regulated by the smallGTPase rab5 and that fusion events are influenced by aninternalized live organism, Listeria monocytogenes(LM). We compare the in vitro fusion of phagosomescontaining heat-killed organisms (dead LM) with that ofphagosomes containing a live nonhemolytic mutant(live LMhly2). Unlike the wild-type organism, LMhly2 re-mains trapped inside the phagosome. Phagosome-endo-some fusion was reconstituted using biotinylated orga-nisms and endosomes containing horseradish peroxi-dase conjugated with avidin. With both live LMhly2 anddead LM preparations, in vitro phagosome-endosomefusion was time-, temperature-, and cytosol-dependent.Live LMhly2 phagosomes exhibited a faster rate of fu-sion. Fusion in both preparations was regulated by rab5and possibly by other GTPases. Anti-rab5 antibodiesand immunodepletion of cytosolic rab5 inhibited fusion.Addition of glutatione S-transferase-rab5 in the GTPform stimulated phagosome-endosome fusion, whereasaddition of a dominant negative mutant of rab5 blockedfusion. Purified live LMhly2 phagosomal membraneswere enriched in rab5 as revealed by Western blotting,compared with dead LM phagosomes. Fusion of endo-somes with dead LM containing phagosomes requiredATP and was inhibited by ATP depletion and by N-eth-ylmaleimide (NEM) and anti-NEM-sensitive factor (NSF)antibodies. Unexpectedly, phagosome-endosome fusionwith live LMhly2-containing phagosomes was not inhib-ited by ATP depletion nor by NEM or anti-NSF antibod-ies. Western blot analysis revealed that live LMhly2-con-taining phagosomes were enriched for membrane-bound NSF, while dead LM containing phagosomescontained low or undetectable quantities. Washing liveLMhly2-containing phagosomes with 0.5 M KCl removedNSF associated with the membranes and rendered themNEM, ATP, anti-NSF antibody sensitive for fusion. We
conclude that rab5 regulates phagosome-endosome fu-sion and that live microorganisms can up-regulate thisprocess by recruiting rab5 to the membrane.
Phagocytosis, an important element of the host-defense sys-tem, is initiated by the attachment of a particle or organism tothe phagocyte via cell surface receptors followed by ingestionand formation of a phagosome. Subsequently, a series of se-quential intracellular membrane fusion and budding eventsoccur that accommodate maturation of the phagosome and thedelivery of hydrolytic enzymes and other molecules importantfor host defense. Proper orchestration of these events isthought to lead to the destruction of the pathogen and to theinitiation of appropriate immunological responses.The Gram-positive bacterium, Listeria monocytogenes (LM),1
is a facultative intracellular parasite capable of causing severedisease in immunocompromised humans and animals (1). LMinvades both phagocytic and nonphagocytic cells. Internaliza-tion of LM is receptor-mediated, and the invasive processseems to involve the bacterial structural protein, internalin(2–6).After internalization, phagosomes containing LM are
thought to fuse with lysosomes to form phagolysosomes (7–11).Studies with heat-killed LM (dead LM) indicate that phago-somes receive material from the trans-Golgi network and fromlysosomes (10). Studies with Staphylococcus aureus phago-somes have rendered similar conclusions (12). Virulent LM hasbeen shown to lyse the phagolysosomal membrane and to es-cape into the cytoplasm where growth flourishes. Escape fromthe phagosome is mediated by listeriolysin O (also known ashemolysin). Listeriolysin O-defective mutants, i.e. nonhemo-lytic mutants (LMhly2) (13–15), are unable to escape from thephagolysosome and are degraded by the phagocyte. In thisrespect, LMhly2 mutants behave as heat-killed bacteria (deadLM). Our goal was to analyze early phagosome-endosome fu-sion events with LM containing phagosomes using an in vitrofusion assay, where the role of different factors and the influ-ence of the live microorganism could be examined.Over the past few years, there has been a substantial in-
crease in our knowledge of vesicle budding and membranefusion events along the endocytic pathway and the role of
* This work has been supported in part by grants from the NationalInstitute for Health (to P. D. S.) and from the Council for TobaccoResearch, U. S. A. (3980) (to A. W-N.) The costs of publication of thisarticle were defrayed in part by the payment of page charges. Thisarticle must therefore be hereby marked “advertisement” in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.§ Recipient of a Postdoctoral Fellowship from the Formacion de Per-
sonal Investigador, Ministerio de Educacion y Ciencia, Madrid, Spain.i Recipient of a CONICET Fellowship, Argentina.‡‡ To whom correspondence should be addressed: Dept. of Cell Biol-
ogy and Physiology, Washington University School of Medicine, Box8228, 660 S. Euclid Avenue, St. Louis, MO 63110. Tel.: 314-362-6950;Fax: 314-362-1490.
1 The abbreviations used are: LM, L. monocytogenes; LMhly2, nonhemo-lytic mutant of L. monocytogenes; GST, glutathione S-transferase; GTPgS,guanosine 59-O-(thiotriphosphate); HBS, Hank’s balanced salt solution;TES, 2-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}ethanesulfonic acid;HRP, horse radish peroxidase; NEM, N-ethylmaleimide; NSF, NEM-sensi-tive factor; PNS, postnuclear supernatant; mAb, monoclonal antibody; BSA,bovine serum albumin; PBS, phosphate-buffered saline; PAGE, polyacryl-amide gel electrophoresis; GDI, GDP dissociation inhibitor.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 271, No. 23, Issue of June 7, pp. 13834–13843, 1996© 1996 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
13834
by guest on May 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

GTP-binding proteins as regulators of these processes (16–20).The GTPase rab5 plays a central role in early endocytic events.Although rab5 was found to be present on phagosomes isolatedfollowing phagocytosis of latex beads (21), a functional role forrab5 in phagocytosis has not yet been described. Here, we showthat in vitro fusion of phagosomes, containing both live anddead bacteria, with endosomes is regulated by rab5. Both fu-sion events require cytosolic proteins. However, the ATP re-quirement and the sensitivity of fusion to NEM are quite dif-ferent for phagosomes containing live and dead organisms.These differences lead us to postulate that rab5-regulated earlyphagosome fusion is dependent on the status of the microor-ganism. Moreover, the data indicate that live L. monocytogenesdirectly affects and modulates the fusion process by recruitingrab5 to the phagosomal membranes.
MATERIALS AND METHODS
Biological Reagents—J774E clone, a mannose receptor-positivemacrophage cell line, was grown as described previously (12). Mono-clonal antibodies (mAb) used include: 4F11, a mouse IgG2ak monoclonalantibody that recognizes mouse rab5 (22); HDP-1, an anti-DNP mousemAb was employed as an irrelevant antibody (23); 6E6, a mouse IgGmAb that recognizes the D1-D2 domains of NSF (a gift from Sidney W.Whiteheart (University of Kentucky, Lexington, KY) (24) was used forWestern blotting; 4A6, a mouse IgM monoclonal antibody that recog-nizes native NSF, was kindly provided by James E. Rothman (Sloane-Kettering Memorial Hospital, NY) and used in the in vitro fusionassays. An irrelevant IgM monoclonal antibody was included as acontrol (Sigma). Polyclonal antibodies specific for rab7 and rab5 weregenerated by immunizing rabbits with GST-rab fusion proteins. Theseantibodies recognize human and mouse rab5 and rab7, respectively.2 Apolyclonal antibody against chicken immunoglobulins made in rabbitswas obtained from Sigma and included as an irrelevant antibody.Horseradish peroxidase (HRP) conjugated with avidin (HRP-avidin)was obtained from Pierce. Cytosol was prepared as described elsewhere(16). Cytosol was gel-filtered through 1 ml of Sephadex G-25 spincolumn just before use in the fusion assay. Protein concentrations weremeasured as described previously (25) using bovine serum albumin asthe standard. All other chemicals were obtained from Sigma. PurifiedGDI was obtained from B. Goud (Pasteur Institute, Paris, France) (26).Purification of Recombinant Rab5 Fusion Proteins, Prenylation, and
Solubility—The rab5 mutants used in this study were described previ-ously (27, 28) and included rab5:S34N and rab5:Q79L. Recombinantrab5 proteins were expressed in large quantity as glutathione S-trans-ferase fusion proteins in Escherichia coli strain JM 101 and wereaffinity-purified by glutathione-Sepharose resin (GST-rab5). Beforeprenylation, rab-geranylgeranyl transferase (REP-1) was partially pu-rified following a protocol previously reported (29, 30). Recombinantrabs were prenylated as described previously (29, 31, 32). Briefly, rab5substrate (10 mM) was incubated with the REP-1 in 50 ml of 50 mM
Hepes/KOH, pH 7, 5 mM MgCl2, 0.5 mM Nonidet-P40, 1 mM DTT (bufferA) containing 12 mM geranylgeranyl pyrophosphate (GGPP) for 30 minat 37 °C. After the reaction, the prenylated rab5 proteins were aliquotedat 4 mg/ml and stored at 280 °C. Under these conditions, 70% of therab5 was prenylated as measured by the incorporation of [3H]GGPP.Rab5 copurifies with rab escort protein as described by Andres et al.(33). Rab escort protein has been shown to mediate rab5 binding toendosomal membranes (34). Solubility of the purified proteins waschecked as follows. Recombinant purified proteins (1 mg) were resus-pended in 200 ml of the following solutions: (a) buffer B (50 mM Hepes/KOH, pH 7, 1 mM MgCl2, 1 mM DTT); (b) buffer B containing 1 mM
Nonident P-40; and (c) buffer B containing 0.1% BSA. The solubility ofrab5 proteins was evaluated by the presence of a visible aggregate after40 min at 37 °C incubation under each of the conditions describedabove. Aggregates were found where the protein was resuspended inbuffer B without detergent or BSA. Purified protein stocks contained1–3% BSA, which resulted in a relatively small amount of BSA in ourfusion reactions (less than 100 mmol of BSA/mol of prenylated rab5).BSA may affect the solubility and the appropriate targetting of preny-lated rabs in the absence of appropriate escort proteins (35).Phagocytic Probes—The nonhemolytic L. monocytogenes mutant
strain used in this study (DP-L2161) (LMhly2) (15) derived from thewild-type strain (10403S) was kindly provided by D. A. Portnoy (Uni-
versity of Pennsylvania, Philadelphia, PA). The bacteria were grown inbrain-heart infusion broth (Difco) at 37 °C with aeration in the presenceof 150 mg/ml streptomycin. The bacteria were harvested in the logarith-mic phase of growth and were heat-killed by treatment at 60 °C for 1 h.Heat-killed bacteria (dead LM) were confirmed dead by plating in bloodagar plates. Bacteria were extensively washed and stored at 4 °C.LMhly2 and dead LM (109 bacteria) were biotinylated with ss-NHS-biotin (Pierce) according to the manufacturer’s recommendations and asdescribed previously (36). Briefly, bacteria were washed three times inPBS (pH 7.4) and resuspended in PBS (pH 8.0) at 4 °C. The bacteriawere treated with ss-NHS-biotin at 0.5 mg/ml for 2 min with gentleshaking. They were washed sequentially in PBS, 50 mM NH4Cl, PBS,0.1 mM CaCl2, PBS, 1 mM MgCl2 to quench free biotin, and resuspendedin PBS. Biotinylation did not affect bacteria viability as determined byplating in blood agar plates. The number of biotin molecules per orga-nism was estimated in every experiment using a colorimetric assaywith the dye 2-(49-hydroxyazobenzene)benzoic acid (Pierce) according tothe manufacturer’s instructions. While the numbers vary from experi-ment to experiment, routinely ;40 mol of ss-NHS-biotin/109 bacteriawere incorporated, and no significant differences were found betweendead and live LM. ss-NHS-biotin/dead LM were stored at 4 °C andss-NHS-biotin/live LMhly2 were stored at 270 °C until use.To identify biotinylated bacterial proteins, 109 biotinylated bacteria
were extracted (36) by resuspension in 100 ml of PBS containing 1%SDS. Following centrifugation, SDS-PAGE, and transfer to nitrocellu-lose, blotting with avidin-HRP indicated that multiple proteins in bothpreparations of bacteria were biotinylated. The blots were essentiallyidentical for the two preparations.Assay for Bacterial Uptake—The assay was performed essentially as
previously reported (37, 38). Briefly, LMhly2 bacteria were grown over-night in brain-heart infusion broth. After extensive washing, bacteriawere resuspended in RPMI (minus methionine and cysteine) with 1 mCiof [35S]methionine (ICN Tran35S-label). The bacteria were incubatedwith rotation for 6 h, washed with cold PBS, and resuspended in PBS.35S-Labeled dead LM were prepared by heating as described above.Labeled bacteria incorporated 1–2 cpm/bacterium. Dead LM were alsosurface-labeled with 125I by the chloramine T method at 4 °C (37). Foruptake assays, live 35S-labeled LMhly2 and 125I- or 35S-labeled dead LMwere added (3 3 105 cpm/well) to 2 3 106 cells (E-clone) in 24-well platesand centrifuged (2,000 rpm, 5 min, 4 °C) to speed adherence and tosynchronize the infection. Following incubation at 37 °C for differenttimes, cells were solubilized with 1% Triton X-100. Experiments wereperformed in duplicate.Preparation of Phagosomes and Endosomes for in Vitro Fusion—
J774E clone macrophages (108 cells) were incubated with ss-NHS-biotin/dead-LM or ss-NHS-biotin/live LMhly2 (109 bacteria) (1 h, 4 °C)and centrifuged to synchronize the infection (2,000 rpm, 5 min, 4 °C).Uptake was initiated by addition of prewarmed HBSA (Hank’s balancedsalt solution buffered with 10 mM HEPES and 10 mM TES, pH 7.4, andsupplemented with 1% BSA). After 10 min at 37 °C, uptake was stoppedby addition of ice-cold HBSA. Cells were sequentially washed withHBSA, PBS-EDTA, and HBE (250 mM sucrose, 0.5 mM EGTA, 20 mM
HEPES-KOH, pH 7.2) by centrifugation (300 3 g for 4 min). Cells wereresuspended with HBE (2 3 108 cells/ml) and homogenized in a ballbearing homogenizer (12). Homogenates were centrifuged at 400 3 g for3 min to eliminate nuclei and intact cells. The postnuclear supernatants(PNS) were quickly frozen in liquid nitrogen and stored at 280 °C.Phagosomal fractions were obtained by diluting a quickly thawed PNSaliquot in 1 ml of HBE and centrifuging at 12,000 3 g in a Microfuge for10 s at 4 °C, as previously reported (12, 39, 40). The supernatant waskept at 4 °C and the pellet resuspended with HBE and centrifugedagain. The resulting supernatants were combined and centrifuged at12,000 3 g for 6 min at 4 °C. This pellet (phagosomal fraction) contain-ing 70% of the total phagosomes in the sample was used for in vitrofusion assay. Early endosomes containing HRP-avidin conjugate wereprepared as described previously with several modifications (41). J774Eclone (108 cells) was incubated with HRP-avidin (200 mg/ml) for 1 h at4 °C. Under these conditions, HRP-avidin is essentially endocytosed viathe mannose receptor (42). Uptake was initiated by the addition ofprewarmed medium (1 ml) at 37 °C and, after 10 min, terminated byadding ice-cold medium. Cells were washed and homogenized, and thePNS fraction was quickly frozen. To prepare endosomes, a thawed PNS(200 ml) was diluted up to 3 ml with HBE and pelleted for 1 min at37,000 3 g at 4 °C. The supernatant was centrifuged for 5 min at 50,0003 g at 4 °C. The pellet of the second centrifugation, enriched in 10-minendosomes (endosomal fraction) was used for in vitro fusion assays. TheHRP-avidin was contained within intact vesicles by the criterion thatgreater than 90% of the HRP activity was sedimented by centrifugation2 G. Li, personal communication.
Rab5 and Phagosome-Endosome Fusion 13835
by guest on May 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

at 100,000 3 g.Fusion Reaction—Phagosomal fractions containing biotinylated dead
LM or biotinylated live LMhly2 and endosomal fractions containingHRP-avidin were mixed in fusion buffer (250 mM sucrose, 0.5 mM
EGTA, 20 mM HEPES-KOH, pH 7.2, 1 mM dithiothreitol, 1.5 mMMgCl2,100 mM KCl, including an ATP-regenerating system, 1 mM ATP, 8 mM
creatine phosphate, 31 units/ml creatine phosphokinase, and 0.25mg/ml avidin as scavenger) supplemented with gel-filtered cytosol. Af-ter incubation for 60 min at 37 °C, the reaction was stopped by chillingon ice. The HRP-avidin/biotin-bacteria complexes were recovered bycentrifugation (10,000 3 g, 6 min, at 4 °C) after solubilization of mem-branes with solubilization buffer (PBS, 0.5% Triton X-100 containing0.25 mg/ml avidin as scavenger). The enzymatic activity of HRP-avidinassociated with the bacteria was then measured. The fusion activitywas quantified as absorbance units/mg of protein (the HRP assay de-scribed below). Protein concentrations were determined in each exper-iment for all samples. Two controls were included in each experimentcorresponding to (a) total activity and (b) background activity. Totalactivity is that which is particle associated when endosomes and pha-gosomes are mixed and then lysed with detergent. Total activity corre-sponds to the total fusion activity that could be achieved per experi-ment. Values around 480 absorbance units/mg were routinely recordedfor both dead and live LM preparations. Background activity corre-sponds to particle-associated HRP when endocytic and phagocytic ves-icles were mixed in fusion buffer lacking cytosolic proteins. These val-ues were quite low and were subtracted from all other values. Formeasuring the fusion activity, each sample is compared to the fusionobserved in the presence of complete fusion buffer containing the high-est cytosol concentration which was normalized to a value of 1 aftersubtracting the background activity. The highest effective cytosol con-centration was standarized for both fusion assays (with dead and liveLM) and observed to be around 1 mg/ml for both assays. This fusionvalue corresponds to as much as 70% of the total activity depending onthe experiment. The values obtained in each experiment are listed intable and figure legends. All reactions were incubated 60 min at 37 °C.Antibodies (mAb 4F11 anti-rab5, anti-rab7, mAb 4A6 anti-NSF, mAb
HDP-1 anti-DNP, and controls) were added at the concentration indi-cated and incubated for 45 min on ice before the fusion assay. Forkinetic studies, samples were incubated for different times at 37 °C andthen cooled on ice.Treatment of Phagocytic Vesicles with GDI—The protocol was essen-
tially as previously reported (43, 44). Briefly, phagocytic vesicles (100mg) were preincubated in fusion buffer plus a protease inhibitor mixture(1 mM phenylmethylsulfonyl fluoride, 20 mg/ml leupeptin, 20 mg/mlaprotinin) for 20 min at 30 °C in the presence or absence of eitherGTPgS (1 mM) or GDP (1 mM). This incubation was followed by additionof 6 mg of purified GDI for 10 min at 30 °C. Samples were washed twicein fusion buffer and resuspended either in sample buffer to be analyzedby SDS-PAGE or in fusion buffer and tested in a fusion reaction assay(the vesicles to be evaluated for fusion were pretreated with GDI in thepresence of GDP).Horseradish Peroxidase Assay—The enzyme assay was conducted in
96-well microplates (Costar Co.) using o-dianisidine as the chromogenicsubstrate (45). Briefly, the reaction was started by adding 20 ml of eachsample to 100 ml of 0.5 N sodium acetate (pH 5.0) containing 0.342 mM
o-dianisidine and 0.003% H2O2. The reaction was conducted at roomtemperature for 20 min and stopped by adding 100 ml of 0.1 N H2SO4.Absorbance was measured at A450 nm in a Bio-Rad microplate reader.Each value was expressed as absorbance units/mg of protein. For quan-tification, a standard curve with different concentrations of HRP-avidinwas included in each experiment.Plastic Embedding and Cryosection Staining with Anti-rab5 Anti-
body—J774E clone cells were grown to confluence on 35-mm tissueculture dishes. 5 3 107 dead or live LM were added per dish andcentrifuged to synchronize the infection. Following 12 min of incubationat 37 °C, unbound bacteria were washed out with PBS, 5 mM EDTA.Cells were fixed in 1% glutaraldehyde, 0.1 M Na-cacodylate buffer andprepared for electron microscopy as described previously (16, 41). Forcryosection staining experiments, cells grown on 35-mm tissue culturedishes were incubated for 6 min with BSA-gold (10 nm), washed, andinfected with dead or live LMhly2 for 10 min, as described above. Thecells were washed, scraped off, and fixed in suspension in 2% paraform-aldehyde, 0.2% glutaraldehyde in PBS, pH 7.2, for 2 h at room temper-ature. Tissue was embedded in 10% gelatin. After pelleting the tissue,the gelatin was solidified on ice. Blocks for ultracryotomy were pre-pared and immunolabeled as described by Slot et al. (46) with thefollowing modifications: 10% goat serum was used in the blocking bufferin place of 1% BSA. Immunolabeling with primary antibody mAb 4F11
(anti-rab5) (65 mg/ml) was carried out overnight at 4 °C. Incubationwith secondary antibody (Jackson ImmunoResearch Labs., West Grove,PA) 18 nm of goat anti-mouse IgG/gold (1:15) was carried out for 1 h.Sections were stained with uranyl acetate and embedded in methylcellulose according to a modification of the Tokuyasu method (47, 48).All electron microscopy specimens were viewed and photographed usinga Zeiss 902 electron microscope.Preparation of Highly Purified Phagosomes—The phagosomal frac-
tions described above were resuspended in HBE containing 5 mM
EDTA, 1 mM phenylmethylsulfonyl fluoride, 20 mg/ml leupeptin, 20mg/ml aprotinin, and filtered through a 5-mm pore filter. The flow-through was loaded on top of a 12% sucrose cushion. Samples werecentrifuged (1,700 rpm, 45 min, 4 °C), and purified phagosomes wererecovered in the last 100 ml at the bottom of the tube. This protocol issimilar to one described previously (49).The purity of the organelles was monitored by two criteria: (i) elec-
tron microscopy observation to assess contamination with other or-ganelles and (ii) biochemical analysis to check contamination with othercellular components. Plasma membrane contamination was assayed aspreviously reported (50). Briefly, after internalization of bacteria andextensive washings, cell surface was labeled with HRP (300 mg/ml) for30 min at 4 °C. J774E clone, a mannose-receptor cell line, binds andinternalizes HRP, a mannosylated protein. Mannan inhibits both HRPbinding and uptake in these cells.3 After several washings in PBS,homogenization and phagosome isolation was performed as describedabove. HRP was then measured in the final purified phagosomes. Thegalactosyltransferase activity was checked as a marker for Golgi con-tamination using [3H]UDP-galactose (50). The endosome contaminationwas recorded by mixing an aliquot of PNS after bacterial uptake and analiquot of a PNS after 5 min of uptake of b-glucuronidase or HRP.Phagosomes were isolated as above, and endosome contamination wasmeasured as the percentage of b-glucuronidase (41), or HRP activityrecovered in the phagosomes was compared to the total activity presentin PNS containing the endosomal probe. Endosomes were marked byallowing a separate set of cells to internalize either HRP or b-glucuron-idase for 5 min. An aliquot of the PNS obtained from such cells wasincluded in the phagosome purification protocol. Recovery of 0.32 and0.25% for b-glucuronidase and HRP, respectively, from a total recoveryof 70% of the bacteria indicated low enrichment of endosomes in thepreparation.Purified phagosomes (30 mg of total protein) were analyzed by SDS-
PAGE in 15% polyacrylamide gels, transferred to nitrocellulose mem-branes, and checked for the presence of rab5 with the mAb 4F11(anti-rab5) (1:5,000); rab7 with a rabbit IgG polyclonal antibody (1:200)or NSF with the mAb 6E6 (1:500) followed by incubation with a HRP-conjugated goat anti-mouse IgG (1:10,000) or HRP-conjugated goatanti-rabbit IgG (1:5,000). Blots were developed by ECL (AmershamCorp.). Typical exposure times were always less than 2 min.Immunodepletion of Cytosolic Rab5—The assay was performed as
described previously (44). In brief, quick thawed cytosol aliquots (200ml) (5 mg protein/ml) were incubated for 4 h at 4 °C with 10 mg/ml mAb4F11 (anti-rab5) or control antibody mAb HDP-1 (anti-DNP). Bothcytosols were separately incubated (1 h, 4 °C) with protein A-Sepharosebeads (a 50% solution prepared in PBS containing 2% BSA). Aftercentrifugation (12,000 3 g, 5 min, 4 °C) supernatants were treated foranother cycle with protein A-Sepharose beads, and 50 mg of each samplewere separated by SDS-PAGE in 15% polyacrylamide gels transferredonto nitrocellulose membranes. The membranes were incubated with arabbit anti-rab5 polyclonal antibody (1:3,000) followed by incubationwith an HRP-conjugated goat anti-rabbit IgG (1:10,000). Blots weredeveloped by ECL (Amersham Corp.).
RESULTS
Reconstitution of Phagosome-Endosome Fusion with DeadLM- and Live LMhly2-containing Phagosomes—Fig. 1A shows atypical in vitro fusion experiment carried out with phagosomescontaining dead LM (filled circles) or live LMhly2 (open circles)with endosomes containing HRP-avidin. These data indicatethat phagosome fusion, using both live and dead organisms, iscytosol-dependent. The two assays were quite similar in theircytosol dependence. The average of a large number of experi-ments indicated essentially no difference between the two prep-arations. To further delineate possible influences of the live
3 G. Li and E. Peters, personal communication.
Rab5 and Phagosome-Endosome Fusion13836
by guest on May 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

organism on fusion, we performed a detailed study of the pa-rameters regulating these fusion events (Table I). GTPgS,when added in the presence of low levels of cytosol, stimulatedfusion implicating one or more GTPases as regulators of pha-gosome fusion, irrespective of whether the microorganism waslive or heat-killed. Previous work revealed the presence of rab5and rab7 on the phagosomal membranes prepared followinglatex bead ingestion (21). To test whether in vitro fusion couldbe regulated by these small GTPases, antibodies specific forrab5 and rab7 were added to the fusion reactions. Neither ofthe phagosome-endosome fusion assays was affected by anti-rab7 antibodies; however, anti-rab5 antibodies strongly inhib-ited both assays. As indicated in Table I, the presence of anti-rab5 in the fusion reactions clearly blocked phagosome-endosome fusion in both assays. The inhibition was observed atall cytosol concentrations tested (data not shown). Inhibitionwas dependent on antibody concentration, and concentrationsas low as 10 ng/ml blocked 75–90% of the fusion reaction (datanot shown). Interestingly, the fusion of live LMhly2-containingphagosomes was particularly sensitive to the rab5 antibody.Both phagosome-endosome fusion assays were temperature-
sensitive and required salt (e.g. KCl) and both cytosolic andmembrane-bound proteins. Trypsin treatment of both phago-somal and endosomal membranes impaired the fusion process.Differential Accumulation of Rab5 on Phagosomal Mem-
branes of Dead and Live LM-containing Phagosomes—Al-though only rab5 seems to regulate both phagosome-endosomefusion assays and rab7 plays no role (as reflected in the dataobtained with specific antibodies), we checked for the presenceof these proteins on the membranes of both dead LM- and liveLMhly2-containing phagosomes. Highly purified phagosomemembranes were analyzed by SDS-PAGE, and the presence ofrab5 or rab7 was detected by Western blot analysis usingspecific antibodies. Measurement of a Golgi membrane marker,galactosyltransferase activity, in this highly purified phago-some preparation detected less than 0.2% of the total (relativeto the marker present in the PNS). HRP bound to the E-clonecell surface (as a measure of cell surface marker) was alwaysless than 0.8%. Contamination of phagosome preparations withendosomes was never more than 0.35%, as measured by theb-glucuronidase or HRP activities recovered in these isolated
phagosomes (see “Materials and Methods”). As shown in Fig.2A, rab5 was detected on both types of phagosomes; however,the time course of rab5 recruitment to the membranes (7 and20 min) was significantly faster for phagosomes containing liveLMhly2. Small amounts of rab7 were found compared to rab5levels but, whereas rab7 was found on the live LMhly2 phago-somes, it was virtually absent from the dead LM phagosomes.Since differences in the enrichment of rab5 could be a conse-quence of faster fusion kinetics with phagosomes containinglive bacteria, we compared the fusion kinetics of both assays.As shown in Fig. 2B, the kinetics obtained with the live LMhly2
phagosomes revealed a much faster fusion rate with significantfusion occurring in just 5 min, while phagosomes containingdead LM required at least 30 min to reach a similar relativefusion activity. Differences in the uptake of dead or live bacte-ria cannot explain these results because the uptake rate for thetwo preparations of bacteria was the same (data shown in Fig.2C). The levels of biotinylation of dead or live bacteria weresimilar, 42 mol of ss-NHS-biotin/109 for dead LM and 41 molss-NHS-biotin/109 for live LMhly2. Moreover, the same proteinswere biotinylated in the dead and live LMhly2 preparations(data not shown) and the maximal signal recovered in thephagosomes was similar for both preparations, around 70% ofthe total signal measured in the PNS. Analysis of the numberof bacteria per phagosome compared to total numbers of bac-teria per cell (data shown in Fig. 3; panel A corresponds to cellsinfected with dead LM, and panel B with live LM) showed that,independent of the status of the bacteria (dead or live), 17% ofinternalized bacteria were in phagosomes containing singlebacteria, 26% in phagosomes containing two to three bacteria,and the remainder, 56%, in phagosomes containing four ormore bacteria. It is possible that in our phagosome purificationprocedure we may have selected those phagosomes containingone, two, or three bacteria instead of those containing morethan four that might be more sensitive to membrane disruptionduring cell lysis.The presence of rab5 in the phagosomes containing live
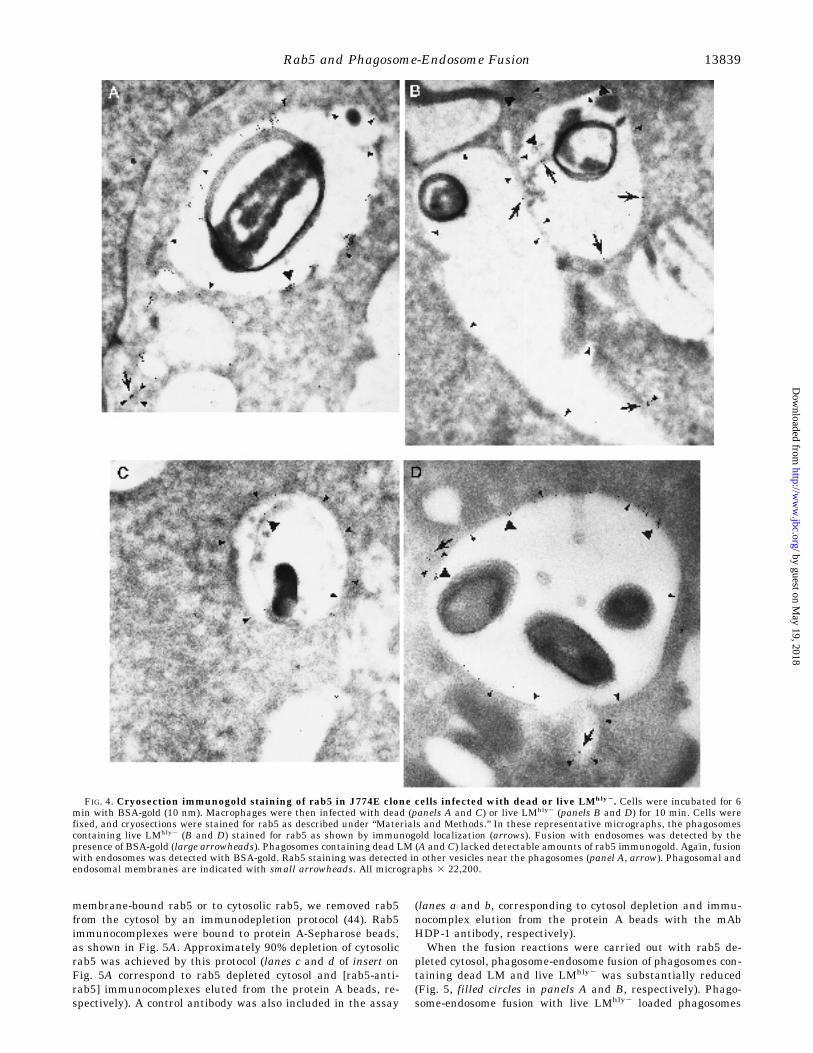
LMhly2 or dead LM was also analyzed using the cryosectionimmunogold technique. Endosomes were labeled with BSA-gold (10 nm) (indicated by large arrowheads, Fig. 4) and rab5
FIG. 1. Effect of cytosol on phagosome-endosome fusion withdead LM- and live LMhly2-containing phagosomes. Phagosome-endosome fusion reactions with phagosomes loaded with dead LM(filled circles) and with phagosomes containing live LMhly2 (open cir-cles). Phagosomal (containing biotinylated dead LM or live LMhly2) andendosomal fractions (containing HRP-avidin) were resuspended in fu-sion buffer and supplemented with different concentrations of gel-filtered cytosol. Fusion was measured as described under “Materialsand Methods.” Background values were 53 and 56, and values at thehighest cytosol concentration were 295 and 306 absorbance units/mg ofprotein for the dead and live assays, respectively.
TABLE ICharacteristics of dead and live LMhly2 phagosome/endosome fusion
Condition: reference (1mg/ml cytosol)
Relative fusion of phagosomes with:
Dead LM Live LMhly2
Controla 1.00 6 0.010 1.00 6 0.0102Cytosolb 0.00 6 0.000 0.00 6 0.000Low cytosol 1 GTPgSb 1.30 6 0.120 1.19 6 0.1302KClb 0.03 6 0.001 0.05 6 0.010Trypsin (10 mg/ml)b 0.04 6 0.002 0.06 6 0.001Anti-rab 7 (100 ng/ml)b 0.95 6 0.020 0.90 6 0.020Anti-rab 5 (100 ng/ml)b 0.25 6 0.005 0.12 6 0.007
a Phagosomes and endosomes (as in Fig. 1) were resuspended incomplete fusion buffer and 1 mg/ml cytosolic proteins, or as otherwiseindicated, and fusion was measured as described under “Materials andMethods.” Background activities were 54 and 57 and control values (1mg/ml cytosolic proteins) were 274 and 287 absorbance units/mg ofprotein for the dead and live fusion assays, respectively.
b The following conditions were tested: 2cytosol, cytosol was omitted;low cytosol 1GTPgS, cytosolic proteins at 0.125 mg/ml and 20 mM
GTPgS added to the fusion assay (values were 356 and 345 absorbanceunits/mg protein for dead and live LM phagosomes); 2KCl, the salt wasnot included in the buffer; trypsin, both sets of vesicles were incubated(1 h, 4 °C) with 10 mg/ml trypsin. To quench the excess trypsin, thesamples were incubated (30 min, 4 °C) with 20 mg/ml soybean trypsininhibitor, 1 mM phenylmethylsulfonyl fluoride, and 1 mM leupeptin;anti-rab7, polyclonal anti-rab7 antibody (0.1 mg/assay) was added to thefusion reaction (control antibody was included); anti-rab5, mAb 4F11anti-rab5 antibody (0.1 mg/assay) (as a control mAb HDP-1) was addedto the fusion reaction. Values are the mean of at least four determina-tions 6 SD.
Rab5 and Phagosome-Endosome Fusion 13837
by guest on May 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

was detected by immunogold staining (18 nm) (indicated byarrows, Fig. 4). As shown in Fig. 4, B and D, rab5 is localized inthose phagosomes containing live LMhly2. Fusion of these pha-gosomes with endosomes was detected by the presence of BSA-gold (large arrowheads). Phagosomes containing dead LM(panels A and C) had also fused with endosomes (labeled withBSA-gold and indicated by large arrowheads) but lacked sig-nificant rab5 staining, even though rab5 could be detected inother vesicles near the phagosomes (panel A, arrow). Phagoso-mal and endosomal membranes are marked with smallarrowheads.Effect of rab5-depleted Cytosol on in Vitro Phagosome-Endo-
some Fusion—To further explore the requirement for rab5 andto determine whether antibody-mediated inhibition was due to
FIG. 2. Kinetics of phagosome-endosome fusion and the accu-mulation of rab5 by dead LM and live LMhly2- containing pha-gosomes. Panel A shows a Western blot analysis of highly purifiedphagosomes containing dead LM (left) or live LMhly2 (right) after dif-ferent uptake times (7 and 20 min). 50 mg of total protein were loadedper lane. Rab5 was detected with mAb 4F11 anti-rab5 (1:5,000), fol-lowed by a HRP-conjugated goat anti-mouse (1:20,000) antibody andrab7 with a rabbit IgG polyclonal antibody (1:200) and HRP-conjugatedgoat anti-rabbit (1:5,000) antibody using ECL. Panel B shows the timecourse of phagosome-endosome fusion with dead LM-or live LMhly2-containing phagosomes. Fusion reactions were performed as describedwith a cytosol concentration of 1 mg/ml. Background values were 56 and53 and values at 1 mg/ml of cytosol concentration were 308 and 298absorbance units/mg of protein for the dead and live assays, respec-tively. Filled circles correspond to phagosomes containing live LMhly2
and open circles to phagosomes containing dead LM. Panel C shows theuptake with 35S-labeled dead LM (open circles) and 35S-labeled LMhly2
(filled circles) by macrophages (J774E clone). Radioactive bacteria (3 3105 cpm/well) were added to 2 3 106 cells (E-clone). Plates were centri-fuged (2,000 rpm, 5 min, 4 °C) and then incubated at 37 °C for differenttimes. At each final time point, cells were lysed with 1% Triton X-100,and radioactivity was measured in a b-counter. Data represent themean of triplicate determinations.
FIG. 3. Electron microscopy observations showing the hetero-geneity of phagosomes containing dead or live LM. J774E clonecells were infected with dead (panel A) or live LM (panel B) for 12 minas described under “Materials and Methods.” Small arrowheads, pha-gosomes containing single bacteria; small arrows, phagosomes contain-ing three bacteria; large arrowheads, phagosomes containing multiplebacteria; and large arrows, phagosomes containing multiple bacteriaconnected to other phagosomes. Bar represents 0.89 mm in panel A and1.7 mm in panel B.
Rab5 and Phagosome-Endosome Fusion13838
by guest on May 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from
membrane-bound rab5 or to cytosolic rab5, we removed rab5from the cytosol by an immunodepletion protocol (44). Rab5immunocomplexes were bound to protein A-Sepharose beads,as shown in Fig. 5A. Approximately 90% depletion of cytosolicrab5 was achieved by this protocol (lanes c and d of insert onFig. 5A correspond to rab5 depleted cytosol and [rab5-anti-rab5] immunocomplexes eluted from the protein A beads, re-spectively). A control antibody was also included in the assay
(lanes a and b, corresponding to cytosol depletion and immu-nocomplex elution from the protein A beads with the mAbHDP-1 antibody, respectively).When the fusion reactions were carried out with rab5 de-
pleted cytosol, phagosome-endosome fusion of phagosomes con-taining dead LM and live LMhly2 was substantially reduced(Fig. 5, filled circles in panels A and B, respectively). Phago-some-endosome fusion with live LMhly2 loaded phagosomes
FIG. 4. Cryosection immunogold staining of rab5 in J774E clone cells infected with dead or live LMhly2. Cells were incubated for 6min with BSA-gold (10 nm). Macrophages were then infected with dead (panels A and C) or live LMhly2 (panels B and D) for 10 min. Cells werefixed, and cryosections were stained for rab5 as described under “Materials and Methods.” In these representative micrographs, the phagosomescontaining live LMhly2 (B and D) stained for rab5 as shown by immunogold localization (arrows). Fusion with endosomes was detected by thepresence of BSA-gold (large arrowheads). Phagosomes containing dead LM (A and C) lacked detectable amounts of rab5 immunogold. Again, fusionwith endosomes was detected with BSA-gold. Rab5 staining was detected in other vesicles near the phagosomes (panel A, arrow). Phagosomal andendosomal membranes are indicated with small arrowheads. All micrographs 3 22,200.
Rab5 and Phagosome-Endosome Fusion 13839
by guest on May 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

was more severely retarded by rab5 depletion than with deadLM containing phagosomes. Addition of exogenous prenylatedGST-rab5 fusion protein restored the activity of this rab5 de-pleted cytosol in both assays (filled squares), suggesting thatthe effect of immunodepletion on fusion was totally dependenton rab5. In some experiments the restorative action of rab5 wasblocked by the addition of rab5 antibody (data not shown).GDI Extraction of Rab5 Bound to Phagosomal Membrane:
Effects on Phagosome-Endosome Fusion—GDI is known to bindto GDP forms of rab proteins putatively blocking GDP/GTPexchange and causing an accumulation of rab proteins in thecytosol in an inactive form (51, 52). To further analyze the roleof cytosolic rab5 in mediating phagosome-endosome fusion, weadded purified GDI to the assays. This protocol has been pre-
viously shown (44) to inhibit transport in reactions that requirerecruitment of prenylated rab proteins from cytosolic GDI com-plexes. As shown in Fig. 5C, fusion was dramatically blocked bypurified GDI (160 mg/ml) in both assays, indicating that cyto-solic rab5 is recruited to the membranes. When differentamounts of GDI were included in the assay, concentrations aslow as 16 mg/ml blocked more than 50% of the fusion (Fig. 5D).The role of membrane bound rab5 on phagosome-endosomefusion was studied by treating phagosomal membranes withGDI which removes GDP-rab5 (43, 44, 51). One reason toextract rab5 from the membranes was to determine whethermembrane bound rab5 provided some residual stimulus tofusion since phagosomes containing dead LM continue to fuseat a slow rate after depletion of cytosolic rab5 (Fig. 5A). Asshown in Fig. 6A, GDI treatment extracts GDP-rab5 from the
FIG. 5. Rab5 immunodepleted cytosol does not support phago-some-endosome fusion. Phagosomes and endosomes (as in Fig. 1)were resuspended in fusion buffer containing rab5-immunodepleted orcontrol cytosols: mAb 4F11 anti-rab5-treated (filled circles) or mAbHDP-1 anti-DNP treated (open circles) cytosols, respectively, as de-scribed under “Materials and Methods.” In some cases, fusion reactionsperformed with rab5-depleted cytosol were supplemented with preny-lated GST-rab5 fusion protein (30 ng/ml) (filled squares). Panel A showsphagosome-endosome fusion of dead LM phagosomes. Background was79 and the highest cytosol value was 348 absorbance units/mg of pro-tein. Panel B shows fusion with phagosomes containing live LMhly2.Background was 73 and the highest cytosol concentration value was346 absorbance units/mg of protein. Inset in panel A confirms thecytosolic depletion of rab5 by Western blot analysis. 50 mg of proteinwere loaded per lane. Nitrocellulose membranes were incubated with apolyclonal rabbit anti-rab5 (1:3000) followed by a HRP-conjugated goatanti-rabbit (1:5000) antibody and detected by ECL. Lane a, cytosolicrab5 after treatment with an irrelevant mAb HDP-1 anti-DNP anti-body; lane b, rab5 eluted from the protein A-Sepharose after incubationwith mAb HDP-1 and adsorption to the beads; lane c, cytosolic rab5after treatment with the mAb 4F11 anti-rab5; and lane d, rab5 elutedfrom protein A-Sepharose after treatment with the mAb 4F11 anti-rab5followed by adsorption onto the beads. Panel C fusion reactions ofcytosol pretreated with purified GDI (160 mg/ml) for 30 min at 4 °Cbefore addition of phagocytic and endocytic vesicles. Cytosol concentra-tion was 1 mg/ml. Background values were 56 and 53 and values for 1mg/ml cytosol concentration were 329 and 332 absorbance units/mg ofprotein for dead and live LM, respectively. Panel D shows fusion reac-tions of cytosol pretreated with different amounts of GDI (1.6–160mg/ml) as in panel C. Background and 1 mg/ml cytosol concentrationvalues were the same as in panel C. Values were the mean of threedeterminations 6 S.D.
FIG. 6.Rab5 and phagosome-endosome fusion: effect of GDI onmembrane bound rab5. The Western blot in panel A shows thepresence of rab5 in phagosomes (containing dead or live LM) incubatedwith (1GDI) or without (2GDI) purified GDI (6 mg/sample) for 20 minat 30 °C under different conditions: in the presence of GTPgS (lanes 1and 4) or GDP (1 mM) (lanes 2 and 5) or with HBE buffer (lanes 3 and6). Panel B shows phagosome-endosome fusion experiments performedwith GDI-treated phagosomes (containing dead LM or live LMhly2)(vesicles treated with GDI in the presence of 1 mM GDP) and untreatedendosomes (containing HRP-avidin). Fusion reactions were carried outin fusion buffer in the presence of untreated cytosol (1 mg/ml) or cytosoldepleted of rab5 (immunodepletion described under “Materials andMethods”). Values are the mean of three determinations 6 S.D. Back-ground values were 80 and 70 and values for 1 mg/ml cytosol concen-tration were 380 and 357 absorbance units/mg of protein for dead andlive fusion reactions, respectively. Panel C shows rab5 stimulation ofphagosome-endosome fusion without GTP hydrolysis. Phagosomes andendosomes (as in Fig. 1) were resuspended in fusion buffer containing0.125 mg/ml cytosol in the presence or absence of different GST-rab5fusion proteins. Minus symbols (2) indicate the absence of GST fusionproteins: rab5:S34N, a dominant negative mutant or rab5:Q79L, amutant defective in GTP hydrolysis (30 ng/ml). Data are representativeof four independent experiments. Solid bars correspond to fusion reac-tions using dead LM containing phagosomes and striped bars corre-spond to live LMhly2 containing phagosomes. Background values were69 and 61 and values for 0.125 mg/ml cytosol concentration were 177and 163 absorbance units/mg of protein for dead and live LM phago-somes, respectively. Panel D indicates the copurification of both rab5fusion proteins with rab escort protein, REP-1 (GGTPase).
Rab5 and Phagosome-Endosome Fusion13840
by guest on May 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

phagosomal membranes. As a control, rab5 was locked in theGTP-bound form by the addition of GTPgS. Under these con-ditions, rab5 was not removed from the membranes. Fusionwas readily measured after rab5 removal from the membranesin the presence of normal cytosol. As indicated above andconfirmed in Fig. 6B, live LMhly2 phagosome-endosome fusionwas completely blocked in the presence of rab5 depleted cytosolwhereas dead LM phagosome-endosome fusion was still meas-urable. To further confirm the action of rab5 in supportingphagosome-endosome fusion, two different prenylated GST-rab5 fusion proteins were added to the in vitro phagosome-endosome fusion assays, the active mutant rab5:Q79L and thedominant negative mutant rab5:S34N. As shown in Fig. 6C,fusion was markedly activated by exogenous GST-rab5:Q79L, amutant defective in GTP hydrolysis and inhibited by GST-rab5:S34N, a dominant negative mutant which is unable to ex-change GDP for GTP. The specificity of rab5:Q79L activation ofphagosome-endosome fusion was checked by including mAb4F11 anti-rab5 in the fusion reaction. mAb 4F11 produced adramatic reversal of the rab5 effect (from 0.88 unit of relativefusion to 0.10 unit in the presence of mAb 4F11 anti-rab5). Thesolubility and purification of the rab5 fusion proteins (de-scribed under “Materials and Methods”) was apparently as-sured by the presence of REP-1, the rab escort protein thatcopurifies in our rab5 preparations (Fig. 6D, lane 1, corre-sponds to purified rab5:Q79L and lane 2 to rab5:S34N).Differences between Dead and Live LMhly2 in Phagosome-
Endosome Fusion—Analysis of the energy requirements of
both assays (Table II) indicate that fusion of dead LM phago-somes with endosomes was sensitive to ATP depletion, whereasfusion with the live LMhly2 phagosomes was not. The liveLMhly2 fusion assay was also insensitive to deoxyglucose treat-ment. Another curious finding is that live LMhly2 phagosome-endosome fusion assay was insensitive to NEM and to anti-NSF antibodies whereas dead LM phagosome-endosomefusions were highly sensitive. Mild washing of phagosomalmembranes with salt (0.5 M KCl) restored sensitivity to ATPdepletion and also restored the NEM and anti-NSF (data notshown) sensitivity. Addition of cytosol in which endogenousNSF was inactivated by prior incubation at 37 °C was unable torestore fusion in salt-washed vesicles. However, addition ofNSF in the presence of inactivated cytosol restored fusion withboth preparations. These findings indicate that, with respect tofusion, salt-washed live LMhly2 phagosomes behave like deadLM phagosomes and phagosomes containing S. aureus parti-cles (39). Therefore, we looked for NSF in both dead and liveLMhly2 phagosomes by Western blotting. As shown in Fig. 7A,NSF appeared to be enriched on phagosomal membranes con-taining live LMhly2, whereas the levels present on dead LMphagosomal membranes was extremely low, often undectable.Mild salt treatment of these phagosomes (0.5 M KCl) removedNSF from the phagosomes containing live LMhly2, but onlyslightly changed levels in dead LM phagosomes. Analysis ofrab5 in the same salt-washed phagosome membranes indicatedno effect of salt treatment on rab5 levels. Thus, recovery ofalmost total fusion activity (around 80%) with purified NSF(His6-NSFmyc) on these salt-washed phagosomes points toNSF as the NEM and ATP-sensitive factor removed by thisprocedure. Interestingly, when NSF was analyzed in thosephagosomal membranes treated with GDI, a surprising findingwas obtained. The amount of NSF on dead LM phagosomalmembranes was not affected by the absence of rab5, whereasNSF associated with live LM phagosomal membranes was sub-stantially reduced by prior GDI treatment (Fig. 7B). These data
FIG. 7. Relationship between NSF and rab5 present on themembranes of dead and live Lmhly2 phagosomes. Panel A showsthe effect of KCl washing on NSF and rab5 present on the phagosomalmembranes. Dead LM- or live LMhly2-containing phagosomes wereincubated (1) or not incubated (2) with 0.5 M KCl as described in TableII. Phagosomes were solubilized in SDS buffer, and 50 mg of proteinwere loaded per lane. Following transfer, nitrocellulose membraneswere incubated with monoclonal anti-NSF (mAb 6E6) (1:500) or withmAb 4F11 anti-rab5 (1:5,000), followed by a HRP-conjugated goat anti-mouse (1:10,000) antibody and detected by ECL. Panel B shows aWestern blot of NSF revealing the effect of GDI treatment of phagoso-mal membrane associated NSF. Purified phagosomes were treated withGDI as in Fig. 6A. The phagosomes were sedimented, resuspended inSDS buffer, and following SDS-PAGE, transferred to nitrocellulose.NSF was detected with mAb 6E6 (1:500) followed by incubation with aHRP-conjugated goat anti-mouse (1:10,000) antibody using ECL.
TABLE IIDifferences between dead and live LMhly2 in
phagosome/endosome fusion
Condition: reference(1 mg/ml cytosol)
Relative fusion of phagosomes with:
Dead LM Live LMhly2
Controla 1.00 6 0.010 1.00 6 0.0102ATPb 0.05 6 0.002 0.85 6 0.020NEM (3 mM)b 0.18 6 0.010 0.98 6 0.010Anti-NSF (100 ng/ml)b 0.07 6 0.001 0.97 6 0.0022-DO-gluc (1 mM)b 0.18 6 0.020 1.00 6 0.0100.5 M KCl wash (1RS)c 0.88 6 0.010 0.87 6 0.0250.5 M KCl wash (1DS)d 0.08 6 0.003 0.20 6 0.0100.5 M KCl wash (1NEM)e 0.01 6 0.005 0.01 6 0.0020.5 M KCl wash (1hiCYT) f 0.03 6 0.005 0.05 6 0.0050.5 M KCl wash (1NSF)g 0.80 6 0.025 0.84 6 0.042
a Phagosomes and endosomes (as in Fig. 1) were resuspended incomplete fusion buffer supplemented with cytosolic proteins (1 mg/ml).Background activities were 73 and 70 and control values were 403 and364 absorbance units/mg of protein for dead and live LM, respectively.
b The following conditions were tested: 2ATP, and ATP-depletingsystem (5 mM glucose, 25 units/ml hexokinase) was substituted for theATP-regenerating system; NEM, vesicles and cytosol in fusion bufferwere incubated (30 min, 4 °C) with 3 mM NEM, and excess NEM wasquenched with 3 mM dithiothreitol before fusion; anti-NSF IgM, mAb4A6 (0.1 mg/assay) was added to the fusion reaction (an irrelevant IgMmAb antibody was used as control); 2-DO-gluc, 2-deoxyglucose at 1 mM
was included in the fusion assay. Values are the mean of at least fourdeterminations 6 S.D.
c Phagosomes were incubated with 0.5 M KCl (4 °C, 10 min). Thevesicles were then sedimented to remove excess salt and resuspended incomplete cytosol with RS (ATP regenerating system). KCl wash re-sulted in an approximately 20% reduction in fusion activity. Valueswere 351 and 328 absorbance units/mg of protein for the dead and livefusion assays, respectively.
d Fusion of salt-washed phagosomes in complete cytosol with an ATPdepleting system (DS).
e Fusion of salt-washed phagosomes in complete cytosol plus 3 mM
NEM.f Fusion of salt-washed phagosomes in heat-inactivated cytosol (hi
CYT) (15 min, 37 °C to inactivate NSF) (54) (hi CYT (1 mg/ml)). Valueswere 80 and 85 OD units/mg protein.
g NSF (100 ng/ml) was added to the membranes in the presence ofheat-inactivated cytosol. Values were 303 and 305 absorbance units/mgprotein for the dead LM and live LM, respectively.
Rab5 and Phagosome-Endosome Fusion 13841
by guest on May 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

reflect a relationship between rab5 and NSF present on pha-gosomal membranes. Removal of rab5 from the membranesaffects the membrane-association of NSF. However, removal ofNSF from the membranes does not appear to affect the mem-brane association of rab5. These data suggest that rab5 regu-lates the binding of NSF to the phagosomal membranes.
DISCUSSION
Upon internalization by phagocytes, L. monocytogenes pha-gosomes have been shown to fuse with lysosomes (10, 11);however, neither the fusion with endosomes nor its regulationhave been investigated. Work from our laboratory has shownthat early phagosome-endosome fusion events can be reconsti-tuted in vitro (12, 39). Here we have developed a similar assayusing biotinylated-bacteria and HRP-conjugated with avidin.Binding of the endosome probe (HRP-avidin) to the biotiny-lated bacteria (contained within phagosomes) was used as ameasure of phagosome-endosome fusion.Our goal was to address the question of whether live micro-
organisms regulate or modulate phagosome-endosome fusionevents. In these studies, we have taken advantage of LMhly2, amutant deficient in hemolysin which remains inside thephagolysosome. Thus, both dead and live organisms used inthis study are ultimately degraded by the host cell. An exten-sive analysis of the parameters regulating in vitro fusion be-tween phagosomes containing dead LM or live LMhly2 withendosomes revealed a dependence on cytosolic proteins, salts(i.e. KCl), and membrane-bound proteins irrespective ofwhether live or dead LM were used. GTPgS stimulated fusionin both assays implicating multiple GTPases, as previouslyreported for endocytosis (16, 31, 53) and phagocytosis (39, 54).GTP-binding proteins such as rab5 and rab7 have been lo-
calized to phagosomal membranes (21). Using specific antibod-ies, our studies indicate that rab5 regulates early phagosome-endosome fusion events. Rab7, while present, appears not toplay a role in these events. The inhibitory effect of anti-rab5antibody was dependent on the concentration of cytosol andantibody. Phagosome-endosome fusion with dead LM-loadedphagosomes was consistently less sensitive to antibody inhibi-tion than fusion with the live LMhly2 phagosomes.To explore more fully the effects of the antibody, we prepared
purified dead LM or live LMhly2 phagosomes and checked forthe presence of rab5. Both preparations contained rab5, butrab5 was enriched on live LMhly2 phagosomal membranes.Interestingly, rab7 was found only on live LMhly2 phagosomes.However, since specific antibodies were without effect rab7appears to play no role in the phagosome-endosome fusion. Afaster fusion rate was observed with live LMhly2-loaded pha-gosomes compared with dead LM phagosomes, although themaximum amount of fusion observed was similar. Substantialphagosome-endosome fusion occurred within 5–10 min whenlive bacteria loaded phagosomes were used while the phago-somes containing dead bacteria required 25–30 min to obtain asimilar relative fusion. These differential rates in the fusionkinetics cannot be explained by (i) differences in uptake, (ii) bydifferential biotinylation of the bacterial proteins, or (iii) bydifferent numbers of bacteria per phagosome, since these pa-rameters were very similar. To further explore the role of rab5,we depleted rab5 from the cytosol (44). Rab5 depleted cytosolwas unable to support fusion of live LMhly2-loaded phago-somes. Dead LM-loaded phagosomes fused poorly after rab5depletion. Total recovery of fusion was achieved by addingGST-rab5 to the rab5-depleted cytosol. Another approach uti-lized GDI. GDI binds rab proteins in the GDP state and therebyextracts them from membranes (35, 44, 52). Addition of GDIblocked phagosome fusion with both preparations, althoughmore inhibition was observed with the live LMhly2-loaded pha-
gosomes. Consistent with earlier results, addition of rab5 in theGTP-bound form was active in promoting fusion, and GTPhydrolysis was not required. Moreover, the dominant negativemutant of rab5 (rab5:S34N) inhibited both phagosome-endo-some fusion assays. These findings indicate that rab5 is re-quired for phagosome-endosome fusion, but that the live prep-aration is consistently more sensitive to the absence of rab5.The remaining fusion found in dead LM-loaded phagosomes inthe absence of cytosolic rab5 could be due the presence ofmembrane-bound rab5. To resolve this point, we removedmembrane-bound rab5 from dead LM phagosomes by GDItreatment. Even in the absence of detectable membrane boundrab5 and rab5-depleted cytosol, dead LM phagosomes were stillmarginally active in the fusion assay while live LMhly2 phago-somes were completely inactive. The mechanism of rab5-inde-pendent phagosome-endosome fusion with dead LM phago-somes remains to be explored.The energy requirements for the two phagosome-endosome
fusion assays are quite different. Similar to earlier resultsusing S. aureus (39, 54), phagosome-endosome fusion with thedead LM preparation was sensitive to ATP depletion. However,fusion of endosomes with phagosomes loaded with live LMhly2
was insensitive to ATP depletion. It is possible that the sourceof ATP in the two fusion assays might be different, e.g. the liveorganism may generate ATP. Alternatively, ATP-sensitive fac-tors indispensable for docking and fusion may have alreadycarried out their function or the need for certain components(e.g. NSF) may have been bypassed. We favor the latter possi-bility supported by the following findings: (i) a lack of sensitiv-ity to NEM and anti-NSF antibodies, (ii) presence of NSF onphagosomes containing live LMhly2 and its virtual absence ondead LM phagosomes, (iii) a mild salt treatment of live LMhly2
phagosomes removes NSF and renders the fusion events sen-sitive to ATP and to NEM treatment, and (iv) fusion withsalt-washed vesicles was restored by the addition of recombi-nant NSF to the assay in the presence of heat-inactivatedcytosol. This is consistent with the findings of Rodriguez et al.(55), who found that fusion in salt-washed endosomes was fullyrecovered by NSF. When the relationship between membrane-bound NSF and rab5 was examined, differences between thelive and dead LM preparations emerged. NSF was removedfrom both preparations with a mild salt wash, a procedure thatdid not affect rab5 associated with the membranes. However,following removal of rab5 with GDI, binding of NSF to live LMphagosomal membranes was lost, while the low levels of NSFassociated with dead LM preparations were unaffected. Thesedata suggest that the enhanced binding of NSF to live LMphagosomal membranes is coupled to rab5 or some other un-known rab. Why does NSF accumulate on the membranes ofLMhly2 phagosomes? It is possible that the point in the tempo-ral sequence of events where NSF acts has already beenpassed, and NSF is no longer needed. Similar conclusions havebeen drawn from experiments with Golgi membranes using anin vitro transport assay (56). It is also possible that other fusionfactors are active here as suggested by Simons and colleagues(57), and the accumulation of NSF is secondary and perhaps, asindicated below, simply a consequence of rab5 accumulation onthe LMhly2 membranes. However, at this point it is not possibleto delineate the role played by NSF in live LM phagosome-endosome fusion.What is the mechanism by which rab5 is recruited to the
phagosomal membrane and why are phagosomes containinglive LMhly2 so much more active in rab5 and NSF recruitment?The mechanism may be related to the guanine nucleotide ex-change rate and the nucleotide status of rab5. The nucleotidestatus of rab5 would affect its membrane accumulation, since
Rab5 and Phagosome-Endosome Fusion13842
by guest on May 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

hydrolysis of GTP triggers release of rab5 to the cytosol andsubsequent binding to GDI. Accordingly, the live organismmayinhibit a rab5 GAP activity, resulting in an accumulation ofrab5:GTP on the membrane. Alternatively, rab5 may hydrolyzeGTP and remain on the membrane as a complex with otherdownstream proteins which in turn may lock NSF on the mem-branes. What could be the reason why LM drives binding ofrab5 and NSF onto the membranes? First, it is clear from ourresults that cytosolic rab5 is required for fusion of live and deadphagosomes. Thus, rab5, and possibly NSF, accumulated onthe membranes may not be functional. A more reasonablepossibility is that the presence of rab5 and NSF on the mem-branes prevents maturation of the phagosome. Perhaps, thesequential binding and dissociation of rab5 precedes in anobligatory way, the binding of other factors that are necessaryfor fusion with elements of the lysosomal compartment. Thus,the live organism may extend its lifetime in the endosomalcompartment from whence it can translocate into the cytosoliccompartment. It might be speculated that the microorganismhas a requirement to access the endocytic compartment of thehost cell, thereby providing maximal surface area available ina relatively nonlethal intracellular compartment from whichtranslocation to the cytosol can occur. LM phagosomes contain-ing live bacteria may be more immature than their counter-parts containing dead bacteria. Thus fusion would be up-regu-lated because maturation was delayed in the phagosomescontaining live Lmhly2. That would be in agreement with thefaster kinetics of their fusion with endosomes and with themore avid binding of rab5. Phagosomes containing dead LMmay have matured faster; they have less avidity for rab5 andfuse more slowly with endosomes. In summary, our resultswith rab5 represent the first documentation of an active par-ticipation of a live microorganism on membrane traffickingalong the phagocytic pathway. The bacteria appear to modulatethe function of rab5 putatively by affecting nucleotide ex-change. Clearly more work is needed to delineate the biochem-ical mechanisms involved.
Acknowledgments—We thank D. A. Portnoy for kindly providing uswith the L. monocytogenes strains, B. Goud for purified GDI, and S. W.Whiteheart and J. E. Rothman for purified NSF and anti-NSF antibod-ies. We also thank M. A. Levy for her excellent technical support withthe cryosection immunostaining experiments, L. La Rose for her assist-ance with electron microscopy preparations, C. Adles for the tissueculture help, and E. Peters for antibody purification and technicalsupport. We are grateful to E. R. Unanue, E. A. Groisman, M. Rabino-vitch, L. S. Mayorga, and M. I. Colombo for critically reading themanuscript and for their helpful suggestions.
REFERENCES
1. Gray, M. L., and Killinger, A. H. (1966) Bacteriol. Rev. 30, 309–3822. Kuhn, M., Kathariou, S., and Goebel, W. (1988) Infect. Immun. 56, 79–823. Weinberg, D. S., and Unanue, E. R. (1981) J. Immunol. 126, 794–7994. Drevets, D. A., and Campbell, P. A. (1991) Infect. Immun. 59, 2645–26525. Alvarez-Dominguez, C., Carrasco-Marin, E., and Leyva-Cobian, F. (1993) In-
fect. Immun. 61, 3664–36726. Mengaud, J., Ohayon, H., Gounon, P., Mege, R-M., and Cossart, P. (1996) Cell
84, 923–9327. Bouvier, G., Benoiel, A. M., Foa, C., and Bongrand, P. (1994) J. Leukocyte Biol.
55, 729–7348. Galliard, J-L., Berche, P., Mounier, J., Richard, S., and Sansonetti, P. (1987)
Infect. Immun. 55, 2822–28299. Tilney, L. G., and Portnoy, D. A. (1989) J. Cell Biol. 109, 1597–160810. Harding, C. V., and Geuze, H. J. (1992) J. Cell Biol. 119, 531–542
11. de Chastellier C., and Berche P. (1994) Infect. Immun. 62, 543–55312. Pitt, A., Mayorga, L. S., Schwartz A. L., and Stahl P. D. (1992) J. Biol. Chem.
267, 126–13213. Portnoy, D. A., Jacks, P. S., and Hinrichs, D. (1988) J. Exp. Med. 167,
1459–147114. Cossart, P., Vicente, M. F., Mengaud J., Baquero, F., Perez-Diaz, J. C., and
Berche, P. (1989) Infect. Immun. 57, 3629–363615. Jones, S., and Portnoy, D. A. (1994) Infect. Immun. 62, 5608–561316. Mayorga, L. S., Diaz, R., Colombo, M. I., and Stahl, P. D. (1989) Cell Regul. 1,
113–12417. Goud, B., and McCaffrey M. (1991) Curr. Opin. Cell Biol. 5, 626–63318. Bomsel, M., and Mostov, K. (1992) Mol. Biol. Cell 3, 1317–132819. Gruenberg, J., and Clague, M. J. (1992) Curr. Opin. Cell Biol. 4, 593–59920. Zerial, M., and Stenmark, H. (1993) Curr. Opin. Cell Biol. 5, 613–62021. Desjardins, M., Huber, H., Parton, R. G., and Griffiths G. (1994) J. Cell Biol.
124, 677–68822. Qiu, Y., Xu, X., Wandinger-Ness, A., Dalke, D. P., and Pierce, S. (1994) J. Cell
Biol. 125, 595–60523. Otsuka, F. L., Welch, M. J., McElvany, K. D., Nicolotti, R. A., and Fleichman,
J. B. (1984) J. Nucl. Med. 25, 1343–134924. Whiteheart, S. W., Rossnagel, K., Buhrow, S. A., Brunner, M., Jaenicke, R.,
and Rothman, J. E. (1994) J. Cell Biol. 126, 945–95425. Bradford, M. M. (1976) Anal. Biochem. 72, 248–25626. Sasaki, T., Kikuchi, A., Araki, S., Hata, Y., Isomura, M., Kuroda, S., and Takai,
Y. (1990) J. Biol. Chem. 265, 2333–233727. Li, G., and Stahl, P. D. (1993) J. Biol. Chem. 268, 24475–2448028. Li, G., Barbieri, M. A., Colombo, M. I., and Stahl P. D. (1994) J. Biol. Chem.
269, 14631–1463529. Seabra, M. C., Goldstein, J. L., Sudhof, T. C., and Brown, M. S. (1992) J. Biol.
Chem. 267, 14497–1450330. Seabra, M. C., Reiss, Y., Casey, P. J., Brown, M. S., and Goldstein, J. L. (1991)
Cell 65, 429–43431. Barbieri, M. A., Li, G., Colombo, M. I., and Stahl P. D. (1994) J. Biol. Chem.
269, 18720–1872232. Shirataki, H., Kaibuchi, K., Yamaguchi, T., Wada, K., Horiuchi, H., and Takai,
Y. (1992) J. Biol. Chem. 267, 10946–1094933. Andres, D. A., Seabra, M. C., Brown, M. S., Armstrong, S. A., Smeland, T. E.,
Cremers, F. P., and Goldstein, S. L. (1993) Cell 73, 1091–109934. Alexandrov, K., Horiuchi, H., Steele-Mortimer, O., Seabra, M. C., Zerial, M.
(1994) EMBO J. 13, 5262–527335. Dirac-Svejstrup, A. B., Soldati, T., Shapiro, A. D., and Pfeffer, S. R. (1994)
J. Biol. Chem. 269, 15427–1543036. Kocks, C., Gouin, E., Tabouret, M., Berche, P., Ohayon, H., and Cossart, P.
(1992) Cell 68, 521–53137. Ziegler, K., and Unanue, E. R. (1981) J. Immunol. 127, 1869–187538. Cluff, C. W., Garcia, M., and Ziegler, H. K. (1990) Infect. Immun. 58,
3601–361239. Mayorga, L. S., Bertini, F., and Stahl, P. D. (1991) J. Biol. Chem. 266,
6511–651740. Pitt, A., Mayorga, L. S., Stahl, P. D., and Schwartz, A. L. (1992) J. Clin. Invest.
90, 1978–198341. Diaz, R., Mayorga, L., and Stahl, P. (1988) J. Biol. Chem. 263, 6093–610042. Lang, T., and de Chastellier, C. (1985) Biol. Cell 53, 149–15443. Garret, M. D., Zahner, J. E., Cheney, C. M., and Novick, P. J. (1994) EMBO J.
13, 1718–172844. Soldati, T., Rieder, M. A., and Pfeffer, S. R. (1993) Mol. Biol. Cell 4, 425–43445. Gruenberg, J., Griffiths, G., and Howell, K. E. (1989) J. Cell Biol. 108,
1301–131646. Slot, J. W., Geuze, H. J., Gigengack, S., Lienhard, G. E., and James, D. E.
(1991) J. Cell Biol. 113, 123–13547. Tokuyasu, K. T. (1980) Histochem. J. 12, 381–40348. Griffiths, G., McDowall, A., Back, R., and Dubochet, J. (1984) J. Ultrastruct.
Res. 89, 65–7849. Sturgill-Koszycki, S., Schlesinger, P. H., Chakraborty, P., Haddix, P. L., Col-
lins, H. L., Fok, A. K., Allen, R. D., Gluck, S. L., Heuser, J., and Russell, D.G. (1994) Science 263, 678–681
50. Desjardins, M., Celis, J.-E., Van Meer, G., Dieplinger, H., Jahraus, A., Grif-fiths, G., and Huber, L. A. (1994) J. Biol. Chem. 269, 32194–32200
51. Elazar Z., Mayer, T., and Rothman, J. E. (1994) J. Biol. Chem. 269, 794–79752. Ullrich, O., Horiuchi, H., Bucci, C., and Zerial, M. (1994) Nature 368, 157–16953. Mayorga, L. S., Diaz, R., and Stahl, P. D. (1989) Science 44, 1475–147754. Beron, W., Colombo, M. I., Mayorga, L., and Stahl, P. D. (1995) Arch. Biochem.
Biophys. 317, 337–34255. Rodriguez, L., Stirling, C. J., and Woodman, P. G. (1994) Mol. Biol. Cell 5,
773–78356. Wattenberg, B. W., Raub, T. J., Hiebsch R. R., and Weidman, P. J. (1992) J.
Cell Biol. 118, 1321–133257. Ikonen, E., Tagaya, M., Ullrich, O., Montecucco, C., and Simons, K. (1995) Cell
81, 571–580
Rab5 and Phagosome-Endosome Fusion 13843
by guest on May 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Wandinger-Ness and Philip D. StahlCarmen Alvarez-Dominguez, Alejandro M. Barbieri, Walter Berón, Angela
Phagosome-Endosome Fusionin Vitro Influences Rab5-regulated Listeria monocytogenesPhagocytosed Live
doi: 10.1074/jbc.271.23.138341996, 271:13834-13843.J. Biol. Chem.
http://www.jbc.org/content/271/23/13834Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/271/23/13834.full.html#ref-list-1
This article cites 57 references, 38 of which can be accessed free at
by guest on May 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from