virtual screening of anti-histamine drugs to hhr1 receptor...
TRANSCRIPT

Journal of Pharmacy Research Vol.5 Issue 5.May 2012
Kranthi Gattu et al. / Journal of Pharmacy Research 2012,5(5),2852-2860
2852-2860
Research ArticleISSN: 0974-6943
Available online throughhttp://jprsolutions.info
*Corresponding author.Kranthi GattuCollege of Pharmacy,Sri Krishnadevaraya University,Anantapur, Andhra Pradesh, India
Virtual screening of anti-histamine drugs to hhr1 receptor by molecular modelling and docking studies
Kranthi Gattu *1 ,Deepika Godugu1, Kavitha G2, Nandini G3
*1 College of Pharmacy, Sri Krishnadevaraya University, Anantapur, Andhra Pradesh, India2 Department of Bio-chemistry, Osmania University, Hyderabad, Andhra Pradesh.
3 Department of Pharmaceutical Chemistry, Osmania University College for Women, Hyderabad, Andhra Pradesh, India
Received on:11-01-2012; Revised on: 17-02-2012; Accepted on:19-04-2012
ABSTRACTAs a part of our search for novel histamine H1 receptor antagonist, we elected HHR1 protein, involved in a variety of physiological actions such asinflammation, gastric acid secretion, neurotransmitter release and mast cell mediated chemotaxis upon binding to the biogenic amine called histamine. In thiswork, we have constructed a 3D model of (P35367) domain, using the SWISS-Model method and obtained a refined model after energy minimization. Thefinal refined model was further assessed by Prosa and PROCHECK program, and the results show that this model is reliable. The stable structure of P35367is further used for docking with the 31 ligand molecule of Histamine H1 antagonist. Docking results indicate that conserved amino-acid residues HumanHHR1 main play an important role in maintaining a functional conformation and are directly involved in donor substrate binding. The interaction betweenthe domain and the ligands proposed in this study are useful for understanding the potential mechanism of domain and the inhibitor binding. As is wellknown, hydrogen bonds play important role for the structure and function of biological molecules. In this study it was found that, TYR 98, LEU 147, TRP148, VAL 149, ILE 150, PRO 151, ILE 152, TRP 155, PHE 180, MET 183, THR 184, and ILE 187 are important for strong hydrogen bonding interactionwith the inhibitors. To the best of our knowledge PHE 180, MET 183 and ILE 187 is conserved in this domain and may be important for structural integrityor maintaining the hydrophobicity of the inhibitor-binding pocket. The molecule Meclozine showed best docking results with target protein. Our resultsmay be results may be helpful for further experimental investigations.
Keywords: Docking Studies, Histamine antagonist, HHR1, Modeling.
INTRODUCTIONAntihistamines are a class of pharmaceutical drugs which antagonize thehistamine effects by competitive inhibition of histamine receptors. Theyhave an affinity for a specific receptor. The histamine receptors have beenclassified into subtypes H1, H2 and more recently, H3. The major allergicresponses are mediated through the H1 receptor and H2 effects include esoph-ageal contraction, gastric acid secretion and increased lower airway secretion.The main function of the H3 receptor seems to be to turn off histaminesecretion but its exact physiologic role is currently not known.
HHR1 is one of the four histamine receptors namely H1, H2, H3 and H4.These receptors involved in a variety of physiological actions such as inflam-mation, gastric acid secretion, neurotransmitter release and mast cell medi-ated chemo taxis upon binding to the biogenic amine called histamine (1, 2). HHR1 belongs to class I of the G protein coupled receptors (GPCRs) and itinteracts with G proteins to activate phospholipaseC (3). GPCRs constitutethe largest family of cell surface proteins involved in signal transduction (4,5). Intrinsically, they are the major targets for the antiallergic drug therapydue to the role of HHR1 in allergic reactions. Among the examples of availablemedicines interacting with GPCRs antihypertensive beta-blockers, opioidreceptor agonists such as morphine, histamine H1 and H2 receptor antago-nists as antiallergic agents and antacids have gained their clinical importance(6).
It is believed that GPCRs activate their associated signal transduction path-ways not only upon agonist activation but also in the absence of agonists,resulting in constitutive receptor activity. Such activity may be calmed downby so-called inverse agonists that were originally classified as antagonists.The concept of constitutively active GPCRs is firmly rooted in receptor
pharmacology. In order to explain all these one has to understand a receptorthat can exist in more than one state, one active (R*) and other inactive (R).Agonists and some drugs prefer the active state R*, while inverse agonists gofor R (7). In this concept ‘neutral antagonists’ would not alter the R/R*distribution. Various human diseases have been developed as results of con-stitutive receptor activity (8). It is evident that inverse agonists are essentialfor the treatment of these diseases (9). Even well known HHR1 antagonistssuch as mepyramine, acrivastine, cetirizine, epinastine, loratadine are inverseagonists. Although an inverse HHR1 agonist would suppress any apparentconstitutive HHR1 activity, long term exposure of cells expressing constitu-tively active GPCRs to inverse agonists may result in receptor up-regulation.The development of novel inverse HHR1 agonists would give a pharmaco-logical tool to study the potential physiological role of constitutive HHR1activity which is not yet clear.
In this study, we focused to develop a valid pharmacophore model for HHR1inverse agonists, using it in virtual screening for new lead compounds andfind their interactions with catalytic residues of HHR1 by molecular docking.We have applied various molecular modeling methodologies to achieve thisgoal. Our study revealed the pharmacophore features that are essential for aHHR1 inverse agonist and led us to identify the molecules with the greateraffinity for the HHR1. Our results were also proved with molecular dockingstudy.
METHODOLOGY
Sequence Alignment and Model Building:The initial model of HHR1 protein (P35367) was built by using homology-modeling methods and the MODELLER9v7. The query sequence from Homosapiens was submitted to DIAL server for HHR1 prediction. The predicteddomain was searched to find out the related protein structure to be used as atemplate by the BLAST (Basic Local Alignment Search Tool) (10, 11) pro-gram against PDB (Protein Data bank). The closest structural homologues

Journal of Pharmacy Research Vol.5 Issue 5.May 2012
Kranthi Gattu et al. / Journal of Pharmacy Research 2012,5(5),2852-2860
2852-2860
were structure of Cholesterol bound form of human beta2 adrenergic recep-tor (58 % sequence identity) with PDB code 3D4S were utilized as referencestructure for building the model. The reference structure was downloadedfrom the PDB (12) and the target sequence was imported in the FASTAformat. Sequence of the reference structures were extracted from the respec-tive structure files and aligned with the target sequence using the defaultparameters in ClustalX (13). The initial alignment was further modifiedmanually to generate the final alignment. The 3D4S structure was used as thetemplates for building the 3D model of the HHR1 receptor usingMODELLAR9v7 (14). Side chains of conserved residues were retained andall side chains were optimized.
Structural Refinement and Validation:In order to refine the developed HHR1 models from Homo sapiens, energyminimization was performed using swiss pdb viewer (spdbv) (15) with aRoot Mean Square Deviation (RMSD) gradient per atom criterion of 4 Ao. Thedeveloped model of HHR1 model from Homo sapiens was superposed on totemplate 3D4s to determine the Ca and back bone RMSD values using onlineserver Superpose (16). The refined molecules were checked with WHAT IF(17), ProSA-Web (18) and Ramachandran plots were constructed utilizingPROCHECK (19) at ADIT.
Secondary Structure Prediction and Domain Analysis:A secondary structural conformation of the developed HHR1 models from
Homo sapiens was predicted by online server PDBsum (20). PDBsum alsopredicts range of conserved residues in the developed model. The predictedmodel of HHR1 models from Homo sapiens was subjected to motif analysisusing Motif scan server (21).
Active site Identification:Active site of HHR1 receptor (P35367) was identified using CastP server. Anew program, CAST, for automatically locating and measuring protein pock-ets and cavities, is based on precise computational geometry methods, in-cluding alpha shape and discrete flow theory. CAST identifies and measurespockets and pocket mouth openings, as well as cavities. The program speci-fies the atoms lining pockets, pocket openings, and buried cavities; the vol-ume and area of pockets and cavities; and the area and circumference ofmouth openings.
Docking MethodIn order to elucidate possible interactions of the protein HHR1 and hista-mine drugs performed docking studies using FRED (OpenEye ScientificSoftware, Santa Fe, NM). The relevant stereo isomers of the compoundswere minimized with the MMFF force field in the Openeye package. Con-formation and minimization of the compounds was performed using Omega(OpenEye Scientific Software, Santa Fe, NM). Fred requires a set of inputconformers for each ligand. The conformers were generated by Omega andstored in a single binary file. After this the output file is used for docking.
Figure1. Multiple alignment of HHR1 from Homo sapiens and thetemplate Cholesterol bound form of human beta2 adrenergic receptor(58 % sequence identity) with PDB code 3D4S.

Journal of Pharmacy Research Vol.5 Issue 5.May 2012
Kranthi Gattu et al. / Journal of Pharmacy Research 2012,5(5),2852-2860
2852-2860
Figure 2: The final 3D structure of HHR1 receptor. The structure isobtained by energy minimizing the average conformation over the last1000 femto seconds of molecular dynamics simulation.
Structural Validation of the Developed ModelThe predicted model of HHR1 from Homo sapiens was examined for valida-tion purpose using different criteria. The RMSD analysis of the developedmodel was evaluated by means of deviation from its template using Super-pose. The Ca RMSD and the back bone RMSD deviations for the model andthe template crystal structure were 0.97 A° (Fig.3). This less RMSD devia-tion from the template reveals that our developed model can be securely usedin the subsequent structural behavior.
Figure 3: Superposition of the developed HHR1 receptor A from Homosapiens (rendered in red) and the highest identity template 3D4S (ren-dered in green).
The final structure was further checked by verify 3D graph and the resultshave been shown in Fig.4. The overall scores indicates acceptable proteinenvironment. In the below plot, the vertical axis (Y-Axis) represents theaverage 3D-1D profile score for residues in a 21-residue sliding window, thecenter of which is at the sequence position indicated by the horizontal axis(X-Axis). A window length of 21 residues strikes a useful balance betweensmoothing fluctuations and localizing the error.
Figure 4: The 3D profiles verified results of HHR1 receptor proteinmodel; overall quality score indicates residues are reasonably folded.
The stereo chemical quality of the predicted model was evaluated usingPROCHECK in ADIT. The Ramachandran plot of the f /? distribution ofeach residue of the developed HHR1 protein model obtained fromPROCHECK revealed 91.1 % residues were in the most favored regions, 8.4% residues in the additionally allowed regions and 0.6% residues in thegenerously allowed region, 0% were residues were in disallowed region (Fig.5).Expect Glycine and Proline there were no residues in the disallowed regions.This high percentage of residues in the most favored regions indicates thatthe predicted HHR1 protein model is well constructed and more reasonable.
Figure 5: Ramachandran plot produced by predicted model of HHR1protein from Homo sapiens
WHAT IF analysis was also performed to evaluate the stereo chemical qual-ity of the developed HHR1 protein model. Standard bond lengths and bonddistances of the predicted model were determined using WHAT IF. Theanalysis revealed RMS Z-scores for bond lengths and bond distances is 0.42and 0.15, respectively, which is close to 1, indicating a high quality of themodeled structure. ProSA-Web (Table 1) analysis of the developed HHR1

Journal of Pharmacy Research Vol.5 Issue 5.May 2012
Kranthi Gattu et al. / Journal of Pharmacy Research 2012,5(5),2852-2860
2852-2860
model reveals that the Z-score value is -7.43 (Fig.6.) and it is in the range ofnative conformations of the templates. The overall residue energies calcu-lated for the predicted HHR1 model by ProSA- Web, reveals that theseenergies were located in the negative regions indicating that the developedmodel was is reliable.
Figure 6: ProSA-Web analysis of the developed HHR1 protein modelreveals that the Z-score value is -7.43
Secondary structure prediction and domain analysis:Secondary structural analysis (Fig.7) of developed HHR1 from Homo sapi-ens was carried out through PDBSUM online reveals the number of second-ary structure conformations like helices, strands, loops and turns. The re-sults predicted that HHR1 model was composed of 8 helices, 10 beta-sheetand other secondary structural conformations i.e., 10 helix-helix interactionsin the model.
Figure 7: secondary structural conformations shown by HHR1 proteinfrom Homo sapiens
Active site Identification of HHR1 Protein:After the final model was built, the possible binding sites of HHR1 fromHomo sapiens was searched based on the structural comparison of templateand the model build and also with CASTp server and was shown in Fig 8.Since, HHR1 and the 3D4S are well conserved in both sequence and struc-ture; their biological function should be identical. In fact from the structure-structure comparison of template, final refined model of HHR1 domain usingSPDBV program and was shown in Fig 3. It was found that secondarystructures are highly conserved and the residues, TYR 98, LEU 147, TRP148, VAL 149, ILE 150, PRO 151, ILE 152, TRP 155, PHE 180, MET 183,THR 184, ILE 187.
Figure 8: Active site of HHR1 protein (Magentacolor indicates active Site Regions)
DOCKING STUDIESFrom the 100 lead molecules the high ranked 31 mol-ecules were chosen based on Lipinski’s Rule-of-Fivefor docking simulations on to HHR1 using Openeye.Each of the lead molecules was set with docking simu-lations of 100 for docking onto HHR1 model and thus was used efficiently tostudy the lead molecule binding processes with the active sites, almost all the31 selected lead molecules which were docked have shown to interact wellwith the active site amino acids. The lead molecule interacts with almost allthe active site residues in the catalytic pocket, the interactions may be broughtabout by vanderwals interactions or hydrogen bonding. The interactionsshown by all the lead molecules were shown in the following Fig 9. Theinhibitors designed are as follows:
Cloperastine (1) Cetrizine (2)

Journal of Pharmacy Research Vol.5 Issue 5.May 2012
Kranthi Gattu et al. / Journal of Pharmacy Research 2012,5(5),2852-2860
2852-2860
Clemastine (3)
Levocetrizine (4)
Chloropheniramine (5) Pheniramine (6)
Diphenhydramine (7) Ebastine (8)
Embramine (9) Fexofidine (10)
Dexchlorpheniramine (11) Doxylamine (12)
Lafutidine (13) Meclozine (14)
Ranitidine (15)
Olopatadine (19)
VUF-6002 (18)
VUF-6002 (18) Olopatadine (19)
Quetiapine (20) Loratidine (21)

Journal of Pharmacy Research Vol.5 Issue 5.May 2012
Kranthi Gattu et al. / Journal of Pharmacy Research 2012,5(5),2852-2860
2852-2860
Desloratidine (22) Dimetidine (23)
ABT-239 (25)
A-349,821 (24)
Cimetidine (26)
Ciproxifan (27)
Clobenprobit (28)
Famotidine (29)
JNJ 7777120(30) Prometazine (31)
Table 1: Chemical Properties of the Ligand Compounds
1 C20H24ClNO 329.86 96.11± 0.3 1.566± 0.02 1.120± 0.06 38.10± 0.510-24
2 C23H30N2O2 366.49 110.04± 0.3 1.575± 0.02 1.101± 0.06 43.62± 0.510-24
3 C21H26ClNO 343.89 100.41± 0.3 1.553± 0.02 1.097± 0.06 39.80± 0.510-24
4 C21H25ClN2O3 388.88 105.94± 0.3 1.589± 0.02 1.237± 0.06 41.99± 0.510-24
5 C21H28ClNO 345.90 102.13± 0.4 1.572± 0.02 1.11± 0.1 40.49± 0.510-24
6 C16H20N2 240.34 75.91± 0.3 1.556± 0.02 1.018± 0.06 30.09± 0.510-24
7 C17H21NO 255.35 79.56± 0.3 1.551± 0.02 1.024± 0.06 31.54± 0.510-24
8 C32H39NO2 469.65 144.65± 0.4 1.590± 0.03 1.09± 0.1 57.34± 0.510-24
9 C18H22BrNO 348.27 91.46± 0.3 1.561± 0.02 1.234± 0.06 36.25± 0.510-24
10 C32H39NO4 501.65 145.86± 0.3 1.596± 0.02 1.171± 0.06 57.82± 0.510-24
11 C16H19ClN2 274.78 80.80± 0.3 1.565± 0.02 1.107± 0.06 32.03± 0.510-24
12 C17H22N2O 270.36 81.86± 0.3 1.554± 0.02 1.043± 0.06 32.45± 0.510-24
13 C22H29N3O4S 431.54 117.69± 0.3 1.598± 0.02 1.252± 0.06 46.65± 0.510-24
14 C25H27ClN2 390.94 118.03± 0.3 1.617± 0.02 1.159± 0.06 46.79± 0.510-24
15 C13H22N4O3S 314.40 85.64± 0.3 1.558± 0.02 1.184± 0.06 33.95± 0.510-24
16 C19H28N2O4 348.43 95.66± 0.3 1.532± 0.02 1.130± 0.06 37.92± 0.510-24
17 C15H24N4S 292.44 84.56± 0.4 1.614± 0.03 1.20± 0.1 33.52± 0.510-24
18 C13H15ClN4O 278.73 75.19± 0.3 1.662± 0.02 1.371± 0.06 29.80± 0.510-24
19 C21H23NO3 337.41 99.62± 0.3 1.640± 0.02 1.221± 0.06 39.49± 0.510-24
20 C21H25N3O2S 383.50 110.15± 0.5 1.652± 0.05 1.27± 0.1 43.66± 0.510-24
21 C22H23ClN2O2 382.88 105.85± 0.3 1.614± 0.02 1.261± 0.06 41.96± 0.510-24
22 C19H19ClN2 310.82 90.05± 0.3 1.625± 0.02 1.221± 0.06 35.69± 0.510-24
23 C20H24N2 292.41 92.23± 0.3 1.587± 0.02 1.065± 0.06 36.56± 0.510-24
24 C26H34N2O3 422.55 123.24± 0.3 1.555± 0.02 1.101± 0.06 48.85± 0.510-24
25 C22H22N2O 330.42 100.29± 0.4 1.641± 0.03 1.18± 0.1 39.75± 0.510-24
26 C9H16N6S 240.32 66.09± 0.5 1.645± 0.05 1.31± 0.1 26.20± 0.510-24
27 C16H18N2O2 270.32 76.43± 0.3 1.613± 0.02 1.231± 0.06 30.30± 0.510-24
28 C14H17ClN4S 308.82 85.45± 0.5 1.645± 0.05 1.31± 0.1 33.87± 0.510-24
29 C8H15N7O2S3 337.44 79.06± 0.5 1.808± 0.05 1.83± 0.1 31.34± 0.510-24
30 C14H16ClN3O 277.74 77.10± 0.3 1.655± 0.05 1.332± 0.06 30.56± 0.510-24
31 C17H20N2S 284.41 87.81± 0.3 1.615± 0.02 1.131± 0.06 34.81± 0.510-24
S.No Molecular Formula Molar Index of Density Polarisabilityformula weight refractivity cm3 refraction g/cm3 cm3

Journal of Pharmacy Research Vol.5 Issue 5.May 2012
Kranthi Gattu et al. / Journal of Pharmacy Research 2012,5(5),2852-2860
2852-2860
Docking of the inhibitors with P35367 was performed using FRED v 2.1,which is based on Rigid Body Shape-Fitting (Open Eye Scientific Software,Santa Fe, NM). This program generates an ensemble of different rigid bodyorientations (poses) for each compound conformer within the binding pocketand then passes each molecule against a negative image of the binding site.Poses clashing with this ‘bump map’ are eliminated. Poses surviving thebump test are then scored and ranked with a Gaussian shape function. Wedefined the binding pocket using the ligand-free protein structure and a boxenclosing the binding site. This box was defined by extending the size of acocrystalized ligand by 4 Å (addbox parameter of FRED). This dimensionwas considered here appropriate to allow, for instance, compounds largerthan the cocrystallized ones to fit into the binding site. One unique pose foreach of the best-scored compounds was saved for the subsequent steps. Thecompounds used for docking was converted in 3D with OMEGA (sameprotocol as above) (OpenEye Scientific Software, Santa Fe, NM). To thisset, the substrate (generation of multiconformer with Omega) correspondingto the modeled protein were added. All docking calculations were carried outusing Open eye and the files generated were analyzed for their binding con-formations. Analysis was based on Free energy of binding; Lowest dockedenergy and calculated RMSD values (Table.2). The total clusters of dockingconformations, with the 31 docked lead molecules showed negative bindingenergies
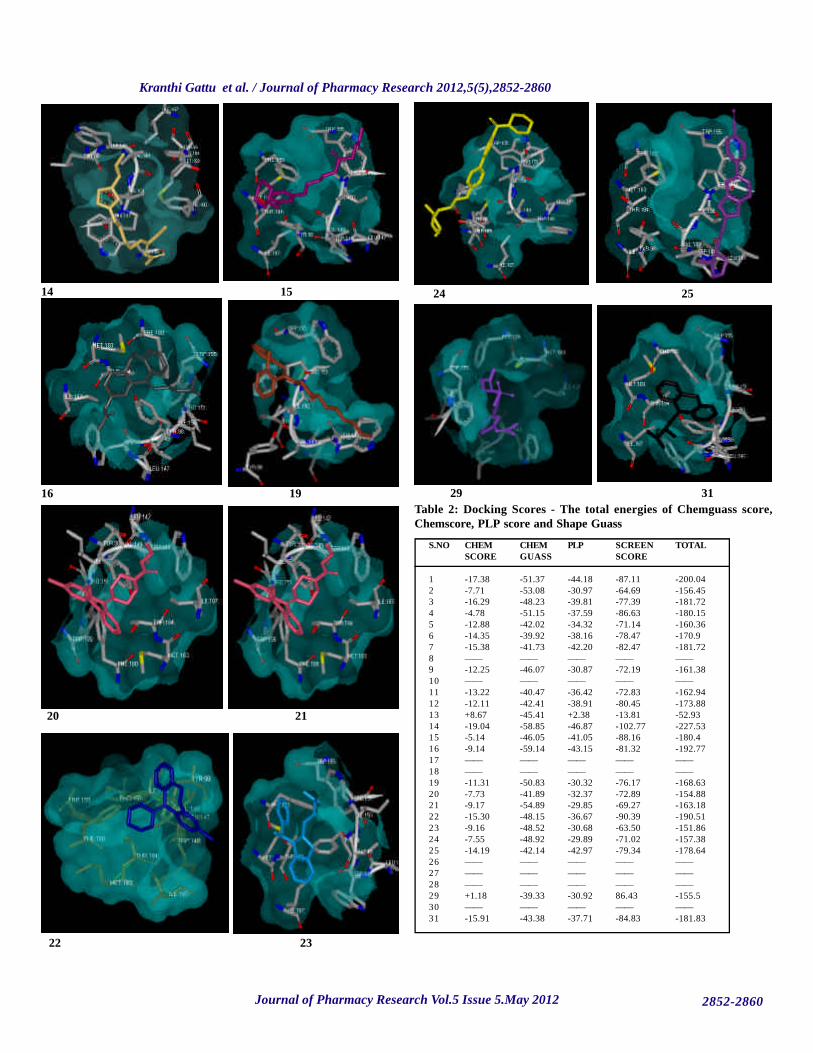
Figure 9: Binding of ligands to the active site of HHR1 protein
1 2
3
4
5 6
7 8
9
10
11 12
Journal of Pharmacy Research Vol.5 Issue 5.May 2012
Kranthi Gattu et al. / Journal of Pharmacy Research 2012,5(5),2852-2860
2852-2860
29 31
24 25
22 23
20 21
16 19
14 15
Table 2: Docking Scores - The total energies of Chemguass score,Chemscore, PLP score and Shape Guass
S.NO CHEM CHEM PLP SCREEN TOTALSCORE GUASS SCORE
1 -17.38 -51.37 -44.18 -87.11 -200.042 -7.71 -53.08 -30.97 -64.69 -156.453 -16.29 -48.23 -39.81 -77.39 -181.724 -4.78 -51.15 -37.59 -86.63 -180.155 -12.88 -42.02 -34.32 -71.14 -160.366 -14.35 -39.92 -38.16 -78.47 -170.97 -15.38 -41.73 -42.20 -82.47 -181.728 —— —— —— —— ——9 -12.25 -46.07 -30.87 -72.19 -161.3810 —— —— —— —— ——11 -13.22 -40.47 -36.42 -72.83 -162.9412 -12.11 -42.41 -38.91 -80.45 -173.8813 +8.67 -45.41 +2.38 -13.81 -52.9314 -19.04 -58.85 -46.87 -102.77 -227.5315 -5.14 -46.05 -41.05 -88.16 -180.416 -9.14 -59.14 -43.15 -81.32 -192.7717 —— —— —— —— ——18 —— —— —— —— ——19 -11.31 -50.83 -30.32 -76.17 -168.6320 -7.73 -41.89 -32.37 -72.89 -154.8821 -9.17 -54.89 -29.85 -69.27 -163.1822 -15.30 -48.15 -36.67 -90.39 -190.5123 -9.16 -48.52 -30.68 -63.50 -151.8624 -7.55 -48.92 -29.89 -71.02 -157.3825 -14.19 -42.14 -42.97 -79.34 -178.6426 —— —— —— —— ——27 —— —— —— —— ——28 —— —— —— —— ——29 +1.18 -39.33 -30.92 86.43 -155.530 —— —— —— —— ——31 -15.91 -43.38 -37.71 -84.83 -181.83

Journal of Pharmacy Research Vol.5 Issue 5.May 2012
Kranthi Gattu et al. / Journal of Pharmacy Research 2012,5(5),2852-2860
2852-2860
To understand the interaction between HHR1 (P) and Histamine antagonistdrugs (L), the HHR1-Histamine antagonist (P-L) complex was generatedusing FRED 2.1, which is based on Rigid Body Shape-Fitting and the binding3D conformation complex of the best docked Histamine antagonist drugs.This figure shows that Histamine antagonist drugs located in the center of theactive site, and is stabilized by hydrogen bonding. Hydrogen bonds play animportant role for structure and function of biological molecules, especiallyfor enzyme catalysis. Among the docked Histamine antagonist drugs,Meclozine is tightly bound by to the active site. The bondings of otherHistamine antagonist drugs with HHR1 were seen from Fig 9.
To determine the key residues that comprise the active site of the model, theinteraction energies of the Histamine antagonist drugs with HHR1 werecalculated. Tab.2 gives deep interaction energies for all Histamine antagonistdrugs. From the Tab.2 we can see that among the P-L complexes Meclozinehas a large favourable total energy of -227.53 Kcal mol-1. From these resultswe can suggest that PHE 180, MET 183, THR 184, ILE 187 are importantresidues in HHR1 and are the main contributors to the inhibitor interaction.TYR-98, LEU-147, and VAL-149 are not involved in the bonding with His-tamine antagonist drugs. However, we think that PHE 180, MET 183 andILE 187 may be important residues because these form hydrogen bonds withother Histamine antagonist drugs. From the alignment result, we can see thatthese four residues are highly conserved, and these are important for enzymecatalysis.
CONCLUSION The article entitled “Modeling and Docking Studies of Anti Histamine Drugs”deals with a Histamine antagonist protein HHR1, involved in a variety ofphysiological actions such as inflammation, gastric acid secretion, neurotrans-mitter release and mast cell mediated chemotaxis upon binding to the biogenicamine called histamine. In this work, we have constructed a 3D model of(P35367) domain, using the SWISS-Model method and obtained a refinedmodel after energy minimization. The final refined model was further as-sessed by ProsA and PROCHECK program, and the results show that thismodel is reliable. The stable structure of P35367 is further used for dockingwith the 31 ligand molecule of Histamine H1 antagonist. Docking resultsindicate that conserved amino-acid residues HHR1 protein main play animportant role in maintaining a functional conformation and are directlyinvolved in donor substrate binding. The interaction between the domain andthe ligands proposed in this study are useful for understanding the potentialmechanism of domain and the inhibitor binding. As is well known, hydrogenbonds play important role for the structure and function of biological mol-ecules. In this study it was found that, TYR 98, LEU 147, TRP 148, VAL149, ILE 150, PRO 151, ILE 152, TRP 155, PHE 180, MET 183, THR 184,and ILE 187. are important for strong hydrogen bonding interaction with theinhibitors. To the best of our knowledge PHE 180, MET 183 and ILE 187 isconserved in this domain and may be important for structural integrity ormaintaining the hydrophobicity of the inhibitor-binding pocket. The mol-ecule Meclozine showed best docking results with target protein.
ACKNOWLEDGEMENT:Authors are thankful to Dr. Kavitha Godugu, Department of Biochemistry,at Osmania University, Hyderabad (AP), India, for providing guidance tocarry out this work. The authors are grateful to Mr. Jayasimha (GlobalInformatics Hyderabad) for providing tools for Molecular Modeling andDocking Studies.
REFERENCES1. Steven, M. F.; Tom, I. B.; Richard, R. N.; Edward, M. R.; Jean
Phillipe, P.; Anthony, P. D.; Michael, S.; Anthony. (2005). J. H.Pharmacol. Rev, 57, 279.
2. Hofstra, C. L.; Desai, P. J.; Thurmond, R. L.; Fung-Leung, W. P.(2003). J.Pharmacol. Exp. Ther, 305, 1212.
3. Hill, S. J.; Ganellin, C. R.; Timmerman, H.; Schwartz, J. C.; Shankley,N. P.; Young, J. M.; Schunack, W.; Levi, R.; Haas, H. L. (1997).Pharmacol. Rev, 49, 253.
4. Maria, J. M.; Silvio, G. (2001). J. TRENDS in PharmacologicalSciences, 221, 368.
5. Louis, M. L. (2008). Mol. Biotechnol, 39, 239.6. Willem, S.; Ineke van, W.; Adriaan, P. I. (2005). Med. Res, 25, 398.7. Prather, L. P. (2004). Sci. STKE, 215.8. Govoni, M.; Bakker, R. A.; Wetering, I.; Smit, J. M.; Menge, M. B.
P.; Timmerman, H.; Elz, S.; Schunack, W.; Leurs, R.(2003). J.Med. Chem, 46, 5812.
9. Tao, Y. (2008) Pharmacol. Ther, 120, 129.10. Altschul, S. F., Gish, W., Miller, W., Myers, E. W. and Lipman, D.
J. (1990). A basic local alignment search tool. J. Mol. Biol. 215:403-410.
11. Altshul, S.F., Madden, T. L., Schaffer, A. A., Zhang, and J., Zhang,W., Millle, W., Lipman, D. Gapped Blast and PSI BLAST: a newgeneration of protein data base search programs, J. Nucleic AcidRes. 25 (1997) 3389-3402
12. Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N.,Weissig, H., Shindyalov, I. N., Bourne, P. E. The Protein DataBank. Nucleic A P.E.: Acids Research. 2000 ,28, 235-242
13. Thompson, J.D., Higgins, D.G., Gibson, T.J., Clustal W: improv-ing the sensitivity of progressive multiple sequence alignmentthrough sequence weighting position specific gap penalties andweight matrix choice, Nucl. Acids. Res. 1994, 22, 4673-4680
14. http://www.salilab.org/modeller/9v7.15. Guex, N., Peitsch, M. C. SWISS MODEL and the Swiss pdb
viewer: an environment for comparative protein modeling, Elec-trophoresis. 18 (1997) 2714-2723.
16. Rajarshi Maiti., Gary H., Van Domselaar., Haiyan Zhang., andDavid S. Wishart SuperPose: a simple server for sophisticatedstructural superposition. Nucleic Acids Res. 2004 ,32, 590-594
17. Vriend, G. Homology modeling with low sequence identity. Meth-ods.1998,14,293-300.
18. Sippl, M. J., Recognition of errors in three-dimensional structuresof proteins. Proteins, 17 (1993) 355-362.
19. Laskowski, R.A., MacArthur, M.W., Moss, D.S., Thronton, J.M.PROCHECK a program to check the stereo chemical quality ofprotein structure, J. Appl. Cryst. 1993, 26, 283-291.
20. Laskowski, R.A., Watson, J.D., Thornton, J.M. ProFunc: a serverfor predicting protein function from 3D structure. Nucleic AcidsRes, 2005, 33, W89-93.
21. Pagni, M., Ioannidis, V., Cerutti, L., Zahn-Zabal, M., Jongeneel,C.V., Hau, J., Martin, O., Kuznetsov,D., Falquet, L., MyHits:improvements to an interactive resource for analyzing protein se-quences. Nucleic Acids Res, 2007, 35, 433-437.
Source of support: Nil, Conflict of interest: None Declared