virtual interface substructure synthesis method for normal mode analysis of super-large molecular...
TRANSCRIPT

Virtual interface substructure synthesis method for normal mode analysis of super-large molecular complexes at atomic resolutionXuehui Chen, Yunxiang Sun, Xiongbo An, and Dengming Ming Citation: The Journal of Chemical Physics 135, 144108 (2011); doi: 10.1063/1.3647314 View online: http://dx.doi.org/10.1063/1.3647314 View Table of Contents: http://scitation.aip.org/content/aip/journal/jcp/135/14?ver=pdfcov Published by the AIP Publishing Articles you may be interested in Equilibration of complexes of DNA and H-NS proteins on charged surfaces: A coarse-grained model point of view J. Chem. Phys. 141, 115102 (2014); 10.1063/1.4895819 Fast and anisotropic flexibility-rigidity index for protein flexibility and fluctuation analysis J. Chem. Phys. 140, 234105 (2014); 10.1063/1.4882258 A molecular thermodynamic model for the stability of hepatitis B capsids J. Chem. Phys. 140, 235101 (2014); 10.1063/1.4882068 Reorientation of the helix of the tryptophan-rich gp41W peptide from HIV-1 at interfaces J. Chem. Phys. 139, 225105 (2013); 10.1063/1.4841795 Monte Carlo study of the molecular mechanisms of surface-layer protein self-assembly J. Chem. Phys. 134, 125103 (2011); 10.1063/1.3565457
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
152.2.176.242 On: Sun, 30 Nov 2014 11:55:23

THE JOURNAL OF CHEMICAL PHYSICS 135, 144108 (2011)
Virtual interface substructure synthesis method for normal mode analysisof super-large molecular complexes at atomic resolution
Xuehui Chen, Yunxiang Sun, Xiongbo An, and Dengming Minga)
Department of Physiology and Biophysics, School of Life Sciences, Fudan University, 220 Handan Road,Shanghai 200433, People’s Republic of China
(Received 31 March 2011; accepted 17 September 2011; published online 12 October 2011)
Normal mode analysis of large biomolecular complexes at atomic resolution remains challenging incomputational structure biology due to the requirement of large amount of memory space and centralprocessing unit time. In this paper, we present a method called virtual interface substructure synthe-sis method or VISSM to calculate approximate normal modes of large biomolecular complexes atatomic resolution. VISSM introduces the subunit interfaces as independent substructures that joincontacting molecules so as to keep the integrity of the system. Compared with other approximatemethods, VISSM delivers atomic modes with no need of a coarse-graining-then-projection proce-dure. The method was examined for 54 protein-complexes with the conventional all-atom normalmode analysis using CHARMM simulation program and the overlap of the first 100 low-frequencymodes is greater than 0.7 for 49 complexes, indicating its accuracy and reliability. We then appliedVISSM to the satellite panicum mosaic virus (SPMV, 78 300 atoms) and to F-actin filament structuresof up to 39-mer, 228 813 atoms and found that VISSM calculations capture functionally importantconformational changes accessible to these structures at atomic resolution. Our results support theidea that the dynamics of a large biomolecular complex might be understood based on the motions ofits component subunits and the way in which subunits bind one another. © 2011 American Instituteof Physics. [doi:10.1063/1.3647314]
INTRODUCTION
Dynamics has proved to be critical for functions ofbiomolecules,1 of particular interest are the dynamic mo-tions of large biomolecular structures such as DNA replica-tion complex, ribosome, F-actin filaments, and virus capsid.2
However, due to their large size and multiple spatio-temporalscales in motion there are big hurdles to the atom-leveldynamics simulations of these structures with the con-ventional computer resources.3 As a result normal modeanalysis (NMA) emerged as an alternative strategy for molec-ular dynamic simulation and was found efficient in iden-tifying functionally important conformational changes ofbiomolecular structures.4, 5 NMA finds vibrational motions bysolving eigenvalue problem through diagonalizing the Hes-sian matrix, whose elements are the mass-weighted second-derivatives of the potential energy of the studied system withrespect to the atomic Cartesian coordinates. Although NMAhas essentially no time-scale limitation, its application tolarge systems inevitably meets the barrier of steeply increasedamount of computation time and memory, two quantities in-creasing, respectively, as a cube function and a square func-tion of the number of atoms in the studied system. This un-favorable situation has stimulated continuous efforts over theyears to develop further simplification methods to calculateapproximate normal modes for larger and larger biomolecularstructures.6–8, 12
Most approximation methods have their roots in reduc-tion of the degrees of freedom (DOF) of the studied systems.
a)Electronic mail: [email protected]. Tel: 8621-5566-5621.
Here two distinct strategies are often applied: the coarse-grained modeling in which a small number of representa-tive atoms are selected to stand for the overall structure ofthe studied system, and the partial Hessian approximationin which the fast and localized motions of the overall struc-ture are filtered out, leading to a problem of lower DOFs.13
A widely used coarse-grained model is the Cα-based elas-tic network model (ENM).8, 14 In ENM a protein is regardedas a network of its Cα-atoms and each Cα is connected toneighboring Cα’s within a cutoff distance rc by linkers withspring constant γ . ENM and its variations have been suc-cessfully applied to the calculations of the motions of manylarge biostructures including ribosome, chaperonin GroEL,and viral capsids.15, 16 A variety of coarse-grained modelsthat use different representative atoms other than Cα atomswere also developed for both NMA and generic molecular dy-namic simulations of very large systems.17, 18 The rotation andtranslation block method is a typical example of the partialHessian method in which fast local atomic fluctuations aresignificantly filtered out by considering only the rigid-bodymotions of pre-defined fragments which are usually one or afew consecutively connected residues.9 The atomic dynamicsof the system are then recovered from the fragment motionsby a projection procedure. Along this direction were recentlydeveloped a variety of methods including the block normalmode,10 the mobile block Hessian model,11 and the vibra-tional subsystem analysis.12 Compared with the conventionalNMA the detailed atomic motions are either ignored as incoarse-grained modeling or frozen to some extent as in partialHessian approximations. However, even for large biomolecu-lar structures accurate descriptions of their motions at atomic
0021-9606/2011/135(14)/144108/10/$30.00 © 2011 American Institute of Physics135, 144108-1
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
152.2.176.242 On: Sun, 30 Nov 2014 11:55:23

144108-2 Chen et al. J. Chem. Phys. 135, 144108 (2011)
resolution by using NMA are still desirable and valuable,especially in cases where the conformational changes ofthe structures are coupled with or regulated by the bind-ing to small molecules or other biomolecules. Here we re-port a method based on the Rayleigh-Ritz principle and showthat it can deliver approximate normal modes of super-largebiomolecular complexes at atomic resolution, with satisfac-tory accuracy when compared with those derived by the con-ventional all-atom NMA. It is called virtual interface sub-structure synthesis method or VISSM.
The substructure synthesis method (SSM) was originallypresented and now broadly used in the field of mechanicalengineering to identify vibrational movements of very com-plicated structures.18 Recently we applied SSM to coarse-grained biomolecular dynamics simulations and developeda method that could deliver normal modes for super largebiomolecular structures at Cα level.19, 20 In SSM, a biomolec-ular complex is regarded as an assemblage of smaller-sizedsubstructures which are usually chosen as the subunits, do-mains, separated chains, etc. The motions of substructuresare combined to deliver the normal modes of the overallstructure using the Rayleigh-Ritz principle,21 which resultsin a reduced eigenvalue problem. The combination is notarbitrary but subject to the so-called geometry-compatibleconditions (GCCs) applied at the substructure boundaries,these conditions essentially glue the substructures up to re-cover the integrity of the overall structure. Unlike its coun-terpart in mechanical engineering the application of SSM inmodeling biomolecular complexes meets the challenge to de-fine the right GCCs since the molecular boundaries or inter-faces often contain subtle atomic contacts. In our previousworks,19, 20 an overlapping GCC was defined in which everyinterface residue is assigned twice, to both of the two adjacentsubstructures, and becomes an interface residue-pair, and thetwo residues of a residue-pair are required to have exactly thesame displacement, making the residue-pair moving as a sin-gle residue. This GCC suffers from the destroy of the integrityof substructures due to the assignment of scattered interfaceresidues from their neighboring substructures, thus make con-ventional all-atom NMA inapplicable to the substructures.Furthermore, the superposition at boundary areas eliminatesthe interface flexibilities that sometimes might be importantto the dynamics of the overall structure. To overcome theseshortcomings here we present a method in which substructureinterfaces are introduced as additional substructures, calledvirtual interface substructures or VISs, to the system. In thismethod, no GCC is required amongst the real substructuresand instead overlapping GCCs are applied only between aVIS and its relevant real substructures. In this way the realsubstructures are combined together through VISs while theyare independent of each other both structurally and dynami-cally. VISs are often composed of scattered atoms from neigh-boring substructures and the conventional NMA are usuallynot applicable to VISs, instead, some atomic elastic networkmodel or relevant models are found being able to describethe dynamics of VISs, introducing certain flexibility to theinterfaces.
To validate VISSM, we calculated all-atom normalmodes of 54 protein complexes selected from a protein
docking test-set22 and evaluated the mode overlaps (F100) us-ing the first 100 VISSM modes and the first 100 modes ob-tained by the conventional NMA using CHARMM simulationprogram.23 The overlap F100 is at least as high as 70% for 49complexes, indicating atomic VISSM modes are relatively ac-curate. We then applied VISSM to two assemblage structures,namely the spherical satellite panicum mosaic virus (SPMV,78 300 atoms) and the linear F-actin filaments. We found thatVISSM can identify a variety of functionally important con-formational changes accessible to these structures in atomicresolution. All the calculations were performed in a 32-bitsworkstation and only cost limited memory and central pro-cessing unit (CPU) time.
METHODS
VISSM for system composed of two substructures
In SSM a molecular complex is described as an assem-blage of a number of smaller sized substructures and the se-lection of substructures is quite natural such as subunits, do-mains, or separated chains. Here we first consider the simplestcase where only two substructures, called, respectively, s andr, exist. This is true for most of the complexes in DOCK-GROUND test-set.22 Denote us(Ps, t) as the time-dependentdisplacement of any atom Ps of s and follow the spirit ofRayleigh-Ritz principle, us(Ps, t) can be expressed as
us (Ps, t) =Ns∑i=1
φs,i (Ps) ζs,i (t), (1a)
or in the matrix form
us (Ps, t) = �s (Ps) ζs(t), (1b)
where φs, i is the ith admissible function that describes allow-able conformational changes of s, ζ s, i is the amplitude of φs, i,and Ns is the number of the admissible functions. φs, i is a 3× ns vector with ns being the number of atoms in s. Thechoice of φs, i’s is very flexible depending on the nature ofthe substructure, here we used the all-atom normal modes ob-tained by the conventional NMA using CHARMM simulationpackage.
To apply the Rayleigh-Ritz principle the kinetic and po-tential energies of substructure s are expressed as following:
Ts = 1
2uT
s Ms us = 1
2ζ
T
s �Ts Ms�s ζ s,
Vs = 1
2uT
s K sus = 1
2ζ T
s �Ts Ks�sζ s,
(2)
where ζ s is the disjoint configuration vector, Ms is themass-matrix, and Ks is the mass-weighted second-derivativematrix of the potential energy or the Hessian matrix. Sincewe choose �s as the normal modes of s, the above equationis simplified as the following:
Ts = 1
2ζ
Ts ζ s,
Vs = 1
2ζ T
s �sζ s,
(3)
where �s is a diagonal matrix whose elements are eigen-values of Ks. We have similar equations as Eqs. (2) and (3)
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
152.2.176.242 On: Sun, 30 Nov 2014 11:55:23

144108-3 Atomic normal mode analysis with VISSM J. Chem. Phys. 135, 144108 (2011)
(a) (b)
(c)
FIG. 1. (a) VISSM defines the VIS of the RAN-NFT2 complex as the inter-face between Ras-family GTPase Ran (red) and the nuclear transport factor2(green), which is composed of the red and green balls. (b) The dependenceof the mode-overlap and the Cα RMSF correlation function on the cutoffdistance rb. (c) The dependence of the mode-overlap and RMSF correlationfunction on the interaction strength γ between VIS heavy atoms. All of thebiomolecular structures presented in this work are prepared with VMD.35
for the kinetic and potential energies of substructure r. Inorder to combine them together we introduce the interfacebetween s and r, denoted as Is, r or simply I, as an addi-tional substructure to the system (see the scattered balls inFig. 1(a)). Is, r can be split into two parts as following:
I s,r = I ss,r ∪ I r
s,r ,
I ss,r = {a|∀a ∈ s and d (a, r) < rb},
I rs,r = {a|∀a ∈ r and d (a, s) < rb},
(4)
where rb is the cutoff distance that determines the size ofthe interface and d(a, o) is the minimum distance betweenatom a and substructure o. The total number of atoms in I isdenoted as nI. Since I is often composed of scattered atomsinstead of continued peptide-chains, the conventional NMAis not applicable to it. Fortunately, atomic elastic networkmodels6, 14 or relevant models24 are able to derive I’s normalmodes, which then are used to define I’s kinetic energy andpotential energy following Eqs. (2) and (3),
TI = 1
2ζ
TI ζ I , VI = 1
2ζ T
I �Iζ I , (5)
where the values of the elements in �I depend on ENMparameters γ and rc.
Consider the assembled structure composed of two realsubstructures s, r and the VIS I, its total kinetic energy andpotential energy can be written as
T = Ts + Tr + TI = 1
2ζ
Ts ζ s + 1
2ζ
Tr ζ r +1
2ζ
TI ζ I = 1
2ζ
Td ζ d,
(6)
V = V s + Vr + VI = 1
2ζ T
s �sζ s + 1
2ζ T
r �rζ r
+1
2ζ T
I �Iζ I = 1
2ζ T
d �dζ d, (7)
where
ζ d (t) = (ζ T
s , ζ Tr , ζ T
I
)T(8)
is the disjoint generalized coordinates describing configura-tion changes of the assembled structure which have the di-mension of N = Ns + Nr + NI, and �d is the N × N block −diag{�s, �r, �I } whose elements reads as following:
�d =
⎛⎜⎝
�s 0 0
0 �r 0
0 0 �I
⎞⎟⎠ . (9)
Let the generalized coordinates be harmonic function of time,
ζ d (t) = ζ d exp(iωt), (10)
where ω is the vibration frequency of the assembled structureand ζ d a time-independent amplitude vector, then the disjointRayleigh quotient for the assembled structure reads
Rd = ζ Td �dζ d/ζ
Td ζ d . (11)
Now in order to keep the structural integrity of the system, thecomponents of the configuration vector ζ d are not indepen-dent of one another but subject to the geometric compatibilityconstraints or GCCs. Considering that the VIS I shares atomswith both real substructures s and r as shown in Eq. (4), wemust have the following equations for every shared atoms:
us (a, t) = uI (a, t) , ∀a ∈ I s,
ur (a, t) = uI (a, t) , ∀a ∈ I r ,(12)
which defines 3 × nI constraints which is exactly the totalnumber of DOFs of I. Let L = N − 3nI and ζ be an L-dimension independent vector, then the solution of Eq. (12)gives an N × L constraint matrix C that associates the vectorζ with the configuration vector ζ d as following:
ζ d = C ζ . (13)
Insert Eq. (13) to Eq. (11), the Rayleigh quotient turnsinto
R = ζ T Hζ/ζ T �ζ , (14)
where
H = CT�dC, � = CTC (15)
are, respectively, the generalized Hessian matrix and thetransfer matrix, both having the dimension of L × L. Follow-ing the Rayleigh-Ritz principle, solving the vibrational mo-tions of the assembled system leads to the calculation of thestationary values of R, which in turn results in the followingL × L generalized eigenvalue problem:
HU′ = �U ′�. (16)
To calculate the atomic displacement of the normal modes ofthe assembled structure, we first construct mode matrix forthe assembled system composed of the three parts: s, r, and I,
�d = block − diag {�s,�r ,�I } , (17)
then derive the atomic displacement matrix U = �dCU′. Byeliminating the virtually interface substructure I from U we
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
152.2.176.242 On: Sun, 30 Nov 2014 11:55:23

144108-4 Chen et al. J. Chem. Phys. 135, 144108 (2011)
finally obtain the normal modes of the two-component struc-ture with the following matrix U:
U = (1n×n, 0n×nI
)U =
((�s 00 �r
), 0n×nI
)C U′, (18)
where 1 is the identity matrix and 0 is the zero matrix, and n= ns + nr is the total number of atoms in the complex. Unlikematrix U′, matrix U might not be mass-weight orthogonalizeddue to a deletion of the VIS I.
VISSM for system composed of more than twosubstructures
Consider a biomolecular complex composed of m(m > 2) real substructures, s = 1, 2,. . . , m, and l distinctinterfaces Iν , ν = 1, 2, . . . , l. Similar to Eq. (8), the disjointgeneralized coordinates reads
ζ d = (ζ T
1 , ζ T2 , ..., ζ T
m, ζ TI1
, ζ TI2
, ..., ζ TI l
)T, (19)
whose dimension is
N =m∑
i=1
Ni +l∑
j=1
NIj, (20)
which is equal to the total number of the admissible functions.Following Eq. (12) the GCCs now read
us (a, t) = uI (a, t) ∀a ∈ I s, I = I1, I2, · · · , I l , (21)
where s runs over all the substructures relevant to the interfaceI. Let nI j be the number of atoms in Ij and N I = ∑l
j=1nI j
the total number of interface atoms, then above equations de-termine NI constraints to the disjoint generalized coordinateζ d. Let L = N − 3nI, then solving Eq. (21) gives an N × Lconstraint matrix C. The harmonic potential energy of the as-sembled structure is defined with the following N × N eigen-value matrix:
�d = block − diag{�1,�2, · · · ,�r ,�I1 ,�I2 , · · · ,�I1}.(22)
Using these new C, �d, and mode matrix �d = block− diag{�1, . . . ,�m,�I1 , . . . ,�I l
} and following a similarprocedure described by Eqs. (15)–(18) we can obtain approx-imate normal modes of the m-mer assembled structure.
The all-atom normal modes as substructureadmissible functions
VISSM needs atomic substructure admissible functionsto deliver atomic normal modes of the assembled structure.Here admissible functions are defined by substructure all-atom normal modes, obtained by using the conventional NMAin the CHARMM simulation package.25 The RMS gradientthreshold of 0.001 kcal/mol/Å2 is used for the structural min-imization, a procedure required by the standard NMA, leav-ing the minimized structure close to its crystal structure (theCα root-mean-square deviation or RMSD is usually about1.0 Å). Although a few imaginary modes might appear whenusing such a relatively loose threshold, calculations on com-plex structures showed that they do not affect the significanceof the positive low-frequency global modes.5, 25, 26 Only posi-tive CHARMM modes are selected as the admissible functions.
6 zero-frequency modes describing rigid-body motions aremanually generated and added to the substructure admissi-ble functions. This protocol was applied to 54 heterodimerprotein complexes selected from a protein docking test set.22
The total number of atoms of every selected protein complexis less than 7500 so that we can calculate the atomic normalmodes of the complexes using the conventional NMA withour 32-bit workstation.
The mode overlap and root-mean-squarefluctuation correlation
The VISSM atomic normal modes and the normal modesderived by conventional NMA are compared with the modeoverlap. Let φssm
i be the ith VISSM normal mode and φnmaj the
jth conventional all-atom normal mode, the overlap betweenthese two modes is defined as
Fn = 1
n
n∑i,j=1
(φssm
i · φnmaj
||φssmi ||∥∥φnma
j
∥∥)2
, (23a)
where ||φ|| is equal to the length of vector φ. Since φssmi s are
not necessarily orthogonal to one another, they are orthogo-nalized to φ
ssmj ’s using the Gram-Schmidt algorithm, and the
above equation is replaced by
Fn = 1
n
n∑i,j=7
(φ
ssmi · φnma
j
||φssmi ||∥∥φnma
j
∥∥)2
. (23b)
For convenient, we chose n = 100. In the harmonic approxi-mation the atomic fluctuation correlations of the system at thetemperature T can be expressed with the following covariancematrix:
R = (〈�r i�r j 〉) = kBT
n∑k=7
�φi,k · �φj,k
ω2k
, (24a)
where ωk is the frequency of the kth normal mode and kB isthe Boltzmann’s constant. Usually the following normalizedmatrix is used so as to eliminate the temperature effect:
R′ =(
Ri,j√Ri,i × Rj,j
), (24b)
whose diagonal elements are equal to 1 and the off-diagonalelements vary between +1 for fully correlated motion to −1for anti-correlated motion. The diagonal elements of R definethe root-mean square fluctuations (RMSFs) for each atoms,
fi = ⟨�r2
i
⟩ = kBT
n∑k=7
|φi,k|2ω2
k
. (25)
It was well known that at ambient temperature the RMSFsvalues are dominantly determined by a small number of low-est frequency modes, here for simplicity we set n = 100. Thecorrelation function between the Cα’s RMSFs obtained byVISSM and those by the conventional NMA reads
CRMSF =∑Nr
i=1 f ssmi × f nma
i
||fssm|| ||fnma|| , (26)
where the index i runs over every Cα’s and Nr is the totalnumber of Cα’s.
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
152.2.176.242 On: Sun, 30 Nov 2014 11:55:23

144108-5 Atomic normal mode analysis with VISSM J. Chem. Phys. 135, 144108 (2011)
RESULTS
Evaluation of the parameters in VISSM
VISSM has two parameters: the boundary cutoff distancerb that determines the size of VISs and the spring constant γ
that defines the interaction strength between atoms in VISs.To examine the effects of these parameters on the quality ofthe synthesized modes, we took the Ran-NTF2 complex asan example. The Ran-NTF2 complex is composed of two sol-uble components of the nuclear protein import machinery –the Ras-family GTPase Ran and the nuclear transport factor2(NTF2) (see Fig. 1(a), the coordinates of the complex weretaken from PDB entry 1A2K (Ref. 27)). The red and greenballs highlight the interface heavy atoms that dominate theinteractions between the two proteins, including the putativeswitch II loop of Ran (residues 65 to 78) that extrudes intothe hydrophobic cavity on the surface of NTF2 and the C-terminal tail of NTF2 packing against the switch I loop of Ran(residues 39 to 43). These interactions were found to accountfor the selectivity of NTF2 binding to GDP-Ran other thanbinding to GTP-Ran, indicating their importance for nuclearprotein import in vivo.28
The first 120 low-frequency all-atom normal modes ofeach protein obtained by CHARMM were used as admissi-ble functions for VISSM calculations. The all-atom mode-overlap F100 and the Cα RMSF correlation CRMSF were eval-uated by comparing the conventional all-atom NMA modeswith a series of VISSM all-atom modes calculated with differ-ent parameters (rb, γ ). Figures 1(b) and 1(c) show the depen-dences of F100 and CRMSF on the two parameters, respectively.The mode-overlap F100 and RMSF correlation CRMSF havehigher values around rb = 3.2–3.4 Å. Both F100 and CRMSF
reach higher values around γ = 1 kcal/mol/Å2. CRMSF is lit-erally constant for γ around 1–2 kcal/mol/Å2 and then beginsto decrease slowly after γ > 2 kcal/mol/Å2. F100 decreasesfast when γ > 1.2 kcal/mol/Å2. Our calculations showed thatthe ENM cutoff distance rc does not have much effect on thesynthesized modes, provided it is large enough – say, 10 Å ormore – to include most pairwise interactions in VISs. Similarparameter dependences were also observed for other proteincomplexes. In the following calculation we chose rb = 3.3 Åand γ = 1 kcal/mol/Å2.
Figures 2(a)–2(c) show the first three VISSM modesof the complex. The 7th VISSM mode is a hinge-motionin which Ran Phe72 sits tightly inside the cavity of NTF2and the vibration can efficiently change the distance betweenthe NTF2 Asp92/Asp94 loop and Ran Lys71 thus might in-volve the creation/elimination of the salt bridge between theseresidues. The 8th VISSM mode is also a hinge motion withits immobile center shifts to the area where NFT2 C-terminalcontacts the switch I loop of Ran, leaving the room for relativemotion between Ran Phe72 and the NTF2 cavity; this motioncan change the binding state between the two proteins. Mode9 shows itself as a torsion motion with the rotation axis pass-ing through the contact area around Ran Phe72 in the cav-ity; this motion helps the two proteins bind to other compo-nents of the import machinery while keeping themselves ingood contact. As a comparison, Figs. 2(d)–2(f) list the cor-responding lowest-frequency normal modes of the complex
(a) (b) (c)
(d) (e) (f)
(g) (g)
FIG. 2. (a), (b), and (c), respectively, show the amplitude and direction of theatomic motions of RAN-NFT2 complex for the two hinge modes (the 7th and8th modes) and the torsion mode (the 9th mode) as obtained by VISSM. (d),(e), and (f), respectively, show the corresponding modes as obtained by theconventional NMA. For clarity, only Cα atoms are attached with arrows todemonstrate their displacements. This is also applied to the following figureswhen arrows are applied. (g) compares the normal mode frequency spectrumobtained by VISSM (filled triangles) with that obtained by conventional all-atom NMA (open circles). (h) compares the atom-atom equal-time cross-correlation functions determined by using VISSM modes (upper triangle) andthose by using the conventional all-atom NMA (lower triangle).
derived by CHARMM. The dot products between the threesets of modes are, respectively, 0.80, 0.70, and 0.89, and themode overlap is F3 = 0.95 for the first three modes and F100
= 0.90 for the first 100 modes. Figure 2(g) compares thefrequency spectrum derived by VISSM and that by the stan-dard NMA and a small deviation of less than 5% is detected.Figure 2(h) compares the equal-time cross-correlation func-tions and tells that VISSM modes can also deliver rea-sonably good approximation of the internal dynamic cor-relations for residues both within protein subunits and inprotein-protein interfaces. These results indicate that VISSMcan deliver functionally important normal mode of the assem-bled structures at atomic resolution.
Comparison with the conventional all-atom NMAfor 54 protein complexes
Table I lists the mode overlaps for 54 protein com-plexes. Except the worst case of proteinase A complexed with
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
152.2.176.242 On: Sun, 30 Nov 2014 11:55:23

144108-6 Chen et al. J. Chem. Phys. 135, 144108 (2011)
TABLE I. The mode-overlaps and Cα RMSF correlation functions for 54 protein complexes.a
PDB code Protein F100 CRMSF Resolution (Å)
1A2K Nuclear transport factor 2 (NTF2) / RAN 0.85 0.97 2.501A2X Troponin C/Troponin I 0.78 0.90 2.301AK4 Cyclophilin A/HIV-1 capsid 0.84 0.92 2.361AY7 Guanyl-specific ribonuclease Sa/Barstar 0.82 0.90 1.701BZQ Rnase A/Antibody cAb-RN05 0.80 0.90 2.801CGJ α-chymotrypsinogen/Pancreatic secretory trypsin
inhibitor (PSTI)0.78 0.95 2.30
1CXZ His-tagged transforming protein RhoA/PKN 0.82 0.97 2.201DF9 Nonstructural protein 1/Bowman-Birk type trypsin
inhibitor0.63 0.90 2.10
1DP5 Proteinase A/Proteinase inhibitor IA3 0.28 0.44 2.201E96 Ras-related C3 botulinum toxin substrate 1
(RAC1)/NCF2 TPR domain (p67phox)0.78 0.95 2.40
1F02 Intimin/Translocated intimin receptor 0.83 0.87 2.901FFG Chemotaxis protein CheY / Chemotaxis protein CheA 0.78 0.76 2.101FQJ Chimera of guanine nucleotide-binding
protein/Regulator of g-protein signaling 9 (RGS9)0.83 0.90 2.02
1GCQ Growth factor receptor-bound protein 2 (GRB2)/Vav 0.74 0.89 1.681I8L T lymphocyte activation antigen CD80 / Cytotoxic
t-lymphocyte protein 4 (CTLA-4)0.80 0.93 3.00
1IRA Interleukin-1 receptor antagonist (IL1RA)/Interleukin-1receptor
0.68 0.65 2.70
1J2Jb ADP-ribosylation factor 1 (ARF1)/ADP-ribosylationfactor binding protein GGA1
0.78 0.90 1.60
1JK9 Superoxide dismutase (SOD1)/Copper chaperone forSOD (CCS)
0.71 0.78 2.90
1JTP Lysozyme C/Single-domain antibody 0.82 0.87 1.901KAC Fiber knob protein/Coxsackie virus and adenovirus 0.79 0.80 2.601KPS Ran-gtpase activating protein 1
(RANGAP1)/Ubiquitin-like protein sumo-1 conjugatingenzyme (UBC9)
0.84 0.96 2.50
1LFD Ralgds/Ras 0.82 0.93 2.101M27 Sh2 domain protein 1A/Proto-oncogene
tyrosine-protein kinase Fyn0.81 0.90 2.50
1MBX ATP-dependent clp protease ATP-bindingsubunit/Protein yljA
0.76 0.58 2.25
1ML0 M3 protein/Small inducible cytokine 0.84 0.95 2.801MQ8 Intercellular adhesion molecule-1/Integrin alpha L 0.74 0.81 3.301MZW U-snRNP-associated cyclophilin (cyclophilin H) / WD
splicing factor Prp40.85 0.87 2.00
1NMU Maltodextrin-binding protein / 60S ribosomal proteinL30
0.90 0.87 2.31
1NPE Entactin / Laminin B2 chain 0.73 0.73 2.301PXV Cysteine protease / Cysteine protease inhibitor 0.72 0.93 1.801QA9 Human CD2 protein / Human CD58 protein 0.75 0.84 3.201QAV α-1 syntrophin / Neuronal nitric oxide synthase 0.80 0.75 1.901R8S ADP-ribosylation factor 1 (ARF1) / ARF
nucleotide-binding site opener (ARNO)0.75 0.92 1.46
1SQ2 Lysozyme C / Novel antigen receptor 0.82 0.94 1.451SYX Spliceosomal U5 snrnp-specific 15 kDa protein / CD2
antigen cytoplasmic tail-binding protein 20.82 0.92 2.35
1TE1 Xylanase inhibitor protein i (XIP-I) / Endo-1,4-xylanase(GH11)
0.84 0.93 2.50
1U0S Chemotaxis protein CheY / Chemotaxis protein CheA 0.79 0.84 1.901UUZ Inhibitor of vertebrate lysozyme (IVY) / Lysozyme C 0.75 0.90 1.801WRDc Target of Myb protein 1 (Tom1) / Ubiquitin 0.75 0.85 1.751XQS Hspbp1 protein / Heat shock 70 kDa protein 1 (HSP70) 0.73 0.95 2.901XU1 Tumor necrosis factor ligand superfamily member 13
(TNFSF13) / Tumor necrosis factor receptorsuperfamily 13B (TNFRSF13B)
0.78 0.88 1.90
1Z0K GTP-binding protein / FYVE-finger-containing Rab5effector protein
0.78 0.85 1.92
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
152.2.176.242 On: Sun, 30 Nov 2014 11:55:23

144108-7 Atomic normal mode analysis with VISSM J. Chem. Phys. 135, 144108 (2011)
TABLE I. (Continued.)
PDB code Protein F100 CRMSF Resolution (Å)
1Z3G Ookinete surface protein Pvs25 / 2A8 Fab light chain 0.79 0.86 3.301Z5Y Cytochrome c biogenesis protein ccmG / Disulfide
interchange protein DsbD0.84 0.85 1.94
2A5D ADP-ribosylation factor 6 / Cholera enterotoxin Asubunit
0.76 0.78 1.80
2B12d Cytochrome c peroxidase, mitochondrial (CCP) /Cytochrome c iso-1
0.69 0.82 3.02
2BH1 Cholera toxin secretion protein / Type II traffic wardenATPase
0.77 0.82 2.40
2C1M Importin-alpha2 subunit / Nucleoporin 50 kDa (NUP50) 0.69 0.74 2.202HRK Glutamate-trna ligase / GU4 nucleic-binding protein 1
(G4p1 protein)0.77 0.90 2.05
2OOBc E3 ubiquitin-protein ligase Cbl-b / Ubiquitin 0.88 0.87 1.902UUY Cationic trypsin / Tryptase inhibitor 0.78 0.89 1.153BP5 Programmed cell death protein 1 (PD-1) / Programmed
cell death 1 ligand 20.75 0.93 1.80
3SIC Subtilisin BPN’ / Streptomyces subtilisin inhibitor 0.79 0.94 1.803YGS Apoptotic protease activating factor 1 / Procaspase 9 0.81 0.82 2.50
aThe coordinates of the listed complexes were taken from the DOCKGROUND database for protein docking. The default parameter for the boundary cutoff distance rb is 3.3 Å andthe spring constant γ is 1.0 kcal/mol/Å2. However, slightly larger cutoff distances were required for a few cases given below.brb = 3.4 Å.crb = 3.5 Å.drb = 3.7 Å.
inhibitor IA3 (PDB entry 1DP5,29 see the discussion for de-tails), the average mode-overlap for the first 100 nonzeromodes is 0.78 with F100 no less than 0.7 for 49 complexes andthe average Cα-RMSF correlation is 0.86 with CRMSF no lessthan 0.75 for the 49 complexes, indicating that VISSM all-atom normal modes are accurate in describing low-frequencymodes of the complexes. We noticed that due to the loose con-tact between component proteins VISSM might not be ableto define effective interfaces using the default boundary cut-off rb = 3.3 Å, in that case slightly larger rb is desired. Wealso examined a series of rb’s from 3.0 to 3.8 Å with fixedγ = 1 kcal/mol/Å2, and found that the average F100 remainsaround 0.74–0.78, indicating that VISSM modes are relativelyindependent on these parameters.
To demonstrate its efficiency in delivering normal modesof large biostructures at atomic resolution, we applied VISSMto a spherical structure of the satellite panicum mosaic virus(SPMV) capsid and a linear structure of the F-actin filamentsat a variety of length.
Atomic normal modes of the satellite panicum mosaicvirus capsid
The motion of viral capsids had recently attracted manyresearch efforts of which the structure and dynamics of satel-lite viruses are of particular interest due to their small size instructure and their natural reproduction capability.16, 30 SPMVis a T = 1 icosahedral virus whose capsid is composed of60 identical coat proteins with single stranded RNA genomeconcealed inside, encoding for the coat protein and another9.4-kDa C-terminal protein.31 Each coat protein folds intoeight-stranded jellyroll β-barrel with its positively chargedN-terminal protruding towards the inner negatively charged
nucleic acid. Around the five-fold axis five coat proteins con-tact one another through loops where Thr95 sits at the top,forming a pentameric subunit (see Fig. 3(a)). The neighbor-ing pentameric subunits interact mostly through the N- andC-terminal ends of the β-barrel. The extended-atom modelof TOPH19 and the polar-hydrogen parameter set for pro-tein, PARAM19, were used for the energy minimization andNMA of the pentameric subunit,32 which leads to 6525 atoms
(a) (b)
(c) (d) (e)
FIG. 3. (a) The monomer, a star-like pentamer (red), is defined as componentsubstructure of the globular structure of SPMV. (b) The frequency degeneracyreveals symmetry inside the virus structure. (c)–(e), respectively, show theamplitudes and directions of lowest-frequency vibration motions accessibleto the virus structure: (c) the 7th mode in which the spherical structure takesa squash-and-stretch along an axis of the virus ball, (d) the second lowest-frequency mode (the 12th mode) in which monomers tend to push out/crowdof the sphere at 3 evenly distributed directions, and (e) the unique centralsymmetric mode (the 24th mode) in which the structure expands/contractsisotropically.
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
152.2.176.242 On: Sun, 30 Nov 2014 11:55:23

144108-8 Chen et al. J. Chem. Phys. 135, 144108 (2011)
for the pentameric subunit and a total of 78 300 atoms forthe full viral capsid. One hundred atomic normal modes ofthe pentameric subunit were obtained using the VIBRANand DIAG modules in CHARMM and used as admissiblefunctions. Eleven new pentameric subunits were iterativelygenerated using the transformation matrices listed in the PDBfile, and their admissible functions are obtained by rotatingthe admissible functions of the first subunit according to thetransformation matrices.
Figure 3(b) shows the frequency spectrum of the vi-brational modes. Due to the symmetric arrangement of thestructure vibrational modes demonstrate highly degenerateespecially in the low-frequency region. The degenerations forthe first 30 vibration modes are 5, 3, 4, 4, 5, 1, 3, and 5. A sim-ilar degeneration number distribution had also been reportedfor the structure of the satellite tobacco mosaic viral capsid.16
An inspection of the motions of lowest frequency modesshows the typical conformation changes of the pentamericsubunit can be one of the following: (A) that one monomermoves close to/apart from the opposite two monomers whileits two neighboring monomers moves apart from/close toeach other, (B) that two adjacent monomers synchronouslyslide back-and-forth again the rest three monomers,(C) that whole subunit rotates as a whole around its 5-foldaxis clockwise/anti-clockwise, (D) the five monomers evenlyexpand/shrink relative to the 5-fold center, and (E) that thewhole subunit moves as a whole close to/apart from the centerof capsid structure. For example, Fig. 3(c) demonstrates the7th mode of the capsid in which the spherical structurestretches/compresses along an axis of the ball structureand at the same time compresses/stretches along a secondaxis that is literally orthogonal to the first one. The confor-mational changes of the 12 subunits were identified to beBAABDAADBAAB. Figure 3(d) shows the 12th mode inwhich the subunits moves along three evenly intersectedaxes, with one axis (called middle axis, for convenience)passing through the centers of the 2nd and 3rd. The se-quence of the conformation changes for the 12 subunit isABBAAAAAAAAA. In this mode, the capsid structurestretches/shrinks along the middle axis in one direction and atthe same time shrinks/stretches along the other two axes in theopposite direction. Figure 3(e) shows the nondegenerate 24thmode which is an isotropic expansion/contraction motion thateffectively changes the diameter of the capsid structure whilekeeping the capsid in perfect spherical shape. In this mode allthe pentameric units were found to take the same conforma-tion changes as a combination of D and E. This mode has thehighest collectivity of 0.81 as measured by the kappa value.
All-atom normal modes of F-actin filaments
Actin, as one of the most highly conserved proteins existsabundantly in eukaryotic cells, can exist in two mutually ex-changeable forms in cell: the free globular monomer (G-actin)and the assembled filamentous actin (F-actin). Over the yearscontinuous efforts were devoted to study the structure and dy-namics of a variety of F-actin filaments.33 Here we considerthe all-atom vibration motions of a recently published F-actinfilament model – the Oda model.34 The coordinates of the
(a) (b) (c)
FIG. 4. The amplitudes and directions of the all-atom motions of the threetypical low-frequency modes of a 13-mer F-actin segment: (a) the bendingmode in which end monomers and middle monomers move in the oppositedirection, resulting from two anti-symmetric local rotations whose rotationaxes are literally perpendicular to the filament axis, (b) the twisting mode inwhich the two halves rotate at opposite direction around the filament axis,and (c) the stretching mode in which the two halves move in the oppositedirection along the filament axis.
actin monomer are taken from PDB entries 2ZWH, and theADP and Ca2+ are kept in the structure in the simulation. In F-actin filament the monomers are arranged into two intertwin-ing right-handed helices that turn out to be a double-strandedhelical structure (see Fig. 4). This helical structure repeats it-self after every 13 monomers (the 13-subunit repeat, 76 271atoms for Oda model) of a length about 37.5 nm and its totallength can be several microns. A total of 55 all-atom normalmodes with a frequency not great than 10.2 cm−1 were usedas admissible functions to describe conformational changesof the first monomer. The follow-up monomers were added tothe structure iteratively by rotating the first monomer accord-ing to a transformation matrix (provided by Oda via personalcommunication). Each monomer has exactly the same set ofadmissible functions as those of the first one, except that arotation operation, which is also defined by the transforma-tion matrix, is applied to the admissible functions. A longestfilament of three13-subunit repeats (228 813 atoms) was ob-tained. Figure 4 shows the atomic displacements of three typ-ical vibrational motions of a segment of filament structureof one 13-subunit repeat (for clarity only Cα’s are shown).These modes show that although the overall vibration of thefilaments can be described as waves of a homogeneous elas-tic rod the atomic displacements vary a lot from monomer tomonomer, indicating that the filament undergoes very inho-mogeneous dynamic fluctuations in its different parts.
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
152.2.176.242 On: Sun, 30 Nov 2014 11:55:23
144108-9 Atomic normal mode analysis with VISSM J. Chem. Phys. 135, 144108 (2011)
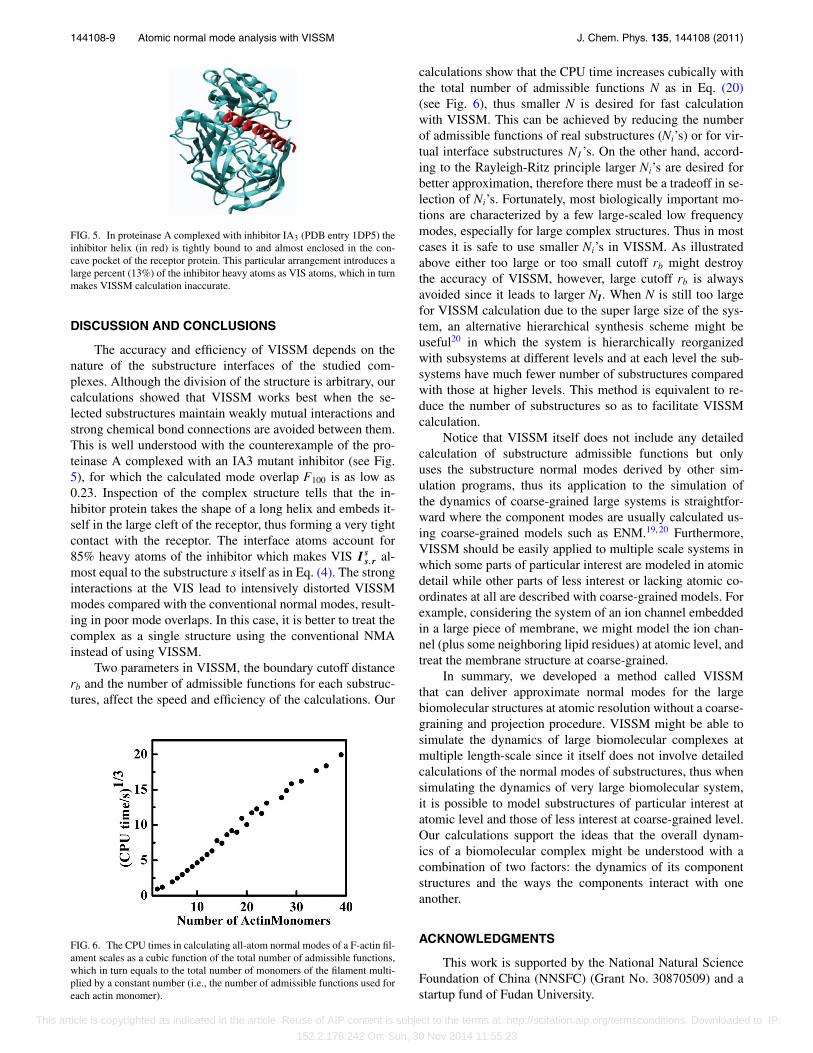
FIG. 5. In proteinase A complexed with inhibitor IA3 (PDB entry 1DP5) theinhibitor helix (in red) is tightly bound to and almost enclosed in the con-cave pocket of the receptor protein. This particular arrangement introduces alarge percent (13%) of the inhibitor heavy atoms as VIS atoms, which in turnmakes VISSM calculation inaccurate.
DISCUSSION AND CONCLUSIONS
The accuracy and efficiency of VISSM depends on thenature of the substructure interfaces of the studied com-plexes. Although the division of the structure is arbitrary, ourcalculations showed that VISSM works best when the se-lected substructures maintain weakly mutual interactions andstrong chemical bond connections are avoided between them.This is well understood with the counterexample of the pro-teinase A complexed with an IA3 mutant inhibitor (see Fig.5), for which the calculated mode overlap F100 is as low as0.23. Inspection of the complex structure tells that the in-hibitor protein takes the shape of a long helix and embeds it-self in the large cleft of the receptor, thus forming a very tightcontact with the receptor. The interface atoms account for85% heavy atoms of the inhibitor which makes VIS I s
s,r al-most equal to the substructure s itself as in Eq. (4). The stronginteractions at the VIS lead to intensively distorted VISSMmodes compared with the conventional normal modes, result-ing in poor mode overlaps. In this case, it is better to treat thecomplex as a single structure using the conventional NMAinstead of using VISSM.
Two parameters in VISSM, the boundary cutoff distancerb and the number of admissible functions for each substruc-tures, affect the speed and efficiency of the calculations. Our
FIG. 6. The CPU times in calculating all-atom normal modes of a F-actin fil-ament scales as a cubic function of the total number of admissible functions,which in turn equals to the total number of monomers of the filament multi-plied by a constant number (i.e., the number of admissible functions used foreach actin monomer).
calculations show that the CPU time increases cubically withthe total number of admissible functions N as in Eq. (20)(see Fig. 6), thus smaller N is desired for fast calculationwith VISSM. This can be achieved by reducing the numberof admissible functions of real substructures (Ni’s) or for vir-tual interface substructures NI ’s. On the other hand, accord-ing to the Rayleigh-Ritz principle larger Ni’s are desired forbetter approximation, therefore there must be a tradeoff in se-lection of Ni’s. Fortunately, most biologically important mo-tions are characterized by a few large-scaled low frequencymodes, especially for large complex structures. Thus in mostcases it is safe to use smaller Ni’s in VISSM. As illustratedabove either too large or too small cutoff rb might destroythe accuracy of VISSM, however, large cutoff rb is alwaysavoided since it leads to larger NI. When N is still too largefor VISSM calculation due to the super large size of the sys-tem, an alternative hierarchical synthesis scheme might beuseful20 in which the system is hierarchically reorganizedwith subsystems at different levels and at each level the sub-systems have much fewer number of substructures comparedwith those at higher levels. This method is equivalent to re-duce the number of substructures so as to facilitate VISSMcalculation.
Notice that VISSM itself does not include any detailedcalculation of substructure admissible functions but onlyuses the substructure normal modes derived by other sim-ulation programs, thus its application to the simulation ofthe dynamics of coarse-grained large systems is straightfor-ward where the component modes are usually calculated us-ing coarse-grained models such as ENM.19, 20 Furthermore,VISSM should be easily applied to multiple scale systems inwhich some parts of particular interest are modeled in atomicdetail while other parts of less interest or lacking atomic co-ordinates at all are described with coarse-grained models. Forexample, considering the system of an ion channel embeddedin a large piece of membrane, we might model the ion chan-nel (plus some neighboring lipid residues) at atomic level, andtreat the membrane structure at coarse-grained.
In summary, we developed a method called VISSMthat can deliver approximate normal modes for the largebiomolecular structures at atomic resolution without a coarse-graining and projection procedure. VISSM might be able tosimulate the dynamics of large biomolecular complexes atmultiple length-scale since it itself does not involve detailedcalculations of the normal modes of substructures, thus whensimulating the dynamics of very large biomolecular system,it is possible to model substructures of particular interest atatomic level and those of less interest at coarse-grained level.Our calculations support the ideas that the overall dynam-ics of a biomolecular complex might be understood with acombination of two factors: the dynamics of its componentstructures and the ways the components interact with oneanother.
ACKNOWLEDGMENTS
This work is supported by the National Natural ScienceFoundation of China (NNSFC) (Grant No. 30870509) and astartup fund of Fudan University.
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
152.2.176.242 On: Sun, 30 Nov 2014 11:55:23

144108-10 Chen et al. J. Chem. Phys. 135, 144108 (2011)
1K. Henzler-Wildman and D. Kern, Nature (London) 450(7172), 964(2007); M. Gerstein and N. Echols, Curr. Opin. Chem. Biol. 8(1), 14(2004).
2D. Russel, K. Lasker, J. Phillips, D. Schneidman-Duhovny, J. A.Velazquez-Muriel, and A. Sali, Curr. Opin. Cell. Biol. 21(1), 97(2009).
3J. L. Klepeis, K. Lindorff-Larsen, R. O. Dror, and D. E. Shaw, Curr. Opin.Struct. Biol. 19(2), 120 (2009).
4B. Brooks and M. Karplus, Proc. Natl. Acad. Sci. U.S.A. 80(21), 6571(1983); N. Go, T. Noguti, and T. Nishikawa, ibid. 80(12), 3696 (1983);M. Levitt, C. Sander, and P. S. Stern, J. Mol. Biol. 181(3), 423 (1985);J. Ma and M. Karplus, Proc. Natl. Acad. Sci. U.S.A. 95(15), 8502 (1998);A. Thomas, K. Hinsen, M. J. Field, and D. Perahia, Proteins 34(1), 96(1999); D. N. Kim, J. Altschuler, C. Strong, G. McGill, and M. Bathe,Nucleic Acids Res. 39, D451 (2011).
5Q. Cui, G. Li, J. Ma, and M. Karplus, J. Mol. Biol. 340(2), 345 (2004).6M. M. Tirion, Phys. Rev. Lett. 77(9), 1905 (1996).7I. Bahar, A. R. Atilgan, and B. Erman, Folding Des. 2(3), 173 (1997);D. Ming, Y. Kong, M. A. Lambert, Z. Huang, and J. Ma, Proc. Natl. Acad.Sci. U.S.A. 99(13), 8620 (2002); T. Haliloglu, I. Bahar, and B. Erman,Phys. Rev. Lett. 79, 4 (1997).
8K. Hinsen, Proteins 33(3), 417 (1998).9F. Tama, F. X. Gadea, O. Marques, and Y. H. Sanejouand, Proteins 41(1), 1(2000).
10G. Li and Q. Cui, Biophys. J. 83(5), 2457 (2002).11A. Ghysels, D. Van Neck, V. Van Speybroeck, T. Verstraelen, and M.
Waroquier, J. Chem. Phys. 126(22), 224102 (2007).12J. Hafner and W. Zheng, J. Chem. Phys. 130(19), 194111 (2009).13I. Bahar and A. J. Rader, Curr. Opin. Struct. Biol. 15(5), 586 (2005);
V. Tozzini, ibid. 15(2), 144 (2005).14A. R. Atilgan, S. R. Durell, R. L. Jernigan, M. C. Demirel, O. Keskin, and
I. Bahar, Biophys. J. 80(1), 505 (2001).15F. Tama, M. Valle, J. Frank, and C. L. Brooks III, Proc. Natl. Acad. Sci.
U.S.A. 100(16), 9319 (2003); O. Keskin, I. Bahar, D. Flatow, D. G. Covell,and R. L. Jernigan, Biochemistry 41(2), 491 (2002).
16Z. Yang, I. Bahar, and M. Widom, Biophys. J. 96(11), 4438 (2009).17K. Eom, S. C. Baek, J. H. Ahn, and S. Na, J. Comput. Chem. 28(8), 1400
(2007); H. Gohlke and M. F. Thorpe, Biophys. J. 91(6), 2115 (2006);
G. Song and R. L. Jernigan, Proteins 63(1), 197 (2006); R. D. Hills,Jr., L. Lu, and G. A. Voth, PLOS Comput. Biol. 6(6), e1000827 (2010);A. Arkhipov, P. L. Freddolino, K. Imada, K. Namba, and K. Schulten,Biophys. J. 91(12), 4589 (2006).
18W. C. Hurty, AIAA Journal 3(4), 678 (1965); A. L. Hale and L. Meirovitch,J. Sound Vib. 69(2), 309 (1980).
19D. Ming, Y. Kong, Y. Wu, and J. Ma, Proc. Natl. Acad. Sci. U.S.A. 100(1),104 (2003).
20D. Ming, Y. Kong, Y. Wu, and J. Ma, Biophys. J. 85(1), 27 (2003).21G. Temple and W. G. Bickley, Rayleigh’s Principle and Its Applications to
Engineering (Dover, New York, 1956).22D. Douguet, H. C. Chen, A. Tovchigrechko, and I. A. Vakser, Bioinformat-
ics 22(21), 2612 (2006).23B. R. Brooks, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus,
J. Comput. Chem. 4, 187 (1983).24M. Lu and J. Ma, Proc. Natl. Acad. Sci. U.S.A. 105(40), 15358 (2008);
J. N. Stember and W. Wriggers, J. Chem. Phys. 131(7), 074112 (2009).25B. R. Brooks, D. Janežic, and M. Karplus, J. Comput. Chem. 16(12), 1522
(1995).26D. Ming, M. E. Wall, and K. Y. Sanbonmatsu, BMC Bioinf. 8, 470 (2007).27M. Stewart, H. M. Kent, and A. J. McCoy, J. Mol. Biol. 277(3), 635 (1998).28B. M. Paschal and L. Gerace, J. Cell. Biol. 129(4), 925 (1995); U. Nehrbass
and G. Blobel, Science 272(5258), 120 (1996).29M. Li, L. H. Phylip, W. E. Lees, J. R. Winther, B. M. Dunn, A. Wlodawer,
J. Kay, and A. Gustchina, Nat. Struct. Biol. 7(2), 113 (2000).30F. Tama and C. L. Brooks III, J. Mol. Biol. 345(2), 299 (2005); H. W. van
Vlijmen and M. Karplus, ibid. 350(3), 528 (2005); G. Vliegenthartand G. Gompper, J. Comput.-Aided Mater. Des. 14(0), 111 (2007);A. Arkhipov, W. H. Roos, G. J. Wuite, and K. Schulten, Biophys. J. 97(7),2061 (2009); E. C. Dykeman and O. F. Sankey, Phys. Rev. E 81(2), 021918(2010).
31N. Ban and A. McPherson, Nat. Struct. Biol. 2(10), 882 (1995).32E. Neria, S. Fischer, and M. Karplus, J. Chem. Phys. 105(5), 1902 (1996).33K. C. Holmes, J. Struct. Biol. 170(2), 184 (2010).34T. Oda, M. Iwasa, T. Aihara, Y. Maeda, and A. Narita, Nature (London)
457(7228), 441 (2009).35W. Humphrey, A. Dalke, and K. Schulten, J. Mol. Graphics 14(1), 33
(1996).
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
152.2.176.242 On: Sun, 30 Nov 2014 11:55:23