viral genome replication || host factors that restrict retrovirus replication
TRANSCRIPT

Chapter 15Host Factors that Restrict RetrovirusReplication
Mark D. Stenglein, April J. Schumacher, Rebecca S. LaRue,and Reuben S. Harris
Abstract/Primer Over the past several decades, it has become clear that a varietyof cellular proteins actively restrict retrovirus replication. Two families of proteinsin particular, the TRIMs and the APOBEC3s, coordinate a robust innate defense toretrovirus infection. The TRIM proteins, led by TRIM5alpha, impose a replicationblock after entry, such that the invading retrovirus is degraded prior to integration.The APOBEC3 proteins, notably APOBEC3G, inhibit the replication of retrovirusesby a mutagenic mechanism that is associated with degradation of viral DNA. Retro-viruses have evolved means of avoiding their host’s TRIM and APOBEC3 defenses.Often, however, this leaves the virus susceptible to TRIMs and APOBECs fromother species. Thus, these restriction systems limit the cross-species mobility ofretroviruses. The prospects of developing new antiviral therapies that exploit theseinnate host defenses are promising.
Abbreviations AIDS, acquired immunodeficiency syndrome; AGM, Africangreen monkey; APOBEC1, apolipoprotein B mRNA editing catalytic subunit 1;APOBEC3G, apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like3G; CA, retroviral capsid protein; CypA, cyclophilin A; Fv, Friend virus; HIV-1,human immunodeficiency virus type 1; Lv1, lentivirus susceptibility factor 1; MLV,murine leukemia virus; Ref1, restriction factor 1; SIV, simian immunodeficiencyvirus; SIVmac, SIV derived from rhesus macaque; TRIM, tripartite motif protein;Vif, virion/viral infectivity factor; Z-motif, APOBEC zinc-binding motif.
R.S. Harris (B)Department of Biochemistry, Molecular Biology and Biophysics,University of Minnesota, 321 Church Street South East,6-155 Jackson Hall, Minneapolis, MN 55455, USAe-mail: [email protected]
C.E. Cameron et al. (eds.), Viral Genome Replication,DOI 10.1007/b135974 15, C© Springer Science+Business Media, LLC 2009
297

298 M.D. Stenglein et al.
Introduction
Overview
Most (if not all) organisms are vulnerable to viral infections and, consequently,many systems have been developed for protection. Many bacteria, for instance,utilize site-specific restriction endonucleases to cleave foreign DNA that is notmarked as ‘self’ (reviewed by Tock and Dryden, 2005). The B and T lymphocytesof vertebrates are capable of specialized adaptive immune responses, recognizingforeign invaders (antigens), and neutralizing them through specific antibody- andcell-mediated responses. In addition to adaptive immunity, vertebrates have innatedefense systems that recognize and eliminate invading pathogens. Key moleculesinclude Toll-like receptors, antimicrobial peptides, interferons, and many others,which are beyond the scope of this chapter (reviewed in Chapter 9 of Fields et al.,2007). Here we focus on mammalian proteins, termed retrovirus restriction factors,which limit the infectivity of a broad and growing number of viruses (reviewed byBieniasz, 2004; Goff, 2004; Mangeat and Trono, 2005; Chiu and Greene, 2006;Hache et al., 2006; Holmes et al., 2007b; Towers, 2007).
Brief History
A longstanding rule is that any given retrovirus can infect only certain cell types (e.g.,Friend, 1957; Lilly, 1967). Cells in which the virus can replicate are termed ‘permis-sive’ and other cells are termed ‘non-permissive’. One obvious reason for this is thatmany non-permissive cells lack machinery that the virus requires for replication. Forinstance, viruses often employ receptors located on the cell surface to facilitate theirentry into the cell. An example of this is the CD4 and the CXCR4 or CXCR5 polypep-tides that the human immunodeficiency virus-1 (HIV-1) envelope protein (gp120)recognizes and uses for particle entry. This in part explains why CD4+ T cells are afavored HIV-1 reservoir, and why HIV-1 cannot efficiently infect CD4–. Many othercellular factors have integral roles in nearly every stage of the retrovirus life cycle [seeChapter 6 in this text and an excellent review by Goff (2007)].
Another decades-old observation is that some cell types that seem to containthe necessary complement of positively acting factors are nevertheless resistant toretroviral infection. One example of such a scenario is the ‘resting’ CD4+ T cell,which is resistant to HIV-1 infection (Stevenson et al., 1990; Zack et al., 1990). Asecond type of resistance can be observed when cross-species viral infections areattempted, even between similar species. For instance, although HIV-1 efficientlyinfects most humans, it is unable to infect several closely related primate species,for example, the rhesus macaque (Shibata et al., 1995; Himathongkham and Luciw,1996; Hofmann et al., 1999). These observations can be explained by hypothesiz-ing that certain cells express dominant factors that interfere with retroviral replica-tion. Several such proteins have now been described and they are called restrictionfactors.

15 Host Factors that Restrict Retrovirus Replication 299
To date, the discovery and characterization of all retroviral restriction factors hasfollowed a similar general storyline. First, an investigator notices that a particularvirus is able to infect some type of cell or organism but is unable to infect a closelyrelated organism or cell type (in the best-case scenario these cell lines are clonallyrelated). Second, the nature of this difference in infectivity is determined. Is the lackof infection due to the lack of a necessary co-factor or the presence of an inhibitor?At what stage of the viral life cycle does replication halt? Is the block to infectiongenetically dominant? Finally, cloning of the gene in question precipitates a majoradvance in the understanding of the molecular mechanism of the restriction andelucidation of additional steps of the viral life cycle.
The First Described Restriction Factors: Fv1 and Fv4
The origin of the hunt for retroviral restriction factors can be traced back to 1956,when Charlotte Friend isolated a virus (now called the Friend murine leukemia virusor Friend MLV) that was able to infect and cause leukemia in some mouse strainsbut not others (Friend, 1957). Over the next several decades, many scientists investi-gated the basis of this difference (for additional reviews on this topic, see Jolicoeur,1979; Goff, 1996; Bieniasz, 2003, 2004; Goff, 2004). These investigations havefocused most extensively on two genes, Fv1 and Fv4 (Lilly, 1967; Suzuki, 1975).
The Fv4 gene is found in a Japanese mouse strain and genetic crosses using thisstrain demonstrated that Fv4 confers dominant resistance to Friend MLV infection(Suzuki, 1975). Cells isolated from Fv4+ mice and cultured in vitro are also resistantto infection, suggesting that resistance due to Fv4 does not depend on a compleximmune response (Kai et al., 1976). Ikeda and coworkers discovered the molecularidentity of Fv4, which is an envelope gene of a truncated, integrated, MLV-likeprovirus (Ikeda et al., 1985). When expressed on the cell surface, this envelope-like protein competes with the envelope proteins of incoming virus particles forbinding with their cognate receptor (Kai et al., 1986; Ikeda and Sugimura, 1989).This prevents the retrovirus from entering the cell, and therefore from replicating.
Fv1 is another locus that dominantly confers resistance to MLV infection (Lilly,1967). Like Fv4, Fv1 confers resistance in cells grown in tissue culture (Hartleyet al., 1970). Fv1 and Fv4 segregate independently in genetic crosses, and thus arenot identical (Suzuki, 1975). Crossing experiments revealed that there are two majorFv1 alleles: Fv1N and Fv1B (Hartley et al., 1970). NIH Swiss mice are homozygousfor the Fv1N allele, and Balb/c mice are homozygous carriers of the Fv1B allele.A mouse’s Fv1 genotype controls its susceptibility to infection by different strainsof MLV. So, N-tropic virus is able to replicate in mice carrying the Fv1N allele(NIH mice), but not on mice with a B allele (Balb/c). Conversely, B-tropic virus canreplicate on mice with the Fv1B allele. The N and B alleles confer resistance dom-inantly; a heterozygous mouse, with genotype Fv1B/N is resistant to both N-tropicand B-tropic virus (Pincus et al., 1971). This dominance suggests that the Fv1 geneproduct is not a necessary co-factor for viral replication but rather an inhibitoryfactor.
300 M.D. Stenglein et al.
Another informative characteristic of Fv1-mediated resistance is that it is sat-urable (Decleve et al., 1975; Pincus et al., 1975). This means that the resistance canbe overcome by increasing the titer of virus used for inoculation. This is true evenif the inoculum consists of replication-defective viral particles (Bassin et al., 1978;Boone et al., 1990). Models to explain this phenomenon propose that saturating viralparticles titrate out the machinery on which Fv1-mediated restriction relies.
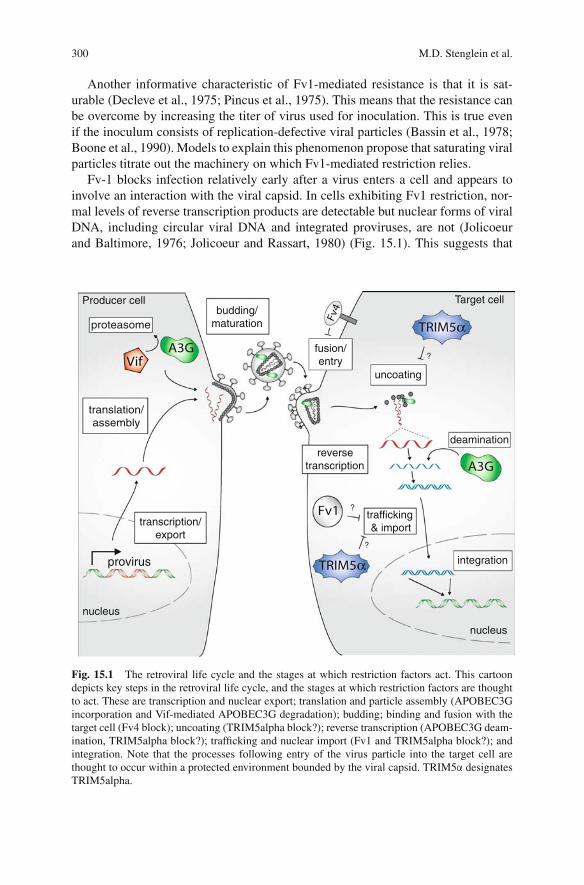
Fv-1 blocks infection relatively early after a virus enters a cell and appears toinvolve an interaction with the viral capsid. In cells exhibiting Fv1 restriction, nor-mal levels of reverse transcription products are detectable but nuclear forms of viralDNA, including circular viral DNA and integrated proviruses, are not (Jolicoeurand Baltimore, 1976; Jolicoeur and Rassart, 1980) (Fig. 15.1). This suggests that
provirus
translation/ assembly
budding/ maturation
fusion/ entry
nucleus
trafficking & import
integration
uncoating
reverse transcription
transcription/ export
proteasome
deamination
?A3G
TRIM5α
Vif
A3G
?
TRIM5α
Fv1 ?
Fv4
Target cellProducer cell
nucleus
Fig. 15.1 The retroviral life cycle and the stages at which restriction factors act. This cartoondepicts key steps in the retroviral life cycle, and the stages at which restriction factors are thoughtto act. These are transcription and nuclear export; translation and particle assembly (APOBEC3Gincorporation and Vif-mediated APOBEC3G degradation); budding; binding and fusion with thetarget cell (Fv4 block); uncoating (TRIM5alpha block?); reverse transcription (APOBEC3G deam-ination, TRIM5alpha block?); trafficking and nuclear import (Fv1 and TRIM5alpha block?); andintegration. Note that the processes following entry of the virus particle into the target cell arethought to occur within a protected environment bounded by the viral capsid. TRIM5α designatesTRIM5alpha.

15 Host Factors that Restrict Retrovirus Replication 301
Fv1-restricted viruses enter the cell and reverse transcribe their genome, but fail totransit to the nucleus and integrate their genome. A single amino acid (110) in theviral capsid protein (CA) defines the difference in Fv1 susceptibility between N- andB-tropic MLV strains, implying that Fv1 targets the viral capsid (DesGroseillers andJolicoeur, 1983; Kozak and Chakraborti, 1996).
The molecular identity of Fv1 was determined in 1996 by the Stoye lab, whoshowed that Fv1 was a gene with sequence similarity to the gag gene of endogenousretroviruses of the HERV-L/MERV-L family (Best et al., 1996). This suggests thatthe Fv1 protein may be directly engaging the retroviral particle, but the details ofthis restriction remain to be determined.
TRIM5alpha and Related Proteins
Retroviral Restriction Factors in Mammalian Cells: Ref1, Lv1,and TRIM5alpha
The first of the two major families of mammalian restriction factors that this chap-ter will focus on is the TRIM5s. As was the case with Fv1 and Fv4, many of thecharacteristics of TRIM5-mediated restriction were described several years beforeits molecular identity was discovered.
As discussed above, the mouse Fv1 and Fv4 genes confer resistance to infectionby certain MLV strains. In 2000, Towers et al. reasoned that other mammalian cellsmight express similar factors that confer resistance to MLV infection (Towers et al.,2000). Indeed, many mammalian cell lines proved resistant to infection by N-tropicMLV (although most remained relatively susceptible to infection by B-MLV). Theauthors attributed this resistance to cellular inhibitors of viral replication similar toFv1, and named the putative factor Ref1 (restriction factor 1).
Around the same time, evidence was mounting that retroviruses other than MLV,including primate lentiviruses were limited in their host range (Shibata et al., 1995;Himathongkham and Luciw, 1996; Hofmann et al., 1999). For example, manynon-human primates could not be productively infected with HIV-1. It was uncer-tain whether this limitation was due to the absence of factors in these cells thatthe viruses required to replicate or to dominant antiviral factors such as Fv1 andRef1. This question was resolved by several key studies in 2002, which showed thatmany primate cells express a dominant factor that, like Ref1, blocks lentiviral infec-tion (Besnier et al., 2002; Cowan et al., 2002; Munk et al., 2002). This factor wasdubbed Lv1 (lentivirus susceptibility factor 1) (Cowan et al., 2002).
The original studies describing Ref1 and Lv1 and those that followed closely ontheir heels began to paint a clearer picture of these restriction factors and their mech-anism of inhibition (Towers et al., 2000; Besnier et al., 2002; Cowan et al., 2002;Munk et al., 2002; Towers et al., 2002; Besnier et al., 2003; Hatziioannou et al.,2003; Kootstra et al., 2003; Berthoux et al., 2004; Hatziioannou et al., 2004b). Mosttested cell lines restricted infection of some retroviruses, especially retroviruses

302 M.D. Stenglein et al.
from other species. For example, human cells restricted infection by N-MLV andequine infectious anemia virus (EIAV) but did not restrict HIV-1, HIV-2, or the rhe-sus macaque simian immunodeficiency virus (SIVmac) (Hatziioannou et al., 2003).
These studies also showed that Ref1 and Lv1 blocked viral infection in a waythat was reminiscent of the Fv1 block in murine cells. Furthermore, several keycharacteristics of Fv1-mediated and Ref1-mediated restriction were identical. First,restriction by Ref1 and Lv1 was dominant in heterokaryon assays, in which per-missive and non-permissive cells are fused (Cowan et al., 2002; Munk et al., 2002).Secondly, for Lv1 and Ref1, as was for Fv1, the viral capsid protein determineswhether a virus is sensitive to restriction (Kozak and Chakraborti, 1996; Towerset al., 2000; Cowan et al., 2002; Hatziioannou et al., 2004b; Ikeda et al., 2004;Dodding et al., 2005; Lassaux et al., 2005). Typically, the capsid of a particularretrovirus confers resistance to restriction by the Ref1 or Lv1 of the virus’s hostspecies. For example, replacing the CA and adjoining P2 region of the SIVmac gaggene with the corresponding HIV-1 sequence results in a virus that is restricted likeHIV-1 (Cowan et al., 2002). Thirdly, Ref1 and Lv1 block replication at a similarstage of the viral life cycle. Judging by the failure to detect reverse-transcribed lin-ear viral DNA, this block occurs after the virus has entered the target cell but beforeit has reverse transcribed its genome (Fig. 15.1) (Towers et al., 2000; Besnier et al.,2002; Cowan et al., 2002; Munk et al., 2002). An important distinction is that thisis slightly earlier than the Fv1-mediated block in mouse cells, which occurs afterreverse transcription but before nuclear import of the viral pre-integration complex(Jolicoeur and Rassart, 1980; Yang et al., 1980; Towers et al., 2000). A final simi-larity between Ref1 and Lv1 was that, as was the case with Fv1, restriction could besaturated, i.e., overwhelmed by increasing the viral titer or by pre-treating the cellswith virus particles (Hartley et al., 1970; Boone et al., 1990; Besnier et al., 2002;Cowan et al., 2002; Munk et al., 2002; Towers et al., 2002; Kootstra et al., 2003;Dodding et al., 2005). Some viruses were even found capable of cross-saturation.For example, African green monkey cells normally restrict HIV-1 and SIVmac, butpretreatment of these cells with HIV-1 particles increases the efficiency of a subse-quent SIVmac infection and vice versa (Cowan et al., 2002). These data suggestedthat a single mechanism inhibits both HIV-1 and SIVmac in these cells.
Because of the striking similarities between Lv1 and Ref1, it was speculated thatthey were species-specific alleles of the same gene (Cowan et al., 2002; Serhanet al., 2002). The cloning of the gene responsible for both activities validated thisprediction.
The Cloning of TRIM5alpha/Ref1/Lv1
As explained above, HIV-1 is capable of infecting human cells but unable to infectmany other primate cells. This inability was attributed to a restriction activity namedLv1, but the molecular identity of Lv1 was unknown. In a breakthrough study in2004, Stremlau and colleagues identified a cDNA from a rhesus macaque librarythat conferred to human cells resistance to HIV-1 infection (Stremlau et al., 2004).

15 Host Factors that Restrict Retrovirus Replication 303
The cDNA contained a rhesus gene named TRIM5alpha. Several characteristics ofTRIM5alpha-mediated restriction indicated that it might be the Lv1/Ref1 gene. Theblock occurred prior to reverse transcription, as evidenced by a severe decrease inthe accumulation of reverse transcription products (Fig. 15.1). Chimeric SIV/HIVviruses showed that the viral determinants of the block mapped to the capsid (CA)gene. Knockdown of endogenous TRIM5alpha in primary rhesus monkey lungfibroblasts resulted in an increase in the efficiency of HIV-1 infection.
It was not long before it was confirmed that species-specific alleles ofTRIM5alpha were identical to (or at least essential for) Lv1 in monkey cells andRef1 in human cells (Hatziioannou et al., 2004a; Keckesova et al., 2004; Perronet al., 2004; Yap et al., 2004). In accordance with this idea, human TRIM5alphaexpression limited infection by N-MLV but not B-MLV. Similarly, expression ofnon-human primate TRIM5alpha variants limited infectivity by a variety of retro-viruses. In all cases, heterologous expression of a TRIM5alpha allele conferred arestriction activity normally associated with cells of the species from which theallele had been derived (Hatziioannou et al., 2004a; Keckesova et al., 2004; Perronet al., 2004; Yap et al., 2004; Nakayama et al., 2005; Saenz et al., 2005; Songet al., 2005a; Ylinen et al., 2005; Kaumanns et al., 2006). Conversely, knockdownof endogenous TRIM5alpha increased cells’ susceptibility to infection by normallyrestricted viruses.
The overarching conclusion from these studies was that many primate cellsexpress TRIM5alpha proteins that block infection by a variety of retroviruses. Typ-ically, however, TRIM5alpha of a given species is ineffective against that species’retroviruses. For instance human TRIM5alpha exhibits only a modest inhibitoryeffect against HIV-1 (Stremlau et al., 2004). This suggests that retroviruses haveevolved ways around the TRIM5alpha-imposed restriction in their host’s cells.
The Mechanism of TRIM5alpha-Mediated Retroviral Restriction
In humans, TRIM5alpha is just one of a large family of TRIM genes (there arenearly 70 in humans) (Reymond et al., 2001; Nisole et al., 2005). The TRIM proteinsare named after their characteristic tripartite motif (Fig. 15.2) (Reddy et al., 1992;Borden, 1998; Reymond et al., 2001). This motif, also known as the RBCC motif,consists of a RING domain, one or two B boxes, and a coiled-coil domain.
The RING domain (really interesting new gene) contains a number of conservedcysteines and histidines that coordinate two zinc atoms (Saurin et al., 1996). SomeRING domains have been reported to have E3 ubiquitin ligase activity, and someTRIM proteins (including TRIM5alpha) have been shown to ubiquitinate them-selves in a RING domain-dependent manner (Xu et al., 2003; Diaz-Griffero et al.,2006a).
The B box is another zinc-binding domain with a three-dimensional structuresimilar to a RING domain, but with a poorly characterized function (Reddy andEtkin, 1991; Reddy et al., 1992; Massiah et al., 2007). TRIM5alpha has a single Bbox, but other TRIMs have one or two of these domains.

304 M.D. Stenglein et al.
4971
Tripartite Motif (TRIM)
RING domain
Zn-binding
Ubiquitin ligase activity
B-box 2 domainZn-binding
Other?
Coiled Coil domain
Trimerization
B30.2 (SPRY) domain
CA recognition/binding
Functionally replaced by Cyclophilin A in TrimCyp
RING BB CC B30.2 (SPRY)
Fig. 15.2 A cartoon depicting the domain organization of rhesus macaque TRIM5alpha.
The coiled-coil domain has been implicated in multimerization of the TRIM pro-teins, and TRIM5alpha forms a trimer (Reymond et al., 2001; Mische et al., 2005;Perez-Caballero et al., 2005; Javanbakht et al., 2006a). Truncated TRIM5alphaproteins lacking the coiled-coil domain fail to multimerize, demonstrating thatthis domain is required for multimerization (Mische et al., 2005). However, theTRIM5gamma isoform, which lacks the carboxy-terminal B30.2(SPRY) domain,forms dimers, not trimers, suggesting that multiple domains influence the oligomericstate of the protein.
In TRIM5alpha, the tripartite (RBCC) motif is followed by a B30.2(SPRY)domain (Fig. 15.2) (Henry et al., 1997; Rhodes et al., 2005; Woo et al., 2006; Jameset al., 2007). SPRY domains have been implicated in protein–protein interactions.
All of the domains of TRIM5alpha are necessary to block retroviral infection.Truncated proteins lacking the amino-terminal RING, B box, or coiled-coil domainshave severely attenuated antiviral activity (Javanbakht et al., 2005; Perez-Caballeroet al., 2005). Mutant proteins with a disrupted coiled-coil domain fail to trimerizeand also fail to block infection (Javanbakht et al., 2006a). A similar inability toblock infection is observed with some point mutants in these domains (Stremlauet al., 2004; Javanbakht et al., 2005, 2006a). Similarly, truncated proteins lackingthe SPRY domain show that the tripartite motif (RBCC domains) by itself is inactiveagainst retroviruses (Stremlau et al., 2004; Perez-Caballero et al., 2005). A model toexplain the contribution of TRIM5alpha’s domains proposes that the B30.2(SPRY)domain provides the capsid interaction surface, the coiled-coil domain promotestrimerization, and the RING and B box domains provide some unknown effectorfunction, perhaps involving the RING domain-associated ubiquitin ligase activity(Fig. 15.2).
Different species’ TRIM5alphas restrict different retroviruses. Four variableloops in the B30.2(SPRY) domain determine this difference in efficacy (Nakayamaet al., 2005; Stremlau et al., 2005; Yap et al., 2005; Ohkura et al., 2006; Perron et al.,2006). Primate species’ TRIM5alpha alleles differ most in these variable regions(Song et al., 2005b). These regions, called V1–V4, are predicted to be surface-exposed loops (Ohkura et al., 2006; Perron et al., 2006; Woo et al., 2006; Jameset al., 2007). It has been proposed that these loops form the capsid interaction

15 Host Factors that Restrict Retrovirus Replication 305
surface, and that different loop configurations enable interactions with differentviral capsids. A striking finding is that the specificity can be determined by aslittle as a single amino acid (Stremlau et al., 2005; Yap et al., 2005; Li et al.,2006b). Amino acid 332 of TRIM5alpha is arginine in humans and proline in rhesusmacaques. Mutating the arginine to proline in human TRIM5alpha confers stronganti-HIV-1 activity to the human protein (Stremlau et al., 2005; Yap et al., 2005;Li et al., 2006b). An intriguing hypothesis is that TRIM5alpha alleles, differing intheir B30.2(SPRY) variable loops, become fixed in populations in response to pan-demic retroviral infections (Kaiser et al., 2007). In this case, the ability of the fixedallele to ward off the exigent pathogen is counterbalanced by its inactivity againstother retroviruses. Such a scenario has been proposed to explain why the humanTRIM5alpha allele is inactive against HIV-1 (Kaiser et al., 2007).
The TRIM5alpha gene is found in a cluster of TRIM genes on chromosome11p15 (Reymond et al., 2001). From this locus six alternatively spliced TRIM5isoforms are expressed (Reymond et al., 2001). TRIM5alpha is the longest of these,and it encodes a protein of 497 amino acids (rhesus macaque TRIM5alpha). OtherTRIM5 isoforms lack at least one of the above-mentioned domains and those testedlack antiviral activity (Stremlau et al., 2004; Perez-Caballero et al., 2005).
TRIM5s are expressed ubiquitously in adult tissues (Reymond et al., 2001).TRIM5alpha localizes to the cytoplasm of cells, in a diffuse cytosolic manner and inbodies that appear as bright dots in fluorescent microscopy (Reymond et al., 2001;Xu et al., 2003; Campbell et al., 2007). This localization pattern does not appear tobe functionally required for blocking retroviral infection, although it is consistentwith the requirement that the protein be able to engage incoming viral particles inthe cytoplasm (Perez-Caballero et al., 2005;, Song et al., 2005c).
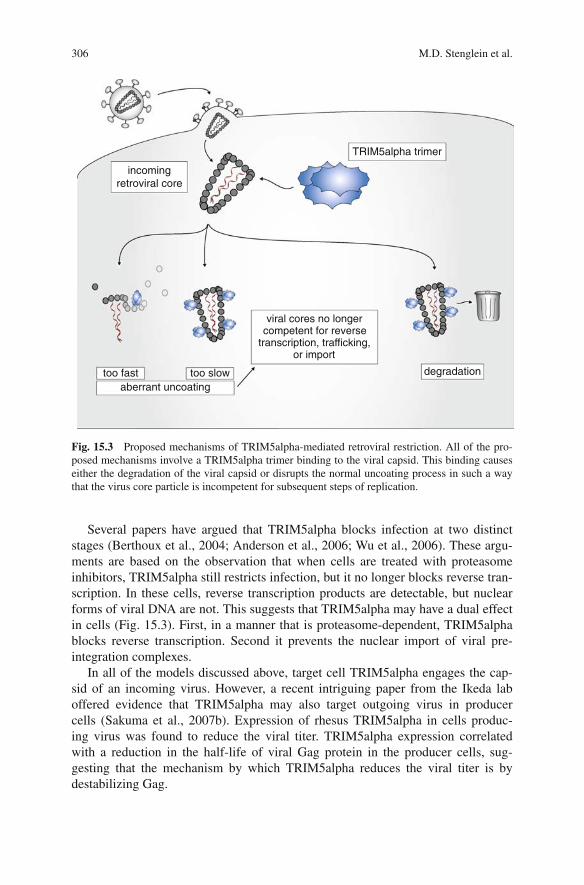
The precise mechanism by which TRIM5alpha acts remains unclear. Severalcandidate mechanisms have been put forward, the general theme of which is thatTRIM5alpha acts by modulating the stability or activity of the viral core (Fig. 15.3).All of the mechanisms involve a TRIM5alpha trimer binding the hexameric lat-tice of the capsid of an incoming virus. Indeed, TRIM5alpha–capsid binding hasbeen demonstrated in vitro, and mutations to either protein that disrupt this interac-tion correlate with a loss of restriction (Sebastian and Luban, 2005; Chatterji et al.,2006; Li et al., 2006b; Stremlau et al., 2006b). The difference between the proposedmechanisms is the fate of the TRIM5-bound viral capsid (Fig. 15.3). In the simplestcase, TRIM5alpha binds to the capsid and renders it incompetent to perform reversetranscription. This could be because TRIM5alpha blocks necessary cellular factorsfrom accessing the viral core. A corollary of this model is that TRIM5alpha cova-lently modifies the capsid, perhaps by ubiquitinating or sumoylating it (Stremlauet al., 2004). In another proposed model, TRIM5alpha binding triggers the degra-dation of the capsid (Chatterji et al., 2006). This destruction of the capsid wouldpreclude the completion of the virus replication cycle. A final model proposes thatTRIM5alpha mediates the faster than normal uncoating of the capsid from the viralcore (Stremlau et al., 2006b; Perron et al., 2007). In this scenario, the accelerateduncoating disrupts the normal progression of events required for a successful infec-tion (Forshey et al., 2002).
306 M.D. Stenglein et al.
viral cores no longercompetent for reverse
transcription, trafficking,or import
TRIM5alpha trimer
degradationtoo fast too slowaberrant uncoating
incoming retroviral core
Fig. 15.3 Proposed mechanisms of TRIM5alpha-mediated retroviral restriction. All of the pro-posed mechanisms involve a TRIM5alpha trimer binding to the viral capsid. This binding causeseither the degradation of the viral capsid or disrupts the normal uncoating process in such a waythat the virus core particle is incompetent for subsequent steps of replication.
Several papers have argued that TRIM5alpha blocks infection at two distinctstages (Berthoux et al., 2004; Anderson et al., 2006; Wu et al., 2006). These argu-ments are based on the observation that when cells are treated with proteasomeinhibitors, TRIM5alpha still restricts infection, but it no longer blocks reverse tran-scription. In these cells, reverse transcription products are detectable, but nuclearforms of viral DNA are not. This suggests that TRIM5alpha may have a dual effectin cells (Fig. 15.3). First, in a manner that is proteasome-dependent, TRIM5alphablocks reverse transcription. Second it prevents the nuclear import of viral pre-integration complexes.
In all of the models discussed above, target cell TRIM5alpha engages the cap-sid of an incoming virus. However, a recent intriguing paper from the Ikeda laboffered evidence that TRIM5alpha may also target outgoing virus in producercells (Sakuma et al., 2007b). Expression of rhesus TRIM5alpha in cells produc-ing virus was found to reduce the viral titer. TRIM5alpha expression correlatedwith a reduction in the half-life of viral Gag protein in the producer cells, sug-gesting that the mechanism by which TRIM5alpha reduces the viral titer is bydestabilizing Gag.

15 Host Factors that Restrict Retrovirus Replication 307
In summary, several mechanisms have been put forth to model TRIM5alpha-mediated retrovirus restriction. Each model has supporting evidence. Moreover, itis important to note that these mechanisms are not necessarily mutually exclusive.Indeed, it would behoove the cell to target the virus at as many points as possi-ble. As data continue to accumulate, the mechanism or mechanisms of TRIMalpha-mediated restriction will become apparent.
Cylclophilin A and TRIM
It is impossible to discuss TRIM5alpha-mediated retroviral restriction without dis-cussing its intimate relationship with cyclophilin A (reviewed by Nisole et al., 2005;Luban, 2007; Towers, 2007). Cyclophilin A (CypA) is a peptidyl–prolyl isomeraseencoded by the PPIA gene (Fischer et al., 1998). It was identified as a HIV-1 capsid-binding protein in a yeast two-hybrid screen (Luban et al., 1993). CypA interactswith a proline-rich surface of the HIV-1 capsid protein, and it catalyzes the iso-merization of the peptidyl–prolyl bond between residues glycine 89 and proline90 (Gamble et al., 1996; Bosco et al., 2002). The CypA–CA interaction has beenshown to be important for HIV-1 infectivity, as disruption of this interaction resultsin a substantial decrease in infectivity (Thali et al., 1994; Braaten et al., 1996c;Braaten et al., 1996a,b; Braaten and Luban, 2001). The target cell (i.e., the cell thatis being newly infected) provides the functionally important CypA (Franke et al.,1994; Sokolskaja et al., 2004; Hatziioannou et al., 2005).
CypA modulates TRIM5alpha’s antiviral activity in certain cell types. In rhesusmacaques and African green monkey cells, TRIM5alpha restriction requires CypA(Berthoux et al., 2005a; Chatterji et al., 2005; Keckesova et al., 2006; Sokolskajaet al., 2006). It has been proposed that in these simian cells, CypA isomerizationof the capsid renders it sensitive to TRIM5alpha. In contrast to this, in human cellsCypA is required for HIV-1 infection. There, CypA binds to the HIV-1 capsid andthis binding is thought to protect the virus from the activity of a restriction factor.This restriction factor was originally thought to be human TRIM5alpha, but nowit is believed that CypA shields the capsid from an as yet to be discovered factor(Sayah and Luban, 2004; Keckesova et al., 2006; Sokolskaja et al., 2006; Stremlauet al., 2006a).
The relevance of CypA to TRIM5-mediated restriction is further highlighted (inastounding fashion) by the existence of a TRIMCyp fusion gene in several pri-mate species. The TRIMCyp gene appears to have arisen when a CypA mRNAwas transposed into the TRIM5 locus by LINE-1 retrotransposon machinery. TheLuban lab originally identified TRIMCyp while investigating an apparent CypA-mediated antiviral activity in owl monkey cells (Sayah et al., 2004). The TRIM-Cyp gene encodes a protein similar to TRIM5alpha but with CypA replacing theB30.2(SPRY) domain (Fig. 15.2). In this fusion protein, CypA supplies the capsid-binding activity previously provided by the B30.2(SPRY) domain (Nisole et al.,2004; Diaz-Griffero et al., 2006b). This allele is obviously an effective functionalreplacement for TRIM5alpha, as it has become fixed in all owl monkey species

308 M.D. Stenglein et al.
(Ribeiro et al., 2005). Since the original discovery of the (new world) owl monkeyTRIMCyp, a nearly equivalent TRIMCyp gene has been described in (old world)macaques (Liao et al., 2007; Brennan et al., 2008; Newman et al., 2008; Virgenet al., 2008; Wilson et al., 2008). These TRIMCyp genes appear to have evolvedindependently in the two primate lineages. This unlikely occurrence represents abeautiful example of convergent evolution and suggests that the TRIMCyp fusionprotein may provide a strong selective advantage.
The Evolution of TRIMs as Antiviral Defenses
In primates, TRIM5alpha is just one member of a large protein family, and severalstudies have assessed the antiviral activity of some of the other primate TRIMs,namely TRIMs 1, 4, 6, 18, 19, 21, 22, 27, and 34 (Yap et al., 2004, 2005; Li et al.,2006a; Zhang et al., 2006; Li et al., 2007). These TRIMs were chosen for studybecause they have exhibited activity against other viruses, because they have themost similar sequence or overall domain structure to TRIM5alpha, or because theirgenes are at the same locus on chromosome 11 as TRIM5alpha. Apart from twomonkey species’ TRIM1s, which restricts N-MLV (Yap et al., 2004, 2005), theseother TRIMs failed to exhibit significant activity against a variety of retroviruses.However, using chimeric proteins, two studies demonstrated that it is possible tofunctionally replace the RBCC domain of TRIM5alpha (or TRIMCyp) with those ofseveral other TRIMs (Li et al., 2006a; Yap et al., 2006). This suggests that the amino-terminal RBCC domains of these TRIMs are competent for restriction but they arenot effectively targeted to the viral capsid by their carboxy-terminal domains.
Truncated TRIM6 and TRIM34 proteins lacking their carboxy-terminalB30.2(SPRY) domain have a dominant negative effect on human TRIM5alpha’sability to restrict N-MLV infection (Zhang et al., 2006). These proteins may hetero-multimerize with TRIM5alpha, thereby preventing it from functioning normally.Various TRIM5 proteins exhibit a similar dominant negative effect on each other,especially in cross-species contexts or when truncated proteins are used (Stremlauet al., 2004; Berthoux et al., 2005b; Perez-Caballero et al., 2005).
It has been proposed that other TRIM proteins play roles in the defense againstviral infection. The promyelocytic leukemia protein, PML, also known as TRIM19,and TRIM22 are such family members (for further discussion, please see Nisoleet al., 2005; Everett and Chelbi-Alix, 2007; Towers, 2007).
In addition to the primate TRIM5alphas, there are orthologs in other mammalianspecies. So far, TRIM5alpha or TRIM5alpha-like TRIM proteins with antiretrovi-ral activity have been described in cows and rabbits (Si et al., 2006; Ylinen et al.,2006; Schaller et al., 2007). There is also a murine TRIM5 ortholog, but its antiviralactivity remains to be confirmed (Hoffman et al., 2006; Noser et al., 2006). It islikely that primate TRIM5s and these other mammalian homologs are derived froman ancestral TRIM with antiretroviral activity.
There is convincing evidence that selective pressures due to pathogenic retro-viruses have driven the evolution of the TRIM5s. As described above, severalvariable capsid-interacting loops in the B30.2(SPRY) domain determine the antiviral

15 Host Factors that Restrict Retrovirus Replication 309
specificity of different species’ TRIM5alpha proteins. Evolutionary analyses haveshown that the regions of the TRIM5 genes encoding these loops bear strong sig-natures of episodic positive selection (Liu et al., 2005; Sawyer et al., 2005; Ortizet al., 2006). Such signatures are attributable to pathogen-induced selection. A com-pelling study showed that two Old World Monkey species (sooty mangabeys andrhesus macaques) are maintaining multiple TRIM5alpha alleles (Newman et al.,2006). Phylogenetic analysis argues that these alleles were present in the commonancestor of these species and that therefore balancing selection has maintained thesealleles for millions of years. The complex evolutionary history of TRIM5s suggeststhat these proteins have been entangled in long-term struggles against pathogensincluding retroviruses.
TRIM Frontiers
Although significant progress has been made toward understanding TRIM5alpha-mediated retroviral restriction, many key questions remain unanswered. One signifi-cant point is that there has been no demonstration yet of TRIM5alpha’s effectivenessin vivo. All of the studies to date have used tissue culture systems (although it shouldbe mentioned that the in vitro results mirror the reality of many viruses’ limited hostranges). Several studies have examined the relationship between naturally occur-ring TRIM5 polymorphisms, the protein’s antiviral activity, and clinical measures ofHIV disease and epidemiology (Goldschmidt et al., 2006; Javanbakht et al., 2006b;Sawyer et al., 2006; Speelmon et al., 2006; Nakayama et al., 2007; Vigano et al.,2007). These studies have not produced convincing associations. Another impor-tant question is whether TRIM5alpha-related antiviral therapies can be developed.Two pilot studies have explored the possible use of TRIM5alpha in gene therapy(Anderson and Akkina, 2005; Sakuma et al., 2007a). The therapeutic expression ofrhesus TRIM5alpha in human cells would be predicted to make them resistant toHIV-1 infection. Drugs that modulate the TRIM5alpha–capsid interaction representanother therapeutic possibility. For example, small molecules that increased humanTRIM5alpha’s affinity for the HIV-1 capsid might empower the protein to betterrestrict the virus. All in all, our understanding of this important antiviral defensesystem has rocketed forward in recent years, but many important questions stillawait an answer.
The APOBEC3 Proteins of Mammals
Discovery
The second of the two major families of mammalian restriction factors thatthis chapter will focus on is the APOBEC3s. In particular, the focus will beon the antiretroviral activity of family member APOBEC3G (apolipoprotein BmRNA editing enzyme, catalytic polypeptide-like 3G, A3G). As was the case withTRIM5alpha, the existence of APOBEC3G was inferred before its identity was

310 M.D. Stenglein et al.
unveiled. In this case, APOBEC3G’s discovery was rooted in the observation thatHIV-1 molecular clones that lacked the virion infectivity factor (Vif) accessory genewere able to replicate only in a subset of human T-cell lines. The Vif-proficientparental virus replicated normally in these cells (e.g., Fisher et al., 1987; Strebelet al., 1987; Nara and Fischinger, 1988; Gabuzda et al., 1992; von Schwedler et al.,1993; Simon and Malim, 1996). Hybrid T-cell lines, derived from fusing permis-sive and non-permissive lines, also failed to replicate Vif-defective viruses (Madaniand Kabat, 1998; Simon et al., 1998). These experiments argued against modelsthat suggested that Vif was required to compensate for a cellular factor lacking innon-permissive cells. Instead, the results were consistent with the existence of adominant cellular factor in the non-permissive cells that prevented replication ofVif-defective HIV-1. The fact that Vif is required for HIV-1 replication on primaryhuman cells and the fact that Vif-deficient SIV fails to replicate or cause disease inrhesus macaques highlight the importance of the non-permissive condition and thelikelihood that it approximates the cellular environment in vivo (Fisher et al., 1987;Strebel et al., 1987; Gabuzda et al., 1992; von Schwedler et al., 1993; Gabuzda et al.,1994; Desrosiers et al., 1998; Victoria and Robinson, 2005).
This non-permissive versus permissive dichotomy was particularly striking forthe permissive CEM T-cell line and its non-permissive derivative, CEM-SS (Simonand Malim, 1996). Malim and coworkers reasoned that the dominant cellular factorwould be more highly expressed in non-permissive cells, and performed subtractivehybridization experiments to isolate messages present in CEM but absent in CEM-SS(Sheehy et al., 2002). Among the many differentially expressed mRNAs so identi-fied, one termed APOBEC3G (initially called CEM15) was expressed in CEM butnot in CEM-SS. Expression of the APOBEC3G gene alone rendered CEM-SS cellsnon-permissive for Vif-defective virus replication (Sheehy et al., 2002). APOBEC3Gshowed sequence similarity to the mRNA cytosine-to-uracil (C-to-U) editing proteinAPOBEC1, which leads to suggestions that APOBEC3G functioned by editing themessage of a cellular protein and thereby endowing it with anti-HIV activity. This wasjust one hypothesis, however, and the discovery by Malim and coworkers inspired anumber of investigations that answered two key questions: (1) what is the molecularmechanism underlying the antiviral effect of APOBEC3G and (2) how does Vif permitHIV-1 to replicate in the presence of this potent antiviral protein?
A Deamination-Dependent Mechanism
As described above, initial speculation on APOBEC3G’s antiviral mechanismfocused on the protein’s potential as an mRNA editor. This speculation wassoon discarded in favor of a model wherein APOBEC3G acts as a DNA muta-tor. Neuberger and colleagues provided evidence in favor of this by demonstrat-ing that APOBEC3G possesses a DNA (rather than an RNA) cytosine deaminaseenzymatic activity (Harris et al., 2002). Prior work from the Neuberger labora-tory had shown that a related protein, activation-induced deaminase (AID), was aDNA cytosine-to-uracil (C-to-U) deaminase. The evidence for this was that AID
15 Host Factors that Restrict Retrovirus Replication 311
expression in E. coli caused an increase in mutation frequencies and a correspond-ing C/G-to-T/A transition mutation bias (Petersen-Mahrt et al., 2002). Moreover,these effects were more pronounced in cells lacking uracil excision repair, strength-ening the conclusion that DNA cytosines were being deaminated (Petersen-Mahrtet al., 2002). The AID-catalyzed uracils would become fixed as C/G-to-T/A muta-tions when they templated the incorporation of adenines during replication. Simi-lar experiments showed that APOBEC3G triggered a mutator phenotype in E. coli,indicating that it is a DNA cytosine deaminase (Harris et al., 2002).
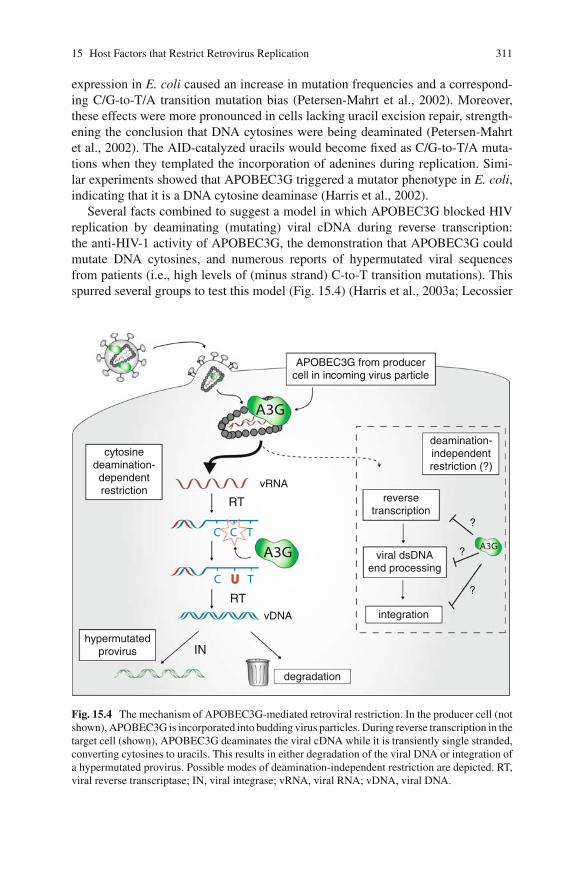
Several facts combined to suggest a model in which APOBEC3G blocked HIVreplication by deaminating (mutating) viral cDNA during reverse transcription:the anti-HIV-1 activity of APOBEC3G, the demonstration that APOBEC3G couldmutate DNA cytosines, and numerous reports of hypermutated viral sequencesfrom patients (i.e., high levels of (minus strand) C-to-T transition mutations). Thisspurred several groups to test this model (Fig. 15.4) (Harris et al., 2003a; Lecossier
APOBEC3G from producer cell in incoming virus particle
A3G
C C T
RT
UUC T
A3G
RT
cytosine deamination-dependent restriction
hypermutated provirus
deamination- independent restriction (?)
integration
reverse transcription
viral dsDNA end processing
A3G
?
?
?
IN
degradation
vRNA
vDNA
Fig. 15.4 The mechanism of APOBEC3G-mediated retroviral restriction. In the producer cell (notshown), APOBEC3G is incorporated into budding virus particles. During reverse transcription in thetarget cell (shown), APOBEC3G deaminates the viral cDNA while it is transiently single stranded,converting cytosines to uracils. This results in either degradation of the viral DNA or integration ofa hypermutated provirus. Possible modes of deamination-independent restriction are depicted. RT,viral reverse transcriptase; IN, viral integrase; vRNA, viral RNA; vDNA, viral DNA.

312 M.D. Stenglein et al.
et al., 2003; Mangeat et al., 2003; Zhang et al., 2003). When APOBEC3G wasexpressed in cells producing virus, the viral titer was not reduced, but the infectiv-ity of the resulting particles was severely attenuated. Also, in strong support of themodel, proviral DNA that accumulated in the presence of APOBEC3G exhibitedmassive increases in plus-strand G-to-A hypermutation. This hypermutation couldbe attributed to the deamination of minus-strand (cDNA) cytosines during reversetranscription (Fig. 15.4) (Harris et al., 2003a; Mangeat et al., 2003; Zhang et al.,2003). Thus, it seemed that a major part of the mechanism of APOBEC3G-mediatedrestriction of Vif-deficient HIV-1 was due to DNA cytosine deamination. Thiswas further supported by experiments indicating that the putative zinc-coordinatingresidues of the active site were required for antiviral activity (Mangeat et al., 2003).
In addition to triggering hypermutation of the viral genome, APOBEC3G alsocauses degradation of viral cDNA (Fig. 15.4) (Goncalves et al., 1996; Simonand Malim, 1996; Bishop et al., 2006; Holmes et al., 2007a; Mbisa et al., 2007).APOBEC3G’s enzymatic activity is required for this (Mbisa et al., 2007). The mech-anism of degradation has not been explained because, despite initial predictions,the most obvious cellular DNA repair pathway – UNG2-dependent base excisionrepair – does not appear to be involved (Harris et al., 2003a,b; Kaiser and Emerman,2006; Mbisa et al., 2007; Schumacher et al., 2008). However, a potentially importantclue was published independently by the Pathak and Yu groups, who showed thatAPOBEC3G interferes with integration by inhibiting integrase and/or by creatingaberrant cDNA ends that cannot be properly engaged by integrase (Luo et al., 2007;Mbisa et al., 2007). Future studies may clarify whether the degradation is an activereaction or simply the result of a failure to produce integration-competent viralDNA structures (which would eventually be degraded by cellular nucleases).
The human APOBEC3G protein is 384 amino acids long. It is composed of twosimilar domains arranged in tandem, each of which contains a characteristic zinc-binding motif (Fig. 15.5). It is impossible to discern by simply examining the aminoacid sequences of these motifs whether one or both harbor the protein’s enzymaticactivity. A number of studies have shown that the two domains provide complemen-tary activities. Studies with chimeric and mutant proteins showed that the carboxy-terminal zinc-binding domain harbors the protein’s enzymatic activity (Hache et al.,2005; Navarro et al., 2005; Newman et al., 2005; Iwatani et al., 2006; Chen et al.,2007, 2008). In contrast, mutations to the conserved residues in the amino-terminal‘pseudo-active’ site demonstrated that it is essential for nucleic acid binding andefficient incorporation into the viral particle (Fig. 15.5). It is thought that this amino-terminal domain allows APOBEC3G to gain access to the viral particle by binding aviral ribonucleoprotein complex that includes the viral genome and the viral nucle-ocapsid protein (e.g., Alce and Popik, 2004; Cen et al., 2004; Luo et al., 2004;Schafer et al., 2004; Svarovskaia et al., 2004; Zennou et al., 2004; Khan et al.,2005; Iwatani et al., 2006; Burnett and Spearman, 2007). Once so incorporated,APOBEC3G travels along in the virus particle to another cell that will be infected.There, APOBEC3G’s carboxy-terminal-mediated enzymatic activity is broughtto bear during reverse transcription. In this manner, both domains contribute toantiviral activity.

15 Host Factors that Restrict Retrovirus Replication 313
HxE-x24-28-PCx2-4C
Zn2+
beta sheetalpha helix
Zinc-binding motif
exon-exon junction
APOBEC3G
Pseudo active site
(b)
(a)
Nucleic acid bindingVirion incorporation
D128Vif interaction surface
Active siteDeaminase activity
3841
Fig. 15.5 Domain organization of the APOBEC3 proteins. (a) A schematic of the carboxy-terminal half of APOBEC3G, representing a typical APOBEC3 domain. Dashed lines delineatethe exon/exon boundaries. The NMR structure-based secondary structure elements are depictedas black (alpha helix) and gray (beta sheet) boxes (Chen et al., 2008). The characteristic zinc-binding motif is shown. (b) The ‘double-domain’ structure of human APOBEC3G, consisting oftwo APOBEC3 domains as depicted in (a).
A Deamination-Independent Mechanism?
A number of reports have suggested that APOBEC3G may also harbor an antivi-ral activity that does not depend on the protein’s enzymatic activity (Shindo et al.,2003; Dutko et al., 2005; Navarro et al., 2005; Newman et al., 2005; Iwatani et al.,2006). One series of studies tested the HIV-1 restriction activity of deaminase-defective APOBEC3G mutants. These mutants exhibited nearly wild-type antiviralactivity in single-cycle infectivity assays (Shindo et al., 2003; Navarro et al., 2005;Newman et al., 2005; Iwatani et al., 2006). APOBEC3G derivatives with analo-gous substitutions in the amino-terminal, pseudo-catalytic zinc-coordinating motifalso appeared capable of HIV-1 restriction. In contrast, the restriction activity ofAPOBEC3G was fully compromised when both the amino- and carboxy-terminaldomains were mutated (Shindo et al., 2003; Newman et al., 2005; Iwatani et al.,2006). These studies concluded therefore that APOBE3G could exert an antiviraleffect via carboxy-terminal-mediated deamination or by an unspecified activity pro-vided by the amino-terminal zinc-binding motif.
These studies have spawned a debate about whether APOBEC3G’s cytosinedeaminase activity is strictly required for HIV restriction. Several lines of evidencesuggest that it is indeed required. One line is based on the fact that many of thestudies purporting to show that APOBEC3G’s enzymatic activity is dispensablefor restriction were carried out under high expression conditions. In contrast to

314 M.D. Stenglein et al.
these, several recent studies have included careful titrations of protein expressionlevels (Holmes et al., 2007a; Mbisa et al., 2007; Miyagi et al., 2007; Schumacheret al., 2008). Under high expression conditions, wild-type and mutant constructsexhibited similar anti-HIV-1 activity. In contrast, under low expression conditions,only wild-type APOBEC3G exerted significant antiviral activity. A high expres-sion level is potentially non-physiological, and under such conditions a nucleicacid-binding protein like APOBEC3G could derail HIV-1 infectivity even when cat-alytically inert. Corroborating the results of these titration experiments is an exper-iment involving the stable expression of near-physiological levels of wild-type andmutant APOBEC3G in CEM-SS T-cell clones (Miyagi et al., 2007; Hache et al.,2008; Schumacher et al., 2008). In this experiment, growth of a Vif-defective viruswas inhibited completely by APOBEC3G (as observed originally by Malim and co-workers; Sheehy et al., 2002), but the same virus preparation replicated normally incells expressing the deaminase-deficient protein (Miyagi et al., 2007; Schumacheret al., 2008). Together, these series of experiments bolster the argument that thecatalytic activity of APOBEC3G is indeed required for HIV-1 restriction.
It should be noted that it is not necessarily possible to generalize data relatedto APOBEC3G’s ability to restrict HIV-1 to other viruses (and vice versa).APOBEC3G has shown clear deaminase-independent activity against hepatitis Bvirus (HBV), against porcine endogenous retrovirus (PERV), and against the retro-transposon Alu (Seppen, 2004; Turelli et al., 2004; Rosler et al., 2005; Suspeneet al., 2005; Hulme et al., 2007; Jonsson et al., 2007; Nguyen et al., 2007). Sim-ilarly, data from other APOBEC3 proteins cannot be generalized to APOBEC3G(and vice versa); other human APOBEC3 proteins have exhibited DNA deaminase-independent activity. The next decade of research will undoubtedly demonstratewhich APOBEC3 proteins target which retroelements (exogenous viruses and/orendogenous retrotransposons) physiologically. Furthermore, it may be revealed thatdifferent APOBEC3 proteins employ different mechanisms to inhibit these vari-ous retroelements. All mechanisms, however, must account for the fact that thezinc-coordinating DNA cytosine deaminase domain is the major defining and evolu-tionarily conserved activity of this protein family (a point highlighted by structuralcomparisons; e.g., Chen et al., 2008).
How Vif Counteracts APOBEC3G
Before APOBEC3G was identified, several groups had demonstrated the necessityof Vif for growth in non-permissive cell lines including primary human T cellsand macrophages (e.g., Fisher et al., 1987; Strebel et al., 1987; Gabuzda et al.,1992; von Schwedler et al., 1993; Simon and Malim, 1996). It was also clear fromtrans-expression experiments that Vif was required in virus-producing cells andnot in target cells. Additional advances were hampered by the fact that Vif is ahighly basic, 23 kDa protein that has repeatedly resisted biochemical and structuralstudies. Despite these technical hurdles, several groups were able to use geneticapproaches to independently converge on a common explanation: that Vif functions

15 Host Factors that Restrict Retrovirus Replication 315
by triggering the proteasome-dependent destruction of human APOBEC3G (Fig.15.1) (Conticello et al., 2003; Marin et al., 2003; Sheehy et al., 2003; Yu et al., 2003;Liu et al., 2004; Mehle et al., 2004). Moreover, the responsible molecules werequickly identified by the Yu group, who used an epitope-tagged HIV-1 Vif proteinto affinity purify an E3 ubiquitin ligase complex consisting of CUL5, ELONGINB, ELONGINC and RBX1 (Yu et al., 2003). Cells depleted for these proteins orexpressing dominant-negative variants preserved APOBEC3G and resisted HIV-1(Vif+) infection (Yu et al., 2003; Mehle et al., 2004).
The importance of the Vif-mediated APOBEC3G degradation is highlightedby the fact that it is conserved. For instance, the Vif from a virus that infectsAfrican green monkeys (SIVagm) degrades that species’ APOBEC3G (Bogerdet al., 2004). Although this mechanism of APOBEC3G neutralization is conserved,the APOBEC3G–Vif interaction is remarkably species specific (Mariani et al.,2003). For instance, Vif derived from viruses that infect monkeys (SIV infecting rhe-sus macaques or African green monkeys) is unable to degrade human APOBEC3G,which is therefore able to inhibit the growth of these viruses (Bogerd et al., 2004;Mangeat et al., 2004; Schrofelbauer et al., 2004). Correspondingly, Vif from HIV-1 does not degrade rhesus macaque or African green monkey APOBEC3G, whichinhibit the human virus. This species specificity can be mapped to a single aminoacid. Mutating residue 128 from aspartic acid to lysine (D128K) enables humanAPOBEC3G to resist degradation by HIV-1 Vif, and sensitizes it to degradation bySIVmac or SIVagm Vif (Bogerd et al., 2004; Mangeat et al., 2004; Schrofelbaueret al., 2004; Xu et al., 2004). This specificity suggests that each retroviral vif genehas evolved to optimally counteract its host species’ APOBEC3 proteins.
It should be noted that Vif might employ more than one mechanism to neutral-ize APOBEC3G (Stopak et al., 2003; Kao et al., 2004; Santa-Marta et al., 2005).One study indicated that Vif could directly inhibit the DNA deaminase activity ofAPOBEC3G (Santa-Marta et al., 2005). A second study suggested that Vif couldimpair APOBEC3G translation (Stopak et al., 2003). A third identified a Vif variantthat restored infectivity of HIV-1 but did not cause obvious APOBEC3G degrada-tion (Kao et al., 2007). In any event, despite the obvious appeal of a direct inhibitionmechanism (in addition to proteosome-dependent degradation), a consensus opinionhas yet to emerge and more research is clearly needed in this area.
Broad Functionality of the Mammalian APOBEC3 Protein Family
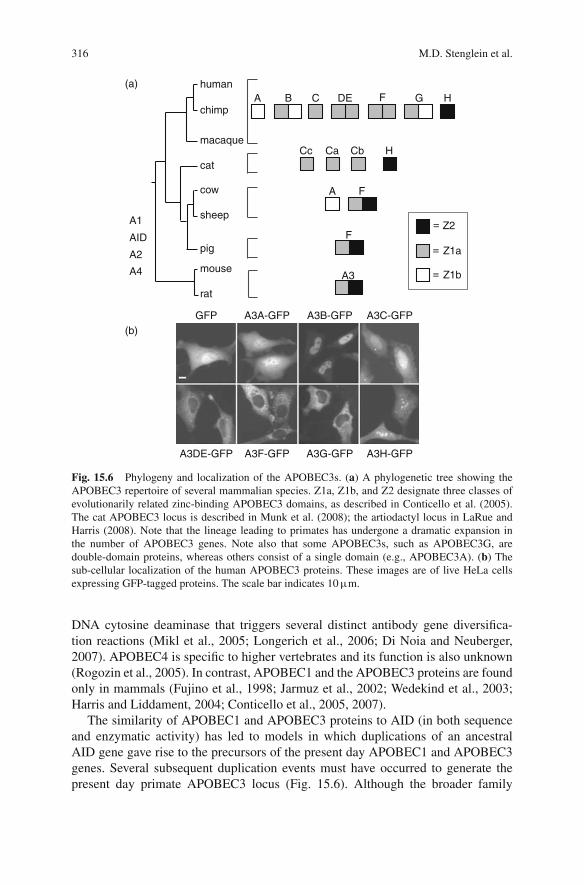
As alluded to above, APOBEC3G is one member of a larger family of related pro-teins, a fact that can be appreciated through a phylogenetic overview (Fig. 15.6).Humans encode a total of 11 family members: seven APOBEC3 proteins,APOBEC3A, -3B, -3C, -3DE, -3F, -3G, and -3H, from a single locus on chro-mosome 22p13, APOBEC4 from 1q25.3, APOBEC2 from 6p21, APOBEC1 from12p13.1, and AID from 12p13. APOBEC2 and AID are evolutionarily the oldest,as they are the only ones found in all vertebrates. The physiological function ofAPOBEC2 remains a mystery, but AID has a pivotal role in B lymphocytes as the
316 M.D. Stenglein et al.
human
chimp
macaque
cat
cow
sheep
pig
mouse
rat
F
FA
Cb H
GDEBA C H
A3
A2
A4
AID
A1
(a)F
(b)GFP A3A-GFP A3B-GFP A3C-GFP
A3DE-GFP A3F-GFP A3G-GFP A3H-GFP
=
=
=
Z1a
Z1b
Z2
CaCc
Fig. 15.6 Phylogeny and localization of the APOBEC3s. (a) A phylogenetic tree showing theAPOBEC3 repertoire of several mammalian species. Z1a, Z1b, and Z2 designate three classes ofevolutionarily related zinc-binding APOBEC3 domains, as described in Conticello et al. (2005).The cat APOBEC3 locus is described in Munk et al. (2008); the artiodactyl locus in LaRue andHarris (2008). Note that the lineage leading to primates has undergone a dramatic expansion inthe number of APOBEC3 genes. Note also that some APOBEC3s, such as APOBEC3G, aredouble-domain proteins, whereas others consist of a single domain (e.g., APOBEC3A). (b) Thesub-cellular localization of the human APOBEC3 proteins. These images are of live HeLa cellsexpressing GFP-tagged proteins. The scale bar indicates 10 μm.
DNA cytosine deaminase that triggers several distinct antibody gene diversifica-tion reactions (Mikl et al., 2005; Longerich et al., 2006; Di Noia and Neuberger,2007). APOBEC4 is specific to higher vertebrates and its function is also unknown(Rogozin et al., 2005). In contrast, APOBEC1 and the APOBEC3 proteins are foundonly in mammals (Fujino et al., 1998; Jarmuz et al., 2002; Wedekind et al., 2003;Harris and Liddament, 2004; Conticello et al., 2005, 2007).
The similarity of APOBEC1 and APOBEC3 proteins to AID (in both sequenceand enzymatic activity) has led to models in which duplications of an ancestralAID gene gave rise to the precursors of the present day APOBEC1 and APOBEC3genes. Several subsequent duplication events must have occurred to generate thepresent day primate APOBEC3 locus (Fig. 15.6). Although the broader family

15 Host Factors that Restrict Retrovirus Replication 317
of polynucleotide cytosine deaminases appears to have expanded gradually dur-ing vertebrate evolution, the APOBEC3s have experienced a much more dramaticand recent expansion in mammals. At the heart of this expansion is the conserved,zinc-coordinating motif (Fig. 15.5). This motif is encoded by a single exon and itconsists of at least four key residues, one histidine, one glutamate, and two cys-teines, H–X1–E–X23–28–C–X2–4–C (X indicates a non-conserved residue) (Jarmuzet al., 2002; Wedekind et al., 2003; Harris and Liddament, 2004; Conticello et al.,2005, 2007). The histidine and the cysteines directly coordinate zinc. The glutamateparticipates indirectly by binding a water molecule, which in turn binds zinc andserves as the nucleophile for cytosine deamination. Some APOBEC3 proteins havetwo conserved zinc-coordinating (Z) motifs, whereas other family members haveonly one.
A recent burst of near-complete mammalian genome sequences has revealed thatprimates encode the greatest number of APOBEC3 zinc-coordinating motifs (11 ofthese so-called Z-motifs encoded by seven genes) and the rodents have the small-est (two Z-motifs encoded by one gene) (Fig. 15.6). These zinc(Z)-coordinatingmotifs segregate phylogenetically into three sub-groups, Z1a, Z1b, and Z2 (Conti-cello et al., 2005). The Z1s and the Z2 differ at many positions, including an obviousserine or threonine that precedes the conserved cysteines, SWSPCX2–4C (Z1s) orTWSPCX2–4C (Z2). The Z1a and Z1b motifs also differ at more than 20 positions,but a key identifier (WF in Z1a domains and X(V/I) in Z1b proteins) can be foundin six residues carboxy-terminal to the conserved HXE motif. Interestingly, mam-mals that branch phylogenetically between rodents and humans have intermediateAPOBEC3 gene numbers (Jonsson et al., 2006; Munk et al., 2008 and Fig. 15.6).For instance, artiodactyls such as sheep and cattle have one A3A-like gene and oneA3F-like gene (three Z-motifs in two genes) (Jonsson et al., 2006; LaRue and Harris,2008). And felines have three A3C-like genes and one A3H-like gene (Munk et al.,2008). These expansions are entirely attributable to the Z1-motif, as the Z2-motif issingle-copy in all mammals. Species within a larger order/family or members of aparticular species may have fewer Z-motifs. For instance, at least one deletion musthave occurred to cause domesticated pigs (another artiodactyl) to have only two Z-motifs (one APOBEC3 gene due to an APOBEC3A-3F deletion) and some humansto have nine Z-motifs (six APOBEC3 genes due to an APOBEC3A-3B deletion)(Kidd et al., 2007). The functional significance of these deletions is not yet clear,but it is certainly worth investigating.
In addition to the dramatic Z-motif expansions, the primate APOBEC3 genescontain signatures of strong positive selection (Sawyer et al., 2004; Zhang andWebb, 2004). These two features combine to indicate that this locus is under a pow-erful and ongoing selective pressure, which appears to intensify in specific mam-malian lineages (e.g., primates). Although the precise selective pressures have notbeen (and may never be) identified, accumulating evidence suggests that retrovirusesand endogenous retrotransposons may provide the major driving forces. This is sup-ported by many reports of APOBEC3 proteins inhibiting endogenous retroelements,observations that endogenized mice retroelements bear scars of APOBEC3-likedeamination events, and the fact that the overall number of active retroelements

318 M.D. Stenglein et al.
appears to be considerably less in humans than mice (Lander et al., 2001; Esnaultet al., 2005; Schumacher et al., 2005; Bogerd et al., 2006a, b; Chiu et al., 2006;Esnault et al., 2006; Stenglein and Harris, 2006; Hulme et al., 2007; Jern et al.,2007; Jonsson et al., 2007; Kinomoto et al., 2007; Schumacher et al., 2008). In otherwords, there is a compelling inverse correlation between the number of APOBEC3genes and retroelement mobility.
APOBEC3 Frontiers
The number of APOBEC3G PubMed occurrences has risen from 4 in 2002 to327 currently, and this rate shows no signs of diminishing. Several frontiers arewide open. First, which APOBEC3 proteins are physiologically relevant to therestriction of HIV-1 and other medically relevant viruses? The answer to thisimportant question could come from a variety of sources, including analyses ofvariations in human APOBEC3 genes, simian experiments, and cell-based experi-ments. Currently, APOBEC3G and APOBEC3F are the leading candidates becausetheir mutational signatures (determined by the nucleotide preceding the deaminatedcytosine) are observed in patient-derived HIV-1 sequences (e.g., Liddament et al.,2004). Moreover, these proteins appear co-expressed and are the only two humanAPOBEC3 proteins that HIV-1 Vif can inhibit significantly (aforementioned refer-ences for APOBEC3G and Bishop et al., 2004; Liddament et al., 2004; Wiegandet al., 2004; Zheng et al., 2004; Simon et al., 2005; Holmes et al., 2007a).
Second, a structural understanding of the APOBEC3G–Vif interaction would beof great benefit. Advances in this area have been hindered by the fact that bothAPOBEC3G and Vif are poorly soluble. Indeed, Vif is the only HIV-1 protein forwhich there is no high-resolution structural information. However, a recent solutionstructure of the APOBEC3G catalytic domain has indicated that single-strand DNAis recognized by a positively charged, arginine-rich brim, which facilitates deami-nation by flipping out the target cytosine base such that it can be accommodated byan active site pocket (Chen et al., 2008). Moreover, this structure has facilitated amodel of the full-length APOBEC3G protein, which offers several testable predic-tions that relate to the Vif-interacting region (defined by D128, Fig. 15.5). Overall,this first structure will provide the foundation for many experiments and will helpanswer important questions, for example, what is the mechanism of Vif binding?What determines nucleic acid substrate specificity? And, what is the stoichiometryof the APOBEC3G–Vif interaction?
Third, not much is known about APOBEC3 gene regulation. APOBEC3G and-3F appear broadly and constitutively expressed, but other APOBEC3 genes likeAPOBEC3B and -3DE appear to be less abundant and tissue restricted (Harrisand Liddament, 2004, Liddament et al., 2004; Wiegand et al., 2004). Moreover,several of the APOBEC3 genes appear to be interferon inducible, suggesting thatAPOBEC3 proteins may play a role in the innate immune response (Rose et al.,2004b; Taylor et al., 2004; Bonvin et al., 2006; Chen et al., 2006; Peng et al., 2006;Sarkis et al., 2006; Tanaka et al., 2006; Komohara et al., 2007; Stopak et al., 2007;Ying et al., 2007). It is therefore highly likely that several of these genes will be

15 Host Factors that Restrict Retrovirus Replication 319
induced by virus infection, which can be a very potent inducer of the interferonresponse.
Finally, moving back to the APOBEC3 proteins themselves. Common sensealone would dictate that such potent DNA mutating enzymes would be subjectto tight post-translational control within the cell. Failure to do so could be catas-trophic and/or contribute to carcinogenesis. APOBEC3G, for instance, appears pre-dominantly cytoplasmic, safely away from the genomic DNA (Fig. 15.6) (Mangeatet al., 2003; Rose et al., 2004a; Wichroski et al., 2005; Jonsson et al., 2006; Kozaket al., 2006; Stenglein and Harris, 2006; Wichroski et al., 2006). Although sev-eral groups have identified a plethora of candidate APOBEC3G-interacting proteins(and RNAs) that may be important for post-translational regulation and retroelementrestriction, the best evidence to date was provided by Greene’s laboratory. Chiuet al. reported that APOBEC3G resides in enzymatically inactive, high-molecular-mass (HMM) ribonucleoprotein complexes in activated CD4+ T cells and in enzy-matically active, low-molecular-mass (LMM) ribonucleoprotein complex in restingCD4+ T cells (Chiu et al., 2005, 2006; Kozak et al., 2006; Gallois-Montbrun et al.,2007). This finding demonstrates that the state of the cell can directly impact thenature of the APOBEC3G-associated factors, and that this in turn can determinethe permissiveness of the cell for virus infection. Moreover, taken together withthe fact that human APOBEC3 proteins can occupy nearly every sub-cellular com-partment, it is likely that these proteins are subject to multiple layers of regulation(Fig. 15.6).
Other Retrovirus Restriction Factors
Although APOBEC3G and TRIM5alpha are the most studied and best understoodrestriction systems, several others have been described, and the existence of undis-covered factors has been inferred. The Goff lab described the zinc finger antiviralprotein (ZAP) (Gao et al., 2002). ZAP is a rat protein that limits retroviral replica-tion by targeting cytoplasmic viral RNA for degradation (Guo et al., 2004, 2007).Another restriction factor that blocks infection by HIV-2, termed Lv2, has beendescribed (Schmitz et al., 2004). Lv2 remains an inferred activity and an Lv2 genehas yet to be cloned. Additionally, as described above, CypA seems to protect theHIV-1 capsid from a restriction factor whose molecular identity remains undeter-mined. Thus, the mammalian repertoire of restriction factors continues to expand.
Despite the increasing number of cellular restriction factors, the central impor-tance of TRIM5alpha and APOBEC3G is highlighted by two recent reports. Thesedemonstrated that simian barriers to HIV-1 infection could be overcome by replac-ing the capsid and vif-coding regions of HIV-1 with the corresponding SIV genes(Hatziioannou et al., 2006; Kamada et al., 2006). Recall that Vif and Capsid arethe primary viral determinants of susceptibility to APOBEC3G and TRIM5alpharestriction, respectively. The resulting chimeric virus (over 90% HIV-1) was ableto replicate in normally non-permissive monkey cells. The prospect of using suchviruses as HIV-1/AIDS disease models is very good. Moreover, these studiesimply that the TRIM- and APOBEC3-mediated barriers are critical cellular barriers

320 M.D. Stenglein et al.
that function to limit the zoonotic transmission of lentiviruses and, perhaps, ofretroviruses in general. Such barriers must be overcome before a TRIM and/orAPOBEC3 susceptible virus can colonize a host and potentially cause disease.
Conclusions and Future Directions
In the past several decades it has become increasingly clear that the natural hostrange of many retroviruses is extremely limited, and that this is due in large part toretrovirus restriction factors. For a retrovirus to be able to infect a particular host, itmust circumvent that host’s restriction factors. Usually, this ability comes at a cost,as it typically means that the virus will be susceptible to restriction by other species’restriction factors. This conflict between virus and host defenses has left traces ofpositive selection on the genes involved. In terms of human health, the ultimate goalof studying restriction factors is to enable the development of novel antiviral thera-pies. These therapies could take several forms, for instance, restriction factors couldbe used in gene therapy. Alternatively, small molecule drugs could tip the balancein favor of the restriction factor, for example, by inhibiting the APOBEC3G–Vifinteraction. As our understanding of restriction factors continues to increase, theprospects of developing such therapies will improve proportionately.
Acknowledgments We thank N. Somia, L. Mansky, M. Huseby, and several laboratory mem-bers for thoughtful comments. Studies in the authors’ laboratory are supported by grants fromthe National Institutes of Health (AI064046 and GM080437), the Medica Foundation (MinnesotaPartnership for Biotechnology and Medical Genomics) and the University of Minnesota (LeukemiaResearch Fund and Cancer Center Brainstorm Program), and the Cancer Biology Training Grant(CA009138).
References
Alce, T. M. and Popik, W. 2004. APOBEC3G is incorporated into virus-like particles by a directinteraction with HIV-1 Gag nucleocapsid protein. J Biol Chem. 279(33): 34083–6.
Anderson, J. and Akkina, R. 2005. TRIM5alpharh expression restricts HIV-1 infection in lentiviralvector-transduced CD34+-cell-derived macrophages. Mol Ther. 12(4): 687–96.
Anderson, J. L., Campbell, E. M., Wu, X., Vandegraaff, N., Engelman, A. and Hope, T. J. 2006.Proteasome inhibition reveals that a functional preintegration complex intermediate can be gen-erated during restriction by diverse TRIM5 proteins. J Virol. 80(19): 9754–60.
Bassin, R. H., Duran-Troise, G., Gerwin, B. I. and Rein, A. 1978. Abrogation of Fv-1b restric-tion with murine leukemia viruses inactivated by heat or by gamma irradiation. J Virol. 26(2):306–15.
Berthoux, L., Sebastian, S., Sokolskaja, E. and Luban, J. 2004. Lv1 inhibition of human immunod-eficiency virus type 1 is counteracted by factors that stimulate synthesis or nuclear translocationof viral cDNA. J Virol. 78(21): 11739–50.
Berthoux, L., Sebastian, S., Sokolskaja, E. and Luban, J. 2005a. Cyclophilin A is required forTRIM5{alpha}-mediated resistance to HIV-1 in Old World monkey cells. Proc Natl Acad SciUSA. 102(41): 14849–53.
Berthoux, L., Sebastian, S., Sayah, D. M. and Luban, J. 2005b. Disruption of human TRIM5alphaantiviral activity by nonhuman primate orthologues. J Virol. 79(12): 7883–8.

15 Host Factors that Restrict Retrovirus Replication 321
Besnier, C., Takeuchi, Y. and Towers, G. 2002. Restriction of lentivirus in monkeys. Proc NatlAcad Sci USA. 99(18): 11920–5.
Besnier, C., Ylinen, L., Strange, B., Lister, A., Takeuchi, Y., Goff, S. P. and Towers, G. J.2003. Characterization of murine leukemia virus restriction in mammals. J Virol. 77(24):13403–6.
Best, S., Le Tissier, P., Towers, G. and Stoye, J. P. 1996. Positional cloning of the mouse retrovirusrestriction gene Fv1. Nature. 382(6594): 826–9.
Bieniasz, P. D. 2003. Restriction factors: a defense against retroviral infection. Trends Microbiol.11(6): 286–91.
Bieniasz, P. D. 2004. Intrinsic immunity: a front-line defense against viral attack. Nat Immunol.5(11): 1109–15.
Bishop, K. N., Holmes, R. K., Sheehy, A. M., Davidson, N. O., Cho, S. J. and Malim, M. H.2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol. 14(15):1392–6.
Bishop, K. N., Holmes, R. K. and Malim, M. H. 2006. Antiviral potency of APOBEC proteinsdoes not correlate with cytidine deamination. J Virol. 80(17): 8450–8.
Bogerd, H. P., Doehle, B. P., Wiegand, H. L. and Cullen, B. R. 2004. A single amino acid differencein the host APOBEC3G protein controls the primate species specificity of HIV type 1 virioninfectivity factor. Proc Natl Acad Sci USA. 101(11): 3770–4.
Bogerd, H. P., Wiegand, H. L., Doehle, B. P., Lueders, K. K. and Cullen, B. R. 2006a. APOBEC3Aand APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. NucleicAcids Res. 34(1): 89–95.
Bogerd, H. P., Wiegand, H. L., Hulme, A. E., Garcia-Perez, J. L., O’Shea, K. S., Moran, J. V. andCullen, B. R. 2006b. Cellular inhibitors of long interspersed element 1 and Alu retrotransposi-tion. Proc Natl Acad Sci USA. 103(23): 8780–5.
Bonvin, M., Achermann, F., Greeve, I., Stroka, D., Keogh, A., Inderbitzin, D., Candinas, D.,Sommer, P., Wain-Hobson, S., Vartanian, J. P. and Greeve, J. 2006. Interferon-inducible expres-sion of APOBEC3 editing enzymes in human hepatocytes and inhibition of hepatitis B virusreplication. Hepatology. 43(6): 1364–74.
Boone, L. R., Innes, C. L. and Heitman, C. K. 1990. Abrogation of Fv-1 restriction by genome-deficient virions produced by a retrovirus packaging cell line. J Virol. 64(7): 3376–81.
Borden, K. L. 1998. RING fingers and B-boxes: zinc-binding protein–protein interaction domains.Biochem Cell Biol. 76(2–3): 351–8.
Bosco, D. A., Eisenmesser, E. Z., Pochapsky, S., Sundquist, W. I. and Kern, D. 2002. Catalysisof cis/trans isomerization in native HIV-1 capsid by human cyclophilin A. Proc Natl Acad SciUSA. 99(8): 5247–52.
Braaten, D., Franke, E. K. and Luban, J. 1996a. Cyclophilin A is required for an early step in thelife cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription.J Virol. 70(6): 3551–60.
Braaten, D., Franke, E. K. and Luban, J. 1996b. Cyclophilin A is required for the replication ofgroup M human immunodeficiency virus type 1 (HIV-1) and simian immunodeficiency virusSIV(CPZ)GAB but not group O HIV-1 or other primate immunodeficiency viruses. J Virol.70(7): 4220–7.
Braaten, D., Aberham, C., Franke, E. K., Yin, L., Phares, W. and Luban, J. 1996c. CyclosporineA-resistant human immunodeficiency virus type 1 mutants demonstrate that Gag encodes thefunctional target of cyclophilin A. J Virol. 70(8): 5170–6.
Braaten, D. and Luban, J. 2001. Cyclophilin A regulates HIV-1 infectivity, as demonstrated bygene targeting in human T cells. Embo J. 20(6): 1300–9.
Brennan, G., Kozyrev, Y. and Hu, S. L. 2008. TRIMCyp expression in Old World primates Macacanemestrina and Macaca fascicularis. Proc Natl Acad Sci USA. 105(9): 3569–74.
Burnett, A. and Spearman, P. 2007. APOBEC3G Multimers Are Recruited to the Plasma Mem-brane for Packaging into Human Immunodeficiency Virus Type 1 Virus-Like Particles in anRNA-Dependent Process Requiring the NC Basic Linker. J Virol. 81(10): 5000–13.

322 M.D. Stenglein et al.
Campbell, E. M., Dodding, M. P., Yap, M. W., Wu, X., Gallois-Montbrun, S., Malim, M. H., Stoye,J. P. and Hope, T. J. 2007. TRIM5 alpha cytoplasmic bodies are highly dynamic structures. MolBiol Cell. 18(6): 2102–11.
Cen, S., Guo, F., Niu, M., Saadatmand, J., Deflassieux, J. and Kleiman, L. 2004. The interactionbetween HIV-1 Gag and APOBEC3G. J Biol Chem. 279(32): 33177–84.
Chatterji, U., Bobardt, M. D., Stanfield, R., Ptak, R. G., Pallansch, L. A., Ward, P. A., Jones, M. J.,Stoddart, C. A., Scalfaro, P., Dumont, J. M., Besseghir, K., Rosenwirth, B. and Gallay, P. A.2005. Naturally occurring capsid substitutions render HIV-1 cyclophilin A independent inhuman cells and TRIM-cyclophilin-resistant in Owl monkey cells. J Biol Chem. 280(48):40293–300.
Chatterji, U., Bobardt, M. D., Gaskill, P., Sheeter, D., Fox, H. and Gallay, P. A. 2006. Trim5alphaaccelerates degradation of cytosolic capsid associated with productive HIV-1 entry. J BiolChem. 281(48): 37025–33.
Chen, K., Huang, J., Zhang, C., Huang, S., Nunnari, G., Wang, F. X., Tong, X., Gao, L., Nikisher,K. and Zhang, H. 2006. Alpha interferon potently enhances the anti-human immunodeficiencyvirus type 1 activity of APOBEC3G in resting primary CD4 T cells. J Virol. 80(15): 7645–57.
Chen, K. M., Martemyanova, N., Lu, Y., Shindo, K., Matsuo, H. and Harris, R. S. 2007. Extensivemutagenesis experiments corroborate a structural model for the DNA deaminase domain ofAPOBEC3G. FEBS Lett. 581(24): 4761–6.
Chen, K. M., Harjes, E., Gross, P. J., Fahmy, A., Lu, Y., Shindo, K., Harris, R. S. and Matsuo, H.2008. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G.Nature. 452(7183): 116–9.
Chiu, Y. L., Soros, V. B., Kreisberg, J. F., Stopak, K., Yonemoto, W. and Greene, W. C. 2005.Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature. 435(7038):108–14.
Chiu, Y. L. and Greene, W. C. 2006. Multifaceted antiviral actions of APOBEC3 cytidine deami-nases. Trends Immunol. 27(6): 291–7.
Chiu, Y. L., Witkowska, H. E., Hall, S. C., Santiago, M., Soros, V. B., Esnault, C., Heidmann, T.and Greene, W. C. 2006. High-molecular-mass APOBEC3G complexes restrict Alu retrotrans-position. Proc Natl Acad Sci USA. 103(42): 15588–93.
Conticello, S. G., Harris, R. S. and Neuberger, M. S. 2003. The Vif protein of HIV triggers degra-dation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol. 13(22): 2009–13.
Conticello, S. G., Thomas, C. J., Petersen-Mahrt, S. and Neuberger, M. S. 2005. Evolution of theAID/APOBEC Family of Polynucleotide (Deoxy)Cytidine Deaminases. Mol Biol Evol. 22(2):367–77.
Conticello, S. G., Langlois, M. A., Yang, Z. and Neuberger, M. S. 2007. DNA deamination inimmunity: AID in the context of its APOBEC relatives. Adv Immunol. 94: 37–73.
Cowan, S., Hatziioannou, T., Cunningham, T., Muesing, M. A., Gottlinger, H. G. andBieniasz, P. D. 2002. Cellular inhibitors with Fv1-like activity restrict human and simianimmunodeficiency virus tropism. Proc Natl Acad Sci USA. 99(18): 11914–9.
Decleve, A., Niwa, O., Gelmann, E. and Kaplan, H. S. 1975. Replication kinetics of N- and B-tropic murine leukemia viruses on permissive and nonpermissive cells in vitro. Virology. 65(2):320–32.
DesGroseillers, L. and Jolicoeur, P. 1983. Physical mapping of the Fv-1 tropism host range deter-minant of BALB/c murine leukemia viruses. J Virol. 48(3): 685–96.
Desrosiers, R. C., Lifson, J. D., Gibbs, J. S., Czajak, S. C., Howe, A. Y., Arthur, L. O. andJohnson, R. P. 1998. Identification of highly attenuated mutants of simian immunodeficiencyvirus. J Virol. 72(2): 1431–7.
Di Noia, J. M. and Neuberger, M. S. 2007. Molecular mechanisms of antibody somatic hypermu-tation. Annu Rev Biochem. 76: 1–22.
Diaz-Griffero, F., Li, X., Javanbakht, H., Song, B., Welikala, S., Stremlau, M. and Sodroski, J.2006a. Rapid turnover and polyubiquitylation of the retroviral restriction factor TRIM5. Virol-ogy. 349(2): 300–15.

15 Host Factors that Restrict Retrovirus Replication 323
Diaz-Griffero, F., Vandegraaff, N., Li, Y., McGee-Estrada, K., Stremlau, M., Welikala, S., Si, Z.,Engelman, A. and Sodroski, J. 2006b. Requirements for capsid-binding and an effector functionin TRIMCyp-mediated restriction of HIV-1. Virology. 351(2): 404–19.
Dodding, M. P., Bock, M., Yap, M. W. and Stoye, J. P. 2005. Capsid processing requirements forabrogation of Fv1 and Ref1 restriction. J Virol. 79(16): 10571–7.
Dutko, J. A., Schafer, A., Kenny, A. E., Cullen, B. R. and Curcio, M. J. 2005. Inhibition of a yeastLTR retrotransposon by human APOBEC3 cytidine deaminases. Curr Biol. 15(7): 661–6.
Esnault, C., Heidmann, O., Delebecque, F., Dewannieux, M., Ribet, D., Hance, A. J., Heidmann, T.and Schwartz, O. 2005. APOBEC3G cytidine deaminase inhibits retrotransposition of endoge-nous retroviruses. Nature. 433(7024): 430–3.
Esnault, C., Millet, J., Schwartz, O. and Heidmann, T. 2006. Dual inhibitory effects of APOBECfamily proteins on retrotransposition of mammalian endogenous retroviruses. Nucleic AcidsRes. 34(5): 1522–31.
Everett, R. D. and Chelbi-Alix, M. K. 2007. PML and PML nuclear bodies: implications in antiviraldefence. Biochimie. 89(6–7): 819–30.
Fields, B. N., Knipe, D. M. and Howley, P. M. 2007. Fields’ virology (5th). Philadelphia: WoltersKluwer Health/Lippincott Williams & Wilkins.
Fischer, G., Tradler, T. and Zarnt, T. 1998. The mode of action of peptidyl prolyl cis/trans iso-merases in vivo: binding vs. catalysis. FEBS Lett. 426(1): 17–20.
Fisher, A. G., Ensoli, B., Ivanoff, L., Chamberlain, M., Petteway, S., Ratner, L., Gallo, R. C. andWong-Staal, F. 1987. The sor gene of HIV-1 is required for efficient virus transmission in vitro.Science. 237(4817): 888–93.
Forshey, B. M., von Schwedler, U., Sundquist, W. I. and Aiken, C. 2002. Formation of a humanimmunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol.76(11): 5667–77.
Franke, E. K., Yuan, H. E. and Luban, J. 1994. Specific incorporation of cyclophilin A into HIV-1virions. Nature. 372(6504): 359–62.
Friend, C. 1957. Cell-free transmission in adult Swiss mice of a disease having the character of aleukemia. J Exp Med. 105(4): 307–18.
Fujino, T., Navaratnam, N. and Scott, J. 1998. Human apolipoprotein B RNA editing deaminasegene (APOBEC1). Genomics. 47(2): 266–75.
Gabuzda, D. H., Lawrence, K., Langhoff, E., Terwilliger, E., Dorfman, T., Haseltine, W. A. andSodroski, J. 1992. Role of Vif in replication of human immunodeficiency virus type 1 in CD4+T lymphocytes. J Virol. 66(11): 6489–95.
Gabuzda, D. H., Li, H., Lawrence, K., Vasir, B. S., Crawford, K. and Langhoff, E. 1994. Essentialrole of vif in establishing productive HIV-1 infection in peripheral blood T lymphocytes andmonocyte/macrophages. J Acquir Immune Defic Syndr. 7(9): 908–15.
Gallois-Montbrun, S., Kramer, B., Swanson, C. M., Byers, H., Lynham, S., Ward, M. andMalim, M. H. 2007. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexesfound in P bodies and stress granules. J Virol. 81(5): 2165–78.
Gamble, T. R., Vajdos, F. F., Yoo, S., Worthylake, D. K., Houseweart, M., Sundquist, W. I. andHill, C. P. 1996. Crystal structure of human cyclophilin A bound to the amino-terminal domainof HIV-1 capsid. Cell. 87(7): 1285–94.
Gao, G., Guo, X. and Goff, S. P. 2002. Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science. 297(5587): 1703–6.
Goff, S. P. 1996. Operating under a Gag order: a block against incoming virus by the Fv1 gene.Cell. 86(5): 691–3.
Goff, S. P. 2004. Retrovirus restriction factors. Mol Cell. 16(6): 849–59.Goff, S. P. 2007. Host factors exploited by retroviruses. Nat Rev Microbiol. 5(4): 253–63.Goldschmidt, V., Bleiber, G., May, M., Martinez, R., Ortiz, M. and Telenti, A. 2006. Role of
common human TRIM5alpha variants in HIV-1 disease progression. Retrovirology. 3: 54.Goncalves, J., Korin, Y., Zack, J. and Gabuzda, D. 1996. Role of Vif in human immunodeficiency
virus type 1 reverse transcription. J Virol. 70(12): 8701–9.

324 M.D. Stenglein et al.
Guo, X., Carroll, J. W., Macdonald, M. R., Goff, S. P. and Gao, G. 2004. The zinc finger antiviralprotein directly binds to specific viral mRNAs through the CCCH zinc finger motifs. J Virol.78(23): 12781–7.
Guo, X., Ma, J., Sun, J. and Gao, G. 2007. The zinc-finger antiviral protein recruits the RNAprocessing exosome to degrade the target mRNA. Proc Natl Acad Sci USA. 104(1): 151–6.
Hache, G., Liddament, M. T. and Harris, R. S. 2005. The retroviral hypermutation specificity ofAPOBEC3F and APOBEC3G is governed by the C-terminal DNA cytosine deaminase domain.J Biol Chem. 280(12): 10920–4.
Hache, G., Mansky, L. M. and Harris, R. S. 2006. Human APOBEC3 proteins, retrovirus restric-tion, and HIV drug resistance. AIDS Rev. 8(3): 148–57.
Hache, G., Shindo, K., Albin, J. S. and Harris, R. S. 2008. Evolution of HIV-1 Isolates that Usea Novel Vif-Independent Mechanism to Resist Restriction by Human APOBEC3G. Curr Biol.18(11): 819–24.
Harris, R. S., Petersen-Mahrt, S. K. and Neuberger, M. S. 2002. RNA editing enzyme APOBEC1and some of its homologs can act as DNA mutators. Mol Cell. 10(5): 1247–53.
Harris, R. S., Bishop, K. N., Sheehy, A. M., Craig, H. M., Petersen-Mahrt, S. K., Watt, I. N.,Neuberger, M. S. and Malim, M. H. 2003a. DNA deamination mediates innate immunity toretroviral infection. Cell. 113(6): 803–9.
Harris, R. S., Sheehy, A. M., Craig, H. M., Malim, M. H. and Neuberger, M. S. 2003b. DNAdeamination: not just a trigger for antibody diversification but also a mechanism for defenseagainst retroviruses. Nat Immunol. 4(7): 641–3.
Harris, R. S. and Liddament, M. T. 2004. Retroviral restriction by APOBEC proteins. Nat RevImmunol. 4(11): 868–77.
Hartley, J. W., Rowe, W. P. and Huebner, R. J. 1970. Host-range restrictions of murine leukemiaviruses in mouse embryo cell cultures. J Virol. 5(2): 221–5.
Hatziioannou, T., Cowan, S., Goff, S. P., Bieniasz, P. D. and Towers, G. J. 2003. Restriction ofmultiple divergent retroviruses by Lv1 and Ref1. Embo J. 22(3): 385–94.
Hatziioannou, T., Perez-Caballero, D., Yang, A., Cowan, S. and Bieniasz, P. D. 2004a. Retrovirusresistance factors Ref1 and Lv1 are species-specific variants of TRIM5alpha. Proc Natl AcadSci USA. 101(29): 10774–9.
Hatziioannou, T., Cowan, S., Von Schwedler, U. K., Sundquist, W. I. and Bieniasz, P. D. 2004b.Species-specific tropism determinants in the human immunodeficiency virus type 1 capsid.J Virol. 78(11): 6005–12.
Hatziioannou, T., Perez-Caballero, D., Cowan, S. and Bieniasz, P. D. 2005. Cyclophilin interac-tions with incoming human immunodeficiency virus type 1 capsids with opposing effects oninfectivity in human cells. J Virol. 79(1): 176–83.
Hatziioannou, T., Princiotta, M., Piatak, M., Jr., Yuan, F., Zhang, F., Lifson, J. D. andBieniasz, P. D. 2006. Generation of simian-tropic HIV-1 by restriction factor evasion. Science.314(5796): 95.
Henry, J., Ribouchon, M. T., Offer, C. and Pontarotti, P. 1997. B30.2-like domain proteins: a grow-ing family. Biochem Biophys Res Commun. 235(1): 162–5.
Himathongkham, S. and Luciw, P. A. 1996. Restriction of HIV-1 (subtype B) replication at theentry step in rhesus macaque cells. Virology. 219(2): 485–8.
Hofmann, W., Schubert, D., LaBonte, J., Munson, L., Gibson, S., Scammell, J., Ferrigno, P. andSodroski, J. 1999. Species-specific, postentry barriers to primate immunodeficiency virus infec-tion. J Virol. 73(12): 10020–8.
Hoffman, B. G., Williams, K. L., Tien, A. H., Lu, V., de Algara, T. R., Ting, J. P. and Helgason, C.D. 2006. Identification of novel genes and transcription factors involved in spleen, thymus andimmunological development and function. Genes Immun. 7(2): 101–12.
Holmes, R. K., Koning, F. A., Bishop, K. N. and Malim, M. H. 2007a. APOBEC3F can inhibitthe accumulation of HIV-1 reverse transcription products in the absence of hypermutation.Comparisons with APOBEC3G. J Biol Chem. 282(4): 2587–95.
Holmes, R. K., Malim, M. H. and Bishop, K. N. 2007b. APOBEC-mediated viral restriction: notsimply editing? Trends Biochem Sci. 32(3): 118–28.

15 Host Factors that Restrict Retrovirus Replication 325
Hulme, A. E., Bogerd, H. P., Cullen, B. R. and Moran, J. V. 2007. Selective inhibition of Aluretrotransposition by APOBEC3G. Gene. 390(1–2): 199–205.
Ikeda, H., Laigret, F., Martin, M. A. and Repaske, R. 1985. Characterization of a molecularlycloned retroviral sequence associated with Fv-4 resistance. J Virol. 55(3): 768–77.
Ikeda, H. and Sugimura, H. 1989. Fv-4 resistance gene: a truncated endogenous murine leukemiavirus with ecotropic interference properties. J Virol. 63(12): 5405–12.
Ikeda, Y., Ylinen, L. M., Kahar-Bador, M. and Towers, G. J. 2004. Influence of gag on humanimmunodeficiency virus type 1 species-specific tropism. J Virol. 78(21): 11816–22.
Iwatani, Y., Takeuchi, H., Strebel, K. and Levin, J. G. 2006. Biochemical activities of highly puri-fied, catalytically active human APOBEC3G: correlation with antiviral effect. J Virol. 80(12):5992–6002.
James, L. C., Keeble, A. H., Khan, Z., Rhodes, D. A. and Trowsdale, J. 2007. Structural basisfor PRYSPRY-mediated tripartite motif (TRIM) protein function. Proc Natl Acad Sci USA.104(15): 6200–5.
Jarmuz, A., Chester, A., Bayliss, J., Gisbourne, J., Dunham, I., Scott, J. and Navaratnam, N. 2002.An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22.Genomics. 79(3): 285–96.
Javanbakht, H., Diaz-Griffero, F., Stremlau, M., Si, Z. and Sodroski, J. 2005. The contribution ofRING and B-box 2 domains to retroviral restriction mediated by monkey TRIM5alpha. J BiolChem. 280(29): 26933–40.
Javanbakht, H., Yuan, W., Yeung, D. F., Song, B., Diaz-Griffero, F., Li, Y., Li, X., Stremlau, M.and Sodroski, J. 2006a. Characterization of TRIM5alpha trimerization and its contribution tohuman immunodeficiency virus capsid binding. Virology. 353(1): 234–46.
Javanbakht, H., An, P., Gold, B., Petersen, D. C., O’Huigin, C., Nelson, G. W., O’Brien, S. J.,Kirk, G. D., Detels, R., Buchbinder, S., Donfield, S., Shulenin, S., Song, B., Perron, M. J.,Stremlau, M., Sodroski, J., Dean, M. and Winkler, C. 2006b. Effects of human TRIM5alphapolymorphisms on antiretroviral function and susceptibility to human immunodeficiency virusinfection. Virology. 354(1): 15–27.
Jern, P., Stoye, J. P. and Coffin, J. 2007. Role of APOBEC3 in Genetic Diversity among Endoge-nous Murine Leukemia Viruses. PLoS Genetics. preprint(2007): e183.eor.
Jolicoeur, P. and Baltimore, D. 1976. Effect of Fv-1 gene product on proviral DNA formation andintegration in cells infected with murine leukemia viruses. Proc Natl Acad Sci USA. 73(7):2236–40.
Jolicoeur, P. 1979. The Fv-1 gene of the mouse and its control of murine leukemia virus replication.Curr Top Microbiol Immunol. 86: 67–122.
Jolicoeur, P. and Rassart, E. 1980. Effect of Fv-1 gene product on synthesis of linearand supercoiled viral DNA in cells infected with murine leukemia virus. J Virol. 33(1):183–95.
Jonsson, S. R., Hache, G., Stenglein, M. D., Fahrenkrug, S. C., Andresdottir, V. and Harris, R. S.2006. Evolutionarily conserved and non-conserved retrovirus restriction activities of artiodactylAPOBEC3F proteins. Nucleic Acids Res. 34(19): 5683–94.
Jonsson, S. R., Larue, R. S., Stenglein, M. D., Fahrenkrug, S. C., Andresdottir, V. and Harris, R. S.2007. The Restriction of Zoonotic PERV Transmission by Human APOBEC3G. PLoS ONE.2(9): e893.
Kai, K., Ikeda, H., Yuasa, Y., Suzuki, S. and Odaka, T. 1976. Mouse strain resistant to N-, B-, andNB-tropic murine leukemia viruses. J Virol. 20(2): 436–40.
Kai, K., Sato, H. and Odaka, T. 1986. Relationship between the cellular resistance to Friend murineleukemia virus infection and the expression of murine leukemia virus-gp70-related glycopro-tein on cell surface of BALB/c-Fv-4wr mice. Virology. 150(2): 509–12.
Kaiser, S. M. and Emerman, M. 2006. Uracil DNA glycosylase is dispensable for human immunod-eficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidinedeaminase Apobec3G. J Virol. 80(2): 875–82.
Kaiser, S. M., Malik, H. S. and Emerman, M. 2007. Restriction of an extinct retrovirus by thehuman TRIM5alpha antiviral protein. Science. 316(5832): 1756–8.

326 M.D. Stenglein et al.
Kamada, K., Igarashi, T., Martin, M. A., Khamsri, B., Hatcho, K., Yamashita, T., Fujita, M.,Uchiyama, T. and Adachi, A. 2006. Generation of HIV-1 derivatives that productively infectmacaque monkey lymphoid cells. Proc Natl Acad Sci USA. 103(45): 16959–64.
Kao, S., Miyagi, E., Khan, M. A., Takeuchi, H., Opi, S., Goila-Gaur, R. and Strebel, K. 2004.Production of infectious human immunodeficiency virus type 1 does not require depletion ofAPOBEC3G from virus-producing cells. Retrovirology. 1(1): 27.
Kao, S., Goila-Gaur, R., Miyagi, E., Khan, M. A., Opi, S., Takeuchi, H. and Strebel, K. 2007. Pro-duction of infectious virus and degradation of APOBEC3G are separable functional propertiesof human immunodeficiency virus type 1 Vif. Virology. 369(2): 329–39.
Kaumanns, P., Hagmann, I. and Dittmar, M. T. 2006. Human TRIM5alpha mediated restrictionof different HIV-1 subtypes and Lv2 sensitive and insensitive HIV-2 variants. Retrovirology.3: 79.
Keckesova, Z., Ylinen, L. M. and Towers, G. J. 2004. The human and African green monkeyTRIM5alpha genes encode Ref1 and Lv1 retroviral restriction factor activities. Proc Natl AcadSci USA. 101(29): 10780–5.
Keckesova, Z., Ylinen, L. M. and Towers, G. J. 2006. Cyclophilin A renders human immunod-eficiency virus type 1 sensitive to Old World monkey but not human TRIM5 alpha antiviralactivity. J Virol. 80(10): 4683–90.
Khan, M. A., Kao, S., Miyagi, E., Takeuchi, H., Goila-Gaur, R., Opi, S., Gipson, C. L., Parslow, T.G., Ly, H. and Strebel, K. 2005. Viral RNA is required for the association of APOBEC3Gwith human immunodeficiency virus type 1 nucleoprotein complexes. J Virol. 79(9):5870–4.
Kidd, J. M., Newman, T. L., Tuzun, E., Kaul, R. and Eichler, E. E. 2007. Population Stratificationof a Common APOBEC Gene Deletion Polymorphism. PLoS Genet. 3(4): e63.
Kinomoto, M., Kanno, T., Shimura, M., Ishizaka, Y., Kojima, A., Kurata, T., Sata, T. andTokunaga, K. 2007. All APOBEC3 family proteins differentially inhibit LINE-1 retrotrans-position. Nucleic Acids Res. 35(9): 2955–64.
Komohara, Y., Suekane, S., Noguchi, M., Matsuoka, K., Yamada, A. and Itoh, K. 2007. Expressionof APOBEC3G in kidney cells. Tissue Antigens. 69(1): 95–8.
Kootstra, N. A., Munk, C., Tonnu, N., Landau, N. R. and Verma, I. M. 2003. Abrogation of pos-tentry restriction of HIV-1-based lentiviral vector transduction in simian cells. Proc Natl AcadSci USA. 100(3): 1298–303.
Kozak, C. A. and Chakraborti, A. 1996. Single amino acid changes in the murine leukemia viruscapsid protein gene define the target of Fv1 resistance. Virology. 225(2): 300–5.
Kozak, S. L., Marin, M., Rose, K. M., Bystrom, C. and Kabat, D. 2006. The anti-HIV-1 editingenzyme APOBEC3G binds HIV-1 RNA and messenger RNAs that shuttle between polysomesand stress granules. J Biol Chem. 281(39): 29105–19.
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., Devon, K.,Dewar, K., Doyle, M., FitzHugh, W., Funke, R., Gage, D., Harris, K., Heaford, A., Howland, J.,Kann, L., Lehoczky, J., LeVine, R., McEwan, P., McKernan, K., Meldrim, J., Mesirov, J. P.,Miranda, C., Morris, W., Naylor, J., Raymond, C., Rosetti, M., Santos, R., Sheridan, A.,Sougnez, C., Stange-Thomann, N., Stojanovic, N., Subramanian, A., Wyman, D., Rogers, J.,Sulston, J., Ainscough, R., Beck, S., Bentley, D., Burton, J., Clee, C., Carter, N., Coulson, A.,Deadman, R., Deloukas, P., Dunham, A., Dunham, I., Durbin, R., French, L., Grafham, D.,Gregory, S., Hubbard, T., Humphray, S., Hunt, A., Jones, M., Lloyd, C., McMurray, A.,Matthews, L., Mercer, S., Milne, S., Mullikin, J. C., Mungall, A., Plumb, R., Ross, M.,Shownkeen, R., Sims, S., Waterston, R. H., Wilson, R. K., Hillier, L. W., McPherson, J. D.,Marra, M. A., Mardis, E. R., Fulton, L. A., Chinwalla, A. T., Pepin, K. H., Gish, W. R.,Chissoe, S. L., Wendl, M. C., Delehaunty, K. D., Miner, T. L., Delehaunty, A., Kramer, J. B.,Cook, L. L., Fulton, R. S., Johnson, D. L., Minx, P. J., Clifton, S. W., Hawkins, T., Branscomb,E., Predki, P., Richardson, P., Wenning, S., Slezak, T., Doggett, N., Cheng, J. F., Olsen, A.,Lucas, S., Elkin, C., Uberbacher, E., Frazier, M., Gibbs, R. A., Muzny, D. M., Scherer, S. E.,Bouck, J. B., Sodergren, E. J., Worley, K. C., Rives, C. M., Gorrell, J. H., Metzker, M. L.,

15 Host Factors that Restrict Retrovirus Replication 327
Naylor, S. L., Kucherlapati, R. S., Nelson, D. L., Weinstock, G. M., Sakaki, Y., Fujiyama, A.,Hattori, M., Yada, T., Toyoda, A., Itoh, T., Kawagoe, C., Watanabe, H., Totoki, Y., Taylor, T.,Weissenbach, J., Heilig, R., Saurin, W., Artiguenave, F., Brottier, P., Bruls, T., Pelletier, E.,Robert, C., Wincker, P., Smith, D. R., Doucette-Stamm, L., Rubenfield, M., Weinstock, K.,Lee, H. M., Dubois, J., Rosenthal, A., Platzer, M., Nyakatura, G., Taudien, S., Rump, A., Yang,H., Yu, J., Wang, J., Huang, G., Gu, J., Hood, L., Rowen, L., Madan, A., Qin, S., Davis, R.W., Federspiel, N. A., Abola, A. P., Proctor, M. J., Myers, R. M., Schmutz, J., Dickson, M.,Grimwood, J., Cox, D. R., Olson, M. V., Kaul, R., Shimizu, N., Kawasaki, K., Minoshima, S.,Evans, G. A., Athanasiou, M., Schultz, R., Roe, B. A., Chen, F., Pan, H., Ramser, J., Lehrach,H., Reinhardt, R., McCombie, W. R., de la Bastide, M., Dedhia, N., Blocker, H., Hornischer, K.,Nordsiek, G., Agarwala, R., Aravind, L., Bailey, J. A., Bateman, A., Batzoglou, S., Birney, E.,Bork, P., Brown, D. G., Burge, C. B., Cerutti, L., Chen, H. C., Church, D., Clamp, M., Copley,R. R., Doerks, T., Eddy, S. R., Eichler, E. E., Furey, T. S., Galagan, J., Gilbert, J. G., Harmon,C., Hayashizaki, Y., Haussler, D., Hermjakob, H., Hokamp, K., Jang, W., Johnson, L. S., Jones,T. A., Kasif, S., Kaspryzk, A., Kennedy, S., Kent, W. J., Kitts, P., Koonin, E. V., Korf, I., Kulp,D., Lancet, D., Lowe, T. M., McLysaght, A., Mikkelsen, T., Moran, J. V., Mulder, N., Pollara,V. J., Ponting, C. P., Schuler, G., Schultz, J., Slater, G., Smit, A. F., Stupka, E., Szustakowski,J., Thierry-Mieg, D., Thierry-Mieg, J., Wagner, L., Wallis, J., Wheeler, R., Williams, A., Wolf,Y. I., Wolfe, K. H., Yang, S. P., Yeh, R. F., Collins, F., Guyer, M. S., Peterson, J., Felsenfeld,A., Wetterstrand, K. A., Patrinos, A., Morgan, M. J., de Jong, P., Catanese, J. J., Osoegawa,K., Shizuya, H., Choi, S. and Chen, Y. J. 2001. Initial sequencing and analysis of the humangenome. Nature. 409(6822): 860–921.
LaRue, R. S., Jonsson, S. R., Silverstein, K. A., Lajoie, M., Bertrand, D., El-Mabrouk, N., Hotzel,I., Andresdottir, V., Smith, T. P., Harris, R. S. 2008. The artiodactyl APOBEC3 innate immunerepertoire shows evidence for a multi-functional domain organization that existed in the ances-tor of placental mammals. BMC Mol. Biol. 9: 104.
Lassaux, A., Sitbon, M. and Battini, J. L. 2005. Residues in the murine leukemia virus capsid thatdifferentially govern resistance to mouse Fv1 and human Ref1 restrictions. J Virol. 79(10):6560–4.
Lecossier, D., Bouchonnet, F., Clavel, F. and Hance, A. J. 2003. Hypermutation of HIV-1 DNA inthe absence of the Vif protein. Science. 300(5622): 1112.
Li, X., Li, Y., Stremlau, M., Yuan, W., Song, B., Perron, M. and Sodroski, J. 2006a.Functional replacement of the RING, B-box 2, and coiled-coil domains of tripar-tite motif 5alpha (TRIM5alpha) by heterologous TRIM domains. J Virol. 80(13):6198–206.
Li, Y., Li, X., Stremlau, M., Lee, M. and Sodroski, J. 2006b. Removal of arginine 332 allows humanTRIM5alpha to bind human immunodeficiency virus capsids and to restrict infection. J Virol.80(14): 6738–44.
Li, X., Gold, B., O’HUigin, C., Diaz-Griffero, F., Song, B., Si, Z., Li, Y., Yuan, W., Stremlau,M., Mische, C., Javanbakht, H., Scally, M., Winkler, C., Dean, M. and Sodroski, J. 2007.Unique features of TRIM5alpha among closely related human TRIM family members. Virol-ogy. 360(2): 419–33.
Liao, C. H., Kuang, Y. Q., Liu, H. L., Zheng, Y. T. and Su, B. 2007. A novel fusion gene, TRIM5-Cyclophilin A in the pig-tailed macaque determines its susceptibility to HIV-1 infection. Aids.21 Suppl 8: S19–26.
Liddament, M. T., Brown, W. L., Schumacher, A. J. and Harris, R. S. 2004. APOBEC3F proper-ties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr Biol. 14(15):1385–91.
Lilly, F. 1967. Susceptibility to two strains of Friend leukemia virus in mice. Science. 155(761):461–2.
Liu, B., Yu, X., Luo, K., Yu, Y. and Yu, X. F. 2004. Influence of primate lentiviral Vif and pro-teasome inhibitors on human immunodeficiency virus type 1 virion packaging of APOBEC3G.J Virol. 78(4): 2072–81.

328 M.D. Stenglein et al.
Liu, H. L., Wang, Y. Q., Liao, C. H., Kuang, Y. Q., Zheng, Y. T. and Su, B. 2005. Adaptive evolutionof primate TRIM5alpha, a gene restricting HIV-1 infection. Gene. 362: 109–16.
Longerich, S., Basu, U., Alt, F. and Storb, U. 2006. AID in somatic hypermutation and class switchrecombination. Curr Opin Immunol. 18(2): 164–74.
Luban, J., Bossolt, K. L., Franke, E. K., Kalpana, G. V. and Goff, S. P. 1993. Human immunodefi-ciency virus type 1 Gag protein binds to cyclophilins A and B. Cell. 73(6): 1067–78.
Luban, J. 2007. Cyclophilin A, TRIM5, and resistance to human immunodeficiency virus type 1infection. J Virol. 81(3): 1054–61.
Luo, K., Liu, B., Xiao, Z., Yu, Y., Yu, X., Gorelick, R. and Yu, X. F. 2004. Amino-terminal regionof the human immunodeficiency virus type 1 nucleocapsid is required for human APOBEC3Gpackaging. J Virol. 78(21): 11841–52.
Luo, K., Wang, T., Liu, B., Tian, C., Xiao, Z., Kappes, J. and Yu, X. F. 2007. Cytidine deaminasesAPOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integraseand inhibit proviral DNA formation. J Virol. 81(13): 7238–48.
Madani, N. and Kabat, D. 1998. An endogenous inhibitor of human immunodeficiency virus inhuman lymphocytes is overcome by the viral Vif protein. J Virol. 72(12): 10251–5.
Mangeat, B., Turelli, P., Caron, G., Friedli, M., Perrin, L. and Trono, D. 2003. Broad antiretroviraldefence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature.424(6944): 99–103.
Mangeat, B., Turelli, P., Liao, S. and Trono, D. 2004. A single amino acid determinant governs thespecies-specific sensitivity of APOBEC3G to Vif action. J Biol Chem. 279(15): 14481–3.
Mangeat, B. and Trono, D. 2005. Lentiviral vectors and antiretroviral intrinsic immunity. HumGene Ther. 16(8): 913–20.
Mariani, R., Chen, D., Schrofelbauer, B., Navarro, F., Konig, R., Bollman, B., Munk, C., Nymark-McMahon, H. and Landau, N. R. 2003. Species-specific exclusion of APOBEC3G from HIV-1virions by Vif. Cell. 114(1): 21–31.
Marin, M., Rose, K. M., Kozak, S. L. and Kabat, D. 2003. HIV-1 Vif protein binds the editingenzyme APOBEC3G and induces its degradation. Nat Med. 9(11): 1398–403.
Massiah, M. A., Matts, J. A., Short, K. M., Simmons, B. N., Singireddy, S., Yi, Z. and Cox, T. C.2007. Solution structure of the MID1 B-box2 CHC(D/C)C(2)H(2) zinc-binding domain:insights into an evolutionarily conserved RING fold. J Mol Biol. 369(1): 1–10.
Mbisa, J. L., Barr, R., Thomas, J. A., Vandegraaff, N., Dorweiler, I. J., Svarovskaia, E. S.,Brown, W. L., Mansky, L. M., Gorelick, R. J., Harris, R. S., Engelman, A. andPathak, V. K. 2007. Human immunodeficiency virus type 1 cDNAs produced in the presenceof APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J Virol. 81(13):7099–110.
Mehle, A., Strack, B., Ancuta, P., Zhang, C., McPike, M. and Gabuzda, D. 2004. Vif overcomesthe innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem. 279(9): 7792–8.
Mikl, M. C., Watt, I. N., Lu, M., Reik, W., Davies, S. L., Neuberger, M. S. and Rada, C. 2005.Mice deficient in APOBEC2 and APOBEC3. Mol Cell Biol. 25(16): 7270–7.
Mische, C. C., Javanbakht, H., Song, B., Diaz-Griffero, F., Stremlau, M., Strack, B., Si, Z.and Sodroski, J. 2005. Retroviral restriction factor TRIM5alpha is a trimer. J Virol. 79(22):14446–50.
Miyagi, E., Opi, S., Takeuchi, H., Khan, M., Goila-Gaur, R., Kao, S. and Strebel, K. 2007. Enzy-matically active APOBEC3G is required for efficient inhibition of human immunodeficiencyvirus type 1. J Virol. 81(24): 13346–53.
Munk, C., Brandt, S. M., Lucero, G. and Landau, N. R. 2002. A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc Natl Acad Sci USA. 99(21):13843–8.
Munk, C., Beck, T., Zielonka, J., Hotz-Wagenblatt, A., Chareza, S., Battenberg, M., Thielebein, J.,Cichutek, K., Bravo, I. G., O’Brien, S. J., Lochelt, M. and Yuhki, N. 2008. Functions, structure,and read-through alternative splicing of feline APOBEC3 genes. Genome Biol. 9(3): R48.

15 Host Factors that Restrict Retrovirus Replication 329
Nakayama, E. E., Miyoshi, H., Nagai, Y. and Shioda, T. 2005. A specific region of 37 aminoacid residues in the SPRY (B30.2) domain of African green monkey TRIM5alpha determinesspecies-specific restriction of simian immunodeficiency virus SIVmac infection. J Virol.79(14): 8870–7.
Nakayama, E. E., Carpentier, W., Costagliola, D., Shioda, T., Iwamoto, A., Debre, P., Yoshimura,K., Autran, B., Matsushita, S. and Theodorou, I. 2007. Wild type and H43Y variant of humanTRIM5alpha show similar anti-human immunodeficiency virus type 1 activity both in vivo andin vitro. Immunogenetics. 59(6): 511–5.
Nara, P. L. and Fischinger, P. J. 1988. Quantitative infectivity assay for HIV-1 and-2. Nature.332(6163): 469–70.
Navarro, F., Bollman, B., Chen, H., Konig, R., Yu, Q., Chiles, K. and Landau, N. R. 2005.Complementary function of the two catalytic domains of APOBEC3G. Virology. 333(2):374–86.
Newman, E. N., Holmes, R. K., Craig, H. M., Klein, K. C., Lingappa, J. R., Malim, M. H. andSheehy, A. M. 2005. Antiviral function of APOBEC3G can be dissociated from cytidine deam-inase activity. Curr Biol. 15(2): 166–70.
Newman, R. M., Hall, L., Connole, M., Chen, G. L., Sato, S., Yuste, E., Diehl, W., Hunter, E.,Kaur, A., Miller, G. M. and Johnson, W. E. 2006. Balancing selection and the evolution of func-tional polymorphism in Old World monkey TRIM5alpha. Proc Natl Acad Sci USA. 103(50):19134–9.
Newman, R. M., Hall, L., Kirmaier, A., Pozzi, L. A., Pery, E., Farzan, M., O’Neil, S. P. and John-son, W. 2008. Evolution of a TRIM5-CypA splice isoform in old world monkeys. PLoS Pathog.4(2): e1000003.
Nguyen, D. H., Gummuluru, S. and Hu, J. 2007. Deamination-independent inhibition of hepatitisB virus reverse transcription by APOBEC3G. J Virol. 81(9): 4465–72.
Nisole, S., Lynch, C., Stoye, J. P. and Yap, M. W. 2004. A Trim5-cyclophilin A fusion proteinfound in owl monkey kidney cells can restrict HIV-1. Proc Natl Acad Sci USA. 101(36):13324–8.
Nisole, S., Stoye, J. P. and Saib, A. 2005. TRIM family proteins: retroviral restriction and antiviraldefence. Nat Rev Microbiol. 3(10): 799–808.
Noser, J. A., Towers, G. J., Sakuma, R., Dumont, J. M., Collins, M. K. and Ikeda, Y. 2006.Cyclosporine increases human immunodeficiency virus type 1 vector transduction of primarymouse cells. J Virol. 80(15): 7769–74.
Ohkura, S., Yap, M. W., Sheldon, T. and Stoye, J. P. 2006. All three variable regions of theTRIM5alpha B30.2 domain can contribute to the specificity of retrovirus restriction. J Virol.80(17): 8554–65.
Ortiz, M., Bleiber, G., Martinez, R., Kaessmann, H. and Telenti, A. 2006. Patterns of evolution ofhost proteins involved in retroviral pathogenesis. Retrovirology. 3: 11.
Peng, G., Lei, K. J., Jin, W., Greenwell-Wild, T. and Wahl, S. M. 2006. Induction of APOBEC3family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J ExpMed. 203(1): 41–6.
Perez-Caballero, D., Hatziioannou, T., Yang, A., Cowan, S. and Bieniasz, P. D. 2005. Humantripartite motif 5alpha domains responsible for retrovirus restriction activity and specificity.J Virol. 79(14): 8969–78.
Perron, M. J., Stremlau, M., Song, B., Ulm, W., Mulligan, R. C. and Sodroski, J. 2004. TRIM5alphamediates the postentry block to N-tropic murine leukemia viruses in human cells. Proc NatlAcad Sci USA. 101(32): 11827–32.
Perron, M. J., Stremlau, M. and Sodroski, J. 2006. Two surface-exposed elements of theB30.2/SPRY domain as potency determinants of N-tropic murine leukemia virus restrictionby human TRIM5alpha. J Virol. 80(11): 5631–6.
Perron, M. J., Stremlau, M., Lee, M., Javanbakht, H., Song, B. and Sodroski, J. 2007. The humanTRIM5alpha restriction factor mediates accelerated uncoating of the N-tropic murine leukemiavirus capsid. J Virol. 81(5): 2138–48.

330 M.D. Stenglein et al.
Petersen-Mahrt, S. K., Harris, R. S. and Neuberger, M. S. 2002. AID mutates E. coli suggesting aDNA deamination mechanism for antibody diversification. Nature. 418(6893): 99–103.
Pincus, T., Hartley, J. W. and Rowe, W. P. 1971. A major genetic locus affecting resistance toinfection with murine leukemia viruses. I. Tissue culture studies of naturally occurring viruses.J Exp Med. 133(6): 1219–33.
Pincus, T., Hartley, J. W. and Rowe, W. P. 1975. A major genetic locus affecting resistance toinfection with murine leukemia viruses. IV. Dose-response relationships in Fv-1-sensitive andresistant cell cultures. Virology. 65(2): 333–42.
Reddy, B. A. and Etkin, L. D. 1991. A unique bipartite cysteine-histidine motif defines a subfamilyof potential zinc-finger proteins. Nucleic Acids Res. 19(22): 6330.
Reddy, B. A., Etkin, L. D. and Freemont, P. S. 1992. A novel zinc finger coiled-coil domain in afamily of nuclear proteins. Trends Biochem Sci. 17(9): 344–5.
Reymond, A., Meroni, G., Fantozzi, A., Merla, G., Cairo, S., Luzi, L., Riganelli, D., Zanaria, E.,Messali, S., Cainarca, S., Guffanti, A., Minucci, S., Pelicci, P. G. and Ballabio, A. 2001. Thetripartite motif family identifies cell compartments. Embo J. 20(9): 2140–51.
Rhodes, D. A., de Bono, B. and Trowsdale, J. 2005. Relationship between SPRY and B30.2 proteindomains. Evolution of a component of immune defence? Immunology. 116(4): 411–7.
Ribeiro, I. P., Menezes, A. N., Moreira, M. A., Bonvicino, C. R., Seuanez, H. N. and Soares, M. A.2005. Evolution of cyclophilin A and TRIMCyp retrotransposition in New World primates. JVirol. 79(23): 14998–5003.
Rogozin, I. B., Basu, M. K., Jordan, I. K., Pavlov, Y. I. and Koonin, E. V. 2005. APOBEC4, a newmember of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases predictedby computational analysis. Cell Cycle. 4(9): 1281–5.
Rose, K. M., Marin, M., Kozak, S. L. and Kabat, D. 2004a. The viral infectivity factor (Vif) ofHIV-1 unveiled. Trends Mol Med. 10(6): 291–7.
Rose, K. M., Marin, M., Kozak, S. L. and Kabat, D. 2004b. Transcriptional regulation ofAPOBEC3G, a cytidine deaminase that hypermutates human immunodeficiency virus. J BiolChem. 279(40): 41744–9.
Rosler, C., Kock, J., Kann, M., Malim, M. H., Blum, H. E., Baumert, T. F. and von Weizsacker,F. 2005. APOBEC-mediated interference with hepadnavirus production. Hepatology. 42(2):301–9.
Saenz, D. T., Teo, W., Olsen, J. C. and Poeschla, E. M. 2005. Restriction of feline immunodefi-ciency virus by Ref1, Lv1, and primate TRIM5alpha proteins. J Virol. 79(24): 15175–88.
Sakuma, R., Noser, J. A., Ohmine, S. and Ikeda, Y. 2007a. Inhibition of HIV-1 replication bysimian restriction factors, TRIM5alpha and APOBEC3G. Gene Ther. 14(2): 185–9.
Sakuma, R., Noser, J. A., Ohmine, S. and Ikeda, Y. 2007b. Rhesus monkey TRIM5alpha restrictsHIV-1 production through rapid degradation of viral Gag polyproteins. Nat Med. 13(5): 631–5.
Santa-Marta, M., da Silva, F. A., Fonseca, A. M. and Goncalves, J. 2005. HIV-1 Vif can directlyinhibit apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3G-mediated cyti-dine deamination by using a single amino acid interaction and without protein degradation.J Biol Chem. 280(10): 8765–75.
Sarkis, P. T., Ying, S., Xu, R. and Yu, X. F. 2006. STAT1-independent cell type-specific regulationof antiviral APOBEC3G by IFN-alpha. J Immunol. 177(7): 4530–40.
Saurin, A. J., Borden, K. L., Boddy, M. N. and Freemont, P. S. 1996. Does this have a familiarRING? Trends Biochem Sci. 21(6): 208–14.
Sawyer, S. L., Emerman, M. and Malik, H. S. 2004. Ancient adaptive evolution of the primateantiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2(9): E275.
Sawyer, S. L., Wu, L. I., Emerman, M. and Malik, H. S. 2005. Positive selection of primateTRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc Natl AcadSci USA. 102(8): 2832–7.
Sawyer, S. L., Wu, L. I., Akey, J. M., Emerman, M. and Malik, H. S. 2006. High-frequency per-sistence of an impaired allele of the retroviral defense gene TRIM5alpha in humans. Curr Biol.16(1): 95–100.

15 Host Factors that Restrict Retrovirus Replication 331
Sayah, D. M. and Luban, J. 2004. Selection for loss of Ref1 activity in human cells releaseshuman immunodeficiency virus type 1 from cyclophilin A dependence during infection. J Virol.78(21): 12066–70.
Sayah, D. M., Sokolskaja, E., Berthoux, L. and Luban, J. 2004. Cyclophilin A retrotranspositioninto TRIM5 explains owl monkey resistance to HIV-1. Nature. 430(6999): 569–73.
Schafer, A., Bogerd, H. P. and Cullen, B. R. 2004. Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology.328(2): 163–8.
Schaller, T., Hue, S. and Towers, G. J. 2007. An active TRIM5 protein in rabbits indicates a com-mon antiviral ancestor for mammalian TRIM5 proteins. J Virol. 81(21): 11713–21.
Schmitz, C., Marchant, D., Neil, S. J., Aubin, K., Reuter, S., Dittmar, M. T. and McKnight, A.2004. Lv2, a novel postentry restriction, is mediated by both capsid and envelope. J Virol.78(4): 2006–16.
Schrofelbauer, B., Chen, D. and Landau, N. R. 2004. A single amino acid of APOBEC3G controlsits species-specific interaction with virion infectivity factor (Vif). Proc Natl Acad Sci USA.101(11): 3927–32.
Schumacher, A. J., Nissley, D. V. and Harris, R. S. 2005. APOBEC3G hypermutates genomic DNAand inhibits Ty1 retrotransposition in yeast. Proc Natl Acad Sci USA. 102(28): 9854–9.
Schumacher, A. J., Hache, G., Macduff, D. A., Brown, W. L. and Harris, R. S. 2008. The DNAdeaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunode-ficiency virus type 1 restriction. J Virol. 82(6): 2652–60.
Sebastian, S. and Luban, J. 2005. TRIM5alpha selectively binds a restriction-sensitive retroviralcapsid. Retrovirology. 2: 40.
Seppen, J. 2004. Unedited inhibition of HBV replication by APOBEC3G. J Hepatol. 41(6):1068–9.
Serhan, F., Jourdan, N., Saleun, S., Moullier, P. and Duisit, G. 2002. Characterization of producercell-dependent restriction of murine leukemia virus replication. J Virol. 76(13): 6609–17.
Sheehy, A. M., Gaddis, N. C., Choi, J. D. and Malim, M. H. 2002. Isolation of a human gene thatinhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 418(6898): 646–50.
Sheehy, A. M., Gaddis, N. C. and Malim, M. H. 2003. The antiretroviral enzyme APOBEC3G isdegraded by the proteasome in response to HIV-1 Vif. Nat Med. 9(11): 1404–7.
Shibata, R., Sakai, H., Kawamura, M., Tokunaga, K. and Adachi, A. 1995. Early replication blockof human immunodeficiency virus type 1 in monkey cells. J Gen Virol. 76(11): 2723–30.
Shindo, K., Takaori-Kondo, A., Kobayashi, M., Abudu, A., Fukunaga, K. and Uchiyama, T. 2003.The enzymatic activity of CEM15/Apobec-3G is essential for the regulation of the infectiv-ity of HIV-1 virion but not a sole determinant of its antiviral activity. J Biol Chem. 278(45):44412–6.
Si, Z., Vandegraaff, N., O’Huigin, C., Song, B., Yuan, W., Xu, C., Perron, M., Li, X., Marasco, W.A., Engelman, A., Dean, M. and Sodroski, J. 2006. Evolution of a cytoplasmic tripartite motif(TRIM) protein in cows that restricts retroviral infection. Proc Natl Acad Sci USA. 103(19):7454–9.
Simon, J. H. and Malim, M. H. 1996. The human immunodeficiency virus type 1 Vif protein mod-ulates the postpenetration stability of viral nucleoprotein complexes. J Virol. 70(8): 5297–305.
Simon, J. H., Gaddis, N. C., Fouchier, R. A. and Malim, M. H. 1998. Evidence for a newly discov-ered cellular anti-HIV-1 phenotype. Nat Med. 4(12): 1397–400.
Simon, V., Zennou, V., Murray, D., Huang, Y., Ho, D. D. and Bieniasz, P. D. 2005. Natural Variationin Vif: Differential Impact on APOBEC3G/3F and a Potential Role in HIV-1 Diversification.PLoS Pathog. 1(1): e6.
Sokolskaja, E., Sayah, D. M. and Luban, J. 2004. Target cell cyclophilin A modulates humanimmunodeficiency virus type 1 infectivity. J Virol. 78(23): 12800–8.
Sokolskaja, E., Berthoux, L. and Luban, J. 2006. Cyclophilin A and TRIM5alpha independentlyregulate human immunodeficiency virus type 1 infectivity in human cells. J Virol. 80(6):2855–62.

332 M.D. Stenglein et al.
Song, B., Javanbakht, H., Perron, M., Park, D. H., Stremlau, M. and Sodroski, J. 2005a. Retrovirusrestriction by TRIM5alpha variants from Old World and New World primates. J Virol. 79(7):3930–7.
Song, B., Gold, B., O’Huigin, C., Javanbakht, H., Li, X., Stremlau, M., Winkler, C., Dean, M. andSodroski, J. 2005b. The B30.2(SPRY) domain of the retroviral restriction factor TRIM5alphaexhibits lineage-specific length and sequence variation in primates. J Virol. 79(10): 6111–21.
Song, B., Diaz-Griffero, F., Park, D. H., Rogers, T., Stremlau, M. and Sodroski, J. 2005c.TRIM5alpha association with cytoplasmic bodies is not required for antiretroviral activity.Virology. 343(2): 201–11.
Speelmon, E. C., Livingston-Rosanoff, D., Li, S. S., Vu, Q., Bui, J., Geraghty, D. E., Zhao, L. P.and McElrath, M. J. 2006. Genetic association of the antiviral restriction factor TRIM5alphawith human immunodeficiency virus type 1 infection. J Virol. 80(5): 2463–71.
Stenglein, M. D. and Harris, R. S. 2006. APOBEC3B and APOBEC3F inhibit L1 retrotranspositionby a DNA deamination-independent mechanism. J Biol Chem. 281(25): 16837–41.
Stevenson, M., Stanwick, T. L., Dempsey, M. P. and Lamonica, C. A. 1990. HIV-1 replication iscontrolled at the level of T cell activation and proviral integration. Embo J. 9(5): 1551–60.
Stopak, K., de Noronha, C., Yonemoto, W. and Greene, W. C. 2003. HIV-1 Vif blocks the antiviralactivity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell.12(3): 591–601.
Stopak, K. S., Chiu, Y. L., Kropp, J., Grant, R. M. and Greene, W. C. 2007. Distinct pat-terns of cytokine regulation of APOBEC3G expression and activity in primary lymphocytes,macrophages, and dendritic cells. J Biol Chem. 282(6): 3539–46.
Strebel, K., Daugherty, D., Clouse, K., Cohen, D., Folks, T. and Martin, M. A. 1987. The HIV ′A′
(sor) gene product is essential for virus infectivity. Nature. 328(6132): 728–30.Stremlau, M., Owens, C. M., Perron, M. J., Kiessling, M., Autissier, P. and Sodroski, J. 2004. The
cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys.Nature. 427(6977): 848–53.
Stremlau, M., Perron, M., Welikala, S. and Sodroski, J. 2005. Species-specific variation in theB30.2(SPRY) domain of TRIM5alpha determines the potency of human immunodeficiencyvirus restriction. J Virol. 79(5): 3139–45.
Stremlau, M., Song, B., Javanbakht, H., Perron, M. and Sodroski, J. 2006a. Cyclophilin A: anauxiliary but not necessary cofactor for TRIM5alpha restriction of HIV-1. Virology. 351(1):112–20.
Stremlau, M., Perron, M., Lee, M., Li, Y., Song, B., Javanbakht, H., Diaz-Griffero, F.,Anderson, D. J., Sundquist, W. I. and Sodroski, J. 2006b. Specific recognition and accelerateduncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc Natl Acad Sci USA.103(14): 5514–9.
Suspene, R., Guetard, D., Henry, M., Sommer, P., Wain-Hobson, S. and Vartanian, J. P. 2005.Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases invitro and in vivo. Proc Natl Acad Sci USA. 102(23): 8321–6.
Suzuki, S. 1975. FV-4: a new gene affecting the splenomegaly induction by Friend leukemia virus.Jpn J Exp Med. 45(6): 473–8.
Svarovskaia, E. S., Xu, H., Mbisa, J. L., Barr, R., Gorelick, R. J., Ono, A., Freed, E. O., Hu, W. S.and Pathak, V. K. 2004. Human apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (APOBEC3G) is incorporated into HIV-1 virions through interactions with viral andnonviral RNAs. J Biol Chem. 279(34): 35822–8.
Tanaka, Y., Marusawa, H., Seno, H., Matsumoto, Y., Ueda, Y., Kodama, Y., Endo, Y., Yamauchi, J.,Matsumoto, T., Takaori-Kondo, A., Ikai, I. and Chiba, T. 2006. Anti-viral protein APOBEC3Gis induced by interferon-alpha stimulation in human hepatocytes. Biochem Biophys Res Com-mun. 341(2): 314–9.
Taylor, M. W., Grosse, W. M., Schaley, J. E., Sanda, C., Wu, X., Chien, S. C., Smith, F., Wu, T. G.,Stephens, M., Ferris, M. W., McClintick, J. N., Jerome, R. E. and Edenberg, H. J. 2004. Globaleffect of PEG-IFN-alpha and ribavirin on gene expression in PBMC in vitro. J InterferonCytokine Res. 24(2): 107–18.

15 Host Factors that Restrict Retrovirus Replication 333
Thali, M., Bukovsky, A., Kondo, E., Rosenwirth, B., Walsh, C. T., Sodroski, J. and Gottlinger,H. G. 1994. Functional association of cyclophilin A with HIV-1 virions. Nature. 372(6504):363–5.
Tock, M. R. and Dryden, D. T. 2005. The biology of restriction and anti-restriction. Curr OpinMicrobiol. 8(4): 466–72.
Towers, G., Bock, M., Martin, S., Takeuchi, Y., Stoye, J. P. and Danos, O. 2000. A con-served mechanism of retrovirus restriction in mammals. Proc Natl Acad Sci USA. 97(22):12295–9.
Towers, G., Collins, M. and Takeuchi, Y. 2002. Abrogation of Ref1 retrovirus restriction in humancells. J Virol. 76(5): 2548–50.
Towers, G. J. 2007. The control of viral infection by tripartite motif proteins and cyclophilin A.Retrovirology. 4: 40.
Turelli, P., Mangeat, B., Jost, S., Vianin, S. and Trono, D. 2004. Inhibition of hepatitis B virusreplication by APOBEC3G. Science. 303(5665): 1829.
Victoria, J. G. and Robinson, W. E., Jr. 2005. Disruption of the putative splice acceptor site forSIV(mac239)Vif reveals tight control of SIV splicing and impaired replication in Vif non-permissive cells. Virology. 338(2): 281–91.
Vigano, A., Saresella, M., Schenal, M., Erba, P., Piacentini, L., Tornaghi, R., Naddeo, V., Gia-comet, V., Borelli, M., Trabattoni, D. and Clerici, M. 2007. Immune activation and normallevels of endogenous antivirals are seen in healthy adolescents born of HIV-infected mothers.Aids. 21(2): 245–8.
Virgen, C. A., Kratovac, Z., Bieniasz, P. D. and Hatziioannou, T. 2008. Independent genesis ofchimeric TRIM5-cyclophilin proteins in two primate species. Proc Natl Acad Sci USA. 105(9):3563–8.
von Schwedler, U., Song, J., Aiken, C. and Trono, D. 1993. Vif is crucial for human immunodefi-ciency virus type 1 proviral DNA synthesis in infected cells. J Virol. 67(8): 4945–55.
Wedekind, J. E., Dance, G. S., Sowden, M. P. and Smith, H. C. 2003. Messenger RNA editing inmammals: new members of the APOBEC family seeking roles in the family business. TrendsGenet. 19(4): 207–16.
Wichroski, M. J., Ichiyama, K. and Rana, T. M. 2005. Analysis of HIV-1 viral infectivity factor-mediated proteasome-dependent depletion of APOBEC3G: correlating function and subcellularlocalization. J Biol Chem. 280(9): 8387–96.
Wichroski, M. J., Robb, G. B. and Rana, T. M. 2006. Human retroviral host restriction factorsAPOBEC3G and APOBEC3F localize to mRNA processing bodies. PLoS Pathog. 2(5): e41.
Wiegand, H. L., Doehle, B. P., Bogerd, H. P. and Cullen, B. R. 2004. A second human antiretro-viral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. Embo J. 23(12):2451–8.
Wilson, S. J., Webb, B. L., Ylinen, L. M., Verschoor, E., Heeney, J. L. and Towers, G. J. 2008.Independent evolution of an antiviral TRIMCyp in rhesus macaques. Proc Natl Acad Sci USA.105(9): 3557–62.
Woo, J. S., Imm, J. H., Min, C. K., Kim, K. J., Cha, S. S. and Oh, B. H. 2006. Structural andfunctional insights into the B30.2/SPRY domain. Embo J. 25(6): 1353–63.
Wu, X., Anderson, J. L., Campbell, E. M., Joseph, A. M. and Hope, T. J. 2006. Proteasomeinhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infec-tion. Proc Natl Acad Sci USA. 103(19): 7465–70.
Xu, L., Yang, L., Moitra, P. K., Hashimoto, K., Rallabhandi, P., Kaul, S., Meroni, G., Jensen,J. P., Weissman, A. M. and D’Arpa, P. 2003. BTBD1 and BTBD2 colocalize to cyto-plasmic bodies with the RBCC/tripartite motif protein, TRIM5delta. Exp Cell Res. 288(1):84–93.
Xu, H., Svarovskaia, E. S., Barr, R., Zhang, Y., Khan, M. A., Strebel, K. and Pathak, V. K. 2004.A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resis-tance to HIV-1 virion infectivity factor-induced depletion. Proc Natl Acad Sci USA. 101(15):5652–7.

334 M.D. Stenglein et al.
Yang, W. K., Kiggans, J. O., Yang, D. M., Ou, C. Y., Tennant, R. W., Brown, A. and Bassin, R.H. 1980. Synthesis and circularization of N- and B-tropic retroviral DNA Fv-1 permissive andrestrictive mouse cells. Proc Natl Acad Sci USA. 77(5): 2994–8.
Yap, M. W., Nisole, S., Lynch, C. and Stoye, J. P. 2004. Trim5alpha protein restricts both HIV-1and murine leukemia virus. Proc Natl Acad Sci USA. 101(29): 10786–91.
Yap, M. W., Nisole, S. and Stoye, J. P. 2005. A single amino acid change in the SPRY domain ofhuman Trim5alpha leads to HIV-1 restriction. Curr Biol. 15(1): 73–8.
Yap, M. W., Dodding, M. P. and Stoye, J. P. 2006. Trim-cyclophilin A fusion proteins can restricthuman immunodeficiency virus type 1 infection at two distinct phases in the viral life cycle. JVirol. 80(8): 4061–7.
Ying, S., Zhang, X., Sarkis, P. T., Xu, R. and Yu, X. 2007. Cell-specific Regulation of APOBEC3Fby Interferons. Acta Biochim Biophys Sin (Shanghai). 39(4): 297–304.
Ylinen, L. M., Keckesova, Z., Wilson, S. J., Ranasinghe, S. and Towers, G. J. 2005. Differen-tial restriction of human immunodeficiency virus type 2 and simian immunodeficiency virusSIVmac by TRIM5alpha alleles. J Virol. 79(18): 11580–7.
Ylinen, L. M., Keckesova, Z., Webb, B. L., Gifford, R. J., Smith, T. P. and Towers, G. J. 2006.Isolation of an active Lv1 gene from cattle indicates that tripartite motif protein-mediated innateimmunity to retroviral infection is widespread among mammals. J Virol. 80(15): 7332–8.
Yu, X., Yu, Y., Liu, B., Luo, K., Kong, W., Mao, P. and Yu, X. F. 2003. Induction of APOBEC3Gubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 302(5647):1056–60.
Zack, J. A., Arrigo, S. J., Weitsman, S. R., Go, A. S., Haislip, A. and Chen, I. S. 1990. HIV-1 entryinto quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure.Cell. 61(2): 213–22.
Zennou, V., Perez-Caballero, D., Gottlinger, H. and Bieniasz, P. D. 2004. APOBEC3G incorpora-tion into human immunodeficiency virus type 1 particles. J Virol. 78(21): 12058–61.
Zhang, H., Yang, B., Pomerantz, R. J., Zhang, C., Arunachalam, S. C. and Gao, L. 2003. Thecytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature.424(6944): 94–8.
Zhang, J. and Webb, D. M. 2004. Rapid evolution of primate antiviral enzyme APOBEC3G. HumMol Genet. 13(16): 1785–91.
Zhang, F., Hatziioannou, T., Perez-Caballero, D., Derse, D. and Bieniasz, P. D. 2006. Antiretroviralpotential of human tripartite motif-5 and related proteins. Virology. 353(2): 396–409.
Zheng, Y. H., Irwin, D., Kurosu, T., Tokunaga, K., Sata, T. and Peterlin, B. M. 2004. HumanAPOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replica-tion. J Virol. 78(11): 6073–6.