ultraefficient homogeneous catalyst for the co -to … · ultraefficient homogeneous catalyst for...
TRANSCRIPT

Ultraefficient homogeneous catalyst for the CO2-to-COelectrochemical conversionCyrille Costentin1, Guillaume Passard, Marc Robert1, and Jean-Michel Savéant1
Université Paris Diderot, Sorbonne Paris Cité, Laboratoire d’Electrochimie Moléculaire, Unité Mixte de Recherche Université—Centre National de la RechercheScientifique, 75205 Paris Cedex 13, France
Contributed by Jean-Michel Savéant, August 31, 2014 (sent for review August 9, 2014; reviewed by Harry B. Gray and Alain Deronzier)
A very efficient electrogenerated Fe0 porphyrin catalyst was ob-tained by substituting in tetraphenylporphyrin two of the oppo-site phenyl rings by ortho-, ortho’-phenol groups while the othertwo are perfluorinated. It proves to be an excellent catalyst of theCO2-to-CO conversion as to selectivity (the CO faradaic yield isnearly quantitative), overpotential, and turnover frequency. Bench-marking with other catalysts, through catalytic Tafel plots, showsthat it is the most efficient, to the best of our knowledge, homo-geneous molecular catalyst of the CO2-to-CO conversion at present.Comparison with another Fe0 tetraphenylporphyrin bearing eightortho-, ortho’-phenol functionalities launches a general strategywhere changes in substituents will be designed so as to optimizethe operational combination of all catalyst elements of merit.
solar fuels | CO2 reduction | electrochemistry | catalysis |contemporary energy challenges
The reductive conversion of CO2 to CO is an important issueof contemporary energy and environmental challenges (1–10).
Several low-oxidation-state transition metal complexes have beenproposed to serve as homogeneous catalyst for this reaction innonaqueous solvents such as N,N’-dimethylformamide (DMF) oracetonitrile (11–23).Among them, electrochemically generated Fe0 complexes
have been shown to be good catalysts, provided they are used inthe presence of Brönsted or Lewis acids (17–19). More recentinvestigations have extended the range of Brönsted acids able toboost the catalysis of the CO2-to-CO conversion by electrogeneratedFe0TPP (Scheme 1) without degrading the selectivity of the reaction.They have also provided a detailed analysis of the reaction mecha-nism (24, 25).This is notably the case with phenol, which gave rise to the
idea of installing prepositioned phenol groups in the catalystmolecule as pictured in Scheme 1 under the heading “CAT.” Theresult was indeed a remarkably efficient and selective catalyst ofthe CO2-to-CO conversion (26). At first blush, the comparisonwith the role of phenol in the case of FeTPP would entail attrib-uting this considerable enhancement of catalysis to a local con-centration of phenol much larger than can be achieved in solution.In fact, as analyzed in detail elsewhere (27), the role played by theinternal phenol moieties is twofold. They indeed provide a verylarge local phenol concentration, favoring proton transfers, butthey also considerably stabilize the initial Fe0–CO2 adductthrough H bonding. Although the favorable effect of pendantacid groups has been noted in several cases (see ref. 27 and ref-erences therein), this was, to our knowledge, the first time theirexact role was deciphered. The difference in the role played by thephenol moieties takes place within the framework of two differentmechanisms (see Scheme 2 for CAT and FCAT and Scheme 3 forFeTPP) (27). With FeTPP, the first step is, as with CAT andFCAT, the addition of CO2 on the electrogenerated Fe0 complex(et1 in Schemes 2 and 3). The strong stabilization of the Fe0–CO2adduct formed according to reaction 1 (in Schemes 2 and 3) in thelatter cases compared with the first has a favorable effect on ca-talysis, but one consequence of this stabilization is that catalysisthen required an additional proton (reactions 21 and 22 in Scheme
2), the final, catalytic loop-closing step being the cleavage of oneof the C–O bonds of CO2 concerted with both electron transferfrom the electrode and proton transfer from one of the localphenol groups (et2 in Scheme 2). In the FeTPP case, the C–Obond-breaking step (reaction 2 in Scheme 3) is different: it in-volves an intramolecular electron transfer concerted with protontransfer and cleavage of the C–O bond. The catalytic loop isclosed by a homogeneous electron transfer step (et2 in Scheme 3)that regenerates the initial FeI complex.The object of the present contribution is to test the idea that
introduction of different substituents on the periphery of theporphyrin ring may improve the efficiency of the catalysis ofCO2-to-CO conversion. In such a venture, we will have to takeinto account both the overpotential at which the reaction takesplace and the catalytic rate expressed as the turnover frequency asdetailed in the following sections. Taking these two aspects si-multaneously into consideration is essential in view of the pos-sibility that substitution may improve one of the two factors anddegrade the other, or vice versa. As a first example, we examinedthe catalytic performances of the FCAT molecule (Scheme 1), inwhich four of the eight phenol groups have been preserved in thesame ortho-, ortho’- positions on two of the opposite phenyl rings,while the two other phenyl rings have been perfluorinated (thesynthesis and characterization of this molecule is described in SIText). A query that first comes to mind is as follows. The inductiveeffect of the fluorine atoms is expected to ease the reduction ofthe molecule to the Fe0 oxidation state, and thus to be favorableto catalysis in terms of overpotential. However, will this benefit beblurred by a decrease of its reactivity toward CO2? Indeed, the
Significance
Conversion of CO2 into liquid fuels is one of the most importantcontemporary energy and environmental challenges. As a firststep in this direction, the electrochemical reduction of CO2 to COrequires catalysts, usually derived from transition metal com-plexes. In this paper, a very efficient, electrogenerated iron-porphyrin catalyst was obtained by introducing both pendantacid groups and fluorine substituents in the molecule. The for-mer stabilize the CO2-catalyst key intermediate via H bondingand provide a high local proton concentration. The latter helpdecrease the energy required to drive catalysis. Benchmarkingthis molecule with other catalysts shows that it is at present themost efficient, to the best of our knowledge, homogeneousmolecular catalyst of the CO2-to-CO conversion in terms of se-lectivity, overpotential, and turnover frequency.
Author contributions: C.C., M.R., and J.-M.S. designed research; G.P. performed re-search; C.C., G.P., M.R., and J.-M.S. analyzed data; and C.C., M.R., and J.-M.S. wrotethe paper.
Reviewers: H.B.G., California Institute of Technology; and A.D., Centre National de laRecherche Scientifique.
The authors declare no conflict of interest.1To whom correspondence may be addressed. Email: [email protected],[email protected], or [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1416697111/-/DCSupplemental.
14990–14994 | PNAS | October 21, 2014 | vol. 111 | no. 42 www.pnas.org/cgi/doi/10.1073/pnas.1416697111
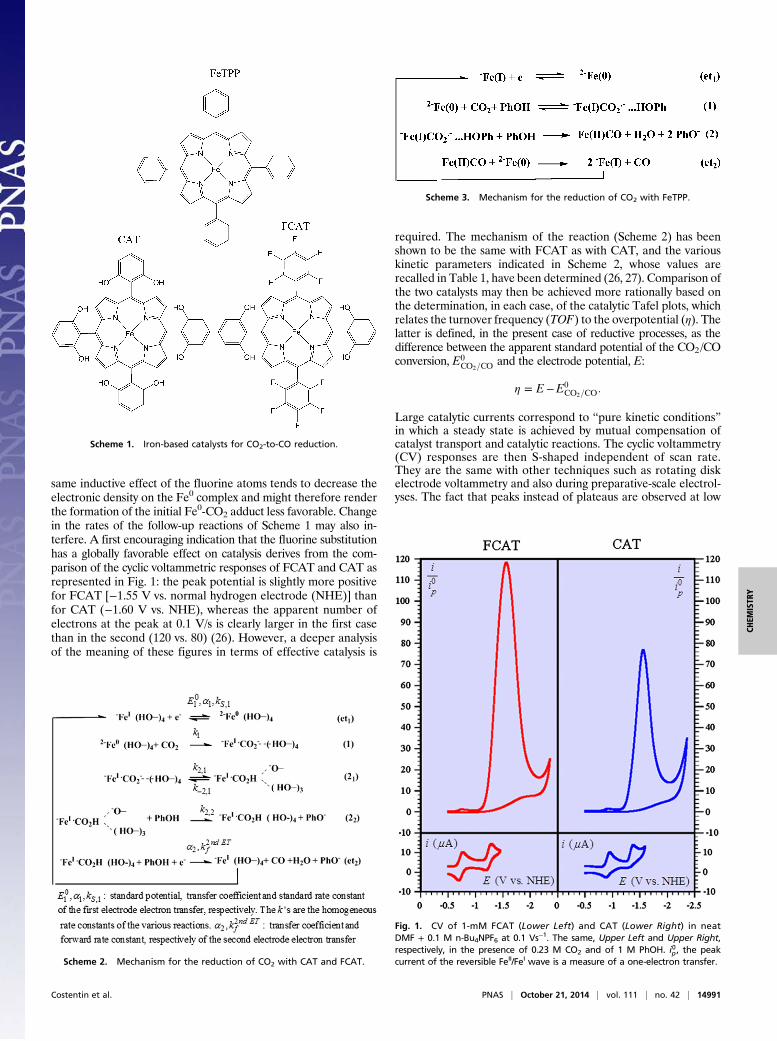
same inductive effect of the fluorine atoms tends to decrease theelectronic density on the Fe0 complex and might therefore renderthe formation of the initial Fe0-CO2 adduct less favorable. Changein the rates of the follow-up reactions of Scheme 1 may also in-terfere. A first encouraging indication that the fluorine substitutionhas a globally favorable effect on catalysis derives from the com-parison of the cyclic voltammetric responses of FCAT and CAT asrepresented in Fig. 1: the peak potential is slightly more positivefor FCAT [−1.55 V vs. normal hydrogen electrode (NHE)] thanfor CAT (−1.60 V vs. NHE), whereas the apparent number ofelectrons at the peak at 0.1 V/s is clearly larger in the first casethan in the second (120 vs. 80) (26). However, a deeper analysisof the meaning of these figures in terms of effective catalysis is
required. The mechanism of the reaction (Scheme 2) has beenshown to be the same with FCAT as with CAT, and the variouskinetic parameters indicated in Scheme 2, whose values arerecalled in Table 1, have been determined (26, 27). Comparison ofthe two catalysts may then be achieved more rationally based onthe determination, in each case, of the catalytic Tafel plots, whichrelates the turnover frequency (TOF) to the overpotential (η). Thelatter is defined, in the present case of reductive processes, as thedifference between the apparent standard potential of the CO2/COconversion, E0
CO2=COand the electrode potential, E:
η = E−E0CO2=CO:
Large catalytic currents correspond to “pure kinetic conditions”in which a steady state is achieved by mutual compensation ofcatalyst transport and catalytic reactions. The cyclic voltammetry(CV) responses are then S-shaped independent of scan rate.They are the same with other techniques such as rotating diskelectrode voltammetry and also during preparative-scale electrol-yses. The fact that peaks instead of plateaus are observed at low
Scheme 1. Iron-based catalysts for CO2-to-CO reduction.
Scheme 2. Mechanism for the reduction of CO2 with CAT and FCAT.
Scheme 3. Mechanism for the reduction of CO2 with FeTPP.
Fig. 1. CV of 1-mM FCAT (Lower Left) and CAT (Lower Right) in neatDMF + 0.1 M n-Bu4NPF6 at 0.1 Vs−1. The same, Upper Left and Upper Right,respectively, in the presence of 0.23 M CO2 and of 1 M PhOH. i0p, the peakcurrent of the reversible FeII/FeI wave is a measure of a one-electron transfer.
Costentin et al. PNAS | October 21, 2014 | vol. 111 | no. 42 | 14991
CHEM
ISTR
Y

scan rate, as in Fig. 1, derives from secondary phenomena relatedto the observed high catalytic efficiencies, such as substrate con-sumption, inhibition by products, and deactivation of the catalyst.These factors and the ways to go around their occurrence to finallyobtain a full characterization of the mechanism and kinetics of thecatalysis process are discussed in detail elsewhere (27). Underpure kinetic conditions, the active catalyst molecules are then
confined within a thin reaction-diffusion layer adjacent to theelectrode surface. During the time where the catalyst remainsstable, the TOF is defined as
TOF = Nproduct�Nactive cat;
where Nproduct is the number of moles of the product, generatedper unit of time, and Nactive cat is the maximal number of moles ofthe active form catalyst contained in the reaction–diffusion layerrather than in the whole electrochemical cell (for more informa-tion on the notions of pure kinetic conditions, reaction–diffusionlayer, and on the correct definition of TOF, see refs. 23, 26, 28).For the present reaction mechanism (Scheme 2) as well as for allreaction schemes belonging to the same category, the TOF–η rela-tionships are obtained from the following equations (28, 29), usingthe notations defined in Scheme 2 and the data listed in Table 1.
TOF =TOFmax8>>>><
>>>>:1+
ipl2FSC0
catk2nd ETf
expðα2 fEÞ
+ipl
2FSC0cat
ffiffiffiffiffiffiffiffiDcat
p ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffik1½CO2�
p exp�FRT
�E−E0
1
��9>>>>=>>>>;
;
with
Table 1. Kinetic characteristics of the reactions in Scheme 2from ref. 27
Parameters for catalysis FCAT CAT
k1ðM�1 · s�1Þ 3 × 105 >5 × 106
ðk21=k−21Þk22ðM�1 · s�1Þ 2.5 × 104 —
k21ðs�1Þ 3 × 104 7 × 103
kap2 ðs�1Þ 2.1 × 104 7 × 103
α2 0.3[PhOH] (M) log k2nd ET
f (cm s−1)0.3 −8.8 −9.40.5 −8.8 −9.41 −8.6 −9.32 −8.4 −9.03 −8.25 −8.8
The kap2 value for FCAT is given for [PhOH] = 3 M. It is independent from
the acid concentration in the case of CAT.
Table 2. Comparison of FCAT and CAT with other catalysts of the CO2/CO conversion
ReferenceSolvent + acid
E0CO2=CO
CatalystE0cat
kap1 ½CO2�kap2 logTOFmax (s
−1) logTOF0 (s−1)*
27; this work DMF +3 M PhOH CAT >5 × 106 3.8 −6.0−0.69 −1.35 See Table 1
27; this work DMF +3 M PhOH FCAT >5 × 106 4.0 −5.5−0.69 −1.28 See Table 1
25 DMF +3 M PhOH Fe0TPP 3.5 × 104 4.5 −8.0−0.69 −1.43 †
21 DMF +0.1 M HBF4 m-(triphos)2Pd2‡ 35 1.5 −7.4
−0.23 −0.76 †
20 CH3CN +0.8 M CF3CH2OH Re(bpy)(CO)3(py) 875 2.9 −8.0-0.65 −1.30 †
12 CH3CN +1.4 M CF3CH2OH Mn(bpytBu)(CO)3Br§ 680 2.8 −9.8
-0.65 −1.40 †
22 CH3CN RuII(tpy−)(bpy−)§ 7.6 0.9 −10.8−0.65 −1.34 †
22 CH3CN RuII(tpy−)(Mebim-py−)§ 59 1.8 −9.9−0.65 −1.34 †
23 CH3CN N2 = Mn(CO)3{ 5 × 103 3.7 −7.0
−0.65 −1.28 †
Potentials in V vs. NHE, first-order or pseudo-first-order rate constants in s−1.*TOF at η = 0.†kap
2 >>kap1 ½CO2�.
‡
§py = pyridine, tpy = 2,2’:6’,2’’-terpyridine, bpy =2,2’-bipyridine, Mebimpy = 2,6-bis(1-methyl benzimidazol-2-yl)pyridine.{
14992 | www.pnas.org/cgi/doi/10.1073/pnas.1416697111 Costentin et al.

TOFmax =1
1k1½CO2�+
1kap2
; kap2 =k21k22½PhOH�
k−21 + k22½PhOH�:
The S-shaped catalytic wave is characterized by a plateau currentipl that may be expressed as
ipl =2FS
ffiffiffiffiffiffiffiffiDcat
pC0
cat
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffik1½CO2�
p1+
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffik1½CO2�
pffiffiffiffiffiffiffiffiffik2;ap
p 1+
ffiffiffiffiffiffiffiffiffik2;ap
pffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffik1½CO2�
p!"1+
k2;apk2;2C0
Z
#; [1]
where S is electrode surface area; C0cat and Dcat are concentration
and diffusion coefficient of the catalyst, respectively.The logTOF–η plots (Fig. 2) move upward as the phenol con-
centration increases. They are more favorable for FCAT than forCAT whatever [PhOH]. A more direct comparison between the twocatalysts at [PhOH] = 3 M is shown in Fig. 3, where the results ofpreparative-scale electrolyses are also displayed within the samelogTOF vs. η framework, pointing to the superiority of FCAT overCAT. This is confirmed by preparative-scale electrolyses. Fixed-potential electrolyses were performed at −1.08 and −1.14 V vs.NHE with 1 mM FCAT and CAT, respectively, using a carboncrucible as working electrode under 1 atm. CO2 (0.23 M) in thepresence of 3 M PhOH. The current density iel=Sel is stable over 3 hwith FCAT and 0.5 h with CAT and the production of CO ispractically quantitative (faradaic yields of 100 ± 10% and 100 ± 5%,respectively, less than 1% H2 in both cases). iel=Sel= 0.5 and 0.3mA/cm2 with FCAT and CAT, respectively; Sel, the workingelectrode surface area of the preparative-scale electrode electrolysis,is much larger (20 cm2) than in CV experiments (0.07 cm2). Thecorresponding TOF value at the operated overpotential is calculatedfrom TOF = ðiel=iplÞTOFmax, in which ipl is the plateau current givenby Eq. 1. The TOF values thus obtained are 240 s−1 (at η = 0.39 V)and 170 s−1 (at η = 0.45 V) for FCAT and CAT, respectively. As seenin Fig. 3, they satisfactorily match the TOF–η relationships derivedfrom CV taking into account inevitable imperfections in cell con-figuration leading to residual ohmic drop. Besides catalytic per-formances evaluated through logTOF–η relationship, durability isimportant in the evaluation of catalyst efficiency. It has beenevaluated through estimation of the catalyst degradation overprolonged electrolysis. This estimation is based on recording CVsin the electrolysis solution during electrolysis. It turns out that (seeSI Text for details) FCAT is more stable than CAT or simple
FeTPP. Complete degradation of the initial 10−5 moles of catalystis observed after the passage of 575, 200, and 290 coulombs forFCAT, CAT, and simple FeTPP, corresponding to 600, 210, and300 catalytic cycles for FCAT, CAT and FeTPP, respectively.How do these modified Fe0TPP catalysts compare with other
catalysts of the CO2/CO conversion described in the literature?Table 2 summarizes the various parameters that we could extractfrom previous reports as detailed in SI Text. Fig. 3 illustrates theensuing benchmarking of all catalysts. In terms of preparative-scale electrolyses, the available information indicates that thestability of the catalysts is of the same order as for the two cat-alysts FCAT and CAT described here.Our conclusion is twofold. (i) The title iron porphyrin generated
electrochemically under its Fe0 form (FCAT) operated in thepresence of 3 M phenol in DMF appears to be the best homoge-neous catalyst of the CO2-to-CO conversion to date. This clearlyappears after benchmarking of presently available catalyst of thisreaction under the form of catalytic Tafel plots relating turnoverfrequency with overpotential (Fig. 3). Such plot allows optimizationof the catalytic reaction by appropriately compromising betweenrapidity and energy costs. A further advantageous feature of FCATis that it relies on one of the cheapest and most earth-abundantmetals. (ii) Fluorine substitution in passing from CAT to FCAT wasdesigned to favor catalysis in terms of overpotential thanks to theinductive effect of the fluorine substituents. At the same time itcould have rendered the follow-up reactions less favorable, possiblyannihilating the initial favorable effect of fluorine sub or evenmaking catalysis globally less efficient than with CAT. The obser-vation that this is not the case, and that the substitution has a globalpositive effect in this case, opens the route to the design and testingof further substituted molecules, which could become even moreefficient catalysts of the CO2/CO conversion. It should be particu-larly fruitful to use the prepositioned phenol functionalities to favorthe formation and proton-coupled transformation of the initialFe0–CO2 adduct and to play with electron withdrawing substituentsto improve the capabilities of the catalyst in terms of overpotential.See SI Text for experimental details and data treatment of the
other catalysts in Table 2.
ACKNOWLEDGMENTS. Partial financial support from the Agence Nationalede la Recherche (ANR 2010 BLAN 0808) is gratefully acknowledged.
Fig. 2. Catalytic Tafel plots for the two catalysts (see text) as a function of theconcentration of phenol in the solution, inM, from bottom to top: 0.3, 0.5, 1, 2, 3.
Fig. 3. Benchmarking of all catalysts based on catalytic Tafel plots derivedfrom CV experiments. See Table 2.
Costentin et al. PNAS | October 21, 2014 | vol. 111 | no. 42 | 14993
CHEM
ISTR
Y

1. Benson EE, Kubiak CP, Sathrum AJ, Smieja JM (2009) Electrocatalytic and homoge-neous approaches to conversion of CO2 to liquid fuels. Chem Soc Rev 38(1):89–99.
2. Rakowski DuBois M, DuBois DL (2009) Development of molecular electrocatalysts forCO2 reduction and H2 production/oxidation. Acc Chem Res 42(12):1974–1982.
3. Windle CD, Perutz RN (2012) Advances in molecular photocatalytic and electro-catalytic CO2 reduction. Coord Chem Rev 256(21-22):2562–2570.
4. Savéant J-M (2008) Molecular catalysis of electrochemical reactions. Mechanistic as-pects. Chem Rev 108(7):2348–2378.
5. Costentin C, Robert M, Savéant J-M (2013) Catalysis of the electrochemical reductionof carbon dioxide. Chem Soc Rev 42(6):2423–2436.
6. Schneider J, Jia H, Muckerman JT, Fujita E (2012) Thermodynamics and kinetics of CO2,CO, and H+ binding to the metal centre of CO2 reduction catalysts. Chem Soc Rev41(6):2036–2051.
7. Appel AM, et al. (2013) Frontiers, opportunities, and challenges in biochemical andchemical catalysis of CO2 fixation. Chem Rev 113(8):6621–6658.
8. Aresta M, Dibenedetto A, Angelini A (2014) Catalysis for the valorization of exhaustcarbon: From CO2 to chemicals, materials, and fuels. technological use of CO2. ChemRev 114(3):1709–1742.
9. Smieja JM, et al. (2012) Kinetic and structural studies, origins of selectivity, and in-terfacial charge transfer in the artificial photosynthesis of CO. Proc Natl Acad Sci USA109(39):15646–15650.
10. Oh Y, Hu X (2013) Organic molecules as mediators and catalysts for photocatalyticand electrocatalytic CO2 reduction. Chem Soc Rev 42(6):2253–2261.
11. Bourrez M, Molton F, Chardon-Noblat S, Deronzier A (2011) [Mn(bipyridyl)(CO)3Br]:An abundant metal carbonyl complex as efficient electrocatalyst for CO2 reduction.Angew Chem Int Ed Engl 50(42):9903–9906.
12. Smieja JM, et al. (2013) Manganese as a substitute for rhenium in CO2 reductioncatalysts: The importance of acids. Inorg Chem 52(5):2484–2491.
13. Keith JA, Grice KA, Kubiak CP, Carter EA (2013) Elucidation of the selectivity ofproton-dependent electrocatalytic CO2 reduction by fac-Re(bpy)(CO)3Cl. J Am ChemSoc 135(42):15823–15829.
14. Froehlich JD, Kubiak CP (2012) Homogeneous CO2 reduction by Ni(cyclam) at a glassycarbon electrode. Inorg Chem 51(7):3932–3934.
15. Hammouche M, Lexa D, Savéant J-M, Momenteau M (1988) Catalysis of the electro-chemical reduction of carbon dioxide by iron(“0”) porphyrins. J Electroanal Chem 249:347–351.
16. Hammouche M, Lexa D, Momenteau M, Savéant J-M (1991) Chemical catalysis ofelectrochemical reactions. Homogeneous catalysis of the electrochemical reduction ofcarbon dioxide by iron (“0”) porphyrins. Role of the addition of magnesium cations.J Am Chem Soc 113:8455–8466.
17. Bhugun I, Lexa D, Saveant J-M (1994) Ultraefficient selective homogeneous catalysisof the electrochemical reduction of carbon dioxide by an iron(0) porphyrin associatedwith a weak Broensted acid cocatalyst. J Am Chem Soc 116:5015–5016.
18. Bhugun I, Lexa D, Saveant JM (1996) Catalysis of the electrochemical reduction ofcarbon dioxide by iron(0) porphyrins: Synergystic effect of weak Bronsted acids. J AmChem Soc 118:1769–1776.
19. Bhugun I, Lexa D, Saveant JM (1996) Catalysis of the electrochemical reduction ofcarbon dioxide by iron(0) porphyrins. Synergistic effect of Lewis acid cations. J PhysChem 100:19981–19985.
20. Wong K-Y, Chung W-H, Lau C-P (1998) The effect of weak Bronsted acids on theelectrocatalytic reduction of carbon dioxide by a rhenium tricarbonyl bipyridyl com-plex. J Electroanal Chem 453:161–169.
21. Raebiger JW, et al. (2006) Electrochemical reduction of CO2 to CO catalyzed by a bi-metallic palladium complex. Organometallics 25:3345–3351.
22. Chen Z, et al. (2011) Electrocatalytic reduction of CO2 to CO by polypyridyl rutheniumcomplexes. Chem Commun (Camb) 47(47):12607–12609.
23. Sampson MD, et al. (2014) Manganese catalysts with bulky bipyridine ligands for theelectrocatalytic reduction of carbon dioxide: Eliminating dimerization and alteringcatalysis. J Am Chem Soc 136(14):5460–5471.
24. Costentin C, Drouet S, Robert M, Savéant J-M (2012) Turnover numbers, turnoverfrequencies, and overpotential in molecular catalysis of electrochemical reactions.Cyclic voltammetry and preparative-scale electrolysis. J Am Chem Soc 134(27):11235–11242.
25. Costentin C, Drouet S, Passard G, Robert M, Savéant J-M (2013) Proton-coupledelectron transfer cleavage of heavy-atom bonds in electrocatalytic processes. Cleav-age of a C-O bond in the catalyzed electrochemical reduction of CO2. J Am Chem Soc135(24):9023–9031.
26. Costentin C, Drouet S, Robert M, Savéant J-M (2012) A local proton source enhancesCO2 electroreduction to CO by a molecular Fe catalyst. Science 338(6103):90–94.
27. Costentin C, Passard G, Robert M, Savéant J-M (2014) Pendant acid–base groups inmolecular catalysts: H-bond promoters or proton relays? Mechanisms of the conver-sion of CO2 to CO by electrogenerated iron(0)porphyrins bearing prepositionedphenol functionalities. J Am Chem Soc 136:11821–11829.
28. Savéant J-M (2006) Elements of Molecular and Biomolecular Electrochemistry: AnElectrochemical Approach to Electron Transfer Chemistry (Wiley, Hoboken, NJ), pp108–111.
29. Costentin C, Savéant J-M (2014) Multielectron, multistep molecular catalysis of elec-trochemical reactions: Benchmarking of homogeneous catalysts. ChemElectroChem 1:1226–1236.
14994 | www.pnas.org/cgi/doi/10.1073/pnas.1416697111 Costentin et al.