tungstate promoted vanadium phosphate catalysts for the gas phase oxidation of methanol to...
TRANSCRIPT

1558 Catal. Sci. Technol., 2013, 3, 1558--1564 This journal is c The Royal Society of Chemistry 2013
Cite this: Catal. Sci. Technol.,2013,3, 1558
Tungstate promoted vanadium phosphate catalystsfor the gas phase oxidation of methanol toformaldehyde†
Gobinda C. Behera,a Kulamani Parida,*a Nicholas F. Dummer,b Gareth Whiting,b
Nruparaj Sahu,a Albert F. Carley,b Marco Conte,b Graham J. Hutchingsb andJonathan K. Bartley*b
Formaldehyde is an important intermediate in the pharmaceutical, plastic and automobile industries
which is produced commercially via methanol oxidation. A series of tungstate promoted vanadium
phosphate catalysts were prepared and characterised using a combination of XRD, UV-vis spectrometry,
XPS and ammonia TPD. The 25 wt% WO3–VPO catalyst exhibited good activity for the selective gas
phase oxidation of methanol with 490% conversion at around 250 1C. This is a considerably lower
temperature than used in the current industrial process using either silver or iron molybdate catalysts.
This increased activity at low temperatures is ascribed to the increase in Brønsted acidity and redox
properties of the tungstate doped materials.
1. Introduction
The oxidation of primary alcohols to aldehydes is a fundamentallyimportant commercial process.1–10 As a consequence of themanufacturing and cost-effective significance of formaldehyde,the partial oxidation of methanol has been subject of compre-hensive studies utilizing a wide variety of kinetic, synthetic,spectroscopic and theoretical methods.11–14 Two processes havebeen commercialized for methanol oxidation to formaldehydeusing either silver or iron molybdate catalysts.13 These catalyststypically operate at high temperatures (ca. 400 1C), and developingoxidation catalysts that work at lower reaction temperatures whilstmaintaining high activity may be useful for a variety of oxidationreactions in the fine chemical industry.
Iron molybdate catalysts have gained market share from thesilver process due to the higher selectivity to formaldehyde. However,the catalysts deactivate as molybdenum reacts with methanol toform volatile species under the reaction conditions, resulting indepletion of molybdenum from the catalyst.15–17 As a consequence,both the activity and selectivity to formaldehyde decreases with time,
and re-condensation of the sublimed molybdenum in colderregions causes an increased pressure drop. Although the industrialcatalyst contains excess molybdenum (Fe : Mo = 1 : 2.2) to avoidformation of the undesirable iron rich phases, the catalyst must bereplaced after 1–2 years of operation.15
The instability of the iron molybdate catalyst has led to thedevelopment of alternative Mo-free catalysts.18 Haggbled et al.19
studied a series of Fe1�x–Alx–V–O catalysts which gave 90%selectivity to formaldehyde, Isaguliants et al.20 reported 97%selectivity to formaldehyde over a Mg2V2O7 catalyst at highertemperature and Niskala et al.21 studied titania and vanadia basedcatalysts and found formaldehyde selectivities of 93% over TiO2,75% over V2O5/SiO2 and 96% over V2O5/(SiO2 + TiO2) respectively.
Vanadium based catalysts have been applied to a wide variety ofselective oxidation reactions, in particular the oxidation of n-butaneto maleic anhydride over vanadium phosphate (VPO) catalysts, aprocess that has been successfully commercialized and intensivelystudied.22–24 The activity of VPO catalysts is related to the redoxpotential of the surface vanadium species and high lattice oxygenmobility. Therefore, wider applications of this material are nowemerging, mainly in selective oxidation, ammoxidation, dehydro-genation and dehydration reactions, as well as the conversion ofbio-renewables and low temperature liquid phase reactions.24–26
Modern day interest in VPO materials stems from their structuraldiversity and potential applications in catalysis due to theirnontoxic nature and ability to promote selective reactions at lowtemperatures. Selective oxidation of methanol to formaldehyde
a CSIR-Institute of Minerals & Materials Technology, Bhubaneswar-751013, Odisha,
India. E-mail: [email protected]; Fax: +91-674-2581637;
Tel: +91-674-2581636 (Ext. 425)b Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, UK
CF10 3AT. E-mail: [email protected]
† Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cy20801j
Received 19th November 2012,Accepted 1st March 2013
DOI: 10.1039/c3cy20801j
www.rsc.org/catalysis
CatalysisScience & Technology
PAPER
Publ
ishe
d on
04
Mar
ch 2
013.
Dow
nloa
ded
by T
he U
nive
rsity
of
Mel
bour
ne L
ibra
ries
on
28/0
9/20
13 1
4:18
:04.
View Article OnlineView Journal | View Issue

This journal is c The Royal Society of Chemistry 2013 Catal. Sci. Technol., 2013, 3, 1558--1564 1559
with molecular oxygen over VPO catalysts offers a sustainable,environmentally benign alternative to traditional processes.
The selective oxidation of methanol to formaldehyde withcatalysts containing vanadium has been commercialised in theFormox process and contributes to a total formaldehyde produc-tion of around 25 MTPA. However, these catalysts comprise mainlyof molybdenum and iron as a mixed metal oxide with vanadium asa promoter. Industrial catalysts for oxidation reactions rarely use asingle bulk phase and often contain promoter elements that canhave a variety of benefits. A number of dopants have beeninvestigated for VPO catalysts that can act purely as texturalpromoters, or enhance the activity and selectivity of the bulkcatalyst.27 In this study we have investigated VPO materials, withthe addition of tungsten oxide as catalysts for methanol oxidationin an effort to increase the yield of formaldehyde at lower reactiontemperatures than the current state of the art.
Tungstate based materials are an interesting class of solids,which was first reported as a strongly acidic system by Hino andArata.28 There is general consensus that the active sites are ofan acidic nature,29,30 although some reports suggest the importanceof redox cycles involving the reduction of metal oxides centres andradical like intermediates.31 Among the transition metal oxides,tungsten oxides give the strongest Brønsted acid sites, either as bulkor supported oxides,32,33 although their structure and catalyticproperties are greatly influenced by the support. Several studieshave explained how the interaction between the support and thetungsten oxide domains affects the catalytic activity. Indeed, for anysupported WO3 system, WOx domains exhibit a range of surfacestructures that depend strongly on the synthetic protocols.34,35
2. Experimental2.1. Materials and methods
2.1.1. Preparation of the VOHPO4�0.5H2O precursor. TheVOHPO4�0.5H2O precursor was prepared according to thefollowing procedure. V2O5 (5.0 g, 98.5%, SBMC) and ortho-H3PO4 (30 mL, 85%, Aldrich) were refluxed in deionised water(120 mL) for 24 h. The yellow solid was recovered by vacuumfiltration, washed with cold water (100 mL) and acetone(100 mL) and dried in air (110 1C, 24 h). Powder X-ray diffrac-tion (not shown) confirmed that the solid was vanadyl phos-phate dihydrate, VOPO4�2H2O.36
The VOPO4�2H2O (4 g) was then refluxed with iso-butanol(80 mL, 99%, Spectrochem) for 21 h and the resulting solid(VOHPO4�0.5H2O) was recovered by filtration, dried in air(110 1C, 16 h), refluxed in deionised water (9 mL H2O g�1 solid)for 2 h, filtered hot and dried in air (110 1C, 16 h).
2.1.2. Preparation of the WO3 promoted VPO. WO3–VPOcatalysts with different loadings were prepared by wet impregnationusing water as solvent. The requisite amount of ammoniummetatungstate (99.9%, Aldrich) was dissolved in 30 mL of deionisedwater, to which VOHPO4�0.5H2O (1 g) was added. The resultingsuspension was vigorously stirred at room temperature until all thesolvent had evaporated, dried at 60 1C and then calcined at 400 1Cfor 4 h. A series of WO3–VPO catalysts were synthesized with 5, 10,15, 20 and 25 wt% of WO3.
2.2. Catalyst characterization
X-Ray powder diffraction patterns (XRPD) were acquired using anX’Pert PANalytical diffractometer operating at 40 kV and 40 mAselecting the Cu-Ka radiation. Analysis of the patterns was carriedout using X’Pert HighScore Plus software. Crystallite sizes forthe metal and metal oxide clusters were determined using theScherrer equation assuming spherical particle shapes and a Kfactor of 0.89. The line broadening was determined using a Voigtprofile function convoluting the Gaussian and Lorentzian profilepart of the reflection peak and the instrumental broadening forthe Bragg–Brentano geometry used was estimated to be 0.061.
The BET surface area and pore volume measurements weredetermined by N2 adsorption at �196 1C using a MicromeriticsASAP2010 instrument. The samples were evacuated for 2 h at 110 1Cto remove physisorbed water prior to surface area measurements.
The acid character of the catalysts was elucidated from theNH3 TPD performed using a Micromeritics AutoChem 2920 auto-mated chemisorption analyzer equipped with a thermal conductivitydetector (TCD). About 0.1 g of the sample was placed in a quartzU-tube. Prior to analysis, the samples were degassed at 110 1C for 2 hunder N2 flow (50 ml min�1). The sample was then cooled to 40 1Cand saturated with NH3 under a flow of 20 vol% NH3 (balanceHe, 25 ml min�1) for 30–40 min. Then N2 gas was flowed overcatalyst (50 ml min�1) for 30 min to remove the physisorbedNH3. The temperature was increased from 40 to 800 1C underthe flow of N2 to obtain the TPD profile. The amount of NH3
consumed was determined using a TCD detector which allowsthe number of Brønsted and Lewis acid sites in the sample tobe determined.
Diffuse reflectance UV-vis spectra were recorded using aVarian Cary 100 UV-Vis spectrophotometer. Each spectrumwas recorded in the range of 200–800 nm using boric acid asthe reflectance standard.
X-Ray photoelectron spectroscopy (XPS) measurements wereperformed using a Kratos Axis Ultra DLD spectrometer usingmonochromatic AlKa radiation. Samples were mounted usingdouble-sided adhesive tape and binding energies referenced tothe C(1s) binding energy of adventitious carbon contamination,taken to be 284.9 eV.
2.3. Catalyst testing
Catalyst testing was carried out using a fixed bed reactor. Thecatalyst (ca. 0.3 g) was placed in the centre of a quartz tube (held inplace using two pieces of quartz wool) and placed in the reactorfurnace (Carbolite). The outlet from the reactor was heated to120 1C to prevent the condensation of reaction products in thelines. Helium was passed through a saturator containing metha-nol (99.5% Aldrich) which was maintained at 7 1C (to achieve5 vol% methanol) using a chilled water bath. Oxygen andmethanol/helium flows were controlled using mass flow controllersto give a total flow rate of 60 ml min�1 (MeOH : O2 : He = 5 : 10 : 85).Data were collected over the range 200–400 1C with the catalystallowed to equilibrate for 45 minutes after each rise in temperature.No hysteresis was observed, indicating the catalysts were stable overthis temperature range.
Paper Catalysis Science & Technology
Publ
ishe
d on
04
Mar
ch 2
013.
Dow
nloa
ded
by T
he U
nive
rsity
of
Mel
bour
ne L
ibra
ries
on
28/0
9/20
13 1
4:18
:04.
View Article Online

1560 Catal. Sci. Technol., 2013, 3, 1558--1564 This journal is c The Royal Society of Chemistry 2013
Product analysis was carried out using an on-line Varian 3400Cx gas chromatograph. Two columns were used in a series/bypassconfiguration: Carbosieve S-11 (3 m) for separation of O2 and COand a Porapak Q (1 m) for the separation of methanol, dimethylether (DME), methyl formate (MF), formaldehyde (FA) and CO2.A TCD was used for the detection of O2, CO and CO2 and an FIDused to analyse methanol, formaldehyde, methyl formate anddimethyl ether.
3. Results and discussion
The impregnation was carried out with ammonium metatung-state which in aqueous solution dissociates according to thefollowing equilibrium:
W12O410� + 21H2O " 12WO4
2� + 14H3O+
The attachment of the W(VI) oxo-species, preferentially in themonomeric form, takes place on the VPO surface when it is addedto the solution. After activation, the polymerization of monomericunits by formation of W–O–W bridges leads to WOx clusters. Whenthese WOx clusters are supported on VPO the resulting system canaccommodate protons due to the existence of a negative chargedelocalized across the extended tungsten oxide network, similar tothe effect observed for heteropoly acids.37,38
3.1. Catalyst characterization
Fig. 1 displays the XRD patterns of the VOHPO4�0.5H2O andWO3–VPO materials. All major diffraction peaks can be attributed toVOHPO4�0.5H2O. The most intense reflection for VOHPO4�0.5H20 isfound at 2y = 15.61 and is assigned to the (001) reflection (JCPDS,4-880). The XRD patterns reveal that there is a slight reduction in theintensity of the (001) reflection in the WO3–VPO compared with theparent precursor. In the tungstate doped materials there is also anadditional peak at 2y = 12.81 which corresponds to the most intensepeak of H2WO4�H2O (PDF# 00-018-1420) which fits with the
decomposition route indicated above. Determination of thecrystallite VOHPO4�0.5H2O size based on the (001) reflectionat 2y = 15.61, showed that the materials doped with 5–20 wt%WO3 were approximately 40 nm (Table 1), while the materialdoped with 25 wt% WO3 was around 60 nm.
XRD analysis of the samples after reaction (Fig. 2) revealed thatthe WO3–VPO materials contained different amounts of VOHPO4�0.5H2O, (VO)2P2O7 (vanadyl pyrophosphate) and VOPO4 dependingon the WO3 loading. The 5 wt% WO3 material comprises (VO)2P2O7
with a small amount of VOPO4, which is what you would expectfrom an undoped catalyst. As the loading of WO3 increased therewas a decrease in the intensities of the (VO)2P2O7 reflections coupledwith an increase in VOPO4 and VOHPO4�0.5H2O reflections. Thesephases are interchangeable with VOHPO4�0.5H2O to (VO)2P2O7 con-trolled by the removal of H2O and V(IV) to V(V) controlled by theredox conditions. Determination of the ratio between the intensitiesof the (200) to (024) reflections (I200/I024) of (VO)2P2O7 at 2y = 22.8and 28.71 respectively, correlate with crystal stacking order.39 In ourcase, this ratio move from 1.52 for the 5 wt% WO3 to 0.45 for the25 wt% WO3 material, indicating there is an increase in defects inthe pyrophosphate structure when the WO3 loading increases.
The surface area analysis of the promoted and unpromotedmaterials was carried out by N2 adsorption–desorption measure-ments (please see ESI†) and the results are summarized in Table 2.VOHPO4�0.5H2O has a surface area of B15 m2 g�1 whilst the WO3–VPO samples exhibit significantly lower values with respect to theundoped VPO. The lower surface area of WO3–VPO is due toobstruction of the pores of the VPO materials. Kim et al. studiedWO3 impregnated onto a number of supports and found thatmonolayer coverage equates to around 4.5 W nm�1.40 In this studyall the loadings are above this value and so multilayer ornanoparticle structures will form on the VPO surface.
It is known that the acidity of the VPO catalysts is an importantaspect in controlling the catalyst performance. Previous IR studies ofthe acid sites using NH3, pyridine and acetonitrile as probe mole-cules showed the existence of Lewis (V(IV)) and Brønsted (P–OH) acidsites.41–43 Fig. 3 displays the NH3–TPD profiles of VPO and WO3–VPO materials. The amount of Brønsted and Lewis acid sites wascalculated from the respective peak area. The bonding betweenBrønsted acid sites and NH3 is found to be weaker and NH3
desorbed at lower temperature (B400 1C), whereas desorptionof NH3 from Lewis acids sites occurs at higher temperature(B575 1C). The analysis of the peak areas to determine the
Fig. 1 XRD patterns of (a) VOHPO4�0.5H2O; (b) WO3 (c) 5 wt% WO3–VPO;(d) 10 wt% WO3–VPO; (e) 15 wt% WO3–VPO; (f) 20 wt% WO3–VPO; (g) 25 wt%WO3–VPO.
Table 1 Crystallite size of VOHPO4�0.5H2O and (VO)2P2O7 phases present in thecatalysts before and after reaction (n.d. = not detected). Catalysts were heated fromroom temperature to 400 1C with 45 minutes stabilisation at each temperature
Sample
Crystallite size (nm)
I200/I024
(VO)2P2O7
VOHPO4�0.5H2O (VO)2P2O7
PrecursorPostreaction Precursor
Postreaction
5 wt% 47 n.d. n.d. 9 1.5210 wt% 40 25 n.d. 6 0.9115 wt% 44 30 n.d. 7 0.4720 wt% 42 36 n.d. 10 0.4525 wt% 61 25 n.d. 5 0.45
Catalysis Science & Technology Paper
Publ
ishe
d on
04
Mar
ch 2
013.
Dow
nloa
ded
by T
he U
nive
rsity
of
Mel
bour
ne L
ibra
ries
on
28/0
9/20
13 1
4:18
:04.
View Article Online
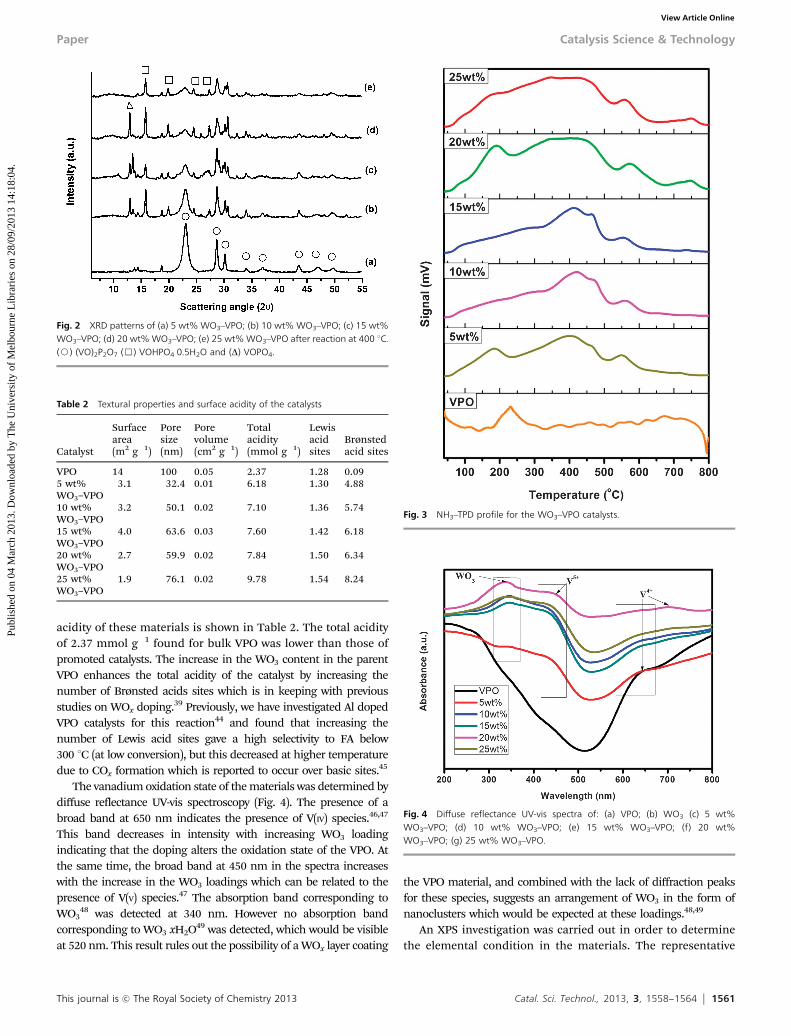
This journal is c The Royal Society of Chemistry 2013 Catal. Sci. Technol., 2013, 3, 1558--1564 1561
acidity of these materials is shown in Table 2. The total acidityof 2.37 mmol g�1 found for bulk VPO was lower than those ofpromoted catalysts. The increase in the WO3 content in the parentVPO enhances the total acidity of the catalyst by increasing thenumber of Brønsted acids sites which is in keeping with previousstudies on WOx doping.39 Previously, we have investigated Al dopedVPO catalysts for this reaction44 and found that increasing thenumber of Lewis acid sites gave a high selectivity to FA below300 1C (at low conversion), but this decreased at higher temperaturedue to COx formation which is reported to occur over basic sites.45
The vanadium oxidation state of the materials was determined bydiffuse reflectance UV-vis spectroscopy (Fig. 4). The presence of abroad band at 650 nm indicates the presence of V(IV) species.46,47
This band decreases in intensity with increasing WO3 loadingindicating that the doping alters the oxidation state of the VPO. Atthe same time, the broad band at 450 nm in the spectra increaseswith the increase in the WO3 loadings which can be related to thepresence of V(V) species.47 The absorption band corresponding toWO3
48 was detected at 340 nm. However no absorption bandcorresponding to WO3�xH2O49 was detected, which would be visibleat 520 nm. This result rules out the possibility of a WOx layer coating
the VPO material, and combined with the lack of diffraction peaksfor these species, suggests an arrangement of WO3 in the form ofnanoclusters which would be expected at these loadings.48,49
An XPS investigation was carried out in order to determinethe elemental condition in the materials. The representative
Fig. 2 XRD patterns of (a) 5 wt% WO3–VPO; (b) 10 wt% WO3–VPO; (c) 15 wt%WO3–VPO; (d) 20 wt% WO3–VPO; (e) 25 wt% WO3–VPO after reaction at 400 1C.(J) (VO)2P2O7 (&) VOHPO4�0.5H2O and (D) VOPO4.
Table 2 Textural properties and surface acidity of the catalysts
Catalyst
Surfacearea(m2 g�1)
Poresize(nm)
Porevolume(cm2 g�1)
Totalacidity(mmol g�1)
Lewisacidsites
Brønstedacid sites
VPO 14 100 0.05 2.37 1.28 0.095 wt%WO3–VPO
3.1 32.4 0.01 6.18 1.30 4.88
10 wt%WO3–VPO
3.2 50.1 0.02 7.10 1.36 5.74
15 wt%WO3–VPO
4.0 63.6 0.03 7.60 1.42 6.18
20 wt%WO3–VPO
2.7 59.9 0.02 7.84 1.50 6.34
25 wt%WO3–VPO
1.9 76.1 0.02 9.78 1.54 8.24
Fig. 3 NH3–TPD profile for the WO3–VPO catalysts.
Fig. 4 Diffuse reflectance UV-vis spectra of: (a) VPO; (b) WO3 (c) 5 wt%WO3–VPO; (d) 10 wt% WO3–VPO; (e) 15 wt% WO3–VPO; (f) 20 wt%WO3–VPO; (g) 25 wt% WO3–VPO.
Paper Catalysis Science & Technology
Publ
ishe
d on
04
Mar
ch 2
013.
Dow
nloa
ded
by T
he U
nive
rsity
of
Mel
bour
ne L
ibra
ries
on
28/0
9/20
13 1
4:18
:04.
View Article Online
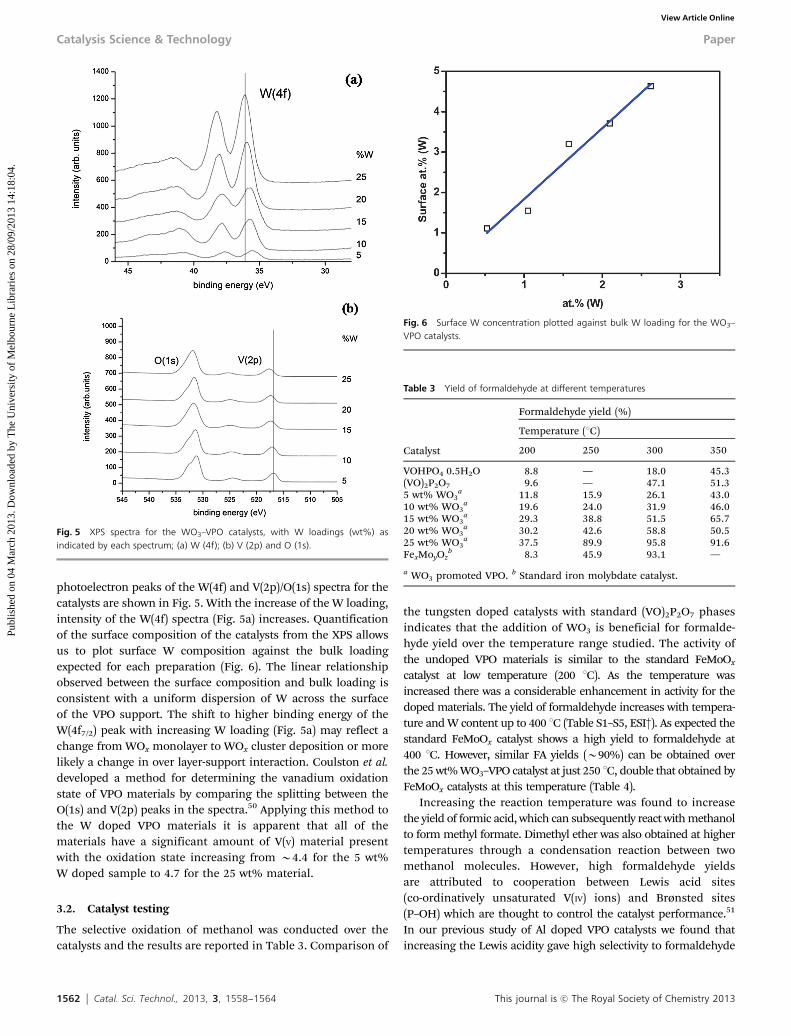
1562 Catal. Sci. Technol., 2013, 3, 1558--1564 This journal is c The Royal Society of Chemistry 2013
photoelectron peaks of the W(4f) and V(2p)/O(1s) spectra for thecatalysts are shown in Fig. 5. With the increase of the W loading,intensity of the W(4f) spectra (Fig. 5a) increases. Quantificationof the surface composition of the catalysts from the XPS allowsus to plot surface W composition against the bulk loadingexpected for each preparation (Fig. 6). The linear relationshipobserved between the surface composition and bulk loading isconsistent with a uniform dispersion of W across the surfaceof the VPO support. The shift to higher binding energy of theW(4f7/2) peak with increasing W loading (Fig. 5a) may reflect achange from WOx monolayer to WOx cluster deposition or morelikely a change in over layer-support interaction. Coulston et al.developed a method for determining the vanadium oxidationstate of VPO materials by comparing the splitting between theO(1s) and V(2p) peaks in the spectra.50 Applying this method tothe W doped VPO materials it is apparent that all of thematerials have a significant amount of V(V) material presentwith the oxidation state increasing from B4.4 for the 5 wt%W doped sample to 4.7 for the 25 wt% material.
3.2. Catalyst testing
The selective oxidation of methanol was conducted over thecatalysts and the results are reported in Table 3. Comparison of
the tungsten doped catalysts with standard (VO)2P2O7 phasesindicates that the addition of WO3 is beneficial for formalde-hyde yield over the temperature range studied. The activity ofthe undoped VPO materials is similar to the standard FeMoOx
catalyst at low temperature (200 1C). As the temperature wasincreased there was a considerable enhancement in activity for thedoped materials. The yield of formaldehyde increases with tempera-ture and W content up to 400 1C (Table S1–S5, ESI†). As expected thestandard FeMoOx catalyst shows a high yield to formaldehyde at400 1C. However, similar FA yields (B90%) can be obtained overthe 25 wt% WO3–VPO catalyst at just 250 1C, double that obtained byFeMoOx catalysts at this temperature (Table 4).
Increasing the reaction temperature was found to increasethe yield of formic acid, which can subsequently react with methanolto form methyl formate. Dimethyl ether was also obtained at highertemperatures through a condensation reaction between twomethanol molecules. However, high formaldehyde yieldsare attributed to cooperation between Lewis acid sites(co-ordinatively unsaturated V(IV) ions) and Brønsted sites(P–OH) which are thought to control the catalyst performance.51
In our previous study of Al doped VPO catalysts we found thatincreasing the Lewis acidity gave high selectivity to formaldehyde
Fig. 5 XPS spectra for the WO3–VPO catalysts, with W loadings (wt%) asindicated by each spectrum; (a) W (4f); (b) V (2p) and O (1s).
Fig. 6 Surface W concentration plotted against bulk W loading for the WO3–VPO catalysts.
Table 3 Yield of formaldehyde at different temperatures
Catalyst
Formaldehyde yield (%)
Temperature (1C)
200 250 300 350
VOHPO4�0.5H2O 8.8 — 18.0 45.3(VO)2P2O7 9.6 — 47.1 51.35 wt% WO3
a 11.8 15.9 26.1 43.010 wt% WO3
a 19.6 24.0 31.9 46.015 wt% WO3
a 29.3 38.8 51.5 65.720 wt% WO3
a 30.2 42.6 58.8 50.525 wt% WO3
a 37.5 89.9 95.8 91.6FexMoyOz
b 8.3 45.9 93.1 —
a WO3 promoted VPO. b Standard iron molybdate catalyst.
Catalysis Science & Technology Paper
Publ
ishe
d on
04
Mar
ch 2
013.
Dow
nloa
ded
by T
he U
nive
rsity
of
Mel
bour
ne L
ibra
ries
on
28/0
9/20
13 1
4:18
:04.
View Article Online

This journal is c The Royal Society of Chemistry 2013 Catal. Sci. Technol., 2013, 3, 1558--1564 1563
at low conversion leading to maximum yields of around 60%.This suggests that the Brønsted acidity we have introduced withW doping is responsible for the high conversion at low tem-perature. The increase in the yield of formaldehyde observedwith higher WO3 loadings may be due to the increase in thenumber of available acid sites on the catalyst surface due to theWOx clusters. When these WOx clusters are supported on VPOthe resulting system can accommodate protons due to theexistence of a negative charge delocalized across the extendedtungsten oxide network.
Acid sites are commonly thought to favour the production ofDME, and this has been observed previously for WO3 bulk andsupported catalysts.41,52 However in this study the combinationof enhanced Brønsted acidity and the redox properties of theVPO seem to give good selectivity and conversion at lowtemperatures. The increase in the amount of V(V) in thecatalysts may also be an advantage as this is the oxidation stateof many vanadium oxidation catalysts.24
The methanol oxidation process can be divided into threeessential steps:53–55
(i) Dissociative adsorption of gaseous methanol takes placeon the surface of the VPO solid catalyst to yield surfacemethoxo-vanadium(V) centres.
(ii) The lattice oxygen of the VPO materials might play amajor role in this mechanism. The activated methoxo ligandsare oxidized by the lattice oxygen and eliminated as formalde-hyde, following in the reduction of the catalyst and the creationof an oxygen vacancy. This step is considered as rate determiningstep and high temperatures are required.
(iii) Re-oxidation of the bulk catalyst by oxygen in the feedcompletes the cycle.
From the UV-vis studies it is clear that the presence of WO3
species increases the number of V(V) ions which are probablyresponsible for the formation of the product, while V(IV) isactive in the formation of by-products.53 Furthermore, thepartial oxidation of methanol is assumed to follow a two stepmechanism identical to that proposed by Van Krevelen andMars for the oxidation of organic compounds over a vanadiumpentoxide catalyst.54 Accordingly, methanol is adsorbed on anactive site and is first oxidized to formaldehyde. In the secondstep the reduced catalyst is transformed into its original form.55
Based on this mechanism it is clear that an ideal catalystshould have lattice oxygen mobility and acid sites in order toachieve good performance. Volta et al. reported that hydrogenabstraction occurs on Lewis acid sites (V(IV)) and C–H bondcleavage results from interactions between Lewis acid sites and
Brønsted acid sites.56 Centi et al. also reported the importantrole of Brønsted acidity in the oxidation reaction.57 Centi offersthree hypotheses for the role of Brønsted acidity: the stabilisa-tion of reaction intermediates, the stabilisation of an adsorbedoxygen species, or the generation of an organic surface speciesthat is involved in oxygen activation or transport. The syner-getic effect between WO3 and VPO makes the catalyst system anefficient material for aldehyde production.
4. Conclusions
We have successfully synthesized a series of WO3 promotedVPO. The selective gas phase oxidation of methanol to formal-dehyde proceeds efficiently at lower temperature than theindustrial process using WO3–VPO as an effective catalyst.The change in redox properties of the catalysts with thetungsten oxides loading enhances the catalytic activity at lowtemperatures without increasing the selectivity to dimethylether which commonly occurs over acid catalysts. Our findingsmay pave the way to the implementation of new systems for thelow temperature synthesis of aldehydes using O2 as the oxidantwith VPO based catalysts.
Acknowledgements
We wish to thank the DST, British Council, and UKIERI forfunding.
References
1 R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations ofOrganic Compounds, Academic Press, New York, 1981.
2 M. Beller and C. Bolm, Transition Metals for Organic Synthesis,Verlag GmbH & Co. KGaA, Weinheim, Germany, 2004, vol. 2.
3 G. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Science,2000, 287, 1636–1639.
4 M. Vazylyev, D. Sloboda-Rozner, A. Haimov, G. Maayan andR. Neumann, Top. Catal., 2005, 34, 93–99.
5 M. Pagliaro, S. Campestrini and R. Ciriminna, Chem. Soc.Rev., 2005, 34, 837–845.
6 K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda,J. Am. Chem. Soc., 2004, 26, 10657–10666.
7 I. E. Marko, P. R. Giles, M. Tsukazaki, S. M. Brown andC. J. Urch, Science, 1996, 274, 2044–2046.
8 M. J. Schultz, R. S. Adler, W. Zierkiewicz, T. Privalov andM. S. Sigman, J. Am. Chem. Soc., 2005, 127, 8499–8507.
9 U. R. Pillai and E. Sahle-Demessie, Appl. Catal., A, 2003, 245,103–109.
10 M. Hudlicky, Oxidations in Organic Chemistry, AmericanChemical Society, Washington, DC, 1990.
11 E. M. McCarron III, R. L. Harlow, Z. G. Li, C. Suto andY. J. Yuen, J. Solid State Chem., 1998, 136, 247–252.
12 (a) D. E. Ardissone, N. G. Valente, L. E. Cadus and L. A. Arrua,Ind. Eng. Chem. Res., 2000, 39, 2902–2909; (b) A. Faliks,R. A. Yetter, C. A. Floudas, S. L. Bernasek, M. Fransson andH. Rabitz, J. Phys. Chem. A, 2001, 105, 2099–2105.
Table 4 Effect of temperature on 25 wt% WO3–VPO. TOF = moles of formalde-hyde per mole of catalyst per minute
Temperature(1C)
Conversion(%)
Formaldehydeselectivity (%)
Formaldehydeyield (%)
TOF(min�1)
200 38 98.9 37.5 40.9250 92 96.8 89.9 99.1300 100 95.8 95.8 107.7350 100 91.6 91.6 107.7400 100 79.6 79.6 107.7
Paper Catalysis Science & Technology
Publ
ishe
d on
04
Mar
ch 2
013.
Dow
nloa
ded
by T
he U
nive
rsity
of
Mel
bour
ne L
ibra
ries
on
28/0
9/20
13 1
4:18
:04.
View Article Online

1564 Catal. Sci. Technol., 2013, 3, 1558--1564 This journal is c The Royal Society of Chemistry 2013
13 (a) R. Radhakrishnan, C. Reed, S. T. Oyama, M. Seman,J. N. Knodo, K. Domen, Y. Ohminami and K. Asakura,J. Phys. Chem. B, 2001, 105, 8519–8530; (b) L. J. Burcham,L. E. Briand and I. E. Wachs, Langmuir, 2001, 17, 6164–6174.
14 (a) R. S. Weber, J. Phys. Chem., 1994, 98, 2999–3005;(b) A. Rahmouni and C. Barbier, THEOCHEM, 1995, 330,359–364.
15 A. Andersson, M. Hernelind and O. Augustsson, Catal.Today, 2006, 112, 40–44.
16 A. P. V. Soares, M. F. Portela, A. Kiennemann and J. M. M.Millet, React. Kinet. Catal. Lett., 2002, 75, 13–20.
17 A. P. V. Soares, M. F. Portela, A. Kiennemann and L. Hilaire,Chem. Eng. Sci., 2003, 58, 1315–1322.
18 I. E. Wachs and L. E. Briand, US Pat., 7 193 117 B2, 2007,assigned to Lehigh University.
19 R. Haggbled, J. B. Wagner, S. Hansen and A. Andersson,J. Catal., 2008, 258, 345–355.
20 G. V. Isaguliants and I. P. Belmestnykh, Catal. Today, 2005,100, 441–445.
21 N. Niskala, T. Laitinen, S. Ojala, S. Pitkaaho, A. Kucerov,R. L. Keiski, Proceedings of the Skypro Conference, Universityof Oulu, Finland, 2010, 70–73.
22 G. Centi, F. Trifiro, J. R. Ebner and V. M. Franchetti, Chem.Rev., 1988, 88, 55–80.
23 B. K. Hodnett, Catal. Today, 1993, 16, 131–138.24 N. F. Dummer, J. K. Bartley and G. J. Hutchings, Adv. Catal.,
2011, 54, 189–247.25 G. C. Behera and K. M. Parida, Appl. Catal., A, 2012,
413–414, 245–253.26 G. C. Behera, K. M. Parida and D. P. Das, J. Catal., 2012, 289,
190–198.27 G. J. Hutchings, Appl. Catal., 1991, 72, 1–32.28 M. Hino and K. Arata, J. Chem. Soc., Chem. Commun., 1988,
1259–1260.29 L. L. Murrell and N. C. Dispenziere Jr., Catal. Lett., 1990, 4,
235–240.30 A. F. Perez-Cadenas, C. Moreno-Castilla, F. J. Maldonado-
Hodar and J. L. G. Fierro, J. Catal., 2003, 217, 30–37.31 J. C. Vartuli, J. G. Santiesteban, P. Traverso, N. Cardona
Martinez, C. D. Chang and S. A. Stevenson, J. Catal., 1999,187, 131–138.
32 G. Busca, Phys. Chem. Chem. Phys., 1999, 1, 723–736.33 V. M. Benitez, C. A. Querini, N. S. Figoli and R. A. Comelli,
Appl. Catal., A, 1999, 178, 205–218.34 D. S. Kim, M. Ostromecki and I. E. Wachs, J. Mol. Catal. A:
Chem., 1996, 106, 93–102.
35 C. Martin, P. Malet, G. Solana and V. Rives, J. Phys. Chem. B,1998, 102, 2759–2768.
36 G. Busca, F. Cavani, G. Centi and F. Trifiro, J. Catal., 1986,99, 400–414.
37 D. G. Barton, S. L. Soled and E. Iglesia, Top. Catal., 1998, 6,87–99.
38 D. G. Barton, M. Shtein, R. Wilson, S. L. Soled and E. Iglesia,J. Phys. Chem., 1999, 103, 630–640.
39 F. Wang, J.-L. Dubois and W. Ueda, Appl. Catal., A, 2010,376, 25–32.
40 T. Kim, A. Burrows, C. J. Kiely and I. E. Wachs, J. Catal.,2007, 246, 370–381.
41 F. Cavani and F. Trifiro, Appl. Catal., A, 1997, 157, 195–221.42 L. M. Cornaglia and E. A. Lombardo, Stud. Surf. Sci. Catal.,
1994, 90, 429–440.43 M. Abon and J. C. Volta, Appl. Catal., A, 1997, 157, 173–193.44 G. C. Behera and K. M. Parida, Chem. Eng. J., 2012, 180,
270–276.45 M. Badlani and I. E. Wachs, Catal. Lett., 2001, 75, 137–149.46 I. W. C. E. Arends and R. A. Sheldon, Appl. Catal., A, 2001,
212, 175–187.47 B. Solsona, V. A. Zazhigalov, J. M. Lopez Nieto,
I. V. Bacherikova and E. A. Diyuk, Appl. Catal., A, 2003,249, 81–92.
48 J. Guo, Y. Li, S. Zhu, Z. Chen, Q. Liu, D. Zhang, W.-J. Moonand D. M. Song, RSC Adv., 2012, 2, 1356–1363.
49 J. Dıaz-Reyes, V. Dorantes-Garcıa, A. Perez-Benıtez andJ. A. Balderas-Lopez, Superficies Vacio, 2008, 21, 12–17.
50 G. W. Coulston, E. A. Thompson and N. Herron, J. Catal.,1996, 163, 122–129.
51 (a) G. Busca, G. Centi and F. Trifiro, J. Am. Chem. Soc., 1985,107, 7757–7758; (b) S. J. Puttock and C. H. Rochester,J. Chem. Soc., Faraday Trans. 1, 1986, 82, 2773–2779;(c) G. Busca, G. Centi, F. Trifiro and V. Lorenzelli, J. Phys.Chem., 1986, 90, 1337–1344.
52 G. Deo and I. E. Wachs, J. Catal., 1994, 146, 335–345.53 V. V. Gulliants, J. B. Bezinger, S. Sundaresan, I. E. Wachs,
J. M. Jehng and J. E. Roberts, Catal. Today, 1996, 28,275–295.
54 M. Sohrabi, B. Dabir and F. Mozaffari, Chem. Eng. Technol.,1991, 14, 96–100.
55 M. Sohrabi and H. Aghdasinia, Chem. Eng. Technol., 2003,26, 69–73.
56 M. Abon and J. C. Volta, Appl. Catal., A, 1997, 157, 173–193.57 G. Centi, G. Golinelli and G. Busca, J. Phys. Chem., 1990, 94,
6813–6819.
Catalysis Science & Technology Paper
Publ
ishe
d on
04
Mar
ch 2
013.
Dow
nloa
ded
by T
he U
nive
rsity
of
Mel
bour
ne L
ibra
ries
on
28/0
9/20
13 1
4:18
:04.
View Article Online