transient arrest in a quiescent state allows ovarian cancer cells to survive suboptimal growth...
TRANSCRIPT

Transient arrest in a quiescent state allows ovarian cancer cellsto survive suboptimal growth conditions and is mediated byboth Mirk/dyrk1b and p130/RB2
Jing Hu, Hassan Nakhla, and Eileen Friedman
Pathology Department, Upstate Medical University, State University of New York, Syracuse, New York
Some ovarian cancer cells in vivo are in a reversible quiescent state where they can contribute to cancer spread under
favorable growth conditions. The serine/threonine kinase Mirk/dyrk1B was expressed in each of seven ovarian cancer cell
lines and in 21 of 28 resected human ovarian cancers, and upregulated in 60% of the cancers. Some ovarian cancer cells
were found in a G0 quiescent state, with the highest fraction in a line with an amplified Mirk gene. Suboptimal culture
conditions increased the G0 fraction in SKOV3 and TOV21G, but not OVCAR4 cultures. Less than half as many OVCAR4 cells
survived under suboptimal culture conditions as shown by total cell numbers, dye exclusion viability studies, and assay of
cleaved apoptotic marker proteins. G0 arrest in TOV21G and SKOV3 cells led to increased levels of Mirk, the CDK inhibitor
p27, p130/Rb2, and p130/Rb2 complexed with E2F4. The G0 arrest was transient, and cells exited G0 when fresh nutrients
were supplied. Depletion of p130/Rb2 reduced the G0 fraction, increased cell sensitivity to serum-free culture and to
cisplatin, and reduced Mirk levels. Mirk contributed to G0 arrest by destabilization of cyclin D1. In TOV21G cells, but not in
normal diploid fibroblasts, Mirk depletion led to increased apoptosis and loss of viability. Because Mirk is expressed at low
levels in most normal adult tissues, the elevated Mirk protein levels in ovarian cancers may present a novel therapeutic
target, in particular for quiescent tumor cells which are difficult to eradicate by conventional therapies targeting dividing cells.
Normal diploid cells such as lymphocytes and fibroblasts gointo a reversible growth arrest called quiescence in responseto anti-proliferative environmental signals. These normal cellsmaintain quiescence through upregulating the CDK inhibitorp27 in part through increasing the stability of the CDK in-hibitor p27 through the kinase Mirk,1–3 upregulate the tran-scriptional repressor HES1 to block senescence or differentia-tion,4,5 and block apoptosis through several mechanisms. Asubpopulation of tumor cells, possibly including stem cells,remains in a nondividing quiescent state that renders themrelatively resistant to chemotherapy and radiation therapywhich target dividing cells.6 By entering a quiescent state, tu-mor cells can resist the nutrient deficiencies, hypoxic andacidic conditions within the tumor mass. Mirk has also beenreported by our group to maintain the viability of quiescentpancreatic cancer cells by reducing intracellular levels of reac-tive oxygen species (ROS).7 Normal cells in quiescence acti-vate pathways that protect them from metabolic stress, and a
subpopulation of tumor cells is likely to utilize similar path-ways to survive within the tumor microenvironment. Theanti-apoptotic proteins BCL2 and BCL-XL have been shownto facilitate G0 quiescence in murine embryonic fibroblastsby decreasing RNA content and cell size, and by upregulatingp27 protein through its stabilization following phosphoryla-tion of p27Ser10 by Mirk.3
Epithelial carcinoma of the ovary is the fifth most fre-quent cause of cancer death in women. Most patients withovarian cancer have widespread disease at presentation, withyearly mortality approximately 65% of the incidence ratebecause of lack of effective treatment. The persistence ofdrug-resistant, quiescent ovarian cancer cells is known todecrease patient survival. Solitary disseminated tumor cellsthat are negative for proliferation markers such as Ki67 arethought to be the source of tumor recurrence. Analysis of 90resected ovarian tumors for the Ki67 antigen which is presentin all phases of the cell cycle except G0 and early G1 showedthat only about 30% of all tumor cells were in cycle.8 Recentstudies demonstrated that membrane PHK-label retainingcells within an ovarian adenocarcinoma-derived line wereslow-cycling, quiescent, with high clonogenic capacity andtumorigenicity in NOD/SCID mice.9
Factors that allow the prolonged survival of quiescent tu-mor cells in vivo are of clinical relevance. These include theras-related tumor suppressor gene ARH1 which inducesautophagy, inhibits the PI3-kinase pathway and regulatesdormancy in ovarian cancer cell xenografts,10,11 the stress-
Key words: Mirk/dyrk1B, ovarian cancer, quiescence
Additional Supporting Information may be found in the online
version of this article.
DOI: 10.1002/ijc.25692
History: Received 4 May 2010; Accepted 7 Sep 2010; Online 20 Sep
2010
Correspondence to: Eileen Friedman, Pathology Department,
Upstate Medical University, Syracuse, NY 13210, USA,
Tel.: 315-464-7148, E-mail: [email protected]
Can
cerCellBiology
Int. J. Cancer: 129, 307–318 (2011) VC 2010 UICC
International Journal of Cancer
IJC

activated protein kinase p38,12 and antioxidant proteins andfactors which control their expression13 such as Mirk, whichdecreases the level of toxic ROS in tumor cells, increasingtheir survival7 and their clonogenic growth.14 Mirk/Dyrk1B isa member of the Minibrain/dyrk family of kinases,15–17 whichmediate survival of differentiating cells in certain normal tis-sues: skeletal muscle (Mirk/dyrk1B),18 neuronal cells(Dyrk1A),15,19 erythropoietic cells (Dyrk3),20,21 and sperm(Dyrk4).22 Dyrk1A is one of the Down Syndrome genes,exhibits a 1.5-fold gene dosage pattern in Down syndromepatients and prevents nuclear occupancy of nuclear factor ofactivated T-cells (NFAT) transcription factors.19,23 AlthoughDyrk1A knockout caused embryonic lethality, knockout ofMirk/dyrk1B caused no evident phenotype in mice,24 suggest-ing that Mirk is not an essential gene for normal cells. Sup-porting this interpretation, normal diploid fibroblasts exhib-ited no alteration in the capacity for colony formation after20-fold depletion of Mirk.14 However, Mirk was found to beamong the four most promigratory genes in the SKOV3ovarian cancer cell line,25 suggesting that Mirk might play arole in ovarian cancer spread. Enhanced tumor cell survivalduring a quiescent period might increase the size of the pop-ulation of solitary cells capable of spread. The role of Mirk inachieving and maintaining G0 quiescence, as a part of thedormant cell phenotype, was examined in ovarian cancer celllines in our study.
Material and MethodsMaterials
All ovarian adenocarcinoma cell lines and the BJ fibroblaststrain were obtained from the ATCC and maintained in Dul-becco’s modified Eagle’s medium (DMEM) supplementedwith 7% heat-inactivated fetal bovine serum. All of the linesremained free of mycoplasma. Affinity-purified polyclonalantibody to a region within the Mirk unique C-terminus wasdescribed previously.26
RNA interference
The synthetic RNAi duplexes siA, siC and siD were directedto different expressed sequences within the Mirk gene. Theseand two RNAi duplexes targeted to the p130/Rb2 proteinwere obtained from Invitrogen. RNAi duplexes andGC-matched control RNAi duplexes were transfected intocells with lipofectamine 2000 in serum-containing growthmedium as described.7
Immunohistochemistry
Eighteen archived blocks of papillary serous ovarian cancerfrom University Hospital from 2000 to 2006, ten slides ofovarian cancer with matched normal ovarian tissue from theNational Resource Center, and archived blocks of first-tri-mester fetal tissue were utilized. Both cancer and fetal tissueswere obtained in accordance with institutional review proce-dures. Immunohistochemical staining for Mirk was per-formed exactly as detailed in Ref. 27. A mean of six determi-
nations were made per section for quantification of staining,with an average standard deviation of 12%.
Methods
Two-parameter flow cytometry, western blotting, Mirk activ-ity assays and substrates, cell number measurement by1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan, thiazolylblue formazan (MTT) assay, and cell viability by dye exclu-sion were described in previous studies.7,26,28,29
ResultsLog phase cultures of each of five ovarian cancer cell lines
contain a fraction of cells in G0 and OVCAR3 cultures with
the highest fraction of G0 cells proliferate the slowest
Normal fibroblasts are known to cycle into a quiescent G0state under suboptimal growth conditions and to activelymaintain quiescence through a defined genetic program.30
However, the presence of quiescent G0 tumor cells has beencontroversial. Quiescent cells degrade their ribosomes allow-ing G0 cells to be identified by their lower RNA content thanG1 cells, but 2N DNA content. Cellular DNA was stainedwith Hoechst 33258, Pyronin Y then added to bind to RNA,and both fluorochromes measured by two parameter flowcytometry. The BJ strain of human normal diploid fibroblastsmaintained in log phase growth had a low fraction (10%) ofcells in G0 while serum-starvation placed the majority of cells(66%) in G0 as noted by others30 (Fig. 1a, data representativeof 2–3 replicates). Five ovarian cancer cell lines were assayedduring log phase growth to determine whether any cellscycled into a G0 state even when the majority of cells wereproliferating. Surprisingly, OVCAR3, SKOV3, TOV21G,OVCAR4 and OV90 cultures each contained a fraction ofcells in G0 (Figs. 1a and 1c). These averaged 17 6 3% (SD)for SKOV3 cultures, 17 6 4% for TOV21G cultures, 38 62% for OVCAR3 cultures and 20 6 2% for OVCAR4 andOV90 cultures (data not shown). Reflecting their lower frac-tion of G0 cells, SKOV3 and TOV21G cultures grew overtwice as fast as OVCAR3 cultures (Fig. 1b), as did OVCAR4and OV90 cultures (data not shown). Thus, the entry of cellsinto G0 lowers the fraction of cycling cells within the culture,increasing the average doubling time.
G0 is not a permanent state and is induced by various
suboptimal culture conditions
Stressing cells by culturing them under suboptimal conditionsincreased the fraction of cells in G0. TOV21G, SKOV3,OVCAR3, OVCAR4 cells and OV90 cells were cultured for 3days either in serum-free DMEM or maintained as log phasecultures in DMEM plus 7% fetal bovine serum (FBS). Cultureunder serum-free conditions increased the fraction of G0cells an average of 3.2 (60.2) fold for SKOV3 cultures, 6.5(60.4) fold for TOV21G cultures, and 1.5 (60.1) forOVCAR3 cultures (with high initial levels of G0 cells, Fig.1a), but did not increase the fraction of G0 cells in OVCAR4(Fig. 1c) or OV90 cultures (data not shown).
Can
cerCellBiology
308 Mirk in ovarian cancer
Int. J. Cancer: 129, 307–318 (2011) VC 2010 UICC

Figure 1. Ovarian cancer cell cultures contain a fraction in G0 which increased in some lines grown in suboptimal conditions. a, Upper
panel: A log phase culture of the BJ strain of normal human diploid fibroblasts in growth medium was analyzed for cell cycle position by
two parameter flow cytometry, first staining for DNA by Hoescht 33258, then staining for RNA using Pyronin Y. Boxes outline cells in G0
(arrow), G1, S and G2þM. G0 and G1 cells both have 2N DNA content so the G1 box is directly over the G0 box, with the G0 cells having
less total RNA. (Middle panel) BJ normal human diploid fibroblasts were serum-starved for 72 hr and then analyzed for cell cycle position
by two parameter flow cytometry using the same gating and staining protocol as above. Lower panels: A log phase culture and a serum-
starved culture of OVCAR3 ovarian cancer cells were analyzed for cell cycle position by two parameter flow cytometry in identical fashion to
the BJ fibroblasts. b, OVCAR3, SKOV3 and TOV21G ovarian cancer cell cultures in log phase growth were replated at similar cell densities,
then allowed to grow for up to 5 days in growth medium. Relative cell number was determined by MTT assay of duplicate cultures 6 SD.
The mean fraction of cells in G0, as determined by two parameter flow cytometry, from two to three experiments is shown. c, SKOV3,
TOV21G and OVCAR4 ovarian cancer cells were plated and allowed to enter cycle by culture in growth medium (DMEM þ 7% FBS) for
24 hr, then switched to serum-free medium for 3 days before analysis for cell cycle position by two parameter flow cytometry.
G0 cells indicated by arrows.
Can
cerCellBiology
Hu et al. 309
Int. J. Cancer: 129, 307–318 (2011) VC 2010 UICC
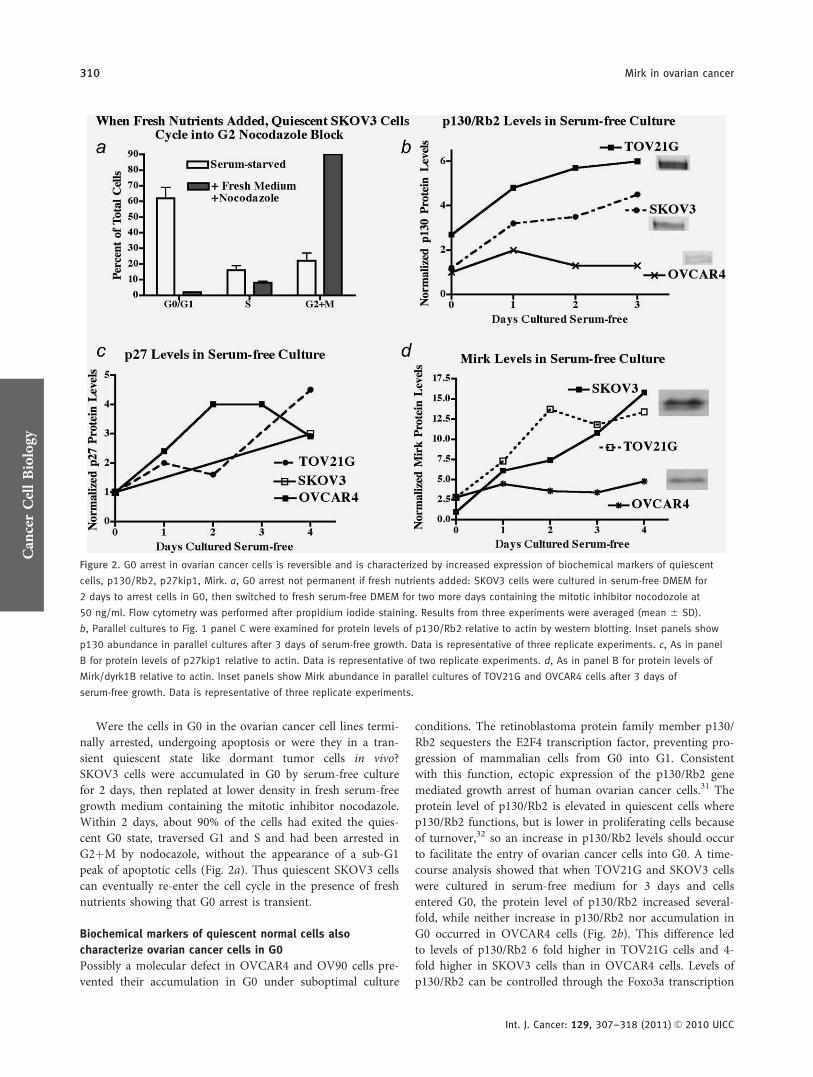
Were the cells in G0 in the ovarian cancer cell lines termi-nally arrested, undergoing apoptosis or were they in a tran-sient quiescent state like dormant tumor cells in vivo?SKOV3 cells were accumulated in G0 by serum-free culturefor 2 days, then replated at lower density in fresh serum-freegrowth medium containing the mitotic inhibitor nocodazole.Within 2 days, about 90% of the cells had exited the quies-cent G0 state, traversed G1 and S and had been arrested inG2þM by nodocazole, without the appearance of a sub-G1peak of apoptotic cells (Fig. 2a). Thus quiescent SKOV3 cellscan eventually re-enter the cell cycle in the presence of freshnutrients showing that G0 arrest is transient.
Biochemical markers of quiescent normal cells also
characterize ovarian cancer cells in G0
Possibly a molecular defect in OVCAR4 and OV90 cells pre-vented their accumulation in G0 under suboptimal culture
conditions. The retinoblastoma protein family member p130/Rb2 sequesters the E2F4 transcription factor, preventing pro-gression of mammalian cells from G0 into G1. Consistentwith this function, ectopic expression of the p130/Rb2 genemediated growth arrest of human ovarian cancer cells.31 Theprotein level of p130/Rb2 is elevated in quiescent cells wherep130/Rb2 functions, but is lower in proliferating cells becauseof turnover,32 so an increase in p130/Rb2 levels should occurto facilitate the entry of ovarian cancer cells into G0. A time-course analysis showed that when TOV21G and SKOV3 cellswere cultured in serum-free medium for 3 days and cellsentered G0, the protein level of p130/Rb2 increased several-fold, while neither increase in p130/Rb2 nor accumulation inG0 occurred in OVCAR4 cells (Fig. 2b). This difference ledto levels of p130/Rb2 6 fold higher in TOV21G cells and 4-fold higher in SKOV3 cells than in OVCAR4 cells. Levels ofp130/Rb2 can be controlled through the Foxo3a transcription
Figure 2. G0 arrest in ovarian cancer cells is reversible and is characterized by increased expression of biochemical markers of quiescent
cells, p130/Rb2, p27kip1, Mirk. a, G0 arrest not permanent if fresh nutrients added: SKOV3 cells were cultured in serum-free DMEM for
2 days to arrest cells in G0, then switched to fresh serum-free DMEM for two more days containing the mitotic inhibitor nocodozole at
50 ng/ml. Flow cytometry was performed after propidium iodide staining. Results from three experiments were averaged (mean 6 SD).
b, Parallel cultures to Fig. 1 panel C were examined for protein levels of p130/Rb2 relative to actin by western blotting. Inset panels show
p130 abundance in parallel cultures after 3 days of serum-free growth. Data is representative of three replicate experiments. c, As in panel
B for protein levels of p27kip1 relative to actin. Data is representative of two replicate experiments. d, As in panel B for protein levels of
Mirk/dyrk1B relative to actin. Inset panels show Mirk abundance in parallel cultures of TOV21G and OVCAR4 cells after 3 days of
serum-free growth. Data is representative of three replicate experiments.
Can
cerCellBiology
310 Mirk in ovarian cancer
Int. J. Cancer: 129, 307–318 (2011) VC 2010 UICC

factor,33 or post-translationally through protein stabilization.One or both of these regulations may be aberrant inOVCAR4 cells. Immunohistochemical analysis by othersrevealed loss or decrease in expression of p130/Rb2 in about40% of 45 resected human ovarian cancers assayed,31 so theOVCAR4 line may reflect this large subgroup of ovarian can-cers with low p130/Rb2 expression.
Mirk protein levels increased 4–7 fold in SKOV3 andTOV21G cells, but not in OVCAR4 cells, cultured underthese suboptimal growth conditions (Fig. 2d). In time-coursestudies, Mirk levels increased within 24 hr of the switch toserum-free culture and then continued to increase. Thus anincrease in level of Mirk protein was found only when ovar-ian cancer cells accumulated in G0. The CDK inhibitorp27kip1 helps to maintain the G0 state by binding to CDK/cyclin complexes. Levels of p27 were increased 20-fold whenSU86.86 pancreatic cancer cells were made quiescent in G0.7
Levels of p27 differed dramatically between the OVCAR4,SKOV3 and TOV21G cell lines, but were increased severalfold in each line by serum-free culture (Fig. 2c). The inabilityof OVCAR4 cells to arrest in G0 could not be ascribed toalterations in abundance or regulation of p27.
SKOV3 and TOV21G ovarian cancer cells accumulate in G0
to survive suboptimal culture conditions
SKOV3, TOV21G and OVCAR4 cells were cultured in se-rum-free conditions for up to 6 days. Measurement of rela-tive cell number by MTT assay showed that both SKOV3and TOV21G cultures grew to a higher cell density than theOVCAR4 cultures that exhibited declining cell numbers after3 days of serum-free culture (Fig. 3a). Analysis of parallelcultures revealed that OVCAR4 cells underwent more apo-ptosis than TOV21G or SKOV3 cells (Fig. 3b). Critical stepsin apoptosis are cleavages of poly(ADP-ribose) polymerase(PARP) and caspase 3, which were prominent only inOVCAR4 cells after 4 and 6 days of serum-free culture. Mea-surement of cell viability by dye exclusion showed that about60% of the OVCAR4 cells were nonviable and unable toexclude dye after 4–6 days of serum-free culture, comparedwith about 30% of SKOV3 and TOV21G cells (Fig. 3c).These nonviable OVCAR4 cells then underwent apoptosis(Fig. 3b), reducing total cell numbers (Fig. 3a). Other subop-timal culture conditions also led TOV21G and SKOV3 cellsto accumulate in G0. Cells were cultured for 3 days either inserum-free DMEM, in normal growth medium to high celldensity or in low glucose DMEM without FBS. In each case,the fraction of G0 cells increased to 60–80% of the culture,whereas no such increase was seen with OVCAR4 cultures(Supporting Information Fig. 1). Thus, ovarian cancer cellsthat could enter a reversible quiescent arrest in G0 weremore protected from suboptimal growth conditions than tu-mor cells that lacked this capacity. In vivo, such quiescentcells could re-enter the cell cycle under favorable clues fromthe microenvironment.
Mirk helps to maintain the viability of ovarian cancer cells
accumulated in G0 by suboptimal culture
The effect of Mirk on viability of ovarian cancer cells undersuboptimal growth conditions was determined by depletionof Mirk by two RNAi duplexes, each added independently toa parallel culture of either TOV21G or OVCAR4 ovariancancer cells, or the BJ strain of normal human diploid fibro-blasts. The Mirk depleted and control-depleted cells werethen maintained in serum-free media for 4 or 6 days. About60% of the OVCAR4 cells, 30% of the TOV21G cells, butonly 10% of the normal fibroblasts died under these condi-tions (average of Ctsi treated cultures, Fig. 3d). However,these three cell types differed in their sensitivity to depletionof Mirk. There was no detectable effect on the viability ofnormal BJ cells on either day, whereas Mirk depletion didnot further decrease OVCAR4 viability after 4 days of serum-starvation with a marginal effect after 6 days (Fig. 3d). Incontrast, Mirk depletion led to a mean 50% increase inTOV21G cell death after either 4 or 6 days of serum starva-tion. In similar studies, Mirk depletion led to increased clea-vages of PARP and caspase 3 in SKOV3 and in TOV21Gcells, not in OVCAR4 cells, indicating that the loss of Mirkled to cell death by apoptosis (Fig. 3b, lower panel). Thus,Mirk helps to maintain TOV21G ovarian cells in a viablequiescent state by blocking apoptosis, whereas having littleprotective effect for OVCAR4 cells which are not accumu-lated in G0 by suboptimal growth conditions. In addition,Mirk protein was expressed at much lower levels in normalBJ fibroblasts compared to either SKOV3 or TOV21G cancercells, and depletion of Mirk in BJ cells led to no detectableincrease in cell death (Fig. 3d). This data is consistent withearlier studies on Mirk knockout in mice24 and colony for-mation assays14 that showed a selective sensitivity of cancercells to Mirk depletion compared with normal diploid cells.
The mechanism for G0 arrest includes the Mirk-mediated
reduction of cyclin D1 to prevent escape into G1
Mirk has been shown to slow the exit of SU86.86 pancreaticcancer cells and HD6 colon cancer cells from G0 quiescenceby phosphorylating their cyclin D isoforms at a conservedubiquitination site that initiates rapid turnover.7,38,39 Mirkwas shown to also alter ovarian cancer cell cycling throughcyclin D1. Efficient depletion of Mirk in SKOV3, OVCAR3and TOV21G cells by either of two synthetic RNAi duplexesled to a 2-fold increase in cyclin D1 levels after 2–3 days ofculture (data not shown). Thus Mirk reduced cyclin D1 levelsin these cells, restricting their entry into G1 and the cellcycle.
A functional p130/Rb2 protein aids in enabling ovarian
cancer cells to enter a quiescent G0 state and to remain
viable when quiescent
The role of p130 in ovarian cancer cell survival under stressconditions was then studied by depleting p130 from
Can
cerCellBiology
Hu et al. 311
Int. J. Cancer: 129, 307–318 (2011) VC 2010 UICC

TOV21G and from OVCAR4 cells, then comparing their via-bility by dye exclusion assay under various test conditions:serum-containing or serum-free growth media, serum-con-taining growth media plus the chemotherapeutic drug cispla-
tin, or serum-free media plus cisplatin (Figs. 4a and 4c). Thetime of exposure was limited to 4 days to limit the amountof cell loss initiated by cisplatin. Depletion of p130/Rb2 hadno effect on the fraction of dead cells in the unstressed
Figure 3. Cultures of OVCAR4 cells with low fractions in G0 under serum-free culture conditions undergo more apoptosis and more cell
death than cells that can arrest in G0. a, SKOV3, OVCAR4 and TOV21G cells were plated, and allowed to enter cycle by culture in growth
medium for 24 hr, then switched to serum-free medium. Relative cell number was measured by the MTT assay, mean 6 SD shown, n ¼ 2.
b, Upper panel: Parallel cultures from panel A were analyzed by western blotting for markers of apoptosis, cleaved PARP and cleaved
caspase 3. Lower panel: TOV21G, OVCAR4 and SKOV3 cells were depleted of Mirk using two different RNAi duplexes independently (siD or
siC) or GC-matched controls, then cultured serum-free for 4 days before western blotting for Mirk, actin, cleaved PARP and cleaved caspase
3. Only data with siD shown. c, The cultures from panel A were sampled for the fraction of dead cells incapable of exclusion of trypan blue
dye. Mean 6 SD shown, with n ¼ 4 for each point. An average of 488 6 40 cells were assayed per point. d, TOV21G, OVCAR4 and BJ
normal human diploid fibroblasts were depleted of Mirk using two different RNAi duplexes independently (siD or siC) or GC-matched
controls as in panel B, then cultured serum-free for 4 and for 6 days before assay of the percentage of dead cells by trypan blue dye
exclusion. Mean 6 SD shown (n ¼ 4, with two measurements per RNAi duplex). Student’s two-tailed t test used to analyze cultures of
TOV21G gave p < 0.001. (lower panel) Western blot showing depletion of Mirk in parallel cultures after 4 days.
Can
cerCellBiology
312 Mirk in ovarian cancer
Int. J. Cancer: 129, 307–318 (2011) VC 2010 UICC

condition of normal serum-containing growth medium, anaverage of 15% for TOV21G cultures and 30% for OVCAR4cultures (data not shown). However, while the fraction ofdead mock-depleted TOV21G cells was doubled by cisplatin,the fraction of dead p130-depleted TOV21G cells wasincreased much more, over 3-fold. In contrast, p130 depletedOVCAR4 cells were as susceptible to cisplatin treatment as
mock-depleted cells (Fig. 4a). A similar sensitization byp130/Rb2 knockdown occurred when cells were treated withcisplatin in serum-free medium. Cisplatin killed 35% morep130-depleted TOV21G cells than controls, whereas cisplatinkilled an equal fraction of p130-depleted and mock-depletedOVCAR4 cells (Fig. 4a, last four lanes). Thus p130-depletiondecreased the capacity of TOV21G cells to survive under the
Figure 4. Depletion of the G0 control protein p130/Rb2 reduces the ability of SKOV3 and TOV21G cells to arrest in G0, and decreases
TOV21G cell viability. a, TOV21G (TOV) and OVCAR4 (OV4) cells were plated as single cells, allowed to attach overnight, then depleted of
p130/Rb2 by transfection with RNAi duplexes or GC-matched controls and culture for 2 days in growth medium. Medium was then changed
to fresh FBS containing growth medium (þFBS) or fresh FBS free growth medium (�FBS) with 3.3 lM cisplatin. Cells were cultured for 4
days, then the fraction of dead cells was determined by trypan blue exclusion, mean 6 SD shown, with an average of 433 6 38 cells
assayed per condition. b, SKOV3, TOV21G and OVCAR4 cells were depleted of p130 and made quiescent by culture in FBS free growth
medium for 3 and 5 days. The fraction of cells in G0 in p130 depleted and control si-depleted cells was determined by 2-parameter flow
cytometry, mean 6 SE shown. c, Western blot showing p130/Rb2, p27 and Mirk levels in TOV21G cells either depleted of Mirk or depleted
of p130/Rb2, each by two different RNAi duplexes, after 2 days in serum-free conditions. Samples were re-run for determination of p27
abundance. Lower panel: Western blot showing p130/Rb2, Mirk and actin levels after depletion of p130/Rb2 by si2 in TOV21G cultures
followed by culture in serum-free conditions to upregulate Mirk for 0–3 days. d, Western blot showing immunoprecipitated p130, E2F4
bound to immunoprecipitated p130/Rb2, and total p130/Rb2 and antibody heavy chain in the respective lysates from SKOV3, TOV21G and
OVCAR4 cells either maintained in log phase or serum-starved for 3 days.
Can
cerCellBiology
Hu et al. 313
Int. J. Cancer: 129, 307–318 (2011) VC 2010 UICC

stress of DNA damage because of cisplatin, whereas havinglittle effect on the survival of OVCAR4 cells.
The role of p130/Rb2 in G0 arrest was examined bydepleting p130 in SKOV3, TOV21G and OVCAR4 cells, thenplacing cells in serum-free medium for 3 and 5 days in anattempt to induce quiescence. Although the p130 depletionwas only partial (Fig. 4c), and also led to a strong increase inp27 levels, fewer p130-depleted SKOV3 and TOV21G cellswere found in G0 (Fig. 4b). In contrast, depletion of p130had almost no effect on the G0 fraction of OVCAR4 cells.These data, taken together, suggest that p130/Rb2 enablesTOV21G cells to remain in G0 as part of a stress response,and loss of this capacity for G0 arrest decreases theirviability.
When normal diploid cells enter G1 from G0 by additionof mitogens, p130/Rb2 is phosphorylated by G1 cyclin/CDKcomplexes and other kinases at up to 22 sites, thus freeingthe transcription factor E2F4.34–36 E2F4 was expressed atsimilar levels in the three cell lines under different culturesconditions (data not shown). However, about 5-fold moreE2F4 was bound to p130/Rb2 immunoprecipitated fromSKOV3 and TOV21G cells arrested in G0 compared withp130/Rb2 from OVCAR4 cells grown under similar condi-tions (Fig. 4d). Thus fewer serum-starved OVCAR4 cellswere found in G0 because they did not express enough p130/Rb2 (Fig. 2b) capable of sequestering E2F4 to block entryinto G1.
Depletion of p130/Rb2 unexpectedly lowered Mirk levels
As well as maintaining cell viability (Fig. 4a), p130/Rb2allowed the upregulation of Mirk expression that occurredwhen cells were placed in suboptimal growth conditions.TOV21G cells were depleted of p130 by two RNAiduplexes, whereas Mirk was depleted by two duplexes inparallel experiments, with each duplex targeting a differentsequence. Although depletion of Mirk had no effect on thelevel of p130, a partial depletion of p130 by either of twoRNAi duplexes also reduced Mirk levels (Fig. 4c). Thedepletion of p130 was short-lived in TOV21G cells, so after3 days in serum-free DMEM p130 levels rebounded, as didMirk levels, showing another positive correlation (Fig. 4c).Mirk transcription is upregulated through Rho family mem-bers RhoA and cdc42 through E boxes within the Mirkpromoter and is inhibited by MEK-erk signaling.37 In addi-tion, Mirk expression is elevated about 10-fold whenNIH3T3 cells become confluent and arrested in G0, com-pared to cells in S phase.1 Possibly, Mirk is preferentiallyexpressed when cells have entered G0 through the functionof p130/Rb2.
The Mirk gene is amplified in the OVCAR3 cell line
Amplicons are maintained in cancers when one or moregenes within the amplicon provide a selective growth or sur-vival advantage. The 19q13 amplicon has been identified inabout 30% of ovarian cancers,40 in 10–20% of pancreatic can-
cers,41,42 as well as in cases of follicular lymphoma, mantlecell lymphoma, Burkitt’s lymphoma, and in both small-celland non small-cell lung cancer. This amplicon is localized toa region around 19q13.1–13.2, with the Mirk/dyrk1B gene at19q13.1.26 The Mirk gene was one of 16 genes comprisingthe consistently amplified 660 kb subregion of the 19q13amplicon in pancreatic cancers, whereas the nearby geneAkt2 was not.43 Possibly selection for the Mirk gene main-tained the 19q13 amplicon because Mirk mediates survival insome pancreatic cancers.28,14 To test the hypothesis that theMirk gene was amplified in some ovarian cancers, Southernblotting was performed on seven ovarian cancer cell linesthat expressed Mirk protein by western analysis (Fig. 5a).Three of these lines, OVCAR3, SKOV3 and OVCAR8, had ahomogenously staining region of amplified genes includingAkt2 at 19q13.40,44 Four lines without this amplification(OV90, OVCAR4, OVCAR5 and TOV21G) were also ana-lyzed for Mirk gene levels, whereas the BJ strain of normalhuman fibroblasts served as the negative control. OVCAR3cells exhibited a 20-fold amplification of the Mirk gene (Fig.5b). Interestingly, many OVCAR3 cells were found in G0,even in growth medium (Fig. 1), consistent with Mirk’scapacity to block cell cycling by destabilizing cyclin D1. Thep21cip1 gene located on chromosome 6 was used as an inter-nal blotting control as this locus was not amplified or deletedin any ovarian cancer case examined in the National Insti-tutes of Health (NIH) database. The EcoR1 digest of theDNAs in the Southern blot after staining with ethidium bro-mide demonstrates that the OVCAR3 lane was notoverloaded.
Mirk protein was detected in about 75% of resected
ovarian cancers
Twenty-eight cases of ovarian cancers were assayed forMirk expression by immunohistochemistry using affinity-purified polyclonal antibody raised to the unique sequenceat Mirk’s C-terminus (Table 1). Mirk protein was identifiedin 21 of the cases (75%). Mirk abundance in the cytoplasmwas quantified by the IPLabGel Program. A wide range ofMirk protein levels was seen (Fig. 5c). In four cases inwhich Mirk was not present in the tumor, Mirk was alsoabsent from the normal ovarian epithelial tissue. In 15 ofthe cancer cases in which Mirk was expressed, normal-appearing ovarian epithelial tissue from the same patientwas analyzed at the same time for Mirk expression. Analy-sis of these paired samples (Fig. 5d) showed a statisticallysignificant difference in 9 of the 15 cases with p ¼ 0.0001by the student’s paired t test. Presence of the 19q13 ampli-con was not determined. These cases included both papil-lary serous cystadenocarcinomas and poorly differentiatedadenocarcinomas. Mirk expression was absent or at lowabundance in the tumor stroma. Mirk was also detected inthree of three fetal ovaries (data not shown). The increasedexpression of Mirk protein in a large subset of ovarian can-cers and in fetal ovarian tissue was consistent with the
Can
cerCellBiology
314 Mirk in ovarian cancer
Int. J. Cancer: 129, 307–318 (2011) VC 2010 UICC

hypothesis that Mirk is an oncofetal protein which mightplay a role in ovarian tumorigenesis.
DiscussionIn our study, Mirk was detected in about 75% of ovariancancers and found to be an active kinase in each of five ovar-ian cancer cell lines examined (Supporting Information Fig.2). Each of these lines in log phase growth had a small sub-population in G0, with the largest G0 fraction observed inthe line with the slowest growth, the OVCAR3 line whichexhibited a 20-fold amplified Mirk gene. Control of G0 wascompared in three ovarian cancer cell lines each with anactive Mirk kinase, but not an amplified Mirk gene so thatMirk levels could be modulated by RNA interference techni-
ques. In SKOV3 and TOV21G cells, the G0 fraction wasincreased if cells were maintained under suboptimal condi-tions, such as culture in serum-free DMEM or low glucose-containing DMEM, and levels of p130/Rb2 and Mirkincreased when the fraction of G0 cells rose. Neitherincreases in the G0 fraction nor increases in p130/Rb2 orMirk abundance occurred when OVCAR4 cells were culturedunder suboptimal conditions. RNA interference studies dem-onstrated that a functional p130/Rb2 protein enabledTOV21G cells to enter a quiescent G0 state and to remainviable when quiescent. OVCAR4 cells could not arrest in G0because they did not express enough functional p130/Rb2capable of sequestering E2F4 to block entry into G1. About5-fold more E2F4 was bound to p130/Rb2 immunoprecipitated
Figure 5. Mirk levels in ovarian cancer cell lines and in resected ovarian cancers. a, Western blot showing steady state levels of Mirk
protein in normal diploid BJ fibroblasts (BJ) and the human ovarian cancer cell lines OV90 (90), OVCAR3 (O3), OVCAR4 (O4), OVCAR5 (O5),
OVCAR8 (O8), SKOV3 (SK) and TOV21G (TOV), with commassie blue stained blot as a loading control. b, Southern blot of the Mirk gene in
diploid fibroblasts and ovarian cancer cell lines. 15 ug of DNA from each of the cell lines in panel A was digested with EcoR1 before
separation on a 0.8% agarose gel, and staining with ethidium bromide (EtBr). A gene not amplified in ovarian cancer, p21cip1, is the
internal blotting control. c, Mirk protein was detected by immunohistochemistry in sections of normal-appearing ovarian epithelium (left
row) and serous ovarian carcinoma (right rows) from three patients. Upper: moderate Mirk protein levels in normal epithelium, none in
normal stroma, with strong focal staining in carcinoma; Middle: moderate Mirk expression in normal epithelium, none in normal stroma,
with strong focal staining in carcinoma; Lowest: weak expression of Mirk both in normal epithelium and in carcinoma. Parallel sections
receiving only preimmune serum showed only blue counterstain. d, Mirk levels compared in paired cases of ovarian cancers and adjacent
normal tissue, n ¼ 15, with 14 cases of papillary serous adenocarcinoma and 1 case of poorly differentiated adenocarcinoma. Mean age of
patients is 63. Nine cases clustered in group A showed a clear increase in Mirk protein level with p ¼ 0.0001 in a two-tailed t test,
whereas no increase in Mirk levels was seen in the six cases in group B.
Can
cerCellBiology
Hu et al. 315
Int. J. Cancer: 129, 307–318 (2011) VC 2010 UICC

from SKOV3 and TOV21G cells arrested in G0 comparedwith p130/Rb2 from OVCAR4 cells grown under similar con-ditions. More SKOV3 and TOV21G cells than OVCAR4 cellsremained viable in the stress condition of serum-free culture,as more OVCAR4 cells underwent apoptosis. Cells enteredG0 under stress conditions, but exited into cycle when freshnutrients were supplied, showing that G0 arrest was reversi-ble. Depletion of p130/Rb2 also decreased expression ofMirk, showing their inter-relationship.
If ovarian cancer cells had expressed high enough levels ofp130/Rb2 to enter G0, Mirk then kept these cells in a quies-cent, reversible G0 state by destabilizing cyclin D1 so cellscould not enter cycle. Mirk controlled cyclin D1 levels inOVCAR3, SKOV3 and TOV21G cells, all of which becamequiescent in G0 under suboptimal culture conditions. Mirkdid not regulate cyclin D1 levels in OVCAR4 or OV90 cellsthat did not become quiescent under parallel conditions.Mirk may play a similar role in vivo. RNA interference stud-ies showed Mirk depletion led to more apoptosis in SKOV3and TOV21G cells, and more cell death as measured by dyeexclusion. Mirk was shown to maintain the viability of quies-cent pancreatic cancer cells by reducing levels of ROSthrough Mirk’s transcriptional coactivator activity for acohort of antioxidant genes.7 Mirk also blocked apoptosis ineach of four ovarian cancer cell lines by reducing ROS lev-els.45 The increase in expression of Mirk in TOV21G andSKOV3 cells made quiescent by suboptimal growth condi-tions, and loss of their viability after Mirk knockdown, sug-gests that their viability was due, at least in part, to Mirk’santioxidant inducing properties.
The mechanism for induction of quiescence varied. Se-rum-free culture eliminates serum growth factors such asIGF-1 needed for cells to synthesize enough cyclin D to enterG1. High cell density can lead to an exhaustion of somenutrients in growth medium. Both culture under limited glu-cose levels and culture to high cell density will raise cAMP
levels leading to the activation of the AMP-activated proteinkinase (AMPK) heterotrimer complex. AMPK is widelyexpressed in ovarian cancers of all subtypes46 and in ovariancancer cell lines where, through TSC1/2, it blocks the activa-tion of mTOR, thus blocking the activation of mTOR’sdownstream substrates needed for cell cycle progressionincluding ribosomal S6 kinase (reviewed in Ref. 47). Thus inSKOV3 and TOV21G cells increased Mirk levels correlatedwith a lengthened G0, regardless of the mechanism of entryinto quiescence, while in the OVCAR3 line, amplification ofthe Mirk gene led to elevated levels of Mirk protein andmore cells in G0.
In OVCAR4 cells p130/Rb2 was not expressed at a highenough level and displayed abnormal phosphorylation, sop130/Rb2 was not able to sequester much E2F4. As a result,OVCAR4 cells in suboptimal culture conditions could notenter G0. The role of p130/Rb2 in controlling growth arrestin ovarian cancer cells has been documented by other investi-gators. All trans-retinoic acid arrested ovarian cancer cellgrowth in G0/G1 by increasing the stability of the p130/Rb2protein.48 Protein phosphatase 2A dephosphorylated p130/Rb2 within its nuclear localization sequence, enabling impor-tin alpha to bind and mediate p130/Rb2 translocation to thenucleus where it suppressed cell cycling.48 In a recent study,38
Mirk was shown to prevent colon carcinoma cells from exit-ing a quiescent G0 state, at least in part, through destabiliza-tion of G1 cyclins. In the absence of Mirk, elevated levels ofG1 cyclins activated the CDK4 kinase that phosphorylatedthe p130/Rb2 protein that blocks exit from G0 by sequester-ing the E2F4 transcription factor. As a result, quiescent coloncancer cells depleted of Mirk began to cycle, similar to theresults of our study.
Many cancer cells in vivo, including a large fraction ofovarian cancers,8 are not cycling and a subset of these cellsmay be in a G0 quiescent state. Quiescent cells degrade theirribosomes allowing G0 cells to be identified by their lowRNA content by two parameter flow cytometry, their 2NDNA content, and by the presence of biochemical markers,including a multiprotein complex containing p130/Rb2 bind-ing to and sequestering the transcription factor E2F4, andelevated levels of the CDK inhibitor p27kip1. In cells arrestedin G0 by serum deprivation many common promoter sitesare bound by a multiprotein DREAM complex containingp130/E2F4.49 In normal fibroblasts, arrest in G0 is an activeprocess maintained by a set of proteins differing from thoseactive in other cell cycle stages. Depletion of Mirk in normalBJ fibroblasts had no effect on their viability under serum-free culture conditions, while increasing apoptotic cell deathin TOV21G cells (Figs. 3b and d). Pushing tumor cells out ofG0 quiescence into the cell cycle by depletion of Mirk or bypharmacological inhibition of Mirk may enhance tumor cellkill by chemotherapeutic drugs or radiation, while having lesseffect on normal tissues in which Mirk levels are quite lowand in which Mirk appears to have little effect on cellsurvival.
Table 1. Analysis of mirk expression in ovarian cancers byimmunohistochemistry
Characteristics No. of patients
Total 28
Age 63 years (614)
Histology
Papillary serous 24
Positive for Mirk 18
Poorly differentiated 3
Positive for Mirk 2
Granulosa 1
Positive for Mirk 1
Mirk expression
25–100% of tumor cells 20
Focal, <25% of cells 1
Can
cerCellBiology
316 Mirk in ovarian cancer
Int. J. Cancer: 129, 307–318 (2011) VC 2010 UICC

References
1. Deng X, Mercer SE, Shah S, Ewton DZ,Friedman E. The Cyclin-dependent KinaseInhibitor p27Kip1 Is Stabilized in G0 byMirk/dyrk1B Kinase. J Biol Chem 2004;279:22498–504.
2. Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ, Roberts JM. Apathway in quiescent cells that controlsp27Kip1 stability, subcellular localization,and tumor suppression. Genes Dev 2006;20:47–64.
3. Janumyan Y, Cui Q, Yan L, Sansam CG,Vanlentin M, Yang E. G0 function ofBCL2 and BCL-xL requires BAX, BAK,and p27 phosphorylation by Mirk,revealing a novel role of BAX and BAK inquiescence regulation. J Biol Chem 2008;283:34108–20.
4. Sang L, Coller HA, Roberts JM. Control ofthe reversibility of cellular quiescence bythe transcriptional repressor HES1. Science2008;321:1095–100.
5. Sang L, Roberts JM, Coller HA. HijackingHES1: how tumors co-opt the anti-differentiation strategies of quiescent cells.Trends Mol Med 2010;16:17.
6. Barnes D, Melo J. Primitive, quiescent anddifficult to kill: the role of non-proliferating stem cells in chronic myeloidleukemia. Cell Cycle 2006;5:2862–6.
7. Deng X, Ewton DZ, Friedman E. Mirk/Dyrk1B maintains the viability of quiescentpancreatic cancer cells by decreasing ROSlevels. Cancer Res 2009;69:3317–24.
8. Schindlbeck C, Hantschmann P, Zerzer M,Jahns B, Rjosk D, Janni W, Rack, BSommer H, Friese K. Prognostic impact ofKI67, p53, human epithelial growth factorreceptor 2, topoisomerase IIalpha,epidermal growth factor receptor, andnm23 expression of ovarian carcinomasand disseminated tumor cells in the bonemarrow. Int J Gynecol Cancer 2007;17:1047–55.
9. Kusumbe AP, Bapat SA. Cancer stem cellsand aneuploid populations withindeveloping tumors are the majordeterminants of tumor dormancy. CancerRes 2009;69:9245–53.
10. Lu Z, Luo R, Lu Y, Zhang X, Yu Q, KhareS, Kondo S, Kondo Y, Yu, Y. Mills GB,Liao S-L, Bast RC, Jr The tumorsuppressor gene ARHI regulates autophagyand tumor dormancy in human ovariancancer cells. J Clin Invest 2008;118:3917–29.
11. Amaravadi RK. Autophagy-induced tumordormancy in ovarian cancer. J Clin Invest2008;118:3837–40.
12. Adam AP, George A, Schewe D, BragadoP, Iglesias BV, Ranganathan AC, KourtidisA, Conklin DS, Aguirre-Ghiso JA.Computational identification of a
p38SAPK-regulated transcription factornetwork required for tumor cell quiescence.Cancer Res 2009;69:5664–72.
13. Chen C, Liu Y, Liu R, Ikenoue T, Guan K-L,Liu Y, Zheng P. TSC-mTOR maintainsquiescence and function of hematopoieticstem cells by repressing mitochondrialbiogenesis and reactive oxygen species.J Exp Med 2008;205:2397–408.
14. Jin K, Park S-J, Ewton D, Friedman E. Thesurvival kinase Mirk/dyrk1B is adownstream effector of oncogenic K-ras.Cancer Res 2007;67:7247–55.
15. Tejedor F, Zhu X, Kaltenbach E,Ackermann A, Baumann A, Canal I,Heisenberg M, Fischbach KF, Pongs O.Minibrain:a new protein kinase familyinvolved in postembryonic neurogenesis inDrosophila. Neuron 1995;14:287–301.
16. Kentrup H, Becker W, Heukelbach J,Wilmes A, Schurman A, Huppertz C,Kainulainen H, Joost H-G. Dyrk, a dualspecificity protein kinase with uniquestructural features whose activity isdependent on tyrosine residues betweensubdomains VII and VIII. J Biol Chem1996;271:3488–95.
17. Becker W, Weber Y, Wetzel K, EirmbterK, Tejedor F, Joost H-G. Sequencecharacteristics, subcellular localization andsubstrate specificity of dyrk-related kinases,a novel family of dual specificity proteinkinases. J Biol Chem 1998;273:25893–902.
18. Mercer SE, Ewton DZ, Deng X, Lim S,Mazur TR, Friedman E. Mirk/Dyrk1BMediates Survival during theDifferentiation of C2C12 Myoblasts. J BiolChem 2005;280:25788–801.
19. Arron JR, Winslow MM, Polleri A, ChangC-P, Wu H, Gao X, Neilson JR, Chen L,Heit JJ, Kim SK, Yamasaki N, Miyakawa T,Francke U, Graef IA, Crabtree GR. NFATdysregulation by increased dosage ofDSCR1 and DYRK1A on chromosome 21.Nature 2006;441:595–600.
20. Geiger JN, Knudsen GT, Panek L, PanditAK, Yoder MD, Lord KA, Creasy CL,Burns BM, Gaines P, Dillon SB,Wojchowski DM. mDYRK3 kinase isexpressed selectively in late erythroidprogenitor cells and attenuates colony-forming unit-erythroid development. Blood2001;97:901–10.
21. Li K, Zhao S, Karur V, Wojchowski DM.DYRK3 activation, engagement of proteinkinase A/cAMP response element-bindingprotein, and modulation of progenitor cellsurvival. J Biol Chem 2002;277:47052–60.
22. Sacher F, Moller C, Bone W, Gottwald U,Fritsch M. The expression of the testis-specific Dyrk4 kinase is highly restricted tostep 8 spermatids but is not required for
male fertility in mice. Mol Cell Endocrinol2007;267:80–8.
23. Gwack Y, Sharma S, Nardone J, Tanasa B,Iuga A, Srikanth S, Okamura H, Bolton D,Feske S, Hogan PG, Rao A. A genome-wide Drosophila RNAi screen identifiesDYRK-family kinases as regulators ofNFAT. Nature 2006;441:646.
24. Leder S, Czajkowska H, Maenz B, de GraafK, Barthel A, Joost H-G, Becker W.Alternative splicing variants of the proteinkinase DYRK1B exhibit distinct patterns ofexpression and functional properties.Biochem J 2003;372:881–8.
25. Collins CS, Hong J, Sapinoso L, Zhou Y,Liu Z, Micklash K, Schultz, PG, HamptonG. A small interfering RNA screen formodulators of tumor cell motility identifiesMAP4K4 as a promigratory kinase. PNAS2006;103:3775–80.
26. Lee K, Deng X, Friedman E. Mirk proteinkinase is a mitogen-activated proteinkinase substrate that mediates survival ofcolon cancer cells. Cancer Res 2000;60:3631–37.
27. Mercer SE, Ewton DZ, Shah S, Naqvi A,Friedman E. Mirk/Dyrk1b mediates cellsurvival in rhabdomyosarcomas. CancerRes 2006;66:5143–50.
28. Deng X, Ewton DZ, Li S, Naqvi A, MercerSE, Landas S, Friedman E. The kinaseMirk/Dyrk1B mediates cell survival inpancreatic ductal adenocarcinoma. CancerRes 2006;66:4149–58.
29. Deng X, Ewton DZ, Mercer SE, FriedmanE. Mirk/dyrk1B decreases the nuclearaccumulation of class II histonedeacetylases during skeletal muscledifferentiation. J Biol Chem 2005;280:4894–905.
30. Coller H, Sang L, Roberts JM. A newdescription of cellular quiescence. PLOSBiol 2006;4:329–49.
31. D’Andrilli G, Masciullo V, Bagella L,Tonini T, Minimo C, Zannoni GF,Giuntoli RL, II, Carlson JA, Jr, SopranoDR, Soprano KJ, Scambia G, Giordano A.Frequent loss of pRb2/p130 in humanovarian carcinoma. Clin Cancer Res 2004;10:3098–103.
32. Smith E, Leone G, Nevins J. Distinctmechanisms control the accumulation ofthe Rb-related p107 and p130 proteinsduring cell growth. Cell Growth Differ1998;9:297–303.
33. Kops GJPL, Dansen TB, Polderman PE,Saarloos I, Wirtz KWA, Coffer PJ, LamEW, Burgering F, Boudewijn MT.Forkhead transcription factor FOXO3aprotects quiescent cells from oxidativestress. Nature 2002;419:316.
34. Canhoto A, Chestukhin A, Litovchick L,DeCaprio JA. Phosphorylation of the
Can
cerCellBiology
Hu et al. 317
Int. J. Cancer: 129, 307–318 (2011) VC 2010 UICC

retinoblastoma-related protein p130 ingrowth-arrested cells. Oncogene 2000;19:5116–22.
35. Farkas T, Hansen K, Holm K, Lukas J,Bartek J. Distinct phosphorylation eventsregulate p130- and p107-mediatedrepression of E2F-4. J Biol Chem 2002;277:26741–52.
36. Smith EJ, Leone G, DeGregori J, Jakoi L,Nevins JR. The accumulation of an E2F-p130 transcriptional repressor distinguishesa G0 cell state from a G1 cell state.Mol Cell Biol 1996;16:6965–76.
37. Deng X, Ewton DZ, Pawlikowski B,Maimone M, Friedman E. Mirk/dyrk1B isa Rho-induced kinase active in skeletalmuscle differentiation. J Biol Chem 2003;278:41347–54.
38. Jin K, Ewton D, Park S, Hu J, Friedman E.Mirk regulates the exit of colon cancercells from quiescence. J Biol Chem 2009;284:22916–25.
39. Zou Y, Ewton D, Deng D, Mercer S,Friedman E. Mirk/dyrk1B kinasedestabilizes cyclin D1 by phosphorylationat threonine 288. J Biol Chem 2004;279:27790–98.
40. Thompson FH, Nelson MA, Trent JM,Guan X-Y, Liu Y, Yang J-M, Emerson J,
Adair L, Wymer J, Balfour C, Massey K,Weinstein R, Alberts DS, Taetle R.Amplification of 19q13.1-q13.2 sequencesin ovarian cancer: G-band, FISH, andmolecular studies. Cancer Genet Cytogenet1996;87:55.
41. Karhu R, Mahlamaki E, Kallioniemi A.Pancreatic adenocarcinoma-genetic portraitfrom chromosomes to microarrays.Genes Chromosomes Cancer 2006;45:721–30.
42. Moniaux N, Nemos C, Schmied BM,Chauhan SC, Deb S, Morikane K,Choudhury A, VanLith M, Sutherlin M,Sikela JM, Hollingsworth MA, Batra SK.The human homologue of the RNApolymerase II-associated factor 1 (hPaf1),localized on the 19q13 amplicon, isassociated with tumorigenesis. Oncogene2006;25:3247–57.
43. Kuuselo R, Savinainen K, Azorsa DO, BasuGD, Karhu R, Tuzmen S, Mousses S,Kallioniemi A. Intersex-like (IXL) is a cellsurvival regulator in pancreatic cancer with19q13 amplification. Cancer Res 2007;67:1943–49.
44. Cheng J, Godwin A, Bellacosa A, TaguchiT, Franke T, Hamilton T, Tsichlis PN,Testa JR. AKT2, a putative oncogene
encoding a member of a subfamily ofprotein-serine/threonine kinases, isamplified in human ovarian carcinomas.PNAS 1992;89:9267–71.
45. Hu J, Friedman E. Mirk kinase is a novelfactor mediating cisplatin resistance inovarian cancer cells. Genes Cancer, in press.
46. Moreno CS, Matyunina L, Dickerson EB,Schubert N, Bowen NJ, Logani S,Benigno BB, McDonald JF. Evidencethat p53 mediated cell-cycle-arrestinhibits chemotherapeutic treatment ofovarian carcinomas. PLoS ONE 2007;2:e441.
47. Shaw RJ. LKB1 and AMP-activated proteinkinase control of mTOR signalling andgrowth. Acta Physiol 2009;196:65–80.
48. Soprano KJ, Purev E, Vuocolo S, SopranoDR. Rb2/p130 and protein phosphatase 2A:key mediators of ovarian carcinoma cellgrowth suppression by all-trans retinoicacid. Oncogene 2006;25:5315.
49. Litovchick L, Sadasivam S, Florens L, ZhuX, Swanson SK, Velmurugan S, ChestukhinA, DeCaprio JA. Evolutionarily conservedmultisubunit RBL2/p130 and E2F4 proteincomplex represses human cell cycle-dependent genes in quiescence. Mol Cell2007;26:539.
Can
cerCellBiology
318 Mirk in ovarian cancer
Int. J. Cancer: 129, 307–318 (2011) VC 2010 UICC