theoretical analysis on magnetic properties of conjugated organic molecules containing borepin
TRANSCRIPT

Chem. Res. Chin. Univ. 2013, 29(5), 962—968 doi: 10.1007/s40242-013-3129-0
——————————— *Corresponding author. E-mail: [email protected] Received March 19, 2013; accepted March 27, 2013. Supported by the National Natural Science Foundation of China(Nos.21003057, 21173096) and the Specialized Research
Fund for the Doctoral Program of Higher Education of China(No.20110061110018). © Jilin University, The Editorial Department of Chemical Research in Chinese Universities and Springer-Verlag GmbH
Theoretical Analysis on Magnetic Properties of Conjugated Organic Molecules Containing Borepin
WEI Wei1, BAI Fu-quan1, XIA Bao-hui1,2, CHEN Hai-bo3 and ZHANG Hong-xing1* 1. State Key Laboratory of Theoretical and Computational Chemistry,
Institute of Theoretical Chemistry, 2. College of Chemistry,
Jilin University, Changchun 130021, P. R. China; 3. Jilin Provincial Institute of Education, Changchun 130023, P. R. China
Abstract Theoretical study about the magnetic properties of conjugated organic molecules containing borepin with π current density was carried out. 1-(2,4,6-Trimethylphenyl)borepin moiety is the center and other different groups are situated on the both β sides, which are named molecules 1—12 as theoretical model in order to establish the rela-tionship between aromaticity and geometry variation of borepin. The optimized molecular structures of molecules 1—12 are almost keeping planar and the C2—C3 bond length of borepin turns longer from molecule 1 to molecule 12. Different borepin-annulated ring could change the conjugated effect of π-electron between borepin and these bore-pin-annulated rings. Moreover, the molecule presents antiaromaticity, in other words, the molecule became unstable when the C2—C3 bond length of borepin extended more than ca. 0.1417 nm. But the β position fragment and substi-tuent groups of borepin are not affected in this case, they are still steady. However, the central borepin ring current is counteracted by symmetrical overlap of it with affiliated borepin-annulated ring current. Hence, the central borepin ring breaking would be liable to occur. These molecules have higher vertical ionization potentials(VIPs) and lower vertical electron affinities(VEAs), which suggests that these molecules could easily exist in anionic form. Keywords Aromaticity; Stability; Vertical ionization potential; Vertical electron affinity
1 Introduction
Boron-containing π-electron systems have received con-siderable attention in organic materials chemistry[1―3] because of their unique optical and structural properties which result from the vacant p-orbital of the boron center[4]. The organobo-ranes have been utilized for the selection of binding fluoride and cyanide anions. In fact, tricoordinated borons have been used to detect biologically or environmentally relevant anions[5]. The inherently electron-deficient in tricoordinated organobo-rane center makes these boron-containing molecules used as electron-accepting units[6]. Tricoordinated boron is introduced into organoborane systems via direct main-chain conjugation[7],
lateral substitution[8], or as an end-capping unit[6,9]. Furthermore, boron is a part of polycyclic aromatic framework(Scheme 1). Borepin(I) is a neutral seven-member heterocycle which is isoelectronic with the tropylium cation. The 6π-electron hete-rocycle borepin(II) is a Hückel-type aromatic compound like tropylium ion based on the experimental and theoretical re-searches. 1,3,5-Trimethylphenyl as the boron substituent could impart sufficient stability to borepin when borepin is exposed to the ambient environments. As reported by Ashe et al.[10], the synthesized substituted 1-(2,4,6-trimethylphenyl)borepin(III) is
moderately stable in air for a short period of time. The diben-zo[b,f]borepin(IV) ring system(DBB) framework has continued to attract considerably more scientific and technological research interests. Piers et al.[11] and Tovar et al.[12] investigated the DBB ring system, in order to better understand the local aromaticity of the central borepin ring. Nucleus-independent chemical shift(NICS) values proclaim that the borepin ring of benzo-annulated molecules[13] possesses a weak degree of aro-matic characteristics. The relative stability calculated by B3LYP shows that borepin is more stable than boranorborna-diene and boranorcaradiene[14]. Borepins can connect easily with other rings[15], which provide a durable influence on the
Scheme 1 Chemical structures of compounds I―IV Compound I: borepin; compound II: heterocycle borepin; compound III: 1-(2, 4, 6-trimethylphenyl) borepin; compound IV: diben-zo[b,f]borepin.

No.5 WEI Wei et al. 963
borepin core structure[16]. These borepin molecules exhibit strong blue fluorescence with rather high quantum yields in some cases, which suggests that borepin molecules have poten-tial application prospect in organic materials.
Aromaticity, a concept generally associated with organic compounds, can be characterized by geometric(bond length and bond order)[17―19], energetic(stabilization energies)[20,21], and magnetic[1H NMR chemical shifts, NICS, aromaticity ring chemical shielding(ARCS) and ring current indices] crite-ria[22―26]. In addition, the anisotropy of the induced current density(AICD)[27], gauge including magnetically induced cur-rent(GIMIC)[28], continuous transformation of origin of the current density(CTOCD)[29] and stagnation graphs[30] belong to the ring-current category. Although the history of this concept began with the isolation of benzene by Faraday in 1825[31], there has been no simple definition to explain what the aroma-ticity really is. The Hückel’s rule, that predicts the aromaticity for [4n+2] annulenes and antiaromaticity for [4n] annulenes lying in a plane, has been the most widely accepted notion for understanding aromaticity. As a topological complement fol-lowing the Hückel rule, Möbius aromaticity predicts that aro-matic characteristics will appear for [4n] annulenes when they are lying on a twisted Möbius strip[32]. Subsequently, nucleus independent chemical shift(NICS) proposed by Schleyer et al.[33] has become the most widely used aromaticity probe due to its simplicity and efficiency. Therefore, aromaticity is just a virtual quantity, rather than a physical observable one[34].
Aromaticity has provided one of the most fruitful interplays between theory and experiment[35]. Arguably, the special mag-netic behavior associated with induced ring currents in aromat-ic compounds is most closely related to the cyclic electron de-localization[36]. Aromaticity can reflect the molecule stability. In order to find out the relationship between aromaticity and geometric structure to serve this kind of molecular materials design, we compared different kinds of borepin molecules. The methods include the changes of the molecular symmetry, ske-letal structure and substituent.
In this work, one kind of borepin ring molecules is se-lected as the theoretical model, in which 1-(2,4,6- trimethylphenyl)borepin moiety is the center. Those rings, which can be preferably used to synthesize and change the molecular characteristics, are situated on both the sides of 1-(2,4,6-trimethylphenyl)borepin moiety. With the interest in the relationship between aromaticity and geometric structure of borepin, molecules 1—12 were chosen to deeply analyze the stability and activity via the NICS, natural bond orbital(NBO), vertical ionizatio potentials(VIPs) and vertical electron Aaffini-ties(VEAs) analyses. We hope these results would provide ideas for synthesizing novel molecules in the future.
2 Computational Details Full geometry optimizations were performed via the den-
sity functional theory(DFT) method based on Becke’s three parameter hybrid functional and gradient-corrected correlation functional of Lee et al.(B3LYP)[37]. The 6-31G(d) basis set was employed in the calculations. Vibrational frequencies, which
characterize all molecule structure as minima on the corres-ponding potential energy surfaces, were calculated at the same theory level. An NBO analysis[38] was also performed to pro-vide insights into the bond order at the B3LYP/6-31G(d) theory level. In our calculations, the energies of the ionic states and the energy differences of them with those of ground states for each molecule were calculated at the restricted open-shell B3LYP/6-31G(d) level.
The nucleus-independent chemical shifts(NICSs)[33] were calculated at the borepin ring center[NICS(0)], and at the points 0.05[NICS(0.05)], 0.15[NICS(0.15)], 0.25 nm[NICS(0.25)] above the borepin ring center and at the (3, +1) ring critical point of 1,3,5-trimethylphenyl associated with borepin- annulated ring, as well as onefold borepin ring center and the single benzene ring center by the Gauge-independent atomic orbital(GIAO) method at the B3LYP/6-31G(d) level. Within the orbital approximation, the total(first-order) current density for a closed shell system can be formally partitioned into molecular- orbital contributions. In the continuous set of gauge transfor-mation(CSGT) method[39] or later described as the continuous transformation of origin of current density diamagnetic zero (CTOCD-DZ) method[29,40], the current density at each point in the molecule was calculated by choosing itself as the origin of the vector potential. The significant advantage of the CTOCD-DZ method is that the total current density is naturally partitioned into molecular orbital contributions, allowing an efficient rationalization of different types of aromaticity(σ, π, etc.).
We presented the results as total density profile with an isosurface value of 0.23. The critical points were analyzed by means of the ‘atoms in molecules’ theory of Bader[41] via the AIM2000 package. All the calculations were achieved with Gaussian 09 program package[42].
3 Design Considerations As we all known, the properties of molecules can be
changed by symmetry, structural adjusting or substituent tuning. In order to better understand how these diverse annulated rings, substituent or symmetry modulation affect the aromaticity of the borepin ring, a series of conjugated organic molecules con-taining borepin ring are designed(see Scheme 2). The borepin, 2,4,6-trimethylphenyl and borepin-annulated rings are named A, B and C rings. The plane, bond length or substituent alternation more or less affects the properties of A ring. The symmetry constraint is applied to symmetrical structure to do geometry optimization with reduced symmetry even C1, and no manifest difference is cognized in bond distance movement. The aromaticity of the whole molecule is not affected by changing the symmetry under the almost identical current density of every molecule. With the aid of the relationship research between the aromaticity and the borepin geometry as well as the whole molecular structure, we found that molecules with analogous borepin structure have similar properties. In order to obtain the underlying principles, we selected molecules 1—12(structures are shown in Scheme 2) as research models.
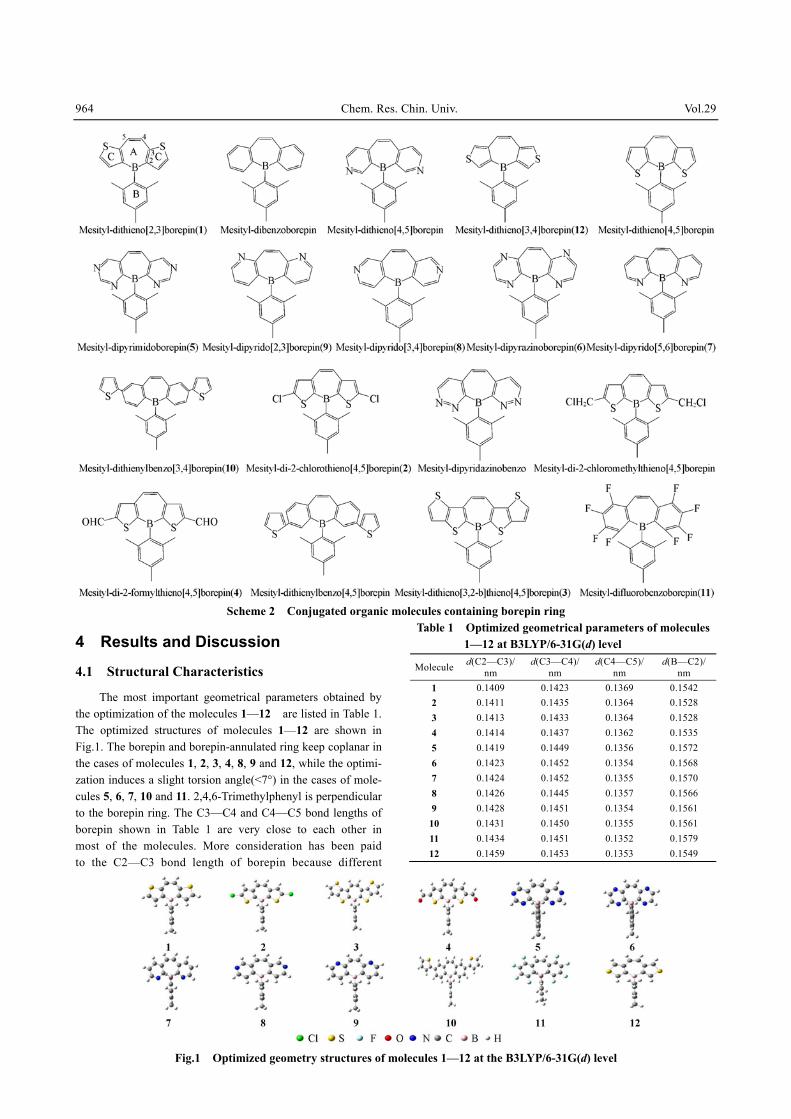
964 Chem. Res. Chin. Univ. Vol.29
Scheme 2 Conjugated organic molecules containing borepin ring
4 Results and Discussion
4.1 Structural Characteristics
The most important geometrical parameters obtained by the optimization of the molecules 1—12 are listed in Table 1. The optimized structures of molecules 1—12 are shown in Fig.1. The borepin and borepin-annulated ring keep coplanar in the cases of molecules 1, 2, 3, 4, 8, 9 and 12, while the optimi-zation induces a slight torsion angle(<7°) in the cases of mole-cules 5, 6, 7, 10 and 11. 2,4,6-Trimethylphenyl is perpendicular to the borepin ring. The C3—C4 and C4—C5 bond lengths of borepin shown in Table 1 are very close to each other in most of the molecules. More consideration has been paid to the C2—C3 bond length of borepin because different
Table 1 Optimized geometrical parameters of molecules 1—12 at B3LYP/6-31G(d) level
Molecule d(C2—C3)/nm
d(C3—C4)/ nm
d(C4—C5)/ nm
d(B—C2)/nm
1 0.1409 0.1423 0.1369 0.1542 2 0.1411 0.1435 0.1364 0.1528 3 0.1413 0.1433 0.1364 0.1528 4 0.1414 0.1437 0.1362 0.1535 5 0.1419 0.1449 0.1356 0.1572 6 0.1423 0.1452 0.1354 0.1568 7 0.1424 0.1452 0.1355 0.1570 8 0.1426 0.1445 0.1357 0.1566 9 0.1428 0.1451 0.1354 0.1561
10 0.1431 0.1450 0.1355 0.1561 11 0.1434 0.1451 0.1352 0.1579 12 0.1459 0.1453 0.1353 0.1549
Fig.1 Optimized geometry structures of molecules 1—12 at the B3LYP/6-31G(d) level

No.5 WEI Wei et al. 965 borepinannulated rings mainly affect the C2—C3 bond length of borepin. The C2—C3 bond length of borepin turns longer from molecule 1 to molecule 12. For example, the bond length of C2—C3 is 0.1409 nm in molecule 1, which is 0.0002 nm shorter than that in molecule 2. However, the calculated C2—C3 bond length changes conspicuously between mole-cules 4 and 5(0.1414 nm for molecule 4, 0.1419 nm for mole-cule 5). This is mainly due to the introduction of nitro-gen-containing ring which reduces the conjugated gated effect of π-electron. Moreover, the C2—C3 bond lengths of mole-cules 11 and 12 have the most variation, and the difference is 0.0025 nm. For molecule 12, the thiophene ring and borepin core display less π-electron conjugated effect, which makes the C2—C3 bond length longer. The B—C2 bond length is longer than C—C bond length of borepin for molecules 1—12, but there is no obvious change from molecule 1 to molecule 12. Boron atom plays a steady effect on B—C bond index and charge distribution.
4.2 Aromaticity
NICS is based on the negative value of the magnetic shielding computed at the geometrical center or at a point above the molecular plane. System with a significant negative NICS value is aromaticity, conversely, it is antiaromaticity. Nonaromaticity cyclic systems have NICS values of zero. The more positive or more negative NICS value means the stronger system antiaromaticity or aromaticity. In this study, the NICS values are evaluated with the GIAO-B3LYP/6-31G(d) method. To assess their aromatic characteristics, NICS values at the A ring center[NICS(0)], 0.05 nm[NICS(0.05)], 0.15 nm [NICS(0.15)], 0.25 nm[NICS(0.25)] above the A ring, and at the B and C ring centers, are obtained(see Table 2).
The C2—C3 bond length increases gradually from mole-cule 1 to molecule 12(see Table 1). The NICS values of A ring at the ring critical points[NICS(0)] in molecules 1(δ –2.3), 2(δ –3.3), 3(δ –2.9), and 4(δ –2.2) are negative, which denote
Table 2 NICS(δ) at A ring center NICS(0) and at points of 0.05[NICS(0.05)], 0.15[NICS(0.15)] and 0.25 nm[NICS(0.25)] above the center of A ring, and at B and C ring centers[B ring NICS(0) and C ring NICS(0)] predicted for mole-cules 1—12 at the B3LYP/6-31G(d) level
Molecule δ
A ring B ring C ring NICS(0) NICS(0.05) NICS(0.15) NICS(0.25) NiCS(0) NiCS(0)
1 –2.3 –3.6 –4.3 –2.3 –8.3 –11.4 2 –3.3 –4.5 –4.6 –2.4 –8.5 –11.7 3 –2.9 –4.1 –4.3 –2.3 –8.3 –10.1 4 –2.2 –3.5 –4.2 –2.3 –8.4 –11.1 5 1.9 –0.8 –2.0 –2.3 –8.7 –6.1 6 0.9 –1.7 –3.1 –0.8 –8.7 –6.1 7 1.1 –1.2 –3.2 –1.4 –8.7 –7.8 8 0.3 –2.1 –3.2 –1.1 –8.0 –7.9 9 0.5 –2.0 –3.0 –1.0 –7.9 –7.8
10 1.0 –0.5 –2.6 –1.8 –7.9 –8.4 11 0.5 –0.8 –2.8 –1.8 –8.8 –14.3 12 3.6 2.3 –0.7 –1.0 –8.3 –11.8
aromaticity. The NICS values of A ring at the ring critical points[NICS(0)] in molecules 5(δ 1.9), 6(δ 0.9), 7(δ 1.1), 8(δ 0.3), 9(δ 0.5), 10(δ 1.0), 11(δ 0.5), and 12(δ 3.6) are positive. It indicates that they possess near antiaromaticity. The calculated results(as listed in Table 2) show that NICS(0) values of B and C rings for all the molecules are negative, which implies that they are aromaticity. The NICS values at the points of 0.05[NICS(0.05)], 0.15[NICS(0.15)] and 0.25 nm[NICS(0.25)] above the center of A ring for most of the molecules are nega-tive except that the NICS(0.05) of A ring of molecule 12 is positive. The relationship between C2—C3 bond length and magnetic index is presented in Fig.2. Fig.2 shows that the NICS values are negative when the C2—C3 bond length of A ring is not more than 0.1414 nm, which demonstrates aromaticity. However, when the C2—C3 bond length of A ring is not less than 0.1419 nm, the NICS values are positive. Moreover, the NICS value of A ring center is zero when the C2—C3 bond length is almost equal to 0.1417 nm. In other words, that the C2—C3 bond length of A ring is 0.1417 nm denotes nonaroma-ticity. These results show that different borepin-annulated rings could change the conjugating effect of π-electron between
Fig.2 Change of molecular NICS values(δ) based on
the different C2—C3 bond lengths a. A ring NICS(0); b. A ring NICS(0.05); c. A ring NICS(0.15); d. A ring NICS(0.25); e. B ring NICS(0); f. C ring NICS(0). Twelve data points represent molecules 1―12 from left to right, respectively. borepin and borepin-annulated rings. And then the aromaticity of borepin can be changed via changing the C2—C3 bond length. Furthermore, the NICS values of B ring for molecules 1 to 12 are about δ –8 which doesn’t change significantly with the increase of C2—C3 bond length. It indicates that the

966 Chem. Res. Chin. Univ. Vol.29
aromaticity of 2,4,6-trimethylphenyl isn’t affected by different borepin-annulated rings and main borepin structure modulating. The NICS values of C ring for molecules 1—12 are negative. This shows that they are aromatic molecules. In addition, NICS value of C ring for molecule 11 shows stronger aromaticity (δ –14.33), which is mainly due to the strong and localization effect of fluorine atom. For molecules 1, 2, 3 and 4, the NICS value at the point of 0.15 nm above A ring center is more than the NICS values at the points of 0.05 and 0.25 nm above A ring(in absolute value). The whole molecule is based on the A ring. So the antiaromaticity of A ring indicates the instability of the entire molecule. The borepin ring is easily to breach. Otherwise it exists in other forms. We found that the C ring current is stable due to the NICS values are rarely changed with the increase of C2—C3 bond length. Furthermore, when the C2—C3 bond length is more than 0.1419 nm but less than 0.1459 nm, the NICS(0.15) value(in absolute value) is larger than NICS(0.05) value and NICS(0.25) value at the corres-pongding points of 0.05 and 0.25 nm above A ring center. In other words, when the critical point is at the point of 0.15 nm above the A ring, NICS value becomes the largest. It is due to the highly sensitive to the distance which is from the conju-gated π orbital of the C and B atoms to the C and B cores. Spe-cially, the NICS(0.05) value at the point of 0.05 nm above A ring for molecule 12 is positive, which shows antiaromaticity. This π current density of π orbital is not destroyed till the NICS(0.05) at the point of 0.05 nm above A ring with C2—C3 bond elongating to 0.1459 nm. It can be concluded that the stability of current density from molecular vertical conjugated π orbital(paratropic) is better than that from crowded π orbi- tal[NICS(0)](diatropic) with some σ orbital contribution. Elec-tron delocalization was assessed by magnetic criteria and current density in cyclic molecules[43]. The calculated results show that the change of substituent(especially for obvious
comparison between molecules 2 and 4) can alter the aroma- ticity of molecules.
NBOs analysis uses one electron density matrix to define the shape of the atomic orbital in the molecular environment, and to drive molecular bonds based on electron density be-tween atoms[44]. It is helpful to have an index that connects quantum mechanical results with the common chemical intui-tion in the theoretical study of molecules, facilitating the ex-traction of chemical knowledge from calculated results[45]. The natural bond order index, based on the natural resonance theory procedure and a part of the natural bond orbital analysis tools, has been proved to yield reliable results for various systems[46].
The bond order indexes were used to analyze the bond charac-ters in this work. As bond length is longer, bond order is small-er. The bond orders of C2—C3 in borepin are presented in Ta-ble 3. The bond order of C2—C3 in molecules 1—12 turns smaller from molecule 1 to molecule 12, while the C2—C3 bond length gets longer from molecule 1 to molecule 12(see Table 3). The bond orders for additional C―C bond are almost changeless for molecules 1—12. The magnitude and the direc-tion of the current density induced by the mobile π electrons in a magnetic field for molecules 1—12 are presented in Fig.3. The direction of magnetic field is perpendicular to the molecu-lar plane. Aromatic ring currents are related to NMR spectrum, as they dramatically influence the chemical shifts of 13C and 1H nuclei in aromatic molecules, as well as in any organic or inor-ganic aromatic molecules. This diminution in the interaction between the C2 and C3 atoms in borepin from molecule 1 to molecule 12 is clearly reflected in the current density map. Fig.3 shows that the presence of different borepin-annulated rings arouses a decrease in the current density via C2 and C3 atoms from molecule 1 to molecule 12 due to their lower mo-bility. It is concluded that the C2—C3 bond is more easy to break when the entire neutral molecule becomes instability.
Table 3 NBO bond order for C2—C3 in borepin at B3LYP/6-31G(d) level Molecule 1 2 3 4 5 6 7 8 9 10 11 12
Bond order 1.372 1.364 1.359 1.348 1.316 1.307 1.305 1.292 1.291 1.276 1.274 1.156
Fig.3 Current density maps for the π electron distribution of molecules 1—12 The NICS values for neat borepin ring(δ –4.6) and ben-
zene ring(δ –9.7) are bigger than corresponding one of A and B rings(and C ring of molecules 10 and 11) of the molecules(in absolute value), respectively. More importantly, the NICS va- lues are all negative when the C2—C3 bond length of neat borepin ring is from 0.1367 nm to 0.1587 nm. The current den-sities of neat borepin with C2—C3 bond lengths of 0.1367, 0.1430 and 0.1587 nm are shown in Fig.4. We can see that the
Fig.4 Current density of neat borepin at C2—C3 bond lengths of 0.1367(A), 0.1430(B) and 0.1587 nm(C)

No.5 WEI Wei et al. 967
current density of C2―C3 bond decreased with the increase of the C2―C3 bond length. But this molecule still keeps its aro-maticity. Therefore, the A ring current factually affected by C ring current for molecules 1 to 12. In other words, the A ring current is counteracted by the symmetrical overlap of it with the C ring current. Moreover, the binate C ring currents show the opposite direction to each other. This is consistent with electronegativity to a certain extent.
4.3 Vertical Ionization Potentials and Vertical Electron Affinities
It is important to investigate the ionization potentials and electron affinities, which can be used to estimate the ener-gy barrier for the injection of holes and electrons, into the molecular layers and the other more stable forms. Ionization
potential(IP) is required to detach an electron from a neutral molecule, while electron affinity(EA) is defined as the differ-ence between the energies of the lowest states of the neutral and the corresponding anion. IPs and EAs are commonly cal-culated via the differences of total energies of the neutral(Eneutral) and ionic systems(Ecation and Eanion) obtained from optimized molecule configuration. The vertical IPs and EAs obtained from absolute energies at ROB3LYP/6-31G(d) level are listed in Table 4. The values of vertical IPs and EAs of molecules 1—12 demonstrate that they are easily to reduce but difficult to oxidize. So even the molecules are stable in ground state, they are easily to reduce or their C―C bond breaking would present under befitting conditions. The density diagram plots of HOMO orbital of cation and anion are depicted in Fig.5.
Table 4 Vertical ionization potentials(VIP, eV) and vertical electron affinities(VEA, eV) for molecules 1—12 at the ROB3LYP/6-31G(d) level
Molecule 1 2 3 4 5 6 7 8 9 10 11 12 VIP 7.13 7.55 7.10 7.77 7.57 7.42 7.75 7.74 7.74 7.00 7.75 7.10 VEA 0.26 0.49 0.55 1.48 1.19 1.00 0.51 0.92 0.56 0.80 0.92 0.18
Fig.5 Density diagram plots of HOMO orbital of cations(A) and anions(B) calculated at the ROB3LYP/6-31G(d) level
Fig.5 shows that the HOMO’s of cation for molecules 3, 6 and 8 only spread over the borepin and borepin-annulated rings. From Table 4, the ionization potential of molecule 3 is lower than those of molecules 6 and 8, which indicates that molecule 3 can easily lose an electron to create a hole. While for mole-cules 10 and 12, the HOMO’s of cation contain the borepin and borepin-annulated rings but not contain boron atom. The VIP of molecule 12(7.10 eV) is bigger than that of molecule 10(7.00 eV). For molecules 1, 2, 4, 5, 7, 9, 11, the HOMOs of cation spread over the whole molecule. For molecules 1 and 2, 2,4,6- trimethylphenyl accounts for 25% and 32%, respectively. However, for molecules 4, 5, 7, 9, 11, 2,4,6-trimethylphenyl accounts for the main part(>70%). VIPs for molecules 1 and 2 are 7.13 and 7.55 eV, respectively. In addition, for molecules 4, 7, 9 and 11, the VIPs are virtually 7.75 eV. Generally, when 2,4,6-trimethylphenyl contribute more to HOMO, the VIPs
value is bigger. The HOMO’s of anion for molecules 6 and 8 displaying bonding character in the borepin and borepin- annulated ring areas are labeled in Fig.5. The VEA for mole-cule 6(1.00 eV) is bigger than that for molecule 8(0.92 eV), which means that molecule 6 is apt to accept an electron com-pared with molecule 8. While for molecules 1, 2, 3, 4, 5, 7, 9, 10, 11, 12, HOMO’s of anion show antibonding character be-tween borepin and borepin-annulated rings. Specially, for mo-lecule 1, the HOMO of anion doesn’t contain boron atom, and the VEA is 0.26 eV. For molecule 12, the HOMO of anion doesn’t contain C3 and C4 atoms of borepin. For molecules 2 and 3, the boron atom contributes 17% and 16%, respectively to HOMO. The VEAs for molecules 2 and 3 are 0.49 and 0.55 eV, respectively. For molecules 4, 5, 7, 9, 10, 11, the boron atom accounts for 13%, 29%, 29%, 28%, 18%, 21%, respec-tively, and the VEA values are 1.48, 1.19, 0.51, 0.56, 0.80 and

968 Chem. Res. Chin. Univ. Vol.29
0.92 eV, respectively. This series of molecules possess different electronic configurations with varied C2—C3 bond length. The VIP and VEA become erratic due to different molecular frontier orbital arrangement and the oxidation and reduction occurred in different location. Different from aromaticity related with cur-rent density, VIP and VEA results can give more information on the difficulty of ionization process.
5 Conclusions We have investigated a series of conjugated organic mo-
lecules 1—12 containing borepin ring. Different borepin- annulated rings could change the conjugating effect of π-electrons between borepin and borepin-annulated rings. This is the direct result of the C2—C3 bond length increase gra- dually from molecule 1 to molecule 12. We drew out the rela-tionship between aromaticity and geometric structure. From the changes in aromaticity and bond length, when the C2—C3 bond length of borepin is longer than ca. 0.1417 nm, the neutral molecules will be hard to maintain their stabilities. The NICS values are negative when the C2—C3 bond length of borepin is not more than 0.1414 nm, which reflects the stability of borepin. Meanwhile, when the C2—C3 bond length of borepin is longer than 0.1417 nm, the NICS values are positive. The values of vertical IPs and vertical EAs of the molecules 1—12 demon-strate that they are more comfortable to exist in anionic form. From NBO analysis, the bond order of C2—C3 in molecules 1—12 turns smaller from molecule 1 to molecule 12, corres-ponding to the C2—C3 bond length of borepin getting longer from molecule 1 to molecule 12. The current density map shows that the presence of different borepin-annulated rings arouses a definite decrease in current density through C2 and C3 atoms from molecule 1 to molecule 12. The central borepin ring current is counteracted by symmetrical overlap of it with affiliated borepin-annulated ring current. Hence, we expect that the result of this work can provide help to designing and syn-thesizing new molecules based on boron ring.
References
[1] Li H., Jäkle F., Angew. Chem. Int. Ed., 2009, 48, 2313 [2] Cheng S. H., Fan F. Y., Xu Y., Li S., Zhu P. W., Li H. D., Liu J. S.,
Chem. Res. Chinese Universities, 2013, 29(4), 816 [3] Tan M., Lian G., Zhong X., Zhang S. J., Cui D. L., Wang Q. L.,
Chem. Res. Chinese Universities, 2012, 28(3), 387 [4] Jakle F., Chem. Rev., 2010, 110, 3985 [5] Hudnall T. W., Gabbai F. P., J. Am. Chem. Soc., 2007, 129, 11978 [6] Liu X. Y., Bai D. R., Wang S., Angew. Chem. Int. Ed., 2006, 45, 5475 [7] Matsumi N., Naka K., Chujo Y., J. Am. Chem. Soc., 1998, 120, 10776 [8] Nagai A., Kokado K., Nagata Y., Chujo Y., Macromolecules, 2008,
41, 8295 [9] Zhou G., Baumgarten M., Müllen K., J. Am. Chem. Soc., 2008, 130,
12477 [10] Ashe III A. J., Klein W., Rousseau R., Organometallics, 1993, 12,
3225 [11] Mercier L. G., Piers W. E., Parvez M., Angew. Chem. Int. Ed., 2009,
48, 6108 [12] Caruso A. Jr., Siegler M. A., Tovar J. D., Angew. Chem. Int. Ed.,
2010, 49, 4213 [13] Subramanian G., Schleyer P. V. R., Jiao H. J., Organometallics, 1997,
16, 2362 [14] Kassaee M. Z., Musavi S. M., Motamedi E., J. Theor. Comput.
Chem., 2010, 9, 379 [15] Jinguji A., Nakazawa R., Yagi T., Murata I., Tetrahedron, 1994, 50,
6495 [16] Schulman J. M., Disch R. L., Organometallics, 2000, 19, 2932 [17] Herndon W. C., J. Am. Chem. Soc., 1973, 95, 2404 [18] Aihara J., J. Org. Chem., 1976, 41, 2488 [19] Jug K., J. Org. Chem., 1983, 48, 1344 [20] Dewar M. J. S., de Llano C., J. Am. Chem. Soc., 1969, 91, 789 [21] Hess B. A. Jr., Schaad L. J., J. Am. Chem. Soc., 1971, 93, 305 [22] Pople J. A., J. Chem. Phys., 1956, 24, 1111 [23] Dauben H. J. Jr., Wilson J. D., Laity J. L., J. Am. Chem. Soc., 1969,
91, 1991 [24] Benson R. C., Flygare W. H., J. Am. Chem. Soc., 1970, 92, 7523 [25] Jusélius J., Sundholm D., Phys. Chem. Chem. Phys., 1999, 1, 3429 [26] Morao I., Lecea B., Cossío F. P., J. Org. Chem., 1997, 62, 7033 [27] Geuenich D., Hess K., Köhler F., Herges R., Chem. Rev., 2005, 105,
3758 [28] Jusélius J., Sundholm D., Gauss J., J. Chem. Phys., 2004, 121, 3952 [29] Lazzeretti P., Malagoli M., Zanasi R., Chem. Phys. Lett., 1994, 220,
299 [30] Pelloni S., Lazzeretti P., Int. J. Quantum Chem., 2011, 111, 356 [31] Faraday M., Philos. Trans. R. London, 1825, 115, 440 [32] Rzepa H. S., Chem. Rev., 2005, 105, 3697 [33] Schleyer P. V. R., Maerker C., Dransfeld A., Jiao H. J., Hommes N. J.
R. V. E., J. Am. Chem. Soc., 1996, 118, 6317 [34] Chen Z. F., Wannere C. S., Corminboeuf C., Puchta R., Schleyer P. V.,
Chem. Rev., 2005, 105, 3842 [35] Bleeke J. R., Chem. Rev., 2001, 101, 1205 [36] Schleyer P. V., Chem. Rev., 2001, 101, 1115 [37] Becke A. D., Phys. Rev. A, 1988, 38, 3098 [38] Reed A. E., Curtiss L. A., Weinhold F., Chem. Rev., 1988, 88, 899 [39] Keith T. A., Bader R. F. W., J. Chem. Phys., 1993, 99, 3669 [40] Zanasi R., J. Chem. Phys., 1996, 105, 1460 [41] Bader R. F. W., Atoms in Molecules: A Quantum Theory, Clarendon
Press, Oxford, 1990 [42] Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M.
A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Peters-son G. A., et al., Gaussian 09, Revision A.02, Gaussian Inc., Wal-lingford CT, 2009
[43] Heine T., Corminboeuf C., Seifert G., Chem. Rev., 2005, 105, 3889 [44] Li X. H., Yin G. X., Zhang X. Z., J. Mol. Struct.(Theochem.), 2010,
957, 61 [45] Becke A. D., Edgekombe K. E., J. Chem. Phys., 1990, 92, 5397 [46] Raúl M. A., Fernando M., Claudio O. A., Sebastián M. R., Patricio F.,
J. Phys. Chem. A, 2011, 115, 4397