· the work described in this thesis was carried out at stratingh intitute for chemistry,...
TRANSCRIPT

University of Groningen
Asymmetric copper-catalyzed alkylations and autocatalysisPellegrini, Tilde
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2019
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Pellegrini, T. (2019). Asymmetric copper-catalyzed alkylations and autocatalysis. [Groningen]: University ofGroningen.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 03-05-2020

Asymmetric copper-catalyzed alkylations and autocatalysis
Tilde Pellegrini

The work described in this thesis was carried out at Stratingh Intitute for Chemistry,
University of Groningen (The Netherlands)
This work was financially supported by Ministry of Education, Culture and Science
(Gravitation program 024.001.035) and NWO
Printed by Ridderprint BV, Ridderkerk, The Netherlands
Cover picture by Giulia Leonetti
Baracoa (Cuba), 2015
ISBN: 978-94-6375-291-6 (Printed Book)
ISBN: 978-94-034-1429-4 (Ebook)

Asymmetric copper-catalyzed alkylations and autocatalysis
PhD thesis
to obtain the degree of PhD at the University of Groningen on the authority of the
Rector Magnificus prof. E. Sterken and in accordance with
the decision by the College of Deans.
This thesis will be defended in public on
Friday 8 March 2019 at 12.45 hours
by
Tilde Pellegrini
born on 10 August 1989 in Florence, Italy

Supervisors Prof. S.R. Harutyunyan
Prof. W.R. Browne
Assessment Committee Prof. A.J. Minnaard
Prof. M. Pineschi
Prof. J.H. van Maarseveen


Table of content List of abbreviations ........................................................................................................ 1
Chapter 1: Introduction .................................................................................................. 3
1.1. Catalysis in asymmetric syntheses ....................................................................... 4
1.2. Asymmetric metallic catalysis .............................................................................. 4
1.3. Asymmetric organocatalysis ................................................................................ 5
1.4. Catalytic (dynamic) kinetic resolution ................................................................. 6
1.5. Non-linear effects in asymmetric catalysis .......................................................... 8
1.5.1. The model ML2 or (ML)2 ............................................................................... 9
1.6. Thesis outline...................................................................................................... 10
1.7. Bibliography ........................................................................................................ 11
Chapter 2: Control of enantioselectivity in the addition of Grignard reagents to
symmetric heteroaryl disubstituted olefins .................................................................. 13
2.1. Introduction ......................................................................................................... 14
2.1.1. Asymmetric addition of organometallic reagents to electron deficient
olefins ....................................................................................................................... 14
2.1.2. Enantioselectivity in the 1,4-addition of nucleophiles to symmetric
disubstituted alkenes ................................................................................................. 15
2.1.3. Copper(I)-catalyzed asymmetric addition of Grignard reagents to (N)-
containing heteroaryl alkenes ....................................................................................19
2.2. Aim ....................................................................................................................... 21
2.3. Results and discussion ........................................................................................ 21
2.3.1. Synthesis of 1,2-disubstituted heteroaryl alkenes ........................................ 21
2.3.2. ACA of Grignard reagents to benzoxazyl alkenes ....................................... 25
2.3.3. ACA of Grignard reagents to symmetric 2-quinoyl alkenes ........................ 31
2.4. Conclusions ......................................................................................................... 32
2.5. Experimental section .......................................................................................... 33
2.5.1. General information .................................................................................... 33
2.5.2. Synthesis of substrates ................................................................................ 34
2.5.3. Catalytic asymmetric addition to 16 ............................................................ 35
2.5.4. Complexes CuBr·L10 and CuBr·L11 ............................................................ 38
2.6. Bibliography ....................................................................................................... 39
Chapter 3: Asymmetric conjugate addition of Grignard reagents to symmetric
bispyridyl alkenes ......................................................................................................... 43

3.1. Introduction ........................................................................................................ 44
3.1.1. Importance of pyridines in medicinal chemistry ........................................ 44
3.1.2. Utilization of bispyridyl compounds in chemistry ...................................... 45
3.1.3. Asymmetric conjugate addition to vinyl pyridines ..................................... 48
3.2. Aim ....................................................................................................................... 51
3.3. Results and discussion ........................................................................................ 51
3.3.1. Asymmetric addition to symmetric 4-pyridyl alkenes ................................. 51
3.3.2. Asymmetric addition to symmetric 2-pyridyl alkenes ................................ 58
3.3.3. Selectivity in the addition to 4-pyridyl, 2-pyridyl and 2-benzoxazyl alkenes
.......................................................................................................................61
3.3.4. Interaction of 1,2-bis(4-pyridyl)ethene (19) with TMSBr ........................... 65
3.4. Conclusions ......................................................................................................... 67
3.5. Experimental section .......................................................................................... 67
3.5.1. General information .................................................................................... 67
3.5.2. Synthesis of substrates ................................................................................ 68
3.5.3. General procedure for the synthesis of 2-(benzoxazol-2-yl)-1-
(pyridinyl)ethan-1-ol ................................................................................................. 69
3.5.4. General procedure for the synthesis of (E)-2-
((pyridinyl)vinyl)benzoxazoles ................................................................................. 70
3.5.5. General procedure for the asymmetric addition to 19 ................................ 70
3.5.6. General procedure for the asymmetric addition to 22 ................................ 73
3.5.7. General procedure for the racemic addition to 22 ...................................... 74
3.5.8. General procedure for the asymmetric addition to 25-27 .......................... 76
3.5.9. Procedure for the NMR studies about interaction between catalyst, 19 and
TMSOTf ......................................................................................................................77
3.5.10. Complexes CuBr·L7 and CuBr·L8 ............................................................ 78
3.6. Bibliography ....................................................................................................... 79
Chapter 4: Autoinductive effects in an asymmetric copper(I)/phosphine catalyzed
reaction ......................................................................................................................... 83
4.1. Introduction ........................................................................................................ 84
4.1.1. Asymmetric autoinduction .......................................................................... 84
4.1.2. Chiral tertiary alcohols ................................................................................ 89
4.2. Aim .......................................................................................................................91
4.3. Results .................................................................................................................91

4.3.1. Enantioselectivity as a function of conversion of the starting material in
1,2-additions of Grignard reagents to carbonyls .......................................................91
4.3.2. Autocatalysis or autoinduction? .................................................................. 94
4.3.3. Asymmetric autoinductive effects: ketones vs. aldehydes .......................... 96
4.3.4. Interaction of an alkoxide with a copper/phosphine complex ................... 98
4.4. Conclusions ....................................................................................................... 102
4.5. Experimental section ........................................................................................ 103
4.5.1. General information .................................................................................. 103
4.5.2. General procedure for the 1,2-addition of Grignard reagents to ketones
(24a,b) .................................................................................................................... 103
4.5.3. General procedure for the 1,2-addition of Grignard reagents to aldehydes
(37a,b) .................................................................................................................... 105
4.5.4. General procedure for monitoring the ee of the product of the reaction . 107
4.5.5. General procedure for the 1,2-addition of Grignard reagents to aldehydes
with the use of additives ......................................................................................... 107
4.5.6. General procedure for the reaction carried out with different Grignard
reagents ................................................................................................................... 107
4.5.7. Procedure for the NMR experiments ........................................................ 108
4.6. Bibliography ..................................................................................................... 109
Chapter 5: Design of an asymmetric organic autocatalytic reaction: the reduction of
ketones and imines with borane ................................................................................... 83
5.1. Introduction .......................................................................................................... 114
5.1.1. Autocatalysis ............................................................................................... 114
5.1.2. Asymmetric autocatalysis ........................................................................... 118
5.1.3. Corey-Bakshi-Shibata reduction and feasibility of asymmetric
autocatalysis…………………………………….…………………………………………………………122
5.2. Aim ................................................................................................................... 124
5.3. Results and discussion ..................................................................................... 124
5.3.1. The Imine Pathway ..................................................................................... 125
5.3.2. The Ketone Pathway .................................................................................. 130
5.3.3. Reduction of the phenyl-(2pyridyl)-ketone .............................................. 130
5.4. Conclusions ........................................................................................................ 133
5.5. Experimental section ........................................................................................ 134
5.5.1. General information .................................................................................. 134
5.5.2. Synthesis of N-Fmoc-38 ............................................................................. 135

5.5.3. Synthesis of the imino-ketone 36 ............................................................... 135
5.5.4. Procedure for the synthesis of 30 .............................................................. 136
5.5.5. Procedure for the reduction of 30 ............................................................. 136
5.5.6. Racemic synthesis of the tert-butyl (S)-2-((R)-
hydroxy(phenyl)methyl)pyrrolidine-1-carboxylate................................................. 137
5.5.7. Asymmetric synthesis of the tert-butyl-2-((R)-
hydroxy(phenyl)methyl)pyrrolidine-1-carboxylate................................................ 140
5.5.8. General procedure for the deprotection of Boc-pyrrolidines ................... 140
5.5.9. General procedure for the reduction of 47 ................................................ 140
5.5.10. CBS-Reduction of 49 .................................................................................. 141
5.6. Bibliography ...................................................................................................... 141
Summary ..................................................................................................................... 146
Samenvatting…………………………………………………………………………………………………150
Aknowledgements…………………………………………………………………………………………..153


1
List of abbreviations ACA: asymmetric conjugate addition
API: active pharmaceutical ingredient
BINAP: 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl
BINOL: 1,1’-bi-2-naphtol
Boc: tert-butyl carbamate
CA: conjugate addition
CBS reduction: Corey-Bakshi-Shibata reduction
CSP-HPLC: chiral solid phase high performance liquid chromatography
DMAE: 2-dimethylaminoethanol
ee: enantiomeric excess
EWG: electron withdrawing group
GC/MS: gas chromatography/mass spectroscopy
HMBC: heteronuclear multiple bond correlation spectroscopy
HMPA; hexamethylphosphoramide
HRMS: high resolution mass spectroscopy
HSQC: heteronuclear single quantum coherence spectroscopy
LA: Lewis Acid
M: metal
MTBE: methyl-tert-butylether
NMR: nuclear magnetic resonance
o.n.: overnight
rt: room temperature
TFA: trifluoroacetic acid
THF: tetrahydrofurane
TLC: thin layer chromatography

2
TM: transition metal
TMEDA: tetramethylethylendiamine
TMSBr: trimethylsilyl bromide
TMSCl: trimethylsilyl chloride
TMSOTf: trimethylsilyl trifluorosulfonate

Chapter 1
Introduction Herein, asymmetric catalysis is introduced. After a general explanation of the
asymmetric syntheses, metal-catalysis and organocatalysis are explained and
examples are given. This introduction touches the topics of kinetic resolution and
dynamic kinetic resolution to obtain enantioenriched compounds. Finally, we
describe asymmetric amplification as a peculiar case of asymmetric catalysis.

Chapter 1
4
1.1. Catalysis in asymmetric syntheses Symmetry is beauty. Symmetric faces result more attractive to people than asymmetric
ones.[1] However, synthetic organic chemists are rather attracted to asymmetry in
molecules. In fact, nature is an efficient asymmetric selector and homochiral
molecules, for example, amino acids and sugars, compose living beings. Consequently,
biological systems will interact differently with the two enantiomers and numerous
pharmacological substances require to be composed by a single enantiomer to be
administered to patients.[2]
Separations of enantiomers from racemic mixtures are commonly used, but their
efficiency is limited by the fact than only half of the product can be utilized.[3–5] For this
reason, copious methods have been developed to obtain enantiopure compounds. The
synthesis with chiral auxiliaries affords enantioenriched products, nevertheless it
requires stoichiometric amount of chiral reagents that are often expensive and require
to be separated from the product.[6] Due to the convenience of using chiral auxiliaries
in substoichiometric amount, enantioselective catalysis was widely developed during
last century.[7,8]
1.2. Asymmetric metallic catalysis Up to date, metallic catalysts are broadly used, both in laboratories and industrial
processes.[9] In fact, these transformations usually require low catalyst loadings (less
than 10 mol%) and are easily tunable thanks to the possibility to vary the metal and to
modify the ligand, carrier of the chiral information.[10,11]
In 2001, the Nobel Prize for chemistry was awarded to Knowles and Noyori
(hydrogenations) and Sharpless (oxidations) for their contribution in asymmetric
catalysis. The group of Knowles developed a convenient synthesis of (L)-DOPA, an
amino acid employed in Parkinson Disease’s treatment via the enantioselective
hydrogenation of 1 with a Rh/chiral phosphine complex (Scheme 1a).[12,13] Noyori et
al. extended the scope of this reaction by using an atropoisomeric phosphine ligand
for the rhodium (BINAP, L2, Scheme 1b).[14,15] On the other hand, asymmetric
oxidations were established by Sharpless and coworkes: among others, the epoxidation
of allylic alcohol by organic peroxides could be performed with Ti(Oi-Pr)4 and diethyl
tartrate (L3) as chiral additive. The use of the natural enantiomer (L)-L3 would have
allowed the oxidation of the double bond from the enantiotopic face below (as drawn
in Scheme 1c), independently from the substitution pattern.[16,17]

Introduction
5
Scheme 1 Asymmetric reductions and oxidations.
1.3. Asymmetric organocatalysis Asymmetric metal-catalysis has enabled the creation of stereocenters in many different
ways. However, some metals are expensive, air sensitive and/or toxic. Inspired by
enzymatic catalysis, organocatalysts are small organic molecules that can catalyze
organic transformations via H-bonding and ionic interactions (Non-covalent catalysis)
or formation of covalent bonds, like NHC carbenes[18] or amines[19] (covalent catalysis).
These catalysts are generally inexpensive as they come from the chiral pool, but often
require higher catalytic loadings.
The term organocatalysis was introduced by McMillan in 2000[20], however, the first
example dates back to 1912, when Bredig and Fiske reported that chichona alkaloids
catalyze the addition of HCN to aldehydes with poor ees.[21] In 1960, O-Acetyl quinine
was used by Pracejus as an efficient catalyst for the addition of methanol to ketenes
with 74% optical yield at -110°C (Scheme 2a). Interestingly, above -40°C, the reaction
afforded the opposite enantiomer of the product.[22] Among the natural amino acids,
proline has been widely used as organic catalyst, for reactions occurring via the

Chapter 1
6
formation of imines or enamines[19] after the pioneering work of Wiechert et al.
concerning proline catalyzed intramolecular aldol reactions (Scheme 2b)[23].
Occasionally, asymmetric organocatalysis is combined with metal-catalysis to enable
reactions that inactive organocatalyst would not be able to catalyze alone.[24]
Scheme 2 Asymmetric organocatalytic transformations.
1.4. Catalytic (dynamic) kinetic resolution Another powerful method to obtain enantiopure compounds using chiral catalysts is
through resolution of racemic mixtures, widely used in industrial processes. We refer
with the term kinetic resolution to an asymmetric reaction where the conversion of the
two enantiomeric substrates to products occurs with different rates (Scheme 3).[25] In
this way, after the reaction, one enantiomer is fully converted into the enantiopure
product, while the other will be left as enantiopure substrate. Similar to other
resolution methods, the upper yield limit for the kinetic resolution of a racemate is
50%. The enantioselectivity will instead depend on the ratio between the kinetic
constants of the two asymmetric processes (s = krel = kfast/ kslow).[26]
Scheme 3 Kinetic resolution according to Pellissier.[25]

Introduction
7
An example is the acylation of benzylic alcohols. (R,R)-Methyl-DUPHOS (17)
promotes the selective benzoylation of (R)-14 and (R)-15 is obtained in 81% ee at 25%
conversion while the unreacted (S)-14 is recovered with 28% ee at 81% conversion
(Scheme 4).[27]
Scheme 4 Kinetic resolution of benzylic alcohol via acylation.[27]
However, if the two enantiomers can interconvert, the product of the resolution can be
collected as a single enantiomer with yields higher than 50%. This process is termed
dynamic kinetic resolution (Scheme 5).[28]
Scheme 5 General scheme for dynamic kinetic resolution.[28]
For instance, chiral substituted acetyl acetates (18) can racemize via keto-enol
tautomery. In the asymmetric hydrogenantion with Nickel Raney and tartaric acid, the
reduction of the (S)-enantiomer is favored and the equilibrium between (S)-18 and
(R)-18 is consequently shifted in this direction (Scheme 6).[29]

Chapter 1
8
Scheme 6 Dynamic kinetic resolution in the hydrogenation of acetyl acetates.[29]
1.5. Non-linear effects in asymmetric catalysis In asymmetric catalysis, the ee of the product (eeprod) is linearly correlated to the one
of the chiral auxiliary or catalyst (eeaux) by a constant that corresponds to the maximum
ee that can be achieved with an enantiopure catalyst/auxiliary (eemax) (Equation 1;
Curve A, Plot 1 Source: review of Kagan and Girard[30]). However, there are cases
where the proportionality between the ee’s of the product and the auxiliary is lost, and
we refer to that as non-linear effects (NLE). If the deviation from linearity is positive
((+)-NLE), the curve will resemble Curve B, Plot 1 while negative deviation ((-)-NLE)
will generate a curve similar to Curve C, Plot 1.
Equation 1: eeprod = eemax*eeaux
Plot 1 Dependence of the eeprod on the eeaux. A) standard enantioselective reaction; B) (+)-NLE; C) (-)-
NLE. Source: Review of Kagan and Girard.[30]
Kagan and coworkers rationalized the non-linearity of enantioselectivity by different
models that involve the formation of diastereomeric complexes or aggregates.[31]
Concerning metallic catalysis, the models are abbreviated with MLn or (ML)n where n
is the number of ligands or complexes that composes the species involved in the
catalysis. The most recurring (and also the simplest) is the model ML2 or (ML)2. Other

Introduction
9
models will not be treated in detail in this thesis, but an extensive explanation can be
found in a fascinating review of Kagan and Girard.[30]
The model ML2 or (ML)2
This model concerns the formation of dimeric complexes and is illustrated in Scheme
7 (Source: H. B. Kagan and T.O. Luukas, Chapter 4, Comprehensive Asymmetric
Catalysis I.[7]). Metal complexes containing two ligands that can have R or S
configuration (LR and LS) can be either homochiral (MLRLR and MLSLS) or heterochiral
(MLRLS). Each of the homochiral complexes catalyzes the reaction enantioselectively
with kinetic constant kRR = kSS, affording the product with a given eemax. MLRLS instead,
catalyzes a racemic reaction with kRS. The final relative concentration of the catalyst
are x, y and z and K is the equilibrium constant between homo- and heterochiral
complexes, while β is the relative amount of heterochiral catalyst compared to the
homochiral one.
Scheme 7 General scheme for the model ML2. Source: H. B. Kagan and T.O. Luukas, Chapter 4,
Comprehensive Asymmetric Catalysis I.[7]
The enantiomeric excess of the product can be expressed as function of the
concentration of the catalytic species and of their kinetic constants in Equation 2.
Equation 2: eeprod = eemax*eeaux*1+𝛽
1+𝑔𝛽
Non-linearity of asymmetric catalysis can be achieved when the following conditions
are satisfied:
The rate of formation of the racemic product is slower than the one of the
enantioenriched one (g ≤ 1)
The formation of the heterochiral complex is thermodynamically favored
(K>>1). In fact, if the racemic complex is not formed (z=0), the value of β is null,
and the correlation between ee of the product and the one of auxiliary is linear.
In this case, the racemic portion of the catalyst is blocked via the formation of
catalytically inactive species. On the other hand, the excess of one of the enantiomers
of the chiral auxiliary catalyzes the enantioselective reaction. We can say that, in the
model ML2, ligands behave like hands (that are the comparison par excellence for
enantiomers).
Chapter 1
10
The positive deviation from linearity in asymmetric catalysis is also referred to as
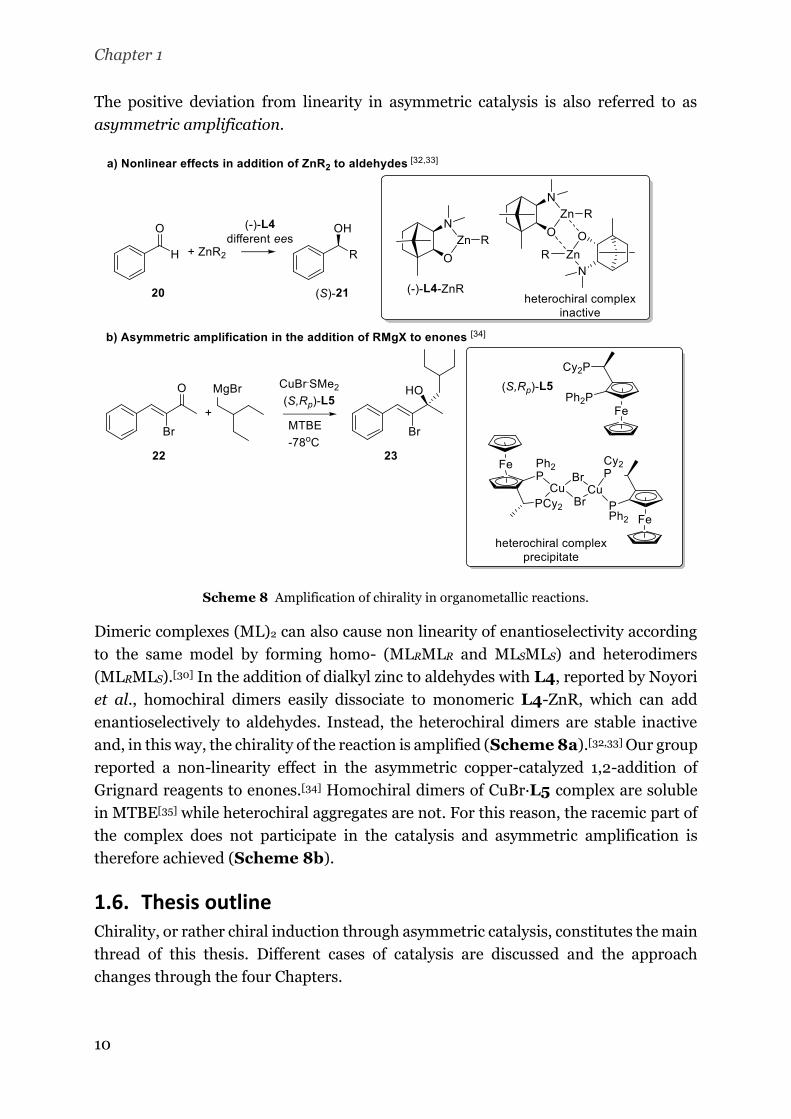
asymmetric amplification.
Scheme 8 Amplification of chirality in organometallic reactions.
Dimeric complexes (ML)2 can also cause non linearity of enantioselectivity according
to the same model by forming homo- (MLRMLR and MLSMLS) and heterodimers
(MLRMLS).[30] In the addition of dialkyl zinc to aldehydes with L4, reported by Noyori
et al., homochiral dimers easily dissociate to monomeric L4-ZnR, which can add
enantioselectively to aldehydes. Instead, the heterochiral dimers are stable inactive
and, in this way, the chirality of the reaction is amplified (Scheme 8a).[32,33] Our group
reported a non-linearity effect in the asymmetric copper-catalyzed 1,2-addition of
Grignard reagents to enones.[34] Homochiral dimers of CuBr·L5 complex are soluble
in MTBE[35] while heterochiral aggregates are not. For this reason, the racemic part of
the complex does not participate in the catalysis and asymmetric amplification is
therefore achieved (Scheme 8b).
1.6. Thesis outline Chirality, or rather chiral induction through asymmetric catalysis, constitutes the main
thread of this thesis. Different cases of catalysis are discussed and the approach
changes through the four Chapters.

Introduction
11
The first two Chapters address the enantioselective addition of Grignard reagents to
symmetric diheteroaryl alkenes, reactive substrates towards the addition of
organometallic reagents. Chapter 2 concerns the asymmetric conjugate addition of
Grignard reagents to generic heteroaryl alkenes. The reactivity of this system and the
consequent issues for enantioselectivity are discussed. Chapter 3 focuses on the
addition of organomagnesium reagents to bispyridyl alkenes. The importance of
pyridines is highlighted and the different behavior of substrates bearing 2-pyridyl or
4-pyridyl moieties is explained. A comparison between the ability of pyridine and
benzoxazole to activate an alkene towards the conjugate addition is made.
The second part of the thesis concerns organic molecules that have a role in their own
enantioselective synthesis. Chapter 4 regards autoinductive effects in the asymmetric
1,2-addition of Grignard reagents to enones. The product, an alkoxide, interacts with
the copper/phosphine catalyst enabling a faster transmetallation that results in a
better enantioselectivity. At the end, Chapter 5 describes the design or an organic
asymmetric autocatalytic reaction inspired by Corey-Bakshi-Shibata reduction of
ketones and imines with borane. The synthesis of the starting materials is reported and
the prospects of asymmetric autocatalysis are discussed.
1.7. Bibliography [1] D. W. Zaidel, S. M. Aarde, K. Baig, Brain Cogn. 2005, 57, 261–263.
[2] L. A. Nguyen, H. He, C. Pham-Huy, Int. J. Biomed. Sci. 2006, 85–100.
[3] S. Ahuja, Chiral Separation Methods for Pharmaceutical and Biotechnological
Products., Wiley, 2013.
[4] F. Toda, Enantiomer Separation: Fundamentals and Practical Methods,
Springer, 2004.
[5] M. Todd, Separation of Enantiomers: Synthetic Methods, Wiley, 2014.
[6] Y. Gnas, F. Glorius, Synthesis (Stuttg). 2006, 1899–1930.
[7] E. N. Jacobsen, A. Pfaltz, H. Yamamoto, Comprehensive Asymmetric Catalysis
I-III, Springer, 1999.
[8] B. M. Trost, Proc. Natl. Acad. Sci. 2004, 101, 5349–5355.
[9] H. U. Blaser, H.-J. Federsel, Asymmetric Catalysis on Industrial Scale:
Challenges, Approaches and Solutions., Wiley, 2010.
[10] H. Pellissier, J. J. Spivey, Chiral Sulfur Ligands: Asymmetric Catalysis, Royal
Society Of Chemistry, 2009.
[11] A. Pfaltz, Chimia (Aarau). 2004, 58, 49–50.
[12] W. S. Knowles, Angew. Chemie - Int. Ed. 2002, 41, 1998–2007.
[13] W. S. Knowles, J. Chem. Ed. 1986, 63, 222–225.
[14] R. Noyori, Angew. Chemie - Int. Ed. 2002, 41, 2008.
[15] D. Glynn, J. Shannon, S. Woodward, Chem. - A Eur. J. 2010, 16, 1053–1060.
[16] K. B. Sharpless, Angew. Chemie - Int. Ed. 2002, 41, 2024.

Chapter 1
12
[17] S. Katsuki, K. B. Sharpless, J. Am. Chem. Soc 1980, 102, 5976–5978.
[18] A. Grossmann, D. Enders, Angew. Chemie Int. Ed. 2012, 51, 314–325.
[19] S. Mukherjee, J. W. Yang, S. Hoffmann, B. List, Chem. Rev. 2007, 107, 5471–
5569.
[20] K. A. Ahrendt, C. J. Borths, D. W. C. MacMillan, J. Am. Chem. Soc 2000, 122,
4243–4244.
[21] G. Bredig, W. S. Fiske, Biochem. Z., 1912, 7–23.
[22] H. Pracejus, Justus Liebigs Ann. Chem. 1960, 634, 9–22.
[23] U. Eder, G. Sauer, R. Wiechert, Angew. Chemie - Int. Ed. 1971, 10, 496–497.
[24] Z.-Y. Han, D.-F. Chen, Y.-Y. Wang, R. Guo, P.-S. Wang, C. Wang, L.-Z. Gong, J.
Am. Chem. Soc. 2012, 134, 6532–6535.
[25] H. Pellissier, Adv. Synth. Catal. 2011, 353, 1613–1666.
[26] J. M. Keith, J. F. Larrow, E. N. Jacobsen, Adv. Synth. Catal. 2001, 343, 5–26.
[27] E. Vedejs, O. Daugulis, S. T. Diver, J. Org. Chem. 1996, 61, 430–431.
[28] R. Noyori, M. Tokunaga, M. Kitamura, Bull. Chem. Soc. Jpn. 1995, 68, 36–56.
[29] A. Tai, H. Watanabe, T. Harada, Bull. Chem. Soc. Jpn. 1979, 52, 1468–1472.
[30] C. Girard, H. B. Kagan, Angew. Chemie - Int. Ed. 1998, 37, 2922–2959.
[31] D. Guillaneux, S.-H. Zhao, O. Samuel, D. Rainford, H. B. Kagan, J. Am. Chem.
Soc 1994, 116, 9430–9439.
[32] E. C. Anthony, M. Kitamura, S. Okada, S. Suga, R. Noyori, J. Am. Chem. Soc.
1989, 111, 4028–4036.
[33] M. Yamakawa ’, R. Noyori, J. Am. Chem. Soc 1995, 117, 6327–6335.
[34] F. Caprioli, A. V. R. Madduri, A. J. Minnaard, S. R. Harutyunyan, Chem.
Commun. 2013, 49, 5450.
[35] F. Caprioli, M. Lutz, A. Meetsma, A. Minnaard, S. Harutyunyan, Synlett 2013,
24, 2419–2422.

Chapter 2
Control of enantioselectivity in the addition of Grignard
reagents to symmetric heteroaryl disubstituted olefins Symmetric olefins bearing two electron withdrawing substituents are very reactive
towards the addition of hard organometallic reagents. For this reason, the
asymmetric addition of organolithium, organomagnesium and organozinc reagents,
the background reaction competes with the catalytic pathway and high
enantioselectivities can hardly be achieved. In this Chapter, we describe conjugate
addition of Grignard reagents to bisheteroaryl olefins promoted by a
copper/phosphine catalyst. The use of a Lewis acid allows selective acceleration of
1,4-addition pathway over side products formation, but is deleterious for the
enantioselectivity. The formation of side products becomes more prominent as the
length of the alkyl chain of Grignard reagents increases. The addition of
methylmagnesium bromide proceeds with excellent enantioselectivity and good
yield.

Chapter 2
14
2.1. Introduction
2.1.1. Asymmetric addition of organometallic reagents to electron deficient
olefins
Electron deficient olefins, such as α,β-unsaturated ketones, (thio)esters, enamides,
nitroalkenes, cyanoalkenes, alkenyl phosphates and sulfones, are important substrates
for the formation of stereodefined C-C bonds. The presence of an electron withdrawing
substituent activates the sp2-carbon in β-position towards the conjugate addition of
nucleophiles (CA, Scheme 1) leading to the formation of a chiral sp3-carbon. Organic
(semi)metallic reagents such as organoboron[1,2], -zinc[3–5], -zirconium[6] –aluminum[7–
11] and organomagnesium[12] reagents are commonly used for the asymmetric Michael
addition in combination with a chiral metallic catalyst (copper(I) or rhodium(I) for
arylboronic acids[2]). This reaction proceeds through transmetallation of the
organometallic reagent on the chiral catalyst, binding to the electron deficient olefin
substrate and subsequent stereoselective addition to the alkene. The order of reactivity
of these reagents is RB(OH)2 << RZrX ≈ R2Zn < R3Al < RMgBr.
Scheme 9 General scheme for the asymmetric conjugate addition of organometallic reagents to
alkenes substituted with electron withdrawing substituents.
The addition of organoboron reagents to electron poor alkenes can occur only if
mediated by a catalyst. Therefore, the chemo- and enantioselectivity of the reaction can
be easily controlled through the choice of the metal salt and the ligand. Instead, the
addition of a highly reactive Grignard reagents to electron deficient olefins, for instance
an enone, can lead to four products (Scheme 2)[13]:
1. The 1,4-addition enolate product, that after aqueous workup affords the ketone
1.
2. The 1,2-addition product, alkoxide 2. The amount of this product increases with
the hardness of the nucleophile.
3. The reduction product 3 caused by a β-hydride transfer from an organometallic
reagent.
4. The enolization product 4, regenerating the starting material upon aqueous
workup.

Control of enantioselectivity in the addition of Grignard reagents to symmetric heteroaryl disubstituted olefins
15
Scheme 10 Chemo- and regioselectivity in the addition of organometallic reagents to enones.
Moreover, the addition of a hard nucleophile to the substrate without mediation of the
chiral catalyst is possible and occurs in a racemic fashion (background reaction). This
decreases the overall enantioselectivity of the reaction. Dialkylzinc reagents display
lower reactivity compared to their organomagnesium counterparts and leading to a
slower background reaction. Grignard reagents, on the other hand, are cheaper, easily
available and more atom economic when comparing to dialkylzinc reagents.
2.1.2. Enantioselectivity in the 1,4-addition of nucleophiles to symmetric
disubstituted alkenes
Enantioselective Micheal additions have been extensively studied and constitute a
useful tool in asymmetric synthesis because of the broad choice of acceptors and
donors. However, the stereoselective conjugate addition of nucleophiles to symmetric
disubstituted electron poor alkenes is still underexplored. Symmetric 1,2-disubstituted
alkenes with the E configuration have a C2h symmetry, and a C2v symmetry when Z
(Scheme 3). The higher symmetry does not represent a challenge for the
enantiocontrol of the reaction. Nevertheless, symmetric alkenes are hardly found
among the plethora of reported methodologies regarding asymmetric conjugate
addition (ACA).
Scheme 11 Symmetry of E and Z symmetric disubstituted alkenes.
However, in biological systems, the ACA these symmetric alkenes occurs with high
enantioselectivity leading to the synthesis of chiral biomolecules. An example is the
biosynthesis of (L)-aspartic acid from fumaric acid and ammonia catalyzed by the
enzyme aspartase[14]. Modification of this enzymatic reaction allows the asymmetric
addition of hydroxylamines and hydrazines with ee’s 97-99% (Scheme 4a).[15] Non-
enzymatic ACA were also achieved, by using unreactive organometallic species like
organoboron reagents: the addition of alkylmalonates (5) to different symmetric

Chapter 2
16
alkenes (6) mediated by the chiral BINOLate 7 affords the product with ees up to 98%
(Scheme 4b).[16]
Scheme 12 Asymmetric addition of nucleophiles to 1,2-diactivated alkenes.
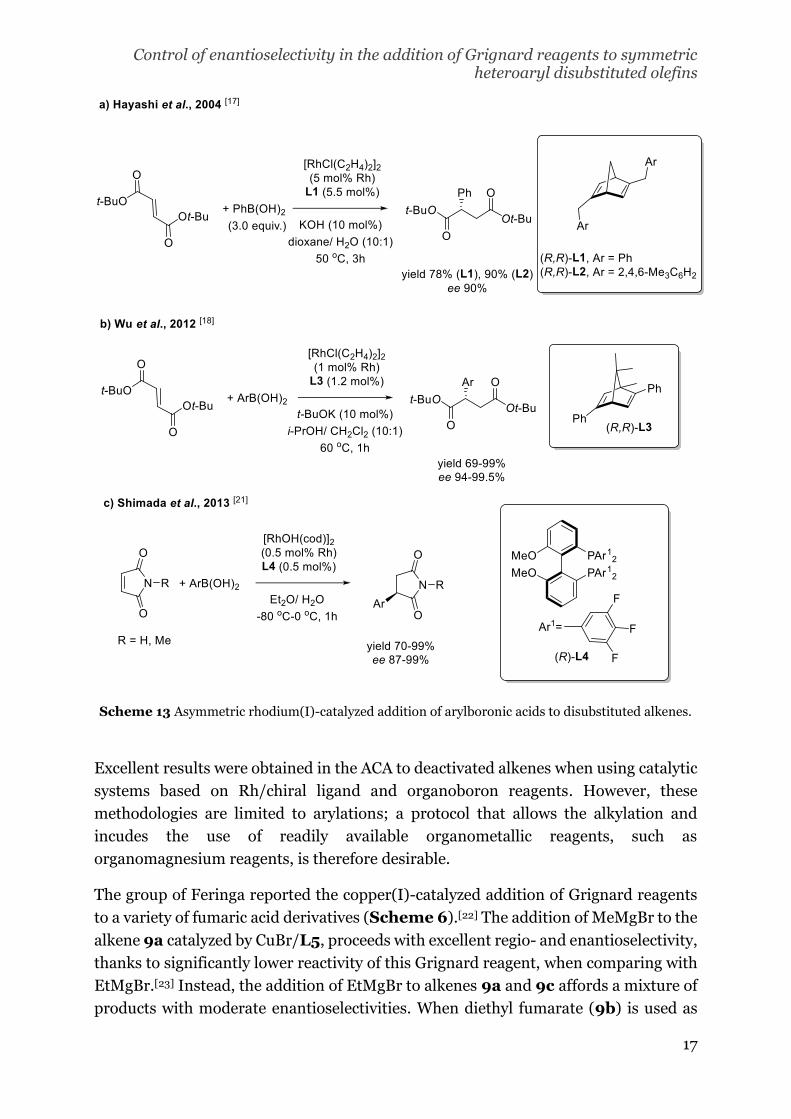
The first contribution using metal catalysis concerns the rhodium(I) catalyzed addition
of arylboronic acids to fumaric esters and maleimide reported by Hayashi and
coworkers.[17] In this case, phosphine ligands, commonly used in rhodium catalysis[2],
afforded the addition reaction with good to excellent yields (94-96%) but poor
enantioselectivities (3-21% ee). Instead, the use of a norbornadiene ligand (L1)
resulted in a decrease of the reactivity (78% yield with L1, 90% with L2) but an
improvement in the enantioselectivity (90% ee, Scheme 5a). In 2012, Wu and
coworkers reported, that among dienes as chiral ligands for the rhodium, L3, can be
efficient in the addition of boronic acids to fumaric esters (Scheme 5b).[18]
Bicyclic[2,2,2]octadienyl ligands were also employed in the construction of C-N chiral
axes via the addition of arylboronic acids to N-substituted maleimide[19] and the
enantioselective cyclopropanation of fumaric esters[20]. Likewise, Rh(I)/diphosphine
catalyst (L4) provided excellent enantioselectivities in the addition of arylboronic acids
to unprotected or N-alkyl maleimides at low temperatures (-0 - 0 oC, Scheme 5c)[21].
Control of enantioselectivity in the addition of Grignard reagents to symmetric heteroaryl disubstituted olefins
17
Scheme 13 Asymmetric rhodium(I)-catalyzed addition of arylboronic acids to disubstituted alkenes.
Excellent results were obtained in the ACA to deactivated alkenes when using catalytic
systems based on Rh/chiral ligand and organoboron reagents. However, these
methodologies are limited to arylations; a protocol that allows the alkylation and
incudes the use of readily available organometallic reagents, such as
organomagnesium reagents, is therefore desirable.
The group of Feringa reported the copper(I)-catalyzed addition of Grignard reagents
to a variety of fumaric acid derivatives (Scheme 6).[22] The addition of MeMgBr to the
alkene 9a catalyzed by CuBr/L5, proceeds with excellent regio- and enantioselectivity,
thanks to significantly lower reactivity of this Grignard reagent, when comparing with
EtMgBr.[23] Instead, the addition of EtMgBr to alkenes 9a and 9c affords a mixture of
products with moderate enantioselectivities. When diethyl fumarate (9b) is used as

Chapter 2
18
Michael-acceptor, the product is obtained with moderate to good ee’s (46% and 65%
using respectively L5 and L6 as ligands for the copper), but the addition of a Grignard
reagent to the olefin 9d is not stereoselective.[22]
Scheme 14 Asymmetric addition of Grignard reagents to various 1,2-disubstituted alkenes.[22]
In the same thesis work, the addition of dialkylzinc reagents to substrates 9a-d is also
described and racemic products were obtained in most cases (10a’ with 21% ee).[22]
From these data, we can evince that the control of both regio- and enantioselectivity
becomes an issue when both Michael-acceptors and donors are particularly reactive.
This fact can be due to two factors:
Due to the higher reactivity, the catalytic process undergoes with lower
enantioselectivity.
The presence of uncatalyzed addition of the Grignard reagent to the alkene as a
racemic background reaction.

Control of enantioselectivity in the addition of Grignard reagents to symmetric heteroaryl disubstituted olefins
19
The interest of this chapter concerns the control of the enantioselectivity in the ACA of
Grignard reagents to symmetric alkenes.
2.1.3. Copper(I)-catalyzed asymmetric addition of Grignard reagents to (N)-
containing heteroaryl alkenes
Heteroarenes are present in drug candidates, with an average of two heteroaryl rings
per candidate drug.[24,25] A feature of around 50% of all Active Pharmaceutical
Ingredients (APIs) is chirality: as the configuration of the drug is often determining its
biological activity, a method to obtain it as a single enantiomer is fundamental.
In 2009, the pioneering work of Lam et al. disclosed the ability of N-containing
heteroarenes to activate olefins towards the enantioselective copper-catalyzed
conjugate addition of hydrides.[26] In the following year, the addition of organoboronic
acids to these substrates, promoted by a chiral rhodium(I) catalyst and microwave
irradiation, was also achieved by the same group. They obtained a cornucopia of chiral
heteroarenes derivatives with excellent yields and enantioselectivities (12, Scheme
7).[27] Instead, the group of Lautens prepared aza-dihydrobenzoezepines with high ees,
via a domino reaction involving the rhodium-catalyzed addition of borates and
palladium-catalyzed C-O coupling.[28] These examples constitute two profitable
methods for the asymmetric arylation of vinyl heteroarenes; nevertheless, procedures
for the alkylation are still underexplored in literature.
Scheme 15 Asymmetric rhodium(I)-catalyzed addition of boronic acids to vinyl heteroarenes.
Our group hypothesized that by using highly reactive Grignard reagents, in the
presence of a chiral copper catalyst, might allow the alkylation of alkenyl heteroarenes.
Early efforts towards the asymmetric addition of phenylmagnesium bromide to vinyl
pyridines, promoted by a nickel(I)-catalyst, were made, but they resulted in poor
enantioselectivities (0-15% ee).[29] In 2016, our group developed the asymmetric
copper(I)-catalyzed addition of Grignard reagents to different alkenyl heteroarenes.[30]
This methodology affords a multitude of chiral heteroaryl derivatives with yields
between 46 and 96% and excellent ee’s (86-99%, Scheme 8). The key to the success
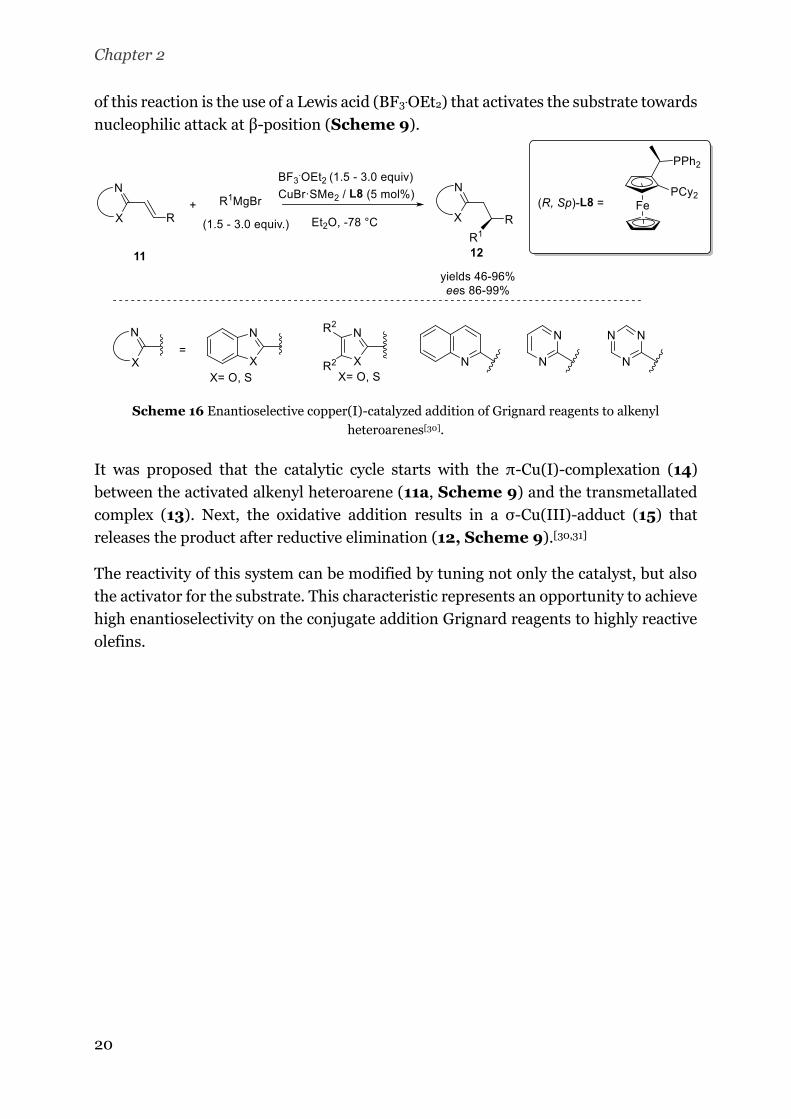
Chapter 2
20
of this reaction is the use of a Lewis acid (BF3.OEt2) that activates the substrate towards
nucleophilic attack at β-position (Scheme 9).
Scheme 16 Enantioselective copper(I)-catalyzed addition of Grignard reagents to alkenyl
heteroarenes[30].
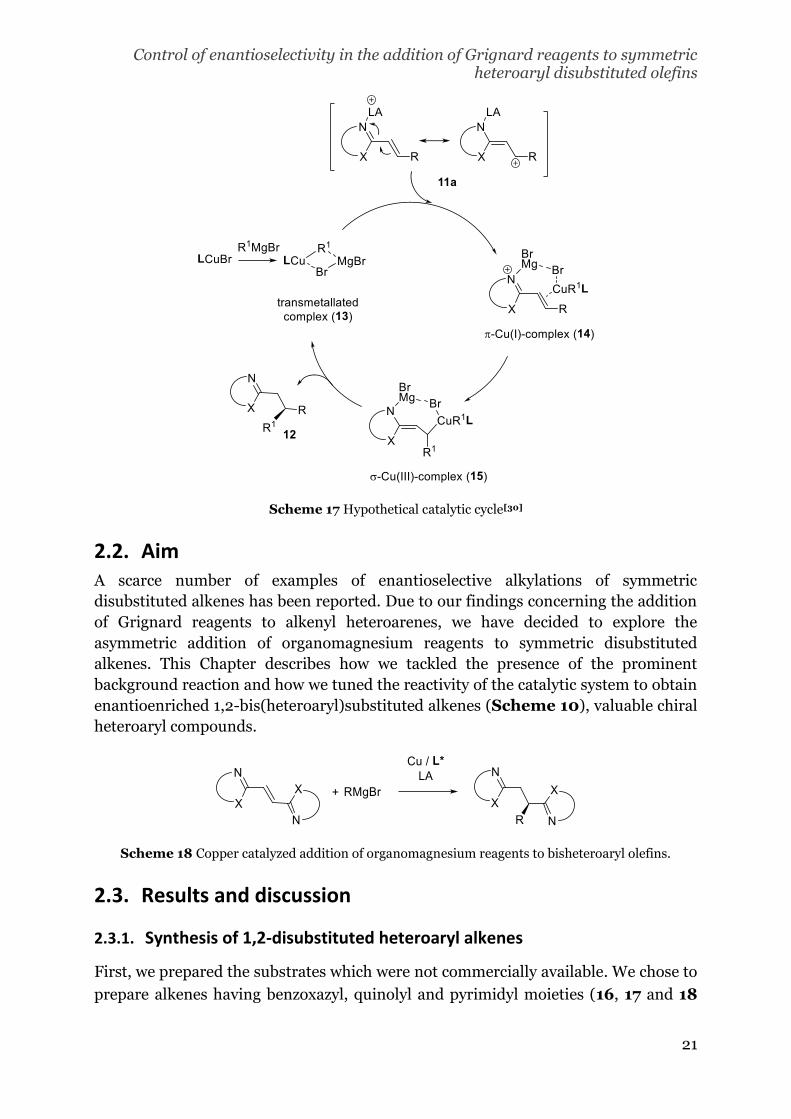
It was proposed that the catalytic cycle starts with the π-Cu(I)-complexation (14)
between the activated alkenyl heteroarene (11a, Scheme 9) and the transmetallated
complex (13). Next, the oxidative addition results in a σ-Cu(III)-adduct (15) that
releases the product after reductive elimination (12, Scheme 9).[30,31]
The reactivity of this system can be modified by tuning not only the catalyst, but also
the activator for the substrate. This characteristic represents an opportunity to achieve
high enantioselectivity on the conjugate addition Grignard reagents to highly reactive
olefins.
Control of enantioselectivity in the addition of Grignard reagents to symmetric heteroaryl disubstituted olefins
21
Scheme 17 Hypothetical catalytic cycle[30]
2.2. Aim A scarce number of examples of enantioselective alkylations of symmetric
disubstituted alkenes has been reported. Due to our findings concerning the addition
of Grignard reagents to alkenyl heteroarenes, we have decided to explore the
asymmetric addition of organomagnesium reagents to symmetric disubstituted
alkenes. This Chapter describes how we tackled the presence of the prominent
background reaction and how we tuned the reactivity of the catalytic system to obtain
enantioenriched 1,2-bis(heteroaryl)substituted alkenes (Scheme 10), valuable chiral
heteroaryl compounds.
Scheme 18 Copper catalyzed addition of organomagnesium reagents to bisheteroaryl olefins.
2.3. Results and discussion
2.3.1. Synthesis of 1,2-disubstituted heteroaryl alkenes
First, we prepared the substrates which were not commercially available. We chose to
prepare alkenes having benzoxazyl, quinolyl and pyrimidyl moieties (16, 17 and 18

Chapter 2
22
respectively, Figure 1), to gain a view over the reactivity of this class of olefins. The
alkenes 16 and 17 were synthesized using modifications of the literature
procedures.[32,33] The synthesis of alkene 18 proved to be challenging because of the
low availability of starting materials containing a 2-pyrimidine moiety, and the olefin
was not succesfully synthesized. However, the attempts for its synthesis will be
discussed.
Figure 1 Symmetric heteroaryl disubstituted alkenes 16-18
Condensation reactions are not commonly used to synthesize 2-benzoxazyl alkenes due
to the high cost of 2-benzoxazyl carbaldehyde.[34] The described synthesis of 16
consists of the formation of the benzoxazole from 2-hydroxilaninile and fumaric acid
using harsh conditions (polyphosphoric acid, PPA, as a solvent).[35] However, because
of the technical difficulties in handling PPA (viscosity and use of high temperatures),
we opted for a microwave assisted synthesis of 16 (Scheme 11)[32]. When fumaric acid
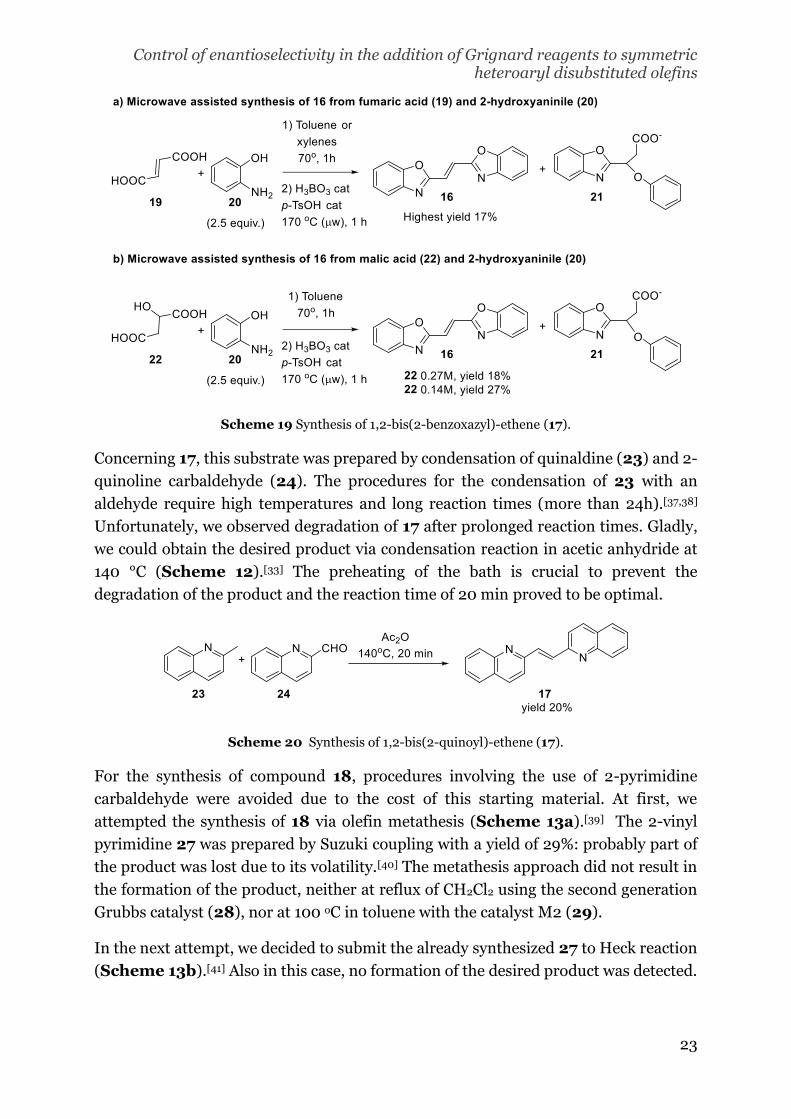
(19) was used as a starting material, 21 was found as a byproduct (Scheme 11a). To
favor the ring closure of the benzoxazyl ring over the conjugate addition, we decided to
use malic acid (22) as precursor.[36] No significant change in the yield was observed
(Scheme 11b). Instead, an increase from 18 to 27% was observed when the
concentration of the reaction was reduced to half, probably due to the better solubility
and stirring.
Control of enantioselectivity in the addition of Grignard reagents to symmetric heteroaryl disubstituted olefins
23
Scheme 19 Synthesis of 1,2-bis(2-benzoxazyl)-ethene (17).
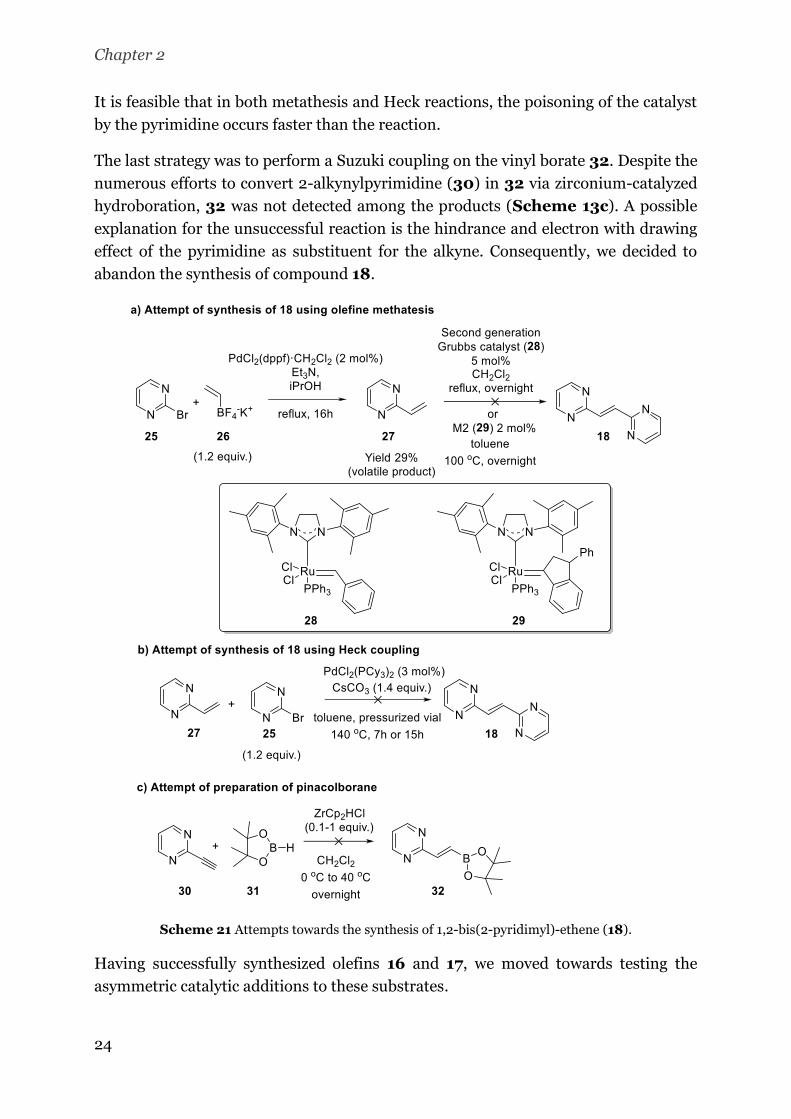
Concerning 17, this substrate was prepared by condensation of quinaldine (23) and 2-
quinoline carbaldehyde (24). The procedures for the condensation of 23 with an
aldehyde require high temperatures and long reaction times (more than 24h).[37,38]
Unfortunately, we observed degradation of 17 after prolonged reaction times. Gladly,
we could obtain the desired product via condensation reaction in acetic anhydride at
140 °C (Scheme 12).[33] The preheating of the bath is crucial to prevent the
degradation of the product and the reaction time of 20 min proved to be optimal.
Scheme 20 Synthesis of 1,2-bis(2-quinoyl)-ethene (17).
For the synthesis of compound 18, procedures involving the use of 2-pyrimidine
carbaldehyde were avoided due to the cost of this starting material. At first, we
attempted the synthesis of 18 via olefin metathesis (Scheme 13a).[39] The 2-vinyl
pyrimidine 27 was prepared by Suzuki coupling with a yield of 29%: probably part of
the product was lost due to its volatility.[40] The metathesis approach did not result in
the formation of the product, neither at reflux of CH2Cl2 using the second generation
Grubbs catalyst (28), nor at 100 oC in toluene with the catalyst M2 (29).
In the next attempt, we decided to submit the already synthesized 27 to Heck reaction
(Scheme 13b).[41] Also in this case, no formation of the desired product was detected.
Chapter 2
24
It is feasible that in both metathesis and Heck reactions, the poisoning of the catalyst
by the pyrimidine occurs faster than the reaction.
The last strategy was to perform a Suzuki coupling on the vinyl borate 32. Despite the
numerous efforts to convert 2-alkynylpyrimidine (30) in 32 via zirconium-catalyzed
hydroboration, 32 was not detected among the products (Scheme 13c). A possible
explanation for the unsuccessful reaction is the hindrance and electron with drawing
effect of the pyrimidine as substituent for the alkyne. Consequently, we decided to
abandon the synthesis of compound 18.
Scheme 21 Attempts towards the synthesis of 1,2-bis(2-pyridimyl)-ethene (18).
Having successfully synthesized olefins 16 and 17, we moved towards testing the
asymmetric catalytic additions to these substrates.

Control of enantioselectivity in the addition of Grignard reagents to symmetric heteroaryl disubstituted olefins
25
2.3.2. ACA of Grignard reagents to benzoxazyl alkenes
We started from 16, as benzoxazole substrate as it was found to be a good model for
addition reactions in the previous work of our group.[30] The addition of
ethylmagnesium bromide on 16 was tested using the optimized methodology,[30] that
involves the use of BF3.OEt2 as Lewis acid and 5 mol% of a catalyst formed by
complexation of CuBr·SMe2 and L8 (Table 1). Unfortunately, substrate 16 was poorly
soluble in commonly used solvents like diethyl ether, MTBE, toluene at room
temperature and -78 oC. Instead, 16 is soluble in CH2Cl2 at room temperature, but not
at -78oC. When ethereal solvents were used as a solvent, racemic product (33a,
entries 1 and 3, Table 1). No conversion to the product was observed in toluene, as
well as in diethyl ether in the absence of BF3.OEt2 (entries 2 and 4). On the contrary,
in CH2Cl2 the reaction reached full conversion and 16 was recovered with 48% yield
and 52% ee in the absence of a Lewis acid (entry 5). This observation denotes an
enhanced reactivity of this double substituted alkene with respect to the
monosubstituted ones.[30] With BF3.OEt2 it was possible to enhance the yield of the
product at the expense of the ee (entries 6-8) that dropped to 27% when 1.5 equiv. of
BF3.OEt2 was used. The strength of the Lewis acid affects these two parameters in a
similar way. In fact, TMSBr (1.5 equiv.) was found to be an efficient Lewis acid
affording the product with 86% yield and 54% ee (entry 9) but the yield was only 36%
when 0.5 equiv. of TMSBr was used. The highest enantioselectivities were achieved
when using TMSCl, but this LA did not allow the reaction to give full conversion (entry
11).

Chapter 2
26
Table 1 Screening of Lewis acids in the addition.
Entry LA (equiv.) Solvent Conversion (%)a Yield (%) b eec
1 BF3.OEt2 (1.5) Et2O 78 n.d. 6
2 None Et2O 0 - -
3 BF3.OEt2 (1.5) MTBE ~50 n.d. racemic
4 BF3.OEt2 (1.5) Toluene 0 - -
5 None CH2Cl2 >95 48 52
6 BF3.OEt2 (1.5) CH2Cl2 >95 58 27
7 BF3.OEt2 (1.0) CH2Cl2 >95 56 44
8 BF3.OEt2 (0.5) CH2Cl2 >95 42 53
9 TMSBr (1.5) CH2Cl2 >95 86 54
10 TMSBr (0.5) CH2Cl2 >95 36 53
11 TMSCl (1.0) CH2Cl2 >95 53 58
a Conversion to 33a determined by 1H-NMR b Isolated yield. c Determined via CSP-
HPLC.
Analysis of the data presented in Table 1, shows that only moderate
enantioselectivities can be achieved in this transformation. This can be explained by a
fast competing background reaction, in both the presence and the absence of Lewis
acid (Scheme 14). It was observed that Lewis acid accelerates the addition of EtMgBr
to the olefin, therefore improve the regio and chemoselectivity of the reaction, in both
the catalyzed and the background reactions.
Scheme 22 Non-catalyzed addition of EtMgBr to 18.
Aiming to improve the enantioselectivity by tuning the structure of the chiral catalyst,
different classes of chiral diphosphines L5-L16 were screened (Table 2) using either

Control of enantioselectivity in the addition of Grignard reagents to symmetric heteroaryl disubstituted olefins
27
BF3.OEt2, TMSCl, BCl3 or no Lewis acid. Ligands L5, L9 and L15 afforded the addition
product with low ees (2-18%). Using ligands L11, L12, L14 and L16, the addition
proceeded with moderate to good yields and moderate enantioselectivities.
Table 2 Ligand screening

Chapter 2
28
a Conversion to 33a was determined by 1H-NMR b Isolated yield. c Determined via CSP-
HPLC. d 10 mol% of the catalyst used e 1.0 equiv. of EtMgBr was used f THF used as
solvent g EtMgBr diluted in CH2Cl2 to 0.6 mL and added over 2h h EtMgBr diluted in
CH2Cl2 and added over 4h
Our interest was caught by L10: with 0.5 equiv. of BF3.OEt2, we could obtain 33a in
42% yield and 69% of ee (entry 5, Table 2). Curiously, the use of 10 mol% of the
catalyst and 1.0 equiv. of the organometallic reagent, caused a decrease of the
enantioselectivity (entries 6 and 7, respective ees 64% and 45%). The change of
reaction solvent from CH2Cl2 to THF resulted in a complete loss of enantiocontrol
(entry 8). BCl3 has a positive effect on the yield and negative on ee (yield 61%, ee 53%).
With L16 we obtained 33a in 81% yield and 59% ee (opposite enantiomer, entry 12).
To our delight, in the absence of BF3.OEt2, the ee was enhanced to 68% which could be
further improved to 76% ee by slow addition of the Grignard reagent over 2h (entries
13 and 14). Again, the yield of the product dropped when the catalyst loading was
doubled (entry 15). Unfortunately, the results of entry 14 could not be repeated. To
guarantee the reproducibility of the reaction, we decided to add the EtMgBr in 4h, as
Entry Ligand LA (equiv.) Conversion
(%)a
Yield (%) b
eec
1 L5 TMSCl (1.0) >95 24 6
2 L5 BF3.OEt2 (0.5) ~90 40 4
3 L9 TMSCl (1.0) >95 37 18
4 L9 BF3.OEt2 (0.5) >95 58 2
5 L10 BF3.OEt2 (0.5) >95 42 69
6d L10 BF3.OEt2 (0.5) 81 47 64
7e L10 BF3.OEt2 (0.5) >95 48 45
8f L10 BF3.OEt2 (0.5) 40 n.d. racemic
9 L10 BCl3 (0.5) >95 61 53
10 L11 BF3.OEt2 (0.5) >95 n.d. 42
11 L12 BF3.OEt2 (0.5) >95 58 26
12 L13 BF3.OEt2 (0.5) >95 81 -59
13 L13 None >95 57 -68
14g L13 None >95 65 -76
15d,g L13 None >95 34 -68
16h L13 None >95 79 -71
17 L14 None >95 n.d. -26
18g L14 None >95 n.d. -38
19 L15 BF3.OEt2 (0.5) >95 n.d. 7
20g L16 None >95 39 40

Control of enantioselectivity in the addition of Grignard reagents to symmetric heteroaryl disubstituted olefins
29
reported in entry 16 (yield and ee, 79 and 71% respectively) and take the conditions of
this entry as the optimized reaction conditions.
To identify the reason for reproducibility issues with respect to the enantioselectivity,
we considered the possibility of the enolization of the product, caused by the free
Grignard in solution in the moment of the quench (Scheme 15a). We tried to
reproduce those conditions by submitting enantioenriched 33a to 2.0 equiv. of
MeMgBr alone or with 1.5 equiv. of BF3.OEt2: no significant change of ee (76% before,
74% and 73% after, Scheme 15b) was observed, thus enolization can be excluded as
possible racemization pathway.
Scheme 23 Possible racemization of the product after the quench upon deprotonation by
organomagnesium reagents.
The scope of the Grignard reagents was evaluated in the optimized conditions (Table
3). The addition of n-HexMgBr affords 33c with similar enantioselectivity to 33a but
with poor yield of 7% (entry 2, Table 3). In fact, the major product derived from 1,2-
addition to the benzoxazole ring. The use of 0.5 equiv. of BF3.OEt2 was beneficial for
the yield for all of the organometallic reagents used (entries 4-7). With 1.0 equiv. the
yield further improved, albeit at the cost of enantioselectivity (entry 7). The trend
observed for linear Grignard reagents shows longer chains lead to lower yields (33a,
yield = 81%, 33b, yield = 65%, 33b, yield = 29%). The enantioselectivity is always
moderate under these conditions. The use of CypMgBr afforded 33d as a racemate in
good yield (entry 8). In the previous screening, we found that 1.5 equiv. of TMSBr as
an additive improved yield and ee, for the reaction (entry 9, Table 1), however in this
reaction it resulted in poor conversion of 14% (entry 11, Table 3). As expected,
MeMgBr was less reactive than the other organomagnesium reagents (entries 9 and

Chapter 2
30
10) and in the presence of BF3.OEt2 (1.5 equiv.) the reaction proceeded with 89%
conversion, 58% yield and moderate ee of 39% (entry 10).[23]
Table 3 Grignard reagents scope.
Entry RMgBr a LA (equiv.) Conversion (%)b Yield (%)c eed
1 EtMgBr None >95 79 71
2 n-HexMgBr None 80 7 70
3 i-BuMgBr None 0 - -
4e EtMgBr BF3.OEt2 (0.5) >95 81 59
5 n-BuMgBr BF3.OEt2 (0.5) >95 65 40
6 n-HexMgBr BF3.OEt2 (0.5) >95 29 45
7 n-HexMgBr BF3.OEt2 (1.5) >95 56 23
8 CypMgBr BF3.OEt2 (0.5) >95 71 6
9 MeMgBr BF3.OEt2 (0.5) 51 n.d. n.d.
10 MeMgBr BF3.OEt2 (1.5) 89 58 39
11 n-HexMgBr TMSBr (1.5) 14 n.d. n.d.
a RMgBr diluted in CH2Cl2 to 0.6 mL and added over 4h b Conversion to 33 was
determined by 1H-NMR. c Isolated yield. d Determined via CSP-HPLC. e Fast addition
of the concentrated RMgBr.
Hoping that higher ees can be achieved with MeMgBr, thanks to its reactivity, we
optimized the reaction conditions further. Changing to ligand L10 did not improve the
enantioselectivity (entry 1, Table 4), and the reaction did not proceed in the absence
of BF3.OEt2 (entry 2). To our delight, the use of L16 allowed the formation of 33e in
59% yield and 97% ee (entry 4) even if a non-catalyzed reaction can occur in the
presence of BF3.OEt2.

Control of enantioselectivity in the addition of Grignard reagents to symmetric heteroaryl disubstituted olefins
31
Table 4 Asymmetric addition of MeMgBr.
Entry Ligand LA (equiv.) Conversion
(%)a
Yield
(%)b
eec
1 L10 BF3.OEt2 (1.5) >95 79 -35
2 L10 None 0 - -
3 d (R)-L16 BF3.OEt2 (1.5) >95 35 -95
4d (S)-L16 BF3.OEt2 (1.5) >95 59 97
5 No catalyst BF3.OEt2 (1.5) >95 - -
a Conversion to 33e was determined by 1H-NMR b Isolated yield. c Determined via CSP-
HPLC. d MeMgBr diluted in CH2Cl2 to 0.6 mL and added over 2h.
In the catalytic addition of Grignard reagents to 16, good enantioselectivities were
achieved only at expense of yield. In fact, the use of a Lewis acid favors the non-
catalyzed addition of the nucleophile over the enantioselective pathway, but is
necessary to prevent the formation of side products. At this point, we were eager to
study the behavior of 17 in this reaction.
2.3.3. ACA of Grignard reagents to symmetric 2-quinoyl alkenes
Next, the reactivity of the alkene 17 was investigated in our reaction. The highest
conversion (58%) was observed when L8 was used as ligand for the copper in the
presence of 1.5 equiv. of BF3.OEt2 (entry 1, Table 5). However, no product 34 could
be isolated by column chromatography. An increase of the temperature to -50oC or an
increase in the amount of Lewis acid entails lower conversion of the starting material
to the product (entries 2 and 3). Likewise, the addition does not occur in the presence
of TMSBr and has a poor conversion with TMSOTf (entries 4 and 5). When L13 is
used the NMR-conversion reaches 45%. In order to obtain a racemic product, we tested
racemic BINAP as ligand but we obtained low or no conversion using BF3.OEt2 or AlCl3
(entries 7 and 8) respectively. Additionally, without copper catalyst, 34 could not be
formed. We presume that this is due to the steric bulk of the TMS group.

Chapter 2
32
Table 5 Reactivity of 17 towards the copper(I)-catalyzed addition of EtMgBr.
Entry Ligand LA (equiv.) Conversion (%)a
1 L8 BF3.OEt2 (1.5) 58
2b L8 BF3.OEt2 (1.5) 0
3 L8 BF3.OEt2 (3.0) 26
4 L8 TMSBr (1.5) 0
5 L8 TMSOTf (1.5) 19
6 L13 BF3.OEt2 (1.5) ~45
7 rac-BINAPc BF3.OEt2 (1.5) 30
8 rac-BINAPc AlCl3 0
9 No catalyst TMSOTf (1.5) 0
a Conversion to 34 was determined by 1H-NMR b Reaction performed at -50oC c10
mol% of catalyst were used.
We suppose that the low reactivity of 17 in this reaction is caused by the excessive steric
hindrance close to the alkene. In fact, once a Lewis acid is coordinated to the nitrogen
atom, it is in proximity of both sides of the alkene. The reactivity of 17 is comparable
to the one of 35 (Figure 2), which is poorly reactive towards the addition of Grignard
reagents[42]. This suggests that 2-(hetero)aryl-substituted vinyl quinolines are
probably too hindered for this type of reaction.
Figure 2 Structure of 2-styrylquinoline.
2.4. Conclusions In this Chapter, the syntheses and applications of two symmetric disubstituted olefins,
substrates in the reactions of ACA of Grignard reagents are described. These
compounds can be obtained via ring closure and subsequent formation of the
heteroarene or via condensation. In both cases, the yields are moderate due to
degradation of the product in the reaction conditions.
The behavior in the asymmetric addition differs for the two substrates. The alkene
bearing two 2-benzoxazyl moieties is very reactive towards the addition of

Control of enantioselectivity in the addition of Grignard reagents to symmetric heteroaryl disubstituted olefins
33
organomagnesium reagents. This reactivity has implications on the enantioselectivity,
because the non-catalyzed addition of the organometallic reagents competes with the
catalytic pathway. The use of Lewis acid improves the yield of the reaction, as it
promotes the conjugate addition rather than the formation of byproducts, but it is
deleterious for the ee. The length of the chain of the Grignard reagent influences greatly
the reaction outcome: a longer chain is leading to lower yields. Again, Lewis acids can
impede side reactions, at expense of enantioselectivity. However, the addition of
methylmagnesium bromide proceeds with excellent enantioselectivity and in good
yield, because of the mild reactivity of this organometallic reagent, allowing the
catalytic reaction to outcompete non catalyzed reaction.
Finally, we found that the symmetric bisquinoyl olefin hardly undergoes conjugate
addition with organomagnesium reagents, probably due to the steric hindrance of the
Lewis acid close to the alkene.
2.5. Experimental section
2.5.1. General information
All reactions using oxygen- and/or moisture-sensitive materials were carried out with
anhydrous solvents (vide infra) under a nitrogen atmosphere using oven dried
glassware and standard Schlenk techniques. Reactions were monitored by 1H NMR.
Purification of the products, when necessary, was performed by flash-column
chromatography using Merck 60 Å 230-400 mesh silica gel, Merck 90 active neutral
or VWR AnalaR NORMAPUR aluminum oxide basic. NMR data was collected on
Bruker Avance NEO 600 (1H at 600.0 MHz; 13C at 150.87MHz), equipped with a
Prodigy Cryo-probe and Varian VXR400 (1H at 400.0 MHz; 13C at 100.58 MHz),
equipped with a 5 mm z-gradient broadband probe. Chemical shifts are reported in
parts per million (ppm) relative to residual solvent peak (CDCl3, 1H: 7.26 ppm; 13C:
77.16 ppm). Coupling constants are reported in Hertz. Multiplicity is reported with the
usual abbreviations (s: singlet, bs: broad singlet, d: doublet, dd: doublet of doublets,
ddd: doublet of doublet of doublets, t: triplet, td: triplet of doublets, q: quartet, m:
multiplet). Exact mass spectra were recorded on a LTQ Orbitrap XL apparatus with
ESI ionization. Enantiomeric excesses (ees) were determined by chiral HPLC analysis
using a Shimadzu LC-10ADVP HPLC equipped with a Shimadzu SPD-M10AVP diode
array detector and by Waters Acquity UPC2 system with PDA detector and QDA mass
detector.
Unless otherwise indicated, reagents and substrates were purchased from commercial
sources and used as received. Solvents not required to be dry were purchased as
technical grade and used as received. Dry solvents were freshly collected from a dry
solvent purification system prior to use. Inert atmosphere experiments were

Chapter 2
34
performed with standard Schlenk techniques with dried (P2O5) nitrogen gas. Grignard
reagents were purchased from Sigma Aldrich and used as received (EtMgBr (3.0M in
Et2O), n-HexMgBr, i-ButMgBr (2.0 M in Et2O), CypMgBr (1.8M in Et2O). All other
Grignard reagents were prepared from the corresponding alkyl bromides and Mg
activated with I2 in Et2O and concentration was determine by NMR titration
method[43]. Chiral ligands (L5, L8, L9 and L12 - L16) were purchased from Sigma
Aldrich and Solvias. Chiral ligands L10 and L11 were prepared according to literature
method.[44] All reported compounds were characterized by 1H and 13C NMR and
compared with literature data. All new compounds were fully characterized by 1H and 13C NMR and HRMS techniques.
2.5.2. Synthesis of substrates
(E)-1,2-bis(2-benzoxazyl)-ethene (16)
In a 35 mL microwave vial, 0.14 g of malic acid (1.35 mmol,
1 equiv.) and 0.38 g of 2-hydroxyaniline (3.33 mmol, 2.5
equiv.) and 10 mL of toluene were added. The suspension
was stirred at 70 oC for 1 h. 10 mg of H3BO3 (0.16 mmol,
0.12 equiv.) and 10 mg of p-toluensulfonic acid (0.06
mmol, 0.04 equiv.) were added. The vial was heated in the microwave at 170 oC (300
W, high stirring) for 1 h. The solvent was removed under reduced pressure and the
crude was purified with a short flash-column chromatography (Al2O3, CH2Cl2).
Compound 16 was obtained as bright orange crystal (0.128 g, 27% yield).
1H-NMR (400 MHz, CDCl3), δ 7.80 (d, J = 9.4 Hz, 2H), 7.70 (s, 2H), 7.60 (d, J = 7.7
Hz, 2H), 7.47 – 7.33 (m, 4H). 13C NMR (101 MHz, Chloroform-d) δ 160.9, 150.8, 142.3, 126.6, 125.1, 123.8, 120.8,
110.9.
HRMS (ESI+): m/z calcd. for C16H10N2O2 ([M+H+]) 263.08150, found 263.08178.
(E)-1,2-bis(2-quinolyl)-ethene (17)
In a Schlenk under dry and inert atmosphere, equipped
with reflux condenser 0.79 g of 2-quinoline
carboxaldehyde (5 mmol, 1 equiv.) and 2-methylquinoline
(5 mmol, 1 equiv.) were dissolved. The Schlenk was placed
in a pre-heated bath at 140 oC and stirred for 20 min. The
reaction mixture was allowed to cool to rt and then poured into ice. A saturated solution
of NaHCO3 was added until pH 9 (gas formed at this stage). The reaction mixture was
extracted with toluene (3x20 mL) and dried with MgSO4. The solvent was removed
under reduced pressure. The crude was purified by flash-column chromatography
(Al2O3, pentane:AcOEt, 4:1) and crystallization from EtOH. Compound 17 was
obtained as bright yellow crystals (0.114 g, 20% yield).

Control of enantioselectivity in the addition of Grignard reagents to symmetric heteroaryl disubstituted olefins
35
1H NMR (400 MHz, Chloroform-d) δ 8.20 (d, J = 8.6 Hz, 2H), 8.12 (d, J = 8.5 Hz,
2H), 7.96 (s, 2H), 7.83 (m, 4H), 7.74 (td, J = 8.5, 1.4 Hz, 2H), 7.54 (td, J = 8.2, 1.2 Hz,
2H). 13C NMR (101 MHz, Chloroform-d) δ 156.0, 148.8, 135.2, 130.4, 130.0, 128.2, 128.1,
127.2, 120.1.
HRMS (ESI+): m/z calcd. for C20H14N2 ([M+H+]) 283.12298, found 283.12319.
2-vinylpyrimidine (27)
A solution of 2-chloropyrimidine (1.27 g, 8.00 mmol), potassium
vinyltrifluoroborate (1.29 g, 9.60 mmol), PdCl2(dppf)·CH2Cl2 (131 mg, 0.16
mmol), and Et3N (1.12 mL, 8.00 mmol) in i-PrOH (125 mL) was heated to
reflux for 16 h. The mixture was cooled to rt and partitioned between
CH2Cl2 (100 mL) and H2O (40 mL). The aqueous layer was separated and extracted
with CH2Cl2 (2 x 50 mL). The combined organic layers were washed with a saturated
solution of NaCl (100 mL), dried over MgSO4, filtered, and the solvent was removed
under reduced pressure. The pure compound 27 was obtained after flash
chromatography (SiO2, pentane:Et2O, 9:1) as a pale yellow oil (0.247 g, 2.3 mmol, 29%
yield)
1H NMR (400 MHz, Chloroform-d) δ 8.70 (d, J = 4.9 Hz, 2H), 7.13 (t, J = 4.9 Hz, 1H),
6.88 (dd, J = 17.3, 10.6 Hz, 1H), 6.62 (dd, J = 17.4, 1.6 Hz, 1H), 5.73 (dd, J = 10.6, 1.6
Hz, 1H). 13C NMR (101 MHz, Chloroform-d) δ 157.0, 144.0, 136.5, 123.8, 119.1.
HRMS (ESI+): m/z calcd. for C13H13N2 ([M+]) 107,06037, found 107.06005.
2.5.3. Catalytic asymmetric addition to 16
General procedure
In a heat dried Schlenk tube equipped with septum and magnetic stirring bar, the CuBr
·SMe2 (5 mol%), and (S,Sp)-L13 (6 mol%) were dissolved in CH2Cl2 (1mL/0.1mmol of
substrate) and stirred under nitrogen atmosphere for 15 min. The substrate (0.1 mmol,
1 equiv.) was added at once. After stirring for 5 min at rt the reaction mixture was
cooled to -78 °C and BF3·OEt2 (0-1.5 equiv.) was added. RMgBr (2.0 equiv) was diluted
in CH2Cl2 (0.6 ml total volume) and added over 4 hours. After stirring at -78 °C for 16h,
the reaction was quenched with MeOH (0.5 mL) followed by saturated aqueous NH4Cl
solution and warmed to rt. The reaction mixture was extracted with CH2Cl2 (3 × 10
mL). Combined organic phases were dried over MgSO4, filtered and solvents were
evaporated under reduced pressure. The oily crude was purified by column
chromatography on neutral Al2O2 using a mixture of pentane and EtOAc (9:1) as
eluent. The configuration of the products was not assigned.

Chapter 2
36
2,2'-(butane-1,2-diyl)bis(benzoxazole) (33a)
The reaction was performed with 16 (26 mg, 0.1 mmol, 1.0
equiv.), EtMgBr (0.2 mmol, 3.0 M in Et2O) diluited in
CH2Cl2 (0.6 mL total volume), CuBr·SMe2 (2.0 mg, 0.010
mmol, 5 mol%), (S,Sp)-L13 (4.2 mg, 0.012 mmol, 6 mol%),
in 1 mL CH2Cl2. Product 33a was obtained as yellow oil
(46.2 mg, 1.6 mmol, yield 79%, ee 71%). The absolute configuration of 33a was not
assigned. 1H NMR (400 MHz, Chloroform-d) δ 7.80 – 7.59 (m, 2H), 7.53 – 7.39 (m, 2H), 7.34
– 7.23 (m, 4H), 3.75 (qd, J = 7.5, 5.7 Hz, 1H), 3.60 (dd, J = 15.6, 7.5 Hz, 1H), 3.40 (dd,
J = 15.6, 7.2 Hz, 1H), 2.08 – 1.84 (m, 2H), 0.99 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 167.8, 164.5, 150.8, 150.7, 144.0, 141.2, 141.1,
124.7, 124.2 (2C), 119.8, 119.7, 110.5, 110.4, 39.2, 31.6, 26.3, 11.2.
HRMS (ESI+): m/z calcd. for C18H16N2O2 ([M+H+]) 293.1285, found 293.1288.
CSP-HPLC: (254nm, Chiralcel OB-H, n-heptane/i-PrOH = 95:5, 40 °C, 0.5 ml/min.),
tR = 13.83 min (major), tR = 12.38 min (minor).
2,2'-(hexane-1,2-diyl)bis(benzoxazole) (33b)
The reaction was performed with 16 (26 mg, 0.1 mmol,
1.0 equiv.), n-BuMgBr (0.2 mmol, 1.8 M in Et2O) diluted
in CH2Cl2 (0.6 mL total volume), CuBr·SMe2 (1.0 mg,
0.005 mmol, 5 mol%), (S,Sp)-L13 (4.1 mg, 0.006 mmol,
6 mol%), BF3·OEt2 (0.006 mL, 0.05 mmol, 0.5 equiv.)
in 1 mL CH2Cl2. Product 33b was obtained as colorless
oil (20.7 mg, 0.065 mmol, 65% yield, 40% ee). The
absolute configuration of 33b was not assigned.
1H NMR (400 MHz, Chloroform-d) δ 7.72 – 7.61 (m, 2H), 7.54 – 7.40 (m, 2H), 7.34 –
7.21 (m, 4H), 3.84 – 3.72 (m, 1H), 3.59 (dd, J = 15.6, 7.7 Hz, 1H), 3.39 (dd, J = 15.6, 7.0
Hz, 1H), 2.06 – 1.82 (m, 2H), 1.39 – 1.22 (m, 4H), 0.91 – 0.76 (m, 3H). 13C NMR (151 MHz, Chloroform-d) δ 168.2, 164.7, 151.0, 150.9, 141.4, 141.3, 124.82,
124.80, 124.3, 120.0, 119.9, 110.7, 110.6, 38.0, 33.3, 32.3, 29.1, 22.6, 14.0.
HRMS (ESI+): m/z calcd. for C20H20N2O2 ([M+H+]) 321,1598, found 321.1602.
CSP-HPLC: (254nm, Chiralcel OZ-H, n-heptane/i-PrOH = 95:5, 40 °C, 0.5
mL/min.), tR = 14.73 min (major), tR = 13.00 min (minor).

Control of enantioselectivity in the addition of Grignard reagents to symmetric heteroaryl disubstituted olefins
37
2,2'-(octane-1,2-diyl)bis(benzoxazole) (33c)
The reaction was performed with 16 (26 mg, 0.1 mmol, 1.0
equiv.), n-HexMgBr (0.2 mmol, 2.0 M in Et2O) diluted in
CH2Cl2 (0.6 mL total volume), CuBr·SMe2 (1.0 mg, 0.005
mmol, 5 mol%), (S,Sp)-L13 (4.1 mg, 0.006 mmol, 6 mol%),
BF3·OEt2 (0.006 mL, 0.05 mmol, 0.5 equiv.) in 1 mL
CH2Cl2. Product 33c was obtained as colorless oil (18.9 mg,
0.054 mmol, 54% yield, 23% ee). The absolute
configuration of 33c was not assigned.
1H NMR (400 MHz, Chloroform-d) δ 7.74 – 7.55 (m, 2H), 7.55 – 7.37 (m, 2H), 7.28
(m, 4H), 3.80 (qd, J = 7.6, 5.6 Hz, 1H), 3.59 (dd, J = 15.6, 7.7 Hz, 1H), 3.39 (dd, J =
15.6, 7.0 Hz, 1H), 1.94 (m, 2H), 1.48 – 1.04 (m, 8H), 0.83 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 168.2, 164.6, 151.0, 150.8, 141.4, 141.3, 124.8,
124.8, 124.3, 120.0, 119.9, 110.7, 110.5, 38.0, 33.5, 32.3, 31.7, 29.1, 26.9, 22.7, 14.1.
HRMS (ESI+): m/z calcd. for C22H24N2O2 ([M+H+]) 349.1911, found 349,1916.
CSP-HPLC: (254nm, Chiralcel AD-H, n-heptane/i-PrOH = 95:5, 40 °C, 0.5
mL/min.), tR = 23.43 min (major), tR = 22.33min (minor).
2,2'-(1-cyclopentylethane-1,2-diyl)bis(benzoxazole) (33d)
The reaction was performed with 16 (26 mg, 0.1 mmol, 1.0
equiv.), CypMgBr (0.2 mmol, 2.0 M in Et2O) diluted in
CH2Cl2 (0.6 mL total volume), CuBr·SMe2 (1.0 mg, 0.005
mmol, 5 mol%), (S,Sp)-L13 (4.1 mg, 0.006 mmol, 6 mol%),
BF3·OEt2 (0.006 mL, 0.05 mmol, 0.5 equiv.) in 1 mL
CH2Cl2. Product 33d was obtained as colorless oil (18.6 mg, 0.056 mmol, 56% yield,
6% ee). The absolute configuration of 33d was not assigned.
1H NMR (400 MHz, Chloroform-d) δ 7.69 – 7.56 (m, 2H), 7.52 – 7.35 (m, 2H), 7.32 –
7.19 (m, 4H), 3.73 – 3.58 (m, 2H), 3.45-3.35 (dd, 1H), 2.48-2.31 (m , 1H), 2.05 – 1.91
(m, 1H), 1.75 – 1.48 (m, 5H), 1.41 (d, J = 10.1 Hz, 2H). 13C NMR (101 MHz, Chloroform-d) δ 168.3, 165.2, 151.2, 151.0, 141.7, 141.6, 125.0,
125.0, 124.5, 120.3, 120.2, 111.0, 110.8, 44.5, 43.3, 32.1, 31.4, 30.8, 25.6, 25.4.
HRMS (ESI+): m/z calcd. for C21H20N2O2 ([M+H+]) 333.1598, found 333.1602.
CSP-HPLC: (254nm, Chiralcel OD-H, n-heptane/i-PrOH = 95:5, 40 °C, 0.5
mL/min.), tR = 16.64 min, tR = 21.86 min.

Chapter 2
38
2,2'-(propane-1,2-diyl)bis(benzoxazole) (33e)
The reaction was performed with 16 (26 mg, 0.1 mmol, 1.0
equiv.), MeMgBr (0.2 mmol, 3.0 M in Et2O) diluted in
CH2Cl2 (0.6 mL total volume), CuBr·SMe2 (1.0mg, 0.005
mmol, 5 mol%), (S)-L16 (4.1 mg, 0.006 mmol, 6 mol%),
BF3·OEt2 (0.02 mL, 0.15 mmol, 1.5 equiv.) in 1 mL CH2Cl2. Product 33e was obtained
as colorless oil (16.1 mg, 0.059 mmol, yield 59%, 97% ee). The absolute configuration
of 33e was not assigned.
1H NMR (400 MHz, Chloroform-d) δ 7.74 – 7.56 (m, 2H), 7.48 (ddd, J = 9.3, 5.5, 3.5
Hz, 2H), 7.31 (dq, J = 6.2, 4.2, 3.7 Hz, 4H), 3.89 (dp, J = 8.3, 6.9 Hz, 1H), 3.68 (dd, J
= 15.6, 6.2 Hz, 1H), 3.33 (dd, J = 15.6, 8.3 Hz, 1H), 1.59 (d, J = 7.0 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 168.7, 164.5, 151.0, 151.0, 141.4, 141.3, 125.0,
124.9, 124.4, 120.0, 119.9, 110.6, 110.6, 33.6, 32.6, 18.5.
HRMS (ESI+): m/z calcd. for C17H14O2N2Na([M+Na+]) 301.0949, found 301.0947.
CSP-HPLC: (254nm, Chiralcel OB-H, n-heptane/i-PrOH = 98:2, 40 °C, 0.5
mL/min.), tR = 21.41 min (major), tR = 18.79 min (minor).
2.5.4. Complexes CuBr·L10 and CuBr·L11
(R)-1-[(SP)-2-(Dicyclohexylphosphino)ferrocenyl]-ethyl-di(3,5-
xylyl)phosphine-CuBr complex (CuBr·L10)
Copper complex CuBr·L10 was synthesized according to the
literature procedure.[44]
1H NMR (CDCl3, 400 MHz):δ 7.33 (d, J = 9.2 Hz, 2H), 7.16 (d, J = 9.1 Hz, 2H), 6.97
(s, 1H), 6.89 (s, 1H), 4.33 (s, 1H), 4.29 (s, 1H), 4.21 (s, 1H), 4.02 (s, 5H), 3.57 (m, 1H),
2.57 (m, 1H), 2.29 (s, 6H), 2.19 (s, 6H), 2.03 – 0.87 (m, 25H).
13C NMR (CDCl3, 100.58 MHz): δ 138.1 (d, J = 9.3 Hz), 137.7 (d, J = 9.6 Hz), 132.5
(dd, J = 19.0, 8.2 Hz), 132.0 (d, J = 16.2 Hz), 131.8, 131.6 (d, J = 16.4 Hz), 131.6 , 130.1
(m), 128.7, 125.6, 93.6 (d, J = 24.4 Hz), 74.4 (d, J = 18.6 Hz), 73.4, 68.9, 39.4 (dd, J =
11.0, 5.7 Hz), 35.5 (m), 33.7 (d, J = 11.1 Hz), 31.8 (d, J = 10.9 Hz), 30.3 (dd, J = 14.3,
6.7 Hz), 29.8 , 28.1 (d, J = 16.5 Hz), 27.3 (d, J = 8.4 Hz), 26.8 (d, J = 12.3 Hz), 26.1 (d,
J = 25.5 Hz), 24.3, 21.4 (d, J = 19.8 Hz), 18.6.
31P NMR (CDCl3, 161.94 MHz): δ 13.47.
HRMS (ESI+): m/z calcd. for C40H52BrCuFeP2 ([M+H+]) 792.13676, found
792.13707.

Control of enantioselectivity in the addition of Grignard reagents to symmetric heteroaryl disubstituted olefins
39
(R)-1-[(SP)-2-(Dicyclohexylphosphino)ferrocenyl]-ethyl-di[3,5-bis-
(trifluoromethyl)phenyl] phosphine-CuBr complex (CuBr·L11)
Copper complex CuBr·L11 was synthesized according to the
literature procedure.[44] The analytical data were found to be in
accordance with those reported in the literature.[44]
1H NMR (CDCl3, 400 MHz) δ 8.28 (s, 2H), 7.89 (s, 2H), 7.85 (s,
1H), 7.37 (s, 1H), 4.30 (s, 1H), 4.23 (s, 1H), 4.18 (s, 5H), 4.12 (s,
1H), 3.86 (q, 1H), 1.0–2.0 (m, 25H).
31P NMR (CDCl3, 161.94 MHz): δ 14.31 (br. d, J = 155.3 Hz), −9.53 (br. d, J = 149.9
Hz). 19F NMR (CDCl3, 376.29 MHz): δ -63.1.
2.6. Bibliography [1] C. Wu, G. Yue, C. Duc-Trieu Nielsen, K. Xu, H. Hirao, J. Zhou, J. Am. Chem.
Soc. 2016, 138, 742–745.
[2] G. Berthon-Gelloz, T. Hayashi, in Boronic Acids, Wiley-VCH Verlag GmbH &
Co. KGaA, Weinheim, Germany, 2011, pp. 263–313.
[3] B. L. Feringa, M. Pineschi, L. A. Arnold, R. Imbos, A. H. M. De Vries, Angew.
Chemie - Int. Ed. 1997, 36, 2620–2623.
[4] B. L. Feringa, Angew. Chemie - Int. Ed. 1996, 35, 2374–2376.
[5] N. Krause, Angew. Chemie - Int. Ed. 1998, 37, 283–285.
[6] Z. Gao, S. P. Fletcher, Chem. Sci. 2016, 8, 641–646.
[7] A. L. Silva, R. A. Toscano, L. A. Maldonado, J. Org. Chem. 2013, 78, 5282–
5292.
[8] C. Hawner, K. Li, V. Cirriez, A. Alexakis, Angew. Chemie - Int. Ed. 2008, 47,
8211–8214.
[9] P. Garcia, N. Germain, S. Woodward, A. Alexakis, Synlett 2015, 26, 901–906.
[10] K. Endo, D. Hamada, S. Yakeishi, T. Shibata, Angew. Chemie - Int. Ed. 2013,
52, 606–610.
[11] T. L. May, M. K. Brown, A. H. Hoveyda, Angew. Chemie - Int. Ed. 2008, 47,
7358–7362.
[12] S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnaard, B. L. Feringa,
Chem. Rev. 2008, 108, 2824–52.
[13] P. Ortiz, F. Lanza, S. R. Harutyunyan, in Top. Organomet. Chem., Springer,
2016, pp. 99–134.
[14] S. Englard, H. C. Berger, J. Biol. Chem. 1958, 233, 1003–1009.
[15] B. Weiner, G. J. Poelarends, D. B. Janssen, B. L. Feringa, Chem. - A Eur. J.

Chapter 2
40
2008, 10094–10100.
[16] M. Sakamoto, T. Kaneko, Y. Orito, M. Nakajima, Synlett 2016, 9, 2477–2480.
[17] R. Shintani, K. Ueyama, I. Yamada, T. Hayashi, Org. Lett. 2004, 6, 3425–
3427.
[18] Y.-C. Chung, D. Janmanchi, H.-L. Wu, Org. Lett. 2012, 14, 2766–2769.
[19] W. L. Duan, Y. Imazaki, R. Shintani, T. Hayashi, Tetrahedron 2007, 63, 8529–
8536.
[20] R. Takechi, T. Nishimura, Chem. Commun. 2015, 51, 8528–8531.
[21] T. Korenaga, A. Ko, K. Shimada, J. Org. Chem 2013, 78, 9975–9980.
[22] B. Weiner, New Methods towards the Synthesis of β-Amino Acids - PhD Thesis,
University of Groningen, 2009.
[23] W. Schlenk, J. Schlenk, Ber. Dt. Chem. Ges. 1929, 62B, 920–924.
[24] J. S. Carey, D. Laffan, Pharm. Process Dev. Curr. Chem. Eng. Challenges 2011,
9, 39–65.
[25] S. D. Roughley, A. M. Jordan, J. Med. Chem. 2011, 54, 3451–3479.
[26] L. Rupnicki, A. Saxena, H. W. Lam, J. Am. Chem. Soc 2009, 131, 10386–
10387.
[27] G. Pattison, G. Piraux, H. Lam, J. Am. Chem. Soc 2010, 132, 14373–14375.
[28] A. A. Friedman, J. Panteleev, J. Tsoung, V. Huynh, M. Lautens, Angew. Chemie
- Int. Ed. 2013, 52, 9755–9758.
[29] I. N. Houpis, J. Lee, I. Dorziotis, A. Molina, B. Reamer, R. P. Volante, P. J.
Reider, Tetrahedron 1998, 54, 1185–1195.
[30] R. P. Jumde, F. Lanza, M. J. Veenstra, S. R. Harutyunyan, Science (80-. ).
2016, 325, 433–437.
[31] S. R. Harutyunyan, F. López, W. R. Browne, A. Correa, D. Peña, R. Badorrey, A.
Meetsma, A. J. Minnaard, B. L. Feringa, J. Am. Chem. Soc. 2006, 128, 9103–
9118.
[32] M. Krull, A. Lerch, Morschhaeaeuser, H. Beye, Norbert; Gethoeffer, H. Ritter,
Schmitz Sara, Espacenet - Bibliographic Data DE102006047618 (B3) ― 2007-
11-15, 2007.
[33] M. A. Fakhfakh, X. Franck, A. Fournet, B. Figadère, Synth. Commun. 2002,
32, 2863–875.
[34] “1,3-benzoxazole-2-carbaldehyde | CAS No. 62667-25-8 | Sigma-Aldrich,” can
be found under
https://www.sigmaaldrich.com/catalog/buildingblock/product/enamine/ena4
65562557?lang=en®ion=NL, 2017.
[35] F. Li, J. Zhuang, G. Jiang, H. Tang, A. Xia, L. Jiang, Y. Song, Y. Li, D. Zhu,
Chem. Mater. 2008, 20, 1194–1196.
[36] V. A. Online, Y. Hao, X. Dong, Y. Chen, 2014, 3468–3472.
[37] Z. Jamal, Y.-C. Teo, Synlett 2014, 25, 2049–2053.
[38] L. Xu, Z. Shao, L. Wang, H. Zhao, J. Xiao, Tetrahedron Lett. 2014, 55, 6856–

Control of enantioselectivity in the addition of Grignard reagents to symmetric heteroaryl disubstituted olefins
41
6860.
[39] A. K. Chatterjee, F. D. Toste, T. Choi, R. H. Grubbs, Adv. Synth. Catal. 2002,
344, 634–637.
[40] A. Saxena, B. Choi, H. W. Lam, J. Am. Chem. Soc. 2012, 134, 8428–8431.
[41] M. Li, R. Hua, Appl. Organometal. Chem. 2008, 22, 397–401.
[42] F. Lanza, Harnessing the Reactivity of Alkenyl Heteroarenes through Copper
Catalysis and Lewis Acids - PhD Thesis, University of Groningen, 2018.
[43] T. R. Hoye, B. M. Eklov, M. Voloshin, Org. Lett. 2004, 6, 2567–2570.
[44] R. Oost, J. Rong, A. J. Minnaard, S. R. Harutyunyan, Catal. Sci. Technol. 2014,
4, 1997–2005.

Chapter 2
42

Chapter 3
Asymmetric conjugate addition of Grignard reagents to
symmetric bispyridyl alkenes
Chiral pyridines are recurrent structural motives in medicinal compounds and
ligands used in organometallic reactions. Herein we report a methodology for the
asymmetric addition of Grignard reagents to Lewis acid activated symmetric
bispyridyl alkenes. While additions to 4-pyridyl substituted alkenes can be achieved
with good yields and excellent ees, additions to 2-pyridyl alkenes can only be achieved
in a racemic fashion. Additionally, a comparison between the abilities of the two
isomeric pyridines and benzoxazole to activate an alkene towards the CA is made.
Part of this chapter has been published: R. P. Jumde, F. Lanza, T. Pellegrini, S. R.
Harutyunyan, Nat. Commun. 2017, 8.

Chapter 3
44
3.1. Introduction
3.1.1. Importance of pyridines in medicinal chemistry
Pyridine derivatives are essential in medicinal chemistry. For instance, in the list of
FDA approved drugs, pyridine occupies the second place of most present nitrogen
containing-heterocycles (62 out of 640 drugs). Furthermore, more than 50% of the
drugs containing pyridine moiety has one substituent on the ring.[1]
Figure 3 Chiral pyridine-containing drugs and natural products.
For their frequent recurrence in pharmaceutical and natural compounds, chiral
molecules containing pyridine motive are very attractive for medicinal chemists
(Figure 1). The stereogenic center is often situated close to the ring: indeed, in many
chiral approved drugs, the chiral center is located in α-position respect to the
heteroarene.[1,2] In particular, molecules containing two or more pyridine rings,
bonded together by a two-carbon atom alkyl spacer, have displayed biological activity
as nervous system agent,[3–5] anti-infective agent[6] and respiratory agents[7] (Figure
2). For these reasons, the development of pharmaceutical compounds would profit
from a methodology capable to afford enantiomerically enriched pyridine derivatives.

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
45
Figure 4 Biologically active molecules containing two or more pyridine rings.
3.1.2. Utilization of bispyridyl compounds in chemistry
Besides their importance in medicinal chemistry, pyridine is a recurring moiety also in
organic and inorganic chemistry. The nitrogen of the pyridine ring can be an electron
donor and, therefore, pyridines have been widely employed as ligands for transition
metal catalysis.[8–13] In literature, multitude of bidentate ligands have been reported,
containing 2-pyridyl units. Among those, bipy (1, Figure 3) is one of the most
remarkable and has been applied in homogeneous catalysis[14–16] and metal extraction
from aqueous media[17]. Moreover, metallic complexes with 1 have been used as dyes
for photovoltaic panels.[18] Also a ligand containing four 2-pyridyl substituent, N4Py
(2), has been widely used to chelate transition metals used in redox reactions.[19–21]
Figure 5 Pyridine chelating ligands.
In the series of pyridine containing ligands, bpe (3, Figure 3) has been employed as
chelating ligand for metals. Pt(II)·3[22] as well as Pd(II)·3 complexes[23] have been
prepared. Bpe has been proven to dissociate easily in square planar complexes
[PtMe2·3]: because of the distortion of 3 caused by the broad bite angle, the complex
has a limited stability and can easily undergo reactions with halogens and HCl.[24]
[PtR2·3] complexes have been tested in the catalysis of hydrophenylation of alkenes
with scarce outcomes.[25]
Nevertheless, the N-oxide of 3, L1, was successfully employed in the copper-catalyzed
N-arylation of imidazoles (Scheme 1).[26] L1 has shown increased reactivity in

Chapter 3
46
comparison with its precursor and bidentate ligands with a shorter divider between the
pyridine moieties.
Scheme 24 Copper-catalyzed N-arylation of imidazoles.[26]
Additionally, 3 is a building block for the synthesis of macrocyclic ligands (for example,
4[27] and 5[28], Figure 4). The tridentate ligand 4 stabilizes d8 metal like Pt(II) and its
oxidative addition intermediate. The complexes Fe(III)Cl·5 and Mn(III)Cl·DMF·5
catalyze the epoxidation of styrenes, displaying higher activities than the ones carrying
other acyclic ligands.
Figure 6 Macrocyclic ligands 4[27] and 5[28].
Instead, 6 (Figure 5), bearing 4-pyridine units, cannot chelate the metals because the
lone pairs on the nitrogen atoms are too far from each other. Nevertheless, this
remarkable molecule can be a linker in binuclear catalysts used in Suzuki coupling[29]
and transfer hydrogenation of ketones[30] (respectively 7 and 9, Figure 5). In the
latter, the higher reactivity of the complex is due to interaction between the two Ru(II)-
NNN moieties.[30]

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
47
Figure 7 1,2-Bis(4-pyridyl)-ethane and catalysts obtained from it.[29,30]
The pyridine 6 is also largely used in the preparation of inorganic-organic
assemblies[31]: metal clusters are used to construct porous solids. In organically
functionalized zinc-substituted polyoxyvanadates, the geometry of the ligand plays a
key role in the determination of the final structure. In fact, chiral R and L helices as
well as a sinuate chain (Figure 6, source: Chang et al., 2007[31]) were prepared and
characterized. Furthermore, this molecule was also utilized in the synthesis of Metal
Organic Frameworks (MOFs)[32].

Chapter 3
48
Figure 8 Assemblies of zinc-substituted polyoxovanadates and zinc organoamine subunits (Source:
Chang et al., 2007[31]).
3.1.3. Asymmetric conjugate addition to vinyl pyridines
Given the interest in chiral pyridines, different methods for their synthesis have been
developed. The most common and straightforward is the direct functionalization of the
pyridyl ring, despite its need of superstoichiometric amounts of chiral reagents. A more
recent method exploit the ability of pyridyl substitutents can activate olefins towards

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
49
the addition of nucleophiles.[33,34] In first instance, Melchiorre et al. achieved the
alkylation of vinyl pyridines via the addition of α-amino radicals thanks to the use of
Brønsted acids, but only with moderate ee of the product (10, Scheme 2a).[34]
Scheme 25 ACA to vinyl pyridines.
In 2009, the group of Lam reported a highly enantioselective addition of hydrides to
β,β-disubstituted pyridines catalyzed by transition metals (Scheme 2b).[35] Vinyl-
pyridines have reduced affinity towards nucleophiles with respect to other α,β-
unsaturated compounds, in fact, they can activate the alkene only by induction and no
further mesomeric electron withdrawing effect is possible.[36] For this reason, even if
non-enantioselective CA have been reported,[37] the asymmetric addition of carbon
nucleophiles are more difficult to achieve. The first examples consist of addition of
arylboronic acids that involves the use of activated electron-deficient pyridines (like
13), rhodium catalysts and high temperature, and proceeds with high
enantioselectivities (Scheme 2c).[38,39]

Chapter 3
50
In 2017, our group reported a novel strategy for the alkylation of β-substituted
heteroaryl olefins via the copper(I)-catalyzed addition of Grignard reagents (Chapter
2, Paragraph 2.1.3.).[40] Although by using of BF3.OEt2 it was possible to activate a
wide range of vinyl heteroarenes, vinyl pyridines proved to be more challenging
substrates. However, we observed that both 2-pyridyl and 4-pyridyl alkenes can
undergo CA in the presence of higher loadings of LA and Grignard reagents (3.0 equiv.
of Lewis acid and 3.0 equiv. of RMgBr, vs . respectively 1.5 and 2.0 equiv. previously
reported for other hetereoarenes[41]). Interestingly, the behavior of the two pyridyl-
alkenes are remarkably different from each other (Scheme 3). 4-pyridyl olefins (15)
can be converted to the chiral alkane in the presence of TMSOTf (Scheme 3a), while
under the same conditions, 2-pyridines (17) require an electron withdrawing
substituent to afford the addition product 18. Moreover 2-pyridines generally prefer
BF3.OEt2 as additive (Scheme 3b), that denotes a lower reactivity of 2-pyridyl alkenes
compared to 15. With both pyridyl alkenes, excellent yields and enantioselectivities are
achieved.[41]
Scheme 26 Reactivity of 4- and 2-pyridyl alkenes towards the asymmetric addition of Grignard
reagents.[41]
In a similar way to what has been reported for the copper-catalyzed asymmetric
addition of Grignard reagents to other vinyl heteroarenes,[40] the proposed mechanism
involves:
1) The transmetallation of the organomagnesium reagent by the copper(I)
complex.
2) The formation of a π-complex between copper and the olefin.
3) The oxidative addition (Cu(I) oxidizes to Cu(III)).

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
51
4) The reductive elimination, where catalyst and the enolate of the product are
released.[41]
3.2. Aim Chiral compounds containing two or more pyridine rings are present in a cornucopia
of commercial drugs and natural products. Pyridines are also capable of coordinating
transition metals and can be found in bidentate and macrocyclic ligands (2-pyridines)
or are utilized in the preparation of multinuclear metallic catalysts, inorganic/organic
assemblies and MOFs. The introduction of a chiral center to these compounds can open
the possibilities for new ligands for asymmetric catalysis and the control of chirality in
supramolecular aggregates.
For this reason, our aim is to develop a methodology for synthesis of highly
enantioenriched pyridine derivatives through the asymmetric copper-catalyzed
addition of Grignard reagents to symmetric 1,2-bispyridyl substituted olefins
(Scheme 4). The achievement of enantioselectivity in the addition of highly reactive
organometallic reagents to 1,2-disubstituted alkenes is troublesome, due to the
presence of background reaction competing with the catalytic asymmetric pathway.
Scheme 27 Asymmetric addition of organomagnesium reagents to bispyridylalkenes.
3.3. Results and discussion
3.3.1. Asymmetric addition to symmetric 4-pyridyl alkenes
Our investigation began with the addition of ethylmagnesium bromide to 1,2-bis(4-
pyridyl)ethene (19). The catalyst CuBr·SMe2/L5 was used first in the initial screening
as it gave the best results in our previous work.[41] In this case, it is important to obtain
the product with good conversion, as it cannot be separated from the starting material
via either column chromatography, prep-TLC or crystallization. From the initial
solvent screening, CH2Cl2 was found to be the most suitable, as it afforded the product
20a with 82% conversion and 40% ee (entry 1, Table 1). On the contrary, reactions
in diethyl ether, MTBE or toluene only led to low conversion (entries 1-6, 8). We
observed that 19 was poorly soluble in these solvents, even at room temperature.
Curiously, increasing the reaction temperature to 50 °C led to a decrease in conversion

Chapter 3
52
(entry 4). The reaction in THF as solvent afforded the product in good yield, albeit as
a racemate (entry 7).
Table 6 Effect of the solvent on the asymmetric addition of ethylmagnesium bromide to 19.
Entry Solvent T (°C) Conversion (%)a ee (%)b
1 CH2Cl2c -78 82 40
2 Et2O -78 30 rac
3 Et2O d -78 0 -
4 Et2O -50 15 -
5 MTBE -78 0 -
6 Toluene -78 26 -
7 THF -78 89 rac
8 CH2Cl2:Et2O (9:1) -78 49 n.d.
a Conversion to 20a was determined by 1H-NMR. b Determined via CSP-HPLC. c
Performed with 1.1 equiv. of TMSOTf. d Reaction was performed with BF3.OEt2 (1.1
equiv.).
Afterwards, the effect of different Lewis acids in the activation of 19 was studied
(Table 2). The strength of the trimethylsilanes used for this purpose was determined
by the leaving group ability: TMSOTf>TMSI>TMSBr>TMSCl.

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
53
Table 7 Effect of the Lewis acid on the asymmetric addition of ethylmagnesium bromide to 19.
Entry LA (equiv.) Equiv.
EtMgBr
Conversion (%)a Yield (%)b ee
(%)c
1 TMSOTf (3.0) 3.0 86 67 9
2d TMSOTf (3.0) 3.0 97 86 rac
3 TMSOTf (1.1) 1.5 89 57 4
4 TMSOTf (2.2) 1.5 27 n.d. n.d.
5 TMSOTf (0.5) 20 0 - -
6 TMSI (3.0) 3.0 83e - -
7 TMSBr (3.0) 3.0 85e 67 33
8f TMSBr (1.5) 2.0 90e 53 46
9 f TMSBr (1.0) 2.0 45e n.d. n.d.
10 TMSCl (3.0) 3.0 48e n.d. 58
11g TMSCl (3.0) 3.0 32e n.d. 71
12 TMSCl (3.0) 1.5 + 1.5 44e n.d. 58
13 TMSCl (1.0) 3.0 <5 - -
14 TMSCl (5.0) 5.0 28e n.d. n.d.
15 BF3.OEt2 (1.1) 2.0 44h n.d. 68
16 BF3.OEt2 (3.0) 3.0 -h - -
17 BF2OTf (1.5) 2.0 14 - -
18 AlMe3 (1.0) 2.0 0 - -
19 Mg(OTf)2 (3.0) 3.0 0 - -
20 None 1.5 0 - -
a Conversion was determined by 1H-NMR. b Isolated yield. c Determined via CSP-HPLC. d EtMgBr was diluited in Et2O and added over 1h. e Product 21 formed f EtMgBr was
diluited in Et2O and added over 2h. g EtMgBr was diluited in Et2O and added over 3h. h Unidentified side products were formed.
Although TMSOTf was the best additive in our previous work[41], its use did not give
good enantioselectivities in this reaction. When using TMSOTf to activate the
substrate, the reaction proceeded with loss of enantioselectivity, either with fast or slow
addition of the organometallic reagent (entries 1-3, Table 2). When the amount of
EtMgBr was lower than the one of TMSOTf, the reaction reached only 27% conversion

Chapter 3
54
(entry 4). This suggests the partial degradation of the Grignard reagent with an excess
of Lewis acid. The use of a substoichiometric amount of TMSOTf was deleterious for
the reaction (entry 5): the substrate required at least one equivalent of Lewis acid to
undergo CA. With TMSI, the reaction did not reach full conversion and the β-hydride
transfer product was detected (ratio 21:20a = 80:20, entry 6). In the presence of
TMSBr, less reduction product was formed (entries 7-9), the least when 3.0 equiv. of
Lewis acid were used (ratio 21:20a = 20:80, entry 7). On the other hand, the increase
of the quantity of TMSBr caused a loss of ee (33% ee entry 7 vs. 46% entry 8). The
low reactivity of TMSCl granted good enantioselectivities (58% when the Grignard was
added in one shot and improved to 71% when added over 3h (entries 10 and 11), in
both cases with low conversions. The addition of 1.5 equiv. of the organometallic
reagent over 2h had no influence on the outcome of the reaction (entry 10 vs. entry
12). No product was obtained when only 1.0 equiv. of TMSCl was used (entry 13). It
was found that a large excess of the Lewis acid could inhibit the reaction (entry 14).
Instead, BF3.OEt2 (1.1 equiv. or 3.0 equiv.) favored the formation of side products
(entries 15 and 16). On the other hand, BF2OTf, AlMe3 or Mg(OTf)2 were not effective
in the activation of the starting material. Note: BF2OTf, obtained by reaction of
stoichiometric amounts of TMSOTf and BF3.OEt2 is reported to have a superior Lewis
acidity with respect to BF3.[42] AlMe3 was proven to be a valid Lewis acid additive in the
addition of organozinc reagents to aldehydes[43] and in the nickel catalyzed aryl-ether
bond cleavage[44].
Given the dependence of the enantioselectivity on the strength and amount of the
Lewis acid, we wondered about the presence of a non-catalyzed reaction. We tested the
reaction between substrate, Grignard reagent and Lewis acid in the absence of a
catalyst. Indeed, both TMSOTf and TMSBr were able to promote the addition of the
Grignard reagent. With TMSBr, the background reaction is slower than with TMSOTf
(48% conversion vs. 92%, entries 1 and 2, Table 3). For this reason, it is possible to
obtain higher enantioselectivities when using TMSBr and the reaction results were
more reproducible as well. Instead, BF3.OEt2 did not favor the racemic addition of
ethylmagnesium bromide to 19 (entry 3).

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
55
Table 8 Background addition of ethylmagnesium bromide to 19.
Entry L.A. (equiv.) Equiv. EtMgBr Conversion (%)a
1 TMSOTf (1.5) 2.0 92
2 TMSBr (1.5) 2.0 48
3 BF3.OEt2 (1.1) 2.0 <10
a Conversion was determined by 1H-NMR.
1.5 Equiv. of TMSBr and 2.0 equiv. of EtMgBr were selected as optimal conditions to
continue our screening. The Grignard reagent was diluted and added to the reaction
over 2h to avoid the uncatalyzed reaction. Sixteen ligands were tested, including
ferrocenyl diphosphines, L3 and L5-15, BINAP (L16), phosphoroamidites (L17-L18)
and Tröger’s base L19 (Table 4).

Chapter 3
56
Table 9 Ligand screening for the addition of ethylmagnesium bromide to 19.

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
57
Entry Ligand Conversion
(%)c
Yield 20a
(%)b
Ratio
20a:21c
ee (%)d
1 L3 88 69 100:0 52
2 L6 89 54 89:11 91
3 L7 66 n.d. 100:0 60
4 L8 <10e - - -
5 L9 83 81 100:0 rac
6 L10 92 76 100:0 -13
7 L11 ~80e 29 100:0 rac
8 L12 <10 - - -
9 L13 0 - - -
10 L14 82 68 100:0 -29
11 L15 <5 - - -
12f L16 15 - - -
13 g L17 92e - 0:100 -
14 g L18 >90 - 0:100 -
15h L19 23e n.d. - -
EtMgBr was diluted in Et2O and added over 2h. a Conversion was determined by 1H-
NMR. b Isolated yield c Ratio was determined by 1H-NMR. d Determined via CSP-HPLC. e Unidentified side products were formed. f Reaction was performed at ˗65 °C. g Reaction was performed with 24 mol% of ligand. h Reaction was performed at rt.
In the presence of the diphosphines L3, L5-8 and L14 the product was formed with
good conversion and yield. Using L6, the product 20a was obtained as an 89:11
mixture with 21 with an excellent 91% ee (entry 2, Table 4). The change of one of the
phosphine substituents from phenyl to cyclohexyl gave a decrease of enantioselectivity
(entry 7). Bulky ligands L9-11 afforded the product either as a racemate or with low
ee (entries 5-7). Complexes containing L12-13 and L15-19 were unable to catalyze
the addition of the Grignard reagent. Surprisingly, when employing monodentate
phosphoroamidites L17 or L18, only the β-hydride transfer product was obtained
(entries 13 and 14). In entries 12 and 15, higher temperatures were applied in order
to improve the solubility of the complexes CuBr·SMe2/L16 and L19 in the reaction
media, but in both cases no significant amount of product was obtained.
Hence, the scope of the Grignard reagents was studied with the catalyst formed by
CuBr·SMe2 and L6, which showed superior performance with respect to the
enantiocontrol of this reaction. To our delight, the addition of organomagnesium
reagents with a linear alkyl chain as well as with alkenyl Grignard reagents and γ-
branched ones proceeded with excellent enantioselectivities (products 20a, 20b, 20d
and 20e Scheme 5). This enantioselectivity is unprecedented for the asymmetric
addition of highly reactive nucleophiles to symmetric acyclic alkenes. Also, the addition

Chapter 3
58
of CypMgBr to 19 proceeded with good, but lower enantioselectivity (20f, 71% ee). The
presence of β-hydride transfer from the organometallic reagent to the substrate as a
side reaction affects the final yields (40-66%). As it is possible to get products of
addition of both α- and γ-branched Grignard reagents, it is surprising that β-branched
i-BuMgBr was found unreactive in this transformation. In addition, methylation and
phenylation of 19 were not successful either.
Scheme 28 Organomagnesium reagents scope. EtMgBr diluted in Et2O and added over 2h.
3.3.2. Asymmetric addition to symmetric 2-pyridyl alkenes
2-Pyridyl olefins are characterized by a lower reactivity with respect those bearing a 4-
pyridyl ring.[41] Correspondingly, substrate 22 displayed significantly different
reactivity compared to 19. In Table 5, it emerges that only selected Lewis acids,
TMSOTf, TMSI and TMSBr, can activate this substrate towards the addition of the
Grignard. Full conversion to the product was achieved using TMSOTF in non-
coordinating solvents, such as toluene or CH2Cl2. However, under these conditions, the
product was obtained with low ees (0-15%, entries 2-4, Table 5). No conversion to
23a was observed. A possible reason for the poor enantiocontrol is the non-catalyzed
addition of the Grignard (entry 5). The use of substoichiometric TMSOTf is not
sufficient to form 23a. The conversion decreased when using silyl-based Lewis acid
with poorer leaving groups (81% with TMSI and 45% with TMSBr, respectively entries
7 and 8). Using TMSBr in the catalytic reaction, 75% ee was achieved (entry 8). With
this Lewis acid it is possible to achieve good enantioselectivity because it is too weak to
activate the substrates towards the background reaction (entry 9). Lewis acid
additives like TMSCl and BF3.OEt2 are not hard enough to activate 22 towards the 1,4-
addition. An increase in the reaction temperature did not cause an increase in the
reactivity of the system, but rather inhibited the reaction. We suppose that this is due

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
59
to a partial degradation of the Grignard reagent caused by the Lewis acid at
temperatures above ˗78 °C (entries 5 and 9).
Table 10 Reactivity of substrate 22 towards the asymmetric addition of ethylmagnesium bromide
Entry L.A. (equiv.) T (°C) Conversion
(%)a
Yield (%)b ee (%)c
1d TMSOTf (1.5) -78 0 - -
2e TMSOTf (1.5) -78 67 n.d. 0
3 TMSOTf (1.5) -78 full 34 15
4 TMSOTf (3.0) -78 full 85 12
5f TMSOTf (3.0) -78 full n.d. -
6 TMSOTf (0.5) -78 0 - -
7 TMSI (3.0) -78 81 53 0
8 TMSBr (3.0) -78 45 19 75
9f TMSBr (3.0) -78 0 - -
10 TMSBr (3.0) -50 0 - -
11 TMSCl (3.0) -78 0 - -
12 BF3.OEt2 (3.0) -78 11 - -
13 BF3.OEt2 (3.0) -50 0 - -
In combination with 1.5 equiv. of LA, 2.0 equiv of EtMgBr were used. With 3.0 equiv.
of LA, 3.0 equiv. of EtMgBr. a Conversion was determined by 1H-NMR. b Isolated yield c Determined via CSP-HPLC. d Reaction was performed in Et2O. e Reaction was
performed in toluene. f Reaction was performed in the absence of catalyst.
We further evaluated the catalytic system by changing the copper(I)-source and the
ligand. Replacing CuBr·SMe2 with (CuOTf)2•toluene afforded product 23a with 51%
conversion and 5% ee (entry 1, Table 6). With complexes of CuTC/L5 (Structure in
Figure 7a; entry 2, Table 5) or CuBr·SMe2 and other chiral ferrocenyl diphosphines
(entries 3-7), no product was formed.

Chapter 3
60
Table 11 Catalyst screening in the addition of ethylmagnesium bromide to 22.
Entry Cu(I) source Ligand Conversion
(%)a
ee (%)b
1 (CuOTf)2•toluene L5 51 5
2 CuTC L5 <5 -
3 CuBr·SMe2 L3 0 -
4 CuBr·SMe2 L6 0 -
5 CuBr·SMe2 L7 0 -
6 CuBr·SMe2 L8 0 -
7 CuBr·SMe2 L14 0 -
a Conversion was determined by 1H-NMR. b Determined via CSP-HPLC.
Figure 9 Structure of CuTc and 1,2-bis(2-quinoyl)ethene.
Our investigations confirmed the difficult reactivity of this compound. In Chapter 2,
Paragraph 2.3.3., we discussed the low reactivity of compound 24 (Figure 7b) in
the copper catalyzed addition of EtMgBr. In both 22 and 24, the alkenyl substituent is
adjacent to the nitrogen on the heteroarene: it is feasible that, the Lewis acid bound to
the nitrogen creates an excessive hindrance to allow the coordination of the catalyst to
the double bond. On the contrary, the Grignard reagent can add to the alkene faster in
a racemic way without a catalyst assistance.
Nonetheless, we had discovered that 23 could be prepared upon simple addition of the
organomagnesium reagent to 22 in the presence of TMSOTf. Thus, we developed a
catalyst free procedure for the synthesis of racemic compounds 23b-d with yields of
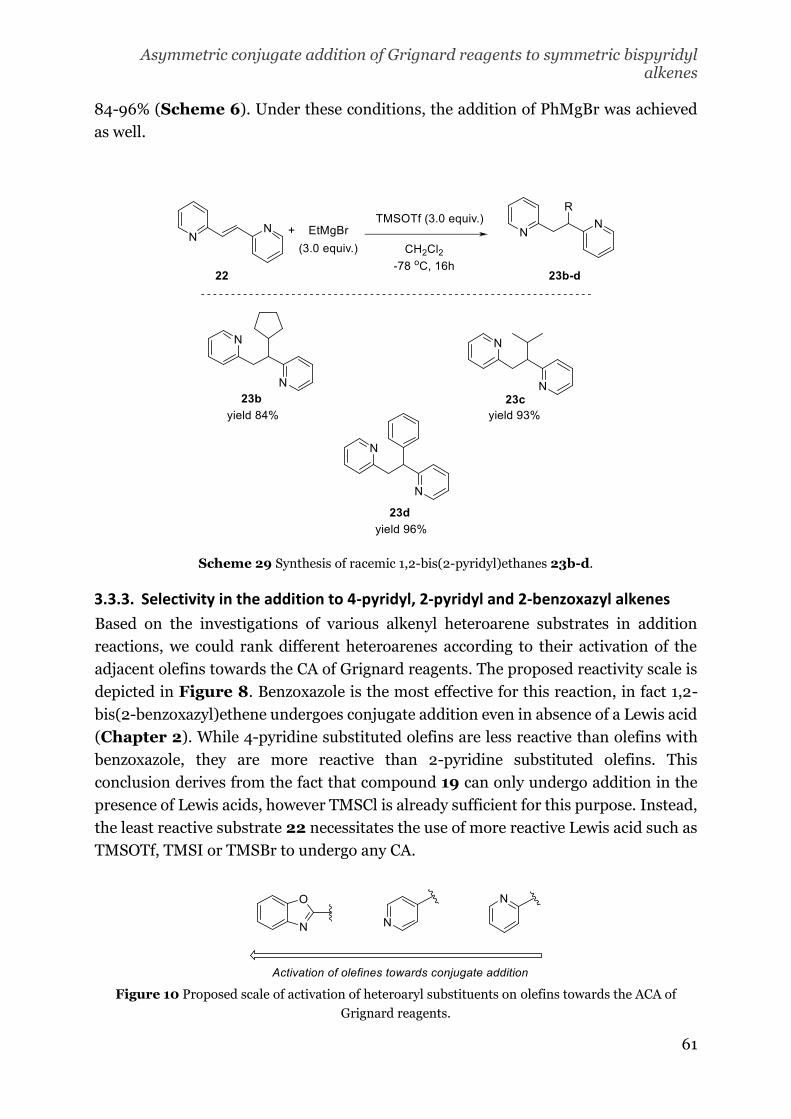
Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
61
84-96% (Scheme 6). Under these conditions, the addition of PhMgBr was achieved
as well.
Scheme 29 Synthesis of racemic 1,2-bis(2-pyridyl)ethanes 23b-d.
3.3.3. Selectivity in the addition to 4-pyridyl, 2-pyridyl and 2-benzoxazyl alkenes
Based on the investigations of various alkenyl heteroarene substrates in addition
reactions, we could rank different heteroarenes according to their activation of the
adjacent olefins towards the CA of Grignard reagents. The proposed reactivity scale is
depicted in Figure 8. Benzoxazole is the most effective for this reaction, in fact 1,2-
bis(2-benzoxazyl)ethene undergoes conjugate addition even in absence of a Lewis acid
(Chapter 2). While 4-pyridine substituted olefins are less reactive than olefins with
benzoxazole, they are more reactive than 2-pyridine substituted olefins. This
conclusion derives from the fact that compound 19 can only undergo addition in the
presence of Lewis acids, however TMSCl is already sufficient for this purpose. Instead,
the least reactive substrate 22 necessitates the use of more reactive Lewis acid such as
TMSOTf, TMSI or TMSBr to undergo any CA.
Figure 10 Proposed scale of activation of heteroaryl substituents on olefins towards the ACA of
Grignard reagents.

Chapter 3
62
However, all these conclusions are based on non-direct experiments. In none of the
experiments prior to this study, the reactivity of these moieties has been directly
compared. We have designed compound 25, 26 and 27 (Figure 9) to make a direct
comparison between the reactivity of different heteroarenes.
Figure 11 Structure of alkenes 25-27, bearing two different heteroaryl substituents in 1,2-position.
Alkene 25 was prepared by condensation reaction of the aldehyde 28 and γ-pinacoline
(29) with a low yield, probably due to the degradation of the product at the reflux
temperature of DMF. Compounds 26 and 24 were synthesized by condensation of 2-
methylbenzoxazol (30) and the pyridine carbaldehyde with LDA at low temperature,
to avoid the thermal decomposition of the product, and subsequent elimination with
MsCl (Scheme 7).
Scheme 30 Syntheses of 2-(2-(pyridin-4-yl)vinyl)pyridine (25), 2-(2-(pyridin-4-yl)vinyl)benzoxazole
(26) and 2-(2-(pyridin-2-yl)vinyl)benzoxazole (27).
The reactivity of alkenes 25, 26 and 27 towards the asymmetric addition of Grignard
was studied. The addition of ethylmagnesium bromide to 25 affords selectively 33 as
only product with all the Lewis acid tested (TMSOTf, TMSBr and BF3·OEt2, entries 1-
4, Table 2). The structure of 33 was confirmed by 1H-13C HMBC NMR spectroscopy
(Figure 10). High enantioselectivities were achieved with TMSBr (90% ee with ligand
L5 and 94% with L6 (entries 2 and 4). 1,4-Conjugate addition has occurred with
respect to the 4-pyridine moiety. The reaction occurred stereoselectively, meaning that
the steric bulk of the 2-pyridine was not sufficient to impede the interaction of the
transmetallated catalyst with of the olefin.

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
63
Table 12 Reactivity of 25 towards the asymmetric conjugate addition of ethylmagnesium bromide.
a Conversion was determined by 1H-NMR. b Isolated yield cDetermined via CSP-
HPLC.
Figure 12 1H-13C-HMBC (1H at 600.0 MHz, 13C at 150.75 MHz, CDCl3), δ. Expansion of the signal
corresponding to 2Je-Hf (blue), 3Je-Hg (blue),3Je-Hk (purple) and 2Jh-Hg (green). The coupling between the
quaternary carbon of the 2-pyridyl (Ce) and the Hk belonging to the ethyl substituent (purple)
confirms that ethyl and 2-pyridine moiety are bonded to the same carbon atom. Ch, instead, has a
coupling only with Hg and Hf.
Next, the reactivity of the alkenes 26 and 27 towards the copper-catalyzed addition of
EtMgBr was studied. A complex mixture of products was obtained when using
CuBr·SMe2/L6 and TMSBr as additive in the additions reactions using both
heteroarene substrates. Interestingly, conjugate addition with respect to the pyridyl
Entry Ligand LA Yield (%) b ee (%)c
1 L5 TMSOTf 66 39
2 L5 TMSBr 40 90
3 L5 BF3·OEt2 57 26
4 L6 TMSBr 70 94

Chapter 3
64
ring is observed when using L14 and TMSBr (entry 2, Table 8). Also here, the
structure of the compound was confirmed by 1H-13C HMBC NMR. Instead, addition of
ethylmagnesium bromide to 27, led to the formation of the isomer 35b both in the
absence and the presence of Lewis acids (TMSBr or BF3·OEt2, entries 3-5, Table 8).
The structure of 35b was determined by NMR. Based on the results illustrated in
Chapter 2 and Chapter 3 (Paragraphs 3.3.1. and 3.3.2.) and on the previous work
reported by our group[40,41], the expected order of reactivity is benzoxazylalkenes > 4-
pyridylalkenes > 2-pyridyl alkenes. A feasible explanation for the formation of
products 34 and 35, is that the pyridine nitrogen is more Lewis basic compared to the
benzoxazole nitrogen and therefore Lewis acid activation occurs preferentially on the
pyridine. However, the reactivity of the 2-pyridyl-alkenes/Lewis acid adduct is
troublesome and the formation of product 35a has been observed only with 3.0 equiv.
of TMSBr and rac-L16 as ligand (entry 6, Table 8).
Table 13 Reactivity of 26 and 27 towards the asymmetric conjugate addition of ethylmagnesium
bromide.
a Isolated yield b Determined via CSP-HPLC.
Entry Substrate Ligand LA Product Yield
(%) a
ee
(%)b
1 26 L6 TMSBr 34a 0 -
2 26 L14 TMSBr 34a 53 6
3 27 L14 TMSBr 35b 60 rac
4 27 L14 BF3·OEt2 35b 39 rac
5 27 L14 None 35b 39 rac
6 27 rac-L16 TMSBr (3.0) 35a:34b
(1:2)
32 -

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
65
Figure 13 a) Product 34a, correlation in 13C HMBC NMR (1H at 400 MHz, 13C at 101 MHz, CDCl3), 2JCc-Hd (blue), 3JCb-Hg (blue),) and
3JCd-Hb (green). The correlation between the Cb and Cc with proton Hd
confirms that the pyridyl moiety is binded to the methylene. Likewise, Cd is correlated with Hb of the
pyridine ring. b) Product 35b, correlation in 13C HMBC NMR (1H at 600.0 MHz, 13C at 150.75 MHz,
CDCl3), 3JCe-Hg (blue),) and 3JCd-Hf (green). The coupling of the quaternary carbon Ce with the protons of
the ethyl group, and the one of Cf with Hd indicate that the pyridine ring and the ethyl are bonded to
the same carbon atom.
3.3.4. Interaction of 1,2-bis(4-pyridyl)ethene (19) with TMSBr
To get more insight in the mode of activation of the substrate by the Lewis acid, the
nature of the activated species in solution was studied. The observations done in the
catalytic reactions were taken into account:
1) The alkene 19 was not soluble in ether and toluene at room temperature, but
soluble in CH2Cl2.
2) A precipitate was formed upon addition of TMSBr to the solution of 19 and the
catalyst at -78°C in CH2Cl2.
The color of the catalyst solution (CuBr·L5) changes from orange to bright red when
the substrate is introduced. The complex CuBr·L3 was chosen as model catalyst,
given that better NMR spectra can be recorded for this complex when compared to
that obtained with CuBr·L5 of CuBr·L6 (Figure 12a), and 19 was chosen as
substrate.
When 10 equiv. of 19 were added to the solution of CuBr·L3, the doublets in the 31P
spectrum corresponding to the phosphines shifted slightly up field (Figure 12b).
To the solution of CuBr·SMe2·L3 and 10 equiv. of 19 in CD2Cl2, 5 equiv. of TMSOTf
were added. The reaction became immediately turbid and the resolution of the
spectrum decreased because of the presence of a solid (Figure 12c). The chemical
shift of the two doublets remained unchanged, meaning that a significant part of
the complex CuBr·L3 was not affected by the addition of TMSOTf, but it rather
reacted with 19. On the contrary, when TMSOTf was added to CuBr·SMe2·L3 in
CD2Cl2, the complex was decomposed (Figure 12d).

Chapter 3
66
Figure 14 Reaction of the complex CuBr·L3 with 19 and TMSOTf, 31P-NMR (162 MHz, CD2Cl2), δ.
From these results, we can assume that substrate 19 protects the catalyst from
decomposition. It is reported in literature, treating 4,4’-bipyridine with 2.0 equiv. of
TMSOTf, the salt [Me3Si(4,4’-bipyridine)SiMe3]2+(OTf-)2 can be isolated.[45] We
suppose that the TMSOTf interacts with the pyridine moiety to form the salt
[Me3Si(19)SiMe3]2+(OTf-)2 or [(19)SiMe3]+OTf- (Figure 14) that precipitates from the
reaction mixture. This salt is a powder, insoluble in organic solvent and crystals could
not be grown. It was also not possible to perform NMR spectra because of its poor
solubility in organic solvents. However, the fact that [(19)SiMe3]+OTf- is less prone
than its precursor to be attacked by TMSBr and that the surnatant only contains free
19, suggests that the precipitate consists of the monosubstituted salt.
Figure 15 Structures of salts formed upon mixing 19 and TMSOTf.

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
67
3.4. Conclusions In summary, we have reported the asymmetric synthesis of alkanes bearing 4-pyridine
moieties with remarkable ees (70-97%). These compounds are highly valuable for
medicinal and supramolecular chemistry. Given the high reactivity of the two Michael-
partners used, the addition of the Grignard reagents proceeds with surprisingly high
enantioselectivity. Nevertheless, suppression of the β-hydride transfer remains a
challenge. The reactivity of an alkene bearing a 2-pyridyl moiety differs significantly
from its 4-pyridyl isomer. In this case, it is not possible to achieve good conversion and
good ees because of the fast non-catalyzed addition of RMgBr that competes with the
catalytic enantioselective pathway. However, it is possible to exploit the high activity
of TMSOTf to prepare racemic alkanes bearing two pyridine rings without the use of a
catalyst. These compounds could be used as ligands in transition metal catalysis.
By submitting alkenes bearing two different heteroaryl substituents to the addition
reaction, we compared directly the reactivity of 2-benzoxazyl, 4-pyridyl and 2-pyridyl
olefins. It is possible to outline that the reactivity of these heteroaryl olefins towards
nucleophilic conjugate addition in the presence of a Lewis acid is 4-pyridyl > 2-
benzoxazyl > 2-pyridyl.
We have tried to clarify the substrate activation mode by the Lewis acid. Upon addition
of TMSX, a precipitate is formed: we believe that this is the salt formed upon reaction
of bis-pyridine substrate and 1.0 equiv. of additive. However, we were not able to
characterize this salt due to its insolubility in organic solvents
3.5. Experimental section
3.5.1. General information
All reactions using oxygen- and/or moisture-sensitive materials were carried out with
anhydrous solvents (vide infra) under a nitrogen atmosphere using oven dried
glassware and standard Schlenk techniques. Reactions were monitored by 1H NMR.
Purification of the products, when necessary, was performed by flash-column
chromatography using Merck 60 Å 230-400 mesh silica gel, Merck 90 active neutral
or VWR AnalaR NORMAPUR aluminum oxide basic. NMR data was collected on
Bruker Avance NEO 600 (1H at 600.0 MHz; 13C at 150.87MHz), equipped with a
Prodigy Cryo-probe and Varian VXR400 (1H at 400.0 MHz; 13C at 100.58 MHz, 19F at
376.50 MHz, 31P at 161.97 MHz), equipped with a 5 mm z-gradient broadband probe.
Chemical shifts are reported in parts per million (ppm) relative to residual solvent peak
(CDCl3, 1H: 7.26 ppm; 13C: 77.16 ppm. CD2Cl2: 1H: 5.32 ppm; 13C: 54.0 ppm). Coupling
constants are reported in Hertz. Multiplicity is reported with the usual abbreviations
(s: singlet, bs: broad singlet, d: doublet, dd: doublet of doublets, ddd: doublet of
doublet of doublets, t: triplet, td: triplet of doublets, q: quartet, m: multiplet). Exact

Chapter 3
68
mass spectra were recorded on a LTQ Orbitrap XL apparatus with ESI ionization.
Enantiomeric excesses (ees) were determined by Chiral HPLC analysis using a
Shimadzu LC-10ADVP HPLC equipped with a Shimadzu SPD-M10AVP diode array
detector and by Waters Acquity UPC2 system with PDA detector and QDA mass
detector.
Unless otherwise indicated, reagents and substrates were purchased from commercial
sources and used as received. Solvents not required to be dry were purchased as
technical grade and used as received. Dry solvents were freshly collected from a dry
solvent purification system prior to use. Inert atmosphere experiments were
performed with standard Schlenk techniques with dried (P2O5) nitrogen gas. Grignard
reagents were purchased from Sigma-Aldrich and used as received (EtMgBr (3.0M in
Et2O), n-HexMgBr, i-BuMgBr (2.0 M in Et2O), CypMgBr (1.8M in Et2O). All other
Grignard reagents were prepared from the corresponding alkyl bromides and Mg
activated with I2 in Et2O and concentration was determine by NMR titration
method[46]. Chiral ligands L3, L5, L6, L9 and L10 - L16 were purchased from Sigma
Aldrich and Solvias. L7-L8 were prepared according to the literature procedure.[47]
L17-L18 were obtained from the laboratory of Ben L. Feringa.[48] L19 was obtained
from the laboratory of Jérôme Lacour.[49] All reported compounds were characterized
by 1H and 13C NMR and compared with literature data. All new compounds were fully
characterized by 1H and 13C NMR and HRMS techniques. The absolute configurations
of products were not assigned.
3.5.2. Synthesis of substrates
(E)-2-(2-(pyridin-4-yl)vinyl)pyridine (25)
In a 250ml three-necked round bottom flask, 4.8 ml (75 mmol, 3
equiv.) of 4-methyl pyridine were dissolved in 65 mL of dry DMF
and 4.2 g of freshly mashed KOH pellets (75 mmol 3 equiv.). The
reaction was stirred at 60 oC for 1 h. 2.38 g of 2-
pyridinecarboxaldehyde (25 mmol, 1 equiv.) were added and the reaction was refluxed
for 30 min. Once the reaction had reached room temperature, 20 mL of water were
added and the aqueous phase was extracted with 50 mL of CH2Cl2. The organic phase
was washed with water (3 x 20 mL). The organic phase was dried with MgSO4 and the
solvent was removed under reduced pressure. The product was obtained after flash-
column chromatography (Neutral Al2O3, pentane:EtOAc, 80:20→ 0:100, v/v) as a
white solid (583 mg, 0.78 mmol, yield 11 %). The NMR data are in agreement with the
ones present in literature.[50] 1H NMR (400 MHz, CDCl3) δ 8.64 (d, J = 4.8 Hz, 1H), 8.60 (d, J = 4.9 Hz, 2H), 7.70
(t, 1H), 7.58 (d, J = 16.1 Hz, 1H), 7.44 – 7.38 (m, 3H), 7.33 (d, J = 16.1 Hz, 1H), 7.22
(dd, 1H). 13C NMR (101 MHz, CDCl3) δ 154.4, 150.3, 149.9, 144.0, 136.8, 132.2, 130.0, 123.1,
123.0, 121.3.

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
69
3.5.3. General procedure for the synthesis of 2-(benzoxazol-2-yl)-1-
(pyridinyl)ethan-1-ol
In a Schlenk under dry and inert atmosphere, 1.1 mL of iPr2NH (7.2 mmol, 1.2 equiv.)
were dissolved in 18 mL of dry THF. A solution of n-BuLi (2.6 mL, 2.6 M in toluene,
6.6 mmol, 1.1 equiv.) were added dropwise at -78 oC. After 5 minutes, a solution of 0.72
mL of 2-methylbenzoxazole (6.0 mmol, 1.0 equiv.) in 2 mL of THF was added dropwise
and the reaction was allowed to stir for 15 min. A solution of the pyridine
carboxaldehyde (7.2 mmol, 1.2 equiv.) in 3 mL THF was added dropwise. The solution
was allowed to reach room temperature and stirred overnight. The reaction was
quenched with 5 mL of saturated aqueous NH4Cl solution and extracted with CH2Cl2
(3 x 10 mL). The organic phase was dried with MgSO4 and the solvent was removed
under reduced pressure. The crude was purified by column chromatography (Neutral
Al2O3, CH2Cl2 -> AcOEt -> EtOH -> MeOH).
2-(benzoxazol-2-yl)-1-(pyridin-4-yl)ethan-1-ol (31)
Product was obtained from 0.72 mL of 2-methylbenzoxaole
(6.0 mmol ) using 4.5 mL of n-BuLi 1.6 M solution in hexanes.
The product was obtained as a yellow solid (1.44 g, 6 mmol,
quantitative yield) without need of further purification.
1H NMR (400 MHz, Chloroform-d) δ 8.57 (d, J = 4.2 Hz, 2H), 7.67 (dd, J = 6.0, 3.2
Hz, 2H), 7.41 – 7.36 (m, 2H), 7.33 (dd, 2H), 5.35 (dd, 1H), 4.76 (bs, 1H), 3.40 – 3.17
(m, 2H). 13C NMR (101 MHz, Chloroform-d) δ 164.2, 151.5, 150.6, 150.1, 140.7, 125.2, 124.7,
120.8, 119.8, 110.7, 69.8, 37.6.
HRMS (ESI+): m/z calcd. for C14H13N2O2 ([M+H+]) 241.09715, found 241.09715.
2-(benzoxazol-2-yl)-1-(pyridin-2-yl)ethan-1-ol (32)
Product was obtained from 0.72 mL of 2-methylbenzoxaole (6.0
mmol ) using a 2.5 mL of n-BuLi 2.6 M solution in toluene and
recovered after column chromatography (Al2O3, CH2Cl2 ->
AcOEt -> EtOH -> MeOH) as brown solid (0.892 g, 3.72 mmol,
67% yield).
1H NMR (400 MHz, Chloroform-d) δ 8.57 (d, J = 4.4 Hz, 1H), 7.75 – 7.61 (m, 2H),
7.53 – 7.41 (m, 2H), 7.36 – 7.13 (m, 3H), 5.39 (m, J = 9.0, 4.7 Hz, 1H), 4.77 (d, J = 5.5
Hz, 1H), 3.51 (dd, J = 15.9, 4.2 Hz, 1H), 3.36 (dd, J = 15.8, 8.4 Hz, 1H). 13C NMR (101 MHz, Chloroform-d) δ 164.7, 160.5, 150.8, 148.8, 137.1, 124.9, 124.4,
122.9, 120.6, 119.8, 110.7, 102.5, 71.0, 37.3.
HRMS (ESI+): m/z calcd. for C14H13N2O2 ([M+H+]) 241.09715, found 241.09753

Chapter 3
70
3.5.4. General procedure for the synthesis of (E)-2-((pyridinyl)vinyl)benzoxazoles
In a round bottom flask, 1.44 g (6.0 mmol) of 2-(benzoxazol-2-yl)-1-(pyridinyl)ethan-
1-ol were dissolved in 25 mL CH2Cl2 and cooled to 0 oC. First 0,56 mL of MsCl (7.2
mmol) and then 2.00 mL of Et3N (14.4 mmol, 2.4 equiv.) were added dropwise
maintaining the reaction at 0 oC. The reaction was allowed to warm up to rt and stirred
overnight. The reaction was quenched with 10 mL of H2O, extracted with CH2Cl2 (3 x
20 mL). The organic phase was dried with MgSO4 and the solvent was removed under
reduced pressure. The crude was purified by column chromatography (Neutral Al2O3,
CH2Cl2 -> AcOEt).
(E)-2-(2-(pyridin-4-yl)vinyl)benzoxazole (26)
Product was obtained from 1.44 g of 31 (6.0 mmol) as a yellow
solid (821 mg, 3.69 mmol, 62% yield).
1H NMR (400 MHz, Chloroform-d) δ 8.71 – 8.65 (m, 2H),
7.78 – 7.73 (m, 2H), 7.71 (d, J = 16.4 Hz, 1H), 7.46 – 7.42 (m,
2H), 7.42 – 7.33 (m, 2H), 7.26 (d, J = 16.4 Hz, 1H). 13C NMR (101 MHz, Chloroform-d) δ 161.7, 150.8, 150.7, 142.4, 142.17, 136.5, 126.1,
125.0, 121.5, 120.5, 118.6, 110.7.
HRMS (ESI+): m/z calcd. for C14H13N2O2 ([M+H+]) 223.08659, found 223.08655.
(E)-2-(2-(pyridin-2-yl)vinyl)benzoxazole (27)
Product was prepared from 0.892 g of 32 (3.72 mmol), 0.34
mL of MsCl (4.4 mmol, 1.2 equiv.), 1.23 mL of Et3N ( 8.8
mmol, 2.4 equiv.) in 20 mL of CH2Cl2. 27 was obtained as a
yellow solid (0.576 g, 2.59 mmol, 70% yield).
1H NMR (400 MHz, Chloroform-d) δ 8.68 (d, J = 4.7 Hz, 1H), 7.81 (d, J = 15.9 Hz,
1H), 7.77 – 7.68 (m, 2H), 7.62 (d, J = 16.0 Hz, 1H), 7.58 – 7.50 (m, 1H), 7.45 (d, J = 7.8
Hz, 1H), 7.39 – 7.29 (m, 2H), 7.29 – 7.20 (m, 1H). 13C NMR (101 MHz, Chloroform-d) δ 162.6, 153.6, 150.7, 150.3, 142.4, 138.3, 136.9,
125.7, 124.7, 123.9, 123.9, 120.3, 118.1, 110.6.
HRMS (ESI+): m/z calcd. for C14H13N2O2 ([M+H+]) 223.08659, found 223.08705.
3.5.5. General procedure for the asymmetric addition to 19
In a heat dried Schlenk tube equipped with septum and magnetic stirring bar, the
CuBr·SMe2 (10 mol%), and (R,Sp)-L6 (12 mol%) were dissolved in CH2Cl2
(1mL/0.1mmol of substrate) and stirred under nitrogen atmosphere for 15 min. The
substrate (0.1 - 0.2 mmol, 1 equiv.) was added at once. After stirring for 5 min. at rt the
reaction mixture was cooled to -78 °C and TMSBr (1.5 equiv.) was added followed by

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
71
RMgBr (2.0 equiv.). After stirring at -78 °C for 16h, the reaction was quenched with
MeOH (0.5 mL) followed by saturated aqueous NH4Cl solution and warmed to RT.
Reaction mixture was extracted with CH2Cl2 (3 × 10 mL). Combined organic phases
were dried over MgSO4, filtered and solvents were evaporated on rotary evaporator.
The oily crude was purified by a small chromatography on neutral Al2O3 using a
mixture of pentane and EtOAc (9:1) as eluent to remove the complex, and subsequently
pure EtOAc. The product was obtained by filtration through basic Al2O3 using
CH2Cl2:EtOAc (1:1) as an eluent.
4-(1-(pyridin-4-yl)butan-4-yl)pyridine (20a) The reaction was performed with 0.1 mmol 1,2-bis(4-
pyridyl)ethene, EtMgBr (0.2 mmol, 3M in Et2O) diluted in Et2O
(0.6 mL total volume), CuBr·SMe2 (2.1 mg, 0.01 mmol, 10 mol%),
(R,Sp)-L6 (7.7 mg, 0.012 mmol, 12 mol%), TMSBr (0.02 mL, 0.15
mmol, 1.5 equiv.) in 1 mL CH2Cl2. Product 20a was obtained as
pale yellow oil (15.4 mg, 0.075 mmol, yield 75%, 91% ee). The absolute configuration
of 20a was not assigned. 1H NMR (400 MHz, Chloroform-d) δ 8.47 (d, J = 6.0 Hz, 2H), 8.41 (dd, J = 5.9 Hz,
2H), 6.99 (d, J = 6.1 Hz, 2H), 6.91 (d, J = 5.8 Hz, 2H), 3.00 – 2.90 (m, 1H), 2.86 – 2.69
(m, 2H), 1.83 – 1.58 (m, 2H), 0.80 (t, J =7.4 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 153.0, 150.0, 149.80, 148.7, 124.5, 123.3, 48.7,
42.0, 28.4, 12.0.
HRMS (ESI+): m/z calcd. for C16H17N2 ([M+H+]) 213.13863, found 213.13841.
CSP-HPLC: (254nm, Chiralcel OD-H, n-heptane:i-PrOH = 90:10, 40 °C, 0.5
mL/min.), tR = 41.96 min (major), tR = 3.21 min (minor).
4,4'-(hexan -1,2-diyl)dipyridine (20b)
The reaction was performed with 0.2 mmol 1,2-bis(4-
pyridyl)ethene, (0.2 mmol, 1.8M in Et2O) diluted in Et2O (1.2 mL
total volume), CuBr·(R,Sp)-L6 (16.9 mg, 0.02 mmol, 10 mol%),
TMSBr (0.04 mL, 0.30 mmol, 1.5 equiv.) in 2 mL CH2Cl2. Product
20b was obtained as pale yellow oil (27.8 mg, 0.12 mmol, yield
60%, 99% ee). The absolute configuration of 20b was not assigned. 1H NMR (400 MHz, Chloroform-d) δ 8.47 (d, J = 5.6 Hz, 2H), 8.41 (d, J = 5.7 Hz,
2H), 6.98 (d, J = 5.5 Hz, 2H), 6.90 (d, J = 5.5 Hz, 2H), 3.00 – 2.86 (m, 1H), 2.80 (d, J
= 8.7 Hz, 2H), 1.77 – 1.62 (m, 2H), 1.36 – 0.98 (m, 4H), 0.82 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 153.3, 150.0, 149.8, 148.7, 124.5, 123.2, 46.9,
42.3, 35.1, 29.6, 22.7, 14.0.
HRMS (ESI+): m/z calcd. for C16H21N2 ([M+H+]) 241.16993, found 241.17016.

Chapter 3
72
CSP-HPLC: (254nm, Chiralcel OJ-H, n-heptane:i-PrOH = 90:10, 40 °C, 0.5
mL/min.), tR = 28.52min (major), tR = 40.76 min (minor).
4-(1-(pyridin-4-yl)octan-4-yl)pyridine (20c)
The reaction was performed with 0.1 mmol 1,2-bis(4-
pyridyl)ethene, n-HexMgBr (0.2 mmol, 2M in Et2O) diluted in
Et2O (0.6 mL total volume), CuBr·SMe2 (2.1 mg, 0.01 mmol, 10
mol%), (R,Sp)-L6 (7.7 mg, 0.012 mmol, 12 mol%), TMSBr (0.02
mL, 0.15 mmol, 1.5 equiv.) in 1 mL CH2Cl2. Product 20c was
obtained as pale yellow oil (17.1 mg, 0.064 mmol, yield 64%, 94%
ee). The absolute configuration of 20c was not assigned. 1H NMR (400 MHz, Chloroform-d) δ 8.46 (d, J = 6.0 Hz, 2H), 8.41 (d, J = 6.1 Hz, 2H),
6.98 (d, J = 5.8 Hz, 2H), 6.90 (d, J = 6.0 Hz, 2H), 2.93 (m, 1H), 2.85 – 2.71 (m, 2H),
1.74 – 1.56 (m, 2H), 1.31 – 1.07 (m, 8H), 0.84 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 153.3, 150.0 (2C), 149.7 (2C), 148.8, 124.5 (2C),
123.3 (2C), 46.9, 42.3, 35.4, 31.7, 29.8, 27.4, 22.7, 14.1.
HRMS (ESI+): m/z calcd. for C18H25N2 ([M+H+]) 269.20123, found 269.20149.
CSP-HPLC: (254nm, Chiralcel OD-H, n-heptane:i-PrOH = 94:6, 40 °C, 0.5
mL/min.), tR = 46.13 min (major), tR min = 42,17 min (minor).
4,4'-(hept-6-ene-1,2-diyl)dipyridine (20d)
The reaction was performed with 0.2 mmol 1,2-bis(4-
pyridyl)ethene, (0.2 mmol, 2M in Et2O) diluted in Et2O (1.2 mL
total volume), CuBr·(R,Sp)-L6 (16.9 mg, 0.02 mmol, 10 mol%),
TMSBr (0.04 mL, 0.30 mmol, 1.5 equiv.) in 2 mL CH2Cl2. Product
20d was obtained as pale yellow oil (19.2 mg, 0.054 mmol, yield
54%, 90% ee). The absolute configuration of 20d was not
assigned. 1H NMR (400 MHz, Chloroform-d) δ 8.47 (d, J = 5.0 Hz, 2H), 8.41 (d, J = 4.8 Hz,
2H), 6.99 (d, J = 4.9 Hz, 2H), 6.89 (d, J = 4.9 Hz, 2H), 5.78 – 5.53 (m, 1H), 5.02 – 4.83
(m, 2H), 2.98 – 2.86 (m, 1H), 2.86 – 2.71 (m, 2H), 2.12 – 1.88 (m, 2H), 1.77 – 1.56 (m,
2H), 1.48 – 1.11 (m, 2H). 13C NMR (101 MHz, Chloroform-d) δ 153.0, 150.1 (2C), 149.8 (2C), 148.6, 138.2 (2C),
124.5, 123.3, 115.1 (2C), 46.9, 42.3, 34.8, 33.6, 26.7.
HRMS (ESI+): m/z calcd. for C17H21N2 ([M+H+]) 253.169938, found 253.17021.
CSP-HPLC: (254nm, Chiralcel OD-H, n-heptane:i-PrOH = 90:10, 40 °C, 0.5
mL/min.), tR = 35.68 min (major), tR = 33.40 min (minor).

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
73
4,4'-(5-methylhexane-1,2-diyl)dipyridine (20e)
The reaction was performed with 0.1 mmol 1,2-bis(4-
pyridyl)ethene, (0.2 mmol, 2M in Et2O) diluted in Et2O (0.6 mL
total volume), CuBr·SMe2 (2.1 mg, 0.01 mmol, 10 mol%), (R,Sp)-
L6 (7.7 mg, 0.012 mmol, 12 mol%), TMSBr (0.04 mL, 0.30 mmol,
1.5 equiv.) in 1 mL CH2Cl2. Product 20e was obtained as pale
yellow oil (10.3 mg, 0.040 mmol, yield 40%, 94% ee). The absolute configuration of
20e was not assigned. 1H NMR (400 MHz, Chloroform-d) δ 8.47 (d, J = 5.2 Hz, 2H), 8.41 (d, J = 5.1 Hz, 2H),
7.04 – 6.92 (m, 2H), 6.92 – 6.84 (m, 2H), 3.03 – 2.86 (m, 1H), 2.86 – 2.67 (m, 2H),
1.79 – 1.54 (m, 2H), 1.46 (m, 1H), 1.10 (m, 1H), 0.97 (m, 1H), 0.81 (dd, J = 6.6, 5.4 Hz,
6H). 13C NMR (101 MHz, Chloroform-d) δ 153.3, 150.0, 149.8, 148.7, 124.5, 123.3, 47.2,
42.4, 36.6, 33.3, 28.1, 22.8, 22.4.
HRMS (ESI+): m/z calcd. for C17H23N2 ([M+H+]) 255.18558, found 255.18597.
CSP-HPLC: (254nm, Chiralcel OD-H, n-heptane:i-PrOH = 95:5, 40 °C, 0.5 mL/min.),
tR = 54.39 min (major), tR = 50.44 min (minor).
4,4'-(5-methylhexane-1,2-diyl)dipyridine (20f)
The reaction was performed with 0.2 mmol 1,2-bis(4-
pyridyl)ethene, CypMgBr (0.2 mmol, 1.8 M in Et2O) diluted in
Et2O (1.2 mL total volume), CuBr·(R,Sp)-L6 (16.9 mg, 0.02 mmol,
10 mol%), TMSBr (0.04 mL, 0.30 mmol, 1.5 equiv.) in 2 mL
CH2Cl2. Product 20f was obtained as pale yellow oil (33.0 mg, 0.13
mmol, yield 66%, 72% ee). The absolute configuration of 20f was
not assigned. 1H NMR (400 MHz, Chloroform-d) δ 8.41 (d, J = 5.0 Hz, 2H), 8.34 (d, J = 4.9 Hz,
2H), 6.91 (t, J = 6.0 Hz, 2H), 6.79 (d, J = 1151.9 Hz, 2H), 3.16 (dd, J = 13.4, 3.9 Hz, 1H),
2.74 (dd, J = 13.4, 10.8 Hz, 1H), 2.57 (td, J = 10.3, 3.9 Hz, 1H), 2.24 – 2.00 (m, 2H),
1.64 – 1.21 (m, 6H), 1.04 – 0.85 (m, 1H). 13C NMR (101 MHz, Chloroform-d) δ 152.8, 149.8, 149.7, 148.9, 124.5, 123.7, 53.6,
45.5, 41.1, 31.9, 31.7, 25.4, 25.2.
HRMS (ESI+): m/z calcd. for C17H21N2 ([M+H+]) 253.16993, found 253.17008.
CSP-HPLC: (254nm, Chiralcel OD-H, n-heptane:i-PrOH = 85:15, 40 °C, 0.5
mL/min.), tR = 15.14 min (major), tR = 35.10 min (minor).
3.5.6. General procedure for the asymmetric addition to 22
In a heat dried Schlenk tube equipped with septum and magnetic stirring bar,
CuBr·SMe2 (10 mol%), and (R,Sp)-L5 (12 mol%) were dissolved in CH2Cl2 (0.1M
solution) and stirred under nitrogen atmosphere for 15 min. The substrate (0.1 - 0.2

Chapter 3
74
mmol, 1 equiv.) was added at once. After stirring for 5 min. at rt the reaction mixture
was cooled to -78 °C and TMSBr or TMSCl (1.5-3.0 equiv.) was added followed by
RMgBr (2.0-3.0 equiv.). After stirring at -78 °C for 16h, the reaction was quenched with
MeOH (0.5 mL) followed by saturated aqueous NH4Cl solution and warmed to RT.
Reaction mixture was extracted with CH2Cl2 (3 × 10 mL). Combined organic phases
were dried over MgSO4, filtered and solvents were evaporated on rotary evaporator.
The oily crude was purified by flash column chromatography on neutral Al2O3 using a
mixture of pentane and EtOAc (9:1 -> 4:1).
(S)-4-(1-(pyridin-2-yl)butan-4-yl)pyridine (23a)
The reaction was performed with 0.1 mmol 1,2-bis(4-
pyridyl)ethene, EtMgBr (0.2 mmol, 3M in Et2O), CuBr·SMe2 (2.1
mg, 0.01 mmol, 10 mol%), (R,Sp)-L5 (7.13 mg, 0.012 mmol, 12
mol%), TMSBr (0.04 mL, 0.3 mmol, 3.0 equiv.) in 1 mL CH2Cl2.
Product 23a was obtained as pale yellow oil (4.0 mg, 0.019 mmol,
yield 19%, 75% ee). The absolute configuration of 23a was not assigned. 1H NMR (400 MHz, Chloroform-d) δ 8.57 (d, J = 4.7 Hz, 1H), 8.51 (d, J = 4.9 Hz, 1H),
7.45 (m, 2H), 7.04 (ddd, 2H), 6.98 (d, J = 7.8 Hz, 1H), 6.89 (d, J = 7.8 Hz, 1H), 3.28 –
3.05 (m, 3H), 1.95 – 1.61 (m, 2H), 0.79 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 164.0, 160.8, 149.5, 149.3, 136.0, 136.0, 123.9,
123.8, 121.3, 121.0, 49.9, 44.2, 28.1, 12.2.
HRMS (ESI+): m/z calcd. for C14H16N2 ([M+H+]) 213.13863, found 213.13893.
CSP-HPLC: (254nm, Chiralcel OZ-H, n-heptane:i-PrOH = 95:5, 40 °C, 0.5 mL/min.),
tR = 17.81 min (major), tR = 19.78 min (minor).
3.5.7. General procedure for the racemic addition to 22
In a heat dried Schlenk tube equipped with septum and magnetic stirring bar, 1,2-
bis(2-pyridyl)-ethylene (0.8-0.89 mmol, 1 equiv) was dissolved in CH2Cl2. After
stirring for 5 min. at rt the reaction mixture was cooled to -78 °C and TMSBr (3.0
equiv.) was added, followed by RMgBr (3.0 equiv.). After stirring at -78 °C for 16h, the
reaction was quenched with MeOH (1 mL) followed by saturated aqueous NH4Cl
solution and warmed to rt. The reaction mixture was extracted with CH2Cl2 (3 × 10
mL). The combined organic phases were dried over MgSO4, filtered and solvents were
evaporated on rotary evaporator. The oily crude was purified by column
chromatography on neutral Al2O3 using a mixture of pentane and EtOAc (4:1-> 0:1).

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
75
2,2'-(3-methylbutane-1,2-diyl)dipyridine (23b)
The reaction was performed with 0.8 mmol 1,2-bis(2-pyridyl)ethene,
i-PrMgBr (2.4 mmol, 3M in Et2O), TMSOTf (0.45 mL, 2.4 mmol, 3.0
equiv.) in 10 mL CH2Cl2. Product 23b was obtained as colorless oil
(167 mg, 0.74 mmol, yield 93%).
1H NMR (400 MHz, Chloroform-d) δ 8.53 (d, J = 4.8 Hz, 1H), 8.45 (d, J = 5.0 Hz, 1H),
7.39 (dd, J = 7.6 Hz, 1H), 7.32 (dd, J = 8.6, 6.7 Hz, 1H), 6.99 (d, J = 5.2 Hz, 1H), 6.94
(d, J = 5.7 Hz, 1H), 6.85 (d, J = 7.8 Hz, 1H), 6.76 (d, J = 7.8 Hz, 1H), 3.39 – 3.14 (m,
3H), 2.22 – 2.03 (m, 1H), 1.08 (d, J = 6.8 Hz, 3H), 0.79 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ (ppm); 163.3, 161.3, 149.21, 149.19, 135.8, 135.6,
124.6, 123.8, 121.1, 120.8, 55.0, 41.1, 32.9, 21.0, 20.9.
HRMS (ESI+): m/z calcd. for C15H19N2 ([M+H+]), 227,15428 found 227,15443.
2,2'-(1-cyclopentylethane-1,2-diyl)dipyridine (23c)
The reaction was performed with 0.89 mmol 1,2-bis(2-
pyridyl)ethene, CypMgBr (2.7 mmol, 1.5M in Et2O), TMSOTf (0.51
mL, 2.7 mmol, 3.0 equiv.) in 10 mL CH2Cl2. Product 23c was
obtained as colorless oil (188 mg, 0.75 mmol, yield 84%).
1H NMR (400 MHz, Chloroform-d) δ (ppm); 8.52 (dd, J = 11.0, 3.9
Hz, 1H), 8.44 (d, J = 4.7 Hz, 1H), 7.37 (td, J = 7.6, 1.9 Hz, 1H), 7.31 (td, J = 7.7, 1.9 Hz,
1H), 7.02 – 6.90 (m, 2H), 6.81 (d, J = 7.8, 1.1 Hz, 1H), 6.71 (d, J = 7.8 Hz, 1H), 3.34 –
3.17 (m, 2H), 3.07 (td, J = 10.1, 4.7 Hz, 1H), 2.40 – 2.25 (m, 1H), 2.05 – 1.95 (m, 1H),
1.71 – 1.28 (m, 6H), 1.11 – 0.99 (m, 1H).
13C NMR (101 MHz, Chloroform-d) δ (ppm); 166.5 , 163.5 , 151.8 , 151.7 , 138.3 , 138.2
, 126.5 , 126.2, 123.6 , 123.3 , 56.8 , 48.1 , 45.8 , 34.1 , 34.0 , 27.9 , 27.7 .
HRMS (ESI+): m/z calcd. for C17H21N2 ([M+H+]) 253.16993, found 253.17041.
2,2'-(1-phenylethane-1,2-diyl)dipyridine (23d)
The reaction was performed with 0.8 mmol 1,2-bis(2-pyridyl)ethene,
CypMgBr (2.4 mmol, 3M in Et2O), TMSOTf (0.45 mL, 2.4 mmol, 3.0
equiv.) in 10 mL CH2Cl2. Product 23d was obtained as colorless oil
(224 mg, 0.86 mmol, yield 96%).
1H NMR (400 MHz, Chloroform-d) δ(ppm); 8.57 (d, J = 4.8 Hz, 1H),
8.50 (d, J = 4.8 Hz, 1H), 7.48 (d, J = 7.8 Hz, 1H), 7.39 (d, J = 7.2 Hz, 1H), 7.34 (d, J =
7.6 Hz, 2H), 7.23 (t, J = 7.6 Hz, 2H), 7.14 (d, J = 7.6 Hz, 2H), 7.05 (t, J = 6.8 Hz, 1H),
7.00 (d, J = 6.0 Hz, 1H), 6.92 (d, J = 7.8 Hz, 1H), 4.72 (d, J = 7.9 Hz, 1H), 3.80 (dd, J
= 13.6, 8.0 Hz, 1H), 3.52 (dd, J = 13.6, 7.7 Hz, 1H).

Chapter 3
76
13C NMR (101 MHz, Chloroform-d) δ 165.4 , 162.8 , 151.8(2C) , 146.0 , 138.9 , 138.5 ,
131.0 (2C), 130.8 (2C) , 129.0 , 126.6 , 126.1 , 123.9 , 123.6 , 55.9 , 46.2 .
HRMS (ESI+): m/z calcd. for C18H17N2 ([M+H+]) 261.13863, found 261.13880.
3.5.8. General procedure for the asymmetric addition to 25-27
In a heat dried Schlenk tube equipped with septum and magnetic stirring bar, the
CuBr·SMe2 (5 mol%), and (R,Sp)-L5, (R,Sp)-L6 or (S,Rp)-L14 (6 mol%) were dissolved
in CH2Cl2 (1mL/0.1mmol of substrate) and stirred under nitrogen atmosphere for 15
min. The substrate (0.1 mmol, 1 equiv.) was added at once. After stirring for 5 min at
rt the reaction mixture was cooled to -78 °C and EtMgBr (2.0 equiv.) was diluted in
CH2Cl2 to a total volume of 0.6 mL and added over 2 h. After stirring at -78 °C for 18
h, the reaction was quenched with MeOH (0.5 mL) followed by saturated aqueous
NaHCO3 solution and warmed to rt. The reaction mixture was extracted with CH2Cl2
(3 × 10 mL). Combined organic phases were dried over MgSO4, filtered and solvents
were evaporated on rotary evaporator. The oily crude was purified by flash column
chromatography on neutral Al2O3 using a mixture of pentane and EtOAc as eluent.
2-(1-(pyridin-4-yl)butan-2-yl)pyridine (33)
The reaction was performed with 0.1 mmol 25, TMSBr (0.15
mmol, 1.5 equiv), EtMgBr (3M in Et2O, 0.2 mmol, 2.0 equiv),
CuBr·SMe2 (0.01 mmol, 10 mol%), ligand (R,Sp)-L6 (0.012
mmol, 12 mol%) in 1mL CH2Cl2. After purification by flash
column chromatography (neutral Al2O3, pentane: EtOAc, 9:1 ->
1:1). Product 33 was obtained as pale-yellow oil (14.9 mg, 0.07 mmol, 70% yield, 92%
ee). The absolute configuration of 33 was not assigned. 1H NMR (400 MHz, CDCl3), δ 8.59 – 8.54 (m, J = 5.5 Hz, 1H), 8.40 – 8.31 (d, 2H),
7.53 – 7.44 (m, 1H), 7.12 – 7.05 (m, 1H), 6.96 – 6.86 (m, 3H), 3.11 – 3.00 (m, 2H), 2.99
– 2.86 (m, 2H), 1.87 – 1.67 (m, 1H), 0.84 – 0.74 (t, 3H). 13C NMR (101 MHz, CDCl3), δ 163.1, 149.9, 149.64, 149.58, 136.2, 124.6, 123.6, 121.6,
50.8, 41.1, 28.2, 12.1.
HRMS (ESI+): m/z calcd. for C14H17N2 ([M+H+]) 213,13863, found 213,13883.
CSP-HPLC: (210 nm, Chiralcel OZ-H, n-heptane:i-PrOH = 95:5, 40 °C, 1.0 mL/min.),
tR = 19.11 min (major), tR = 21.65 min (minor).
2-(1-(pyridin-4-yl)butan-2-yl)benzoxazole (34a)
The reaction was performed with 0.1 mmol 26, EtMgBr (0.2
mmol, 3.0 M in Et2O) diluted in CH2Cl2 (0.6 mL total volume),
CuBr·SMe2 (1.0 mg, 0.005 mmol, 5 mol%), (S,Rp)-L14 (4.1 mg,
0.006 mmol, 6 mol%), TMSBr (0.02 mL, 0.15 mmol, 1.5
equiv.) in 1 mL CH2Cl2. After purification by flash column

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
77
chromatography (neutral Al2O3, pentane:EtOAc, 4:1) and filtration on K2CO3, the
product 34a was obtained as colorless oil (13.3 mg, 0.053 mmol, 53% yield, 65% ee).
1H NMR (400 MHz, Chloroform-d) δ 8.44 (d, J = 6.0 Hz, 2H), 7.70 – 7.59 (m, 1H),
7.51 – 7.35 (m, 1H), 7.33 – 7.26 (m, 2H), 7.07 (d, J = 6.0 Hz, 2H), 3.33 – 3.18 (m, 2H),
3.05 (dd, J = 13.1, 6.1 Hz, 1H), 1.99 – 1.76 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 168.3, 150.7, 150.0, 148.2, 141.2, 124.8, 124.4,
124.3 (2C), 119.9, 110.5, 102.5, 42.7, 38.7, 26.6, 11.8.
HRMS (ESI+): m/z calcd. for C16H17N2O ([M+H+]) 253.1335, found 253.1338.
CSP-HPLC: (254nm, Chiralcel AD-H, n-heptane/i-PrOH = 98:2, 40 °C, 0.5
mL/min.), tR = 58.97 min (major), tR = 64.05 min (minor).
2-(2-(pyridin-2-yl)butyl)benzoxazole (35b)
The reaction was performed with 0.1 mmol 27, EtMgBr (0.2
mmol, 3.0 M in Et2O) diluted in CH2Cl2 (0.6 mL total volume),
CuBr·SMe2 (1.0 mg, 0.005 mmol, 5 mol%), (S,Rp)-L14 (4.1 mg,
0.006 mmol, 6 mol%), TMSBr (0.02 mL, 0.15 mmol, 1.5 equiv.)
in 1 mL CH2Cl2. After purification by flash column
chromatography (neutral Al2O3, pentane:EtOAc, 25:1 -> 9:1) and filtration on K2CO3,
the product 35b was obtained as colorless oil (13.3 mg, 0.060 mmol, 60% yield,
racemic). 1H NMR (400 MHz, Chloroform-d) δ 8.57 (d, J = 4.2 Hz, 1H), 7.66 – 7.59 (m, 1H),
7.55 (td, J = 7.7, 1.8 Hz, 1H), 7.49 – 7.35 (m, 1H), 7.38 – 7.22 (m, 2H), 7.15 (d, J = 7.8
Hz, 1H), 7.10 (ddd, J = 7.5, 4.8, 1.2 Hz, 1H), 3.48 – 3.22 (m, 3H), 1.98 – 1.72 (m, 2H),
0.83 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 166.3, 162.7, 150.7, 149.7, 141.5, 136.3, 124.5,
124.1, 123.4, 121.7, 119.7, 110.4, 47.3, 34.1, 28.3, 11.9.
HRMS (ESI+): m/z calcd. for C16H17N2O ([M+H+]) 253.1335, found 253.1336.
CSP-HPLC: (254nm, Chiralcel OB-H, n-heptane:i-PrOH = 99:1, 40 °C, 0.5 ml/min.),
tR = 21.44 min, tR = 28.70 min.
3.5.9. Procedure for the NMR studies about interaction between catalyst, 19 and
TMSOTf
a) Preparation of Sample A: under inert atmosphere, 7.4 mg of complex Cu·L3
(0.010 mmol) were dissolved in 0.6 mL of CD2Cl2. The complex was transferred
in a dry NMR tube (Sample A) and 1H, 13C and 31P NMR spectra were recorded
(1H at 400.0 MHz; 13C at 100.6 MHz, 31P at 161.9 MHz).
b) Preparation of Sample B: to Sample A, 18.2 mg of 19 (0.1 mmol) were added
and 1H, 13C and 31P NMR spectra were recorded (1H at 400.0 MHz; 13C at 100.6
MHz, 31P at 161.9 MHz).

Chapter 3
78
c) Preparation of Sample C: to Sample B, 9 μL of TMSOTf (0.05 mmol) were
added. 1H, 13C and 31P NMR spectra were recorded (1H at 400.0 MHz; 13C at
100.6 MHz, 31P at 161.9 MHz).
d) Preparation of Sample D: under inert atmosphere, 7.4 mg of complex Cu/L3
(0.010 mmol) were dissolved in 0.6 mL of CD2Cl2. The complex was transferred
in a dry NMR tube and 18 μL of TMSOTf (0.12 mmol) were added. 1H, 13C and 31P NMR spectra were recorded (1H at 400.0 MHz; 13C at 100.6 MHz, 31P at 161.9
MHz).
3.5.10. Complexes CuBr·L7 and CuBr·L8
(R)-1-[(SP)-2-(Dicyclohexylphosphino)ferrocenyl]-ethyl-di(3,5-
xylyl)phosphine-CuBr complex (CuBr·L7)
Copper complex CuBr·L7 was synthesized according to the
literature procedure.[47]
1H NMR (CDCl3, 400 MHz):δ 7.33 (d, J = 9.2 Hz, 2H), 7.16 (d, J
= 9.1 Hz, 2H), 6.97 (s, 1H), 6.89 (s, 1H), 4.33 (s, 1H), 4.29 (s, 1H),
4.21 (s, 1H), 4.02 (s, 5H), 3.57 (m, 1H), 2.57 (m, 1H), 2.29 (s, 6H), 2.19 (s, 6H), 2.03 –
0.87 (m, 25H).
13C NMR (CDCl3, 100.58 MHz): δ 138.1 (d, J = 9.3 Hz), 137.7 (d, J = 9.6 Hz), 132.5
(dd, J = 19.0, 8.2 Hz), 132.0 (d, J = 16.2 Hz), 131.8, 131.6 (d, J = 16.4 Hz), 131.6 , 130.1
(m), 128.7, 125.6, 93.6 (d, J = 24.4 Hz), 74.4 (d, J = 18.6 Hz), 73.4, 68.9, 39.4 (dd, J =
11.0, 5.7 Hz), 35.5 (m), 33.7 (d, J = 11.1 Hz), 31.8 (d, J = 10.9 Hz), 30.3 (dd, J = 14.3,
6.7 Hz), 29.8 , 28.1 (d, J = 16.5 Hz), 27.3 (d, J = 8.4 Hz), 26.8 (d, J = 12.3 Hz), 26.1 (d,
J = 25.5 Hz), 24.3, 21.4 (d, J = 19.8 Hz), 18.6.
31P NMR (CDCl3, 161.94 MHz): δ 13.47.
HRMS (ESI+): m/z calcd. for C40H52BrCuFeP2 ([M+H+]) 792.13676, found
792.13707.
(R)-1-[(SP)-2-(Dicyclohexylphosphino)ferrocenyl]-ethyl-di[3,5-bis-
(trifluoromethyl)phenyl] phosphine-Cu complex (CuBr·L8)
Copper complex CuBr·L8 was synthesized according to the
literature procedure.[47] The analytical data were found to be in
accordance with those reported in the literature.11
1H NMR (CDCl3, 400 MHz): δ 8.28 (s, 2H), 7.89 (s, 2H), 7.85 (s, 1H), 7.37 (s, 1H),
4.30 (s, 1H), 4.23 (s, 1H), 4.18 (s, 5H), 4.12 (s, 1H), 3.86 (q, 1H), 1.0–2.0 (m, 25H).

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
79
31P NMR (CDCl3, 161.94 MHz): δ 14.31 (br. d, J = 155.3 Hz), −9.53 (br. d, J = 149.9
Hz). 19F NMR (CDCl3, 376.29 MHz): δ -63.1.
3.6. Bibliography
[1] E. Vitaku, D. T. Smith, J. T. Njardarson, J. Med. Chem. 2014, 57, 10257–10274.
[2] J. S. Carey, D. Laffan, Pharm. Process Dev. Curr. Chem. Eng. Challenges 2011,
9, 39–65.
[3] A. Gomtsyan, R. G. Schmidt, E. K. Bayburt, G. A. Gfesser, E. A. Voight, J. F.
Daanen, D. L. Schmidt, M. D. Cowart, H. Liu, R. J. Altenbach, et al., J. Med.
Chem. 2016, 50, 4926–4947.
[4] E. A. Voight, J. F. Daanen, R. G. Schmidt, A. Gomtsyan, M. J. Dart, P. R. Kym, J.
Org. Chem 2016, 81, 12060–12064.
[5] S. Kang, W. Tang, H. Li, G. Chreifi, P. Martaek, L. J. Roman, T. L. Poulos, R. B.
Silverman, J. Med. Chem. 2014, 57, 4382–4392.
[6] A. Usta, A. Yaşar, N. Yayli, Ş. A. Karaoǧlu, N. Yayli, Turkish J. Chem. 2009, 33,
621–632.
[7] P. J. Choi, H. S. Sutherland, A. S. T. Tong, A. Blaser, S. G. Franzblau, C. B.
Cooper, M. U. Lotlikar, A. M. Upton, J. Guillemont, M. Motte, et al., Bioorg. Med.
Chem. Lett. 2017, 27, 5190–5196.
[8] A. Hayashi, M. Okazaki, F. Ozawa, R. Tanaka, Organomet. 2007, 26, 5246–
5249.
[9] K. M. Engle, J.-Q. Yu, J. Org. Chem. 2013, 78, 8927–8955.
[10] G. Chelucci, Chem. Soc. Rev. 2006, 35, 1230–1243.
[11] R. Chinchilla, C. Naera, Chem. Soc. Rev. Chem. Soc. Rev 2011, 40, 5084–5121.
[12] D. M. Peloquin, T. A. Schmedake, Coord. Chem. Rev. 2016, 323, 107–119.
[13] A. J. Pardey, C. Longo, Coord. Chem. Rev. 2010, 254, 254–272.
[14] C. Schneider, A. R. Sreekanth, E. Mai, Angew. Chemie - Int. Ed. 2004, 43, 5691–
5694.
[15] X. Wang, Y. Huang, Y. Xu, X. Tang, W. Wu, H. Jiang, J. Org. Chem. 2017, 82,
2211–2218.
[16] Y. Hayashi, S. Santoro, Y. Azuma, F. Himo, T. Ohshima, K. Mashima, J. Am.
Chem. Soc. 2013, 135, 6192–6199.
[17] K. Kavallieratos, J. M. Rosenberg, J. C. Bryan, Inorg. Chem. 2005, 44, 2573–
2575.
[18] A. Adeloye, P. Ajibade, Molecules 2014, 19, 12421–12460.
[19] Q. Li, W. R. Browne, G. Roelfes, Inorg. Chem. 2011, 50, 8318–8325.
[20] D. Wang, M. Zhang, P. Bü, L. Que, J. Am. Chem. Soc 2010, 132, 7638–7644.
[21] K. J. Young, M. K. Takase, G. W. Brudvig, Inorg. Chem. 2013, 52, 7615–7622.

Chapter 3
80
[22] W. Kleibhoemer, B. Krebs, A. T. M. Marcelis, J. Reedelijk, J. L. van der Veer,
Inorg. Chem. Acta 1983, 75, 45–50.
[23] G. R. Newkome, V. K. Gupta, H. C. R. Taylor, F. R. Fronczek, Organomet. 1984,
3, 1549–1554.
[24] M. Safa, M. C. Jennings, R. J. Puddephatt, Organomet. 2011, 2, 5625–5635.
[25] B. A. Mckeown, H. E. Gonzalez, T. Michaelos, T. B. Gunnoe, T. R. Cundari, R. H.
Crabtree, M. Sabat, Organomet. 2013, 32, 3903–3913.
[26] L. Liang, Z. Li, X. Zhou, Org. Lett. 2009, 11, 3294–3297.
[27] A. N. Vedernikov, M. Pink, K. G. Caulton, J. Org. Chem. 2003, 68, 4806–4814.
[28] L. Yang, Z. Wu, L. Liang, X. Zhou, J. Organomet. 2009, 694, 2421–2426.
[29] S. Kuan Yen, L. Koh, H. V. Huynh, T. S. A. Hor, Chem. Asian. J. 2008, 3, 1649–
1656.
[30] T. Liu, H. Chai, L. Wang, Z. Yu, Organomet. 2017, 36, 2914–2921.
[31] Y. Qi, Y. Li, C. Qin, E. Wang, H. Jin, D. Xiao, X. Wang, S. Chang, Inorg. Chem.
2007, 46, 3217–3230.
[32] B. Gole, U. Sanyal, R. Banerjee, P. S. Mukherjee, Inorg. Chem. 2016, 55, 2345–
2354.
[33] D. Best, H. Wai Lam, J. Org. Chem. 2014, 79, 831–845.
[34] H. B. Hepburn, P. Melchiorre, Chem. Commun. 2016, 3520, 3520–3523.
[35] L. Rupnicki, A. Saxena, H. W. Lam, J. Am. Chem. Soc 2009, 131, 10386–10387.
[36] G. Pattison, G. Piraux, H. Lam, J. Am. Chem. Soc 2010, 132, 14373–14375.
[37] D. A. Klumpp, Synlett. 2012, 23, 1590–1604.
[38] A. Saxena, H. Wai Lam, Chem. Sci 2011, 2, 2326–2331.
[39] A. A. Friedman, J. Panteleev, J. Tsoung, V. Huynh, M. Lautens, Angew. Chemie
- Int. Ed. 2013, 52, 9755–9758.
[40] R. P. Jumde, F. Lanza, M. J. Veenstra, S. R. Harutyunyan, Science (80-. ). 2016,
325, 433–437.
[41] R. P. Jumde, F. Lanza, T. Pellegrini, S. R. Harutyunyan, Nat. Commun. 2017, 8.
[42] E. L. Myers, C. P. Butts, V. K. Aggarwal, Chem. Commun 2006, 4434–4436.
[43] J. Shannon, D. Bernier, D. Rawson, S. Woodward, Chem. Commun. 2007,
3945–3947.
[44] P. Kelley, G. A. Edouard, S. Lin, T. Agapie, Chem. - A Eur. J. 2016, 22, 17173–
17176.
[45] A. P. M. Robertson, S. S. Chitnis, S. Chhina, H. J. Cortes, B. O. Patrick, H. A.
Jenkins, N. Burford, Can. J. Chem. 2016, 94, 424–429.
[46] T. R. Hoye, B. M. Eklov, M. Voloshin, Org. Lett. 2004, 6, 2567–2570.
[47] R. Oost, J. Rong, A. J. Minnaard, S. R. Harutyunyan, Catal. Sci. Technol. 2014,
4, 1997–2005.
[48] L. A. Arnold, R. Imbos, A. Mandoli, A. Â. H. M. De Vries, R. Naasz, B. L. Feringa,
Tetrahedron 2000, 56, 2865–2878.
[49] A. Sharma, L. Guénée, J. V. Naubron, J. Lacour, Angew. Chemie - Int. Ed. 2011,

Asymmetric conjugate addition of Grignard reagents to symmetric bispyridyl alkenes
81
50, 3677–3680.
[50] “trans-1-(2-Pyridyl)-2-(4-pyridyl)-ethylene 97% | Sigma-Aldrich,” can be found
under
https://www.sigmaaldrich.com/catalog/product/aldrich/197459?lang=en®i
on=NL, 2017.

Chapter 3
82

Chapter 4
Autoinductive effects in asymmetric copper(I)/phosphine
catalyzed addition reactions The phenomenon of asymmetric autoinduction is studied for the copper(I) catalyzed
addition of Grignard reagent to enones and enals. The interaction of the product, an
alkoxide, with the catalyst is studied by monitoring the change in the ee of the product
with the conversion of the substrate and by different experiments where the alkoxides
are used as additives in the reaction. The formation of a copper/alkoxide/phosphine
complex is proposed based on NMR experiments.

Chapter 4
84
4.1. Introduction
4.1.1. Asymmetric autoinduction
Conventional asymmetric catalysis produces an enantioenriched compound thanks to
the presence of a chiral catalyst. The purpose of a chiral catalyst is to increase the
reaction rate and to create an enantiomeric bias in the product (Scheme 1a).
Therefore, the enantiomeric excess of the product is proportional to the ee of the
catalyst itself (eecat) and to the ability of the catalyst to discriminate the synthesis of the
two enantiomers (eemax). Nevertheless, there are cases where product itself affects the
performances of the catalyst in terms of both turnover and enantioselection: we refer
to these cases as asymmetric autoinduction (Scheme 1b and c).
Scheme 31 Asymmetric catalysis and asymmetric autoinduction.
The definition of asymmetric autoinduction concerns two cases:[1]
The product can interact with an achiral catalyst and induce chirality in its own
synthesis in absence of any other chiral entity (for instance a chiral catalyst or
ligand, Scheme 1b).
The product interacts with a chiral complex to improve its performance in terms
of enantioselectivity, but it is not able to transfer the chiral information without
another chiral species (Scheme 1c).
In this Chapter, we will refer with the term autoinduction specifically to the second
case, where the product itself is not a sufficient chiral source for the enantioselectivity.
The first case will be discussed in detail in Chapter 5.
A consequence of the interaction of the product with the catalyst is the change of
enantioselectivity during the course of the reaction. In fact, in standard asymmetric
catalysis, the ee of the product is constant during the reaction (Chart 1, blue). If the

Autoinductive effects in asymmetric copper(I)/phosphine catalyzed addition
reactions
85
presence of a product has a positive effect on the enantioselectivity, the ee grows with
the conversion (red): instead, if the product poisons or affect the catalyst negatively,
the ee decreases with the conversion (green).
Chart 1 Variation of the enantioselectivity in standard and autoinductive asymmetric catalysis.
Monitoring the ee of a reaction at different conversions gives clear indication of the
type of catalytic reaction observed. For the same reason, the use of the enantioenriched
product from the beginning of the reaction should improve the enantioselectivity of the
isolated product . In this way, the induction period (initial time where the amount of
product is not sufficient to contribute to the catalysis) is avoided and, at the end, the
product is collected with higher ee.
Danda et al. reported that the enantioselectivity of the hydrocyanation of 1, catalyzed
by 3 increases during the course of the reaction (Scheme 2) and upon addition of (S)-
2 from the beginning of the reaction, a more enantioenriched product is isolated.[2]
Scheme 32 Asymmetric autoinduction in the hydrocyanation of substituted benzaldehyde.[2]
50
55
60
65
70
75
80
85
90
95
100
0 20 40 60 80 100
Maj
or
en
anti
om
er%
Conversion%
no auto (+)-auto (-)-auto

Chapter 4
86
Blackmond and coworkers studied the effect of the product in the asymmetric α-
amination of aldehydes by adding fresh reagents to the reaction once it is completed.
They report that in the first reaction, the ee of (R)-6 is 46%, while, after three further
cycles, is becomes 71% (Scheme 3).[3]
Scheme 33 Asymmetric autoinduction in the α-amination of aldehydes.[3]
However, the literature about asymmetric autoinduction concerns mostly
organometallic reactions, because the metallic center favors the interaction between
multiple components of the reaction. An example is the aldol condensation catalyzed
by Ti(IV) BINOLate, which forms chiral alkoxides that can participate to their own
catalysis. In the reaction between silyloxyfurans (7, Scheme 4a) and benzaldehyde,
the presence of (S)-9 causes an increment of the ee from 40% to 96%.[4] A similar case
is the condensation of Chan’s diene (10), where the use of the product ((R)-11)
increases significantly the enantioselectivity (Scheme 4b).[5] The increase in the ee of
the product was also reported for the addition of diethylzinc to ketones mediated by
titanium(IV)-isopropoxide (Scheme 4c).[6]

Autoinductive effects in asymmetric copper(I)/phosphine catalyzed addition
reactions
87
Scheme 34 Asymmetric autoinduction in titanium mediated reactions.
Organozinc reagents can be involved in symmetric autoinduction also in the absence
of Ti(IV). Curiously, in the aldol condensation between 13 and 14, the catalyst obtained
upon reaction of L2 and ZnEt2, affords 15 with 6% ee in the opposite enantiomer
respect to the product at full conversion (Scheme 5a). The initial addition of 15
increases the ee to 88% (same enantiomer at full conversion).[7]
Likewise, the enantioselectivity of the addition of diethyl zinc to trifluoromethyl
ketones, reported by the group of Espinet, increases with the conversion of the
substrate to alcohol 17 (55% ee to 88%, Scheme 5b).[8]

Chapter 4
88
Scheme 35 Asymmetric autoinduction in the addition of dialkylzinc to aldehydes.
Examples of asymmetric autoinductive Diels-Alder cycloadditions, catalyzed by
aluminum based Lewis acids, are also present in literature.[9,10] Recently, a dependence
of the ee on the product concentration was observed on the palladium-mediated [3+3]-
annulation of 4-hydroxycoumarins (Scheme 6a).[11] In this case, the enantioinduction
is inversed with the course of the reaction (the ee changes from 18% to 64% of the
opposite enantiomer).
A different, but very interesting, case of autoinduction regards the oxidative kinetic
resolution of benzylic alcohols catalyzed by manganese(II). Kinetic resolutions are
reactions where the two enantiomers of a mixture are converted to the product with
different reaction rates.[12] If the reaction of one enantiomer is significantly slower than
its antipode (high krel), this will be collected at the end of the reaction as enantiopure.
As the reaction implies a change in the ee, the presence of autoinduction cannot be
detected in the above-mentioned ways. Bryliakov et al. measured the kinetics of
oxidation of the two enantiomers of 21. They reported that the selectivity in the
oxidation to ketones increases drastically when 22 reaches highest enantiomeric
excesses (Scheme 6b).[13]

Autoinductive effects in asymmetric copper(I)/phosphine catalyzed addition
reactions
89
Scheme 36 Asymmetric autoinductive metal catalyzed reactions: a) inversion of the sense of the
chiral induction in the annulation of cumarins; b) product induced increase of the reaction rate in
chiral resolution.
In many of the above mentioned examples, product and catalyst have similar
functionalities (for instance, Ti(Oi-Pr)4/BINOL catalyzing aldol reactions).[4,5] Instead,
in this Chapter, the influence of an alkoxide (product) on a copper/phosphine based
catalytic system will be discussed.
4.1.2. Chiral tertiary alcohols
Chiral tertiary alcohols are widely used as building blocks in pharmaceutical industry
and their synthesis in an enantioselective fashion is a challenge for organic chemist.[14–
16] In the recent past, these compounds were obtained by asymmetric addition of
organometallic reagents to ketones using a chiral catalyst.[17–24] As well as other highly
reactive organometallic reagents like organoaluminum[25–30] and organozinc
reagents,[31–38] Grignard reagents have been employed for this purpose.[39–45]
Copper(I)-phosphine complexes had previously been found to afford the product of
conjugated addition on enones in an enantioselective way. In 2012, the groups of
Harutyunyan and Minnaard developed the asymmetric copper(I) catalyzed addition of
a Grignard reagents to α-methyl-substituted enones[46], in which allylic tertiary alcohol

Chapter 4
90
were obtained in excellent yields and enantiopurities when a β-branched Grignard was
employed, while the use of linear organometallic reagents generally comported a
decrease of the enantiopurity of the product (25aa and 25ab, Scheme 7a). The use
of the chiral ferrocenyl diphosphine L4, ethereal solvents and low temperatures were
the keys to overcome the 1,4-addition, β-hydride transfer and enolization. [46–49]
Scheme 37 Asymmetric addition of Grignard reagents to electrophiles.
The scope was extended to α-bromo-substituted enones (25)[49], aryl-alkyl ketones
(26)[[50], and aryl-heteroaryl ketones (27)[51], silyl ketones(28)[52], silyl imines (29)[53]
and imines[54] (30, Scheme 7b). NMR studies revealed that the Grignard
transmetallates on the dimeric CuBr/phosphine complex, and that the transmetallated
complex is a monomer (Scheme 7c). The transmetallated complex coordinates the
ketone forming a π-complex. Finally, the alkyl group is transferred to the ketone.[55,56]

Autoinductive effects in asymmetric copper(I)/phosphine catalyzed addition
reactions
91
Curiously, during the study of the addition to ketones, it was observed that the use of
small amounts of isopropanol caused an increase of the ee of the product.[57] Alcohols,
in the presence of organometallic reagents, are converted to alkoxides that can interact
with the metallic center of the catalyst and modify its activity. Copper(I) can form
complexes with alkoxides: in 1994 Yamamoto et al. proved that a transmetallated
(Ph3P)2CuMe (31) reacts with diphenyl methanol (32) to form the copper(I) alkoxide
complex (33, Scheme 8a).[58] A similar complex could also be obtained upon
treatment of (Ph3P)3CuCl (34) with NaOPh (35), that replaces the chloride and
generates (Ph3P)3CuOPh (36, Scheme 8b).[58] Alkoxides are also used in
homogeneous metallic catalysis to improve the transmetallation rate.[59–62]
Scheme 38 Formation of copper(I) complexes containing alkoxides.[58]
4.2. Aim During the development of a methodology for the asymmetric addition of Grignard
reagents to aldehydes, it was discovered that the presence of isopropanol caused a
small increase in the ee of the product.[57] This phenomenon could be due to an
interaction of the copper-catalyst with an alkoxide, formed by reaction of isopropanol
with the Grignard reagent. As the product of the reaction is an alkoxide, it could have
a positive influence in the asymmetric catalysis. The aim of this work is to get further
insight into this observation. The extent of this phenomenon and the mechanistic
implications will be studied.
4.3. Results
4.3.1. Enantioselectivity as a function of conversion of the starting material in 1,2-
additions of Grignard reagents to carbonyls
First, we decided to monitor the enantiomeric ratio of the product as a function of the
substrate conversion in two different Cu(I)-catalyzed 1,2-addition of Grignard reagents

Chapter 4
92
using ketones 24a and 24b (Scheme 9). The reaction using 24a, were performed at
-78°C. In the case of 24b, it was necessary to decrease the temperature until -105°C to
be able to detect the change in the conversion. For these kind of reactions, that involve
heterogeneous mixture and are carried out at cryogenic temperature, continuous
kinetic monitoring in situ is rather challenging. In order to have samples at different
conversions, we set up the reaction in several flasks at different times. Slow addition,
used in the standard procedure to improve the enantiodiscrimination[46,49], was
avoided in order not to interfere with real conversions at given time.
Scheme 39 Scheme of addition of i-BuMgBr to enones 24a and 24b.
Chart 2 Increase of the enantioselectivity during the course of the reaction. The conversion to 25a-b
was determined via GC/MS chromatography. The percentage of major enantiomer of the product was
determined by CSP-HPLC.
As depicted in Chart 2, for both 24a and 24b the enantioselectivity increases during
the reaction. However, due to the high reactivity of the Grignard reagents, the collected
data were quite dispersed and could only be used them as qualitative proof of
asymmetric autoinduction. Specifically, in the case of ketone 24a, the addition of the
isobutyl magnesium bromide at the beginning of the reaction (conversion 5%) gives
the ketone in only 56% ee, while at full conversion it reaches 80%. Similarly, the alcohol
25b has 42% ee at 1% conversion, while at 73% conversion the measured ee is 76%.
50
55
60
65
70
75
80
85
90
95
100
0 20 40 60 80 100
maj
or
en
anti
om
er
%
Conversion %
• 24a •24b

Autoinductive effects in asymmetric copper(I)/phosphine catalyzed addition
reactions
93
Two further experiments were carried out in order to explore the potential of the
alkoxide to improve the ee of the product. Tipically, enals undergo enantioselective 1,2-
addition of organomagnesium reagents with lower enantioselectivities than enones
(Scheme 10) and a change in the enantioselectivity would be more evident than on
the addition to ketones. Thus we decided to study the increase of enantioselectivity
using additives in the addition reactions of aldehydes. The structure of the
organometallic reagent has a strong effect on the enantioselectivity. The addition of i-
BuMgBr to aldehyde 37a affords 38a with ee of 60%, but the use of EtMgBr entails the
formation of 38ab with 17% ee.
Scheme 40 Addition of i-BuMgBr bromide to α-β-unsaturated aldehydes.
α,β-Unsaturated aldehydes are very reactive towards this reaction and it was not
possible to monitor the ee of the product at different conversions. For this reason, the
alkoxide 38ab-MgBr was formed in situ to study its effect on the synthesis of 38aa
(Scheme 11). First, to the solution of catalyst and 37a in MTBE, 0.3 equiv. of EtMgBr
were added. In this way, 0.3 equiv. of 38ab-MgBr were formed and 0.7 equiv. of 37a
were left in the reaction mixture. At this point, i-BuMgBr was added. After the quench,
38aa had 88% ee, which is significantly higher than the one previously reported and
implies that the presence of 38ab-MgBr influences the enantioselectivity of the
reaction.

Chapter 4
94
Scheme 41 Reaction with in situ formation of the alkoxide by addition of different organomagnesium
reagents. a The ratio between 38aa and 38ab was determined via 1H-NMR. b The ee of the product was
determined by CSP-HPLC.
The reverse experiment was also performed (Scheme 12), by adding in sequence 0.3
equivalents of i-BuMgBr and then EtMgBr. Likewise, the ee of 38ab was significantly
improved from 17% to 34%, confirming that the presence of 38aa-MgBr from the
beginning of the formation of the product is improving its final enantiomeric excess.
Scheme 42 In situ formation of 38aa-MgBr by addition of different organomagnesium reagents. a
The ratio between 38aa and 38ab was determined via 1H-NMR. b The ee of the product was
determined by CSP-HPLC.
4.3.2. Autocatalysis or autoinduction?
An asymmetric autocatalytic reaction proceeds in an enantioselective fashion when the
chiral product is the only catalyst. Instead, the case where the products cooperates with
the chiral catalyst to change its activity is defined asymmetric autoinduction

Autoinductive effects in asymmetric copper(I)/phosphine catalyzed addition
reactions
95
(Paragraph 4.1.1.). In the previous experiment, it emerged that the product alkoxide
influences the enantioselectivity of the reaction. Our interest was to understand if the
reaction was autocatalytic or autoinductive. For this purpose, the addition of
isobutylmagnesium bromide to 37b was performed in the absence of the complex
CuBr.L4.
The alkoxide 38ba-MgBr without complex CuBr-L4 afforded the product with
complete conversion but no enantioselectivity (Scheme 13a). Similarly, the reaction
using an organocuprate as organometallic reagent gave 38ba as a racemate, therefore
asymmetric autocatalysis was excluded. In the reaction a chiral copper-phosphine
complex was used. For this reason, it could not be excluded that 38ba-MgBr could
induce asymmetry in combination with an achiral copper-phosphine complex. Hence,
copper bromide and the achiral phosphine ligand n-Bu3P were used with the chiral
alkoxide 22ba (Scheme 13b). The reaction provided racemic product, so it was
confirmed that L4 is necessary to guarantee the enantioselectivity of the addition.
Scheme 43 Influence of alkoxide 38ba on enantioselectivity of 1,2-addition of organometallic
reagents to 37b. a The conversion to 38ba was determined via GC/MS chromatography. b The ee of the
product was determined by chiral HPLC. The ee was corrected for the one of 38ba used as additive.
Next, the influence of the amount of alkoxide and its configuration on the reaction were
studied. At first, the alcohol 38aa was added to the solution of CuBr·L4 and 37a
followed by addition of an excess of Grignard reagent. Excess of Grignard reagent was

Chapter 4
96
added to ensure both the deprotonation of the alcohol and the conversion of the
reaction. We observed that, under these conditions, either using 38aa or its
enantiomer in 20 mol%, no increment of the ee was observed (entries 1 and 3, Table
1). After increasing the amount of alcohol to 50 mol%, a significant improvement was
noticed (76% ee, entry 2). However, with 20 mol% of the preformed alkoxide (38aa-
MgBr, entry 4), the ee was comparable to the one in entry 2. Interestingly, an achiral
alcohol as i-PrOH could also improve the ee of the product to 74% (a similar result was
reported by the group of Minnaard.[63] These results indicate that the positive effect of
the alkoxide on the enantioselectivity is not specific for the product of the reaction, but
for any alkoxide present in the reaction mixture.
Table 14 Effect of an alkoxide on the enantioselectivity of the asymmetric 1,2-addition of Grignard
reagents to aldehydes.
Entry Additive (x mol%, ee%) Conversiona eeb
1 c,d 38aa (20 mol%, 62) 81 61
2 c,d 38aa (50 mol%, 62) 54 76
3 c,d 38aa (20 mol%, -60) 55 60
4 c 38aa-MgBr (20 mol%, 62) 75 76
5 d i-PrOH (20 mol%) 90 74
a The conversion to 38aa was determined via GC/MS chromatography. b The ee was
determined by chiral HPLC. c The conversion and ees were corrected for the amount of
38aa used. d 2.0 equiv. of i-BuMgBr were used. The excess of organometallic reagent
was needed to deprotonate the alcohol.
4.3.3. Asymmetric autoinductive effects: ketones vs. aldehydes
The addition of Grignard reagents to the enals proceeds with an increase of asymmetric
induction when alkoxides are used as additives. An experiment was designed to
understand when or whether this phenomenon occurs in addition reactions to ketones.
The addition of EtMgBr to enone 24a is known to be less enantioselective when
compared to β-branched organomagnesium reagents.[46] For this reason, it is easier to
detect an increase the ee of the product obtained from ethylmagnesium bromide. The
asymmetric 1,2-addition to 24a was performed with the additive 25aa-MgBr. The
alkoxide was formed in situ upon addition of 0.7 equiv. of i-BuMgBr to 24a. The
remaining ketone was converted into 25ab-MgBr upon addition of EtMgBr. The

Autoinductive effects in asymmetric copper(I)/phosphine catalyzed addition
reactions
97
enantiomeric excess of 25ab (41%) was comparable to the one obtained in the reported
methodology.[46]
Scheme 44 In situ formation of 25aa-MgBr and subsequent addition of EtMgBr to 24a. a The ratio
between 25aa and 25ab was determined via 1H-NMR. b The ee of the product was determined by CSP-
HPLC.
To detect an increase of ee in the addition of EtMgBr to 24a, 1.0 equiv. of both 24a
and EtMgBr were added to the reaction mixture after performing the reaction with the
standard procedure. Also in this case, the ee of the product collected after the reaction
quench was comparable to the reported one[46] (Scheme 14). This means that in the
reactions with low enantioselectivity, the change of the ee cannot be noticed or does
not occur.
Scheme 45 1,2-addition of EtMgBr to 24a in the presence of 1.0 equivalent of 25ab and 5 mol% of
CuBr·L4. a The conversion to 25ab was determined via GC/MS chromatography. b The ee of the
product was determined by CSP-HPLC.

Chapter 4
98
From the results of our experiments it emerged that the enantioselective 1,2-additions
to aldehydes are more sensitive to an alkoxide additives with respect to additions to
ketones. This could be explained by a more prominent background reaction of addition
of Grignard to aldehydes when compared to ketones. In fact, at -78 oC, the Grignard
reagent adds to aldehydes also in absence of catalyst. Moreover, we performed the
addition of i-BuMgBr to 37ab with a stoichiometric amount of complex CuBr·L4 to
ensure conditions in which no background reaction can occur. This experiment
afforded 38ab with 76% ee, same ee observed with the use of an alkoxide as additive
(Scheme 16).
Scheme 46 Addition of i-BuMgBr to 37a with stoichiometric catalyst.
Consequently, we hypothesized that the role of the alkoxide, is to form a more active
catalyst upon coordination with CuBr·L4, and thus to compete more efficiently with
noncatalysed pathway. This also would explain why presence of alkoxide is most
beneficial for the enantioselectivity in the reactions where fast non-catalysed reactions
occur as well (as contrasted to ketones).
4.3.4. Interaction of an alkoxide with a copper/phosphine complex
In order to verify our hypothesis of the complex formation between alkoxide and CuBr
L4 complex and its catalytic activity we studied the interaction between the Cu(I)-
phosphine catalyst and an alkoxide using NMR spectroscopy.
As L4 gives broad peaks in 31P-NMR spectra, the system was simplified by substituting
L4 with another ligand from the same family, L5. To reduce the complexity of the study
further, i-PrOH and i-PrOMgBr were chosen respectively as an alcohol and alkoxide.
The Grignard reagent used was MeMgBr. In the first experiment (Scheme 17a), the
complex CuBr·L5 (39, Figure 1a) was treated with MeMgBr leading to
transmetallated species 40. Formation of this species is confirmed by two doublets
appearing at 7.45 ppm and -25.89 ppm in the 31P-NMR spectrum, (Figure 1b).
Moreover, two doublets with minor intensity appeared at 14.63 and -18.08 ppm. The
presence of these signals was already reported for this catalyst system but their nature
is still unclear. For simplicity we will refer to the most intense peaks that represent the
catalytically active species, as A-species, and to the less intense ones as B-species. [55]
Isopropanol was added to species 40 and two new doublets at 2.00 ppm and -23.33
ppm were observed at this point (Figure 1c).We hypothesized that the new signals
could belong to 41 (copper/alkoxide) or 42 (transmetallated copper/alkoxide species).
To support our hypothesis we designed and performed a second experiment (Scheme

Autoinductive effects in asymmetric copper(I)/phosphine catalyzed addition
reactions
99
17b). We were able to form species 41 by direct reaction between complex 39 (Figure
17a) and the alkoxide. 31P NMR spectrum (Figure 2b) of this reaction mixture was
matching with the one observed in the previous experiment (Figure 1c). Moreover,
upon addition of MeMgBr to this reaction mixture with the species 41, it was converted
back to the transmetallated species 40 (Figure 2c) and no B-species was observed as
it was in Figure 1b. This fact suggests that, probably the rate of 41 to undergo
transmetallation in the presence of a Grignard reagent is equal or superior to 39.
Scheme 47 Experiments 1 and 2. Each step was characterized by 1H, 31P and 1H-31P-HMBC techniques

Chapter 4
100
Figure 16 Experiment 1: a) 31P-NMR (161 MHz, CD2Cl2) spectrum of 39 at -60°C. b) 31P-NMR (161
MHz, CD2Cl2) spectrum of 40 at -60°C. c) 31P-NMR (161 MHz, CD2Cl2) spectrum of 41 at -60°C, obtained
from reaction of 40 and i-PrOH.

Autoinductive effects in asymmetric copper(I)/phosphine catalyzed addition
reactions
101
Figure 17 Experiment 2: a) 31P-NMR (161 MHz, CD2Cl2) spectrum of the complex 39 at -60°C. b) 31P-
NMR (161 MHz, CD2Cl2) spectrum of the complex 41, obtained from the reaction of 39 with
magnesiumbromide isopropoxide at -60°C. c) 31P-NMR (161 MHz, CD2Cl2) spectrum of the complex 40
at -60°C upon addition of MeMgBr to 41.
These experiments suggest that after the reaction begins, the actual catalyst is the
CuOR/phosphine complex. Based on these new data and the catalytic cycle previously
proposed for 1,2-addition of carbonyl compounds,[64] we can propose a new feasible
catalytic cycle that can explain the role of the alkoxide in these reactions. The Grignard
reagent and the catalyst form the transmetallated complex (I, Scheme 18), that
coordinates the carbonyl to afford a π-complex (II) and the alkyl group is transferred
(III). The product is a CuOR/phosphine complex. Upon transmetallation with the
alkyl magnesium reagent (IV), the transmetallated complex is formed. We propose
that the transmetallation of this complex occurs faster than with CuBr/phosphine.
Next, the transmetallated complex obtained from CuOR/phosphine, forms a π-
complex with the carbonyl moiety of the substrate (V) and, after the reductive
elimination and alkylation of the substrate, the product is eliminated and the catalyst
is regenerated (VI).

Chapter 4
102
Scheme 48 Catalytic cycle of asymmetric copper-catalyzed addition of Grignard reagents to ketones.
4.4. Conclusions In the asymmetric copper(I)-catalyzed addition of Grignard reagents to carbonyl
compounds, the formation of the complex CuOR/phosphine was observed. This
complex undergoes transmetallation faster than its precursor (CuBr/phosphine) and
the effect of the presence of an alkoxide is more relevant in reactions with fast
background addition of the organometallic reagent present (addition to α,β-
unsaturated aldehydes). As the product of 1,2-addition reaction is an alkoxide, it has a
role on its own enantioselective synthesis (asymmetric autoinduction). In fact, adding
the product at the beginning of the reaction causes an enhancement of the
enantioselectivity simply allowing the catalytic reaction to outcompete the background
reaction. However the enhancement is not specific for to the addition product alkoxide
but is applicable to an alkoxide derived from any alcohol.

Autoinductive effects in asymmetric copper(I)/phosphine catalyzed addition
reactions
103
4.5. Experimental section
4.5.1. General information
All reactions using oxygen- and/or moisture-sensitive materials were carried out with
anhydrous solvents (vide infra) under a nitrogen atmosphere using oven dried
glassware and standard Schlenk techniques. Reactions were monitored by 1H NMR.
Purification of the products, when necessary, was performed by flash-column
chromatography using Merck 60 Å 230-400 mesh silica gel, Merk 90 active neutral or
VWR AnalaR NORMAPUR aluminum oxide basic. NMR data was collected on Bruker
Avance NEO 600 (1H at 600.0 MHz; 13C at 150.87MHz), equipped with a Prodigy Cryo-
probe and Varian VXR400 (1H at 400.0 MHz; 13C at 100.58 MHz, 19F at 376.50 MHz, 31P at 161.97 MHz), equipped with a 5 mm z-gradient broadband probe. Chemical shifts
are reported in parts per million (ppm) relative to residual solvent peak (CDCl3, 1H:
7.26 ppm; 13C: 77.16 ppm. CD2Cl2: 1H: 5.32 ppm; 13C: 54.0 ppm). Coupling constants
are reported in Hertz. Multiplicity is reported with the usual abbreviations (s: singlet,
bs: broad singlet, d: doublet, dd: doublet of doublets, ddd: doublet of doublet of
doublets, t: triplet, td: triplet of doublets, q: quartet, m: multiplet). Exact mass spectra
were recorded on a LTQ Orbitrap XL apparatus with ESI ionization. Enantiomeric
excesses (ees) were determined by Chiral HPLC analysis using a Shimadzu LC-10ADVP
HPLC equipped with a Shimadzu SPD-M10AVP diode array detector and by Waters
Acquity UPC2 system with PDA detector and QDA mass detector.
Unless otherwise indicated, reagents and substrates were purchased from commercial
sources and used as received. Solvents not required to be dry were purchased as
technical grade and used as received. Dry solvents were freshly collected from a dry
solvent purification system prior to use. Inert atmosphere experiments were
performed with standard Schlenk techniques with dried (P2O5) nitrogen gas. Grignard
reagents were purchased from Sigma-Aldrich and used as received (EtMgBr (3.0 M in
Et2O), i-BuMgBr (2.0 M in Et2O). i-BuMgBr in MTBE was prepared from the
corresponding alkyl bromide and Mg activated with I2 in MTBE and concentration was
determined by titration with I2 in a solution of LiCl in THF (0.5 M).[65] Chiral ligands
L1 and L1 were purchased from Sigma Aldrich and Solvias. Ketones 24a and 24b were
prepared according to literature procedures.[66,67] All reported compounds were
characterized by 1H and 13C NMR and compared with literature data. The absolute
configurations of products were attributed by comparison with our previous work.[46,49]
4.5.2. General procedure for the 1,2-addition of Grignard reagents to ketones
(24a,b)
A flame dried Schlenk tube equipped with septum and stirring bar was charged with
CuBr·SMe2 (0.015 mmol, 3.08 mg, 5 mol%) and L4 (L1, 0.018 mmol, 10.70 mg, 6

Chapter 4
104
mol%). Dry MTBE (3 mL) was added and the solution was stirred at room temperature
for 15 minutes. Then the ketone (0.3 mmol in 1 mL MTBE) was added and the resulting
solution was cooled to -78 ⁰C. The Grignard reagent (0.36 mmol, 0.12 in 1 mL MTBE)
was added dropwise over 15 minutes. The reaction was left overnight and quenched by
addition of 1 ml MeOH and saturated aqueous solution of NH4Cl (2 mL) and the
mixture was warmed to room temperature, diluted with Et2O and the two layers were
separated. The aqueous layer was extracted with Et2O (3 x 5 mL) and the combined
organic phases were dried with anhydrous Na2SO4, filtered and the solvent was
removed under reduced pressure. The crude product was purified by flash column
chromatography (SiO2, pentane:Et2O, 9:1).
(R,E)-2,3,5-Trimethyl-1-phenylhex-1-en 3-ol (25aa)
The reaction was performed with 48.0 mg of 24a (0.3 mmol, 1.0
equiv.), i-BuMgBr (0.36 mmol, 1.2 equiv. in 1 mL MTBE), CuBr·L4
(12.2 mg, 0.015 mmol, 5 mol%), in 3 mL MTBE. Note: it is crucial
for the outcome of the reaction to perform the addition of the
diluted Grignard manually. Product 25aa was obtained as
transparent oil (20.1 mg, 0.09, yield 31%, ee 75%). The NMR data are in agreement
with the ones present in literature. [46] The absolute configuration was assigned
according to previous literature.[46] 1H NMR (400 MHz, CDCl3) δ 7.32 (t, 2H), 7.29 – 7.15 (m, 3H), 6.69 (s, 1H), 1.83 (d, J
= 1.4 Hz, 3H), 1.81 – 1.63 (m, 2H), 1.40 (d, J = 1.4 Hz, 3H), 0.95 (dd, 6H)
13C NMR (101MHz, CDCl3) δ 143.5, 138.6, 129.0, 128.0, 126.1, 123.2, 76.6, 48.9, 29.0,
24.4, 24.4, 15.1.
CSP-HPLC: (254nm, Chiralcel OD-H, n-heptane:i-PrOH = 99:1, 40 °C, 0.5
mL/min.), tR = 15.71 min (major), tR = 18.41 min (minor).
(R,Z)-2-Bromo-3,5-dimethyl-1-phenylhex-1-en 3-ol (25ba)
The reaction was performed with 67.5 mg of 24b (0.3 mmol, 1.0
equiv.), i-BuMgBr (0.36 mmol, in 1 mL MTBE), CuBr·L4 (12.2 mg,
0.015 mmol, 5 mol%), in 15 mL MTBE. Product 25ba was obtained
as transparent oil (37.5 mg, 0.13 mmol, yield 44%, ee 79%). The
NMR data are in agreement with the ones present in literature.[49]
The absolute configuration was assigned according to previous literature.[49] 1H NMR (400 MHz, CDCl3) δ 7.55 (2H, m), 7.40 – 7.33 (2H, m), 7.32-7.27 (2H, m),
7.24 (1H, s), 2.00 (1H, s) 1.89 (1H, dd), 1.85 – 1.75 (2H, m), 1.65 (1H, dd), 1.58 (3H, s),
0.99 (6H, dd). 13C NMR (101MHz, CDCl3) δ 136.2, 134.7, 129.0, 128.1, 127.7, 126.3, 77.6, 49.1, 28.8,
24.5, 24.3, 24.3.
CSP-HPLC: (254nm, Chiralcel AD-H, n-heptane:i-PrOH = 98:2, 40 °C, 0.5
mL/min.), tR = 18.15 min (major), tR = 21.06 min (minor).

Autoinductive effects in asymmetric copper(I)/phosphine catalyzed addition
reactions
105
(R,E) -2,3-dimethyl-1-phenylpent-1-en 3-ol (25ab)
The reaction was performed with 48.0 mg of 24a (0.3 mmol, 1.0
equiv.), EtMgBr (0.36 mmol, 3M in Et2O, diluted in MTBE until 1
mL), CuBr·L4 (12.2 mg, 0.015 mmol, 5 mol%), in 3 mL MTBE. After
stirring for 2h, 48.0 mg of 24a (0.3 mmol, 1 equiv.), EtMgBr (0.36
mmol 3M in Et2O, diluted in MTBE until 1 mL) were added again. Product 25ab was
obtained as transparent oil (80.7 mg, 3.1 mmol, yield 53%, ee 38%). The NMR data are
in agreement with the ones present in literature. [46] The absolute configuration was
assigned according to previous literature.[46] 1H NMR (400 MHz, CDCl3) δ 7.32-7.27 (2H, m), 7.26-7.17 (3H, m), 6.65 (H, s), 1.83
(3H, s), 1.78-1.60 (2H, m), 1.57 (1H, s), 1.40 (3H, s), 0.87 (3H, t). 13C NMR (101MHz, CDCl3) δ 143.0, 138.5, 129.0, 128.0, 126.1, 123.7, 76.2, 33.0, 27.4,
14.6, 8.1.
CSP-HPLC: (254nm, Chiralcel AD-H, n-heptane:i-PrOH = 99:1, 40 °C, 0.5
mL/min.), tR = 31.33 min (major), tR = 34.17 min (minor).
4.5.3. General procedure for the 1,2-addition of Grignard reagents to aldehydes
(37a,b)
A flame dried Schlenk tube equipped with septum and stirring bar was charged with
CuBr·SMe2 (0.015 mmol, 3.08 mg, 5 mol%) and L4 (0.018 mmol, 10.70 mg, 6 mol%).
Dry MTBE (3 mL) was added and the solution was stirred at room temperature for 15
minutes. Then the aldehyde (0.3 mmol, 1.0 equiv.) was added and the resulting
solution was cooled to -78 ⁰C. The Grignard reagent (0.36 mmol in 1 mL MTBE) was
added over 3 h with the use of a syringe pump. The reaction was quenched after 3
hours by addition of 1 ml MeOH and saturated aqueous solution of NH4Cl (2 mL) and
the mixture was warmed to room temperature, diluted with Et2O and the two layers
were separated. The aqueous layer was extracted with Et2O (3 x 5 mL) and the
combined organic phases were dried with anhydrous Na2SO4, filtered and the solvent
was removed under reduced pressure. The crude product was purified by flash column
chromatography (SiO2, pentane:Et2O, 9:1).
(R,E)-2,3,5-Trimethyl-1-phenylhex-1-en-3-ol (38aa)
The reaction was performed with 14.6 mg of 37a (0.1 mmol, 1.0
equiv.), i-BuMgBr (0.12 mmol, in 0.3 mL MTBE), CuBr·SMe2 (1.0
mg, 0.005 mmol, 5 mol%), L4 (3.6 mg, 0.006 mmol, 6 mol%), in
1 mL MTBE. Product 38aa was obtained as transparent oil (13.2
mg, 0.066 mmol, yield 66%, ee 65%). The NMR data are in
agreement with the ones present in literature.[57] The absolute configuration was
assigned according to previous literature.[57]

Chapter 4
106
1H NMR (400 MHz, CDCl3), δ 7.36-7.25 (4H, m), 7.34-7.19 (1H, tt), 6.30 (1H, s), 4.28-
4.24 (1H, t), 1.87 (3H, s). 13C NMR (101MHz, CDCl3), δ (ppm): 140.7, 137.6, 128.9, 128.1, 125.6, 76.4, 44.4, 24.9,
23.1, 22.5, 13.1.
CSP-HPLC: (254nm, Chiralcel OD-H, n-heptane:i-PrOH = 99:1, 40 °C, 0.5
mL/min.), tR = 41.04 min (major), tR = 34.69 min (minor).
(R, Z)-2-Bromo-5-methyl-1-phenylhex-1-en-3-ol (38ba)
The reaction was performed with 63.3 mg of 37b (0.3 mmol, 1.0
equiv.), i-BuMgBr (0.36 mmol, in 1 mL MTBE), CuBr·SMe2 (3.1
mg, 0.015 mmol, 5 mol%), L4 (10.7 mg, 0.018 mmol, 6 mol%), in 1
mL MTBE. Product 38ba was obtained as transparent oil (57.4 mg,
0.21 mmol, yield 71%, ee 61%). The NMR data are in agreement with the ones present
in literature.[57] The absolute configuration was assigned according to previous
literature.[57] 1H NMR (400 MHz, CDCl3), δ 7.61 (1H, d), 7.39-7.30 (m, 3H), 7.05 (s, 1H), 4.33 (1H,
m), 1.95 (d, 1H), 1.82-1.69 (1H, m), 169-1.50 (2H, m), 0.98 (6H, m)., 1.79-1.68 (2H, m),
1.59-1.42 (4H, m), 0.97 (6H, d). 13C NMR (101MHz, CDCl3), δ (ppm): 130.9, 129.1, 128.8, 128.1, 128.1, 127.9, 76.0,
45.0, 24.6, 22.9, 22.5.
CSP-HPLC: (254nm, Chiralcel AD-H, n-heptane:i-PrOH = 99:1, 40 °C, 0.5
mL/min.), tR = 35.80 min (major), tR = 37.56 min (minor).
(S,E)-2,3-Dimethyl-1-phenylpent-1-en-3-ol (38ab)
The reaction was performed with 14.6 mg of 37a (0.1 mmol, 1.0
equiv.), EtMgBr (0.12 mmol, 3M in Et2O, diluted in MTBE until 1 mL),
CuBr·SMe2 (1.0 mg, 0.005 mmol, 5 mol%), L1 (3.6 mg, 0.006 mmol,
6 mol%), in 1 mL MTBE. Product 38aa was obtained as transparent
oil (15.3 mg, 0.087 mmol, yield 87%, ee 17%). The absolute configuration was assigned
according to previous literature.[57] 1H NMR (400 MHz, CDCl3), δ 7.39-7.19 (5H, m), 6.49 (3H, s), 4.11, (1H, t), 1.86 (3H,
s), 1.79-1.60 (2H, m), 1.63 (1H, bs), 1.35-1.17 (2H, m), 0.95 (3H, t). 13C NMR (101 MHz, CDCl3) δ (ppm): 140.0, 137.6, 129.0, 128.1, 126.4, 126.0, 79.6,
27.9, 13.1, 10.1.
HRMS (ESI+): m/z calcd. for C12H15 ([M-H2O]) 159.11683, found 159.11680.
CSP-HPLC: (254nm, Chiralcel AD-H, n-heptane:i-PrOH = 99:1, 40 °C, 0.5
mL/min.), tR = 58.31 min (major), tR = 51.92 min (minor).

Autoinductive effects in asymmetric copper(I)/phosphine catalyzed addition
reactions
107
4.5.4. General procedure for monitoring the ee of the product of the reaction
A flame dried Schlenk tube equipped with septum and stirring bar was charged with
CuBr·SMe2 (0.005 mmol, 1.03 mg, 5 mol%) and the L4 (0.006 mmol, 3.57 mg, 6
mol%). Dry MTBE (1 mL) was added and the solution was stirred at room temperature
for 15 minutes. Then the ketone (0.1 mmol, 1.0 equiv. in 0.3 mL MTBE) was added and
the resulting solution was cooled to -78 ⁰C (Substrate 24a) or -105⁰C (Substrate 24b).
The Grignard reagent (0.12 mmol in Et2O, 1.2 equiv.) was added in one shot. The
reaction was quenched at different times by addition of 1 ml MeOH and saturated
aqueous solution of NH4Cl (2 mL) and the mixture was warmed to room temperature,
diluted with Et2O and the two layers were separated. The aqueous layer was extracted
with Et2O (3 x 5 mL) and the combined organic phases were dried with anhydrous
Na2SO4, filtered and the solvent was removed under reduced pressure. The conversion
was measured by GC/MS and the ee by CSP-HPLC.
4.5.5. General procedure for the 1,2-addition of Grignard reagents to aldehydes
with the use of additives
A flame dried Schlenk tube equipped with septum and stirring bar was charged with
CuBr·SMe2 (0.015 mmol, 3.08 mg, 5 mol%) and L4 (0.018 mmol, 10.70 mg, 6 mol%).
Dry MTBE (3 mL) was added and the solution was stirred at room temperature for 15
minutes. Then the aldehyde (0.3 mmol, 1.0 equiv.) and the additive were added and
the resulting solution was cooled to -78 ⁰C. The Grignard reagent (0.36 mmol in 1 mL
MTBE, 1.2 equiv.) was added over 3 hours with the use of a syringe pump. The reaction
was quenched after 3 hours by addition of 1 ml MeOH and saturated aqueous solution
of NH4Cl (2 mL) and the mixture was warmed to room temperature, diluted with Et2O
and the two layers were separated. The aqueous layer was extracted with Et2O (3 x 5
mL) and the combined organic phases were dried with anhydrous Na2SO4 , filtered and
the solvent was removed under reduced pressure. The crude product was purified by
flash column chromatography (SiO2, pentane:Et2O, 9:1). The conversion was measured
by GC/MS and the ee by CSP-HPLC.
4.5.6. General procedure for the reaction carried out with different Grignard
reagents
A flame dried Schlenk tube equipped with septum and stirring bar was charged with
CuBr·SMe2 (0.015 mmol, 3.08 mg, 5 mol%) and L4 (0.018 mmol, 10.70 mg, 6 mol%).
Dry MTBE (3 mL) was added and the solution was stirred at room temperature for 15
minutes. Then 24a or 37a (0.3 mmol in 1 mL MTBE, 1.0 equiv.) was added and the
resulting solution was cooled to -78 ⁰C. The first Grignard reagent (0.21 mmol, 0.7
equiv. diluted to 0.3 mL with MTBE for 24a or 0.09 mmol, 0.3 equiv. diluted to 0.3

Chapter 4
108
mL with MTBE, for 37a) was added over 1 hour with the use of a syringe pump. The
solution was allowed to stir at -78 ⁰C overnight (24a) or for 30 min (37a). The second
Grignard (, 0.3 equiv. diluted to 0.3 mL with MTBE for 24a 0.21 mmol, 0.7 equiv.
diluted to 0.3 mL with MTBE, for 37a or 0.09 mmol) was added over 3 hours with the
use of a syringe pump. The reaction was quenched after overnight stirring (for 24a) or
after 3 hours (37a) by addition of 1 ml MeOH and saturated aqueous solution of NH4Cl
(2 mL) and the mixture was warmed to room temperature, diluted with Et2O and the
two layers were separated. The aqueous layer was extracted with Et2O (3 x 5 mL) and
the combined organic phases were dried with anhydrous Na2SO4 , filtered and the
solvent was removed under reduced pressure. The ratio of the two products was
measured by 1H NMR and the ee by CSP-HPLC.
4.5.7.Procedure for the NMR experiments
Preparation of the complex CuBr·L5 (39)
A Schenk tube equipped with septum and stirring bar was charged with CuBr·SMe2
(30.8mg, 0.15 mmol) and L5·EtOH (96.1 mg, 0.15 mmol). Dry CH2Cl2 (8mL) was
added and the solution was stirred for 30 min. The solvent was removed under reduced
pressure. 39 was obtained as orange crystals (110.6 mg, 0.15 mmol, quant. yield). The
NMR data are in agreement with the ones present in literature.[55,68]
Experiment 1
An oven dried NMR tube maintained under nitrogen atmosphere and equipped with a
septum was charged with 0.04mL of MeMgBr in Et2O (0.11 mmol, 10 equiv., 3M in
Et2O). The solvent was removed in vacuo and the MeMgBr was dried with the use of a
heat gun until the formation of a foam. The solid was cooled to -78 ⁰C. A solution of
CuBr·L5 (8 mg, 1.0 equiv., 0.011 mmol) in CD2Cl2 (0.6 mL) was added and the solution
was stirred with a vortex stirrer until maximum solubility of the Grignard reagent and
change in the color of the solution from orange to yellow. 1H and 31P spectra were
recorded at -60 ⁰C using a Varian NMR spectrometer, operating at 400.0 MHz for the
proton, 161.0 for the phosphorous nuclei). To the solution was added i-PrOH (7µL,
0.09mmol mmol, 8 equiv.) and 1H and 31P spectra were recorded at -60 ⁰C.
Experiment 2
An oven dried NMR tube maintained under nitrogen atmosphere and equipped with a
septum was charged with 0.03 mL MeMgBr in Et2O of (0.088 mmol, 8 equiv., 3M in
Et2O) and i-PrOH (7µL, 0.09 mmol, 8 equiv.). The Et2O was removed in vacuo and i-
PrOMgBr was dried with the use of a heat gun until the formation of a foam. The solid
was cooled to -78 ⁰C. A solution of CuBr·L5 (8 mg, 1.0 equiv., 0.011 mmol) in CD2Cl2
(0.6 mL) was added and the solution was stirred with a vortex stirrer until maximum
solubility of the alkoxide and change in the color of the solution from orange to yellow. 1H and 31P spectra were recorded at -60 ⁰C using a Varian NMR spectrometer,
operating at 400.0 MHz for the proton, 161.0 for the phosphorous nuclei).

Autoinductive effects in asymmetric copper(I)/phosphine catalyzed addition
reactions
109
Subsequently, 0.04mL of MeMgBr in Et2O (0.11 mmol, 10 equiv., 3M in Et2O) were
added to the NMR tube and 1H and 31P spectra were recorded at -60 ⁰C.
4.6. Bibliography [1] S. E. Denmark, Topics in Stereochemistry. Volume 22, J. Wiley, 1999.
[2] H. Danda, H. Nishikawa, K. Otaka, J. Org. Chem. 1991, 56, 6740–6741.
[3] H. Iwamura, S. P. Mathew, D. G. Blackmond, J. Am. Chem. Soc. 2004, 126,
11770–1771.
[4] M. Szlosek, B. Figadère, Angew. Chem. Int. Ed. Engl. 2000, 39, 1799–1801.
[5] R. Villano, M. De Rosa, C. Salerno, A. Soriente, A. Scettri, Tetrahedron:
Asymmetry 2002, 13, 1949–1952.
[6] A. M. Costa, C. García, P. J. Carroll, P. J. Walsh, Tetrahedron 2005, 61, 6442–
6446.
[7] B. M. Trost, A. Fettes, B. T. Shireman, J. Am. Chem. Soc. 2004, 126, 2660–
2661.
[8] M. Calvillo-Barahona, J. A. Casares, C. Cordovilla, M. N. Genov, J. M.
Martínez-Ilarduya, P. Espinet, Chem. - A Eur. J. 2014, 20, 14800–14806.
[9] F. Rebiere, O. Riant, H. B. Kagan, Tetrahedron: Asymmetry 1990, 1, 199–214.
[10] D. P. Heller, D. R. Goldberg, W. D. Wulff, J. Am. Chem. Soc. 1997, 119, 10551–
10552.
[11] S. Wang, J. Xiao, J. Li, H. Xiang, C. Wang, X. Chen, R. G. Carter, H. Yang,
Chem. Commun. 2017, 53, 4441–4444.
[12] H. Pellissier, Tetrahedron 2018, 74, 3459–3468.
[13] E. P. Talsi, D. G. Samsonenko, K. P. Bryliakov, ChemCatChem 2017, 9, 2599–
2607.
[14] J. L. Stymiest, V. Bagutski, R. M. French, V. K. Aggarwal, Nature 2008, 456,
778–82.
[15] H. K. Scott, V. K. Aggarwal, Chem. - A Eur. J. 2011, 17, 13124–13132.
[16] E. J. Corey, A. Guzman-Perez, Angew. Chemie - Int. Ed. 1998, 37, 388–401.
[17] M. Shibasaki, M. Kanai, Chem. Rev. 2008, 108, 2853–2873.
[18] K. Soai, S. Niwa, Chem. Rev. 1992, 92, 833–856.
[19] M. Hatano, K. Ishihara, Chem. Rec. 2008, 8, 143–155.
[20] L. Pu, H.-B. Yu, Chem. Rev. 2001, 101, 757–824.
[21] C. M. Binder, B. Singaram, Org. Prep. Proced. Int. 2011, 43, 139–208.
[22] M. Yus, J. C. González-Gómez, F. Foubelo, Chem. Rev. 2011, 111, 7774–7854.
[23] H. Pellissier, Tetrahedron 2015, 71, 2487–2524.
[24] O. Riant, J. Hannedouche, Org. Biomol. Chem. 2007, 5, 873–888.
[25] C.-A. Chen, K.-H. Wu, H.-M. Gau, Angew. Chemie - Int. Ed. 2007, 46, 5373–6.
[26] C. Chen, K. Wu, H. Gau, Adv. Synth. Catal. 2008, 350, 1626–1634.

Chapter 4
110
[27] A. S. C. Chan, F.-Y. Zhang, C.-W. Yip, J. Am. Chem. Soc. 1997, 119, 4080–
4081.
[28] B. L. Pagenkopf, E. M. Carreira, Tetrahedron Lett. 1998, 39, 9593–9596.
[29] L. Zhang, B. Tu, M. Ge, Y. Li, L. Chen, W. Wang, S. Zhou, J. Org. Chem. 2015,
80, 8307–8313.
[30] Y.-X. Yang, Y. Liu, L. Zhang, Y.-E. Jia, P. Wang, F.-F. Zhuo, X.-T. An, C.-S. Da,
J. Org. Chem. 2014, 79, 10696–10702.
[31] M. Yus, D. J. Ramón, O. Prieto, Tetrahedron: Asymmetry 2003, 14, 1103–
1114.
[32] M. Yus, D. J. Ramón, O. Prieto, Tetrahedron: Asymmetry 2002, 13, 2291–
2293.
[33] E. F. Di Mauro, M. C. Kozlowski, J. Am. Chem. Soc. 2002, 124, 12668–12669.
[34] C. García, L. K. LaRochelle, P. J. Walsh, J. Am. Chem. Soc. 2002, 124, 10970–
10971.
[35] D. J. Ramón, M. Yus, Angew. Chemie - Int. Ed. 2004, 43, 284–287.
[36] S.-J. Jeon, H. Li, C. García, L. K. LaRochelle, P. J. Walsh, J. Org. Chem. 2005,
70, 448–55.
[37] M. Hatano, T. Mizuno, K. Ishihara, Chem. Commun. 2010, 46, 5443–5445.
[38] Y. Kinoshita, S. Kanehira, Y. Hayashi, T. Harada, Chem. - A Eur. J. 2013, 19,
3311–4.
[39] Y. Muramatsu, T. Harada, Angew. Chemie 2008, 47, 1088–1090.
[40] Y. Muramatsu, T. Harada, Chem. - A Eur. J. 2008, 14, 10560–10563.
[41] Y. Muramatsu, S. Kanehira, M. Tanigawa, Y. Miyawaki, T. Harada, Bull. Chem.
Soc. Jpn. 2010, 83, 19–32.
[42] D. Itakura, T. Harada, Synlett 2011, 2011, 2875–2879.
[43] E. Fernández-Mateos, B. Maciá, M. Yus, Adv. Synth. Catal. 2013, 355, 1249–
1254.
[44] E. Fernández-Mateos, B. Maciá, D. J. Ramón, M. Yus, European J. Org. Chem.
2011, 2011, 6851–6855.
[45] E. Fernández-Mateos, B. Maciá, M. Yus, European J. Org. Chem. 2014, 2014,
6519–6526.
[46] A. V. R. Madduri, A. J. Minnaard, S. R. Harutyunyan, Chem. Commun. (Camb).
2012, 48, 1478–80.
[47] J. Rong, T. Pellegrini, S. R. Harutyunyan, Chemistry 2015, DOI
10.1002/chem.201503412.
[48] A. V. R. Madduri, S. R. Harutyunyan, A. J. Minnaard, Drug Discov. Today.
Technol. 2013, 10, e21-7.
[49] A. V. R. Madduri, A. J. Minnaard, S. R. Harutyunyan, Org. Biomol. Chem.
2012, 10, 2878–84.
[50] A. V. R. Madduri, S. R. Harutyunyan, A. J. Minnaard, Angew. Chem. Int. Ed.
Engl. 2012, 51, 3164–7.

Autoinductive effects in asymmetric copper(I)/phosphine catalyzed addition
reactions
111
[51] P. Ortiz, A. M. del Hoyo, S. R. Harutyunyan, European J. Org. Chem. 2015,
2015, 72–76.
[52] J. Rong, R. Oost, A. Desmarchelier, A. J. Minnaard, S. R. Harutyunyan, Angew.
Chemie - Int. Ed. 2015, 127, 3081–3085.
[53] J. Rong, J. F. Collados, P. Ortiz, R. P. Jumde, E. Otten, S. R. Harutyunyan, Nat.
Commun. 2016, 7, 13780.
[54] P. Ortiz, J. F. Collados, R. P. Jumde, E. Otten, S. R. Harutyunyan, Angew.
Chemie - Int. Ed. 2017, 56, 3041–3044.
[55] S. R. Harutyunyan, F. López, W. R. Browne, A. Correa, D. Peña, R. Badorrey, A.
Meetsma, A. J. Minnaard, B. L. Feringa, J. Am. Chem. Soc. 2006, 128, 9103–
9118.
[56] M. Pérez, M. Fañanás-Mastral, P. H. Bos, A. Rudolph, S. R. Harutyunyan, B. L.
Feringa, Nat. Chem. 2011, 3, 377–381.
[57] A. V. R. Madduri, Total Synthesis of (-)-Borrelidin and (-)-Doliculide and the
Development of the Catalytic Asymmetric Addition of Grignrd Reagents to
Ketones, University of Groningen, 2012.
[58] K. Osakada, T. Takizawa, M. Tanaka, T. Yamamoto, J. Organomet. Chem.
1994, 473, 359–369.
[59] S. L. Shi, M. Kanai, M. Shibasaki, Angew. Chemie - Int. Ed. 2012, 51, 3932–
3935.
[60] Y. Yang, I. B. Perry, S. L. Buchwald, J. Am. Chem. Soc 2016, 138, 9787–9790.
[61] B. H. Lipshutz, J. M. Servesko, B. R. Taft, J. Am. Chem. Soc. 2004, 126, 8352–
8353.
[62] J.-X. Chen, J. F. Daeuble, J. M. Stryker, Tetrahedron 2000, 56, 2789–2798.
[63] M. Hanstein, Asymmetric Catalysis in the Synthesis of Cis -Cyclopropryl
Containing Fatty Acids and the Addition of Grignard Reagents to Carbonyl
Compounds - PhD Thesis, University of Groningen, 2014.
[64] P. Ortiz, F. Lanza, S. R. Harutyunyan, in Top. Organomet. Chem., Springer,
2016, pp. 99–134.
[65] A. Krasovskiy, P. Knochel, Synthesis (Stuttg). 2006, 890–891.
[66] R. Moser, Z. V. Boskovic, C. S. Crowe, B. H. Lipshutz, J. Am. Chem. Soc.
2010, 132, 7852–7853.
[67] S.-M. Lu, C. Bolm, Angew. Chemie Int. Ed. 2008, 47, 8920–8923.
[68] M. Rodríguez-Fernández, X. Yan, J. F. Collados, P. B. White, S. R.
Harutyunyan, J. Am. Chem. Soc. 2017, 139, 14224–14231.

Chapter 4
112

Chapter 5
Design of an asymmetric organic autocatalytic reaction: the
reduction of ketones and imines with borane After an overview regarding asymmetric autocatalysis, in this chapter the design of
an organic autocatalytic reaction is discussed. Our approach is inspired by Corey-
Bakshi-Shibata reduction of ketones and imines with borane. The syntheses of
substrates and (auto)catalysts are reported and the potential of this reaction for
asymmetric autocatalysis is evaluated.

Chapter 5
114
5.1. Introduction How was life originated in the prebiotic world? The way life was formed from animated
matter is one of the most intriguing mysteries that scientists are trying to solve. The
key resides in the feature of living beings to make exact copies of themselves, called
self-replication. Over the past fifty years, scientists aimed to build artificial systems
able to mimic the self-reproduction observed in living systems.[1] RNA and DNA are
able to replicate themselves, acting as templates for new ribonucleotide chains. This
feature appeared at first to be a unique feature of enzymatic catalysis,[2,3] but in 1986
examples of enzyme-free replication of hexadeoxynucleotides from
trideoxynucleotides was achieved by von Kiedrowski et al..[1,4] In this system, the self-
replication was based on the recognition via H-bond of trideoxynucleotides by the
template, that could facilitate the formation of the hexamer. The strong interaction
between template and product was facilitating the formation of the product but, on the
other hand, impeding the turnover of the template/catalyst. The low reaction rate
through the linkage of the two trimers via the formation of a phosphoroamidate in the
presence of a condensing agent to achieve the typical autocatalytic kinetic pattern.[5]
Later, various examples of self-replicating systems were reported[1,6,7].
5.1.1. Autocatalysis
Autocatalysis is the easiest form of molecular self-replication and this concerns a
reaction where the products acts as catalyst for its own synthesis (Scheme 1a).[7] In
fact, reactions with this behavior are characterized by a typical sigmoidal kinetic curve
(time vs. product), caused by the dependence of the reaction rate on the concentration
of the product/catalyst (Scheme 1b, source of the plots: review of Bissette and
Fletcher)[7]. After an initial induction period, where the concentration of the
product/catalyst is low, the reaction rate dramatically increases as an effect of the
autocatalysis. If the product is an efficient catalyst for its own production, the kinetic
curve will be exponential, while a more sluggish autocatalysis will result in a parabolic
curve.[7] Consequently, the presence of autocatalysis can be detected by two features:
A sigmoidal curve time/product
An acceleration of the reaction by initial addition of the product to avoid the
initial induction period.

Design of an asymmetric organic autocatalytic reaction: the reduction of ketones and imines with borane
115
Scheme 49 Source of the plots: review of Bissette and Fletcher.[7]
Autocatalysis is only the simplest case of how a product can affect its own synthesis.
Blackmond offers a precise classification of the cases and the implications on the
kinetics.[8] Autocatalysis is the simplest case, where in a system there is only one
catalytic cycle. However there is the possibility that the product of a reaction can act as
catalyst for a second reaction, and that the product of the second reaction works as
catalyst for the first one, and this can potentially occur also for a larger number of
catalytic reactions. We refer to a cycle of two (or more) reactions related because the
product of one can catalyzed the other, as cross catalysis.[6] This phenomenon has been
proposed to be a valid explanation of the origin of life.[9,10] Another case is
autoinduction, which is extensively discussed in the Chapter 4 of this thesis. The
following pages will focus on autocatalysis of small molecules.
Several efforts were made toward the design of a non-enzymatic autocatalytic reaction
involving small molecules, to achieve a simple artificial self-replicating system. Many
examples involve template based autocatalysis.[7,11,12] For this purpose, the following
aspects must be kept into account:
The binding of the template must be specific for the couple of reagents rather
than for the product or molecules of the same species. The interaction of the
substrates with the template has to be sufficiently strong, in order to catalyze
efficiently their reaction.

Chapter 5
116
The interaction of the template with the product (himself) must be weak to
guarantee the release of the catalyst
The recognition sites must be sufficiently far from each other to avoid self
binding[7]
These factors make the design of an autocatalytic reaction quite challenging.
Nevertheless, besides providing a feasible explanation for the emergence of life, this
peculiar type of catalysis has some intrinsic advantages: [13]
The efficiency of the process is high, as it is an automultiplication
The amount of catalyst increases during the course of the reaction and, for this
reason, the rate of the reaction drastically increases with the conversion.
As product and catalyst coincide, no separation of the product from the catalyst
is needed.
In the 90s, the group of Rebek designed an autocatalytic reaction involving
nitrogeneous bases.[11] The replication involved recognition of an ester (1) and an
amine (2) by adenine-[11,14] or thymine like moieties[15] and the subsequent linkage
through transamidation (Scheme 2a). The length of the spacers between these two
recognition sites is critical for the autocatalytic mechanism as it could influence the
non-catalyzed association of the two reagents (Scheme 2b). The addition of further
functional groups cause an excessive strength of ligation between two molecule of the
templates, poisoning of the catalyst.[16] The hypothesis of template catalysis was then
proved by kinetic studies by Reinhoudt e al..[12]

Design of an asymmetric organic autocatalytic reaction: the reduction of ketones and imines with borane
117
Scheme 50 Autocatalysis in Rebek’s system. [11,14]
Inspired by the recognition by H-bonding, autocatalytic cycloadditions, including
Diels-Alder reaction and addition of azides to maleimide have been developed by the
groups of Sutherland[17] and Philp[18]. The complexity of these reactions, where
multiple diastereomers of the products can be formed (therefore multiple feasible
catalytic cycles), required thorough kinetic analyzes to be rationalized.[19]
Autocatalysis was detected also in organometallic reactions: Collum et al. noticed a rate
enhancement by the product in the ortho-lithiation of phenyl amides and other arenes.
This effect was most important at high concentration, where the formation of
aggregates affects the rate limiting step of the catalytic cycle. [20,21]

Chapter 5
118
More recent studies on autocatalysis concern the use of organometallic reagents and
physical autocatalysis proceeding via the formation of micelles.[7,22,23]
5.1.2. Asymmetric autocatalysis
If the product of an autocatalytic reaction is chiral and enantiomerically enriched, it is
possible that it transfers this chiral information to the new molecules formed. This
event, called asymmetric autocatalysis, has been adopted as one of the feasible
explanations for homochirality on Earth (the consistency of configuration between
natural amino acids (L), sugars (D) and other natural molecules)[24], via the
enhancement of a small imbalance between the two enantiomers. However,
autocatalysis is not sufficient to explain how a small imbalance of two enantiomers can
be amplified to obtain one pure enantatiomer. It was hypothesized that the major
enantiomer must inhibit the synthesis of its antipode to allow the propagation of
chirality.[25]
In this Chapter, we will focus on the ability of the product alone to induce asymmetry
in its own production. We will include both asymmetric autocatalysis and those cases
of asymmetric autoinduction where the product is the only chiral auxiliary in the
reaction (the catalyst is achiral). Instead, the cases of autoinduction where the product
interact with a chiral catalyst to improve his own production has been discussed in
Chapter 4.
The first example of a chiral molecule inducing enantioselectivity in its own production
dates back to 1989 and is described by Alberts and Wynberg.[26] In both addition of
ethyllithium or diethylzinc to benzaldehyde catalyzed by Ti(IV), the (+)-product-
alkoxide can favor the synthesis of the same enantiomer (Scheme 3) with significant
enantioselectivity.
Scheme 51 Asymmetric induction in addition of ethyllithium to benzaldehyde by Alberts and
Wynberg.[26]
Few years later, Soai and coworkers also reported asymmetric autocatalysis in a similar
way in the addition of diethylzinc to ferrocenylcarboxaldehyde.[27] Simultaneously,
they discovered an autocatalyic system that represents until now a unique example of
asymmetric autocatalysis: the asymmetric addition of diethylzinc to
pyrimidinaldehyde.

Design of an asymmetric organic autocatalytic reaction: the reduction of ketones and imines with borane
119
At first, it was reported that 7 can favor its own formation with a moderate ee (Scheme
4a).[28] But in 1995, Soai described that a highly enantioenriched product could be
obtained, utilizing almost racemic 9 (ee ~ 0.00005%) as chiral initiator[29]. This
phenomenon was consequently named asymmetric amplification. Moreover, also
small amounts of other chiral organic molecules[30,31], circularly polarized light
(CPL)[32], chiral inorganic[33–35] and organic crystals[36] could be used as chiral source.
Impressively, even in the absence of any chiral entity, the synthesis of 7 still occurred
with enantioselectivity, which indicates that the amplification is so efficient that even
small statistical imbalances between the enantiomers can be enhanced thus leading to
absolute enantioselective synthesis.[37]
The non-linear correlation between the ee of the catalyst and of the product ((+)-NLE),
is due to the aggregation of the catalyst. Molecules of the minor enantiomer of the
catalyst are inactivated by the formation of inactive oligomers with stoichiometric
amount of the major enantiomer. In this way, the excess of major enantiomer is able
to catalyze the reaction towards its own synthesis.[29,38,39] Numerous studies aimed to
explain the mechanism of this amplification[40] and the nature of the aggregates formed
in the reaction. The isolation of tetramers or higher oligomeric aggregates
aggregates[41] suggested, multiple oligomeric species participate to the amplification
mechanism. Kinetic studies confirmed this hypothesis.[42] The understanding of the
mechanism behind absolute asymmetric synthesis in Soai’s reactions paves the way to
the design of further autocatalytic reactions whit amplification of chirality.
Scheme 52 Soai’s asymmetric autocatalytic reaction.[29]

Chapter 5
120
Soai’s asymmetric autocatalytic system has been included in more complicated cross-
catalytic cycles by the group of Amedjkouh. Soai’s autocatalyst 9 can serve as a
asymmetric catalyst for the addition of diisopropylzinc reagents to a second aldehyde
and the product of this reaction would simultaneously enhance the chiral amplification
of Soai’s reaction (Scheme 5).[43,44]
Scheme 53 Amedjkouh’s cross catalytic system.[45]
Carreira and coworkers also described asymmetric autoinduction in synthesis of the
Efavirenz intermediate 15 via the addition of a diorganozinc reagent to the
trifluoromethyl ketone 13.[46] By the simple use of (S)-15 from the beginning of the
reaction, further product is obtained with 76% ee. The use of the chiral ligand L1 in
combination with the product increased the ee to 91% (Scheme 6).
Scheme 54 Autocatalysis in the synthesis of (S)-15.[46]
When designing an asymmetric autocatalytic cycle it is convenient to involve
organometallic reactions that are generally characterized by high rates and high
turnover of the catalyst. Furthermore, metallic catalyst, having multiple coordination

Design of an asymmetric organic autocatalytic reaction: the reduction of ketones and imines with borane
121
sites, can easily form aggregates and give rise to propagation of chirality. However,
when it comes to explaining the origin of life and of homochirality on earth, it is hard
to find an explanation involving artificial water- and air-sensitive reagents, such as
dialkyl zinc.
Mauksch and Tsogoeva were the first to report asymmetric autocatalysis and (+)-NLE
in the Mannich reaction between α-immino esters and acetone.[47] The authors claim
that, in the presence of 1-5 mol% of enantiopure 19, ee values up to 27% can be
obtained, but with higher catalytic loading the enantioselectivity improves up to 96%
ee (Scheme 7a). The recognition of the two reactants by the template occurs via H-
bonding. However, few controversies arose about the determination of the mechanism
of autocatalytic and asymmetric amplification: while the authors proposed that
heterodimers of 19 are labile and regenerate the reactants,[48,49] Blackmond et al. argue
that this concept violates the principle of microscopic reversibility[50]. Moreover, in the
original study, the enantioenriched product used as catalyst was prepared via natural
proline catalyzed Mannich reaction. The group of Feringa decribes that 17, synthesized
in an alternative way, has no catalytic activity and they question if residual proline can
act as real catalyst for this reaction.[51] Mauksch and Tsogoeva also report spontaneous
symmetry breaking in this reaction (Scheme 7b).[52]
Scheme 55 Aymmetric autocatalysis in Mannich condensation.[52]
Asymmetric autocatalysis in the Mannich reactions attracted interest by several groups
in the last years,[53,54] as the development of a purely organic reaction would constitute
a valuable proof for the origin of homochirality, while Soai’s reaction can only be
referred to as a proof of concept for an autocatalytic mechanism. Mannich reaction can
be performed autocatalytically using a limited scope of ketones and it can occur also in

Chapter 5
122
water[53], but the mechanism must be clarified further to understand if this can be
called proper autocatalysis.
5.1.3. Corey-Bakshi-Shibata reduction and feasibility of asymmetric autocatalysis
Our group aims to develop a novel asymmetric autocatalytic reaction. We chose Corey-
Bakshi-Shibata reduction as possible target for our design, that concerns the reduction
of ketones or imines with borane by mean of a chiral oxazaborolidine.[55,56]
In 1981 Itsuno et al. reported the reduction of ketones using stoichiometric amounts
of oxazaborolidine derived from (L)-valine (20, Scheme 8).[57] Depending on the
difference between the steric hindrances of the two substituents, optical yields up to
79% could be achieved with this methodology. The selectivity could be improved
further with the use of 21 as precursor for the reagent.[58]
Scheme 56 Itsuno’s reduction of ketones.[57,58]
In 1987 Corey, Bakshi and Shibata published a highly enantioselective methodology for
the reduction of ketones with borane, upon use of catalytic amounts of the
oxazaborolidine 24, prepared from the natural amino acid (L)-proline.[55,56,59]
Generally, this reactions requires moderately low temperatures (0-25°C) and short
reaction times (0.5-3h). With only 10 mol% catalytic loading, excellent
enantioselectivities were achieved in the asymmetric reduction of acetylbenzene 22
(Scheme 9). As the oxazaborolidine was air and moisture sensitive, the use of its more
stable analog, B-Me-24, has become more frequent.[59]
Scheme 57 CBS reduction of acetylbenzene 22.[56]
The ketone scope of the CBS reduction is broad and the difference in the steric bulk of
the two substituents plays an important role in the enantiodiscrimination. The
mechanism involves the formation of a complex between the oxazaborolidine and the
borane, where the nitrogen atom coordinates the borane activating it towards the
donation of hydride. The ketone instead coordinates to the boron atom of 24, in such
Design of an asymmetric organic autocatalytic reaction: the reduction of ketones and imines with borane
123
a way to minimize the steric repulsions between the more hindered substituents and
the catalyst (Figure 1).[56] However, electronic factors also play a role, as it was
demonstrated by Corey via the reduction of substituted benzophenones.[60]
Figure 18 Transition state of CBS-reduction.[56]
Similarly to ketones, also imines can be reduced, but necessitate longer reaction times
and higher temperatures (Scheme 10).[61] In this case, the oxazaborolidine formed
from 21 assures the best enantioselectivity, but requires higher catalytic loadings than
24.
Scheme 58 CBS-reduction of imines.[61]
The group of Feringa inquired whether the reduction of ketones with borane can
undergo with an asymmetric autocatalytic mechanism. They used (S)-28 having an
enantiomeric excess of 10% in the reduction of 27, hypothesizing that the
oxaborolidine 29 can be formed in situ (Scheme 11). The reaction, even in the absence
of 28, proceeded fast but unfortunately the product had an ee of 5.8% which is within
the error margin of the chiral HPLC.[62]
Scheme 59 Reduction of 27 in the presence of its enantioenriched product 28.[62]
Using CBS-reduction, optically active alcohols and amines can be prepared. Alcohol
and amines are functional groups that can form the catalyst oxazaborolidine upon
reaction with borane. For this reason, this reaction has a good potential to become the
next example of asymmetric autocatalysis.

Chapter 5
124
5.2. Aim As discussed in the introduction, asymmetric autocatalysis is a rare event and concerns
metal-catalyzed reactions. The development of an organic asymmetric autocatalytic
reaction would shed light on the way homochirality on Earth was originated. Therefore,
the aim of this chapter is to develop an organic autocatalytic and enantioselective
reaction. Our design of such a reaction is based on the modification of the Corey-
Bakshi-Shibata reduction of ketones and imines with borane, where the organocatalyst
is the oxazaborolidine 24 derived from diphenylprolinol 31. With the idea of attaining
asymmetric autocatalysis in mind, we hypothesised that diphenylprolinol (31) could
be obtained by CBS-reduction of the iminoalcohol 30 (Scheme 12). Furthermore, we
expected that the phenylprolinol 35 is a potential catalyst for the CBS reduction and
can function as autocatalyst in the reduction of the ketone 33 or the imine 34 with
borane (Scheme 12). Herein we describe our efforts to to test this hypothesis.
Scheme 60 Design of asymmetric autocatalytic CBS-reduction.
5.3. Results and discussion Using CBS-reductions, it is possible to reduce ketones to alcohols or imines to amines.
In our proposed system, two pathways are outlined. The first involves the reduction of
the prochiral imine 30, bearing an alcoholic functionality. The catalyst 34 can be
obtained from the product 31 (Scheme 13a). In the following pages, we will refer to
this pathway as the imine pathway. The second possibility is that the oxazaborolidine
35 is obtained from the amino alcohol 35, that is the product of either reduction of
imine 33 or ketone 34 (Scheme 13b). Both of these starting materials bear a chiral
center, meaning that the diastereoselectivity of their reduction should be evaluated in
place of the enatioselectivity. This second pathway will be called the ketone pathway.

Design of an asymmetric organic autocatalytic reaction: the reduction of ketones and imines with borane
125
Scheme 61 Syntheses of 24 and 35.
With this in mind, we proceeded towards the synthesis of the substrates required for
our designed autocatalytic reactions
5.3.1. The Imine Pathway
First, we focused on the synthesis of the iminoalcohol 30 (Scheme 14) that could be
obtained upon addition of an organometallic reagent to the iminoketone 36.[63] The
aminoketone 33, when exposed to the air, easily oxidizes to 36, because of the
stabilization of the product by conjugation.[63,64] We planned to prepare 33 from the
natural amino acid proline (37).
Scheme 62 Retrosynthesis of 30.
At first, we attempted the arylation of a Boc-protected proline (N-Boc-37) with PhLi in
the presence of HMPA but Boc-33 was not formed under these conditions (Scheme
15a). Then, we tried to obtain Boc-33 by formation of a mixed anhydride with
diphenylchlorophosphite and subsequent reaction with PhMgBr but unfortunately,
this strategy did not lead to the formation of the product either (Scheme 15b).
Therefore, we decided to synthesize 33 by Friedel-Craft alkylation (Scheme 16a).

Chapter 5
126
Scheme 63 Attempts towards the preparation of N-Boc-33.
The proline was converted in the acylchloride 38·HCl by using PCl5 as chlorinating
agent. Upon addition of AlCl3 and benzene 33·HCl was formed. Even if this salt does
not undergo oxidation as fast as the free ketoamine 33, its purification revealed to be
tedious and we could not isolate the pure compound. We decided therefore to subject
the crude to oxidation by oxygen in a basic environment directly after the reaction. In
this way, the product 36 was obtained in poor yield (6%) (Scheme 16a). This
compound is relatively unstable and undergoes radical polymerization; it must be
isolated rapidly, stored at 4oC and used within few weeks.
To improve the efficiency of this synthesis, we protected the amine as carbamate (Boc
and Fmoc). The chlorination of N-Boc-37 using SOCl2 or PCl5 did not afford the
corresponding acyl chloride (Scheme 16b): we attributed the unsuccessful result to
the limited stability of the Boc to the acidic conditions. For this reason, we changed the
proline protecting group to Fmoc. In this case, it was possible to obtain N-Fmoc-38
with SOCl2 in toluene (Scheme 16c). Unfortunately, after Friedel-Craft acylation, no
product was isolated, probably due to the acylation of the aromatic rings on the Fmoc
moiety. Our last approach concerned the reaction of the Fmoc-38 with PhMgBr[65]. In
this case also, the synthesis of N-Fmoc-33 was not achieved.

Design of an asymmetric organic autocatalytic reaction: the reduction of ketones and imines with borane
127
Scheme 64 Attempts towards the preparation of N-protected-33.
From these results, we envisioned that the unprotected amine 33 is considerably
unstable and we concentrated our efforts on the direct preparation of the imino-ketone
36. Helquist et al. reported an elegant procedure for the synthesis of this
compound[66], which involves the silver-catalyzed hydroamination and benzylic
oxidation of compound 39. The reported synthesis of the precursor requires two
synthetic steps[67] from compound 42, prepared from lithium phenyl acetylide (40)
and 1-bromo-3-chloropropane (41)[68]. In our hands, this procedure resulted on a yield
of 50% of 42 and 15% in the preparation of 39. Compound 36 was obtained in 13%
yield (Scheme 17a).a *Experiments performed by Dr. Francesco Lanza. NMR-Spectra
match with the reported.[66–68] Convenient procedure reported in 2017 by Cheng et
al.[69]

Chapter 5
128
In 2018, 36 was obtained by Yu et al. by acyl migration of α-azidyl tertiary ketones
with excellent yield (Scheme 17b).[70] Because this methodology was published very
recently, this synthetic route was not explored further.
Scheme 65 Alternative methodologies for the syntheses of 36.
Given that 33 is oxidized by oxygen to imino-ketone, we decided to prepare first the
amino alcohol 35 and subsequently treat it with a classic oxidizing agent for secondary
alcohols. Starting the ortho lithiation of the N-Boc-pyrroliyne (44) with s-BuLi and
TMEDA and condensation with benzaldehyde, we obtained N-Boc-35 as a mixture of
the syn and anti diastereomer in 79%.[71] The product was removed in acidic conditions
to afford 35 in 92% yield (Scheme 18a). The Swern oxidation afforded pure 36 in
22% yield. The low yield is due to the difficulty to remove the Et3N avoiding any acidic
workup that could hydrolize the imine. Using the Dess-Martin oxidation with
hypervalent-iodine as oxidizing agent (45), the starting material was converted
completely to an unidentified product.

Design of an asymmetric organic autocatalytic reaction: the reduction of ketones and imines with borane
129
Scheme 66 Synthesis of 36 by oxidation of 35.
With 36 in our hands, we could prepare 30 by simple addition of PhLi that afforded
the product of addition to the ketone at -78 oC. The chemoselective addition of the less
reactive MeLi was reported on 2-acetyl-2,4-dihydropyrrol is described in literature;[63]
it is remarkable that the addition of a more reactive organolithium reagent also
proceeds with an elevated chemoselectivity (Scheme 19).
Scheme 67 Synthesis of the imino-alcohol 30.
Once 30 became available, the moment had come to test the potential for autocatalysis
in its reduction. The reduction of 30 with borane in the presence of the catalyst B-Me-
24 took place to afford 31 in toluene as solvent (entries 1 and 2, Table 1), while only
a dirty crude was obtained in THF. The imino alcohol 30 could be reduced also in the
absence of catalyst. The reduction rates with or without catalyst are comparable. It is
feasible that the alcohol functional group can activate the borane towards the hydride
transfer, as hypothesized by the group of Feringa in the reduction of 27[62]. The amine
was subsequently protected with Boc to allow the separation of the two enantiomers
on the chiral HPLC. Unfortunately, we discovered that the reduction occurs with low

Chapter 5
130
or no enantioselectivity (Table 1). This fact, together with the nearly similar rates of
the catalyzed and uncatalyzed reduction pathways, leave few hopes for a successful
asymmetric autocatalysis. Consequently, this research line was abandoned.
Table 15 CBS reduction of 30.
Entry Solvent (x mol%) yield Boc-31 (%) ee (%)a
1 Toluene 25 78 13
2 Toluene 40 38 racemic
3 THF 25 n.d. b n.d.
a Calculated with the formula eeprod = eemeas-(eecat*x/100). eemeas was determined via
CSP-HPLC b Dirty crude, no Boc-2 could be isolated.
5.3.2. The Ketone Pathway
At this point of our travel towards the design of an asymmetric auto-organocatalyzed
reaction, we concentrated our efforts on the second pathway. Here we planned to
achieve asymmetric induction in the reduction of 33 or 34 catalyzed by 35 (Scheme
12). As discussed in Paragraph 5.3.1., the amino ketone 33 is an unstable compound
that spontaneously oxidizes to 36. A comparable behavior can be expected for
compound 34, and to the best of our knowledge, no synthesis of the latter has been
reported so far. Moreover, both compound 33 and 34 bear a preformed stereogenic
center and therefore, they cannot be employed for the evaluation of the
enantioselectivity of their reduction. Then we planned to submit 36 directly to the
CBS-reduction with syn-35 or anti-35 as catalyst.

Design of an asymmetric organic autocatalytic reaction: the reduction of ketones and imines with borane
131
Scheme 68 Asymmetric synthesis of syn- and anti-phenylprolinol (35).
The Boc-protected amino alcohols were prepared with the procedure reported by
Gilday et al.[72], that involves the enantioelective α-deprotonation of the Boc-
pyrrolidine (44) with catalytic (+)-sparteine (46) and the subsequent trapping of the
lithiated 44 with benzaldehyde (Scheme 20a). This reaction proceeds with syn-
diastereoselectivity and (S,S)-syn-N-Boc-35 and (S,R)-anti-N-Boc-35 were obtained
with respective ees of 76% and 68%. The amino alcohol 35 was recovered after
deprotonation with TFA (Scheme 20b).
The catalytic activity of (S,S)-syn-35 and (R,S)-anti-35 was tested in the CBS
reduction, taking the acetyl ferrocene (47) as model ketone molecule. Both of the
diastereoisomers of 35 were found to be active as asymmetric catalysts, even though
their performance doesn't match to that of Me-34 in terms of enantioselectivity (entry
1, Table 2). Interestingly, anti-35 (with configuration R,S) provided (R)-48 in 97%
yield and an enantiomeric excess of 86% (when normalized with respect to one of the
catalyst; entry 3, Table 2). On the contrary, syn-35 afforded the opposite enantiomer
of the product with a lower enantioselectivity in 77% yield (entry 2, Table 2),
demonstrating that it’s the configuration of the alcohol that directs the hydride trasfer.
In anti-35, the stereocenters are favorably combined and form the matched catalyst.
The syn is instead the mismatched catalyst.

Chapter 5
132
Table 16 Asymmetric reduction of 47
Entry Catalyst Yield (%)a ee (%)b
1 c B-Me-24 96 98, (R)
2 d (S,S)-syn-35/BH3·SMe2 (ee 78%) 77 31 (40e), (S)f
3 d (R,S)-anti-35/BH3·SMe2 (ee 66%) 97 57 (86e), (R)f
a Isolated yield b Determined via CSP-HPLC. c reaction was performed following the
literature procedure.[73] d 35 and BH3·SMe2 (0.5 equiv.) were stirred at 40oC for 45 min
prior to the addition of 47. e ee of the product normalized with the ee of the catalyst.
Calculated with the formula eemeas/eecat = eeeff. f Configuration assigned by comparison
with the literature.[73]
At this point, the anti-diastereomer of 35 should have been tested in the asymmetric
reduction of 36. Unfortunately, due to the instability of the imino-ketone 36 and the
tedious synthesis, it was not possible for us to perform further studies concerning its
autocatalytic reduction and we renounced to this research line.
5.3.4. Reduction of the phenyl-(2-pyridyl)-ketone
Our last effort towards the design of asymmetric autocatalysis inspired by CBS-
reduction concerned the reduction of the phenyl 2-pyridyl ketone (49). We imagined
that pyridine moiety could substitute the pyrroldine moiety of 34 and coordinate to
the borane (Scheme 21). A catalyst where the nitrogen does not form covalent bonds
with the borane can be generated faster than the oxazaborolidine and therefore is a
better candidate for an autocatalytic reaction. Nevertheless the reduction of 49 by the
Me-24 and borane proceeded with a complete lack of enantioselectivity due to the poor
differentiation in the steric bulk between the two substituents of the ketone, so we
cannot expect any asymmetric autocatalysis using CBS method to reduce this
substrate.

Design of an asymmetric organic autocatalytic reaction: the reduction of ketones and imines with borane
133
Scheme 69 Autocatalysis in the reduction of phenyl-(2-pyridyl)-ketone.
5.4. Conclusions We discussed the design of an asymmetric autocatalytic reaction inspired by CBS-
reduction of ketones and imines. Two possible autocatalytic cycles were envisioned:
the first concerning the reduction of an imine, the second of a ketone. The starting
material for both pathways towards autocatalysis were prepared, with low yields due
to the instability of one of the intermediates.
For what concerned the reduction of the imine, the reaction rate in the presence of the
autocatalyst shows no significant improvement with respect to the not catalyzed one.
Moreover we observed no clear influence of the product on the enantioselectivity.
For the pathway involving the reduction of the ketone, two diastereomeic (syn and
anti) enantioeriched precursors for the catalyst were prepared. They were tested in the
reduction of a model ketone. The catalyst generated from the anti diastereomer
showed good performance (matched catalyst). However, we did not evaluate the
potential of this reaction for the autocatalysis because the substrate could only be
obtained in low yields and is unstable.
Our a posteriori considerations about the possibility of autocatalysis on this system
are:
In both pathways, the product of the reaction is an amino alcohol, which is the
precursor of the catalyst (oxazaborolidine). Consequently, the rate of formation
of the oxazaborolidine is crucial for an efficient autocatalysis.

Chapter 5
134
In an efficient asymmetric CBS reduction, the difference between the
dimensions of the substituents of the ketone or imine is important. In the case
of autocatalysis, a less hindered substrate would generate a less hindered
product that would be a worst catalyst for this reaction. The similarity of the
product/catalyst with the starting material constitutes a limitation for the chiral
induction.
As in CBS reduction, the product has no possibility to form oligomeric species,
this reaction has no potential for asymmetric amplification.
5.5. Experimental section
5.5.1. General information
All reactions using oxygen- and/or moisture-sensitive materials were carried out with
anhydrous solvents (vide infra) under a nitrogen atmosphere using oven dried
glassware and standard Schlenk techniques. Reactions were monitored by 1H NMR.
Purification of the products, when necessary, was performed by flash-column
chromatography using Merck 60 Å 230-400 mesh silica gel or VWR AnalaR
NORMAPUR aluminum oxide basic. NMR data was collected on Bruker Avance NEO
600 (1H at 600.0 MHz; 13C at 150.87MHz), equipped with a Prodigy Cryo-probe and
Varian VXR400 (1H at 400.0 MHz; 13C at 100.58 MHz), equipped with a 5 mm z-
gradient broadband probe. Chemical shifts are reported in parts per million (ppm)
relative to residual solvent peak (CDCl3, 1H: 7.26 ppm; 13C: 77.16 ppm). Coupling
constants are reported in Hertz. Multiplicity is reported with the usual abbreviations
(s: singlet, bs: broad singlet, d: doublet, dd: doublet of doublets, t: triplet, , q: quartet,
m: multiplet). Exact mass spectra were recorded on a LTQ Orbitrap XL apparatus with
ESI ionization. Enantiomeric excesses (ees) were determined by Chiral HPLC analysis
using a Shimadzu LC-10ADVP HPLC equipped with a Shimadzu SPD-M10AVP diode
array detector and by Waters Acquity UPC2 system with PDA detector and QDA mass
detector.
Unless otherwise indicated, reagents and substrates were purchased from commercial
sources and used as received. Solvents not required to be dry were purchased as
technical grade and used as received. Dry solvents were freshly collected from a dry
solvent purification system prior to use. The DMAE was distilled on KOH and stored
on MS4A. Inert atmosphere experiments were performed with standard Schlenk
techniques with dried (P2O5) nitrogen gas. All reported compounds were characterized
by 1H and 13C NMR and compared with literature data. All new compounds were fully
characterized by 1H and 13C NMR and HRMS techniques. The absolute configurations
of products were attributed by comparison with previous literature.

Design of an asymmetric organic autocatalytic reaction: the reduction of ketones and imines with borane
135
5.5.2. Synthesis of N-Fmoc-38
(9H-fluoren-9-yl)-methyl (S)-2-(chlorocarbonyl)pyrrolidine-1-
carboxylate
Under inert atmosphere, 0.26 mL of SOCl2 (3.5 mmol, 1.3 equiv.) were
added dropwise at 70°C to a suspension of N-Fmoc-37 (1.0 g, 2.7 mmol,
1.0 equiv.). The reaction was stirred at 70°C for 8 h. Dry toluene (6 mL)
were added and the azeotrope SOCl2/toluene was distilled under reduced
pressure (procedure repeated 3 times). When about 2 mL of toluene were
left, the solution was cooled to -20°C. The crystals were filtered and dried under
reduced pressure an pale yellow crystals of N-Fmoc-38 were obtained (0.66 g, 1.9
mmol, 69% yield). The NMR data are in agreement with the ones present in
literature[74]. 1H NMR (400 MHz, Chloroform-d) δ 7.77 (dd, J = 7.6, 3.4 Hz, 4H), 7.60 (t, J = 7.8
Hz, 1H), 7.55 (d, J = 7.5 Hz, 1H), 7.41 (t, J = 7.4 Hz, 2H), 7.37 – 7.23 (m, 2H), 4.69
(dd, J = 8.8, 4.0 Hz, 1H), 4.60 – 4.32 (m, 2H), 4.23 (dt, J = 29.9, 6.5 Hz, 1H), 3.70 –
3.48 (m, 2H), 2.40 – 2.18 (m, 2H), 2.10 – 1.85 (m, 2H).
13C NMR (101 MHz, Chloroform-d) δ 174.4, 155.4, 143.9, 143.8, 143.5, 141.4, 127.8,
127.7, 127.2, 127.1, 127.0, 125.1, 125.0, 124.8, 124.7, 120.1, 120.0, 67.9, 67.8, 67.6, 67.3,
47.2, 47.1, 46.6, 30.5, 29.3, 24.1, 23.0.
5.5.3. Synthesis of the imino-ketone 36
(3,4-dihydro-2H-pyrrol-5-yl)(phenyl)methanone
Under inert atmosphere, 2.0 g of (L)-proline (9) (17.4 mmol, 1.0
equiv.) were added to a suspension of 3.6 g of PCl5 (17.4 mmol, 1.0
equiv.) in CH2Cl2 (100 mL) at 0oC. the reaction was allowed to warm
to rt and stir for 2h. The solvent was removed under reduced pressure,
then the crude was dissolved in benzene (70 mL) and 6.9 g of AlCl3
were added to the reaction mixture (52 mmol, 3.0 equiv.), the reaction was warmed up
to 60oC. After 3 h, the reaction mixture was quenched into crushed ice and a 1M HCl
aqueous solution. The organic phase was separated and the acqueous phase was
washed with 20 mL of AcOEt. The organic phase was neutralized with NaHCO3 sat in
H2O and aluminates were filtrated. The aqueous phase was extracted with CH2Cl2 (3 x
20 mL). 1.5 mL of HCl (37% in H2O) were added and the solvent was removed under
reduced pressure. The crude was dissolved in H2O (100mL) and the pH was adjusted
to ~7 with NaHCO3. A balloon containing O2 was attached to the reaction vessel.
Subsequently, the reaction was warmed to 50oC and stirred for 1h. The water phase
was extracted with CH2Cl2 (3 x 20 mL). The joint organic phases where dried on
Na2SO4, filtered and the solvent was removed under reduced pressure. The crude was
stored under inert atmosphere at 4oC and cleaned the day after by column
chromatography (SiO2, pentane:AcOEt, 4:1). 36 was obtained as a dark brown oil (117

Chapter 5
136
mg, 0.68 mmol, yield 4%) and stored under inert atmosphere at 4oC for a maximum
time of 4 weeks. The spectral data match those reported in literature[75]
1H NMR (400 MHz, Chloroform-d) δ 8.17 (d, J = 8.2, 1.1 Hz, 2H), 7.62 – 7.50 (m, 1H),
7.46 (t, J = 7.6 Hz, 2H), 4.23 (tt, J = 7.6, 2.5 Hz, 2H), 2.96 (tt, 2H), 2.01 (p, 2H). 13C NMR (101 MHz, Chloroform-d) δ 191.1, 174.3, 135.7, 133.5, 130.6, 128.4, 63.4, 35.8,
21.8.
5.5.4. Procedure for the synthesis of 30
(3,4-dihydro-2H-pyrrol-5-yl)diphenylmethanol
In a heat dried Schlenk under inert atmosphere, 116.5 mg (0.67 mmol,
1.0 equiv.) of 36 were dissolved in 10 mL of Et2O and the solution was
cooled to -60oC. 0.35 mL of a solution of s-BuLi in n-Bu2O (1.9 M,
0.67 mmol, 1.0 equiv.) were added over 1h. The solution was allowed
to warm to -15oC over 3h. The reaction was quenched after 2.5 hours
with 0.7 mL of MeOH and 4 mL saturated aqueous NH4Cl solution and extracted with
Et2O (3 x 5mL). The combined organic phases were dried on MgSO4, filtrated and the
solvent was removed under reduced pressure. The product was purified by column
chromatography (SiO2, pentane : EtOAc, 9:1 -> 8:2) obtained as a colorless solid (129
mg, 0.51 mmol, 77% yield) 1H NMR (400 MHz, Chloroform-d) δ 7.41 – 7.21 (m, 10H), 5.67 (broad s, 1H), 3.93 (t,
J = 7.4 Hz, 2H), 2.50 (t, J = 8.2 Hz, 2H), 2.05 (p, J = 7.7 Hz, 2H). 13C NMR (101 MHz, Chloroform-d) δ 181.4, 143.8, 128.3, 127.9, 127.7, 80.2, 59.6, 35.1,
24.5.
HRMS (ESI+): m/z calcd. for C16H23NO2Na ([M+Na+]) 250.12264, found 250.12385
5.5.5. Procedure for the reduction of 30
N-Boc-diphenylprolinol (Boc-31)
In a heat dried Schlenk maintained under inert atmosphere, 10.2 mg
of B-Me-CBS-oxazaborolidine (B-Me-24) (0.045 mmol, 0.25 equiv.)
were dissolved in toluene (0.6 mL) and 0.05 mL of BH3.SMe2 (2M in
THF, 0.10 mmol, 0.7 equiv.) were added and allowed to stir at rt for
15 min. 0.065 mL of BH3.SMe2 (2M in THF, 0.13 mmol, 0.9 equiv.)
were added amd a solution of imine (37.7 mg, 0.15 mmol, 1.0 equiv.)
in 0.4 mL of toluene was added over 20 min. The reaction was stirred at 45oC for 17
hours, monitored by TLC (SiO2, pentane:AcOEt, 4:1) and quenched with MeOH (0.5
mL). The crude was dissolved in CH2Cl2 (4.5mL) and 105 mg of Boc2O (0.48 mmol, 1.1
equiv.) were added at 0oC. After stirring at rt for 3h, the solvent was removed under
reduced pressure and the crude was purified by column chromatography (SiO2,
pentane:AcOEt, 9:1). N-Boc-31 was obtained as a white solid (151 mg, 35.1 mmol, 78%
yield).

Design of an asymmetric organic autocatalytic reaction: the reduction of ketones and imines with borane
137
1H NMR (400 MHz, Chloroform-d) δ 7.42 – 7.34 (m, 4H), 7.34 – 7.22 (m, 6H), 4.89
(dd, J = 8.9, 3.7 Hz, 1H), 3.35 (q, J = 9.3 Hz, 1H), 2.86 (m, 1H), 2.09 (dq, J = 13.3, 8.8
Hz, 1H), 1.92 (m, 1H),1.51-1.37 (m, 2H), 1.43 (s, 9H), 0.78 (broad s, 1H). 13C NMR (101 MHz, Chloroform-d) δ 146.6 (quaternary), 143.9 (quaternary), 128.4,
128.0, 127.8, 127.5, 127.2, 127.2, 81.9, 80.8, 48.0, 29.9, 28.5, 23.1.
HRMS (ESI+): m/z calcd. for C22H26NO3 ([M+H+]) 352.19072, found 352.19161
CSP-HPLC: (254nm, Chiralcel OD-H, n-heptane:i-PrOH = 99:1, 40 °C, 0.5 ml/min.),
tR = 33.20 min, tR = 26.74 min
5.5.6. Racemic synthesis of the tert-butyl (S)-2-((R)-
hydroxy(phenyl)methyl)pyrrolidine-1-carboxylate
tert-butyl pyrrolidine-1-carboxylate (N-Boc-44)
In a 100 mL round bottom flask, 2.0 g of Boc2O (9.16 mmol, 1.0 equiv.)
were dissolved in 30 mL of CH2Cl2 at 0oC. To the solution, 0.96 mL of
pyrrolidine (44) (11.5 mmol, 1.26 equiv.) were added dropwise. The
reaction was allowed to warm up to rt and stirred for 2h. The solvent and
the residual pyrrolidine were removed under reduced pressure. N-Boc-
44 was obtained after a short column chromatography (SiO2, pentane : AcOEt, 4.1) as
transparent oil (1.42 g, yield 91%). The NMR data are in agreement with the ones
present in literature. [76] 1H NMR (400 MHz, Chloroform-d) δ 3.43 – 3.27 (d, 4H), 1.88 (t, J = 4.5, 2.2 Hz, 4H),
1.50 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 193.9, 79.0, 46.1, 45.8, 28.7, 25.9, 25.1.
(Rac)-tert-butyl-2-(hydroxy(phenyl)methyl)pyrrolidine-1-carboxylate (N-
Boc-35)
In a heat dried 50 mL Schlenk kept undert inhert and
anhydrous atmosphere, 5.57 mL of a 1.4 M solution of s-BuLi
in cyclohexane (7.8 mmol, 1.3 equiv.) were added to a
solution of 5.57 mL of anhydrous TMEDA (7.8 mmol, 1.3
equiv.) in 12 mL of Et2O at -78oC. The solution was stirred at
-78oC for 30 min and a solution of 1.04g of N-Boc-44 (6.0
mmol, 1.0 equiv.) in 1 mL of Et2O was added over 15 min. the
reaction was stirred for 4 h and 0.81 mL of benzaldehyde (849 mg, 8.0 mmol, 2.0
equiv.) in 1 mL of Et2O was added dropwise. The reaction was allowed to warm up to
rt and stir overnight. It was quenched with 5 mL of a saturated aqueous NH4Cl solution
and extracted with Et2O (3 x 10 mL) and the combined organic phases were dried on
Na2SO4, filtrated and the solvent was removed under reduced pressure. The crude was
purified by column chromatography (SiO2, CH2Cl2 : acetone = 97:3) and the product
was obtained as a colorless oil as a mixture of syn and anti isomer (10:1) with a

Chapter 5
138
combined yield of 79% (4.8 mmol). The spectral data match those reported in
literature[71,72]
syn-tert-butyl-2-(hydroxy(phenyl)methyl)pyrrolidine-1-carboxylate (syn-
N-Boc-35)
Pale yellow oil, 709 mg (3,69 mmol, 61% yield). The NMR data are in
agreement with the ones present in literature [71,72] 1H NMR (400 MHz, Chloroform-d) δ 7.43 – 7.25 (m, 5H), 5.80 (broad
s, 1H), 4.53 (d, 1H), 4.09 (td, 1H), 3.56 – 3.15 (m, 2H), 1.79 – 1.55 (m,
1H), 1.46 (s, 1H). 13C NMR (151 MHz, Chloroform-d) δ 154.9, 141.0, 128.7, 128.5, 127.8,
127.1, 80.9, 65.5, 45.9, 28.7, 25.5.
HRMS (ESI+): m/z calcd. for C16H23NO2Na ([M+Na+]) 300.15822, found 300.15823
anti-tert-butyl-2-(hydroxy(phenyl)methyl)pyrrolidine-1-carboxylate
(anti-N-Boc-35)
Pale yellow oil, 294 mg (1.06 mmol, 18% yield). The NMR data are in
agreement with the ones present in literature. [71,72]
1H NMR (600 MHz, Chloroform-d) δ 7.41 – 7.19 (m, 5H), 4.95 (broad
s, 1H), 4.24 (broad s, 1H), 3.37 (broad s, 1H), 2.93 (broad s, 1H), 1.83 (s,
3H), 1.52 (s, 10H), 1.48 – 1.38 (m, 1H). 13C NMR (151 MHz, Chloroform-d) δ 141.4, 128.2, 127.4, 126.9, 80.4,
63.5, 47.95, 28.7, 28.6, 28.6, 27.1, 23.7. Quaternary signals of the Boc-
group were not detected.
HRMS (ESI+): m/z calcd. for C16H23NO2Na ([M+Na+]) 300.15823, found 300.15823
5.5.7. Asymmetric synthesis of the tert-butyl-2-((R)-
hydroxy(phenyl)methyl)pyrrolidine-1-carboxylate
In a heat dried 50 mL Schlenk, kept undert inhert
and anhydrous atmosphere, 9.6 mL of a 1.4 M
solution of s-BuLi in cyclohexane (13.5 mmol, 2.3
equiv.) were added to 17 mL of Et2O at -78oC. A
solution of 0.42 mL of (+)-sparteine (423 mg, 1.8
mmol, 0.3 equiv.) in 1 mL of Et2O was added
dropwise, followed by a solution of 0.60 mL of
DMAE (535 mg, 6 mmol, 1.0 equiv.) in 1 mL of Et2O. The solution was stirred at -78oC
for 10 min and a solution of 0.98 mg of N-Boc-44 (6.0 mmol, 1.0 equiv.) in 1.0 mL of
Et2O was added over 15 min. the reaction was stirred for 3h and 0.81 mL of
benzaldehyde (849 mg, 8.0 mmol, 2.7 equiv.) in 1 mL of Et2O was added dropwise. The
reaction was allowed to warm up to rt and stir overnight. It was quenched with 5 mL

Design of an asymmetric organic autocatalytic reaction: the reduction of ketones and imines with borane
139
of a saturated aqueous NH4Cl solution and extracted with Et2O (3 x 10 mL) and the
combined organic phases were dried on Na2SO4, filtrated and the solvent was removed
under reduced pressure. The crude was purified by column chromatography (SiO2,
CH2Cl2 : acetone = 97:3) and the product was obtained as a colorless oil as a mixture of
syn and anti isomer (10:1) with a combine yield of 59% (3.5 mmol).
tert-butyl-(S)-2-((S)-hydroxy(phenyl)methyl)pyrrolidine-1-carboxylate
((S,S)-syn-N-Boc-35)
Pale yellow oil, 709 mg (2.56 mmol, 43% yield, 76% ee). The NMR
data are in agreement with the ones present in literature [71,72] 1H NMR (400 MHz, Chloroform-d) δ 7.43 – 7.25 (m, 5H), 5.80
(broad s, 1H), 4.53 (d, 1H), 4.09 (td, 1H), 3.56 – 3.15 (m, 2H), 1.79
– 1.55 (m, 1H), 1.46 (s, 1H). 13C NMR (151 MHz, Chloroform-d) δ 154.9, 141.0, 128.7, 128.5,
127.8, 127.1, 80.9, 65.5, 45.9, 28.7, 25.5.
HRMS (ESI+): m/z calcd. for C16H23NO2Na ([M+Na+]) 300.15822, found 300.15823
CSP-HPLC: (190 nm, Chiralcel OD-H, n-heptane/i-PrOH = 90:10, 40 °C, 0.5
ml/min.), tR = 13.83 min (major), tR = 12.38 min (minor)
tert-butyl-(S)-2-((R)-hydroxy(phenyl)methyl)pyrrolidine-1-carboxylate
((S,R)-anti-N-Boc-35)
Pale yellow oil, 260 mg (0.94 mmol, 16% yield, 68% ee) The NMR
data are in agreement with the ones present in literature. [71,72] 1H NMR (600 MHz, Chloroform-d) δ 7.41 – 7.19 (m, 5H), 4.95
(broad s, 1H), 4.24 (broad s, 1H), 3.37 (broad s, 1H), 2.93 (broad s,
1H), 1.83 (s, 3H), 1.52 (s, 10H), 1.48 – 1.38 (m, 1H). 13C NMR (151 MHz, Chloroform-d) δ 141.4, 128.2, 127.4, 126.9,
80.4, 63.5, 47.95, 28.7, 28.6, 28.6, 27.1, 23.7. Quaternary signals
of the Boc-group were not detected.
HRMS (ESI+): m/z calcd. for C16H23NO2Na ([M+Na+]) 300.15823, found 300.15823
CSP-HPLC: (190 nm, Chiralcel OD-H, n-heptane/i-PrOH = 99:1, 40 °C, 0.5 ml/min.),
tR = 47.10 min (major), tR = 44.24 min (minor)
5.5.8. General procedure for the deprotection of Boc-pyrroldines
Under inert atmosphere, Boc-35 (1.0 equiv.) was dissolved in CH2Cl2 and TFA (4.0-4.1
equiv.) was added dropwise at 0oC. The reaction was allowed to stir for 20h at rt and
was quenched with NH3 (25% in H2O). The organic layer was separated and the
aqueous phase was extracted with CH2Cl2 (3 x 5 mL). The joined organic phases were
dried with MgSO4, filtered and the solvent was removed under reduced pressure. The
product was purified with a short column chromatography (SiO2, pentane:AcOEt, 9:1
-> MeOH)

Chapter 5
140
(S)-phenyl((S)-pyrrolidin-2-yl)methanol ((S,S)-syn-35)
Reaction performed on 400 mg (1.44 mmol, 1.0 equiv.) of (S,S)-syn-N-
Boc-35 and 0.85 mL of TFA (6.32 mmol, 4.4 equiv.). The (S,S)-syn-35
was obtained as a yellow oil (101 mg, 0.97 mmol, yield 67%). The NMR
data are in agreement with the ones present in literature.[71] 1H NMR (400 MHz, Chloroform-d) δ 7.43 – 7.28 (m, 4H), 7.29 – 7.18
(m, 1H), 4.34 (d, J = 6.8 Hz, 1H), 3.85 (broad s, 2H), 3.42-3.30 (m, 1H), 2.99 (t, 2H),
1.98 – 1.34 (m, 4H). 13C NMR (101 MHz, Chloroform-d) δ 142.8, 128.5, 127.7, 126.7, 75.5, 65.1, 46.3, 28.4,
26.0.
(R)-phenyl((S)-pyrrolidin-2-yl)methanol ((R,S)-anti-35)
Reaction performed on 177 mg (0.64 mmol, 1.0 equiv.) of (S,R)-anti-
N-Boc-35 and 0.20 mL of TFA (2.60, 4.1 equiv.). (R,S)-anti-35 was
obtained as a yellow oil (78.1 mg, 0.44 mmol, yield 66%). The NMR
data are in agreement with the ones present in literature.[71] 1H NMR (400 MHz, Chloroform-d) δ 7.48 – 6.91 (m, 5H), 4.78 (d,
1H), 3.56 (broad s, 2H), 3.41 (m, 1H), 3.11 – 2.79 (m, 2H), 1.82 – 1.52 (m, 3H), 1.52 –
1.34 (m, 1H). 13C NMR (101 MHz, Chloroform-d) δ 142.1, 128.4, 127.3, 126.0, 73.7, 64.2, 46.8, 25.5,
24.9.
5.5.9. General procedure for the reduction of 47
(R)-2-ferrocenyl-ethanol (48)
In a heat dried Schlenk maintained under inert atmosphere, 0.09 mL
of a solution of BH3.SMe2 (2M in THF, 0.18 mmol, 0.6 equiv.) were
added at rt to 15.9 mg of (R,S)-anti-35 (0.09 mmol, 0.25 equiv.)
dissolved in 1 mL THF. The solution was stirred at 40oC for 40 min and
subsequently it was allowed to cool to rt in 20 min. A solution of 68 mg
of 47 (0.3 mmol, 1 equiv.) in 0.3 mL of THF and 0.11 mL of a solution of BH3.SMe2 (2M
in THF, 0.21 mmol, 0.7 equiv.) were added simultaneously at rt over 15 minutes. The
reaction was quenched after 1h with MeOH (0.3 mL). The solvent was removed under
reduced pressure and the crude was purified by column chromatography (SiO2,
pentane:AcOEt, 4:1) to obtain the alcohol 48 as an orange solid (66 mg, 0.29 mmol,
yield 97%, ee 57%). Spectral data match with the ones reported in literature[77]. 1H NMR (400 MHz, Chloroform-d) δ 4.55 (qd, J = 6.4, 4.7 Hz, 1H), 4.31 – 4.01 (m,
9H), 1.90 – 1.78 (m, 1H), 1.44 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 95.0, 71.0, 68.4, 68.1, 68.0, 66.3, 66.2, 65.7,
23.9.
CSP-HPLC: (254 nm, Chiralcel AD-H, n-heptane/i-PrOH = 95:5, 40 °C, 0.5 ml/min.),
tR = 28.15 min (major), tR = 29.23 min (minor)

Design of an asymmetric organic autocatalytic reaction: the reduction of ketones and imines with borane
141
5.5.10. CBS-Reduction of 49
Phenyl-(2-pyridyl)-methanol (51)
Product 51 was obtained following the literature procedure[73] from 49
(183 mg, 1 mmol, 1.0 equiv.), Me-1 (45 mg, 0.2 mmol, 20 mol%),
BH3.SMe2 (0.73, 2M in THF, 0.15 mmol, 1.5 equiv.) in 1 ml THF, as a
white solid (123mg, 0.66 mmol, yield 66%, racemic). Spectral data
match with the ones reported in literature.[78] 1H NMR (400 MHz, Chloroform-d) δ 8.56 (d, J = 4.9, 1H), 7.61 (td, J = 7.7, 1.7 Hz,
1H), 7.42 – 7.34 (m, 2H), 7.39 – 7.25 (m, 3H), 7.29 – 7.12 (m, 2H), 5.75 (s, 1H). 13C NMR (101 MHz, Chloroform-d) δ 161.0, 147.9, 143.3, 137.0, 128.7, 128.0, 127.2,
122.6, 121.5, 75.1.
CSP-HPLC: (254 nm, Chiralcel AD-H, n-heptane/i-PrOH = 95:5, 40 °C, 0.5 ml/min.),
tR = 28.65 min, tR = 34.86 min
5.6. Bibliography [1] D. H. Lee, K. Severin, M. R. Ghadiri, Curr. Opin. Chem. Biol. 1997, 1, 491–496.
[2] G. F. Joyce, Nature 1989, 338, 217–224.
[3] L. E. Orgel, Crit. Rev. Biochem. Mol. Biol. 2004, 39, 99–123.
[4] B. G. Bag, G. von Kiedrowski, Pure Appl. Chem. 1996, 68, 2145–2152.
[5] G. von Kiedrowski, B. Wlotzka, J. Helbing, M. Matzen, S. Jordan, Angew.
Chemie - Int. Ed. 1991, 30, 423–426.
[6] H. Duim, S. Otto, Beilstein J. Org. Chem 2017, 13, 1189–1203.
[7] A. J. Bissette, S. P. Fletcher, Angew. Chemie - Int. Ed. 2013, 52, 12800–12826.
[8] D. G. Blackmond, Angew. Chemie - Int. Ed. 2009, 48, 386–390.
[9] V. Vasas, C. Fernando, M. Santos, S. Kauffman, E. Szathmáry, Biol. Direct
2012, 7, 1.
[10] O. Markovitch, D. Lancet, Artif. Life 2012, 18, 243–266.
[11] T. Tjivikua, P. Ballester, J. Rebek, J. Am. Chem. Soc. 1990, 112, 1249–1250.
[12] D. N. Reinhoudt, D. M. Rudkevich, F. de Jong, J. Am. Chem. Soc. 1996, 118,
6880–6889.
[13] K. Soai, T. Kawasaki, Chirality 2006, 26, 469–478.
[14] J. S. Nowick, Q. Feng, T. Tjivikua, P. Ballester, J. Rebek, J. Am. Chem. Soc.
1991, 113, 8831–8839.
[15] T. K. Park, Q. Feng, J. J. Rebek, J. Am. Chem. Soc. 1992, 114, 4529–4532.
[16] E. A. Wintner, M. M. Conn, J. Rebek, J. Am. Chem. Soc 1994, 116, 8877–8884.
[17] B. Wang, I. O. Sutherland, Chem. Commun 1997, 1495–1496.
[18] A. Vidonne, D. Philp, European J. Org. Chem. 2009, 2009, 593–610.
[19] I. Stahl, G. von Kiedrowski, J. Am. Chem. Soc. 2006, 128, 14014–14015.
[20] A. C. Hoepker, L. Gupta, Y. Ma, M. F. Faggin, D. B. Collum, J. Am. Chem. Soc.
2011, 133, 7135–7151.

Chapter 5
142
[21] K. J. Singh, A. C. Hoepker, D. B. Collum, J. Am. Chem. Soc. 2008, 130,
18008–18017.
[22] A. J. Bissette, S. P. Fletcher, Orig. Life Evol. Biosph. 2015, 45, 21–30.
[23] J. Ortega-Arroyo, A. J. Bissette, P. Kukura, S. P. Fletcher, Proc. Natl. Acad. Sci.
U. S. A. 2016, 113, 11122–11126.
[24] B. Feringa, R. A. van Delden, Angew. Chem. Int. Ed. Engl. 1999, 38, 3418–
3438.
[25] D. G. Blackmond, Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5732–5736.
[26] A. H. Alberts, H. Wynberg, J. Am. Chem. Soc 1989, 111, 7265–7266.
[27] K. Soai, T. Hayase, K. Takai, Tetrahedron: Asymmetry 1995, 6, 637–638.
[28] K. Soai, S. Niwa, H. Hori, J. Chem. Soc. Chem. Commun. 1990, 982–983.
[29] K. Soai, T. Shibata, H. Morioka, K. Choji, Nature 1995, 378, 767–768.
[30] I. Sato, S. Osanai, K. Kadowaki, T. Sugiyama, T. Shibata, K. Soai, Chem. Lett.
2002, 31, 168–169.
[31] S. Tanji, A. Ohno, I. Sato, K. Soai, Org. Lett. 2001, 3, 287–289.
[32] T. Kawasaki, M. Sato, S. Ishiguro, T. Saito, Y. Morishita, I. Sato, H. Nishino, Y.
Inoue, K. Soai, J. Am. Chem. Soc. 2005, 127, 3274–3275.
[33] K. Soai, S. Osanai, K. Kadowaki, S. Yonekubo, T. Shibata, I. Sato, J. Am. Chem.
Soc. 1999, 11235–11236.
[34] I. Sato, K. Kadowaki, K. Soai, Angew. Chemie - Int. Ed. 2000, 39, 1510–1512.
[35] I. Sato, K. Kadowaki, H. Urabe, J. H. Jung, Y. Ono, S. Shinkai, K. Soai,
Tetrahedron Lett. 2003, 44, 721–724.
[36] T. Kawasaki, K. Jo, H. Igarashi, I. Sato, M. Nagano, H. Koshima, K. Soai,
Angew. Chemie - Int. Ed. 2005, 44, 2774–2777.
[37] K. Soai, I. Sato, T. Shibata, S. Komiya, M. Hayashi, Y. Matsueda, H. Imamura,
T. Hayase, H. Morioka, H. Tabira, et al., Tetrahedron: Asymmetry 2003, 14,
185–188.
[38] C. Girard, H. B. Kagan, Angew. Chemie - Int. Ed. 1998, 37, 2922–2959.
[39] T. Shibata, T. Hayase, J. Yamamoto, K. Soai, Tetrahedron: Asymmetry 1997,
8, 1717–1719.
[40] T. Gehring, M. Quaranta, B. Odell, D. G. Blackmond, J. M. Brown, Angew.
Chemie - Int. Ed. 2012, 51, 9539–9542.
[41] A. Matsumoto, T. Abe, A. Hara, T. Tobita, T. Sasagawa, T. Kawasaki, K. Soai,
Angew. Chemie - Int. Ed. 2015, 54, 15218–15221.
[42] M. N. Oble-Terán, J.-M. Cruz, J.-C. Micheau, T. Buhse, Chem. Cat. Chem.
2018, 10, 642–648.
[43] C. Romagnoli, B. Sieng, M. Amedjkouh, European J. Org. Chem. 2015, 2015,
4087–4092.
[44] M. Funes-Maldonado, B. Sieng, M. Amedjkouh, Org. Lett. 2016, 18, 2536–
2539.
[45] M. Funes-Maldonado, B. Sieng, M. Amedjkouh, European J. Org. Chem. 2015,

Design of an asymmetric organic autocatalytic reaction: the reduction of ketones and imines with borane
143
4081–4086.
[46] N. Chinkov, A. Warm, E. M. Carreira, Angew. Chemie - Int. Ed. 2011, 50,
2957–2961.
[47] M. Mauksch, S. B. Tsogoeva, I. M. Martynova, S. Wei, Angew. Chemie - Int. Ed.
2007, 46, 393–396.
[48] M. Mauksch, S. B. Tsogoeva, ChemPhysChem 2008, 9, 2359–2371.
[49] M. Mauksch, S. Wei, M. Freund, A. Zamfir, S. B. Tsogoeva, Orig. Life Evol.
Biosph. 2010, 40, 79–91.
[50] D. G. Blackmond, Angew. Chemie - Int. Ed. 2009, 48, 2648–2654.
[51] P. H. Bos, Novel Asymmetric Copper-Catalyzed Transformations, University of
Groningen, 2012.
[52] M. Mauksch, S. B. Tsogoeva, S. Wei, I. m. Martynova, Chirality 2007, 19, 816–
825.
[53] M. Amedjkouh, M. Brandberg, Chem. Commun. 2008, 3043–3045.
[54] X. Wang, Y. Zhang, H. Tan, Y. Wang, P. Han, D. Z. Wang, J. Org. Chem 2010,
75, 2403–2406.
[55] E. J. Corey, C. J. Helal, Angew. Chemie - Int. Ed. 1998, 37, 1986–2012.
[56] E. J. Corey, R. K. Bakshi, S. Shibata, J. Am. Chem. Soc 1987, 109, 5551–5553.
[57] A. Hirao, S. Itsuno, S. Nakahama, N. Yamazaki, J. Chem. Soc. Chem. Commun.
1981, 315.
[58] S. Itsuno, Y. Sakurai, K. Ito, A. Hirao, S. Nakahama, Bull. Chem. Soc. Jpn.
1987, 60, 395–396.
[59] E. J. Corey, R. K. Bakshi, S. Shibata, C.-P. Chen, V. K. Singh, J. Am. Chem. Soc
1987, 109, 7925–7926.
[60] E. J. Corey, C. J. Helal, Tetrahedron Lett. 1995, 36, 9153–9156.
[61] B. T. Cho, Y. S. Chun, Tetrahedron: Asymmetry 1992, 3, 1583–1590.
[62] S. Guduguntla, Exploring Asymmetric Catalytic Transformations, University of
Groningen, 2017.
[63] N. G. De Kimpe, C. V. Stevens, M. A. Keppens, J. Agric. Food Chem. 1993, 41,
1458–1461.
[64] J. Deblander, S. Van Aeken, A. Adams, N. De Kimpe, K. A. Tehrani, FOOD
Chem. 2015, 168, 327–331.
[65] R. Vícha, M. Potáček, Tetrahedron 2005, 61, 83–88.
[66] J. M. Carney, P. J. Donoghue, W. M. Wuest, O. Wiest, P. Helquist, Org. Lett.
2008, 10, 3903–3906.
[67] T. E. Mü, M. Grosche, E. Herdtweck, A.-K. Pleier, E. Walter, Y.-K. Yan,
Organometallics 2000, 19, 170–183.
[68] Y. Ashikari, T. Nokami, J.-I. Yoshida, Org. Lett. 2012, 14, 938–941.
[69] N. S. Upadhyay, J. Jayakumar, C.-H. Cheng, Chem. Commun. 2017, 53, 2491–
2494.

Chapter 5
144
[70] T. Yang, X. Fan, X. Zhao, W. Yu, n.d., DOI 10.1021/acs.orglett.8b00409.
[71] F. Bächle, I. Fleischer, A. Pfaltz, Adv. Synth. Catal. 2015, 357, 2247–2254.
[72] J. L. Bilke, S. P. Moore, P. O’Brien, J. Gilday, Org. Lett. 2009, 11, 1935–1938.
[73] M. Woltersdorf, R. Kranich, H. G. Schmalz, Tetrahedron 1997, 53, 7219–7230.
[74] J. Cianci, J. B. Baell, A. J. Harvey, Tetrahedron Lett. 2007, 48, 5973–5975.
[75] T. Yang, X. Fan, X. Zhao, W. Yu, Org. Lett. 2018, 20, 1875–1879.
[76] M. Liniger, K. Estermann, K.-H. Altmann, J. Org. Chem. 2013, 78, 11066–
11070.
[77] M. Noji, Y. Konno, K. Ishii, J. Org. Chem. 2007, 72, 5161–5167.
[78] H. Yang, N. Huo, P. Yang, H. Pei, H. Lv, X. Zhang, Org. Lett. 2015, 17, 4144–
4147.

145

146
Summary
The interest of scientists for chirality is related to the recurrence of this feature in
nature. On one side, it is necessary to develop methods to achieve chiral drugs in only
one enantiomer, given that the configuration of the drugs influences its biological
activity. Asymmetric catalysis represents a convenient solution to this problem, as it
allows for the use of expensive chiral auxiliaries in substoichiometric amount. On the
other side, the scientific community is intrigued by emulating the way chirality is
generated in living systems: with this purpose, reactions are designed where a molecule
can orchestrate its own formation via asymmetric autocatalysis or autoinduction of
chirality. This thesis addresses both topics: the first part concerns the enantioselective
C-C bond formation through the conjugate addition of Grignard reagents to symmetric
heteroaryl alkenes.
Specifically, Chapter 2 describes the asymmetric copper(I)-catalyzed addition of
Grignard reagents to symmetric heteroaryl disubstituted alkenes. The reactivity of
these substrates is rather problematic, given that the non-catalyzed addition of
organomagnesium reagents competes with the catalyzed one. Additionally, the
variation of the heteroaryl substituent strongly affects the outcome of the reaction, as
well as the length of the alkyl chain of the nucleophile. For this reason, each substrate
requires specific reaction conditions. The use of a Lewis acid promotes the conjugate
addition, but, simultaneously decreases the enantioselecivity. After optimization, we
could obtain the product in good to excellent ees. Good yields are obtained when small
chain Grignard reagents were employed, but decreases for longer alkyl chains.
Chapter 3 regards the synthesis of chiral molecules bearing two pyridyl moieties via
the addition of organomagnesium reagents to bispyridyl alkenes. Substrates bearing 4-
pyridine require the activation of a Lewis acid to undergo conjugate addition, but, at
the same time, a too strong activation results in the lack of enantioselectivity. After the
optimization of the reaction conditions, the products were obtained in good yields and
excellent enantioselectivities. Instead, alkanes with 2-pyridyl substituents cannot be
prepared stereoselectively. However, a catalyst-free protocol was developed that could
afford the CA product in excellent yields. In this Chapter, the reactivity of activated
heteroaryl alkenes towards the CA of Grignard reagents is also compared.

Summary
147
The last half of this work turns to reactions where the product influences the
enantioselectivity in its own synthesis. The two cases discussed are substantially
different from each other: one regards asymmetric autoinduction in metal-catalysis,
while the other autocatalysis in an organocatalytic reaction.
In Chapter 4, the 1,2-addition of a Grignard reagent to enals and enones entails the
formation of an alkoxide, which interacts with the Cu(I)/chiral phosphine complex
(catalyst). As the new copper complex undergoes transmetallation faster than its
precursor, the formation of the product has a positive influence on the
enantioselectivity. This process is more prominent for reactions where the background
addition of the organometallic reagent competes with the catalytic pathway. The
presence of asymmetric autoinduction was studied by monitoring the ee in the course
of the reaction and by using the product as an additive for its own formation.
The thesis ends with Chapter 5 that narrates the design of an asymmetric organic
autocatalyzed reaction. This project is inspired by CBS reduction of ketones and
imines. In order to achieve autocatalysis, we synthesized ketones and imines that
afford an amino alcohol, which can form an oxazaborolidine upon reaction with
borane. We divided the study in two branches: one that regards the reduction of an
imine, the second of a ketone. Both substrates are synthesized in low yields due to the
instability of the product or of the intermediates. In the case of the imine reduction,
the autocatalytic reaction occurs with lack of enantioselectivity. For what concerns the
second pathway, testing the autocatalyst on a model ketone revealed a
matched/mismatched behavior. However, their activity in the autocatalytic reaction
could not be studied due to the small amount of substrate available and its
decomposition. We conclude that this system is not suitable for asymmetric
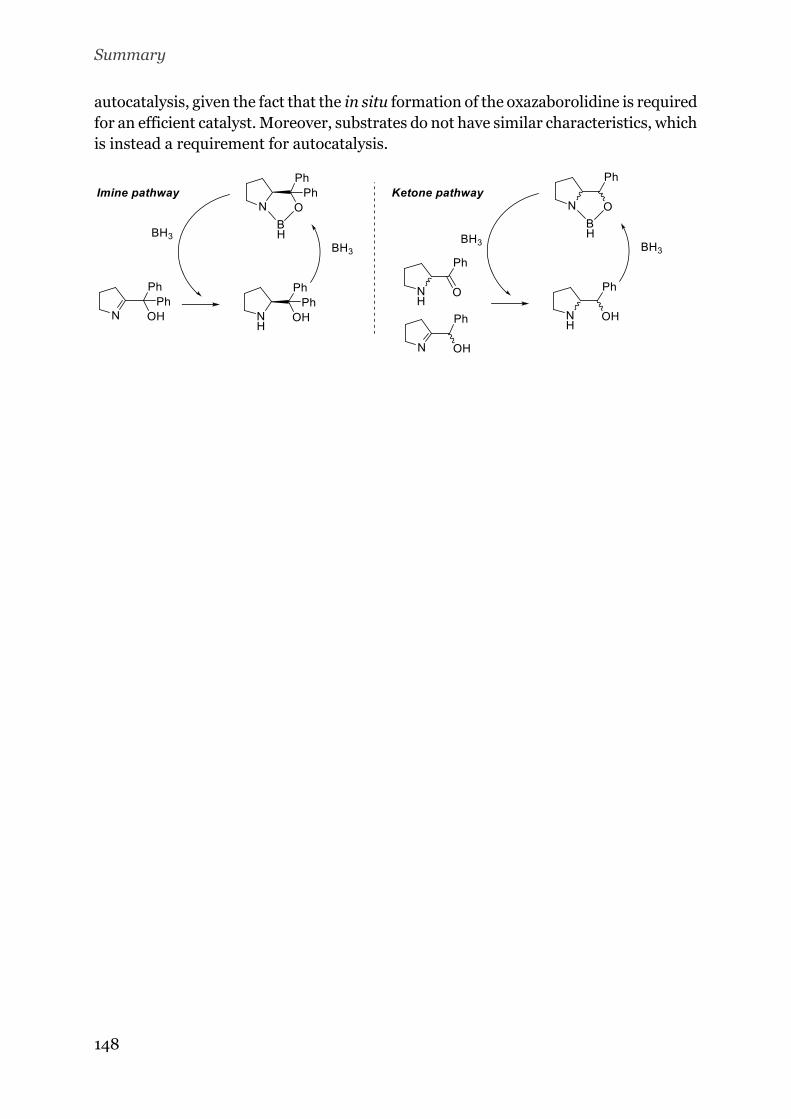
Summary
148
autocatalysis, given the fact that the in situ formation of the oxazaborolidine is required
for an efficient catalyst. Moreover, substrates do not have similar characteristics, which
is instead a requirement for autocatalysis.


150
Samenvatting
De interesse van wetenschappers voor chiraliteit is gerelateerd aan het veelvuldig
voorkomen van dit kenmerk in de natuur. Enerzijds is het noodzakelijk om methoden
te ontwikkelen voor het verkrijgen van chirale geneesmiddelen in slechts één
enantiomeer, gezien het feit dat de configuratie van invloed is op de biologische
activiteit. Asymmetrische katalyse vormt een handige oplossing voor dit probleem,
omdat hierdoor dure chirale hulpstoffen in substoichiometrische hoeveelheden
gebruikt kunnen worden. Aan de andere kant is de wetenschappelijke gemeenschap
geïntrigeerd door de manier waarop chiraliteit wordt gegenereerd in levende systemen.
Hierdoor worden nieuwe reacties ontworpen waarbij een molecuul zijn eigen formatie
kan bevorderen via asymmetrische autokatalyse of auto-inductie van chiraliteit. Dit
proefschrift behandelt beide onderwerpen: het eerste deel betreft de enantioselectieve
C-C-bindingsformatie door de geconjugeerde additie van Grignard-reagentia aan
symmetrische heteroaryl-alkenen.
Hoofdstuk 2 beschrijft de asymmetrische koper(I)-gekatalyseerde additie van
Grignard-reagentia aan symmetrische heteroaryl digesubstitueerde alkenen. De hoge
reactiviteit van deze substraten is tamelijk problematisch, aangezien hierdoor de niet-
gekatalyseerde additie van organomagnesiumreagentia concurreert met de
gekatalyseerde. Bovendien beïnvloedt de variatie van de heteroarylsubstituent sterk de
uitkomst van de reactie, evenals de lengte van de alkylketen van het nucleofiel.
Hierdoor vereist elk substraat specifieke reactieomstandigheden. Het gebruik van een
Lewiszuur bevordert de geconjugeerde additie, maar leidt tot lagere
enantioseleciviteiten. Na enige optimalisatie konden we het product in goede tot
uitstekende ees verkrijgen. Goede opbrengsten worden verkregen wanneer Grignard-
reagentia met een kleine keten werden gebruikt, maar neemt af met langere
alkylketens.
Hoofdstuk 3 betreft de synthese van chirale moleculen met twee pyridylgroepen door
de additie van organomagnesiumreagentia aan bispyridylalkenen. Substraten met 4-
pyridinegroepen vereisen de activering met een Lewiszuur om de geconjugeerde
additie te laten plaatsvinden. Aan de andere kant, een te sterke activering leidt tot
vermindering van enantioselectiviteit. Na de optimalisatie van de
reactieomstandigheden konden de producten worden verkregen in goede opbrengsten
en uitstekende enantioselectiviteiten. Alkanen met 2-pyridylsubstituenten kunnen met
deze methode niet stereoselectief worden gesynthetiseerd. Echter, een katalysatorvrij
protocol kon worden ontwikkeld die toegang geeft tot het CA-product in uitstekende

Samenvatting
151
opbrengsten. In dit hoofdstuk wordt ook de reactiviteit van geactiveerde
heteroarylalkenen vergeleken in de geconjugeerde additie van Grignard-reagentia.
De tweede helft van dit proefschrift draait om reacties waarbij het product de
enantioselectiviteit in zijn eigen synthese beïnvloedt. Twee concepten worden
besproken die wezenlijk van elkaar verschillen. Eerst beschrijven we een
asymmetrische auto-inductie door middel van metaalkatalyse en het tweede concept
gaat over autokatalyse bij een organokatalytische reactie.
Hoofdstuk 4 omvat de 1,2-additie van Grignard-reagentia aan enals en enones. Het
gevormde alkoxide kan een interactie aangaan met het Cu(I) / chirale fosfinecomplex
(katalysator). Omdat dit nieuwe kopercomplex sneller transmetallatie ondergaat dan
de originele katalysator, heeft de vorming van het product een positieve invloed op de
enantioselectiviteit. Dit proces is meer prominent voor reacties waarbij de
achtergrondreactie van het organometallische reagens concurreert met de katalytische
route. De aanwezigheid van asymmetrische autoinductie werd bestudeerd door de
enantiomere overmaat te volgen tijdens de reactie en door het product als een additief
voor zijn eigen formatie te gebruiken.
Het proefschrift eindigt met hoofdstuk 5 dat een asymmetrische organische
autokatalyse beschrijft. Dit project is geïnspireerd door CBS-reductie van ketonen en
iminen. Om autokatalyse te bereiken, hebben we ketonen en iminen gesynthetiseerd
die een aminoalcohol opleveren. Deze producten kunnen een oxazaborolidine vormen
na reactie met boraan. Het onderzoek is verdeelt in twee takken: een die uitgaat van de
reductie van een imine en de tweede die een keton reduceert. Beide substraten konden
worden gesynthetiseerd, hoewel in lage opbrengsten vanwege de instabiliteit van het

Samenvatting
152
product of van de tussenproducten. In het geval van de iminereductie treedt de
autokatalytische reactie op maar volstrekt racemisch. Wat de tweede route betreft,
bleek uit het testen van de autokatalysator op een modelketon een
matched/mismatched gedrag. De activiteit in de autokatalytische reactie kon echter
niet worden onderzocht vanwege de kleine hoeveelheid substraat die beschikbaar was
en de instabiliteit ervan. We concluderen dat dit systeem niet geschikt is voor
asymmetrische autokatalyse, gezien het feit dat de in situ vorming van het
oxazaborolidine vereist is voor een efficiënte katalysator. Bovendien hebben substraten
geen vergelijkbare kenmerken, wat een vereiste is voor autokatalyse.

153
Aknowledgements
The last, but not the least important chapter of this thesis is dedicated to people that
were part of my life in these four years in Groningen.
Syuzi, thank you for making me come to the Netherlands! I am grateful for this
opportunity and for your wise and critical guidance. You are truly a good chemist and
you have reached many important goals in your life. I wish you all the best.
I also want to thank Wesley for being so caring towards all the people in Stratingh
Institute.
Prof. Adri Minnaard, Prof. Jan van Maarseveen and Prof. Mauro Pineschi, many
thanks for accepting to be in the assessment committee of this thesis, giving your
feedback and approving it.
This thesis would not have been written without the precious help of Pieter and Johan
for what concerns the NMR measurements, Theodora for HRMS and Monique for
HPLC and GC analysis. Thank you very much.
Annette, your work goes beyond the secretarial support. Thank you for the help with
the most disparate issues. Tineke, I would also like to thank you for all the good words
and your assistance.
During my PhD, I was very lucky to be part of a group where very different people were
perfectly matching together. Few months after we met, we became very good friends
and, as a sort of magic, the Sublime Group was assembled.
I would start by thanking my paranymphs, Simone and Ciccio for being on my side
during these years, one in the lab and the other in the office: you have really colored
my life! Simo, I really miss you scaring me with the heat gun and improvising new tools
and decorations for the lab. Ciccione, thanks for always being there to help and for
finding always the right words… to make me laugh.
Pablo, during our PhDs, you have worked very hard both to write an excellent thesis
and to build strong relationships. A big thank for all the initiatives you had and for the
constant enthusiasm in enjoying life together. Ravi, during these years you became my
guide, or even a sort of older brother. I will always be grateful for your support and I
know you will realize all your dreams. Johnny, for how you keep changing a lot, you
never lose your originality. Thank you for your friendship that goes beyond the long
distance. Mamen, my fellow sublime girl, thank you for fighting with me for women’s
right during lunchtime and for fitness during pilates, yoga and African dance classes.
Juan Fer, thank you for being the cool grandfather of the group. With your joyfulness
and knowledge, you have motivated me a lot.

Aknowledgements
154
Moreover, I would like to thank Juani for looking after anyone in the lab, Kirill for the
very interesting talks and Russian poem and Marieke, for introducing our group to the
Dutch life. Then all the people met in the group: Xingchen, Yafei, Manas, Paula, Ilayda,
Luo, Daan, Danijel, Eduarda, Sofiya, Hovo, Filip, Emilio, Johanan, Joost, Natasha and
Dima (to whom I acknowledge the idea of autocatalysis in CBS reduction). I would like
to especially thank my two students, Renske and Bouke.
At this point, I would like to mention an unofficial member of our group: Giulia, you
had a very big impact on my life in Groningen and you really made me love this city.
You are a very powerful person and I hope we will keep on discovering new things and
places together in the coming years. I would also like to thank you for the beautiful
picture that you took during our travel to Cuba and that is now on the cover of this
thesis.
When moving to a new country, you risk to lose some old friends. But fortunately, some
of my friends from Pisa moved to Groningen few months later than I. My dear Varsha,
you feel like a sister to me. Dhanyawad for being sweet and supportive. In Pisa, I also
met a special boy with a lot of crazy ideas. I am happy that one of these was coming for
his PhD in Groningen, just six months later than I. Marcotti, these were definitely “the
ten best years of your life” and also of mine!
Patrizio, thanks for the thousands of running jokes and for being an awesome salsa
partner. Alessandra, even if the last year of PhD is the most critical, meeting you at this
point really cheered me up. Milocco, thank you for your friendship and also for always
being up for a drink. Matea, you are an explosion of energy and creativity. Thank you
for involving me in all kinds of crazy activity and surprising me every time we meet. I
wish all the best to you and to your lovely family. Gerda, I have really enjoyed the girls
talks and the cultural trips together. I hope we will organize many more in the next
years! I would alse like to say thank to all the Italians in Groningen for the dinners,
discussions and the beautiful moments spent together.
Since I arrived in the Stratingh Institute, there were many good and bad weeks, but
fortunately they all ended with a Friday borrel. I would like to thank all the people that
crushed on those dusty sofas and animated the Friday evenings. I would also like to
thank all the members of the Stratingh Institute and biochemistry group for the
friendly working atmosphere.
After the positive experience at the Stratingh, I was very lucky to end up at Fontys in
Eindhoven, where I have met new nice colleagues and friends. Bedankt voor een warm
welkom en voor de gezelligheid!

Aknowledgements
155
I would like to express my love for Giulia and Marialaura, who kept being my good
friends from Italy and thank Prof. Anna Iuliano for preparing me to this PhD and for
her affection.
Thanks also to Sara, Moira and my uncles and aunts for their love and the regular
delivery of jams and chocolate.
Stefano, thank you for the help with the stellingen. Your English sounds very elegant!
At my arrival in Groningen, I was welcomed by two precious boys, Guy and Arjen.
Hartelijk dank voor de twee jaar samen in Rivierenhof! I also want to express my
gratitude to Koos and Hetty to be always available to help. Van harte bedankt, jullie
waren erbij in een moment van nood, zonder dat jullie mij kenden. Jullie zijn een
geweldige familie!
Ina, Bert en Mark, thank you for welcoming me in your family. Ik ben blij dat we elkaar
via Rik hebben gevonden. En bovenal, bedankt dat jullie mij hebben gered elke keer
dat ik door NS in de steek werd gelaten!
Rik, even if we met at the beginning of my PhD, we have lived these four years in two
different countries. All the difficulties of the long distance were worth it, to finally be
together. Bedankt voor jouw geloof in onze relatie gedurende deze jaren en dat jij me
blij maakt. Ti amo tanto.
I feel allowed to be emotional thanking my family. Babbo e Mamma, grazie per avermi
sempre incoraggiato a fare quello che mi rendeva felice. Il vostro amore incondizionato
mi ha supportato in tutti questi anni e mi ha fatto arrivare fino a questo punto. Finché
dura fa verdura. Vi voglio tanto bene. E ne voglio anche a te, Giangi. Anche se non te
lo dico mai, hai contribuito enormemente nel rendermi la persona che sono e te ne
sono molto grata.