the role of protein folding and protein … · to my dear mother, marie tower able, who instilled...
TRANSCRIPT

THE ROLE OF PROTEIN FOLDING AND PROTEIN TRAFFICKING IN HUMAN DISEASE
by
CRISTY TOWER-GILCHRIST
ELIZABETH S. SZTUL, COMMITTEE CHAIR JAMES COLLAWN
KEVIN KIRK ANNE BURTON THEIBERT
BRADLEY K. YODER
A DISSERTATION
Submitted to the graduate faculty of The University of Alabama at Birmingham, in partial fulfillment of the requirements for the degree of
Doctor of Philosophy
BIRMINGHAM, ALABAMA
2011

ii
THE ROLE OF PROTEIN FOLDING AND PROTEIN TRAFFICKING IN HUMAN DISEASE
CRISTY TOWER-GILCHRIST
CELL BIOLOGY
ABSTRACT
This dissertation documents my findings in two unrelated projects.
Project 1:
Expansion of CAG repeats encoding glutamine in huntingtin and ataxin 3 causes
the neurodegenerative diseases Huntington's disease (HD) and spinocerebellar ataxia 3
(SCA3), respectively. Both poly-glutamine (polyQ) expanded proteins misfold and ag-
gregate within the cell. Preventing aggregation of polyQ proteins through molecular or
pharmacological approaches provide therapeutic advantage in animal models of HD and
SCA3. I hypothesized that the UL97 kinase encoded by the human cytomegalovirus
(HCMV) may be able to prevent the aggregation of polyQ proteins.
Initially, I showed that the UL97 kinase prevents the deposition of aggregates of
two non-polyQ proteins: the Golgi protein GCP-170 (GFP170*); and the nuclear Werner
Syndrome protein (WRN). Subsequently, I uncovered that UL97 prevents the deposition
of aggregates of both polyQ huntingtin and ataxin 3. UL97 dispersed nuclear PML bodies
and decreased p53-mediated transcription. These results identify UL97 as a novel tool to
probe the cellular mechanisms that contribute to the formation of aggregates in polyQ
disorders.
Project 2:

iii
A major challenge in the field is to understand the mechanisms involved in the
trafficking of transmembrane proteins to cilia. I hypothesized that a network of proteins
functions within the secretory and the endosomal pathways to regulate the delivery of
signaling proteins to cilia. To identify pathways and components of cellular machinery
involved in ciliary trafficking, I tracked the transport of a ciliary cargo somatostain recep-
tor 3 (SSTR3).
I showed that SSTR3 localizes to cilia of inner medullary collecting duct cells
(IMCDs). SSTR3 is also found in the early and recycling endosomes identified by labe-
ling with the endosomal markers Rab5, Rab21, Rab4, and Rab11. Using time-lapse imag-
ing, I observed the delivery of SSTR3 from endosomal compartments to the base of cilia.
SSTR3 segregates within Rab21- and Rab4-containing subdomains of early endosomes
and expression of dominant inactive mutants of Rab21, Rab4, and Rab11 severely im-
pairs SSTR3 trafficking. My findings defined a novel role for Rab21 and Rab4 in ciliary
trafficking. My work will pave the way towards better understanding of the mechanisms
that regulate cilia traffic.

iv
DEDICATION
To Christopher L. Tower

v
ACKNOWLEDGMENTS
First and foremost, I would like to thank my mentor, Dr. Elizabeth Sztul for ac-
cepting me into her lab. From the first day I sat in her office heart pounding with antici-
pation and asked about rotating in the lab, she has showed me so much kindness and re-
spect. She set out to ensure that my knowledge of science was greatly expanded. She was
confident in me allowing me to freely learn and explore in my own way. Throughout my
years in the lab when different situations arose in my life that could have possibly af-
fected my work she was supportive and there to console me and for that I am grateful.
Life in lab entails going to lab every single day and encountering people who are
very different. These people are the ones who make time in lab endurable and fun. I thank
all past and present members of the Sztul lab. From the past I thank Dr. Robert Grabski,
Dr. Lianwu Fu, Dr. Melanie Styers, and Dr. Tomasz Szul for answering the thousand of
questions I asked during their time in lab. From the present I thank Dr. Marlene Winkel-
bauer, Dr. Paulina Wyrozumska, Jason Lowery, Eunjoo Lee, Helen Lin, John Wright,
and Jay Bhatt. Each lab member in their own way gave support and more importantly
made me laugh when my experiments were not working. With that being said, the tech-
nical advice and many scientific discussions made lab life enjoyable. It was truly a plea-
sure and honor to work with each of them. I wish them all the best in the future.
I also extend a very special thanks to my committee members: Dr. Elizabeth
Sztul, Dr. James Collawn, Dr. Kevin Kirk, Dr. Anne Burton-Theibert, and Dr. Bradley
Yoder for offering constructive criticism to better my science and the great advice I re-
ceived over the years. I thank Dr. Yoder for providing reagents on many occasions that
helped with getting my project up and running.

vi
In the department of Vision Science I thank Clifford Kennon who has been a very
strong shoulder to lean on and made sure to report to my Mom if I stepped out of line.
If not for my family and friends being a support system giving love and under-
standing to help me throughout graduate school, I would not have made it this far. To my
husband, Melvin Gilchrist, who endured the long nights up with me practicing for my
many presentations throughout graduate school and for agreeing to stay up with me while
I write this dissertation, you are amazing and I love you. Melvin you took up my slack
with our beautiful daughter, Christinia Gilchrist, when I had to go away to present my
research at conferences, I am grateful. Thank you God for blessing me with Christina
Gilchrist, she came into my life when I needed her most, it is truly a joy to be your Chris-
tina. To my dear mother, Marie Tower Able, who instilled in me that education was im-
portant; I thank her for the many years of guidance and molding me into a wonderful per-
son with a bright future. She believed in me when all else failed and stood by me when
all was dark around. She encouraged me to go all the way no matter the obstacles that
stood in my way. I am also thankful that God blessed me with wonderful siblings who
were also my best friends. My brother, Christopher Tower even though he departed this
life while I was still on my journey to getting a Ph.D. I dedicate this dissertation to you.
To my beloved sisters, Jackie Tower, Savannah Tower, and Roberta Tower-Hatcher, I
could not have asked for greater friends and love. You made me so happy and thanks for
celebrating with me anytime I accomplished a goal. I am thankful to my many nephews
and nieces you keep me on my toes and never ceased to amaze me the questions they
asked about me being in school ‘my entire life.’ To my sister through friendship, Sha-
wanda Payne, I appreciate you for always being there.

vii
Finally but not least I am thankful to my friends who understood from my stand
point what it was like to be a graduate student and made me laugh when I wanted to cry
and pull my hair out and helped me make it through my first years in graduate school, Dr.
Tori Matthews and Erin White. For the friends I encountered during the later part of my
graduate career, Dr. Melanie Styers and Dr. Marlene Winkelbauer, when I was in the
tunnel and could see the light, you kept me from stressing out. Thanks Melanie and Mar-
lene for taking the time to care and make the transition easier.

viii
TABLE OF CONTENTS Page
ABSTRACT ........................................................................................................................ ii DEDICATION ................................................................................................................... iv ACKNOWLEDGMENTS ...................................................................................................v LIST OF TABLES ...............................................................................................................x LIST OF FIGURES ........................................................................................................... xi LIST OF ABBREVIATIONS .......................................................................................... xiii INTRODUCTION ...............................................................................................................1 Protein Misfolding and Aggregation ..............................................................................1 Protein Folding Diseases: Polyglutamine Disorders .......................................................3 Anti-Aggregation Approaches as Therapeutics ..............................................................5 UL97 Kinase of Human Cytomegalovirus as an Anti-Aggregation Factor ....................7 Purpose of Research in Project 1: using UL97 as an anti-aggregation agent in polyQ neurodegenerative disease ............................................................................................10 Cilia ...............................................................................................................................10 Trafficking of Proteins within Cilia: Intraflagellar Transport ......................................13 Trafficking of Proteins to Cilia: Route of Transmembrane Proteins ............................17 Role of Rab GTPases in Ciliogenesis and Ciliary Trafficking .....................................19 Rab21 as Possible Regulator of Ciliary Trafficking .....................................................24 Purpose of Research in Project 2: Characterize Ciliary Delivery of SSTR3 ................26 HUMAN CYTOMEGALOVIRUS UL97 KINASE PREVENTS THE DEPOSITION OF MUTANT PROTEIN AGGREGATES IN CELLULAR MODELS OF HUNTINGTON’S DISEASE AND ATAXIA…………………………………………..27 THE CILIARY G PROTEIN-COUPLED RECEPTOR SOMATOSTATIN RECEPTOR 3 CYCLES BEWTEEN ENDOSOMES AND CILIA THROUGH A RAB21, RAB4, AND RAB11-REGULATED PATHWAY….…………………………………………..66 DISCUSSION ..................................................................................................................104 Novel Means to Combat Aggregation Diseases .........................................................104 HCMV UL97 as Anti-Aggregation Factor ............................................................104 Molecular Mechanism of UL97 action ..................................................................105 Novel Pathways and Mechanisms for Delivery of Proteins to Cilia ..........................106

ix
TABLE OF CONTENTS (Continued) Page
SSTR3 Transport to Cilia.......................................................................................107 Small GTPases in Ciliopathies ..............................................................................108 Future Directions ........................................................................................................110 Using UL97 to Probe for Anti-Aggregation Therapies .........................................110 Identifying Ciliary Trafficking Machinery ............................................................113 GENERAL LIST OF REFERENCES .............................................................................115 APPENDIX: SUPPLEMENTAL FIGURES FOR HUMAN CYTOMEGALOVIRUS UL97 KINASE PREVENTS THE DEPOSITION OF MUTANT PROTEIN AGGREGATES IN CELLULAR MODELS OF HUNTINGTON’S DISEASE AND ATAXIA .........................................................................................................................130

x
LIST OF TABLES
Table Page
INTRODUCTION 1 Aggresomal Diseases .....................................................................................................5 2 Ciliary Targeting Sequences ........................................................................................18

xi
LIST OF FIGURES
Figure Page
INTRODUCTION 1 Model of IFT and targeting of protein to the ciliary compartment ..............................15 2 Localization and Function of Rab GTPases .................................................................21 3 The Rab switch and its circuitry ..................................................................................22 HUMAN CYTOMEGALOVIRUS UL97 KINASE PREVENTS THE DEPOSITION OF
MUTANT PROTEIN AGGREGATES IN CELLULAR MODELS OF HUNTINGTON’S DISEASE AND ATAXIA
1 UL97 prevents deposition of nuclear aggregates of WRN protein ..............................40 2 UL97 prevents aggregation of ataxin-3 containing expanded polyQ track ..................42 3 UL97 prevents aggregation of the polyQ expanded HttExon1-82Q ...........................45 4 UL97 disrupts PML bodies ..........................................................................................47 5 Disruption of PML bodies by UL97 is linked to inhibition in AT3-72Q aggregation ...................................................................................................................49 6 Disruption of PML bodies by UL97 is linked to inhibition in HttExon1-82Q aggregation ...................................................................................................................51 7 UL97 decreases p53-mediated transcription .................................................................54
THE CILIARY G PROTEIN-COUPLED RECEPTOR SOMATOSTATIN RECEPTOR
3 CYCLES BEWTEEN ENDOSOMES AND CILIA THROUGH A RAB21, RAB4, AND RAB11-REGULATED PATHWAY

xii
LIST OF FIGURES (Continued)
Figure Page
1 SSTR3 localizes to primary cilia and endosomes ........................................................74 2 SSTR3 trafficking is regulated by Rab21 ....................................................................76 3 SSTR3 trafficking is regulated by Rab4 ......................................................................80 4 SSTR3 trafficking is regulated by Rab11 ....................................................................83 5 SSTR3-containing vesicles traffic directly to cilia ......................................................85 6 SSTR3-containing vesicles traffic from Rab21- and Rab4-containing endosomes directly to cilia ...........................................................................................87 7 Rab11-containing endosomal compartments give rise to SSTR3 ciliary vesicles .........................................................................................................................88

xiii
LIST OF ABBREVIATIONS ADPKD Autosomal Dominant Polycystic Kidney Disease Arf ADP ribosylation factor Arl Arf like proteins ARPKD Autosomal Recessive Polycystic Kidney Disease BBS Bardet-Biedl Syndrome
C. elegans Caenorhabditis elegans
GAP GTPases Activating Protein
GEF Guanine Nucleotide Exchange Factor
GFP Green Fluorescent Protein
GDP Guanosine Diphosphate
GTP Guanosine Triphosphate
GPCR G Protein-Coupled Receptor
HCMV Human Cytomegalovirus
HD Huntington’s disease
Htt Huntingtin
IFT Intraflagellar Transport
IFTA IFT-associated
IMCD Inner Medullary Collecting Duct

xiv
LIST OF ABBREVIATIONS (Continued)
um micrometer
polyQ polyglutamine
PKD Polycystic Kidney Disease
RABL Rab-like
RP Retinitis Pigmentosa
SCA3 Spinocerebellar Ataxia type 3
SSTR3 Somatostatin receptor 3
WRN Werner protein

1
INTRODUCTION
Protein Misfolding and Aggregation
Newly synthesized proteins must be correctly folded and modified to ensure
proper trafficking and correct functioning once the protein reaches its final destination.
Protein folding in the cell occurs either in the cytoplasm (for cytoplasmic proteins) or
within the secretory pathway in the ER (for transmembrane proteins and secretory pro-
teins) (Cohen and Kelly, 2003).
The ultimate outcome in the life of a protein is either correct folding or misfolding
that often leads to aggregation (reviewed in Garcia-Mata et al., 2002). The causes of pro-
tein misfolding can be due to incomplete protein synthesis, postsynthetic damage, muta-
tions within the protein such as amino acid misincorporation during translation, outside
stresses including pH, temperature, ionic strength, or overexpression of proteins. The cell
is equipped with a number of ‘quality control’ mechanisms to minimize misfolding and
dispose of misfolded proteins prior to aggregation (Wickner et al., 1999; Olzmann et al.,
2008). The two major defenses against misfolded proteins are molecular chaperones and
the proteasome (Goldberg, 2003; Muchowski and Wacker, 2005; Nalepa et al., 2006).
Chaperones are ATP-dependent unfoldases that occur ubiquitously in all cellular
compartments that sustain protein synthesis and folding reactions. Chaperones in the cy-
toplasm and the lumen of the ER bind to exposed hydrophobic domains of proteins to
allow the newly synthesized polypeptide chains several opportunites to fold. Chaperones

2
also mediate refolding of proteins damaged by stress, and retrotranslocation of misfolded
proteins back into the cytoplasm where they are degraded by the proteasome (reviewed in
Cohen and Kelly, 2003).
Misfolded proteins are targeted for degradation in the proteasome. The 26S pro-
teasome is composed of a barrel-shaped 20S catalytic core that digest proteins into short
peptides and capped on either end by 19S regulatory caps responsible for substrate rec-
ognition and transport into the core. In the ubiquitin-proteasome system, a protein is
tagged for degradation by covalent linkage of ubiquitin monomers which are recognized
by the 19S regulatory cap of the proteasome. The substrate protein must be partially un-
folded for translocation into the 20S core particle to be degraded.
Failure of proteins to fold correctly or remain correctly folded or the production
of misfolded proteins that exceed the cell’s degradative capacity leads to interactions of
these misfolded proteins with other unfolded or partially folded proteins and results in the
formation of intracellular aggregates (reviewed in Garcia-Mata et al., 2002; Dobson,
2003). Aggregation of proteins most likely occurs co-translationally while the nascent
peptide chains are synthesized on polyribosomes. These small aggregresome particles
form throughout the cell and are quickly transported to the microtubule organizing center
where they coalesce to form aggresomes (Johnston et al., 1998; Garcia-Mata et al., 1999;
Fu et al., 2005). The active recruitment of chaperones and proteasome components sug-
gests that the formation of aggresomes is a dynamic process that the cell uses to cope
with misfolded proteins (Wigley et al., 1999; Garcia-Mata et al., 1999; Garcia-Mata et
al., 2002; Fu et al., 2005).

3
Protein Folding Diseases: Polyglutamine repeat disorders
A number of inherited neurodegenerative disorders are directly associated with
the deposition of misfolded proteins in aggresomes in neurons and are collectively termed
aggresomal diseases (Table 1) (Garcia-Mata et al., 2002). A specific group of protein-
folding diseases of the central nervous system are the poly glutamine (polyQ) repeat dis-
orders. These diseases are caused by an expansion of CAG trinucleotide repeats that en-
code glutamine within a specific cellular protein that lead to altered misfolded domain in
the mutated protein (Trottier et al., 1995; Scherzinger et al., 1997; Perez et al., 1998).
Misfolding leads to aggregate formation, altered protein function and altered protein-
protein interactions ultimately leading to neuronal dysfunction and degeneration (Paulson
et al., 2000).
One of the most common polyQ diseases is Huntington’s disease (HD). HD is an
autosomal dominant neurodegenerative disease that occurs in 3-10 cases per 100,000 in-
dividuals as a result of mutation in the huntingtin gene. The protein product, huntingtin,
is a cytosolic protein that is ubiquitously expressed and shown to function in protein traf-
ficking, vesicle transport, postsynaptic signaling, transcriptional regulation, and apotosis
(Liu et al., 2000; Sun et al., 2001; Marcora et al., 2003; Gauthier et al., 2004; Huang et
al., 2004; Metzler et al., 2007). The associated mutation is a stretch of CAG repeats near
the 5’-end in exon 1 of the HD gene coding sequences (MacDonald et al., 1993).
The age of onset is between the ages of 35 and 50 years with the number of CAG
repeats affecting the progression of the disease. Individuals with a repeat length of 6 to
35 appear to be unaffected, however if the CAG repeats expansions are greater than 38 it
causes disease (Gusella and MacDonald, 1995). The regions of the brain affected by neu-

4
ronal loss and the appearance of neuronal intranuclear inclusions of the mutant huntingtin
are the striatum and cerebral cortex (Vonsattel et al., 1985; Gil and Rego, 2008). HD is
characterized by irrepressible motor dysfunction, cognitive decline and psychiatric dis-
turbances, which lead to progressive dementia and death approximately 15-20 years after
disease onset (Bates, 2001; Landles and Bates, 2004).
Another debilitating polyQ disease is spinocerebellar ataxia type 3 (SCA3) also
known as Machado-Joseph disease. SCA3 is an autosomal dominant disorder and the
highest prevalence of ataxias is caused by the expansion of CAG repeats in the protein
ataxin 3. The mutant ataxin 3 aggregates to form intranuclear inclusions as well as cytop-
lasmic inclusions within neurons (review Ross, 1997; Sun et al., 2007). Studies show
that wild-type ataxin3 functions in de-ubiquitination and transcriptional regulation (Riess
et al., 2008). The onset of SCA3 is typically between the ages of 30 and 50 years, al-
though early onsets in childhood have been reported. SCA3 affects the spinal cord, and
causes loss of myelination of fibres in spinal tract and posterior funiculi, substantia nigra,
nuclei pontis as well as the nuclei of the vestibular and cranial nerves, column of Clark
and anterior horn. SCA3 is characterized by parkinsonism, dystonia, resteless legs, lower
motor neuron impairment, REM behavior disorder, neuropsychiatric symptoms similar to
HD (reviewed in Matilla-Dueñas et al., 2010). The death of individuals with SCA3 is
generally 10 years after onset.

5
Note: Table 1 from “Hassles with taking out the garbage” by R. Garcia-Mata, Y. S. Gao, and E. Sztul, 2002, Traffic, 3(6), p. 388-396. Copyright 2002 by Elsevier. Reprinted with permission.
Anti-Aggregation Approaches as Therapeutics
Preventing aggregation through pharmacological, molecular, and biochemical ap-
proaches that target processes leading to aggregation or oligomerization appears to be a
viable strategy to inhibit polyQ-induced neurodegeneration. Disaccharides that inhibit
polyQ aggregation in vitro also prevent neurodegeneration in vivo (Tanaka et al., 2004).
In a transgenic mouse HD model oral administration of the disaccharide trehalose was
effective in preventing polyQ aggregation in cerebrum neurons. Furthermore there was a
significant decrease in neurodegeneration in these mice as evident by improvement in
motor function and extended lifespan (Tanaka et al., 2004). A high-throughput screen of
3 libraries using a FRET-based assay identified over 4,000 biologically active small

6
compounds including an inhibitor of caspase 1, an inhibitor of EGFR tyrosine kinase, and
an inhibitor of of rho-associated p160ROCK kinase that reduced polyQ aggregation in
cellular and Drosophila models of HD neurodegeneration (Pollitt et al., 2003; Desai et
al., 2006).
In addition to pharmacogical strategies, modulations of cellular targets have been
shown to prevent aggregation and reverse neurotoxicity (Muchowski and Wacker, 2005).
Molecular chaperones are essential and the first line of defense again protein misfolding.
Overexpression of molecular chapersones, heat shock protein 40 (HSP40) and heat shock
protein 70 (HSP 70) in cells has been shown to prevent the formation of spherical and
annular oligomeric structures by a polyQ Httex1 (Wacker et al., 2004) while promoting
monomeric conformations (Wacker et al., 2004). More importantly, in SCA1 transgenic
mice, overexpression of HSP 70 decreased neurodegeneration and improved motor coor-
dination (Cummings et al., 2001). Likewise, overexpressing HSP 70 (Warrick et al.,
1999; Warrick et al., 2005) and HSP 40 (Chan et al., 2000) inhibits polyQ aggregation
and rescues neurodegeneration in Drosophilia models of SCA3.
Cellular signaling pathways have been implicated in preventing polyQ aggrega-
tion and neurodegeneration. Specifically, the IGF-1/Akt pathway and arfaptin2 substrate
of Akt have been shown to prevent polyQ aggregation and prevent neurodegeneration
(Humbert et al., 2002; Peters et al., 2002; Colin et al., 2005). One mechanism by which
Akt serine/threonine kinase may mediate its anti-aggregation and cytoprotective function
is by directly phosphorylating Htt serine 421 (in human Htt with 23 glutamines) which in
turn prevents the aggregation of phosphorylated Htt and Htt-induced cell death in cellular
models of HD (Humbert et al., 2002). Additionally, there is an Htt independent mechan-

7
ism for Akt action in which Akt directly phosphorylates arfaptin2 on serine 260. This
prevents aggregation of pathogenic polyQ htt fragments and is neuroprotective in primary
striatal neurons expressing the polyQ Htt fragment (Peters et al., 2002).
Select proteins and peptides also have been shown to prevent polyQ aggregation.
In an in vitro study, a monoclonal antibody (IC2) that recognizes only the polyQ ex-
panded portion of the mutated Htt protein inhibited polyQ aggregation (Heiser et al.,
2000). A bivalent peptide that binds Htt was shown to prevent Htt aggregation in cells
while a polyglutamine binding peptide 1 (QBP1) was shown to bind polyQ stretches and
significantly decrease polyQ aggregation (Kazantsev et al., 2002; Nagai et al., 2003).
More importantly, QBP1 was shown to inhibit polyQ aggregation and polyQ-induced
neurodegeneration in Drosophilia models of polyQ diseases (Nagai et al., 2003). Addi-
tional work using an inducible cell-culture model system to screen for loss of aggregates
showed that compounds that inhibit polyQ aggregation in cells also suppress neurodege-
neration in Drosophilia models of polyQ diseases (Apostol et al., 2003). Importantly, all
these findings strongly support the model of polyQ aggregation as conferring the cytotox-
icty of mutated proteins and indicate that preventing aggregation represents important
therapeutic strategies for neurodegenerative diseases such as HD and SCA3. They also
provide the rationale for further identification of novel means of preventing polyQ aggre-
gation.
UL97 Kinase of Human Cytomegalovirus as an Anti-Aggregation Factor
All human herpesviruses encode at least one well conserved serine/threonine pro-
tein kinase that is important in viral infection (Michel et al., 1998). In HCMV, the UL97
protein has been identified as essential for efficient replication and virion production

8
(Prichard et al., 1999). The UL97 kinase is a ~80 kDa tegument protein composed of 707
amino acids. UL97 is localized within the nucleus in infected or transfected cells and
UL97 contains a nuclear localization signal within its N-terminal domain (Prichard et al.,
2005). UL97 shows sequence homology with cellular serine/threonine kinases and con-
tains conserved subdomains involved in substrate and ATP binding (Michel et al., 1998).
Previous studies have shown that purified UL97 autophosphorylates at least three serine
and threonine sites (~65% of incorporated phosphate comigrates with serines and ~35%
with threonines) at sites contained within its N-terminal 200 residues (Baek et al., 2002a;
Cui et al., 2006). UL97 kinase activity requires an invariant lysine at position 355 within
the highly conserved subdomain II, and a K355M mutant of UL97 is catalyticaly inactive
(Marschall et al., 2001). It is likely that K355 aligns the phosphates of ATP since the ana-
logous lysine in cyclic AMP-dependent kinase performs this function (Knighton et al.,
1991). The availability of the active and the catalytically inactive UL97 provides excel-
lent control for functional studies.
UL97 phosphorylates the UL44 viral protein in vitro and in vivo (Krosky et al.,
2003). In addition to viral proteins UL97 also can phosphorylate a number of cellular
proteins that are thought to be targets of cdc2 (cyclin dependent kinase) specifically mye-
lin basic protein, histones H1, H2B and H3, lamins A, B, and C, the largest subunit of
RNA polymerase II (Baek et al., 2002a; Baek et al., 2002b; Baek et al., 2004; Marschall
et al., 2005) and the retinoblastoma protein (Hume et al., 2008; Prichard et al., 2008).
The UL97-mediated phosphorylation of these substrates has been shown only in vitro,
and might not represent true cellular targets. This is suggested by the finding that phos-
phorylation of the RNA polymerase does not appear to be mediated by UL97 in vivo

9
(Baek et al., 2004). The identity of cellular proteins that UL97 phosphorylates in vivo is
unknown. The motif that UL97 phosphorylates does not appear to be conserved, although
arginine or lysine residues 5 positions downstream of the phosphorylated serine appear to
be common (but not absolutely required) (Baek et al., 2002b). Interestingly, the active
and inactive UL97 have been shown to directly bind to cellular p32, a lamina-associated
protein that interacts with the nuclear lamin B receptor (Marschall et al., 2005) and the
SF2 splicing factor (Krainer et al., 1991). Whether UL97 phosphorylates p32 in vivo and
what functional consequences this might have is unknown (Marschall et al., 2005)
The UL97 protein is important for viral replication, and a recombinant virus with
a large deletion in UL97 replicates poorly (Prichard et al., 1999). Important to our study,
virion assembly in the nucleus appears to be significantly impaired in cells infected with
virus lacking UL97 and this results in the inappropriate aggregation and the sequestration
of viral proteins in large nuclear aggregates (Prichard et al., 2005). One of the aggregated
proteins is the viral pp65 tegument protein. Thus, it appears that UL97 prevents aggrega-
tion of viral components. The anti-aggregation function of UL97 during viral assembly is
also reflected in a transient expression system. Specifically, pp65 transfected alone into
cells forms aggregates, but co-expression of UL97 with pp65 prevents pp65 aggregation
(Prichard et al., 2005). The inhibition of aggregate formation requires kinase activity
since the K355M mutant of UL97 doesn't prevent pp65 aggregation (Prichard et al.,
2005). These findings suggest that UL97 prevents pp65 aggregation in a kinase-
dependent manner and has anti-aggregation activity that may be used to prevent cellular
protein aggregation.

10
Purpose of Research in Project 1: Using UL97 as an anti-aggregation agent in polyQ
neurodegenerative diseases
Intensive research has led to great advances in our understanding of how protein
misfolding and mislocalization can lead to human diseases. Similarly, studies on the mo-
lecular underpinnings of the polyQ neurodegenerative diseases have shown that ap-
proaches to decrease the formation of aggregates are neuro-protective. However, the task
remains to uncover the cellular mechanisms that participate in aggregate deposition and
to identify novel strategies to prevent aggregate deposition. Previous work has suggested
that the UL97 kinase of HCMV can prevent aggregation of viral proteins. Thus, I ex-
plored the possibility that UL97 might be a general anti-aggregation factor. Chapter 2 of
this dissertation documents my findings that UL97 can be used as a novel tool to dissect
aggregation mechanisms and that UL97 can prevent deposition of aggregated polyQ pro-
teins.
Cilia
Cilia are important organelles involved in the reception and transduction of extra-
cellular signals. Cilia are hair-like appendages formed by a nine-fold bundle of doublet
microtubules sheathed by a specialized plasma membrane that protrudes from the cellular
plasma membrane (Davenport and Yoder, 2005). Cilia originate from the centriole-
related structures called basal bodies by the coordinated recruitment of soluble and
transmembrane ciliary proteins. Cilia are microtubule based and are classified according
to microtubule components and function (Davenport and Yoder, 2005) into three major
classes: motile cilia, non-motile cilia termed primary cilia, and nodal cilia.

11
Motile cilia have been extensively studied and are comprised of a 9+2 microtu-
bule doublet consisting of 9 pairs of microtubules surrounding two central single micro-
tubules (central pair complex) (Berbari et al., 2009 ). This class of cilia is generally
found on epithelial cells of trachea, ependymal cells in the brain, and on cells lining the
oviduct (Afzelius, 1995). Motile cilia normally concentrate in large numbers and beat in
an orchestrated wavelike fashion (Porter and Sale, 2000; Wemmer and Marshall, 2004).
Primary non-motile cilia and nodal cilia are composed of a 9+0 microtubule ar-
rangement without a central pair of microtubules (Figure 1) (Berbari et al., 2009 ). Pri-
mary cilia are found on the vast majority of eukaryotic cells, usually singly on cells.
Primary cilia are immotile and play a role in mechanosensation, chemosensation and vi-
sion (Praetorius et al., 2003; Praetorius and Spring, 2003; Christensen et al., 2007; Chris-
tensen et al., 2008). Nodal cilia lack the central pair of microtubules but have dynein
arms with a 9+0 structure and have the ability to move in propeller-like fashion distinct
from primary cilia. An essential role for nodal cilia has been indicated in establishing
left-right body asymmetry (Nonaka et al., 1998).
Many human diseases have been associated with absence or dysfunction of cilia.
These diseases have been collectively termed ‘ciliopathies’ and a variety of phenotypes
are associated with ciliopathies including fluid filled cysts in the kidneys, situs invertus,
left-right asymmetry, obesity, blindness, and infertility (Morgan et al., 1998; Nonaka et
al., 1998; Marszalek et al., 1999; Mykytyn et al., 2001; Stratigopoulos et al., 2008).
One of the most common ciliopathies is Polycystic kidney disease (PKD) a genet-
ic disorder characterized by the growth of numerous fluid-filled cysts, primarily in the
kidneys. PKD is the fourth leading cause of kidney failure in the U.S. About half of

12
people with the most common form, autosomal dominant polycystic kidney disease
(ADPKD), progress to irreversible kidney failure, termed end-stage renal disease
(ESRD). The second form of PKD is known as autosomal recessive PKD (ARPDK).
ADPKD occurs in 1:400 live births and affects an estimated 600,000 people while
ARPKD is a much rarer condition, occurring in 1 in 20,000 live births but generally re-
sults in a severe and rapidly progressing disease phenotype (http://www2.niddk.nih.gov;
Torres and Harris, 2007). There are no effective treatments for the underlying causes of
PKD; therefore, patients are usually prescribed pain-relieving drugs, antibiotics to treat
infections, and medications to control blood pressure that are aimed at preserving or
slowing the decline in kidney function.
The majority (~85%) of ADPKD cases are due to mutations in the Pkd1 gene,
which encodes polycystin-1 (PC1), and the remainder due to mutations in Pkd2, which
encodes polycystin-2 (PC2). ARPKD is caused by mutations in fibrocystin (Yoder et al.,
2002; Zhang et al., 2004) (Pazour et al., 2002). The molecular basis of the ADPKD has
been tracked to ciliary dysfunction in a flow sensing calcium channel mechanotransduc-
ing complex composed of PC1 and PC2 (Qian et al., 1997; Tsiokas et al., 1997; Hanaoka
et al., 2000; Nauli et al., 2003; Nauli and Zhou, 2004). The defect is exemplified in pkd-/-
mice which are defective in flow sensing and develop polycystic disease (Nauli et al.,
2003; Liu et al., 2005; Siroky et al., 2006).
Additional evidence from mammalian and other model systems has led to a gen-
eral acceptance that the primary defect in ADPKD relates to the ciliary misfunction of
PC1/2. The molecular nature of ADPKD-causing mutations is largely unknown, but
many encode full-length or only partially truncated proteins. It is postulated that missense

13
mutations appear to cause mislocalization of the polycystins altering ciliary trafficking
(Xu et al., 2007). This suggests that correct delivery of PC1/2 to cilia is essential for cor-
rect kidney function. However the mechanism by which polycystins and other transmem-
brane proteins gain access to the cilium is poorly understood. Gaining an understanding
of the complete network of proteins that facilitate the delivery of sensory proteins to cilia
is essential for designing therapeutic strategies to either inhibit trafficking of pathogenic
mutants or to promote the delivery of endogenous or engineered functional proteins to
cilia.
Trafficking of Proteins within Cilia: Intraflagellar Transport
Primary cilia are devoid of ribosomes and the cell must transport proteins that as-
semble and maintain the cilium from the cytoplasm (for soluble proteins) or the endop-
lasmic reticulum (ER) (for transmembrane proteins). Transport within the cilium is ac-
complished by intraflagellar transport (IFT) (Rosenbaum and Witman, 2002). IFT was
first observed by differential interference contrast microscopy in Chlamydomonas flagel-
la. IFT is an evolutionarily conserved process and also has been observed in C. elegans,
Drosophila and mice (Kozminski et al., 1993). IFT particle movement in cilia of C. ele-
gans sensory neurons and in primary cilia of cultured IMCD and LLC-PK1 kidney cells
showed a slower rate than that observed in Chlamydomonas (Orozco et al., 1999; Snow
et al., 2004; Follit et al., 2006). IFT is the bidirectional movement of cargo proteins from
the base of the cilium to the tip (anterograde) by heterotrimeric kinesin II motor proteins
and back from the tip compartment to the base (retrograde) via cytoplasmic dynein pro-
teins (Figure 1) (Kozminski et al., 1993; Pedersen and Rosenbaum, 2008). Ciliary precur-
sors, cargo, and motor proteins concentrate around basal body where they assemble into

14
complexes called IFT particles that carry proteins into cilia axoneme and deliver signals
from cilia into cytoplasm in response to environmental stimuli (Wang et al., 2006).
IFT particles biochemically purified from Chlamydomonas are organized into two
distinct complexes with four proteins in complex A (IFT144, 140, 139,122A) and 12 pro-
teins in complex B (IFT172, 88, 81, 80, 74/72, 57/55, 52, 46, 27, 20) (Cole, 2003; Follit
et al., 2009). Two additional subunits IFT121/122B and IFT43 have been added to Com-
plex A (reviewed in (Cole, 2003). Complex B is involved in anterograde traffic while
Complex A involved in retrograde traffic (reviewed in (Pedersen et al., 2008). Both are
essential for intra-ciliary trafficking since mutations in Complex B result in very short
cilia, while mutations or depletions of Complex A components result in cilia with a bulb-
like top containing accumulated IFT rafts (Qin et al., 2001). Interestingly, some IFT mu-
tants such as IFT140/CHE-11, IFT81 and IFT74 have only modest defects in cilia (Ko-
bayashi et al., 2007). It is possible that those IFT components have more specialized
functions that do not involve ciliogenesis per-se, but rather facilitate the delivery of spe-
cific components to the cilium.

15
Figure 1. Model for IFT and targeting of proteins to the ciliary compartment. The nine peripheral microtubule doublets of the axoneme form the backbone of the cilium while the basal body at the base is utilized as a template. The axoneme is sheathed in the cilia membrane, which is distinct from the cell membrane. Structures at the base of the cilium such as the transition fibers and the basal body are important for regulating the protein content of the cilia membrane.
Proteins destined for the ciliary compartment (membrane proteins as well as axonemal components) are transported in Golgi-derived vesicles to the base of the cilium where the vesicles are exocytosed and the ciliary proteins associate with IFT particles. This Golgi-to-cilium-mediated vesicle transport, which is proposed to involve cytoplasmic dynein 1 MT-based movement, depends on the IFT complex B proteins IFT20 and DYF-11, the small G protein Rab8 and associated GEFs (e.g., Rabin 8, Rabaptin 5), FAPP2, and adap-ter proteins such as AP-1. BBS proteins and other proteins localized in the pericentro-somal region (e.g., PCM-1, EB1, p150Glued) may provide a link between the Golgi-derived vesicles and the transition fibers at the ciliary base and may also serve to anchor MTs at the basal body. At the ciliary base, only proteins (or protein complexes) contain-ing specific ciliary targeting motifs are allowed access through the zone defined by the transition fibers. Selective entry of proteins into the ciliary compartment probably in-volves specific G proteins and GEFs that are associated with NPHPs, MKS, and B9 do-main-containing proteins. Following entry into the ciliary compartment, these proteins,

16
along with inactive cytoplasmic dynein 2, are transported anterogradely along the axo-neme by kinesin-II-mediated IFT. At the ciliary tip IFT particles are remodeled, kinesin-II is inactivated, and cytoplasmic dynein 2 is activated. Ciliary turnover products (e.g., inactive receptors) are transported retrogradely along ciliary axonemes by cytoplasmic dynein 2 for recycling or degradation in the cytoplasm. Recycling or turnover of ciliary membrane receptors may involve ubiquitination (e.g., via BBS proteins) and/or dephos-phorylation of the receptors as well as binding to endosomal vesicle adapter proteins such as STAM-1/Hrs (Bae and Barr, 2008) or β-arrestin (Kovacs et al., 2008). Figure based on references (Azimzadeh and Bornens, 2007; Leroux, 2007; Rosenbaum and Witman, 2002) as well as references cited in the text. Abbreviations: EV, endocytic vesicle; MT, microtubule; PCM, pericentriolar material. Figure generated by Jacob M. Schroder, Uni-versity of Copenhagen.
Note: Figure and legend from “Intraflagellar transport (IFT): Role of ciliary assembly, resoprtion and signaling” by L. B. Pendersen and J. L. Rosenbaum, 2008, Current Topics in Developmental Biology, 85, p. 23-61. Copyright 2008 by Elsevier. Reprinted with permission.

17
Trafficking of Proteins to Cilia: Route of Transmembrane Proteins
Proteins localized to distinct cellular compartments contain sequences that serve
as “cellular codes” to direct them to a particular organelle. These signals are recognized
by specific cellular machinery (Pazour and Bloodgood, 2008). A number of known ci-
liary targeting motifs are listed in (Table 2). The first ciliary targeting sequence motif was
defined in rhodopsin, a seven transmembrane G protein-coupled receptor (GPCR) con-
centrated in rods outer segments and involved in phototransduction in photoreceptor cells
(Hargrave and McDowell, 1992; Tam et al., 2000; Nachury et al., 2010). The ciliary tar-
geting sequence of rhodopsin is found in the C-terminal cytoplasmic tail and when added
to a membrane green fluorescent protein (GFP) was shown to be sufficient for targeting
of GFP to the outer segment in transgenic frogs (Tam et al., 2000). Furthermore, muta-
tions in the last 5 amino acids of rhodopsin lead to retinitis pigmentosa (RP) (Deretic et
al., 1998) and this region is most affected in RP patients with single amino acids substitu-
tions, suggesting that QVSPA is the ciliary targeting motif (Pazour and Bloodgood,
2008). Strikingly, the ciliary trafficking motif in polycystin-2 trafficking is the same mo-
tif (RVQP) in the N-terminal cytoplasmic domain of the protein (Geng et al., 2006). The
motif acts in an autonomous fashion since the first 15 amino acids of polycystin-2 that
include the RVQP motif is sufficient to target a heterologous protein (the transferrin re-
ceptor) to cilia. This motif is also used by the odorant responsive cyclic nucleotide gated
channel CNGB1B (Jenkins et al., 2006) but is absent from polycystin-1 and several other
cilia transmembrane proteins, indicating that RVQP is not a universal code (Pazour and
Bloodgood, 2008). Polycysin-1 is thought to contain a ciliary localization sequence in its
C-terminal 112 residues (Xu et al., 2007), however boundaries of the targeting sequence

18
were not further examined and therefore it is possible that a smaller motif may exist in
this region that targets protein to cilia (Pazour and Bloodgood, 2008).
Additionally, another ciliary targeting signal AX[S/A]XQ was identified in the
third intracellular loop of GPCRs that target to cilia including somatostatin receptor 3
(SSTR3), serotonin receptor 6 (5HT6), and melanocortin concentrating hormone receptor
(MCHR1) (Berbari et al., 2008). Likewise, this motif is found in known ciliary mem-
brane proteins such as rhodopsin, opsin, and multiple olfactory receptors. Lipid modifi-
cations are abundant in ciliary membrane proteins and found in a number of ciliary tar-
geting signals (Godsel and Engman, 1999; Tam et al., 2000; Geng et al., 2006; Tao et al.,
2009; Follit et al., 2010{Tao, 2009 #3373). Lipidation is thought to allow the incorpora-
tion of proteins into lipid rafts through myristoylation and/or palmitoylation (Nachury et
al., 2010). Work by Follit et al 2010 demostrated that mutations in fibrocystin ciliary
targeting sequences prevented lipidation and abolished ciliary targeting (Follit et al.,
2010). Lipid rafts have been shown to direct apical targeting in mammalian cells and one

19
can speculate that a similar process is important in the sorting of proteins to the ciliary
membrane.
Proposed models for ciliary membrane trafficking include targeted delivery and
lateral transport. The direct trafficking route from the Golgi to the base of cilium is the
most studied of the models (Deretic and Papermaster, 1991; Emmer et al., 2010). The
direct model suggests that vesicles of different lipid composition and containing ciliary
cargo form in the Golgi by a mechanism that recognizes ciliary targeting sequences
(Klemm et al., 2009; Emmer et al., 2010). It is proposed that IFT20 loads onto ciliary-
bound vesicles and might serve as an adaptor to recruit other ciliary cargo. Vesicles des-
tined for the cilia interact with GTP-Rab8 which is activated by Rabin8 and the BBSome
(octameric complex of Bardet-Biedl syndrome (BBS) proteins). This interaction is
thought to facilitate the movement of vesicles to the base of the cilium near the zone of
transition (Hao and Scholey, 2009; Jin et al., 2010; Nachury et al., 2010). Vesicles then
fuse with the periciliary membrane and the ciliary lipids and proteins enter the cilium.
Upon reaching the cilium, cargo is assembled onto IFT complexes and is transport into
cilium by IFT. DYF-11 is a key component of IFT-B and might promote a separate
membrane association of the IFT complex through Rabaptin5 and GTP-Rab8 (Omori et
al., 2008). In the lateral transport model proteins are transported laterally from the plasma
membrane into the membrane of the cilium breaching the diffusion barrier that exists at
the base of the cilium (Hunnicutt et al., 1990; Milenkovic et al., 2009).
Role of Rab GTPases in Ciliogenesis and Ciliary Trafficking
Rab proteins are the largest subfamily of the Ras superfamily of monomeric small
GTPases. To date there are more than 70 human Rab GTPases (Stenmark and Olkkonen,

20
2001; Colicelli, 2004). There are 14 subfamilies of Rabs based on distinct subfamily-
specific sequence motifs (Pereira-Leal and Seabra, 2000; Schwartz et al., 2007). Rabs are
small proteins ranging in size from 24 to 25 kDa in molecular weight. As shown in Fig-
ure 2, Rab GTPases are present on all intracellular compartments. Rabs are known to
regulate the biogenesis of membrane compartments and to facilitate protein movement
between distinct compartments (Zerial and McBride, 2001). Rabs regulate many steps of
membrane transport by facilitating motor recruitment, vesicle formation, and ve-
sicle/organelle fusion (Zerial and McBride, 2001; Colicelli, 2004). Previous studies show
that like all GTPases, Rabs cycle between a GTP bound state (active) and a GDP bound
state (inactive) (Figure 3) which imposes temporal and spatial regulation of membrane
transport (Zerial and McBride, 2001; Stenmark, 2009). Rab membrane targeting is de-
pendent on the addition of a prenyl group to the C-terminus, proteolytic processing, and
carboxymethylation (Casey and Seabra, 1996). The prenylation takes place on the SH
groups of cysteines within the sequence CXXX, CC, CXC, CCXX, or CCXX found at
the very C-terminus (Stenmark and Olkkonen, 2001; Brighouse et al., 2010).
Rab activation is mediated by guanine nucleotide exchange factors (GEFs) that
promote the GDP/GTP exchange on the Rab. Rab activation is accompanied by the
translocation of the Rab from the cytosol onto a membrane where it performs its function.
Rab deactivation is mediated by GTPase-activating proteins (GAPs) that promote the hy-
drolysis of bound GTP to GDP (Pfeffer, 2001; Segev, 2001). Deactivation results in the
release of the Rab from membranes back into the cytosol. Activated Rabs are believed to
function by interacting with specific effectors that mediate downstream signaling events
(Munro, 2002; Wang et al., 2008).

21
Figure 2. Localization and function of Rab GTPases. A diagram of an epithelial cell with transport pathways and the localizations of selected Rab GTPases. RAB1, located at endoplasmic reticulum (ER) exit sites and the pre-Golgi intermediate compartment (IC), mediates ER-Golgi trafficking. RAB2, located at the IC, might also regulate Golgi–ER trafficking. The Golgi-localized RAB6, RAB33 and RAB40 mediate intra-Golgi trafficking. RAB33, together with RAB24, also regulates the formation of autophagosomes. RAB8 mediates constitutive biosynthetic trafficking from the trans-Golgi network (TGN) to the plasma membrane and also participates in GLUT4 vesicle translocation (with RAB10 and RAB14) and ciliogenesis (with RAB17 and RAB23). RAB3, RAB26, RAB27 and RAB37 mediate various types of regulated exocytic events and RAB27 also mediates the translocation of melanosomes to the cell periphery. RAB32 and RAB38 are involved in the biogenesis of melanosomes and RAB32 also controls mitochondrial fission. RAB13 regulates the assembly of tight junctions between

22
epithelial cells. RAB18 controls the formation of lipid droplets. RAB22 mediates trafficking between the TGN and early endosomes and vice versa. RAB5, which is localized to early endosomes, phagosomes, caveosomes and the plasma membrane, mediates endocytosis and endosome fusion of clathrin-coated vesicles (CCVs), macropinocytosis (with RAB34) and maturation of early phagosomes (with RAB14 and RAB22). RAB21 mediates integrin endocytosis. RAB11 and RAB35 mediate slow endocytic recycling through recycling endosomes, whereas RAB4 mediates fast endocytic recycling directly from early endosomes. RAB15 is involved in the trafficking from early endosomes to recycling endosomes and in the trafficking from apical recycling endosomes to the basolateral plasma membrane. RAB17 and RAB25 control trafficking through the apical recycling endosomes to the apical plasma membrane. The late endosome-associated RAB7 mediates maturation of late endosomes and phagosomes, and their fusion with lysosomes. Another late endosomal GTPase, RAB9, mediates trafficking from late endosomes to the TGN.
Note: Figure and legend reprinted from “Rab GTPases as coordinators of vesicle traffic” by Harald Stenmark, 2009, Nature Reviews Molecular Cell Biology, 10(8), p. 513-525. Copyright 2009 by Macmillan Publishers. Reprinted with permission.
Figure 3. The Rab switch and its circuitry. Conversion of the GDP-bound into the GTP-bound form occurs through the exchange of GDP for GTP, which is catalysed by guanine nucleotide exchange factor (GEF) and causes conformation change. The GTP-bound ‘ac-tive’ conformation is recognized by multiple effector proteins and converted back to the GDP-bound ‘inactive’ form through hydrolysis of GTP, which is stimulated by a GTPase-activating protein (GAP) and release of an inorganic phosphate (Pi). Newly syn-thesized Rabs, in GDP-bound form, are recognized by a Rab escort protein (REP). The REP presents the Rab to a geranylgeranyl transferase (GGT) which geranylgeranylates the Rab on one or two carboxy-terminal Cys residues. The geranylgeranylated, GDP-bound Rab is recognized by Rab GDP dissociation inhibitor (GDI), which regulates the membrane cycle of Rab. Targeting of Rab GDI complex to specific memebranes is me-diated by interaction with a membrane-bound GDI displacement factor GDF.

23
Note: Figure and legend reprinted from “Rab GTPases as coordinators of vesicle traffic” by Harald Stenmark, 2009, Nature Reviews Molecular Cell Biology, 10(8), p. 513-525. Copyright 2009 by Macmillan Publishers. Reprinted with permission.
Several Rabs have been implicated in cilia formation and/or function based on
proteomic and genetic studies. Rab GTPases that have been implicated in ciliogenesis
include Rab8a, Rab10, Rab11, Rab17, and Rab23 (Deretic et al., 1995; Nachury et al.,
2007; Yoshimura et al., 2007; Babbey et al., 2010; Knödler et al., 2010). Rab8, Rab17,
and Rab23 were implicated in ciliogenesis in a study that screened 39 RabGAPs for their
involvement in cilia formation. Three RabGAPs prevented primary cilium formation in
cells, and further analysis indicated that these RabGAPs act on Rab 8a, -17, and -23. The
requirement for Rab8 was further confirmed by showing that a dominant negative Rab8
mutant inhibits cilia formation. Rab8 is activated by its guanine nucleotide exchange fac-
tor (GEF) Rabin8 and this allows its entry into the primary cilium (Nachury et al., 2007).
In a recent study Rab11 was localized to the base of cilia and appears essential in cilioge-
nesis because depletion of endogenous Rab11 results in loss of cilia while the expression
of a Rab11 dominant negative mutant leads to formation of shorter cilia (Knödler et al.,
2010). Rab10 localizes to the basal body and along the length of cilia, but its role in cili-
ogenesis is controversial because knockdown of endogenous Rab10 resulted in varying
defects in ciliogenesis (Babbey et al., 2010). Additionally, two Rab-like proteins, Rab-
like5 (IFTA-2) and Rab-like4 (IFT27) were shown to play a role in primary cilium func-
tion in C. elegans (Shafer et al., 2006; Qin et al., 2007). While the molecular mechanism
of IFTA-2 action is unclear, studies of IFT27 suggest that it plays a role in cell cycle-
control and thus may influence ciliogenesis indirectly (Qin et al., 2007).

24
Additionally, Rab3, Rab6, Rab8a, and Rab11 have been identified as components
of vesicles containing rhodopsin, suggesting that these Rabs may regulate protein trans-
port to the base of the cilium (Deretic, 1997). Rab8 and its GEF Rabin 8 (Moritz et al.,
2001; Nachury et al., 2007; Knödler et al., 2010) have been shown to mediate the dock-
ing and fusion of the BBSome. The BBSome consist of seven BBS proteins, BBS1,
BBS2, BBS4, BBS5, BBS7, BBS8 and BBS9 and BBIP10 (Nachury et al., 2007; Loktev
et al., 2008). The BBSome is an effector for the Arf-like GTPase Arl6/BBS3 and aids in
the recruitment of the BBSome to cilia. The BBSome sorts transmembrane proteins by
recognizing the ciliary targeting sequence in the proteins to mediate the delivery of the
protein to cilia (Nachury et al., 2007; Jin et al., 2010). Trafficking of fibrocystin through
the endomembrane system to primary cilia is mediated by Rab8 (Follit et al., 2010). Re-
cently it was shown in frog retina that Rab11, along with FIP3 (Rab11 effector), Arf4,
and the ArfGTPase-activating protein ASAP1, participate in the formation of a complex
involved in the selection and packaging of cargoes designated to the cilia (Mazelova et
al., 2009a). In a study by Boehlke et al., 2010, ciliary fluorescence recovery after pho-
tobleaching was used to quantitatively compare the turnover of ciliary transmembrane
proteins in which depletion of Rab23 or expression of the dominant-negative Rab23 de-
creased the ciliary steady state of hedgehog-associated transmembrane receptor Smoo-
thened (Boehlke et al., 2010). Rab10 colocalizes with the exocyst complex at the base of
primary cilia and physically interacts in a protein complex with Sec8 suggesting a role for
Rab10 and the exocyst in membrane transport to primary cilia (Babbey et al., 2010).
Rab21 as Possible Regulator of Ciliary Traffic

25
Previous studies have identified endosomal Rab11 and Rab17 as regulators of ci-
liogenesis and ciliary traffic. This has raised the possibility that other endosomal Rabs
also might regulate ciliary trafficking. Rab21 is related to Rab17, belongs to subfamily 9
of Rabs and localizes to early endosomes with some localization at the plasma membrane
(Simpson et al., 2004; Brighouse et al., 2010). Rab21 appears to function in endocytosis,
and cells expressing the inactive GDP bound Rab21T33N mutant have defects in endocy-
tosis of epidermal growth factor and transferrin receptors and fail to deliver both recep-
tors to endosomes and lysosomes for degradation (Simpson et al., 2004). In addition,
Rab21 functions in endocytosis of integrins and cell-extracellular matrix adhesion pro-
teins (Pellinen et al., 2006). Rab21-mediated regulating integrin trafficking has been
shown to be essential for normal cell division (Pellinen et al., 2008). The activity of
Rab21 is regulated by the VPS9-ankyrin-repeat protein (VARP) that acts as Rab21 gua-
nine nucleotide exchange factor and activates Rab21 by facilitating the GDP/GTP ex-
change. Depletion of cellular VARP leads to deactivation of Rab21 activity and disrupts
endosome dynamics (Zhang et al., 2006).
Another endosomal Rab is Rab4 that localizes on early and recycling endosomes
(van der Sluijs et al., 1992; Trischler et al., 1999). Rab4 occupies distinct domains of ear-
ly endosomes and is segregated from Rab5 that localizes in adjacent micro-domains
(Sönnichsen et al., 2000). Studies show that Rab4 controls early sorting events and func-
tions in endocytosis of transferrin and neonatal Fc receptors (Bucci et al., 1992; Ward et
al., 2005). Fast recycling of internalized transferrin receptor and glycosphingolipids from
early endosomes to the plasma memebrane is mediated by Rab4 (van der Sluijs et al.,

26
1992; Choudhury et al., 2004; Maxfield and McGraw, 2004). Recently Rab4 has been
shown to be important for adherens junction disassembly (Mruk et al., 2007).
Purpose of Research for Project 2: Characterize Ciliary Delivery Pathway of SSTR3
Correct deployment of sensing protein to the cilium is essential for organismal
development and homeostasis. Recent inquiry has identified targeting information re-
quired for ciliary delivery of proteins and has begun to characterize the cellular mechan-
isms that ensure correct trafficking of proteins to and within the cilium. However, the
pathways taken by proteins to cilia and the identity of all cellular components that regu-
late trafficking of ciliary proteins remains unknown. Thus, we explored the pathways by
which the ciliary protein SSTR3 reaches the cilium and assessed the function of various
Rabs in SSTR3 trafficking. Chapter 3 of this dissertation documents my findings that
SSTR3 is packaged into vesicles that bud off distinct early and recycling endosomal
compartments, tranlsocated towards the cilium and directly fuse at the base of the cilium.
Furthermore, my results indicate that Rab21, Rab4 and Rab11 are novel regulators of
SSTR3 deployment to the cilium.

27
HUMAN CYTOMEGALOVIRUS UL97 KINASE PREVENTS THE DEPOSITION OF
MUTANT PROTEIN AGGREGATES IN CELLULAR MODELS OF HUNTINGTON’S DISEASE AND ATAXIA
CRISTY TOWER, LIANWU FU, RACHEL GILL, MARK PRICHARD, MATHIEU LESORT, and ELIZABETH SZTUL
Neurobiology of Disease Vol. 41, Issue 1, 11-22, January 2011
Copyright 2011 by
Elsevier
Used by permission
Format adapted and errata corrected for dissertation

28
ABSTRACT
The presence of aggregates of abnormally expanded polyglutamine (polyQ)-
containing proteins are a pathological hallmark of a number of neurodegenerative diseas-
es including Huntington’s disease (HD) and spinocerebellar Ataxia-3 (SCA3). Previous
studies in cellular, Drosophila, and mouse models of HD and SCA have shown that neu-
rodegeneration can be prevented by manipulations that inhibit polyQ aggregation. We
have shown that the UL97 kinase of the human cytomegalovirus (HCMV) prevents ag-
gregation of the pp71 and pp65 viral tegument proteins. To explore whether UL97 may
act as a general antiaggregation factor, we examined whether UL97 prevents aggregation
of cellular non-polyQ and polyQ proteins. We report that UL97 prevents the deposition
of aggregates of two non-polyQ proteins: a protein chimera (GFP170*) composed of the
green fluorescent protein and a fragment of the Golgi Complex protein (GCP-170), and a
chimera composed of the red fluorescent protein (RFP) fused to the Werner syndrome
protein (WRN), a RecQ helicase and exonuclease involved in DNA repair. Furthermore,
we show that UL97 inhibits aggregate deposition in cellular models of HD and SCA3.
UL97 prevents the deposition of aggregates of the mutant huntingtin exon 1 containing
82 glutamine repeats (HttExon1-Q82) or full length ataxin-3 containing a 72 polyQ track
(AT3-72Q). The kinase activity of UL97 appears critical, as the kinase-dead UL97 mu-
tant (K335M) fails to prevent aggregate formation. We further show that UL97 disrupts
nuclear PML bodies and decreases p53-mediated transcription. The universality of the
antiaggregation effect of UL97 suggests that UL97 targets a key cellular factor that regu-
lates cellular aggregation mechanisms. Our results identify UL97 as a novel means to
modulate polyQ aggregation and suggest that UL97 can serve as a novel tool to probe the

29
cellular mechanisms that contribute to the formation of aggregates in polyglutamine dis-
orders.
INTRODUCTION
The expansion of trinucleotide CAG repeats encoding glutamines within specific
cellular proteins is the cause of inherited neurodegenerative diseases termed polygluta-
mine disorders. Huntington’s disease, spinobulbar muscular atrophy (SBMA), dentatoru-
bral-pallidoluysian atrophy (DRPLA), and spinocerebellar ataxias (SCA) 1, 2, 3, 6, 7, and
17 are all caused by an abnormally expanded polyQ domain (reviewed in Cummings and
Zoghbi, 2000; McCampbell et al., 2001; Orr and Zoghbi, 2007). A striking neuropatho-
logical hallmark of these polyQ diseases is the presence of neuronal insoluble cytoplas-
mic aggregates and nuclear insoluble inclusions (NIIs) formed by mutant polyQ proteins
(reviewed in Ross, 2002). The role NIIs and cytoplasmic aggregates in the pathological
processes of polyQ diseases remains contentious. While some consider the
NIIs/cytoplasmic aggregates as direct toxic intermediates, some have proposed that
NIIs/cytoplasmic aggregates are a point of sequestration of a toxic product and thereby
beneficial to a neuron (Arrasate et al., 2004; Ross et al., 1999).
Multiple lines of evidence support the view that protein aggregation is a complex
process that is initiated by the accumulation of misfolded polyQ-containing proteins into
a variety of higher-order intermediate conformational assemblies that ultimately form in-
soluble inclusion bodies. Conformational rearrangements of these mutated proteins like-
ly change their biological activities and contribute to their toxicities. Importantly, recent
reports suggest that the toxicity of mutant polyQ-containing proteins might be not related
to the insoluble inclusion bodiesbut rather to soluble oligomeric or other intermediate

30
conformations (Kayed et al., 2003; Kayed et al., 2004). In support of this idea, polyQ-
induced neurodegeneration can be prevented by pharmacological and molecular manipu-
lations that target processes leading to oligomerization and aggregation. For instance, in-
hibition of polyglutamine oligomerization in a transgenic mouse model of HD has a
marked protective effect on survival, weight loss, and motor function (Sanchez et al.,
2003), supporting the idea that oligomerization of expanded polyglutamine may play a
pivotal role in the protein’s toxicity. Similarly, the green tea polyphenol epi-
gallocatechin-3-gallate (EGCG) inhibits aggregation of polyQ Httex1 in vitro and reduces
polyQ-induced cytotoxicity in yeast cells and Drosophila models of the disease
(Ehrnhoefer et al., 2006).
In addition to pharmacological approaches, modulations of cellular pathways that
prevent aggregation also reverse neurotoxicity. Molecular chaperones are the first line of
defense against protein aggregation, and numerous studies have shown that overexpres-
sion of chaperones prevents polyQ aggregation and reduces the pathology of neurodege-
nerative diseases (Muchowski and Wacker, 2005). In cultured cells, the chaperones heat
shock protein 40kDa (Hsp40) and 70kD (Hsp70) have been found to prevent the forma-
tion of spherical and annular oligomeric structures by a polyQ Httex1 (Wacker et al.,
2004). Significantly, overexpression of Hsp70a in SCA1 transgenic mice decreases neu-
rodegeneration and improves motor coordination (Cummings et al., 2001). Similarly, in-
hibiting polyQ aggregation by overexpressing Hsp70 (Warrick et al., 1999; Warrick et
al., 2005) and Hsp40 (Chan et al., 2000) rescues neurodegeneration in Drosophila models
of SCA3. Altogether, these findings strongly suggest that specific polyQ conformational
structures confer the cellular toxicity of the mutated proteins and that the identification of

31
novel pharmacological, molecular, or genetic means to prevent polyQ aggregation is like-
ly to have direct impact on developing therapeutic strategies for neurodegenerative dis-
eases such as HD and SCA3.
We have recently documented that the UL97 kinase of HCMV prevents the ag-
gregation of viral components during infection (Prichard et al., 2008). UL97 is a ~80-kDa
tegument protein composed of 707 amino acids that is expressed during HCMV infection
(Michel et al., 1998). UL97 contains a nuclear localization signal within its N-terminal
domain and is targeted to the nucleus in infected or transfected cells (Prichard et al.,
2005). UL97 is a kinase and shows homology to cellular serine/threonine kinases within
conserved subdomains involved in substrate and ATP binding (Michel et al., 1998). The
kinase activity of UL97 requires the invariant lysine at position 355, and the K355M mu-
tant is inactive (Marschall et al., 2001). UL97 is important for viral replication, and a re-
combinant virus with a large deletion in UL97 replicates poorly and contains abnormal
aggregates of viral proteins within the nuclei (Prichard et al., 1999); (Prichard et al.,
2005). One of the aggregated proteins is the pp65 viral tegument protein, which also
forms nuclear aggregates when expressed in transfected mammalian cells. Importantly,
UL97, but not the catalytically inactive UL97 K355M, prevents pp65 aggregation (Pri-
chard et al., 2005), suggesting that UL97 has antiaggregation activity and that the antiag-
gregation effect of UL97 is dependent on its kinase activity.
In addition to viral proteins, UL97 also prevents aggregation of GFP170*, a pro-
tein chimera formed by fusing an internal segment (amino acids 566- 1375) of the Golgi
protein GCP-170 to the C-terminus of GFP (Misumi et al., 2001; Hicks and Machamer,
2002; Prichard et al., 2008). We have shown previously that GFP170* forms nuclear ag-

32
gregates similar in structure to those formed by the viral pp65 and also deposits in large
ribbon-like aggregates within the cytoplasm (Fu et al., 2005a). UL97 prevents the forma-
tion of both the nuclear and the cytoplasmic GFP170* aggregates (Prichard et al., 2008).
As with pp65, the catalytically inactive UL97/K355M mutant is unable to prevent
GFP170* aggregation. Thus, UL97 prevents the aggregation of both a viral and a cellular
protein. Herein, we examined the possibility that UL97 may possess a general antiaggre-
gation activity and may serve as a tool for understanding and inhibiting the mechanisms
that contribute to aggregation in polyQ diseases.
We report that UL97 has a strong antiaggregation effect on non-polyQ proteins as
well as polyQ-expanded proteins associated with HD and SCA3. We show that UL97
prevents the deposition of aggregates of the non-polyQ Werner protein (WRN) that caus-
es the premature aging disease Werner syndrome. We also show that UL97 prevents ag-
gregation of a pathogenic construct that encodes the full-length ataxin-3 containing a 72-
glutamine expansion (AT3-72Q), and of a pathogenic N-terminal huntingtin domain cor-
responding to the exon1of this protein and containing an expanded track of 82 glutamine
residues (HttExon1-82Q). In all cases, the catalytically inactive UL97/K355M mutant
does not prevent aggregation. The similarity of the UL97 effect on the viral pp65 protein,
the non-polyQ GFP170* and WRN proteins, and the polyQ AT3-72Q and HttExon1-82Q
proteins suggests that UL97 has general antiaggregation effect. This similarity is also
consistent with the hypothesis that aggregation of diverse proteins may occur through a
common mechanism that is targeted by the UL97 kinase. In agreement, we show that
UL97 disperses nuclear PML bodies and causes a decrease in p53-mediated transcription.

33
Our results identify UL97 as a novel means to inhibit the aggregation of polyQ
proteins. They also designate UL97 as a new molecular tool to further examine the cellu-
lar mechanisms that lead to polyQ aggregation and neurodegeneration in HD and SCA3.
MATERIALS AND METHODS
Antibodies and reagents
Monoclonal antibody to the V5-epitope was purchased from Invitrogen (catalogue
# R960-25; Carlsbad, CA). Polyclonal anti-GFP antibody was purchased from Abcam
(catalogue no. ab290-50; Cambridge, MA). Anti-myc monoclonal antibody was pur-
chased from Covance (catalogue no. PRB-150B; Denver, PA). Anti-myc (A-14) (catalo-
gueno. sc-789) polyclonal antibody, anti-PML (PG-M3) (catalogue no sc-966) monoc-
lonal antibody, and anti-PML (H-238) (sc-5621) polyclonal antibody were from Santa
Cruz (Santa Cruz, CA). Fugene 6 transfection reagent was purchased from Roche (cata-
logue no. 11814443001; Indianapolis, IN), and was used in luciferase experiments, Mirus
IT-LTI tranfection reagent (catalogue no. MIR2300; Madison, WI) was used for transfec-
tion of cells for immunoflourescence microscopy. BCA protein assay kit was purchased
from Thermo Scientific (catalogue no. 23225; Rockford, IO). Alexa Fluor 594-labeled
goat anti-rabbit, Alexa Fluor 488-labeled goat anti-mouse, and Hoechst 33258 were from
Invitrogen Molecular Probes, Inc (Eugene, OR).
DNA constructs
The construction of the GFP-GCP170* chimera has been previously described
(Fu et al., 2005b). UL97 and K355M V5-epitope tagged plasmids have been previously
described (Prichard et al., 2005). Plasmids encoding pGL2-p21A luciferase have been

34
previously described (Chinery et al., 1997) and were a generous gift from Dr. Xinbin
Chen (UC Davis School of Veterinary Medicine). HttExon1-8299Q-GFP plasmid has
been described in Chun et al. (2001). The AT3-72Q plasmid was generously provided by
Dr. Randall Pittman (University of Pennsylvania School of Medicine). The mRFP-
Werner construct has been described previously (Vaitiekunaite et al., 2007) and was a
gift from Dr. Marek Rusin (Maria Sklodowska-Curie Memorial Institute, Gliwice, Pol-
and) .
Cell culture and transfections
HeLa cells were purchased from ATCC (Manassas, VA). HeLa cells were cul-
tured in minimum essential medium (MEM) supplemented with glucose, and glutamine
(Mediatech, Comprehensive Cancer Center of the University of Alabama at Birmingham,
AL). Media were supplemented with 10% fetal bovine serum (FBS, Life Technologies,
Grand Island, NY), 100 U/ml of penicillin and streptomycin (Invitrogen Corporation,
Grand Island, NY), nonessential amino acids, and 1mM sodium pyruvate. HT1080 cells
were a gift from Dr. Susan Nozell (University of Alabama at Birmingham). HT1080 cells
were maintained in MEM plus L-glutamine and 10% fetal bovine serum. These cells ex-
press endogenous wild-type p53 and were used for luciferase assays. All cells were
grown in incubator with 95% air in the atmosphere and 5% carbon dioxide (CO2) at
37°C.
HeLa cells were grown in monolayers on coverslips in six-well plates, and cells
were transfected with TransLTI (Mirus) or FuGENE 6 (Roche) reagents or with Lipofec-
tin (Invitrogen), according to the manufacturer’s protocols.

35
Cell viability assays were performed as follows: cells were cotransfected with
AT3-72Q and either a vector lacking UL97, a vector encoding UL97, or a vector encod-
ing UL97/K355M. After 24 or 48 h of culture, cells were treated with 15mg/ml of fluo-
rescein diacetate for 5min, washed 1x in phosphate-buffered saline (PBS) and processed
for immunofluorescence microscopy.
Immunofluorescence microscopy and quantification
At 48 to 72 h after transfection, cells were washed three times in PBS, fixed in 3%
paraformaldehyde for 10 min and then quenched with 10 mM ammonium chloride. The
cells were permeabilized in 0.2% Triton-X 100 in PBS for 7 min and washed 3 times in
PBS. The coverslips were blocked in PBS, 2.5% goat serum, 0.2% Tween-20 for 5 min
followed by blocking in PBS, 0.4% fish skin gelatin and 0.2% Tween-20. Cells were in-
cubated with primary antibody for 1 h at room temperature. Coverslips were washed with
PBS, 0.2% Tween-20 5 times for 5min and incubated with secondary antibodies for
45 min. Coverslips were washed as described above and the nucleus was stained with
Hoechst 33258. The coverslips were mounted on slides in 9:1 glycerol/PBS with 0.1% p-
phenylenediamine (Sigma-Aldrich; St. Louis, MO).
Fluorescence patterns were visualized with a Leitz Orthoplan microscope with
epifluorescence and Hoffman Modulation Contrast optics from Chroma Technology
(Brattleboro, VT, USA). Optical sections were captured with a CCD high-resolution
camera from Roper Scientific (Tucson, AZ, USA) equipped with a camera/computer in-
terface. Images were analyzed with a power Mac using IPLab Spectrum software (Scana-
lytics, Fairfax, VA, USA). A Perkin Elmer ERS6FE FRAP Spinning Disc Confocal was

36
used to capture images to visualize the association of polyQ aggregates with PML Bo-
dies. Images were analyzed in Volocity software.
For quantitation of aggresome patterns (diffuse staining versus aggresome locali-
zation), 100 cells were counted from randomly selected fields. Counting was performed
in a “blind” manner by different members of the Sztul laboratory for GCP170*, mRFP-
Werner, AT3-72Qmyc, and HttExon1-82Q-GFP. Data are presented as mean±SD. The
Student’s t-test was used to determine statistical significance, and a p-value of less than
0.05 was considered significant.
Luciferase reporter assays
HT1080 cells were seeded in two 12-well plates and were grown for 24 h before
transfection. The cells were cotransfected in triplicate with 500 ng of pGL2-p21A lucife-
rase reporter construct and 350 ng of pcDNA3 (empty vector), or UL97-V5, or K355M-
V5. Cells were washed once in PBS and lysed in 200 ul of luciferase lysis buffer (25 mM
Tris-phosphate (pH 7.8), 2 mM DTT, 2 mM 1,2-diaminocyclohexane-N,N,N´,N´-
tetraacetic acid, 10% glycerol, 1% Triton X-100) with rocking for 5 min at room tem-
perature. Then , 40 ul of lysate was added to 100 ul of luciferin substrate (20 mM Tri-
cine, 1 mM MgCO3, 2.67 mM MgSO4, 0.1 mM EDTA, 33.3 mM DTT, 270 uM coen-
zyme A, 470 uM Luciferin, and 530 uM ATP). The lucifirase activity in each sample was
measured in a Luminometer from Promega. The settings for the luminometer were a de-
lay time of 3 sec with an integration time of 10 sec. Each experiment was done in tripli-
cate.
RESULTS

37
UL97 prevents aggregation of the non-polyQ proteins GFP170* and WRN
We have shown previously that GFP170* is an aggregation-prone protein that can
be used to explore cellular mechanisms involved in the formation of aggresomes (Fu et
al., 2005b). To test whether UL97 might affect deposition of GFP170* aggregates, we
compared GFP170* localization in cells transfected with GCP170* in the presence of ei-
ther an empty plasmid, a plasmid encoding UL97 or a plasmid encoding the catalytically
inactive UL97/K355M. Consistent with our previous findings, cellular expression of
GFP170* in the presence of an empty plasmid resulted in the presence of large ribbon-
like aggregates within the cytoplasm, and large spherical inclusions within the nucleus
(Supplemental Fig. 1A). In clear contrast with these observations, when GFP170* was
expressed in the presence of UL97, large cytoplasmic and nuclear GFP170* aggregates
were rarely observed, and instead, GFP170* appeared diffuse throughout the cytoplasm
and the nucleoplasm (Supplemental Fig. 1B). To determine whether the effect of UL97
on the formation of GFP170* aggregates was dependent on its kinase activity, HeLa cells
were transfected with plasmids encoding GFP170* and the kinase-dead UL97/K355M.
Expression of the kinase dead UL97 did not prevent the formation of cytoplasmic or nuc-
lear GFP170* aggregates (Supplemental Fig. 1C). Interestingly, UL97/K355M co-
localizes with cytoplasmic GCP170* aggregates but does not associate with nuclear in-
clusions. This is consistent with the predominantly cytoplasmic localization of
UL97/K355M observed in transfected cells (Prichard et al., 2005 and Supplemental Fig.
2). It is unknown why the kinase-dead UL97/K355M remains within the cytoplasm while
the wild-type UL97 shuttle into the nucleus.

38
Quantitative analysis revealed that the frequency of cells with large nuclear
GFP170* aggregates is significantly decreased in cells transfected with UL97 (~10%) as
compared to cells transfected with the empty plasmid (~79%) (Supplemental Fig. 1D).
Expression of the kinase-dead UL97/K355M did not significantly affect the frequency of
cells with aggregates (~73%) when compared to cells transfected with the empty plasmid
(Supplemental Fig. 1D). These observations suggest that UL97 prevents GFP170* aggre-
gation by a mechanism dependent on its kinase activity.
To further examine UL97 effects on protein aggregation, we tested whether UL97
can prevent aggregation of the WRN protein that when mutated causes the adult progeria
disease Werner syndrome. WRN is a RecQ helicase and exonuclease (Gray et al., 1997;
Huang et al., 1998) involved in DNA repair and telomere maintenance (Kamath-Loeb et
al., 2000; Kamath-Loeb et al., 2004). Endogenous WRN exhibits a diffuse nuclear pat-
tern, sometimes interspersed with nuclear or nucleolar foci (Marciniak et al., 1998;
Opresko et al., 2003). When WRN is tagged at the N-terminus with monomeric red fluo-
rescent protein (WRN-RFP), the protein forms multiple nuclear aggregates (Vaitiekunaite
et al., 2007).
To test the effect of UL97 on the formation of WRN aggregates, cells were trans-
fected with WRN-RFP in the presence of either an empty plasmid, a plasmid encoding
UL97 or a plasmid encoding the kinase dead UL97/K355M. Consistent with previous
studies (Vaitiekunaite et al., 2007), expression of the WRN-RFP protein resulted in the
presence of numerous “donut-shaped” nuclear inclusions (Fig. 1A). In clear contrast to
these observations, when WRN-RFP was expressed in the presence of UL97, nuclear
WRN-RFP aggregates were rarely observed but instead WRN-RFP fluorescence ap-

39
peared diffuse throughout the nucleoplasm (Fig. 1B). Expression of UL97 had no effect
on the nuclear localization of WRN-RFP, suggesting that the mechanism preventing the
formation of WRN-RFP aggregates does not involve its nuclear import and/or retention.
Expression of the kinase dead UL97/K355M does not prevent the formation of nuclear
WRN-RFP aggregates (Fig. 1C). Thus, UL97 prevents the formation of WRN-RFP nuc-
lear aggregates in a kinase-dependent manner.
Quantitative analysis revealed that in the absence of UL97 a majority (~80%) of
cells expressing WRN-RFP contained nuclear aggregates, whereas expression of UL97
resulted in significantly decreased frequency of cells with aggregates (~7%) (Fig. 1D).
Expression of the kinase-dead UL97/K355M did not significantly affect the frequency of
cells with aggregates (~70%) when compared to cells transfected with the empty plasmid
(Fig1D). However, we did notice that the size of WRN-RFP aggregates was consistently
reduced in the presence of the UL97/K355M kinase-dead mutant.
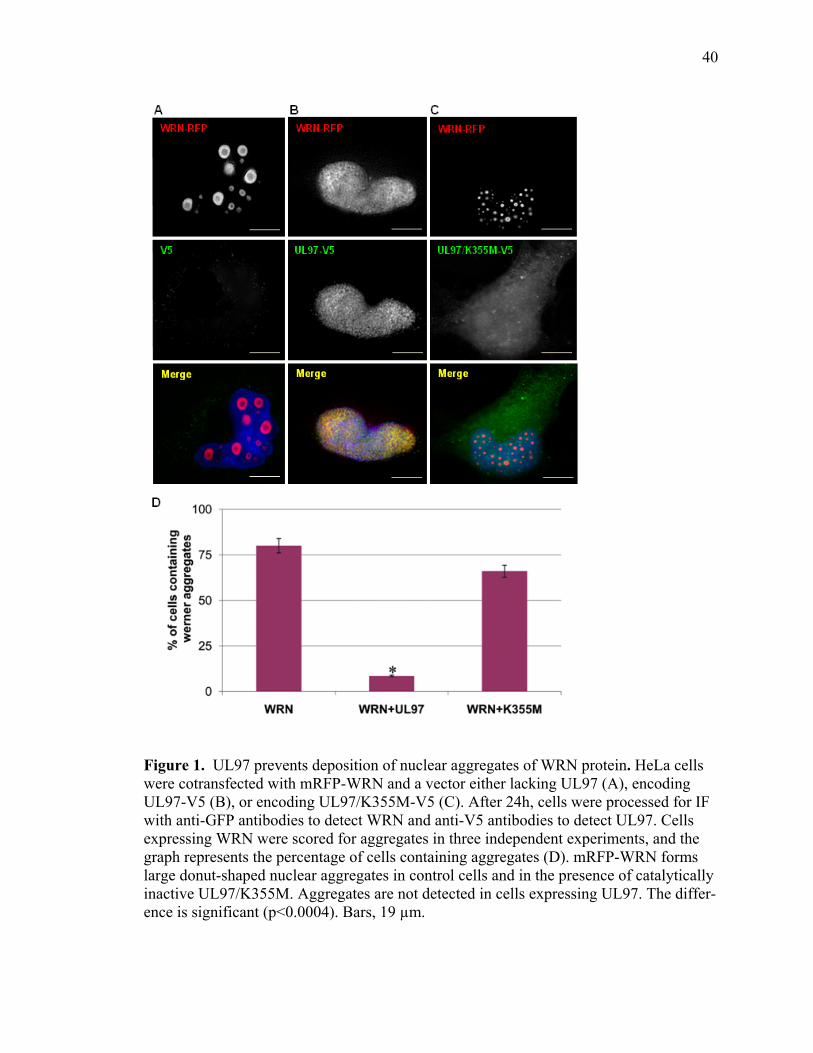
40
Figure 1. UL97 prevents deposition of nuclear aggregates of WRN protein. HeLa cells were cotransfected with mRFP-WRN and a vector either lacking UL97 (A), encoding UL97-V5 (B), or encoding UL97/K355M-V5 (C). After 24h, cells were processed for IF with anti-GFP antibodies to detect WRN and anti-V5 antibodies to detect UL97. Cells expressing WRN were scored for aggregates in three independent experiments, and the graph represents the percentage of cells containing aggregates (D). mRFP-WRN forms large donut-shaped nuclear aggregates in control cells and in the presence of catalytically inactive UL97/K355M. Aggregates are not detected in cells expressing UL97. The differ-ence is significant (p<0.0004). Bars, 19 µm.

41
The finding that UL97 exerts antiaggregation effect on the pp65 and pp71 viral
proteins (Prichard et al., 2008) and on two nonviral proteins, GFP170* and WRN (this
study), suggests that UL97 has a general antiaggregation property. To test this hypothe-
sis, we next explored the possibility that UL97 prevents the aggregation of polyQ-
expanded proteins linked to the neurodegenerative diseases SCA3 and HD.
UL97 prevents the aggregation of Ataxin-3 containing an expanded polyQ domain
We used a HeLa cell-based assay to examine the formation of aggregates after
transfecting cells with a plasmid encoding full-length ataxin-3 containing 72 glutamine
residues (AT3-72Q) and tagged at its C-terminus domain with myc (Zhong and Pittman,
2006). To assess the effect of UL97 on AT3-72Q aggregation, cells were transfected with
AT3-72Q in the presence of either an empty plasmid, a plasmid encoding UL97, or a
plasmid encoding the catalytically inactive UL97/K355M. In agreement with previous
studies (Burnett et al., 2003; Burnett and Pittman, 2005), expression of AT3-72Q and the
empty vector resulted in the presence of large aggregates that were localized within the
nucleus (Fig. 2A). In contrast, expression of AT3-72Q in the presence of UL97 prevented
the formation of aggregates, and AT3-72Q localized in small punctate structures scattered
throughout the cytoplasm and the nucleoplasm (Fig. 2B). Expression of AT3-72Q in the
presence of the kinase dead UL97/K355M did not prevent the formation of large AT3-
72Q aggregates (Fig. 2C). The kinase-dead mutant UL97/K355M colocalizes with cytop-
lasmic AT3-72Q aggregates but not with the nuclear aggregates. This is consistent with
the cytoplasmic localization of UL97/K355M (Supplemental Fig. 1 and 2).
Quantitative analysis reveals that the frequency of cells with AT3-72Q aggregates
is significantly decreased in cells transfected with UL97 (~6%) as compared to cells
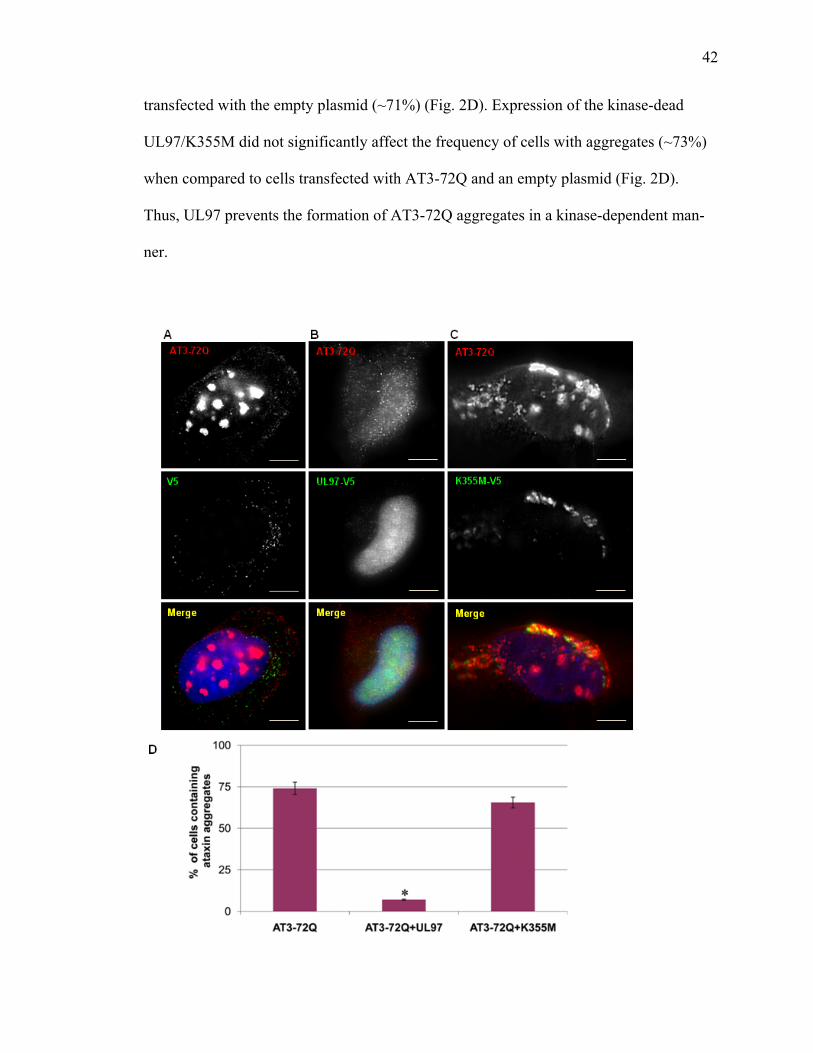
42
transfected with the empty plasmid (~71%) (Fig. 2D). Expression of the kinase-dead
UL97/K355M did not significantly affect the frequency of cells with aggregates (~73%)
when compared to cells transfected with AT3-72Q and an empty plasmid (Fig. 2D).
Thus, UL97 prevents the formation of AT3-72Q aggregates in a kinase-dependent man-
ner.

43
Figure 2. UL97 prevents aggregation of ataxin-3 containing expanded polyQ track. HeLa cells were cotransfected with AT3-72Q-myc and either a vector lacking UL97 (A), a vec-tor encoding UL97-V5 (B), or a vector encoding UL97/K355M-V5 (C). After 72h, cells were processed for IF with anti-myc antibodies to detect AT3-72Q and anti-V5 antibo-dies to detect UL97. Cells expressing AT3-72Q were scored for aggregates in three inde-pendent experiments, and the graph represents the percentage of cells containing aggre-gates (D). AT3-72Q-myc forms large cytoplasmic and nuclear aggregates in control cells and in cells expressing the catalytically inactive UL97/K355M. Aggregates are not de-tected or greatly reduced in cells that express UL97. The difference is significant (p<0.0007). Bars, 19
UL97 prevents the aggregation of the huntingtin exon-1 fragment containing an ex-
panded polyQ domain
We next examined the effects of UL97 on the formation of polyQ aggregates in
an HD cellular model. HeLa cells were transfected with a plasmid encoding the hunting-
tin exon1 containing 82 glutamines residues and a GFP tag at its N-terminus domain
(HttExon1-82Q) (Chun et al., 2001). In agreement with our previous studies (Chun, Le-
sort et al. 2001), expression of HttExon1-82Q resulted in the formation of large aggre-
gates in the perinuclear area of cells expressing HttExon1-82Q and the empty vector (Fig.
3A). In contrast, aggregates were not detected in cells expressing HttExon1-82Q in the
presence of UL97, and instead HttExon1-82Q presented a diffuse cytoplasmic staining
(Fig. 3B). UL97 prevents the formation of HttExon1-82Q in a kinase dependent manner
as the dead kinase UL97/K355M did not prevent the formation of Htt Exon1-82Q aggre-
gates (Fig. 3C). In cells coexpressing Htt Exon1-82Q aggregates and UL97/K355M, the
mutant kinase colocalize with Htt Exon1-82Q cytoplasmic aggregates (analysis of mul-
tiple confocal planes indicate all structures containing UL97/K355M are outside the nuc-
leus). This is consistent with the cytoplasmic distribution of UL97/K355M and the prefe-

44
rential association of this mutant with cytoplasmic GCP170* and AT-372Q aggregates
(Supplemental Fig. 1 and 2; Fig. 2C).
Quantification of these results reveals that the number of HttExon1-82Q aggre-
gates is significantly lower in cells transfected with UL97 (~10%) when compared to
cells transfected with the empty vector (~69%) (Fig. 3D). In contrast, expression of the
kinase-dead UL97/K355M did not significantly affect the frequency of cells with aggre-
gates (~67%) when compared to cells transfected with HttExon1-82Q and an empty
plasmid (Fig. 3D).
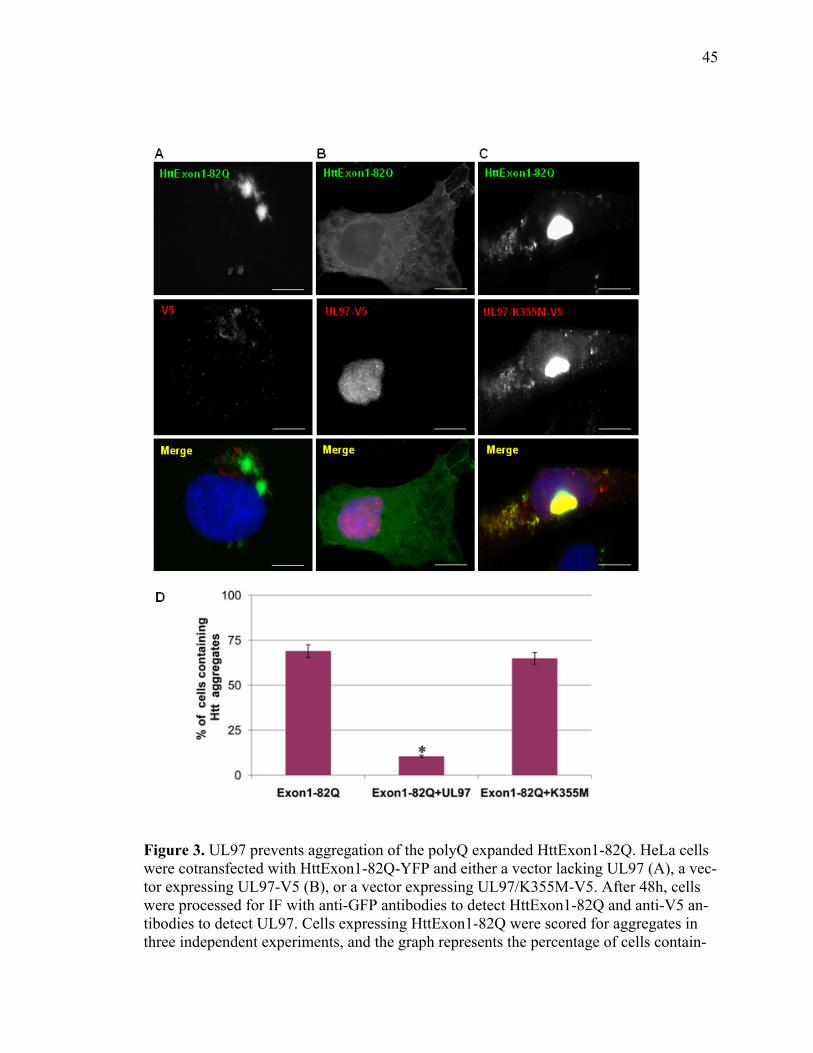
45
Figure 3. UL97 prevents aggregation of the polyQ expanded HttExon1-82Q. HeLa cells were cotransfected with HttExon1-82Q-YFP and either a vector lacking UL97 (A), a vec-tor expressing UL97-V5 (B), or a vector expressing UL97/K355M-V5. After 48h, cells were processed for IF with anti-GFP antibodies to detect HttExon1-82Q and anti-V5 an-tibodies to detect UL97. Cells expressing HttExon1-82Q were scored for aggregates in three independent experiments, and the graph represents the percentage of cells contain-

46
ing aggregates (D). Bars, 19 µm. HttExon1-82Q-YFP forms large cytoplasmic and nuc-lear aggregates in the absence of UL97 or in the presence of catalytically inactive UL97/K355M. Aggregates are not detected or greatly reduced in cells that express UL97. The difference is significant (p<0.004).
UL97 coordinately disrupts PML bodies and prevents polyQ aggregation
PML bodies have been linked to the aggregation of various viral and cellular pro-
teins (Bernardi and Pandolfi, 2007; Bonilla et al., 2002; Nichol et al., 2009). We have
reported recently that UL97 disrupt PML bodies, as monitored by the dispersion of the
Sp100 component of PML bodies (Prichard et al., 2008). This suggests that the UL97 an-
tiaggregation activity may be linked to PML disruption. To test this possibility, we ex-
amined the effect of UL97 on the integrity of PML bodies by following the localization
of the defining component of PML bodies, the PML protein (pPML). pPML is required
for nucleating and maintaining PML bodies, and cells devoid of pPML do not assemble
PML bodies (Shen et al., 2006). In control cells, pPML is detected in 15-30 discrete nuc-
lear foci (Figure 4A). Expression of UL97 has a dramatic effect and causes the disap-
pearance of PML bodies, with the number of PML bodies decreasing to only few (some-
times larger) structures (Fig. 4B). UL97 localizes in a diffuse nuclear pattern in trans-
fected cells, as previously reported (Prichard et al., 2008). Interestingly, although UL97
localizes to the nucleus, it does not appear to target to PML bodies, even when they be-
come enlarged. In contrast, the inactive UL97/K355M mutant does not cause pPML mis-
localization (Fig. 4C). UL97/K355M is detected in the cytoplasm and appears largely re-
stricted from the nucleus. It is likely that the mutant can nott enter or be retained within
the nucleus. This might explain the inability of UL97/K355M to disrupt PML bodies.
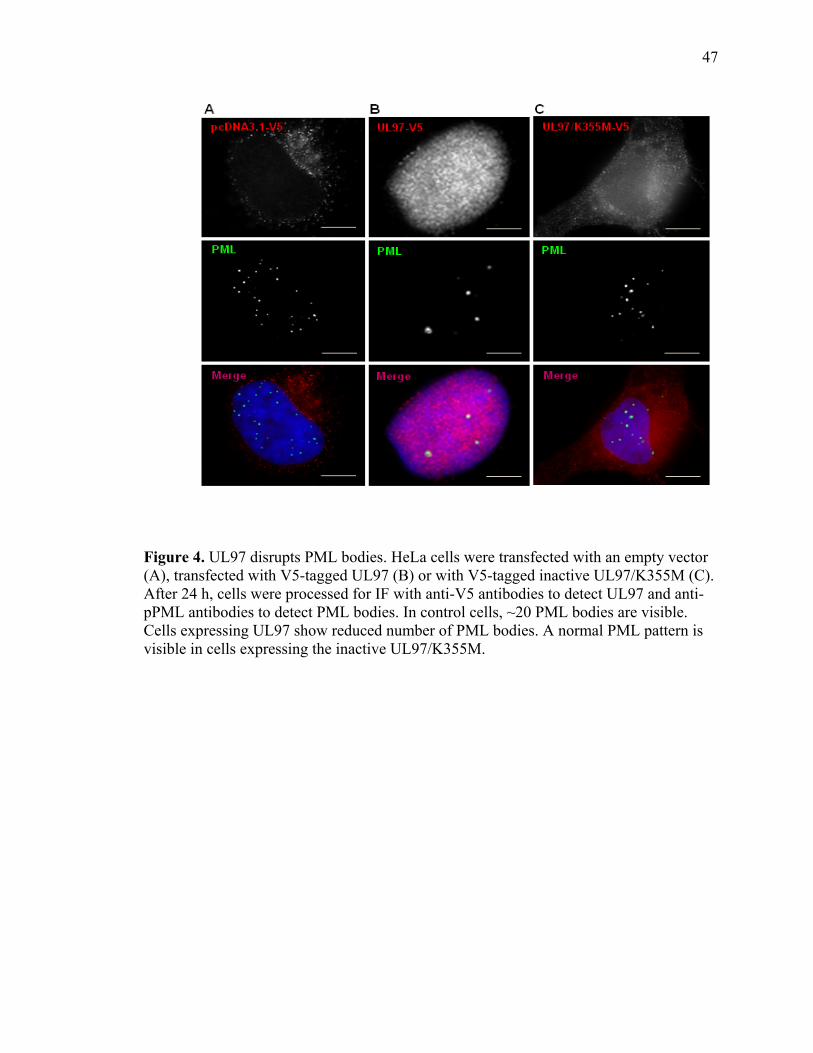
47
Figure 4. UL97 disrupts PML bodies. HeLa cells were transfected with an empty vector (A), transfected with V5-tagged UL97 (B) or with V5-tagged inactive UL97/K355M (C). After 24 h, cells were processed for IF with anti-V5 antibodies to detect UL97 and anti-pPML antibodies to detect PML bodies. In control cells, ~20 PML bodies are visible. Cells expressing UL97 show reduced number of PML bodies. A normal PML pattern is visible in cells expressing the inactive UL97/K355M.

48
The disruptive effect of UL97 on PML bodies might be linked to its antiaggrega-
tion property. To test this hypothesis, we explored the relationship between UL97 effects
on PML bodies and UL97 antiaggregation property in cells expressing AT3-72Q or
HttExon1-82Q. The integrity of PML bodies was assessed in cells expressing mutant
AT3-72Q and either a vector lacking UL97, a vector encoding UL97, or a vector encod-
ing the catalytically inactive UL97/K355M. Cells expressing AT3-72Q and the empty
vector contain multiple nuclear and cytoplasmic AT3-72Q aggregates (Fig. 5A). The
cells also contain multiple PML bodies within the nucleus. There appears to be a close
spatial association between the AT3-72Q aggregates and PML bodies, as most PML bo-
dies lie adjacent to an aggregate (arrowheads). Cells coexpressing AT3-72Q and UL97
show diffuse cytoplasmic and nuclear staining of AT3-72Q, confirming the antiaggrega-
tion effect of UL97 (Fig.5B). The cells contain drastically reduced number of PML bo-
dies. The few remaining PML bodies colocalize with the small AT3-72Q aggregates de-
tected in the nucleus (arrowheads). Cells coexpressing AT3-72Q and the inactive
UL97/K355M contain a number of large nuclear and cytoplasmic AT3-72Q aggregates
(Fig. 5C). PML bodies are present in these cells and appear to be extensively remodeled.
The PML bodies are recruited to the aggregates to form a “beaded necklace” pattern
around the aggregates (arrowheads). Thus, UL97 appears to coordinately disrupt PML
bodies and prevents aggregation of AT3-72Q.
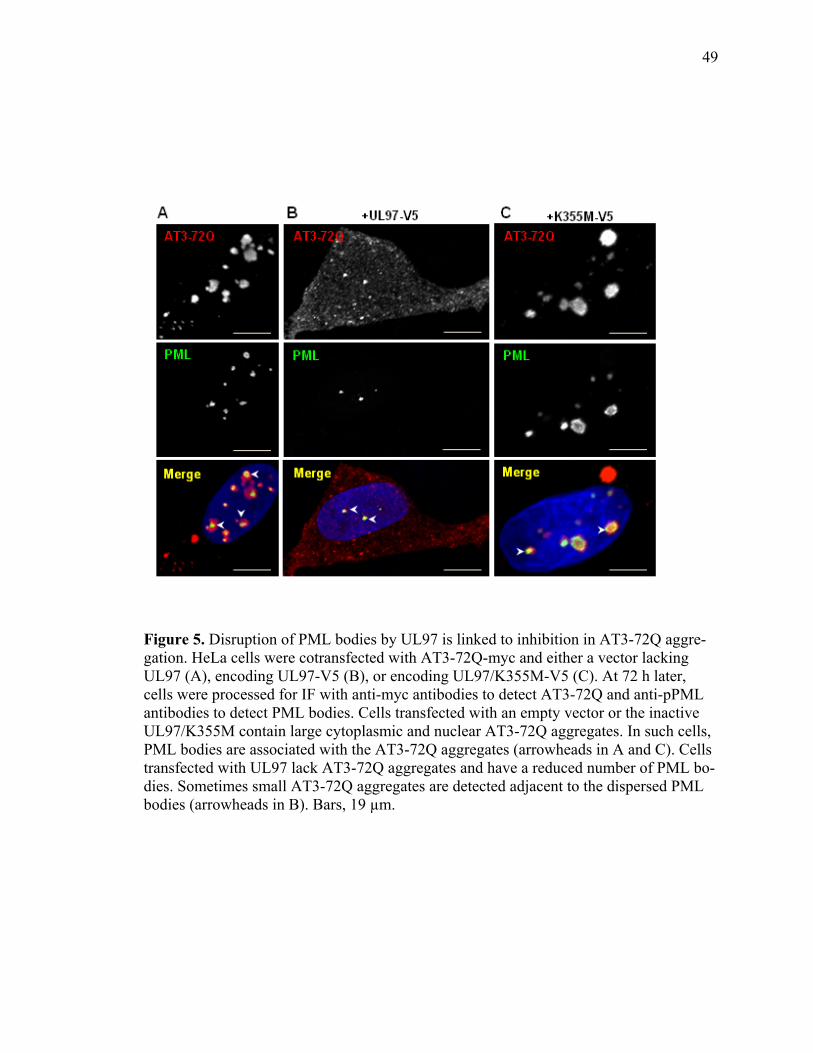
49
Figure 5. Disruption of PML bodies by UL97 is linked to inhibition in AT3-72Q aggre-gation. HeLa cells were cotransfected with AT3-72Q-myc and either a vector lacking UL97 (A), encoding UL97-V5 (B), or encoding UL97/K355M-V5 (C). At 72 h later, cells were processed for IF with anti-myc antibodies to detect AT3-72Q and anti-pPML antibodies to detect PML bodies. Cells transfected with an empty vector or the inactive UL97/K355M contain large cytoplasmic and nuclear AT3-72Q aggregates. In such cells, PML bodies are associated with the AT3-72Q aggregates (arrowheads in A and C). Cells transfected with UL97 lack AT3-72Q aggregates and have a reduced number of PML bo-dies. Sometimes small AT3-72Q aggregates are detected adjacent to the dispersed PML bodies (arrowheads in B). Bars, 19 µm.

50
We also explored the relationship between PMLs and inhibition of HttExon1-82Q
aggregation. Cells were cotransfected with HttExon1-82Q and either a vector lacking
UL97, a vector encoding UL97, or a vector encoding the catalytically inactive
UL97/K355M. Cells expressing HttExon1-82Q and the empty vector contain large cytop-
lasmic HttExon1-82Q aggregates (Fig. 6A). The cells show multiple PML bodies within
the nucleus in a pattern indistinguishable from that in control cells (compare to Fig. 4A).
In contrast, cells co-expressing HttExon1-82Q and UL97 show diffuse staining of HttEx-
on1-82Q throughout the cytoplasm (Fig. 6B). The lack of HttExon1-82Q aggregates con-
firms the antiaggregation effect of UL97. The cells contain dramatically reduced number
of PML bodies, and the remaining PMLs appear smaller than in control cells. Cells ex-
pressing HttExon1-82Q and the inactive UL97/K355M contain large, irregularly shaped
cytoplasmic aggregates of HttExon1-82Q (Fig. 6C). The cells contain a normal comple-
ment of PML bodies within the nucleus. Thus, expression of UL97 causes the disruption
of PML bodies and prevents aggregation of HttExon1-82Q.
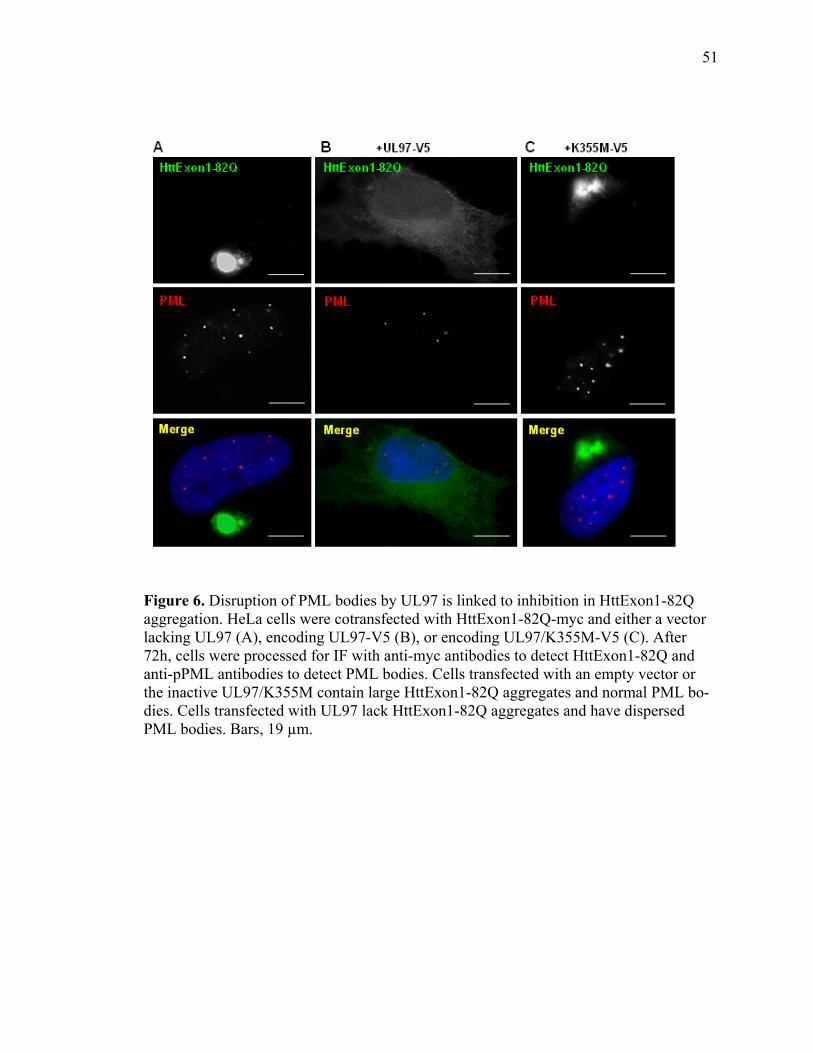
51
Figure 6. Disruption of PML bodies by UL97 is linked to inhibition in HttExon1-82Q aggregation. HeLa cells were cotransfected with HttExon1-82Q-myc and either a vector lacking UL97 (A), encoding UL97-V5 (B), or encoding UL97/K355M-V5 (C). After 72h, cells were processed for IF with anti-myc antibodies to detect HttExon1-82Q and anti-pPML antibodies to detect PML bodies. Cells transfected with an empty vector or the inactive UL97/K355M contain large HttExon1-82Q aggregates and normal PML bo-dies. Cells transfected with UL97 lack HttExon1-82Q aggregates and have dispersed PML bodies. Bars, 19 µm.

52
The commonality of the regulatory effect of UL97 on PML integrity suggests that
the aggregation processes of polyQ proteins could be functionally coupled to the status of
PML bodies and that the dispersion of PML bodies by UL97 could be linked to antiag-
gregation. The coordinated actions of UL97 in dispersal of PML bodies and prevention of
aggregation both require the kinase activity of UL97.
UL97 inhibits p53-dependent transcription
PML bodies have been implicated in transcription, and they contain the transcrip-
tional regulators Sp100, pPML, CBP, and p53 (de Stanchina et al., 2004; Hofmann et al.,
2002; Moller et al., 2003; Pampin et al., 2006; Salomoni et al., 2006; reviewed in Bernar-
di and Pandolfi, 2007). Thus, we explored the link between UL97 effects on PML bodies
and p53-mediated transcription. p53 activity was assessed in HT1080 fibrosarcoma cells
by measuring transcription from a reporter construct composed of firefly luciferase fused
to the p21 promoter that contains two p53 response elements (pGL2-p21A). Comparisons
were made between cells cotransfected with pGL2-p21A and either the empty vector
(control), a vector containing UL97 (UL97), or a vector containing UL97/K355M
(UL97/K355M). Basal transcription levels were quantified in cells transfected with
pGL2-p21A and the empty vector (Fig. 7A, control), and these are consistent with pre-
viously reported results. Cells co-transfected with pGL2-p21A and UL97 showed a 30%
reduction in p53 activity (Fig. 7A, UL97). The inhibitory effect was dependent upon
UL97 kinase activity, as cells cotransfected with pGL2-p21A and the inactive
UL97/K355M showed no changes in the levels of p53-dependent transcription (Fig. 7A,
UL97/K355M).

53
The HT1080 cells used in these experiments contain endogenous p53. To increase
p53-dependent transcription and thus increase the experimental signal, we performed ad-
ditional experiments in HT1080 cells transfected with exogenous p53. Comparisons were
made between cells cotransfected with p53, pGL2-p21A and either a vector lacking
UL97 (p53+control), a vector encoding UL97 (p53+UL97), or a vector encoding
UL97/K355M (p53+UL97/K355M). As expected, cells expressing exogenous p53
showed a significantly higher basal level of transcription (Figure 7A, p53 and
p53+control). Coexpression of UL97 decreased p53-mediated transcription by 50 %
(Fig.7A, p53+UL97). Coexpression of the inactive UL97/K355M had no effect on p53-
mediated transcription (Fig. 7A, p53+UL97/K355M). These results indicate that UL97
represses the transcriptional activity of p53 in a kinase-dependent manner, and suggest a
potential mechanism that links the effects of UL97 on aggregation, integrity of PML bo-
dies, and p53-mediated transcription.
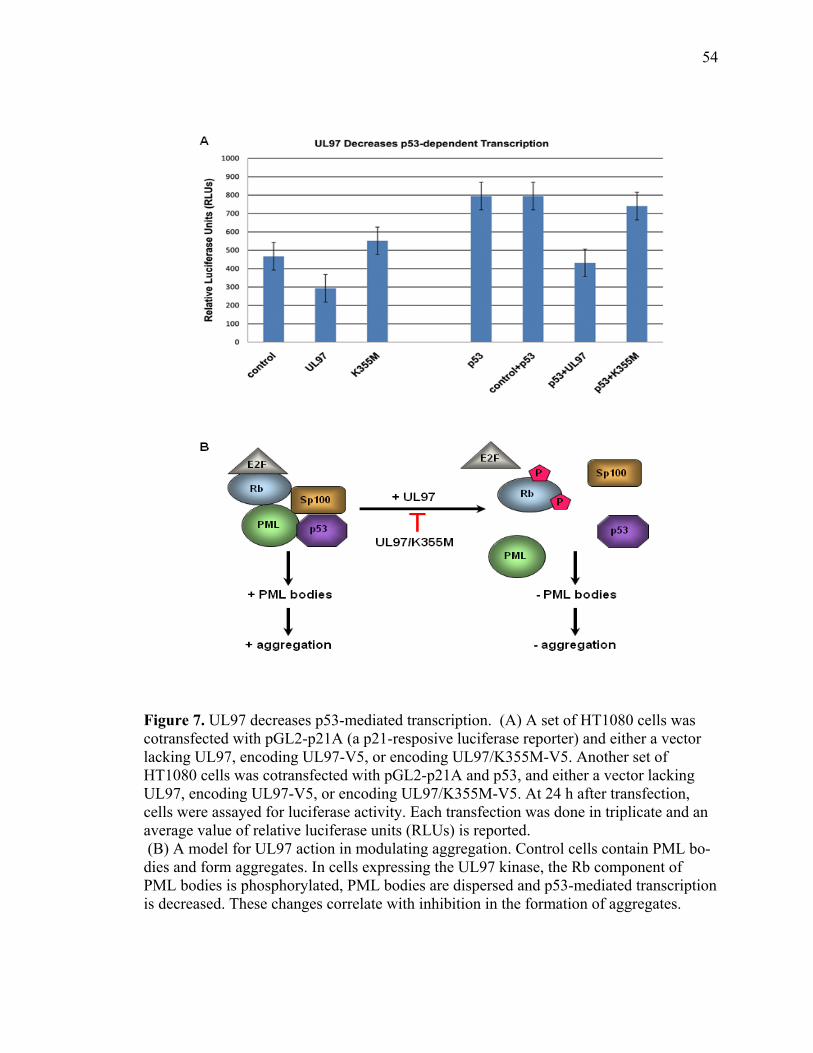
54
Figure 7. UL97 decreases p53-mediated transcription. (A) A set of HT1080 cells was cotransfected with pGL2-p21A (a p21-resposive luciferase reporter) and either a vector lacking UL97, encoding UL97-V5, or encoding UL97/K355M-V5. Another set of HT1080 cells was cotransfected with pGL2-p21A and p53, and either a vector lacking UL97, encoding UL97-V5, or encoding UL97/K355M-V5. At 24 h after transfection, cells were assayed for luciferase activity. Each transfection was done in triplicate and an average value of relative luciferase units (RLUs) is reported. (B) A model for UL97 action in modulating aggregation. Control cells contain PML bo-dies and form aggregates. In cells expressing the UL97 kinase, the Rb component of PML bodies is phosphorylated, PML bodies are dispersed and p53-mediated transcription is decreased. These changes correlate with inhibition in the formation of aggregates.

55
We assessed whether the UL97 kinase might rescue the cytotoxicity associated
with AT3-72Q polyQ aggregates. In these experiments cells were cotransfected with
AT3-73Q and either a vector lacking UL97, a vector encoding UL97, or a vector encod-
ing UL97/K355M. After 24 or 48 h of culture, cell viability was measured by the uptake
of fluorescein diacetate. UL97 appears to increase the percentage of viable cells express-
ing AT3-72Q (data not shown).
DISCUSSION
The results of this study provide strong evidence for an antiaggregation effect of
the UL97 viral kinase. UL97 prevents the aggregation of GFP170* and WRN proteins,
and of two polyQ-containing proteins, AT3-72Q and HttExon1-82Q that are associated
with SCA3 and Huntington disease, respectively. Remarkably, UL97 is able to prevent
the formation of both cytoplasmic and nuclear aggregates formed by these proteins. Con-
sistent with previous studies revealing that UL97 possesses a nuclear localization signal
(Prichard et al., 2005), we find that UL97 localizes preferentially within the nucleus. This
suggests that the antiaggregation properties of UL97 are likely to be mediated through
events occurring within the nucleus. Within the nucleus, the UL97 kinase may interact
and phosphorylate nuclear substrates to prevent aggregation. This model is consistent
with our findings that the UL97/K355M mutant defective in kinase activity is essentially
restricted from the nucleus and is unable to prevent protein aggregation. Further work
will be required to dissect the relative contributions of the nuclear localization and the
kinase activity towards the antiaggregation function of UL97. Together, our studies doc-
ument that UL97 localizes within the nucleus and prevents protein aggregation in a ki-
nase-dependent manner.

56
UL97 shows kinase activity towards a number of cellular substrates. For example,
UL97 phosphorylates the myelin basic protein; histones H1, H2B, and H3; lamins A, B,
and C; and the largest subunit of RNA polymerase II in vitro (Baek et al., 2002a,b, 2004;
Marschall et al., 2005). Whether any of these known UL97 substrates are linked to the
antiaggregating property of UL97 remains to be elucidated. However, recent studies sug-
gest a role for the retinoblastoma (Rb) protein, a recently identified UL97 substrate, in
UL97-mediated antiaggregation. Rb is a tumor-suppressor that controls cell cycle pro-
gression through G1 phase (Hume et al., 2008; Prichard et al., 2008). Importantly, Rb lo-
calizes to PML bodies, where it forms stable complexes with unphosphorylated pPML
(Alcalay et al., 1998). The identification of Rb as UL97 substrate seems especially rele-
vant in the light of the apparent relationship between PML bodies and aggregate forma-
tion, and the coordinate ability of UL97 to disrupt PML bodies and prevent aggregation.
The mechanisms by which UL97 causes disruption of PML bodies remains to be further
characterized. However, our findings are consistent with a model (Fig. 7B) in which
UL97 causes changes in the architecture of PML bodies through a phosphorylation de-
pendent mechanism. Remodeling and disuption of PML bodies would then lead to
changes in the transcription of p53-responsive genes and affect the formation of aggre-
gates. In support of this model, we provide evidence that GFP170*, WRN and AT3-72Q
deposit adjacent to PML bodies and cause the rearrangement of PML bodies around the
aggregates. Further, we show that UL97 disrupts PML bodies. The UL97-mediated dis-
ruption of PML is likely to have important consequences on transcriptional activity. In
support, our study provides clear evidence that UL97-mediated disruption of PML bodies
results in the inhibition of p53-mediated transcription. Whether the antiaggregation action

57
of UL97 is linked to alterations in a larger transcriptional repertoire remains to be ex-
plored. Importantly, UL97-induced disruption of PML bodies and alteration in p53-
dependent transcription are linked to UL97 ability to prevent aggregation of AT3-72Q
and HttExon1-82Q in cellular models of SCA3 and HD, respectively.
Huntington’s disease, spinocerebellar ataxia 3, and several other polyglutamine
disorders are characterized by the accumulation of cytoplasmic and/or nuclear aggregates
within the brain of affected individuals (Davies et al., 1997; DiFiglia et al., 1997). In
these diseases, a mutated protein misfolds to undergo an alternative conformation that in
most cases results in its aggregation and accumulation as inclusion bodies in neurons.
The role of these aggregates in the etiology of misfolded diseases has been the subject of
intense debate, with models that consider the aggregates as harmful, benign or beneficial
(Ross and Poirier, 2005; Sisodia, 1998). A body of emerging evidence suggests that ag-
gregates might be cytoprotective. Initial clues came from postmortem studies that re-
vealed a relatively poor correlation between the neurons in which inclusion bodies are
present and the neurons preferentially vulnerable in specific neurodegenerative disorders
(Kuemmerle et al., 1999; reviewed in Ross, 2002). Further temporal studies of the ap-
pearance of aggregates and the onset of clinical pathology suggest that tissue damage and
pathology can manifest before detection of aggregates (Saudou et al., 1998), raising the
possibility that aggregates may actually be protective. Additional studies by Arrasate et
al. (2004) provided evidence that the formation of inclusion bodies reduces the risk of
neuronal death. A potential explanation is that inclusion bodies and visible aggregates
constitute a point of sequestration of intermediate conformational assemblies that corre-
late more directly to the neuronal vulnerability than the aggregates (Poirier et al.,

58
2002,2005; Ross et al., 2003). Indeed, accumulating evidence supports the view that pro-
tein aggregation is a complex process, thought to be initiated by the interactions between
the misfolded proteins to generate a variety of higher-order intermediate assemblies that
ultimately form insoluble inclusion bodies (Poirier et al., 2002; Wetzel, 2006).
Using recombinant mutant huntingtin exon1 with an expanded polyQ track in in
vitro studies, a number of studies have characterized the morphological and structural
features of aggregation and provided evidence of globular and protofibrillar intermediates
rich in β-structure prior to the formation of fibrils and aggregates (Chen et al., 2001,
2002; Poirier et al., 2002, 2005). Similar intermediate conformational assemblies have
been identified for other proteins capable of forming aggregates in various neurodegener-
ative disorders, suggesting common mechanism of aggregation and potentially toxicity
(Kayed et al., 2003). In line with this model, accumulating evidence suggests that the tox-
icity of amyloidogenic proteins involved in several neurodegenerative disorders may not
be related to the insoluble protein aggregates but rather to the soluble oligomeric or other
intermediate assemblies. For instance, soluble oligomeric or protofibrillar forms of amy-
loids are more potent toxins than the mature fibrils of Aβ (Dahlgren et al., 2002), α-
synuclein (Conway et al., 2000), IAPP (Anguiano et al., 2002) and polyQ (Kayed et al.,
2003; Kayed et al., 2009; Kayed et al., 2004). In effect, the formation of aggregates might
be a protective mechanism to sequester and isolate these toxic intermediate conforma-
tional species (Ross and Poirier, 2005).
In this study, we used cellular models of aggregation to show the efficacy of the
UL97 kinase in preventing the deposition of non-polyQ and polyQ aggregates. Although
it is clear that UL97 targets cellular pathways that control aggregation, it remains to be

59
determined whether UL97 affects the formation and accumulation of potentially toxic
soluble oligomeric or protofibrillar conformations of the mutated proteins. The ability of
UL97 to inhibit aggregation of polyQ proteins in cellular models of diseases as diverse as
HD and SCA3 raises the possibility that UL97 might be a tool to dissect the aggregation
mechanisms in diseases associated with polyQ protein deposits. Thus, characterizing the
molecular mechanisms of UL97 action in preventing protein aggregation may reveal a
novel rational approach to prevent polyQ-mediated neurodegeneration.
ACKNOWLEDGMENTS
We thank Dr. Susan Nozelle for assisting with luciferase assays and thoughtful
insight into transcriptional aspects of this work. We are also grateful to Dr. Melanie
Styers for critically reviewing the manuscript.
FOOTNOTES
Article published online ahead of print. Neurobiol Dis. 2010 Aug 20. PMID: 207
32421. Article and publication date are at http://dx.doi.org/10.1016/j.nbd.2010.08.013.
Online version of this article contains supplemental figures. Online version is
available at http://www.sciencedirect.com/science/journal/09699961.
Corresponding author. E-mail addresses: [email protected]

60
REFERENCES
Alcalay, M., L. Tomassoni, E. Colombo, S. Stoldt, F. Grignani, M. Fagioli, L. Szekely, K. Helin, and P.G. Pelicci. 1998. The promyelocytic leukemia gene product (PML) forms stable complexes with the retinoblastoma protein. Mol Cell Biol. 18:1084-93.
Anguiano, M., R.J. Nowak, and P.T. Lansbury, Jr. 2002. Protofibrillar islet amyloid po-lypeptide permeabilizes synthetic vesicles by a pore-like mechanism that may be relevant to type II diabetes. Biochemistry. 41:11338-43.
Arrasate, M., S. Mitra, E.S. Schweitzer, M.R. Segal, and S. Finkbeiner. 2004. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Na-ture. 431:805-10.
Baek, M.C., P.M. Krosky, and D.M. Coen. 2002a. Relationship between autophosphory-lation and phosphorylation of exogenous substrates by the human cytomegalovirus UL97 protein kinase. J Virol. 76:11943-52.
Baek, M.C., P.M. Krosky, Z. He, and D.M. Coen. 2002b. Specific phosphorylation of exogenous protein and peptide substrates by the human cytomegalovirus UL97 protein kinase. Importance of the P+5 position. J Biol Chem. 277:29593-9.
Baek, M.C., P.M. Krosky, A. Pearson, and D.M. Coen. 2004. Phosphorylation of the RNA polymerase II carboxyl-terminal domain in human cytomegalovirus-infected cells and in vitro by the viral UL97 protein kinase. Virology. 324:184-93.
Bernardi, R., and P.P. Pandolfi. 2007. Structure, dynamics and functions of promyelocyt-ic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 8:1006-16.
Bonilla, W.V., D.D. Pinschewer, P. Klenerman, V. Rousson, M. Gaboli, P.P. Pandolfi, R.M. Zinkernagel, M.S. Salvato, and H. Hengartner. 2002. Effects of promyelocytic leu-kemia protein on virus-host balance. J Virol. 76:3810-8.
Burnett, B., F. Li, and R.N. Pittman. 2003. The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum Mol Genet. 12:3195-205.
Burnett, B.G., and R.N. Pittman. 2005. The polyglutamine neurodegenerative protein ataxin 3 regulates aggresome formation. Proc Natl Acad Sci U S A. 102:4330-5.
Chan, H.Y., J.M. Warrick, G.L. Gray-Board, H.L. Paulson, and N.M. Bonini. 2000. Me-chanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum Mol Genet. 9:2811-20.

61
Chen, S., V. Berthelier, J.B. Hamilton, B. O'Nuallain, and R. Wetzel. 2002. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry. 41:7391-9.
Chen, S., V. Berthelier, W. Yang, and R. Wetzel. 2001. Polyglutamine aggregation beha-vior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol. 311:173-82.
Chinery, R., J.A. Brockman, M.O. Peeler, Y. Shyr, R.D. Beauchamp, and R.J. Coffey. 1997. Antioxidants enhance the cytotoxicity of chemotherapeutic agents in colorectal cancer: a p53-independent induction of p21WAF1/CIP1 via C/EBPbeta. Nat Med. 3:1233-41.
Chun, W., Lesort M., Tucholski J., Faber P.W., MacDonald M.E., Ross C.A., Johnson G.V., 2001. Tissue transglutaminase selectively modifies proteins associated with trun-cated mutant huntingtin in intact cells. Neurobiol Dis. 8:391-404.
Conway, K.A., J.D. Harper, and P.T. Lansbury, Jr. 2000. Fibrils formed in vitro from al-pha-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry. 39:2552-63.
Cummings, C.J., Y. Sun, P. Opal, B. Antalffy, R. Mestril, H.T. Orr, W.H. Dillmann, and H.Y. Zoghbi. 2001. Over-expression of inducible HSP70 chaperone suppresses neuropa-thology and improves motor function in SCA1 mice. Hum Mol Genet. 10:1511-8.
Cummings, C.J., and H.Y. Zoghbi. 2000. Trinucleotide repeats: mechanisms and patho-physiology. Annu Rev Genomics Hum Genet. 1:281-328.
Dahlgren, K.N., A.M. Manelli, W.B. Stine, Jr., L.K. Baker, G.A. Krafft, and M.J. LaDu. 2002. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neu-ronal viability. J Biol Chem. 277:32046-53.
Davies, S.W., M. Turmaine, B.A. Cozens, M. DiFiglia, A.H. Sharp, C.A. Ross, E. Scher-zinger, E.E. Wanker, L. Mangiarini, and G.P. Bates. 1997. Formation of neuronal intra-nuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 90:537-48.
de Stanchina, E., E. Querido, M. Narita, R.V. Davuluri, P.P. Pandolfi, G. Ferbeyre, and S.W. Lowe. 2004. PML is a direct p53 target that modulates p53 effector functions. Mol Cell. 13:523-35.
DiFiglia, M., E. Sapp, K.O. Chase, S.W. Davies, G.P. Bates, J.P. Vonsattel, and N. Aro-nin. 1997. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 277:1990-3.
Ehrnhoefer, D.E., M. Duennwald, P. Markovic, J.L. Wacker, S. Engemann, M. Roark, J. Legleiter, J.L. Marsh, L.M. Thompson, S. Lindquist, P.J. Muchowski, and E.E. Wanker.

62
2006. Green tea ( - )-epigallocatechin-gallate modulates early events in huntingtin mis-folding and reduces toxicity in Huntington's disease models. Hum Mol Genet. 15:2743-51.
Fu, L., Y.S. Gao, and E. Sztul. 2005a. Transcriptional repression and cell death induced by nuclear aggregates of non-polyglutamine protein. Neurobiol Dis. 20:656-65.
Fu, L., Y.S. Gao, A. Tousson, A. Shah, T.L. Chen, B.M. Vertel, and E. Sztul. 2005b. Nuclear aggresomes form by fusion of PML-associated aggregates. Mol Biol Cell. 16:4905-17.
Gray, M.D., J.C. Shen, A.S. Kamath-Loeb, A. Blank, B.L. Sopher, G.M. Martin, J. Oshima, and L.A. Loeb. 1997. The Werner syndrome protein is a DNA helicase. Nat Ge-net. 17:100-3.
Hicks, S.W., and C.E. Machamer. 2002. The NH2-terminal domain of Golgin-160 con-tains both Golgi and nuclear targeting information. J Biol Chem. 277:35833-9.
Hofmann, T.G., A. Moller, H. Sirma, H. Zentgraf, Y. Taya, W. Droge, H. Will, and M.L. Schmitz. 2002. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat Cell Biol. 4:1-10.
Huang, S., B. Li, M.D. Gray, J. Oshima, I.S. Mian, and J. Campisi. 1998. The premature ageing syndrome protein, WRN, is a 3'-->5' exonuclease. Nat Genet. 20:114-6.
Hume, A.J., J.S. Finkel, J.P. Kamil, D.M. Coen, M.R. Culbertson, and R.F. Kalejta. 2008. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science. 320:797-9.
Kamath-Loeb, A.S., E. Johansson, P.M. Burgers, and L.A. Loeb. 2000. Functional inte-raction between the Werner syndrome protein and DNA polymerase delta. Proc Natl Acad Sci U S A. 97:4603-8.
Kamath-Loeb, A.S., P. Welcsh, M. Waite, E.T. Adman, and L.A. Loeb. 2004. The enzy-matic activities of the Werner syndrome protein are disabled by the amino acid polymor-phism R834C. J Biol Chem. 279:55499-505.
Kayed, R., E. Head, J.L. Thompson, T.M. McIntire, S.C. Milton, C.W. Cotman, and C.G. Glabe. 2003. Common structure of soluble amyloid oligomers implies common mechan-ism of pathogenesis. Science. 300:486-9.
Kayed, R., A. Pensalfini, L. Margol, Y. Sokolov, F. Sarsoza, E. Head, J. Hall, and C. Glabe. 2009. Annular protofibrils are a structurally and functionally distinct type of amy-loid oligomer. J Biol Chem. 284:4230-7.

63
Kayed, R., Y. Sokolov, B. Edmonds, T.M. McIntire, S.C. Milton, J.E. Hall, and C.G. Glabe. 2004. Permeabilization of lipid bilayers is a common conformation-dependent ac-tivity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem. 279:46363-6.
Kuemmerle, S., C.A. Gutekunst, A.M. Klein, X.J. Li, S.H. Li, M.F. Beal, S.M. Hersch, and R.J. Ferrante. 1999. Huntington aggregates may not predict neuronal death in Hun-tington's disease. Ann Neurol. 46:842-9.
Marciniak, R.A., D.B. Lombard, F.B. Johnson, and L. Guarente. 1998. Nucleolar locali-zation of the Werner syndrome protein in human cells. Proc Natl Acad Sci U S A. 95:6887-92.
Marschall, M., A. Marzi, P. aus dem Siepen, R. Jochmann, M. Kalmer, S. Auerochs, P. Lischka, M. Leis, and T. Stamminger. 2005. Cellular p32 recruits cytomegalovirus kinase pUL97 to redistribute the nuclear lamina. J Biol Chem. 280:33357-67.
Marschall, M., M. Stein-Gerlach, M. Freitag, R. Kupfer, M. van Den Bogaard, and T. Stamminger. 2001. Inhibitors of human cytomegalovirus replication drastically reduce the activity of the viral protein kinase pUL97. J Gen Virol. 82:1439-50.
McCampbell, A., A.A. Taye, L. Whitty, E. Penney, J.S. Steffan, and K.H. Fischbeck. 2001. Histone deacetylase inhibitors reduce polyglutamine toxicity. Proc Natl Acad Sci U S A. 98:15179-84.
Michel, D., P. Schaarschmidt, K. Wunderlich, M. Heuschmid, L. Simoncini, D. Muhl-berger, A. Zimmermann, I. Pavic, and T. Mertens. 1998. Functional regions of the human cytomegalovirus protein pUL97 involved in nuclear localization and phosphorylation of ganciclovir and pUL97 itself. J Gen Virol. 79 ( Pt 9):2105-12.
Misumi, Y., M. Sohda, A. Tashiro, H. Sato, and Y. Ikehara. 2001. An essential cytoplas-mic domain for the Golgi localization of coiled-coil proteins with a COOH-terminal membrane anchor. J Biol Chem. 276:6867-73.
Moller, A., H. Sirma, T.G. Hofmann, H. Staege, E. Gresko, K.S. Ludi, E. Klimczak, W. Droge, H. Will, and M.L. Schmitz. 2003. Sp100 is important for the stimulatory effect of homeodomain-interacting protein kinase-2 on p53-dependent gene expression. Oncogene. 22:8731-7.
Muchowski, P.J., and J.L. Wacker. 2005. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 6:11-22.
Nichol, J.N., L.A. Petruccelli, and W.H. Miller, Jr. 2009. Expanding PML's functional repertoire through post-translational mechanisms. Front Biosci. 14:2293-306.

64
Opresko, P.L., W.H. Cheng, C. von Kobbe, J.A. Harrigan, and V.A. Bohr. 2003. Werner syndrome and the function of the Werner protein; what they can teach us about the mole-cular aging process. Carcinogenesis. 24:791-802.
Orr, H.T., and H.Y. Zoghbi. 2007. Trinucleotide repeat disorders. Annu Rev Neurosci. 30:575-621.
Pampin, M., Y. Simonin, B. Blondel, Y. Percherancier, and M.K. Chelbi-Alix. 2006. Cross talk between PML and p53 during poliovirus infection: implications for antiviral defense. J Virol. 80:8582-92.
Poirier, M.A., H. Jiang, and C.A. Ross. 2005. A structure-based analysis of huntingtin mutant polyglutamine aggregation and toxicity: evidence for a compact beta-sheet struc-ture. Hum Mol Genet. 14:765-74.
Poirier, M.A., H. Li, J. Macosko, S. Cai, M. Amzel, and C.A. Ross. 2002. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J Biol Chem. 277:41032-7.
Prichard, M.N., W.J. Britt, S.L. Daily, C.B. Hartline, and E.R. Kern. 2005. Human cyto-megalovirus UL97 kinase is required for the normal intranuclear distribution of pp65 and virion morphogenesis. J Virol. 79:15494-502.
Prichard, M.N., N. Gao, S. Jairath, G. Mulamba, P. Krosky, D.M. Coen, B.O. Parker, and G.S. Pari. 1999. A recombinant human cytomegalovirus with a large deletion in UL97 has a severe replication deficiency. J Virol. 73:5663-70.
Prichard, M.N., E. Sztul, S.L. Daily, A.L. Perry, S.L. Frederick, R.B. Gill, C.B. Hartline, D.N. Streblow, S.M. Varnum, R.D. Smith, and E.R. Kern. 2008. Human cytomegalovirus UL97 kinase activity is required for the hyperphosphorylation of retinoblastoma protein and inhibits the formation of nuclear aggresomes. J Virol. 82:5054-67.
Ross, C.A. 2002. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington's disease and related disorders. Neuron. 35:819-22.
Ross, C.A., and M.A. Poirier. 2005. Opinion: what is the role of protein aggregation in neurodegeneration? Nat Rev Mol Cell Biol. 6:891-8.
Ross, C.A., M.A. Poirier, E.E. Wanker, and M. Amzel. 2003. Polyglutamine fibrillogene-sis: the pathway unfolds. Proc Natl Acad Sci U S A. 100:1-3.
Ross, C.A., J.D. Wood, G. Schilling, M.F. Peters, F.C. Nucifora, Jr., J.K. Cooper, A.H. Sharp, R.L. Margolis, and D.R. Borchelt. 1999. Polyglutamine pathogenesis. Philos Trans R Soc Lond B Biol Sci. 354:1005-11.

65
Salomoni, P., I. Guernah, and P.P. Pandolfi. 2006. The PML-nuclear body associated protein Daxx regulates the cellular response to CD40. Cell Death Differ. 13:672-5.
Sanchez, I., C. Mahlke, and J. Yuan. 2003. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature. 421:373-9.
Saudou, F., S. Finkbeiner, D. Devys, and M.E. Greenberg. 1998. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuc-lear inclusions. Cell. 95:55-66.
Shen, T., Lin, H., Scaglioni, P. Yung, T., Pandolfi, P, 2006. The mechanisms of PML-nuclear body formation. Mol Cell. 24 (3), 331-339.Sisodia, S.S. 1998. Nuclear inclusions in glutamine repeat disorders: are they pernicious, coincidental, or beneficial? Cell. 95:1-4.
Vaitiekunaite, R., D. Butkiewicz, M. Krzesniak, M. Przybylek, A. Gryc, M. Snietura, M. Benedyk, C.C. Harris, and M. Rusin. 2007. Expression and localization of Werner syn-drome protein is modulated by SIRT1 and PML. Mech Ageing Dev. 128:650-61.
Wacker, J.L., M.H. Zareie, H. Fong, M. Sarikaya, and P.J. Muchowski. 2004. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by parti-tioning monomer. Nat Struct Mol Biol. 11:1215-22.
Warrick, J.M., H.Y. Chan, G.L. Gray-Board, Y. Chai, H.L. Paulson, and N.M. Bonini. 1999. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 23:425-8.
Warrick, J.M., L.M. Morabito, J. Bilen, B. Gordesky-Gold, L.Z. Faust, H.L. Paulson, and N.M. Bonini. 2005. Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol Cell. 18:37-48.
Wetzel, R. 2006. Kinetics and thermodynamics of amyloid fibril assembly. Acc Chem Res. 39:671-9.
Zhong, X., and R.N. Pittman. 2006. Ataxin-3 binds VCP/p97 and regulates retrotranslo-cation of ERAD substrates. Hum Mol Genet. 15:2409-20.

66
THE CILIARY G PROTEIN-COUPLED RECEPTOR SOMATOSTATIN RECEPTOR
3 CYCLES BETWEEN ENDOSOMES AND CILIA THROUGH A RAB21, RAB4, AND RAB11-REGULATED PATHWAY
CRISTY TOWER-GILCHIRST, TOMASZ SZUL, EUNJOO LEE, AND ELIZABETH SZTUL
Submitted to the Journal of Molecular Biology of the Cell Format adapted for dissertation

67
ABSTRACT
The pathways and the molecular machinery that regulate delivery of G-
protein coupled receptors (GPCR) to cilia are under investigation. We show that the
GPCR somatostatin receptor 3 (SSTR3) traffics between the cilia and intracellular com-
partments identified as early endosomes by their content of Rab4, Rab5, and Rab21, and
recycling endosomes by their content of Rab11. We document that SSTR3 preferentially
segregates into the Rab21- and Rab4-containing subdomains of early endosomal mem-
branes. SSTR3 trafficking through each endosomal compartment is regulated by the cog-
nate Rab since expression of inactive mutants of Rab21, Rab4, and Rab11 inhibits
SSTR3 trafficking. Using dual color live imaging we show the formation of vesicles
containing SSTR3 from Rab21-, Rab4- and Rab11-containing endosomes and show that
these vesicles traffic directly to the base of the cilium where they fuse. The highest level
of direct delivery of SSTR3 to the cilium occurs from Rab11 endosomes. Our findings
suggest a novel route for direct delivery of ciliary proteins from endosomal compartments
to the cilium.
INTRODUCTION
Primary cilia are found on the vast majority of eukaryotic cells and are involved
in the reception and transduction of extracellular signals during mechanosensation, che-
mosensation, and vision (Christensen et al., 2007). Defects in ciliary assembly or the de-
livery of proteins to cilia underlie many human diseases collectively called ciliopathies
that include polycystic kidney disease, Bardet-Beidl Syndrome, and retinitis pigmentosa
(Pazour and Bloodgood, 2008; Marshall, 2008 ; Berbari et al., 2009 ). Cilia are formed by

68
a nine-fold bundle of doublet microtubules sheathed by a specialized subdomain of the
plasma membrane (PM) that contains ciliary proteins and excludes non-ciliary compo-
nents (Pazour and Bloodgood, 2008; Nachury et al., 2010). Cilia are formed by the coor-
dinate recruitment of soluble and transmembrane ciliary proteins. The molecular mechan-
isms that regulate assembly of cilia and mediate protein traffic within cilia have been ex-
tensively studied and the roles played by basal body (BB) and intraflagellar transport sys-
tem (IFT) components are emerging. In contrast, less is known about the trafficking
pathways and mechanisms that selectively deliver transmembrane ciliary proteins to the
base of the cilia prior to entry into the cilium.
Previous studies described membranous vesicles in the area at the base of the ci-
lium, suggesting that ciliary proteins are delivered to the base of the cilia via vesicular
carriers. In the mammalian retina, photo-transduction is mediated by a specialized prima-
ry cilium that contains the GPCR rhodopsin (Hargrave and McDowell, 1992; Berbari et
al., 2009 ). Extensive studies suggest that rhodopsin is sorted and packaged into vesicles
at the Golgi and that these vesicles are targeted to the base of cilium where they fuse
(Follit et al., 2006; Maerker et al., 2008). In addition, recent work has implicated the
trans-Golgi network (TGN) and recycling endosomes in the trafficking of rhodopsin
transport carriers to cilia in photoreceptor-rod outer segments in retinal cells (Mazelova et
al., 2009a; Mazelova et al., 2009b; Knödler et al., 2010). However not all ciliary GPCRs
may utilize the direct vesicular pathway. Smoothened, a transmembrane receptor in-
volved in hedgehog signaling (Corbit et al., 2005; Rohatgi et al., 2007) appears to deviate
from the direct pathway (vesicular transport from post-Golgi out to cilia) and is trans-

69
ported laterally from the plasma membrane to the ciliary membrane (Milenkovic et al.,
2009).
Members of the Rab family of small GTPases are known to regulate the biogene-
sis of membrane compartments and to facilitate protein movement between distinct com-
partments (Zerial and McBride, 2001). More recently, numerous Rabs have been shown
to have roles in ciliary trafficking. A screen of 39 RabGAPs for their involvement in cilia
formation in retinal pigment epithelial (RPE) cells implicated Rab8a, Rab17, and Rab23
in ciliogenesis (Yoshimura et al., 2007). Rab8 requirement was further confirmed by
showing that a dominant negative Rab8 mutant inhibits cilia formation and that the BBS1
subunit of the BBSome (an octameric complex composed of highly conserved Bardet-
Biedl syndrome proteins) associates with Rabin8. Rabin8 is the guanine nucleotide ex-
change factor (GEF) for Rab8 that promotes its activation and entry into the cilium (Na-
chury et al., 2007). The cilia-localized Rab8 may regulate ciliogenesis by promoting the
docking and fusion of vesicles bearing transmembrane proteins (Pazour and Bloodgood,
2008). Additionally, Rab3, Rab6, Rab8a, and Rab11 have been identified as components
of vesicles containing rhodopsin, suggesting that these Rabs may regulate protein trans-
port to the base of the cilium (Deretic, 1997). In addition to Rabs, two Rab-like proteins,
Rab-like5 (IFTA-2) and Rab-like4 (IFT27) are known to play a role in primary cilium
function in C. elegans (Shafer et al., 2006; Qin et al., 2007). Thus, a number of Rab and
Rab-like GTPases have emerged as key players in cilia assembly and ciliary trafficking.
Whether additional Rabs may participate in ciliary trafficking remains under investiga-
tion.

70
In this study, we used SSTR3 to provide novel insight into the trafficking path-
ways of ciliary GPCRs and to document the involvement of additional Rabs in ciliary
trafficking. Endogenous SSTR3 has been localized to neuronal cilia in many regions of
the rodent brain (Handel et al., 1999) and in cultured hippocampal neurons and also tar-
gets to cilia when expressed exogenously in inner-medullary collecting duct (IMCD)
cells (Berbari et al., 2007; Berbari et al., 2008a; Berbari et al., 2008b).
SSTR3 is regulated by its ligand somatostatin, a neuropeptide involved in neuro-
transmission and hormone secretion from the anterior pituitary gland, pancreas, and ga-
strointestinal tract (Jacobs and Schulz, 2008). Binding of somatostatin induces SSTR3
internalization from the PM via clathrin coated vesicles in a beta-arrestin 2- and adaptor
protein 2-dependent process (Tulipano et al., 2004; Jacobs and Schulz, 2008). Interna-
lized SSTR3 is delivered to endosomes containing Rab11 and endocytosed transferin,
two markers of recycling endosomes (Kreuzer et al., 2001). Additionally, internalized
SSTR3 also localizes to early endosomal compartments lacking transferrin. Upon ligand
washout, SSTR3 recycles from the endosomal compartments back to the plasma mem-
brane (Kreuzer et al., 2001; Tulipano et al., 2004). The pathway(s) taken by SSTR3 to
reach cilia after exit from the endosomal compartments is currently unknown. Protein
traffic between endosomes and the cell surface is known to be regulated by specific Rabs,
but the role of specific Rabs in SSTR3 trafficking through endosomal compartments and
delivery to cilia is unknown.
We used IMCD cells stably expressing SSTR3 to examine its trafficking and to
define the role of endosomal Rabs in ciliary delivery of SSTR3. We show that SSTR3
continuously cycles between the cilium and endosomal compartments via small vesicular

71
carriers. We identify the endosomal compartments as early endosomes based on their
content of Rab5, Rab21, and Rab4 and also recycling endosomes based on their content
of Rab11. We show that SSTR3 preferentially traffics through Rab21- and Rab4-
containing subdomains of early endosomes and largely bypasses the Rab5 subdomain.
Further, we show that SSTR3 moves rapidly through the early endosomes and undergoes
slower movement through the recycling endosome. We find that sorting of SSTR3 within
early and recycling endosomes requires the function of the cognate Rabs because expres-
sion of dominant negative forms of Rab21, Rab4, and Rab11 inhibit SSTR3 trafficking.
In addition, we show that SSTR3 can be delivered to cilia from each of these endosomal
compartments in a direct process that involves the formation of vesicles from the endo-
somal compartment, translocation to the base of the cilium and fusion therein. SSTR3 is
preferentially delivered to cilia from the Rab11-positive recycling endosome but can also
traffic directly to the cilia from the Rab21 and Rab4 early endosomal compartments. Our
findings document the existence of a direct pathway from the endosome to the base of the
cilium for trafficking of G-protein coupled receptors.
RESULTS
1. SSTR3 localizes to cilia and endosomes in IMCD cells
Kidney-derived IMCD cells readily polarize and form cilia in culture, making the
cells an ideal model system to analyze ciliary trafficking. GFP-tagged SSTR3 stably ex-
pressed in IMCD cells targets to cilia as shown by co-localization with acetylated tubulin
(Figure 1A, arrowheads) and adenylyl cyclase 3 (Supplemental Figure 1B), both estab-
lished ciliary markers. A proportion of SSTR3 can also be detected at the plasma mem-

72
brane (Supplemental Figure 1A) consistent with previous reports (Tulipano et al., 2004;
War et al., 2011). Analysis of multiple focal planes through the cell also shows localiza-
tion of SSTR3 to numerous punctate structures dispersed throughout the cell (Figure 1B).
The size and shape of these structures is consistent with endosomal compartments; there-
fore we assessed the nature of these compartments by determining their Rab content.
Rab GTPases show exquisite selectivity in their subcellular localization making
them ideal compartment markers. SSTR3 expressing cells were transfected with tagged
Rabs and the level of co-localization between SSTR3 and each Rab was measured.
SSTR3 was detected in a proportion of early endosomes marked with Rab5 (Figure 1C).
The quantitation of SSTR3 co-localization with Rab5 was assessed by Pearson’s coeffi-
cient (where a 0 value indicates no co-localization and a value of 1 indicates complete co-
localization) and revealed a relatively low coefficient of .512, suggesting that SSTR3
might bypass or does not remain within many of the Rab5-containing structures (Figure
1H). In addition to Rab5, early endosomes also contain Rab21 (Simpson et al., 2004;
Zhang et al., 2006). Interestingly, Rab21 contains an unorthodox X-box (a transcriptional
motif present in multiple ciliary proteins), suggesting that Rab21 might be found in the
cilium and possibly involved in trafficking of ciliary proteins (Efimenko et al., 2006).
SSTR3 extensively co-localized with Rab21 (Figure 1D), with a co-localization coeffi-
cient of 0.823 (Figure 1H). Early endosomes are dynamic structures that remodel their
membrane by the loss of Rab5 and recruitment of Rab4 (van der Sluijs et al., 1992; Daro
et al., 1996; Spang, 2009). SSTR3 exhibited excellent co-localization with Rab4 (Figure
1E). The majority of SSTR3-containing structures show extensive co-localization with

73
Rab4. Quantification indicates a correspondingly high co-localization coefficient of 0.861
(Figure 1H).
Early endosomes differentiate into late endosomes by the loss of Rab5 and Rab4
and recruitment of Rab7 (Dunn and Maxfield, 1992; Rink et al., 2005; Vonderheit and
Helenius, 2005; Poteryaev et al., 2010). SSTR3 shows almost no co-localization with
Rab7 (Figure 1F and 1H), with a co-localization coefficient of 0.027. Rab7 positive struc-
tures define a degradative pathway destined to fuse with lysosomes; the miniscule levels
of SSTR3 in these endosomes suggest that under steady state only a small proportion of
SSTR3 is destined for degradation. Previous data show that following internalization only
a small fraction of SSTR3 is targeted for degradation in non-polarized RIN cells and
HEK 293 cells (Roosterman et al., 1997; Kreuzer et al., 2001; Tulipano et al., 2004; Ja-
cobs and Schulz, 2008; Roosterman et al., 2008). The low levels of SSTR3 in the Rab7
pathway suggest that the vast majority of SSTR3 recycles.
To assess whether SSTR3 is found in recycling endosomes, we analyzed coloca-
lization with Rab11, a previously characterized marker of recycling endosomes (Satoh et
al., 2005). Strikingly, SSTR3 shows extensive co-localization with Rab11 (Figure 1G),
with the highest co-localization coefficient of 0.910 (Figure 1H). Thus, SSTR3 localizes
extensively to Rab11-containing recycling endosomes as well as Rab21 and Rab4 early
sorting endosomes. The localization of SSTR3 to endosomal compartments known to be
involved in recycling to the plasma membrane raised the possibility that SSTR3 may traf-
fic through early and recycling endosomes in a process regulated by these Rabs.
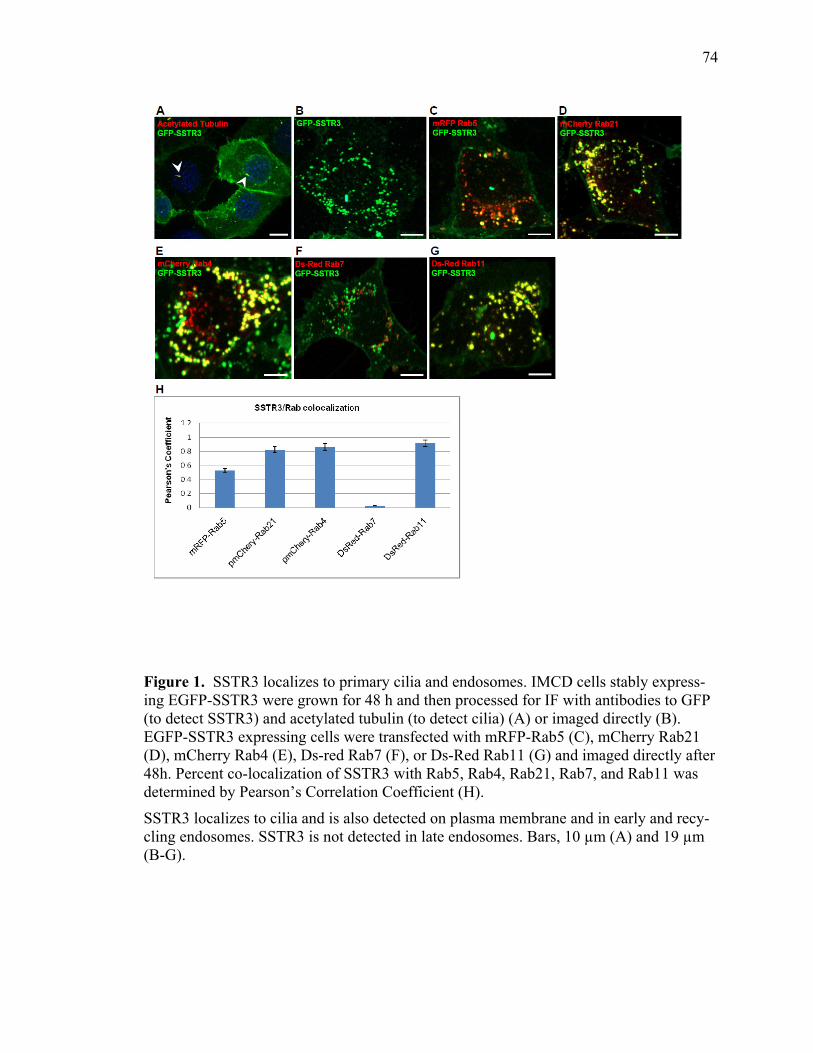
74
Figure 1. SSTR3 localizes to primary cilia and endosomes. IMCD cells stably express-ing EGFP-SSTR3 were grown for 48 h and then processed for IF with antibodies to GFP (to detect SSTR3) and acetylated tubulin (to detect cilia) (A) or imaged directly (B). EGFP-SSTR3 expressing cells were transfected with mRFP-Rab5 (C), mCherry Rab21 (D), mCherry Rab4 (E), Ds-red Rab7 (F), or Ds-Red Rab11 (G) and imaged directly after 48h. Percent co-localization of SSTR3 with Rab5, Rab4, Rab21, Rab7, and Rab11 was determined by Pearson’s Correlation Coefficient (H).
SSTR3 localizes to cilia and is also detected on plasma membrane and in early and recy-cling endosomes. SSTR3 is not detected in late endosomes. Bars, 10 µm (A) and 19 µm (B-G).

75
2. SSTR3 traffic is regulated by Rab21
To assess the role of Rab21 in trafficking of SSTR3, we analyzed SSTR3 traffick-
ing through Rab21-containing endosomes by two-color live imaging (Figure 2A). Analy-
sis of multiple SSTR3/Rab21 endosomes shows them to be extremely dynamic and un-
dergo continuous movements between the cell center and periphery (Movie 1). In addi-
tion, analysis of consecutive movie frames shows that SSTR3/Rab21 endosomes conti-
nuously fuse and fragment. This is consistent with previous describing showing the dy-
namic nature of early endosomes. Careful analysis of SSTR3 co-localization with Rab21
shows clearly separate subdomains containing only SSTR3 (green) or only Rab21 (red) in
continuity with regions containing both proteins (yellow) (Figure 2A, inset, arrowheads).
Such segregation suggested that SSTR3 moves through the Rab21 endosome. Indeed we
observed the fusion of elements containing only SSTR3 with Rab21-containing endo-
somes (Figure 2B, arrowheads). We also detected the formation of SSTR3-containing
elements from Rab21-labeled endosomes (Figure 2C arrowheads). Segregation of cargo
proteins within early endosomes has been observed previously (Sönnichsen et al., 2000;
Barbero et al., 2002; Stenmark, 2009). However, sorting of GPCRs within early endo-
somal sub-domains has not been previously described (Jean-Alphonse and Hanyaloglu,
2011).
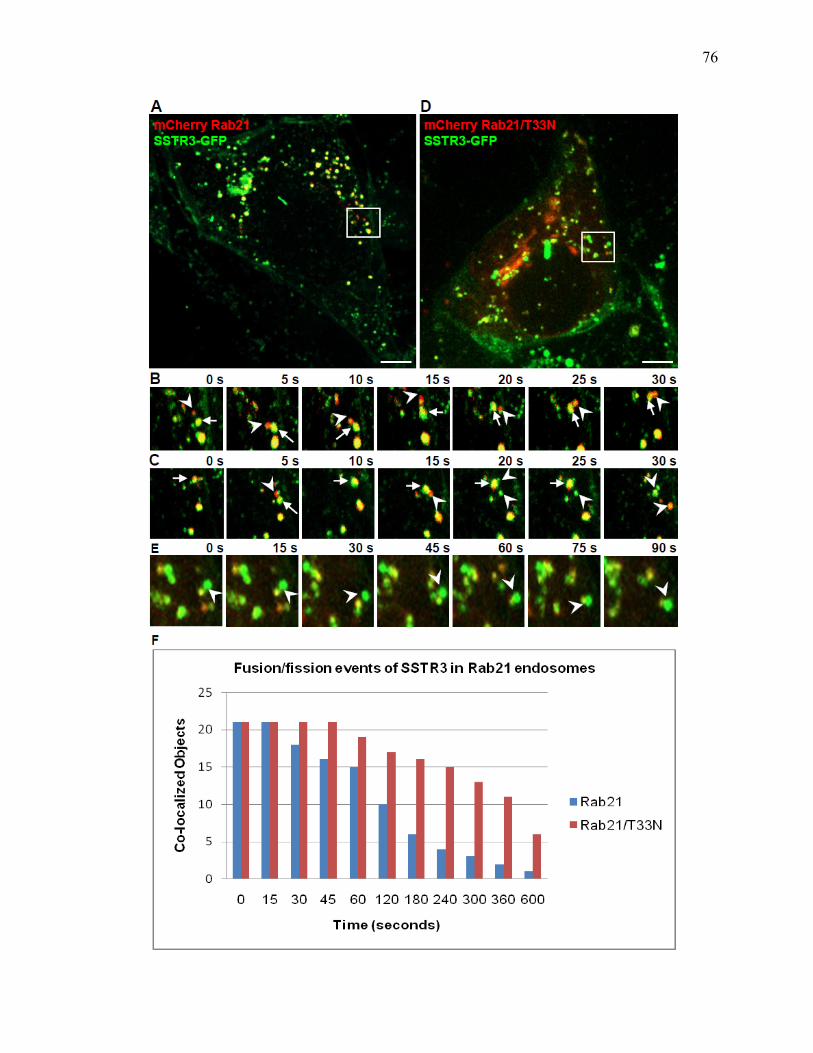
76

77
Figure 2. SSTR3 trafficking is regulated by Rab21. EGFP-SSTR3 expressing IMCD cells were transfected with mCherry-Rab21 (A-C) or mCherry-Rab21/T33N inactive mu-tant (D-E). After 48 hours cells were imaged and a series of consecutive images focusing on cell regions outlined in A and D is presented. Box in A and C outlines regions of A and C depicted in B and D. Arrows denote SSTR3-containing Rab21 or Rab21/T33N-positive endosomes while arrowheads point to fusion or fission events (B,C, E). Fusion and fission events were quantified by analyzing 21 randomly selected SSTR3/Rab21 or SSTR3/Rab21/T33N endosomes and measuring the co-localization coefficient between SSTR3 and Rab21 or Rab21/T33N as a function of time (F). SSTR3 rapidly sorts out of Rab21-containing endosomes but is retained within Rab21/T33N-containing endosomes. Bars, 19 µm.
To provide a measure of SSTR3 passage through the Rab21 endosome we per-
formed a time-dependent co-localization coefficient analysis. We analyzed 21 individual
endosomes over 10 minutes and determined the SSTR3 and Rab21 co-localization coeffi-
cient of each structure every 15 seconds. The initial co-localization level for each struc-
ture was set at 1 and we counted endosomes as no longer having SSTR3 when their co-
localization coefficient fell below 0.100. Our rationale for this approach was that sorting
and exit of SSTR3 (green) from the Rab21 endosome should cause a measurable decrease
in the co-localization coefficient. Indeed, SSTR3 rapidly segregated from Rab21, and
more than half of Rab21 endosomes lost SSTR3 within 120 seconds (Figure 2F). The ma-
jority of SSTR3 protein sorted out from the parent endosome within 360 seconds. This
observation suggests that SSTR3 rapidly moves through Rab21 endosomes.
Kinetic analysis of SSTR3 behavior in control cells defined the baseline for as-
sessing the function of Rab21 in SSTR3 trafficking. Like all small GTPases, Rab21
cycles between the active GTP-bound and the inactive GDP-bound form. The
Rab21/T33N mutant assumes a conformation analogous to that of the inactive Rab21 and
acts in a dominant negative manner when expressed in cells (Simpson et al., 2004). Cells

78
expressing Rab21/T33N have defects in endocytosis of epidermal growth factor and
transferrin receptors and fail to deliver both receptors to endosomes and lysosomes for
degradation (Simpson et al., 2004). In addition, Rab21/T33N inhibits endocytosis and
recycling of integrins and cell-extracellular matrix adhesion proteins (Simpson et al.,
2004; Pellinen et al., 2006; Pellinen et al., 2008).
Expression of Rab21/T33N in our system decreased the level of SSTR3 co-
localization in membrane structures scattered throughout the cell (Figure 2D). Many of
these structures exhibit regions containing only SSTR3, and these domains appear rela-
tively stable and don't undergo extensive fusions or fissions (Figure 2E, arrowheads).
This observation suggested that Rab21/T33N may inhibit movement of SSTR3 through
Rab21/T33N-positive endosomes. Indeed, when Rab21/T33N endosomes were moni-
tored in live cells, SSTR3 (green) sub-domains exhibited limited budding (Figure 2E, ar-
rows). Quantitation of SSTR3 movement through the Rab21/T33N endosome shows that
SSTR3 remains within Rab21/T33N structures for a prolonged time period (Figure 2F).
SSTR3 is still detected in more than half of Rab21/T33N endosomes even after 360
seconds, at a time when SSTR3 is almost completely cleared from Rab21 wild-type en-
dosomes. Thus, it appears that SSTR3 trafficking through early endosomes requires func-
tional Rab21.
3. SSTR3 traffic is regulated by Rab4
SSTR3 trafficking through Rab4-containing endosomes was also assessed by two-
color live imaging (Figure 3A). Similar to Rab21-positive endosomes, SSTR3/Rab4 en-
dosomes are extremely dynamic and undergo continuous fusions and fissions (Movie 2),
consistent with previous reports (Daro et al., 1996; McCaffrey et al., 2001). SSTR3 ap-

79
pears to continuously sort within Rab4 endosomes as evidenced by dynamic separation of
small SSTR3-containing elements continuous with membranes containing Rab4 (Figure
3A, inset). These elements may represent vesicles that either fuse with or bud from Rab4-
containing endosomes. We probed SSTR3 passage through the Rab4 endosomes by ex-
amining SSTR3 localization relative to Rab4-containing elements. We routinely observed
the separation of a Rab4 element from a structure containing Rab4 and SSTR3 (Figure
3B arrowheads). Often it appeared as if the same endosome gave rise to numerous Rab4-
positve elements from its edges in rapid succession. We also observed many instances of
SSTR3/Rab4 endosomes giving rise to elements containing exclusively SSTR3 (Figure
3B, arrows). Often, numerous SSTR3-containing elements were seen forming from a sin-
gle endosome at the same time (Figure 3B, open arrowheads).
The kinetics of SSTR3 passage through the Rab4 endosomes were determined by
measuring the co-localization coefficient of SSTR3 and Rab4 in 25 individual endosomes
every 15 seconds over 6 minutes. SSTR3 rapidly segregated from Rab4 endosomes and
more than half of Rab4 endosomes lost SSTR3 within 60 seconds (Figure 3E). SSTR3
was almost completely sorted out from the parent endosome within 240 seconds. These
data suggests that SSTR3 rapidly moves through Rab4 endosomes. SSTR3 passage
through Rab4 endosomes appears significantly faster than through Rab21 endosomes.

80

81
Figure 3. SSTR3 trafficking is regulated by Rab4. EGFP-SSTR3 expressing cells were transfected with mCherry Rab4 (A) or mCherry Rab4/S22N inactive mutant (C). After 48 hours cells were imaged and a series of consecutive images focusing on cell regions out-lined in A and C is presented. Box in A and C outlines regions of A and C depicted in B and D. Arrows denote SSTR3-containing Rab4 or Rab4/S22N -positive endosomes while arrowheads point to fusion or fission events (B, D). Fusion and fission events were quan-tified by analyzing 25 randomly selected SSTR3/Rab4 or SSTR3/Rab4/S22N endosomes and measuring the co-localization coefficient between SSTR3 and Rab4 or Rab4/S22N as a function of time (E). SSTR3 rapidly sorts out of Rab4-containing endosomes but is re-tained within Rab4/S22N-containing endosomes. Bars, 19 µm.
To determine the role of Rab4 in SSTR3 trafficking we expressed the Rab4/S22N
mutant that acts in a dominant negative manner to inhibit endosomal trafficking of MHC
class I-related receptor, neonatal Fc (Bucci et al., 1992; Bucci et al., 1994; Ward et al.,
2005), and transferrin receptor (van der Sluijs et al., 1992). SSTR3 localizes to numerous
elements containing Rab4/S22N, but many of these elements show extensive regions con-
taining only SSTR3 (Figure 3C, inset). This observation suggested that Rab4/S22N might
inhibit exit of SSTR3 from endosomes. Indeed, SSTR3 (green) sub-domains did not sepa-
rate from Rab4/S22N-containing endosomes (Figure 3D, arrows). Even after 120
seconds, a bud containing sorted SSTR3 didn't disconnect from the globular endosome.
Quantitation of SSTR3 movement through the Rab4/S22N endosome shows that SSTR3
remains within Rab4/S22N structures for prolonged time periods (Figure 3E). SSTR3 is
still detected in more than half of Rab4/S22N-positive endosomes after 360 seconds, at a
time when SSTR3 is completely cleared from Rab4 wild-type endosomes. Thus, SSTR3
trafficking through early endosomes appears to require functional Rab4.
4. SSTR3 traffic is regulated by Rab11

82
SSTR3 trafficking through Rab11-containing endosomes was assessed by two-
color live imaging (Figure 4A). Live imaging of SSTR3/Rab11 endosomes shows them
to be dynamic and undergo rapid short- and long-range movements (Figure 4A and Mov-
ie 3). Fusion of SSTR3/Rab11 endosomes with other Rab11/SSTR3 endosomes and their
fragmentation are often observed.
SSTR3 and Rab11 show sub-domain segregation, with SSTR3 only (green) re-
gions and Rab11-only (red) regions in continuity with SSTR3/Rab11 (yellow) elements
(Figure 4A, inset). SSTR3 traffics extensively through Rab11 compartments, as shown by
multiple sorting events of SSTR3 out of Rab11 endosomes (Figure 4B). In the series
shown, fission of SSTR3 from Rab11 endosomes at 60, 90, and 105 seconds can be ob-
served (Figure 4B and C, arrow heads). SSTR3 passage through Rab11 endosomes was
then assessed by kinetic co-localization measurements. As shown in Figure 3E, SSTR3
rapidly clears from parent Rab11 endosomes with more than half of the original Rab11
endosomes having lost SSTR3 within 180 seconds. This rate is slightly slower than
SSTR3 passage through Rab21 and Rab4 endosomes. The slower SSTR3 movement is
consistent with previous data showing slower sorting in recycling endosomes and faster
sorting in early endosomal compartments (Sheff et al., 1999; Maxfield and McGraw,
2004; Ward et al., 2005).
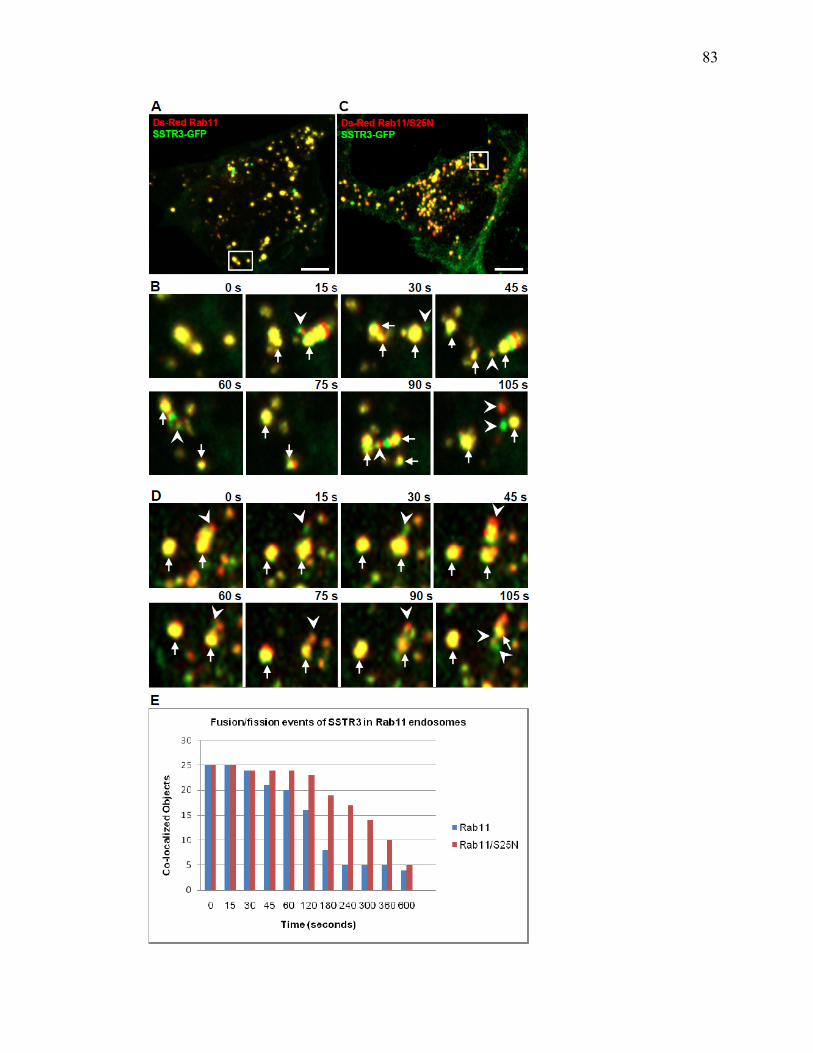
83

84
Figure 4. SSTR3 trafficking is regulated by Rab11. EGFP-SSTR3 expressing cells were transfected with DsRed Rab11 (A, B) or DsRed Rab11/S25N inactive mutant (C, D). Af-ter 48 hours cells were imaged and a series of consecutive images focusing on cell re-gions outlined in A and C is presented. Box in A and C outline regions of A and C de-picted in B and D. Arrows denote SSTR3-containing Rab11 or Rab11/S25N -positive endosomes while arrowheads point to fusion or fission events (B, D). Fusion and fission events were quantified by analyzing 25 randomly selected SSTR3/Rab11 or SSTR3/Rab11/S25N endosomes and measuring the co-localization coefficient between SSTR3 and Rab11 or Rab11/S25N as a function of time (E). SSTR3 rapidly sorts out of Rab11-containing endosomes but is retained within Rab11/S25N -containing endosomes. Bars, 19 µm.
The role of Rab11 in SSTR3 trafficking was explored by expressing the dominant
negative Rab11/S25N that inhibits trafficking of transferrin receptor (Choudhury et al.,
2002) and neonatal Fc receptor (Ward et al., 2005). Expression of Rab11/S25N leads to
slight fragmentation of recycling endosomes (Figure 4C). Kinetic analysis indicates that
SSTR3 sorts out of Rab11/S25N endosomes and budding of SSTR3 elements from
Rab11/S25N endosomes can be observed (Figure 4D, arrowheads), but that the number
of SSTR3 elements budding from Rab11/S25N endosomes is decreased. More than half
of Rab11/S25N endosomes still contain SSTR3 after 400 seconds, while the vast majority
of Rab11 wild-type endosomes lose SSTR3 within 180 seconds (Figure 4E). Thus,
SSTR3 traffic through Rab11/S25N endosomes is inhibited, suggesting that trafficking of
SSTR3 requires functional Rab11.
5. SSTR3-containing vesicles traffic to cilia
Previous electron microscopic analyses described membranous vesicles in the
area at the base of the cilium, suggesting that ciliary proteins are delivered to the base of
the cilia via vesicular carriers (Molla-Herman A, 2010). Movement of SSTR3 to cilia was
tracked in live cells focusing on the region of the cell containing the cilium. As shown in

85
Figure 5A, SSTR3 is detected in the cilium and in punctate structures immediately adja-
cent to the ciliary base. SSTR3 traffics extensively between these compartments through
tubules protruding from the punctate structures and smaller vesicles that originate from
these structures (Figure 5B and 5C). .
Figure 5. SSTR3-containing vesicles traffic directly to cilia. IMCD cells were trans-fected with EGFP-SSTR3-GFP and cultured for 24 hours to allow cilia formation and then imaged. A series of consecutive images focusing on cell regions outlined in A is pre-sented at different times starting at time 0 (panels in B) and starting 120 seconds later (panels in C). A vesicle containing SSTR3 moves from a larger structure to the base of the cilium where it fuses (B, yellow arrows). A vesicle containing SSTR3 buds from the base of the cilium and moves into the cell center (C, yellow arrows). Bar, 19 µm.
Significantly, numerous small vesicles are seen ferrying SSTR3 to and from the cilium
and in the short time span of 90 seconds, more than six SSTR3-containing vesicles were
observed moving to and from the cilium in this particular cell (Supplemental movie 4).
Traffic of SSTR3 to the cilium can be observed in the top panel, where a vesicle contain-
ing SSTR3 moves from the periphery to the vicinity of the cilium and fuses at the base of
the cilium (Figure 5B, yellow arrow). A single event of SSTR3 traffic from the cilium is

86
depicted in Figure 5C where a vesicle containing SSTR3 buds from the base of the cilium
at 6 seconds, is completely detached by 24 seconds and moves toward cell center at 36
seconds. The detection of vesicles carrying SSTR3 directly to the cilium raised the possi-
bility that these vesicles may originate from endosomes. Thus, we assessed the origin of
the SSTR3-carrying vesicles by monitoring the Rab content of the parental compartment.
6. Multiple endosomal compartments give rise to SSTR3-containing ciliary vesicles
We used two-color live imaging to simultaneously monitor SSTR3 and each Rab
implicated in SSTR3 trafficking as described above. We tracked SSTR3- and Rab-
containing endosomes in 10 cells over 10 minutes to quantify the number of cells in
which SSTR3-containing vesicles budded from a Rab endosomes and directly reached
the cilium. The rationale for this approach, rather than quantitating the number of fusions
within a single cell, was that the movement of SSTR3 vesicles to the ciliary base was
very dynamic and it was impossible to quantitate the number of fusion events occurring
within the cell. Thus, we decided to score the number of cells in which a fusion event oc-
curred within a defined time interval. SSTR3/Rab21 endosomes were often observed
moving from the periphery of the cell to the base of the cilium (Figure 6A). Often, such
endosomes give rise to SSTR3 vesicles that move to the base of the cilium and fuse there-
in (arrows). Quantitative analysis of SSTR3 ciliary delivery from Rab21 endosomes re-
vealed that in 7 out of the 10 cells examined, SSTR3 vesicles budded from SSTR3/Rab21
structures and fused at the base of the cilium (Figure 7B).
Similarly, SSTR3/Rab4 endosomes were seen to move into close proximity to the
base of the cilium. Seconds later, an SSTR3-containing vesicle budded from one endo-
some and fused at the base of the cilium (Figure 6B, arrows). Of the 10 cells examined, 8

87
cells contained SSTR3/Rab4 endosomes that moved to the base of the cilium and gave
rise to SSTR3 vesicle that then fused with the base of the cilium (Figure 7B).
Figure 6. SSTR3-containing vesicles traffic from Rab21- and Rab4-containing endo-somes directly to cilia. EGFP-SSTR3 expressing cells were transfected with mCherry-Rab21 (A) or mCherry Rab4. After 72 h, cells were treated with HEPES buffer and live cell imaging was performed. Single frames from the corresponding time sequence shown with the first frame set to zero. The arrow depicts single Rab21/SSTR3-containing (A) or Rab4/SSTR-containing endosome (B). Rab21-positive endosome containing SSTR3 moves to the base of the cilium where it fuses (A). Also a Rab4 positive endosome con-taining SSTR3 moves to the base of the cilium and SSTR3-only containing vesicle buds off and traffics to the cilium (B). Bars, 19 µm.
Movie sequences of SSTR3/Rab11 endosomes show these structures moving
from the cell periphery to the base of the cilium where they gave rise to SSTR3 vesicles
that move to and fuse at the base of the cilium (Figure 7A). Formation of SSTR3 vesicles
from Rab11 endosomes was observed in 9 of the 10 cells analyzed (Figure 7B). Our find-
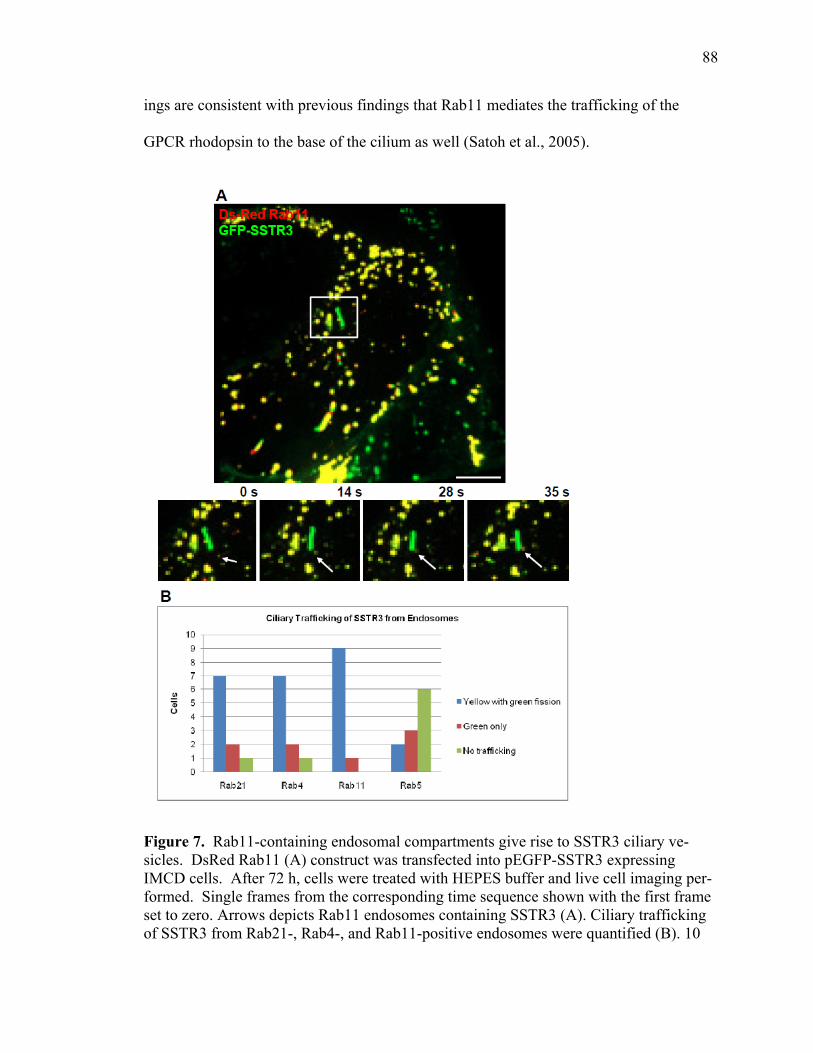
88
ings are consistent with previous findings that Rab11 mediates the trafficking of the
GPCR rhodopsin to the base of the cilium as well (Satoh et al., 2005).
Figure 7. Rab11-containing endosomal compartments give rise to SSTR3 ciliary ve-sicles. DsRed Rab11 (A) construct was transfected into pEGFP-SSTR3 expressing IMCD cells. After 72 h, cells were treated with HEPES buffer and live cell imaging per-formed. Single frames from the corresponding time sequence shown with the first frame set to zero. Arrows depicts Rab11 endosomes containing SSTR3 (A). Ciliary trafficking of SSTR3 from Rab21-, Rab4-, and Rab11-positive endosomes were quantified (B). 10

89
cells were analyzed for delivery of SSTR3 from endosomes to cilia throughout the dura-tion of a specified time series. Bars, 19 µm. Ciliary vesicles originate from Rab11 posi-tive endosomes, move to the base of the cilium and fuse (A). Ciliary vesicles originated more often from Rab11-positive endosomes than from other endosomes assessed, includ-ing SSTR3 traffic from Rab21 and Rab4-positive endosomes (B).
Taken together, our data suggests that SSTR3 is trafficked in a direct route from
endosomes to the primary cilium. SSTR3 trafficking to cilium occurs from Rab11 recy-
cling endosomes and also from Rab21 and Rab4 early endosomes.
Discussion
Trafficking of SSTR3 from endosomes to cilia
This study documents real time trafficking of a cilia receptor (SSTR3) contained
in vesicles budding from early and recycling endosomes and fusing at the ciliary base. In
our work, we show that early endosomes characterized by their content of Rab21 and
Rab4 and recycling endosomes containing Rab11 mediate large amount of SSTR3 trans-
port to cilia. Inactivation of Rab21, Rab4, and Rab11 by expression of dominant inactive
mutants of each Rab inhibited SSTR3 sorting from the cognate endosome, consistent
with multiple pathways of SSTR3 delivery to the cilium. Our data suggest that SSTR3 is
able to be trafficked to cilia from multiple endosomal compartments and from subdo-
mains of early endosomes through events mediated by distinct Rabs (Stenmark, 2009).
Role of Rab11 in ciliary trafficking
Rhodopsin transport to rod outer segments requires the small GTPase Rab11 (also
Rab6 and Rab8) (Deretic and Papermaster, 1991; Deretic, 1997; Satoh et al., 1997; Mo-
ritz et al., 2001; Yoshimura et al., 2007; Boehlke et al., 2010). Endogenous Rab11 can be

90
found at the base of cilia in RPE cells and appears essential since depletion of endogen-
ous Rab11 causes loss of cilia and expression of a dominant negative Rab11 leads to
formation of shorter cilia (Knödler et al., 2010).
We show that SSTR3 traffics through the Rab11 recycling endosome and that
Rab11 endosomes give rise to SSTR3-containing vesicular carriers that directly fuse at
the base of cilia. Rab11 has been shown to mediate protein transport from recycling en-
dosomes to the plasma membrane and to the TGN (Stenmark, 2009). Our findings add
another destination for Rab11-derived carriers - directly to the base of the cilium.
A molecular link between Rab11, Rab8, and the BBSome has been recently de-
fined. Rab11 localized to base of cilium forms a complex with Rabin8, the GEF for Rab8,
(Knödler et al., 2010) to activate Rab8 and promote Rab8 association with the BBS1
component of the BBSome. This Rab11-Rab8-BBsome cascade is proposed to mediate
docking and fusion at the ciliary base for ciliary proteins transported from the recycling
endosome to the cilium. It is likely that this cascade is involved in SSTR3 traffic to the
cilium, based on the requirement for Rab11 (this study) and the BBSome (Jin et al.,
2010) in SSTR3 trafficking. Previous studies described the BBSome as being able to
form a coat and recognize the ciliary sorting signal in SSTR3. The possible relationship
between the BBSome coat and the additional Rabs described in this study (Rab4 and
Rab21) remains to be determined. Similarly, the downstream effector proteins of Rab21,
Rab4, and Rab11 that mediate SSTR3 trafficking to cilia remain to be identified.
It is possible that ciliary proteins other than rhodopsin and SSTR3 also utilize the
Rab11 endosomal pathway and that the recycling endosome represents a general "hold-
ing" compartment for ciliary proteins. The level of receptors within cilia at any given

91
time has to be tightly regulated and it is possible that recycling endosomes represent a
reservoir for holding ciliary proteins. The Rab11 compartment has been shown to
represent a reservoir for proteins such as transferrin receptor that are internalized from
non-ciliary subdomains of the plasma membrane. Our data suggest that it may also
represent a reservoir for ciliary proteins.
Rab21 involvement in ciliary trafficking
Many plasma membrane proteins recycle from early endosomes in so-called rapid
recycling pathway. In this study we document that SSTR3 traffics from Rab21- and
Rab4-containing endosomes directly to cilia. Our findings provide a novel function for
Rab21 in trafficking of ciliary GPCRs. Rab21 was not identified as a Rab affecting cili-
ogenesis in a screen of different Rab GTPases (Yoshimura et al., 2007) nor has the func-
tion of Rab21 in trafficking of rhodopsin or other ciliary proteins been examined. Rab21
has been shown to localize to early endosomes (Simpson et al., 2004) and functions in
endocytosis of integrins and cell-extracellular matrix adhesion proteins (Pellinen et al.,
2006). The activity of Rab21 is regulated by the VPS9-ankyrin-repeat protein (VARP)
that acts as a GEF for Rab21 and activates Rab21 by facilitating GDP/GTP exchange.
Depletion of cellular VARP leads to deactivation of Rab21 and disrupts endosome dy-
namics (Zhang et al., 2006). Similarly, expression of the inactive Rab21/T33N mutant
inhibits endosomal trafficking of transferrin (Simpson et al., 2004). Here, we show that
the Rab21 endosomes traffic ciliary SSTR3 and that active Rab21 is required for sorting
of SSTR3 from the Rab21 endosome. In some preliminary experiments we observed that
expression of the dominant inactive Rab21/T33N in cells resulted in shortened cilia when
compared to wild type Rab21 expressing cells (data not shown). These preliminary find-

92
ings suggested that cells expressing inactive Rab21 may have shorter cilia and suggested
a possible role for Rab21 in cilia assembly. However, subsequent studies didn't show
consistent changes in ciliary length and we decided not to pursue this area of research
further. The available data don't allow a statistically significant conclusion to be drawn
on the role of Rab21 in ciliary assembly.
Rab proteins show spatial and temporal separation within subdomains of single
endosomal compartments (Sönnichsen et al., 2000; de Renzis et al., 2002; Stenmark,
2009) and Rab21 only partially co-localizes with Rab5 (Simpson et al., 2004; Efimenko
et al., 2006). We show that SSTR3 co-localizes much better with Rab21 than with Rab5
within early endosomes. The molecular mechanisms that govern SSTR3 preferential pas-
sage through the Rab21 subdomain remain to be investigated. SSTR3 also localized to
the Rab4-containing subdomain of early endosomes. Whether SSTR3 traffics through
Rab21- and Rab4-containing compartment sequentially or partitions into both at the same
time is unknown. However, SSTR3 sorting and exit from both compartments requires
active forms of Rab21 and Rab4.
The relationships between Rabs and other trafficking regulators in SSTR3 deli-
very to cilia are largely unknown. Recent studies suggest that SSTR3 localization to cilia
is mediated by BBS proteins since SSTR3 is not found in cilia of hippocampal neurons
from BBS2-/- and BBS4-/- mice (Berbari et al., 2008b). SSTR3 has been shown to bind
the BBS2 subunit of the BBSome, thus implicating the BBSome in SSTR3 transport to
cilia (Jin et al., 2010). However, the subcellular compartment in which the BBSome as-
sociates with SSTR3 to mediate its ciliary trafficking hasn't been identified. BBSome
components have been localized to the base of the cilium. One can speculate that SSTR3

93
traffics from Rab21 and Rab4 early endosomes and Rab11 recycling endosomes by ve-
sicles that bud from those compartments and move to the base of the cilium where
SSTR3 becomes associated with the BBSome that then mediates SSTR3 transport across
the diffusion barrier into the cilium.
This paper describes new cilia trafficking regulators: Rab21and Rab4 while con-
firming the role of Rab11. It will be interesting to determine whether Rab21, Rab4, and
Rab11 may be affected in ciliopathies. While Rabs have not been identified as ciliopathy
genes, they have been shown to regulate many aspects of cilia formation through their
role in vesicle trafficking, membrane fusion at the ciliary base, and the regulation of in-
traflagellar transport (Li and Hu, 2011). Interestingly, other small GTPases potentially
involved in trafficking have been linked to ciliopathies. For example, ARL13B has been
implicated in Joubert Syndrome (Caspary et al., 2007; Cantagrel et al., 2008). Another
potential candidate is Rabl5 shown to play a role in flagella formation of trypanosome
(Adhiambo et al., 2009). Thus, it is likely that mutations in other small GTPases involved
in ciliary trafficking may cause ciliopathies. Understanding the cellular machinery in-
volved in ciliary trafficking will not only provide potentially novel therapeutic targets for
the treatment of rare genetic ciliopathy syndromes but also some of their more common
clinical features such as obesity, blindness, anosmia or cognitive deficits.
MATERIALS AND METHODS
Antibodies and Reagents
Polyclonal anti-GFP antibody was purchased from Abcam (catalogue # ab290-50; Cam-
bridge, MA). Monoclonal anti-tubulin acetylated was purchased from Sigma (catalogue#

94
T6793; Saint Louis, Missouri). Anti-adenyl cyclase III (C-20) (catalogue # sc-588) polyc-
lonal antibody was purchased from Santa Cruz (Santa Cruz, CA). Secondary antibodies
used in immunofluorescence were Alexa Fluor 594-labeled goat anti-rabbit, Alexa Fluor
488-labeled goat anti-mouse, and Hoechst 33258 were all from Invitrogen Molecular
Probes, Inc (Eugene, OR). Clones, primer sequences, and PCR conditions are available
on request. Geneticin (G418) was purchased from Gibco/Invitrogen Corporation (cata-
logue number 11811-031; Carlsbad, CA) and was used in cell culture media to keep cells
under selection.
DNA Constructs
Construction of the pEGFP-SSTR3 plasmid was from Dr. Gregory Pazour (University of
Massachusetts Medical School Worcester, MA). Constructs pEGFP-Rab21 wild-type,
Rab21/T33N, and Rab21/Q78L were obtained from Dr. Jeremy Simpson (UCD Dublin
Conway Institute of Biomolecular & Biomedical Research (Simpson et al., 2004) and
subcloned into pmCherry-C1 (Clonetech) using BgIII and SacII. The primers employed
to generate pmCherry-C1-tagged Rab21 wild-type, Rab21/T33N, and Rab21/Q78L were
5’-GATCGAAGATCTATGCGACGACGA-3’ and 5’-
GATCCCGCGGTTATCCAGAAGA-3’. DNA sequencing was performed by the UAB
Genomics Core Facility of the Heflin Center for Human Genetics to confirm insertion
into pmCherry-C1 vector. The construct pmCherry-C2-Rab4a wild-type was a gift from
Dr. James Goldenring. To generate the point mutant Rab4/S22N dominant negative con-
struct site-directed mutagenesis by polymerase chain reaction of the Rab4a wild-type
construct was performed using primers 5’-
GGAAATGCAGGAACTGGCAAGAATTGCTTACTTCATCAGTTTATT-3’ and 5’-

95
CAATAAACTGATGAAGTAAGCAATTCTTGCCAGTTCCTGCATTTCC-3’. The
clone was verified by sequencing. Constructs DsRed-Rab11wild-type, DsRed-
Rab11/S25N, mRFP-Rab5wild-type, and DsRed-Rab7 were from Addgene “Addgene
plasmids 12679, 12680, 14437, and 12661 respectively” and previously described
(Choudhury et al., 2002; Vonderheit and Helenius, 2005).
Cell culture and transfections
Mouse kidney IMCD3 cells were grown in Dulbecco’s modified of Eagles Me-
dium/Ham’s F12 50/50 (Mediatech, Comprehensive Cancer Center of the University of
Alabama at Birmingham, AL ) supplemented with 10% fetal bovine serum, 100 unit/ml
penicillin/streptomycin, 1.2 g/L sodium bicarbonate, and 0.5 mM sodium pyruvate (Invi-
trogen). Stable IMCDs over expressing EGFP-SSTR3 clone 1 (SSTR3#1 IMCD-3) were
a gift from Dr. Bradley Yoder (University of Alabama at Birmingham) and cultured in
same media as described above but supplemented with 200ug/ml of Geneticin to keep the
cells under selection. All cells were grown in an incubator with 95% air in the atmos-
phere and 5% carbon dioxide (CO2) at 37°C. For transfections cell were electroporated
using BioRad Gene pulser II (Hercules, CA) and the electroporation conditions for a
4mm cuvette (Fisher, Freemont, CA ) was a voltage set to 320 with a capacitance of 950.
Immunofluorescence microscopy and live cell imaging
Immunofluorescence was performed as described previously (Tower et al., 2011). For
live cell imaging, SSTR3#1 IMCD3 cells were grown in 10-cm plates until 100% conflu-
ence was reached then the cells were trypsinized, washed in 10 ml of media, and spun at
2000 rpm for 4 min. The cells were resuspended in 400ml of cytomix (120 mM KCl; 0.15
mM CaCl2; 10mM K2HPO4/KH2PO4, 2.5 mM Hepes, 2 mM EGTA, 5mM MgCl2; pH

96
adjusted with KOH to pH 7.6) and supplemented with fresh 2 mM ATP and 5mM gluta-
thione before adding cytomix to cells with 10-20ug DNA. Cells (n= 5x106) were electro-
porated as previously described (Berbari et al., 2008a), transferred to glass bottom plates
(Warner Instruments) and allowed to grow for 48 to 72 hours. Time-lapse confocal imag-
ing of cells transiently expressing pEGFP-SSTR3 were performed on a Leica DMRXE
upright, epifluorescence/ Nomarski microscope outfitted with Leica TCS SP2 laser con-
focal optics (Leica; Exton, PA). Optical sections through the Z axis were generated using
a computer-controlled focus step motor. Flattened projections of image stacks and 3D
renderings were prepared using proprietary confocal imaging software from Leica. Dual
color time-lapse imaging of cells expressing SSTR3#1 with different Rabs was per-
formed on the PerkinElmer Confocal System UltraVIEWERS 6FE-US equipped with
100X oil objective and processed and analyzed with Volocity 5.2. For all live cell imag-
ing, media containing 2uM Hepes was added to cells and cells were maintained at 37° on
a temperature controlled chamber and time-lapse series exposure times were 200 to 300
ms for each channel, images were acquired every 6 seconds on Leica confocal and for
dual color imaging on Perkin Elmer confocal images were acquired every 5 or 7 seconds
10 to 20 minutes using Volocity software (Perkin Elmer), roughly. Movies were then
exported into Quicktime (Adobe Systems, Inc., San Jose, CA) and sequential still frames
were taken from Quicktime movies. Further processing of images was done using Photo-
shop 6.0 and QuickTime (Adobe Systems, Inc., San Jose, CA) movies were created at a
rate of four to five frames per second.
For quantification of co-localization, Pearson’s correlation coefficients were cal-
culated by Volocity software of z-stacked images. Co-localization was assessed based on

97
a scale of 0 to 1, where values below 0 meant no co-localization and a 1 meant complete
co-localization. A coefficient below 0.100 was also considered not co-localized with val-
ues above that being co-localized the reason for this boundary is because when two fluo-
rophores were separated but close the coefficient were 0 to 0.094. To quantify fusion and
fission events, 21 to 25 fluorescent objects randomly selected from different fields of the
cell were analyzed and the Pearson’s correlation coefficients were recorded throughout
the duration of time-lapse sequences. The changes in co-localization as measured by
Pearson’s coefficient were used to determine the time proteins remained associated and
plotted on a graph.
Acknowledgements
We gratefully acknowledge Drs. Nicolas Berbari, Melanie Styers, Marlene Winkle-
bauer, and Amber O’Connor for thoughtful insight and critical reading of this manuscript
and Dr. Jon Lehman, Zach Kosan and Mandy Croyle for assistance in time-lapse imaging.
We are grateful to Dr. Bradley Yoder for providing IMCD cells stably expressing SSTR3
and reagents. This work was supported by P30 DK074038-03.

98
REFERENCES
Adhiambo, C., Blisnick, T., Toutirais, G., Delannoy, E., and Bastin, P. (2009). A novel function for the atypical small G protein Rab-like 5 in the assembly of the trypanosome flagellum. J Cell Sci. 122, 834-841.
Barbero, P., Bittova, L., and Pfeffer, S.R. (2002). Visualization of Rab9-mediated vesicle transport from endosomes to the trans-Golgi in living cells. J Cell Biol. 156, 511-518.
Berbari, N.F., Bishop, G.A., Askwith, C.C., Lewis, J.S., and Mykytyn, K. (2007). Hippo-campal neurons possess primary cilia in culture. J Neurosci Res 85, 1095-1100.
Berbari, N.F., Johnson, A.D., Lewis, J.S., Askwith, C.C., and Mykytyn, K. (2008a). Iden-tification of ciliary localization sequences within the third intracellular loop of G protein-coupled receptors. Mol Biol Cell 19, 1540-1547.
Berbari, N.F., Lewis, J.S., Bishop, G.A., Askwith, C.C., and Mykytyn, K. (2008b). Bar-det-Biedl syndrome proteins are required for the localization of G protein-coupled recep-tors to primary cilia. Proc Natl Acad Sci U S A 105, 4242-4246.
Berbari, N.F., O'Connor, A.K., Haycraft, C.J., and Yoder, B.K. (2009 ). The primary ci-lium as a complex signaling center. Curr Biol 19, R526-535.
Boehlke, C., Bashkurov, M., Buescher, A., Krick, T., John, A.K., Nitschke, R., Walz, G., and Kuehn, E.W. (2010). Differential role of Rab proteins in ciliary trafficking: Rab23 regulates smoothened levels. J Cell Sci. 123, 1460-1467.
Bucci, C., Parton, R.G., Mather, I.H., Stunnenberg, H., Simons, K., Hoflack, B., and Zerial, M. (1992). The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell 70, 715-728.
Bucci, C., Wandinger-Ness, A., Lütcke, A., Chiariello, M., Bruni, C.B., and Zerial, M. (1994). Rab5a is a common component of the apical and basolateral endocytic machinery in polarized epithelial cells. Proc Natl Acad Sci U S A 91, 5061-5065.
Cantagrel, V., Silhavy, J.L., Bielas, S.L., Swistun, D., Marsh, S.E., Bertrand, J.Y., Audol-lent, S., Attié-Bitach, T., Holden, K.R., Dobyns, W.B., Traver, D., Al-Gazali, L., BR, A., Lindner, T.H., Caspary, T., Otto, E.A., Hildebrandt, F., IA, G., Logan, C.V., Johnson, C.A., Bennett, C., Brancati, F., Group, I.J.S.R.D.S., Valente, E.M., Woods, C.G., and Gleeson, J.G. (2008). Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet. 83, 170-179.
Caspary, T., Larkins, C.E., and Anderson, K.V. (2007). The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell. 12, 767-778.

99
Choudhury, A., Dominguez, M., Puri, V., Sharma, D.K., Narita, K., Wheatley, C.L., Marks, D.L., and Pagano, R.E. (2002). Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J Clin Invest. 109, 1541-1550.
Christensen, S.T., Pedersen, L.B., Schneider, L., and Satir, P. (2007). Sensory cilia and integration of signal transduction in human health and disease. Traffic 8, 97-209.
Corbit, K.C., Aanstad, P., Singla, V., Norman, A.R., Stainier, D.Y., and Reiter, J.F. (2005). Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018-1021.
Daro, E., van der Sluijs, P., Galli, T., and Mellman, I. (1996). Rab4 and cellubrevin de-fine different early endosome populations on the pathway of transferrin receptor recy-cling. oc Natl Acad Sci U S A. 93, 9559-9564.
de Renzis, S., Sönnichsen, B., and Zerial, M. (2002). Divalent Rab effectors regulate the sub-compartmental organization and sorting of early endosomes. Nat Cell Biol. 4, 124-133.
Deretic, D. (1997). Rab proteins and post-Golgi trafficking of rhodopsin in photoreceptor cells. Electrophoresis. 18, 2537-2541.
Deretic, D., and Papermaster, D.S. (1991). Polarized sorting of rhodopsin on post-Golgi membranes in frog retinal photoreceptor cells. J Cell Biol 113, 1281-1293.
Dunn, K.W., and Maxfield, F.R. (1992). Delivery of ligands from sorting endosomes to late endosomes occurs by maturation of sorting endosomes. J Cell Biol. 117, 301-310.
Efimenko, E., Blacque, O.E., Ou, G., Haycraft, C.J., Yoder, B.K., Scholey, J.M., Leroux, M.R., and Swoboda, P. (2006). Caenorhabditis elegans DYF-2, an orthologue of human WDR19, is a component of the intraflagellar transport machinery in sensory cilia. Mol Biol Cell 17, 4801-4811.
Follit, J.A., Tuft, R.A., Fogarty, K.E., and Pazour, G.J. (2006). The intraflagellar trans-port protein IFT20 is associated with the Golgi complex and is required for cilia assem-bly. Mol Biol Cell 17, 3781-3792.
Handel, M., Schulz, S., Stanarius, A., Schreff, M., Erdtmann-Vourliotis, M., Schmidt, H., Wolf, G., and Höllt, V. (1999). Selective targeting of somatostatin receptor 3 to neuronal cilia. Neuroscience 89, 909-926.
Hargrave, P.A., and McDowell, J.H. (1992). Rhodopsin and phototransduction: a model system for G protein-linked receptors. FASEB J. 6, 2323-2331.
Jacobs, S., and Schulz, S. (2008). Intracellular trafficking of somatostatin receptors. Mol Cell Endocrinol. 286, 58-62

100
Jean-Alphonse, F., and Hanyaloglu, A.C. ( 2011). Regulation of GPCR signal networks via membrane trafficking. Mol Cell Endocrinol 331, 205-214.
Jin, H., White, S.R., Shida, T., Schulz, S., Aguiar, M., Gygi, S.P., Bazan, J.F., and Na-chury, M.V. (2010). The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 141, 1208-1219.
Knödler, A., Feng, S., Zhang, J., Zhang, X., Das, A., Peränen, J., and Guo, W. (2010). Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci U S A. 107, 6346-6351.
Kreuzer, O.J., Krisch, B., Déry, O., Bunnett, N.W., and Meyerhof, W. (2001). Agonist-mediated endocytosis of rat somatostatin receptor subtype 3 involves beta-arrestin and clathrin coated vesicles. J Neuroendocrinol. 13 279-287
Li, Y., and Hu, J. (2011). Small GTPases and cilia. Protein Cell 2, 13-25.
Maerker, T., van Wijk, E., Overlack, N., Kersten, F.F., McGee, J., Goldmann, T., Sehn, E., Roepman, R., Walsh, E.J., Kremer, H., and Wolfrum, U. (2008). A novel Usher pro-tein network at the periciliary reloading point between molecular transport machineries in vertebrate photoreceptor cells. Hum Mol Genet. 17, 71-86.
Marshall, W. (2008 ). The cell biological basis of ciliary disease. J Cell Biol. 180, 17-21.
Maxfield, F.R., and McGraw, T.E. (2004). Endocytic recycling. Nat Rev Mol Cell Biol. 5, 121-132.
Mazelova, J., Astuto-Gribble, L., Inoue, H., Tam, B.M., Schonteich, E., Prekeris, R., Mo-ritz, O.L., Randazzo, P.A., and Deretic, D. (2009a). Ciliary targeting motif VxPx directs assembly of a trafficking module through Arf4. Embo J 28, 183-192.
Mazelova, J., Ransom, N., Astuto-Gribble, L., Wilson, M.C., and Deretic, D. (2009b). Syntaxin 3 and SNAP-25 pairing, regulated by omega-3 docosahexaenoic acid, controls the delivery of rhodopsin for the biogenesis of cilia-derived sensory organelles, the rod outer segments. J Cell Sci. 122, 2003-2013.
McCaffrey, M.W., Bielli, A., Cantalupo, G., Mora, S., Roberti, V., Santillo, M., Drum-mond, F., and Bucci, C. (2001). Rab4 affects both recycling and degradative endosomal trafficking. FEBS Lett. 495, 21-30.
Milenkovic, L., Scott, M.P., and Rohatgi, R. (2009). Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. Cell Biol. 187, 365-374.

101
Molla-Herman A, G.R., Blisnick T, Meunier A, Serres C, Silbermann F, Emmerson C, Romeo K, Bourdoncle P, Schmitt A, Saunier S, Spassky N, Bastin P, Benmerah A. (2010). The ciliary pocket: an endocytic membrane domain at the base of primary and motile cilia. J Cell Sci. 123, 1785-1795.
Moritz, O.L., Tam, B.M., Hurd, L.L., Peranen, J., Deretic, D., and Papermaster, D.S. (2001). Mutant rab8 Impairs docking and fusion of rhodopsin-bearing post-Golgi mem-branes and causes cell death of transgenic Xenopus rods. Mol Biol Cell 12, 2341-2351.
Nachury, M.V., Loktev, A.V., Zhang, Q., Westlake, C.J., Peranen, J., Merdes, A., Slu-sarski, D.C., Scheller, R.H., Bazan, J.F., Sheffield, V.C., and Jackson, P.K. (2007). A Core Complex of BBS Proteins Cooperates with the GTPase Rab8 to Promote Ciliary Membrane Biogenesis. Cell 129, 1201-1213.
Nachury, M.V., Seeley, E.S., and Jin, H. (2010). Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu Rev Cell Dev Biol. 26, 59-87.
Pazour, G.J., and Bloodgood, R.A. (2008). Targeting proteins to the ciliary membrane. Curr Top Dev Biol 85, 115-149.
Pellinen, T., Arjonen, A., Vuoriluoto, K., Kallio, K., Fransen, J.A., and Ivaska, J. (2006). Small GTPase Rab21 regulates cell adhesion and controls endosomal traffic of beta1-integrins. J Cell Biol 173, 767-780.
Pellinen, T., Tuomi, S., Arjonen, A., Wolf, M., Edgren, H., Meyer, H., Grosse, R., Kitz-ing, T., Rantala, J.K., Kallioniemi, O., Fässler, R., Kallio, M., and Ivaska, J. (2008). Inte-grin trafficking regulated by Rab21 is necessary for cytokinesis. Dev Cell. 15,371-385.
Poteryaev, D., Datta, S., Ackema, K., Zerial, M., and Spang, A. (2010). Identification of the switch in early-to-late endosome transition. Cell. 141, 497-508.
Qin, H., Wang, Z., Diener, D., and Rosenbaum, J. (2007). Intraflagellar transport protein 27 is a small G protein involved in cell-cycle control. Curr Biol 17, 193-202.
Rink, J., Ghigo, E., Kalaidzidis, Y., and Zerial, M. (2005). Rab conversion as a mechan-ism of progression from early to late endosomes. Cell 122, 735-749.
Rohatgi, R., Milenkovic, L., and Scott, M.P. (2007). Patched1 regulates hedgehog signal-ing at the primary cilium. Science 317, 372-376.
Roosterman, D., Brune, N.E., Kreuzer, O.J., Feld, M., Pauser, S., Zarse, K., Steinhoff, M., and Meyerhof, W. (2008). Intracellular degradation of somatostatin-14 following somatostatin-receptor3-mediated endocytosis in rat insulinoma cells. FEBS J. 275, 4728-4739.

102
Roosterman, D., Roth, A., Kreienkamp, H.J., Richter, D., and Meyerhof, W. (1997). Dis-tinct agonist-mediated endocytosis of cloned rat somatostatin receptor subtypes expressed in insulinoma cells. J Neuroendocrinol. 9, 741-751
Satoh, A., Tokunaga, F., Kawamura, S., and Ozaki, K. (1997). In situ inhibition of vesicle transport and protein processing in the dominant negative Rab1 mutant of Drosophila. J Cell Sci 110 ( Pt 23), 2943-2953.
Satoh, A.K., O'Tousa, J.E., Ozaki, K., and Ready, D.F. (2005). Rab11 mediates post-Golgi trafficking of rhodopsin to the photosensitive apical membrane of Drosophila pho-toreceptors. Development 132, 1487-1497.
Shafer, J.C., Winkelbauer, M.E., Williams, C.L., Haycraft, C.J., Desmond, R.A., and Yo-der, B.K. (2006). IFTA-2 is a conserved cilia protein involved in pathways regulating longevity and dauer formation in Caenorhabditis. J Cell Sci. 119, 4088-4100.
Sheff, D.R., Daro, E.A., Hull, M., and Mellman, I. (1999). The receptor recycling path-way contains two distinct populations of early endosomes with different sorting func-tions. J Cell Biol. 145, 123-139.
Simpson, J.C., Griffiths, G., Wessling-Resnick, M., Fransen, J.A., Bennett, H., and Jones, A.T. (2004). A role for the small GTPase Rab21 in the early endocytic pathway. J Cell Sci 117, 6297-6311.
Sönnichsen, B., De Renzis, S., Nielsen, E., Rietdorf, J., and Zerial, M. (2000). Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor im-aging of Rab4, Rab5, and Rab11. J Cell Biol. 149, 901-914.
Spang, A. (2009). On the fate of early endosomes. Biol Chem. 390, 753-759.
Stenmark, H. (2009). Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 10, 513-525.
Tower, C., Fu, L., Gill, R., Prichard, M., Lesort, M., and Sztul, E. (2011). Human cyto-megalovirus UL97 kinase prevents the deposition of mutant protein aggregates in cellular models of Huntington's disease and ataxia. Neurobiol Dis 41, 11-22.
Tulipano, G., Stumm, R., Pfeiffer, M., Kreienkamp, H.J., Höllt, V., and Schulz, S. (2004). Differential beta-arrestin trafficking and endosomal sorting of somatostatin recep-tor subtypes. J Biol Chem. 279, 21374-21382.
van der Sluijs, P., Hull, M., Webster, P., Mâle, P., Goud, B., and Mellman, I. (1992). The small GTP-binding protein rab4 controls an early sorting event on the endocytic pathway. Cell 70 729-740.

103
Vonderheit, A., and Helenius, A. (2005). Rab7 associates with early endosomes to me-diate sorting and transport of Semliki forest virus to late endosomes. PLoS Biol. 3, e233.
War, S.A., Somvanshi, R.K., and Kumar, U. (2011). Somatostatin receptor-3 mediated intracellular signaling and apoptosis is regulated by its cytoplasmic terminal. Biochim Biophys Acta. 1813, 390-402.
Ward, E.S., Martinez, C., Vaccaro, C., Zhou, J., Tang, Q., and Ober, R.J. (2005). From sorting endosomes to exocytosis: association of Rab4 and Rab11 GTPases with the Fc receptor, FcRn, during recycling. Mol Biol Cell. 16, 2028-2038.
Yoshimura, S., Egerer, J., Fuchs, E., Haas, A.K., and Barr, F.A. (2007). Functional dis-section of Rab GTPases involved in primary cilium formation. J Cell Biol 178, 363-369.
Zerial, M., and McBride, H. (2001). Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2, 107-117.
Zhang, X., He, X., Fu, X.Y., and Chang, Z. (2006). Varp is a Rab21 guanine nucleotide exchange factor and regulates endosome dynamics. J Cell Sci 119, 1053-1062.

104
DISCUSSION
Novel Means to Combat Aggregation Diseases
Misfolding and aggregation of proteins have been shown to cause numerous pa-
thologies collectively called aggregation diseases. Among them, diseases caused by the
aggregation of proteins with polyQ repeats have been studied in cellular and organismal
systems. Importantly to the focus of this thesis, pharmacological, molecular and genetic
approaches that prevent aggregation of polyQ proteins show strong protective function
against the pathological manifestations of polyQ diseases. Thus, novel means of prevent-
ing proteins aggregation have the potential to identify novel therapeutic approaches to
combat polyQ diseases. We entered this field by focusing on the anti-aggregation proper-
ties of the UL97 kinase of HCMV.
HCMV UL97 kinase as an anti-aggregation factor
UL97 is a viral kinase that prevents aggregation of the pp71 and pp65 viral tegu-
ment proteins. To determine whether UL97 may act as a general anti-aggregation factor,
UL97 effects on cellular non-polyQ and polyQ proteins were examined. I uncovered that
UL97 prevents the deposition of aggregates of two non-polyQ proteins: GFP170* and
WRN-RFP. The expression of the UL97/K355M catalytically inactive mutant fails to
prevent aggregation. Furthermore, nuclear and cytoplasmic aggregates form in the pres-
ence of the inactive mutant. More importantly, UL97 inhibits deposition of aggregates of
the polyQ containing mutant proteins huntingtin (HttExon1-Q82) and the full length atax-

105
in-3 containing a 72 polyQ track (AT3-72Q). UL97 localizes to the nucleus and disrupts
the architecture of PML bodies. There appears to be a link in the disruption of PML bo-
dies by UL97 to the inhibition of AT3-72Q aggregate formation. PML bodies have been
implicated in transcriptional function as they contain such transcriptional regulators as
sp100, PML, CBP, and p53. Thus I explored whether UL97 influences transcription and
showed that UL97 decreased p53-mediated transcription. In addition, UL97 was shown
to be cytoprotective as it significantly increased cell viability of cells expressing AT3-
72Q. These results identify UL97 as a novel means to modulate polyQ aggregation, and
suggest that UL97 can be used to probe cellular anti-aggregation mechanisms.
Molecular mechanism of UL97 action
UL97 has been shown to phosphorylate a number of cellular proteins in vitro
while the identity of cellular targets that UL97 phosphorylates in vivo is unknown. Cellar
targets include histones, lamins, E2F splicing factor, p32, and retinoblastoma protein
(Rb). Recently, UL97 kinase activity to Rb has been linked to PML body disruption.
PML bodies have been linked to the aggregation of various viral and cellular proteins
(Bonilla et al., 2002; Bernardi and Pandolfi, 2007; Nichol et al., 2009). Recent reports
and data presented in this dissertation show that UL97 disrupts PML bodies as monitored
by dispersion of Sp100 component (Prichard et al., 2008) and decrease in the number of
PML bodies (this dissertation), suggesting that UL97 anti-aggregation activity may be
linked to PML disruption. The model that I propose is that UL97 inhibits polyQ aggrega-
tion by phosphorylating a target protein. In this model the relationship between PML bo-
dies disruption and the anti-aggregational function of UL97 is as follows. In the absence

106
of the kinase PML bodies are formed (core proteins associate with one another) and ag-
gregates are formed. However when UL97 is present, the kinase phosphorylates Rb (Pri-
chard et al., 2008) leading to the dissociation of Rb from E2F and pPML ultimately caus-
ing disruption of PML bodies. UL97 may act through a previously identified target or
through pathways shown to prevent polyQ aggregation in the cell to upregulate the tran-
scription/protein levels/activity of proteins or pathway to clear/prevent general aggrega-
tion.
Another potential target of UL97 that may aid in preventing aggregation is p32.
p32 binds UL97 directly in vitro and in vivo (Marschall et al., 2005). p32 directly binds
lamin B receptor and appears to link UL97 to the lamin B receptor. P32 is proposed to
regulate nuclear import and hence its phosphorylation may impact on nucleocytoplasmic
shuttling of transcriptional regulators or other molecules. UL97 has been shown to local-
ize to the nucleus of cells and it possible that this localization is critical for UL97 to exert
its anti-aggregation function. The inactive mutant of UL97 largely localizes to the cytop-
lasmic with some nuclear localization suggesting that nuclear proteins can be possible
targets of the kinase. Alternatively UL97 may directly regulate independent cellular tar-
gets involved in transcription, apoptosis, cell death, and degradation.
Novel Pathways and Mechanisms for Delivery of Proteins to Cilia
Cilia are hair-like projections found on eukaryotic cells that play important roles
in chemo- and mechanosensation. Absence or dysfunction of cilia leads to a number of
human diseases collectively called ciliopathies. Ciliopathies also can result from misloca-
lization of ciliary proteins. All ciliary proteins are synthesized in the cytoplasm and must

107
be efficiently delivered to the cilium by as yet incompletely characterized pathways and
mechanisms. Our goal was to provide additional information on pathways and molecular
mechanisms of ciliary trafficking by focusing on the delivery of SSTR3 to cilia.
SSTR3 Transport to Cilia
SSTR3 is one of six somatostain receptors found in mammals. The protein con-
sists of seven transmembrane domains with an extracellular N-terminus and a cytoplas-
mic C-terminus. We used an IMCD cell line stably expressing SSTR3 to monitor its traf-
ficking to cilia. Initial analysis of static subcellular localization of SSTR3 showed that
SSTR3 is found predominantly in the primary cilium (based on co-localization with ace-
tylated tubulin and the ciliary marker adenylate cyclase). In addition, SSTR3 is also
found in the early and recycling endosomes where it co-localized with endosomal mark-
ers Rab4, Rab5, Rab21, and Rab11. SSTR3 shows extensive co-localization with Rab4,
Rab21, and Rab11, but less overlap with Rab5. Almost no co-localization of SSTR3 with
Rab7 was observed suggesting that SSTR3 is not detected in late endosomes.
Using time-lapse imaging, I observed GFP-SSTR3 rapidly budding from endo-
somes in small vesicles that traversed the cell to the base of the cilium, and then fused
with the base of the cilium. "Reverse" vesicles that budded from the base of the cilium,
traveled towards the endosomes and fused with endosomes were also observed. Tracking
of SSTR3 indicates that SSTR3 is extremely dynamic and continuously traffics between
the cilium and the endosomes via small vesicles. Thus, we uncovered a direct cellular
pathway to the cilium from early endosomal structures and we confirmed a pathway from
recycling endosomes to the cilium.

108
Early endosomes contain Rab5, Rab4, and Rab21. The role of these Rabs in regu-
lating delivery of SSTR3 to cilia was analyzed by using dual color live imaging. SSTR3
vesicles originating from Rab21, Rab4, and Rab11 endosomes trafficked to the base of
the cilium where the vesicles fused.
The early endosomal Rab21 has been shown to have an unorthodox X-box
(present in multiple ciliary proteins), suggesting that it might be involved in trafficking of
ciliary proteins. I uncovered that Rab21 co-localizes extensively with SSTR3 within
punctate endosomal structures but not within the cilium axoneme (only Rab8 has been
shown to localize to cilia). The role of Rab21 in SSTR3 trafficking was assessed by tran-
siently expressing a dominant negative mutant of Rab21. Expressing Rab21/T33N se-
verely impaired sorting of SSTR3.
SSTR3 was found on subdomains of early endosomes positive for Rab21 and
Rab4. Similar to Rab21, SSTR3 was sorted from Rab4 positive endosomes and express-
ing the dominant negative mutant Rab4/S22N severely impaired sorting of SSTR3. In
contrast to Rab21 and Rab4, SSTR3 was not extensively co-localized with Rab5 and the
sorting of SSTR3 from these endosomes was much slower and not impaired by expres-
sion of the Rab5/I133N mutant. SSTR3 also co-localiaed with Rab11 and we showed that
SSTR3 is not properly sorted in cells expressing the Rab11/S25N inactive mutant. Thus,
SSTR3 cycling through the early endosomal compartment is regulated by Rab21 and
Rab4 and in the recycling endosome by Rab11. Together my data indicate that SSTR3 is
trafficked directly from endosomes to cilia and this transport is Rab-dependent.
Small GTPAses in Ciliopathies

109
Specific members of the ARF and the related Arf like proteins (Arls) subfamilies
of small GTPases have been shown to be involved in ciliopathies, possibly by regulating
key stages of ciliary trafficking. Arl 6 (also called BBS3) has been characterized as the
gene mutated in Bardet Biedl Syndrome 3(BBS3) patients (Chiang et al., 2004; Fan et al.,
2004). BBS is characterized by obesity, mental retardation, renal cysts, retinal degenera-
tion, and polydactyly (Zaghloul and Katsanis, 2009). Further work shows that overex-
pression of GDP- or GTP-locked forms of Arl6 affects cilia assembly and Wnt signaling
in vivo (Wiens et al., 2010). Similarly, mutations in Arl13B that inhibit GTP binding ac-
tivity of ARL13B cause Joubert syndrome characterized by congenital cerebellar ataxia,
hypotonia, oculomotor apraxia, mental retardation, cystic kidney and polydactyly. Addi-
tionally, ARL13B mutant mice have defects in cilia structure and hedgehog signaling
similar to phenotypes observed in human patients suffering from the disease (Caspary et
al., 2007).
Arf4 is the only reported Arf protein to show cilia related function (Mazelova et
al., 2009b). Arf4 binds to the ciliary targeting sequence of rhodopsin and forms a com-
plex consisting of Arf4, ASAP (Arf4 GAP), FIP 2 (Arf 4 effector), and Rab11 to mediate
transport of rhodopsin from the TGN to the primary cilium (Mazelova et al., 2009a).
A number of Rabs have been shown to play a role in ciliogenesis and cilia func-
tion. Analysis of photoreceptor cells linked Rab8 to cilia (Deretic et al., 1995). Since then
Rab8 has been shown to be required for post-Golgi trafficking of rhodopsin (Moritz et
al., 2001). Rab8 plays a role in ciliogenesis as evident by the fact that depletion of Rab8
inhibits cilia formation (Nachury et al., 2007; Yoshimura et al., 2007). Recently Rab8 has
been shown to regulate cilia entry of fibrocystin (Follit et al., 2010) and smoothened re-

110
ceptor (Boehlke et al., 2010). In addition to Rab8, previous work revealed that Rab23 is
present in cilia and mutations in Rab23 cause Carpenters’ syndrome which has pheno-
types that overlap with other ciliopathies (Jenkins et al., 2007; Boehlke et al., 2010). Fur-
thermore, Rab23 has been shown to play a role in trafficking of smoothened, hedgehog
signaling receptor, to cilia (Boehlke et al., 2010). Another Rab that regulates ciliogenesis
and localizes to the cilia base and acts as a redundant player to Rab8 is Rab10 (Babbey et
al., 2010). Even though Rabs have not been identified as ciliopathy genes so far, they
play major roles in ciliogenesis, membrane trafficking, vesicle formation and regulation
of IFT machinery (Li and Hu, 2011).
Future Directions
Using UL97 to Probe for Anti-Aggregation Therapies
Nine neurodegenerative diseases arise through the expansion of polyQ tracks in
unrelated proteins. PolyQ expanded proteins are aggregation prone and inclusions are
hallmarks of these diseases. The striking discovery that the UL97 viral kinase prevents
aggregation of polyQ proteins in cellular models of Huntington's disease and ataxia-3
identifies a novel means to prevent polyQ aggregation as a possible therapeutic approach
to combat the cytopathology of neurodegenerative diseases. Importantly, UL97
represents a means to understand cellular pathways that regulate aggregation and deposi-
tion of polyQ inclusions. Future studies will focus on exploring the molecular mechanism
of UL97 action and some are outlined below.

111
We have shown that UL97 kinase disperses PML bodies, suggesting that UL97
may modulate the structure or function of PML components. The cellular proteins, PML,
CBP, Daxx, p53, Sp100, and Rb are core components of PML bodies may be altered to
affect PML formation. The requirement for PML bodies in the anti-aggregation action of
UL97 could be examined in PML null cell models in which the PML protein is absent
and PML bodies don't assemble. It would be interesting to assess whether UL97 could
still prevent the formation of polyQ aggregates in absence of intact PML bodies. Eluci-
dating the exact mechanism by which UL97 disrupts PML bodies will lead to a better un-
derstanding of the link between PML bodies, UL97, and protein aggregation.
Additionally, studies need to be conducted to identify molecular targets of
UL97 and characterize their role in polyQ aggregation. This work uncovered that the an-
ti-aggregation ability of UL97 requires its kinase activity, suggesting that UL97 acts by
phosphorylating target proteins. One approach to identify kinase targets would be to test
whether UL97 prevents polyQ aggregation by impacting on proteins known to be phos-
phorylated by UL97 in vitro, or on cellular pathways known to inhibit polyQ aggregation.
Whether UL97 phosphorylates any of these substrates in vivo will be tested by monitor-
ing their phosphorylation status in UL97 expressing cells as compared to cells expressing
the inactive UL97/K355M mutant. The importance of UL97-mediated phosphorylation of
target proteins will be assessed by siRNA depleting cells of endogenous proteins and re-
placing with mutants that can’t be phosphorylated by UL97 and assessing the effect of
such replacement on polyQ aggregation. A second approach would be to use a radiolabe-
ling screen in which radiolabeled ATP (which acts as a donor for tyrosine and se-
rine/threonine phosphorylation of cellular proteins) will be added to control cells, UL97

112
expressing cells, and cells expressing the inactive UL97/K355M mutant to identify pro-
teins that are specifically phosphorylated in response to UL97 levels. All cellular proteins
phosphorylated in response to UL97 would then be assessed for their possible role in pre-
venting polyQ aggregation. Exploring the molecular mechanism of UL97 action and
identifying the cellular targets of UL97 will identify the molecular pathways and cellular
components that regulate polyQ aggregation.
PolyQ aggregation in HD and SCA3 has been linked to dysregulation of cellular
pathways mediating protein folding/degradation, transcriptional activity, and cell death
and apoptosis, all of which are likely to contribute to neurodegeneration. Future work
would be directed at characterizing the ability of UL97 to reverse polyQ-induced changes
in protein folding and protein degradation, alterations in transcriptional activity, and cell
death and apoptosis. To assess whether UL97 influences polyQ-induced protein
degradation, cells expressing polyQ protein alone, polyQ protein and UL97, or polyQ
protein and the inactive UL97/K355M mutant will be lysed and proteasomal activity will
be measured by cleavage of a reporter fluorescent substrate Suc-LLVY-AMC, In
addition, proteasomal activity can be assayed in intact cells by co-transfecting cells with a
4UB-LUCiferase reporter together with polyQ protein alone or with polyQ proteins and
either UL97 or the inactive UL97/K355M mutant. The effect of UL97 on cell death will
be measured by fluorescent-associated cell sorting (FACS) after loading with an indicator
fluorescent dye, by LDH release and MTT assay kit. Cell death will be measured in
control cells, cells expressing polyQ protein only, cells expressing UL97 only, cells
expressing the inactive UL97/K355M mutant alone and in cells co-expressing polyQ
protein and UL97 or polyQ protein and the UL97/K355M mutant. The effect of UL97 on

113
apoptosis will be measured by the production of cleaved fragments of caspase-3 and
PARP, and by TUNEL. The findings presented in this dissertation suggest that UL97
rescues cytotoxicity associated with polyQ aggregates. Further examination of UL97
cytoprotective role in animal models will be informative By expressing UL97 in a
Drosophilia model of HD and determining if the kinase is able to prevent aggregation of
the mutated protein and reduce neurodegeneration or be cytoprotective will extend the
findings described in this dissertation.
Identifying Ciliary Trafficking Machinery
During the last decade, there has been an explosion in the cilia field to find out
how signaling molecules are transported to cilia. A number of key regulators include
small GTPases such as Rabs, Arfs, and Arls. This dissertation described novel insights
into the cellular and molecular machinery that ensure correct trafficking of proteins to
cilia. My study shows that a GPCR is delivered to the primary cilium in a Rab-dependent
fashion and that endosomes represent key stages in delivery of such protein to cilia. Spe-
cific Rabs that have not been previously implicated in ciliary trafficking have been identi-
fied, Rab21 and Rab4. However, there are many future inquiries that would further broa-
den our knowledge of cellular machinery that plays a role in delivery of proteins to pri-
mary cilia.
In this work we examined the role of Rabs in mediating ciliary transport of
SSTR3 by assessing the effect of dominant negative Rabs on SSTR3 trafficking. To con-
firm the role the Rabs play in SSTR3 trafficking siRNA-mediated depletion of the Rabs
identified in this work should be used to assess their roles in ciliary trafficking. Rabs are
regulated by Rab GEFS (activates Rab) and GAPs (inactivates). With the identification

114
of specific Rabs in targeting SSTR3 to cilia it would be worthwhile to determine if the
GEFs and GAPs of the identified Rabs also regulate SSTR3 trafficking. Future direction
would encompass determining if the identified Rabs mediate trafficking of disease rele-
vant proteins such as smoothened or polycystin-1 or polycystin-2 and will inform on the
nature of vesicles that originate from endosomes and destined for the ciliary membrane.
Arfs are another family of small GTPases that regulate membrane trafficking.
Arf4 has been implicated in regulating trafficking of rhodopsin, suggesting that Arfs play
a role in ciliary trafficking. Thus, future studies should assess the role of Arf 4 and its
regulatory GEFs and GAPs in trafficking of other ciliary proteins including SSTR3. This
could be done by assessing the effect of dominant inactive mutants of Arf4-GEFs and
Arf4-GAPs on SSTR3 trafficking as well as the assessment of siRNA depletion of Arf-
GEFs and Arf-GAPs on SSTR3 trafficking. The players identified may be utilized by
other ciliary proteins. The participation of specific members of Rabs, Arf-GEFs, and Arfs
could create a cascade of events that would generate vesicles from the TGN, endosomes,
and plasma membrane that target cargo-containing vesicles to cilia.

115
GENERAL LIST OF REFERENCES
Afzelius, B.A. (1995). Role of cilia in human health. Cell Motil Cytoskeleton 32, 95-97. Apostol, B.L., Kazantsev, A., Raffioni, S., Illes, K., Pallos, J., Bodai, L., Slepko, N., Bear, J.E., Gertler, F.B., Hersch, S., Housman, D.E., Marsh, J.L., and Thompson, L.M. (2003). A cell-based assay for aggregation inhibitors as therapeutics of polyglutamine-repeat disease and validation in Drosophila. Proc Natl Acad Sci U S A 100, 5950-5955. Babbey, C.M., Bacallao, R.L., and Dunn, K.W. (2010). Rab10 associates with primary cilia and the exocyst complex in renal epithelial cells. Am J Physiol Renal Physiol 299, F495-506. Baek, M.C., Krosky, P.M., and Coen, D.M. (2002a). Relationship between autophospho-rylation and phosphorylation of exogenous substrates by the human cytomegalovirus UL97 protein kinase. J Virol 76, 11943-11952. Baek, M.C., Krosky, P.M., He, Z., and Coen, D.M. (2002b). Specific phosphorylation of exogenous protein and peptide substrates by the human cytomegalovirus UL97 protein kinase. Importance of the P+5 position. J Biol Chem 277, 29593-29599. Baek, M.C., Krosky, P.M., Pearson, A., and Coen, D.M. (2004). Phosphorylation of the RNA polymerase II carboxyl-terminal domain in human cytomegalovirus-infected cells and in vitro by the viral UL97 protein kinase. Virology 324, 184-193. Bates, G.P. (2001). Huntington's disease. Exploiting expression. Nature 413, 691, 693-694. Berbari, N.F., Johnson, A.D., Lewis, J.S., Askwith, C.C., and Mykytyn, K. (2008). Iden-tification of ciliary localization sequences within the third intracellular loop of G protein-coupled receptors. Mol Biol Cell 19, 1540-1547. Berbari, N.F., O'Connor, A.K., Haycraft, C.J., and Yoder, B.K. (2009 ). The primary ci-lium as a complex signaling center. Curr Biol 19, R526-535. Bernardi, R., and Pandolfi, P.P. (2007). Structure, dynamics and functions of promyelo-cytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 8, 1006-1016. Boehlke, C., Bashkurov, M., Buescher, A., Krick, T., John, A.K., Nitschke, R., Walz, G., and Kuehn, E.W. (2010). Differential role of Rab proteins in ciliary trafficking: Rab23 regulates smoothened levels. J Cell Sci. 123, 1460-1467.

116
Bonilla, W.V., Pinschewer, D.D., Klenerman, P., Rousson, V., Gaboli, M., Pandolfi, P.P., Zinkernagel, R.M., Salvato, M.S., and Hengartner, H. (2002). Effects of promyelocytic leukemia protein on virus-host balance. J Virol 76, 3810-3818. Brighouse, A., Dacks, J.B., and Field, M.C. (2010). Rab protein evolution and the history of the eukaryotic endomembrane system. Cell Mol Life Sci 67, 3449-3465. Bucci, C., Parton, R.G., Mather, I.H., Stunnenberg, H., Simons, K., Hoflack, B., and Zerial, M. (1992). The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell 70, 715-728. Casey, P.J., and Seabra, M.C. (1996). Protein prenyltransferases. J Biol Chem 271, 5289-5292. Caspary, T., Larkins, C.E., and Anderson, K.V. (2007). The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell. 12, 767-778. Chan, H.Y., Warrick, J.M., Gray-Board, G.L., Paulson, H.L., and Bonini, N.M. (2000). Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum Mol Genet 9, 2811-2820. Chiang, A.P., Nishimura, D., Searby, C., Elbedour, K., Carmi, R., Ferguson, A.L., Secr-ist, J., Braun, T., Casavant, T., Stone, E.M., and Sheffield, V.C. (2004). Comparative ge-nomic analysis identifies an ADP-ribosylation factor-like gene as the cause of Bardet-Biedl syndrome (BBS3). Am J Hum Genet 75, 475-484. Choudhury, A., Sharma, D.K., Marks, D.L., and Pagano, R.E. (2004). Elevated endosom-al cholesterol levels in Niemann-Pick cells inhibit rab4 and perturb membrane recycling. Mol Biol Cell 15, 4500-4511. Christensen, S.T., Pedersen, L.B., Schneider, L., and Satir, P. (2007). Sensory cilia and integration of signal transduction in human health and disease. Traffic 8, 97-209. Christensen, S.T., Pedersen, S.F., Satir, P., Veland, I.R., and Schneide, r.L. (2008). The primary cilium coordinates signaling pathways in cell cycle control and migration during development and tissue repair. Curr Top Dev Biol., 261-301. Cohen, F.E., and Kelly, J.W. (2003). Therapeutic approaches to protein-misfolding dis-eases. Nature 426, 905-909. Cole, D.G. (2003). The intraflagellar transport machinery of Chlamydomonas reinhardtii. Traffic 4, 435-442. Colicelli, J. (2004). Human RAS superfamily proteins and related GTPases. Sci STKE 2004, RE13.

117
Colin, E., Regulier, E., Perrin, V., Durr, A., Brice, A., Aebischer, P., Deglon, N., Hum-bert, S., and Saudou, F. (2005). Akt is altered in an animal model of Huntington's disease and in patients. Eur J Neurosci 21, 1478-1488. Cui, L., Jeong, H., Borovecki, F., Parkhurst, C.N., Tanese, N., and Krainc, D. (2006). Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127, 59-69. Cummings, C.J., Sun, Y., Opal, P., Antalffy, B., Mestril, R., Orr, H.T., Dillmann, W.H., and Zoghbi, H.Y. (2001). Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet 10, 1511-1518. Davenport, J.R., and Yoder, B.K. (2005). An incredible decade for the primary cilium: a look at a once-forgotten organelle. Am J Physiol Renal Physiol 289, F1159-1169. Deretic, D. (1997). Rab proteins and post-Golgi trafficking of rhodopsin in photoreceptor cells. Electrophoresis. 18, 2537-2541. Deretic, D., Huber, L.A., Ransom, N., Mancini, M., Simons, K., and Papermaster, D.S. (1995). Rab8 in retinal photoreceptors may participate in rhodopsin transport and in rod outer segment disk morphogenesis. J Cell Sci 108 ( Pt 1), 215-224. Deretic, D., and Papermaster, D.S. (1991). Polarized sorting of rhodopsin on post-Golgi membranes in frog retinal photoreceptor cells. J Cell Biol 113, 1281-1293. Deretic, D., Schmerl, S., Hargrave, P.A., Arendt, A., and McDowell, J.H. (1998). Regula-tion of sorting and post-Golgi trafficking of rhodopsin by its C-terminal sequence QVS(A)PA. Proc Natl Acad Sci U S A 95, 10620-10625. Desai, U.A., Pallos, J., Ma, A.A., Stockwell, B.R., Thompson, L.M., Marsh, J.L., and Di-amond, M.I. (2006). Biologically active molecules that reduce polyglutamine aggregation and toxicity. Hum Mol Genet 15, 2114-2124. Dobson, C.M. (2003). Protein folding and misfolding. Nature 426, 884-890. Emmer, B.T., Maric, D., and Engman, D.M. (2010). Molecular mechanisms of protein and lipid targeting to ciliary membranes. J Cell Sci 123, 529-536. Fan, Y., Esmail, M.A., Ansley, S.J., Blacque, O.E., Boroevich, K., Ross, A.J., Moore, S.J., Badano, J.L., May-Simera, H., Compton, D.S., Green, J.S., Lewis, R.A., van Haelst, M.M., Parfrey, P.S., Baillie, D.L., Beales, P.L., Katsanis, N., Davidson, W.S., and Le-roux, M.R. (2004). Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet-Biedl syndrome. Nat Genet 36, 989-993. Follit, J.A., Li, L., Vucica, Y., and Pazour, G.J. (2010). The cytoplasmic tail of fibrocys-tin contains a ciliary targeting sequence. J Cell Biol. 188, 21-28.

118
Follit, J.A., Tuft, R.A., Fogarty, K.E., and Pazour, G.J. (2006). The intraflagellar trans-port protein IFT20 is associated with the Golgi complex and is required for cilia assem-bly. Mol Biol Cell 17, 3781-3792. Follit, J.A., Xu, F., Keady, B.T., and Pazour, G.J. (2009). Characterization of mouse IFT complex B. Cell Motil Cytoskeleton 66, 457-468. Fu, L., Gao, Y.S., Tousson, A., Shah, A., Chen, T.L., Vertel, B.M., and Sztul, E. (2005). Nuclear aggresomes form by fusion of PML-associated aggregates. Mol Biol Cell 16, 4905-4917. Garcia-Mata, R., Bebok, Z., Sorscher, E.J., and Sztul, E.S. (1999). Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J Cell Biol 146, 1239-1254. Garcia-Mata, R., Gao, Y.S., and Sztu, l.E. (2002). Hassles with taking out the garbage: aggravating aggresomes. Traffic 3, 388-396. Gauthier, L.R., Charrin, B.C., Borrell-Pagès, M., Dompierre, J.P., Rangone, H., Corde-lières, F.P., De Mey, J., MacDonald, M.E., Lessmann, V., Humbert, S., and Saudou, F. (2004). Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118, 127-138. Geng, L., Okuhara, D., Yu, Z., Tian, X., Cai, Y., Shibazaki, S., and Somlo, S. (2006). Po-lycystin-2 traffics to cilia independently of polycystin-1 by using an N-terminal RVxP motif. J Cell Sci 119, 1383-1395. Gil, J.M., and Rego, A.C. (2008). Mechanisms of neurodegeneration in Huntington's dis-ease. Eur J Neurosci 27, 2803-2820. Godsel, L.M., and Engman, D.M. (1999). Flagellar protein localization mediated by a calcium-myristoyl/palmitoyl switch mechanism. EMBO J. 18, 2057-2065. Goldberg, A.L. (2003). Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895-899. Gusella, J.F., and MacDonald, M.E. (1995). Huntington's disease: CAG genetics expands neurobiology. Curr Opin Neurobiol 5, 656-662. Hanaoka, K., Qian, F., Boletta, A., Bhunia, A.K., Piontek, K., Tsiokas, L., Sukhatme, V.P., Guggino, W.B., and Germino, G.G. (2000). Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 408, 990-994.

119
Hao, L., and Scholey, J.M. (2009). Intraflagellar transport at a glance. J Cell Sci 122, 889-892. Hargrave, P.A., and McDowell, J.H. (1992). Rhodopsin and phototransduction: a model system for G protein-linked receptors. FASEB J. 6, 2323-2331. Heiser, V., Scherzinger, E., Boeddrich, A., Nordhoff, E., Lurz, R., Schugardt, N., Le-hrach, H., and Wanker, E.E. (2000). Inhibition of huntingtin fibrillogenesis by specific antibodies and small molecules: implications for Huntington's disease therapy. Proc Natl Acad Sci U S A 97, 6739-6744. Huang, K., Yanai, A., Kang, R., Arstikaitis, P., Singaraja, R.R., Metzler, M., Mullard, A., Haigh, B., Gauthier-Campbell, C., Gutekunst, C.A., Hayden, M.R., and El-Husseini, A. (2004). Huntingtin-interacting protein HIP14 is a palmitoyl transferase involved in palmi-toylation and trafficking of multiple neuronal proteins. Neuron 44, 977-986. Humbert, S., Bryson, E.A., Cordelieres, F.P., Connors, N.C., Datta, S.R., Finkbeiner, S., Greenberg, M.E., and Saudou, F. (2002). The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev Cell 2, 831-837. Hume, A.J., Finkel, J.S., Kamil, J.P., Coen, D.M., Culbertson, M.R., and Kalejta, R.F. (2008). Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 320, 797-799. Hunnicutt, G.R., Kosfiszer, M.G., and Snell, W.J. (1990). Cell body and flagellar agglu-tinins in Chlamydomonas reinhardtii: the cell body plasma membrane is a reservoir for agglutinins whose migration to the flagella is regulated by a functional barrier. J Cell Biol 111, 1605-1616. Jenkins, D., Seelow, D., Jehee, F.S., Perlyn, C.A., Alonso, L.G., Bueno, D.F., Donnai, D., Josifova, D., Mathijssen, I.M., Morton, J.E., Orstavik, K.H., Sweeney, E., Wall, S.A., Marsh, J.L., Nurnberg, P., Passos-Bueno, M.R., and Wilkie, A.O. (2007). RAB23 muta-tions in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am J Hum Genet 80, 1162-1170. Jenkins, P.M., Hurd, T.W., Zhang, L., McEwen, D.P., Brown, R.L., Margolis, B., Ver-hey, K.J., and Martens, J.R. (2006). Ciliary targeting of olfactory CNG channels requires the CNGB1b subunit and the kinesin-2 motor protein, KIF17. Curr Biol 16, 1211-1216. Jin, H., White, S.R., Shida, T., Schulz, S., Aguiar, M., Gygi, S.P., Bazan, J.F., and Na-chury, M.V. (2010). The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 141, 1208-1219. Johnston, J.A., Ward, C.L., and Kopito, R.R. (1998). Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 143, 1883-1898.

120
Kazantsev, A., Walker, H.A., Slepko, N., Bear, J.E., Preisinger, E., Steffan, J.S., Zhu, Y.Z., Gertler, F.B., Housman, D.E., Marsh, J.L., and Thompson, L.M. (2002). A bivalent Huntingtin binding peptide suppresses polyglutamine aggregation and pathogenesis in Drosophila. Nat Genet 30, 367-376. Klemm, R.W., Ejsing, C.S., Surma, M.A., Kaiser, H.J., Gerl, M.J., Sampaio, J.L., de Ro-billard, Q., Ferguson, C., Proszynski, T.J., Shevchenko, A., and Simons, K. (2009). Se-gregation of sphingolipids and sterols during formation of secretory vesicles at the trans-Golgi network. J Cell Biol 185, 601-612. Knighton, D.R., Zheng, J.H., Ten Eyck, L.F., Ashford, V.A., Xuong, N.H., Taylor, S.S., and Sowadski, J.M. (1991). Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253, 407-414. Knödler, A., Feng, S., Zhang, J., Zhang, X., Das, A., Peränen, J., and Guo, W. (2010). Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci U S A. 107, 6346-6351. Kobayashi, T., Gengyo-Ando, K., Ishihara, T., Katsura, I., and Mitani, S. (2007). IFT-81 and IFT-74 are required for intraflagellar transport in C. elegans. Genes Cells 12, 593-602. Kozminski, K.G., Johnson, K.A., Forscher, P., and Rosenbaum, J.L. (1993). A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc Natl Acad Sci U S A 90, 5519-5523. Krainer, A.R., Mayeda, A., Kozak, D., and Binns, G. (1991). Functional expression of cloned human splicing factor SF2: homology to RNA-binding proteins, U1 70K, and Drosophila splicing regulators. Cell 66, 383-394. Krosky, P.M., Baek, M.C., Jahng, W.J., Barrera, I., Harvey, R.J., Biron, K.K., Coen, D.M., and Sethna, P.B. (2003). The human cytomegalovirus UL44 protein is a substrate for the UL97 protein kinase. J Virol 77, 7720-7727. Landles, C., and Bates, G.P. (2004). Huntingtin and the molecular pathogenesis of Hun-tington's disease. Fourth in molecular medicine review series. EMBO Rep 5, 958-963. Li, Y., and Hu, J. (2011). Small GTPases and cilia. Protein Cell 2, 13-25. Liu, W., Murcia, N.S., Duan, Y., Weinbaum, S., Yoder, B.K., Schwiebert, E., and Satlin, L.M. (2005). Mechanoregulation of intracellular Ca2+ concentration is attenuated in col-lecting duct of monocilium-impaired orpk mice. Am J Physiol Renal Physiol 289, F978-988. Liu, Y.F., Dorow, D., and Marshall, J. (2000). Activation of MLK2-mediated signaling cascades by polyglutamine-expanded huntingtin. J Biol Chem 275, 19035-19040.

121
Loktev, A.V., Zhang, Q., Beck, J.S., Searby, C.C., Scheetz, T.E., Bazan, J.F., Slusarski, D.C., Sheffield, V.C., Jackson, P.K., and Nachury, M.V. (2008). A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev Cell. 15, 854-865. MacDonald, M.E., Ambrose, C.M., Duyao, M.P., Myers, R.H., Lin, C., Srinidhi, L., Barnes, G., Taylor, S.A., James, M., Groot, N., MacFarlane, H., Jenkins, B., Anderson, M., Wexler, N.S., Gusella, J.F., Bates, G.P., Baxendale, S., Hummerich, H., Kirby, S., North, M., Youngman, S., Mott, R., Zehetner, G., Sedlacek, Z., Poustka, A., Frischauf, A., Lehrach, H., Buckler, A.J., Church, D., Doucette-Stamm, L., O'Donovan, M.C., Riba-Ramirez, L., Shah, M., Stanton, V.P., Strobel, S.A., Draths, K.M., Wales, J.L., Peter Der-van, P., Housman, D.E., Altherr, M., Shiang, R., Thompsom, L., Fielder, T., Wasmuth, J.J., Tagle, D., Valdes, J., Elmer, L., Allard, M., Castilla, L., Swaroop, M., Blanchard, K., S. Collins, F.S., Snell, R., Holloway, T., Gillespie, K., Datson, N., Shaw, D., and Harper, P. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72, 971. Marcora, E., Gowan, K., and Lee, J.E. (2003). Stimulation of NeuroD activity by hun-tingtin and huntingtin-associated proteins HAP1 and MLK2. Proc Natl Acad Sci U S A 100, 9578-9583. Marschall, M., Marzi, A., aus dem Siepen, P., Jochmann, R., Kalmer, M., Auerochs, S., Lischka, P., Leis, M., and Stamminger, T. (2005). Cellular p32 recruits cytomegalovirus kinase pUL97 to redistribute the nuclear lamina. J Biol Chem 280, 33357-33367. Marschall, M., Stein-Gerlach, M., Freitag, M., Kupfer, R., van Den Bogaard, M., and Stamminger, T. (2001). Inhibitors of human cytomegalovirus replication drastically re-duce the activity of the viral protein kinase pUL97. J Gen Virol 82, 1439-1450. Marszalek, J.R., Ruiz-Lozano, P., Roberts, E., Chien, K.R., and Goldstein, L.S. (1999). Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc Natl Acad Sci U S A 96, 5043-5048. Matilla-Dueñas, A., Sánchez, I., Corral-Juan, M., Dávalos, A., Alvarez, R., and Latorre, P. (2010). Cellular and molecular pathways triggering neurodegeneration in the spinoce-rebellar ataxias. Cerebellum 9, 148-166. Maxfield, F.R., and McGraw, T.E. (2004). Endocytic recycling. Nat Rev Mol Cell Biol. 5, 121-132. Mazelova, J., Astuto-Gribble, L., Inoue, H., Tam, B.M., Schonteich, E., Prekeris, R., Mo-ritz, O.L., Randazzo, P.A., and Deretic, D. (2009a). Ciliary targeting motif VxPx directs assembly of a trafficking module through Arf4. Embo J 28, 183-192. Mazelova, J., Ransom, N., Astuto-Gribble, L., Wilson, M.C., and Deretic, D. (2009b). Syntaxin 3 and SNAP-25 pairing, regulated by omega-3 docosahexaenoic acid, controls

122
the delivery of rhodopsin for the biogenesis of cilia-derived sensory organelles, the rod outer segments. J Cell Sci. 122, 2003-2013. Metzler, M., Gan, L., Wong, T.P., Liu, L., Helm, J., Liu, L., Georgiou, J., Wang, Y., Bis-sada, N., Cheng, K., Roder, J.C., Wang, Y.T., and Hayden, M.R. (2007). NMDA receptor function and NMDA receptor-dependent phosphorylation of huntingtin is altered by the endocytic protein HIP1. J Neurosci. 27, 2298-2308. Michel, D., Schaarschmidt, P., Wunderlich, K., Heuschmid, M., Simoncini, L., Muhl-berger, D., Zimmermann, A., Pavic, I., and Mertens, T. (1998). Functional regions of the human cytomegalovirus protein pUL97 involved in nuclear localization and phosphoryla-tion of ganciclovir and pUL97 itself. J Gen Virol 79 ( Pt 9), 2105-2112. Milenkovic, L., Scott, M.P., and Rohatgi, R. (2009). Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. Cell Biol. 187, 365-374. Morgan, D., Turnpenny, L., Goodship, J., Dai, W., Majumder, K., Matthews, L., Gard-ner, A., Schuster, G., Vien, L., Harrison, W., Elder, F.F., Penman-Splitt, M., Overbeek, P., and Strachan, T. (1998). Inversin, a novel gene in the vertebrate left-right axis path-way, is partially deleted in the inv mouse. Nat Genet 20, 149-156. Moritz, O.L., Tam, B.M., Hurd, L.L., Peranen, J., Deretic, D., and Papermaster, D.S. (2001). Mutant rab8 Impairs docking and fusion of rhodopsin-bearing post-Golgi mem-branes and causes cell death of transgenic Xenopus rods. Mol Biol Cell 12, 2341-2351. Mruk, D.D., Lau, A.S., Sarkar, O., and Xia, W. (2007). A GTPase catenin interactions are involved in cell junction dynamics in the testis. J Androl 28, 742-754 Muchowski, P.J., and Wacker, J.L. (2005). Modulation of neurodegeneration by molecu-lar chaperones. Nat Rev Neurosci 6, 11-22. Munro, S. (2002). Organelle identity and the targeting of peripheral membrane proteins. Curr Opin Cell Biol 14, 506-514. Mykytyn, K., Braun, T., Carmi, R., Haider, N.B., Searby, C.C., Shastri, M., Beck, G., Wright, A.F., Iannaccone, A., Elbedour, K., Riise, R., Baldi, A., Raas-Rothschild, A., Gorman, S.W., Duhl, D.M., Jacobson, S.G., Casavant, T., Stone, E.M., and Sheffield, V.C. (2001). Identification of the gene that, when mutated, causes the human obesity syndrome BBS4. Nat Genet 28, 188-191. Nachury, M.V., Loktev, A.V., Zhang, Q., Westlake, C.J., Peranen, J., Merdes, A., Slu-sarski, D.C., Scheller, R.H., Bazan, J.F., Sheffield, V.C., and Jackson, P.K. (2007). A Core Complex of BBS Proteins Cooperates with the GTPase Rab8 to Promote Ciliary Membrane Biogenesis. Cell 129, 1201-1213.

123
Nachury, M.V., Seeley, E.S., and Jin, H. (2010). Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu Rev Cell Dev Biol. 26, 59-87. Nagai, Y., Fujikake, N., Ohno, K., Higashiyama, H., Popiel, H.A., Rahadian, J., Yama-guchi, M., Strittmatter, W.J., Burke, J.R., and Toda, T. (2003). Prevention of polygluta-mine oligomerization and neurodegeneration by the peptide inhibitor QBP1 in Drosophi-la. Hum Mol Genet 12, 1253-1259. Nalepa, G., Rolfe, M., and Harper, J.W. (2006). Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov 5, 596-613. Nauli, S.M., Alenghat, F.J., Luo, Y., Williams, E., Vassilev, P., Li, X., Elia, A.E., Lu, W., Brown, E.M., Quinn, S.J., Ingber, D.E., and Zhou, J. (2003). Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33, 129-137. Nauli, S.M., and Zhou, J. (2004). Polycystins and mechanosensation in renal and nodal cilia. Bioessays 26, 844-856. Nichol, J.N., Petruccelli, L.A., and Miller, W.H., Jr. (2009). Expanding PML's functional repertoire through post-translational mechanisms. Front Biosci 14, 2293-2306. NIDDK Recent Advances & Emergung Opportunities: Kidney, Urologic, Hematologic Disease http://www2.niddk.nih.gov/NR/rdonlyres/1456527C-5E95-461C9F8E29EAE04125FC/0/Advance2008_SOD_Polycystic.pdf Nonaka, S., Tanaka, Y., Okada, Y., Takeda, S., Harada, A., Kanai, Y., Kido, M., and Hi-rokawa, N. (1998). Randomization of left-right asymmetry due to loss of nodal cilia ge-nerating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell 95, 829-837. Olzmann, J.A., Li, L., and Chin, L.S. (2008). Aggresome formation and neurodegenera-tive diseases: therapeutic implications. Curr Med Chem 15, 47-60. Omori, Y., Zhao, C., Saras, A., Mukhopadhyay, S., Kim, W., Furukawa, T., Sengupta, P., Veraksa, A., and Malicki, J. (2008). Elipsa is an early determinant of ciliogenesis that links the IFT particle to membrane-associated small GTPase Rab8. Nat Cell Biol 10, 437-444. Orozco, J.T., Wedaman, K.P., Signor, D., Brown, H., Rose, L., and Scholey, J.M. (1999). Movement of motor and cargo along cilia. Nature 398, 674. Paulson, H.L., Bonini, N.M., and Roth, K.A. (2000). Polyglutamine disease and neuronal cell death. Proc Natl Acad Sci U S A 97, 12957-12958.

124
Pazour, G.J., Baker, S.A., Deane, J.A., Cole, D.G., Dickert, B.L., Rosenbaum, J.L., Wit-man, G.B., and Besharse, J.C. (2002). The intraflagellar transport protein, IFT88, is es-sential for vertebrate photoreceptor assembly and maintenance. J Cell Biol 157, 103-113. Pazour, G.J., and Bloodgood, R.A. (2008). Targeting proteins to the ciliary membrane. Curr Top Dev Biol 85, 115-149. Pedersen, L.B., and Rosenbaum, J.L. (2008). Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol 85, 23-61. Pedersen, L.B., Veland, I.R., Schrøder, J.M., and Christensen, S.T. (2008). Assembly of primary cilia. Dev Dyn 237, 1993-2006. Pellinen, T., Arjonen, A., Vuoriluoto, K., Kallio, K., Fransen, J.A., and Ivaska, J. (2006). Small GTPase Rab21 regulates cell adhesion and controls endosomal traffic of beta1-integrins. J Cell Biol 173, 767-780. Pellinen, T., Tuomi, S., Arjonen, A., Wolf, M., Edgren, H., Meyer, H., Grosse, R., Kitz-ing, T., Rantala, J.K., Kallioniemi, O., Fässler, R., Kallio, M., and Ivaska, J. (2008). Inte-grin trafficking regulated by Rab21 is necessary for cytokinesis. Dev Cell. 15,371-385. Pereira-Leal, J.B., and Seabra, M.C. (2000). The mammalian Rab family of small GTPases: definition of family and subfamily sequence motifs suggests a mechanism for functional specificity in the Ras superfamily. J Mol Biol 301, 1077-1087. Perez, M.K., Paulson, H.L., Pendse, S.J., Saionz, S.J., Bonini, N.M., and Pittman, R.N. (1998). Recruitment and the role of nuclear localization in polyglutamine-mediated ag-gregation. J Cell Biol 143, 1457-1470. Peters, P.J., Ning, K., Palacios, F., Boshans, R.L., Kazantsev, A., Thompson, L.M., Woodman, B., Bates, G.P., and D'Souza-Schorey, C. (2002). Arfaptin 2 regulates the ag-gregation of mutant huntingtin protein. Nat Cell Biol 4, 240-245. Pfeffer, S.R. (2001). Rab GTPases: specifying and deciphering organelle identity and function. Trends Cell Biol 11, 487-491. Pollitt, S.K., Pallos, J., Shao, J., Desai, U.A., Ma, A.A., Thompson, L.M., Marsh, J.L., and Diamond, M.I. (2003). A rapid cellular FRET assay of polyglutamine aggregation identifies a novel inhibitor. Neuron 40, 685-694. Porter, M.E., and Sale, W.S. (2000). The 9 + 2 axoneme anchors multiple inner arm dyneins and a network of kinases and phosphatases that control motility. J Cell Biol. 151, F37-42.

125
Praetorius, H.A., Frokiaer, J., Nielsen, S., and Spring, K.R. (2003). Bending the primary cilium opens Ca2+-sensitive intermediate-conductance K+ channels in MDCK cells. J Membr Biol 191, 193-200. Praetorius, H.A., and Spring, K.R. (2003). The renal cell primary cilium functions as a flow sensor. Curr Opin Nephrol Hypertens 12, 517-520. Prichard, M.N., Britt, W.J., Daily, S.L., Hartline, C.B., and Kern, E.R. (2005). Human cytomegalovirus UL97 Kinase is required for the normal intranuclear distribution of pp65 and virion morphogenesis. J Virol 79, 15494-15502. Prichard, M.N., Gao, N., Jairath, S., Mulamba, G., Krosky, P., Coen, D.M., Parker, B.O., and Pari, G.S. (1999). A recombinant human cytomegalovirus with a large deletion in UL97 has a severe replication deficiency. J Virol 73, 5663-5670. Prichard, M.N., Sztul, E., Daily, S.L., Perry, A.L., Frederick, S.L., Gill, R.B., Hartline, C.B., Streblow, D.N., Varnum, S.M., Smith, R.D., and Kern, E.R. (2008). Human cyto-megalovirus UL97 kinase activity is required for the hyperphosphorylation of retinoblas-toma protein and inhibits the formation of nuclear aggresomes. J Virol 82, 5054-5067. Qian, F., Germino, F.J., Cai, Y., Zhang, X., Somlo, S., and Germino, G.G. (1997). PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet 16, 179-183. Qin, H., Rosenbaum, J.L., and Barr, M.M. (2001). An autosomal recessive polycystic kidney disease gene homolog is involved in intraflagellar transport in C. elegans ciliated sensory neurons. Curr Biol 11, 457-461. Qin, H., Wang, Z., Diener, D., and Rosenbaum, J. (2007). Intraflagellar transport protein 27 is a small G protein involved in cell-cycle control. Curr Biol 17, 193-202. Riess, O., Rüb, U., Pastore, A., Bauer, P., and Schöls, L. (2008). SCA3: neurological fea-tures, pathogenesis and animal models. Cerebellum 7, 125-137. Rosenbaum, J.L., and Witman, G.B. (2002). Intraflagellar transport. Nat Rev Mol Cell Biol 3, 813-825. Ross, C.A. (1997). Intranuclear neuronal inclusions: a common pathogenic mechanism for glutamine-repeat neurodegenerative diseases? Neuron 19, 1147-1150. Scherzinger, E., Lurz, R., Turmaine, M., Mangiarini, L., Hollenbach, B., Hasenbank, R., Bates, G.P., Davies, S.W., Lehrach, H., and Wanker, E.E. (1997). Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90, 549-558. Schwartz, S.L., Cao, C., Pylypenko, O., Rak, A., and Wandinger-Ness, A. (2007). Rab GTPases at a glance. J Cell Sci 120, 3905-3910.

126
Segev, N. (2001). Ypt and Rab GTPases: insight into functions through novel interac-tions. Curr Opin Cell Biol 13, 500-511. Shafer, J.C., Winkelbauer, M.E., Williams, C.L., Haycraft, C.J., Desmond, R.A., and Yo-der, B.K. (2006). IFTA-2 is a conserved cilia protein involved in pathways regulating longevity and dauer formation in Caenorhabditis. J Cell Sci. 119, 4088-4100. Simpson, J.C., Griffiths, G., Wessling-Resnick, M., Fransen, J.A., Bennett, H., and Jones, A.T. (2004). A role for the small GTPase Rab21 in the early endocytic pathway. J Cell Sci 117, 6297-6311. Siroky, B.J., Ferguson, W.B., Fuson, A.L., Xie, Y., Fintha, A., Komlosi, P., Yoder, B.K., Schwiebert, E.M., Guay-Woodford, L.M., and Bell, P.D. (2006). Loss of primary cilia results in deregulated and unabated apical calcium entry in ARPKD collecting duct cells. Am J Physiol Renal Physiol 290, F1320-1328. Snow, J.J., Ou, G., Gunnarson, A.L., Walker, M.R., Zhou, H.M., Brust-Mascher, I., and Scholey, J.M. (2004). Two anterograde intraflagellar transport motors cooperate to build sensory cilia on C. elegans neurons. Nat Cell Biol 6, 1109-1113 Sönnichsen, B., De Renzis, S., Nielsen, E., Rietdorf, J., and Zerial, M. (2000). Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor im-aging of Rab4, Rab5, and Rab11. J Cell Biol. 149, 901-914. Stenmark, H. (2009). Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 10, 513-525. Stenmark, H., and Olkkonen, V.M. (2001). The Rab GTPase family. Genome Biol 2, REVIEWS3007. Stratigopoulos, G., Padilla, S.L., LeDuc, C.A., Watson, E., Hattersley, A.T., McCarthy, M.I., Zeltser, L.M., Chung, W.K., and Leibel, R.L. (2008). Regulation of Fto/Ftm gene expression in mice and humans. Am J Physiol Regul Integr Comp Physiol 294, R1185-1196. Sun, J., Xu, H., Negi, S., Subramony, S.H., and Hebert, M.D. (2007). Differential effects of polyglutamine proteins on nuclear organization and artificial reporter splicing. J Neu-rosci Res 85, 2306-2317. Sun, Y., Savanenin, A., Reddy, P.H., and Liu, Y.F. (2001). Polyglutamine-expanded hun-tingtin promotes sensitization of N-methyl-D-aspartate receptors via post-synaptic densi-ty 95. J Biol Chem. 276, 24713-24718.

127
Tam, B.M., Moritz, O.L., Hurd, L.B., and Papermaster, D.S. (2000). Identification of an outer segment targeting signal in the COOH terminus of rhodopsin using transgenic Xe-nopus laevis. J Cell Biol 151, 1369-1380. Tanaka, M., Machida, Y., Niu, S., Ikeda, T., Jana, N.R., Doi, H., Kurosawa, M., Nekooki, M., and Nukina, N. (2004). Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med 10, 148-154. Tao, B., Bu, S., Yang, Z., Siroky, B., Kappes, J.C., Kispert, A., and Guay-Woodford, L.M. (2009). Cystin localizes to primary cilia via membrane microdomains and a target-ing motif. J Am Soc Nephrol 20, 2570-2580. Torres, V. E., Harris, P. C., and Pirson, Y. (2007). Autosomal dominant polycystic kid-ney disease. Lancet 369, 1287-301 Trischler, M., Stoorvogel, W., and Ullrich, O. (1999). Biochemical analysis of distinct Rab5- and Rab11-positive endosomes along the transferrin pathway. J Cell Sci 112 ( Pt 24), 4773-4783. Trottier, Y., Lutz, Y., Stevanin, G., Imbert, G., Devys, D., Cancel, G., Saudou, F., Weber, C., David, G., Tora, L., Agid, Y., Brice, A., and Mandel, J.L. (1995). Polyglutamine ex-pansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature 378, 403-406. Tsiokas, L., Kim, E., Arnould, T., Sukhatme, V.P., and Walz, G. (1997). Homo- and he-terodimeric interactions between the gene products of PKD1 and PKD2. Proc Natl Acad Sci U S A 94, 6965-6970. van der Sluijs, P., Hull, M., Webster, P., Mâle, P., Goud, B., and Mellman, I. (1992). The small GTP-binding protein rab4 controls an early sorting event on the endocytic pathway. Cell 70 729-740. Vonsattel, J.P., Myers, R.H., Stevens, T.J., Ferrante, R.J., Bird, E.D., and Richardson, E.P.J. (1985). Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44, 559-577. Wacker, J.L., Zareie, M.H., Fong, H., Sarikaya, M., and Muchowski, P.J. (2004). Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by par-titioning monomer. Nat Struct Mol Biol 11, 1215-1222. Wang, F., Zhang, H., Zhang, X., Wang, Y., Ren, F., Zhang, X., Zhai, Y., and Chang, Z. (2008). Varp interacts with Rab38 and functions as its potential effector. Biochem Bio-phys Res Commun 372, 162-167. Wang, Q., Pan, J., and Snell, W.J. (2006). Intraflagellar transport particles participate di-rectly in cilium-generated signaling in Chlamydomonas. Cell 125, 549-562.

128
Ward, E.S., Martinez, C., Vaccaro, C., Zhou, J., Tang, Q., and Ober, R.J. (2005). From sorting endosomes to exocytosis: association of Rab4 and Rab11 GTPases with the Fc receptor, FcRn, during recycling. Mol Biol Cell. 16, 2028-2038. Warrick, J.M., Chan, H.Y., Gray-Board, G.L., Chai, Y., Paulson, H.L., and Bonini, N.M. (1999). Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet 23, 425-428. Warrick, J.M., Morabito, L.M., Bilen, J., Gordesky-Gold, B., Faust, L.Z., Paulson, H.L., and Bonini, N.M. (2005). Ataxin-3 suppresses polyglutamine neurodegeneration in Dro-sophila by a ubiquitin-associated mechanism. Mol Cell 18, 37-48. Wemmer, K.A., and Marshall, W.F. (2004). Flagellar motility: all pull together. Curr Bi-ol. 14, R992-993 Wickner, S., Maurizi, M.R., and Gottesman, S. (1999). Posttranslational quality control: folding, refolding, and degrading proteins. Science 286, 1888-1893. Wiens, C.J., Tong, Y., Esmail, M.A., Oh, E., Gerdes, J.M., Wang, J., Tempel, W., Ratt-ner, J.B., Katsanis, N., Park, H.W., and Leroux, M.R. (2010). Bardet-Biedl syndrome-associated small GTPase ARL6 (BBS3) functions at or near the ciliary gate and mod-ulates Wnt signaling. J Biol Chem 285, 16218-16230. Wigley, W.C., Fabunmi, R.P., Lee, M.G., Marino, C.R., Muallem, S., DeMartino, G.N., and Thomas, P.J. (1999). Dynamic association of proteasomal machinery with the centro-some. J Cell Biol. 145, 481-490. Xu, C., Rossetti, S., Jiang, L., Harris, P.C., Brown-Glaberman, U., Wandinger-Ness, A., Bacallao, R., and Alper, S.L. (2007). Human ADPKD primary cyst epithelial cells with a novel, single codon deletion in the PKD1 gene exhibit defective ciliary polycystin locali-zation and loss of flow-induced Ca2+ signaling. Am J Physiol Renal Physiol 292, F930-945. Yoder, B.K., Hou, X., and Guay-Woodford, L.M. (2002). The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol 13, 2508-2516. Yoshimura, S., Egerer, J., Fuchs, E., Haas, A.K., and Barr, F.A. (2007). Functional dis-section of Rab GTPases involved in primary cilium formation. J Cell Biol 178, 363-369. Zaghloul, N.A., and Katsanis, N. (2009). Mechanistic insights into Bardet-Biedl syn-drome, a model ciliopathy. J Clin Invest 119, 428-437. Zerial, M., and McBride, H. (2001). Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2, 107-117.

129
Zhang, C., Williams, E.H., Guo, Y., Lum, L., and Beachy, P.A. (2004). Extensive phos-phorylation of Smoothened in Hedgehog pathway activation. Proc Natl Acad Sci U S A 101, 17900-17907. Zhang, X., He, X., Fu, X.Y., and Chang, Z. (2006). Varp is a Rab21 guanine nucleotide exchange factor and regulates endosome dynamics. J Cell Sci 119, 1053-1062.

130
APPENDIX A
SUPPLEMENTAL FIGURES FOR HUMAN CYTOMEGALOVIRUS UL97 KINASE PREVENTS THE DEPOSITION OF MUTANT PROTEIN AGGREGATES IN CEL-
LULAR MODELS OF HUNTINGTON’S DISEASE AND ATAXIA
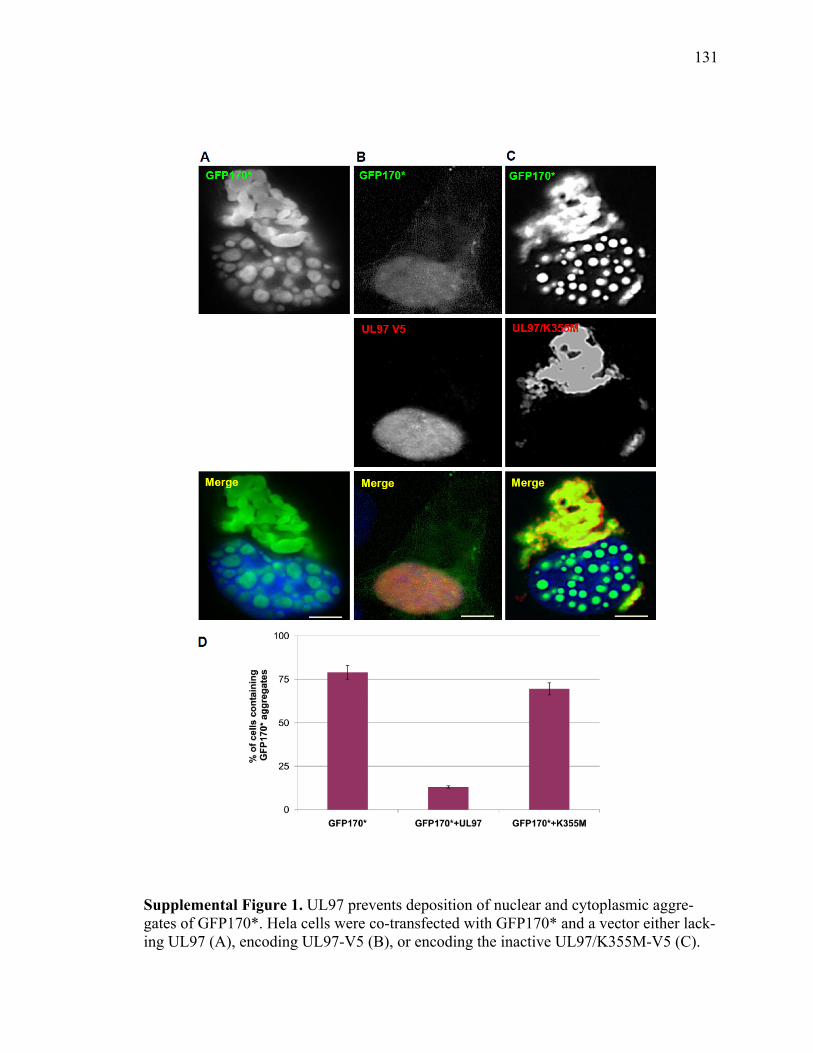
131
Supplemental Figure 1. UL97 prevents deposition of nuclear and cytoplasmic aggre-gates of GFP170*. Hela cells were co-transfected with GFP170* and a vector either lack-ing UL97 (A), encoding UL97-V5 (B), or encoding the inactive UL97/K355M-V5 (C).
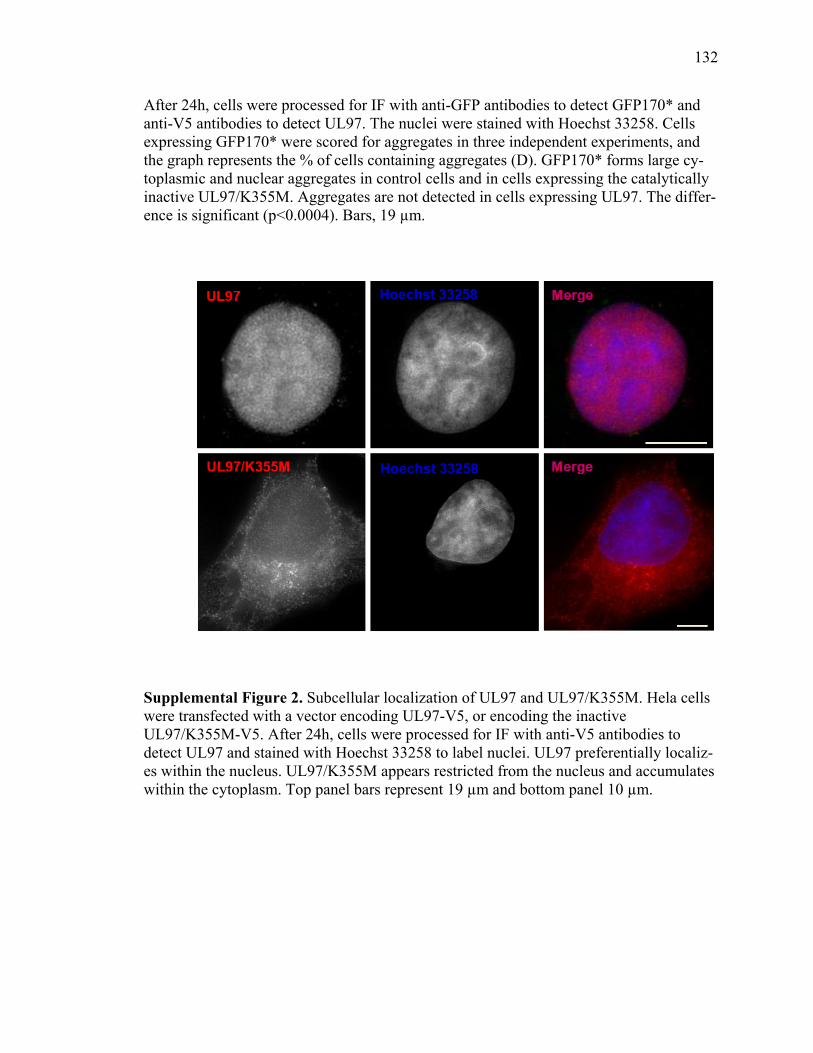
132
After 24h, cells were processed for IF with anti-GFP antibodies to detect GFP170* and anti-V5 antibodies to detect UL97. The nuclei were stained with Hoechst 33258. Cells expressing GFP170* were scored for aggregates in three independent experiments, and the graph represents the % of cells containing aggregates (D). GFP170* forms large cy-toplasmic and nuclear aggregates in control cells and in cells expressing the catalytically inactive UL97/K355M. Aggregates are not detected in cells expressing UL97. The differ-ence is significant (p<0.0004). Bars, 19 µm.
Supplemental Figure 2. Subcellular localization of UL97 and UL97/K355M. Hela cells were transfected with a vector encoding UL97-V5, or encoding the inactive UL97/K355M-V5. After 24h, cells were processed for IF with anti-V5 antibodies to detect UL97 and stained with Hoechst 33258 to label nuclei. UL97 preferentially localiz-es within the nucleus. UL97/K355M appears restricted from the nucleus and accumulates within the cytoplasm. Top panel bars represent 19 µm and bottom panel 10 µm.