the rgk family: a regulatory tail of small gtp-binding proteins
TRANSCRIPT

Update TRENDS in Cell Biology Vol.15 No.12 December 2005640
The RGK family: a regulatory tail of small GTP-bindingproteins
Kathleen Kelly
Cell and Cancer Biology Branch, Center for Cancer Research, NCI, NIH Building 37, Room 1068, Bethesda, MD 20892, USA
RGK proteins are small Ras-related GTP-binding pro-
teins that function as potent inhibitors of voltage-
dependent calcium channels, and two members of the
family, Gem and Rad, modulate Rho-dependent remo-
deling of the cytoskeleton. Within the Ras superfamily,
RGK proteins have distinct structural and regulatory
characteristics. It is an open question as to whether RGK
proteins catalyze GTP hydrolysis in vivo. Binding of
calmodulin and the 14-3-3 protein to RGK proteins
controls downstream pathways. Here, we discuss the
structural and functional properties of RGK proteins and
highlight recent work by Beguin and colleagues addres-
sing the mechanism of Gem regulation by calmodulin
and 14-3-3.
Introduction
RGK (Rad, Gem, Kir) proteins are Ras-related GTP-binding proteins. The family comprises four members,Gem (the mouse homolog is often referred to as Kir), Rad,Rem and Rem2. The basic structure of RGK proteinsconsists of a Ras-like core, albeit with crucial substitutions(see below), a conserved C-terminal extension, and anN-terminal extension that is not conserved among familymembers (see Figure 1). RGK proteins are expressed in atissue-specific manner and are subject to both transcrip-tional and posttranscriptional mechanisms that dynami-cally regulate protein levels relative to environmentalcues [1–3]. RKG family members suppress voltage-gatedcalcium channel activity upon heterologous expression [4]and interact with and inhibit the Rho/Rho kinase pathwayto function in cytoskeletal remodeling [3]. Recently,Beguin et al. [5] have provided important clues to aidour understanding of how these events are mutuallyregulated by beginning to biochemically unravel themechanism of control of Gem by competinginteracting proteins.
RGK structure
Ras-family GTPases share a set of conserved elements,designated G1 through G5, involved in GDP/GTP bindingand GTP hydrolysis. Ras-family GTPases possess high-affinity guanine-nucleotide binding activity and relativelylow, but easily detectable, intrinsic GTP hydrolysisactivities. Other cellular regulatory proteins, known asGTPase-activating proteins (GAPs), accelerate the intrin-sic GTPase activity to promote formation of the GDP-
Corresponding author: Kelly, K. ([email protected]).Available online 19 October 2005
www.sciencedirect.com
bound form of Ras-family proteins, whereas GDP–GTPexchange factors (GEFs) promote formation of the GTP-bound forms [6].
RGK proteins contain crucial substitutions in aminoacids that participate in GTP hydrolysis, including aminoacids at positions equivalent to the G2 (T35) and G3(DXXG60) elements of Ras. In addition, recombinant Gem[1] and Rem [2] display exceedingly low levels of intrinsicGTPase activity. It is quite possible that RGK familymembers are not GTPases but guanine nucleotide-bindingproteins, predominantly found in the GTP-bound state asa result of the approximately ten-fold higher concen-trations of GTP relative to GDP in most cells. Analternative hypothesis is that the RGK GTPase activityis highly dependent upon GAPs and a unique catalyticmechanism, although there is no evidence for this.
Another notable characteristic of RGK proteins is theirconserved 40-residue C-terminal extension, which lacksmotifs directing fatty acylation, a biochemical modifi-cation that mediates the membrane association of themajority of Ras family proteins. Features of the RGKC-terminus include its high fraction (25%) of basic aminoacid residues, and the possession of crucial serinephosphorylation sites and binding regions for Ca2C/cal-modulin (CaM) and for 14-3-3 [7].
RGK inhibition of VDCC
Voltage-dependent calcium channels (VDCCs) regulateentry of extracellular calcium into electrically excitablecells, which initiates excitation coupling to crucialprocesses including neurotransmitter release, secretion,contraction and transcription [8]. Calcium channels aremultisubunit complexes comprising a main pore-forminga1 subunit, which determines the channel ‘type’, com-plexed with b, a2d and g subunits. Cavb subunits arerequired for proper membrane trafficking of a1 and areinvolved in modulating the electrophysiological propertiesof the channel [9]. Various signaling pathways can acutelymodulate calcium entry occurring via VDCCs. One suchpathway, CaM, can function to alternatively inhibit orfacilitate L-type channels through interaction with theCava subunit.
Gem was identified as an interacting protein thatbinds the Cavb3 subunit through its Ras-like core [4].Overexpression of Gem dramatically inhibits the elec-trical current density of various ectopically andendogenously expressed VDCCs, including L-, P/Q-and N-, but not b-deficient T-types [4,7]. Other RGKfamily members including Rad, Rem and Rem2 interact

14-3-3 bindingRegulatory phosphorylations
264 288CAM Binding
RAS Core (GTP-bindingNH2
GemRadRem1Rem2 40%52%
(% identical/conserved changes)7%
S261 S289
TRENDS in Cell Biology
Figure 1. Diagrammatic representation of the human Gem protein with the
positions of regulatory phosphorylations, 14-3-3 and Ca2C/CaM binding, and the
Ras-related core indicated. The percentage of identical or conserved amino acid
residues among the family members (hGem, hRad, hRem1,and hRem2) in the core
and the NH2- and COOH-termini is shown.
Update TRENDS in Cell Biology Vol.15 No.12 December 2005 641
with Cavb subunits and have been shown to inhibitL-type channels, suggesting a conserved structure–function relationship with respect to Cavb interactionswithin the family [10–12].
There appear to be at least two mechanisms thatcontribute to VDCC inhibition. Overexpression of Gem orRem can interfere with de novo calcium channel assemblyby competing for the binding surface on Cavb thatinteracts with Cava [4,13]. This results in a reduction ofplasma membrane VDCC expression and consequently aninhibition of channel activity, and might be relevant fordevelopmental modulation of VDCC expression byRKG proteins.
Of significance for dynamic regulation of VDCCfunction is the question of how RKG proteins affect pre-existing channels. Although relatively little has been donethat directly addresses the half-lives of calcium channels,one prior study suggests that VDCC half-lives in ratsympathetic ganglion (RSG) neurons are greater than 36–48 h [14]. If these data are correct, RGK inhibition ofchannel trafficking alone would be expected to inhibitVDCC function over a time-frame of days. Yet, endogen-ously expressed channels are completely inhibited within18 h of introducing DNA encoding Gem into RSG neurons[7]. Importantly, it has been directly demonstrated in HITT15 pancreatic b cells infected with adenovirus carryingRem2 that the surface expression of endogenous Cava1
receptors is unchanged despite a virtually completeinhibition of the VDCC current [12]. Thus, an interestinginference is that that Rem2, and possibly Gem, mightacutely produce nonconducting channel species as a resultof interacting with Cavb.
What regulatory controls act on the RGK proteins tomodulate their VDCC-inhibitory function? GTP binding toGem appears to be required, as suggested by assaysmeasuring recombinant Gem interaction with Cavbin vitro [4]. A similar conclusion is inferred by the lackof in vivo VDCC inhibitory activity for a Gem pointmutation (S89N) whose recombinant form demonstratesreduced GTP binding affinity. A functional Ca2C/CaMbinding site is also necessary for full inhibition of VDCCsby transfected Gem [4,5,7], although the CaM-binding site
www.sciencedirect.com
does not appear to be required for recombinant Gembinding to Cavb [4]. In transfection systems, mutation ofthe CaM-binding site results in predominant localizationof Gem to the nucleus, suggesting that Gem mutantslacking CaM binding are inactive owing to their mis-localization and loss of availability at the plasmamembrane [4]. An important but unresolved question iswhether Ca2C/CaM binding regulates Gem localization inphysiological situations. Ca2C/CaM exists in both thecytoplasmic and nuclear compartments, although therelative concentrations of free Ca2C/CaM in the twocompartments might vary with cell type. One possibilityis that Ca2C/CaM binding to Gem promotes the inter-action of Gem with a cytoplasmic and or membrane-associated scaffolding protein. Other RGK proteinssimilarly require cognate Ca2C/CaM-binding regions inorder to demonstrate maximal VDCC-inhibitory functions[10–12]. In contrast to Ca2C/CaM binding, loss of 14-3-3binding does not affect Gem VDCC function [5,7].
RGK regulation of cytoskeletal remodeling
Ectopic expression of Gem leads to remodeling of thecytoskeleton in a manner consistent with Gem inhibitionof ROK [3]. Gem or Rad expression leads to neuriteextension in neuroblastoma cells and disassembly ofstress fibers and focal adhesions in fibroblasts or epithelialcells. ROK is a major effector of the Rho GTPase, and theformation of a ROKb–Gem complex blocks the interactionof ROK with specific substrates including myosin lightchain (MLC) and myosin phosphatase [3]. Similarly, Radinteracts with and inhibits the ROKa isoform, but Remand Rem2 do not appear to regulate this specific form ofcytoskeletal remodeling. Other forms of morphologicalalterations, such as ‘dendritic processes’, have beendescribed following overexpression of RGK familymembers. It is not clear whether or in what cell typesdendritic processes are normally dependent upon ROK.
Gem-mediated reorganization of the cytoskeleton isregulated by mechanisms that are different from thosethat mediate Gem VDCC inhibition. Phosphorylation ofserines 261 and 289, located in the C-terminal extension,is required for Gem-mediated remodeling of the cytoske-leton [7]. In addition to regulating cytoskeletal reorgan-ization, phosphorylation of Ser289 in conjunction withSer23 results in bi-dentate 14-3-3 binding. Two conse-quences of 14-3-3 binding are stabilization of thephosphorylation levels of Ser261 and Ser289 andincreased Gem protein half-life [7]. As the Gem core issufficient for complex formation with ROKb, one interpret-ation of these data is that C-terminal phosphorylationsmediate a conformational change to the core, which isstabilized by formation of a 14-3-3 complex, and isnecessary for ROK binding. Alternatively, as ROK andits substrates are localized to membranes and theassociated contractile apparatus in cells [15], perhapsappropriate Gem colocalization with ROK is dependent onphosphorylation. Gem proteins bearing mutations in theCaM-binding region or with reduced GTP-binding affinityare fully active with respect to cytoskeletal remodeling.
Update TRENDS in Cell Biology Vol.15 No.12 December 2005642
Recent advances
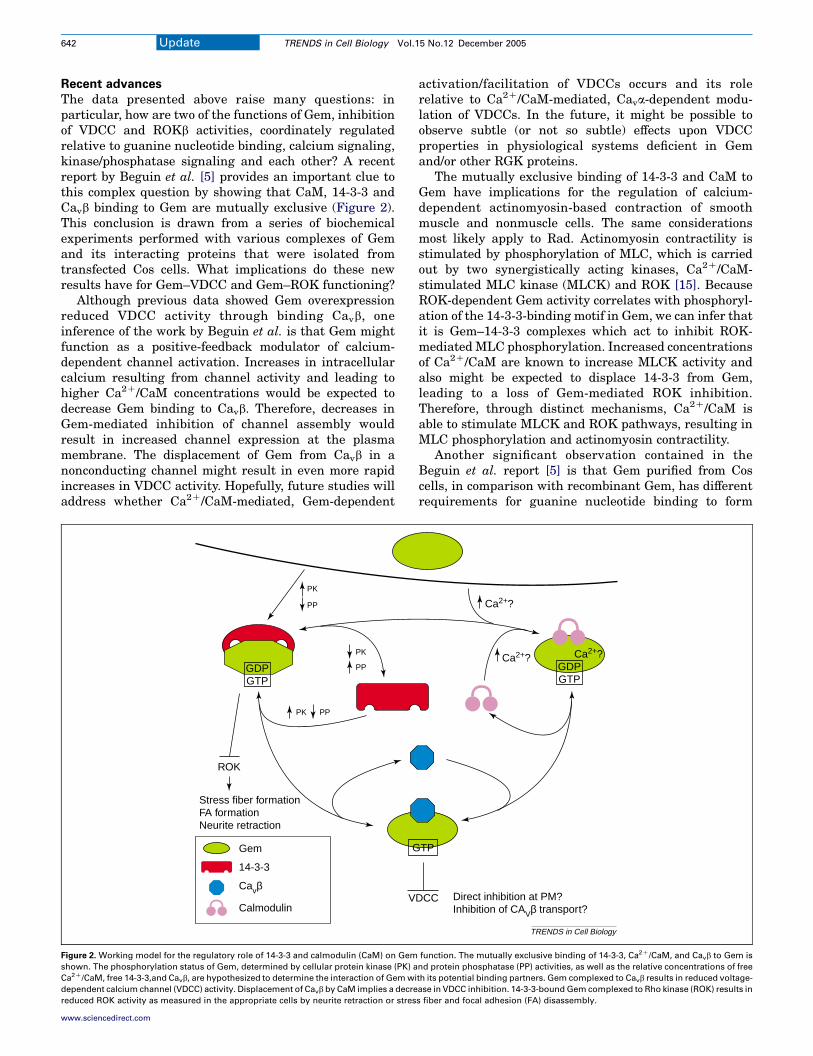
The data presented above raise many questions: inparticular, how are two of the functions of Gem, inhibitionof VDCC and ROKb activities, coordinately regulatedrelative to guanine nucleotide binding, calcium signaling,kinase/phosphatase signaling and each other? A recentreport by Beguin et al. [5] provides an important clue tothis complex question by showing that CaM, 14-3-3 andCavb binding to Gem are mutually exclusive (Figure 2).This conclusion is drawn from a series of biochemicalexperiments performed with various complexes of Gemand its interacting proteins that were isolated fromtransfected Cos cells. What implications do these newresults have for Gem–VDCC and Gem–ROK functioning?
Although previous data showed Gem overexpressionreduced VDCC activity through binding Cavb, oneinference of the work by Beguin et al. is that Gem mightfunction as a positive-feedback modulator of calcium-dependent channel activation. Increases in intracellularcalcium resulting from channel activity and leading tohigher Ca2C/CaM concentrations would be expected todecrease Gem binding to Cavb. Therefore, decreases inGem-mediated inhibition of channel assembly wouldresult in increased channel expression at the plasmamembrane. The displacement of Gem from Cavb in anonconducting channel might result in even more rapidincreases in VDCC activity. Hopefully, future studies willaddress whether Ca2C/CaM-mediated, Gem-dependent
PK
PP
GDPGTP
G
V
PK
PP
ROK
Stress fiber formationFA formationNeurite retraction
Gem
14-3-3
Cavβ
Calmodulin
PK PP
Figure 2. Working model for the regulatory role of 14-3-3 and calmodulin (CaM) on Gem
shown. The phosphorylation status of Gem, determined by cellular protein kinase (PK) a
Ca2C/CaM, free 14-3-3,and Cavb, are hypothesized to determine the interaction of Gem wi
dependent calcium channel (VDCC) activity. Displacement of Cavb by CaM implies a decre
reduced ROK activity as measured in the appropriate cells by neurite retraction or stres
www.sciencedirect.com
activation/facilitation of VDCCs occurs and its rolerelative to Ca2C/CaM-mediated, Cava-dependent modu-lation of VDCCs. In the future, it might be possible toobserve subtle (or not so subtle) effects upon VDCCproperties in physiological systems deficient in Gemand/or other RGK proteins.
The mutually exclusive binding of 14-3-3 and CaM toGem have implications for the regulation of calcium-dependent actinomyosin-based contraction of smoothmuscle and nonmuscle cells. The same considerationsmost likely apply to Rad. Actinomyosin contractility isstimulated by phosphorylation of MLC, which is carriedout by two synergistically acting kinases, Ca2C/CaM-stimulated MLC kinase (MLCK) and ROK [15]. BecauseROK-dependent Gem activity correlates with phosphoryl-ation of the 14-3-3-binding motif in Gem, we can infer thatit is Gem–14-3-3 complexes which act to inhibit ROK-mediated MLC phosphorylation. Increased concentrationsof Ca2C/CaM are known to increase MLCK activity andalso might be expected to displace 14-3-3 from Gem,leading to a loss of Gem-mediated ROK inhibition.Therefore, through distinct mechanisms, Ca2C/CaM isable to stimulate MLCK and ROK pathways, resulting inMLC phosphorylation and actinomyosin contractility.
Another significant observation contained in theBeguin et al. report [5] is that Gem purified from Coscells, in comparison with recombinant Gem, has differentrequirements for guanine nucleotide binding to form
Ca2+?
Ca2+? Ca2+?GDPGTP
TP
DCC Direct inhibition at PM?Inhibition of CAVβ transport?
TRENDS in Cell Biology
function. The mutually exclusive binding of 14-3-3, Ca2C/CaM, and Cavb to Gem is
nd protein phosphatase (PP) activities, as well as the relative concentrations of free
th its potential binding partners. Gem complexed to Cavb results in reduced voltage-
ase in VDCC inhibition. 14-3-3-bound Gem complexed to Rho kinase (ROK) results in
s fiber and focal adhesion (FA) disassembly.

Update TRENDS in Cell Biology Vol.15 No.12 December 2005 643
complexes with Cavb. Specifically, immunopurified Gembinds recombinant Cavb, and the interaction is neitherstimulated by incubation with GTP-gS nor inhibited byincubation with GDP-bS. By contrast, GTP-gS- versusGDP-bS-bound recombinant Gem interacts significantlymore avidly with immunopurified Cavb. The latter dataand virtually identical prior experiments [4] are the onlydirect observations suggesting that GTP binding to Gem isrequired for complex formation with Cavb. One interpret-ation of the recent data is that Gem exists in aconstitutively active form in Cos cells, and GDP-bS cannotdisplace endogenously bound GTP. Different scenarios arealso plausible. For example, other cellular proteins mightexist that mediate formation of complexes containing bothGem and Cavb, and such complexes do not have specificguanine-nucleotide binding requirements. AdditionalRGK mutants deficient in guanine-nucleotide bindingshould be considered in future studies. Because only asingle cognate guanine-nucleotide binding-deficientmutant (e.g. GemS89N) has been used in the variousstudies described to date, it is possible that the functionaleffects observed for VDCC might be related to a structuralmodification other than GDP/GTP binding.
Concluding remarks
RGK family proteins are fascinating for their uniquestructure, their means of regulation and their function asmodulators of VDCC and cytoskeletal organization.Although much has been learned from overexpression ofRGK proteins, systems that explore RGK proteins in aphysiological context and at physiological concentrationswill be an important direction for the future. For example,almost nothing is known about the localization ofendogenous RGK proteins and whether their locationshifts with respect to changes in environmental signals. Inaddition, the RGK field has been hampered by a lack ofinformation about the three-dimensional structure ofthese molecules. Such structures might help predictpotential mechanisms of GTP catalysis and identifypotential protein-interaction domains, which in turn willlead to the development of mutants that can be used infunctional analyses.
Reproduction of material
Interested in reproducing part or all of an article published by ElGlobal Rights Department with details of how and where the req
on-line, plea
http://www.elsevier.com/wps/find/obtainpermi
Alternatively, ple
ElseviGlobal Rights D
PO BoxOxford OX5
Phone: (+44) 18Fax: (+44) 186
permissions@e
www.sciencedirect.com
AcknowledgementsThe author acknowledges support by the intramural research program ofthe NIH, National Cancer Institute, Center for Cancer Research. I wouldlike to thank Andy Cheng for producing the illustrations and UlrichSiebenlist for his comments. I apologize to those authors whose work isnot specifically referenced owing to length restrictions.
References
1 Cohen, L. et al. (1994) Transcriptional activation of a ras-like gene(kir) by oncogenic tyrosine kinases. Proc. Natl. Acad. Sci. U. S. A. 91,12448–12452
2 Finlin, B.S. et al. (2000) Rem2, a new member of the Rem/Rad/Gem/-Kir family of Ras-related GTPases. Biochem. J. 347, 223–231
3 Ward, Y. et al. (2002) The GTP binding proteins Gem and Rad arenegative regulators of the Rho-Rho kinase pathway. J. Cell Biol. 157,291–302
4 Beguin, P. et al. (2001) Regulation of Ca2C channel expression at thecell surface by the small G-protein kir/Gem. Nature 411, 701–706
5 Beguin, P. et al. (2005) 14-3-3 and calmodulin control subcellulardistribution of Kir/Gem and its regulation of cell shape and calciumchannel activity. J. Cell Sci. 118, 1923–1934
6 Wennerberg, K. et al. (2005) The Ras superfamily at a glance. J. CellSci. 118, 843–846
7 Ward, Y. et al. (2004) Phosphorylation of critical serine residues inGem separates cytoskeletal reorganization from down-regulation ofcalcium channel activity. Mol. Cell. Biol. 24, 651–661
8 Maier, L.S. and Bers, D.M. (2002) Calcium, calmodulin, and calcium-calmodulin kinase II: heartbeat to heartbeat and beyond. J. Mol. Cell.Cardiol. 34, 919–939
9 Dolphin, A.C. (2003) Beta subunits of voltage-gated calcium channels.J. Bioenerg. Biomembr. 35, 599–620
10 Beguin, P. et al. (2005) Roles of 14-3-3 and calmodulin binding insubcellular localization and function of the small G protein Rem2.Biochem. J. 390, 67–75
11 Finlin, B.S. et al. (2003) Regulation of voltage-gated calcium channelactivity by the Rem and Rad GTPases. Proc. Natl. Acad. Sci. U. S. A.100, 14469–14474
12 Finlin, B.S. et al. Regulation of L-type Ca2C channel activity andinsulin secretion by the Rem2 GTPase. J. Biol. Chem. (in press)
13 Sasaki, T. et al. (2005) Direct inhibition of the interaction betweenalpha-interaction domain and beta-interaction domain of voltage-dependent Ca2C channels by Gem. J. Biol. Chem. 280, 9308–9312
14 Moss, F.J. et al. (2002) The novel product of a five-exon stargazin-related gene abolishes Ca(V)2.2 calcium channel expression. EMBO J.21, 1514–1523
15 Fukata, Y. et al. (2001) Rho–Rho-kinase pathway in smooth musclecontraction and cytoskeletal reorganization of non-muscle cells.Trends Pharmacol. Sci. 22, 32–39
0962-8924/$ - see front matter Published by Elsevier Ltd.
doi:10.1016/j.tcb.2005.10.002
from Elsevier articles
sevier, or one of our article figures? If so, please contact ouruested material will be used. To submit a permission requestse visit:
ssionform.cws_home/obtainpermissionform
ase contact:
erepartment
800,1DX, UK.65-8438305-853333lsevier.com