the neurology · of neurology, johns hopkins university, baltimore, maryland, and bernd kieseier,...
TRANSCRIPT

v o l u m e 6 n u m b e r 1 | W I n T e r 2 0 1 4
Selected reports from the
29th Congress of the european Committee for Treatment and research
in multiple Sclerosis (eCTrImS 2013)
Peter A. Calabresi, mDGuest editor
CONTINUING EDUCATION FOR PHYSICIANS AND NURSES: 2.5 CREDITS AVAILABLE
NeurologyThe
REPORT
This activity is supported by an educational grant from Biogen Idec.

Guest Editor: Peter A. Calabresi, MD
The opinions or views expressed in this publication are those of the authors and do not necessarily reflect the opinions or recommendations of Biogen Idec, the University of Cincinnati, or the publisher, Direct One Communications, Inc. Please consult the full prescribing information before using any medication mentioned in this publication.
This publication was made possible through an educational grant from Biogen Idec.
Copyright © 2014 by Direct One Communications, Inc. All rights reserved. Printed in the USA.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 1
V O L U M E 6 N U M B E R 1 | W I N T E R 2 0 1 4
Selected Reports from the 29th Congress of the European Committee for Treatment
and Research in Multiple Sclerosis (ECTRIMS 2013)
Peter A. Calabresi, MD, Guest Editor
NeurologyThe
REPORT
4 IntroductionPeter A. Calabresi, MDJohns Hopkins Multiple Sclerosis Center and School of Medicine, Baltimore, Maryland
6 B-CellModulationinMultipleSclerosisRiley M. Bove, MDPartners Multiple Sclerosis Center, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts
12 TheCurrentClinicalArenaofProgressiveMultipleSclerosisCarrie M. Hersh, DOMellen Center for Multiple Sclerosis Treatment and Research, Neurological Institute, Cleveland Clinic, Cleveland, Ohio
17 AdvancesinImmunomodulatoryTherapyforMultipleSclerosisSona Narula, MDChildren’s Hospital of Philadelphia, Philadelphia, Pennsylvania
23 ANewEraofTherapyinMultipleSclerosis:BalancingtheOptionsandChallengesAheadJennifer L. Orthmann-Murphy, MD, PhDHospital of the University of Pennsylvania, Philadelphia, Pennsylvania
30 NatalizumabandDimethylFumarate:AFreshTakeonPivotalTrialsandReportsfromOngoingMonitoringCarolyn Bevan, MD, MSMultiple Sclerosis Center, University of California, San Francisco, School of Medicine, San Francisco, California
35 CME/CNEPostTestandEvaluation

2 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
RATIONALE AND PURPOSEA flood of recent approvals of novel immunotherapies for treating the relapsing-remitting form of multiple sclerosis (MS) has greatly expanded the options available to clinicians and has made life more bearable for the majority of patients with MS. However, comparatively little or no improvement has been made in understanding the pathologic development of MS, identifying potential therapeutic targets, or treating patients whose disease progresses.
This edition of The Neurology Report focuses on the etiology and immunopathogenesis of MS, the search for immunologic targets for pharmacologic suppression, and the results of recent clinical studies of novel biologic agents that appear to interrupt the course of this disease from many different directions and promise to change forever the way we now approach the management of MS.
The authors of this report also delve into the complexities of deciding when and how aggressively to initiate disease-modifying therapy, choosing appropriate therapy for individual patients, anticipating and managing the adverse effects of immunomodulatory drugs, and transitioning patients from one agent to another. This report is based upon presentations delivered at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS 2013), held October 2–5, 2013, in Copenhagen, Denmark.
The articles in this edition, written from the academic perspective of physicians-in-training at leading medical institutions, summarize the import of these new findings and place them into clinical context. This
About This CME/CNE Activityactivity has been developed and approved by a planning committee of nationally recognized thought leaders to meet a perceived educational need to provide neurologists, other physicians, and nurses with diagnostic and therapeutic strategies to help them perform their clinical roles.
LEARNING OBJECTIVESAfter studying this issue of The Neurology Report, participants in this educational activity should be able to:
• Discuss efforts to redefine the clinical course of MS, develop new clinical outcome assessment tools, and identify key research areas and molecular therapeutic targets.
• Summarize the development of pharmacologic therapies for relapsing-remitting MS and their relative advantages and disadvantages.
• Explore the tools and clinical evidence now available for choosing initial therapy for individual patients, monitoring their progress, and transitioning them from one disease-modifying therapy to another.
• Describe the role of B cells in the pathogenesis of MS and current research into the potential role of B-cell modulation in its treatment.
• Review current research in understanding the pathogenesis of disease progression in MS and efforts to prevent or arrest further deterioration in patients with progressive forms of the disease.
TARGET AUDIENCENeurologists, other physicians, and nurses significantly involved in the diagnosis and management of MS should find participating in this educational activity valuable.
ACCREDITATION AND CREDIT DESIGNATIONPhysicians: This activity has been planned and
implemented in accordance with the Essential Areas and Policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint sponsorship of the University of Cincinnati and Direct One Communications, Inc. The University of Cincinnati is accredited by the ACCME to provide continuing medical education for physicians.
The University of Cincinnati designates this Enduring Material Activity for a maximum of 2.5 AMA PRA Category 1 Credits ™. Physicians should only claim credit commensurate with the extent of their participation in the activity.
Nurses: A total of 2.5 continuing education contact hours for nurses are approved by the Ohio Board of Nursing through the OBN Approver Unit at the University of Cincinnati College of Nursing, Continuing Education Program (OBN-011-93). Contact hours are valid in most states. Program #140107-1.
CREDIT AVAILABILITYActivity release date: January 5, 2014 Expiration date: January 6, 2015
METHOD OF PARTICIPATIONThis Enduring Material Activity is available in print and online at www.NeurologyReport.com and consists of an introduction, five articles, a postactivity assessment, and an evaluation. Estimated time to complete the activity is 2.5 hours.
To receive credit, participants must read the CME information on these two pages, including the learning objectives and disclosure statements, as well as the full content of this

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 3
monograph, and then complete the post test and evaluation form online at www.NeurologyReport.com. Upon successful completion of the post test (80% correct) and evaluation form, a CME certificate of participation will be awarded automatically. The certificate may be printed directly from the Web site or e-mailed and printed later.
There are no fees for participating in or receiving credit for this activity.
CME REVIEWERRick Ricer, MD Adjunct Professor of Family Medicine University of Cincinnati Cincinnati, Ohio
CME ACCREDITATIONSusan P. Tyler, MEd, CMP, CCMEP Director, Continuing Medical Education University of Cincinnati Cincinnati, Ohio
FACULTY DISCLOSURESAll faculty members (or anyone else in a position to control content, such as activity planners) are required to complete a Disclosure of Commercial Interest and Resolution form and to cooperate with identified methods for resolving conflict of interest prior to participating in the activity. The University of Cincinnati requires disclosure to the learners of all relevant financial relationships and adheres strictly to the ACCME Standards for Commercial Support.
Peter A. Calabresi, MD, is Professor of Neurology and Director of the Division of Neuroimmunology at Johns Hopkins School of Medicine and Director of the Johns Hopkins Multiple Sclerosis Center, Baltimore, Maryland. Dr. Calabresi has served as an advisor to Vaccinex and Vertex Pharmaceuticals and as a consultant to AbbVie, MedImmune, and Prothena; he has received grant support from Biogen Idec and Novartis.
Riley M. Bove, MD, Associate Neurologist, Partners Multiple Sclerosis
Center, Brigham and Women’s Hospital, and Instructor in Neurology, Harvard Medical School, Boston, Massachusetts, has nothing to disclose.
Carrie M. Hersh, DO, a Neuroimmunology Fellow in the Mellen Center for Multiple Sclerosis Treatment and Research, Neurological Institute, Cleveland Clinic, Cleveland, Ohio, has nothing to disclose.
Sona Narula, MD, a Pediatric Multiple Sclerosis Fellow in the Department of Neurology, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, has nothing to disclose.
Jennifer L. Orthmann-Murphy, MD, PhD, a Senior Neurology Resident at the Hospital of the University of Pennsylvania, Philadelphia, Pennsylvania, has nothing to disclose.
Carolyn Bevan, MD, MS, a Clinical Fellow in Neurology at the Multiple Sclerosis Center, University of California, San Francisco, School of Medicine, San Francisco, California, has nothing to disclose.
Rick Ricer, MD, has nothing to disclose.
Susan P. Tyler, MEd, CMP, CCMEP, has nothing to disclose.
Jacqueline Keenan and Edwin Geffner of Direct One Communications, Inc., have nothing to disclose.
DISCLAIMERThis activity is an independent educational activity under the direction of the University of Cincinnati. The activity was planned and implemented in accordance with the Essential Areas and Policies of the ACCME, the Ethical Opinions/Guidelines of the American Medical Association, the US Food and Drug Administration (FDA), the Office of Inspector General of the US Department of Health and Human Services, and the Pharmaceutical Research and Manufacturers of America Code on Interactions With Healthcare Professionals, thus assuring the highest degree of independence, fair balance, scientific rigor, and objectivity.
However, the planning committee, faculty, University of Cincinnati, Biogen Idec, and Direct One Communications, Inc. shall in no way be liable for the currency of information or for any errors, omissions, or inaccuracies in this activity. The opinions and recommendations presented herein are those of the faculty and do not necessarily reflect the views of the provider, producer, or grantor. Participants in this activity are encouraged to refer to primary references or full prescribing information resources.
DISCLOSURE OF UNAPPROVED/OFF-LABEL USEDiscussions concerning drugs, dosages, devices, and procedures may reflect the clinical experience of the planning committee or faculty, may be derived from the professional literature or other sources, or may suggest uses that are investigational and not approved labeling or indications.
In this edition of The Neurology Report, Dr. Bove describes the results of two randomized, placebo-controlled, clinical trials of rituximab in treating patients with RRMS and primary progressive MS; however, the use of rituximab for either indication has not been approved by the FDA. Other immunologic agents mentioned in this report that have not been approved by the FDA for use in treating MS include ocrelizumab, riluzole, amiloride, ibudilast, alemtuzumab, peginterferon beta-1a, daclizumab high-yield process, and laquinimod.
CONTACT INFORMATIONWe would like to hear your comments regarding this or other educational activities produced by Direct One Communications, Inc. In addition, suggestions for future activities are welcome. Contact us at:
Direct One Communications, Inc. 1424 Ridge Road Syosset, NY 11791 Phone: 516-364-1020 Fax: 516-364-4217 Website: www.CMEdirect.net
AboutThisCME/CNEActivity

4 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
Introduction Selected Reports from the 29th Congress of the European Committee for Treatment and Research in Multiple SclerosisPeter A. Calabresi, MD, Guest Editor
Johns Hopkins Multiple Sclerosis Center and School of Medicine, Baltimore, Maryland
Dr. Calabresi is Professor of Neurology and Director of the Division of Neuroimmunology at Johns Hopkins School of Medicine and Director of the Johns Hopkins Multiple Sclerosis Center, Baltimore, Maryland.
Our understanding of multiple sclerosis (MS) and the abil-ity to treat it effectively have grown tremendously over
the past 20 years. At least 10 different types of drugs for treating the relapsing-remitting form of the disease (RRMS) have been approved by the US Food and Drug Administration (FDA), and many intriguing new therapies currently are in the research pipeline. However, the intricacies of the immunopathogenesis of MS and identification of potential thera-peutic targets remain elusive, as does effective treatment of patients whose disease progresses.
During the 29th Congress of the Euro-pean Committee for Treatment and Re-search in Multiple Sclerosis (ECTRIMS), held October 2–5, 2013, in Copenhagen, Denmark, researchers, clinicians, and other health professionals representing a variety of disciplines convened to share their knowledge on the causes, identifi-cation, treatment, and outcomes of MS. Some 7,000 participants attended poster and abstract sessions, workshops, sym-posia, and other sessions to learn of new discoveries and about targeted therapies that can slow and even halt the course of this disease. To create these articles
for The Neurology Report, five talented fellows and residents in neurology from five prominent teaching centers in the United States traveled to Copenhagen, Denmark, to attend sessions at the meet-ing that covered the medical management of MS and described novel treatments that attack the pathology of the disease at different points.
n B-CELL MODULATION IN MS
The exploration of MS is relatively new, compared with that of other chronic dis-eases, and the achievements of particular individuals reflect the extensive efforts of the research teams who work with them. Riley M. Bove, MD, from Brigham and Women’s Hospital in Boston, describes a lecture given at ECTRIMS by Prof. Stephen L. Hauser, the 2013 winner of the prestigious Charcot Award. The author follows Dr. Hauser’s journey in MS research, touching upon his work in genetics, in investigating the impor-tance of B cells to the disease process, and in studying the safety and efficacy of B-cell depletion by targeting the CD20 antigen on the surface of B lymphocytes as a promising new approach to treating RMMS. Above all, the goals expressed by Dr. Hauser in his lecture are shared by all in MS research—a more thorough understanding of the pathogenesis of MS and finding new and potentially more ef-fective ways to arrest or reverse its effects.
n THE CURRENT CLINICAL ARENA OF PROGRESSIVE MS
Some phases of MS are less aggres-sively treated than are others because of a lack of effective therapies. Carrie M.
Hersh, DO, from the Cleveland Clinic, provides an overview of efforts to rede-fine the clinical course of MS, develop and adopt new clinical outcome assess-ment tools, and identify key research areas. In particular, she describes current research on RRMS, primary progressive MS (PPMS), and secondary progressive MS (SPMS), including clinical trials of new therapeutic agents and examination of factors that could impede the disease process. Among active areas of research are studies investigating the link between inflammation and damage to the central nervous system, the role of early damage to spinal neurons as a predictor of PPMS, and clinical and molecular factors that herald progression. Dr. Hersh delves into ways that relapses affect neurologic func-tion in patients with progressive MS and describes studies evaluating the ability of natalizumab to reduce the progression of disability in MS.
n ADVANCES IN IMMUNOMODULATORY THERAPY FOR MS
A variety of new and emerging mo-lecular therapeutics are reaching the market or emerging from late-stage clini-cal trials, and they could result in more convenient therapy, better efficacy, and greater safety for patients with MS. Sona Narula, MD, from the Children’s Hospital of Philadelphia, describes recent research on such newly approved or investigational treatments as alemtuzumab, peginterferon beta-1a, and daclizumab high-yield pro-cess, describing the benefits and risks of each of these drugs. Dr. Narula examines their mechanisms of action and potential

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 5
PeterA.Calabresi,MD Introduction
clinical use and discusses how they may change the management of MS.
n BALANCING THE OPTIONS AND CHALLENGES AHEAD
Jennifer L. Orthmann-Murphy, MD, PhD, from the Hospital of the University of Pennsylvania, provides an overview of the broad spectrum of therapeutic agents approved over the past 25 years to treat RRMS and the challenges that lie ahead. She describes the development of natali-zumab, fingolimod, teriflunomide, and di-methyl fumarate, taking special note of the rationale for their use and the patient and disease considerations that must be taken into account when they are prescribed. Dr. Orthmann-Murphy also delves into the need for a universal treatment algorithm to guide clinicians in choosing therapy for individual patients and transitioning them from one disease-modifying thera-peutic to another; in the absence of such an algorithm, she discusses the tools and
evidence now available for making treat-ment decisions. Finally, she describes an exciting line of current research involv-ing the use of anti–LINGO-1 antibodies to protect axons in MS lesions through remyelination, prevent axonal degenera-tion, and ameliorate the progressive form of the disease.
n NATALIZUMAB AND DIMETHYL FUMARATE: A FRESH LOOK
In the last article in this issue of The Neurology Report, Carolyn Bevan, MD, MS, from the University of California, San Francisco, reviews the evidence from the pivotal clinical trials that led up to the FDA’s approval of natalizumab and dimethyl fumarate, post hoc safety and efficacy analyses of the data emerging from those trials, and results from ongo-ing postmarketing monitoring studies of both drugs. In the 2-year AFFIRM trial of natalizumab, for example, more treated patients showed no evidence of disease
activity either clinically or on magnetic resonance imaging than did patients who were given placebo. Subanalyses of this trial were encouraging—relapses in treated patients were less severe than in the placebo group. Other studies uncov-ered patient groups that achieved better outcomes when placed on natalizumab or were switched to it. Likewise, 4-year follow-up of the patients enrolled in the pivotal dimethyl fumarate clinical trials demonstrated the sustained clinical and radiographic efficacy and safety of this new oral medication over the long term.
We are grateful to the authors of this report for providing us with their insights into the pathology and current treatment of MS, as well as opening a window into the potential future of MS therapeutics. Future editions of The Neurology Report certainly will describe breakthroughs in the treat-ment of this chronic and devastating dis-ease, as well as further our ability to make wise treatment decisions for our patients.

6 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
The Charcot Award, inaugurated in 1969 and given every 2 years by the Multiple Sclerosis Inter-national Federation (MSIF),
recognizes lifetime achievement in out-standing research into our understand-ing or treatment of multiple sclerosis (MS). The winner is invited to give the Charcot Lecture at the annual congress of the European Committee for Treat-ment and Research in Multiple Sclerosis (ECTRIMS) and at the biennial MSIF Council Meeting.
The award is given in commemoration of Jean-Martin Charcot (1825–1893), the founder of modern neurology. In 1882, he established the first neurology clinic in Europe at the Salpêtrière Hospital in Paris.
Among his many discoveries, Charcot described MS in 1868, calling it sclérose en plaques.
n 2013 CHARCOT AWARD WINNER
Stephen L. Hauser, MD, is Chair of the Department of Neurology at the University of California, San Francisco (UCSF). Dr. Hauser graduated from the Massachusetts Institute of Technol-ogy and Harvard Medical School and trained in neurology at the Massachusetts General Hospital. He moved to UCSF in 1992. Among his many academic leadership positions, Dr. Hauser is a past president of the American Neurological Association and editor-in-chief of Annals of Neurology. In addition to the Charcot Award, Dr. Hauser has received the Jacob Javits Neuroscience Investigator Award and the 2008 John Dystel Prize for Mul-tiple Sclerosis Research. In April 2010, Dr. Hauser was appointed by President Barack Obama to the Presidential Com-mission for the Study of Bioethical Issues.
Dr. Hauser’s first major contribution to the field of MS was advancing our understanding of the genetic basis of MS.
His efforts to uncover genes that confer increased susceptibility to MS—includ-ing the identification of specific genes in the major histocompatibility complex (MHC) human leukocyte antigen (HLA) system—culminated in the founding of the International Multiple Sclerosis Genetics Consortium (IMSGC) in 2002. The IMSGC led to the identification of the first two non-HLA genes involved in MS susceptibility: IL2RA (CD25) and IL7RA (CD127). Since then, the IMSGC has helped in identifying over 100 non–HLA-risk alleles, most of which are associated with immune function.1
Dr. Hauser’s laboratory has published the complete genome sequences and epi-genome of twins discordant for MS and has established the first national DNA repository for MS. His second major contribution to the field of MS, advancing our understanding of the role of B cells in MS pathogenesis and its therapeutic implications, was the basis of his 2013 Charcot Lecture.
n FROM THE BENCH TO THE BEDSIDE AND BACK
Dr. Hauser began his address with words memorializing Dr. Christian Con-favreux, a leading MS researcher in Lyon, France, and then acknowledged previous Charcot Award winners. He introduced his lecture as a story common to many translational scientists, “from the bench-side and back,” highlighting moments of “incremental advance,” frequent “disap-pointment,” and “occasional transforma-tional experiences” along the way.
TraditionalViewofMSImmunologyA simplified model of the immune
pathophysiology of MS has emerged
B-Cell Modulation in Multiple SclerosisRiley M. Bove, MDPartners Multiple Sclerosis Center, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts
Abstract Over the past decade, a role for B cells has emerged in the patho-physiology of multiple sclerosis (MS), which has traditionally been considered a T cell–mediated disease. At the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis, Charcot Award recipient Stephen L. Hauser, MD, discussed the emerging role of B cells in MS and its treatment. This article reviews evidence from clinical trials of the therapeutic role of rituximab, a chimeric monoclonal antibody that targets CD20 receptors on the surface of B cells. When compared with placebo, rituximab decreases the number of gadolinium-enhancing lesions on magnetic resonance imaging and frequency of clinical relapse. The therapeutic potential of ocrelizumab, a humanized anti-CD20 monoclonal antibody currently being tested in patients with MS, is also discussed. Targeting inflammation resulting from B-cell activity may only be one component of a longer-term strategy for halting disease progression in MS.
Dr. Bove is Associate Neurologist, Partners Multiple Sclerosis Center, Brigham and Women’s Hospital, and Instructor in Neurology, Harvard Medical School, Boston, Massachusetts.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 7
RileyM.Bove,MD B-Cell Modulation in Multiple Sclerosis
from studies of immune responses in adult-onset MS, lessons from therapeutic trials targeting the immune response, and data generated from animal models. According to this model, the initial step in pathogenesis involves the peripheral activation of CD4+ T helper (Th) type 1 cells in response to a stimulating antigen. The assortment of molecules released, including costimulatory signals and cy-tokines, influences the response profile of the activated immune cells.2
The initial stimulating antigen is un-known, but the presence of an infectious antigen is likely; this antigen subsequently cross-reacts with a central nervous system (CNS) antigen, a process known as “mo-lecular mimicry.” Subsequently, activated T cells transmigrate across the blood-brain barrier by various steps involving adhesion, chemoattraction, and active infiltration into the CNS. This leads to reactivation of infiltrating cells within the CNS, which contributes to perivascular inflammation and injury. Release of ad-ditional CNS antigens may result in the recruitment of T cells with specificity for additional CNS antigens, called “epitope spreading,” which may further propagate a chronic immune response. Further modifications to this model include the appreciation of other factors, most im-portantly ThIL-17.
BacktotheBenchIn trying to understand why aspects of
chronic relapsing, remitting MS (RRMS) were not adequately modeled in typical rodent models of MS or experimental autoimmune encephalomyelitis (EAE). Dr. Hauser acknowledged the advice of his mentor, Dr. Raymond D. Adams (1911–2008), Chief of Neurology at Mas-sachusetts General Hospital, who encour-aged his protégés to look to new models. In 1992, in collaboration with Drs. Nor-man Letvin and Luca Massacesi, macro-phage-mediated vesicular demyelination was replicated in New World monkeys (Callithrix jacchus; marmosets),3 who developed chronic relapsing-remitting and sometimes progressive disease, with evidence of remyelination. Dr. Claude Genain, among others, demonstrated
that both encephalitogenic T cells and pathogenic antibodies were needed to replicate the demyelinating phenotype.4 In collaboration with Dr. Cedric Raine, Dr. Hauser and coworkers found that autoantibodies that recognize the immu-nizing antigen were deposited within the vesiculated myelin sheaths in this animal model of MS and that similar antibodies against diverse antigens appeared in hu-man MS lesions.5 This discovery implied that humoral immunity might be a key factor in MS pathogenesis. Subsequently, Dr. Christian von Büdingen and col-leagues recognized that CD20+ B cells were present in MS lesions.6 This finding would be instrumental down the road in the development of rituximab and the rationale for its use in patients with MS.
n A ROLE FOR B CELLS
Over the past decade, several lines of evidence led Dr. Hauser and others to
question the role of autoimmune B cells and humoral mechanisms in the immu-nology of MS. First, a puzzling aspect of the traditional model is that therapies based upon this theory, such as interferon beta-1a and glatiramer acetate, did not fully prevent relapse or the accumulation of disability. Second, an element included in the diagnostic criteria for MS was an elevation in immunoglobulin-G (IgG) synthesis in the cerebrospinal fluid (CSF) relative to peripheral blood IgG levels and the presence of oligoclonal bands. Third, most MS lesions demonstrate deposition of antibody and activation of complement, vesicular disintegration of the myelin membrane, and detectable autoantibodies targeting diverse antigens in the CSF.
Memory B cells, which cross the blood-brain barrier, are believed to undergo restimulation, antigen-driven affinity maturation, clonal expansion, and differentiation into antibody-secreting plasma cells within the CNS to trigger cellular-dependent and complement-dependent cytotoxic effects. B cells can influence the priming of effector T cells by functioning as antigen-presenting cells. Further, abnormalities in B-cell cytokine responses have been reported in MS pa-tients, and production of cytokines and chemokines by B cells may be involved in the formation of ectopic lymphoid-like structures. Finally, B cells may be the reservoir for Epstein-Barr virus (EBV), and infection with EBV is a known risk factor for MS.7,8 Altogether, these observations suggest that B cells exert both antibody-dependent and antibody-independent effects in MS, and targeting B cells might disrupt critical processes in MS pathogenesis.
n ENTER RITUXIMAB
The story of rituximab and one of its champions, Dr. Lee Marshall Nadler, stands as a legend of the modern era of targeted therapies. Rituximab is a geneti-cally engineered, chimeric monoclonal antibody targeting the CD20 antigen ex-pressed on B lymphocytes from the pre–B-cell stage through differentiation into mature B cells but excluding plasma cells.
Key Points
• Anti-CD20 trials have revealed that B cells are central players in the pathogenesis of focal lesions in multiple sclerosis (MS).
• The mechanism of action likely involves blocking the activation of pathogenic T cells by B cells through a function of antigen-presenting cells, but it cannot exclude bystander cytokine effects or autoantibodies. Many questions remain:— Are the “culprit” B cells in the central
nervous system (CNS), peripheral circulation, or both?
— What explains the sustained protection against disease activity after B-cell depletion?
• Not all approaches based on B-cell manipulation are likely to be effective in treating MS; some may potentially worsen the disease.
• These clinical trials also set the stage for even more selective approaches for treating relapsing-remitting MS:— Focus on subsets of B cells or
pathogenic B-cell clones• Targeting resident B cells in the CNS may
soon be feasible:— BCR signaling pathway inhibitors: Btk
(ibrutinib), Syk, Pl3Kδ— Follicle inhibitors: LTβ receptor
• A properly designed clinical trial could elucidate the role of B cells in the pathogenesis of progressive MS.
Adapted, with permission, from Dr. Stephen Hauser's Charcot Lecture at ECTRIMS 2013.
8 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
RileyM.Bove,MD B-Cell Modulation in Multiple Sclerosis
In the 1980s, Dr. Nadler and colleagues employed the methods of monoclonal antibody research to focus on the dis-covery of molecules uniquely expressed on human B cells. Eventually, all known human B-cell–specific antigens (CD19, CD20, CD21, and CD22) were discov-ered in his laboratory. He used these monoclonal antibodies to classify human B-cell leukemia and lymphomas, and he was the first investigator to administer a monoclonal antibody to a human. Even-tually, the anti-B1 monoclonal antibody he developed led to the discovery of the CD20 cell-surface antigen on B cells and the development of rituximab.
Rituximab depletes CD20+ B cells by activating both cell-mediated and com-plement-dependent cytotoxic processes and by promoting apoptosis. Rituximab currently is used in the treatment of a range of diseases, including non-Hodg-kin’s lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, Wegener’s granulomatosis, and microscopic poly-angiitis and off-label in systemic lupus erythematosus and idiopathic thrombo-cytopenic purpura.9
HERMESTrial:RituximabPromisinginRRMS
In considering the therapeutic poten-tial of rituximab in MS, Dr. Hauser and
colleagues hypothesized that B-cell deple-tion might decrease antibody production, reduce cytokine networks, and limit B-cell–mediated antigen presentation and activation of T cells and macrophages.
In 2008, Dr. Hauser and others7 re-ported their findings from the HERMES trial, a phase 2, double-blind, 48-week trial involving 104 patients diagnosed with RRMS who were randomized to receive rituximab or placebo. Participants were 18–55 years of age, had experi-enced at least one relapse, and had an Expanded Disability Status Scale (EDSS) score of 0–5. Treated patients received 1,000 mg of rituximab intravenously (IV) on days 1 and 15 with premedication (acetaminophen and diphenhydramine hydrochloride) to minimize infusion-related reactions.
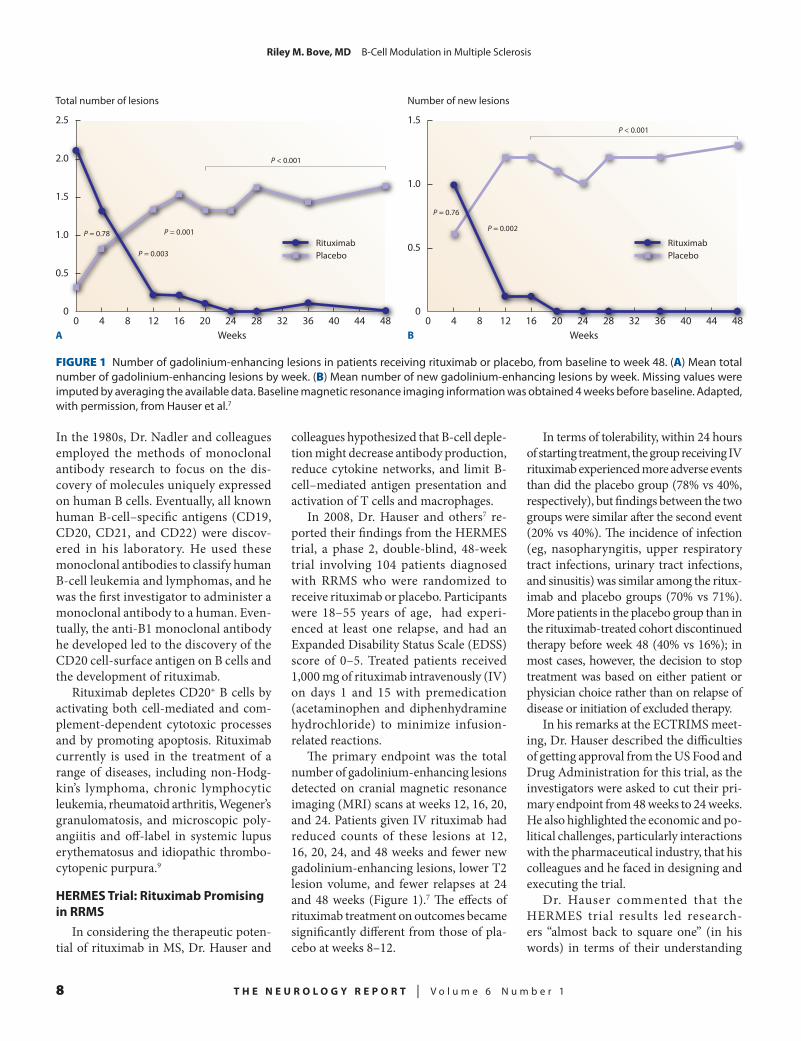
The primary endpoint was the total number of gadolinium-enhancing lesions detected on cranial magnetic resonance imaging (MRI) scans at weeks 12, 16, 20, and 24. Patients given IV rituximab had reduced counts of these lesions at 12, 16, 20, 24, and 48 weeks and fewer new gadolinium-enhancing lesions, lower T2 lesion volume, and fewer relapses at 24 and 48 weeks (Figure 1).7 The effects of rituximab treatment on outcomes became significantly different from those of pla-cebo at weeks 8–12.
In terms of tolerability, within 24 hours of starting treatment, the group receiving IV rituximab experienced more adverse events than did the placebo group (78% vs 40%, respectively), but findings between the two groups were similar after the second event (20% vs 40%). The incidence of infection (eg, nasopharyngitis, upper respiratory tract infections, urinary tract infections, and sinusitis) was similar among the ritux-imab and placebo groups (70% vs 71%). More patients in the placebo group than in the rituximab-treated cohort discontinued therapy before week 48 (40% vs 16%); in most cases, however, the decision to stop treatment was based on either patient or physician choice rather than on relapse of disease or initiation of excluded therapy.
In his remarks at the ECTRIMS meet-ing, Dr. Hauser described the difficulties of getting approval from the US Food and Drug Administration for this trial, as the investigators were asked to cut their pri-mary endpoint from 48 weeks to 24 weeks. He also highlighted the economic and po-litical challenges, particularly interactions with the pharmaceutical industry, that his colleagues and he faced in designing and executing the trial.
Dr. Hauser commented that the HERMES trial results led research-ers “almost back to square one” (in his words) in terms of their understanding
12840 2416 20 28 3632 48WeeksA
Total number of lesions
0.5
1.0
1.5
2.0
2.5
04440
RituximabPlacebo
P = 0.78
P = 0.003
P = 0.001
P < 0.001
12840 2416 20 28 3632 48WeeksB
Number of new lesions
0.5
1.0
1.5
04440
P = 0.76
P = 0.002
P < 0.001
RituximabPlacebo
FIGURE 1 Number of gadolinium-enhancing lesions in patients receiving rituximab or placebo, from baseline to week 48. (A) Mean total number of gadolinium-enhancing lesions by week. (B) Mean number of new gadolinium-enhancing lesions by week. Missing values were imputed by averaging the available data. Baseline magnetic resonance imaging information was obtained 4 weeks before baseline. Adapted, with permission, from Hauser et al.7

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 9
RileyM.Bove,MD B-Cell Modulation in Multiple Sclerosis
of the underlying immunopathology of MS, as the “immediate effect” implied a prominent role for B cells. In terms of the mechanism of action of rituximab in MS, Dr. Hauser and his coworkers postulated that rituximab treatment led to the lysis of memory B cells located in the peripheral blood and lymphoid tissues. Addition-ally, they hypothesized that rituximab interfered with antigen presentation by B cells and with activation of T cells or macrophages by pro-inflammatory B-cell cytokines. Because CD20 is not expressed on stem cells or plasma cells, median im-munoglobulin levels were not affected during the trial. Dr. Hauser suggested that oligoclonal immunoglobulin might be a surrogate marker for B-cell clones.
OLYMPUSTrial:RituximabLessPromisinginPrimaryProgressiveMS(PPMS)
The results of the OLYMPUS trial, in which patients with PPMS received IV rituximab or placebo on a 2:1 basis, revealed less potential for rituximab in patients with primary, progressive dis-ease.10 In this double-blind, randomized clinical trial, patients received rituximab or placebo every 24 weeks over a period of 96 weeks (a total of four courses of treat-ment). No significant reduction in time to clinically definite progression (defined as an increase in EDSS score sustained over 12 weeks) was found between rituximab and placebo. Patients given rituximab had less of a decrease in T2 lesion volume, but total brain volume was similar in both patient groups.
Interestingly, in subgroup analysis, rituximab therapy was associated with delayed time to clinically definite progres-sion in patients under 51 years of age, in those with gadolinium-enhancing lesions on MRI, and in patients who were both under 51 years old and had gadolinium-enhancing lesions. These analyses sug-gested that (1) some PPMS patients have evidence of inflammation early in the disease course, which influences the rate of progression; (2) early, aggressive treatment of inflammation in PPMS may be beneficial; and (3) age-related neuro-biologic changes, such as immunosenes-
cence, occur in MS and carry implications for therapeutic decisions.
Ocrelizumab:ABetterBenefit-to-RiskRatio?
Given concerns about the immuno-genic effect of repeated rituximab infu-sions, as well as political difficulties, Dr. Hauser and his colleagues hypothesized that ocrelizumab, a recombinant human-ized monoclonal antibody that selectively targets CD20+ B lymphocytes, might offer similar therapeutic benefits to rituximab in MS with less risk of immunogenicity and infusion-site reactions.8 Ocrelizumab is biosimilar but not bioidentical to ritux-imab. In vitro, ocrelizumab demonstrates more antibody-dependent, cell-mediated cytotoxicity than rituximab and less complement-dependent cytotoxicity.11 Thus, by increasing antibody-dependent, cell-mediated, cytotoxic effects, ocreli-zumab may modulate tissue-dependent mechanisms of pathogenic response more effectively than rituximab.
In 2011, Dr. Hauser joined Dr. Ludwig Kappos and coworkers8 in publishing the results of a phase 2, placebo-controlled trial involving 220 patients with RRMS who were randomly assigned to receive 600 or 2,000 mg of ocrelizumab, inter-feron beta-1a, or placebo. Patients with RRMS were included in the study if they were 18–55 years of age and had at least
two relapses within the prior 3 years (including at least one in the prior year), an EDSS score of 1–6 at baseline, and evidence of inflammatory disease (at least six T2 lesions on MRI or two relapses in the prior year). Among exclusion criteria was an EDSS score ≤ 2 in patients who had had the disease for more than 15 years. Patients received four treatment cycles of 24 weeks followed by a 1-year treatment-free observation period.
In the primary analysis at 24 weeks, when compared with the placebo group, patients given 600 or 2,000 mg of ocreli-zumab had 89% or 96% fewer T1 gado-linium-enhancing lesions, respectively (Figure 2).8 Both doses of ocrelizumab were superior to interferon beta-1a in reducing these lesions.
Two phase 3 pivotal trials in RRMS patients and the first phase 2 pivotal trial in PPMS patients (the ORCHESTRA trial) are ongoing. Twists and turns in this re-search program included the halting of an ocrelizumab program in rheumatoid ar-thritis patients in 2010 because of the ap-pearance of opportunistic infections. Dr. Hauser noted that RA treatment involves polypharmacy in an older population.
n RECOVERY PERIOD AFTER B-CELL DEPLETION
The prolonged benefits of B-cell depletion after exposure to rituximab
40 128 16 24Weeks
Mean number of T1 gadolinium-enhancing lesions
1.0
2.0
3.0
4.0
5.0
020
600 mg Ocrelizumab (n = 51)2,000 mg Ocrelizumab (n = 52)Interferon beta-1a (n = 52)Placebo (n = 54)
FIGURE 2 Number of T1 gadolinium-enhancing lesions by week in patients receiving ocreli-zumab, interferon beta-1a, or placebo. Vertical bars = 95% confidence interval. Adapted, with permission, from Kappos et al.8

10 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
RileyM.Bove,MD B-Cell Modulation in Multiple Sclerosis
suggest that protection may extend be-yond the period of B-cell depletion. Dr. Hauser cited the contributions of many of his colleagues to investigation of the immunologic changes occurring in the peripheral blood and CSF during this “recovery” period. In the peripheral cir-culation, immunologic changes include dominance of naïve and immature B cells, an increase in the numbers of in-terleukin 10-secreting B regulatory cells and CD25+FoxP3+ T regulatory cells, and a decline in Th1 and Th17 proinflamma-tory responses. In the CSF, the number of T and B cells decreases, and resting CD19+ bright B cells predominate. None-theless, the effect of rituximab treatment on oligoclonal bands is incompletely understood. Early evidence suggests no decrease after just one course of B-cell depletion. Repeated treatment courses may be important, as they have proven to be in the treatment of rheumatoid arthritis.
n EMERGING INVESTIGATIONS INTO B-CELL FUNCTION IN MS
Myelin oligodendrocyte glycoprotein (MOG) is a glycoprotein that appears to be involved in either completion or maintenance of the myelin sheath. It has emerged as a potential antigen involved in the pathogenesis of MS.
At the University of California, San Francisco, researchers in Dr. Scott Zamvil’s laboratory immunized both healthy mice and MHC II-deficient mice with EAE. B-cell class II-deficient mice repopulated with normal B cells developed normal EAE to extracellular mouse MOG but were protected against extracellular human MOG.12 This observation suggested that a simple substitution may have induced a conformational change and led the hu-man MOG antigen to be completely B-cell dependent; this protection could not be restored by injecting MOG antibody. Thus, there may be a repertoire of B-cell–depen-dent antigens that might help to elucidate underlying triggers in MS.
Dr. Hauser also described recent ge-netic studies that “solved how heritable our antibody repertoire is.”13 Twin stud-ies have revealed identical expression of
heavy-chain variable and D segments on antibodies, but the hypervariable comple-mentarity-determining regions that bind antigens are “absolutely environmental,” Dr. Hauser said.
Next, he reviewed the IgG sequences identified by Dr. Christian von Büdingen, among others, to identify “clonotypes” of CSF and peripheral-blood B cells and oligoclonal bands.14 These studies revealed oligoclonal B cells in CSF that are finger-prints for MS. In lineage analyses, multiple different oligoclonal bands belong to the same clone and may be responding to a smaller number of antigenic determinants than the bands would suggest. Members of these oligoclonal bands could be iden-tified both in the CSF and in peripheral blood mononuclear cells. In addition, some B cells found only in the peripheral blood were associated with oligoclonal bands found only in the CSF, suggesting some exchange of B cells across the blood-brain barrier (Figure 3).14 B-cell therapy may disrupt this circuitry.
Finally, Dr. Hauser mentioned genetic studies that revealed many single nucleo-
tide polymorphisms involved in B-cell activation and B-cell receptor signaling pathways that may influence susceptibility to a “hyperpolarized, proinflammatory B-cell state,” concluding that the genetics “needs to link to function to understand how inheritance predisposes to this B-cell problem.”
n UNFINISHED BUSINESS
In his closing remarks, Dr. Hauser related the story of a “leading citizen of California” with long-standing RRMS who had experienced ongoing inflam-matory activity despite adherence to many disease-modifying therapies. He was treated with rituximab off label since 2003. To date, he has demonstrated an excellent response to rituximab in terms of inflammatory activity. Still, his disease continues to progress clinically. Over the past 10 years, he has gone from an EDSS score of 3 to requiring bilateral crutches. MRI tractography has revealed ongoing atrophy of the motor cortex. Dr. Hauser commented, “I think that the best ques-tion here is, are there B cells in lymphoid
A MS-1 B MS-5
C MS-1
D MS-6
9
6
2
22
2
2
3
3
2
25
3
5 2
52
3
7
2 2
3 2 2 6
2 2 2
13
FIGURE 3 Lineage trees of multiple sclerosis (MS) IgG-VH sequences suggest ongoing B-cell exchange across the blood-brain barrier. IgG-VH lineages suggestive of ongoing B-cell exchange across the blood-brain barrier for patient MS-1 (A and C), MS-5 (B), and MS-6 (D). These lineages could also reflect affinity maturation occurring in both compartments in paral-lel. Blue nodes represent cerebrospinal fluid-derived IgG-VH sequences; red nodes represent PB-derived IgG-VH sequences; and green nodes represent identical sequences found in both compartments. Reproduced, with permission, from von Büdingen et al.14

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 11
RileyM.Bove,MD B-Cell Modulation in Multiple Sclerosis
follicles that are contributing to percolat-ing [of disease activity]?” This anecdote highlights our incomplete understanding of MS.
REFERENCES
1. Beecham AH, Patsopoulos NA, Xifara DK, et al; International Multiple Sclerosis Genetics Con-sortium (IMSGC). Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. 2013;45:1353–1360.
2. Chitnis T. The role of CD4 T cells in the pathogenesis of multiple sclerosis. Int Rev Neurobiol. 2007;79:43–72.
3. Massacesi L, Joshi N, Lee-Parritz D, Rombos A, Letvin NL, Hauser SL. Experimental allergic en-cephalomyelitis in cynomolgus monkeys: quantita-tion of T cell responses in peripheral blood. J Clin Invest. 1992;90:399–404.
4. von Büdingen HC, Hauser SL, Ouallet JC,
Tanuma N, Menge T, Genain CP. Epitope recognition on the myelin/oligodendrocyte glycoprotein differ-entially influences disease phenotype and antibody effector functions in autoimmune demyelination. Eur J Immunol. 2004;34:2072–2083.
5. Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med. 1999;5:170–175.
6. von Büdingen HC, Bar-Or A, Zamvil SS. B cells in multiple sclerosis: connecting the dots. Curr Opin Immunol. 2011;23:713–720.
7. Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–688.
8. Kappos L, Li D, Calabresi PA, et al. Ocreli-zumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicen-tre trial. Lancet. 2011;378:1779–1787.
9. Nadler LM, Roberts WC. Lee Marshall Nadler, MD: a conversation with the editor. Proc
(Baylor Univ Med Cent). 2007;20:381–389.10. Hawker K, O’Connor P, Freedman MS, et
al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66:460–471.
11. Ocrelizumab [data on file]. South San Fran-cisco, CA: Genentech; 2003.
12. Weber MS, Prod’homme T, Patarroyo JC, et al. B-cell activation influences T-cell polariza-tion and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol. 2010; 68:369–383.
13. Baranzini SE, Mudge J, van Velkinburgh JC, et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464:1351–1356.
14. von Büdingen HC, Kuo TC, Sirota M, et al. B cell exchange across the blood-brain barrier in multiple sclerosis. J Clin Invest. 2012;122:4533–4543.

12 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
The definition of progressive mul-tiple sclerosis (MS) is unique to different disciplines. The neurologist characterizes it as
progressive myelopathy or cognitive im-pairment. The imager judges the extent of disease based upon progressive atrophy on conventional magnetic resonance im-aging (MRI), decreasing magnetization transfer ratio (MTR) or N-acetyl aspartate (NAA) levels, or the results of fractional anisotropy. The pathologist describes it as axonal or oligodendrocyte pathology. The physiatrist defines it as loss of function or worsening symptoms. And the patient understands it as loss of independence and function and the inability to work. Historically, the majority of untreated
patients with relapsing-remitting multiple sclerosis (RRMS) eventually develop sec-ondary progressive MS (SPMS).1
Currently, strong imaging biomarkers to screen new anti-inflammatory thera-pies and as many as 10 different disease-modifying therapies for treating patients with relapsing MS are available. However, the treatment of progressive MS remains a crucial, unmet challenge.2
n DEFINING AND UNDERSTANDING PROGRESSIVE MS
Many international research teams are working to better understand progressive MS and to develop targeted treatment options. Efforts to redefine the clinical course are being led by the European Committee for Treatment and Research in Multiple Sclerosis/National Multiple Sclerosis Society (ECTRIMS/NMSS) International Advisory Committee on Clinical Trials in Multiple Sclerosis. The Multiple Sclerosis Outcome Assessments Consortium (MSOAC), a conglomerate of industry, academic, regulatory, and patient representatives, strives to develop
and support the adoption of new clinical outcome assessment tools for use in future MS clinical trials.
The International Collaborative on Progressive MS3 has identified five key priority areas for research:
1. Discovery of experimental models that reproduce the key clinical and patho-logic features of primary progressive MS (PPMS) and SPMS.
2. Identification and validation of biologic targets and opportunities to repurpose existing MS medications for use in treating progressive MS.
3. Recognition and validation of proof-of-concept clinical trial outcomes.
4. Development of precise, reproduc-ible, and broad-based clinical outcome measures that are both sensitive to change and predictive over time.
5. Optimization of symptom-man-agement and rehabilitation strategies that can help to reduce the impact of disability and improve the quality of life of patients affected by progressive and relapsing MS.
At present, there are no validated phase 2 outcomes that identify adequate models in defining progressive MS. Whole-brain atrophy currently is recommended, but it is limited by one measure per brain; further, imaging quality varies from scan to scan, is affected by a low signal-to-noise ratio, and changes slowly. Some alternative measures that have been proposed are seg-mented atrophy (eg, cortical gray matter atrophy), MTR, diffusion tensor imag-ing, spectroscopy, functional MRI, and optical coherence tomography; however, their usefulness in defining or measuring progressive MS remains unknown. Limi-tations and possible confounders include the degree of reliability, responsiveness over time, predictive ability of measur-
The Current Clinical Arena of Progressive Multiple SclerosisCarrie M. Hersh, DOMellen Center for Multiple Sclerosis Treatment and Research, Neurological Institute, Cleveland Clinic, Cleveland, Ohio
Abstract The majority of patients with multiple sclerosis (MS) develop a progres-sive phase of disease characterized by the insidious accumulation of neurologic disability. More therapies for relapsing-remitting MS are becoming available; however, treatment options for primary progressive MS (PPMS) and secondary progressive MS (SPMS) are still limited. Global efforts have addressed this crucial unmet challenge via diverse collaborative efforts, such as the International Pro-gressive MS Alliance and the MS Outcome Assessments Consortium. Ongoing phase 3 clinical trials of potential therapies for progressive forms of MS include the ASCEND study of natalizumab in patients with SPMS; other studies are in-vestigating early neuronal damage in the spinal cord of patients with PPMS and potential predictive markers of progressive MS. The strides we have made and will continue to make in evaluating the pathogenesis of PPMS and SPMS will gauge the ongoing efforts to develop safe and effective treatments for progressive MS.
Dr. Hersh is a Neuroimmunology Fellow in the Mellen Center for Multiple Sclerosis Treatment and Research, Neurological Institute, Cleveland Clinic, Cleveland, Ohio.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 13
CarrieM.Hersh,DO The Current Clinical Arena of Progressive Multiple Sclerosis
ing disability and treatment response, and multicenter implementation in both cross-sectional and longitudinal studies.
IdentifyingValidatedModelsandDrugTherapies
A myriad of efforts currently are ad-dressing the identification of good, vali-dated models and potential drug therapies for progressive forms of MS.
The International Progressive MS Al-liance is an expanding global alliance of MS organizations that seeks to expedite the development of therapies for effective modification and symptom management of progressive MS. This alliance currently involves the United States, the United Kingdom, Italy, Denmark, Spain, and Canada and is in its early stages, but its status is rapidly progressing. Since April 2012, this collaboration has been orga-nized via working groups in priority ar-eas, the development of research support mechanisms, and an international call in September 2013 for research proposals (www.EndProgressiveMS.org).
n POTENTIAL THERAPEUTICS FOR PROGRESSIVE MS
Despite the increasing availability of novel and effective treatments for RRMS, therapeutic options for patients with PPMS and SPMS are still lacking.4 Most MS patients initially are diagnosed with a disease course defined by periodic re-lapses with full or partial recovery of neu-rologic function; immunopathologically, MS is defined by active inflammation. The majority of patients subsequently develop a progressive accumulation of disability dominated by neurodegeneration and fewer superimposed relapses.
The pathology of progressive MS can involve widespread, compartmentalized inflammation; cortical lesions reflecting meningeal inflammation; and normal-appearing inflammation of the white matter. These pathologic changes cor-relate with neuronal and axonal damage and disease progression.5–7 The results of histopathologic studies suggest that inflammation and neurodegeneration occur simultaneously throughout the disease course, not in a stepwise fashion.
Such findings further complicate the classification of progressive MS forms as RRMS with incomplete remissions versus SPMS with superimposed relapses versus a gradual progression of disease from the outset (PPMS). Disease progression over time tends to be smooth, and the dividing line between these three phases is often blurred.
ClinicalTrialsDisease-modifying therapies for RRMS
prevent disability and neurologic impair-ment, but their ability to prevent insidi-ous disease progression remains unclear. Limited data from randomized, controlled studies support the effectiveness of any therapy currently approved for RRMS or
SPMS in slowing the progression of dis-ability in SPMS.8
The OLYMPUS study examined the usefulness of rituximab-induced B-cell depletion in patients with PPMS.9 Analysis of the data from this clinical trial suggested that placebo-treated patients experienced faster disease progression than did those given rituximab, although this finding was not statistically signifi-cant. A subgroup analysis of younger pa-tients with PPMS showed that those with gadolinium-enhancing lesions seemed to benefit from rituximab therapy; however, it remains unknown whether the mecha-nism of action involves peripherally cir-culating B cells or those within the central nervous system (CNS).
Other clinical trials have focused on
various factors that could impede the MS process. A recent study measured cerebro-spinal fluid (CSF) levels of osteopontin, a marker for neurofilament damage, to further characterize the role of intrathecal immune activation in progressive MS.10 Neuroprotective trials involving treatment with lamotrigine11 or tetrahydrocannabi-nol12 in patients with progressive MS did not meet their primary endpoints. Statin treatment slowed the development of brain atrophy and progression of the Expanded Disability Status Scale (EDSS) in the ECTRIMS MS-STAT trial,13 although the underlying mechanism remains unknown.
Another recent study showed an im-provement in visual function resulting from the intravenous (IV) infusion of autologous mesenchymal stem cells in a small cohort of patients affected with SPMS.14 A study of the potassium-sparing diuretic amiloride, which has been shown in experimental models of MS to block pH-dependent neurodegeneration in inflammatory lesions, demonstrated a neuroprotective effect in a few PPMS pa-tients, as revealed by diffusion-weighted imaging.15
TreatmentsinthePipelineMS-SMART (www.ms-smart.org), a
placebo-controlled phase 2 trial based in the United Kingdom, is studying three potential therapies (riluzole, amiloride, and ibudilast) that have shown promise in patients with SPMS. Primary outcome measures are the degree of cortical at-rophy (as measured by clinical observa-tions and advanced imaging studies in a subgroup of patients) and CSF changes.
SPRINT-MS (www.neuronext.org/nn102-sprint-ms) is a phase 2 clinical trial based in the United States. Investigators are using the National Institutes of Health-sponsored phase 2 trial network Neuro-NEXT to compare the use of ibudilast versus placebo in patients with PPMS and SPMS. Outcomes, including the degree of cortical atrophy, are being assessed via standardized advanced imaging modali-ties at all sites and considering all clinical measures. Investigators are using head-to-head comparisons of imaging measures and longitudinal validation of clinical
Despite the increasing availability of novel and effective treatments for RRMS, therapeutic options for patients with progressive forms of the disease are still lacking.

14 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
CarrieM.Hersh,DO The Current Clinical Arena of Progressive Multiple Sclerosis
outcomes to predict treatment response and to determine the reliability of therapy in slowing the progression of disability.
SummaryInvestigators have shown a relationship
between systemic and intrathecal immune activation and inflammation in progres-sive MS, and the relationship between inflammation and neuronal and axonal damage is being explored in clinical trials. Positive trial results are beginning to emerge from this research effort, with three ongoing phase 3 studies currently investigating the usefulness of ocreli-zumab, fingolimod, and natalizumab in treating progressive MS. Now that we have a strong foothold on RRMS treatment, we must refocus attention on neuroprotec-tive treatments and target the reduction of neuronal and axonal loss. Significant strides in developing regenerative treat-ments that improve remyelination are being made. Mesenchymal stem cells may be useful in treating progressive MS, and we must focus on the role of neural stem cells in modifying disease progression.
n EARLY SPINAL NEURONAL DAMAGE AS A POTENTIAL PREDICTOR OF PPMS
Most patients with PPMS reach a high level of disability in the first years after disease onset, and the rate of progression may vary from one patient to the next. Pathologic changes in the gray and white matter are associated with disability and predict disease progression.16 The spinal cord has been studied less often with ad-vanced MRI technology.
Abdel-Aziz and colleagues17 used spinal-cord magnetic resonance spectros-copy in a small cohort of patients with progressive MS. They investigated the differences in metabolite concentrations between healthy controls and individuals with early PPMS, observing the relation-ship between metabolites and disability in the latter. Ultimately, they hoped to provide insights into the pathologic changes underlying disability in PPMS. The investigators used total NAA levels as a marker of axonal integrity and meta-bolic function, myoinositol as a marker
of astrocytic activation and proliferation, and glutamate plus glutamine (Glx) lev-els as a marker of neuronal integrity and neurotransmitter pool. Clinical measures were assessed using the EDSS, Nine-Hole Peg Test (9HPT), Timed 25-Foot Walk (T25-FW), 12-Item MS Walking Scale (MSWS-12), Ashworth spasticity scale, grip strength, vibration sense testing, and static posturography. Linear-regression models were used to investigate differenc-es between groups, and regression models (EDSS, T25-FW, 9HPT) and multivariate analysis (static posturography) were used to explore correlations. Covariates were corrected for gender, age, cord area, brain white- and gray-matter fractions, and T2 lesion load.
Results showed lower total NAA (P = 0.03) and Glx peaks (P = 0.03) on mag-netic resonance spectroscopy in PPMS patients when compared with placebo-controlled participants, with the former correlating with higher EDSS scores (P = 0.03); greater rolling on static posturog-raphy (P = 0.005); and worse posterior column sensory function, as measured by vibration sense testing (P = 0.003). Lower total NAA and Glx levels in this cohort of PPMS patients suggested a role for neuro-nal loss and metabolic dysfunction in the glutamatergic pathway in the spinal cord. Additionally, the association between total NAA and Glx levels indicated that these pathologic abnormalities may contribute to disability.
n RADIOLOGICALLY ISOLATED SYNDROME AND PROGRESSIVE FORMS OF MS
Previous studies suggested that the on-set of progressive MS is age-sensitive and independent of the disease course prior to disease progression.18,19 Cranial and spinal cord MRI findings at the time of PPMS diagnosis can be remarkably similar to those of SPMS, and the asymptomatic pre-progression disease course in PPMS is difficult to study systematically.
Kantarci and colleagues20 sought to de-fine the pre-progression period in PPMS, hypothesizing that MRI characteristics of the pre-progression phase would be similar to that of bout-onset progressive
MS (SPMS + progressive MS following a single clinical attack of progressive MS, or SAPMS). They reported PPMS develop-ment after a longitudinal follow-up of a robust multicenter radiologically isolated syndrome (RIS) cohort. Investigators defined RIS as “incidental white matter changes on MRI that suggest[ed] demy-elinating disease and fulfill[ed] three out of four Barkhof criteria”21; symptomatic conversion as an “acute or progressive demyelinating event following the RIS course”; and progression from the onset of the event following the RIS course as the “development of a clinical symptom with the temporal profile revealing at least a 12-month history of neurologic worsen-ing.” Among 20 multicenter databases, in-vestigators retrospectively identified and prospectively followed 451 participants.
In all, 34% of the initial cohort devel-oped symptomatic MS after 5 years, with 9% of the initial cohort ultimately being classified as having PPMS, as indicated by the 2010 Revised Multiple Sclerosis criteria. The reasons for obtaining an ini-tial MRI varied widely among the PPMS cohort and included the investigation of a primary headache disorder (n = 5); trauma (n = 4); low back/radicular pain (n = 2); and one instance each of “spell,” tumor screen, and childhood epilepsy.
Overall, the incidence of PPMS (9%) and sex ratio (43% female) in this RIS co-hort mirrored population-based studies. The CSF findings were positive in 83% of RIS patients who developed PPMS. These patients had a notably higher cervical and thoracic cord lesion load than did other symptomatic patients. Further, pre-pro-gression MRI findings in PPMS patients appeared to be similar to pre-progression MRI findings in patients with SPMS, but a comparative MRI study is needed to better qualify these observations.
n IMPACT OF RELAPSES ON NEUROLOGIC DISABILITY IN PROGRESSIVE MS
Approximately 75% of patients with MS experience disease progression, char-acterized by an insidious accumulation of neurologic disability that may be present from the time of clinical onset and may

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 15
CarrieM.Hersh,DO The Current Clinical Arena of Progressive Multiple Sclerosis
follow a relapsing-remitting phase. The determinants of MS progression are in-completely understood: not all patients develop the progressive phase of disease, age distribution at the onset of disease progression varies, and the accumulation of neurologic disabilities ranges widely among patients. The onset of progression is age-dependent and has an equivalent mean and distribution across clinical phe-notypes.22 An EDSS score of 6.0 serves as a robust disability milestone. These findings provide a uniform starting point for fac-tors that affect the cadence of progression.
Paz Soldan and coworkers23 investigat-ed the impact of relapses on post-progres-sion disability accrual. They hypothesized that patients experiencing clinical relapses accumulate disability faster during the progressive phase of MS. The researchers studied a population- and clinic-based cohort of patients that fulfilled the 2010 McDonald diagnostic criteria for MS. Dis-ease progression was defined as an insidi-ous, irreversible worsening of neurologic disease lasting ≥ 1 year. This definition included brain, brainstem-cerebellar, and spinal cord syndromes and excluded pro-gressive, purely sensory symptoms. The progressive disease course was classified as PPMS, SPMS, or SAPMS. Disability was assigned based on the patient’s Kurtzke EDSS score. Kaplan-Meier analysis was used to generate survival curves from the onset of progressive MS to reaching an EDSS score of 6.0.
The presence of pre-progression re-lapses predicted a shorter time from onset of disease progression to an EDSS score of 6.0. Post-progression disability accumula-tion was slowest in patients with PPMS (50% of patients in 10 years) and SAPMS (50% of patients in 7 years) and fastest in those with SPMS (50% of patients in 4 years; P < 0.0001). Post-progression relapses were more common in patients with SPMS (29.5%) than in those with SAPMS (10.7%) or PPMS (3.1%), reflect-ing the pre-progression relapse status in these groups. Ongoing relapses following the onset of progressive disease inde-pendently predicted a shorter time (~ 2 years) from onset of disease progression to an EDSS score of 6.0 (P = 0.0005). Most
post-progression relapses occurred within 5 years (91.6%) after the onset of progres-sion and before age 55 years (95.2%).
This study showed that relapses prior to or following the onset of disease pro-gression increase the rate of accumulation of post-progression disability and that gender and age at the onset of progres-sive disease have minor influences on disability accumulation. In this context, continued immunomodulation 5 years after the onset of progressive MS, or at least until age 55 years, may be a rea-sonable approach for managing SPMS. However, due to a paucity of findings, these approaches may not be indicated in SAPMS or PPMS.
n EFFICACY OF NATALIZUMAB ON REDUCING DISABILITY PROGRESSION IN SPMS
Natalizumab, a recombinant human-ized anti-α4 integrin antibody, is an approved therapy for relapsing forms of MS. It reduces CNS inflammation by preventing the migration of mononuclear leukocytes across the blood-brain barrier. Additionally, natalizumab may suppress chronic compartmentalized CNS inflam-mation, as indicated by a reduction in the levels of proinflammatory mediators in the CSF, including C-X-C motif chemokine 13 and osteopontin.10,24 Results of recent retrospective studies of clinical data have suggested that natalizumab therapy may reduce disease progression in SPMS.25
The ASCEND trial is an international, multicenter, randomized, double-blinded, placebo-controlled, phase 3 study of the efficacy of natalizumab in reducing disability progression in patients with SPMS.26 Interim data as of August 2013 included over 800 enrolled patients from various countries with baseline functional disability test scores consistent with MS progression. About 62% of the study participants were classified as “low EDSS” (EDSS score of 3.0–5.5), and 38% were classified as “high EDSS” (EDSS score of 6.0–6.5). The patients were randomized 1:1 to receive 300 mg of natalizumab or placebo IV every 4 weeks for 2 years. The primary endpoint of the study is the percentage of patients experiencing con-
firmed progression of disability in one or more of the following measures: EDSS, T25-FW, or 9HPT.
Once completed, ASCEND will pro-vide data on the effect of natalizumab on progression of disability in patients having SPMS not attributable to relapses. ASCEND substudies will explore the ef-fects of natalizumab on cognitive impair-ment using MS-COG, a novel composite measure to assess cognitive function in patients with SPMS.27 The use of com-posite measures, as opposed to a single modality, offers many potential advan-tages: lower error rates, improved sensi-tivity and reliability of collected data, and greater simplicity in assessing treatment effects and clinical meaningfulness of the observed data.28–31 The components of the MS-COG assess two domains: learning/memory and processing speed.
Baseline data are currently available for 112 participants, most of whom have changed their occupation because of MS. Overall, average performances on tests of memory and processing speed and on the total MS-COG composite were lower than were those found among the patients given placebo. Approximately 75% of study participants had at least mild cognitive impairment (z score ≤ –0.5), and about 20% had evidence of severe cogni-tive impairment (z score ≤ –2.0). At the conclusion of the study, the researchers intend to provide evidence supporting the validity and feasibility of the MS-COG to measure treatment effects on cognitive function in MS clinical trials.
n CONCLUSION
Furthering our understanding of the aspects of human pathology relevant to disease progression in MS will necessitate validation of a preclinical model that emulates human pathology and devel-opment of high-throughput screening tools. It is also crucial to develop and validate an outcome biomarker; we can accomplish this critical task by using clinical trials to advance methodologies. Finally, we must develop accepted clinical outcome measures to better understand progressive MS pathology and to guide development of targeted disease-mod-

16 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
CarrieM.Hersh,DO The Current Clinical Arena of Progressive Multiple Sclerosis
ifying and symptomatic treatments. In the current clinical arena of imminently manageable relapsing MS, we must focus on expanding and unifying international collaborations to better understand and treat progressive MS.
REFERENCES
1. Weinshenker BG. The natural history of multiple sclerosis. Neurol Clin. 1995;13:119–146.
2. Fox R. From relapsing-remitting to secondary progressive MS. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark.
3. Fox R, Thompson A, Baker D, et al. Setting a research agenda for progressive multiple sclerosis: the International Collaborative on Progressive MS. Mult Scler. 2012;18:1534–1540.
4. Sellebjerg F. Therapeutic opportunities for progressive MS. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Abstract 196.
5. Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128:2705–2712.
6. Frischer JM, Bramow S, Dal-Bianco A, et al. The relation between inflammation and neu-rodegeneration in multiple sclerosis brains. Brain. 2009;132:1175–1189.
7. Magliozzi R, Howell OW, Reeves C, et al. A gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol. 2010;68:477–493.
8. Fizner D, Simons M. Chronic progressive multiple sclerosis—pathogenesis of neurodegenera-tion and therapeutic strategies. Curr Neuropharma-col. 2010;8:305–315.
9. Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive mul-tiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. OLYMPUS Trial Group. Ann Neurol. 2009;66:460–471.
10. Romme Christensen J, Börnsen L, Khademi M, et al. CSF inflammation and axonal damage are increased and correlate in progressive multiple sclerosis. Mult Scler. 2013;19:877–884.
11. Kapoor R, Furby J, Hayton T, et al. La-motrigine for neuroprotection in secondary progres-sive multiple sclerosis: a randomised, double-blind, placebo-controlled, parallel-group study. Lancet
Neurol. 2010;9:681–688.12. Zajicek JP, Hobart JC, Slade A, Barnes
D, Mattison PG. Multiple sclerosis and extract of cannabis: results of the MUSEC trial. MUSEC Research Group. J Neurol Neurosurg Psychiatry. 2012;83:1125–1132.
13. Chataway J, Alsanousi A, Chan D, et al. ECTRIMS MS-STAT Trial. A treatment for pro-gressive MS. Multiple Sclerosis Research Web site. October 11, 2012. http://multiple-sclerosis-research.blogspot.com/2012/10/ectrims-ms-stat-trial.html. Accessed October 17, 2013.
14. Connick P, Kolappan M, Crawley C, et al. Autologous mesenchymal stem cells for the treat-ment of secondary progressive multiple sclerosis: an open-label phase 2a proof-of-concept study. Lancet Neurol. 2012;11:150–156.
15. Arun T, Tomassini V, Shardella E, et al. Tar-geting ASIC1 in primary progressive multiple scle-rosis: evidence of neuroprotection with amiloride. Brain. 2013;136:106–115.
16. Sastre-Garriga J, Ingle JT, Chard DT, Ramió-Torrentà L, Miller DH, Thompson AJ. Grey and white matter atrophy in early clinical stages of primary progressive multiple sclerosis. Neuroimage. 2004;22:353–359.
17. Abdel-Aziz K, Solanky BS, Wheeler-Kingshott CAM, et al. Evidence for early neuro-nal damage in the cervical cord of patients with primary progressive multiple sclerosis. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Abstract 197.
18. Confavreux C, Vukusic S. Age at dis-ability milestones in multiple sclerosis. Brain. 2006;129:595–605.
19. Koch M, Mostert J, Heersema D, De Key-ser J. Progression in multiple sclerosis: further evidence of an age dependent process. J Neurol Sci. 2007;255:35–41.
20. Kantarci OH, Okuda DT, Siva A, et al. First report of the pre-progression prospective follow-up in a series of patients with primary progressive multiple sclerosis evolving from radiologically isolated syndrome. Radiologically Isolated Syn-drome Consortium (RISC); Club Francophone de la Sclérose en Plaques (CFSEP). Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Abstract 198.
21. Okuda DT, Mowry EM, Behestian A, et al. Incidental MRI anomalies suggestive of multiple sclerosis: the radiologically isolated syndrome.
Neurology. 2009;72:800–805.22. Tutuncu M, Tang J, Zeid NA, et al. On-
set of progressive phase is an age-dependent clinical milestone in multiple sclerosis. Mult Scler. 2013;19:188–198.
23. Paz Soldan MM, Novotna M, Crusan DJ, Atkinson EJ, Kantarci OH. Pre- and post-progression relapses impact disability in progressive multiple sclerosis. Presented at the 29th Congress of the Eu-ropean Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Abstract 199.
24. Sellebjerg F, Börnsen L, Khademi M, et al. Increased cerebrospinal fluid concentrations of the chemokine CXCL13 in active MS. Neurology. 2009;73:2003–2010.
25. Cadavid D, Jurgensen S, Lee S. Impact of natalizumab on ambulatory improvement in sec-ondary progressive and disabled relapsing-remitting multiple sclerosis. PLoS One. 2013;8:e53297.
26. Mikol D, Freedman MS, Goldman MD, et al. ASCEND study of natalizumab efficacy on reducing disability in patients with secondary progressive multiple sclerosis: baseline demographics and dis-ease characteristics. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 1087.
27. Cadavid D, Brochet B, Mancardi GL, et al. The MS-COG, a novel endpoint for measurement of cognitive function in multiple sclerosis clini-cal trials: baseline characteristics of the cognitive substudy of the ASCEND natalizumab secondary progressive multiple sclerosis study. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 1088.
28. Kleist P. Composite endpoints for clini-cal trials: current perspectives. Int J Pharm Med. 2007;21:187–198.
29. Doraiswamy PM, Bieber F, Kaiser L, Krishnan KR, Reuning-Scherer J, Gulanski B. The Alzheimer’s Disease Assessment Scale: patterns and predictors of baseline cognitive performance in multicenter Alzheimer’s disease trials. Neurology. 1997;48:1511–1117.
30. Kern RS, Nuechterlein KH, Green MF, et al. The MATRICS consensus cognitive battery, part 2: co-norming and standardization. Am J Psychiatry. 2008;165:214–220.
31. Nuechterlein KH, Green MF, Kern RS, et al. The MATRICS consensus cognitive battery, part 1: test selection, reliability, and validity. Am J Psychiatry. 2008;165:203–213.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 17
Over the past several years, the number of therapies de-veloped to treat multiple sclerosis (MS) has increased
significantly. It is critically important that physicians understand the potential adverse effects, drug interactions, and im-munologic effects of these agents before they recommend particular treatments for individual patients.
Many of the posters exhibited at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) in Copenhagen, Denmark, presented current data on im-munomodulatory therapies that are either newly approved or in phase 3 clinical
trials; these data also identified some of the risks related to the use of these treat-ments. Other key posters described the potential risks and benefits of switching between these medications and proposed algorithms for doing so. Results of these studies likely will impact clinical practice and standards for MS therapy in the near future.
n ALEMTUZUMAB
Alemtuzumab is a humanized mono-clonal antibody recently approved in the European Union to treat active relapsing-remitting MS (RRMS). In the United States, it is approved only for use in the treatment of B-cell chronic lymphocytic leukemia.
Alemtuzumab selectively targets the CD52 antigen on mature lymphocytes to effectively deplete circulating T and B lymphocytes. Use of this drug reduced the annualized relapse rate in one phase 2 study (CAMMS223)1 and two phase 3 studies (CARE-MS I and CARE-MS II)2,3 when compared with subcutaneous (SC) administration of interferon beta-1a.
A number of posters at ECTRIMS 2013 described the potential adverse effects, immunogenicity, and pattern of lymphocyte depletion and repopulation associated with alemtuzumab therapy of RRMS. The two CARE-MS studies were randomized, rater-blinded, active-controlled phase 3 trials of 24 months’ duration that enrolled patients between September 2007 and April 2009.
CARE-MS I enrolled treatment-naïve patients with RRMS who were believed to have active disease (as determined by the occurrence of at least two relapses over the past 2 years and at least one relapse within the past year) and who were between 18 and 50 years of age.2 Participants in this study needed to have a baseline Expanded Disability Status Scale (EDSS) score ≤ 3 and MS symptom onset within the past 5 years. They could not have been previ-ously treated with MS disease-modifying therapy, immunosuppressants, investiga-tional agents, or monoclonal antibodies.
CARE-MS II enrolled patients who relapsed despite therapy for RRMS.3 Study participants had to be 18–55 years of age and have a baseline EDSS score ≤ 5, MS symptom onset within 10 years, and currently active disease (at least two relapses over the past 2 years and at least one relapse within the past year) with disease activity despite treatment (one or more relapses during treatment with interferon beta-1a or glatiramer acetate for at least 6 months).
In both studies, patients were random-ized to receive intravenous (IV) infusions of alemtuzumab (12 mg/d on 5 consecu-tive days at study entry and on 3 consecu-tive days 12 months later) or interferon beta-1a (44 µg SC 3 times weekly). Pri-mary endpoints were annualized relapse
Advances in Immunomodulatory Therapy for Multiple SclerosisSona Narula, MDChildren’s Hospital of Philadelphia, Philadelphia, Pennsylvania
Abstract Significant research has been directed toward the development of more convenient and effective immunomodulatory therapy for relapsing-remitting multiple sclerosis (RRMS). During the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis in Copenhagen, Denmark, investigators presented information on emerging immunomodula-tory therapies, including alemtuzumab, peginterferon beta-1a, and daclizumab high-yield process. With the number of treatment options rapidly expanding, the need to fully understand the potential risks and benefits of these novel therapies and how to safely transition between them is of utmost importance. While many new immunomodulatory therapies have been shown to be effec-tive in decreasing the frequency of clinical relapse and reducing MRI lesion burden in patients with RRMS, identifying the patient subgroup that would most benefit from these therapies and the optimal time for their introduction have yet to be determined.
Dr. Narula is a Pediatric Multiple Sclerosis Fellow in the Department of Neurology, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania.

18 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
SonaNarula,MD Advances in Immunomodulatory Therapy for Multiple Sclerosis
rate and time to accumulation of disability sustained over 6 months.
In both CARE-MS studies, alemtu-zumab demonstrated superior efficacy over interferon beta-1a in patients with active RRMS over a span of 2 years. In CARE-MS I, the relapse rate was reduced by 55% with no significant reduction in sustained disability. In CARE-MS II, alemtuzumab therapy was associated with a 49% reduction in relapse rate and a 42% reduction in the risk of 6-month sustained disability, when compared with interferon beta-1a. Adverse events associated with alemtuzumab treatment in both trials included infusion-associated reactions, infections of mild-to-moderate severity, and subsequent autoimmune disorders (eg, immune thrombocytopenia, thyroid disorders).
LymphocyteCountsandEfficacyOutcomeswithAlemtuzumabBased on a presentation by Per Soelberg Sørensen, MD, University of Copenhagen and Department of Neurology, Rigshospitalet, Copenhagen, Denmark.
Alemtuzumab selectively depletes B and T lymphocytes after each treatment course, with the cell counts reaching a nadir about 1 month after treatment.4,5 Within weeks after depletion, B cells start to repopulate, with mean values approach-ing the normal range about 3 months after a treatment course.5 T-cell reconstitution usually is slower, with mean CD4 and CD8 cell counts reaching the lower limit of normal within about 12 and 9 months, respectively, after treatment.5 Whether the rate and degree of lymphocyte repopula-tion after treatment with alemtuzumab was associated with a change in clinical disease activity was unknown. Therefore, Sørensen and colleagues6 performed an analysis to detect whether patterns of lymphocyte depletion or reconstitution were associated with disease activity in patients enrolled in the CARE-MS studies.
Lymphocyte counts at months 1 and 13 (after initial treatment) and lympho-cyte repopulation rates were compared with subsequent relapse rates and risk for 6-month sustained disability in all sub-jects. There was no significant difference between lymphocyte counts at months
1 and 13 among study participants who did or did not experience a subsequent relapse or accrual of sustained disability. There also was no significant difference in the calculated lymphocyte repopulation rates among patients who experienced a relapse or showed sustained disability as compared with those who did not. Thus, the rate of lymphocyte repopulation af-ter alemtuzumab administration cannot be used to predict the return of clinical disease activity.
EvaluatingtheImmunogenicityofAlemtuzumabBased on a presentation by Tjalf Ziemssen, MD, University Clinic Carl Gustav Carus, Dresden, Germany.
Anti-drug antibodies may reduce treat-ment efficacy in some patients with MS.
Anti-alemtuzumab antibodies develop after treatment; therefore, Ziemssen and others7 investigated the effect of these antibodies on the efficacy and safety of alemtuzumab in patients enrolled in the CARE-MS I trial.
Screening for anti-alemtuzumab antibodies was accomplished in these treatment-naïve patients via enzyme-linked immunosorbent assay (ELISA) just prior to infusion and at 1, 3, and 12 months after every treatment course. If anti-alemtuzumab antibodies were de-tected, an assay was performed to assess for inhibitory antibodies, and a titer was determined. These values were then cor-related with safety and efficacy data and total lymphocyte counts to determine an association.
In the course of this 2-year study, 87% of patients had detectable anti-alemtu-zumab antibodies at least once, and 81% of patients also had at least one positive inhibitory antibody test. The highest proportion of patients with positive anti-alemtuzumab antibodies following the initial infusion was observed soon after treatment (months 1 and 3) and slowly declined until the second treatment was administered 1 year later. Identification of both binding and inhibitory antibodies was further increased (with higher peak titers) after the second treatment course of alemtuzumab, with the highest proportion of patients having a positive antibody test at month 13.
Although they were commonly de-tected, neither anti-alemtuzumab nor inhibitory antibodies were associated with significant changes in the annualized relapse rate, magnetic resonance imaging (MRI) outcome measures, or accrual of sustained disability over the 2-year study period. Further, antibody status did not affect the overall incidence of adverse effects or infusion-associated reactions during the study.
InfectionRiskwithAlemtuzumabinPatientswithRRMSBased on a presentation by Eva Havrdova, MD, Charles University in Prague, First Medical Faculty, Prague, Czech Republic.
Havrdova and colleagues8 studied in-fection risk in patients treated with alem-tuzumab versus interferon beta-1a in the CARE-MS studies. Throughout both CARE-MS studies, safety assessments for infections, including the incidence, grade, relationship to study drug, and outcome, were recorded. Additionally, lymphocyte and neutrophil counts before and after treatment with alemtuzumab were investigated as possible predictors of infection risk following treatment. Of note, patients were ineligible for the CARE-MS studies if they had an active serious infection or were at high risk for a serious infection; if they were infected with human immunodeficiency virus (HIV) or hepatitis B or C virus; or if they had active tuberculosis, a history of fungal infection, or cervical human
In both CARE-MS studies, alemtuzumab demonstrated superior efficacy over interferon beta-1a in patients with active RRMS over a span of 2 years.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 19
SonaNarula,MD Advances in Immunomodulatory Therapy for Multiple Sclerosis
TABLE 1Treatment-Emergent Infections with Alemtuzumab in the CARE-MS Studies
Interferonβ-1a,44μg Alemtuzumab,12mg (n=389) (n=811)
Patients with infections, n (%) 221 (56.8) 589 (72.6) Related 36 (9.3) 303 (37.4) Unrelated 212 (54.5) 503 (62.0) Grade 1 (mild) 116 (29.8) 371 (45.7) Grade 2 172 (44.2) 480 (59.2) Grade 3 6 (1.5) 30 (3.7) Grade 4 0 1 (0.1) Grade 5 (fatal) 0 0Infections leading to treatment 0 0 withdrawal, n (%)Infections leading to study 0 0 discontinuation, n (%)Serious infections, n (%) 5 (1.3) 23 (2.8)
Source: Havrdova et al8
papillomavirus positivity with a high-risk strain.2,3
A total of 1,200 patients were included in this safety analysis. The frequency of infection was higher in the alemtuzumab-treated group than in the group that re-ceived interferon beta-1a (72.6% vs 56.8%, respectively). The majority of infections were mild to moderate in both groups, with 2.8% of the alemtuzumab cohort and 1.3% of the group given interferon beta-1a expe-riencing a serious infection (Table 1).8 The incidence of infection was highest during the first month after the first treatment with alemtuzumab and did not increase after the second treatment a year later.
Of note, herpetic infections were noted in 16% of the alemtuzumab group and only 2.8% of the interferon beta-1a–treated group. Beginning in late-2008, alemtuzumab-treated patients received prophylactic acyclovir starting on the first day of each treatment course and continuing for the following 28 days. The incidence of herpetic infections was lower in these patients than in those who did not receive prophylaxis (Figure 1).8 Finally, as lymphocyte counts were routinely moni-tored, patients who developed infections were not more lymphopenic than were those who did not.
n PEGINTERFERON BETA-1A
Over the past several years, interferon beta-1a has been one of the first-line im-munomodulatory treatments for MS. Use
of this immunologic agent reduces the relapse rate, disease progression, and le-sion burden in a subset of patients, but its efficacy and tolerability may be improved.
Pegylation, the process of attaching one or more molecules of polyethylene glycol to a therapeutic agent, can boost the stability, half-life, and efficacy of small-protein molecules like interferon beta-1a.9 Pegylation also can reduce renal clearance and protect the molecule from proteolytic degradation, so its circulation time is pro-longed and half-life is extended—all of which may allow for less-frequent dosing.10 Lastly, pegylation can reduce the impact of receptor- and antibody-mediated clear-ance mechanisms and may also diminish antigenicity and immunogenicity.11
Pegylated interferon beta-1a, or peg-interferon beta-1a, was developed by attaching a 20-kDa methoxy-peg-O-2-methylpropionaldehyde group to the alpha-amino group of the N-terminus of interferon beta-1a.11 It has now completed preclinical evaluations and phase 1 testing and currently is the subject of a 2-year randomized, phase 3 clinical trial known as ADVANCE. This ongoing, multicenter, double-blinded, parallel-group trial with a 1-year placebo-controlled period is de-signed to evaluate the safety and efficacy of peginterferon beta-1a injected SC once every 2 or 4 weeks in patients with RRMS. Study participants must be 18–65 years old, have a confirmed diagnosis of RRMS with an EDSS score ≤ 5, and have had at
least two relapses within the preceding 3 years and at least one relapse within the preceding year.9,12
The primary goal of the ADVANCE study is to determine the treatment effi-cacy of peginterferon beta-1a by measur-ing the change in the annualized relapse rate at 1 year. Secondary goals include determining the effect of peginterferon beta-1a on the total number of new or newly enlarging T2 hyperintense lesions on MRI, the proportion of patients with relapses, health-related quality of life, and sustained disability progression. Some of the key data from year 1 of the ADVANCE study are summarized below.
EfficacyofPeginterferonBeta-1aBased on presentations by Peter Calabresi, MD, Department of Neurology, Johns Hopkins University, Baltimore, Maryland, and Bernd Kieseier, MD, Department of Neurology, Heinrick-Heine University, Dusseldorf, Germany.
In terms of efficacy, the reduction in the annualized relapse rate with peginterferon beta-1a was statistically significant in both treatment groups (dosing every 2 weeks, 36%; dosing every 4 weeks, 28%) as com-pared with the placebo group.11 In terms of secondary endpoints, peginterferon beta-1a given either every 2 weeks or ev-ery 4 weeks significantly reduced the risk of relapse within 1 year when compared with the administration of placebo; like-
Course 1 Course 2
0.05
Incidence of herpes viral infections
0.04
0.03
0.02
0.01
0
No prophylaxis
Prophylaxis
No prophylaxisProphylaxis
606205
268521
Patients
FIGURE 1 Incidence of herpes viral infec-tions after treatment with alemtuzumab: effects of acyclovir prophylaxis during the first month. Adapted, with permission, from Havrdova et al.8

20 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
SonaNarula,MD Advances in Immunomodulatory Therapy for Multiple Sclerosis
wise, the active-treatment groups showed a significantly reduced risk of disability progression (38%) when compared with the group receiving placebo. A reduction in the number of new or newly-enlarging T2 hyperintense lesions was also seen in the every-2-week dosing group (67%) and every-4-week dosing group (28%) when compared with those receiving placebo. When given every 2 weeks, peginterferon beta-1a significantly reduced the number of new gadolinium-enhancing lesions and T1 hypointense lesions when compared with placebo administration.
In a post hoc analysis by Calabresi and colleagues,12 the proportion of patients deemed to be free of overall disease ac-tivity, as determined by the absence of clinical and radiologic disease activity over 48 weeks, was significantly higher in the every-2-week treatment group than in the every-4-week dosing group or the group given placebo.
In terms of disability, when compared with the placebo group, both active-treatment groups showed a statistically significant reduction of 38% over 48 weeks in the risk of 12-week sustained disability progression, as measured by the change in EDSS scores (more than a one-point in-crease in EDSS score in patients having an EDSS score ≥ 1 at baseline or a 1.5-point increase in EDSS score in patients with an EDSS score of 0 at baseline that was sustained over 12 weeks) in both treat-ment groups.13
ImmunogenicityofPeginterferonBeta-1aBased on a presentation by Joleen White, PhD, Principal Scientist, Biogen Idec Inc., Cambridge, Massachusetts.
The presence of neutralizing anti-drug antibodies may negatively impact the therapeutic efficacy of all immunomodu-latory therapies. This phenomenon has been seen with interferon beta in three clinical studies, where the presence of neutralizing antibodies was associated with a reduction in the drug’s clinical and radiologic efficacy.14–16
The immunogenicity of peginter-feron beta-1a and the impact of this immunogenicity on the safety, efficacy, and pharmacodynamics of the drug was
determined in patients enrolled in the ADVANCE trial. Immunogenicity was assessed by evaluating for antibodies that bind to interferon beta-1a, neutral-izing antibodies to interferon beta-1a, and antibodies that bind to polyethylene glycol. Overall, the incidence of binding antibodies, neutralizing antibodies, and anti-polyethylene glycol antibodies was low in both treatment groups, and the ma-jority of treatment-emergent antibodies appeared to be transient.17 The presence of antibodies was not associated with adverse safety events or hypersensitivity reactions (although the analysis was limited by the low incidence of treatment-emergent antibodies). There also was no discernible impact of antibody status on efficacy as evidenced by the annualized relapse rate, MRI endpoints, or disability progression.
MRIResultsfromtheFirstYearoftheADVANCEStudyBased on a presentation by Douglas Arnold, MD, Montreal Neurological Institute, McGill University, and NeuroRx Research, Montreal, Quebec, Canada.
Treatment with peginterferon beta-1a reduced the number of new or newly en-larging T2 hyperintense lesions on brain MRI scans by 67% in the every-2-week dosing group and by 28% in the every-4-week dosing group at 48 weeks, both of which were statistically significant when compared with those given placebo.12,18 Additionally, peginterferon beta-1a given every 2 weeks significantly reduced the number of new T1 hypointense lesions, gadolinium-enhancing lesions, and new active lesions at 24 and 48 weeks when compared with placebo administration. Peginterferon beta-1a given every 4 weeks also significantly reduced the number of new active lesions when compared with the use of placebo at 24 and 48 weeks. The proportion of patients free of MRI disease activity at 48 weeks was signifi-cantly higher in both the every-2-week and every-4-week treatment groups as compared with the placebo cohort.
SafetyandTolerabilityofPeginterferonBeta-1aBased on a presentation by Bernd Kieseier, MD, Department of Neurology, Heinrick-Heine University, Dusseldorf, Germany.
The overall incidence of adverse events in both peginterferon beta-1a treatment groups was 94% (compared with 83% in the placebo group), with most of these events being mild or moderate.19 The most frequent adverse events were ery-thema at the injection site, influenza-like illness, pyrexia, and headache (Table 2).19 Influenza-like illness was the most com-mon adverse event leading to treatment discontinuation. There was no increased risk of serious infection or malignancy in the peginterferon beta-1a–treated groups or the placebo group.
Similar to the changes seen in hema-tologic parameters with the use of un-modified interferon beta-1a, reductions in white blood cell counts were noted in pa-tients treated with peginterferon beta-1a. Elevations in liver transaminase levels also occurred, but they were not associated with increases in hepatic adverse events.
n DACLIZUMAB HIGH-YIELD PROCESS
Daclizumab high-yield process (DAC HYP) is a humanized monoclonal anti-body that modulates interleukin 2 (IL-2)-receptor signaling by targeting the alpha chain (CD25) of the IL-2 receptor.20
The efficacy and safety of DAC HYP in adults with RRMS were evaluated in the SELECT trial,20 a 52-week, randomized, double-blind, placebo-controlled study. Patients were randomized to receive 150 or 300 mg of DAC HYP or placebo every 4 weeks for 52 weeks. A significant decrease in the annualized relapse rate, slowing of disability progression, and reduction in the number of new MRI lesions were noted among patients treated with DAC HYP as compared with the placebo group. Patients who completed the SELECT trial were eligible for SELECTION, a 52-week extension study.
ReductionofBrainAtrophywithExtendedTreatmentwithDACHYPBased on a presentation by Ernst-Wilhelm Radue, MD, Medical Image Analysis Center, University Hospital Basel, Basel, Switzerland.
A retrospective study previously showed use of DAC HYP to reduce the rate of brain volume loss in patients with

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 21
SonaNarula,MD Advances in Immunomodulatory Therapy for Multiple Sclerosis
RRMS.21 Radue and colleagues22 evaluated the rate of brain atrophy in patients who received 2 years of treatment with DAC HYP as part of the SELECT and SELEC-TION studies.
During the first 6 months of the study, patients treated with DAC HYP (150 mg) showed a greater loss of whole-brain volume than those given placebo; this finding was considered to be secondary to pseudoatrophy, or reduced brain vol-ume related to lessened inflammation-associated edema. This phenomenon of pseudoatrophy was thought to resolve by the end of year 1; however, patients receiving DAC HYP or placebo exhibited a similar rate of decline in brain volume at that point. During year 2, however, the percentage change in whole-brain volume was 27% lower in the DAC HYP treatment groups than in the placebo group at year 1 and 24% less than in the DAC HYP combined-treatment group at year 1.
These promising results showed that treatment with DAC HYP for 2 years may result in a reduction of brain atrophy in patients with RRMS. However, it would be helpful to correlate this finding with disability progression, quality of life, and cognitive outcomes to further determine its clinical relevance.
n TRANSITIONING PATIENTS WITH RRMS TO IMMUNOMODULATORY THERAPY
With the ongoing development of new immunomodulatory therapies for MS, decisions about when and how to switch a patient’s medication are becoming increasingly complex. Additionally, the safety and efficacy of treatment escala-tion in patients who do not respond to first-line therapies have not been well established.
FIRSTStudyBased on a presentation by Giancarlo Comi, MD, Department of Neurology, Vita-Salute San Raffaele University, San Raffaele Scientific Institute, Milan, Italy.
Comi and colleagues23 completed a post hoc analysis of patients enrolled in the Fingolimod Initiation and Cardiac Safety Trial (FIRST). This study compared the tolerability, efficacy, and safety of fingolimod, a once-daily oral sphingo-sine 1-phosphate receptor modulator, in patients given glatiramer acetate or any interferon-beta formulation during the preceding 6 months versus treatment-naïve patients. The three patient groups studied had received glatiramer acetate or interferon beta in the 6 months prior to
the study or were treatment-naïve.The calculated annualized relapse rate
for each patient group during the 4-month study was lower than the calculated an-nualized relapse rate for that group in the year before the study, regardless of whether patients had been treatment-naïve or treated with interferon beta or glatiramer acetate. There were no signifi-cant differences in the overall incidence of adverse events or serious adverse events among the three groups.
This relatively short study evaluated only a limited subgroup of patients pre-viously treated with disease-modifying therapy, but the authors concluded that fingolimod could be used as an effective and safe escalation therapy for patients previously treated with either interferon beta or glatiramer acetate. These results are useful, but a longer study with an ad-ditional control group and more complete information on the patients’ treatment history could be more informative.
EPOCTrialBased on a presentation by Bruce Cree, MD, PhD, University of California, San Francisco, School of Medicine, San Francisco, California.
Cree and colleagues24 sought to de-scribe patient- and physician-reported satisfaction after switching from first-line disease-modifying therapy to fingolimod. Specifically, an open-label phase 4 study (EPOC) evaluated patient-reported outcomes and physician-reported assess-ments in patients with relapsing forms of MS who switched to fingolimod versus remained on glatiramer acetate, the stan-dard of care. All patients enrolled were deemed to be candidates for a therapy change if their physicians so identified them or if they evidenced poor tolerance or an inadequate response to standard disease-modifying therapy.
Patients were randomized to receive fingolimod or standard-of-care therapy with glatiramer acetate for 6 months. A post hoc analysis evaluated the change from baseline using the Treatment Satis-faction Questionnaire for Medication, the Beck Depression Inventory-II, the Fatigue Severity Scale, and the physician-assessed Clinical Global Impression of Improve-
TABLE 2ADVANCE Trial: Number and Frequency of Adverse Events in ≥ 10% of Patients in Each Treatment Group at Year 1
Peginterferonβ-1a
Placebo Every4weeks Every2weeksEvent,n(%) (n=500) (n=500) (n=512)
Injection-site erythema 33 (7) 282 (56) 315 (62)Influenza-like illness 63 (13) 234 (47) 239 (47)Pyrexia 76 (15) 218 (44) 228 (45)Headache 165 (33) 204 (41) 224 (44)Relapse of multiple sclerosis 159 (32) 111 (22) 96 (19)Myalgia 30 (6) 97 (19) 97 (19)Chills 23 (5) 92 (18) 88 (17)Injection-site pain 15 (3) 67 (13) 77 (15)Asthenia 38 (8) 70 (14) 68 (13)Back pain 57 (11) 64 (13) 61 (12)Injection-site pruritus 6 (1) 56 (11) 68 (13)Nasopharyngitis 77 (15) 69 (14) 53 (10)Arthralgia 35 (7) 54 (11) 57 (11)Fatigue 49 (10) 55 (11) 51 (10)Pain in extremity 49 (10) 54 (11) 44 (9)
Source: Kieseier et al19

22 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
SonaNarula,MD Advances in Immunomodulatory Therapy for Multiple Sclerosis
ment. After 6 months of treatment, both patients and physicians believed that a switch to fingolimod provided benefit. In particular, the investigators found a significant improvement in patient treat-ment satisfaction, especially with regard to convenience and patient-perceived effectiveness.
n CONCLUSION
Many of the posters presented at ECTRIMS 2013 highlighted the breadth of research dedicated to developing im-munomodulatory therapies. Many studies featured drugs with novel mechanisms of action and alternative routes of adminis-tration for patients with RRMS. Ongoing research, data collection, and analysis will help to determine whether these new therapies are truly safe, effective, and ready for routine clinical use.
REFERENCES
1. Coles AJ, Fox E, Vladic A, et al. Alemtu-zumab more effective than interferon β 1a at 5-year follow-up of CAMMS223 Clinical Trial. Neurology. 2012;78:1069–1078.
2. Cohen JA, Coles AJ, Arnold DL, et al. Alem-tuzumab versus interferon β 1a as first-line treatment for patients with relapsing-remitting multiple scle-rosis: a randomized controlled phase 3 trial. Lancet. 2012;380:1819–1828.
3. Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing mul-tiple sclerosis after disease-modifying therapy: a randomized controlled phase 3 trial. Lancet. 2012;380:1829–1839.
4. Hartung HP, Arnold DL, Cohen JA. Lympho-cyte subset dynamics following alemtuzumab treat-ment in the CARE-MS 1 study. Presented at the 28th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 10–13, 2012; Lyon, France. Poster P 935.
5. Kovarova I. Alemtuzumab pharmacokinetics and pharmacodynamics in comparison of alemtu-zumab and Rebif efficacy in multiple sclerosis I. Presented at the 22nd Meeting of the European Neu-rological Society; June 9–12, 2012; Prague, Czecho-slovakia. Poster P 341.
6. Sørensen PS, Arnold DL, Hartung HP, et al. Lymphocyte counts and efficacy outcomes af-ter alemtuzumab in relapsing remitting multiple
sclerosis patients: the CARE-MS studies. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 515.
7. Ziemssen T, Arnold DL, Cohen JA, et al. Immunogenicity of alemtuzumab does not impact safety and efficacy in relapsing remitting multiple sclerosis patients in the CARE-MS I study. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 523.
8. Havrdova E, Arnold DL, Cohen JA, et al. Infection risk with alemtuzumab in patients with relapsing remitting multiple sclerosis: pooled re-sults from the CARE-MS I and CARE-MS II trials. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copen-hagen, Denmark. Poster P 603.
9. Reuss R. PEGylated interferon β-1a in the treatment of multiple sclerosis—an update. Biologics: Targets and Therapy. 2013;7:131–138.
10. Kieseier BC, Calabresi PA. PEGylation of interferon b-1a: a promising strategy in multiple sclerosis. CNS Drugs. 2012;26:205–214.
11. Baker DP, Pepinsky RB, Brickelmaier M. PEGylated interferon β-1a: meeting an unmet medical need in the treatment of relapsing multiple sclerosis. J Interfer Cytokine Res. 2010;30:777–785.
12. Calabresi PA, Kieseier BC, Arnold DL, et al. Peginterferon β-1a provides improvements in clinical and radiological disease activity in relapsing-remitting multiple sclerosis: year 1 findings from the phase 3 ADVANCE study. Presented at the 29th Con-gress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 514.
13. Kieseier BC, Calabresi PA, Liu S, et al. Effect of peginterferon β-1a on disability progression in patients with relapsing-remitting multiple sclerosis: year 1 data from the pivotal phase 3 ADVANCE study. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copen-hagen, Denmark. Poster P 540.
14. Polman CH, Bertolotto A, Deisenhammer F, et al. Recommendations for clinical use of data on neutralising antibodies to interferon-β therapy in multiple sclerosis. Lancet Neurol. 2010;9:740–750.
15. Bertolotti A, Deisenhammer F, Gallo P, et al. Immunogenicity of interferon β: differences among products. J Neurol. 2004;251:II15–II24.
16. Paolicelli D, D’Onghia M, Pellegrini F, et al. The impact of neutralizing antibodies on the risk of disease worsening in interferon b-treated relapsing
multiple sclerosis: a 5 year post-marketing study. J Neurol. 2013;260:1562–1568.
17. White JT, Calabresi PA, Zhu Y, et al. Immu-nogenicity with peginterferon β-1a in patients with relapsing-remitting multiple sclerosis: data from the pivotal phase 3 ADVANCE study. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 600.
18. Arnold DL, Calabresi PA, Kieseier BC, et al. Magnetic resonance imaging results from the first year of the ADVANCE study, a pivotal phase 3 trial of peginterferon β-1a in patients with relapsing-re-mitting multiple sclerosis. Presented at the 29th Con-gress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 989.
19. Kieseier BC, Calabresi PA, Song T, et al. Safety and tolerability of peginterferon β-1a in patients with relapsing-remitting multiple sclerosis: data from the pivotal phase 3 ADVANCE study. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copen-hagen, Denmark. Poster P 1061.
20. Gold R, Giovannoni G, Selmaj K, et al. Daclizumab high-yield process in relapsing-remit-ting multiple sclerosis (SELECT): a randomised, double-blind, placebo-controlled trial. Lancet. 2013;381:2167–2175.
21. Borges IT, Shea CD, Ohayon J, et al. The effect of daclizumab on brain atrophy in relapsing-remitting multiple sclerosis. Mult Scler Relat Disord. 2013;2:133–140.
22. Radue EW, Stefoski D, Gold R, et al. Reduc-tion in brain atrophy with extended daclizumab hyp treatment: results of SELECT and the SELECT extension study. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 977.
23. Comi G, Gold R, Kappos L, et al. Relapse and safety outcomes in patients who transitioned from glatiramer acetate or interferon β to fingolimod in the open-label FIRST study. Presented at the 29th Con-gress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 513.
24. Cree BAC, Kantor D, Steingo BM, et al. Patient- and physician-reported outcomes after therapy switch from glatiramer acetate to fingolimod versus remaining on glatiramer acetate. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 1010.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 23
Multiple sclerosis (MS) is an inflammatory, demyelin-ating disease defined by lesions in the central ner-
vous system (CNS) that are disseminated in time and space. The disease course is heterogeneous and characterized by a symptomatic spectrum ranging from relapsing-remitting episodes of disability to the development of progressive disease.
This article reviews currently available disease-modifying therapies approved for relapsing-remitting MS (RRMS) in the context of current experience and evidence for intervention, and it offers tools for clinicians and patients who are navigating the therapeutic options. It is based upon
A New Era of Therapy in Multiple Sclerosis: Balancing the Options and Challenges AheadJennifer L. Orthmann-Murphy, MD, PhDHospital of the University of Pennsylvania, Philadelphia, Pennsylvania
Abstract The recent explosion in regulatory approvals of disease-modifying medications for patients with relapsing-remitting multiple sclerosis makes the choice of therapy increasingly complicated. It is not yet known which medication is best for particular patients or at specific points of the disease process. Based on nearly two decades of experience with beta interferons, early intervention clearly modifies the disease course; however, no current treatment has been shown to prevent or reverse disability accumulation in the long term. At two Biogen Idec–sponsored symposiums held during the 2013 Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS), experts discussed available tools to help clinicians and patients make these decisions and the evidence supporting new and future therapeutic options.
Dr. Orthmann-Murphy is a Senior Neurology Resident at the Hospital of the University of Pennsylvania, Philadelphia, Pennsylvania.
presentations delivered at two industry-sponsored symposiums held during the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) in Copenhagen.
n A DIVERSE MENU FOR TREATING RRMS
TheTriedandTrueTwenty years of experience have shown
that the treatment of RRMS with inter-feron beta is safe, delays disability, reduces mortality, and delays conversion from clinically isolated syndrome to RRMS.1,2 Figure 1 summarizes the clinical develop-ment of this immunomodulatory agent in RRMS over the past 25 years.3
The mechanism of action of interferon beta in patients with MS is unknown, but it may cause a shift to anti-inflammatory cytokines and prevent leukocytes from crossing the blood-brain barrier.4 Table 1 summarizes the currently available for-mulations of interferon beta, along with administration options, adverse effects, and the pivotal clinical trials that evalu-ated each formulation.
Ongoing trials are striving to improve the efficacy of interferon beta therapy and patient adherence to this treatment. ADVANCE is a phase 3, randomized, controlled trial comparing administra-tion of subcutaneous (SC) pegylated interferon (peginterferon) beta-1a every 2 or 4 weeks with placebo. Pegylation, the addition of polyethylene glycol (PEG), increases the potency and half-life of interferon beta and decreases its immu-nogenicity.5 At 1 year, there was a signifi-cant decrease in the annualized relapse rate, number of gadolinium-enhancing lesions, and production of neutralizing antibodies among treated patients, sug-gesting that peginterferon beta will be an attractive and effective alternative to current formulations.6 Elsewhere in this issue of The Neurology Report, Dr. Sona Narula summarizes the key data from year 1 of the ADVANCE study.
The other injectable option, glat-iramer acetate, is a copolymer compris-ing glutamic acid, lysine, alanine, and tyrosine, which originally was designed to mimic myelin basic protein and induce experimental autoimmune encephalitis (EAE); instead, it ameliorated disease in both animal models7 and humans.8 The mechanism of action is not completely clear; however, much like the interferon-beta treatments, glatiramer acetate may cause a shift toward anti-inflammatory cytokines. Many years of experience has proven that glatiramer acetate is also a safe and effective treatment of RRMS that delays the conversion from a clinically isolated syndrome to RRMS (Table 1).
Mitoxantrone is a type II topoisom-

24 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
JenniferL.Orthmann-Murphy,MD,PhD A New Era of Therapy in Multiple Sclerosis
erase inhibitor that was approved by the US Food and Drug Administration in 2000 to treat rapidly worsening RRMS or secondary progressive MS. However, due to significant cardiotoxicity and a grow-ing list of safer therapeutic alternatives, recent use of mitoxantrone in MS has been limited.9
n TARGETED IMMUNOMODULATION
NatalizumabNatalizumab is the most potent thera-
peutic option available for treating RRMS, but it also carries the highest risk (Table 1). This monoclonal antibody binds α-4 integrin on activated T cells; this interac-tion prevents these cells from binding with VCAM1 on endothelial cells of the blood-brain barrier, thereby blocking entry into the CNS.10
Figure 2,11 a summary timeline of natalizumab development milestones, highlights the accelerated approval of the drug and its eventual withdrawal from the market after three patients developed pro-
gressive multifocal leukoencephalopathy (PML).12 Fortunately, it is now possible to stratify patient risk for developing PML based on the presence of anti–JC virus antibodies as a marker of prior infection, prior or current immunosuppression, and duration of natalizumab treatment (Figure 3).13
The ability to stratify risk for PML is critical. Post hoc analyses of the AFFIRM and SENTINEL trials continued to show the significant efficacy of natalizumab, not only in reducing the relapse rate and disease activity, as found on magnetic resonance imaging (MRI), but also in ef-fecting remission (or “disease-free activity” by both clinical and radiologic measures) in some patients.14,15
Interestingly, patients having an Ex-panded Disability Status Scale (EDSS) score < 3 were more likely to return to baseline after a relapse on natalizumab than were those with an EDSS score > 3, suggesting that this is a critical threshold for repair reserve.16 These results set the stage for a new standard for treatment-
outcome goals and primary endpoints for future clinical trials.
FingolimodFingolimod, another potent immu-
nomodulator, was the first approved oral drug for RRMS (Table 1). Fingolimod binds to sphingosine-1-phosphate (S1P) receptors on lymphocytes, sequestering these cells in lymphatic tissue so that they may not access the CNS.17 Fingolimod was significantly more effective than was interferon beta-1a for reducing annual-ized relapse rates and disease activity on MRI.18 However, this therapy requires more baseline and continued monitoring, given the risk of skin neoplasm, macular edema, bradycardia, and infection related to its use (Table 1).
TeriflunomideTeriflunomide was the next oral im-
munomodulator to be approved (Table 1).19 Its prodrug form, leflunomide, is an effective treatment for another autoim-mune disorder, rheumatoid arthritis.
FIGURE 1 Timeline of development of interferon beta over the past 25 years. RRMS = relapsing-remitting multiple sclerosis; CIS = clinically isolated syndrome; SC = subcutaneous; IM = intramuscular; IFN β-1a = interferon beta-1a; IFN β-1b = interferon beta-1b; NF = new formulation (serum free); IFNB MS = Interferon Beta in Multiple Sclerosis; MSCRG = Multiple Sclerosis Collaborative Research Group; PRISMS = Prevention of Relapses and Disability by Interferon Beta-1a Subcutaneously in Multiple Sclerosis; CHAMPS = Controlled High-Risk Avonex Multiple Scle-rosis Prevention Study; ETOMS = Early Treatment of Multiple Sclerosis; BENEFIT = Betaseron in Newly Emerging Multiple Sclerosis for Initial Treatment; REFLEX = Rebif Flexible Dosing in Early Multiple Sclerosis; EU = European Union. Adapted from Calabresi.3
1995
SC IFN β-1b approved for RRMS
1997
IM IFN β-1a approved for RRMS
2007
SC IFN β-1a NF approved for RRMS in EU
1988 1990 1992 2014201020062002 2012200820041998 20001994 1996
1998
SC IFN β-1a approved for RRMS
2002
IM IFN β-1a approved for CIS
2006
SC IFN β-1b approved for CIS
2012
SC IFN β-1a approved for CIS
SC IFN β-1b in RRMS(IFNB MS Group)
IM IFN β-1a in RRMS (MSCRG) SC IFN β-1a in CIS (REFLEX)
SC IFN β-1a in RRMS(PRISMS)
IM IFN β-1a in CIS(CHAMPS)
SC IFN β-1b in CIS(BENEFIT)
IM IFN β-1a inRRMS (Pen)
SC IFN β-1a in CIS (ETOMS)
Study startPaper publishedApproved in EU

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 25
JenniferL.Orthmann-Murphy,MD,PhD A New Era of Therapy in Multiple Sclerosis
TABLE 1Summary of Currently Approved Disease-Modifying Drugs for Relapsing-Remitting Multiple Sclerosis in the United States
Drug Administration Majorandminoradverseevents Pivotalclinicaltrialevidencea
Interferon beta-1b SC injection every other day • Pyrexia IFNB MS Group37 (1993) • Depression CIS: BENEFIT38 (2006) • Injection-site reactions • Neutralizing antibodies • Hematologic, thyroid, and liver abnormalities Interferon beta-1a IM or SC pen injection once • Pyrexia MSCRGA39 (1996): Annualized relapse rate a week • Depression versus placebo (0.61 vs 0.90) • Injection-site reactions CIS: CHAMPS40 (2000) • Neutralizing antibodies • Hematologic, thyroid, and liver abnormalities Interferon beta-1a SC injection 3 times weekly • Pyrexia PRISMS41 (1998): ~30% in relapse rate • Depression compared with placebo • Injection-site reactions CIS: ETOMS42 (2001) and REFLEX43 (2012) • Neutralizing antibodies • Hematologic, thyroid, and liver abnormalitiesGlatiramer acetate Daily SC injection • Injection-site reactions or lipoatrophy CP1MSSG8 (1995): Annualized relapse rate • Post-injection flushing reaction versus placebo (0.59 vs 0.84) REGARD44 (2008) CIS: PreCISE45 (2009)Mitoxantrone IV infusion every 3 months • Congestive heart failure Edan et al46 (1997) • Leukemia, lymphoma MIMS47 (2002): Annualized relapse rate • Lymphopenia, neutropenia versus placebo (0.35 vs 1.02) • Liver dysfunction • Discolored urine • Alopecia • Nausea • Congenital defects Fingolimod PO once daily • Bradycardia/AV block (first-dose monitoring) TRANSFORMS18 (2010): Annualized relapse • Macular edema rate versus interferon beta-1b (~0.20 vs 0.30) • Infection (disseminated zoster, HSV encephalitis) FREEDOMS48 (2010): Annualized relapse • Transaminitis rate versus placebo (~0.17 vs 0.40) • Skin cancer Natalizumab Monthly IV infusion • PML (requires JC virus antibody testing and risk AFFIRM49 (2006): Annualized relapse rate stratification) versus placebo (0.26 vs 0.81) • Neutralizing antibodies SENTINEL12 (2006): Annualized relapse rate • Infection versus placebo (0.34 vs 0.75) • Allergic reaction Teriflunomide PO once daily • Infection (check PPD prior to starting) TEMSO19 (2011): Annualized relapse rate • Transaminitis versus placebo (0.37 vs 0.54) • Gastrointestinal symptoms (nausea) • Alopecia • Peripheral neuropathy • Pregnancy Category X Dimethyl fumarate PO twice daily • Flushing DEFINE50 (2012): Annualized relapse rate • Gastrointestinal symptoms (nausea, diarrhea) versus placebo (~0.18 vs 0.36) • Lymphopenia CONFIRM22 (2011): Annualized relapse rate • Proteinuria versus placebo (~0.21 vs 0.4), but similar to that of glatiramer acetate
Abbreviations: SC = subcutaneous; MS = multiple sclerosis; IM = intramuscular; IV = intravenous; PO = orally; AV = atrioventricular; HSV = herpes simplex virus; PML = progressive multifocal leukoencephalopathy; PPD = purified protein derivativea Pivotal randomized controlled trials leading to approval or showing efficacy in delaying conversion from clinically isolated syndrome (CIS) to relapsing-remitting multiple sclerosis. Results are noted for the trials that report annualized relapse rates. For further information on these therapies, please consult the full prescribing information and a recent review on safety monitoring.9
Teriflunomide inhibits dihydrooro-tate dehydrogenase, preventing de novo pyrimidine nucleotide synthesis, which may suppress the effector function of acti-vated lymphocytes; however, it is not clear whether this mechanism is responsible for its efficacy in autoimmune disease.20 Treatment with teriflunomide showed a
modest effect versus placebo in reduc-ing the annualized relapse rate (Table 1) and was at least equivalent to interferon beta-1a.19,21
DimethylFumarateThe most recent MS drug approved
in the United States is dimethyl fumarate
(Table 1), a drug that specifically targets and activates the Nrf2-signalling pathway. This leads to the upregulated expression of antioxidant response elements, and it may alter cytokine signaling.22 Dimethyl fumarate is also one of the components of fumaric acid esters used to treat psoriasis; it has an excellent safety profile.22 Results

26 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
JenniferL.Orthmann-Murphy,MD,PhD A New Era of Therapy in Multiple Sclerosis
from a phase 3 clinical trial showed that dimethyl fumarate is at least as good as glatiramer acetate in reducing the annual-ized MS relapse rate.23 Further such head-to-head comparisons will likely guide treatment decisions in the future. In an ongoing, long-term safety extension study known as ENDORSE,24 efficacy has been sustained, and there have been no new or worse safety events than those originally reported in the DEFINE and CONFIRM trials (Table 1).
OtherDrugsOther immunomodulating, disease-
modifying drugs currently in develop-ment or being tested in ongoing phase 3 clinical trials include laquinimod, ritux-imab, and alemtuzumab.22,23,25
n CURRENT TREATMENT CHALLENGES: IN NEED OF A TREATMENT ALGORITHM
These many treatment options and their potential for causing significant adverse events are making RRMS man-agement increasingly complex, but some tools and lines of evidence are available to clinicians to guide management.26
First, early identification and initiation of therapy may improve the course of
RRMS.1,2 The currently available therapies outlined here target immune activity, which is more active early in the course of disease and is evident by the number of relapses that occur and the accumula-tion of gadolinium-enhancing lesions on MRI.27 With the modified 2010 McDonald criteria for diagnosis of RRMS, it is now possible to diagnose and intervene earlier in the disease course.28 However, this shift means that newly diagnosed patients are not directly comparable to those treated in the original clinical trials; instead, they may be more similar to a subgroup of patients from these early trials who had a clinically isolated syndrome. In addition, the patients recruited to modern trials will not be directly comparable to those participating in the original trials. This heterogeneity is evident in Table 1, in which the reported annualized relapse rate in the placebo arm of many of the seminal trials ranged from 0.3 to 1.02.
Second, certain patient characteristics are associated with more aggressive dis-ease than others. These factors include relapse severity, degree of recovery, gen-der, race, age, and disease activity on MRI and are summarized in Table 2.29 Patients with these more aggressive characteristics probably should be treated with more
highly effective therapies earlier in their disease course.
To initiate therapy, the clinician and the patient together must make treatment decisions based on prognosis, treatment goals, patient preference, and analysis of benefit and risk of the options. The clini-cian can navigate the currently available first-line therapies (interferon beta or glat-iramer acetate) and second-line therapies (all others) or rank them as low, moderate, and highly effective. Over time, and with further head-to-head clinical trials, it may be easier to develop a universal algorithm for treatment initiation. In the meantime, the clinician and patient should balance treatment goals (eg, reduction of relapses vs freedom from disease) with potential adverse events.
Once patients are on therapy, response must be aggressively monitored with follow-up MRI and reports of relapse occurrence to determine whether dis-ability is accumulating or adverse effects or neutralizing bodies are developing. At this point, we have limited tools to char-acterize and follow disease severity. They include the EDSS score, accumulation of MRI lesion burden, and clinical report of relapse. The modified Rio Score is an evidence-based algorithm that incor-
2000
Biogen and Elan announce collaboration on the development and commercialization of natalizumab for both MS and Crohn’s disease
2006
Natalizumab returns to the US market with pharmacovigilance plan in place; EMEA approves natalizumab
2005
Natalizumab voluntarily suspended from the US market on the basis of three PML cases
2011
European label lists three con�rmed risk factors for PML
1992
Alpha-4 integrin discovered to be key molecule involved in homing to the brain
2004
FDA approves natalizumab for relapsing forms of MS
1997
Phase 1 research begins
2001
Phase 3 trial of natalizumab in RRMS begins
2012
US label lists three con�rmed risk factors for PML
1990 201420102006200219981994
FIGURE 2 Milestones in the development of natalizumab over the past 20 years. MS = multiple sclerosis; RRMS = relapsing-remitting mul-tiple sclerosis; PML = progressive multifocal leukoencephalopathy; FDA = US Food and Drug Administration; EMEA = European Medicines Agency. Adapted from Rudick et al.11

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 27
JenniferL.Orthmann-Murphy,MD,PhD A New Era of Therapy in Multiple Sclerosis
FIGURE 3 Estimates of the incidence of progressive multifocal leukoencephalopathy (PML) among patients with multiple sclerosis treated in the postmarketing setting with natalizumab, stratified according to prior or no prior use of immunosuppressants and duration of natalizumab treatment (A) and according to positive or negative status with respect to anti–JC virus antibodies, prior or no prior use of immunosuppres-sants, and duration of natalizumab treatment (B). Adapted from Bloomgren et al.13
Number of PML cases
Number of patients treated
Incidence of PML per1,000 patients (95% CI)
1 (hypothetical)
11,625
≤ 0.09 (0–0.48)
Number of PML cases
Number of patients treated
Incidence of PML per1,000 patients (95% CI)
1–24 monthsof natalizumab
exposure
25
44,721
0.56 (0.36–0.83)
25–48 monthsof natalizumab
exposure
94
20,362
4.6 (3.7–5.6)
1–24 monthsof natalizumab
exposure
16
10,043
1.6 (0.91–2.6)
25–48 monthsof natalizumab
exposure
52
4,681
11.1 (8.3–14.5)
1–24 monthsof natalizumab
exposure
25
81,310
0.31 (0.20–0.45)
25–48 monthsof natalizumab
exposure
94
37,024
2.5 (2.1–3.1)
1–24 monthsof natalizumab
exposure
16
18,261
0.88 (0.50–1.4)
25–48 monthsof natalizumab
exposure
52
8,509
6.1 (4.6–8.0)
No Yes
Prior immunosuppressant use?A
B Anti–JC virus antibody status
No Yes
Prior immunosuppressant use?
Negative Positive
porates all three of these tools; this tool accurately predicted responders from the PRISMS patient population.30 Future biomarkers of response to therapy such as brain atrophy on imaging studies and analyses of cerebrospinal fluid (CSF) and serum markers, as well as tools that can consistently identify physical and cogni-tive changes over time, are needed.
n PROGRESSION IN MS: AN UNMET NEED
Despite these exciting new therapeutic choices for patients with RRMS, there is still no disease-modifying intervention for
the progressive form of the disease. The basic pathogenic mechanisms underlying brain atrophy in MS are unknown, and many questions remain unanswered: Is progression a primary neurodegenerative disease occurring simultaneously with demyelination? Is neuronal loss second-ary to loss of oligodendrocytes? What role does the immune response play in progressive disease?
Anti–LINGO-1AntibodiesOne disease-modifying therapy cur-
rently in development, anti-LINGO-1 an-tibodies, specifically targets remyelination
in MS lesions. The goal is to protect axons in MS lesions through remyelination, po-tential prevention of axonal degeneration, and amelioration of the progressive form of the disease.
LINGO-1 is a leucine-rich repeat transmembrane domain protein initially identified by Mi and colleagues as a novel component of the Nogo receptor complex involved in blocking CNS axon regenera-tion.31 LINGO-1 expression is restricted to the CNS and is found in neurons, oligodendrocytes, and oligodendrocyte precursor cells (OPCs).32 Genetic dele-tion or antibody-mediated blocking of

28 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
JenniferL.Orthmann-Murphy,MD,PhD A New Era of Therapy in Multiple Sclerosis
TABLE 2Factors Linked to a Higher Risk of More Aggressive Disease in Patients with Multiple Sclerosis at Diagnosis
Relapseseverity• One moderate or severe attack• Need for corticosteroids and/or
hospitalization • Severe effect on activities of daily living• Effect on more than one functional system • Severe involvement of motor function/
cerebellum/brainstemIncompleterecoveryfromrelapseMagneticresonanceimaging• Two gadolinium-enhancing/new T2
lesions or more than two T1 hypointense lesions
• Two spinal cord lesions• Brain atrophyOlderageMalegenderAfrican-Americanethnicity
Adapted, with permission, from Freedman et al29
LINGO-1 has demonstrated that it is a negative regulator of both oligoden-drocyte differentiation and myelination in vitro and slice culture.32,33 Given that blocking LINGO-1 with anti–LINGO-1 antibody led to OPC differentiation, remyelination, and improvement in func-tional recovery in several mouse models of demyelination33,34 and that chronic MS lesions contained premyelinating oligo-dendrocytes,35 anti–LINGO-1 antibody appears to be an excellent candidate for treatment in MS.
A phase 1 study sponsored by Biogen Idec involving intravenous administration of anti–LINGO-1 antibodies36 to both healthy controls and patients with RRMS and secondary progressive MS provided evidence of the safety of this therapeutic approach, and CSF was collected at con-centrations predicted to be efficacious. Two phase 2 trials currently are enrolling patients with RRMS and relapsing second-ary progressive MS (SYNERGY) or a first episode of optic neuritis (RENEW).
n CONCLUSION
This is an exciting time to be treating patients with RRMS, as many options are available to alter the course of disease.
Further clinical studies that directly com-pare therapies or that identify tools and biomarkers for predicting and evaluating response to therapy are needed before evidence-based algorithms for manage-ment of this disease can be developed. In addition, understanding the pathogenesis of progression in MS and identifying novel approaches for neuroprotection and prevention of disability are important future goals.
REFERENCES
1. Goodin DS, Reder AT, Ebers GC, et al. Sur-vival in MS: a randomized cohort study 21 years after the start of the pivotal IFNbeta-1b trial. Neurology. 2012;78:1315–1322.
2. Trojano M, Pellegrini F, Paolicelli D, et al. Real-life impact of early interferon beta ther-apy in relapsing multiple sclerosis. Ann Neurol. 2009;66:513–520.
3. Calabresi P. Reinventing MS care—evolution: advancing interferons for people with MS. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark.
4. Kieseier BC. The mechanism of action of interferon-beta in relapsing multiple sclerosis. CNS Drugs. 2011;25:491–502.
5. Bailon P, Won CY. PEG-modified biophar-maceuticals. Expert Opin Drug Deliv. 2009;6:1–16.
6. Biogen Idec. New data analyses show sig-nificant clinical and MRI improvements with PLEGRIDY™ (peginterferon beta-1a) [press release]. October 1, 2013. http://www.biogenidec.com/press_release_details.aspx?ID=5981&ReqId=1860099. Accessed October 23, 2013.
7. Teitelbaum D, Webb C, Meshorer A, Arnon R, Sela M. Protection against experimental allergic encephalomyelitis. Nature. 1972;240:564–566.
8. Johnson KP, Brooks BR, Cohen JA, et al. Co-polymer 1 reduces relapse rate and improves disabil-ity in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45:1268–1276.
9. Rommer PS, Zettl UK, Kieseier B, et al. Requirement for safety monitoring for approved MS therapies—an overview. Clin Exp Immunol. September 17, 2013 [Epub ahead of print].
10. Yednock TA, Cannon C, Fritz LC, et al. Prevention of experimental autoimmune encephalo-myelitis by antibodies against alpha 4 beta 1 integrin. Nature. 1992;356:63–66.
11. Rudick R, Polman C, Clifford D, Miller D, Steinman L. Natalizumab: bench to bedside and beyond. JAMA Neurol. 2013;70:172–182.
12. Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354:911–923.
13. Bloomgren G, Richman S, Hotermans C, et al. Risk of natalizumab-associated progressive
multifocal leukoencephalopathy. N Engl J Med. 2012;366:1870–1880.
14. Hutchinson M, Kappos L, Calabresi PA, et al. The efficacy of natalizumab in patients with relapsing multiple sclerosis: subgroup analyses of AFFIRM and SENTINEL. J Neurol. 2009;256:405–415.
15. Havrdova E, Galetta S, Hutchinson M, et al. Effect of natalizumab on clinical and radiological disease activity in multiple sclerosis: a retrospective analysis of the natalizumab safety and efficacy in relapsing-remitting multiple sclerosis (AFFIRM) study. Lancet Neurol. 2009;8:254–260.
16. Giovannoni G. Reinventing MS care—evo-lution: silencing the disease from the start. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark.
17. Brinkmann V, Davis MD, Heise CE, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277:21453–21457.
18. Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–415.
19. O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365:1293–1303.
20. Claussen MC, Korn T. Immune mechanisms of new therapeutic strategies in MS: teriflunomide. Clin Immunol. 2012;142:49–56.
21. Vermersch P, Czlonkowska A, Grimaldi LM, et al. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult Scler. October 14, 2013. Epub ahead of print.
22. Gold R. Oral therapies for multiple scle-rosis: a review of agents in phase III development or recently approved. CNS Drugs. 2011;25:37–52.
23. Lulu S, Waubant E. Humoral-targeted im-munotherapies in multiple sclerosis. Neurotherapeu-tics. 2013;10:34–43.
24. Montalban X. Reinventing MS care—revolu-tion: defending the brain. Presented at the 29th Con-gress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark.
25. Biogen Idec. New TECFIDERA® (dimethyl fumarate) data show sustained efficacy and long-term safety in a broad range of multiple scle-rosis patients [press release]. October 4, 2013. http://www.biogenidec.com/press_release_details.aspx?ID=5981&ReqId=1861556. Accessed October 23, 2013.
26. Fox EJ, Rhoades RW. New treatments and treatment goals for patients with relapsing-remitting multiple sclerosis. Curr Opin Neurol. 2012;25:S11–S19.
27. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurol-ogy. 1996;46:907–911.
28. Polman CH, Reingold SC, Banwell B, et

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 29
JenniferL.Orthmann-Murphy,MD,PhD A New Era of Therapy in Multiple Sclerosis
al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292–302.
29. Freedman MS, Selchen D, Arnold DL, et al. Treatment optimization in MS: Canadian MS work-ing group updated recommendations. Can J Neurol Sci. 2013;40:307–323.
30. Sormani MP, De Stefano N. Defining and scoring response to IFN-beta in multiple sclerosis. Nat Rev Neurol. 2013;9:504–512.
31. Mi S, Lee X, Shao Z, et al. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci. 2004;7:221–228.
32. Mi S, Miller RH, Lee X, et al. LINGO-1 nega-tively regulates myelination by oligodendrocytes. Nat Neurosci. 2005;8:745–751.
33. Mi S, Miller RH, Tang W, et al. Promotion of central nervous system remyelination by induced differentiation of oligodendrocyte precursor cells. Ann Neurol. 2009;65:304–335.
34. Mi S, Hu B, Hahm K, et al. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experi-mental autoimmune encephalomyelitis. Nat Med. 2007;13:1228–1233.
35. Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med. 2002;346:165–173.
36. Sandrock A. Reinventing MS care—revolu-tion: the future is repair. Presented at the 29th Con-gress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark.
37. The IFNB Multiple Sclerosis Study Group.
Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology. 1993;43:655–661.
38. Kappos L, Polman CH, Freedman MS, et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology. 2006;67:1242–1249.
39. Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progres-sion in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group. Ann Neurol. 1996;39:285–294.
40. Jacobs LD, Beck RW, Simon JH, et al. Intramuscular interferon beta-1a therapy initi-ated during a first demyelinating event in multiple sclerosis. CHAMPS study group. N Engl J Med. 2008;343:898–904.
41. PRISMS (Prevention of Relapses and Dis-ability by Interferon-Β 1a Subcutaneously in Multiple Sclerosis) Study Group. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. Lancet. 1998;352:1498–1504.
42. Comi G, Filippi M, Barkhof F, et al. Effect of early interferon treatment on conversion to defi-nite multiple sclerosis: a randomised study. Lancet. 2001;357:1576–1582.
43. Comi G, De Stefano N, Freedman MS, et al. Comparison of two dosing frequencies of subcutane-ous interferon beta-1a in patients with a first clinical demyelinating event suggestive of multiple sclerosis (REFLEX): a phase 3 randomised controlled trial. Lancet Neurol. 2012;11:33–41.
44. Mikol DD, Barkhof F, Chang P, et al. Com-parison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs glatiramer acetate in relaps-ing MS disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol. 2008;7:903–914.
45. Comi G, Martinelli V, Rodegher M, et al. Ef-fect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet. 2009;374:1503–1511.
46. Edan G, Miller D, Clanet M, et al. Thera-peutic effect of mitoxantrone combined with meth-ylprednisolone in multiple sclerosis: a randomized multicentre study of active disease using MRI and clinical criteria. J Neurol Neurosurg Psychiatry. 1997;62:112–118.
47. Hartung HP, Gonsette R, Konig N, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360:2018–2025.
48. Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401.
49. Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910.
50. Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367:1098–1107.

30 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
During the 1990s, the first significant advances in mul-tiple sclerosis (MS) therapy involved the introduction of
the beta interferons and glatiramer acetate. Clinicians now have used these drugs for many years; they are comfortable with monitoring parameters related to their use, and they can readily explain their efficacy and side-effect profiles to their patients.
Much has changed, however, since these platform therapies were first in-troduced. Discussions about therapeutic options for MS are much more complex, and they often stretch the limitations of a single office visit. And whereas these
newer therapies show promise in terms of both efficacy and safety, they have not yet been used for prolonged periods.
Natalizumab and dimethyl fumarate represent important advances in our abil-ity to control MS, but their safety profile is still evolving, as is a full understanding of their efficacy. During the 29th Congress of the European Committee for Treat-ment and Research in Multiple Sclerosis (ECTRIMS), researchers and clinicians discussed the safety and efficacy of na-talizumab and dimethyl fumarate therapy in post hoc analyses of pivotal trials and in phase 4 monitoring cohorts that came from the original study groups. These data provide continued insight into the efficacy of these new medications, while further exploring important safety endpoints.
n NATALIZUMAB
IntroducingaNewEndpoint:DiseaseActivity-FreeStatus
Disease activity-free status (DAFS) is a composite endpoint defined by
freedom from relapse, zero progression of the Expanded Disability Status Scale (EDSS) score over 12 weeks, and lack of gadolinium-enhancing or new/enlarging lesions over 2 years. The DAFS endpoint is an adaptation of a similar outcome measure used in clinical research studies of rheumatoid arthritis. It was introduced when tumor-necrosis factor inhibitors made complete remission of rheumatoid arthritis a realistic possibility; its ap-plication in MS research carries similar significance in terms of advancement of the ability to control the disease.1
AFFIRMStudyHavrdova et al1 evaluated the emerging
endpoint of DAFS in the phase 3 AFFIRM study, which compared use of natalizumab with that of placebo in a 2-year, placebo-controlled, multicenter, randomized clini-cal trial involving patients with relapsing-remitting MS (RRMS).
The AFFIRM data set consisted of 600 patients given natalizumab and 304 given placebo. Patients were grouped according to baseline characteristics and evaluated for DAFS over 2 years. Study participants were divided into subgroups according to age, disease duration, number of relapses experienced in the past year, EDSS score, and total gadolinium-enhancing lesions detected. In all subgroups, a significantly greater proportion of patients given natalizumab than those given placebo achieved freedom from disease activ-ity over 2 years (P ≤ 0.0005). In a sub-
Natalizumab and Dimethyl Fumarate: A Fresh Take on Pivotal Trials and Reports from Ongoing MonitoringCarolyn Bevan, MD, MSMultiple Sclerosis Center, University of California, San Francisco, School of Medicine, San Francisco, California
Abstract Natalizumab was approved after results from the pivotal AFFIRM and SENTINEL trials were released in 2006. Dimethyl fumarate was approved after results of the DEFINE and CONFIRM trials were published in 2012. At the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS), pivotal trials of natalizumab and dimethyl fumarate were revisited, post hoc safety and efficacy analyses were discussed, and results from ongoing postmarketing monitoring studies were reported. Both natalizumab and dimethyl fumarate continue to be effective treatment options for patients with multiple sclerosis who are appropriately screened and monitored.
Dr. Bevan is a Clinical Fellow in Neurology at the Multiple Sclerosis Center, University of California, San Francisco, School of Medicine, San Francisco, California.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 31
CarolynBevan,MD,MS Natalizumab and Dimethyl Fumarate
group analysis of natalizumab-treated patients, a greater proportion of people who were younger or who had shorter disease duration, fewer relapses in the prior year, lower EDSS scores, and fewer gadolinium-enhanced lesions at study baseline reached DAFS.
In subgroup analyses of patients treated with placebo, only the group with fewer gadolinium-enhanced lesions achieved a higher proportion of DAFS. The odds ratios of achieving DAFS fa-vored natalizumab in all subgroups and were significantly greater in patients with an EDSS score < 3.0 than among those having an EDSS score ≥ 3.0 at baseline in the second year.2
TysabriObservationalProgramThe Tysabri Observational Program
(TOP) is an ongoing, open-label, 10-year prospective study of DAFS among RRMS patients given natalizumab therapy in clinical settings in Europe, Australia, Canada, and Argentina.3 The TOP popu-lation involved 2,595 natalizumab-treated patients who were followed for at least 2 years. Investigators used logistic regres-sion to determine proportions of patients with DAFS in subgroups according to the same baseline characteristics used to analyze the AFFIRM data. The major-ity of patients in each subgroup reached DAFS at 2 years of natalizumab therapy. As observed in the AFFIRM population, a greater proportion of patients with lower EDSS scores, fewer relapses, and fewer gadolinium-enhanced lesions at baseline reached DAFS at 2 years of natalizumab treatment.
These results confirmed that the benefits of natalizumab observed in the AFFIRM trial persist in clinical practice, with over 50% of the TOP population achieving freedom from clinical disease activity across all subgroups at 2 years. Im-portantly, patients earlier in their disease state who were younger and had shorter disease duration, fewer relapses in the prior year, lower EDSS scores, and fewer gadolinium-enhanced lesions were more likely to achieve DAFS on natalizumab. This pattern suggested that treating patients who are earlier in their disease
course and less disabled may increase the odds of achieving DAFS.
RelapsesinPatientsonNatalizumabLublin et al4 reported on a post hoc
analysis of data from the AFFIRM study, showing that natalizumab-treated pa-tients experienced a reduction in relapse severity and in post-relapse disability. Relapses were defined as new or recur-rent symptoms without fever that lasted at least 24 hours and that were associated with objective findings on neurologic examination. In all, relapses occurred in 183 patients using natalizumab and 176 patients given placebo during the trial period. Of this group, 143 natalizumab-treated patients and 140 patients given placebo met the criteria for inclusion in the study. Relapse severity was defined by the change between pre-relapse EDSS score and EDSS score recorded during the relapse. Residual relapse-induced dis-ability was defined as the persistence of change in EDSS score recorded at least 30 days after the assessment at relapse. Com-plete recovery was defined as a return to an EDSS score at or below that measured before the relapse and sustained for at least 12 weeks. Importantly, an equal propor-tion of patients in the natalizumab and placebo groups received corticosteroids to help manage relapse.
The natalizumab-treated group dem-onstrated significantly lower relapse severity when compared with the pla-cebo group. Residual relapse-induced disability was also significantly lower in the natalizumab group than in the placebo cohort. Natalizumab-treated patients with an increase in EDSS score ≥ 0.5 point during relapse had a 55% increased cumulative probability of complete recovery from relapse rela-tive to the placebo group. In addition, when compared with the placebo group, natalizumab-treated patients with an in-crease in EDSS score of ≥ 1 point during relapse had a 67% increased cumulative probability of full recovery.
Based upon these observations, the investigators concluded that natalizumab therapy does more than decrease re-lapse frequency as demonstrated in the
AFFIRM trial—it also reduces clinical severity in relapses and improves the likelihood of complete recovery.4
StartingandStoppingNatalizumabKallmann and others5 reported on
data from the TYSTART trial, in which German patients with RRMS starting na-talizumab were monitored for clinical and magnetic resonance imaging (MRI) find-ings and laboratory parameters (including JC virus antibody testing) prospectively in a routine clinical setting at baseline and at 3-month intervals for 12 months.
Of the 258 patients captured at base-line, 168 completed the 12-month follow-up. The mean EDSS score remained stable (3.6 ± 1.7 vs 3.5 ± 1.7 at baseline), and the mean Fatigue Severity Score (FSS) improved from 4.1 ± 1.7 to 2.2 ± 2.1. Two patients experienced a relapse of MS on treatment, which contributed a total of four serious adverse events (SAEs). Of 228 JC virus-antibody tests, 28.1% were positive. None of the patients in the cohort developed progressive multifocal leukoencephalopathy (PML). During the monitoring period, 4.7% of patients discontinued treatment with natalizumab. These findings confirmed that natalizum-ab therapy is effective during the first 12 months after treatment initiation and that it results in stabilization of the EDSS score, improvement of the FSS, and lowering of the frequency of SAEs.
NatalizumabDiscontinuationandDiseaseActivity
Initiation of natalizumab therapy has shown clear benefit in select populations, but there has been some concern about recurrence or worsening of disease activ-ity, either clinically or by MRI, after use of the drug is discontinued. Although this phenomenon has been reported in the literature, it may be subject to selection or reporting bias.
In Denmark, patients are offered na-talizumab only if they are classified as “highly active,” if they have breakthrough disease on platform therapies, or if they experience an aggressive relapse result-ing in a permanent EDSS score increase of 2 points. Sørensen et al6 reviewed data

32 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
CarolynBevan,MD,MS Natalizumab and Dimethyl Fumarate
on all Danish patients who discontinued natalizumab therapy and followed these individuals for 12 months after discon-tinuation.
Of the 1,017 patients in the Danish registry given natalizumab for longer than 24 weeks, 392 discontinued therapy. Of this group, 338 patients were studied. Within the first 1–3 months after natali-zumab discontinuation, patients returned to their pretreatment relapse rate. These individuals subsequently returned to their on-treatment relapse rate, presum-ably because they began using another disease-modifying therapy. It does not appear that this group experienced re-lapse rates or MRI activity to a greater degree than they did before they started natalizumab. These findings suggested that the “immune overshoot” described in earlier reports reflected the exception rather than the rule.
n DIMETHYL FUMARATE
PostHocAnalysisoftheDEFINEandCONFIRMTrials
Results from the phase 3 DEFINE and CONFIRM studies showed that dimethyl fumarate therapy had a significant impact on clinical and imaging parameters and resulted in approval of the drug for relaps-ing MS in the United States, Australia, and Canada.7,8 Havrdova et al9 performed a post hoc analysis of DAFS in patients treated with dimethyl fumarate in the DEFINE and CONFIRM trials.
Again, patients were considered dis-ease activity free if they experienced no relapses, had no EDSS score progression, and developed no new or newly enlarging T2 lesions or gadolinium-enhanced le-sions on MRI. The proportion of patients with no measured overall disease activity at 2 years was 23% in both the twice- and three-times-daily dosing groups, as com-pared with 11% in the placebo group. Pa-tients treated with dimethyl fumarate were twice as likely to have no measured overall disease activity at 6 months, 1 year, and 2 years. These findings reflected promising advances in MS treatment.
Hutchinson et al10 reported on another post hoc analysis of the clinical efficacy
of dimethyl fumarate in the DEFINE and CONFIRM study groups, this time evaluating subgroups of patients strati-fied according to previous treatment with interferon beta-1a or glatiramer acetate. Use of dimethyl fumarate was effective in all subgroups of patients, regardless of the previous therapeutic regimen used.
The same group also evaluated the clin-ical efficacy of dimethyl fumarate among European participants in the DEFINE and CONFIRM studies, this time integrating data from the two trials in a post hoc analysis. 11 The results showed a similar benefit of dimethyl fumarate therapy in the integrated population and its compo-nent studies, with an adjusted annualized relapse rate of about 50%, an approxi-mately 30% reduction in confirmed dis-ability progression, an 85% reduction in gadolinium-enhanced lesion activity, and an 80% reduction in the number of new or enlarging T2 hyperintense lesions in the group using twice-daily dosing.
These post hoc analyses of the DEFINE and CONFIRM trials added a new per-spective about the efficacy of dimethyl fumarate and confirmed the findings of the original trials.
Four-YearFollow-UpofPatientsTreatedwithDimethylFumarate
Both clinical and MRI outcomes are being reported as part of a 4-year exten-sion of the DEFINE, CONFIRM, and ENDORSE studies. In ENDORSE, a 5-year extension study of the DEFINE and CONFIRM trials, patients who completed the phase 3 studies are continuing the same dimethyl fumarate dosage (given in two or three daily doses); the placebo or glatiramer acetate treatment groups are being randomized 1:1 to receive active treatment with dimethyl fumarate given twice or three times daily.12,13 Endpoints include the annualized relapse rate; pro-portion of patients who relapsed; and disability progression, as measured by the EDSS score every 6 months.
Clinical and imaging data from the first 2 years of the ENDORSE study were reported at ECTRIMS.12 Among 1,736 patients enrolled in ENDORSE who were treated with dimethyl fumarate for 4
years (on active treatment with dimethyl fumarate in DEFINE or CONFIRM and then on dimethyl fumarate for 2 years in the extension study), the relapse rate remained low, as did the proportion of patients who relapsed at 4 years (36.2%). Among the group treated with dimethyl fumarate twice daily for 4 years, 15.4% experienced disease progression. The groups of patients initially on placebo or glatiramer acetate and transitioned to di-methyl fumarate for the first 2 years of the ENDORSE study showed similar clinical outcomes when compared with the group treated for 4 years. These data reinforce the long-term safety and efficacy of di-methyl fumarate as a disease-modifying therapy in MS.
In addition to clinical outcomes, MRI disease activity among the ENDORSE cohort was assessed. In patients treated with dimethyl fumarate in the DEFINE and CONFIRM trials and who continued on treatment in the ENDORSE study, the adjusted mean numbers of new/enlarging T2 hyperintense lesions were 1.3 in the group given twice-daily dosing and were similar among those who took the drug three times daily. Numbers of new non-enhancing T1 hypointense lesions were low (0.6) in patients given twice-daily dos-ing and were similar to findings in those dosed three times daily. The numbers of new non-enhancing T1 hypointense le-sions and gadolinium-enhanced lesions also remained low. In patients switched from placebo or glatiramer acetate to dimethyl fumarate, MRI measures ap-proached those of the group treated with dimethyl fumarate in the phase 3 stud-ies.13 These results confirmed the efficacy of dimethyl fumarate in MRI endpoints.
DimethylFumarateSafetyMonitoringintheENDORSETrial
As with all newly approved therapies, the safety profile of dimethyl fumarate continues to evolve as years of treatment increase. Phillips et al14 reported on all available safety data from the ENDORSE cohort through June 2013 according to treatment received in the parent and extension studies. The total number of patient years ranged from 212.8 in the

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 33
CarolynBevan,MD,MS Natalizumab and Dimethyl Fumarate
group initially given glatiramer acetate and then given dimethyl fumarate three times daily to 1,187.4 in the group that started and continued on dimethyl fumarate given twice daily. The overall incidence of adverse events, SAEs, and discontinuations of therapy due to adverse events was similar among those given di-methyl fumarate in the parent studies and continued on treatment in the ENDORSE trial and patients previously treated with placebo and glatiramer acetate in the parent studies and switched to dimethyl fumarate in the ENDORSE trial.
Flushing and adverse gastrointestinal (GI) events occurred more commonly among individuals treated with dimethyl fumarate. Approximately 10% of patients continued on dimethyl fumarate in the ENDORSE study, and 29% of patients switched from placebo to dimethyl fu-marate experienced flushing. Diarrhea occurred in 8% of patients who contin-ued on dimethyl fumarate and 13% of those who were switched from placebo to dimethyl fumarate. Interestingly, there was a slightly lower rate of these events in patients switched from glatiramer acetate to dimethyl fumarate, with 21% expe-riencing flushing and 9% experiencing diarrhea. The incidence of these events peaked during the first month and then decreased in the parent studies.
MS relapse also was observed in the treated groups, with 27% experiencing MS relapse in the continuing-therapy group as compared with 27% in the prior-placebo group and 20% in the prior-glatiramer acetate group. Other adverse events oc-curring in ≥ 10% of patients in any treat-ment group included nasopharyngitis, urinary tract infections, headache, upper respiratory tract infections, back pain, and upper abdominal pain.
In the ENDORSE trial, 4%–6% of patients who continued on dimethyl fu-marate from the parent studies discontin-ued therapy due to adverse events, as did 14%–23% of patients who were switched to dimethyl fumarate. Among the latter group, flushing and GI upset were the leading reasons for discontinuation. No-tably, MS relapse was the most commonly reported SAE. Other SAEs occurring in
no more than four patients in any treat-ment group included urinary tract infec-tion (nine patients); appendicitis (four patients); cellulitis (three patients); and breast cancer, gastritis, falls, and uterine leiomyoma (one patient each). There were no confirmed opportunistic infections.
The incidence of malignancies in the group continued on dimethyl fumarate was 1%, with no evidence of increased risk of malignancy. In terms of hematologic changes, patients who were switched to dimethyl fumarate in the ENDORSE trial developed decreases in mean white blood cell (WBC) and lymphocyte counts simi-lar to those seen in the parent studies. In patients continued on dimethyl fumarate, mean WBC and lymphocyte counts re-mained stable as compared with levels in the parent studies. In the ENDORSE group
who remained on dimethyl fumarate, there was a 6%–7% incidence of WBC counts < 3.0 × 109/L and a 7%–10% incidence in those switched to dimethyl fumarate therapy. There was a 6%–8% incidence of lymphocyte counts < 0.5 × 109/L among patients continued on dimethyl fumarate; there was a 5%–9% incidence among those switched to the drug. Elevated aspartate aminotransferase and alanine amino-transferase levels at least three times the upper limit of normal were noted among < 2% of patients, and there were no cases of drug-induced liver injury. Proteinuria, microalbuminuria, and hematuria were the most commonly noted renal adverse events, occurring in at least 3% of patients in all treatment groups.14
The effect of dimethyl fumarate on lymphocyte counts in treated patients was analyzed in more detail by Fox et
al15 based on integrated analysis of the placebo-controlled phase 2b, DEFINE, and CONFIRM studies. In these trials, treatment-discontinuation rules were developed with WBC criteria and were not based on lymphocyte counts. In the phase 2b trial, dimethyl fumarate was discontinued if WBC counts dropped to < 1.5 × 109/L at any time during the study or if WBC counts < 2.0 × 109/L were sustained for 4 weeks. In the DEFINE and CONFIRM trials, use of dimethyl fuma-rate was discontinued if WBC counts < 2.0 × 109/L were sustained for 4 consecutive weeks after study treatment was withheld or a WBC count < 2.0 × 109/L was con-firmed on retesting.
In patients treated with dimethyl fumarate, integrated analysis showed that mean WBC counts fell by 11% and mean lymphocyte counts fell by 30% through week 48 of treatment; they later plateaued but remained within normal limits through the remainder of the 96 weeks. Grade 3/4 lymphopenia (defined as a lymphocyte count < 0.5 × 109/L in one or more consecutive assessments) was observed in a higher percentage of patients in the dimethyl fumarate group than in the placebo group. As in previ-ous studies, the incidence of grade 3/4 lymphopenia increased through week 48 and then stabilized. Importantly, there was no increased incidence of infections or serious infections associated with development of lymphopenia during the monitoring period, and no opportunistic infections were observed. In patients with lymphopenia who discontinued dimethyl fumarate, lymphocyte counts rose after discontinuation, but they did not return to pretreatment baseline levels 4 weeks after treatment discontinuation.15 The sig-nificance of the duration of lymphopenia after drug discontinuation is unclear, but it could influence decisions about switching to other medications that also may cause lymphopenia.
n CONCLUSION
Both natalizumab and dimethyl fuma-rate represent important advancements in treating RRMS. Natalizumab contin-ues to demonstrate consistent efficacy
Both natalizumab and dimethyl fumarate represent important advancements in treating RRMS.

34 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
CarolynBevan,MD,MS Natalizumab and Dimethyl Fumarate
in both clinical and imaging outcomes, with risk stratification facilitated by JC virus antibody testing. Its influence on recovery after an active relapse is still more intriguing. Although natalizumab is a powerful and effective drug, the risk of PML requires careful monitoring. In addition, patients should be closely monitored for recurrence of their previous level of aggressive disease activity after discontinuing the drug.
Meanwhile, dimethyl fumarate is a meaningful new addition to the growing list of oral therapies for MS. It effectively treats both clinical and radiographic dis-ease. However, it has just recently entered the market; continued surveillance by investigators affiliated with the ENDORSE study will provide important information about its safety profile over time.
There was a time when clinicians could offer few options to MS patients. Now, the landscape of MS therapeutics has changed drastically. However, clinicians must en-sure that patients understand the complex framework of treatment choice, including both the benefits and risks entailed in each approach.REFERENCES
1. Havrdova E, Galetta S, Stefoski D, Comi G. Freedom from disease activity in multiple sclerosis. Neurology. 2010;74:S3–S7.
2. Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of na-talizumab for relapsing multiple sclerosis. AFFIRM Investigators. N Engl J Med. 2006;354:899–910.
3. Kappos L, Belachew S, Butzkueven H, et al. Long-term safety and efficacy and association be-tween baseline treatment history and post-baseline relapses in multiple sclerosis patients treated with
natalizumab in the TYSABRI® Observational Pro-gram (TOP). Presented at the 64th Annual Meeting of the American Academy of Neurology; April 21–28, 2012; New Orleans, Louisiana. Poster P 04.133.
4. Lublin FD, Cutter G, Giovannoni G, et al. Natalizumab reduces the disabling amplitude of multiple sclerosis relapses and improves post-relapse residual disability. Presented at the 29th Congress of the European Committee for Treatment and Re-search in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P524.
5. Kallmann B, Hoffmann R, Walter U, et al. Monitoring and management of multiple sclerosis patients starting natalizumab therapy in Germany: final study results of TYSTART. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 1066.
6. Sørensen PS, Oturai A, Sellebjerg F, Magyari M, Koch-Henriksen N. Recurrence or rebound of clinical relapses after discontinuation of natalizumab therapy. Presented at the 29th Congress of the Eu-ropean Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 1058.
7. Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. DEFINE Study Investigators. N Engl J Med. 2012;367:1098–1107.
8. Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. CONFIRM Study Investigators. N Engl J Med. 2012;367:1087–1097.
9. Havrdova E, Gold R, Fox RJ, et al. Effect of BG-12 (dimethyl fumarate) on freedom from measured clinical and neuroradiologic disease ac-tivity over time in patients with relapsing-remitting multiple sclerosis: results from the phase 3 studies. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copen-hagen, Denmark. Poster P 521.
10. Hutchinson M, Gold R, Fox RJ, et al. Clinical efficacy of BG-12 (dimethyl fumarate) for relapsing-remitting multiple sclerosis according to prior therapy: an integrated analysis of the phase
3 DEFINE and CONFIRM studies. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 563.
11. Hutchinson M, Fox R, Bar-Or A, et al. Ef-ficacy of BG-12 (dimethyl fumarate) in relapsing-remitting multiple sclerosis in patients from Europe: an integrated analysis of the phase 3 DEFINE and CONFIRM studies. Presented at the 29th Congress of the European Committee for Treatment and Re-search in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 1013.
12. Gold R, Phillips JT, Bar-Or A, et al. 4-Year follow-up of oral BG-12 (dimethyl fumarate) treat-ment in relapsing-remitting multiple sclerosis: integrated clinical efficacy data from the DEFINE, CONFIRM, and ENDORSE studies. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 538.
13. Miller DH, Fox RJ, Gold R, et al. 4-Year follow-up of oral BG-12 (dimethyl fumarate) treat-ment in relapsing remitting multiple sclerosis: inte-grated magnetic resonance imaging outcomes from DEFINE, CONFIRM, and ENDORSE. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 1004.
14. Phillips JT, Fox RJ, Selmaj K, et al. Safety profile of BG-12 (dimethyl fumarate) in relapsing-remitting multiple sclerosis: long-term interim re-sults from the ENDORSE extension study. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 996.
15. Fox RJ, Gold R, Phillips JT, et al. Lymphocyte count reductions in relapsing-remitting multiple sclerosis patients treated with oral BG-12 (dimethyl fumarate): integrated analysis of the placebo-con-trolled studies. Presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); October 2–5, 2013; Copenhagen, Denmark. Poster P 1018.

T H E N E U R O L O G Y R E P O R T | W i n t e r 2 0 1 4 35
CME/CNE Post TestUsing this page as a worksheet, select the best answer to each question based on your reading of the articles in this edition of The Neurology Report, then complete the evaluation on page 36 and see the instructions below it to obtain CME or CNE credit.
1. Infection with ___________ is a known risk factor for multiple sclerosis (MS).a. Varicella zoster virusb. Hepatitis C virusc. Epstein-Barr virusd. Cytomegalovirus
2. In the HERMES trial, treatment of MS with rituximab led to:a. Fewer new gadolinium-enhancing
lesionsb. A reduction in the total number of
gadolinium-enhancing lesionsc. A reduction in T2 lesion volumed. All of the above
3. Which of the following statements about ocrelizumab is true?a. Ocrelizumab is biosimilar to rituximab.b. Ocrelizumab is bioidentical to rituximab.c. Ocrelizumab targets the CD4 receptor
on B cells.d. Unlike rituximab, ocrelizumab can be
given orally.
4. Histopathologic studies of progressive MS suggest that inflammation and neuro-degeneration occur:a. In a stepwise fashionb. Sporadicallyc. Simultaneouslyd. Completely independently of one
another
5. Results from the OLYMPUS study in patients with primary progressive MS sug-gested that:a. Younger patients with gadolinium-
enhancing lesions seemed to benefit the most from treatment with rituximab.
b. Significantly more patients given pla-cebo experienced faster disease progres-sion than did those given rituximab.
c. Neither rituximab nor placebo halted disease progression after 1 year.
d. Rituximab, but not placebo, slowed the development of brain atrophy.
6. Paz Soldan et al, investigating the im-pact of relapses on post-progression dis-ability, found that disability accumulated fastest in patients with:
a. Relapsing-remitting MSb. A single clinical attack of progressive
MSc. Primary progressive MSd. Secondary progressive MS
7. Notable adverse events associated with alemtuzumab therapy in the CARE-MS trials included:a. Dysphoriab. Hypertensionc. Thyroid abnormalities and other auto-
immune disordersd. Severe infections
8. In the ADVANCE study of pegylated interferon beta-1a versus placebo, which of the following was observed among patients treated with the drug?a. No significant reduction in the risk of
relapse at 1 yearb. Fewer new or newly enlarging T2 hy-
perintense lesionsc. No significant reduction in the risk of
disability progression over 48 weeksd. A significantly higher proportion of
patients free of disease activity in the group treated every 4 weeks compared with those treated every 2 weeks
9. The most common adverse event lead-ing to treatment discontinuation in the ADVANCE study was:a. Pyrexiab. Erythemac. Influenza-like illnessd. Headache
10. Despite its efficacy, the use of mitoxan-trone is limited in MS because of concerns over its:a. Cardiotoxicityb. Hepatoxicityc. Renal toxicityd. Neurotoxicity
11. The most potent therapeutic option for treating MS currently available that also carries the highest risk is:a. Fingolimodb. Natalizumabc. Mitoxantroned. Teriflunomide
12. Which of the following investigational therapies specifically targets remyelination in MS lesions, potentially preventing axo-nal degeneration?a. Alemtuzumabb. Daclizumab high-yield processc. Laquinimodd. Anti–LINGO-1 antibodies
13. In the AFFIRM study, which of the fol-lowing baseline characteristics predicted a poorer chance of being free of disease activ-ity at 2 years among MS patients receiving natalizumab?a. An Expanded Disability Status Scale
(EDSS) score > 3.0b. Fewer gadolinium-enhanced lesionsc. Shorter disease durationd. Youthful age
14. A long-term, prospective, multinational study of nearly 2,600 MS patients receiving natalizumab in clinical settings has shown that _____ achieve freedom from disease activity at 2 years.a. 20%b. 30%c. 40%d. 50% or more
15. Long-term follow-up of MS patients treated with dimethyl fumarate revealed that:a. Among the group treated with di-
methyl fumarate twice daily for 4 years, more than 20% experienced disease progression.
b. The numbers of new or newly enlarg-ing T2 hyperintense lesions and new, nonenhancing T1 hypointense lesions were lower in patients who took the drug three times daily compared with twice daily.
c. Mean WBC and lymphocyte counts fell during the first 48 weeks of treatment but then plateaued and remained within normal limits for up to 96 weeks.
d. A higher percentage of patients switched from placebo or glatiramer acetate to dimethyl fumarate relapsed, compared with those who continued on dimethyl fumarate (27%).

36 T H E N E U R O L O G Y R E P O R T | V o l u m e 6 N u m b e r 1
Your candid and thorough completion of this evaluation will help the University of Cincinnati improve the quality of its continu-ing education activities. Thank you for your participation. Strongly agree Agree Disagree1. As a result of this activity, I am more knowledgeable about …
a. Efforts to redefine the clinical course of multiple sclerosis (MS), develop ❑ ❑ ❑new clinical outcome assessment tools, and identify key research areas and molecular therapeutic targets.
b. The development of pharmacologic therapies for relapsing-remitting MS ❑ ❑ ❑and their relative advantages and disadvantages.
c. The tools and clinical evidence now available for choosing initial therapy for ❑ ❑ ❑individual patients, monitoring their progress, and transitioning them from one disease-modifying therapy to another.
d. The role of B cells in the pathogenesis of MS and current research into the ❑ ❑ ❑potential role of B-cell modulation in its treatment.
e. Current research in understanding the pathogenesis of disease progression ❑ ❑ ❑in MS and efforts to prevent or treat it.
Strongly agree Agree Disagree2. I found the content of this educational activity …
a. Clearly written and well organized. ❑ ❑ ❑
b. Accurate and timely. ❑ ❑ ❑
c. Related to its overall objectives. ❑ ❑ ❑
d. Free from commercial bias. ❑ ❑ ❑
e. Relevant to my own clinical practice. ❑ ❑ ❑
Yes No Don’t know3. Did the information you received from this CME/CNE activity:
a. Confirm the way you currently manage your patients? ❑ ❑ ❑
b. Suggest new options for managing your patients that you might apply ❑ ❑ ❑in the future?
Patient Board CME/CNE management review credit
4. I used the information in this issue for … (check all that apply) ❑ ❑ ❑
5. Approximately how long (in minutes) did it take you to complete this activity, minutes including this evaluation?
Evaluation
Instructions for Obtaining CME or CNE CreditTo receive credit for this free educational activity and a certificate of participation from the University of Cincinnati:
• Study the educational material presented in this issue of The Neurology Report.• Using page 35 as a worksheet, answer all of the post-test questions based on the content of the articles.• Visit www.NeurologyReport.com on the Web by January 6, 2015, select this issue of The Neurology Report, and
click “CME/CNE Credit” to apply for credit online and complete the post test and evaluation.• Complete the registration form, enter your post-test answers from the worksheet on page 35, and respond to all of the
questions on the evaluation form, then click the button to submit your answers. The full text of each article may be accessed at www.NeurologyReport.com, should you need to refer to it again.
• If you answer correctly at least 12 (80%) of the 15 post-test questions, you will immediately receive credit for completing this educational activity and can access your CME or CNE certificate online by clicking the “Certificate” button at the bottom of the evaluation form. Follow the on-screen instructions to print or e-mail your certificate.

