the membrane domain of 3-hydroxy-3-methylglutaryl-coenzyme a
TRANSCRIPT

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1988 by The American Society for Biochemistry and Molecular Biology, Inc.
Vol. 263, No. 14, Issue of May 15, pp. 6836-6841,1988 Printed in U. S. A .
The Membrane Domain of 3-Hydroxy-3-methylglutaryl-coenzyme A Reductase Confers Endoplasmic Reticulum Localization and Sterol-regulated Degradation onto ,&Galactosidase*
(Received for publication, September 21, 1987)
David G. Skalnik$$, Hiroshi Naritall, Claudia Kent 11, and Robert D. Simoni**$ From the $Department of Biological Sciences, Stanford University, Stanford, California 94305
A hybrid gene has been constructed consisting of coding sequence for the membrane domain of the en- doplasmic reticulum protein 3-hydroxy-3-methylglu- taryl-coenzyme A (HMG-CoA) reductase linked to the coding sequence for the soluble enzyme Escherichia coli @-galactosidase. Expression of the hybrid gene in transfected Chinese hamster ovary cells results in the production of a fusion protein (HMGal) which is local- ized in the endoplasmic reticulum. The fusion protein contains the high-mannose oligosaccharides character- istic of HMG-CoA reductase. Importantly, the @-galac- tosidase activity of HMGal decreases when low density lipoprotein is added to the culture media. Therefore, the membrane domain of HMG-CoA reductase is suf- ficient to determine both correct intracellular localiza- tion and sterol-regulation of degradation. Mutant fu- sion proteins which lack 64, 85, or 98 amino acid residues from within the membrane domain of HMG- CoA reductase are found to be localized in the endo- plasmic reticulum and to retain @-galactosidase activ- ity. However, sterol-regulation of degradation is abol- ished.
The enzyme 3-hydroxy-3-methylglutaryl-coenzyme A re- ductase (HMG-CoA’ reductase) catalyzes the conversion of HMG-CoA to mevalonate and is the major regulatory enzyme in sterol biosynthesis (1, for review). It is a 97-kDa integral membrane glycoprotein localized in the endoplasmic reticu- lum. We have demonstrated that it is inserted into the mem- brane cotranslationally in a signal recognition particle-de- pendent manner and that it lacks a cleavable signal sequence (2).
The determination of the primary structure of the enzyme from the nucleotide sequence has led to a predicted secondary structure and topological model of the enzyme (3). HMG-CoA
* The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
f Present address: Div. of Hematology and Oncology, The Chil- dren’s Hospital, Boston, MA 02115.
llThis work was done while H. N. was on leave from The Research Institute for Food Science, Kyoto University, which is his current address.
IIThis work was done while C. K. was on leave from the Biochem- istry Dept., Purdue University, which is her current address.
**TO whom correspondence should be addressed. The abbreviations used are: HMG-CoA, 3-hydroxy-3-methylglu-
taryl coenzyme A; LDL, low density lipoprotein; CHO, Chinese hamster ovary; LPS, lipid poor serum; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; Endo H, endoglycosidase H.
reductase contains seven putative membrane-spanning re- gions which are highly conserved between species (4, 5). Comparison of the deduced amino acid sequences from differ- ent species also reveals a divergent linker region between the highly conserved membrane domain and the cytoplasmic cat- alytic domain of HMG-CoA reductase.
The regulation of the level of HMG-CoA reductase is com- plex and includes regulation of both transcription of the gene and degradation of the protein (6, 7). In Chinese hamster ovary (CHO) cells, grown in the presence of exogenous cho- lesterol, the half-life of HMG-CoA reductase is 2-4 h. How- ever, when HMG-CoA reductase activity is induced, the half- life of the protein is extended (7-9). Under conditions of abundant cellular sterol, the synergistic effect of reduced transcription of the HMG-CoA reductase gene and enhanced degradation of the protein results in a rapid reduction in the level of this key enzyme, thereby dramatically reducing the cells ability to synthesize mevalonic acid.
It has been shown that a 52-kDa proteolytic fragment, representing the cytoplasmic portion of HMG-CoA reductase, is sufficient for enzymatic activity (3, 10). A major question has been the function of the noncatalytic membrane domain of the enzyme. The striking conservation of sequence through- out the 40-kDa membrane domain suggests that it may be important for more than membrane anchorage. Insight into this question comes from the finding that a soluble form of HMG-GOA reductase, synthesized from a modified cDNA clone, no longer exhibits sterol-regulation of degradation (11). This suggests that the membrane domain of HMG-CoA re- ductase may interact with regulatory molecules. The mem- brane domain of the protein also undoubtedly provides the signal for localization of this protein to the endoplasmic reticulum.
A fusion protein consisting of the membrane domain of HMG-CoA reductase linked to the amino terminus of p - galactosidase has been constructed in order to directly study the function of the membrane domain of HMG-CoA reduc- tase. Analysis of the behavior of this chimeric molecule (HMGal) in vivo has allowed the identification of HMG-CoA reductase characteristics which are conferred onto the heter- ologous ,&galactosidase protein. Such characteristics are hence determined solely by the noncatalytic membrane do- main of HMG-CoA reductase. The behavior of mutant fusion proteins (AHMGal) was then observed in an attempt to localize the primary sequences required for these membrane domain functions.
EXPERIMENTAL PROCEDURES
Materials-All materials, unless specified, were readily available from commercial sources. All restriction endonucleases and DNA- modifying enzymes were obtained from New England Biolabs. The plasmids pGHlOl and pMLB1113 were the generous gifts of Dr. Gail
6836

Regulation of Degradation HMG-CoA Reductase 6837
Herman and Dr. Michael Berman, respectively. Anti-@-galactosidase monoclonal antibody was purchased from Boehringer Mannheim. Low density lipoprotein (LDL) was isolated from freshly drawn blood and purified as previously described (12). Geneticin was obtained from Gibco.
Cell Culture-Chinese hamster ovary (CHO-K1) cell monolayers were grown in minimal essential media supplemented with either 5% fetal calf serum or 5% lipid poor serum (LPS) which was prepared by the method of Rothblat et d. (13). The culture media was changed every 2 days.
Construction of Fusion Genes-The plasmid pSV2-HMGal was constructed by cloning the 5’ EcoRI-PstI 1.4-kb fragment of HMG- CoA reductase cDNA from pDGS6 into the EcoRI and PstI polylinker sites of pMLB1113 (Fig. 1). The resulting HMG-CoA reductase/& galactosidase fusion gene was excised by digestion with EcoRI and BalI, followed by the addition of EcoRI linkers and ligation into the expression vector pSV2 (14). The plasmid pSV2-Gal was constructed by excising the @-galactosidase sequence from pGHlOl with SalI, followed by a blunt-end ligation into pSV2.
In order to produce mutations within the membrane domain of HMG-CoA reductase, the cDNA insert from ADS11 (15) was cloned into the EcoRI site of pBR322 to generate pDGS2. The majority of the 3”untranslated region was removed by digestion with Aat2 and a partial EcoRI digestion followed by ligation, producing pDGS4. This plasmid was digested with ApaI (Fig. 2), which cuts uniquely at nucleotide 495 (where position +1 is the site of translation initiation (4)), corresponding to the fifth putative membrane span of HMG- CoA reductase. The sample was then digested with the exonuclease Bal3I and ligated. The cloning mix was transformed into competent Escherichia coli and selected for resistance to ampicillin. Plasmid was isolated from each of 18 recovered colonies as described (16), and the Hind111 fragment surrounding the deletion was subcloned into the M13-sequencing vector mplO. The DNA sequence surrounding each deletion junction was determined by the method of di-deoxy chain termination (17). Three of the 18 clones were identified as retaining correct translational reading frame across the deletion junction. These clones are lacking nucleotides 294-587,359-613, and 418-609, and hence lack amino acid residues 99-196 (A98), 121-205 (A85), and 140-203 (A64), respectively. Each mutant membrane domain was fused to 8-galactosidase and cloned into pSV2 as described above.
DNA Transfection into CHO-Kl Cells and Isolation of Clones Ex- pressing p-Galactosidase Activity-Approximately 2 pg of pSV2-neo DNA (18) and 20-200 pg of pSV2-HMGal, pSV2-AHMGal, or pSV2- Gal DNA were introduced by calcium phosphate coprecipitation (19) into subconfluent monolayers of CHO-K1 cells growing in medium containing 5% fetal calf serum. Transfectants were selected with 750 pg/ml of active geneticin. Colonies resistant to geneticin were cloned and maintained in 500 pg/ml of geneticin.
Clones resistant to geneticin were grown in 24-well dishes and tested for @-galactosidase activity in situ. Cells were washed once with phosphate-buffered saline then made permeable with 200 pl of 50 pg/ml digitonin for 10 min at room temperature (20), and @- galactosidase activity was measured by 0-nitrophenyl galactoside hydrolysis (21). 800 pl of buffer Z (0.1 M sodium phosphate buffer, pH 7.0, 10 mM potassium chloride, 1 mM magnesium sulfate, and 50 mM 8-mercaptoethanol) containing 1 mg/ml 0-nitrophenyl galacto- side was added directly to the permeable cells. After the development of yellow color, the reaction was stopped by the addition of the 1 ml of reaction volume to 0.5 ml of 1 M sodium carbonate. Samples were quantitated by measuring optical density a t 420 nm. The assay was shown to be linear with respect to protein (from 5-80 pg) and time (as long as 8 h). The adherent cells were then washed with phosphate- buffered saline to remove traces of the reaction mixture and soluhi- lized with 0.5 ml of 1 M sodium hydroxide. Protein concentrations were then determined by the method of Lowry et al. (22).
Determination of the Presence of Transfected Genes-Cells grown to confluence in medium containing 5% fetal calf serum in 150-cm2 dishes were washed once with phosphate-buffered saline, then lysed with 3 ml of 200 mM Tris-HC1, pH 8.3, 10 mM sodium chloride, 100 mM EDTA, 1% sodium dodecyl sulfate (SDS), and 50 pg/ml protein- ase K. The lysate was incubated a t 37 “C overnight. DNA was then isolated through a series of extractions as previously described (15).
Identification of the presence of transfected DNA sequences was determined essentially by the method of Southern (23). DNA samples were digested with either EcoRI (HMGal and AHMGal clones) or BamHI (Gal clones), resolved by electrophoresis on a 1% agarose gel, blotted onto nitrocellulose, and probed with a 32P-labeled lac 2 DNA fragment.
Subcellular Fractionation of Cells Expressing Fusion Proteim- Cells growing in 150-cm2 dishes were incubated for 4 h in 3 ml of media containing 5% dialyzed LPS lacking methionine and contain- ing 1200 pCi of [35S]methionine. Cells were harvested and extracts fractionated into soluble and microsomal components essentially as described (24). The level of immunoprecipitable fusion protein was determined using an anti-@-galactosidase monoclonal antibody. Im- munoprecipitations were performed and analyzed by SDS-polyacryl- amide gel electrophoresis (SDS-PAGE) as described (25, 26). Addi- tionally, @-galactosidase activity of similarly prepared fractions was also determined by measuring 0-nitrophenyl galactoside hydrolysis essentially as described (21).
Resolution of individual subcellular compartments was carried out by sucrose gradient fractionation of cellular homogenates (27). HMG- CoA reductase activity was determined by a slight modification of the method of Shapiro et al. (28). HMG-CoA reductase activity in fractions was used as a marker for the endoplasmic reticulum. Cho- lesterol content of fractions was determined by the method of Solow et al. (29) and was used as a marker for the plasma membrane.
Detection of Oligosaccharide Modification of Fusion Protein.-Cells growing in 5% LPS medium in 150-cm2 dishes were labeled with [35S] methionine, and extracts were prepared as described (25). Immuno- precipitated samples were tested for endoglycosidase H sensitivity as described (2). In addition, cells were labeled with 5 mCi of 13H] mannose in 3 ml of medium containing 5% LPS and 0.1% glucose (10% of the glucose in the normal growth medium) and similarly analyzed by immunoprecipitation followed by Endo H treatment and
Effect of LDL on p-Galactosidase Activity in Transfected Cells- Cells growing in media containing 5% LPS were seeded at subcon- fluency in 24-well dishes. Fresh media was added to all wells at the start of the experiment. LDL (125 pg/ml) was then added to wells in quadruplicate in a staggered fashion, such that cells exposed to LDL for 0, 6, 12, 24, 36, or 48 h were harvested simultaneously for assay. @-Galactosidase specific activity was determined in situ as described above.
SDS-PAGE.
RESULTS
Construction and Expression of HMGal and AHMGal Fusion Genes-The procedures used to construct fusion genes be- tween the membrane domain of HMG-CoA reductase and @- galactosidase and the construction of mutant proteins are shown in Figs. 1 and 2 and are described under “Experimental Procedures.” Plasmids containing fusion genes were cotrans- fected into CHO-K1 cells along with pSV2-neo, and clones
EcoRl .%“A
LCOKl
Sal I
FIG. 1. Construction of a fusion gene between HMG-CoA reductase and &galactosidase. The cloning procedure is described under “Experimental Procedures.” Thin line, pBR322; solid segment, HMG-CoA reductase; open segment, lac Z; hatched segment, SV40; I ,
HMGR, HMG-CoA reductase; Amp‘, p-lactamase gene. lac repressor gene; Z, @-galactosidase gene; Y, lactose permease gene;

6838 Regulation of Degradation HMG-CoA Reductase Apo I
I 2 3 4 ’ 5 6 NH2
7 / F - C O O H
oa 50 1 0 0 150 200 250 300 350 400 889
!-A644 +A85 1
-A98 4
FIG. 2. Generation of deletions in the membrane domain of HMG-CoA reductase. The procedure is described under “Experi- mental Procedures.” The restriction enzyme ApaI cuts at the site indicated and the deletions designated 64,85, and 98 were constructed by digestion with Ba131. The numbered regions indicate the seven putative membrane domains.
TABLE I ~-Galactosidase-specific activities for geneticin-resistant cell lines
cotransfected with HMGal or AHMGal fusion genes
Clone @-Galactosidase activity
A.m nmlmglh pSV2-Gal pSV2-HMGal pSV2-A64HMGal pSV2-A85HMGal pSV2-A98HMGal
6-8 6-8 7-9 2-3 2-3
1 2 3 4 5 6
4822 bp - 3675bp - 2323bp - 1929bp-
FIG. 3. Presence of cotransfected DNA sequences in gene- ticin-resistant clones. High molecular weight DNA was isolated from cells and analyzed by Southern blotting as described under “Experimental Procedures.” The nitrocellulose filter was probed with a nick-translated lae 2 DNA fragment. DNA was isolated from cells transfected with lane 1, parental CHO cells; lane 2, pSV2-Gal; lane 3, pSV2-HMGal; lane 4, pSV2-A64HMGal; lane 5, pSV2-A85HMGal; lane 6, pSV2-A98HMGal.
resistant to the antibiotic geneticin were isolated. The deter- mination of @-galactosidase activity in individual clones in- dicated that the percentage of geneticin-resistant clones which exhibited significant @-galactosidase activity varied widely between transfections. Transfection with pSV2-Gal gave the highest frequency, with the majority of clones exhib- iting high P-galactosidase activity. On the other hand, it was necessary to screen 160 clones in order to obtain a suitable clone for pSV2-A85HMGal. Transfected cell lines exhibiting relatively high levels of @-galactosidase activity were activity matched as well as possible and used for the studies reported here (Table I). ..
The transfected @-galactosidase or fusion genes were de- tected by DNA blot analysis using a lac 2 probe. The presence of lac 2-specific bands in geneticin-resistant clones is dem- onstrated in Fig. 3. No signal was detected in DNA isolated from the parental CHO-K1 cell line (lane 1). Lanes 2 and 3 correspond to clones expressing high levels of @-galactosidase or HMGal protein, while lanes 4-6 correspond to clones expressing the three AHMGal constructions. The different size of lac 2-hybridizing bands reflects the size of the trans- fected construction. A wide range of @-galactosidase activity
was observed among geneticin-resistant clones. There was not a good correlation between the copy number of transfected genes and the measured activity of @-galactosidase (data not shown). This is presumably due to differences in the rate of transcription between clones depending on the site of integra- tion in the host genome and/or rearrangements of the trans- fected DNA.
Subcellular Localization of Fusion Proteins-Cell extracts were prepared as described under “Experimental Procedures’’ and fractionated into soluble and microsomal components by centrifugation. Both @-galactosidase activity and immunopre- cipitable 35S-labeled protein are found largely in the soluble fraction of a cell line transfected with pSV2-Gal (Fig. 4, lanes 1 and 2). Conversely, both @-galactosidase activity and im- munoprecipitable protein are found in the microsomal frac- tion of extracts prepared from cells expressing HMGal, A64HMGa1, A85HMGa1, and A98HMGal (lanes 3-10). It was necessary to perform this analysis on a pSV2-Gal clone ex- hibiting @-galactosidase activity approximately 30-fold greater than that seen in the pSV2-HMGal and pSV2-AHMGal clones in order to detect immunoprecipitable 35S-labeled pro- tein. This may be due to inefficient tetramerization of the HMGal and AHMGal fusion proteins in the endoplasmic reticulum relative to the soluble @-galactosidase protein, in- dicating low specific activity for the fusion proteins. The small amount of soluble HMGal and AHMGal @-galactosidase ac- tivity is likely due to contamination between fractions.
Cell homogenates were subjected to sucrose gradient frac- tionation in order to resolve subcellular compartments and further define the localization of the fusion proteins. As shown in Fig. 5 (panel A ) , the @-galactosidase activity for the HMGal fusion protein is found largely in the endoplasmic reticulum fraction, cofractionating with the endogenous HMG-CoA re- ductase activity. This demonstrates that the membrane do- main of HMG-CoA reductase is conferring “reductase” tar- geting upon the HMGal fusion protein. Each mutant fusion protein is also found to be localized in the endoplasmic reticulum fractions (Fig. 5, panels B-D), indicating that the sorting properties of these three deletion mutants appear normal. Attempts to localize the various fusion proteins by immunofluorescence microscopy were unsuccessful, probably due to the low levels of the fusion proteins.
The glycosylation of the fusion proteins was examined to further analyze the behavior of the membrane domain of HMG-CoA reductase in these constructions. As demonstrated in Fig. 6, HMGal is labeled by [3H]mannose (panel A, lane 2), indicating that it is glycosylated. Furthermore, the radioactiv- ity in this band is lost upon treatment with Endo H (panel A,
86 14 7 93 5 95 II 89 12 88
p galactosidase Activity (%)
FIG. 4. Crude fractionation of extracts from cells express- ing &galactosidase and HMGal fusion proteins. Cell extracts were prepared and separated into soluble and microsomal fractions as described under “Experimental Procedures.’’ Proteins labeled with [35S]methionine were analyzed by immunoprecipitation followed by SDS-PAGE. At the bottom of each lane is the percentage of p- galactosidase activity found in similarly prepared samples. Extracts were prepared from cells expressing: lanes 1 and 2, @-galactosidase; lanes 3 and 4, HMGal; lanes 5 and 6, A64HMGal; lanes 7 and 8, A85HMGa1, lanes 9 and 10, A98HMGal. Odd numbers are the soluble fractions and even numbers are the microsomal fractions.

Regulation of Degradation HMG-CoA Reductase 6839
0.6-
0.4 -
0.2-
- I ,X 0.4”
a 2 Ln
.-
._ > %
2 0.2-.2
’ u g
- c
o w
f! 0.6-% I r , o
2 0.4-8 o w 0.2-Q I
0.6 -
0.4 -
0.2 - 6otd X,
400
Loo
Gradient Fraction FIG. 5. Sucrose gradient fractionation of extracts prepared
from cells expressing HMGal and AHMGal proteins. Fraction- ation was performed as described under “Experimental Procedures.” 0, P-galactosidase activityA,,/ml/h; 0, HMG-CoA reductase activity nmol/ml/min; 0, cholesterol level, (mg/ml).
A, B’ Gal HMGal A64HMGal A98HMGol Endo H Endo H + - - + - + - + - + - + - + - + - +
. c
P
100-
FIG. 6. Glycosylation state of fusion proteins. Cells were la- beled with either [35S]methionine or [3H]mannose and extracts pre- pared as described under “Experimental Procedures.” Immunoprecip- itated protein was treated with either 0.3 M citrate, 0.1% SDS buffer or Endo H, then analyzed on SDS-PAGE. Panel A, 8% gel of immu- noprecipitate of [3H]mannose-labeled protein. Lane 1, treatment with Endo H; lane 2, no Endo H treatment. Panel B, 5% gel of [35S] methionine-labeled protein. Lanes 1-4, P-galactosidase lanes 5-8, HMGal; lanes 9-12, A64HMGal; lanes 13-16, A98HMGal. Odd-num- bered lanes, no Endo H treatment; even-numbered lanes, treatment with Endo H. Only the bottom portion of the gel is shown for panel B. Duplicate alternating samples +/- Endo H are presented in order to make the small difference in mobility more apparent.
lane 1 ) , demonstrating that the oligosaccharide is in the high- mannose form which is characteristic of the oligosaccharide seen on native HMG-CoA reductase (2). Additionally, [35S] methionine-labeled HMGal exhibits a small mobility shift on SDS-PAGE after treatment with Endo H (panel B, lanes 5- 8). This also indicates that the protein is glycosylated in a high-mannose form. No such mobility shift is seen when 35S-
labeled P-galactosidase is treated with Endo H (panel B, lanes 1-4). The results presented in Fig. 6 indicate that fusion proteins A64HMGal and A98HMGal are also glycosylated normally, as indicated by a small mobility shift following Endo H treatment (lanes 9-16). This suggests that these mutant HMG-CoA reductase membrane domains are func- tioning normally with respect to targeting to the endoplasmic reticulum membrane. It was not possible to perform this experiment with A85HMGal due to the low level of labeling with [35S]methionine in this clone.
Regulation of the Degradation Rate of Fusion Protein by LDL-Since the half-life of HMG-CoA reductase is respon- sive to the level of sterols (8), it was determined if the degradation rate of the fusion proteins also varied in response to the presence of LDL. The results of an experiment in which &galactosidase activity was measured in cells growing either in delipidated media or media containing 125 pg/ml LDL is presented in Fig. 7. A 75% decrease in HMGal activity is observed within 24 h of the addition of LDL (panel A) . This is very similar to the observed decrease in HMG-CoA reduc- tase activity under similar conditions (8). No decrease in p- galactosidase activity in response to LDL was seen when cells expressing soluble 6-galactosidase were tested. None of the mutant fusion proteins exhibit a decrease in P-galactosidase activity in response to the addition of LDL to delipidated media (panels B-D). This may indicate that sequences in- volved in the binding of regulatory molecules have been disrupted in these clones.
Since the SV40 early promoter in pSV2 is not affected by LDL (8), any change in 8-galactosidase activity is a reflection of a change in the half-life of the chimeric protein. In addition, the kinetics and extent of HMGal repression in response to LDL are identical to that seen with HMG-CoA reductase (8). This indicates that the decrease in HMGal activity observed after the addition of LDL is due to an increase in the rate of enzyme degradation.
DISCUSSION
The function of the membrane domain of HMG-CoA re- ductase has been examined by constructing a fusion with p- galactosidase. The membrane domain is found to confer both endoplasmic reticulum localization and sterol-regulation of degradation onto this chimeric protein. Previous work has demonstrated that a soluble form of HMG-CoA reductase is not subject to sterol-regulated degradation (11). The current data demonstrate that the membrane domain of HMG-CoA reductase is not only necessary but is sufficient for both localization to the endoplasmic reticulum and sterol-regula- tion of degradation.
Analysis of fusion proteins lacking 64,85, or 98 amino acid residues from the HMG-CoA reductase membrane domain indicates that these mutant proteins are still targeted to the endoplasmic reticulum membrane. However, the degradation rates of these proteins are no longer regulated in response to the presence of sterols.
Recent work by Jingami et al. (24) supports these findings. They analyzed a mutant version of HMG-CoA reductase which lacked the fourth and fifth putative membrane spans (3) and also found a loss of sterol-regulation of protein deg- radation.
The chimeric fusion proteins described in this work are enzymatically active in spite of the requirement of tetramer- ization for @-galactosidase function. It seems remarkable that an attachment of 40 kDa of HMG-CoA reductase membrane domain does not prevent this assembly. Silhavy et al. (30) found that a fusion between P-galactosidase and the product
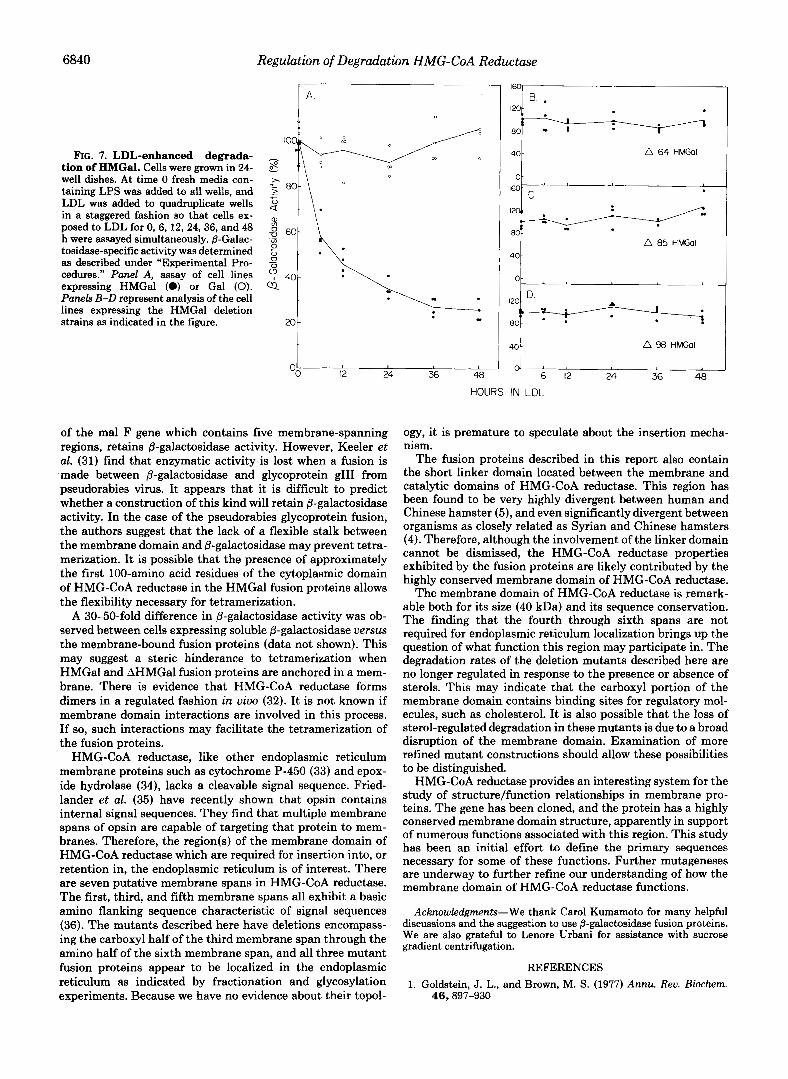
6840 Regulation of Degradation HMG-CoA Reductase
l e
FIG. 7. LDL-enhanced degrada- - - tion of HMGal. Cells were grown in 24- 8 well dishes. At time 0 fresh media con- A
taining LPS was added to all wells, and .z 80- LDL was added to quadruplicate wells 2 in a staggered fashion so that cells ex- posed to LDL for 0,6,12,24,36, and 48 ! 60- h were assayed simultaneously. @-Galac- .a tosidase-specific activity was determined 3 as described under “Experimental Pro- _o cedures.” Panel A, assay of cell lines 40- expressing HMGal (0) or Gal (0). Q Panels B-D represent analysis of the cell lines expressing the HMGal deletion strains as indicated in the figure. x)-
40 ! A 64 HMGol
.1 I60 C
I A 85 HMGal
401
4 4 A 98 HMGol
H 6 12 24 36 48
of the mal F gene which contains five membrane-spanning regions, retains @-galactosidase activity. However, Keeler et al. (31) find that enzymatic activity is lost when a fusion is -made between @galactosidase and glycoprotein gIII from pseudorabies virus. It appears that it is difficult to predict whether a construction of this kind will retain @-galactosidase activity. In the case of the pseudorabies glycoprotein fusion, the authors suggest that the lack of a flexible stalk between the membrane domain and &galactosidase may prevent tetra- merization. It is possible that the presence of approximately the first 100-amino acid residues of the cytoplasmic domain of HMG-CoA reductase in the HMGal fusion proteins allows the flexibility necessary for tetramerization.
A 30-50-fold difference in 8-galactosidase activity was ob- served between cells expressing soluble @-galactosidase uersw the membrane-bound fusion proteins (data not shown). This may suggest a steric hinderance to tetramerization when HMGal and AHMGal fusion proteins are anchored in a mem- brane. There is evidence that HMG-CoA reductase forms dimers in a regulated fashion in uiuo (32). It is not known if membrane domain interactions are involved in this process. If so, such interactions may facilitate the tetramerization of the fusion proteins.
HMG-CoA reductase, like other endoplasmic reticulum membrane proteins such as cytochrome P-450 (33) and epox- ide hydrolase (34), lacks a cleavable signal sequence. Fried- lander et al. (35) have recently shown that opsin contains internal signal sequences. They find that multiple membrane spans of opsin are capable of targeting that protein to mem- branes. Therefore, the region(s) of the membrane domain of HMG-CoA reductase which are required for insertion into, or retention in, the endoplasmic reticulum is of interest. There are seven putative membrane spans in HMG-CoA reductase. The first, third, and fifth membrane spans all exhibit a basic amino flanking sequence characteristic of signal sequences (36). The mutants described here have deletions encompass- ing the carboxyl half of the third membrane span through the amino half of the sixth membrane span, and all three mutant fusion proteins appear to be localized in the endoplasmic reticulum as indicated by fractionation and glycosylation experiments. Because we have no evidence about their topol-
HOURS IN LDL
ogy, it is premature to speculate about the insertion mecha- nism.
The fusion proteins described in this report also contain the short linker domain located between the membrane and catalytic domains of HMG-CoA reductase. This region has been found to be very highly divergent between human and Chinese hamster (5), and even significantly divergent between organisms as closely related as Syrian and Chinese hamsters (4). Therefore, although the involvement of the linker domain cannot be dismissed, the HMG-CoA reductase properties exhibited by the fusion proteins are likely contributed by the highly conserved membrane domain of HMG-CoA reductase.
The membrane domain of HMG-CoA reductase is remark- able both for its size (40 kDa) and its sequence conservation. The finding that the fourth through sixth spans are not required for endoplasmic reticulum localization brings up the question of what function this region may participate in. The degradation rates of the deletion mutants described here are no longer regulated in response to the presence or absence of sterols. This may indicate that the carboxyl portion of the membrane domain contains binding sites for regulatory mol- ecules, such as cholesterol. It is also possible that the loss of sterol-regulated degradation in these mutants is due to a broad disruption of the membrane domain. Examination of more refined mutant constructions should allow these possibilities to be distinguished.
HMG-CoA reductase provides an interesting system for the study of structure/function relationships in membrane pro- teins. The gene has been cloned, and the protein has a highly conserved membrane domain structure, apparently in support of numerous functions associated with this region. This study has been an initial effort to define the primary sequences necessary for some of these functions. Further mutageneses are underway to further refine our understanding of how the membrane domain of HMG-CoA reductase functions.
Acknowledgments-We thank Carol Kumamoto for many helpful discussions and the suggestion to use P-galactosidase fusion proteins. We are also grateful to Lenore Urbani for assistance with sucrose gradient centrifugation.
REFERENCES 1. Goldstein, J. L., and Brown, M. S. (1977) Annu. Reu. Biochern.
46,897-930

Regulation of Degradation HMG-CoA Reductase 684 1
2. Brown, D. A., and Simoni, R. D. (1984) Proc. Natl. Acad. Sci. U. 19. Graham, F. C., and Van der Eb, A. J. (1973) Virology 52, 456-
3. Liscum, L., Finer-Moore, J., Stroud, R. M., Luskey, K. L., Brown, 20. Leonard, D. A., and Chen, H. W. (1987) J. Biol. Chem. 262,
4. Skalnik, D. G., and Simoni, R. D. (1985) DNA 4,439-444 21. Pardee, A. B., Jacob, F., and Monod, J. (1959) J. Mol. Biol. 1, 5. Luskey, K. L., and Stevens, B. (1985) J. Biol. Chem. 260,10271- 165-178
22. Lowry, 0. H., Rosebrough, N. J., Farr, A. L., and Randall, R. J. 6. Faust, J. R., Luskey, K. L., Chin, D. J., Goldstein, J. L., and (1951) J. Biol. Chem. 193,265-275
S. A. 81,1674-1678 467
M. S., and Goldstein, J. L. (1985) J. Biol. Chem. 260,522-530 7914-7919
10277
Brown, M. S. (1982) Proc. Natl. Acad. Sci. U. S. A. 79, 5205- 23. Southern, E. M. (1975) J. Mol. Biol. 98, 503-517 5209 24. Jingami, H., Brown, M. S., Goldstein, J. L., Anderson, R. G. W.,
7. Edwards, P. A., Lan, S.-F., Tanaka, R. D., and Fogelman, A. M. and Luskey, K. L. (1987) J. Cell. Biol. 104, 1693-1704 (1983) J. Biol. Chem. 258, 7272-7275 25. Hardeman, E. C., Jenke, H. S., and Simoni, R. D. (1983) Proc.
8. Chin, D. J., Gil, G., Faust, J . R., Goldstein, J. L., and Brown, M. Natl. Acad. Sei. U. S. A. 80, 1516-1520 S. (1985) Mol. Cell. Biol. 5,634-641 26. Laemmli, U. K. (1970) Nature 227, 680-685
9. Hardeman, E. C., Endo, A., and Simoni, R. D. (1984) Arch. 27. Kaplan, M. R., and Simoni, R. D. (1985) J. Cell. Biol. 101,446- Biochem. Biophys. 232,549-561 453
10. Edwards, P. A., Lemongello, D., Kane, J., Schechter, I., and 28. Shapiro, D. J., Nordstrom, J. L., Mitschelen, J. J., Rodwell, V. Fogelman, A. M. (1980) J. Bwl. Chem. 255,3715-3725 W., and Schimke, R. T. (1974) Biochim. Biophys. Acta 370,
11. Gil, G., Faust, J. R., Chin, D. J., Goldstein, J. L., and Brown, M. 369-377 S. (1985) Cell 41,249-258 29. Solow, E. B., and Freeman, L. W. (1970) Clin. Chem. 16, 472-
12. Goldstein, J. L., Basu, S. K., and Brown, M. S. (1983) Methods 476 Enzymol. 98, 241-260 30. Silhavy, T. J., Casadaban, J. J., Shuman, H. A., and Beckwith,
13. Rothblat, G. H., Arbogast, L. V., Ouellette, L., and Howard, B. J. R. (1976) Proc. Natl. Acad. Sci. U. S. A. 73, 3423-3427 V. (1976) In Vitro, (Rockville) 12,554-557 31. Keeler, C. L., Whealy, M. E., and Enquist, L. W. (1986) Gene
14. Mulligan, R. C., and Berg, P. (1980) Science 209, 1422-1427 (Amst.) 50, 215-224 15. Skalnik, D. G., Brown, D. A., Brown, P. C., Friedman, R. C., 32. Ness, G. C . , McCreery, M. J., Sample, C. E., Smith, M., and
Biol. Chem. 260, 1991-1994 Hardeman, E. C., Schimke, R. T., and Simoni, R. D. (1985) J. Pendleton, L. C. (1985) J. Biol. Chem. 260, 16395-16399
33. Bar-Nun, S., Kreibich, G., Adesnik, M., Alterman, L., Negishi, 16. Birnboim, H. C., and Doly, J. (1979) Nucleic Acids Res. 7, 1513- M., and Sabatini, D. D. (1980) Proc. Natl. Acad. Sci. U. S. A.
1523 77,965 17. Sanger, J., Nicklen, S., and Coulson, A. R. (1977) Proc. Natl. 34. DuBois, G. C., Appella, E., Armstrong, R., Levin, W., Lu, A. Y.
Acad. Sci. U. S. A. 74,5463-5467 H., and Jerina, D. M. (1979) J. Bwl. Chem. 264,6240 18. Southern, P. J., and Berg, P. (1982) J. Mol. Appl. Gen. 1, 327- 35. Friedlander, M., and Blobel, G. (1985) Nature 318,338-342
341 36. Von Heijne, G. (1984) EMBO J. 3,2315-2318