the j b c vol. 272, no. 32, issue of august 8, pp. 19993 ... · the cell to exit from mitosis and...
TRANSCRIPT

A Novel Protein, Psp1, Essential for Cell Cycle Progression ofSchizosaccharomyces pombe Is Phosphorylated by Cdc2-Cdc13upon Entry into G0-like Stationary Phase of Cell Growth*
(Received for publication, April 4, 1997, and in revised form, May 23, 1997)
Young-Joo Jang, Misun Won, Kyung-Sook Chung, Dong-Uk Kim, Kwang-Lae Hoe,Chankyu Park‡, and Hyang-Sook Yoo§
From the Cell Cycle & Signal Research Unit, Korea Research Institute of Bioscience & Biotechnology, KIST, P.O.Box 115, Yusong, Taejon, 305-600 and the ‡Department of Biological Sciences, KAIST, Yusong, Taejon, 305-608, Korea
A novel gene, psp11, which functionally complementsa temperature-sensitive mutant defective in cell cycleprogression both in G1/S and G2/M has been isolatedfrom the genomic and cDNA libraries of Schizosaccha-romyces pombe. Disruption of this gene is lethal for cellgrowth at 30 °C indicating that it is an essential gene forvegetative cell growth. Western analysis of the proteinby polyclonal antibody made from glutathione S-trans-ferase-Psp1 fusion protein indicated that the Psp1 pro-tein exists in two different molecular weight forms de-pending on the growth state of the cell. In vitroexperiments with a phosphatase showed that this differ-ence is due to phosphorylation. The dephosphorylatedform of the protein is dominant in actively growing cellswhereas the phosphorylated form becomes the majorspecies when cells enter the stationary phase. The Cdc2-Cdc13 complex is shown to phosphorylate the GST-Psp1fusion protein in vitro, and site-directed mutagenesisand phosphoamino acid analysis indicated that the ser-ine residue at position 333 in the carboxyl-terminal re-gion is required for phosphorylation. In situ fluoresceinisothiocyanate-conjugated antibody staining showedthat this protein tends to be localized to both ends of thecell upon entry into the stationary phase of cell growth.However, overexpression of the novel protein Psp1 inactively growing cells inhibits cell growth causing accu-mulation of DNA (4n or 8n). Thus we speculate that Psp1can function at both G1/S and G2/M phases complement-ing the defect of the new mutant we have isolated. It islikely that Psp1 is required both for proper DNA repli-cation and for the process of mitosis.
DNA replication (S) and mitosis (M) are the two major eventsin the eukaryotic cell division cycle and are proceeded by thetwo gap periods, G1 and G2, respectively. Cell cycle transitionfrom G1 to S phase (G1/S) and from G2 to mitosis (G2/M) occursin a strict sequence. Blocking S phase prevents onset of thesubsequent mitosis, which would be lethal if chromosome rep-lication had not been completed, and blocking mitosis preventsinitiation of the subsequent S phase which would otherwiselead to increase in ploidy. Dependences of S phase and mitosis
are examples of checkpoints that ensure orderly progressionthrough the cell cycle. Many genes are thought to be involved inthis process, and tight regulation of function or expression ofthe relevant genes is required for proper cell cycle progression.The checkpoint control at the two transition points, G1/S andG2/M, ensures either progression or blocking of cell cycle ac-cording to the state of the cell. Several regulators such ascyclin-dependent kinases (CDKs)1 function at these points (1,2). CDK activity is subject to regulation by association withpositive regulatory subunits known as cyclins, negative regu-lators known as CDK inhibitors (CKIs), and by phosphoryla-tion (reviewed by Pines (3), Morgan (4), Elledge and Harper (5),and Harper and Elledge (6)). The balanced function of thesefactors controls CDK activity and serves to integrate signalsintended to coordinate cell cycle transitions. The levels of theseproteins are tightly regulated both transcriptionally and post-translationally as the cell cycle progresses. In yeast a singleCDK, Cdc2 in fission yeast and Cdc28 in budding yeast, isrequired at the first checkpoint G1/S (7–12), while in multicel-lular eukaryotes, several different CDKs, including CDK2,CDK4, and CDK6, are involved at different stages of G1 and Sphase (13–17). Different G1 cyclins such as CLN1, CLN2, andCLN3 of budding yeast or cyclin D and cyclin E of mammaliancells and S phase cyclins such as CLB5, CLB6, or cyclin E andcyclin A are associated with these CDKs to ensure kinasefunction of the CDKs (18–22) at G1 to S phase progression. Thephosphorylation state of the CDK itself and proper timing ofdestruction of the cyclins are important for regulation of CDKactivities (4). Thus checkpoint control at G1/S by these multipleelements is important for the cell to commence DNA replica-tion, to initiate the cell cycle, and to integrate the positive andnegative signals of the cell cycle. At the second checkpoint,G2/M, p34cdc2, the prototypic member of cyclin-dependent ki-nase, is known to be the major regulator both in yeast andmammalian cells. Association with G2 cyclins such as cyclin Bis required to function as an active protein kinase and to induceactive mitosis (23, 24). The phosphorylation state of Cdc2 di-rectly affects its kinase function and cell cycle progression tomitosis (4, 25, 26). Cdc25 dephosphorylates Cdc2 at serine/threonine residues, and this activates the kinase function ofthe Cdc2-Cdc13 (cyclin B) complex (27–29). Meanwhile, phos-phorylation of Cdc2 by Wee1 kinase results in inactivation ofCdc2 kinase function at G2/M (30–33), and destruction of G2
cyclin by the ubiquitin-associated cyclosome complex signals* This work was supported by Grants N80880, N81310, HS030M, and
HS1030 from the Ministry of Science and Technology of Korea. Part ofthe grants was supported by Dae-oung Pharmaceutical Co. The costs ofpublication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The nucleotide sequence(s) reported in this paper has been submittedto the GenBankTM/EBI Data Bank with accession number(s) L36906.
§ To whom correspondence should be addressed. Tel.: 82-42-860-4170; Fax: 82-42-860-4597; E-mail: [email protected].
1 The abbreviations used are: CDK, cyclin-dependent kinase; CKI,CDK inhibitor; EMM, Edinburgh minimal medium; MMP, phosphate-free minimal medium; EMS, ethyl methanesulfonate; kb, kilobase(s);FITC, fluorescein isothiocyanate; DAPI, 4,6-diamidino-2-phenylindole;PCR, polymerase chain reaction; PAGE, polyacrylamide gelelectrophoresis.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 272, No. 32, Issue of August 8, pp. 19993–20002, 1997© 1997 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org 19993
by guest on June 1, 2018http://w
ww
.jbc.org/D
ownloaded from

the cell to exit from mitosis and to prepare for the next roundof G1 to S phase progression (34). The CKIs such as p21, p27, orp15, which have negative function to inhibit CDK activity (5,35), counteract the cyclins and are required to prevent cell cycleprogression when cells are damaged. Mutations in CKI genescause unregulated cell cycle progression, and this results inabnormal cell growth. In particular, deregulation of START atG1/S may allow cell growth and division to become insensitiveto external cues (36–40). This can be a consequence of eitherthe aberrant expression of positive regulators, such as thecyclins, or the loss of negative regulators, such as the CKIs.Thus proper regulation of cyclin-CDK-CKI complex formationis critical for normal cell cycle progression.
Even though the global cell cycle controls operating at the S(22, 41–45) and M phase transitions (46–48) have been exten-sively analyzed, unknown relevant factors still remain to beidentified. Fission yeast Schizosaccharomyces pombe has beena useful model organism to study cell cycle control in eukary-otic cells. The relative ease of genetic manipulation of yeast hasallowed detailed analysis of gene function in vivo, which hasproduced a paradigm for cell cycle control applicable to higherorganisms. In an attempt to isolate factors involved in controlof cell cycle progression, we searched for new cell cycle-relatedgenes in S. pombe. In this paper we describe isolation of a novelgene, psp11 (a gene encoding phosphoprotein at stationaryphase of S. pombe cell), essential for progression of cell cycle.We used a strategy to isolate a new conditional mutant defec-tive in cell cycle progression by chemical mutagenesis and tofind a clone functionally complementing this mutant pheno-type. Genetic and biochemical studies have permitted us toidentify the function of the newly isolated psp11 in S. pombecells.
EXPERIMENTAL PROCEDURES
Strains and Media—The newly isolated temperature-sensitive (ts2)mutant was derived from a haploid strain, ED665 (h2, ade6-M210,ura4-D18, leu1-32). For gene disruption analysis and back-crossingtest, a S. pombe diploid strain, SP286 (h1/h1, ade6-M210/ade6-M216,ura4-D18/ura4-D18, leu1-32/leu1-32) and a haploid strain, ED668 (h1,ade6-M216, ura4-D18, leu1-32) were used. For genetic complementa-tion tests and comparative flow cytometric analysis, the known cellcycle mutants, cdc22, cdc102, cdc202, and cdc222 and wee12 were used.YE-glucose (YEPD) agar medium and Edinburgh minimal medium(EMM) were used as described by Gutz et al. (49). When necessary, 75mg/ml each of adenine and uracil or 250 mg/ml leucine was added as asupplement. Phloxin B (Sigma) was added to the YEPD and minimalagar medium at a concentration of 20 mg/liter after autoclave. 50–500mM thiamine was added to EMM to repress the nmt1 promoter function,and it was excluded from EMM when induction of the nmt1 promoterfunction is necessary. For in vivo cell labeling with ortho[32P]phosphate, phosphate-free minimal medium (MMP) in the pres-ence of 1 mM phosphate (added from a 0.5 M NaH2PO4 stock solution) or50 mM phosphate was used (50).
Isolation of Temperature-sensitive Mutants of S. pombe Defective inCell Cycle Progression—To isolate ts2 mutants of S. pombe, ED665 cellsgrown in YEPD medium were mutagenized with ethylmethanesulfon-ate (EMS) to 40% survival as described previously (51, 52). The mu-tagenized cells were spread on YEPD plates and incubated at 23 °C for4–6 days. The cells grown on the plates were replica-plated onto YEPDplates containing phloxin B, and the plates were incubated at 23, 36,and 39 °C for 3–4 days. The cells that grew at 23 °C but did not grow at36 or 39 °C were selected. Out of 2.5 3 105 cells mutagenized, 180candidate colonies were first selected and examined for their morpho-logical changes at the restrictive temperature (36 °C) under a lightmicroscope. The 51 colonies that showed defects in cell division werefurther characterized by back-crossing to the parent strain ED665 orED668 and by genetic complementation test with the known cell divi-sion cycle mutants. Their DNA contents at 36 °C were determined byflow cytometrical analysis as described in Jang et al. (52). One mutant,cyj92, that showed an elongated morphology at 36 °C and a defect in cellcycle progression both at G1/S and after G2, was selected and used toclone a gene recovering the mutated phenotype.
Flow Cytometric Analysis—To measure DNA content in each cellcycle point, the cells were prepared by a procedure adapted from Lew etal. (53) and Costello et al. (54). The mutant cells were first grown inYEPD liquid medium at 23 °C to midlog phase (A580 5 0.5) and trans-ferred either directly to fresh YEPD medium or to nitrogen-deficientEMM (0.003% NH4Cl) which leads to cell cycle arrest at G1 in S. pombe.The cells transferred to EMM were incubated at 23 °C for 24–36 h toinduce cell cycle arrest at G1, and were then collected, washed, andresuspended in YEPD medium. The cells transferred to YEPD mediumwere incubated at 36 °C for 4–8 h to induce mutant phenotype. Aliquotsof these cells (1 3 107 cells) were collected, fixed with 70% ethanol forat least 12 h at 4 °C, and treated with RNase A (Sigma, 0.1 mg/ml) for2 h at 37 °C. After staining the cells with propidium iodide solution(0.005% propidium iodide, 0.1% sodium citrate, and 0.03% TritonX-100), the DNAs in the cells were analyzed with Becton DickinsonFACScan System (FACScanTM). Analysis was based on the accumula-tion of 8,000–10,000 cells. As a reference, flow cytometric analysis ofthe mutant cdc22 or cdc102 was carried out simultaneously. To meas-ure DNA content in cells overexpressing psp11, the cells containingpnmt1-psp1 plasmid were first grown in EMM containing 100 mM thi-amine and then transferred to EMM devoid of thiamine and grown for12 h. Aliquots of the cells were reinoculated into fresh EMM lackingthiamine to a cell density A580 5 0.1 and incubated at 30 °C. Cellsamples were collected thereafter every 3 h and subjected to flowcytometric analysis.
Synchronous Cell Cultures—To examine the expression pattern ofthe psp11 gene during cell cycle progression, synchronized cell cultureswere used. The cells arrested at S phase by hydroxyurea or M phase bythiabendazole were used (55–57). ED665 cells were first grown in EMMto a cell density of A580 5 0.5 (1 3 107 cells/ml) and then in the presenceof 12 mM hydroxyurea or 100 mM thiabendazole at 30 °C for an addi-tional 4–5 h to arrest them at S or M phase, respectively. The cells werethen collected by filtration, washed, transferred to fresh EMM, andincubated at 30 °C to release them from S or M phase arrest. Aliquotsof the cells were collected every 20 min thereafter and counted. TotalRNA was prepared from these cell samples.
Cloning of a Gene Complementing the Elongated Mutant Phenotypeof cyj92—To identify a gene responsible for blockage of cell cycle pro-gression when mutated as shown in the mutant cyj92, a functionalcomplementation test of the mutant with a S. pombe genomic librarywas carried out. The mutant cyj92, showing an elongated morphologyand defect in cell cycle progression at 36 °C, was transformed with a S.pombe genomic library prepared by ligating genomic DNA partiallydigested with Sau3AI into the BclI site of pWH5 vector (58, 59).2 Thetransformants showing Leu1 at 23 °C were first selected, replica-platedonto the plates containing phloxin B, and incubated at 36 °C. The pinkcolonies that grew up and showed wild type morphology at 36 °C wereselected. Individual plasmid isolated from these transformants wasretransformed to test its ability to suppress the elongated ts2 pheno-type of cyj92 (50). Two such plasmids, containing 8.5-kb and 6.9-kbinsert DNA, were isolated and characterized by restriction enzymemapping and Southern hybridization. The 4.2-kb Sau3AI-PvuII frag-ment present in both of these plasmids and possessing the suppressivefunction was subcloned (Fig. 2a, pYJ4) and sequenced by the dideoxymethod of Sanger et al. (60) with USB Sequenase (U. S. BiochemicalCorp.). cDNA clones were also isolated by screening a S. pombe cDNAlibrary constructed in the l-ZAP vector (61) with the labeled 1.5-kbPvuII-BamHI fragment encoding most of the coding sequences. TheDNA containing open reading frame sequences was designated aspsp11 (phosphoprotein of stationary phase of S. pombe). The disruptionconstruct of the psp1 gene, with the ura41 as the selectable marker, wasmade as follows (Fig. 2a, pYJ5). The PvuII-EcoRV fragment containingmost of the Psp1 coding sequences, including 59-nontranslating regionDNA, was deleted and replaced with the 1.8-kb ura41 fragment. Theresulting 3.5-kb BamHI-ApaI fragment was used to transform a diploidstrain SP286. Transplacement of the genomic psp1 gene with the ura41
disrupted copy was confirmed by Southern analysis. The disrupteddiploid strain was then sporulated according to the methods of Gutz etal. (49), and viability of the resulting meiotic products was analyzed(Fig. 2c).
Primer Extension Analysis—To determine the transcription start siteof psp11, the primer extension method was used (62). The antisenseprimer sequence that corresponds to the sequence from 1150 to 1169(59-GAGCAATTTCATTATCATG-39) of the amino-terminal coding re-
2 Jang, Y. J., Chung, K. S., Park, C. K., and Yoo, H. S. (1997) Biochim.Biophys. Acta, in press.
A Novel Protein Phosphorylated at Stationary Phase of S. pombe19994
by guest on June 1, 2018http://w
ww
.jbc.org/D
ownloaded from
gion was synthesized and labeled with T4 polynucleotide kinase (NewEngland Biolabs) and [g-32P]ATP. The labeled primer was then hybrid-ized with 5–10 mg of poly(A) RNAs isolated from the ED665 straingrown in EMM. Moloney murine leukemia virus reverse transcriptase(New England Biolabs) was added to the hybridization mixture andincubated at 37 °C for 30 min. The position of transcription start sitewas determined by comparing the size of the extended fragment withthe sequenced DNA using the same primer.
Preparation of Membrane and Cytosolic Fractions—To determine thelocation of the Psp1 protein in S. pombe cells, membrane and cytosolicfractions of the cells were prepared (63). Cells were broken by the glassbead method (64) in bead buffer (20 mM Tris-HCl, pH 7.5, 10 mM EGTA,2 mM EDTA, 0.25 M sucrose, 20 mg of leupeptin/ml, and 10 mM phenyl-methylsulfonyl fluoride) and centrifuged at 100,000 3 g for 1 h at 4 °C.The supernatant was used as a cytosolic fraction. The pellet fractionwas treated with 1% Triton X-100 for 30 min at 4 °C and used as amembrane fraction.
Production of GST-Psp1 Fusion Proteins—To prepare the proteinproduct of the psp1 gene, a coding region DNA of psp11 was fused to theisopropyl-1-thio-b-D-galactopyranoside-inducible glutathione S-trans-ferase (GST) of fusion vector pGEX-3X. The GST-psp1 fusion constructsthat encode different portions of Psp1 were prepared as follows (Fig. 4c,A). The DNA fragment containing the whole coding region (w), amino-terminal sequence 1–307 (Nw), or carboxyl-terminal sequence 323–408(Cw) was cloned into the BamHI site of plasmid pGEX-3X. The DNAfragments encoding carboxyl-terminal sequences 323–408 (Cm1), 330–408 (Cm2), and 338–408 (Cm3), bearing substitutions of serine resi-dues at 327, 333, and 341 with alanine, respectively, were fused into thesame vector. Escherichia coli strain DH5a transformed with thesefusion plasmids was grown in LB medium containing ampicillin, andproduction of GST-Psp1 fusion proteins was induced by growing cells inthe presence of 0.1 mM isopropyl-1-thio-b-D-galactopyranoside at 30 °C.Each GST-Psp1 fusion protein was purified on glutathione-agarosebeads as described previously from E. coli crude cell extracts (65). Forin vitro phosphorylation of Psp1 protein by Cdc2 kinase, GST andGST-Psp1 fusion proteins purified on glutathione-agarose beads wereeluted with 10 mM reduced glutathione in 50 mM Tris-HCl, pH 8.0.
Immunochemical Analysis—To generate polyclonal antibody againstPsp1 protein, affinity-purified GST-Psp1(w) protein was treated withfactor Xa enzyme and separated on SDS-polyacrylamide gel (65). ThePsp1 protein band on the gel was eluted, mixed with Freund’s adjuvantsolution, and used to inject a rabbit. After two more successive injec-tions at 2-week intervals, serum was collected and used as a source ofantibody for Western blot analysis and immunocytological experiments(74). For determining cellular localization of Psp1 protein, FITC-conju-gated anti-rabbit IgG (Sigma, Catalog No. F7512) was used. Cellsgrown at log or stationary phase were collected, washed, and fixed withmethanol (56, 66, 67). Antiserum prepared against Psp1 was mixed firstwith the fixed cell suspension and then treated with FITC-conjugatedanti-rabbit IgG in a concentration of 1/50 dilution. Nuclei of the cellswere counterstained with 49,69-diamidino-2-phenylindole (DAPI) (56).
Expression of psp1 Gene in S. pombe Cells—Transcriptional expres-sion of psp11 during cell cycle progression was examined by Northernanalysis of total RNA from cells grown synchronously by the arrest-release method with hydroxyurea. Total RNA was prepared from thecells collected every 20 min by the method of Carlson and Botstein (68)and analyzed by hybridizing them with the 1.5-kb PvuII-EcoRV DNAfragment radioactively labeled by the random priming method. Theexpression level of Psp protein was also examined with the same syn-chronized cells released from the hydroxyurea arrest. Cell extracts wereprepared, and the level of Psp1 protein in both soluble and particulatefractions was examined by Western analysis using the polyclonal anti-body prepared from the GST-Psp1 fusion. The effect of overexpression ofPsp1 protein in S. pombe cells was examined using the pnmt1-psp1fusion construct. The coding region DNA of psp11 was amplified from S.pombe chromosomal DNA with the 59- and 39-end primer sequences andligated into the SalI site of plasmid vector pREP1 containing thiamine-regulated nmt1 promoter (69). The resulting plasmid pnmt1-psp1 wastransformed into S. pombe cells, and expression of Psp1 protein wasinduced by growing cells in the absence of thiamine for more than 16 h.
Site-directed Mutagenesis Using PCR Methods—To introduce muta-tions substituting serine residues at the putative Cdc2 substrate sites,324LVQSPSC, 330CPPSPKN, and 338AHLSPGS to alanine, we used PCRmethods with mutated primer sequences (70). Change of the first T ofthe triplet sequence encoding serine (TCA, TCC, TCT) to G allowssubstitution of serine with alanine. Thus the following oligonucleotides,corresponding to the amino acid sequences LVQAPSC (m1), CPPAPKN(m2), and AHLAPGS (m3), were synthesized: the sense strands of m1,
59-CTTGTGCAGGCACCTTCTTGC-39, m2, 59-TGCCCTCCTGCCCCA-AAAAAT-39, and m3, 59-GCACATCTTGCCCCTGGATCC-39 and ant-isense strands of m1, 59-GCAAGAAGGTGCCTGCACAAG-39, m2, 59-A-TTTTTTGGGGCAGGAGGGCA-39, and m3, 59-GGATCCAGGGGCAA-GATGTGC-39. Also, the sense strand of the amino terminus, 59-ATGC-CTTTGTCAACTCAATCG-39, and the antisense strand of the carboxylterminus, 59-TCACCGACGTGGTGTATCTAC-39 were synthesized. Tointroduce point mutation at the sequence encoding serine 327 (Ser-327–Ala), the two primer pairs, sense strand of amino terminus and anti-sense strand of m1 and sense strand of m1 and antisense strand ofcarboxyl terminus were used for the first PCR. Then the second PCRwas carried out using the first PCR product as a template and sense
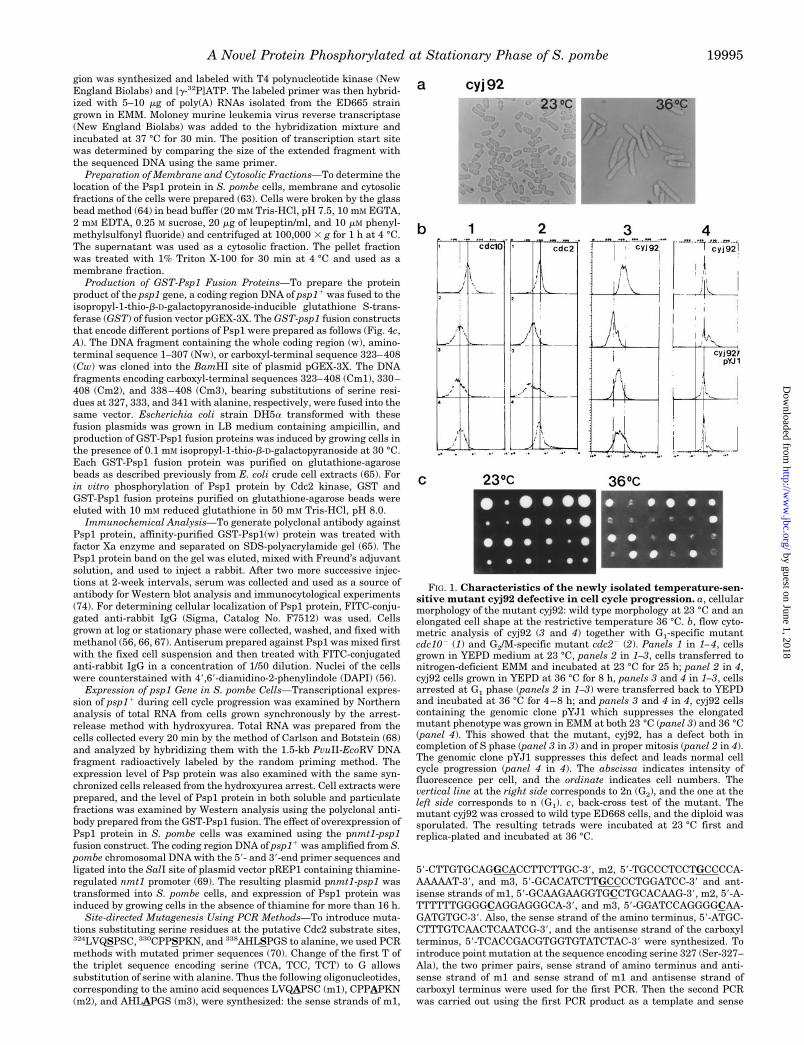
FIG. 1. Characteristics of the newly isolated temperature-sen-sitive mutant cyj92 defective in cell cycle progression. a, cellularmorphology of the mutant cyj92: wild type morphology at 23 °C and anelongated cell shape at the restrictive temperature 36 °C. b, flow cyto-metric analysis of cyj92 (3 and 4) together with G1-specific mutantcdc102 (1) and G2/M-specific mutant cdc22 (2). Panels 1 in 1–4, cellsgrown in YEPD medium at 23 °C, panels 2 in 1–3, cells transferred tonitrogen-deficient EMM and incubated at 23 °C for 25 h; panel 2 in 4,cyj92 cells grown in YEPD at 36 °C for 8 h, panels 3 and 4 in 1–3, cellsarrested at G1 phase (panels 2 in 1–3) were transferred back to YEPDand incubated at 36 °C for 4–8 h; and panels 3 and 4 in 4, cyj92 cellscontaining the genomic clone pYJ1 which suppresses the elongatedmutant phenotype was grown in EMM at both 23 °C (panel 3) and 36 °C(panel 4). This showed that the mutant, cyj92, has a defect both incompletion of S phase (panel 3 in 3) and in proper mitosis (panel 2 in 4).The genomic clone pYJ1 suppresses this defect and leads normal cellcycle progression (panel 4 in 4). The abscissa indicates intensity offluorescence per cell, and the ordinate indicates cell numbers. Thevertical line at the right side corresponds to 2n (G2), and the one at theleft side corresponds to n (G1). c, back-cross test of the mutant. Themutant cyj92 was crossed to wild type ED668 cells, and the diploid wassporulated. The resulting tetrads were incubated at 23 °C first andreplica-plated and incubated at 36 °C.
A Novel Protein Phosphorylated at Stationary Phase of S. pombe 19995
by guest on June 1, 2018http://w
ww
.jbc.org/D
ownloaded from

strand sequence of amino terminus and antisense strand sequence ofcarboxyl terminus as the primers. Amplified DNA was then cloned intovector plasmid pTZ18U. The change in the sequence at residue 327 wasconfirmed by sequencing the amplified fragment (Fig. 4c, B). Mutationsat Ser-333 and Ser-341 were introduced by similar PCR methods. Theprimer pairs of the sense strand of amino terminus and antisensestrand of m2 or m3 were used for the first PCR and followed by the
second PCR with the amino- and carboxyl-terminal sequence pairs. Foroverexpression of these mutated Psp1 in ED665, amplified DNAs werecloned into plasmid vector pREP1 containing the thiamine-induciblenmt1 promoter (pnmt1-m1, pnmt1-m2, and pnmt1-m3).
Phosphatase Treatment and Phosphorylation of Psp1 Protein by theCdc2-Cdc13 Complex—To examine the phosphorylation state of Psp1protein at different stages of cell growth, cellular proteins were treated
FIG. 2. Sequence of psp11 gene com-plementing the mutant phenotype ofa novel ts2 mutant cyj92. a, map ofgenomic clones and their subclones com-plementing the mutant phenotype ofcyj92 and a disruption clone. 1 indicatessuppression of the mutant phenotype. b,nucleotide and deduced amino acid se-quence of psp11 gene. Lowercase and up-percase letters indicate genomic andcDNA sequences, respectively. The ar-rowheads indicate the start sites and theend of two cDNAs. The short intron se-quence at the 59-nontranslating region isabsent in the cDNA clone 1. The potentialCdc2 substrate sequences, 327SPSC,333SPKN, and 341SPGS were underlined.c, effect of disruption of psp1 on cell via-bility. Diploid SP286 strain containingpsp1 gene disrupted with ura41 as evi-denced by the Southern analysis of itschromosomal DNA (lane 2 in 1) producedtwo viable and two nonviable spores at30 °C on tetrad analysis (2). These se-quence data are available from Gen-BankTM under accession number L36906and were deposited in August, 1994.
A Novel Protein Phosphorylated at Stationary Phase of S. pombe19996
by guest on June 1, 2018http://w
ww
.jbc.org/D
ownloaded from

in vitro with phosphatase and analyzed. Cells were grown to log orstationary phase, collected, and broken in phosphatase buffer (50 mM
Tris-HCl, pH 7.8, 5 mM dithiothreitol, 2 mM MnCl2, 100 mg/ml bovineserum albumin) by the glass beads method (50). After removing theglass beads by centrifugation at 500 3 g, the whole cell extract wascentrifuged at 100,000 3 g for 1 h. The supernatant was separated, andthe pellet fraction was solubilized in phosphatase buffer containing 1%Triton X-100. 5–10 ml of supernatant, solubilized pellet fraction, orwhole cell extract was mixed with phosphatase buffer and incubated at30 °C for 30 min in the presence of 400 units of l protein phosphatase(l-PPase, New England Biolabs) (71, 72). The reaction mixture wasboiled in SDS-polyacrylamide gel (SDS-PAGE) loading dye for 5 min.The proteins were then separated on SDS-PAGE and electrotransferredto a nitrocellulose paper for Western analysis with the polyclonal anti-body against Psp1. To determine whether Psp1 could be phosphorylatedin vitro, 1–2 mg of purified GST-Psp1 protein was mixed with 10 unitsof mitotic Cdc2-Cdc13 complex (New England Biolabs) and incubated at30 °C for 30 min in the presence of 40 mCi of [g-32P]ATP. The resultingprotein was then resolved in SDS-PAGE and analyzed by autoradiog-raphy. As a standard substrate of Cdc2 kinase, 2 mg/ml histone H1(Boehringer Mannheim) was used (73). In vivo labeling of the proteinwas carried out with ortho[32P]phosphate according to the procedure ofMoreno et al. (50). Cells were first grown to late log phase (A580 52.5–3.0) in 5–10 ml of low phosphate medium (MMP plus 1 mM phos-phate) and then transferred to the same volume of fresh MMP contain-ing 1 mCi of ortho[32P]phosphate (Amersham, Catalog No. PBS13).After incubation at 30 °C for 5–6 h, cell extracts were prepared from theharvested cells as described above. For immunoprecipitation, 800–1,000-ml aliquots of the cell extracts were mixed with 20 ml of antiseraof Psp1 (final 1/40 dilute) and 50 ml of protein A-agarose (Sigma). Theimmunoprecipitate was analyzed on SDS-PAGE and by autoradiogra-phy (74).
Phosphoamino Acid Analysis—To determine whether the phospho-rylated amino acid is serine as expected for phosphorylation by theCdc2-Cdc13 complex, phosphoamino acid analysis of Psp1 was carriedout. The protein labeled in vivo by ortho[32P]phosphate was immuno-precipitated with Psp1 antibody and separated by SDS-PAGE. At thesame time, GST-Psp1 protein phosphorylated in vitro by the Cdc2-Cdc13 complex with [g-32P]ATP was also separated by SDS-PAGE.After being transferred to a polyvinylidene difluoride membrane (Mil-lipore) and localized by autoradiography, the phosphorylated Psp1 bandwas excised and subjected to phosphoamino acid analysis (64, 75). Theprotein on the membrane was eluted and hydrolyzed in 6 N HCl for 1 h
at 110 °C. After being dried in a Speed Vac evaporator, the sample wasdissolved in 6–10 ml of water containing 100 mg/ml each of phospho-serine, phosphothreonine, and phosphotyrosine. The sample was sub-jected to two-dimensional thin layer electrophoresis (HTLE 7000 appa-ratus, CBS Scientific). The first dimension was electrophoresed for 20min at 1.5 kV in pH 1.9 buffer (50 ml of 88% formic acid, 156 ml ofglacial acetic acid, and 1794 ml of water). The second dimension waselectrophoresed for 16 min at 1.3 kV in pH 3.5 buffer (100 ml of glacialacetic acid, 50 ml of pyridine, and 1880 ml of water). 32P-Labeledindividual phosphoamino acids were identified by aligning the phos-phoamino acid markers stained with ninhydrin to the signals obtainedby autoradiography.
RESULTS
Characteristics of a Novel Temperature-sensitive (ts2) Mu-tant Defective in Cell Cycle Progression and a Gene Comple-menting This ts2 Phenotype—Through EMS mutagenesis of S.pombe cells, several new temperature-sensitive mutants defec-tive in cell cycle progression were obtained (52). Morphologicalanalysis of one of the mutants, cyj92, showed a normal cellshape at 23 °C but showed an elongated cellular morphology at36 °C (Fig. 1a). DNA content analysis of this mutant cell indi-cated a defect in cell cycle progression (Fig. 1b). When cyj92cells were grown in rich medium at 23 °C, most of the cells wereat G2 phase as was the case for wild type S. pombe cells (Fig. 1b,4, panel 1). But when they were shifted to a restrictive temper-ature 36 °C, DNA content increased and in the majority of cellsthe DNA content was 4n (Fig. 1b, 4, panel 2) indicating block-age of cell cycle after G2 phase. When the mutant cells weregrown in nitrogen-deficient medium at 23 °C instead, cellgrowth was arrested at G1 phase as shown in wild type cells(Fig. 1b, 3, panel 2). However, if these G1-arrested cyj92 cellswere shifted to YEPD medium and incubated at 36 °C, unlikewild type cells, they could not progress completely to G2 phase,and cell cycle progression stopped at S phase (Fig. 1b, 3, panels3 and 4). A known G1/S mutant, cdc102, and G2/M mutant,cdc22, showed cell cycle arrest at G1 after nitrogen starvationat permissive temperature 23 °C also (Fig. 1b, 1 and 2, panels2) when they were tested simultaneously with cyj92. They
FIG. 2—continued
A Novel Protein Phosphorylated at Stationary Phase of S. pombe 19997
by guest on June 1, 2018http://w
ww
.jbc.org/D
ownloaded from
showed their mutant phenotypes at the restrictive temperature(36 °C); that is, blockage of cell cycle progression to G1/S phaseand after G2 phase, respectively, as expected (Fig. 1b, 1 and 2,panels 3 and 4). Back-cross of the mutant strain cyj92 to wildtype strain ED668 revealed 21:22 segregation of the mutantphenotype at 36 °C which indicates that the mutation shown incyj92 is a single gene mutation (Fig. 1c). Genetic complemen-tation tests of cyj92 with known cdc mutants of G1 and S suchas cdc102, cdc202, or cdc222 and G2/M phase mutants cdc22
and wee12 showed that the mutation in strain cyj92 was notallelic with any of them. Thus we concluded that the mutant wehad isolated is a new ts2 mutant possessing a defect in cellcycle progression after the G2 phase at restrictive conditionsand also at G1/S phase when cells pass the G2 phase normally.It is likely that once the mutant cells progressed to G2 phase,the mutation in cyj92 can cause blockage of further progressionto M or cytokinesis. If mutant cells were in G1, this progressionthrough the next S phase was also adversely affected. It isalternatively possible that the mutation carried in strain cyj92causes blockage of cell cycle progression both at G1/S and G2/M.
Functional complementation of this mutant with a S. pombegenomic library identified the two overlapping clones of 8.5 kband 6.9 kb (pYJ1 and pYJ2). Subcloning and retransformationdata confirmed that the 4.2-kb PvuII-Sau3A fragment (pYJ4)was sufficient to suppress the ts2 phenotype (Fig. 2a). cyj92mutant cells harboring plasmids pYJ1 or pYJ4 showed normalcell morphology at 36 °C, and their DNA content analysis in-dicated normal cell cycle progression at the restrictive temper-ature, 36 °C. Increased ploidy seen in cyj92 mutant cells (Fig.1b, 4, panel 2) also disappeared in the transformed cells (Fig.1b, 4, panel 4); transformants were mainly 2n like the wildtype. Sequence analysis of this genomic DNA revealed an openreading frame sequence of 408 amino acids (Fig. 2b). Mean-while, sequence analysis of several cDNA clones indicated thepresence of two different transcripts depending on whether ornot a short intron at the 59-terminus was processed. As shownin Fig. 2b, one cDNA clone, which started at 290 (clone 1) didnot contain sequences between 257 and 22 (intron). However,the other cDNA clone, which started at 288 (clone 2) containedthese sequences. Primer extension experiments using mRNAisolated from cells grown in EMM also revealed two transcriptstart sites correlating with the two cDNA clones that differedby the presence or absence of the intron sequence (data notshown). Hydropathy plot analysis of the deduced amino acidsequence of psp11 showed that it is very hydrophobic andcontains a putative transmembrane domain at its carboxylterminus (amino acids 353–369). Sequence homology compari-son of this protein showed that no known protein sequences inthe GenBankTM data base exhibited high homology with it.Thus we concluded this gene to be a newly identified gene anddesignated it psp11 (phosphoprotein of stationary phase of S.pombe). Sporulation and tetrad analysis of a diploid straincontaining one copy of psp1 transplaced with ura41 (Fig. 2a,pYJ5) showed 21:22 segregation for cell viability at 30 °C (Fig.2c). All viable spores were Ura2 indicating that psp1 is essen-tial for vegetative cell growth at 30 °C.
Expression of Psp1—Northern analysis of total RNA isolatedfrom the wild type cells grown in EMM showed a 1.3-kb tran-script, and its level did not fluctuate much throughout the cellcycle. The level of psp1 transcript in the synchronized cellsreleased from the S phase arrest was almost same as that inthe cells progressing through G2 and M phase (data not shown).The level of Psp1 protein detected by Western analysis in thesame synchronized cells was also constant throughout the cellcycle (data not shown). Thus, in actively growing cells theamount of Psp1 protein as well as its transcript is likely to be
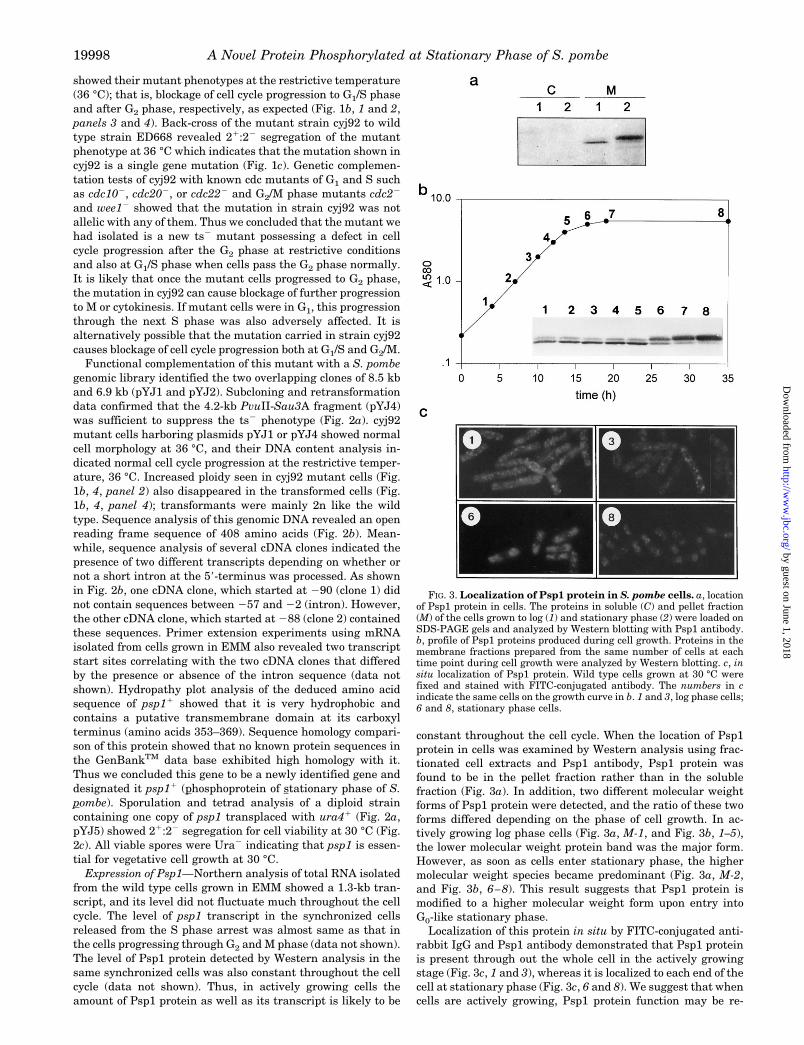
constant throughout the cell cycle. When the location of Psp1protein in cells was examined by Western analysis using frac-tionated cell extracts and Psp1 antibody, Psp1 protein wasfound to be in the pellet fraction rather than in the solublefraction (Fig. 3a). In addition, two different molecular weightforms of Psp1 protein were detected, and the ratio of these twoforms differed depending on the phase of cell growth. In ac-tively growing log phase cells (Fig. 3a, M-1, and Fig. 3b, 1–5),the lower molecular weight protein band was the major form.However, as soon as cells enter stationary phase, the highermolecular weight species became predominant (Fig. 3a, M-2,and Fig. 3b, 6–8). This result suggests that Psp1 protein ismodified to a higher molecular weight form upon entry intoG0-like stationary phase.
Localization of this protein in situ by FITC-conjugated anti-rabbit IgG and Psp1 antibody demonstrated that Psp1 proteinis present through out the whole cell in the actively growingstage (Fig. 3c, 1 and 3), whereas it is localized to each end of thecell at stationary phase (Fig. 3c, 6 and 8). We suggest that whencells are actively growing, Psp1 protein function may be re-
FIG. 3. Localization of Psp1 protein in S. pombe cells. a, locationof Psp1 protein in cells. The proteins in soluble (C) and pellet fraction(M) of the cells grown to log (1) and stationary phase (2) were loaded onSDS-PAGE gels and analyzed by Western blotting with Psp1 antibody.b, profile of Psp1 proteins produced during cell growth. Proteins in themembrane fractions prepared from the same number of cells at eachtime point during cell growth were analyzed by Western blotting. c, insitu localization of Psp1 protein. Wild type cells grown at 30 °C werefixed and stained with FITC-conjugated antibody. The numbers in cindicate the same cells on the growth curve in b. 1 and 3, log phase cells;6 and 8, stationary phase cells.
A Novel Protein Phosphorylated at Stationary Phase of S. pombe19998
by guest on June 1, 2018http://w
ww
.jbc.org/D
ownloaded from

quired, and it is located throughout the whole cell body. How-ever when growth of cells almost ceases at stationary phase,this protein may function only in specialized areas of the cel-lular compartment or as a complex.
Psp1 Protein Is Phosphorylated at Stationary Phase of theCell Cycle—Detection of the different molecular weight formsof the Psp1 protein indicated that the two proteins are eitherproduced from two different size transcripts or modified aftertranslation. Therefore we examined the size of psp1 transcriptboth by Northern analysis and determining the start site of thetranscript using the primer extension method. Total RNA pre-pared from the cells grown in EMM showed only one majorhybridizing band. However, the primer extension experimentindicated that there are at least two transcription start sitesand the length of the 59-nontranslated region is different de-pending on the processing of a short sequence from 257 to 22(Fig. 2b). Even though these two transcripts differ by a 56-basepair sequence in the 59-nontranslated region, they containedthe same ATG codon, and the proteins translated from thesetwo transcripts seem to be the same. Thus we examined thepossibility of modification after translation by phosphorylation.We tested whether the high molecular weight Psp1 protein instationary phase cells contained a phosphate residue(s). West-ern analysis of proteins in the membrane fraction treated withphosphatase in vitro showed a reduction in the amount of thehigh molecular weight Psp1 protein (Fig. 4a, lane 4) and aconcomitant increase in the low molecular weight band instead.In contrast, the lower molecular weight form of Psp1 presentmainly in the log phase cells remained unchanged after phos-phatase treatment (Fig. 4a, lane 2). This result suggests thatPsp1 protein can be phosphorylated. To test this possibility,GST-Psp1 fusion protein was prepared and tested in vitro withthe mitotic Cdc2-Cdc13 complex or with mitogen-activated pro-tein kinase. When the Cdc2-Cdc13 complex was incubated withGST-Psp1 protein in the presence of [g-32P]ATP in vitro, GST-Psp1 protein was phosphorylated as was histone H1 controlwhich is a known substrate of Cdc2-Cdc13 (Fig. 4b, lanes 2 and3). Phosphorylation did not occur in the GST portion of thefusion protein but only in Psp1. However, when mitogen-acti-vated protein kinase was mixed with GST-Psp1 protein, phos-phorylation did not occur at all (data not shown). This suggeststhat phosphorylation of the Psp1 protein by the Cdc2-Cdc13complex was specific.
Cdc2-Cdc13 Complex Phosphorylates a Serine Residue at theCarboxyl-terminal Domain of Psp1—Phosphorylation of GST-Psp1 protein by the Cdc2-Cdc13 complex in vitro indicates thepresence of a specific sequence(s) recognized by Cdc2 in Psp1.Thus the potential Cdc2 substrate sequences, (T/S)PX(K/R), (9,76) were searched. Three such sequences were found in thecarboxyl-terminal region of Psp1 (327SPSC, 333SPKN, and341SPGS) (Fig. 2b). When we used a GST-Psp1 fusion constructdevoid of these sequences (Nw), no phosphorylation of thetruncated Psp1 was observed (Fig. 4c, A, lane 2). This suggeststhat the carboxyl-terminal region of Psp1 is important for phos-phorylation by Cdc2 kinase. The GST-Psp1 containing only thecarboxyl-terminal region sequence (Cw) including those poten-tial Cdc2 kinase substrate sites was enough to serve as the
FIG. 4. Phosphorylation state of Psp1. a, phosphatase treatmentof Psp1 protein in vitro. Proteins in the membrane (M) and total fraction(T) of the cells from log and stationary phase (sta) were treated in vitrowith l-phosphatase (PPase), separated on SDS-PAGE gels, and ana-lyzed by Western blotting with Psp1 antibody. 2 and 1 indicate theuntreated and phosphatase-treated proteins, respectively. b, in vitrophosphorylation of Psp1 with the Cdc2-Cdc13 kinase complex. TheGST-Psp1 fusion protein purified from E. coli was mixed with themitotic Cdc2-Cdc13 complex in the presence of [g-32P]ATP and analyzedby SDS-PAGE and autoradiography. The bands in lanes 1–3 showed thephosphorylated proteins, and those in lanes 4–6 are the same amountof the proteins as in lanes 1–3 stained with Coomassie Blue. c, identi-fication of the serine residue phosphorylated in vitro by Cdc2-Cdc13 (A)and in vivo (B). Three serine residues (327, 333, and 341) at thepotential Cdc2 substrate sites in the carboxyl-terminal region of Psp1protein were mutated to alanine and tested for phosphorylation byCdc2-Cdc13. Lanes 1–6 in A showed the phosphorylated part of theGST-Psp1 fusion protein. The whole Psp1 protein (w), carboxyl-termi-nal protein (Cw), and carboxyl-terminal protein mutated at serine 327(Cm1) showed phosphorylation by Cdc2, but the carboxyl-terminal pro-tein mutated at serine 333 (Cm2, lane 5) did not show phosphorylationby Cdc2. Lanes 0–4 in B showed the phosphorylated Psp1 proteinproduced in vivo from the pnmt1-psp1 plasmid. The entire psp1 genemutated at the base encoding serine residue was fused to pnmt1 vectorand was expressed in wild type ED665 cells. The cells grown to late logphase in the absence of thiamine were labeled in vivo as describedunder “Experimental Procedures.” Cell extracts were prepared andimmunoprecipitated with Psp1 antibody and analyzed on SDS-PAGEgels. The open arrowhead indicates the protein produced from thechromosomal copy of the psp11 gene, and the filled arrowhead indicatesthat from pnmt1-psp1. The asterisk indicates the mutation point. d,phosphoamino acid analysis of Psp1 protein labeled in vitro by Cdc2(left) and in vivo in the presence of ortho[32P]phosphate (right). Sindicates phosphoserine detected by autoradiography; T and Y corre-
spond to the positions of phosphothreonine and phosphotyrosine, re-spectively, detected by ninhydrin staining as shown in 2. e, directinteraction of Psp1 with Cdc2 kinase shown by the yeast two-hybridmethod. psp11 fused to the Gal4 binding domain sequence (pGBT9) andcdc21 fused to the Gal4 activation domain sequence (pGAD) wereintroduced into yeast S. cerevisiae strain SFY526, and expression of thelacZ gene in yeast was examined. 1 and 2, cells containing pGBT9-psp1and pGAD-cdc2 alone, respectively; 3, cells containing both pGBT9-psp1 and pGAD-cdc2; 4, cells containing pCL1 of the whole Gal4 gene.
A Novel Protein Phosphorylated at Stationary Phase of S. pombe 19999
by guest on June 1, 2018http://w
ww
.jbc.org/D
ownloaded from
substrate molecule phosphorylated by Cdc2 (Fig. 4c, A, lane 3).When we substituted each serine residue at 327, 333, and 341
in the carboxyl-terminal region with alanine (Cm1, Cm2, andCm3) and examined the phosphorylation state of the truncatedPsp1 protein by Cdc2-Cdc13 in vitro, the serine at position 333was the only critical one for phosphorylation by Cdc2 kinase(Fig. 4c, A-lanes 4–6). Experiments with in vivo labeled proteinalso indicated that the serine residue at 333 is required forphosphorylation (Fig. 4c, B). PCR-mediated site-directed mu-tagenesis of psp1 generated a base change at the sequencesencoding serines 327, 333, or 341, respectively, and resulted intheir being changed to alanine. When this mutated full-lengthpsp1 was introduced in wild type cells as pnmt1-psp1 fusions(pnmt1-m1, pnmt1-m2, pnmt1-m3) (Fig. 4c, B, m1, m2, m3) andthe phosphorylation state of the mutated Psp1 by in vivo phos-phate labeling at stationary phase was examined, serine 333was shown to be essential for phosphorylation in stationaryphase. The cells containing the unmutated pnmt1-psp1 (w)produced more phosphorylated Psp1 than cells containing onlythe chromosomal copy of psp1 (lanes 1 and 0). This indicatedthat the Psp1 produced from the overexpression plasmidpnmt1-psp1 is phosphorylated like that in the chromosome.However, when the cells containing pnmt1-m2 (mutation atserine 333) was used, in vivo phosphate labeling of the Psp1greatly decreased (lane 3). This decrease was not observedwhen pnmt1-m1 and pnmt1-m3 (containing mutations at 327and 341, respectively) were analyzed instead (lanes 2 and 4).This confirms the importance of serine 333 for phophorylationin vivo. Phosphoamino acid analysis of the Psp1 protein phos-phorylated both in vivo and in vitro confirmed that phospho-rylation actually occurred at a serine residue (Fig. 4d).
To examine whether phosphorylation of Psp1 by Cdc2 oc-curred through direct binding of Cdc2 to Psp1, a yeast two-hybrid system was employed. When the plasmids containingGal4 binding domain-cdc2 and Gal4 activation domain-psp1were introduced into a S. cerevisiae strain SFY526 and b-ga-lactosidase production was examined, b-galactosidase produc-tion was observed. This indicates interaction between Psp1 andCdc2 (Fig. 4e). The level of b-galactosidase produced was not asgreat as that from GAL4 itself (lanes 3 and 4). However thereis strong indication that Cdc2 and Psp1 interact with eachother.
Growth-inhibitory Effect of Overexpressed Psp1 Protein onActively Growing S. pombe Cells—Since a gene disruptionstudy indicated that psp11 is essential for cell growth (Fig. 2c),we examined the multicopy gene dosage effect of this essentialprotein on cell growth. This was done by introducing multicopyplasmid pnmt1-psp1 into wild type cells and measuring theirgrowth rate in the presence and absence of thiamine. A wildtype culture grown to log phase in the presence of thiamine,which represses the nmt1 promoter function, was split in half.
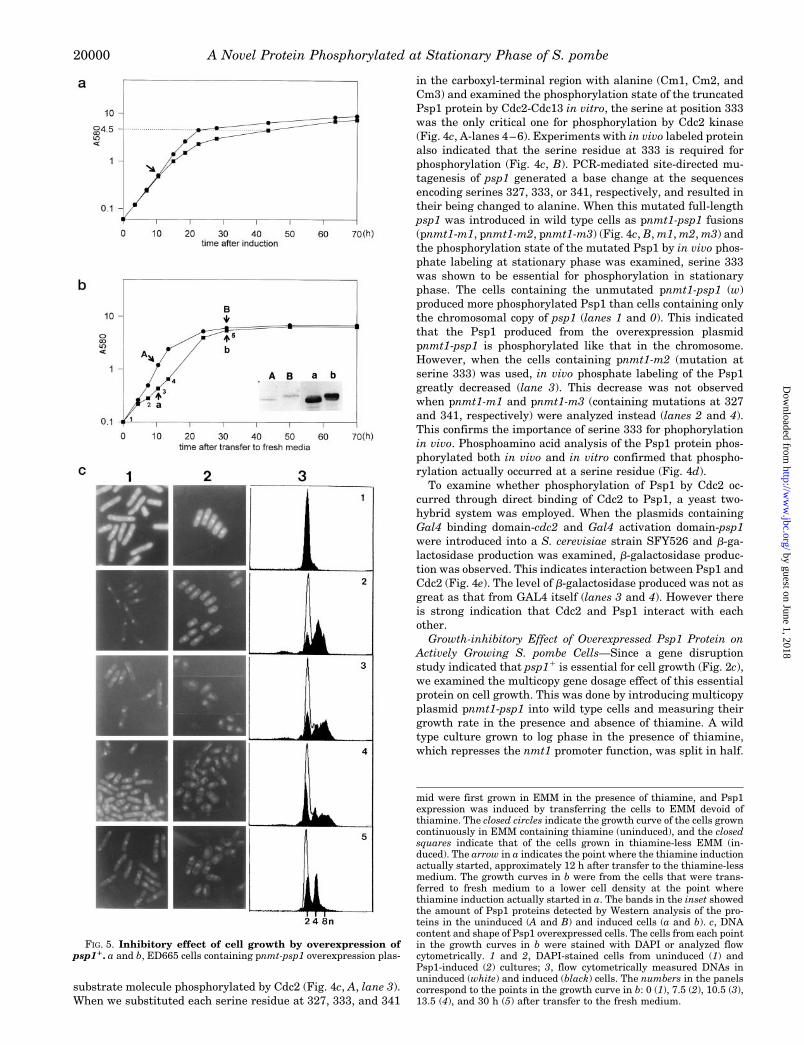
FIG. 5. Inhibitory effect of cell growth by overexpression ofpsp11. a and b, ED665 cells containing pnmt-psp1 overexpression plas-
mid were first grown in EMM in the presence of thiamine, and Psp1expression was induced by transferring the cells to EMM devoid ofthiamine. The closed circles indicate the growth curve of the cells growncontinuously in EMM containing thiamine (uninduced), and the closedsquares indicate that of the cells grown in thiamine-less EMM (in-duced). The arrow in a indicates the point where the thiamine inductionactually started, approximately 12 h after transfer to the thiamine-lessmedium. The growth curves in b were from the cells that were trans-ferred to fresh medium to a lower cell density at the point wherethiamine induction actually started in a. The bands in the inset showedthe amount of Psp1 proteins detected by Western analysis of the pro-teins in the uninduced (A and B) and induced cells (a and b). c, DNAcontent and shape of Psp1 overexpressed cells. The cells from each pointin the growth curves in b were stained with DAPI or analyzed flowcytometrically. 1 and 2, DAPI-stained cells from uninduced (1) andPsp1-induced (2) cultures; 3, flow cytometrically measured DNAs inuninduced (white) and induced (black) cells. The numbers in the panelscorrespond to the points in the growth curve in b: 0 (1), 7.5 (2), 10.5 (3),13.5 (4), and 30 h (5) after transfer to the fresh medium.
A Novel Protein Phosphorylated at Stationary Phase of S. pombe20000
by guest on June 1, 2018http://w
ww
.jbc.org/D
ownloaded from

We continued incubating one half at 30 °C in the same mediumas before. The other half was transferred to medium devoid ofthiamine which induces nmt1 promoter function. With theonset of induction psp1 at 12 h after transfer to thiamine-lessmedium, growth of the cells in thiamine-less medium becameslower than those in thiamine-containing medium (Fig. 5a).When cells grown for 12 h in the above conditions were pas-saged into fresh medium at a lower cell density, the growthdifference between the two cultures was greater in early logphase than that in late log phase (Fig. 5b). Cells overexpressingPsp1 protein in early log phase, as evidenced by Western anal-ysis of the total protein with Psp1 antibody (Fig. 5b, inset, Aand a), showed half the growth rate of the uninduced cells (Fig.5b, A and a). However differences in growth rate, caused bypsp1 overexpression, decreased as cells entered stationaryphase (Fig. 5b, B and b). The protein produced in the cells frompnmt1-psp1 in log phase was mainly the low molecular weightform whereas that in stationary phase was the phosphorylatedhigh molecular weight form (Fig. 5b, inset, a and b) as for thatproduced from the chromosomal copy of psp11 (Fig. 5b, inset, Aand B). The cells with retarded growth following overexpres-sion of Psp1 also exhibited an altered cellular morphology (Fig.5c). These cells rounded up and DAPI staining of nuclear DNArevealed that the area occupied by nuclear DNA enlarged, thatis, the compactness of the nuclear DNA was lost (Fig. 5c, 2,panels 3 and 4). Flow cytometric observation of DNA content inthe Psp1 overexpressed cells indicated doubled or tripled DNAcontents (Fig. 5c, 3, panels 2–4). The changes in DNA contentand cellular morphology were maximum at 13.5 h after trans-fer to fresh medium (4n or 8n, Fig. 5c, 3, panel 4). Growth rateat this hour was about one-fourth of that in uninduced cells(Fig. 5b, 4). However, by 24 h after transfer to fresh media, thegrowth difference became relatively small, and DNA content ofthe induced cells tended to return to amounts found in unin-duced cells (2n or 4n). After 30 h, at which time cells enteredthe stationary phase (Fig. 5b, 5), DNA content and morphologyof the cells overexpressing Psp1 were similar to those of thecells containing only one copy of the psp1 gene (Fig. 5c, 3, panel5). Thus maximal growth inhibition and changes in DNA con-tent on overproducing Psp1 was observed in the actively grow-ing cells rather than in the slow growing cells. However, thedoubling time of the cells in which Psp1 was overexpressed wasprolonged.
DISCUSSION
Temperature-sensitive mutants of S. pombe defective in nor-mal cell cycle progression provide a convenient system to iden-tify the elements involved in cell cycle regulation. Randommutagenesis of S. pombe cells enabled one to isolate such amutant. Using the same mutagenic approach as Nurse et al.(51), we isolated one mutant, cyj92, which possesses an elon-gated cellular morphology similar to G1/S mutants of Nurse etal. (51) at 36 °C. Comparative DNA content analysis of themutant in rich and nitrogen-starved media indicated that themutation in cyj92 hindered progression of S. pombe cell from G1
to S phase and perhaps also from G2 to M phase. The terminal
phenotype of the mutation is accumulation of abnormal DNAwhich is either incapable of completing DNA replication (Fig.1b, 3) or overreplicated in the absence of separation into daugh-ter cells (Fig. 1b, 4). It is likely that the mutation causes bothblocking of S phase which leads to the prevention of onset of thesubsequent mitosis and a defect in mitosis which preventsreinitiation of the subsequent S phase and leads to an increasein ploidy.
The characteristics of the mutant phenotype enabled us toisolate a novel gene, psp11, which suppresses the elongatedmutant phenotype of cyj92 at 36 °C and made cell cycle pro-gression normal. The finding that psp1 gene function is re-quired for normal cell growth at 30 °C suggests that this geneis essential for vegetative growth. However, the fact that itsoverexpression in the actively growing cells (log phase cells) isdetrimental to cell growth indicates that excessive functioningof this gene also causes a defect in normal cell growth. From theflow cytometric DNA analysis data we speculate that this isdue either to accumulation of abnormal DNA or to inhibition ofproduction of the elements necessary for normal cell division.Since we were not able to observe the accumulation of undi-vided cells, that is the cells attached together without septum,we consider that overproduction of Psp1 protein does not affectthe cytokinesis process itself but rather affects the normalprocess of DNA replication. The accumulation of the 2n or 4nDNA content in the actively growing cells by overproduction ofPsp1 protein suggests that the signal for completion of DNAreplication may not be transferred correctly to the elementsworking in G2 or M. Alternatively, it may be possible thatoverexpression of psp1 may lead to bypass or delay of themitosis process and thereby allow continued replication ofDNA. The fact that Psp1 protein is required for normal DNAreplication but excessive function of this protein causes delay ofthe cell cycle suggests that this Psp1 may act at two differentpoints in the cell cycle. Identification of target molecules withwhich Psp1 protein interacts will be necessary to elucidate thedetailed function of this protein. The finding that the inhibitoryeffect on cell growth becomes less severe upon entry into thestationary phase indicated that this protein is in its active formin actively growing cells but is inactivated when cell growthslows down. However, we do not know whether inactivation ofthis protein is a prerequisite for entry into the stationary phaseor conversely entry into stationary phase leads to inactivationof this protein.
The results that Psp1 is a phosphoprotein and that its de-phosphorylated form is the major one in actively growing cellsindicated that the function of this protein depends on phospho-rylation. If this protein functions as a positive element, re-quired in the cell cycle progression, the dephosphorylated formis active in actively growing cells and the phosphorylated Psp1,which is the major form in stationary phase cells, is inactive.However, if Psp1 functions as a negative element, the dephos-phorylated form is inactive in actively growing cells, and thephosphorylated form is its active form in the stationary phase.Although we don’t know where in the cell cycle Psp1 acts, theresult that this protein is phosphorylated by the Cdc2-Cdc13complex in vitro suggests that this protein could be one of thesubstrates for the Cdc2-Cdc13 complex and hence Cdc2 kinasemay be associated with its function.
The carboxyl-terminal domain of this novel protein is phos-phorylated at serine 333 by Cdc2 kinase, and alteration of thisserine to alanine abolishes phosphorylation by Cdc2 kinase invitro. However, in vivo we were not able to detect a defect inphosphorylation in the cdc2 mutant at the restrictive temper-ature. Because it showed the mutant phenotype within 4–5 hafter being shifted to restrictive temperature, this is not long
FIG. 6. Working model for Psp1 function in actively growingand stationary cells.
A Novel Protein Phosphorylated at Stationary Phase of S. pombe 20001
by guest on June 1, 2018http://w
ww
.jbc.org/D
ownloaded from

enough for cdc22 cells to enter the stationary phase to show itskinase function. It is only in the stationary phase of the cellgrowth that Psp1 protein is phosphorylated and cdc22 cells dieafter 4–5 h at the restrictive temperature long before we wereable to detect the defect in Cdc2 kinase function in the mutant.Thus it was not possible to determine whether mutation inCdc2 kinase affected the function of Psp1 in actively growingcells. We consider that phosphorylation of Psp1 protein uponentering the stationary phase is one of the major events forslowing cell growth, and Cdc2 kinase controls this process. Theyeast two-hybrid method confirmed that Cdc2 and Psp1 inter-act directly in vivo. Thus we can suggest that when cell cycleprogresses actively in log phase, Psp1 protein is in its dephos-phorylated state, and Cdc2 function is not required. However,when cell cycle progression slows at the stationary phase, Cdc2kinase phosphorylates Psp1 protein, and the phosphorylatedform of Psp1 facilitates entry into the G0 phase of the cell cycleor inactivates elements required for active cell growth. Cdc2protein function is required for modification of Psp1 protein atthis stage and there must be phosphatase(s), which removesthe phosphate from Psp1 protein when cells begins activegrowth. We did not find yet what gene product is involved indephosphorylation of Psp1 upon entry into active cell growthcycle. It will be interesting to find a specific phosphatase forPsp1, and function of this phosphatase may be a key regulatoryelement for activation of Psp1 protein function. Localization ofthis Psp1 toward each tip of the cell upon entry into the sta-tionary phase may also be related to phosphorylation of thisprotein. It could be suggested that the phosphorylated Psp1may have the property to form protein complexes in the cellsleading to translocation of the protein in a certain compart-ment of cells and the dephosphorylated Psp1 may exist inmonomeric form and is localized throughout cells as an activemonomeric form. The following model for Psp1 protein functioncan be drawn (Fig. 6). Psp1 protein is activated by dephospho-rylation with a phosphatase when the cells enter G1 to S phaseof the cell cycle and is inactivated by phosphorylation withCdc2 kinase upon entry into the stationary or G0 phase. It is anessential protein for cell growth, and regulation of this proteinfunction by phosphorylation could be the key feature in cellcycle progression from G1 to S. In addition to Cdc2 protein, theother element(s) which interact directly with this Psp1 proteinshould exist, and finding this element(s) will be essential forfurther characterization of Psp1 protein function.
Acknowledgments—We thank Dr. Kyu-chung Hur at Ewha Woman’sUniversity for his kind help in phosphoamino acid analysis and Dr.Chun-Jeih Ryu at KRIBB for his help in preparing polyclonal antibodyfor Psp1. We appreciate Dr. Hyang-Bae Kim at Korea University forproviding us with the known cdc mutants we used in this study. We alsothank Dr. Terrance Cooper at the University of Tennessee, Memphis,for his critical reading of this manuscript.
Addendum—While we were preparing this manuscript, Ishii et al.(77) reported the sds231 gene which has the same DNA sequence as thepsp11 gene. The sds231 gene is a multicopy suppressor for mutation inpp1 and 20 S cyclosome/anaphase promoting complex.
REFERENCES
1. Nigg, E. A. (1995) BioEssays 17, 471–4802. Doree, M. & Galas, S. (1994) FASEB J. 8, 1114–11213. Pines, J. (1995) Adv. Cancer Res. 66, 181–2124. Morgan, D. O. (1995) Nature 374, 131–1345. Elledge, S. J. & Harper, J. W. (1994) Curr. Opin. Cell Biol. 6, 847–8526. Harper, J. W. & Elledge, S. J. (1996) Curr. Opin. Genet. Dev. 6, 56–647. Cross, F. R. (1988) Mol. Cell Biol. 8, 4675–46848. Hadwiger, J. A., Wittenberg, C., Richardson, H. E., de Barres Lopes, M. &
Reed, S. I. (1989) Proc. Natl. Acad. Sci. U. S. A. 86, 6255–62599. Pines, J. & Hunter, T. (1990) New Biol. 2, 389–401
10. Norbury, C. & Nurse, P. (1992) Annu. Rev. Biochem. 61, 441–47011. Reed, S. I. (1992) Annu. Rev. Cell Biol. 8, 529–56112. Nasmyth, K. (1993) Curr. Opin. Cell Biol. 5, 166–17913. Meyerson, M., Enders, G. H., Wu, C. L., Su, L. K., Gorka, C., Nelson, C.,
Harlow, E. & Tsai, L. H. (1992) EMBO J. 11, 2909–2917
14. Matsushime, H., Ewen, M. E., Strom, D. K., Kato, J. Y., Hanks, S. K., Roussel,M. F. & Sherr, C. J. (1992) Cell 71, 323–334
15. Pagano, M., Pepperkok, J., Baldin, L. V., Bartek, A. J. & Draetta, G. (1993) J.Cell Biol. 121, 101–111
16. van den Heuvel, S. & Harlow, E. (1993) Science 262, 2050–205417. Meyerson, M. & Harlow, E. (1994) Mol. Cell Biol. 14, 2077–208618. Koff, A., Cross, F., Fisher, A., Schumacher, J., Leguellec, K., Philippe, M. &
Roberts, J. M. (1991) Cell 66, 1217–122819. Sherr, C. J. (1993) Cell 73, 1059–106520. Baldin, V., Bonetta, L., Marcote, M. J., Pagano, M. & Draetta, G. (1993) Genes
Dev. 7, 812–82121. Bates, S., Bonetta, L., MacAllan, D., Parry, D., Holder, A., Dickson, C. &
Peters, G. (1994) Oncogene 9, 71–7922. Hunter, T. & Pines, J. (1994) Cell 79, 573–58223. Knoblich, J. A. & Lehner, C. F. (1993) EMBO J. 12, 65–7424. Hayles, J., Fisher, D., Woollard, A. & Nurse, P. (1994) Cell 78, 813–82225. Draetta, G. & Beach, D. (1988) Cell 54, 17–2626. Atherton-Fessler, A., Parker, L. L., Geahlen, R. L. & Piwnica-Worms, H. (1993)
Mol. Cell Biol. 13, 1675–168527. Russell, P. & Nurse, P. (1986) Cell 45, 145–15328. Galaktionov, K. & Beach, D. (1991) Cell 67, 1181–119429. Gautier, J., Solomon, M. J., Booher, R. N., Bazan, J. F. & Kirschner, M. W.
(1991) Cell 67, 197–21130. Russell, P. & Nurse, P. (1987) Cell 49, 559–56731. Russell, P., Moreno, S. & Reed, S. I. (1989) Cell 57, 295–30332. Parker, L. L., Atherton-Fessler, S., Lee, M. S., Ogg, S., Falk, F. L., Swenson, K.
I. & Piwnica-Worms, H. (1991) EMBO J. 10, 1255–126333. Parker, L. L., Atherton-Fessler, S. & Piwnica-Worms, H. (1992) Proc. Natl.
Acad. Sci. U. S. A. 89, 2917–292134. Murray, A. (1995) Cell 81, 149–15235. Mendenhall, M. D. (1993) Science 259, 216–21936. Harper, J. W., Adami, G. R., Wei, N., Keyomarsi, K. & Elledge, S. J. (1993) Cell
75, 805–81637. Xiong, Y., Hannon, G. J., Zhang, H., Casso, D., Kobayashi, R. & Beach, D.
(1993) Nature 366, 701–70438. Toyoshima, H. & Hunter, T. (1994) Cell 78, 67–7439. Polyak, K. (1994) Cell 78, 59–6640. Hannon, G. J. & Beach, D. (1994) Nature 371, 257–26141. Laskey, R. A., Fairman, M. P. & Blow, J. J. (1989) Science 246, 609–61342. Hunter, T. & Pines, J. (1990) Nature 346, 760–76343. Nurse, P. (1994) Cell 79, 547–55044. Sherr, C. J. (1994) Cell 79, 551–55545. Wuarin, J. & Nurse, P. (1996) Cell 85, 785–78746. Hunt, T. (1989) Curr. Opin. Cell Biol. 1, 268–27447. Nurse, P. (1990) Nature 344, 503–50848. King, R. W., Jackson, P. K. & Kirschner, M. W. (1994) Cell 79, 563–57149. Gutz, H., Heslot, H., Leupold, U. & Loprieno, N. (1974) in Handbook of
Genetics (King, R. C., ed) Vol. 1, pp. 395–446, Plenum Press, New York50. Moreno, S., Klar, A. & Nurse, P. (1991) Methods Enzymol. 194, 795–82351. Nurse, P., Thuriaux, P. & Nasmyth, K. (1976) Mol. Gen. Genet. 146, 167–17852. Jang, Y. J., Kim, D. U., Lee, H. J. & Yoo, H. S. (1994) Mol. Cell 4, 419–42853. Lew, D. J., Dulic, V. & Reed, S. I. (1991) Cell 66, 1197–120654. Costello, G., Rodgers, L. & Beach, D. (1986) Curr. Genet. 11, 119–12555. Mitchison, J. M. & Creanor, J. (1971) Exp. Cell Res. 69, 244–24756. Toda, T., Yamamoto, M. & Yanagida, M. (1981) J. Cell Sci. 52, 271–28757. Macneill, S. A. & Fantes, P. A. (1993) in The Cell Cycle: A Practical Approach
(Fantes, P. A., and Brooks, R., eds) pp. 93–125, IRL Press, Oxford58. Wright, A., Maundrell, K., Heyer, W. D., Beach, D. & Nurse, P. (1986) Plasmid
15, 156–15859. Beach, D. & Nurse, P. (1981) Nature 290, 140–14260. Sanger, F., Nicklen, S. & Coulson, A. R. (1977) Proc. Natl. Acad. Sci. U. S. A.
74, 5463–546761. Park, S. K., Chon, S. K. & Yoo, H. S. (1995) Biochim. Biophys. Acta 1262, 87–9062. McKnight, S. L. & Kingsbury, R. (1982) Science 217, 316–32463. Disatnik, M. H., Hernandez-Sotomayor, S. M., Jones, G., Carpenter, G. &
Mochly-Rosen, D. (1994) Proc. Natl. Acad. Sci. U. S. A. 91, 559–56364. Ausubel, F. M., Brent, R., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith,
J. A. & Struhl, K. (1995) Short Protocols in Molecular Biology, pp. 13.51,17.1–17.14, Wiley, New York
65. Smith, D. B. & Johnson, K. S. (1988) Gene 67, 31–4066. Alfa, C. E. & Hyams, J. S. (1990) J. Cell Sci. 96, 71–7767. Alfa, C. E., Ducommun, D., Beach, D. & Hyams, J. S. (1990) Nature 347,
680–68268. Carlson, M. & Botstein, D. (1982) Cell 28, 145–15469. Maundrell, K. (1990) J. Biol. Chem. 265, 10857–1086470. Clackson, T., Gussow, D. & Jones, P. T. (1991) in PCR: A Practical Approach
(McPherson, M. J., Quirke, P., and Taylor, G. R., eds) pp. 201–214, IRLPress, Oxford
71. Zhuo, S., Clemens, J. C., Hakes, D. J., Barford, D. & Dixon, J. E. (1993) J. Biol.Chem. 268, 17754–17761
72. Gordon, J. A. (1991) Methods Enzymol. 201, 477–48273. Correa-Bordes, J. & Nurse, P. (1995) Cell 83, 1001–100974. Harlow, E. & Lane, D. (1988) Antibodies: A Laboratory Manual, pp. 53–137,
Cold Spring Harbor Laboratory, Cold Spring Harbor, NY75. Kamps, M. P. & Sefton, B. M. (1989) Anal. Biochem. 176, 2–2776. Hanks, S. K. & Quinn, A. M. (1991) Methods Enzymol. 200, 38–6277. Ishii, K., Kumada, K., Toda, T. & Yanagida, M. (1996) EMBO J. 15,
6629–6640
A Novel Protein Phosphorylated at Stationary Phase of S. pombe20002
by guest on June 1, 2018http://w
ww
.jbc.org/D
ownloaded from

Chankyu Park and Hyang-Sook YooYoung-Joo Jang, Misun Won, Kyung-Sook Chung, Dong-Uk Kim, Kwang-Lae Hoe,
of Cell Growth -like Stationary Phase0 Is Phosphorylated by Cdc2-Cdc13 upon Entry into GpombeSchizosaccharomycesA Novel Protein, Psp1, Essential for Cell Cycle Progression of
doi: 10.1074/jbc.272.32.199931997, 272:19993-20002.J. Biol. Chem.
http://www.jbc.org/content/272/32/19993Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/272/32/19993.full.html#ref-list-1
This article cites 73 references, 18 of which can be accessed free at
by guest on June 1, 2018http://w
ww
.jbc.org/D
ownloaded from