the international hapmap project: a rich resource of genetic information julia krushkal 04/15/2010...
TRANSCRIPT

Julia Krushkal
04/15/2010
Lecture in Bioinformatics

The International HapMap Project
• Population-specific sequence variation• Allele frequencies• Linkage disequilibrium patterns• Haplotype information• Tag SNPs• Structural genome variation• Better understanding of human population dynamics and of
the history of human populations• Cell lines available from Coriell Inst. for Medical Research• A rich resource for biomedical genetic analysis
“…Determine the common patterns of DNA sequence variation in the human genome, by characterizing sequence variants, their frequencies, and correlations between them, in DNA samples from populations with ancestry from parts of Africa, Asia and Europe.” Nature (2003)

HapMap Population Samples
Project launched in 2002 to provide a public resource for accelerating medical genetic research
270 Individuals from 4 Geographically Diverse PopulationsYRI: 90 Yorubans from Ibadan, Nigeria 30 parent-offspring trios
CEU: 90 northern and western European-descent living in Utah, USA from the Centre d’Etude du Polymorphisme Humain (CEPH) collection
30 parent-offspring trios
CHB: 45 unrelated Han Chinese from Beijing,China
JPT: 45 unrelated Japanese from Tokyo, Japan
http://www.hapmap.org/ HapMaphttp://www.genome.gov/page.cfm?pageID=10001688 NHGRI
Combined in many analyses

International HapMap Project Papers•The Int. HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851-861. 2007•The Int. HapMap Consortium.A Haplotype Map of the Human Genome. Nature 437:1299-1320.2005•The Int. HapMap Consortium. The International HapMap Project. Nature 426, 789-796.. 2003•The Int. HapMap Consortium. Integrating Ethics and Science in the International HapMap Project. Nature Reviews Genet 5, 467 -475. 2004•Thorisson et al. The International HapMap Project Web site. Genome Res 15:1591-1593. 2005
HapMap-related papers •Sabeti et al. Genome-wide detection and characterization of positive selection in human populations. Nature 449, 913-918. 2007. •Clark et al. Ascertainment bias in studies of human genome-wide polymorphism. Genome Res, 15:1496-1502. 2005 •Clayton et al. Population structure, differential bias and genomic control in a large-scale, case-control association study. Nature Genet 37(11):1243-1246. 2005 •de Bakker et al. Efficiency and power in genetic association studies. Nature Genet 37:1217-1223. 2005•Goldstein, Cavalleri. Genomics: Understanding human diversity. Nature 437:1241-1242. 2005. •Hinds et al. Whole genome patterns of common DNA variation in three human populations. Science 307:1072-1079. 2005. •Myers et al. A fine-scale map of recombination rates and hotspots across the human genome. Science, 310:321-324. 2005•Nielsen et al. Genomic scans for selective sweeps using SNP data.Genome Res 15:1566-1575. 2005•Smith et al. Sequence features in regions of weak and strong linkage disequilibrium. Genome Res 15: 1519-1534. 2005•Weir et al. Measures of human population structure show heterogeneity among genomic regions. Genome Res 15: 1468-1476. 2005.

Nature (2003)

Human Chromosomes
• Contain DNA• 22 pairs of autosomes +
sex-chromosomes (X and Y) + mitochondrial genome
• Contain functional units (genes) and other DNA
Human genome sequence is available as a reference, as a result of the Human Genome Project
A significant amount of inter-individual variation exists

Chromosomes are sets of continuously linked genetic loci
Example:Integrated mapof chromosome 5 from the International HapMap Project,
http://www.hapmap.org

Genetic Variation
DNA
RNA
Transcription
Protein
Translation
Phenotype
ENVIRONMENT
DNA
RNA
Transcription
Protein
Translation
Phenotype
ENVIRONMENT
• Some DNA loci vary among individuals• Linked genetic loci are inherited non-independently• Loci may change with time (mutation, selection, genetic drift)•Some DNA changes lead to quantitative changes in RNA expression and to quantitative or qualitative changes in protein production• Some genetic changes, even small, may lead to disease• A large amount of natural variation occurs in healthy individuals, i.e., many changes are neutral•Loci genetically linked to the disease-causing locus can be used as genetic markers to search for the disease locus
SNP1 SNP2
Sequence variation
AAAC/TGGCTA
There are many types of DNA variation, e.g.
Microsatellite repeats
…AATG AATG AATG AATG…

Polymorphic Site
A locus with common DNA variation 2 alleles in a population Shows difference in DNA sequence among individuals
In most definitions: the most common allele with frequency < 99%, or minor allele frequency (MAF) 1%, or MAF 2%, or at least two alleles have frequencies 1%.
A rare allele that occurs in <1% of the population is usually non considered a polymorphic site.
90%of sequence variation among individuals is due to common variation (MAF 1%, ); 10% are rare variants
Not all disease-predisposing variants are common

SNP=Single Nucleotide Polymorphism
A and C are alleles at SNP locus rs6870660
SNP locus rs6870660
CAAATTCCATG[A or C]AGAAGGAAATACAT
http://www.ensembl.org
A SNP locus on the distal end of the long arm of human chromosome 5 (data from Ensembl)

A SNP locus on the distal end of the long arm of chromosome 5
SNP locus rs6870660
http://www.hapmap.org

2 alleles, A and Bfrequencies p and q p+q =1
The allele frequencies remain constant through time.
A B
A AA AB
B AB BB
p q
p p2 pq
q pq q2F1
Hardy-Weinberg Equilibrium
Egg
Sperm
PAA=p2 PAB=2pq PBB =q2
Under Hardy-Weinberg equilibrium, the relative genotype frequencies are:
F1: (p+q )2
Departures can be characterized by disequilibrium
In autosomal genes, and in absence of disturbing influences, this proportion is maintained through all subsequent generations.

Linkage Disequilibrium
D=pA1B1-pA1pB1
D = Linkage disequilibrium coefficient Coefficient of association
Locus A Locus B
A1 B1
A2 B2
Associations among alleles at different loci
D’=D/|D|max
|D| max = | min(pA1pB2, pA2pB1)|-1 D’ 1 r2 =D2/(pA1pA2pB1pB2)
Normalized disequilibrium coefficient
Squared Correlation coefficientAlso ranges from 0 to 11 – absolute or perfect linkage; 0.8 is the cutoff often used
Extended to multiallelic markers

Regulatory Interactions: The ENCODE Project
2003-Pilot project launched (1% of the genome)2007- Pilot project completed; production phase launched on the entire genome
<>
Production Scale Effort Pilot Scale Effort Data Coordination Center Technology Development Effort
High-through-put experimental and computational approaches to studies of DNA regulatory sites, regulatory interactions, and DNA modification


Genome SNP VariationSize of human genome 3.2 109
bp99.9% identical9-10 mln SNPs may have MAF 5% 30,000 genes
•Phase I (published in 2005)931,340 SNPs passed quality control1 SNP / 3000 bp11,500 nsSNP10 ENCODE regions, 500 kb each
17,944 SNPs1 SNP / 279 bp
•Phase II (published in 2007) Consensus data set:3,107,620 SNPs, QC+ in all panels, polymorphic in 1 panel 1 SNP / 875bp 25-30% of all SNPs with MAF 5%
HapMap SNP Density Coverage
The cumulative # of non-redundant SNPs is shown as a solid line, the # of SNPs validated by genotyping as a dotted line, and double-hit status as a dashed line.

HapMap Phase II

HapMap Phase II•21,177 SNPs from Phase I that had ambiguous position or other low reliability feature were not included in Phase II•Chimpanzee, rhesus macaque used for comparisons and to infer ancestral states of SNPs•3,107,620 SNPs, QC+ in all panels, polymorphic in 1 panel 1 SNP / 875bp 1.14 SNP/kb25-30% of all SNPs with MAF 5%•98.6% of the genome is within 5 kb of the nearest polymorphic SNP•Better representation of rare variation/ SNPs with MAF 1%•Phase II marker data capture overwhelming majority of genome SNP variation, mean r2 of 0.9-0.96 for different populations

http://hapmap.ncbi.nlm.nih.gov/

SNP Differences among Individuals Far Exceed Differences among Populations
Phase 1: Autosomes: Across the 1 million SNPs genotyped, only 11 have fixed differences between CEU and YRI, 21 between CEU and CHB/JPT, and 5 between YRI and CHB/JPT.X chromosome 123 SNPs were completely differentiated between YRI and CHB/JPT, but only 2 between CEU and YRI and 1 between CEU and CHB/JPT.

Importance of Understanding Patterns of Human Genetic Variation
• Without knowing the patterns of correlation, one would need to analyze millions of SNPs and other polymorphisms in the genome
• Alleles at nearby loci occur non-independently
• Knowledge about correlations among polymorphisms allows to us significantly reduce the number of genetic tests, while surveying extensively for variation patterns
• Patterns of correlation are complex
• Need to know local patters of genetic variation rather than simply use SNPs at regular intervals

Patil et al. 2001, Blocks of Limited Haplotype Diversity Revealed by High-Resolution Scanning of Human Chromosome 21. Science 294(5547):1719-23
Genome regions decomposedinto discrete haplotypeblocks, which capture similarity in haplotype organization
Haplotype Maps of the Human Genome

Haplotype Maps Generated by The International HapMap
Project 3 steps of the HapMap
construction
(a) SNPs are identified in DNA samples from multiple individuals.
(b) Adjacent SNPs that are inherited together are compiled into haplotypes.
(c)"Tag" SNPs are identified within haplotypes that uniquely describe those haplotypes. Source: The International HapMap Project

Haplotype Maps of the Human Genome
Haplotypes were inferred for the HapMap project from trios data and from unrelated individuals using Phase (Stephens 01; Stephens and Donnely 03)
Helmuth 2001, Science 293:583-585
Find correlations among groups of SNPs

Haplotype Block Partition Results for Three Populations
Population Blocks Average size, kb* Required SNPs
African-American 235,663 8.8 570,886
European-American 109,913 20.7 275,960
Han Chinese 89,994 25.2 220,809
* Average distance spanned by segregating sites in each block.
Minimum number of SNPs required to distinguish common haplotype patterns with frequencies of 5% or higher.
Hinds et al. 2005 Science
1,586,383 (SNPs) genotyped in 71 Americans of European, African, and Asian ancestry

Hinds et al 2005
Extended LD bin and haplotype block structure around the CFTR gene. LD bins, where each bin has at least one SNP with r2 > 0.8 with every other SNP, are depicted as light horizontal bars, with the positions of constituent SNPs indicated by vertical tick marks as well as the extreme ends of the bars. Isolated SNPs are indicated by plain tick marks. Haplotype blocks, within which at least 80% of observed haplotypes could be grouped into common patterns with frequencies of at least 5%, are depicted as dark horizontal bars. Unlike haplotype blocks that are by design sequential and nonoverlapping, SNPs in one LD bin can be interdigitated with SNPs in multiple other overlapping bins
Population differences in local bin structureDifferences in allele and haplotype frequencies“Although analysis panels are characterized both by differenthaplotype frequencies and, to some extent, different combinations ofalleles, both common and rare haplotypes are often shared acrosspopulations” (The Int. HapMap Project, Nature, 2005)

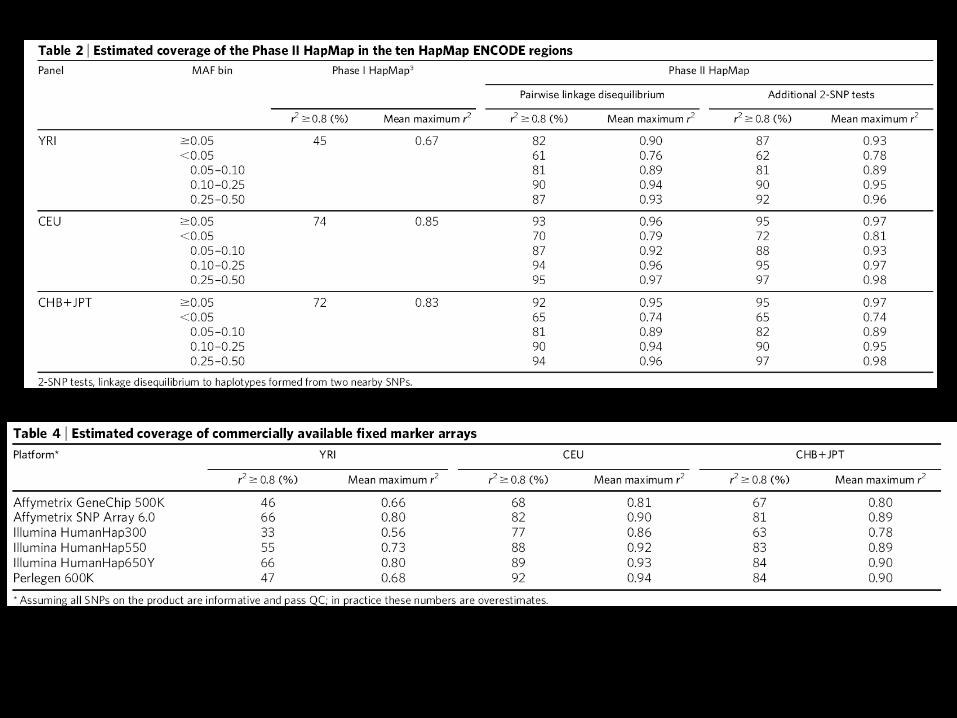
Amount of Captured Sequence Variation in HapMap Phase II
For common variants (MAF 0.05) the mean maximum r2 of any SNP to a typed one is 0.90 in YRI, 0.96 in CEU and 0.95 in CHB/JPT.
1.09 million SNPs capture all common Phase II SNPs with r2 0.8 in YRI.
Very common SNPs with MAF 0.25 are captured extremely well (mean maximum r2 of 0.93 in YRI to 0.97 in CEU)
Rarer SNPs with MAF<0.05 are less well covered (mean maximum r2 of 0.74 in CHB/JPT to 0.76 in YRI).

Amount of Captured Sequence Variation in HapMap Phase II
Additional tag SNPs are unlikely to capture large groups of additional SNPs
Can use to phase new data using HapMap haplotype information, missing data imputation

DNA Chips and Resequencing: High-through-put Analysis of Sequence
VariationAn easy way to access genome-wide variationBoth Affymetrix and Illumina DNA chips contain representative SNP and
CNV probes
Affymetrix GeneChip 6.0: 1.8 million markers for genetic variation, including 906,000 SNPs and 946,000 copy number probes.
Illumina 1M Bead Chip and 1M-duo Bead Chip: ~950,000 genome-spanning tag SNPs;~100,000 additional non-HapMap SNPs, >565,000 SNPs in and near coding regions such as nsSNPs, promoter regions, 3’ and 5’ UTRs; dense coverage in ADME and MHC regions. ~260,000 markers located in novel and reported copy number polymorphic regions.
Sequenom mass arrays (based on Maldi-TOF)
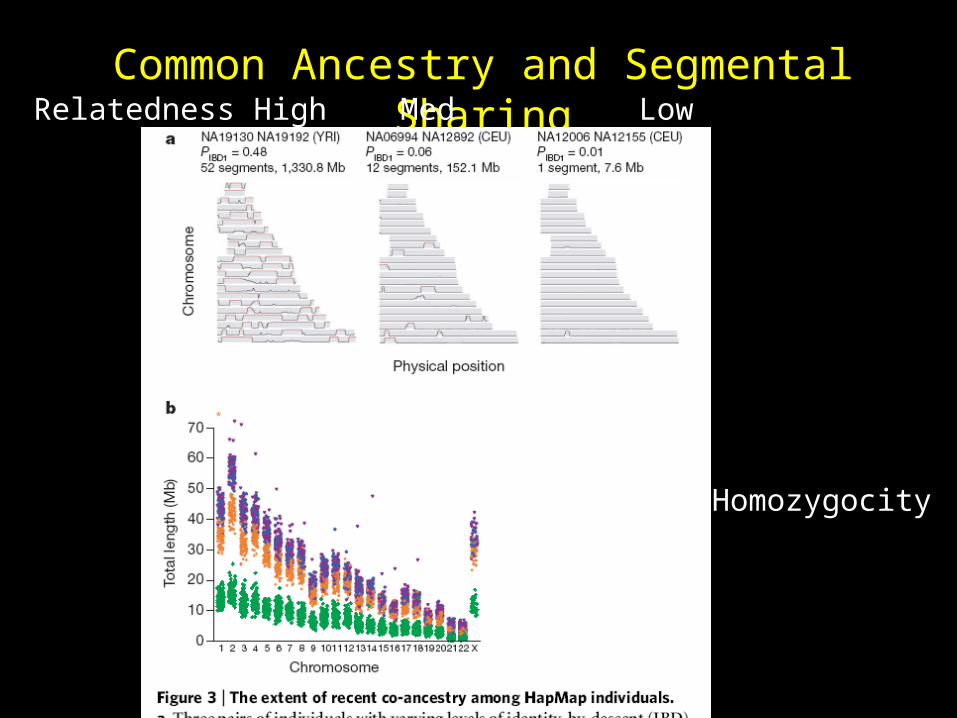
Common Ancestry and Segmental SharingRelatedness High Med Low
Homozygocity

Recombination Hotspots
32,996 recombination hotspots60% of genome recombination, 6% sequence

Recombination and tagSNPs
•Recombination hotspots are frequently insufficient to break down allelic associations
•Common haplotypes often span recombination hotspots
•0.5-1% of SNPs are untaggable: no SNPs with r2 0.2 within 100 kb
•Untaggable SNPs are not in segmental duplications
•They often are in recombination hotspots; some may be due to genealogical structure, mutation hotspots, or gene conversion

Demographic History of Human PopulationsGenealogical History and Allelic Associations
The genealogy for the 13 haplotypes observed in a 40-kb region of Chromosome 1 (between SNPs rs12085605 and rs932087) where there is no evidence for recombination. Location of polymorphic mutations is indicated by circles.
Relative frequency of each haplotype in the sample from each of the three panels (with white indicating 0% and black indicating 100%).
The dotted line in the genealogy indicates a branch of the tree that is not present in theCEU sample and whose removal results in perfect association between SNPs rs12085824 and rs11205476.
Can track genealogical history
McVean et al., 2005. PLoS Genetics 1:e54
Complex patterns of stochastic mutation, recombination, selection, genetic drift in evolutionary history shape the patterns of genome variation
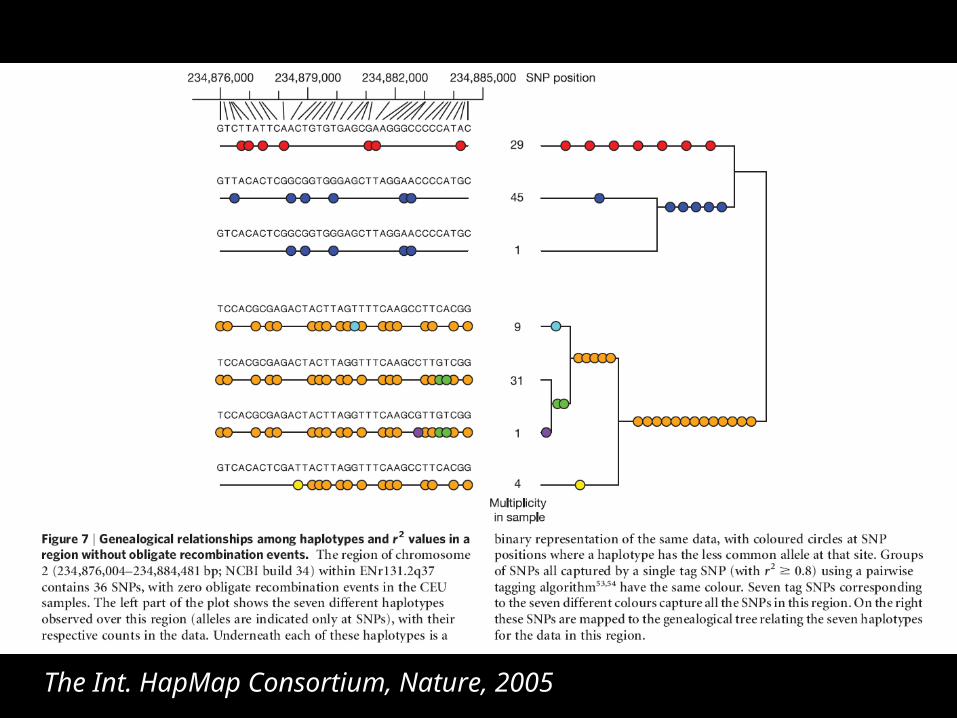
The Int. HapMap Consortium, Nature, 2005

Mirrors at Sanger Center and Baylor College of Medicine
FUNDING AGENCIESNational Institutes of Health – National Human Genome Research Institute (NHGRI)Wellcome Trust
HapMap 3

Hardy-Weinberg p>0.000001 (per population) missingness <0.05 (per population) <3 Mendel errors (per population; only applies to YRI, CEU, ASW, MEX, MKK) SNP must have a rsID and map to a unique genomic location
The "consensus" data set contains data for 1115 individuals (558 males, 557 females; 924 founders and 191 non-founders), only keeping SNPs that passed QC in all populations (overall call rate is 0.998). The "consensus|polymorphic" data set has 35023 monomorphic SNPs (across the entire data set) removed.
QC in HapMap 3


HapMap 3 samples

Data ContentSNP GENOTYPE DATA
label # samples # QC+ SNPs # polymorphic QC+ SNPsASW 71 1632186 1536247CEU 162 1634020 1403896CHB 82 1637672 1311113CHD 70 1619203 1270600GIH 83 1631060 1391578JPT 82 1637610 1272736LWK 83 1631688 1507520MEX 71 1614892 1430334MKK 171 1621427 1525239TSI 77 1629957 1393925YRI 163 1634666 1484416consensus 1115 1525445 1490422


PCR RESEQUENCING DATA •“The sequence-based variant calls were generated by tiling with PCR primer sets spaced approximately 800 bases apart across the ENCODE 3 regions. Following filtering low-quality reads the data were analyzed with SNP Detector version 3, for polymorphic site discovery and individual genotype calling. Various QC filters were then applied. Specifically, we filtered out PCR amplicons with too many SNPs, and SNPs with discordant allele calls in mutliple amplicons. “•Also filtered out were SNPs with low completeness in samples, or with too many conflicting genotype calls in two different strands. •“In the QC+ data set, …filtered out samples which had low completeness, and filtered out SNPs with low call rate in each population (<80%) and not in HWE (p<0.001). In the QC+ data set, the overall false positive rate is ~3.2%, based on a limited number of validation assays.”
http://www.broadinstitute.org/~debakker/p3.html

PCR RESEQUENCING DATA label number of samplesASW 55CEU 119CHB 90CHD 30GIH 60JPT 91LWK 60MEX 27MKK 0TSI 60YRI 120total 712
Data Content


HapMap Project is a Unique Resource for Genome-Wide
Association Studies• Resource for selection of representative tag SNPs
from low diversity haplotype blocks or from highly correlated SNPs
• Tag SNPs with r2 0.8 chosen for popular SNP chips• Resource for selecting custom SNPs for dense
genotyping in candidate regions, determined from genetic pathways of the 1st stage of multistage GWAS
• LD and haplotype information utilized for missing SNP imputation for genotypic problems or in meta-analyses

http://www.genome.gov/26525384
As of 04/15/10, this table includes 543 publications and 2658 SNPs.

Published Genome-Wide Associations through 6/2009, 439 published GWA at p < 5 x 10-8
NHGRI GWA Catalogwww.genome.gov/GWAStudies


Genotype Imputation Using HapMap Information

Genotype Imputation Using HapMap InformationSoftware from Jonathan Marchini’s group
http://www.stats.ox.ac.uk/~marchini/software/gwas/gwas.html

Imputation Common HapMap panel & tagSNPs
Use of HapMap Resources in Meta-Analyses

Figure 9. A schematic overview of structural genome variation data in the genome region containing CFH, CFHR3, and CFHR1, obtained from the Database of Genomic Variants
Structural Genome Variation
• Large number of copy number variants (CNVs) and other genome rearrangements found among individuals
• Some variation is assumed normal, other may cause disease
• Genome databases, e.g. Database of Genomics Variants at the TCAG of the Toronto Hospital of Sick Children, the Copy Number Variation Project Map at the Sanger Center
HapMap samples are also used as a resource for CNV analysis

• Segmental duplications are recombination hotspots, causing global genome rearrangements


Ethical, Legal, and Social ImplicationsIssues
•Patterns of variation can be compared among individuals and populations, e.g.Genetic profiling Racial profiling Population history studies•Extensive genetic information on donors publicly available•Generalization of biomedical results/stigmatization/ genetic determinism•Limitation of population identifiers Loose populations, self-described Limited number of representatives, e.g., Individuals samples from residential community in Bejing Normal University do not represent all 56 officially recognized ethnicities in China•Future use of cell line samples from same donors, they will not be able to withdraw their samples
Nature Reviews Genet. 2004 5:467-475

Ethical, Legal, and Social ImplicationsApproaches•Ethical considerations incorporated from the inception of the
HapMap project•Choice of several world populations•New samples obtained with appropriate consent, rather than use of previously stored samples•No personal identifiers included(CEPH samples have links to individuals, strictly confidential)•No medical information included•Population and gender information included•Community engagement, taking into account international and local ethical guidelines•Community Advisory Groups established/can withdraw community samples•Sensitivity to cultural issues
Nature Reviews Genet. 2004 5:467-475

HapMap Genome Browserhttp://hapmap.ncbi.nlm.nih.gov/

HapMap Genome Browser







UCSC Genome Browser http://genome.ucsc.edu/

Beyond the HapMap1000 Genomes
• An international research consortium formed to create the most detailed and medically useful picture to date of human genetic variation. The project involves sequencing the genomes of approximately 1200 people from around the world and receives major support from the Wellcome Trust Sanger Institute in Hinxton, England, the Beijing Genomics Institute Shenzhen in China and the National Human Genome Research Institute (NHGRI), part of the National Institutes of Health (NIH).

1000 Genomes• An international research consortium formed to create
the most detailed and medically useful picture to date of human genetic variation. The project involves sequencing the genomes of approximately 1200 people from around the world and receives major support from the Wellcome Trust Sanger Institute in Hinxton, England, the Beijing Genomics Institute Shenzhen in China and the National Human Genome Research Institute (NHGRI), part of the National Institutes of Health (NIH).

www.1000genomes.org
The goal of the 1000 Genomes Project is to find most genetic variants that have frequencies of at least 1% in the populations studied




Both Affymetrix and Illumina’s newest genotyping platforms include variants discovered in the 1000 genomes project.

Illumina HumanOmni1-Quad BeadChip Kits• 4-sample•Contains aggressively selected SNP and CNV probes•Markers derived from the 1,000 Genomes Project, all three HapMap phases, and recently published studies. •>1 Mln available assays per sample, containing carefully selected content that delivers dense coverage of the human genome and targets regions known to play a role in human disease.•SNP selection optimized to maintain comprehensive genomic coverage, while reducing tag SNP redundancy. •This has enabled the inclusion of additional content carefully chosen to target high-value regions of the genome and new coding variants identified by the 1000 Genomes Project including:•ABO blood typing SNPs, cSNPs, disease-associated SNPs, eSNPs, SNPs in mRNA splice sites, Absorption, Distribution, Metabolism and Excretion (ADME genes), Ancestry-Informative Markers (AIMs), HLA complexes, indels, introns, MHC regions, miRNA binding sites, mitochondrial DNA, pseudoautosomal region (PAR), promoter regions, and Y-chromosome

Illumina HumanOmni1-Quad BeadChip Kits
•Includes 10,000 SNPs targeting four 1Mb regions known to be associated with three or more human diseases•> 31,000 SNPs predicted to be non-synonymous; 40,000 SNPs covering an additional 100 intervals surrounding published peak markers from the NHGRI GWAS database; and the remaining top single-marker associated SNPs from the GWAS database.•High density markers with a median spacing of 1.2 kb ensure the highest level of resolution for CNV identification in the industry•A complete optimization of the BeadChip design increases the available complexity, while reducing the amount of required DNA to 200 ng