the influence of cyp2d6 polymorphism and quinidine on the disposition and antitussive effect of...
TRANSCRIPT

The influence of CYP2D6 polymorphism and quinidine on the disposition and antitussive effect of dextromethorphan in humans
Objectives: We studied the disposition of dextromethorphan in extensive and poor metabolizer subjects, as well as the effect of this polymorphism on the antitussive action of dextromethorphan. Methods: Six extensive metabolizers were studied on four occasions: (1) after 30 mg dextromethorphan, (2) after 30 mg dextromethorphan 1 hour before 50 mg quinidine, (3) after placebo, and (4) after 50 mg quinidine. Six poor metabolizers were studied on two occasions: (1) after 30 mg dextromethorphan and (2) after placebo. Blood and urine were collected over 168 hours and assayed for dextromethorphan, total (conjugated and unconjugated) dextrorphan, 3-methoxymorphinan, and total 3-hydroxymorphinan. On each occasion at each blood sampling time, capsaicin was administered as an aerosol to provoke cough. Reszhs: Dextromethorphan area under the plasma concentration-time curve (AUC) was 150-fold greater in the poor metabolizers than in the extensive metabolizers, and quinidine increased the AUC in extensive metabolizers 43-fold. The median dextrometborphan half-life was 19.1 hours in poor metabolizers, 5.6 hours in extensive metabolizers given quinidine, and 2.4 hours in extensive metabolizers. For dextrorphan (as total), the AUC was reduced 8.6-fold in poor metabolizers; quinidine had no effect on the AUC. The median half-life was 10.1 hours in poor metabolizers, 6.6 hours in extensive metabolizers given quinidine, and 1.4 hours in extensive metabolizers. The apparent partial clearance of dextromethorphan to dextror- phan was 1.2 L/hr in poor metabolizers, 78.5 L/h r in extensive metabolizers given quinidine, and 970 L/hr in extensive metabolizers. There was a strong (2 = 0.82) and significant (p < 0.01) positive correlation between the prestudy urinary metabolic ratios and the partial clearances of dextromethorphan to dextrorphan. There was very large intersubject variability in responsiveness to capsaicin. There was no difference in the capsaicin-induced cough frequency in the three groups. Dextromethorphan had no antitussive effect in this experimental cough model. Conclusion: The disposition of dextromethorphan was substantially influenced by CYP2D6 status. Cap- saicin may not be an ideal agent in experimental cough studies. (Clin Pharmacol Ther 1996;60:295-307.)
Deborah A. Capon, BSc (I-Ions), Felix Bochner, MD, FRACP, Nicole Kerry, BSc (Hons), Gerd Mikus, MD,” Catherine Danz, RN, and Andrew A. Somogyi, PhD Adelaide, Sotith Amralia, Am-tm&z
From the Department of Clinical and Experimental Pharmacol- ogy, University of Adelaide.
Supported by the National Health and Medical Research Council of Australia. Dr. Mikus was a recipient of a research fellowship of the Deutsche Forschungsgemeinschaft, Bonn, Germany.
Received for publication Nov. 20, 1995; accepted April 26, 1996. Reprint requests: Felix Bochner, MD, Department of Clinical
and Experimental Pharmacology, University of Adelaide, Ade- laide, Australia 5005.
aPresent affiliation: Dr. Margarete Fischer-Bosch-Institut fiir Kli- nische Pharmakologie, Stuttgart, Germany.
Copyright 0 1996 by Mosby-Year Book, Inc. 0009-9236/96/$5.00 + 0 13/l/74604
Dextromethorphan is a widely used, over-the- counter antitussive agent often found in combina- tion with other compounds. Its antitussive efficacy has been documented in patients with spontaneous cough?’ The doses of dextromethorphan found to be effective ranged from 4 mg four times daily’ to 60 mg as a single dose.5
Dextromethorphan is metabolized by 0- and N-demethylation to dextrorphan and 3-methoxy- morphinan, respectively, which are subsequently N-(dextrorphan) and 0-(3-methomorphinan) de- methylated to 3-hydroxymorphinan (Fig. 1).6-1o The
295

296 Capon et al. CLINICAL P HARMACOLOGY &THERAPEUTICS
SEPTEMBER 1996
CYP2D6
dextromethorphan
&methoxymorphinan
J-hydroxymorphinan
Fig. 1. Primary and secondary oxidative pathways for dextromethorphan.
O-demethylation of dextromethorphan to dextrorphan differences in dextromethorphan metabolism result- is catalyzed by the polymorphic CYP2D6,*-l2 which ing from the genetic polymorphism on cough sup- also catalyzes the O-demethylation of 3- pression has not been thoroughly studied. A pilot methoxymorphinan to 3-hydroxymorphinan’2 and study suggested that poor metabolizer subjects therefore cosegregates with the debrisoquin- achieved greater cough suppression than extensive sparteine type oxidative polymorphism.13’14 The ex- metabolizer subjects after a single 30 mg dose of pression of CYP2D6 is genetically controlled and, as dextromethorphan.22 The pharmacodynamic conse- a result of gene deletion or gene mutations, this quences of an interaction between quinidine and enzyme is absent in 5% to 10% of the white popu- dextromethorphan have also not been addressed. lation.14’15 Persons with this deficiency are described The aims of this study were (1) to describe the as poor metabolizers and the remainder as extensive pharmacokinetics of dextromethorphan in human metabolizers. Dextromethorphan is commonly used extensive metabolizer and poor metabolizer sub- as a phenotyping agent to differentiate these two jects, (2) to determine the effect of quinidine on the groups. Quinidine has been shown to be a potent pharmacokinetics of dextromethorphan in extensive inhibitor of CYP2D6,16,17 and single oral doses of 50 metabolizer subjects, and (3) to determine the effect to 100 mg quinidine sulfate can temporarily convert of metabolizer status on the antitussive efficacy of a phenotypic extensive metabolizer subject into a dextromethorphan in healthy volunteers exposed to poor metabolizer subject.17-21 an experimental cough challenge (capsaicin).
The pharmacokinetics of dextromethorphan in humans has not been well documented. Pfaff et a1.7 were able to characterize three subgroups: one with high plasma dextrorphan and low plasma dextro- methorphan (half-life [t1,2] of 2 hours) concentra- tions, another with low plasma dextrorphan and high plasma dextromethorphan (t,,, of 45 hours) concentrations, and an intermediate group. How- ever, they did not phenotype their subjects. No ac- curate characterization of the disposition of a single dose of dextromethorphan in relation to CYP2D6 phenotype has been performed because of method- ologic shortcomings.22’23 In addition, the effect of
METHODS Subjects
The study was approved by the Committee on the Ethics of Human Experimentation of the University of Adelaide and the Human Ethics Committee of the Royal Adelaide Hospital. Subjects gave written consent to participate in the study. All subjects had been previously phenotyped with dextromethorphan as the test agent.24 Four healthy male and two fe- male poor metabolizer subjects (urinary metabolic ratio [total dextrorphan/dextromethorphan], 0.06 to 0.93) and the same number of age-, weight- (within

CLINICAL P HARMACOLOGY &THERAPEUTICS VOLUME 60, NUMBER 3 Capon et al. 297
lo%), and sex-matched extensive metabolizer sub- jects (metabolic ratio, 59 to 3402) were studied. Their ages ranged from 20 to 26 years (mean age, 22.4) and weights ranged from 49 to 86 kg (extensive metabolizers; mean weight, 70 kg) and 45 to 84 kg (poor metabolizers; mean weight, 71 kg). The sub- jects were nonsmokers and did not take any medi- cations for 1 week before and throughout the study. Alcohol was prohibited for 2 days before the study and for the duration of the study. Good health was based on normality of medical history, physical ex- amination, and blood and urine for markers of kid- ney, liver, and bone marrow function. In addition, an ECG was performed for extensive metabolizers, who were to receive quinidine.
Drugs Identical-looking capsules that contained either
30 mg dextromethorphan hydrobromide (Roche Products Pty. Ltd., Sydney, Australia), 50 mg quin- idine sulfate (Sigma Chemical Co., St. Louis, MO.), or placebo were manufactured in the Pharmacy De- partment of the Royal Adelaide Hospital. A stock solution (3.6 x 1O-3 mol/L) of capsaicin (Sigma Chemical Co.) in ethanol was prepared by the Phar- macy Department of the Royal Adelaide Hospital.
Cough challenge Aliquots of the capsaicin stock solution were di-
luted in 0.9% sodium chloride (Baxter Healthcare Pty. Ltd., Sydney, New South Wales, Australia) to yield capsaicin concentrations that varied from 0.1 to 20 p,mol/L. One milliliter of each dilution was placed in a nebulizer bowl (model 1700 Up-Draft Neb-U-Mist, Hudson Oxygen Therapy Sales Com- pany, Temecula, Calif.). The nebulizer was activated by oxygen (8 L/min). The volunteers inhaled the aerosol over 5 seconds. Each concentration was ad- ministered three times, with a l-minute interval be- tween each inhalation. Each subject was challenged by five different capsaicin concentrations and one saline aerosol, administered in random order. The number of coughs in the lo-second interval after the completion of each inhalation was recorded. This allowed the assessment of (1) cough threshold dose, defined as the capsaicin concentration that pro- duced one or more coughs in two of the three replicate concentrations, and (2) the maximum dose, defined as the capsaicin concentration that produced five or more coughs.
This procedure was carried out before drug ad- ministration and 2,4,6,8, 12,24,48,72,96, 120,144,
and 168 hours after drug administration (poor me- tabolizer subjects and extensive metabolizer subjects in phase l), as well as before drug administration and 2, 4, 6, 8, 12, and 24 hours after drug adminis- tration in extensive metabolizer subjects in phase 2.
The subjects and the investigators who adminis- tered the cough challenge and collected the blood and urine samples were blinded with respect to the identity of the treatments.
Experimental protocols
Poor metabolizer subjects. Each subject was studied on two occasions separated by at least 21 days but not more than 42 days. On one occasion, subjects received a placebo capsule and on the other a dex- tromethorphan hydrobromide capsule. The order of administration was randomized. Venous blood (10 ml) was collected into heparinized tubes through a Teflon 18gauge placement unit (Jelco, Critikon Inc., Tampa. Fla.), kept patent with a stylet (Jelco). Collection times were predose and 1,1X, 2,4,8, and 12 hours after dosing (through the catheter) and 24, 48, 72, 96, 120, 144, and 168 hours after dosing (by venipuncture). Plasma was separated from red cells by centrifugation and stored at -20” C until assay. All urine was collected to span the intervals from 0 to 12 hours and 12 to 24 hours and then each 24 hours until 168 hours after drug ingestion. Urine volume and pH were recorded and an aliquot from each sample stored at -20” C until assay. Cough challenges were administered before dosing and ev- ery 2 hours until 12 hours, then after each blood collection as above.
Extensive metabolizer subjects. Each subject was studied on four occasions, with occasions 1 and 2 constituting phase 1 (to evaluate the effect of quin- idine on dextromethorphan pharmacokinetics and pharmacodynamics) and occasions 3 and 4 consti- tuting phase 2 (to determine if quinidine exerted an antitussive effect in its own right).
In phase 1, each occasion was separated by at least 20 days but not more than 42 days. On one occasion, subjects received placebo 1 hour before dextromethorphan hydrobromide and on the other occasion subjects received quinidine sulfate 1 hour before dextromethorphan hydrobromide. The order of drug administration was randomized. The re- mainder of the protocol was the same as that de- scribed for the poor metabolizer subjects, except that additional blood samples were collected Y2, 3, and 6 hours after dosing.
In phase 2, each occasion was separated by 7 days.

298 Capon et al. CLINICAL P HARMACOLOGY & THERAPEUTICS
SEPTEMBER 1996
On one occasion, subjects ingested placebo 1 hour before placebo, and on the other occasion subjects took quinidine sulfate before placebo. The order of drug treatments was randomized. Cough challenges were administered before dosing and 2, 4, 6, 8, 12, and 24 hours after dosing.
HPLC assays Concentrations of dextromethorphan and the
three metabolites (see below) were quantified by the method of Chen et a1.:4 with minor modifications (the hydrolyzing enzyme was @glucuronidase from Escherichia coli, type VII-A, Sigma Chemical Com- pany). Minimum quantifiable concentrations in urine and plasma, respectively, were 0.1 mgJL and 0.39 u&L dextromethorphan, 0.1 mg/L and 1.17 kg/L dextrorphan, 0.1 mg/L and 0.78 kg/L 3-methoxymorphinan, and 0.1 mg/L and 0.78 I&L 3-hydrorrymorphinan.
In urine, the coefficients of variation for interday precision were less than 20% for dextromethorphan and the three metabolites at concentrations of 0.1 mg/L and below 10% at 1.0 mg/L and 10.0 mg/L. The coefficients of variation for intraday precision for low (0.1 mg/L), medium (1.0 mg/L), and high (lo.0 mgn> 4 ua i 1 ty control samples for all four ana- lytes ranged from 2.0 to 10.0% (n = 8 to 9). Accu- racy data for the above concentrations of substances ranged from 1% to 21% as estimated by the percent deviation from the nominal concentrations.
In plasma, the coefficients of variation for intra- day precision for low, medium and high quality con- trol samples, respectively, were dextromethorphan: 1.25 kg/L (4.9%), 5 pg/L (2.1%), and 20 l&L (2.4%); dextrorphan: 3.75 l~,g/L (1.8%), 15 l~,g/L (2.1%), and 60 kg/L (8.3%); 3-methoxymorphinan: 1.25 pg/L (8.5%) 5 pg/L (2.6%), and 20 &L (2.4%); 3-hydroxymorphinan: 2.5 &L (8.0%) 10 kg/L (4.0%), and 40 F~/L (13.1%) (all IZ = 6). The coefficients of variation for interday precision were less than 13% for the low quality control samples (1.2 pg/L or less for each compound), less than 6.2% for the medium quality control samples (3 kg/L for 3-methoxymorphinan and dextromethorphan, 6 &L for 3-hydroxymorphinan, and 9 t&L for dextrorphan) and less than 1.0% for the high quality control samples (50 IJ&L for dextromethor- phan and 3-methoxymorphinan, 100 &L for 3-hydroxymorphinan, and 150 &L for dextror- phan). Accuracy data for the above concentrations of substances were less than 11% as estimated by the percent deviation from the nominal concentrations.
Data analysis The pharmacokinetic parameters peak plasma
concentration (C,,), time to reach C,, (&ax>, area under the plasma concentration-time curve from zero to infinity (AUC) and t,,, were calculated by standard methods. The apparent partial metabolic clearance of dextromethorphan to dextrorphan was calculated as the amount of total (conjugated plus unconjugated) dextrorphan in urine divided by the AUC of dextromethorphan. Intrinsic clearance of dextromethorphan was calculated as dose of dextro- methorphan (as the base) divided by the AUC of dextromethorphan. Renal clearance of dextro- methorphan was calculated as amount of dextro- methorphan in urine divided by the AUC for dex- tromethorphan. The O-demethylation metabolic ratio was calculated as amount of dextrorphan (con- jugated plus unconjugated) excreted in urine over 168 hours divided by the amount of dextromethor- phan excreted during the same period.
Statistics Nonparametric statistical tests were used to ana-
lyze the data. The pharmacokinetic and pharmaco- dynamic data for extensive metabolizers treated with and without quinidine were compared with use of the Wilcoxon matched-pairs signed-rank test. When results of either of these two groups were compared with the results of the poor metabolizer subjects, the Mann-Whitney U test was used. Fried- man tests were used to determine whether tachyphy- laxis to capsaicin had occurred. If significant, the multiple pairwise comparison test was used. A p value of ~0.05 was considered to be statistically significant. All data are reported as the median and range, except in Fig. 4 in which the data are repre- sented as mean values + SD.
RESULTS Pharmacokinetics
The plasma concentration-time profiles and de- rived pharmacokinetic parameters for dextrometh- orphan and total dextrorphan were markedly and significantly different between extensive metaboliz- ers, poor metabolizers, and extensive metabolizers given quinidine (Table I; Figs. 2 and 3).
Extensive metabolizers versus poor metabolizers. The C Inax, AUC, and t,,, values for dextromethorphan were significantly (p < 0.005) larger in the poor metabolizer than in the extensive metabolizer group by an average of 16-fold (C,,), 150-fold (AUC), and eightfold (tr,J. In poor metabolizers, the C,,

CLINICAL PHARMA COLOGY &THERAPEUTICS VOLUME 60, NUMBER 3 Capon et al. 299
Table I. Pharmacokinetic data of dextromethorphan and metabolites in six extensive metabolizers (EM), six poor metabolizers (PM), and in six EM given quinidine (EM + quinidine) from a single oral dose of 30 mg dextromethorphan hydrobromide
Dextromethorphan EM PM EM + quinidine Significance*
Dextrorphan (total) EM PM EM + quinidine Significance
3-HyJroqnorphinan (total) EM PM EM + quinidine Significance
34fethoxymorphinan EM PM EM + quinidine Significance
1.4 (0.7-13.7) 23.0 (18.5-25.7) 24.9 (16.6-40.9)
a3, b4
396 (250-722) 1.7 (1.0-2.1) 9.1 (6.7-12.4) 4.0 (2.0-4.0)
113 (37-129) 2.5 (2.0-4.0) a3, b4, c3 a3, bl
104 (83-135) 4.6 (2.2-5.3)
50 (19-97) a3, b4, c3
2.5 (1.6-3.0) 4.0 (4-12) 3.0 (3.0-4.0) a3, b2, c3
0 (O-0.5) 2.5 (1.6-3.2) 0.8 (O-1.3) a3, b2, c3
0 (O-2.0) 60 (48-97) 4.1 (O-23) a3, b2, c3
2.4 (1.0-4.2) 4.0 (4.0-8.1) 3.0 (1.5-4.0)
a2, c2
9.0 (1.2-160) 1362 (1126-1605) 383 (200-1214)
a3, b4, c3
1701(1420-5650) 198 (146-239)
1669 (1242-5852) a3, c3
648 (621-984) 213 (20-433) 873 (693-1092)
a3, b3, c3
0 (O-2.9) 256 (171-609)
13.8 (O-69) a3, b2, c3
2.4 (1.3-4.0) 19.1 (17.2-22.3) 5.6 (3.8-6.6)
a3, c3
1.4 (1.2-1.9) 10.1 (4.6-13.3) 6.6 (3.6-13.6)
a3, b4
2.0 (1.1-3.3) 20 (3-74) 6.4 (3.7-17.1)
a2, b4, c2
Data are median values; range is given in parentheses. C lllaXl Peak plasma concentration; t,,, time to reach C,,; AUC (O-m), area under the plasma concentration-time curve from zero to infinity; t,,,, half-life. *a, EM versus PM; b, EM versus EM + quinidine; c, PM versus EM + quinidine; 1, p < 0.05; 2, p < 0.01; 3, p < 0.005; 4, p i 0.001. tInsufficient data to calculate tl,z
and AUC values for dextrorphan (conjugated plus unconjugated) were significantly reduced: (C,,, 44- fold and AUC, 8.6-fold) compared with extensive metabolizers (p < 0.005) whereas the terminal t,,, was increased sevenfold (p < 0.005) and t,, was prolonged by an average of 2 hours. For plasma concentrations of 3-hydroxymorphinan (conjugated plus unconjugated), in poor metabolizers the C,, (23-fold) and AUC (threefold) values were signifi- cantly (p < 0.005) reduced, whereas the terminal t,,, was 10 times longer (p < 0.01). Plasma concentra- tions of 3-methoxymorphinan could be quantified in only one extensive metabolizer subject, whereas in all poor metabolizer subjects it was readily quanti- fied and hence the AUC and Cm, values were significantly (p < 0.005) greater in the poor metabo- lizers.
Extensive metabolizers versus extensive metabolizers given quinidine. The Cm, and AUC values for dex- tromethorphan were significantly (p < 0.001) larger in the extensive metabolizer group when quinidine was coadministered by an average of B-fold (Cm,) and 43-fold (AUC), respectively. The Cm, values for dextrorphan (conjugated plus unconjugated) were reduced 3.5-fold (p < O.OOl), and the terminal
t,,, values were fivefold longer (p < 0.001). There was no difference in AUC values. For plasma con- centrations of 3-hydroxymorphinan, quinidine sig- nificantly reduced the C,, twofold (p < O.OOl), increased the AUC 1.3-fold, and prolonged the t 1,2 threefold. Plasma concentrations of 3- methoxymorphinan could be quantified in only one extensive metabolizer subject, whereas when quinidine was coadministered it was quantified in five of the six subjects and hence the AUC and C max values were significantly (p < 0.01) greater.
Extensive metabolizers given quinidine versus poor metabolizers. The t,,, AUC, and tin values for dex- tromethorphan were significantly (p < 0.01) larger in the poor metabolizers by an average of 1 hour (tmZ), fourfold (AUC), and threefold (t&, respec- tively, than in the extensive metabolizer group co- administered quinidine. The Cm, and AUC values for dextrorphan (conjugated and unconjugated) were 1Zfold (Cm,) and eightfold (AUC) higher, respectively, in the extensive metabolizer group co- administered quinidine @ < 0.005) compared with the poor metabolizers. There was no difference in the t,,, between the two groups. For 3-hydroxymorphinan, C,, and AUC values were
300 Cuponetal. CLINICAL PHARMA COLOGY &THERAPEUTICS
SEPTEMBER 1996
+ Dextromethorphan - EM
+ Quinidine + Dextromethorphan - EM
0.1 1 I I I I I I 1 0 25 50 75 100 125 150 175
Time (hr)
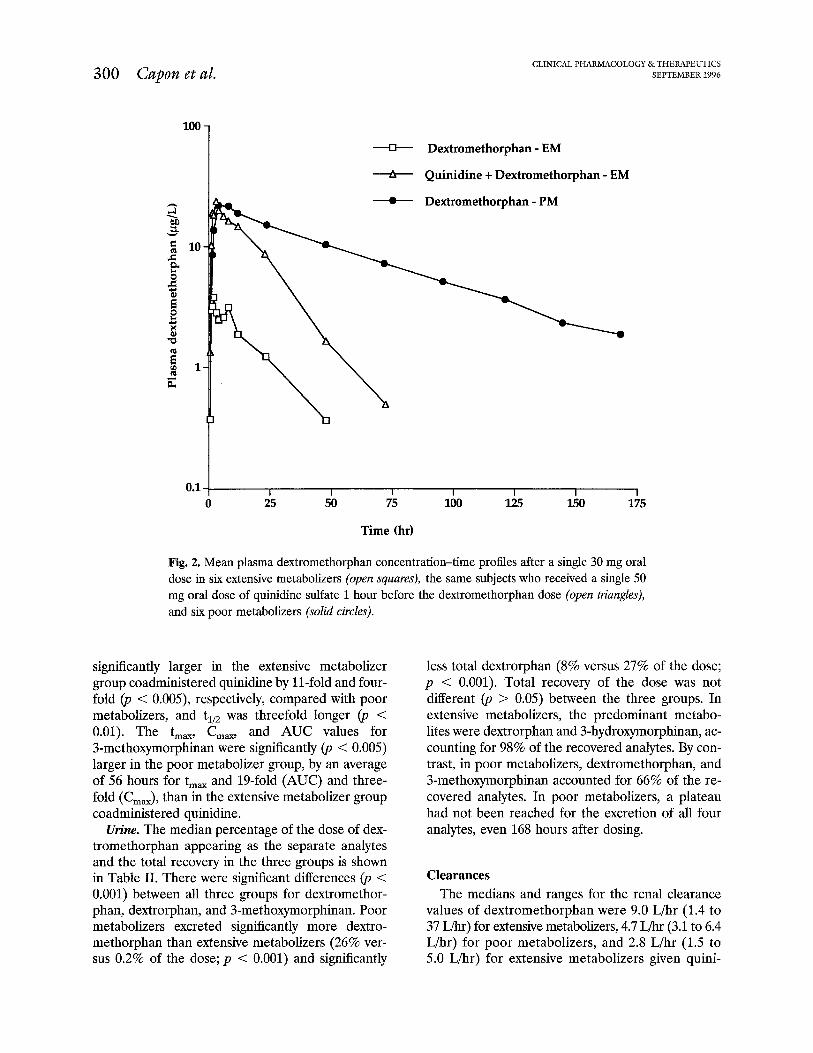
Fig. 2. Mean plasma dextromethorphan concentration-time profiles after a single 30 mg oral dose in six extensive metabolizers (open squares), the same subjects who received a single 50 mg oral dose of quinidine sulfate 1 hour before the dextromethorphan dose (open triangles), and six poor metabolizers {solid circles).
significantly larger in the extensive metabolizer group coadministered quinidine by 11-fold and four- fold (‘JJ < O.OOS), respectively, compared with poor metabolizers, and t,,, was threefold longer (p < 0.01). The t,,, C,,, and AUC values for 3-methoxymorphinan were significantly (p < 0.005) larger in the poor metabolizer group, by an average of 56 hours for t,, and 19-fold (AUC) and three- fold (C,,), than in the extensive metabolizer group coadministered quinidine.
Urine. The median percentage of the dose of dex- tromethorphan appearing as the separate analytes and the total recovery in the three groups is shown in Table II. There were significant differences (p < 0.001) between all three groups for dextromethor- phan, dextrorphan, and 3-methoxymorphinan. Poor metabolizers excreted significantly more dextro- methorphan than extensive metabolizers (26% ver- sus 0.2% of the dose; p < 0.001) and significantly
less total dextrorphan (8% versus 27% of the dose; p < 0.001). Total recovery of the dose was not different (p > 0.05) between the three groups. In extensive metabolizers, the predominant metabo- lites were dextrorphan and 3-hydroxymorphinan, ac- counting for 98% of the recovered analytes. By con- trast, in poor metabolizers, dextromethorphan, and 3-methoxymorphinan accounted for 66% of the re- covered analytes. In poor metabolizers, a plateau had not been reached for the excretion of all four analytes, even 168 hours after dosing.
Clearances The medians and ranges for the renal clearance
values of dextromethorphan were 9.0 L/hr (1.4 to 37 L/hr) for extensive metabolizers, 4.7 L/hr (3.1 to 6.4 L/hr) for poor metabolizers, and 2.8 L/hr (1.5 to 5.0 Whr) for extensive metabolizers given quini-
CLINICAL PHARMA COLOGY & THERAPEUTICS VOLUME 60, NUMBER 3 Capon et al. 301
Dextromethorphan - EM
Quinidine + Dextromethorphan - EM
Dextromethorphan - PM
Time (hr)
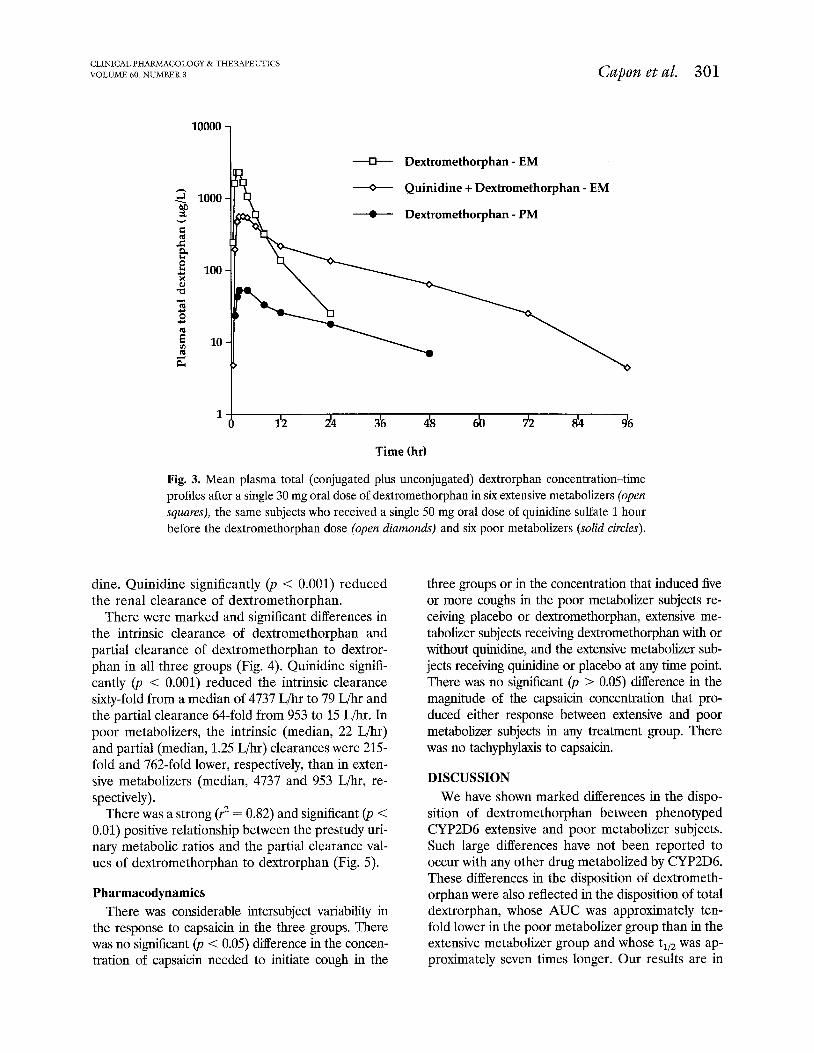
Fig. 3. Mean plasma total (conjugated plus unconjugated) dextrorphan concentration-time profiles after a single 30 mg oral dose of dextromethorphan in six extensive metabolizers (open squares), the same subjects who received a single 50 mg oral dose of quinidine sulfate 1 hour before the dextromethorphan dose (open diamonds) and six poor metabolizers (solid circles).
dine. Quinidine significantly (p < 0.001) reduced the renal clearance of dextromethorphan.
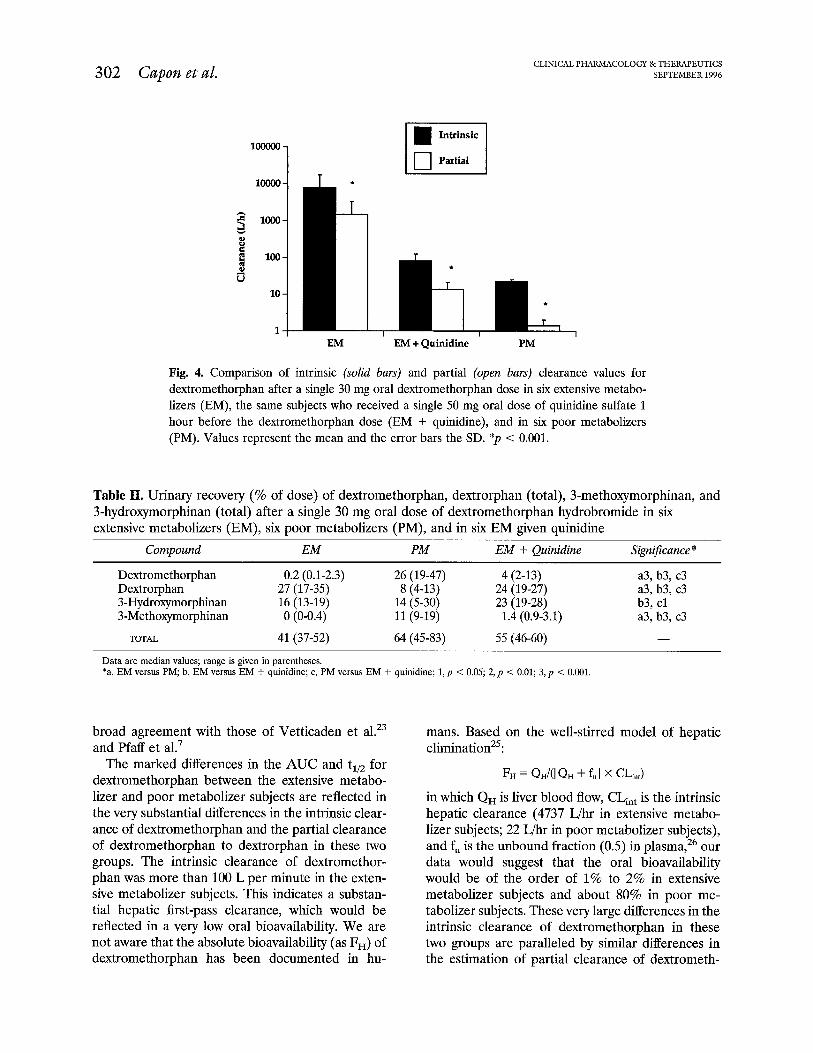
There were marked and significant differences in the intrinsic clearance of dextromethorphan and partial clearance of dextromethorphan to dextror- phan in all three groups (Fig. 4). Quinidine signifi- cantly (p < 0.001) reduced the intrinsic clearance sixty-fold from a median of 4737 L/hr to 79 L/hr and the partial clearance 64-fold from 953 to 15 L/hr. In poor metabolizers, the intrinsic (median, 22 L/hr) and partial (median, 1.25 L/hr) clearances were 215 fold and 762-fold lower, respectively, than in exten- sive metabolizers (median, 4737 and 953 L/hr, re- spectively).
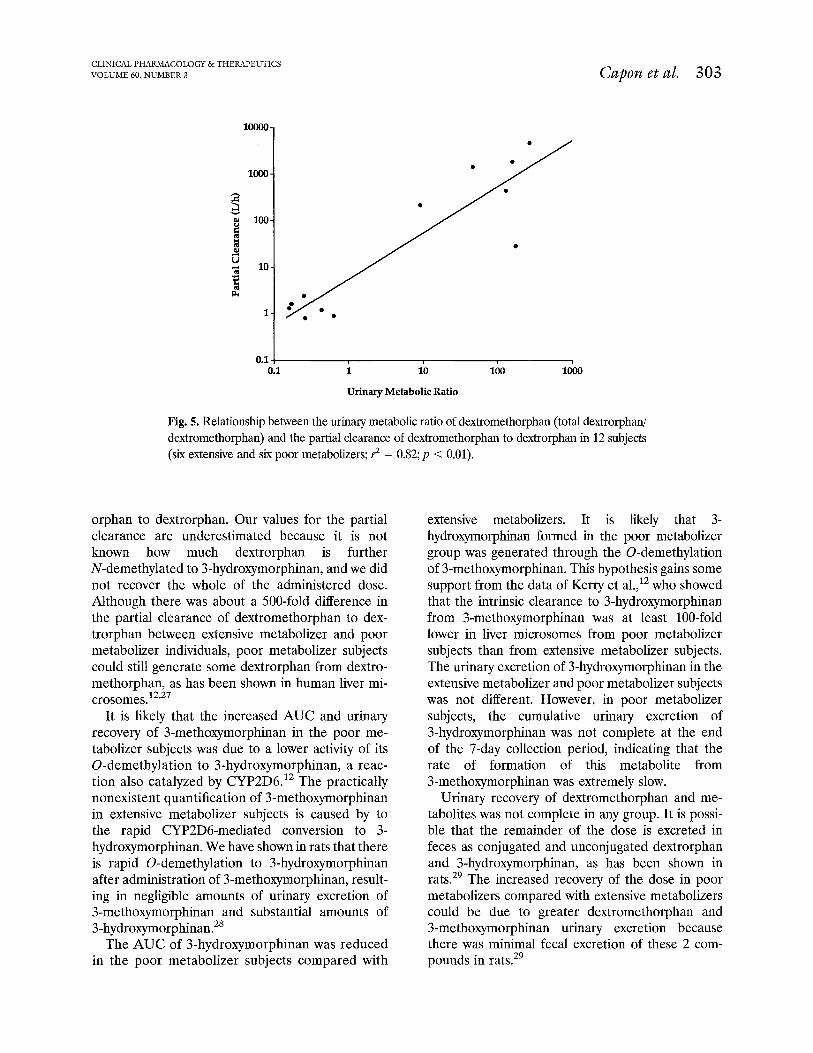
There was a strong (3 = 0.82) and significant (JJ < 0.01) positive relationship between the prestudy uri- nary metabolic ratios and the partial clearance val- ues of dextromethorphan to dextrorphan (Fig. 5).
Pharmacodynamics There was considerable intersubject variability in
the response to capsaicin in the three groups. There was no significant (p < 0.05) difference in the concen- tration of capsaicin needed to initiate cough in the
three groups or in the concentration that induced five or more coughs in the poor metabolizer subjects re- ceiving placebo or dextromethorphan, extensive me- tabolizer subjects receiving dextromethorphan with or without quinidine, and the extensive metabolizer sub- jects receiving quinidine or placebo at any time point. There was no significant (p > 0.05) difference in the magnitude of the capsaicin concentration that pro- duced either response between extensive and poor metabolizer subjects in any treatment group. There was no tachyphylaxis to capsaicin.
DISCUSSION
We have shown marked differences in the dispo- sition of dextromethorphan between phenotyped CYP2D6 extensive and poor metabolizer subjects. Such large differences have not been reported to occur with any other drug metabolized by CYP2D6. These differences in the disposition of dextrometh- orphan were also reflected in the disposition of total dextrorphan, whose AUC was approximately ten- fold lower in the poor metabolizer group than in the extensive metabolizer group and whose t,,, was ap- proximately seven times longer. Our results are in
302 Cuponetal. CLINICAL P HARMACOLOGY & THERAPEUTICS
SEPTEMBER 1996 * iI
EM
Intrinsic rl 0 Partial
l I L--k EM + Quinidiie PM
Fig. 4. Comparison of intrinsic (solid bars) and partial (open bars) clearance values for dextromethorphan after a single 30 mg oral dextromethorphan dose in six extensive metabo- lizers (EM), the same subjects who received a single 50 mg oral dose of quinidine sulfate 1 hour before the dextromethorphan dose (EM + quinidine), and in six poor metabolizers (PM). Values represent the mean and the error bars the SD. *p < 0.001.
Table II. Urinary recovery (% of dose) of dextromethorphan, dextrorphan (total), 3-methoxymorphinan, and 3-hydroxymorphinan (total) after a single 30 mg oral dose of dextromethorphan hydrobromide in six extensive metabolizers (EM), six poor metabolizers (PM), and in six EM given quinidine
Compound EM PM EM f Quinidine Significance *
Dextromethorphan Dextrorphan 3-Hydroxymorphinan 3-Methoxymorphinan
TOTAL
0.2 (0.1-2.3) 27 (17-35) 16 (13-19) 0 (O-0.4)
41 (37-52)
26 (19-47) 4 (2-13) a3, b3, c3 8 (4-13) 24 (19-27) a3, b3, c3
14 (5-30) 23 (19-28) b3, cl 11 (9-19) 1.4 (0.9-3.1) a3, b3, c3 64 (45-83) 55 (46-60) -
Data are median values; range is given in parentheses. *a, EM versus PM, b, EM versus EM + quinidine; c, PM versus EM + quinidine; 1, p < 0.05; 2, p < 0.01; 3, p < 0.001.
broad agreement with those of Vetticaden et a1.23 and Pfaff et a1.7
mans. Based on the well-stirred model of hepatic elimination25:
The marked differences in the AUC and t,,, for dextromethorphan between the extensive metabo- lizer and poor metabolizer subjects are reflected in the very substantial differences in the intrinsic clear- ance of dextromethorphan and the partial clearance of dextromethorphan to dextrorphan in these two groups. The intrinsic clearance of dextromethor- phan was more than 100 L per minute in the exten- sive metabolizer subjects. This indicates a substan- tial hepatic first-pass clearance, which would be reflected in a very low oral bioavailability. We are not aware that the absolute bioavailability (as FH) of dextromethorphan has been documented in hu-
FH = Qd([QH + ful x cbn,)
in which QH is liver blood flow, CL,,, is the intrinsic hepatic clearance (4737 L/hr in extensive metabo- lizer subjects; 22 L/hr in poor metabolizer subjects), and f, is the unbound fraction (0.5) in plasma,26 our data would suggest that the oral bioavailability would be of the order of 1% to 2% in extensive metabolizer subjects and about 80% in poor me- tabolizer subjects. These very large differences in the intrinsic clearance of dextromethorphan in these two groups are paralleled by similar differences in the estimation of partial clearance of dextrometh-
CLINICAL PHABMACOLOGY &THERAPEUTICS VOLUME 60, NUMBER 3 Capon etal. 303
Urinary Metabolic Ratio
Fig. 5. Relationship between the urinary metabolic ratio of dextromethorphan (total dextrorphw dextromethorphan) and the partial clearance of dextromethorphan to de&orphan in 12 subjects (six extensive and six poor metabolizers; ? = 0.82; p < 0.01).
orphan to dextrorphan. Our values for the partial clearance are underestimated because it is not known how much dextrorphan is further N-demethylated to 3-hydroxymorphinan, and we did not recover the whole of the administered dose. Although there was about a 500-fold difference in the partial clearance of dextromethorphan to dex- trorphan between extensive metabolizer and poor metabolizer individuals, poor metabolizer subjects could still generate some dextrorphan from dextro- methorphan, as has been shown in human liver mi- crosomes.12,27
It is likely that the increased AUC and urinary recovery of 3-methoxymorphinan in the poor me- tabolizer subjects was due to a lower activity of its 0-demethylation to 3-hydroxymorphinan, a reac- tion also catalyzed by CYP2D6.r2 The practically nonexistent quantification of 3-methoxymorphinan in extensive metabolizer subjects is caused by to the rapid CYP2D6-mediated conversion to 3- hydroxymorphinan. We have shown in rats that there is rapid 0-demethylation to 3-hydroxymorphinan after administration of 3-methoxymorphinan, result- ing in negligible amounts of urinary excretion of 3-methoxymorphinan and substantial amounts of 3-hydroxymorphinan.28
The AUC of 3-hydroxymorphinan was reduced in the poor metabolizer subjects compared with
extensive metabolizers. It is likely that 3- hydroxymorphinan formed in the poor metabolizer group was generated through the 0-demethylation of 3-methoxymorphinan. This hypothesis gains some support from the data of Kerry et a1.,12 who showed that the intrinsic clearance to 3-hydroxymorphinan from 3-methoxymorphinan was at least loo-fold lower in liver microsomes from poor metabolizer subjects than from extensive metabolizer subjects. The urinary excretion of 3-hydroxymorphinan in the extensive metabolizer and poor metabolizer subjects was not different. However, in poor metabolizer subjects, the cumulative urinary excretion of 3-hydroxymorphinan was not complete at the end of the 7-day collection period, indicating that the rate of formation of this metabolite from 3-methoxymorphinan was extremely slow.
Urinary recovery of dextromethorphan and me- tabolites was not complete in any group. It is possi- ble that the remainder of the dose is excreted in feces as conjugated and unconjugated dextrorphan and 3-hydroxymorphinan, as has been shown in rats.29 The increased recovery of the dose in poor metabolizers compared with extensive metabolizers could be due to greater dextromethorphan and 3-methoxymorphinan urinary excretion because there was minimal fecal excretion of these 2 com- pounds in rats.29

304 Capon etal. CLINICAL I’ HARMACOLOGY &THERAPEUTICS
SEPTEMBER 1996
Although quinidine is not a substrate for CYP2D6,30 it is a very potent inhibitor of the oxidative metabolism of a number of substrates that are catalyzed by this enzyme.16,17,20,21,31”8 We have confirmed that a single 50 mg oral dose of quinidine was able to partially inhibit the for- mation of dextrorphan in the extensive metabo- lizer group, although not to values that character- ized the poor metabolizer subjects. These findings are at variance with those of others, who were able to change the phenotype to that characteristic of poor metabolizers in the majority of subjects with use of substrates debrisoquin,35”7 sparteine,17,33S34 encainide,38 metoprolol,20 and dextromethorphan. 21 The doses of quinidine in these studies varied from 5 to 800 mg daily. Prob- able reasons for the different observations be- tween our results and those of others are that the Michaelis-Menten constant values for sparteine (50 pmol/L3’), debrisoquin (120 pmol/L4’), and metoprolol (40 to 70 pmol/L41) are substantially higher than the value for dextromethorphan (5 pmol/L12).
The strong correlation between the partial clearance of dextromethorphan to dextrorphan and the prestudy metabolic ratio was not unex- pected, but to our knowledge it has not been previously documented. Our results provide the experimental justification for the use of the uri- nary metabolic ratio as an index of functional CYP2D6 activity in humans.
The most surprising and unexpected finding of this study was that dextromethorphan appeared to be inactive as an antitussive agent, even in the presence of very high and sustained concentra- tions of the drug in the poor metabolizer and quinidine-treated groups. There is ample evi- dence for efficacy of dextromethorphan in patients with spontaneous cough.‘-’ However, evidence for the antitussive effect of dextromethorphan in ex- perimentally induced cough is much less convinc- ing. The two most frequently used cough chal- lenges in the past 30 years have been inhaled citric acid and capsaicin. With citric acid, an antitussive effect has been reported by some researchers42-45 but not by others. 46,47 We are aware of only one study in which capsaicin was used to evaluate the effect of dextromethorphan, and in that report dextromethorphan (30 mg) suppressed cough for 150 minutes after dosing.48 Thus we would have expected that a measurable and prolonged effect
should have occurred in the poor metabolizer subjects and a lesser effect in the extensive me- tabolizers.
An explanation for our results is that experi- mental cough challenges, and in particular capsa- icin, might be inappropriate to evaluate antitus- sive drugs because they do not necessarily mimic spontaneous cough. There is a large intersubject variability in the responsiveness to both inhaled citric acid47,4g-51 and capsaicin,52-53 and up to 50% of subjects are either totally or partly insensitive to the tussive effect of citric acid.47,50,51 Although nonresponders to capsaicin have not been re- ported, the large interindividual variability may have contributed to our negative findings, and it may be appropriate to include only those subjects who are the most responsive to this challenge in future studies in an attempt to reduce the large variability.
Capsaicin was chosen because citric acid has been associated with the development of tachy- phylaxis49,51 in the short term (minutes after chal- lenge), a property not shared by capsaicin.52,54S55 The site of the tussive effect of capsaicin in the respiratory tract is not clear. The larynx has been suggested as the main site of stimulation for in- haled capsaicin,52 which acts on C-fiber endings,52 whereas citric acid is thought to also act in the larynx49 through rapidly adapting receptors.56,57 The notion that C-fiber stimulation causes cough has been challenged,58 and it has been suggested that pulmonary C-fiber stimulation by capsaicin in cats actually exerts an inhibitory influence on cough.5g Thus the role of capsaicin as a reliable experimental cough challenge in humans in more prolonged studies is not yet clear, and we would urge caution in the use of this agent until some of these doubts have been resolved.
The role of genetic polymorphism in drug me- tabolism on the pharmacologic effect of a drug could be clinically very important. This will de- pend on whether only the parent drug is active, as has been reported with debrisoquin6’; if only a metabolite is active, as occurs with codeine61,62; or if both the parent compound and a metabolite are active, as has been described in the guinea pig with dextromethorphan.63 This question may be able to be answered only in patients with chronic stable cough, and such a study would be difficult to implement because large numbers of patients would need to be recruited to obtain even a small

CLINICAL P HARMACOLOGY &THERAPEUTICS VOLUME 60, NUMBER 3
number of poor metabolizers. A valid and repro- ducible experimental cough challenge would need to be developed before this study is repeated in healthy subjects.
References 1.
2.
3.
4.
5.
6.
7.
8.
9.
Cass LJ, Frederik WS. Evaluation of a new antitussive agent. N Engl J Med 1953;249:132-6. Cass LJ, Frederik WS, Andosca JB. Quantitative comparison of dextromethorphan hydrobromide and codeine. Am J Med Sci 1954;227:291-6. Ralph N. Evaluation of a new cough suppressant. Am J Med Sci 1954;227:297-303. Matthys H, Bleicher B, Bleicher U. Dextromethor- phan and codeine: objective assessment of antitussive activity in patients with chronic cough. J Int Med Res 1983;11:92-100. Aylward M, Maddock J, Davies D, Protheroe D, Leideman T. Dextromethorphan and codeine: com- parison of plasma kinetics and antitussive effects. Eur J Resp Dis 1984;65:283-91. Barnhart JW. The urinary excretion of dextrometh- orphan and three metabolites in dogs and humans. Toxic01 Appl Pharmacol 1980$5:43-g. Pfaff G, Briegel P, Lamprecht I. Inter-individual vari- ation in the metabolism of dextromethorphan. Int J Pharm 1983;14:173-89. Schmid B, Bircher J, Preisig R, Kiipfer A. Polymor- phic dextromethorphan metabolism: co-segregation of oxidative 0-demethylation with debrisoquin hy- droxylation. Clin Pharmacol Ther 1985;38:618-24. Kiipfer A, Schmid B, Pfaff G. Pharmacogenetics of dextromethorphan 0-demethylation in man. Xenobi- otica 1986;16:421-33.
10. Larrey D, Amouyal G, Tine1 M, Letteron P, Berson A, Labbe G, et al. Polymorphism of dextromethor- phan oxidation in a French population. Br J Clin Pharmacol 1987;24:676-9.
11. Henthorn TK, Benitez J, Avram MJ, Martinez C, LLerena A, Cobaleda J, et al. Assessment of the debrisoquin and dextromethorphan phenotyping tests by gaussian mixture distributions analysis. Clin Phar- macol Ther 1989;45:328-33.
12. Kerry NL, Somogyi AA, Bochner F, Mikus G. The role of CYP2D6 in primary and secondary oxidative metabolism of dextromethorphan: in vitro studies us- ing human liver microsomes. Br J Clin Pharmacol 1994;38:243-8.
13. Eichelbaum M, Gross AS. The genetic polymorphism of debrisoquine/sparteine metabolism-clinical as- pects. Pharmacol Ther 1990;46:377-94.
14. Meyer UA, Skoda RC, Zanger UM. The genetic poly- morphism of debrisoquine/sparteine metabolism-
1.5.
16.
17.
18.
19.
20.
21.
22.
23.
Capon etal. 305
molecular mechanisms. Pharmacol Ther 1990;46:297- 308. Gonzalez FJ, Meyer UA. Molecular genetics of the debrisoquine-sparteine polymorphism. Clin Pharma- co1 Ther 1991;50:233-8. Otton SV, Inaba T, Kalow W. Competitive inhibition of sparteine oxidation in human liver by beta- adrenoceptor antagonists and other cardiovascular drugs. Life Sci 1984;34:73-80. Nielsen MD, Brersen K, Gram LF. A dose-effect study of the in vivo inhibitory effect of quinidine on sparteine oxidation in man. Br J Clin Pharmacol 1990; 29:299-304. Broly F, Vandamme N, Caron J, Libersa C, Lhermitte M. Single dose quinidine treatment inhibits mexil- etine oxidation in extensive metabolisers of debriso- quine. Life Sci 1991;48:123-8. Inaba T, Otton SV, Kalow W. Deficient metabolism of debrisoquine and sparteine. Clin Pharmacol Ther 1980;27:547-9. Leeman T, Dayer P, Meyer UA. Single dose quinidine treatment inhibits metoprolol oxidation in extensive metabolisers. Eur J Clin Pharmacol 1986;29:739-41. Zhang Y, Britto MR, Valderhaug KL, Wedlund PJ, Smith RA. Dextromethorphan: enhancing its systemic availability by way of low-dose quinidine-mediated inhibition of cytochrome P4502D6. Clin Pharmacol Ther 1992;51:647-55. Chen ZR, Somogyi AA, Bochner F. Dextromethor- phan: pharmacogenetics and a pilot study to deter- mine its disposition and antitussive effects in poor and extensive metabolisers. Eur J Clin Pharmacol 1990; 183:1573-4. Vetticaden SJ, Cabana BE, Prasad VK, Purich ED, Jo&man JHJ, Zeeuw R, et al. Phenotypic ditferences in dextromethorphan metabolism. Pharm Res 1989;6:13-9.
24. Chen ZR, Somogyi AA, Bochner F. Simultaneous determination of dextromethorphan and three me- tabolites in plasma and urine using high- performance liquid chromatography with applica- tion to their disposition in man. Ther Drug Monit 1990;12:97-104.
25. Rowland M, Tozer TN. Clinical Pharmacokinetics: concepts and applications. 2nd ed. Philadelphia: Lea & Febiger, 1989.
26. Kalant H, Roschlau WHE. Principles of medical pharmacology. 5th ed. Toronto: BC Decker, 1989:750.
27. Dayer P, Leeman T, Striberni R. Dextromethorphan 0-demethylation in liver microsomes as a prototype reaction to monitor cytochrome P-450 db, activity. Clin Pharmacol Ther 1989;45:34-40.
28. Bochner F, Somogyi AA, Chen ZR. Dextromethor- phan metabolism in rat: interstrain differences and the fate of individually administered oxidative metab- olites. Xenobiotica 1994;24:543-52.

306 Caponetal. CLINICAL PHARMA COLOGY & THERAPEUTICS
SEPTEMBER 1996
29. Kamm JJ, Taddeo AB, van Loon EJ. Metabolism and excretion of tritiated dextromethorphan by the rat. J Pharmacol Exp Ther 1967;158:437-44.
30. Otton SV, Brinn R, Gram LF. In vitro evidence against the oxidation of quinidine by the sparteinel debrisoquine monooxygenase of the liver. Drug Metab Dispos 1988;16:15-7.
31. Otton SV, Inaba T, Kalow W. Inhibition of sparteine oxidation in human liver by tricyclic antidepressants and other drugs. Life Sci 1983;32:795-800.
32. Guengerich FP, Muller-Enoch D, Blair IA. Oxidation of quinidine by human liver cytochrome P450. Mol Pharmacol 1986;30:287-95.
33. Brirm R, Brosen K, Gram LF, Haghfelt T, Otton SV. Sparteine oxidation is practically abolished in quinidine treated patients. Br J Clin Pharmacol 1986;22:194-7.
34. Inaba T, Tyndale RE, Mahon WA. Quinidine: potent inhibition of sparteine and debrisoquine oxidation in vivo. Br J Clin Pharmacol 1986;22:199-200.
35. Speirs CJ, Murray S, Boobis AR, Seddon CE, Davies DS. Quinidine and the identification of drugs whose elimination is impaired in subjects classified as poor metabolisers of debrisoquine. Br J Clin Pharmacol 1986;22:739-43.
36. Bresen K, Gram LF, Haghfelt T, Bertilsson L. Exten- sive metabolisers of debrisoquine become poor me- tabolisers during quinidine treatment. Pharmacol Toxic01 1987;60:312-4.
37. Kobayashi S, Speirs CJ, Watson D, Murray S, Sesardic D, Boobis AR. Time course of inhibition by quinidine of debrisoquine 4-hydroxylase activity in man. Br J Clin Pharmacol 1988;25:673P-4P.
38. Funck-Brentano C, Turgeon J, Woosley RL, Roden DM. Effect of low dose quinidine on encainide phar- macokinetics and pharmacodynamics. Influence of ge- netic polymorphism. J Pharmacol Exp Ther 1989;249: 134-42.
39. Eichelbaum M, Reetz KP, Schmidt EK, Zekorn C. The genetic polymorphism of sparteine metabolism. Xenobiotica 1986;16:465-81.
40. Boobis AR, Murray S, Kahn GC, Robertz GM, Davies DS. Substrate specificity of the form of cyto- chrome P-450 catalyzing the 4-hydroxylation of de- brisoquine in man. Mol Pharmacol 1983;23:474-81.
41. Otton SV, Crewe HK, Lennard MS, Tucker GT, Woods HF. Use of quinidine inhibition to define the role of the sparteine/debrisoquine cytochrome P450 in metoprolol oxidation by human liver chromosomes. J Pharmacol Exp Ther 1988;247:242-7.
42. Bickerman HA, German E, Cohen BM, Itkin SE. The cough response of healthy human subjects stimulated by citric acid aerosol; part II: evaluation of antitussive agents. Am J Med Sci 1957;234:191-205.
43. Calesnick B, Christensen JA. Latency of cough re- sponse as a measure of antitussive agents. Clin Phar- macol Ther 1966;8:374-80.
44. Karttunen P, Tukiainen H, Silvasti M, Kolonen S. Antitussive effect of dextromethorphan and dextromethorphan-salbutamol combination in healthy volunteers with artificially induced cough. Respiration 1987;52:49-53.
45. Grattan TJ, Marshall AE, Higgins KS, Morice AH. The effect of inhaled and oral dextromethorphan on citric acid induced cough in man. Br J Clin Pharmacol 1995;39:261-3.
46. Gravenstein JS, Devloo RA, Beecher HK. Effect of antitussive agents on experimental and pathological cough in man. J Appl Physiol 1954;7:119-39.
47. Empey DW, Laitinen LA, Young GA, Bye CE, Hughes DTD. Comparison of the antitussive effects of codeine phosphate 20 mg, dextromethorphan 30 mg, and noscapine 30 mg using citric acid-induced cough in normal subjects. Eur J Clin Pharmacol 1979;16:393-7.
48. Fuller RW, Haase G, Choudry NB. The effects of dextromethorphan cough syrup on capsaicin-induced cough in normal volunteers [abstract]. Am Rev Resp Dis 1989;A11:739.
49. Barros MJ, Zammattio SL, Rees PJ. Effect of changes in inspiratory flow rate on cough responses to inhaled capsaicin. Clin Sci 1991;81:539-42.
50. Rees PJT, Clark JH. Assessment of antitussive ef- fects by citric acid threshold. Br J Dis Chest 1983; 77:94-7.
51. Pounsford JC, Birch MJ, Saunders KB. Effect of bron- chodilators on the cough response to inhaled citric acid in normal and asthmatic subjects. Thorax 1985; 40:662-7.
52. Collier JG, Fuller RW. Capsaicin inhalation in man and the effects of sodium cromoglycate. Br J Pharma- co1 1984;81:113-7.
53. Fuller RW, Choudry NB. Increased cough reflex as- sociated with angiotensin converting enzyme inhibitor cough. Br Med J 1987;295:1025-6.
54. Fuller RW, Dixon CMS, Barnes PJ. Bronchoconstric- tor response to inhaled capsaicin in humans. J Appl Physiol 1985;58:1080-4.
55. Fuller RW, Karlsson JA, Choudry NB, Pride NB. Effect of inhaled and systemic opiates on responses to inhaled capsaicin in humans. J Appl Physiol 1988;65: 1125-30.
56. Widdicombe JG. Receptors in the trachea and bron- chi of the cat. J Physiol 1954;123:71-104.
57. Lowry RH, Wood AM, Higenbottam TW. Effects of pH and osmolarity on aerosol-induced cough in nor- mal volunteers. Clin Sci 1988;74:373-6.
58. Adcock JJ. Peripheral opioid receptors and the cough reflex. Resp Med 1991;85 (suppl A):43-6.
59. Tatar M, Webber SE, Widdicombe JG. Lung C-fibre activation and defensive reflexes in anaesthetized cats. J Physiol 1988;402:411-20.
60. Idle JR, Mahgoub A, Lancaster R, Smith RL. Hypo-

CLINICAL PHABMA COLOGY &THERAPEUTICS VOLUME 60, NUMBER 3 Cupor, et al. 307
tensive response to debrisoquine and hydroxylation thresholds to copper vapor laser stimuli in extensive phenotype. Life Sci 1978;22:979-84. but not poor metabolizers of sparteine. Clin Pharma-
61. Desmeules J, Gascon MP, Dayer P, Magistris M. Impact co1 Ther 1990;48:686-93. of environmental and genetic factors on codeine anal- 63. Braga PC, Fossati A, Vimercati MG, Caputo R, Guf- gesia. Eur J Clin Pharmacol 1991;41:23-6. fanti EE. Dextrorphan and dextromethorphan: com-
62. Sindrup SH, Brosen K, Bjerring P, Arendt-Nielsen L, parative antitussive effects on guinea pigs. Drugs Exp Larsen U, Angelo HR, et al. Codeine increases pain Clin Res 1994;20:199-203.
01 N THE MOVE? . 8 * v.8 * . .‘ -- , *, 0 Don’t miss a single issue of the journal! To ensure prompt service when you change your address, please photocopy and complete the form below.
Please send your change of address notification at least six weeks before your move to ensure continued service. We regret we cannot guarantee replacement of issues missed due to late notification.
JOURNAL TITLE: Fill in the title of the journal here.
OLD ADDRESS: NEW ADDRESS: Affix the address label from a recent issue of the journal here. Clearly print your new address here.
Name
Address
City/State/ZIP
COPY AND MAIL THIS FORM TO: Journal Subscription Services Mosbv-Year Book, Inc. 11830’Westline Industrial Dr. St. Louis, MO 63146-3318
OR FAX TO: 314-432-1158
OR PHONE: l-800-453-4351 Outside the U.S., call 314-453-4351