the human genome at ten - lehigh universityinbios21/pdf/fall2010/marzillier_120310.pdf · the race...
TRANSCRIPT

The Human Genome at Ten
Jutta Marzillier, Ph.DLehigh University
Biological SciencesBiological Sciences
December 3rd , 2010

The Human Genome at 10 H did i h i d dHow did it start, how was it done, and
what is its Impact on Science and Medicine?
Jutta Marzillier, Ph.DLehigh University
Biological Sciencesg
December 3rd , 2010

10 years ago in June 2000 at the White House:First rough draft of the “book of life” announced
Clinton:Clinton:Completion of the Genome Project would“…revolutionize the diagnosis, preventionand treatment of most, if not all, human
Craig Venter Bill Clinton Francis Collins
diseases……”
Collins:Collins:…”personalized medicine is likely to emergeby the year 2010…..”

10 years ago at the White House:First rough draft of the “book of life” announced
2010 Th t 102010: The next 10 years
Craig Venter Bill Clinton Francis Collins

YearYearofof
2010 21 Jan 2010 11 Feb 2010 18 Feb 2010
……the genomic Era………..
4 Mar 20101 Apr 2010
29 Apr 2010 7 May 2010

O liOutline
•Rationale for Genome Sequencingq g
•Method of DNA sequencing
•DNA sequencing technologies
•Major insights and achievements
•What’s next?

Rationale: Why is the Knowledge about the Human Genome interesting?
http://www.genomenewsnetwork.org/articles/06_00/sequence_primer.shtml
The human body has about 100 trillion cells.Each cell harbors the same genetic information in its nucleus in form of
http://www.pharmainfo.net/files/images/stories/article_images/
Each cell harbors the same genetic information in its nucleus in form of DNA containing chromosomes.Depending on cellular, developmental, and functional stage of a cell only a subset of genes is expressed. g pModifications in the genetic information may result in disease.

Why is Genome Sequencing Important?
• To obtain a ‘blueprint’ – DNA directs all the instructions needed for cell development and function
• To study gene expression in a specific tissue, organ or tumorTo study gene expression in a specific tissue, organ or tumor• To study how humans relate to other organisms • DNA underlies almost every aspect of human health, both, in
function and dis-function•To find correlations how genome information relates to development
of cancer, susceptibility to certain diseases (personalized medicine) and drug metabolism (pharmacogenomics)
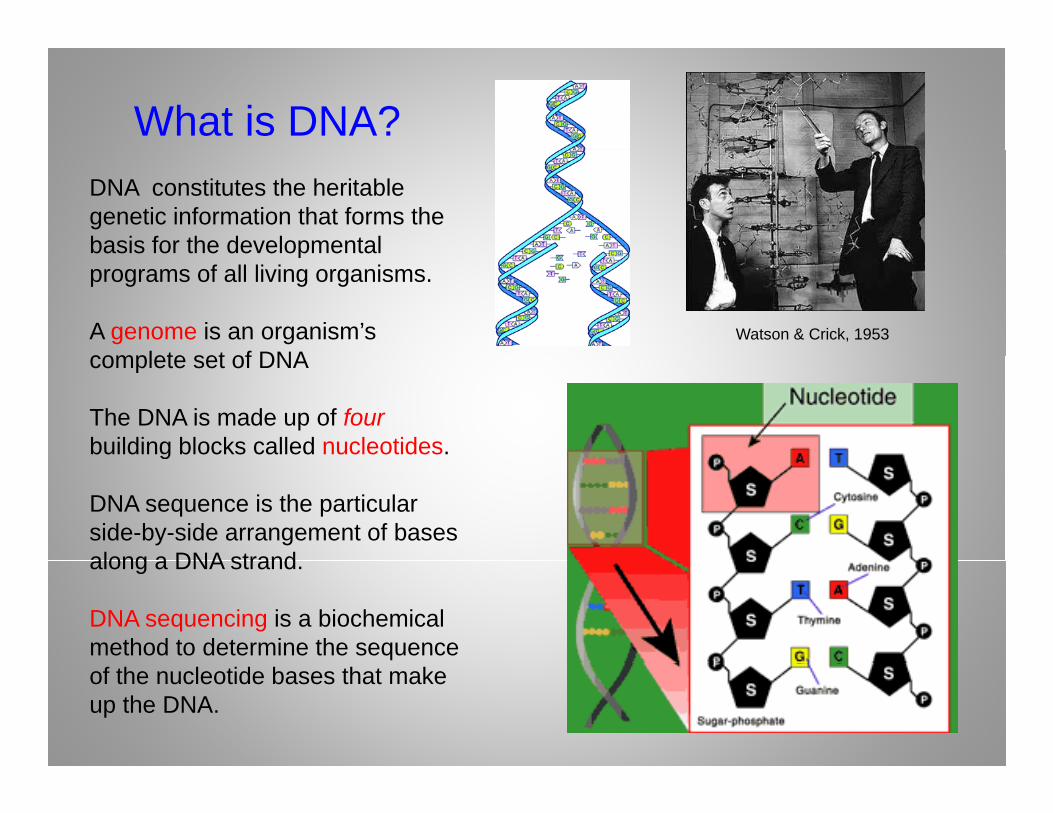
What is DNA?DNA constitutes the heritablegenetic information that forms the basis for the developmentalbasis for the developmental programs of all living organisms.
A genome is an organism’s complete set of DNA
Watson & Crick, 1953
complete set of DNA
The DNA is made up of fourbuilding blocks called nucleotides.g
DNA sequence is the particular side-by-side arrangement of basesalong a DNA strandalong a DNA strand.
DNA sequencing is a biochemical method to determine the sequence of the nucleotide bases that make up the DNA.

Method of DNA sequencing:Principle of DNA Synthesis
Arthur Kornberg demonstrated DNA replication in aArthur Kornberg demonstrated DNA replication in acell-free (in vitro) bacterial extract (Nobel prize, 1959)
Di d DNA l (P l1) t f ilit t DNA th i-Discovered DNA polymerase (Pol1) to facilitate DNA synthesis- Unraveled the mechanism of DNA synthesis
- building blocks (Adenine, Cytosine, Guanine, Thymine)g ( , y , , y )- a single DNA strand serves as a template- can only extend a pre-existing chain (primer)- a free 3’ Hydroxyl end is required
Watson et al., MGB, 2008

Reaction Mechanism for DNA synthesisReaction Mechanism for DNA synthesis
The 3’ hydroxyl group of the primer attacks the -phosphoryl group of the incoming nucleotide thereby forming a phosphodiester bond (SN2 reaction).nucleotide thereby forming a phosphodiester bond (SN2 reaction).
W t t l MBG 2008Watson et al., MBG, 2008

Did S iDideoxy Sequencing according to Sanger
Both nucleotide typescan be incorporated into
Frederick SangerNobel Prize (1980)
ca be co po ated togrowing DNA chain.
Presence of dideoxy-cytosine in growing chain blocks further additionf i i l tidof incoming nucleotides.
Watson et al., MBG, 2008

Dideoxy Method of Sequencing (Sanger, 1975)
‘Normal’ DNA synthesis:DNA strand as templatePrimerDeoxynucleotidesPolymerase enzymeUse several cycles to amplify
DNA sequencingDNA synthesis is carried out in
the presence of limitingthe presence of limiting amounts of dideoxyribonucleosidetriphosphates that results in chain termination
• Through chain termination• Through chain termination fragments of distinct sizes are generated that can be separated by gel electrophoresisOriginal method sed radio GCTACCTGCACCA
GCTACCTGCACCAGA
• Original method used radio-labeled primers or dideoxynucleotides GCTA
GCTACCTGCA
GCTACCTGCACCA

Dideoxy Method of Sequencing
Original method sedOriginal method sed radio labeled primerOriginal method usedOriginal method used radio-labeled primer or dideoxynucleotidesdideoxynucleotides
This method required four separate DNA This method required four separate DNA synthesis reactions to be separated by synthesis reactions to be separated by electrophoresis in four parallel laneselectrophoresis in four parallel laneselectrophoresis in four parallel lanes. electrophoresis in four parallel lanes. The gel needs to be dried, exposed to film, The gel needs to be dried, exposed to film, developed and manually read.developed and manually read.
Approx. Approx. 150150-- 300 300 bases read lengthbases read length
Summary – Sequencing Method Established
Improvements-Use of fluorescently labeled dideoxy-
- Need of four reactions in parallel - Heat labile polymerase- Use of radioactivity- Low resolution on gels
y ynucleotides
-One-lane electrophoresis-Introduction of capillary electrophoresis to
increase resolution (up to 1,000 ntes)g- Approx. 150 nucleotides read length- time consuming
( p )-Use of heat stable polymerase (Taq)- Automation

Fluorescently labeled ddNTPs used for uo esce t y abe ed dd s used osequencing reaction allow one lane analysis
ddNTPsddNTPs used for used for automated sequencing automated sequencing are labeled with are labeled with differentdifferent fluorescent fluorescent dyes representing each dyes representing each of the 4 basesof the 4 bases
dnaseq.genomics.com.cn/dnaseq/en/tech/images
Allows separation of DNA productsin same lane of gel

Capillary Gel Electrophoresis IncreasesCapillary Gel Electrophoresis Increases Resolution
Capillary gel electrophoresis:
Sequence ladder by
Capillary gel electrophoresis:Samples passing a detection window are excited by laser and emitted fluorescence is read by CCD camera. Fluorescent signals are converted intoSequence ladder by
radioactive sequencing compared to fluorescent peaks
Fluorescent signals are converted intobasecalls.
AdvantagegHigh resolutionRead length up to 1,000 nucleotides

ABI 310 Genetic AnalyzerOne-capillary Instrumentp y
Capillary and electrode: Negatively charged DNA migrates towards anode
DNA fragments labeled with different fluorescent dyes migrate according to their size past a laser

Sequencing Electropherogram•• Laser excites dyes, causing them to emit light at longer Laser excites dyes, causing them to emit light at longer
wavelengthswavelengths•• Emitted light is collected by a CCD cameraEmitted light is collected by a CCD camera•• Software converts pattern of emissions into colored peaksSoftware converts pattern of emissions into colored peaks
www.contexo.info/DNA_Basics/dna_sequencing.htmPlot of colors detected in sequencing sample scanned from smallest to largest fragment

Finishing Draft Sequences using Assembly g g ySoftware
Primary sequencing reads:

Finishing Draft Sequences using Assembly g g ySoftware
Primary sequencing reads:
Align sequences for homologiesAlign sequences for homologies
Green: 3-fold coverage
Yellow: 2-fold coverage
Fragment assembly to contig Goal for Genome Sequencing:Sanger: 8-fold coverage454: 30 fold coverage

Sequencing Technologies:Lif S i Th i T h th i C l b tiLife Sciences Thrive Trough their Co-laboration
with Engineering and BioInformatics
BiologistBioInformatics
ww
.itb.
cnr.i
t
Engineers
ww

How is Genome Sequencing Done?How is Genome Sequencing Done?
Clone by Clone Shotgun sequencingClone by Clone Shotgun sequencing
Break genome into random fragments,
fsequence each of the fragments andassemble fragments based on sequence overlaps
Create a crude physical mapof the whole genome by restrictionmapping before sequencing
on sequence overlaps
Break the genome into overlappingfragments and insert them into BACsand transfect into E.coli
Break these inserts in even smallerfragments and sequence

High-Throughput Whole GenomeHigh-Throughput Whole Genome Sequencing
Analysis of 384 sequencing reactions in parallel
George Church, Scientific American, January 2006, pp47-54

JGI Sequencing Facility(Joint Genome Institute, US Department of Energy)
Assume more than 20 384-capillarysequencers running simultaneouslyapprox. 700 bp per capillary runpp p p p yapprox. 3 hours per run
Approx 40 Million bases per dayApprox. 40 Million bases per dayin one facility

What Challenge Was it to gSequence the Human Genome?It took more than 6 years to determine the complete base sequence of the E coli genomecomplete base sequence of the E. coli genome (4.6 Mb). The Human Genome has 3200 Mb.
At this rate it would have taken a single lab more gthan 4,000 years to sequence the entire human genome
The average fragment read length is aboutThe average fragment read length is about500 bases.
It would take a minimum of six million (3
International Co-laborationeffort started
billion/500) to sequence the human genome(no overlap, 1-fold coverage).
Develop procedural and computational
Initial sequencing and analysis of the human genome Nature 409, 860 - 921 (15 February 2001) International Human Genome Sequencing Consortium(headed by Francis Collins)
The Sequence of the Human Genome , Science, Vol 291, I 5507 1304 1351 16 F b 2001Develop procedural and computational
algorithms and efficient database managementIssue 5507, 1304-1351 , 16 February 2001Venter et al. (Celera Genomics)

In 2001 the First Draft of the Human Genomeis Published
How many nucleotide pairs doesthe Human Genome have?
How many genes does it take
3 x 106 or 3 x 109 ?
to make a human?
Roundworm: 6,000Fruitfly: 14,000
How different are genomes from one individual to another?
y
0.1% or 1% or 10 % ?

What did we learn?What did we learn?
Less than 2% of the Human Genome codesfor proteinfor proteinThe human genome encodes for approx. 21,000 protein-coding genesThe human genome sequence is almost exactlythe same (99 9%) in all peoplethe same (99.9%) in all peopleNon-coding DNA segments may have regulatoryfunctions on gene expressionThe genome size does not correlate withthe number of estimated genesthe number of estimated genes
Organism Genome Size Est. Gene #E. coli 4.6 Mb 4,400
Yeast 12.1 Mb 6,200
Humans have only about twice thenumber of genes as a fruit fly andbarely more genes than a worm!
Roundworm 97 Mb 19,700
Fruit Fly 180 Mb 13,600
Rice 389 Mb 37,500
Human 3200 Mb 25,000,
www.ornl.gov/hgmis/publicat/primer

Who’s sequence was used for the ‘Human Genome Project’Genome Project
• Different sources of DNA were used for original sequencing• Celera: 5 individuals; HGSC: ‘many’y• The term ‘genome’ is used as a reference to describe a
composite genome• The many small regions of DNA that vary among
individuals are called polymorphisms:individuals are called polymorphisms:– Mostly single nucleotide polymorphisms (SNPs)– Insertions/ deletions (indels)– Copy number variation– Inversions
http://scienceblogs.com/digitalbio/2007/09/genetic_variation_i_what_is_a.php#more
SNPs: the human genome has at least 20 million SNPsmost of these SNPs contribute to human variation
f th i fl d l t f di tibilitsome of them may influence development of diseases, susceptibilityto certain drugs, toxins, infectious agents
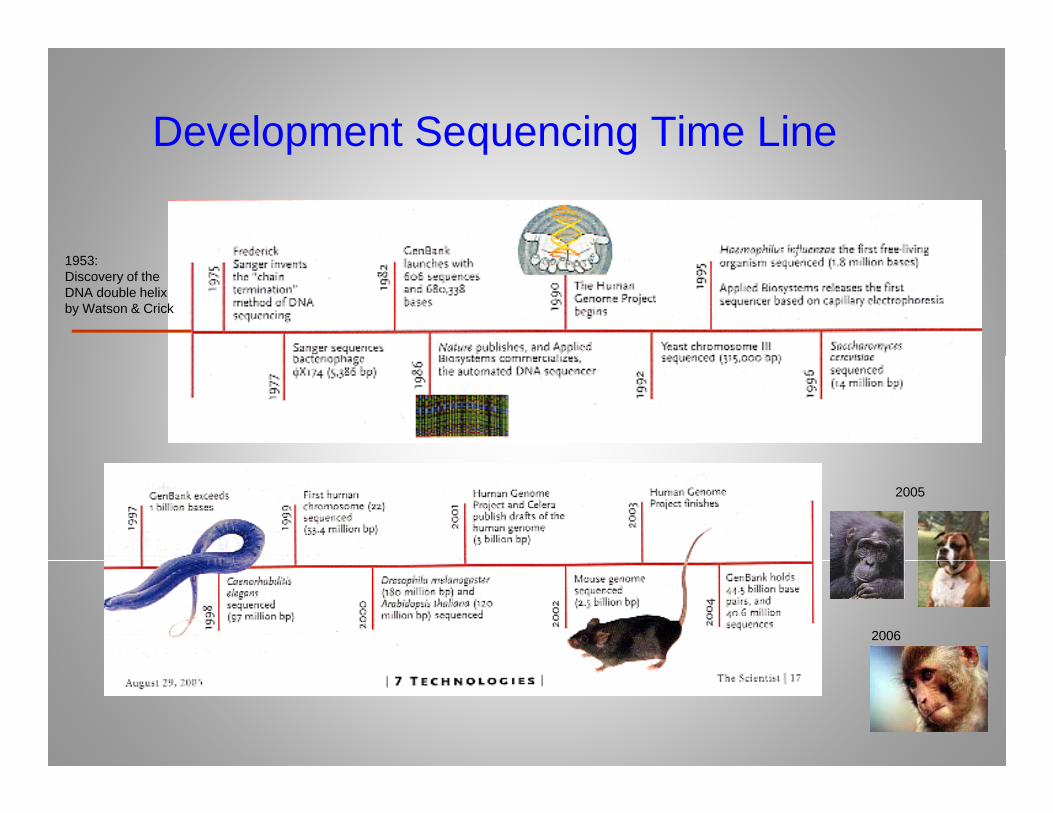
Development Sequencing Time Linep q g
1953:Discovery of theDNA double helixby Watson & Crick
2005
2006

The Race for the $1 000 GenomeThe Race for the $1,000 Genome
Human Genome Project (2001 initial draft):Human Genome Project (2001, initial draft):> $ 3 billion (includes development of technology)“raw” expenses estimated at $300 million
Rhesus macaque (2006)$ 22 million
By end of 2007:$ 1-2 million for full mammalian genome sequence
Wanted: “!!!!!! The $ 1,000 Genome !!!!!!!!!”- low cost- high-throughput - high accuracyg y

454 Pyrosequencing Method454 – Pyrosequencing Method
Flowgram:Flowgram:
Add one kind of dNTP per cycle. Sulfurylase converts PPi to ATP in ypresence of APSLuciferin conversion results in light emission
Intensity of light emission is directlyProportional to amount of releasedPPi l lPPi moleculesSequencing is recorded ‘live’. Read length up to 400 basesProblem: homopolymer detection

Genome Sequencing Milestones
May 2007: James Watson’s genome deposited454 technology (pyrosequencing)2 month, $ 1-2 million, $
Sept 2007: Craig Venter’s genome depositedSanger technology, 4 years, 70 million
*Diploid genomes
*Approx. 3.5% of Watson’s genome could not be matched to the reference genome
Appliedbiosystems.com
pp g g
*Venter’s genome had 4.1 million DNA variants comprising 12.3 Mb- 3.2 million SNPs- non-SNP variants
Personal Genome Project (PGP): initiated by George Church (Harvard) seeking for volunteers to share their DNA sequence and medical records

Next Generation Sequencing (NGS) Focuses onMiniaturization and Parallelization
Sanger Cyclic Array Sequencing
Generate dense planar arrayf DNA f tof DNA features
Apply cycles of enzymatic driven biochemistryy
Imaging-based data collection

Sequence Mate Pairs Provide an AdditionalSequence Mate-Pairs Provide an Additional Layer of Positional Information
DNA fragmentation innebuilizer, end polishing,size selection by DNA gel
l t h ielectrophoresisAdapter ligation and sequencing
-
1000
500
200
Sequence mate-pairs separated by a known distanceAssemble contigs into larger scaffolds using information about
00
+
sequence similarity and distance between mate-pairs

Next generation sequencing – 454 method(P i )(Pyrosequencing)
-Shear DNA into 300 to 400 nte fragments
-Ligate to heterobifunctional adapters
-Capture DNA strands on beads and amplifiyby emulsion PCRby emulsion PCR
-Place beads with amplified DNA fragmentsinto picotiter plate (1.7 Mio wells)
- Perform pyrosequencing reaction
44 m

Solexa/ Illumina SequencingTechnology
reversibleSangerIllumina
Use a set of deoxynucleotides that carry
reversible
Use a set of deoxynucleotides that carry
- each a fluorescent label that can be cleaved off
- a reversibly terminating moiety at the 3’ hydroxyl position

Solexa/ Illumina SequencinggTechnology

Comparison
Sanger Sequencing:In vitro construction of library
454 Sequencing:Direct in-vitro amplificationIn vitro construction of library
Limited parallelization‘large’ volumes (microliter)Expensive
Direct in vitro amplificationMassively parallelReduction of reagent volume to pico or femto-liters per DNA feature
R l ti l h
Long reads (up to 1000 bases)
Relatively cheap
Shorter read length (300-400 bases)g ( p )High accuracy (0.1% error)
4 x 105 bases/run
g ( )Lower raw accuracy (1% error)
6 x 108 bases/run
Illumina sequencingRead length 36 – 50 bases
2 x 109 bases/run

BGI – The Sequencing FactoryBGI – The Sequencing Factory(Beijing Genome Institute)
Featured in Nature issue March 4th, 2010
Purchased 128 HiSeq2000seq encers from Ill mina in Jan ar 2010
,
sequencers from Illumina in January 2010each of which can produce 25billion base pairs of sequence a day

Speed Reading1987: ABI sequencer – 4800 basepairs a day2010: Life Technologies – 100 billion basepairs a day
C. Venter, nature, 464, pg676, 2010

All published fully sequenced human genomesi 2000since 2000

Outlook: Single Molecule Sequencing through Graphene nanoporeGraphene nanopore
•Graphene (Geim & Novoselov, Nobel Prize 2010)is a planar sheet of carbon just one atom thickis a planar sheet of carbon just one atom thick•Strongest material known, electricallyconductive
As a DNA chain passesAs a DNA chain passes through the nanopore, the nucleobases, which are the letters of the genetic code, can be identified. The nanopore in graphene is the first nanopore short enough to distinguishenough to distinguish between two closely neighboring nucleobases

Achievements:The duck billed platypus: part birdThe duck-billed platypus: part bird, part reptile, part mammal – and the genome to prove itgenome to prove it
http://news.nationalgeographic.com/news/bigphotos/4891235.html
Th Pl t i t di t t l l ti- The Platypus is our most distant mammal relative- Has retained a large overlap between two very different classifications- The platypus shares 82% of its genes with human,
d d hi kmouse, dog, opossum and chicken - Decoding the platypus genome helps to understand the origins of mammal evolution
Nature, May 7, 2008

Decoding of a Acute Myeloid Leukemia Genomeg y
* AML i f hit bl d ll hi h l t i th b d* AML is a cancer of white blood cells, which accumulate in the bone marrow andinterfere with the production of normal white and red blood cells.
* Sequenced and compared the genome of cancerous bone marrow cells withnormal skin cells from the same patient using the next generation technologies.
* F d t t l f 10 i t ti i th ll* Found a total of 10 genes carrying mutations in the cancerous cells. * Two of those (FLT3 and NPM1) had already been implicated in the process of
progression to AML.
Proof of concept that whole genome sequencingf t ll i f l t t t
Ley et al., nature 456:66, 2008
of tumor cells is a very useful strategy to uncovergenes implicated in the disease.
Ley et al., nature 456:66, 2008

Twin study surveys genome for cause of Multiple Sclerosis
MS causes the body’s own immune cells to attack the myelin sheath around nerve cells
MS has a genetic component
No clear genetic reason found to explain whyone twin developed MS while the other did not
Both twins carry genetic variants that are linkedto a higher risk getting MS
Baranzini et al
to a higher risk getting MS
But those genetic factors seem to have been insufficient to cause disease of their ownBaranzini et al.
April 29th, 2010 Environmental triggers, epigenetics?

Common Gene Variant Strategygy
Assumption:pTo predict human diseases orsusceptibility common DNA variations (~5% frequency)would be at a faultwould be at a fault.
Many SNP alleles have been uncoveredrelated to specific diseases. however, these variants have onlyaccounted for a small fractionof disease risk.
“A gene is vexed with multiple layersof complexity” (Joseph Nadeau)
Hall. Revolution Postponed. Scientific American, Oct. 2010, pg 60-67

In Contrast:Look at Rare Variants and Beyond the Genes
Other mechanisms ofgene expression regulation:
Inhibitory RNA (RNAi)
Chemical DNA modification, e.g. methyl groups

Lessons learned• Data sharing
• 2000: 4 eukaryotic genomes• 2010: 250 eukaryotic genomes many hundreds of human genomes2010: 250 eukaryotic genomes, many hundreds of human genomes
• Fewer protein-coding genes than thought
S f• Studying of genetic variation and evolutionary origins
• Cost of sequencing has fallen 100,000 fold; high trough-put sequencing
• HapMap project: Catalog more than 20 million SNPs
• Cancer genome atlas to map genomic changes observed in every major type of hhuman cancer
• Obtaining a genome ‘blueprint’ may be sufficient to explain the susceptibility to disease ,, but not the likelyhood to develop the disease. p y ,, y p
• The combination of m a n y factors needs to be taken into account

Ethic Aspects of Genome Sequencing
-How can patient privacy be protected?-Will insurance companies and employersp p y
use genetic information to screen outthose at high risk for disease?
-Genetic engineering of bioterrorism agents
nhs.needham.k12.ma.us

What’s next?What s next?
Conference Topics:• Genetic basis of common diseases• Intricacies of gene regulation• Role of non coding portions of the genome• Role of non-coding portions of the genome• Medical advances• Technological and ethical challenges of human genomics• Personalized genomics• Aspects of variation in the human genome