the dna damage response induces ifn - jimmunol.org journal of immunology the dna damage response...
TRANSCRIPT

of June 28, 2018.This information is current as
The DNA Damage Response Induces IFN
and Nancy C. ReichSabrina Brzostek-Racine, Chris Gordon, Sarah Van Scoy
http://www.jimmunol.org/content/187/10/5336doi: 10.4049/jimmunol.1100040October 2011;
2011; 187:5336-5345; Prepublished online 17J Immunol
MaterialSupplementary
0.DC1http://www.jimmunol.org/content/suppl/2011/10/18/jimmunol.110004
Referenceshttp://www.jimmunol.org/content/187/10/5336.full#ref-list-1
, 33 of which you can access for free at: cites 83 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2011 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 28, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on June 28, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
The DNA Damage Response Induces IFN
Sabrina Brzostek-Racine, Chris Gordon, Sarah Van Scoy, and Nancy C. Reich
This study reveals a new complexity in the cellular response to DNA damage: activation of IFN signaling. The DNAdamage response
involves the rapid recruitment of repair enzymes and the activation of signal transducers that regulate cell-cycle checkpoints and
cell survival. To understand the link between DNA damage and the innate cellular defense that occurs in response to many viral
infections, we evaluated the effects of agents such as etoposide that promote dsDNAbreaks. Treatment of human cells with etoposide
led to the induction of IFN-stimulated genes and the IFN-a and IFN-l genes. NF-kB, known to be activated in response to DNA
damage, was shown to be a key regulator of this IFN gene induction. Expression of an NF-kB subunit, p65/RelA, was sufficient for
induction of the human IFN-l1 gene. In addition, NF-kB was required for the induction of IFN regulatory factor-1 and -7 that are
able to stimulate expression of the IFN-a and IFN-l genes. Cells that lack the NF-kB essential modulator lack the ability to induce
the IFN genes following DNA damage. Breaks in DNA are generated during normal physiological processes of replication,
transcription, and recombination, as well as by external genotoxic agents or infectious agents. The significant finding of IFN
production as a stress response to DNA damage provides a new perspective on the role of IFN signaling. The Journal of
Immunology, 2011, 187: 5336–5345.
An effective DNA damage response is critical for main-taining genomic integrity and preventing mutations thatcan lead to cancer. Double-strand breaks are the most
severe lesions, and they can occur during DNA replication, lym-phocyte V(D)J gene rearrangement, meiosis, viral infection, andin response to naturally occurring ionizing radiation (1–5). TheseDNA breaks are sensed rapidly, and accurate repair is essential toprevent permanent genomic damage. However, the cellular re-sponse to DNA damage engages more than just DNA repair ma-chinery; it engages complex signaling pathways that can promotecell survival or cell death (6, 7). In this report, the activation of anadditional network is revealed: the IFN signal pathway.A primary transducer of the response to double-strand breaks is
the nuclear kinase ataxia-telangiectasia mutated (ATM) (8). ATMbelongs to a family of PI3K-related kinases, several of which areinvolved in the DNA damage response, including ATM and Rad3related (ATR) and DNA-dependent protein kinase (DNA-PK) (9).ATM transduces the DNA damage response signal by phosphor-ylating downstream effectors such as the checkpoint kinases Chk1and Chk2 and the p53 tumor suppressor. These effectors in turnestablish cell-cycle arrest to allow repair of damaged DNA orpromote damage-induced apoptosis. Major alterations in gene ex-
pression occur during this time, and this reflects the action of notonly p53, but also other transcription factors (10). One tran-scription factor that is activated in response to DNA damage isNF-kB (11). NF-kB regulates the expression of diverse genesinvolved in cellular responses that include survival, proliferation,tissue remodeling, inflammation, immunity, and stress. We havefound that NF-kB activation in response to DNA damage directsthe induction of the IFN system, a stress pathway best known forits ability to confer viral resistance.IFNs play vital roles in both innate and adaptive immunity and
consist of three families of cytokines that bind to distinct cell-surface receptors and are designated types I, II, and III (12).The genes encoding type I IFN (primarily a and b) and type III(l) IFN are induced in response to viral or bacterial infection (13).The single type II (g) IFN gene is induced primarily followingreceptor activation of T cells and NK cells. The regulated ex-pression of the IFN-b gene in response to viral infection is aparadigm for cooperativity of DNA binding factors (14). NF-kBand IFN regulatory factors (IRFs) function along with activatingtranscription factor-2/c-Jun in the IFN-b enhancer. The IRFs werefirst characterized as regulators of type I IFN genes and IFN-stimulated genes (ISGs) and are now known to have diverseroles in immunity (15). Activation of ubiquitous IRF-3 duringviral infection supports induction of a subset of ISGs and theIFN-b and IFN-a genes (16, 17). The IRF-1 and IRF-7 genes areinduced in response to secreted IFN and can play a role in thesecondary response to IFN (18). IFNs bind to cell-surface recep-tors that activate Janus kinases and the tyrosine phosphorylation ofSTAT1 and STAT2 (19, 20). We report in this study that signalingvia the DNA damage response in human cells primarily inducesthe IFN-l and IFN-a genes. The promoters of the IFN-l geneshave been found to possess both IRF and NF-kB binding sites (21,22). We demonstrate that NF-kB activation in response to DNAdamage is sufficient and necessary to induce human IFN-l.This study identifies IFN signaling as part of the DNA damage
response. IFNs are essential components of innate immunity andare well recognized for their ability to inhibit viral infection andactivate immune effector cells (23). In addition, they are known fortheir antitumor effects by inhibiting proliferation of cancerouscells and promoting apoptosis (24–26). The IFN arm of the DNA
Department of Molecular Genetics and Microbiology, Stony Brook University, StonyBrook, NY 11794
Received for publication January 5, 2011. Accepted for publication September 12,2011.
This work was supported by National Institute of Allergy and Infectious DiseasesGrants R21AI067885 and PO1AI0555621 and National Institutes of Health GrantT32GM008444.
Address correspondence and reprint requests to Dr. Nancy C. Reich, Department ofMolecular Genetics and Microbiology, Life Sciences Building, Stony Brook Univer-sity, Nicolls Road, Stony Brook, NY 11794-5200. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: ATM, ataxia-telangiectasia mutated; ATR, ataxia-telangiectasia mutated and Rad3 related; CBP, CREB-binding protein; DNA-PK,DNA-dependent protein kinase; HA, hemagglutinin; His, polyhistidine; IKK, IkBkinase; IRF, IFN regulatory factor; ISG, IFN-stimulated gene; MEF, Murine embryofibroblast; NDV, Newcastle disease virus; NEMO, NF-kB essential modulator; wt,wild-type.
Copyright� 2011 by TheAmericanAssociation of Immunologists, Inc. 0022-1767/11/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1100040
by guest on June 28, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

damage response may have evolved as an antiviral mechanism inreaction to DNA damage induced by viruses, as a mechanism thatreduces cellular proliferation to allow DNA repair, or as a mech-anism to promote the death of cells with irreparable damage.
Materials and MethodsCell culture, transfections, and infections
HeLa S3, HT1080, and THP-1 cells were obtained from American TypeCulture Collection. THP-1 cells were maintained in RPMI 1640 with 8%FBS; other cells were maintained in DMEM with 8% FBS. Primary humanmonocytes were isolated from blood cells of healthy donors (Long IslandBlood Services) using the Monocyte Isolation Kit II (Miltenyi Biotec,Auburn, CA) and maintained in RPMI 1640 with 10% FBS. StableHT1080 transfectants with tetracycline-inducible expression of IRF-7 weregenerated according to the manufacturer’s instructions (T-Rex system;Invitrogen, Carlsbad, CA), and the gene was induced with 2 mg/mldoxycycline. Murine embryo fibroblasts (MEFs) from NF-kB essentialmodulator (NEMO)/IkB kinase (IKK) g knockout mice were a gift ofDr. Kenneth Marcu (Stony Brook University) (27), MEFs from IRF3 knock-out mice were a gift of Dr. Tadatsugu Tanaguchi (University of Tokyo) (16,17), MEFs from IRF1 knockout mice were a gift of Dr. Tak Mak (Uni-versity of Toronto) (28), and MEFs from IRF7 knockout mice were a giftof Dr. Michael Gale (University of Washington) (29). DNA transfectionswere performed using FuGENE 6 (Roche Diagnostics, Indianapolis, IN)or TransIt (Mirus, Madison, WI). Newcastle disease virus (NDV) (NJ-LaSota-1946) was a gift from Dr. Paula Pitha-Rowe (Johns Hopkins Uni-versity, Baltimore, MD) and was propagated as described previously (30).Infections were performed at 200 hemagglutination units/ml.
Plasmids and luciferase assays
IRF-3, STAT1, and STAT2 constructs have been described (31–33). Thedominant-negative IkBa plasmid (S32A/S36A) was a gift of Dr. DeanBallard (Vanderbilt University, Nashville, TN) (34). The hemagglutinin(HA)-tagged ubiquitin–K63-only (HA-Ub0R63K) plasmid was a gift ofDr. Dafna BarSagi (New York University) (35). The reporter plasmidencoding the IFN-l1 promoter regulating expression of the firefly lucif-erase gene (pl1[2554/+14]Luc) was a gift of Dr. Takashi Fujita (KyotoUniversity, Kyoto, Japan) (21). The human IRF-7A gene was obtainedfrom Dr. Joseph S. Pagano (University of North Carolina, Chapel Hill, NC)(36) and subcloned into the pcDNA4/TO/Myc-polyhistidine (His) vector(T-Rex system; Invitrogen). The Dual-Luciferase reporter assay systemwas used for luciferase assays with the Renilla luciferase construct pRL-null as an internal control (Promega, Madison, WI).
Reagents
Etoposide was used at 40 mg/ml, and camptothecin, adriamycin, and mi-tomycin were used at the concentrations indicated (Sigma-Aldrich, St.Louis, MO). NF-kB inhibitor BAY 11-7085 was obtained from AlexisBiochemicals (San Diego, CA) and used at 5 mM, and the IKKb inhibitorML120B was a gift from Millennium Pharmaceuticals (Cambridge, MA)and used at 20 mM. The ATM inhibitor AZ12622702/KU55933 was a giftfrom AstraZeneca (Cheshire, U.K.) and used at 10 mM. Abs recognizingSTAT2 (C-20), c-Myc epitope (sc-40), CREB-binding protein (CBP; A-22), and normal rabbit IgG were purchased from Santa Cruz Biotech-nology (Santa Cruz, CA). Ab to STAT2 phosphorylated on tyrosine 689was obtained from Upstate Biotechnology (Lake Placid, NY). Abs to p65phosphorylated on serine 536 and STAT1 phosphorylated on tyrosine 701were obtained from Cell Signaling Technology (Beverly, MA). Ab againstHA epitope (12CAS) was purchased from Roche (Indianapolis, IN). Absderived against IRF-3 and STAT1 were described previously (30, 33). Abto IRF-7 was raised in rabbits against the 246–432 aa region of IRF-7.Anti-mouse or anti-rabbit secondary Abs conjugated to IRDye800 or700 were obtained from Rockland Immunochemicals (Gilbertsville, PA).TNF-a was obtained from Invitrogen.
PCR
RNAwas isolated from cells using SV Total RNA Isolation Kit (Promega),and cDNAwas generated using random hexamer primers and SuperScriptIIReverse Transcriptase (Invitrogen) according to the manufacturer’s pro-tocol. RT-PCR was performed using the indicated primers and Taq poly-merase (Invitrogen). Alternatively, quantitative real-time PCR wasperformed using the indicated primers at their optimal conditions as sug-gested by the manufacturer’s instructions for the LightCycler-FastStartDNA Master SYBR Green I kit (Roche Molecular Biochemicals). Data
was analyzed using the LightCycler software (Roche Molecular Bio-chemicals), and values were normalized to actin mRNA levels. Humanprimers used in the studies corresponded to: IFN-l1 (37), IFN-a6 (38),IFN-a1/13 (38), actin (39), and pan–IFNa 59-CACACAGGCTTCCA-GGCATTC-39 and 59-TCTTCAGCACAAAGGACTCATCTG-39; ISG54(+1608) 59-ATTCTATCACCAAGCCCGTGG-39 and (+1370) 59-TGG-AGTCTGGAAGCCTCATCC-39; IFN-a7 (+194) 59-TCCTCCTCCGGG-AATCTGAAT-39 and (+106) 59-AGGGCCTTGATACTCCTGG-39; IFN-a14 (+315) 59-TAGGAGGGTCTCATCCCAAGC-39 and (+65) 59-TG-GGCTGTAATCTGTCTCAAAC-39; IFN-b (+27) 59-TGCTCTCCTGT-TGTGCTTCTCCAC-39 and (+243) 59-ATAGATGGTC AATGCGGCG-TCC-39; IRF-7 (+1476) 59-GATGTCGTCATAGAGGCT GTTGG-39 and(+1343) 59-TGGTCCTGGTGAAGCTGGAA-39; IRF-1 (+397) 59-TCTT-AGCATCTCGGCTGGACTTC-39 and (+190) 59-CGATACAAAGCAGG-GGAAAAGG-39; and GAPDH (+1007) 59-TACTCCTTGGAGGCCA-TGTG-39 and (+528) 59-CACAGTCCATGCCATCACTG-39. Murine pri-mers corresponded to: pan-murine IFN-a 59-CCTGAGAGAGAAGAAA-CAC AGCC-39 and 59-TCTGCTCTGACCACYTCCCAG -39 (40); murineIFN-l2 (41); and murine Actin (42).
Immunoprecipitation and Western blot
For immunoprecipitation, cells were lysed in 50 mM HEPES (pH 7.2), 250mM NaCl, 0.5% Nonidet P-40, 5 mM EDTA, 1 mM DTT, 1 mM NaF, 0.1mM Na3VO4, and protease inhibitor mixture (Sigma-Aldrich, St. Louis,MO). Lysates were cleared by centrifugation for 5 min at 15,000 3 g andreacted with Abs for 3 h at 4˚C. Immunocomplexes were collected withProtein G-conjugated agarose (Invitrogen). For direct Western blot, cellswere lysed in 50 mM Tris (pH 7.5), 400 mM NaCl, 0.5% Nonidet P-40, 5mM EDTA, 10% glycerol, 50 mM NaF, 0.1 mM Na3VO4, and proteaseinhibitors. The lysates were cleared by centrifugation and directly addedto SDS sample buffer. Proteins were separated on 8% SDS-PAGE andtransferred to nitrocellulose membrane (Pierce Biotechnology, Rockford,IL). Membranes were reacted with indicated Abs, and images weredetected using the Odyssey infrared imaging system (Li-COR Biosciences,Lincoln, NE). Alternatively, secondary anti-rabbit or anti-mouse Abslinked to HRP (Amersham/GE Healthcare, Piscataway, NJ) were used, andthe membrane was incubated in ECL reagents and exposed to film.
Fluorescence imaging
Cells were seeded on coverslips, fixed in 4% paraformaldehyde, and eithervisualized directly for GFP fluorescence or permeabilized in 0.2% TritonX-100 before reaction with anti-Myc Ab. Secondary Abs were conjugatedto rhodamine (Jackson ImmunoResearch Laboratories, West Grove, PA.).Coverslips were mounted in anti-fade solution (Vectashield; Vector Lab-oratories, Burlingame, CA). Images were captured with a Zeiss Axiovert200 M digital deconvolution microscope or Zeiss LSM 510 META NLOtwo-photon laser scanning confocal microscope (Carl Zeiss).
Ubiquitination assay
The HA-Ub0R63K expression plasmid was transfected into a stable cell lineexpressing tetracycline repressor and responsive Myc-His–IRF-7 gene asdescribed above. Doxycycline was added 24 h posttransfection with orwithout etoposide as indicated. Cells were lysed by sonication in 6 Mguanidine HCl, 0.1 M Na2HPO4/NaH2PO4 (pH 8), and 10 mM imidazole.Lysates were incubated with Ni-NTA agarose beads (Qiagen, Chatsworth,CA), and proteins were eluted with SDS loading buffer and analyzed byWestern blot (43).
ResultsEtoposide activation of IFN signaling
Cells respond to viral infection with the induction of ISGs, eitherdirectly by activation of the IRF-3 transcription factor or as anindirect response to autocrine IFN and STATactivation (15, 19, 30).Because the genes induced by IRF-3 or STATs can promote cel-lular apoptosis, we examined gene expression during the DNAdamage response to identify shared proapoptotic-induced genes(24, 44–46). Etoposide, an anticancer drug, was used to initiatethe DNA damage response because it inhibits the ability of top-oisomerase II to religate cleaved DNA (47, 48). The inhibitionresults in an accumulation of double-stranded breaks in DNA,particularly during DNA replication and S/G2 phases of the cellcycle. The increase of dsDNA breaks with time leads to the DNA
The Journal of Immunology 5337
by guest on June 28, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
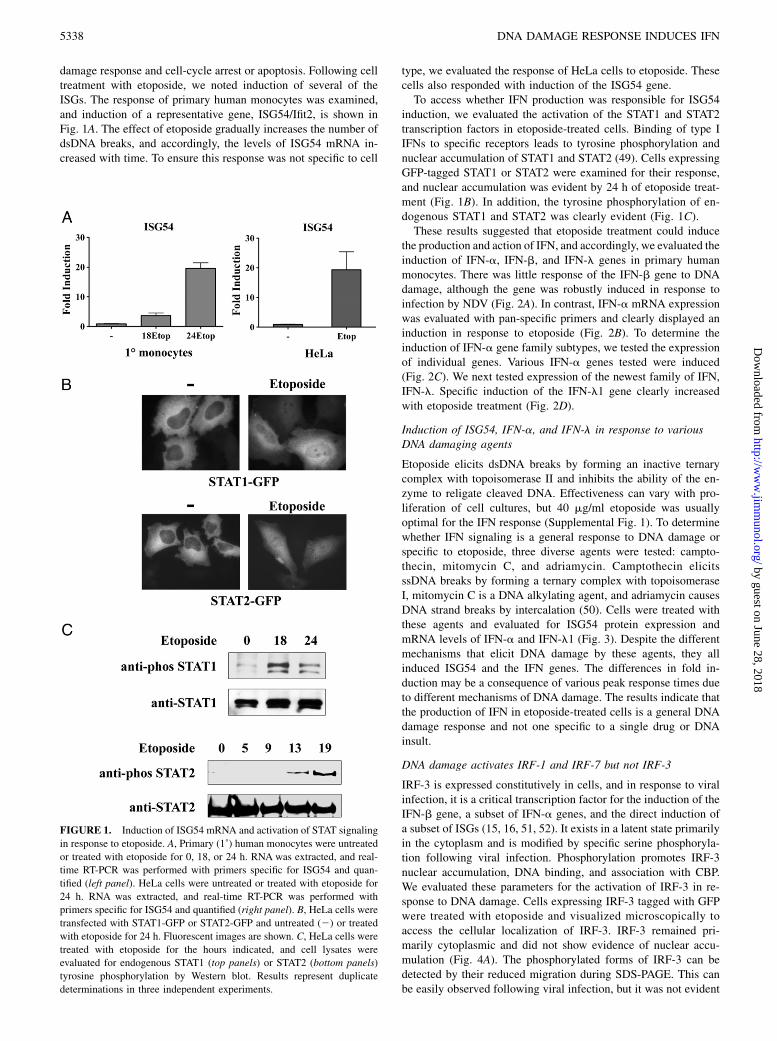
damage response and cell-cycle arrest or apoptosis. Following celltreatment with etoposide, we noted induction of several of theISGs. The response of primary human monocytes was examined,and induction of a representative gene, ISG54/Ifit2, is shown inFig. 1A. The effect of etoposide gradually increases the number ofdsDNA breaks, and accordingly, the levels of ISG54 mRNA in-creased with time. To ensure this response was not specific to cell
type, we evaluated the response of HeLa cells to etoposide. Thesecells also responded with induction of the ISG54 gene.To access whether IFN production was responsible for ISG54
induction, we evaluated the activation of the STAT1 and STAT2transcription factors in etoposide-treated cells. Binding of type IIFNs to specific receptors leads to tyrosine phosphorylation andnuclear accumulation of STAT1 and STAT2 (49). Cells expressingGFP-tagged STAT1 or STAT2 were examined for their response,and nuclear accumulation was evident by 24 h of etoposide treat-ment (Fig. 1B). In addition, the tyrosine phosphorylation of en-dogenous STAT1 and STAT2 was clearly evident (Fig. 1C).These results suggested that etoposide treatment could induce
the production and action of IFN, and accordingly, we evaluated theinduction of IFN-a, IFN-b, and IFN-l genes in primary humanmonocytes. There was little response of the IFN-b gene to DNAdamage, although the gene was robustly induced in response toinfection by NDV (Fig. 2A). In contrast, IFN-a mRNA expressionwas evaluated with pan-specific primers and clearly displayed aninduction in response to etoposide (Fig. 2B). To determine theinduction of IFN-a gene family subtypes, we tested the expressionof individual genes. Various IFN-a genes tested were induced(Fig. 2C). We next tested expression of the newest family of IFN,IFN-l. Specific induction of the IFN-l1 gene clearly increasedwith etoposide treatment (Fig. 2D).
Induction of ISG54, IFN-a, and IFN-l in response to variousDNA damaging agents
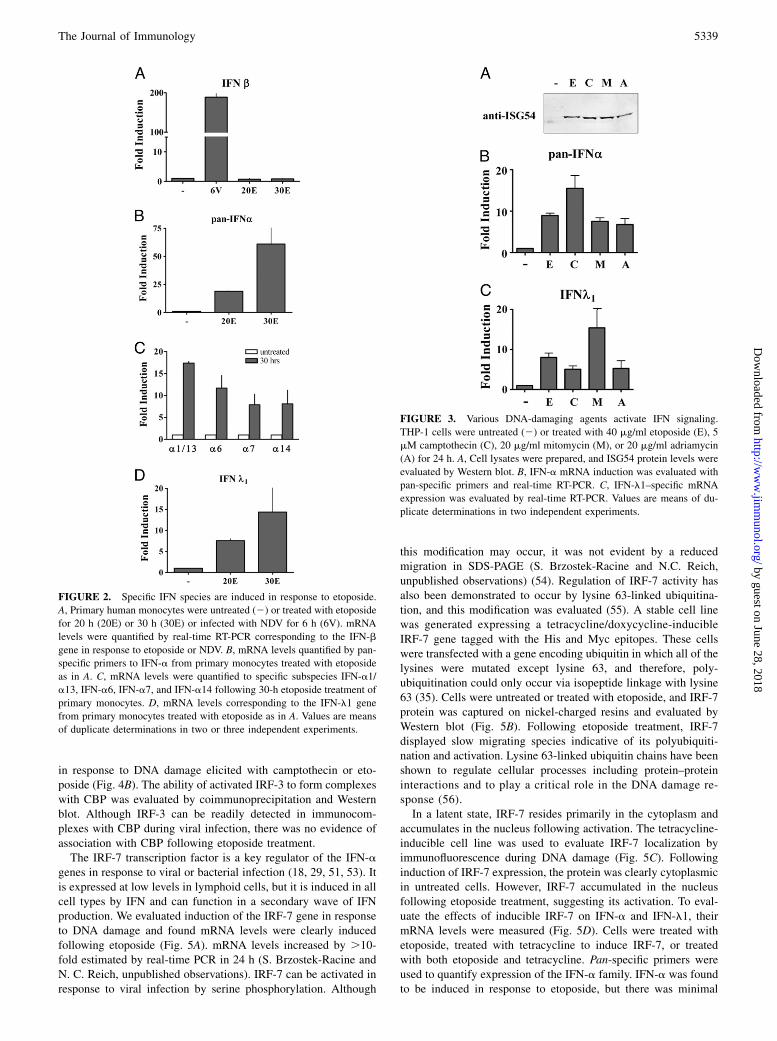
Etoposide elicits dsDNA breaks by forming an inactive ternarycomplex with topoisomerase II and inhibits the ability of the en-zyme to religate cleaved DNA. Effectiveness can vary with pro-liferation of cell cultures, but 40 mg/ml etoposide was usuallyoptimal for the IFN response (Supplemental Fig. 1). To determinewhether IFN signaling is a general response to DNA damage orspecific to etoposide, three diverse agents were tested: campto-thecin, mitomycin C, and adriamycin. Camptothecin elicitsssDNA breaks by forming a ternary complex with topoisomeraseI, mitomycin C is a DNA alkylating agent, and adriamycin causesDNA strand breaks by intercalation (50). Cells were treated withthese agents and evaluated for ISG54 protein expression andmRNA levels of IFN-a and IFN-l1 (Fig. 3). Despite the differentmechanisms that elicit DNA damage by these agents, they allinduced ISG54 and the IFN genes. The differences in fold in-duction may be a consequence of various peak response times dueto different mechanisms of DNA damage. The results indicate thatthe production of IFN in etoposide-treated cells is a general DNAdamage response and not one specific to a single drug or DNAinsult.
DNA damage activates IRF-1 and IRF-7 but not IRF-3
IRF-3 is expressed constitutively in cells, and in response to viralinfection, it is a critical transcription factor for the induction of theIFN-b gene, a subset of IFN-a genes, and the direct induction ofa subset of ISGs (15, 16, 51, 52). It exists in a latent state primarilyin the cytoplasm and is modified by specific serine phosphoryla-tion following viral infection. Phosphorylation promotes IRF-3nuclear accumulation, DNA binding, and association with CBP.We evaluated these parameters for the activation of IRF-3 in re-sponse to DNA damage. Cells expressing IRF-3 tagged with GFPwere treated with etoposide and visualized microscopically toaccess the cellular localization of IRF-3. IRF-3 remained pri-marily cytoplasmic and did not show evidence of nuclear accu-mulation (Fig. 4A). The phosphorylated forms of IRF-3 can bedetected by their reduced migration during SDS-PAGE. This canbe easily observed following viral infection, but it was not evident
FIGURE 1. Induction of ISG54 mRNA and activation of STAT signaling
in response to etoposide. A, Primary (1˚) human monocytes were untreated
or treated with etoposide for 0, 18, or 24 h. RNA was extracted, and real-
time RT-PCR was performed with primers specific for ISG54 and quan-
tified (left panel). HeLa cells were untreated or treated with etoposide for
24 h. RNA was extracted, and real-time RT-PCR was performed with
primers specific for ISG54 and quantified (right panel). B, HeLa cells were
transfected with STAT1-GFP or STAT2-GFP and untreated (2) or treated
with etoposide for 24 h. Fluorescent images are shown. C, HeLa cells were
treated with etoposide for the hours indicated, and cell lysates were
evaluated for endogenous STAT1 (top panels) or STAT2 (bottom panels)
tyrosine phosphorylation by Western blot. Results represent duplicate
determinations in three independent experiments.
5338 DNA DAMAGE RESPONSE INDUCES IFN
by guest on June 28, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
in response to DNA damage elicited with camptothecin or eto-poside (Fig. 4B). The ability of activated IRF-3 to form complexeswith CBP was evaluated by coimmunoprecipitation and Westernblot. Although IRF-3 can be readily detected in immunocom-plexes with CBP during viral infection, there was no evidence ofassociation with CBP following etoposide treatment.The IRF-7 transcription factor is a key regulator of the IFN-a
genes in response to viral or bacterial infection (18, 29, 51, 53). Itis expressed at low levels in lymphoid cells, but it is induced in allcell types by IFN and can function in a secondary wave of IFNproduction. We evaluated induction of the IRF-7 gene in responseto DNA damage and found mRNA levels were clearly inducedfollowing etoposide (Fig. 5A). mRNA levels increased by .10-fold estimated by real-time PCR in 24 h (S. Brzostek-Racine andN. C. Reich, unpublished observations). IRF-7 can be activated inresponse to viral infection by serine phosphorylation. Although
this modification may occur, it was not evident by a reducedmigration in SDS-PAGE (S. Brzostek-Racine and N.C. Reich,unpublished observations) (54). Regulation of IRF-7 activity hasalso been demonstrated to occur by lysine 63-linked ubiquitina-tion, and this modification was evaluated (55). A stable cell linewas generated expressing a tetracycline/doxycycline-inducibleIRF-7 gene tagged with the His and Myc epitopes. These cellswere transfected with a gene encoding ubiquitin in which all of thelysines were mutated except lysine 63, and therefore, poly-ubiquitination could only occur via isopeptide linkage with lysine63 (35). Cells were untreated or treated with etoposide, and IRF-7protein was captured on nickel-charged resins and evaluated byWestern blot (Fig. 5B). Following etoposide treatment, IRF-7displayed slow migrating species indicative of its polyubiquiti-nation and activation. Lysine 63-linked ubiquitin chains have beenshown to regulate cellular processes including protein–proteininteractions and to play a critical role in the DNA damage re-sponse (56).In a latent state, IRF-7 resides primarily in the cytoplasm and
accumulates in the nucleus following activation. The tetracycline-inducible cell line was used to evaluate IRF-7 localization byimmunofluorescence during DNA damage (Fig. 5C). Followinginduction of IRF-7 expression, the protein was clearly cytoplasmicin untreated cells. However, IRF-7 accumulated in the nucleusfollowing etoposide treatment, suggesting its activation. To eval-uate the effects of inducible IRF-7 on IFN-a and IFN-l1, theirmRNA levels were measured (Fig. 5D). Cells were treated withetoposide, treated with tetracycline to induce IRF-7, or treatedwith both etoposide and tetracycline. Pan-specific primers wereused to quantify expression of the IFN-a family. IFN-a was foundto be induced in response to etoposide, but there was minimal
FIGURE 3. Various DNA-damaging agents activate IFN signaling.
THP-1 cells were untreated (2) or treated with 40 mg/ml etoposide (E), 5
mM camptothecin (C), 20 mg/ml mitomycin (M), or 20 mg/ml adriamycin
(A) for 24 h. A, Cell lysates were prepared, and ISG54 protein levels were
evaluated by Western blot. B, IFN-a mRNA induction was evaluated with
pan-specific primers and real-time RT-PCR. C, IFN-l1–specific mRNA
expression was evaluated by real-time RT-PCR. Values are means of du-
plicate determinations in two independent experiments.
FIGURE 2. Specific IFN species are induced in response to etoposide.
A, Primary human monocytes were untreated (2) or treated with etoposide
for 20 h (20E) or 30 h (30E) or infected with NDV for 6 h (6V). mRNA
levels were quantified by real-time RT-PCR corresponding to the IFN-b
gene in response to etoposide or NDV. B, mRNA levels quantified by pan-
specific primers to IFN-a from primary monocytes treated with etoposide
as in A. C, mRNA levels were quantified to specific subspecies IFN-a1/
a13, IFN-a6, IFN-a7, and IFN-a14 following 30-h etoposide treatment of
primary monocytes. D, mRNA levels corresponding to the IFN-l1 gene
from primary monocytes treated with etoposide as in A. Values are means
of duplicate determinations in two or three independent experiments.
The Journal of Immunology 5339
by guest on June 28, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

effect of tetracycline-induced IRF-7 alone. However, etoposideand tetracycline potently increased etoposide-induced IFN-a ex-pression. The results indicate modification of IRF-7 protein inresponse to DNA damage contributes to induction of the IFN-agenes. Expression of IFN-l1 mRNA was similarly evaluated, andIRF-7 activation by etoposide also was found to contribute toinduction of the IFN-l1 gene.Another member of the IRF family, IRF-1, has diverse roles in
response to pathogens, development of the immune system, growtharrest, and apoptosis (15, 57, 58). The protein is constitutivelynuclear but only expressed following induction by cytokines suchas IFN. We evaluated IRF-1 mRNA expression and found mRNAlevels increased by .12-fold estimated by real-time PCR fol-lowing 24 h of etoposide treatment (Fig. 6A) (S. Brzostek-Racineand N.C. Reich, unpublished observations). To evaluate thepossible role of IRF-1 in induction of the IFN genes during DNAdamage, we tested its effect on expression of the IFN-l1 promoterdriving a luciferase reporter gene. The promoter of IFN-l1 pos-sesses IRF binding sites and a bona fide NF-kB site (21, 22). TheIFN-l1 promoter activity was induced following etoposide treat-ment or by cotransfection with a plasmid encoding IRF-1 (Fig.6B). Combined IRF-1 expression with etoposide induced signifi-cant expression of the IFN-l1 promoter. IRF-1 could also bedemonstrated to bind to a site in the IFN-l1 promoter, suggestingdirect action on the IFN-l1 gene (Supplemental Fig. 2).dsDNA breaks that occur in response to etoposide lead to the
recruitment and activation of the ATM kinase. To evaluate thepotential role of ATM in the induction of IFN gene expression, wetested the effects of a pharmacological inhibitor, AZ12622702.Treatment of cells with the ATM inhibitor decreased the ability ofetoposide to induce the IFN-l1 gene and the IFN-a genes (Fig. 6C).The results indicate ATM is a significant signaling kinase upstreamof IFN gene induction, and the residual response may be due toother PI3K-related kinases that are activated during DNA damage.
Role of NF-kB in the response of IFN genes to DNA damage
]IRF-1 and IRF-7 are both induced in response to IFN signaling andto DNA damage. Studies have also indicated that the promoters ofboth of these genes can be activated by the NF-kB transcription
factor (59–61). Because NF-kB activation is a well-characterizedresponse to DNA damage and ATM activity, we determined
whether induction of IRF-1 and IRF-7 by etoposide is a directresponse to NF-kB (11, 62). Inactive NF-kB is localized in the
cytoplasm due to binding IkB (63). IkB is released following itsphosphorylation by an IKK complex composed of IKKa, IKKb,
and NEMO/IKKg. To determine the role of NF-kB in induction ofIRF-1 and IRF-7 by DNA damage, we tested the effect of several
specific IKKb inhibitors (Fig. 6D). Cells were treated with eto-poside in the absence or presence of BAY117085 or ML120B, and
real-time PCR was used to quantify the endogenous expression ofIRF-1 and IRF-7 mRNAs. Both inhibitors effectively blocked the
induction of IRF-1 and IRF-7 expression, indicating activation ofNF-kB in response to DNA damage is necessary to induce these
genes.A major species of NF-kB is the p50 and p65/RelA heterodimer
(63). Because serine phosphorylation of the C terminus of p65 hasbeen shown to increase its transcriptional activity and stability, we
evaluated the phosphorylation of p65 following etoposide treat-ment (64). Etoposide stimulated the serine 536 phosphorylation
of p65, indicating the NF-kB is transcriptionally active (Fig. 7A).The activation of NF-kB can also be detected by its ability to
accumulate in the nucleus. For this reason, we performed immu-nofluorescence staining of the endogenous p65 subunit before
and after etoposide treatment. Cells treated with etoposide clearlydisplayed p65 nuclear accumulation (Fig. 7B). These properties
accompanied the ability of NF-kB to bind a consensus DNA sitein EMSAs (S. Brzostek-Racine and N.C. Reich, unpublished ob-
servations). The data support NF-kB activation in our system ofDNA damage, as reported previously (11).To evaluate the role of NF-kB in the induction of IFN genes
by etoposide, we tested the effects of IKKb inhibition. Cells were
treated with etoposide in the absence or presence of the IKKbinhibitor ML120B, and IFN-a mRNA levels were quantified by
real-time RT-PCR (Fig. 7C). Inhibition of IKKb was found toblock expression of the IFN-a genes, indicating that NF-kB is
required for induction. This is a significant finding because theIFN-a genes are not directly regulated by NF-kB and do not
possess NF-kB binding sites, although they do possess IRF bind-ing sites (51).Induction of the IFN-l1 gene by DNA damage was also was
found to be dependent on NF-kB. Real-time RT-PCR was used toquantify IFN-l1 mRNA levels following etoposide in the presence
or absence of ML120B. Expression of IFN-l1 was blocked with
the inhibition of IKKb and NF-kB. The effect of NF-kB inhibitioncould also be demonstrated with the IFN-l1 promoter driving the
luciferase gene. The IFN-l1 promoter reporter was expressedalone or with a dominant-negative IkB mutant that lacks serine
target phosphorylation sites (S32A/S36A) (34). Etoposide stimu-lated the expression of the IFN-l1 luciferase reporter, but this in-
duction was completely inhibited with coexpression of the NF-kBrepressor IkB (S32A/S36A) (Fig. 7D).Because the promoter of the IFN-l1 gene contains an NF-kB
binding site, the more direct question is whether NF-kB in theabsence of viral infection or DNA damage can induce the IFN-l1
gene (21). To determine whether NF-kB regulates induction of theIFN-l1 gene, we tested the response of the IFN-l1 promoter re-
porter to expression of p65/RelA, a potent activator of NF-kB targetsites. The IFN-l1 gene was induced directly by p65 coexpression,
FIGURE 4. IRF-3 is not activated in response to DNA damage. A, HeLa
cells were transfected with IRF-3–GFP and left untreated (2) or treated
with etoposide for 24 h. Cellular localization was evaluated by fluores-
cence microscopy. Images represent random sampling from three in-
dependent experiments. B, Cells were untreated or infected with NDV for
6 or 12 h, treated with camptothecin (Camp) for 12 or 20 h, or treated with
etoposide (Etop) for 20 or 40 h. Endogenous IRF-3 was detected in cell
lysates by Western blot. C, CBP was immunoprecipitated (IP) from lysates
of untreated cells or cells infected with NDV for 6 h or treated with
camptothecin or etoposide for 36 and 50 h. Western blots (WB) were
performed with Abs to IRF-3.
5340 DNA DAMAGE RESPONSE INDUCES IFN
by guest on June 28, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

indicating NF-kB is sufficient to induce the human IFN-l1 geneindependent of viral infection or DNA damage (Fig. 7D). Thisresult has obvious implications for the involvement of IFN actionin the many signaling pathways that activate NF-kB.
Response of murine embryo fibroblasts
Our studies with human primary cells or established human celllines provide clear evidence that the IFN-l and IFN-a genes areinduced in response to DNA damage by etoposide and that NF-kBis requisite for the induction. Although murine cells may not ac-curately reflect the human response, the murine system affords theability to test cells from animals with specific gene knockouts.For this reason, we obtained MEFs from wild-type (wt) or geneknockout animals and tested their response to DNA damage.Wild-type MEFs or MEFs that lack NEMO were treated withetoposide, and IFN gene induction was evaluated by RT-PCR(Fig. 8). Because murine IFN-l1 is a pseudogene, we assayed ex-pression of murine IFN-l2. Results showed etoposide treatmentinduced the genes encoding IFN-l2 and IFN-a in wt cells, but notin NEMO knockout cells. The results are in accordance with ourstudies in human cells that demonstrated a requirement of NF-kBactivation. In addition, MEFs from IRF3 knockout animals treatedwith etoposide induced the IFN-l2 and IFN-a genes, supportingour studies with human cells that showed IRF3 did not play amajor role in the DNA damage response. Analyses of MEFs thatlack the IRF-1 gene indicated it was critical for IFN-l2 geneexpression, but not for IFN-a expression, whereas MEFs that lackthe IRF-7 gene had a profound defect in the induction of bothIFN-l2 and IFN-a genes in response to etoposide. The promotersof the murine IFN-l genes are not well characterized, and there-
fore, they may respond differently from the human IFN-l genesduring the DNA damage response. The levels of IFN inductionin these spontaneously immortalized MEFs were modest but re-producible following etoposide treatment.
Expression of human IRF and IFN genes with time during theDNA damage response
Our studies with human cells indicate that NF-kB activated byDNA damage stimulates induction of the IFN-l1, IRF-1, and IRF-7 genes. To determine the time course of expression of thesegenes, human THP-1 cells were treated with etoposide, and real-time PCR was used to quantify IRF and IFN mRNA levels. IRF-1and IRF-7 mRNA levels displayed an initial peak of expressionat 4 h of etoposide treatment and reached steady-state levels by∼12 h (Fig. 9A). Expression of the IFN-l1 gene showed a smallincrease at 4 h of etoposide treatment and peaked at 15 h witha kinetic profile similar to that of the IRFs. Expression of IFN-amRNA trailed that of IFN-l1 by 3 to 4 h, possibly indicatinga greater dependency on IRF-7 induction and activation (Fig. 9B).
DiscussionThe DNA damage response rapidly engages multimeric proteincomplexes to repair DNA and activate transcriptional programs thatregulate cell-cycle checkpoints and cell survival (6, 8, 65, 66). Therecruitment of ATM, ATR, and DNA-PK to DNA breaks initiatesphosphorylation and ubiquitination events that lead to specifictranscription factor activation and gene expression. ATM is acti-vated primarily in response to dsDNA breaks, followed by ATR andDNA-PK in response to DNA single strands and ends generated
FIGURE 5. Evidence for a role of IRF-7 in the IFN response to DNA damage. A, THP-1 cells were untreated (2) or treated with etoposide (+) for 24 h.
IRF-7 mRNA induction was evaluated by RT-PCR and displayed on agarose gels. Faint band in untreated sample is nonspecific. mRNA levels of GAPDH
are shown as controls. B, HT1080 stable cell line expressing tetracycline-inducible IRF-7–Myc-His was transfected with the HA-Ub0R63K ubiquitin
(K63Ub), and doxycycline (Dox) was used to induce IRF-7 expression in the absence or presence of etoposide for 24 h. IRF-7 was collected on nickel-
charged resins, and samples were analyzed by Western blot with Ab to IRF-7. Lower panel shows input before resin with anti-myc Ab. C, Expression of
IRF-7–Myc-His was induced with doxycycline in the stable cell line in the absence or presence of etoposide for 15 h before immunostaining with Abs to
Myc. Imaging analysis of three independent experiments indicate nuclear accumulation of IRF-7 at this time in .50% of the cells. D, Pan-specific primers
were used to quantify IFN-a mRNA levels (top panel), and specific primers were used to assess IFN-l1 mRNA levels (bottom panel) in the IRF-7–Myc-
His–inducible cells by real-time PCR. Cells were untreated (2) or treated with etoposide (E), doxycycline (D), or doxycycline and etoposide (D/E) for 24 h.
Values are means of duplicate determinations in two independent experiments.
The Journal of Immunology 5341
by guest on June 28, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

during break resolution. One of the known substrates of ATM isNEMO, and phosphorylation promotes its ubiquitination, nuclearexport, and activation of IKK complexes (62, 67, 68) (Fig. 9C).IKK phosphorylation of IkB leads to release of NF-kB dimers andtheir ability to translocate to the nucleus and bind DNA targets.Our studies demonstrate that NF-kB is sufficient to induce thehuman IFN-l1 gene during the response to DNA damage. Thepromoter of the human IFN-l1 gene has a bona fide NF-kBbinding site as well as an IRF binding site (21). NF-kB alsoinduces the IRF-1 and IRF-7 genes that can influence expressionof the IFN-a and IFN-l genes. The induced IFNs can additionallyamplify expression of the IRFs.These findings add a new dimension to the complexity of the
DNA damage response. ATM appears to be primarily accountablefor initial signal pathways that lead to human IFN gene expres-sion by etoposide. Inhibition of ATM significantly reduces the in-duction of IFN-a and IFN-l1 genes (Fig. 6). The ATM deficiencyresponsible for the development of ataxia-telangiectasia results
in an array of clinical manifestations including cerebellar ataxia,oculocutaneous telangiectasia, immunodeficiency, and suscepti-bility to cancer (69, 70). The lack of IFN production in responseto DNA damage that occurs through physiological processes inataxia-telangiectasia speculatively may contribute to the immu-nodeficiency and tumor formation in the disease. Results of a fewstudies have suggested a potential role of IFN signaling duringDNA damage (71–74). One study observed ISG expression cor-related with resistance to radiation therapy (74), and another
FIGURE 7. NF-kB is activated and required for IFN gene induction in
response to etoposide. A, HeLa cells were treated with etoposide for 15 h
or 5 ng/ml TNF-a for 1 h, and p65 was immunoprecipitated (IP) from
lysates. Specific Ab to p65 phospho-serine 536 was used for the Western
blot (WB). Lower panel displays Western blot with Ab to p65. B, HeLa
cells were untreated or treated with etoposide for 2 h before immuno-
staining with Abs to p65. C, HeLa cells were untreated or treated with
etoposide (E) in absence or presence of ML120B. Left panel, Real-time
PCR was used with pan-specific primers to quantify the endogenous levels
of IFN-a mRNA. Right panel, Real-time PCR was used to quantify the
endogenous levels of IFN-l1 mRNA. D, Left panel, The IFN-l1 luciferase
reporter plasmid was cotransfected with empty vector or with a plasmid
encoding the dominant-negative IkBS32A/S36A (IkBSS/AA) gene. Cells
were untreated or treated with etoposide for 24 h prior to the luciferase
assays. Right panel, The IFN-l1 luciferase reporter plasmid was cotrans-
fected with empty vector (c) or with a plasmid encoding the p65/RelA
gene. Cells were untreated or treated with etoposide for 24 h prior to lu-
ciferase assays. Quantitative results are means of duplicate determinations
in three independent experiments.
FIGURE 6. Activation and inhibition of IRF and IFN genes. A, THP-1
cells were untreated (2) or treated with etoposide (+) for 24 h. IRF-1
mRNA levels were evaluated by RT-PCR and displayed on agarose gels.
mRNA levels of actin are shown as controls. B, The IFN-l1 luciferase
reporter was expressed in HeLa cells untreated or treated with etopo-
side (Etop) for 24 h. Empty vector (c) or IRF-1 expression plasmid (IRF1)
were cotransfected where indicated with or without etoposide treatment
and luciferase activity was measured. C, Effects of the ATM inhibitor
AZ12622702 (AZ) on IFN-a and IFN-l gene expression. HeLa cells were
untreated or treated with AZ for 1 h followed by etoposide (E) for 24 h
as indicated. Real-time PCR was used to quantify IFN-a (left panel) or
IFN-l (right panel) mRNA expression. D, Effects of IKKb inhibitors
BAY117085 (BAY) or ML120B (ML120) on IRF-1 and IRF-7 gene ex-
pression. HeLa cells were untreated or treated with the inhibitors for 1 h
followed by etoposide for 5 h as indicated. Real-time PCR was used to
quantify endogenous IRF-1 (left panel) or IRF-7 (right panel) mRNA
expression. Quantitative results are means of duplicate determinations in
three independent experiments.
5342 DNA DAMAGE RESPONSE INDUCES IFN
by guest on June 28, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

study reported that STAT1 facilitated cell-cycle checkpoint fol-lowing DNA damage (72). IFN pathways not only stimulate Januskinases and STAT factors but also elicit a broad range of effectson transcription and translation. The contribution of IFN signal-ing in the response to DNA breaks may have multifaceted con-sequences.The novel observation of the convergence of the DNA damage
response with IFN signaling stimulates speculation as to thepossible function of IFN in reaction to genotoxic stress. In ourexperimental system, the addition of IFN did not block the apo-ptotic effects of etoposide or significantly contribute to cell death(S. Brzostek-Racine and N.C. Reich, unpublished observations).But IFNs are well characterized for their ability to inhibit viralinfection, and this may reflect the evolutionary link. Many viralinfections are known to stimulate DNA damage response pathways.Viruses like HIV have RNA genomes but integrate viral DNA intothe host genome, creating DNA strand breaks (4). Viruses withDNA genomes can generate ss- and dsDNA breaks during lyticreplication. EBV, HSV1, and adenovirus are a few examples ofDNA viruses that have been documented to activate a DNAdamage response (4, 75, 76). More significantly, some of theseviruses have evolved mechanisms to inhibit activation or down-stream function of the DNA damage response (77–79). Virusesmay inhibit this pathway not only to block cell cycle arrest andapoptosis, but also to block the antiviral functions of IFNs thatare produced by DNA damage.Etoposide and IFN both have been used clinically for years for
their antitumorigenic effects. IFNs produced in response to DNAdamage may contribute to the antitumorigenic effects of etoposide.IFNs are recognized for their ability to cause growth arrest and/orapoptosis in neoplastic cells, although they can stimulate prolif-
eration of healthy cells (24, 25, 46, 80–83). They also have vi-tal immunoregulatory functions that include direct and indirecteffects on activation of NK cells, macrophages, dendritic cells,T cells, and B cells (84, 85). The antiproliferative effects of IFNand the potential enhanced clearance of tumor cells may playa role in the in vivo DNA damage response pathway.The profile of IFN gene expression in response to DNA damage
in human cells is distinct from that induced by viral infection. Theexistence of multiple IFN genes with distinct promoter elementsappears to have evolved as a response to different cellular stresses.A significant finding of our study is the ability of NF-kB to stim-ulate the expression of IFN genes in the absence of viral infec-tion. NF-kB is required for the induction of IRF-1, IRF-7, IFN-l,and IFN-a in response to etoposide. Although the promoter ofthe human IFN-l1 gene possesses both an NF-kB binding site andan IRF binding site, the promoters of the IFN-a genes possessonly IRF binding sites. Results with the knockout MEFs indicatea critical function of IRF-7 activated in response to etoposide forinduction of both murine IFN-l2 and IFN-a genes. Future studiesare needed to provide additional insight on the impact of IRF-1and IRF-7 on the IFN genes during the DNA damage response andwhether there are significant mechanistic differences in the humanversus murine response. NF-kB is activated not only by DNAdamage but also by a wide array of cellular stimuli, and it playsa major role in inflammation, immunity, cell survival, and cancer(63). The intimate link of NF-kB to IFN gene induction during theDNA damage response may reflect a potential role of IFN in otherbiological responses to NF-kB in the absence of infections. Ourstudy adds a significant finding of IFN signaling to the complexityof pathways that are orchestrated by the response to genotoxicstress.
FIGURE 8. Effect of etoposide on MEFs. MEFs isolated from wt mice
or mice with targeted gene knockouts in NEMO (NEMO2/2), IRF-3
(IRF32/2), IRF-1 (IRF12/2), or IRF-7 (IRF72/2) were untreated or treated
with etoposide for 24 h. Real-time RT-PCR was used to quantify endog-
enous levels of murine IFN-l2 (A) or pan–IFN-a (B). Quantitative results
are means of duplicate determinations in three independent experiments.
FIGURE 9. Time course of IRF and IFN mRNA expression in the re-
sponse to etoposide treatment. THP-1 cells were treated with etoposide,
and RNA was isolated from cells during a 24-h period at the times in-
dicated. Real-time RT-PCR with specific primers was used to quantify
IRF-1 and IRF-7 mRNA (A) or IFN-a and IFN-l1 mRNA (B). Results are
means of two experiments performed in duplicate. C, Conceptual model of
dsDNA break response signaling to IFN gene expression. ATM phos-
phorylation of NEMO promotes its ability to bind and activate the IKK
complexes. IKKs phosphorylate IkB, releasing NF-kB to activate gene
targets IFN-l1, IRF-1, and IRF-7. The IRFs induce IFN-a genes and en-
hance IFN-l1 expression.
The Journal of Immunology 5343
by guest on June 28, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

AcknowledgmentsWe thank all of the members of the Reich laboratory for support. We also
thank Dr. Martha Furie, Gregory Sabino, and Indralatha Jayatilaka for
helpfulness in preparation of primary monocytes.
DisclosuresThe authors have no financial conflicts of interest.
References1. Vilenchik, M. M., and A. G. Knudson. 2003. Endogenous DNA double-strand
breaks: production, fidelity of repair, and induction of cancer. Proc. Natl. Acad.Sci. USA 100: 12871–12876.
2. Fugmann, S. D., A. I. Lee, P. E. Shockett, I. J. Villey, and D. G. Schatz. 2000.The RAG proteins and V(D)J recombination: complexes, ends, and trans-position. Annu. Rev. Immunol. 18: 495–527.
3. Richardson, C., N. Horikoshi, and T. K. Pandita. 2004. The role of the DNAdouble-strand break response network in meiosis. DNA Repair (Amst.) 3: 1149–1164.
4. Sinclair, A., S. Yarranton, and C. Schelcher. 2006. DNA-damage responsepathways triggered by viral replication. Expert Rev. Mol. Med. 8: 1–11.
5. Mahaney, B. L., K. Meek, and S. P. Lees-Miller. 2009. Repair of ionizingradiation-induced DNA double-strand breaks by non-homologous end-joining.Biochem. J. 417: 639–650.
6. Jackson, S. P., and J. Bartek. 2009. The DNA-damage response in human biologyand disease. Nature 461: 1071–1078.
7. Su, T. T. 2006. Cellular responses to DNA damage: one signal, multiple choices.Annu. Rev. Genet. 40: 187–208.
8. Shiloh, Y. 2006. The ATM-mediated DNA-damage response: taking shape.Trends Biochem. Sci. 31: 402–410.
9. Abraham, R. T. 2004. PI 3-kinase related kinases: ‘big’ players in stress-inducedsignaling pathways. DNA Repair (Amst.) 3: 883–887.
10. Elkon, R., S. Rashi-Elkeles, Y. Lerenthal, C. Linhart, T. Tenne, N. Amariglio,G. Rechavi, R. Shamir, and Y. Shiloh. 2005. Dissection of a DNA-damage-induced transcriptional network using a combination of microarrays, RNA in-terference and computational promoter analysis. Genome Biol. 6: R43.
11. Li, N., S. Banin, H. Ouyang, G. C. Li, G. Courtois, Y. Shiloh, M. Karin, andG. Rotman. 2001. ATM is required for IkappaB kinase (IKKk) activation inresponse to DNA double strand breaks. J. Biol. Chem. 276: 8898–8903.
12. Fensterl, V., and G. C. Sen. 2009. Interferons and viral infections. Biofactors 35:14–20.
13. Yoneyama, M., and T. Fujita. 2010. Recognition of viral nucleic acids in innateimmunity. Rev. Med. Virol. 20: 4–22.
14. Panne, D., T. Maniatis, and S. C. Harrison. 2007. An atomic model of theinterferon-beta enhanceosome. Cell 129: 1111–1123.
15. Tamura, T., H. Yanai, D. Savitsky, and T. Taniguchi. 2008. The IRF familytranscription factors in immunity and oncogenesis. Annu. Rev. Immunol. 26:535–584.
16. Andersen, J., S. VanScoy, T. F. Cheng, D. Gomez, and N. C. Reich. 2008. IRF-3-dependent and augmented target genes during viral infection. Genes Immun. 9:168–175.
17. Sato, M., H. Suemori, N. Hata, M. Asagiri, K. Ogasawara, K. Nakao, T. Nakaya,M. Katsuki, S. Noguchi, N. Tanaka, and T. Taniguchi. 2000. Distinct and es-sential roles of transcription factors IRF-3 and IRF-7 in response to viruses forIFN-alpha/beta gene induction. Immunity 13: 539–548.
18. Marie, I., J. E. Durbin, and D. E. Levy. 1998. Differential viral induction ofdistinct interferon-alpha genes by positive feedback through interferon regula-tory factor-7. EMBO J. 17: 6660–6669.
19. Stark, G. R., I. M. Kerr, B. R. Williams, R. H. Silverman, and R. D. Schreiber.1998. How cells respond to interferons. Annu. Rev. Biochem. 67: 227–264.
20. Levy, D. E., and J. E. Darnell, Jr. 2002. Stats: transcriptional control and bi-ological impact. Nat. Rev. Mol. Cell Biol. 3: 651–662.
21. Onoguchi, K., M. Yoneyama, A. Takemura, S. Akira, T. Taniguchi, H. Namiki,and T. Fujita. 2007. Viral infections activate types I and III interferon genesthrough a common mechanism. J. Biol. Chem. 282: 7576–7581.
22. Osterlund, P. I., T. E. Pietila, V. Veckman, S. V. Kotenko, and I. Julkunen. 2007.IFN regulatory factor family members differentially regulate the expression oftype III IFN (IFN-lambda) genes. J. Immunol. 179: 3434–3442.
23. Borden, E. C., G. C. Sen, G. Uze, R. H. Silverman, R. M. Ransohoff,G. R. Foster, and G. R. Stark. 2007. Interferons at age 50: past, current and futureimpact on biomedicine. Nat. Rev. Drug Discov. 6: 975–990.
24. Clemens, M. J. 2003. Interferons and apoptosis. J. Interferon Cytokine Res. 23:277–292.
25. Li, W., A. Lewis-Antes, J. Huang, M. Balan, and S. V. Kotenko. 2008. Regu-lation of apoptosis by type III interferons. Cell Prolif. 41: 960–979.
26. Miller, C. H., S. G. Maher, and H. A. Young. 2009. Clinical Use of Interferon-gamma. Ann. N. Y. Acad. Sci. 1182: 69–79.
27. Makris, C., V. L. Godfrey, G. Krahn-Senftleben, T. Takahashi, J. L. Roberts,T. Schwarz, L. Feng, R. S. Johnson, and M. Karin. 2000. Female mice hetero-zygous for IKK gamma/NEMO deficiencies develop a dermatopathy similar tothe human X-linked disorder incontinentia pigmenti. Mol. Cell 5: 969–979.
28. Kimura, T., K. Nakayama, J. Penninger, M. Kitagawa, H. Harada, T. Matsuyama,N. Tanaka, R. Kamijo, J. Vilcek, T. W. Mak, et al. 1994. Involvement of the IRF-
1 transcription factor in antiviral responses to interferons. Science 264: 1921–1924.
29. Honda, K., H. Yanai, H. Negishi, M. Asagiri, M. Sato, T. Mizutani, N. Shimada,Y. Ohba, A. Takaoka, N. Yoshida, and T. Taniguchi. 2005. IRF-7 is the masterregulator of type-I interferon-dependent immune responses. Nature 434: 772–777.
30. Weaver, B. K., K. P. Kumar, and N. C. Reich. 1998. Interferon regulatory factor 3and CREB-binding protein/p300 are subunits of double-stranded RNA-activatedtranscription factor DRAF1. Mol. Cell. Biol. 18: 1359–1368.
31. Kumar, K. P., K. M. McBride, B. K. Weaver, C. Dingwall, and N. C. Reich.2000. Regulated nuclear-cytoplasmic localization of interferon regulatory factor3, a subunit of double-stranded RNA-activated factor 1. Mol. Cell. Biol. 20:4159–4168.
32. Banninger, G., and N. C. Reich. 2004. STAT2 nuclear trafficking. J. Biol. Chem.279: 39199–39206.
33. McBride, K. M., C. McDonald, and N. C. Reich. 2000. Nuclear export signallocated within theDNA-binding domain of the STAT1transcription factor. EMBOJ. 19: 6196–6206.
34. Scherer, D. C., J. A. Brockman, Z. Chen, T. Maniatis, and D. W. Ballard. 1995.Signal-induced degradation of I kappa B alpha requires site-specific ubiquiti-nation. Proc. Natl. Acad. Sci. USA 92: 11259–11263.
35. Jura, N., E. Scotto-Lavino, A. Sobczyk, and D. Bar-Sagi. 2006. Differentialmodification of Ras proteins by ubiquitination. Mol. Cell 21: 679–687.
36. Zhang, L., and J. S. Pagano. 1997. IRF-7, a new interferon regulatory factorassociated with Epstein-Barr virus latency. Mol. Cell. Biol. 17: 5748–5757.
37. Ank, N., H. West, C. Bartholdy, K. Eriksson, A. R. Thomsen, and S. R. Paludan.2006. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses andIFNs and displays potent antiviral activity against select virus infections in vivo.J. Virol. 80: 4501–4509.
38. Loseke, S., E. Grage-Griebenow, A. Wagner, K. Gehlhar, and A. Bufe. 2003.Differential expression of IFN-alpha subtypes in human PBMC: evaluation ofnovel real-time PCR assays. J. Immunol. Methods 276: 207–222.
39. Medhurst, A. D., D. C. Harrison, S. J. Read, C. A. Campbell, M. J. Robbins, andM. N. Pangalos. 2000. The use of TaqMan RT-PCR assays for semiquantitativeanalysis of gene expression in CNS tissues and disease models. J. Neurosci.Methods 98: 9–20.
40. Gadberry, M. D., S. T. Malcomber, A. N. Doust, and E. A. Kellogg. 2005.Primaclade—a flexible tool to find conserved PCR primers across multiplespecies. Bioinformatics 21: 1263–1264.
41. Ank, N., M. B. Iversen, C. Bartholdy, P. Staeheli, R. Hartmann, U. B. Jensen,F. Dagnaes-Hansen, A. R. Thomsen, Z. Chen, H. Haugen, et al. 2008. An im-portant role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviralactivity. J. Immunol. 180: 2474–2485.
42. Cheng, T. F., S. Brzostek, O. Ando, S. Van Scoy, K. P. Kumar, and N. C. Reich.2006. Differential activation of IFN regulatory factor (IRF)-3 and IRF-5 tran-scription factors during viral infection. J. Immunol. 176: 7462–7470.
43. Tansey, W. P. 2006. Detection of ubiquitylated proteins in mammalian cells.Cold Spring Harb. Protoc. Available at: http://cshprotocols.cshlp.org/content/2006/6/pdb.prot4616.abstract.
44. Weaver, B. K., O. Ando, K. P. Kumar, and N. C. Reich. 2001. Apoptosis ispromoted by the dsRNA-activated factor (DRAF1) during viral infection in-dependent of the action of interferon or p53. FASEB J. 15: 501–515.
45. Heylbroeck, C., S. Balachandran, M. J. Servant, C. DeLuca, G. N. Barber,R. Lin, and J. Hiscott. 2000. The IRF-3 transcription factor mediates Sendaivirus-induced apoptosis. J. Virol. 74: 3781–3792.
46. Chawla-Sarkar, M., D. J. Lindner, Y. F. Liu, B. R. Williams, G. C. Sen,R. H. Silverman, and E. C. Borden. 2003. Apoptosis and interferons: role ofinterferon-stimulated genes as mediators of apoptosis. Apoptosis 8: 237–249.
47. Baldwin, E. L., and N. Osheroff. 2005. Etoposide, topoisomerase II and cancer.Curr. Med. Chem. Anticancer Agents 5: 363–372.
48. Meresse, P., E. Dechaux, C. Monneret, and E. Bertounesque. 2004. Etoposide:discovery and medicinal chemistry. Curr. Med. Chem. 11: 2443–2466.
49. Reich, N. C., and L. Liu. 2006. Tracking STAT nuclear traffic. Nat. Rev.Immunol. 6: 602–612.
50. Aoki, R., and J. J. Kavanagh. 2004. Antineoplastic agents: classification andmechanisms of action. In Chemotherapy for Gynecological Neoplasms: CurrentTherapy and Novel Approaches, 1st Ed. P. B. P. R. Angioli, J. J. Kavanagh,S. Pecorelli, and M. Penalver, eds. Marcel Dekker, New York, p. 1–12.
51. Genin, P., R. Lin, J. Hiscott, and A. Civas. 2009. Differential regulation of hu-man interferon A gene expression by interferon regulatory factors 3 and 7. Mol.Cell. Biol. 29: 3435–3450.
52. Hiscott, J. 2007. Triggering the innate antiviral response through IRF-3 activa-tion. J. Biol. Chem. 282: 15325–15329.
53. Au, W. C., P. A. Moore, D. W. LaFleur, B. Tombal, and P. M. Pitha. 1998.Characterization of the interferon regulatory factor-7 and its potential role in thetranscription activation of interferon A genes. J. Biol. Chem. 273: 29210–29217.
54. Caillaud, A., A. G. Hovanessian, D. E. Levy, and I. J. Marie. 2005. Regulatoryserine residues mediate phosphorylation-dependent and phosphorylation-independent activation of interferon regulatory factor 7. J. Biol. Chem. 280:17671–17677.
55. Huye, L. E., S. Ning, M. Kelliher, and J. S. Pagano. 2007. Interferon regulatoryfactor 7 is activated by a viral oncoprotein through RIP-dependent ubiquitina-tion. Mol. Cell. Biol. 27: 2910–2918.
56. Bennett, E. J., and J. W. Harper. 2008. DNA damage: ubiquitin marks the spot.Nat. Struct. Mol. Biol. 15: 20–22.
57. Tamura, T., M. Ishihara, M. S. Lamphier, N. Tanaka, I. Oishi, S. Aizawa,T. Matsuyama, T. W. Mak, S. Taki, and T. Taniguchi. 1995. An IRF-1-dependent
5344 DNA DAMAGE RESPONSE INDUCES IFN
by guest on June 28, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

pathway of DNA damage-induced apoptosis in mitogen-activated T lympho-cytes. Nature 376: 596–599.
58. Tanaka, N., M. Ishihara, M. S. Lamphier, H. Nozawa, T. Matsuyama, T. W. Mak,S. Aizawa, T. Tokino, M. Oren, and T. Taniguchi. 1996. Cooperation of thetumour suppressors IRF-1 and p53 in response to DNA damage. Nature 382:816–818.
59. Sims, S. H., Y. Cha, M. F. Romine, P. Q. Gao, K. Gottlieb, and A. B. Deisseroth.1993. A novel interferon-inducible domain: structural and functional analysis ofthe human interferon regulatory factor 1 gene promoter.Mol. Cell. Biol. 13: 690–702.
60. Pine, R. 1997. Convergence of TNFalpha and IFNgamma signalling pathwaysthrough synergistic induction of IRF-1/ISGF-2 is mediated by a composite GAS/kappaB promoter element. Nucleic Acids Res. 25: 4346–4354.
61. Lu, R., P. A. Moore, and P. M. Pitha. 2002. Stimulation of IRF-7 gene expressionby tumor necrosis factor alpha: requirement for NFkappa B transcription factorand gene accessibility. J. Biol. Chem. 277: 16592–16598.
62. Wu, Z. H., Y. Shi, R. S. Tibbetts, and S. Miyamoto. 2006. Molecular linkagebetween the kinase ATM and NF-kappaB signaling in response to genotoxicstimuli. Science 311: 1141–1146.
63. Vallabhapurapu, S., and M. Karin. 2009. Regulation and function of NF-kappaBtranscription factors in the immune system. Annu. Rev. Immunol. 27: 693–733.
64. Perkins, N. D. 2006. Post-translational modifications regulating the activity andfunction of the nuclear factor kappa B pathway. Oncogene 25: 6717–6730.
65. Harrison, J. C., and J. E. Haber. 2006. Surviving the breakup: the DNA damagecheckpoint. Annu. Rev. Genet. 40: 209–235.
66. Nyberg, K. A., R. J. Michelson, C. W. Putnam, and T. A. Weinert. 2002. Towardmaintaining the genome: DNA damage and replication checkpoints. Annu. Rev.Genet. 36: 617–656.
67. Perkins, N. D. 2007. Integrating cell-signalling pathways with NF-kappaB andIKK function. Nat. Rev. Mol. Cell Biol. 8: 49–62.
68. Stilmann, M., M. Hinz, S. C. Arslan, A. Zimmer, V. Schreiber, andC. Scheidereit. 2009. A nuclear poly(ADP-ribose)-dependent signalosome con-fers DNA damage-induced IkappaB kinase activation. Mol. Cell 36: 365–378.
69. Chun, H. H., and R. A. Gatti. 2004. Ataxia-telangiectasia, an evolving pheno-type. DNA Repair (Amst.) 3: 1187–1196.
70. Lavin, M. F. 2008. Ataxia-telangiectasia: from a rare disorder to a paradigm forcell signalling and cancer. Nat. Rev. Mol. Cell Biol. 9: 759–769.
71. Liu, M., B. T. Hummer, X. Li, and B. A. Hassel. 2004. Camptothecin induces theubiquitin-like protein, ISG15, and enhances ISG15 conjugation in response tointerferon. J. Interferon Cytokine Res. 24: 647–654.
72. Townsend, P. A., M. S. Cragg, S. M. Davidson, J. McCormick, S. Barry,K. M. Lawrence, R. A. Knight, M. Hubank, P. L. Chen, D. S. Latchman, andA. Stephanou. 2005. STAT-1 facilitates the ATM activated checkpoint pathwayfollowing DNA damage. J. Cell Sci. 118: 1629–1639.
73. Tsai, M. H., J. A. Cook, G. V. Chandramouli, W. DeGraff, H. Yan, S. Zhao,C. N. Coleman, J. B. Mitchell, and E. Y. Chuang. 2007. Gene expression pro-filing of breast, prostate, and glioma cells following single versus fractionateddoses of radiation. Cancer Res. 67: 3845–3852.
74. Weichselbaum, R. R., H. Ishwaran, T. Yoon, D. S. Nuyten, S. W. Baker,N. Khodarev, A. W. Su, A. Y. Shaikh, P. Roach, B. Kreike, et al. 2008. Aninterferon-related gene signature for DNA damage resistance is a predictivemarker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci.USA 105: 18490–18495.
75. Shirata, N., A. Kudoh, T. Daikoku, Y. Tatsumi, M. Fujita, T. Kiyono, Y. Sugaya,H. Isomura, K. Ishizaki, and T. Tsurumi. 2005. Activation of ataxiatelangiectasia-mutated DNA damage checkpoint signal transduction elicited byherpes simplex virus infection. J. Biol. Chem. 280: 30336–30341.
76. Lilley, C. E., M. S. Chaurushiya, and M. D. Weitzman. 2010. Chromatin at theintersection of viral infection and DNA damage. Biochim. Biophys. Acta 1799:319–327.
77. Evans, J. D., and P. Hearing. 2005. Relocalization of the Mre11-Rad50-Nbs1complex by the adenovirus E4 ORF3 protein is required for viral replication. J.Virol. 79: 6207–6215.
78. Stracker, T. H., C. T. Carson, and M. D. Weitzman. 2002. Adenovirus onco-proteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418:348–352.
79. Lilley, C. E., M. S. Chaurushiya, C. Boutell, S. Landry, J. Suh, S. Panier,R. D. Everett, G. S. Stewart, D. Durocher, and M. D. Weitzman. 2010. A viral E3ligase targets RNF8 and RNF168 to control histone ubiquitination and DNAdamage responses. EMBO J. 29: 943–955.
80. Moiseeva, O., F. A. Mallette, U. K. Mukhopadhyay, A. Moores, and G. Ferbeyre.2006. DNA damage signaling and p53-dependent senescence after prolongedbeta-interferon stimulation. Mol. Biol. Cell 17: 1583–1592.
81. Takaoka, A., S. Hayakawa, H. Yanai, D. Stoiber, H. Negishi, H. Kikuchi,S. Sasaki, K. Imai, T. Shibue, K. Honda, and T. Taniguchi. 2003. Integration ofinterferon-alpha/beta signalling to p53 responses in tumour suppression andantiviral defence. Nature 424: 516–523.
82. Thyrell, L., S. Erickson, B. Zhivotovsky, K. Pokrovskaja, O. Sangfelt, J. Castro,S. Einhorn, and D. Grander. 2002. Mechanisms of Interferon-alpha inducedapoptosis in malignant cells. Oncogene 21: 1251–1262.
83. Gomez, D., and N. C. Reich. 2003. Stimulation of primary human endothelialcell proliferation by IFN. J. Immunol. 170: 5373–5381.
84. Le Bon, A., and D. F. Tough. 2002. Links between innate and adaptive immunityvia type I interferon. Curr. Opin. Immunol. 14: 432–436.
85. Prchal, M., A. Pilz, O. Simma, K. Lingnau, A. von Gabain, B. Strobl, M. Muller,and T. Decker. 2009. Type I interferons as mediators of immune adjuvants forT- and B cell-dependent acquired immunity. Vaccine 27(Suppl. 6): G17–G20.)
The Journal of Immunology 5345
by guest on June 28, 2018http://w
ww
.jimm
unol.org/D
ownloaded from