the clinical spectrum of adults with tetralogy of fallot ... · pdf filethe clinical spectrum...
TRANSCRIPT

The Clinical Spectrum of Adults with Tetralogy of Fallot: Cardiac and Extra-cardiac Features and Late Outcomes
by
Sara Piran
A thesis submitted in conformity with the requirements for the degree of Master’s of Science
Institute of Medical Science University of Toronto
© Copyright by Sara Piran 2010

ii
The Clinical Spectrum of Adults with Tetralogy of Fallot: Cardiac and Extra-cardiac Features and Late Outcomes
Sara Piran
Master’s of Science
Institute of Medical Science University of Toronto
2010
Abstract
Tetralogy of Fallot (TOF) is a form of complex congenital heart disease (CHD) with clinical and
genetic heterogeneity. Of the few known causes, 22q11.2 deletion syndrome is most common.
We sought to define other clinical subgroups by focusing on congenital cardiac and extra-cardiac
features, and cardiac outcome. Patients were prospectively categorized as “syndromic” if they
had at least two of three features: dysmorphic facies, learning difficulties or voice abnormalities.
We compared cardiac and extra-cardiac characteristics, and late cardiac outcomes between the
syndromic group and a nonsyndromic control group. The syndromic group had a more complex
cardiac disease, was at elevated risk for developing later onset conditions including
neuropsychiatric disorders, endocrine disorders, and hearing deficits, and had a higher mortality
rate compared to the nonsyndromic group. Increased awareness of this subgroup with a
multisystem condition may be helpful for optimizing management and identifying individuals for
referral to medical genetics.

iii
Acknowledgments
First and foremost I would like to thank my kind and amazing supervisor, Dr. Candice
Silversides, for all her support, guidance, wisdom, and invaluable learning experience throughout
my graduate studies.
I would also like to thank my exceptional advisory committee, Drs. Anne Bassett and Peter Liu,
for their continuing support and contribution.
Very special thanks to Dr. Jasmine Grewal for her limitless support and help.
Many thanks to the members of the CGRP for their friendship, help, and all the fun moments we
shared! To Dr. Jodi-Ann Swaby for her invaluable friendship, support, and all the fun talks and
times we had.
To my dearest friends, Zeynep and Shahad, for always being there for me no matter what; I will
always cherish our wonderful memories!
To all the staff and fellows at the TCCCA, thank you for your advice and help; working with all
of you was very enjoyable!
Last but not least, I would like to sincerely thank my beautiful family for all their support and
endless love, and for always believing in me!

iv
Table of Contents
Acknowledgments .......................................................................................................................... iii
Table of Contents ........................................................................................................................... iv
List of Figures .............................................................................................................................. viii
List of abbreviations ....................................................................................................................... ix
Overview ......................................................................................................................................... 1
Introduction ..................................................................................................................................... 5
1.1 Tetralogy of fallot................................................................................................................ 5
1.1.1 Overview ................................................................................................................. 5
1.1.2 Basic TOF cardiac anatomy .................................................................................... 5
1.1.3 Anatomic variations of TOF ................................................................................... 6
1.1.4 Diagnosis ................................................................................................................. 8
1.2 TOF in childhood ................................................................................................................ 9
1.2.1 Surgical management ............................................................................................ 10
1.2.2 Palliative surgery ................................................................................................... 10
1.2.3 Intracardiac repair of TOF..................................................................................... 10
1.3 TOF in adulthood .............................................................................................................. 12
1.3.1 Cardiac features in adulthood late after repair ...................................................... 12
1.3.2 Exercise Limitations .............................................................................................. 13
1.3.3 Reoperation ........................................................................................................... 20
1.4 The non-cardiac phenotype in adults with TOF ................................................................ 22
1.4.1 Congenital (structural) extra-cardiac anomalies ................................................... 22
1.4.2 Later onset manifestations ..................................................................................... 25
1.4.3 Neurodevelopmental outcomes ............................................................................. 26

v
1.5 Genetics of TOF ................................................................................................................ 27
1.5.1 Heritability ............................................................................................................ 27
1.5.2 Genetic causes of TOF .......................................................................................... 28
1.6 Genetic determinants of cardiac outcome ......................................................................... 31
1.7 22q11.2 deletion syndrome (reference group for Study 1 and Study 2) ........................... 33
1.7.1 Characteristic features of 22q11DS....................................................................... 34
1.7.2 Cardiovascular malformations in 22q11DS .......................................................... 34
1.7.3 Ear, nose and throat in 22q11DS ........................................................................... 35
1.7.4 Immune system in 22q11DS ................................................................................. 35
1.7.5 Endocrine system in 22q11DS .............................................................................. 36
1.7.6 Cognitive function, neuropsychiatric and central nervous system abnormalities in 22q11DS ............................................................................................................ 37
1.7.7 Musculoskeletal system in 22q11DS .................................................................... 38
1.7.8 Genitourinary system in 22q11DS ........................................................................ 39
2 Aims/Hypotheses ...................................................................................................................... 41
3 Study 1 ...................................................................................................................................... 44
3.1 Objectives .......................................................................................................................... 45
3.2 Methods ............................................................................................................................. 46
3.3 Results ............................................................................................................................... 49
3.3.1 Congenital cardiovascular anomalies .................................................................... 50
3.3.2 Congenital extra-cardiac anomalies ...................................................................... 51
3.3.3 Late-onset manifestations ...................................................................................... 52
3.4 Discussion ......................................................................................................................... 54
3.5 Clinical Implications ......................................................................................................... 58
3.6 Study limitations and advantages ...................................................................................... 59
3.7 Conclusion ......................................................................................................................... 60

vi
4 Study 2. ..................................................................................................................................... 66
4.1 Objectives .......................................................................................................................... 67
4.2 Methods ............................................................................................................................. 68
4.3 Primary outcomes .............................................................................................................. 70
4.4 Secondary outcomes .......................................................................................................... 70
4.5 Statistical analysis ............................................................................................................. 71
4.6 Results ............................................................................................................................... 72
4.7 Screening characteristics ................................................................................................... 72
4.7.1 Baseline characteristics in childhood .................................................................... 72
4.8 Primary outcomes .............................................................................................................. 73
4.8.1 All-cause mortality ................................................................................................ 73
4.9 Secondary outcomes .......................................................................................................... 75
4.9.1 Adverse Nonfatal Cardiac Events ......................................................................... 75
4.9.2 Cardiac interventions............................................................................................. 76
4.10 Discussion ......................................................................................................................... 77
4.11 Clinical Implications ......................................................................................................... 80
4.12 Study limitations and advantages ...................................................................................... 80
4.13 Conclusion ......................................................................................................................... 81
5 Summary of Thesis and Future Directions ............................................................................... 91
5.1 Future directions ................................................................................................................ 93
6 Appendices ............................................................................................................................... 95
7 References .............................................................................................................................. 101

vii
List of Tables
Table I. Late postoperative complications in patients with TOF
Table II. Potential causes of death in patients with TOF
Table III. Congenital extra-cardiac anomalies associated with TOF
Table IV. Extra-cardiac abnormalities associated with 22q11.2 deletions
Table V. Baseline characteristics in 207 adults with TOF
Table VI. Congenital cardiovascular anomalies in adults with TOF
Table VII. Congenital extra-cardiac anomalies in adults with TOF
Table VIII. Late-onset extra-cardiac conditions in adults with TOF
Table IX. Childhood cardiac and extra-cardiac characteristics in adults with TOF Table X. Pediatric cardiac interventions and adverse cardiac events Table XI. Extra-cardiac characteristics in the five deceased subjects in the syndromic group Table XII. Cardiac characteristics in the five deceased subjects in the syndromic group Table XIII. Adverse nonfatal cardiac events in adulthood Table XIV. Cardiovascular interventions in adulthood

viii
List of Figures
Figure. 1. Intracardiac anatomy of tetralogy of Fallot
Figure 2. Intracardiac repair of TOF
Figure 3. Example of a subject from the syndromic group
Figure 4. Categorization of patients into the nonsyndromic group, syndromic group, and 22q11DS reference group.
Figure 5. Proportion of adult TOF subjects with one or more congenital extra-cardiac anomaly
Figure 6. Differences in survival in adults with TOF
Figure 7. Freedom from adverse cardiac events in adults with TOF Figure 8. Freedom from cardiac intervention in adults with TOF

ix
List of abbreviations
22q11DS 22q11.2 deletion syndrome
ADD/ADHD attention deficit disorder/attention deficit hyperactivity disorder
APVS absent pulmonary valve syndrome
ASD atrial septal defect
AICD automatic implantable cardioverter defibrillator
BT Blalock-Taussig
CAFS conotruncal anomaly face syndrome
CHD congenital heart disease
CHF congestive heart failure
CNV copy number variation
DGS DiGeorge syndrome
ECG electrocardiogram
MAPCA major aorto-pulmonary collateral arteries
MSK musculoskeletal
PVR pulmonary valve replacement
RVOTO right ventricular outflow tract obstruction
RV-PA right ventricle-pulmonary artery
SVC superior vena cava
TOF tetralogy of Fallot
VCFS velocardiofacial syndrome
VSD ventricular septal defect

1
Overview
Tetralogy of Fallot (TOF) is the most common cyanotic congenital heart disease
(Hoffman 1995) with both clinical and genetic heterogeneity. The most common underlying
genetic anomaly in patients with TOF is 22q11.2 deletion syndrome (22q11DS), occurring in 10-
16% of cases (Goldmuntz, Clark et al. 1998, Botto, May et al. 2003). This syndrome is
associated with auxiliary cardiac and extra-cardiac anomalies and phenotype and genotype
characteristics have been extensively studied. In most cases, however, the genetic etiology of
TOF is unknown.
Brief clinical genetic screening can help to identify adults with 22q11DS and other
genetic syndromes (Fung, Chow et al. 2008). Identification of these patients is important because
there are important genetic and clinical issues in this patient population. Among patients with
TOF, there is substantial phenotypic variability and there likely exist other, not yet identified
syndromes, with unique clinical features. Understanding the clinical phenotypes and genetic
variants in patients with TOF is important for both clinical management and genetic counseling.
The overriding goal of this thesis, presented in two parts (STUDY 1 and STUDY 2), was
to attempt to identify clinical features of a subgroup of adults with TOF, identified on clinical
genetic screening, who had features suggestive of a genetic syndrome. Ultimately, clustering
patients into meaningful categories based on phenotypic features may help identify homogenous
groups of patients who may have relevance to the pathogenesis of TOF.

2
STUDY 1.
TOF is a complex cardiac disease with a number of potential cardiac variations including
the most severe form, pulmonary atresia/ventricular septal defect, to a milder form with minimal
outflow tract obstruction. Patients may also have venous and arterial vascular anomalies. Extra-
cardiac defects are also common with prevalence rates varying widely between 6-39%
(Francannet, Lancaster et al. 1993, Gucer, Ince et al. 2005, Song, Hu et al. 2009, Lurie,
Kappetein et al. 1995)(Ferencz, Rubin et al. 1987, Karr, Brenner et al. 1992). These extra-cardiac
features may be particularly important, influencing health status and quality of life. Details
pertaining to extra-cardiac features are less studied when compared to the cardiac manifestations
of the disease, especially in the adult. Earlier studies on extra-cardiac features included patients
with 22q11DS and other genetic syndromes, which are frequently associated with extra-cardiac
anomalies thus biasing the prevalence estimates. In addition, previous studies have not focused
on extra-cardiac characteristics that manifest later in life in TOF patients who do not have known
genetic syndromes. Therefore, our first objective (STUDY 1) was to examine, in detail,
variability in auxiliary cardiac and extra-cardiac features, including later onset extra-cardiac
conditions, in adults with TOF without 22q11.2 deletions or other known generic syndromes.
We compared these clinical characteristics in two groups of adults with TOF; those meeting
criteria suggestive of a possible genetic syndrome, identified by clinical genetic screening, and
those who do not meet such criteria (Fung, Chow et al. 2008). Cardiac and extra-cardiac features
of adults with 22q11DS, a group already extensively studied by our group and others, was used
as a reference (Driscoll, Spinner et al. 1992, McDonald-McGinn, Kirschner et al. 1999, Bassett,
Chow et al. 2005). As the most common genetic disorder associated with TOF, 22q11DS
provides a basis for understanding the relation between a genetic syndrome and TOF, and for

3
increasing awareness of some of the most common non-cardiac abnormalities in patients with
TOF.
STUDY 2.
Cardiac complications in adults with TOF late after repair are well described with
significant morbidity and mortality. While there have been multiple studies examining cardiac
predictors of late outcomes, (Norgaard, Lauridsen et al. 1999, Nollert, Dabritz et al. 2003,
Murphy, Gersh et al. 1993, Hickey, Veldtman et al. 2009), very few studies have focused on the
impact of genetic syndromes on cardiac outcomes (Kyburz, Bauersfeld et al. 2008, Michielon,
Marino et al. 2006) (Bassett, Chow et al. 2009). Although data is limited, outcome is worse in
patients with genetic syndromes (Kyburz, Bauersfeld et al. 2008)(Anaclerio, Di Ciommo et al.
2004b)(Michielon, Marino et al. 2006)(Bassett, Chow et al. 2009). One pediatric study
demonstrated that genetic syndromes such as VACTERL were associated with poor early
surgical outcomes in children with TOF (Michielon, Marino et al. 2006). Other studies which
have found high mortality and morbidity (i.e. cardiac reinterventions) used a mixed population of
patients with congenital heart defects, many of whom had extra-cardiac features (Kyburz,
Bauersfeld et al. 2008, Anaclerio, Di Ciommo et al. 2004b).
The few available studies examining outcomes as they relate to genetic syndromes have
focused on pediatric populations (Kyburz, Bauersfeld et al. 2008)(Anaclerio, Di Ciommo et al.
2004b)(Michielon, Marino et al. 2006). However, late-onset features of disease are not seen in
pediatric studies and these may impact outcome. Indeed, our group has recently reported on
premature death in adults with 22q11DS (Bassett, Chow et al. 2009). Therefore, our second
objective (STUDY 2) was to determine the impact of phenotypic variation on late outcomes by

4
comparing late outcomes in our previously defined adult syndromic group (enriched for extra-
cardiac characteristics) to those without syndromic features and a reference 22q11DS group.

5
Introduction
1.1 Tetralogy of fallot
1.1.1 Overview
Congenital heart disease (CHD) is the most common congenital birth defect and the
leading cause of death in infants and children (Boneva, Botto et al. 2001). TOF is the most
common of the complex cyanotic heart defects with a prevalence of 2.2 per 10,000 live births,
and is a major cause of death in infants (Francannet, Lancaster et al. 1993, Marelli, Mackie et al.
2007, Thom, Haase et al. 2006). Although Niels Stensen first described an abnormal heart,
Etienne-Louis Arthur Fallot is credited for this disease, based on his description in 1888 of a
lesion with pulmonary artery stenosis, ventricular septal communication, rightward deviation of
the aorta’s origin, and right ventricular hypertrophy (Evans 2008). Subsequently, TOF was
described by many other clinicians and pathologists. In 1924 the eponym tetralogy of Fallot was
coined by Maude Abbott. It was Maude Abbott’s 1936 Atlas that widely spread the use of this
term and increasing recognition of TOF, which led to improvements in diagnosis and treatment
of this condition (Evans 2008).
1.1.2 Basic TOF cardiac anatomy
TOF is characterized by four anatomic abnormalities, which result from the anterior and
cephalad deviation of the infundibular septum: ventricular septal defect (VSD), right ventricular
outflow tract obstruction (RVOTO), overriding aorta (deviation of the aorta to the right), and
secondary right ventricular hypertrophy (Figure 1). The VSD usually occurs in the
perimembranous region; however, in some cases it can also extend to the muscular septum. As a

6
result of the septation defect between the two ventricles, most infants born with TOF appear blue
due to the mixing of oxygenated and deoxygenated blood.
1.1.3 Anatomic variations of TOF
Although the hallmark of TOF is the four anatomic abnormalities previously described,
there can also be variations in the cardiac features within TOF (Rao, Anderson et al. 1971). The
anatomic variations in TOF may involve differences at the level of RVOTO, complexity, the
Figure 1. Intracardiac anatomy of Tetralogy of Fallot (TOF). a) Normal heart anatomy. b) TOF is characterized by
four anatomic abnormalities which arise from the anterior and cephalad deviation of the infundibular septum: overriding
aorta (*), ventricular septal defect, right ventricular outflow tract obstruction and secondary right ventricular
hypertrophy. RA: right atrium; RV: right ventricle; LA: left atrium; LV: left ventricle; RVOT: right ventricular outflow
tract; VSD: ventricular septal defect; RVH: right ventricular hypertrophy. Adapted from Greenway et al., Nat Genet; 41:
931-935.

7
branching of the aortic arch, pulmonary artery anatomy as well as other associated cardiac
anomalies (Dabizzi, Teodori et al. 1990, Marino, Digilio et al. 1996).
The RVOTO can occur at multiple levels. Most frequently, the pulmonary valve in TOF
is stenotic and bicuspid and usually hypoplastic, all of which contribute to the mechanical
obstruction to pulmonary blood flow (Perloff, 1987). However, RVOTO can also occur at the
subvalvar and supravalvar levels as a result of infundibular stenosis and hypoplasia or stenosis of
the pulmonary arteries, respectively.
Some patients with TOF have pulmonary atresia with ventricular septal defect, a more
severe variant of TOF. This may be characterized by valvar pulmonary atresia with normal
pulmonary arteries and duct dependent pulmonary circulation, or others may have diminutive
pulmonary arteries with dependence on major aortopulmonary collateral arteries (MAPCAs) for
blood supply to the lungs (Rome, Mayer et al. 1993).
Another uncommon yet severe condition associated with TOF is absent pulmonary valve
syndrome (APVS) occurring in about 3-5% of cases of TOF (Perloff, 1987). Patients with APVS
have variable degrees of RVOTO, dysplastic or entirely absent pulmonary valve leaflets, as well
as aneurysmal (dilated) pulmonary arteries (Kirshbom, Jaggers et al. 1999). The variation in
anatomy and complexity of TOF is important when these patients are undergoing reparative
cardiac surgery.
Associated congenital cardiac anomalies are common in TOF, with an estimated
prevalence of approximately 40%. Aortic arch anomalies are the most common cardiac anomaly
associated with TOF, occurring in about 25% of cases (Tozzi, Hernanz-Schulman et al. 1989,
Zidere, Tsapakis et al. 2006). In TOF, aberrant right and left subclavian arteries are typically
found in 8% and 5% of cases, respectively (Rao, Anderson et al. 1971).

8
Atrial septal defects (ASD) exists in approximately 15% of cases of TOF (Lev, Eckner
1964) (Perloff), although patent foramen ovale is the most common form of interatrial
communication, typically occurring in half the cases of TOF (Lev, Eckner 1964). Less common
is atrioventricular septal defect, present in <5% of cases of TOF (Lev, Eckner 1964).
Approximately 5- 9% of patients with TOF have coronary anomalies (Dabizzi, Caprioli
et al. 1980). Coronary anomalies may include anterior descending artery from right coronary
artery/sinus and crossing the right ventricular outflow tract, accessory anterior descending artery
from right coronary artery/sinus, single coronary artery from left/right coronary sinus or less
common anomalies (Gupta, Saxena et al. 2001).
A left superior vena cava is present in approximately 11% of cases of TOF, almost
always draining into the coronary sinus (Rao, Anderson et al. 1971). Other associated anomalies
that have been described include anomalies of the mitral valve (hooding or clefting of one of the
leaflets), tricuspid valve (clefting in one of the leaflets and an accessory pouch), prolapse of the
right aortic cusp with aortic insufficiency, subaortic stenosis, accessory orifice of the mitral
valve, dextroversion, septal hypertrophy causing left ventricular outflow tract obstruction, left
umbilical vein draining directly into the coronary sinus, and anomalous muscle bundle of the
right ventricle causing subpulmonary stenosis (Rao, Anderson et al. 1971).
1.1.4 Diagnosis
TOF is frequently diagnosed during fetal life by the detection of the above described
symptoms in association with a systolic ejection murmur, and from the turbulent blood flow
across the narrowed right ventricular outflow tract (Apitz, Webb et al. 2009) (Gersony, 2002).
The intensity of the murmur decreases with increasing severity of RVOTO.

9
The chest x-ray of a patient with TOF shows a heart of normal size, but with a boot-
shaped appearance due to right ventricular hypertrophy and the diminished size of the main
pulmonary artery shadow (Gersony, 2002). In patients with TOF, the electrocardiogram (ECG)
usually displays sinus rhythm with right axis deviation and right ventricular hypertrophy. In
postoperative adults, however, QRS prolongation of the right bundle branch block type is
expected (Perloff, 1987). Echocardiography allows for the visualization of the distinctive
anatomic features of TOF, most notably details of the anteriorly displaced infundibular septum,
the malaligned VSD and the outflow tract obstruction of the right ventricular outflow tract
(Perloff, 1987).
1.2 TOF in childhood
The VSD in TOF is almost always single, large and non-restrictive (Apitz, Webb et al.
2009). The direction and magnitude of blood flow through the defect depends on the severity of
the RVOTO. In cases when the RVOTO is severe or in pulmonary atresia (i.e. complete absence
of the pulmonary valve), a right-to-left shunt occurs, lowering arterial oxygen saturations thus
causing these infants to present with cyanosis (Apitz, Webb et al. 2009). Infants with more mild
forms of pulmonary stenosis may initially present with congestive heart failure secondary to left-
to-right shunting through the VSD (Gersony, 2002). However, infants with minimal or no
cyanosis at birth often develop cyanosis during the first few weeks to months of life, when the
RVOTO increases (Apitz, Webb et al. 2009, Starr 2010). In addition to cyanosis, infants with
TOF can also present with exertional dyspnea and hypercyanotic spells, known as “Tet spells”
(Gersony, 2002).

10
1.2.1 Surgical management
Prior to the advent of open-heart surgery, TOF was managed by palliation with a shunt
from the subclavian artery or aorta to the pulmonary artery. However, the evolution of
cardiopulmonary bypass technique opened the era to open-heart surgery, which made possible
the definitive repair of TOF by closing the VSD and relieving RVOTO. In older cohorts, infants
were initially palliated with shunts for many years, before the transition to early repair was made
(Fraser, McKenzie et al. 2001). The age of corrective surgery has gradually deceased, and in the
current era it is between 3-6 months (Apitz, Webb et al. 2009)(Van Arsdell, Maharaj et al. 2000).
1.2.2 Palliative surgery
In 1944, the first palliative treatment for TOF was introduced by Drs. Blalock and
Taussig, known as the Blalock-Taussig (BT) shunt (Harlan, 1995). During the BT shunt, the
subclavian artery is anastomosed to the pulmonary artery, which increases pulmonary blood
flow, improves exercise capacity and reduces cyanosis (Gersony, 2002).
The Potts and Waterston shunts consist of an anastomosis between the descending and
ascending thoracic aorta and pulmonary artery, respectively. This procedure, however; is rarely
used today because it results in excessive pulmonary blood flow and development of pulmonary
vascular disease (Kirklin, 1993).
1.2.3 Intracardiac repair of TOF
In 1954, the first open heart surgery using cardiopulmonary bypass to repair TOF was
performed by Dr. C Walton Lillehei and colleagues (Neill, Clark 1994). The repair of TOF,
which is typically performed in childhood, involves closing the VSD and relieving RVOTO.

11
Surgical interventions performed to relieve the RVOTO may include pulmonary valvotomy,
resection of the infundibular muscle, right ventricular outflow tract patch, transannular patch
(Figure 2), pulmonary valve implantation, an extracardiac conduit placed between the right
ventricle and pulmonary artery, and angioplasty or patch augmentation of central pulmonary
arteries (Gatzoulis, 2003). In the right ventricular outflow tract patch approach, a patch is placed
across the right ventricular outflow tract (over the infundibulum) without disrupting the integrity
of the pulmonary valve annulus. Transannular patching involves placing a patch across the
pulmonary valve annulus (Harlan, 1995)(Gatzoulis, 2003). However, a transannular patch
renders the pulmonary valve incompetent and creates potential for hemodynamic abnormalities
including pulmonary regurgitation (Gatzoulis, 2003). An alternative procedure to transannular
patching (when the pulmonary valve annulus is restrictive) is an extracardiac conduit from the
right ventricle to the distal main pulmonary artery. However, this approach is more frequently
used in patients with TOF-pulmonary atresia, and there is still the possibility of
stenosis/calcification of the conduit (Gatzoulis, 2003). In patients with restrictive supravalvar
anatomy (hypoplastic main pulmonary trunk and/or stenosis of the central pulmonary arteries),
angioplasty (dilation) or patch augmentation (aterioplasty) can be preformed (Gatzoulis, 2003).

12
Figure 2. Intracardiac repair of TOF. 1) Patch closure of the ventricular septal defect, 2) Transanular patch to relieve right ventricular outflow tract obstruction. Adapted from Nevil Thomas Adult Congenital Heart Library; www.achd-library.com.
1.3 TOF in adulthood
1.3.1 Cardiac features in adulthood late after repair
Prior to the advent of cardiac surgery, roughly half of the patients with TOF died within
the first year of life (Apitz, Webb et al. 2009). Without surgical intervention, 40% die by three
years of age, 70% by ten years of age and 95% by fourty years of age (Starr 2010). The evolution
of congenital heart disease surgery has made it possible for most children born with TOF to live
up to adulthood (Karamlou, McCrindle et al. 2006). Nevertheless, late morbidity and mortality
are not uncommon. Although the anatomic and physiologic abnormalities in TOF can be

13
surgically repaired with excellent early outcomes in survival, residual problems and sequelae still
persist in the majority of patients. Residual cardiac problems after repair of TOF include exercise
limitations, pulmonary regurgitation/RVOTO, tricuspid regurgitation, right ventricular
dysfunction/heart failure, arrhythmias, left ventricular dysfunction/heart failure, residual VSD,
aortic insufficiency and conduction abnormalities (Table I).
Table I. Late postoperative complications in patients with TOF
Complications
Exercise capacity Exercise limitations
Hemodynamic lesions Pulmonary regurgitation, tricuspid
regurgitation, aortic insufficiency
Ventricular dysfunction Right ventricular dysfunction, left ventricular
dysfunction, heart failure
Arrhythmias Ventricular tachyarrhythmias, atrial
tachyarrhythmias, bradyarrhythmia
Residual cardiac defects Residual ventricular septal defect, right
ventricular outflow tract obstruction
Conduction abnormalities Right bundle branch block
1.3.2 Exercise Limitations
Several studies have reported impaired exercise capacity in TOF patients after repair
(Wessel, Cunningham et al. 1980, Sarubbi, Pacileo et al. 2000, Yetman, Lee et al. 2001, Rowe,
Zahka et al. 1991). It has been previously shown that residual pulmonary regurgitation

14
negatively affects duration of exercise and maximal heart rate thus limiting exercise capacity
(Yetman, Lee et al. 2001, Rowe, Zahka et al. 1991, Carvalho, Shinebourne et al. 1992).
Impairments in exercise capacity post repair are independent of age at operation, as children who
had surgery as infants (<18 months of age) still presented with impairments in aerobic capacity
(Yetman, Lee et al. 2001). Furthermore, in the same study by Yetman et al, peak VO2 (measure
of oxygen consumption) was significantly less in patients with right ventricular dysfunction.
Nevertheless, the cause of impaired exercise capacity in postoperative TOF patients is likely
multifactorial and is of great value for the long-term assessment of these patients (Yetman, Lee
et al. 2001).
1.3.2.1 Residual RVOTO/pulmonary regurgitation
Residual RVOTO in postoperative TOF patients may occur at the infundibular, valvar
and/or supravalvar levels (Karamlou, McCrindle et al. 2006) (Gersony, 2002). Reintervention
following repair is required if there is residual pulmonary stenosis with right ventricular pressure
two-thirds or more than systemic pressure (Gatzoulis, 2003). In cases where there is mechanical
obstruction of the pulmonary arteries (such as that created from a previous Potts shunt or
Waterston shunt), the most widely used procedure is stenting to relieve the obstruction (Gersony,
2002).
The use of transannular patch and intracardiac repair via a ventriculotomy approach may
contribute to pulmonary valve regurgitation, which in the long term can have devastating
consequences such as progressive right ventricular dilatation and failure, tricuspid valve
regurgitation, exercise intolerance, arrhythmia, and sudden death (Gatzoulis, 2003). Atrial
arrhythmias may also ensue in patients with pulmonary regurgitation (Gatzoulis, 2003).
Pulmonary regurgitation can be well tolerated in severe cases; however, right ventricular

15
volume-overload results in right ventricular dilatation and dysfunction, which worsens with
secondary tricuspid regurgitation (Hickey, Veldtman et al. 2009). A dilated right ventricle
creates potential for electrical instability, as Gatzoulis et al have shown that risk of symptomatic
arrhythmias is greater in patients with right ventricular dilation and QRS prolongation
(Gatzoulis, Balaji et al. 2000, Gatzoulis, Till et al. 1995). Management of pulmonary
regurgitation in patients late after repair may include pulmonary valve replacement, often in
combination with atrial or ventricular cryoablation. However, further clarification is needed
regarding the optimal timing of pulmonary valve replacement (Hickey, Veldtman et al. 2009).
1.3.2.2 Tricuspid regurgitation
Tricuspid valve regurgitation occurs frequently in postoperative TOF patients with right
ventricular enlargement (Uretzky, Puga et al. 1982). In 2003, Mahle et al investigated the factors
contributing to tricuspid regurgitation and the relation between tricuspid regurgitation and right
ventricular dilation (Mahle, Parks et al. 2003). They found that not only is tricuspid regurgitation
common (present in 32% of patients), but it is related to structural valve abnormalities and
dilatation of the tricuspid annulus (most likely related to previous surgery). Furthermore,
tricuspid regurgitation was significantly correlated with right ventricular volume, suggesting that
tricuspid regurgitation may also contribute significantly to right ventricular dilatation in
postoperative TOF patients. These results are in agreement with the study by Kobayashi et al
who found that tricuspid regurgitation is significantly associated with pulmonary regurgitation,
residual VSD, and right ventricular end-systolic and end-dialstolic pressures (Kobayashi,
Kawashima et al. 1991).

16
1.3.2.3 Right ventricular dysfunction and right heart failure
Right ventricular function has been extensively assessed in the pre- and postoperative
patient with TOF and is influenced by many factors including age of the patient, the size of right
ventriculotomy and the extent of muscle resection, adequacy of myocardial protection (and thus
likely the era in which repair was carried out), and severity of pulmonary regurgitation
(Freedom, 2004). In addition to residual right ventricular outflow tract obstruction and
pulmonary regurgitation, Davlouros et al reported a negative influence of RVOT aneurysms
and/or RVOT akinesia on right ventricular dysfunction (Davlouros, Kilner et al. 2002).
Furthermore, infundibular resection and ischemic insult have been hypothesized to worsen right
ventricular function. This is supported by the work of Atallah et al, who reported less right
ventricular dilation and preserved right ventricular systolic function late after repair of TOF by
using a modified approach for relieving RVOT obstruction and avoiding extensive myectomy
(Atallah-Yunes, Kavey et al. 1996). Preservation of the pulmonary valve, less employment of
RVOT or transannular patching and avoidance of extensive myectomy would likely help in
preserving long-term right ventricular systolic function (Davlouros, Kilner et al. 2002).
1.3.2.4 Left ventricular dysfunction and left heart failure
Although most of the focus has been on right ventricular dysfunction as a late
complication after repair of TOF, there is evidence of biventricular dysfunction and it has been
shown that left ventricular dysfunction is a risk factor for sudden death late after repair (Ghai,
Silversides et al. 2002). Davlourost et al have previously identified the length of time a patient
remains palliated, aortic regurgitant fraction, and right ventricular ejection fraction as
independent predictors of left ventricular ejection fraction (Davlouros, Kilner et al. 2002). The
results from this study and others suggest right ventricular-to-left ventricular interaction,

17
highlighting the importance of preserving right ventricular function for multiple long-term
benefits (Davlouros, Kilner et al. 2002).
Symptomatic TOF patients post repair often present with congestive heart failure (CHF)
(Lillehei, Levy et al. 1964). In the study by Rocchini et al determining the causes of CHF in
postoperative TOF patients, CHF was reported in 35% (36/102) of subjects who were followed
up for ten years (Rocchini, Rosenthal et al. 1977). The major factor associated with CHF in this
study was a large residual VSD. However, RVOTO alone was not identified as a common cause
of CHF in these patients. Other major causes identified included tricuspid regurgitation,
pulmonary stenosis, and persistent systemic to pulmonary artery shunts when there was a large
residual VSD present. In the study by Nollert et al, CHF was identified as the second most
common cause of death in TOF patients post repair (Nollert, Fischlein et al. 1997b), thus making
it important to carry out thorough invasive investigations to identify additional muscular VSDs
or residual VSDs prior to (re)intervention (Rocchini, Rosenthal et al. 1977).
1.3.2.5 Aortic insufficiency
Symptomatic aortic insufficiency is uncommon in TOF patients after repair; however,
some of the causes include dilation of the aortic root, bacterial endocarditis, or injury to the
arotic valve at the time of repair (Gersony, 2002). Patients with aortic regurgitation resulting
from progressive aortic root dilation may eventually require aortic valve replacement (Niwa, Siu
et al. 2002).
1.3.2.6 Residual intracardiac shunts
A residual VSD is usually related to an inadequate VSD patch, but may also be related to
additional VSDs (muscular) that were not initially diagnosed during surgery (Gersony, 2002).

18
1.3.2.7 Supraventricular arrhythmias
Atrial flutter and atrial fibrillation are relatively common in postoperative patients with
TOF, with a reported prevalence between 12% and as high as 34% (Harrison, Siu et al.
2001)(Gatzoulis)(Roos-Hesselink, Perlroth et al. 1995). The reported prevalence is likely a factor
of the age of the cohort studied. There is, however, a relationship between hemodynamic
abnormalities and atrial arrhythmias (Gatzoulis, Balaji et al. 2000, Harrison, Siu et al. 2001,
Karamlou, Silber et al. 2006). In a multicentre study assessing risk factors for arrhythmias,
Gatzoulis et al identified tricuspid regurgitation as the predominant hemodynamic lesion in
patients with atrial fibrillation/flutter (Gatzoulis, Balaji et al. 2000). Harrison et al identified
structural abnormalities, specifically a larger mean right atrial volume and a higher frequency of
significant pulmonary regurgitation in patients with atrial arrhythmias (Harrison, Siu et al. 2001).
Hemodynamic lesions (i.e. tricuspid regurgitation and pulmonary regurgitation) can result in
right atrial dilation and increase predisposition to atrial arrhythmogenesis. It has also been
reported that the QRS threshold for artial arrhythmias is 160 msecs (Karamlou, Silber et al.
2006). Gatzoulis et al have found that patients with atrial flutter/fibrillation have an early
increase in QRS duration, followed by a slower QRS rate of change late after repair (Gatzoulis,
Balaji et al. 2000). This suggests that monitoring the rate of QRS change is of greater prognostic
value than reporting absolute QRS duration values at any given time (Gatzoulis, Balaji et al.
2000).
1.3.2.8 Conduction Abnormalities
Right bundle branch block is a very common phenomenon after surgical repair of TOF.
Late onset of complete heart block after repair of TOF, however, is rare. The incidence of

19
complete heart block has been reported as 0.6% up to 1.3% in patients with TOF post repair
(Freedom, 2004). In cases of late onset complete heart block or transient complete heart block,
pacemaker implantation becomes mandatory (Freedom, 2004)(Gatzoulis, 2003).
1.3.2.9 Ventricular Arrhythmias and Sudden Death
Ventricular arrhythmia is an adverse seqeulae in postoperative patients with TOF, which
may serve as a substrate for sudden cardiac death (Yap, Harris 2009). Nonsustained ventricular
arrhythmia (lasting <30 seconds) is very common, occurring in up to 60% of patients with TOF
following repair (Gatzoulis, 2003). Sustained ventricular arrhythmias (lasting >30 seconds),
however, are uncommon (Gatzoulis 2003). It has been reported that prolonged QRS duration
(≥180 msecs) serves as a risk factor for ventricular arrhythmias (Gatzoulis, Balaji et al. 2000,
Gatzoulis, Till et al. 1995). Furthermore, pulmonary regurgitation has been identified as the
predominant hemodynamic lesion predicting ventricular arrhythmias and sudden death. It has
also been shown that the mechanical and electrical properties of the right ventricle are
interrelated; ventricular dilation and slowed conduction (demonstrated by a QRS≥180 msecs)
after repair of TOF poses severe risk to ventricular arrhythmias and sudden cardiac death
(Gatzoulis, Balaji et al. 2000).
Usually, the focus where the arrhythmia originates is in the right ventricular outflow tract, in the
area of previous infundibulectomy or VSD closure (Gatzoulis, 2003). However, in 20% of the
time the reentry focus may be multiple, involving the entire body of the right ventricle
(Gatzoulis, 2003).
Table II demonstrates some of the most common causes of death in patients with TOF.
The most common cause of death after repair of TOF is sudden cardiac death caused by
ventricular tachycardia and fibrillation (Bricker 1995). Although sudden death is uncommon post

20
repair of TOF (Gernosy, 2002), the incidence of arrhythmic sudden death in late follow-up
series is reported to be between 0.5-6%, accounting for roughly one-third to one-half of late
deaths in this patient population (Gatzoulis, 2003). Indeed, in a study by Nollert et al, the risk of
sudden cardiac death was found to increase after 10 years from 0.06% /year to 0.20%/year
(Nollert, Fischlein et al. 1997b). The risk factors associated with late cardiac events include
older age at repair, a high mean ratio of peak systolic right-to-left ventricular pressures
immediately after repair, and the presence of a Potts anastomosis (Murphy, Gersh et al. 1993,
Hickey, Veldtman et al. 2009, Karamlou, McCrindle et al. 2006). In a study by Hickey et al,
pulmonary branch stenosis, arioventricular septal defect and pulmonary atresia variants were
reported as risk factors influencing late hazard for death, with a three-fold higher late risk for
death in the pulmonary atresia variant than classic TOF (Hickey, Veldtman et al. 2009). The rate
of increase of sudden cardiac death in TOF patients after repair warrants long-term follow-up by
cardiologists to identify possible risk factors and improve overall management.
1.3.3 Reoperation
Although most patients have favourable long-term outcomes after repair of TOF (Hickey,
Veldtman et al. 2009, Karamlou, McCrindle et al. 2006), roughly 5-7% of patients require
reoperation (Norgaard, Lauridsen et al. 1999, Nollert, Fischlein et al. 1997a, Oechslin, Harrison
et al. 1999). Indications for reoperation may include pulmonary insufficiency, pulmonary
stenosis, residual RVOTO, residual VSD, aortic valve regurgitation, aortic valve stenosis,
conduit degeneration, and tricuspid regurgitation (Gatzoulis, 2003). Reoperation procedures,
which commonly include pulmonary valve replacement, tricuspid valve replacement/repair and
pulmonary arterioplasty, are usually done in adulthood. In a study by Oechslin et al, the mean
age at reoperation was 33 years and Karamlou et al reported a median age of 23 years in TOF

21
patients undergoing reoperation after initial repair of TOF (Karamlou, Silber et al. 2006,
Oechslin, Harrison et al. 1999). Mid-term survival after reoperation of TOF is excellent, where
the ten-year actuarial survival has been reported as 92-93% (Karamlou, Silber et al. 2006,
Oechslin, Harrison et al. 1999). However, there is currently limited knowledge about the timing
of reoperation to increase long-term survival, especially relating to pulmonary valve replacement
(PVR) in order to avoid irreversible right ventricle damage from pulmonary insufficiency
(Uretzky, Puga et al. 1982).
Table II. Potential causes of death in patients with TOF
Cardiac causes
Operative mortality
Reoperation or interventions
Arrhythmias – ventricular tachycardia and ventricular fibrillation
Heart failure – related to ventricular dysfunction, valve lesions or residual
shunts
Thromboembolic complications – strokes
Myocardial infarction – secondary to coronary anomalies
Aortic root dilation and dissection or rupture
Acquired heart disease – coronary artery disease
Non-cardiac causes
Non-cardiac surgery
Other diseases – asthma, diabetes
Secondary to congenital extra-cardiac anomalies - i.e. renal failure
Infection/sepsis
Thromboembolic complications – stroke or pneumonia
Suicide
Accidents
Side effects of medications

22
1.4 The non-cardiac phenotype in adults with TOF
1.4.1 Congenital (structural) extra-cardiac anomalies
While there have been extensive studies examining the cardiac features of TOF, much
less has been published on the extra-cardiac features of this condition, except in the case of a few
specific conditions such as trisomy 21 or 22q11DS (McDonald-McGinn, Kirschner et al. 1999,
Bassett, Chow et al. 2005, McDonald-McGinn, Gripp et al. 2005, Driscoll, Salvin et al.
1993)(Jones). The prevalence of congenital extra-cardiac anomalies in TOF has been estimated
to range from 6 to 39% (Francannet, Lancaster et al. 1993, Gucer, Ince et al. 2005, Song, Hu et
al. 2009, Lurie, Kappetein et al. 1995, Ferencz, Rubin et al. 1987, Karr, Brenner et al. 1992,
Calzolari, Garani et al. 2003, Meberg, Hals et al. 2007, Marden, Smith et al. 1964). However, the
reported prevalence depends on the inclusion criteria specific to each study, as some studies
consider a genetic syndrome as an extra-cardiac anomaly (Eskedal, Hagemo et al. 2004) and
others have a different classification of what constitutes a major and minor anomaly and to
which organ system an anomaly belongs to (Francannet, Lancaster et al. 1993, Song, Hu et al.
2009, Marden, Smith et al. 1964). The congenital extra-cardiac anomalies associated with TOF
include malformations affecting any organ system, and may occur as part of a genetic or non-
genetic syndrome (Table III). Depending on the severity of the anomaly, surgery may be
required.
The most commonly reported extra-cardiac anomalies in patients with TOF include
musculoskeletal, gastrointestinal, and genitourinary anomalies (Francannet, Lancaster et al.
1993, Song, Hu et al. 2009). However, these studies included individuals with 22q11DS and
other genetic syndromes. In the study by Francannett et al, patients with TOF had the highest
prevalence of extra-cardiac anomalies compared to two other CHDs, hypoplastic left heart

23
syndrome and transposition of the great vessels. In the same study, gastrointestinal anomaly was
most common, with esophageal atresia and anal atresia predominating. Genitourinary anomalies
were the second most common anomaly, with renal agenesis as the most common defect
(Francannet, Lancaster et al. 1993).
More recently, Rauch et al studied genotype-phenotype relations in 230 children (103
female, 127 male, median age 9.9 years) with TOF (Rauch, Hofbeck et al. 2010). They reported
genetic aberrations in 18% (42/230) of these patients who also had associated extra-cardiac
abnormalities. These genetic aberrations included 22q11.2 deletion, trisomy 21, single gene
disorders (involving JAG1, NKX2.5, and TBX1), and other chromosomal aberrations ((Rauch,
Hofbeck et al. 2010). For example, one patient with chromosome 21q22.3 deletion had cleft
palate, inguinal hernias, and hip dysplasia. Another patient with chromosome 1p32.2p31.1
deletion (14 Mb in size) had hydrocephaly, club foot, renal dysplasia, and agenesis of corpus
callosum. Even in those patients with no identified genetic aberration, extra-cardiac anomalies
were common, and in some cases similar compared to those with 22q11DS (Rauch, Hofbeck et
al. 2010).
Results from these studies highlight the clinical and genetic heterogeneity that exists
within TOF. However, most of these studies have not comprehensively reviewed phenotypic
variability in adult patients with TOF in the absence of major genetic anomalies. Identifying
extra-cardiac features is important as multiple organ malformations often form recognizable
patterns that may be indicative of a particular genetic syndrome (Jones, 2006). Furthermore, the
number and pattern of extra-cardiac features can help in identifying possible subgroups of TOF
that may have increased homogeneity relevant to the genetic pathogenesis of TOF (Scutt, Chow
et al. 2001).

24
Table III. Congenital extra-cardiac anomalies associated with TOF
Organ system Examples of extra-cardiac anomalies
Examples of co-existing genetic syndromes
Central nervous system Hydrocephaly, microcephaly, neural tube defects (e.g. spina bifida)
22q11DS, Alagille syndrome
Pharyngeal arch related anomalies
Cleft lip/palate, thyroid agenesis, thymus anomalies, laryngeal stenosis
22q11DS, CHARGE
Gastrointestinal Tracheoesophageal fistula, esophageal atresia, omphalocele, imperforate anus, duodenal atresia, biliary duct atresia
22q11DS, VACTERL, Alagille syndrome, trisomy 21
Musculoskeletal Anomalous thumb, hemivertebrae, polydactyly, syndactyly, hip dislocation, talipes
22q11DS, Holt-Oram syndrome, Alagille syndrome, VACTERL, Klippel-Feil syndrome, Cornelia de Lange syndrome
Genitourinary Duplex kidney, horseshoe kidney, renal ectopy, hydronephrosis, ureter agenesis/stenosis, hypoplastic ovaries, hypospadias
22q11DS, VACTERL, Klinefelter syndrome
Respiratory Agenesis of lung, congenital diaphragmatic hernia, bronchial malformation
22q11DS
Other Agenesis of ears, craniofacial abnormalities, situs inversus
22q11DS, Goldenhar anomaly, Distichiasis-lymphedema syndrome, Williams syndrome, Treacher Collins syndrome, Alagille syndrome
References: (Francannet, Lancaster et al. 1993, Rauch, Hofbeck et al. 2010, Eldadah, Hamosh et al. 2001, Momma,
Takao et al. 2001, Greenway, Pereira et al. 2009, Costain, Silversides et al. 2010, Lammer, Chak et al. 2009,
Ferencz, Neill et al. 1989, Wells, Barker et al. 1994, McElhinney, Krantz et al. 2002, Blake, Prasad 2006, Wyse, al-
Mahdawi et al. 1993, Rittler, Paz et al. 1996, Zhang, Sun et al. 1986a, Bruni, Angeletti et al. 1996, Cascos 1971,
Greenwood, Rosenthal et al. 1975) (Jones, 2006)

25
1.4.2 Later onset manifestations
One of the advantages to studying adults is that it provides information on lifetime
features that manifest later in life, such as psychiatric illnesses (Bassett, Chow et al. 2005). Apart
from 22q11DS and many other known genetic syndromes, there is limiting data on late
manifesting extra-cardiac conditions in adults with TOF. Most studies to date have looked at
neurodevelopmental outcomes in children after repair of TOF (Hovels-Gurich, Konrad et al.
2006) (Zeltser, Jarvik et al. 2008a). However, there are some reports of late manifesting extra-
cardiac conditions in children with TOF both in the presence and absence of specific genetic
aberrations (Momma, Takao et al. 2001). 22q11DS, an extensively studied and well recognized
genetic syndrome, provides an example of a syndrome co-existing with TOF, and serves as a
template for studying extra-cardiac abnormalities in TOF patients without 22q11.2 deletions. In
patients with TOF and 22q11DS, hypoparathyroidism, schizophrenia, major depression, anxiety,
and hearing loss are well documented (Bassett, Chow et al. 2005, Bassett, Chow et al. 2005,
Momma, Takao et al. 2001, Bassett, Hodgkinson et al. 1998). In 2005, Bassett et al reported
58.1% prevalence for psychiatric disorders (including schizophrenia, major depression, anxiety
disorders, attention deficit hyperactivity disorder, impulse control disorders, and substance use
disorders) in an ascertainment subgroup of adults with TOF (Bassett, Chow et al. 2005).
Similarly, other late-onset conditions reported in TOF patients with or without genetic anomalies
include seizures, mental retardation, frequent infections, impaired hearing, and hypernasality
(commonly associated with velopharyngeal insufficiency) (Bassett, Chow et al. 2005, Rauch,
Hofbeck et al. 2010). Velopharyngeal insufficiency, impaired hearing, seizures, and frequent
infections are less prevalent in TOF patients with no genetic aberration and are higher in those
with 22q11DS and other chromosomal aberrations (Rauch, Hofbeck et al. 2010). In 2009,

26
Kovacs et al interviewed 58 adult CHD patients (including a subset with TOF) and reported 50%
(29/58) prevalence for lifetime mood or anxiety disorder (Kovacs, Saidi et al. 2009). Although
the CHD population in this study was not homogenous, the results highlight the need for the
development of psychosocial interventions and the need to raise awareness of the cardiologist to
the high prevalence of psychiatric disorders in adult CHD patients (Kovacs, Saidi et al. 2009).
Despite the above mentioned studies, there is currently limited data on the prevalence of late-
onset extra-cardiac conditions in adult patients with TOF (with no identified genetic anomalies).
As well, most studies have used a pediatric cohort, which makes it limiting to study
neuropsychiatric disorders such as schizophrenia that manifest in adulthood.
1.4.3 Neurodevelopmental outcomes
Neurodevelopmental outcomes in CHD have been extensively studied; however, most of
these studies used patients with hypoplastic left heart syndrome and transposition of the great
arteries (Hovels-Gurich, Konrad et al. 2006, Zeltser, Jarvik et al. 2008a)(Wernovsky 2006,
Wernovsky, Shillingford et al. 2005, Licht, Wang et al. 2004). In a study by Hovels-Gurich et al
assessing neurodevelopmental outcome in 40 school-age children (with normal chromosomal
status and without 22q11.2 deletions) following repair of TOF or VSD, perioperative hypoxemia
was identified as a significant risk factor for neurodevelopmental abnormalities, including mild
cognitive impairment with additional deficits in language and motor function (Hovels-Gurich,
Konrad et al. 2006). In 2008, Zeltser et al studied neurodevelopmental outcomes in sixty 1-year
old children with classic TOF and TOF-pulmonary atresia (excluding patients with major
recognizable genetic defects and those with genetic and phenotypic syndromes, except for
22q11DS) and found an association between genetic variants and worse neurodevelopmental
outcomes (Zeltser, Jarvik et al. 2008a). Although initially not detected, 18.3% of patients were

27
diagnosed with a genetic syndrome later in the study. Zeltser et al found that in addition to
genetic variants, children with TOF and co-existing genetic syndromes had poor
neurodevelopmental outcomes. In a study by DeMaso et al, 22% of children with TOF (most of
whom had undergone reparative cardiac surgery) were reported to have scored less than 1.5
standard deviations on standardized intelligence quotient testing (although no reference was
made as to whether patients had normal chromosomal status) (DeMaso, Campis et al. 1991). The
results from these studies highlight the multifactorial influence of both genetic factors and the
environment in determining neurodevelopmental outcomes in patients with TOF.
1.5 Genetics of TOF
1.5.1 Heritability
TOF is considered to be a complex disease with a multifactorial mode of inheritance.
Familial cases have been described, where a Mendelian inheritance pattern may be present
(Eldadah, Hamosh et al. 2001, Pitt 1962). In the majority of cases, however, TOF occurs
sporadically (i.e. non-familial). In cases with 2 or 3 affected siblings, autosomal recessive
transmission has been hypothesized (Cassidy, Allen 1991). Although there is a tendency for
CHD (and TOF) to run in families, transmission does not follow a Mendelian mode of
inheritance in the majority of cases. Therefore, a multifactorial model of inheritance, i.e. the
interaction of several genetic loci with environmental factors, has been proposed as the most
likely mechanism (Cassidy, Allen 1991, Nora, Nora 1978)(Nora 1968). Reported recurrence
risks vary from 0.5% to as high as 16% for first degree relatives of individuals with TOF (Nora,
Nora 1978, Nora 1968, Digilio, Marino et al. 1997, Burn, Brennan et al. 1998, Whittemore,
Wells et al. 1994). However, most of these studies did not exclude probands with 22q11.2
deletions and other anomalies; thus, recurrence risks reported from these studies are biased by

28
the inclusion of such patients. In 1997, Digilio et al reported a recurrence estimate of 3% in
siblings, 0.5% in parents, and 0.2% in aunts or uncles in families of 102 nonsyndromic probands
with TOF, but without 22q11DS and other anomalies (Digilio, Marino et al. 1997). Further
studies, after excluding probands with genetic anomalies, would be helpful in identifying
unknown genetic aberrations such as copy number variations (CNVs) that could potentially
segregate within families of probands with TOF.
1.5.2 Genetic causes of TOF
In most instances, the exact molecular mechanisms responsible for the formation of TOF
are unknown. There is, however, evidence suggesting that genetic factors and to some extent
environmental factors are important determinants of this disease. Environmental factors account
for only 2% of all congenital heart defects (Kuciene, Dulskiene 2008). Examples of potential
environmental causes of congenital heart malformations, including TOF, are teratogens such as
rubella (Gibson, Lewis 1952), phenylketonuria (Jenkins, Correa et al. 2007), and maternal
insulin-dependent diabetes mellitus (Abu-Sulaiman, Subaih 2004). In approximately one fifth of
patients with CHD, there is a co-existing syndrome or chromosomal aberration (Perloff, 1991).
22q11DS, the most common genetic disorder associated with TOF (occurring in up to 16% of
cases) will be discussed further below (Goldmuntz, Clark et al. 1998, Botto, May et al. 2003).
The second most common chromosomal anomaly associated with TOF is trisomy 21, occurring
in up to 8% of patients with TOF (Ferencz, Neill et al. 1989, Wells, Barker et al. 1994). Other
genetic syndromes co-occurring with TOF are rare, and include Alagille syndrome, Treacher
Collins syndrome, Klippel-Feil syndrome, Cornelia de Lange syndrome, trisomy 13, trisomy 18,
Holt-Oram syndrome, Okihiro syndrome and Townes-Brocks syndrome (Lammer, Chak et al.
2009, Ferencz, Neill et al. 1989, Weismann, Gelb 2007).

29
In addition to genetic and chromosomal anomalies, haploinsufficiency of transcription
factors (NKX2.5, TBX1, TBX5, GATA4), transmembrane receptors NOTCH1 and NOTCH2 and
their ligand JAG1 altering gene dosage have been implicated in TOF (Eldadah, Hamosh et al.
2001, Greenway, Pereira et al. 2009, Goldmuntz, Geiger et al. 2001). Structural genomic changes
or CNVs have also been implicated in CHD (Greenway, Pereira et al. 2009, Costain, Silversides
et al. 2010). Recently, Greenway et al showed that approximately 10% of nonsyndromic cases of
TOF may result from de novo CNVs (Greenway, Pereira et al. 2009).
In addition to those with identified genetic or chromosomal abnormalities, there may be
important subgroups of TOF that could potentially have relevance to a genetic pathogenesis of
this disease (Scutt, Chow et al. 2001). Determining the genetic etiology of TOF will not only
help in understanding the mechanism of this disease but also in identifying genotype-phenotype
correlations.
1.5.2.1 Single gene disorders
Recurrence of CHD in families of patients with TOF suggests that single gene mutations
may play a role in disease pathogenesis. In 2001, Goldmuntz et al identified heterozygous
mutations in the NKX2.5 gene in 6/114 (5%) nonsyndromic TOF patients without 22q11DS
(Goldmuntz, Geiger et al. 2001). Five of the six patients with NKX2.5 mutations had right aortic
arch, and none had any extra-cardiac anomaly.
In a single large kindred with autosomal dominant TOF and reduced penetrance, Eldadah
et al identified missense mutations in the JAG1 gene without features of Alagille syndrome
(Eldadah, Hamosh et al. 2001). Nine of the eleven (82%) mutation carriers manifested cardiac
disease, including TOF, ventricular septal defect with aortic dextroposition, and isolated
peripheral pulmonic stenosis. Furthermore, all mutation carriers had characteristic but variable

30
facial features distinct from Alagille syndrome, and had no pulmonary, skeletal, or
gastrointestinal abnormalities as is commonly present in this syndrome.
VEGF, an endothelial cell-specific mitogen and angiogenic inducer, has been proposed as a
possible modifier gene for TOF that in addition to other factors could increase risk of CHD. In
2005, Lambrechts et al showed that a 3-SNP haplotype, known to lower VEGF levels, was
associated with an increased risk of TOF (Lambrechts, Devriendt et al. 2005). They found that
the low-VEGF haplotype (AAG) was overtransmitted to affected children (p=0.008). Thus,
VEGF is the first modifier gene identified in TOF. However, these results have not been
replicated (Griffin, Hall et al. 2009).
In a study using 47 subjects (ranging in age from birth to 16.5 years) with sporadic TOF,
2 patients were found to have missense mutations in the ZFPM2/FOG2 gene, suggesting that
ZFPM2/FOG2 mutations may contribute to TOF (Pizzuti, Sarkozy et al. 2003). However, no
extra-cardiac features were reported in this study. Furthermore, of the two patients with the
hemizygous mutations, one had TOF-pulmonary atresia with MAPCAs and the other had classic
TOF.
In 2007, Karkera et al demonstrated that loss of function mutations in the Growth
Differentiation Factor-1 (GDF1) is associated with congenital heart lesions ranging from TOF to
transposition of the great arteries (Karkera, Lee et al. 2007). Three out of eight subjects with
GDF1 mutations had TOF, where one had additional VSD, aortic root dilation, and bicuspid
stenotic pulmonary valve stenosis. However, there were no reports on extra-cardiac findings in
patients with the mutations.
In a study investigating genotype-phenotype relations in 230 children with syndromic and
non-syndromic cases of TOF, Rauch et al reported a polyalanine stretch expansion within a

31
region in the TBX1 gene. This mutation resulted in the loss of transcriptional activity due to
cytoplasmatic aggregation of the mutated protein (Rauch, Hofbeck et al. 2010). Cardiac and
extra-cardiac features in this patient included scoliosis, facial asymmetry, upslanting palpebral
fissures, absent pulmonary vein, and isolated left pulmonary artery. The results from this study
suggest that intragenic mutations in the TBX1 gene (resulting in cytoplasmic aggregation of the
mutant protein) may cause nonsyndromic cases of TOF.
1.5.2.2 Copy Number Variants
There has only been one study to investigate high resolution genome-wide CNVs in
patients with TOF. By studying 114 TOF trios (TOF proband and unaffected parents), Greenway
et al identified 11 de novo CNVs at 10 unique loci that were absent or rare in a control sample of
98 trios (Greenway, Pereira et al. 2009). Seven loci identified in this study were new, and were
found to increase risk for sporadic nonsyndromic TOF by about 9-fold. However, in study
subjects with 22q11.2 deletions, no extra-cardiac features were found, as are typically found in
22q11DS. Furthermore, one control subject had a large 22q11.2 deletion. In one subject with
gain of function CNV on chromosome 3p25.1, extra-cardiac features including subtle
craniofacial abnormalities and hyperactivity were reported. Together, the identified de novo
CNVs accounted for 10% of cases of TOF in this study.
1.6 Genetic determinants of cardiac outcome
Few studies have previously examined the effects of genetic syndromes on immediate
surgical or long term outcomes in patients with CHD (Lin, Basson et al. 2008). In 2006,
Michielon et al retrospectively examined the impact of genetic syndromes and congenital extra-
cardiac anomalies on surgical outcomes in pediatric TOF patients without pulmonary atresia

32
(Michielon, Marino et al. 2006). They identified hypoplastic pulmonary arteries and intervention
for extra-cardiac anomalies as independent predictors of mortality. Furthermore, the ten-year
actuarial survival was worse in syndromic patients (i.e. those with a confirmed diagnosed genetic
syndrome) compared to nonsyndromic patients. Poor outcome in pediatric CHD patients with
22q11.2DS has been reported by Kyburz et al (Kyburz, Bauersfeld et al. 2008). In this study, just
under half of the causes of death were non-cardiac related (either due to multiorgan failure,
immunologic causes, pulmonary complications and multiple malformations), suggesting that
extra-cardiac anomalies associated with 22q11.2DS can affect morbidity and mortality in CHD
patients. However, while morbidity was elevated, none of the deaths occurred in TOF patients.
Anaclerio et al have previously shown that in 350 children with conotruncal anomalies, mortality
was higher in syndromic patients than in nonsyndromic patients, although this was at the trend
level (p=0.06) (Anaclerio, Di Ciommo et al. 2004a). Immediate surgical mortality was
significantly higher in 22q11.2 deleted patients with TOF-pulmonary atresia and 22q11.2 deleted
patients with interrupted aortic arch.
In a study investigating mortality in adults with 22q11DS, Bassett et al showed that
patients with 22q11DS (with or without CHD) have significantly diminished life expectancy
(deaths in the 22q11.2 deleted group occurred at 47.5 years vs. 80.4 years in the Canadian
general population) and are at increased risk of sudden death compared to their unaffected
siblings (Bassett, Chow et al. 2009). Of the 12 deceased patients, four had TOF (two with TOF-
pulmonary atresia, one with TOF-absent pulmonary valve syndrome, and one with classic TOF).
Only one patient with TOF had history of arrhythmia (atrial arrhythmia). Two deaths were
reported as sudden, presumably from arrhythmia. Two patients with non-sudden related death
had clinical heart failure. Although no unifying factor could be attributed to the premature

33
deaths, this study emphasizes the importance of genetic background in determining (cardiac)
outcome.
1.7 22q11.2 deletion syndrome (reference group for Study 1 and
Study 2)
The most common underlying genetic anomaly in patients with TOF is 22q11DS,
occurring in 10-16% of cases (Goldmuntz, Clark et al. 1998, Botto, May et al. 2003). We used
patients with 22q11DS as a reference group because extra-cardiac features in this group of
patients are well characterized (McDonald-McGinn, Kirschner et al. 1999, Bassett, Chow et al.
2005). 22q11DS is a multisystem disorder associated with chromosome 22q11.2 interstitial
deletions (Bassett, Chow et al. 2005) with a prevalence of 1 in 4,000 live births (Goodship, Cross
et al. 1998, Liling, Cross et al. 1999). About 90% of deletions are de novo; however, the mode of
inheritance is autosomal dominant so that an offspring of an affected parent has a 50% chance of
inheriting the deletion (Swillen, Vogels et al. 2000). 22q11DS is phenotypically variable and
includes velocardiofacial syndrome (VCFS), DiGeorge syndrome (DGS), and conotruncal
anomaly face syndrome (CTAFS) (Cohen, Chow et al. 1999). 22q11DS is known to affect
organs derived from the third and fourth pharyngeal pouches, namely, the heart, great vessels,
the thymus gland, parathyroid gland, face and branchial arch arteries (Scambler 2010). In
addition to these pharyngeal pouch-related abnormalities, patients with 22q11DS may also
develop many other anomalies and neurobehavioural problems, many of which may manifest in
childhood and adulthood (Bassett, Chow et al. 2005, Cuneo 2001). There is phenotypic
variability even within a family or between identical twins (Yamagishi, Ishii et al. 1998,
Goodship, Cross et al. 1995). This has also been observed consistently in experimental mouse

34
models of the deletion 22q11.2, even when background strain and environment are strictly
controlled (Merscher, Funke et al. 2001).
1.7.1 Characteristic features of 22q11DS
Common characteristic features in patients with 22q11DS include congenital heart
defects, hypocalcemia, palatal anomalies (e.g. cleft palate), renal anomalies (e.g. absent kidney),
neurological conditions e.g. (seizure disorder), lower limb anomalies and neuropsychiatric
conditions (Table IV) (Bassett, Chow et al. 2005, Lindsay 2001). Phenotypic features in patients
with 22q11DS vary by age group, as some characteristics become more noticeable or manifest
later in life. Neonates with 22q11DS can present with feeding difficulties (occurring in 40-90%
of cases) and seizures (possibly secondary to hypocalcemia) (Cuneo 2001). Many children with
22q11DS experience recurrent ear and upper respiratory infections, velopharyngeal insufficiency
(which can manifest as hypernasal speech), developmental delay, learning difficulties and
behavioural problems (Bassett, Chow et al. 2005, Cuneo 2001). In contrast, psychiatric features
usually manifest in later in life ((Bassett, Chow et al. 2005).
1.7.2 Cardiovascular malformations in 22q11DS
Cardiovascular malformations are very common in patients with 22q11DS, with an
estimated prevalence of 80% (Momma 2010). Several multi-center studies have identified TOF
as the most common CHD in patients with 22q11DS, with prevalence varying between 13-39%
(McDonald-McGinn, Kirschner et al. 1999, Ryan, Goodship et al. 1997, Park, Ko et al. 2007,
Matsuoka, Kimura et al. 1998, Oskarsdottir, Persson et al. 2005). TOF-pulmonary atresia with
MAPCAs has been documented in as many as 25% of patients with 22q11DS (Matsuoka,
Kimura et al. 1998). Although most congenital heart defects in 22q11DS are associated with

35
right-sided heart lesions such as conotruncal and aortic arch anomalies, left-sided cardiac defects
have also been observed, although rarely (Goldmuntz, Clark et al. 1998).
1.7.3 Ear, nose and throat in 22q11DS
Palatal abnormalities are a very common finding in 22q11DS (Bassett, Chow et al. 2005,
Cuneo 2001), where the prevalence of velopharyngeal incompetence has been reported in as
many as 70% of patients. (McDonald-McGinn, Kirschner et al. 1999). Submucous and overt
cleft palate are frequently associated with 22q11.2 deletions with prevalence of 16% and 11%,
respectively (McDonald-McGinn, Kirschner et al. 1999).
Craniofacial findings in patients with 22q11.2 deletions include auricular abnormalities,
nasal abnormalities (such as protruding or microtic ears with over-folded or squared helixes),
ptosis (hooded eyelids), ocular hypertelorism (widely spaced out eyes), bulbous tip nose, narrow
palperbral fissures, small mouth, as well as some other variable features including long, narrow
face and malar flatness (Fung, Chow et al. 2008, McDonald-McGinn, Gripp et al. 2005, Cuneo
2001).
1.7.4 Immune system in 22q11DS
The prevalence of immunodeficiency in patients (which are usually present from infancy)
with 22q11DS has been reported in 40-93% of cases (McDonald-McGinn, Kirschner et al. 1999,
Smith, Driscoll et al. 1998). Immunodeficiency is a risk factor for recurrent infections, which are
seldom life-threatening but common in young children with 22q11DS (Ryan, Goodship et al.
1997, Jawad, McDonald-Mcginn et al. 2001). In cases where immunodeficiency is severe (i.e. in
patients with thymic aplasia or absent T-cells), aggressive immunodeficiency treatment and
possibly thymic transplantation may be required (Cuneo 2001). Autoimmune disorders are also

36
very common in 22q11D, some of which include juvenile rheumatoid arthritis, idiopathic
thrombocytopenia purpura, hyperthyroidism, hypothyroidism, and hemolytic anemia (Jawad,
McDonald-Mcginn et al. 2001, Kawame, Adachi et al. 2001, Keenan, Sullivan et al. 1997).
1.7.5 Endocrine system in 22q11DS
Hypocalcemic hypoparathyroidism, caused by absent or small parathyroid glands is
found in 40-60% of patients with 22q11.2 deletions and is typically most serious in the neonatal
period (McDonald-McGinn, Kirschner et al. 1999, Bassett, Chow et al. 2005, Ryan, Goodship et
al. 1997). Neonatal hypocalcemia can either resolve spontaneously and completely or recur later
in life (Greig, Paul et al. 1996). Hypocalcemia requires treatment as untreated patients may
develop secondary seizures, and is clinically significant in the case of the adult or adolescent
with a repaired conotruncal defect who is already at an increased risk of arrhythmias, syncope
and sudden death (Cuneo 2001, Kao, Mariani et al. 2004).
Another endocrine abnormality associated with the 22q11.2 deletion is hypothyroidism.
In a study by Driscoll et al, hypothyroidism was noted in 7% of subjects with VCFS, and Wilson
et al reported a prevalence of 5% in patients with DGS (Driscoll, Budarf et al. 1992, Wilson,
Burn et al. 1993). In 2005, Bassett et al studied cardiac and extra-cardiac features in 78 adults
with 22q11DS, including late-onset features, and documented a prevalence of 20.5% for
hypothyroidism (Bassett, Chow et al. 2005).
Short stature, typically in part due to growth hormone insufficiency, is also very common
in the 22q11.2 deletion, reported in 10-40% of patients (Cuneo 2001).

37
1.7.6 Cognitive function, neuropsychiatric and central nervous system
abnormalities in 22q11DS
Learning disability and psychiatric problems are common in patients diagnosed with
22q11.2DS (Goldberg, Motzkin et al. 1993, Shprintzen, Goldberg et al. 1992, Shprintzen,
Goldberg et al. 1978). Cognitive disabilities are documented in 40-60% of subjects who have
22q11.2 deletions (Cuneo 2001). Cognitive functioning of children with 22q11.2DS represents a
spectrum: intelligence may vary from moderate mental retardation to average intelligence, with
an average IQ of about 70 (Bassett, Chow et al. 2005, Swillen, Devriendt et al. 1997, Moss,
Batshaw et al. 1999). Patients in early childhood typically present with impairments of language
and motor development (Swillen, Devriendt et al. 1997, Gerdes, Solot et al. 1999) and verbal IQ
scores are typically higher than nonverbal or performance IQ scores (Cuneo 2001, McDonald-
McGinn, LaRossa et al. 1997, Swillen, Devriendt et al. 1999). Autism/autistic spectrum disorder
is also common in children with 22q11.2 deletions, with a reported prevalence of 20% (Fine,
Weissman et al. 2005).
In the study by Kao et al, the incidence of unprovoked seizures (precipitating events such
as hypocalcemia, fever and recent surgery) was reported as 7%, suggesting that seizure is a
primary manifestation of the 22q11.2DS and not a secondary result of other features seen in this
syndrome (Kao, Mariani et al. 2004).
Attention deficit with hyperactivity disorder, attention deficit disorder, emotional
instability and anxiety have also been reported during childhood (Goldberg, Motzkin et al. 1993).
Obsessive compulsive disorder has also been described, ranging from 8-33% (Gothelf,
Presburger et al. 2004, Papolos, Faedda et al. 1996). Schizophrenia, one of the most common

38
psychiatric abnormalities diagnosed in 22q11.2DS, is reported in roughly one fifth of patients
(Murphy, Jones et al. 1999, Chow, Zipursky et al. 2002).
Magnetic resonance imaging studies have been conducted to shed light on the
neurobiological basis of 22q11DS and to find a relationship between neuroanatomic
abnormalities and the cognitive/psychiatric anomalies that commonly afflict patients with this
syndrome (Swillen, Vogels et al. 2000). Specifically, structural abnormalities in the central
nervous system have been identified and related to cognitive and neuropsychiatric conditions in
patients with 22q11DS (Eliez, Schmitt et al. 2000, Kates, Burnette et al. 2001, Eliez, Antonarakis
et al. 2001, Campbell, Daly et al. 2006, Eliez, Barnea-Goraly et al. 2002, Kates, Miller et al.
2006, Simon, Ding et al. 2005)(Chow, Zipursky et al. 2002, Bearden, van Erp et al. 2004). For
example, in the study by Chow et al, 22q11.2 deleted patients with schizophrenia were reported
to have significantly smaller total gray matter volume and larger lateral ventricles than controls
(Chow, Zipursky et al. 2002). Given that approximately 25% of patients with 22q11.2DS
develop schizophrenia, these findings suggest that structural brain differences may predispose
some 22q11.2 deleted patients to psychotic disorders (Chow, Zipursky et al. 2002).
1.7.7 Musculoskeletal system in 22q11DS
Musculoskeletal (MSK) anomalies are also very common in patients with 22q11DS, with
a reported prevalence varying from 36% to 48% (Bassett, Chow et al. 2005, Ming, McDonald-
McGinn et al. 1997). Ming et al found upper limb anomalies (e.g. preaxial polydactyly) in 6%
and lower limb anomalies (e.g. club foot) in 15% of subjects with 22q11.2DS. Other MSK
anomalies identified include vertebral (e.g. butterfly vertebrae), and rib anomalies (e.g. rib
fusion), the former of which is highly associated with conotruncal heart defects (Ming,
McDonald-McGinn et al. 1997). In the study by Bassett and colleagues, approximately 48% of

39
adults with 22q11DS were reported to have scoliosis, of which 6.4% required surgical treatment
(Bassett, Chow et al. 2005). The pathogenesis of MSK anomalies in this syndrome remains to be
identified (Ming, McDonald-McGinn et al. 1997).
1.7.8 Genitourinary system in 22q11DS
In 1996, Devriendt et al reported renal anomalies in 4 out of 39 (10%) cases with
22q11DS, which included bilateral obstructive megaureter, 2 cases of unilateral renal agenesis,
and a right multicystic kidney. In 2002, Wu and colleagues identified a structural urinary tract
anomaly in 31% of patients with 22q11.2DS, which included duplex kidney, unilateral
hypertrophied left kidney, multicystic left kidney, echogenic kidney, bilateral small kidneys,
renal cysts, nephrolithiasis, as well as ureteral and bladder anomalies (Wu, Rusnack et al. 2002).
Of 149 male patients, 6% and 8.1% had an undescended testis and hypospadias, respectively. In
the study by Bassett et al, renal agenesis and hydronephrosis/hydroureter were reported in
roughly 6% and 8% of adults with 22q11DS, respectively (Bassett, Chow et al. 2005). The exact
molecular mechanism of genitourinary anomalies in patients with 22q11.2DS has yet to be
elucidated, although it is suspected that a gene(s) in the deleted region may play a role in ureter
bud development during embryogenesis (Devriendt, Swillen et al. 1996).

40
Table IV. Extra-cardiac abnormalities associated with 22q11.2 deletions.
References: (McDonald-McGinn, Kirschner et al. 1999, Bassett, Chow et al. 2005, McDonald-McGinn, Gripp et al.
2005, Murphy, Jones et al. 1999, Ming, McDonald-McGinn et al. 1997, McDonald-McGinn, Tonnesen et al. 2001)
Abnormality Frequency of finding
Cardiac anomalies
Tetralogy of Fallot
Interrupted aortic arch
Ventricular septal defect
Truncus arteriosus
75%
17-22%
14-15%
13-14%
7-9%
Hypocalcemia 64%
Hypothyroidism 21%
Palatal abnormalities
Submucosal and overt cleft palate
Bifid uvula
Velopharyngeal insufficiency
69%
11-16%
5%
27%
Renal anomalies
Agenesis
Hydronephrosis/hydroureter
36-37%
6.4%
7.7%
Recurrent seizures 40%
Lower limb anomalies 15%
Intellectual disability 92%
Attention Deficit Hyperactivity Disorder 35-46%
Schizophrenia 20-30%

41
2 Aims/Hypotheses
Chromosomal aberrations, single gene disorders, and structural genomic or copy number
variations (mostly large) changes have been implicated in the genetic etiology of TOF. However,
these account for only a minority of cases and in most cases the genetic etiology of TOF is
unknown. In addition to genetic heterogeneity, there is substantial phenotypic variability in
patients with TOF. Phenotypic variability may relate to genetic causation and outcome, and may
be represented by clinical characteristics such as auxiliary cardiac and extra-cardiac
abnormalities (i.e. congenital structural defects present from birth or conditions that manifest or
become detectable later in life). For example, atrioventricular septal defects are commonly
associated with trisomy 21 in patients with TOF (Goldmuntz, Clark et al. 1998, Botto, May et al.
2003)(Rauch, Hofbeck et al. 2010). Anomalies of the aortic arch laterality or branching are a
typical finding in TOF patients with 22q11DS (Rauch, Hofbeck et al. 2010) (Perloff, 1991).
Similarly, extra-cardiac anomalies have been reported in association with TOF in the presence of
specific genetic syndromes. Patients with TOF and Holt-Oram syndrome often have anomalies
affecting the limb, and TOF patients with Alagille syndrome consistently present with hepatic
anomalies. However, these only account for a minority of cases. In the vast majority with no
identified genetic cause, there is inconsistency of extra-cardiac features from patient to patient.
Clustering patients into meaningful categories based on phenotypic features may help identify a
homogenous group of patients who may have relevance to the pathogenesis of TOF and late
outcomes.
Most studies that have examined extra-cardiac features associated with TOF have
focused on infants and children (Kyburz, Bauersfeld et al., 2008, Michielon, Marino et al. 2006).
Except in a few select genetic conditions, there are no studies of later onset extra-cardiac features

42
in adults and these are important as they may aid in pattern recognition of specific genetic
syndromes associated with specific anomalies. While the pediatric studies have been important,
more information on the extra-cardiac features and late-onset disease in the adult population is
needed, as these features may impact outcome. Furthermore, the pattern of congenital and later
onset extra-cardiac features (i.e. phenotypic variability) may help in identifying possible
subgroups of TOF that may be enriched for genetic cause(s) of TOF. Emerging genomic
disorders and future genetic discoveries will help identify more molecular genetic causes of
TOF, and recognition of phenotypic features can help.
Although cardiac morbidity and mortality in TOF are well described, there is less
information available examining the relationship between genetic variability and cardiac
outcomes. Available data suggests that patients with genetic syndromes have worse outcomes
(Kyburz, Bauersfeld et al. 2008, Michielon, Marino et al. 2006, Bassett, Chow et al. 2009,
Anaclerio, Di Ciommo et al. 2004a). For example, substantial morbidity (e.g. the need for
cardiac reinterventions, multiorgan failure, and prolonged hospital stay), mortality and premature
death have been reported in patients with 22q11DS, with or without CHD (Kyburz, Bauersfeld et
al. 2008, Bassett, Chow et al. 2009). However, similar to studies on extra-cardiac features in
TOF, most studies on the genetic determinants of outcome have been done in children or have
not specifically focused on TOF (i.e. studies included mixed population of patients with CHD)
((Kyburz, Bauersfeld et al. 2008, Anaclerio, Di Ciommo et al. 2004a). In adults, apart from
studies on 22q11DS or trisomy 21, the relation between genetic variability and late outcomes
have not been well studied (Bassett, Chow et al. 2009, Bell, Rankin et al. 2003). Differences in
cardiac outcomes between “syndromic” and nonsyndromic subjects may relate to underlying
genetic factors and help with identifying non-cardiac risk factors of cardiac outcome.

43
We therefore sought to investigate phenotypic variability in adults with TOF by
identifying a “syndromic group” of adults with TOF, but without 22q11.2 deletions or other
major known genetic syndromes. In STUDY 1, we compared the lifetime prevalence of
congenital cardiac and extra-cardiac anomalies, and later onset extra-cardiac conditions between
the syndromic group and a nonsyndromic control group. We further sought to determine if the
syndromic group has worse adverse outcomes compared to the nonsyndromic comparison group
(STUDY 2). As reference, we also recorded information on patients with TOF and 22q11DS, as
this group is well characterized (Bassett, Chow et al. 2005)(McDonald-McGinn, Kirschner et al.
1999, Driscoll, Salvin et al. 1993)
We hypothesized that the syndromic group, who may represent a more homogenous
group of patients enriched for genetic causes of TOF, would have a greater prevalence of
congenital cardiac and extra-cardiac anomalies, later onset manifestations, and worse adverse
outcomes compared to the nonsyndromic comparison group. Identifying this group of patients
using brief clinical genetic screening would have great relevance to clinical care. In addition, this
subgroup of adults with TOF may have relevance to a genetic pathogenesis of TOF.

44
3 Study 1
Differentiating Patterns of Cardiac and Extra-cardiac Anomalies
in Adults with Tetralogy of Fallot

45
3.1 Objectives
It is known that approximately 10-16% of patients with TOF have 22q11DS (Goldmuntz, Clark
et al. 1998, Botto, May et al. 2003). However, in most cases, the genetic basis is unknown and
among these patients there is substantial phenotypic variability. Using a previously established
screening criterion to select for patients with 22q11DS (Fung, Chow et al. 2008) we have noted a
subgroup of individuals without 22q11DS, but with features suggestive of a genetic syndrome.
We aimed to determine if TOF patients with syndromic features have a different clinical
presentation compared to those without syndromic features with specific focus on the following
characteristics:
1. Congenital cardiovascular anomalies
2. Congenital extra-cardiac anomalies
3. Late-onset manifestations
Clinical features of a reference population of patients with TOF and 22q11DS were also
collected.

46
3.2 Methods
This study was approved by the institutional ethics boards. Adults (≥18 years) with TOF
seen at the Toronto Congenital Cardiac Centre for Adults and who had undergone prospective
clinical screening (Fung, Chow et al. 2008) from 1998 to 2008 [n=447, mean age 39±13 years,
239 males (53%)] comprised the sample population. All patients with TOF seen at the clinic and
who consented were screened. Individuals with major genetic anomalies such as Down
syndrome were not included in the initial screening study. Of these 447 patients, 88% (n=395,
12%) had TOF and the remainder (n=52) had TOF-pulmonary atresia.
Subjects were categorized into three groups: syndromic, nonsyndromic, and 22q11DS.
Subjects were classified as syndromic if they had at least two of the three criteria previously
established to identify adults with 22q11DS: an overall impression of dysmorphic facial features
(e.g. narrow palpebral fissures), cognitive impairment (e.g. self-reported childhood learning
difficulties), and speech problems (predominantly hypernasal speech) (Fung, Chow et al. 2008,
Bassett, Chow et al. 2005). Figure 3 represents an example of a syndromic patient in the study
with dysmoprhic facial features, learning difficulties, and hypernasal speech. Subjects with
22q11DS who met the syndromic criteria and had a confirmed 22q11.2 deletion (n=39) were
used as a reference group (Fung, Chow et al. 2008, Fung, Chow et al. 2008, Bassett, Chow et al.
2005)(Fung, Chow et al. 2008). Of the initial 447 subjects, 15% (n=66) were classified as
syndromic, 9% (n=39) had 22q11DS and the remainder (n=342) formed the total nonsyndromic
group, not meeting syndromic screening criteria on clinical genetic screening (Figure 4).
Excluded from the syndromic group were five subjects with other known genetic syndromes
(CHARGE, Kartagener, Klippel-Feil, Klinefelter’s and distichiasis-lymphedema syndromes) and
five patients with a history of stroke and acquired speech and/or learning difficulties. Subjects in

47
the remaining syndromic group (n=56) were matched randomly by age (within 5 years) and
gender in a 1:2 ratio with subjects who did not meet the syndromic criteria. The matched
nonsyndromic comparison group thus comprised 112 of 342 available subjects (Figure 4). Most
(n=50, 89%) of the subjects in the syndromic group had clinical genetic testing to rule out
22q11DS and other major genetic anomalies.
In addition to the prospectively collected screening data, comprehensive chart reviews
using records from the adult hospital (Toronto General Hospital, Toronto, Canada) and the local
referring pediatric hospital (Hospital for Sick Children, Toronto, Canada) provided lifetime
information on cardiac and minor and major extra-cardiac features. Congenital cardiovascular
abnormalities other than TOF were recorded (e.g. atrial septal defect). Greater emphasis was
placed on major congenital extra-cardiac anomalies because they have the potential to affect the
functioning of the affected individual (Marden, Smith et al. 1964). Based on previous definitions,
major congenital extra-cardiac anomalies included any of the following: central nervous system
(e.g. spina bifida), gastrointestinal (e.g. esophageal atresia), renal (e.g. renal agenesis), genital
(e.g. bicornuate uterus), major ear deformities requiring surgery (e.g. absent left ear lobe), and
major musculoskeletal (e.g. club foot) (Marden, Smith et al. 1964) (Jones, 2006). Minor
congenital anomalies (e.g. syndactyly) were described separately. Short stature (<3rd percentile)
was defined as a height at last follow up of less than 150 cm and 161 cm for females and males,
respectively (http://www.cdc.gov/nchs/about/major/nhanes/growthcharts/datafiles.htm).
We also recorded late-onset extra-cardiac conditions (including thyroid disorders,
disorders of calcium metabolism, and neuropsychiatric disorders) commonly found in 22q11DS
and other genetic syndromes (Jones, 2006). Neuropsychiatric disorders included seizures,
Attention Deficit Disorder/Attention Deficit Hyperactivity Disorder (ADD/ADHD),

48
schizophrenia, and anxiety and/or depression. Treatments for these conditions were also
recorded. Other late-onset conditions frequently found in the general population and/or that were
infrequent in this study were not reported (e.g. gastroesophageal reflux).
Subjects with genetic testing had standard karyotype and fluorescence in-situ
hybridization (FISH) testing for 22q11.2 deletions using a TUPLE 1 (Vysis) or N25 (ONCOR)
probe (Fung, Chow et al. 2008, Bassett, Chow et al. 2005, Driscoll, Salvin et al. 1993).
Statistical analysis
The statistical analysis was performed using SPSS software (version 16, SPSS, Inc.,
Chicago, Illinois). Continuous data are presented as mean ± standard deviation. Differences
between groups (i.e. syndromic and nonsyndromic groups) were determined using Chi-square,
Fisher exact, Kruskal-Wallis or Student’s t tests as appropriate. A p values <0.05 (2-tailed) was
considered statistically significant.
Figure 3. Example of a subject from the syndromic group. This patient has dysmoprhic facial features, learning difficulties, and hypernasal speech. Used with permission.

49
Figure 4. Categorization of patients into the nonsyndromic group, syndromic group, and 22q11DS reference group. Patients with known syndromes (except for 22q11DS) and a history of stroke and acquired speech and/or learning difficulties were excluded.
3.3 Results Table V presents the characteristics of the three TOF groups (n=207, 106 males (51%),
mean age 36 ±10 years). Most subjects had intracardiac repair of TOF (nonsyndromic 98% vs.
syndromic 91% vs. 22q11DS 92%, p=0.08). In the syndromic group (n=56), 62% (35/56) met
two and 38% (21/56) met three of the predefined clinical screening criteria (Fung, Chow et al.
2008). Baseline clinical characteristics are shown in Table V. Results for all subjects in the
syndromic and nonsyndromic groups who underwent genetic testing were negative (Table V).
There were no significant differences between the syndromic and nonsyndromic groups in
regards to age, sex, or family history of CHD (Table V). The 22q11DS reference group was
younger (p=0.007) and had a higher proportion of subjects with a family history of CHD

50
(p=0.02) compared to the main study groups. As expected, given the screening criteria which
emphasized hypernasality, the syndromic group had a significantly greater prevalence of overt
and submucous cleft palate compared to the nonsyndromic group (11% vs. 0, p=0.001).
There was a trend for proportionately more subjects with short stature in the syndromic
compared to the nonsyndromic group (n=8, 14% vs. n=7, 6%, p=0.09). Of the 15 subjects in the
syndromic and nonsyndromic groups with short stature, 6 (40%) had scoliosis.
3.3.1 Congenital cardiovascular anomalies
Table VI demonstrates the prevalence of congenital cardiac and vascular anomalies in the
study sample. The overall prevalence of individuals with cardiovascular anomalies was lowest in
the nonsyndromic group (62%), followed by the syndromic group (70%) and the 22q11DS
reference group (85%). Of the individual anomalies studied the syndromic group had a
significantly higher proportion of subjects with pulmonary atresia and/or major aorto-pulmonary
collateral arteries (MAPCAs) compared to the nonsyndromic group (Table VI). In the syndromic
group, there were 7 subjects (13%) with MAPCAs and 11 subjects (20%) with pulmonary
atresia. In the nonsyndromic group, 7 subjects (6%) had MAPCAs and 12 (11%) had pulmonary
atresia.
The most common venous anomaly besides left superior vena cava draining into coronary
sinus was bilateral/left superior vena cava. Coronary artery anomalies were the most common
arterial anomaly besides aberrant subclavian artery. Aberrant subclavian artery was much more
common in the 22q11DS reference group compared to the main study groups. Other uncommon
cardiac findings included sub-aortic stenosis (n=2), cardiomyopathy (n=2), semilunar valve

51
stenosis (n=1), mitral valve prolapse (n=1), straddling of tricuspid valve (n=1), mesocardia
(n=1), double chambered right ventricle (n=2), and cleft mitral valve leaflet (n=1).
3.3.2 Congenital extra-cardiac anomalies
3.3.2.1 Major anomalies
The overall prevalence of one or more congenital extra-cardiac anomaly in subjects in the
syndromic and nonsyndromic groups was 17% (29/168). There was a trend toward a higher
prevalence of subjects in the syndromic group with one or more major extra-cardiac anomaly
compared to the nonsyndromic group (25% vs. 13%, p=0.06) (Figure 5). Four percent (8/207) of
subjects had two or more congenital extra-cardiac anomalies; 4 subjects in the syndromic group,
2 in the nonsyndromic group, and 2 in the 22q11DS reference group. Other anomalies were
uncommon (Table VII).
The prevalence of congenital anomalies involving organ systems other than
cardiovascular, was similar between the syndromic group and the 22q11DS group (25% vs. 36%,
p=0.25). Interestingly, we found that when the syndromic and nonsyndromic groups were
combined, subjects with these congenital extra-cardiac anomalies were more common in the 28
subjects with pulmonary atresia and/or MAPCAs compared to the 140 without these cardiac
features (n=9, 32% vs. n=20, 14%; p=0.02), with musculoskeletal anomalies predominating in
the former group, present in seven (78%) subjects.
In the syndromic and nonsyndromic groups combined, genitourinary anomalies were the
most common extra-cardiac anomaly followed by musculoskeletal anomalies, with overall
prevalence of 9% (15/168) and 8% (14/168), respectively. However, neither of these
distinguished the syndromic from nonsyndromic groups (Table VII).

52
3.3.2.2 Minor anomalies
The syndromic group had a significantly greater prevalence of minor congenital extra-
cardiac anomalies compared to the nonsyndromic group (23% vs. 9%, p=0.01), but lower than
that in the 22q11DS group (23% vs. 62%). Musculoskeletal anomalies were the most common
minor congenital extra-cardiac anomaly in this study group. These were mostly prevalent in the
22q11DS group (17/39, 44%), followed by the syndromic group (5/56, 9%) and the
nonsyndromic group (4/112, 4%). Minor musculoskeletal anomalies were camptodactyly (n=9),
syndactyly (n=3), torticollis (n=4), tapering fingers (n=8), hypoplastic nails/fingers (n=6),
clindodactyly (n=2), cubitus valgus (n=1), and simian/palmar crease (n=2).
Hernias were the second most common minor congenital anomaly (n=24). There were nine cases
of inguinal hernias, one hiatus, one diaphragmatic, and one umbilical; the remaining were
unspecified. The prevalence of hernias was similar between the syndromic and nonsyndromic
groups (5% vs. 13%, p=0.10); however, it was significantly more prevalent in the 22q11DS
group (p=0.001).
Strabismus was the most common ocular anomaly in all three groups. The 22q11DS had
the highest prevalence of ocular abnormalities (p=0.001), which included strabismus in every
case (one subject also had Duane syndrome). One subject in the syndromic group and one in the
nonsyndromic group had strabismus and anisocoria, respectively. There was one case of
congenital lower limb lymphedema in the syndromic group.
3.3.3 Late-onset manifestations
Overall, the prevalence of subjects with any neuropsychiatric disorder was high in the
syndromic and nonsyndromic groups combined (22%) and was significantly greater in the

53
syndromic group compared to the nonsyndromic group (32% vs. 17%, p=0.03) (Table VIII).
This appeared to be attributable to seizures, conditions with neurodevelopmental origins
including schizophrenia, and ADD/ADHD (Table VIII). ADD/ADHD, first onset of seizure
activity, recurrent otitis media, and in some cases hearing deficits developed in childhood, while
thyroid dysfunction (except for one case in the syndromic group with congenital
hypothyroidism), schizophrenia, anxiety and depression were diagnosed in adulthood. Not
surprisingly, 14 of the 15 subjects in the syndromic group and 2 of the 19 subjects in the
nonsyndromic group with ADD/ADHD, schizophrenia, and/or seizures had learning difficulties.
In the 22q11DS reference group, all but one subject with ADD/ADHD, schizophrenia, and/or
seizures had learning difficulties. Although the prevalence of reported anxiety and/or depression
was similar between the two groups, anxiolytic medications were more commonly used in the
syndromic than in the nonsyndromic group (9% vs. 1%, p=0.02).
The high prevalence of endocrine disorders in the syndromic group was mainly related to
thyroid dysfunction (Table VIII). Of the 23 subjects in the sample with hypothyroidism, 70%
were female. Two subjects in the 22q11DS reference group had hypogonadotropic
hypogonadism in addition to hypocalcemia; one also had a hypoplastic pituitary gland. The high
prevalence of hypocalcemia in the 22q11DS group was likely related to our routine practice to
check ionized calcium levels in this group of patients (Bassett, Chow et al. 2005).
Although there was a greater proportion of subjects with hearing deficits in the
syndromic group compared to the nonsyndromic group, this appeared to be at least partly
accounted for by a history of recurrent otitis media (Table VIII). Myringotomy tube procedure
was performed in 41% (17/41) of all subjects with a history of otitis media. Of the 21 subjects
with hearing deficits, three subjects in the syndromic group (one with congenital deafness and

54
two with congenital ear deformities requiring surgery) and one subject in the 22q11DS group
(with congenital ear deformity requiring surgery) had a congenital cause accounting for their
hearing loss.
When we reanalyzed the data after excluding the six subjects from the syndromic group
who have not yet had genetic testing, along with the corresponding 12 age and gender-matched
subjects from the nonsyndromic group, the results showed little change. Compared to the
nonsyndromic group, the syndromic group`s higher prevalence of subjects with seizures (n=8,
16% vs. n=6, 6%; p=0.047), schizophrenia (n=2, 4% vs. 0%; p=0.04), gastrointestinal anomalies
(n=2, 4% vs. 0%; p=0.04) and ear deformities (m=2, 4% vs. 0%; p=0.04) reached significance,
while pulmonary atresia and/or MAPCAs (n=12, 24% vs. n=12, 12%; p=0.059) were at the trend
level.
3.4 Discussion This is the first study to describe phenotypic variability in adults with TOF. The results
from this study demonstrate that in a substantial proportion of subjects, TOF represents a
multisystem disorder, even when those with 22q11DS and chromosomal anomalies are excluded.
We previously used a brief screening protocol to delineate criteria that successfully identified
adults with 22q11DS. Using this criteria, we have also identified a subgroup of patients without
22q11.2 deletions or other known genetic anomalies, but with syndromic features suggestive of a
genetic syndrome. We found that this group of subjects had evidence of a more severe cardiac
disease, and were at elevated risk for developing later onset neuropsychiatric and endocrine
disorders compared to a gender and age-matched nonsyndromic group. These findings are novel
and have potential implications for clinical management.

55
We found that one in four subjects in the syndromic group had complex cardiac disease
(pulmonary atresia and/or MAPCAs), cardiac features that can be associated with worse
outcomes (Hickey, Veldtman et al. 2009). In some instances, genetic variants have been related
to these more complex cardiac lesions including mutations in NKX2.5 or 22q11.2 deletions
(Goldmuntz, Geiger et al. 2001) (Chessa, Butera et al. 1998).
The overall prevalence of one or more major congenital extra-cardiac anomaly in our
study sample, excluding the 22q11DS group, was 17%. Previous reports have documented
prevalence varying from 6-39% (Francannet, Lancaster et al. 1993, Gucer, Ince et al. 2005,
Lurie, Kappetein et al. 1995, Ferencz, Rubin et al. 1987, Karr, Brenner et al. 1992, Calzolari,
Garani et al. 2003, Meberg, Hals et al. 2007). However, some samples used to determine
previously reported prevalence of comparable anomalies for TOF would have included subjects
with 22q11.2 deletions and other genetic syndromes. Furthermore, the proportion of extra-
cardiac anomalies reported is dependent on the inclusion criteria, as we chose to focus mostly on
major extra-cardiac anomalies. The most common major anomalies encountered in the present
study were genitourinary and musculoskeletal, in keeping with some previous reports
(Francannet, Lancaster et al. 1993, Song, Hu et al. 2009). However, we did not find a high
proportion of TOF subjects with gastrointestinal anomalies, as previously documented
(Francannet, Lancaster et al. 1993, Song, Hu et al. 2009). This may be due to differences in
inclusion criteria and age of the cohort studied.
The overall prevalence of minor congenital extra-cardiac anomalies in both the
syndromic and nonsyndromic groups was 14%, compared to 62% in the 22q11DS group. While
minor anomalies may not be clinically significant, Marden et al have previously reported that
minor anomalies are common in infants with major embryonic defects (Marden, Smith et al.

56
1964). This suggests that minor anomalies may be used as indicators of more severe embryonic
defects, possibly involving a major genetic anomaly (Marden, Smith et al. 1964).
The prevalence of congenital extra-cardiac anomalies reported in our study and previous
reports is substantially higher than general population expectations (Francannet, Lancaster et al.
1993, Lurie, Kappetein et al. 1995)(Song, Hu et al. 2009)(Jung, Kim et al. 1999). For example,
the prevalence of genitourinary anomalies in the syndromic and nonsyndromic groups combined
(9%) is significantly greater than that reported in the general population (0.096%) (Jung, Kim et
al. 1999). Associated extra-cardiac anomalies with TOF encountered in subjects from the
syndromic and nonsyndromic groups may represent new syndromes, a random association, result
from independent disturbances, or occur secondary to the morphogenetic abnormality that caused
TOF (Lurie, Kappetein et al. 1995). For example, since musculoskeletal and cardiac
development occur at different periods of fetal development, one mechanism to explain co-
existing structural defects in both systems may relate to genetic factors regulating both heart and
limb development. TBX5 mutations in Holt-Oram syndrome (Basson, Bachinsky et al. 1997)
provide a model for such mechanisms. Similarly, the higher prevalence of extra-cardiac
anomalies (predominantly involving the musculoskeletal system) in TOF patients with
pulmonary atresia and/or MAPCAs compared to patients without more severe variant forms of
TOF may suggest a more pleiotropic and potentially earlier developmental genetic origin such as
transcription factors.
A novel finding of our study is the high prevalence of several late-onset conditions in the
syndromic group, comparable to those in 22q11DS. Our reported prevalence is greater than that
reported in the general population for seizures (4-10%), attention deficit disorder (5%) and
thyroid dysfunction (up to 1%) (Friedman, Sharieff 2006, Bilous, Tunbridge 1988, Polanczyk, de

57
Lima et al. 2007). Similar to 22q11DS, the syndromic group had a high prevalence of thyroid
disorders. The etiology of thyroid dysfunction in our sample is unknown and may be related to
several causes, including commonly encountered autoimmune causes, congenital hypothyroidism
(which can be caused by missense mutations in the NKX2.5 gene) or faulty neural crest-derived
thyroid glandular cells (Dentice, Cordeddu et al. 2006) (Burke, Johnson et al. 1987)(Weetman,
McGregor 1994).
Almost all subjects in the syndromic and nonsyndromic groups had reparative cardiac
surgery, performed at a similar age (i.e. at a similar era) in both groups. Although multifactorial
in nature, the elevated prevalence of neuropsychiatric conditions observed in the syndromic and
22q11DS groups may be related to genetic and/or other prenatal influences than environmental
factors related to open heart surgery (Fung, Chow et al. 2008, Bassett, Chow et al. 2005, Zeltser,
Jarvik et al. 2008b). Patients with TOF secondary to Down syndrome and 22q11DS have been
shown to have worse neurodevelopmental outcomes compared to those without genetic
syndromes, suggesting that genetic factors, not simply environmental factors related to open
heart surgery, are important (Atallah, Joffe, et al. 2007).
As in the 22q11DS group, we found a strong overlap between learning difficulties and
neuropsychiatric disorders involving ADD/ADHD, seizures, and/or schizophrenia in the
syndromic group, features that may be related to genetic variations. For example, copy number
variations (CNV) on chromosome 1q21.1 have been associated with neurocognitive and
neuropsychiatric disorders including schizophrenia, autism spectrum disorder and mental
retardation, as well as CHD (Mefford, Sharp et al. 2008b, International Schizophrenia
Consortium 2008)(Greenway, Pereira et al. 2009). CNV on chromosome 16p12.1 have also been
associated with learning disability, craniofacial and skeletal abnormalities, psychiatric disorders,

58
growth retardation and congenital heart defects (Girirajan, Rosenfeld et al. 2010). We would
predict that this patient population would be enriched for CNVs. In addition to high penetrant
CNVs causing syndromal features, there still may be low penetrant CNVs in either the
syndromic or nonsyndromic group or both (Bassett, Scherer et al. 2010).
The syndromic group identified represents an initial step towards delineating more
homogenous populations with TOF that could help identify those enriched for specific genetic
causes of this disease. The largest identifiable subgroup, with 22q11.2 deletions, suggests that
other CNVs may play a role in the etiology of TOF, and this is supported by recent studies
(Greenway, Pereira et al. 2009)(Costain, Silversides et al. 2010). The multisystem nature of the
syndromic group would be consistent with CNVs. However, single gene mutations in signaling
factors regulating transcription may also cause multisystem genetic anomalies such as Holt-
Oram syndrome, which can be associated with TOF (Zhang, Sun et al. 1986b). Many other
factors may contribute to the etiology of TOF, including epistatic interactions (Clark, Yutzey et
al. 2006) and teratogenic factors (Jenkins, Correa et al. 2007). Delineating the genetic causes of
TOF is important not only for understanding the disease mechanism but also for determining
genotype phenotype relations.
3.5 Clinical Implications
The high prevalence of congenital extra-cardiac defects and late-onset conditions in
adults with TOF should alert physicians to the potential multisystem nature of this disorder. An
awareness of these phenotypes is important in the clinical setting; the common association of
certain extra-cardiac defects with TOF provides patterns for diagnosis and allows for early
detection and treatment (Greenwood, Rosenthal et al. 1975). Multidisciplinary management with
the endocrinologist, orthopedic surgeon, psychiatrist, geneticists and other specialties may be

59
necessary. Consultation with a clinical geneticist may help identify recognizable genetic
syndromes, allowing for appropriate genetic counseling and anticipatory guidance. Since certain
extra-cardiac conditions are present from birth, some develop in childhood, and others develop
later, more emphasis on extra-cardiac conditions should be made on transition from pediatric to
adult care to ensure that care is being followed through from the pediatric to the adult setting.
3.6 Study limitations and advantages
This was largely a retrospective study that relied on chart review to identify features. The
prevalence of cardiac and extra-cardiac anomalies reported may therefore be underestimated. To
attempt to minimize this bias, we comprehensively reviewed records from both adult and
paediatric clinics. Also, we chose to focus on extra-cardiac anomalies and late-onset conditions
that are common in subjects with 22q11DS and that we have extensively studied and are well
defined (Bassett, Chow et al. 2005). Most subjects in the syndromic group have not yet had a
direct clinical genetics evaluation as an adult to attempt to identify an underlying genetic
etiology. Many had previous genetic evaluation in childhood, when only limited genetic testing
was available. The genetic testing we performed would only have missed copy number changes
below the resolution of the karyotype (approximately 5-10 Mb) and FISH clinical probes used.
These probes detect over 95% of 22q11.2 deletions. Although screening of patients was not done
by a geneticist, the research personnel who screened the patients in clinic were trained at the
same centre, thus minimizing inter-rater bias. As this study examined an adult cohort, paediatric
subjects who did not survive to adulthood were not captured. This may have resulted in the
underrepresentation of extra-cardiac conditions relevant to early mortality. While it is routine
practice to check for other cardiac anomalies in TOF patients using magnetic resonance imaging,
angiography, or catheter lab procedures, extra-cardiac conditions (especially non-overt) can be

60
under-diagnosed, thus the prevalence of some conditions may be underestimated. As our focus
was on determining the prevalence of cardiac and extra-cardiac features, we did not record the
frequency of imaging done for non-cardiac anomalies. It is therefore important for the clinician
to be aware of the multisystem involvement in many patients with TOF in order to make timely
diagnoses and appropriate referrals. For this study, our focus was on adult outcomes and thus we
did not record pediatric pre and post operative courses. An important advantage of this study,
however, was that our sample included adult subjects, which made it possible to study late-onset
conditions with developmental origins, including neuropsychiatric and endocrine disorders.
3.7 Conclusion
Brief clinical screening in the clinical setting can identify a syndromic group of TOF
subjects, in addition to those with 22q11DS (10), who have multisystem disease involving
congenital extra-cardiac features, late-onset conditions, as well as elevated prevalence of severe
cardiac disease. An awareness of these multisystem phenotypic variations is important for the
clinician in cardiac clinics because of their potential impact on management. These extra-cardiac
abnormalities warrant regular monitoring in patients with TOF in order to take timely
preventative measures and improve management. These findings may also provide for a means
of identifying a subset of patients enriched for certain genetic causes of TOF that warrant referral
to medical genetics. Genome-wide microarrays and other specific genetic testing may delineate
the underlying molecular anomalies in some, or eventually many cases.

61
Table V. Baseline Characteristics in 207 adults with TOF
Nonsyndromic group (n=112)
Syndromic group (n=56)
22q11DS reference group
(n=39)
P-value (Nonsyndromic vs. Syndromic groups)
Mean age, y 37 ± 10 37 ± 11 31 ± 10 0.71
Males 56 (50%) 28 (50%) 22 (56%) 1.0
Family history of CHD 15 (13%) 6 (11%) 12 (31%) 0.62
First degree relatives 11 (10%) 5 (9%) 8 (21%) 0.85
Second degree relatives 6 (5%) 1 (2%) 6 (15%) 0.43
Ethnicity-Caucasian (n=203) 87/108 (81%) 48 (86%) 38 (97%) 0.22
Genetic testing results available*
9 (8%) 50 (89%) 39 (100%) <0.001
*Clinical karyotype and fluorescence in-situ hybridization (FISH) testing using probes for common 22q11.2 deletions

62
Tab
le VI. Con
genital cardiovascular an
omalies in adu
lts with TOF
Non
synd
romic
grou
p
(n=1
12)
Synd
romic
grou
p
(n=5
6)
22q1
1DS
referenc
e grou
p
(n=3
9)
P-value
(N
onsynd
romic vs.
Synd
romic group
s)
Any
cardiovascular ano
maly
Pu
lmon
ary
atre
sia
and/
or M
APC
A
69 (6
2%)
14 (1
3%)
39 (7
0%)
14 (2
5%)
33 (8
5%)
10 (2
6%)
0.31
0.04
A
tria
l sep
tal d
efec
t 11
(10%
) 11
(20%
) 6
(15%
) 0.
08
R
ight
aor
tic a
rch
28
(25%
) 21
(38%
) 18
(46%
) 0.
09
Le
ft S
VC
dra
inin
g in
to c
oron
ary
sinu
s 5
(4%
) 4
(7%
) 2
(5%
) 0.
48
A
berr
ant s
ubcl
avia
n ar
tery
9
(8%
) 6
(11%
) 14
(36%
) 0.
57
H
ypop
last
ic p
ulm
onar
y ar
tery
20
(18%
) 12
(21%
) 12
(31%
) 0.
58
O
ther
art
eria
l ano
mal
ies*
17 (1
5%)
7 (1
3%)
6 (1
5%)
0.64
A
bsen
t pul
mon
ary
valv
e
1 (1
%)
1 (2
%)
2 (5
%)
1.00
O
ther
ven
ous
anom
alie
s† 8
(7%
) 4
(7%
) 1
(3%
) 1.
00
MA
PCA
: Maj
or a
orto
-pul
mon
ary
colla
tera
l art
erie
s; S
VC
: Sup
erio
r Ven
a C
ava.
* Art
eria
l ano
mal
ies
incl
uded
ano
mal
ous
orig
in o
f cor
onar
y ar
teri
es, v
erte
bral
art
ery
anom
alie
s, in
nom
inat
e ar
tery
ano
mal
ies,
dou
ble
aort
ic a
rch,
su
bcla
vian
art
ery
anom
alie
s, a
berr
ant c
oron
ary
arte
ry c
ross
ing
over
the
righ
t ven
tric
ular
out
flow
trac
t or p
ulm
onar
y va
lve,
abs
ent l
eft c
oron
ary
arte
ry, h
emitr
uncu
s ar
teri
osus
, and
bitr
uncu
s br
achi
ocep
halic
art
erie
s.
† Ven
ous
anom
alie
s in
clud
ed le
ft s
ided
SV
C, b
ilate
ral S
VC
, inn
omin
ate
vein
ano
mal
ies,
hem
itran
sect
ion
of th
e ri
ght u
pper
pul
mon
ary
vein
, an
omal
ous
righ
t sup
erio
r pul
mon
ary
vein
dra
inin
g in
to th
e ri
ght a
triu
m, a
nd p
artia
l ano
mal
ous
pulm
onar
y ve
in d
rain
age
to th
e SV
C.

63
Tab
le VII. C
ongenital extra-cardiac ano
malies in adu
lts with TOF
Non
synd
romic
grou
p
(n=1
12)
Synd
romic
grou
p (n=5
6)
22q1
1DS
referenc
e grou
p (n=3
9)
P-value
(Non
synd
romic
vs. S
yndr
omic group
s)
Muscu
loskeletal ano
maly
Maj
or m
uscu
losk
elet
al a
nom
aly*
7 (6
%)
5 (4
%)
7 (1
3%)
5 (9
%)
6 (1
5%)
4 (1
0%)
0.17
0.25
Scol
iosi
s re
quir
ing
surg
ery
Gen
itou
rina
ry ano
maly
Ren
al†
2 (2
%)
9 (8
%)
7 (6
%)
3 (5
%)
6 (1
1%)
4 (7
%)
2 (5
%)
4 (1
0%)
3 (8
%)
0.34
0.57
1.00
Gen
ital‡
Gastrointestina
l ano
maly§
Ear ano
maly||
Cen
tral nervo
us system ano
maly¶
2 (2
%)
0 0
1 (1
%)
3 (5
%)
2 (4
%)
2 (4
%)
2 (4
%)
2 (5
%)
3 (8
%)
1 (3
%)
3 (8
%)
0.34
0.11
0.11
0.26
* Maj
or m
uscu
losk
elet
al a
nom
alie
s w
ere
poly
dact
yly,
bif
id te
rmin
al p
hala
nx o
f thu
mb,
dig
italiz
ed th
umb,
clu
b fo
ot, h
emi a
nd b
utte
rfly
ver
tebr
ae,
and
dysp
last
ic ri
bs
† Ren
al a
nom
alie
s w
ere
rena
l age
nesi
s, h
orse
shoe
kid
ney,
ect
opic
kid
ney,
col
lect
ing
duct
ano
mal
y, a
nd c
onge
nita
l hyd
rone
phro
sis
‡ Gen
ital a
nom
alie
s w
ere
unde
scen
ded
test
es, h
ypos
padi
as, c
hord
ee, a
nd b
icor
nuat
e ut
erus
§ G
astr
oint
estin
al a
nom
alie
s in
clud
ed e
soph
agea
l atr
esia
, ana
l atr
esia
/ste
nosi
s, p
ylor
ic s
teno
sis
and
Mec
kel’
s di
vert
icul
um
|| Maj
or e
ar a
nom
alie
s in
clud
ed a
bsen
t lef
t ear
lobe
, abs
ent l
eft e
xter
nal e
ar c
anal
and
con
geni
tal l
op e
ar
def
orm
ity
¶ Cen
tral
ner
vous
sys
tem
ano
mal
ies
incl
uded
spi
na b
ifid
a, h
ydro
ceph
aly,
hyp
opla
stic
pitu
itary
gla
nd, a
nd d
iffu
se c
ereb
ral a
nd c
ereb
ella
r atr
ophy

64
Tab
le VIII. Late-on
set e
xtra-cardiac con
dition
s in adu
lts with TOF
Non
synd
romic
grou
p
(n=1
12)
Synd
romic
grou
p
(n=5
6)
22q1
1DS
referenc
e grou
p (n=3
9)
P-value
(N
onsynd
romic vs.
Synd
romic group
s)
Any
neu
ropsychiatric disord
er
Seiz
ures
19 (1
7%)
7 (6
%)
18 (3
2%)
8 (1
4%)
20 (5
1%)
8 (2
1%)
0.03
0.09
AD
D/A
DH
D*
0 6
(11%
) 2
(5%
) 0.00
1
Schi
zoph
reni
a 0
2 (4
%)
4 (1
0%)
0.11
Anx
iety
and
/or d
epre
ssio
n
Any
end
ocrine
disorde
r
Thy
roid
dys
func
tion
12 (1
1%)
4 (4
%)
4 (4
%)
5 (9
%)
12 (2
1%)
11 (2
0%)
11 (2
8%)
19 (4
9%)
8 (2
1%)
0.72
<0.001
0.00
1
Hyp
ocal
cem
ia a
nd/o
r hyp
opar
athy
roid
ism
Recur
rent otitis med
ia
Hearing
deficit
In th
e ab
senc
e of
otit
is m
edia
0
13 (1
2%)
0 0
1 (2
%)
13 (2
3%)
11 (2
0%)
3 (5
%)
17 (4
4%)
15 (3
8%)
10 (2
6%)
2 (5
%)
0.33
0.05
<0.001
0.04
* AD
D/A
DH
D: A
ttent
ion
Def
icit
Dis
orde
r/A
ttent
ion
Def
icit
Hyp
erac
tivity
Dis
orde
r

65
Figur
e 5. Propo
rtion of adu
lt TOF sub
jects with on
e or m
ore cong
enital extra-cardiac ano
maly

66
4 Study 2.
Late Outcomes in Adults with Tetralogy of Fallot:
Comparison of Syndromic and Nonsyndromic Groups

67
4.1 Objectives
In STUDY 1, we identified a subgroup of adults with TOF with syndromic features but
without 22q11DS or other major genetic anomalies (syndromic group). This group had more
complex cardiac disease and a greater prevalence of later onset conditions. Limited studies on
pediatric patients with genetic syndromes suggest that syndromic patients are at a high risk for
adverse events including mortality (from both cardiac and non-cardiac causes), cardiac
reinterventions, infections, and multiorgan failure after repair of TOF (Kyburz, Bauersfeld et al.
2008, Michielon, Marino et al. 2006). We hypothesized that our newly classified syndromic
group of adults with TOF would have worse outcomes when compared a group of subjects
without these features (nonsyndromic group). Late outcomes in a reference population of
subjects with TOF and 22q11DS (22q11DS group) were studied for comparison.
The objectives of this study were to investigate differences in late outcomes between
syndromic and nonsyndromic adults with TOF. The primary outcome of interest was all-cause
mortality in adulthood (≥18 years of age). Secondary outcomes included: a) adverse nonfatal
cardiac events and b) cardiac interventions in adulthood.

68
4.2 Methods This study was approved by the institutional ethics boards. This was a retrospective study
of adults (≥18 years) with TOF seen at the Toronto Congenital Cardiac Centre for Adults who
had undergone prospective clinical screening (Fung, Chow et al. 2008) between 1998 and 2008
[n=447, mean age 39±13 years, 239 males (53%)]. Individuals with major genetic anomalies
such as Down syndrome were not included in the initial screening study. Of these 447 patients,
88% (n=395, 12%) had TOF and the remainder (n=52) had TOF-pulmonary atresia.
Subjects were categorized into three groups: syndromic, nonsyndromic, and 22q11DS, as
previously described. Using brief clinical genetic screening, subjects were classified as
syndromic if they had at least two of the three criteria previously established to identify adults
with 22q11DS: an overall impression of dysmorphic facial features (e.g. narrow palpebral
fissures), cognitive impairment (e.g. self-reported childhood learning difficulties), and speech
problems (predominantly hypernasal speech) (Fung, Chow et al. 2008, Bassett, Chow et al.
2005). Of the initial 447 subjects, 15% (n=66) were classified as syndromic, 9% (n=39) had
22q11DS and the remainder (n=342) formed the entire nonsyndromic group, not meeting
syndromic screening criteria on clinical genetic screening (Figure 4). Excluded from the
syndromic group were ten subjects with other known genetic syndromes or a history of stroke
and acquired speech and/or learning difficulties. Subjects in the remaining syndromic group
(n=56) were matched by age (within 5 years) and gender in a 1:2 ratio with subjects who did not
meet the syndromic criteria (n=112).
In addition to the prospectively collected screening data, comprehensive chart reviews
were performed. Subjects with clinical genetic testing had standard karyotype and fluorescence
in-situ hybridization (FISH) testing for 22q11.2 deletions using a TUPLE 1 (Vysis) or N25

69
(ONCOR) probe (Fung, Chow et al. 2008, Bassett, Chow et al. 2005, Driscoll, Salvin et al.
1993).
Baseline characteristics included: cardiac anatomy, associated extra-cardiac anomalies,
and pediatric (prior to age 18 years) surgical and interventional procedures (palliative shunts,
intracardiac repairs, pulmonary arterioplasty or pulmonary angioplasty and other cardiac
surgeries). Details pertaining to the type of surgeries and the year of surgery were obtained from
operative notes. Adverse cardiac events in childhood were recorded including heart failure and
arrhythmias.
Follow up details were obtained from medical chart review of clinic notes from the
Toronto Congenital Cardiac Centre for Adults and from referring physicians (cardiac and non-
cardiac specialists). To determine the cause of death, medical records and post mortem results
were reviewed, when available. In addition, results from the last available (or last visit prior to
death in deceased subjects) electrocardiography and transthoracic echocardiogram (valvular and
ventricular function) were recorded. Functional status at last follow up was based on chart
review and graded according to the New York Heart Association (NYHA) functional
classification. Electrocardiographic parameters included: rhythm, heart rate, QRS duration,
corrected QT interval (QTc), presence or absence of ventricular hypertrophy, and right bundle
branch block (RBBB) morphology. Echocardiographic parameters included: a) severity of
valvular regurgitation based on Doppler echocardiographic assessment, classified as none, mild,
moderate or severe, b) systolic function of the systemic and subpulmonic ventricles, classified as
none, mild, moderate, or severe dysfunction, and c) size of the systemic and subpulmonic
ventricles, classified as none, mild, moderate, or severe dilation. Medications at last follow up
were recorded and categorized as cardiac or non-cardiac.

70
4.3 Primary outcomes
All-cause mortality was the primary endpoint of interest. Death was further classified as
cardiac or non-cardiac. Cardiac deaths included sudden death or death secondary to heart failure,
endocarditis, or thromboembolic events (systemic and pulmonary). Sudden cardiac death was
defined as unexpected death from a presumed cardiac cause, occurring without any prior
condition (Braunwald, 2007). All deaths were adjudicated by two people, SP and CS. Age at
death was recorded.
4.4 Secondary outcomes
The secondary outcomes were: a) adverse nonfatal cardiac events and b) cardiac
interventions in adulthood. Adverse nonfatal cardiac events were defined as any of the following:
resuscitated cardiac arrest, ventricular tachyarrhythmias, supraventricular tachyarrhythmias,
complete heart block, heart failure, stroke or transient ischemic event. We included any sustained
(lasting ≥30 seconds) or asymptomatic tachyarrhythmias requiring treatment. Heart failure was
defined as a) pulmonary edema documented on chest radiograph or b) documentation of heart
failure diagnosis and treatement by a physician. Stroke was defined as a thromboembolic or
ischemic neurologic event diagnosed by computer tomography or magnetic resonance imaging.
Cardiac interventions during adulthood included any surgical, catheter or electrophysiologic
procedures. Surgical cardiac interventions included: primary intracardiac repair of TOF,
pulmonary valve replacement, aortic valve replacement, tricuspid valve replacement/ or
annuloplasty, RV-PA conduit, VSD closure, ASD closure, pulmonary arterioplasty, and
unifocalization of MAPCAs. Cardiac catheter procedures included pulmonary angioplasty or
percutaneous coronary artery angioplasty and/or stenting. Electrophysiology procedures included

71
automatic implantable cardioverter/defibrillator (AICD) implantation, pacemaker implantation or
ablation procedures (including MAZE procedures and cryoablation).
4.5 Statistical analysis Statistical analysis was performed using SPSS software (version 16.0, SPSS, Inc.,
Chicago, Illinois). Continuous variables are reported as mean ± standard deviation. Comparisons
of continuous and categorical variables were performed using Student’s t-test, Wilcoxon Rank
Sum, Chi-square or Fisher exact tests, as appropriate. Age at last follow up or death was used for
the survival analysis. Time to first event (nonfatal cardiovascular event or cardiovascular
intervention, as appropriate) after age 18 or time to last follow up was used in the analysis of
secondary outcomes. If more than one adverse cardiac event/cardiac procedure occurred, the date
of the first event was used in the analysis. Kaplan Meier curves were used to depict: a) all-cause
mortality, b) adverse nonfatal cardiac events and c) cardiac interventions in adulthood between
the syndromic and nonsyndromic groups. A log-rank test was used to test differences between
groups. After confirmation of the proportional hazard assumptions were verified, a Cox
regression was used to determine hazards ratios for all cause mortality, adverse nonfatal cardiac
events and cardiac interventions in the syndromic group compared to the nonsyndromic group.
Hazards ratios could not be computed for all-cause mortality as there were no deaths (n=0) in the
nonsyndromic group. Similarly, hazards ratios could not be calculated for RV-PA conduit and
unifocalization procedures. P values <0.05 (2-tailed) were considered statistically significant.

72
4.6 Results
4.7 Screening characteristics
Table V presents the characteristics of the three TOF groups (n=207, 106 males (51%),
mean age 36 ±10 years). In the syndromic group (n=56), 62% (35/56) met two and 38% (21/56)
met three of the predefined clinical screening criteria (Fung, Chow et al. 2008). All patients with
22q11DS had a positive FISH test. The majority (89%) of subjects from the syndromic group,
and a minority (8%) from the nonsyndromic group had karyotype and FISH testing for 22q11.2
deletions, with negative results.
4.7.1 Baseline characteristics in childhood Tables IX and X and show the baseline anatomic and surgical characteristics (prior to 18
years of age) of the three groups; nonsyndromic, syndromic and 22q11DS. Pulmonary atresia
and/or MAPCAs were more common in the syndromic group compared to the nonsyndromic
group (25% vs. 13%, p=0.04). The majority (~90 %) of subjects in each of the three groups had
had reparative cardiac surgery for TOF. The age at repair was similar between all three groups
(p=0.89). The use of transannular patching in childhood was similar in all three groups (p=0.12).
The syndromic group did not differ from the nonsyndromic group with respect to history of heart
failure (p=0.40), ventricular tachyarrhythmias (p=0.26) or atrial tachyarrhythmias (p=0.26) prior
to 18 years of age. All cases of ventricular tachyarrhythmias occurred in the late postoperative
period (2-15 years after reparative cardiac surgery). In 4 cases atrial tachyarrhythmias occurred
in the late postoperative period (1-3 years after reparative cardiac surgery) and in another case
the event occurred in the perioperative period. 22q11DS group had a higher prevalence of
perioperative and non-perioperative heart failure in childhood compared to the two main
comparison groups (23% vs. 4%, p<0.001).

73
4.8 Primary outcomes
4.8.1 All-cause mortality
In total, there were five deaths in the syndromic group, six deaths in the 22q11DS group,
and none in the nonsyndromic group (Tables XI and XII) (Figure 6). Post mortem was available
for only one patient in the 22q11DS group. The median age of death was 44 years; 46 years
(range 18-56) in the syndromic group and 31 years (range 22-53) in the 22q11DS group. The
median age at last follow up or death was similar between the syndromic and nonsyndromic
groups [median 34 (range 19-63) years vs. 32 (18-60) years), p=0.86].
Mortality in the syndromic group. Four of the five deaths in the syndromic group were
cardiac-related, although variable in nature (Table XII). None of deaths were sudden. A 44 year
old female (case 1) with repaired TOF, replacement of the aortic and pulmonary valves and
severe biventricular systolic dysfunction died 14 hours after complicated heart transplant
surgery. A 56 year old female (case 2) with repaired TOF and significant residual pulmonary and
tricuspid regurgitation, right-sided heart failure and chronic atrial fibrillation/flutter and multiple
sclerosis died of an acute respiratory infection and concomitant right-sided heart failure. An 18
year old male (case 3) with repaired TOF and severe pulmonary regurgitation, a VSD and
moderate left ventricular dysfunction died after a cardioversion for new onset atrial flutter. Soon
after his cardioversion he developed progressive bradycardia, atrioventricular block and then had
a cardiac arrest. During the arrest, he developed ventricular fibrillation and could not be
successfully resuscitated. A 49 year old female (case 4) with repaired TOF and a pulmonary
valve implant died of complications (recurrent heart failure, renal failure, gastrointestinal
bleeding and hypothyroidism) after a prolonged hospital admission for endocarditis. She died,

74
expectedly during sleep, after discontinuation of therapy for endocarditis. A 46 year old female
(case 5) with unrepaired TOF and concomitant interstitial lung disease died of pneumonia.
Deceased subjects had more evidence of end stage heart disease compared to the living
subjects in the syndromic group. A history of heart failure prior to death was more frequent in
deceased subjects compared to living subjects in the syndromic group (n=4, 80% vs. n=5, 10%,
p=0.002). All deceased subjects had a history of atrial tachyarrhythmias, which were more
common in the deceased subjects compared to the living subjects (n=5, 100% vs. n=7, 14%,
p<0.001). Moderate or severe left ventricular dysfunction and tricuspid regurgitation were more
common in deceased subjects compared to living subjects in the syndromic group (ventricular
dysfunction; n=2, 40% vs. n=1, 2%, p=0.02, and tricuspid regurgitation; n=4, 80% vs. n=7, 14%,
p=0.004).
Mortality in the 22q11DS group. Six (15%; 2 F, 4 M) subjects in the 22q11DS group
died at a median age of 31 (range 22-53) years. Four of these deaths have been previously
reported (Bassett, Chow et al, 2009). Two of the six deceased subjects in the 22q11DS group had
pulmonary atresia with MAPCAs; one had absent pulmonary valve syndrome. In three cases
death was sudden and unexpected, presumably from arrhythmia. In the other three subjects,
causes of death differed: one subject died of heart failure, one died of stroke, and one died of
pulmonary hemorrhage (unrepaired TOF with pulmonary atresia and MAPCAs).
Survival. The 30-year survival from the date of first clinic visit (survival at age 48 years)
was 100% in the nonsyndromic group, 78± 14% in the syndromic group, and 46± 21% in the
22q11DS group (Figure 6). All-cause mortality was significantly higher in the syndromic group
compared to the nonsyndromic group (p=0.002).

75
There was little difference in the clinical, electrocardiographic or echocardiographic
parameters between the syndromic and nonsyndromic groups at the last follow up visit (prior to
death in deceased subjects) to explain mortality differences between groups. Most adults had
good reported functional capacity and few subjects in either group had NYHA functional class
III or IV symptoms (syndromic 7% vs. nonsyndromic 3%, p=0.14). The average heart rate in the
syndromic group was higher compared to the nonsyndromic group (74 ±14 vs. 69 ± 12, p=0.04).
The QRS duration on the electrocardiogram was similar between both groups (457 ± 59 vs. 468
± 33, p=0.18); however, QTc interval was greater in the 22q11DS group (489 ± 58, p=0.02).
Moderate or severe biventricular dysfunction was similar between the syndromic and
nonsyndromic groups (n=11, 20% vs. n=17, 15%, p=0.46). The syndromic group had a higher
prevalence of moderate or severe tricuspid regurgitation compared to the nonsyndromic group
(n=11, 20% vs. n=6, 5%, p=0.004).
4.9 Secondary outcomes
4.9.1 Adverse Nonfatal Cardiac Events Overall, 23% (47/207) of subjects had at least one adverse nonfatal cardiac event in adulthood
(incidence rate for any cardiac event was 3.0 adverse cardiac events/year). The mean age at the
time of the first adverse cardiac event was 34 ± 11 years. The most common adverse cardiac
event was atrial tachyarrhythmia. Kaplan-Meier curves showing the freedom from adverse
nonfatal cardiac events in adulthood are shown in Figure 7. Overall, freedom from adverse
cardiac events was similar in the syndromic and nonsyndromic groups [HR 1.16, 95% CI 0.60-
2.25, p=0.65) (Table XIII). Heart failure was more common in the syndromic group compared to
the nonsyndromic group [HR 3.91, 95% CI 1.28-11.93, p=0.01]. There were no differences in
mean age for heart failure between the syndromic and nonsyndromic groups (31 ± 12 vs. 35 ±

76
11, p=0.64). Similarly, there was a trend toward higher rates of atrial tachyarrhythmias in the
syndromic group compared to the nonsyndromic group [HR 2.12, 95% CI: 0.91-4.96, p=0.07]
(Table XIII). Mean age at first atrial tachyarrhythmic event was similar between the main
comparison groups (35 ± 12 vs. 36 ± 14, p=0.81). Ventricular arrhythmias were similar between
the two main study groups (p=0.79) (Table XIII). There were no differences in mean age for
ventricular tachyarrhythmias between the syndromic and nonsyndromic groups (32 ± 11 vs. 33 ±
17, p=0.91)
4.9.2 Cardiac interventions Overall, 42% (86/207) of subjects had at least one cardiac intervention in adulthood
(incidence rate for any cardiac intervention was 5.5 interventions/year) (Table XIV). The mean
age at the time of the intervention was 30 ± 9 years. The most common cardiovascular
intervention in adulthood was pulmonary valve replacement (n=30-33% for all groups) (Table
XIV). Kaplan-Meier curves showing the freedom from cardiac interventions in adulthood are
shown in Figure 8. Overall, there were no differences in cardiac interventions between the
syndromic and nonsyndromic groups [HR 0.68, 95% CI: 0.40-1.16, p=0.15]. Pulmonary artery
angioplasty was significantly more common in the syndromic group compared to the
nonsyndromic group (7% vs. 1%, p=0.04).

77
4.10 Discussion
In this study, we found that adults with TOF with readily recognizable syndromic
features, but without 22q11DS or other known genetic syndromes, have a higher mortality rate in
adulthood compared to those without syndromic features. This group had similar mortality rates
to those observed in patients with 22q11DS (Kyburz, Bauersfeld et al. 2008, Bassett, Chow et al.
2009). Deaths occurred at a relatively young age and were primarily cardiac-related, although
variable in nature. While underlying cardiac anatomy and childhood surgical interventions did
not explain differences in mortality, clinical heart failure, tricuspid regurgitation, and atrial
tachycarrhythmias in adulthood were more common in the syndromic group, particularly in those
subjects that died. Mortality differences between clinical subgroup of patients with TOF require
further studies in larger populations.
Deaths in both the syndromic and 22q11DS groups occurred much earlier than expected
compared to the general population. The median age at death in the syndromic group was 46
years and survival at 30 years (from date of first clinic visit at age 18) was only 78± 14%.
Premature death in adults with 22q11DS has been previously reported by Bassett et al, with a
median age of 26 years in deceased subjects with TOF (Bassett, Chow et al. 2009). By
comparison, the average expected lifespan of Canadians is 80 years; 77 years for males, and 82
years for females (http://www40.statcan.gc.ca/l01/cst01/health26-eng.htm). The increased risk of
premature death in both the syndromic and 22q11DS groups is important clinical information for
patients and for the treating physician. Additional surveillance may be useful in some patients.
Atrial tachyarrhythmias and significant tricuspid regurgitation were more common in the
syndromic group, particularly in those subjects who died (all five subjects who died had atrial
tachycarrhythmias and four had significant tricuspid regurgitation). These two findings are likely

78
interrelated and this association has been previously described (Gatzoulis, 2003). Similarly, heart
failure was more common in the syndromic group compared to the nonsyndromic group,
especially in those subjects who died. While 2 of the subjects had significant left ventricular
systolic dysfunction that would predispose to heart failure, the remaining 2 subjects did not have
ventricular dysfunction, but had atrial tachyarrhythmias and tricuspid regurgitation. Previous
studies have documented a relationship between heart failure and atrial arrhythmias in TOF
patients late after repair (Harrison, Siu et al. 2001). In the study by Harrison et al, almost half of
the patients with atrial arrhythmias were reported to have heart failure as a comorbid cardiac
event (Harrison, Siu et al. 2001). Despite a frequent focus on pulmonary regurgitation and
ventricular arrhythmias and their relationship with adverse outcomes, particularly sudden death,
tricuspid regurgitation and atrial arrhythmias, in combination with syndromic features, appear to
be important determinants of late outcomes.
We did not find an association between more complex cardiac disease (i.e. pulmonary
atresia and/or MAPCA) and death in the syndromic group, a lesion that has been previously
identified as a risk factor for death in patients with TOF (Hickey, Veldtman et al. 2009).
However, the study by Hickey et al had a larger sample size (n=1181 with TOF, n=88 with TOF-
pulmonary atresia). The small number of adults with TOF-pulmonary atresia and/or MAPCAs
(n=28, 14 syndromic and 14 nonsyndromic) may have limited our ability to detect this
association. In addition, we included adult patients with TOF in our study, which may have
biased the results; some patients with 22q11DS and complex CHD (i.e. pulmonary atresia and/or
MAPCAs) will not survive to adulthood (Kyburz, Bauersfeld et al, 2008).
A QRS duration ≥160ms has been associated with an increased risk of atrial arrhythmias,
ventricular arrhythmias and sudden cardiac death (Gatzoulis, Balaji et al. 2000, Karamlou, Silber

79
et al. 2006)(Khairy, Aboulhosn et al. 2010). However, we did not find a relationship between
QRS duration and death in the syndromic group. This may reflect the nature of deaths in this
group, which were related to advanced heart disease along with other complications and not to
sudden death.
In the series by Bassett et al, a five of the twelve deaths occurred unexpectedly during
sleep in patients with 22q11DS (including one patient with TOF) (Bassett, Chow et al. 2009).
Findings from that study suggest that premature death in some adults with 22q11DS may be
arrhythmic in origin, perhaps related to long QT, as is often found in the 22q11DS population. In
contrast, there were no sudden unexpected deaths in the syndromic group despite three subjects
having significantly prolonged corrected QT intervals (>500 msec).
Besides anatomic differences, underlying genetic factors may contribute to adverse
cardiac outcomes. The syndromic group identified in this study may be enriched for genetic
causes of TOF such as CNVs (Costain, Silversides et al, 2010, Greenway, Pereira et al, 2009).
Previous studies investigating the impact of genetic syndromes on cardiac outcome in pediatric
patients with TOF and CHD have demonstrated higher mortality rates in TOF patients with
genetic syndromes. In those studies, mortality was related to non-cardiac complications (e.g.
multi-organ failure, pneumonia, surgery for extra-cardiac anomalies) (Kyburz, Bauersfeld et al.
2008, Michielon, Marino et al. 2006). In contrast, non-cardiac related causes accounted for only
a minority of cases of the deaths in our study sample. This may be due to a survival bias as
children with complex extra-cardiac defects may die early of non-cardiac related complications
and thus not survive to adulthood.

80
4.11 Clinical Implications
Although some patients do well after repair of TOF (Norgaard, Lauridsen et al. 1999,
Murphy, Gersh et al. 1993), late morbidity remains an important issue in adulthood. This is
consistent with our study in which we document high rates of adverse nonfatal cardiac events
and cardiac interventions in both the syndromic and nonsyndromic groups, highlighting the need
for careful long term follow up. By using a previously established screening protocol to select for
patients with 22q11DS, we found a clinically important subgroup of adults with TOF with
syndromic features who had a higher mortality rate. This finding is important and suggests that,
in addition to the known cardiac risk factors associated with poor survival, there are other non-
cardiac variables that can help to identify the high risk adult with TOF.
We would anticipate that the syndromic group is enriched for genetic causes of TOF such
as CNVs. Differentiating syndromic from nonsyndromic outcomes, as well as 22q11DS, is of
clinical relevance as it may provide some insight into the potential role that genetic variation
plays in determining cardiac outcome. Post-mortem molecular and tissue studies could be helpful
in identifying potential underlying mechanisms of premature death in the TOF population.
4.12 Study limitations and advantages
The retrospective design of this study, which relied heavily on chart review as the
available source of data, limits the quality of information regarding the clinical course of the
patient. The retrospective nature of this study is especially relevant in the case where the patient
is deceased, when the cause of death is not clear and no post-mortem exam is performed. Of the
11 deaths, only 1 subject had a post-mortem examination. However, in the majority of cases, we
were able to obtain reasonable clinical information pertaining to the cause of death. As this study

81
only focused on adults, a survival bias exists since individuals who died prior to 18 years of age
were not included. However, an important advantage to this study is the focus on the adult
population, enabling us to study late outcomes and predictors of late outcomes. Most subjects in
the syndromic group have not yet had a direct clinical genetics evaluation as an adult to attempt
to identify an underlying genetic etiology. Undiagnosed genetic syndromes in the syndromic
group may inflate the morbidity and mortality estimates as some known syndromes, such as
22q11DS are known to be associated with worse outcomes. We did not record the age at first
clinic visit, but did record all events that had occurred since the age of 18 years. .
4.13 Conclusion In addition to patients with 22q11DS, we have identified a new clinically important
subgroup of adults with syndromic features who are at risk for premature mortality. The cause of
death in this group is likely multifactorial, but seems, at least in part, to be associated with
advanced heart disease. Further, larger studies are required to confirm this observation.

82
Tab
le IX. C
hildho
od cardiac and
extra-cardiac cha
racteristics in
adu
lts with TOF
Tab
le X. P
ediatric cardiac in
terven
tion
s an
d ad
verse card
iac even
ts
Non
synd
romic
grou
p
(n=1
12)
Synd
romic
grou
p (n=5
6)
22q1
1DS
referenc
e grou
p (n=3
9)
P-value
(N
onsynd
romic vs.
Synd
romic group
s)
Cardiac ana
tomy
Pu
lmon
ary
atre
sia
and/
or
MA
PCA
14
(13%
) 14
(25%
) 10
(26%
) 0.04
A
tria
l sep
tal d
efec
t 11
(10%
) 11
(20%
) 6
(15%
) 0.
08
R
ight
aor
tic a
rch
28
(25%
) 21
(38%
) 18
(46%
) 0.
09
Le
ft S
VC
dra
inin
g in
to
coro
nary
sin
us
5 (4
%)
4 (7
%)
2 (5
%)
0.48
A
berr
ant s
ubcl
avia
n ar
tery
9
(8%
) 6
(11%
) 14
(36%
) 0.
57
H
ypop
last
ic p
ulm
onar
y ar
tery
20
(18%
) 12
(21%
) 12
(31%
) 0.
58
O
ther
art
eria
l ano
mal
ies
17 (1
5%)
7 (1
3%)
6 (1
5%)
0.64
A
bsen
t pul
mon
ary
valv
e
1 (1
%)
1 (2
%)
2 (5
%)
1.00
O
ther
ven
ous
anom
alie
s
Associated cong
enital extra-
card
iac an
omalies
8 (7
%)
15 (1
3%)
4 (7
%)
14 (2
5%)
1 (3
%)
14 (3
6%)
1.00
0.06
MA
PCA
: Maj
or a
orto
-pul
mon
ary
colla
tera
l art
ery
SV
C: S
uper
ior v
ena
cava
R
V-P
A: R
ight
ven
tric
le-p
ulm
onar
y ar
tery
Pu
lmon
ary
plas
ty in
clud
ed p
ulm
onar
y ar
tery
art
erio
plas
ty a
nd a
ngio
plas
ty
Ass
ocia
ted
extr
a-ca
rdia
c de
fect
s w
ere
maj
or m
uscu
losk
elet
al (i
nclu
ding
sco
liosi
s re
quir
ing
surg
ery)
, gen
itour
inar
y,
cent
ral n
ervo
us s
yste
m, g
astr
oint
estin
al, a
nd e
ar d
efor
miti
es re
quir
ing
surg
ery.

83
Tab
le X. P
ediatric cardiac in
terven
tion
s an
d ad
verse card
iac even
ts in
207
adu
lts with TOF
Non
synd
romic
grou
p
(n=1
12)
Synd
romic
grou
p (n=5
6)
22q1
1DS
referenc
e grou
p (n=3
9)
P-value
(N
onsynd
romic vs.
Synd
romic group
s)
Ped
iatric cardiac surgery
Sh
unts
Bla
lock
-Tau
ssig
Potts
and
Wat
erst
on
Intracardiac rep
air
Tra
nsan
nula
r pat
ch
56 (5
0%)
48 (4
3%)
10 (9
%)
104
(93%
) 51
(46%
)
35 (6
3%)
31 (5
5%)
6 (1
1%)
50 (8
9%)
19 (3
4%)
15 (3
8%)
15 (3
8%)
2 (5
%)
35 (9
0%)
21 (5
4%)
0.13
0.
23
0.71
0.
43
0.15
R
V-P
A c
ondu
it
10 (9
%)
6 (1
1%)
7 (1
8%)
0.71
Age
at i
ntra
card
iac
repa
ir
5.9
± 3.
4 5.
6 ±
3.2
5.6
± 3.
4 0.
58
Pulmon
ary arteriop
lasty
37 (3
3%)
21 (3
8%)
16 (4
1%)
0.57
Peri- and
non
periop
erative
even
ts in
childho
od
H
eart
failu
re
3 (3
%)
3 (5
%)
9 (2
3%)
0.40
V
entr
icul
ar a
rrhy
thm
ias
1
(1%
) 2
(4%
) 1
(3%
) 0.
26
A
tria
l tac
hyar
rhyt
hmia
s
1 (1
%)
2 (4
%)
2 (5
%)
0.26
RV
-PA
: rig
ht v
entr
icle
-pul
mon
ary
arte
ry
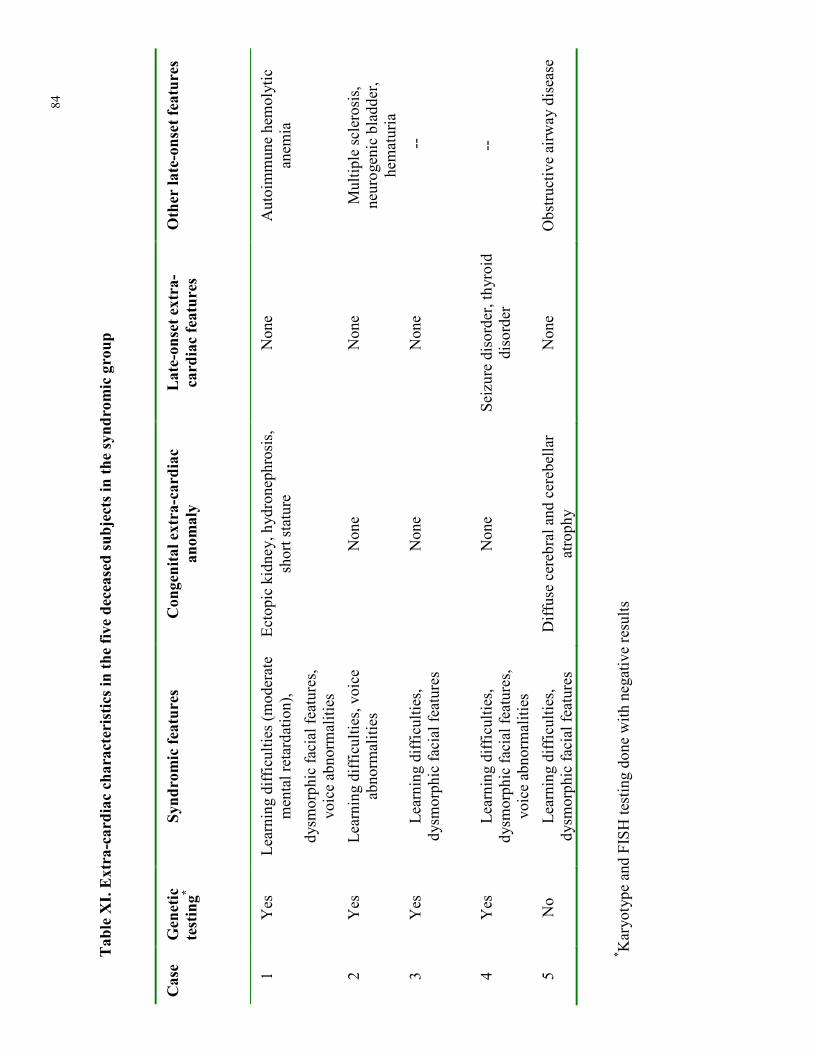
84
Tab
le XI. Extra-cardiac cha
racteristics in
the five deceased subjects in
the synd
romic group
* Kar
yoty
pe a
nd F
ISH
test
ing
done
with
neg
ativ
e re
sults
Case
Gen
etic
testing*
Synd
romic fe
atures
Con
genital extra-cardiac
anom
aly
Late-on
set e
xtra-
card
iac featur
es
Other la
te- onset fe
atur
es
1 Y
es
Lea
rnin
g di
ffic
ultie
s (m
oder
ate
men
tal r
etar
datio
n),
dysm
orph
ic fa
cial
feat
ures
, vo
ice
abno
rmal
ities
Ect
opic
kid
ney,
hyd
rone
phro
sis,
sh
ort s
tatu
re
Non
e A
utoi
mm
une
hem
olyt
ic
anem
ia
2 Y
es
Lea
rnin
g di
ffic
ultie
s, v
oice
ab
norm
aliti
es
Non
e N
one
Mul
tiple
scl
eros
is,
neur
ogen
ic b
ladd
er,
hem
atur
ia
3 Y
es
Lea
rnin
g di
ffic
ultie
s,
dysm
orph
ic fa
cial
feat
ures
N
one
Non
e --
4 Y
es
Lea
rnin
g di
ffic
ultie
s,
dysm
orph
ic fa
cial
feat
ures
, vo
ice
abno
rmal
ities
Non
e Se
izur
e di
sord
er, t
hyro
id
diso
rder
--
5 N
o L
earn
ing
diff
icul
ties,
dy
smor
phic
faci
al fe
atur
es
Dif
fuse
cer
ebra
l and
cer
ebel
lar
atro
phy
N
one
Obs
truc
tive
airw
ay d
isea
se

85
Tab
le XII. C
ardiac cha
racteristics in
the five deceased subjects in
the synd
romic group
Tab
le XIII. Adv
erse non
fatal cardiac events in adu
ltho
Case
Sex
Age at
repa
ir
(yrs)
Cardiac ana
tomy
PVR
Heart
failu
re
Arrhy
thmias
QTc interval
(msec)
QRS du
ration
(m
sec)
Ech
o find
ings
Age at
death (yrs)
Cau
se of death
1 F
9 H
ypop
last
ic
pulm
onar
y ar
tery
, le
ft S
VC
Yes
Y
es
SVT
50
0
75
Mod
erat
e M
R a
nd
TR
, sev
ere
bi-
vent
ricu
lar
dysf
unct
ion
44
Ear
ly p
ost-
op
deat
h af
ter
com
plic
ated
hea
rt
tran
spla
nt
2 F
3 L
eft S
VC
N
o Y
es
Chr
onic
atr
ial
fl
utte
r 43
2
156
Mod
erat
e PR
and
T
R
56
Infe
ctio
n an
d ri
ght h
eart
failu
re
3 M
4
--
No
Yes
A
tria
l flu
tter,
VT
50
0
117
Seve
re P
R,
mod
erat
e T
R,
mod
erat
e le
ft
vent
ricu
lar
dysf
unct
ion
18
Post
-ca
rdio
vers
ion
brad
ycar
dia
and
subs
eque
nt
card
iac
arre
st
4 F
10
ASD
Y
es
Yes
R
ecur
rent
at
rial
fibr
illat
ion
513
138
Mod
erat
e T
R
49
End
ocar
ditis
5 F
Non
e Pu
lmon
ary
atre
sia
with
MA
PCA
, hy
popl
astic
pu
lmon
ary
arte
ry,
righ
t aor
tic a
rch
No
No
Rec
urre
nt
atri
al
flut
ter
416
92
--
46
Pneu
mon
ia
SVC
; sup
erio
r ven
a ca
va
ASD
: atr
ial s
epta
l def
ect
MA
PCA
: maj
or a
orto
-pul
mon
ary
colla
tera
l art
ery
PVR
: pul
mon
ary
valv
e re
plac
emen
t SV
T: s
upra
vent
ricu
lar t
achy
card
ia
VT
: ven
tric
ular
tach
ycar
dia
PR
: pul
mon
ary
regu
rgita
tion
T
R: t
ricu
spid
regu
rgita
tion
M
R: m
itral
regu
rgita
tion

86
Tab
le XIII. Adv
erse non
fatal cardiac events in adu
ltho
od
Non
synd
romic
grou
p
(n=1
12)
Synd
romic
grou
p
(n=5
6)
22q1
1DS
grou
p (n=3
9)
Hazards ratio
(95%
CI)
(Syn
drom
ic vs.
Non
synd
romic
grou
ps)
P-value
(Syn
drom
ic vs.
Non
synd
romic
grou
ps)
Any
cardiac event
25 (2
1%)
15 (2
7%)
7 (1
8%)
1.16
(0.6
0-2.
25)
0.65
Res
usci
tate
d ca
rdia
c ar
rest
2
(2%
) 1
(2%
) 0
1.00
(0.0
9-11
.07)
1.
00
V
entr
icul
ar ta
chya
rrhy
thm
ias
10 (9
%)
5 (9
%)
4 (1
0%)
0.86
(0.2
7-2.
76)
0.79
Atr
ial t
achy
arrh
ythm
ias
11 (1
0%)
12 (2
1%)
4 (1
0%)
2.12
(0.9
1-4.
96)
0.07
Hea
rt fa
ilure
5
(4%
) 9
(16%
) 2
(5%
) 3.
91 (1
.28-
11.9
3)
0.01
Stro
ke/T
IA
3 (3
%)
1 (2
%)
0 0.
78 (0
.08-
7.49
) 0.
83
C
ompl
ete
hear
t blo
ck
5 (4
%)
1 (2
%)
0 0.
39 (0
.05-
3.36
) 0.
37
TIA
: tra
nsie
nt is
chem
ic a
ttack

87
87
Tab
le XIV
. Cardiovascu
lar interven
tion
s in adu
ltho
od
* Interven
tion
s are no
t mutua
lly exclusive
Non
synd
romic
grou
p
(n=1
12)
Synd
romic
grou
p
(n=5
6)
22q1
1DS
grou
p (n=3
9)
Hazards ratio
(95%
CI)
(Syn
drom
ic vs.
Non
synd
romic
grou
ps)
P-value
(Syn
drom
ic vs.
Non
synd
romic
grou
ps)
Any
interven
tion
* 50
(45%
) 19
(34%
) 17
(44%
) 0.
68 (0
.40-
1.16
) 0.
15
Su
rgical in
terven
tion
*
Intr
acar
diac
repa
ir in
adu
lthoo
d
6 (5
%)
1 (2
%)
1 (3
%)
0.23
(0.0
3-1.
94)
0.14
Pu
lmon
ary
valv
e re
plac
emen
t 37
(33%
) 17
(30%
) 13
(33%
) 0.
82 (0
.46-
1.46
) 0.
49
A
ortic
val
ve re
plac
emen
t/val
voto
my
1
(1%
) 1
(2%
) 0
1.78
(0.1
1-30
.11)
0.
69
T
ricu
spid
val
ve re
plac
emen
t/ann
ulop
last
y
8 (7
%)
4 (7
%)
2 (5
%)
0.89
(0.2
7-2.
96)
0.84
RV
-PA
con
duit
4
(4%
) 0
1(3%
) --
0.
15
P
ulm
onar
y ar
teri
opla
sty
22 (2
0%)
10 (1
8%)
8 (2
1%)
0.79
(0.3
7-1.
68)
0.54
VSD
clo
sure
12
(11%
) 4
(7%
) 2
(5%
) 0.
58 (0
.19-
1.81
) 0.
34
A
SD c
losu
re
3 (3
%)
4 (7
%)
0 2.
26 (0
.50-
10.0
0)
0.28
Uni
foca
lizat
ion
of M
APC
As
0 0
1 (3
%)
--
--
Cathe
ter labo
ratory procedu
re*
Pul
mon
ary
arte
ry a
ngio
plas
ty
Electroph
ysiology
procedu
re*
1 (1
%)
4 (7
%)
1 (3
%)
7.5
(0.8
3-67
.56)
0.04
A
ICD
5
(4%
) 4
(7%
) 0
1.42
(0.3
8-5.
30)
0.60
Abl
atio
n 13
(12%
) 6
(11%
) 4
(10%
) 0.
85 (0
.32-
2.24
) 0.
74
P
acem
aker
impl
anta
tion
8
(7%
) 4
(7%
) 2
(5%
) 0.
87 (0
.26-
2.92
) 0.
82
RV
-PA
: rig
ht v
entr
icul
ar-p
ulm
onar
y ar
tery
VSD
: ven
tric
ular
sep
tal d
efec
t
ASD
: atr
ial s
epta
l def
ect
MA
PCA
: maj
or a
orto
-pul
mon
ary
colla
tera
l art
ery
AIC
D: a
utom
atic
impl
anta
ble
card
iove
rter
def
ibri
llato
r
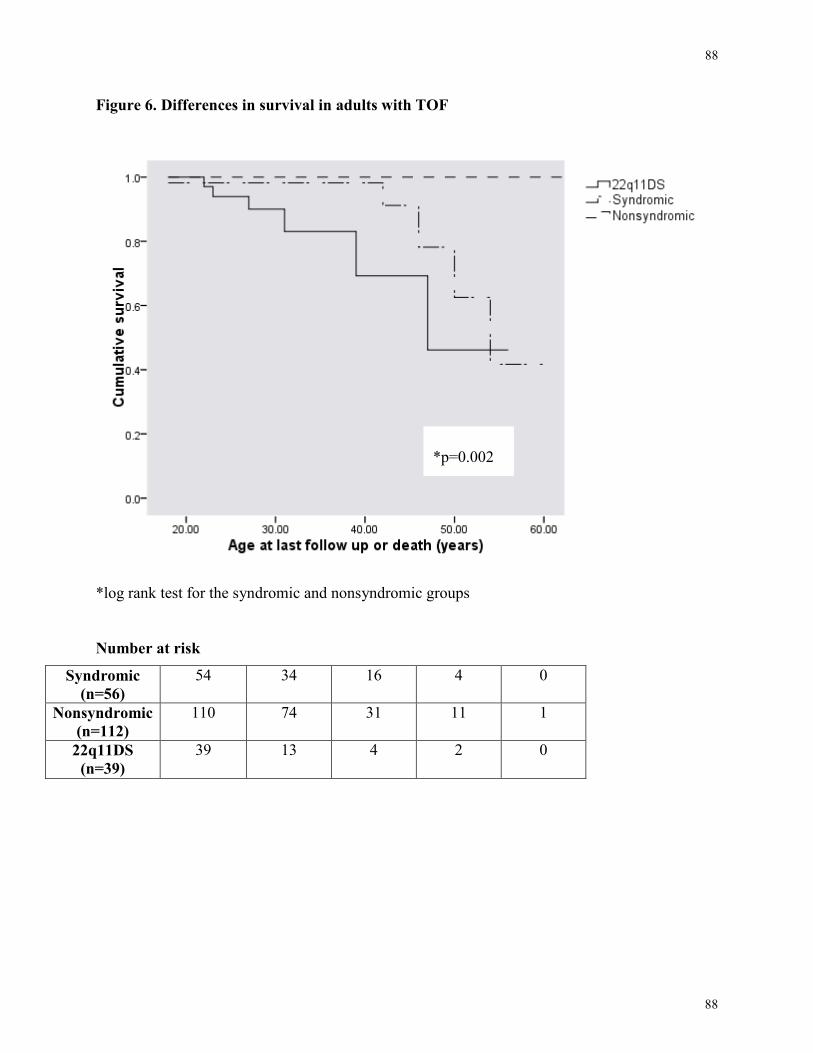
88
88
Figure 6. Differences in survival in adults with TOF
*log rank test for the syndromic and nonsyndromic groups Number at risk
Syndromic (n=56)
54 34 16 4 0
Nonsyndromic (n=112)
110 74 31 11 1
22q11DS (n=39)
39 13 4 2 0
*p=0.002

89
89
Figure 7. Freedom from adverse cardiac events in adults with TOF
*log-rank for the syndromic and nonsyndromic groups
Number at risk
Syndromic (n=56)
53 33 19 1 0 0
Nonsyndromic (n=112)
111 74 27 8 0 0
22q11DS (n=39)
33 16 2 0 0 0
*p=0.65

90
90
Figure 8. Freedom from cardiac intervention in adults with TOF
*log-rank for the syndromic and nonsyndromic groups Number at risk
Syndromic (n=56)
54 31 16 2 0
Nonsyndromic (n=112)
108 63 19 4 0
22q11DS (n=39)
36 13 3 2 0
*p=0.15

91
91
5 Summary of Thesis and Future Directions
Using simple clinical genetic screening, we identified a clinically important subgroup of
adults with TOF who have a more complex cardiac disease, are at elevated risk for developing
neuropsychiatric and endocrine disorders, and have a higher mortality rate compared to an age
and gender matched control group. We show that in the absence of major genetic anomalies and
syndromes, the prevalence of extra-cardiac features are high in patients with TOF, and in some
cases, similar to that found in patients with 22q11DS. The high prevalence of extra-cardiac
features in TOF should alert the clinician to the potential multisystem nature of this disease, and
prompt a more vigorous approach for early recognition and treatment of extra-cardiac
abnormalities. Routine ultrasound (e.g. abdominal ultrasound for gastrointestinal and
genitourinary anomalies), magnetic resonance imaging of the brain and spinal cord, and
laboratory screening for endocrine abnormalities would be of great diagnostic value for
identifying these extra-cardiac features.
While certain extra-cardiac conditions may be present from birth, some develop in late
childhood or adulthood, more emphasis on extra-cardiac conditions is required for transition
from pediatric to adult care to ensure that care is being followed through from the pediatric to the
adult setting. This is important since extra-cardiac anomalies may have important health
consequences.
In agreement with previous studies, we found that adults with 22q11DS have a high
mortality rate (Bassett, Chow et al. 2009). We further showed that patients with syndromic
features, but without 22q11DS or other major known genetic anomalies, have a significantly

92
92
higher mortality rate compared to an age and gender matched control group with TOF. All five
deceased subjects in the syndromic group had learning difficulties, and three had extra-cardiac
abnormalities. Although not proven, the presence of learning difficulties, CHD, and extra-cardiac
anomalies may be due to underlying structural genomic changes altering gene dosage. More
recently, copy number variations have been shown to cause developmental disorders such as
autism spectrum disorder and CHDs (Greenway, Pereira et al. 2009, Costain, Silversides et al.
2010, Mefford, Sharp et al. 2008b, International Schizophrenia Consortium 2008, Girirajan,
Rosenfeld et al. 2010). In particular, CNVs on chromosome 1q21.1 have previously been
implicated in cognitive impairment, CHD, and other congenital anomalies (Greenway, Pereira et
al. 2009)(International Schizophrenia Consortium 2008, Mefford, Sharp et al. 2008a). This is one
mechanism that may explain the connection between cardiac and extra-cardiac anomalies and
learning difficulties. CNVs can also increase susceptibility to early mortality, as Bassett et al
have shown that adults with 22q11.2 deletions (large CNV) are at a higher risk of death
compared to their unaffected siblings (Bassett, Chow et al. 2009). These extra-cardiac features
such as learning difficulties, may serve as clinical markers of more complex genetic diseases
with an increased risk of premature death in patients with TOF. More studies are needed to
examine this relationship.
Studies focusing on phenotype (i.e. clustering patients into meaningful categories),
similar to ours (Scutt, Chow et al, 2001), have great clinical relevance and can help in
discoveries of genetic pathways to TOF. The majority of patients in our study sample, apart from
the 22q11DS group, have not been seen by a clinical geneticist and thus the presence of extra-
cardiac features in those patients may be indicative of an unknown or undiagnosed genetic
syndrome. In fact, there are current reports that 22q11DS is under-recognized in patients with

93
93
CHD (van Engelen, Topf et al. 2010). This signifies the importance of the need for a more
vigorous approach to identify patients with 22q11DS and other genetic syndromes in the clinic,
as these syndromes have reproductive and clinical implications. Classifying patients with TOF
into more homogenous groups, in the absence of major genetic anomalies, may help identify
patterns of cardiac and extra-cardiac features that are commonly associated with TOF and which
may have relevance to underlying chromosomal or cytogenetic aberrations. Molecular genetic
studies in these patients will further our understanding of the genetic causes of TOF.
Recent recommendations suggest that patients with syndromic features should have
genome-wide microarray testing to identify pathogenic structural genomic changes (Miller,
Adam et al. 2010). These include individuals with syndromic features such as developmental
delay, mental retardation, autism and/or multiple congenital anomalies. However, the cost and
challenges in interpreting results from genome-wide microarrays suggest that such testing may
not be justified for patients who do not have syndromic and/or neurodevelopmental features as
they would be expected to have lower yields compared to syndromal patients (Miller, Adam et
al. 2010). Genome-wide studies in TOF patients, especially in those with multiple extra-cardiac
anomalies, may shed some light on potential pathogenic genes involved in disease formation.
Genetic evaluation and hopefully genome-wide testing in families of probands with TOF,
especially in cases where there is a positive family history, would also help in our understanding
of inheritance and genetic causes of this disease.
5.1 Future directions This study identified a newly defined clinical subgroup of patients within TOF with more
complex CHD, higher prevalence of late-onset conditions who are at greater risk of death
compared to a nonsyndromic control group. The syndromic group identified in this study

94
94
represents an initial first step towards identifying a more homogenous group of patients who may
be enriched for certain genetic causes of TOF. Greenway et al and Costain et al have recently
identified CNV(s) in patients with TOF (Greenway, Pereira et al. 2009, Costain, Silversides et al,
2010); genome-wide molecular studies could shed light on the underlying genetic etiology of
TOF and help explain the phenotypic variability that exists within this patient population. In
addition, further larger studies are needed to determine mechanisms that may contribute to
pathologic cause of death in patients late after repair. With the advancement of medical
technology, especially genome wide microarrays, genomic medicine will help with surveillance
to identify high risk patient groups to optimize clinical management. Further studies will be
needed to prospectively follow up with a large number of patients, especially those with
syndromic features, to better understand the possible risk factors for death in this patient
population. Larger longitudinal studies with appropriate control groups will be required to
investigate the impact of genetic aberrations on cardiac and non-cardiac outcomes. Studies using
hearts of patients with TOF with extra-cardiac anomalies may shed some light on common
genetic factors (e.g. transcription factors) that regulate the development of different organ
systems.

95
95
6 Appendices
Data abstraction form:
Syndromic Patients with Tetralogy of Fallot-The Clinical Spectrum of Disease
Study ID #: Evaluated by a geneticist? � Yes � No
A. DEMOGRAPHICAS Gender: � Male �Female Age________ Ethnicity: � White � Aboriginal (e.g. North American Indian, Metis, Inuit, Eskimo) � Chinese � Black � Filipino � Latin American � Southeast Asian (e.g. Cambodian, Indonesian, Laotian, Vietnames) � Arab � West Asian (e.g. Afghan, Iranian) � Japanese � Korean � South Asian (e.g. East Indian, Pakistani, Sri Lankan � Other (please specify): _____________________________ � Not available Date and age at last follow-up: ______________________________________________ Current height (cm)__________weight (kg): _____________________ Demographics at birth: Gestational age (wks): ____________________________________________________ Weight (kg)/ Height (cm) at birth: _______________________________________ B. CARDIAC ANATOMY
���� Pulmonary stenosis ���� Mild ���� Moderate ���� Severe ______________________________ � Sub-pulmonary obstruction: ________________________________________________ ���� Pulmonary Atresia ______________________________________________ ���� Absent pulmonary valve ______________________________________________ � Ventricular septal defect: ____________________________________ � ASD: ______________________________________________________ ���� Patent ductus arteriosis � Major aorto-pulmonary collateral artery (MAPCA) _______________________________________________________________________________ � Aortic abnormality:
Right aortic arch: __________________________________________________________ Hypoplasia of aorta: ________________________________________________________ Aortic Override: ___________________________________________________________

96
96
Aortic Coarctation: _________________________________________________________ Other: ___________________________________________________________
� Aberrent Subclavian Artery: _____________________________________________________ � Other Vascular abnormalities: __________________________________________________________________________________________________________________________________________________ � Other cardiac anomalies: ______________________________________________________________________________________________________________________________________________________________ C. OTHER CONGENITAL ANOMALIES � Facial feature: � Ocular hypertelorism
� narrow palpebral fissures/ � bloated eyelids � low nasal bridge � small mouth � deformed earlobe
Other: ________________________________________________ � Otolaryngeal abnormality: � cleft palate (hard/soft/submucous)
� velopharyngeal insufficiency Other: _______________________________________________________________________ � Developmental delay (Intelligence quotient) : _______________________________________________________________________________ � Visual/Hearing Problems (please specify what): _______________________________________________________________________________ � Central nervous system/ Psychiatric illness (please specify what): _______________________________________________________________________________ � Endocrine (please specify what):
� Hyperthyroid � Hypothyroid � Hypoparathyroidism ���� Hypocalcemia
_______________________________________________________________________________ � GI (please specify what): _______________________________________________________________________________ � Renal (please specify what): _______________________________________________________________________________ � Pulmonary disease (please specify what): _______________________________________________________________________________ � Muskoskeletal (please specify what): _______________________________________________________________________________ � Hematologic / Immunologic (please specify what): _______________________________________________________________________________ � Reproductive (please specify what): _______________________________________________________________________________ � Other (please specify): __________________________________________________________ D. CARDIAC SURGERY � BT Shunt: Year

97
97
� Waterston shunt: Year � Potts shunt: Year � VSD patch: Year � Annular patch/pulmonary artery patch: Year � RVOT Infundibulectomy: Year � PVR: Year � Cryoablation: Year � Maze procedure: Year �EP ablation: Year �ICD implantation: Year �Pacemaker (type): Year �RV to PA conduit (type): Year �Left pulmonary artery angioplasty (type): Year �Left pulmonary artery stent (type): Year �Other: Year Type:
Year Type:
Year Type:
Year Type: E. NONCARDIAC SURGERY
� Palatal flap year__________ � Scoliosis year__________ � Choleyslectomy year__________ � Hernias year__________ � Other year__________ TYPE_____________________________________________________
year__________ TYPE_____________________________________________________

98
98
F. MEDICATIONS AT LAST FOLLOW UP
� No � Yes If YES: Details ACE inhibitors………………….. �
Antiarrhythmics ………………..... (specify)……………………...........
�
ASA……………………………….. � Beta blocker……………………... � Coumadin ……………………….. � Digoxin……………………………. � Diuretics ..................................... � Heparin sc................................. � Nitrates ....................................... � Synthroid………………………….. �
Calcium supplement……………… �
Magensium supplement…………... �
Vitamin D analog…………………. �
Antidepressant……………………. �
Anxiolytic………………………………… � Antipsychotic………………………. �
� Other cardiac (specify):
� Other non-cardiac (specify):
G. FUNCTIONAL STATUS AT LAST FOLLOW UP Peak VO2max………………….
NYHA 1 2 3 4 Percent predicted: ……………. � � � �
H. ECG AT LAST FOLLOW UP
1. ECG available: � No � Yes Normal sinus rhythm: � No � Yes If no, specify: _________________________________________________ QRS duration:

99
99
� No � Yes If yes, specify: _________________________________________________ QT interval: � No � Yes If yes, specify: _________________________________________________ Ventricular hypertrophy: � No � Yes If yes, specify: _________________________________________________ Other: __________________________________________________________________ I. OXYGEN SATS AT LAST VISIT Oxygen saturation: ______________% J. ECHOCARDIOGRAM AT LAST VISIT
1. Ventricular size and function. Systemic ventricle size: � Normal � Mildly dilated � Moderately dilated � Severely dilated Systemic ventricular function: � Normal � mildly reduced � moderately reduced � severely reduced Sub-pulmonary ventricle size:�Normal �Mildly dilated �Moderately dilated �Severely dilated Sub-pulmonary ventricle function:�Normal �mildly reduced � moderately reduced �severely reduced 2. Systemic AV regurgitation: � None � Mild � Moderate � Mod-severe � Severe Venous AV regurgitation: � None � Mild � Moderate � Mod-severe � Severe Aortic regurgitation: � None � Mild � Moderate � Mod-severe � Severe Pulmonic regurgitation: � None � Mild � Moderate � Mod-severe � Severe 3. Systemic AV stenosis: � None � Mild � Moderate � Severe Mean gr…….. Peak gr……. Venous AV stenosis: � None � Mild � Moderate � Severe Mean gr…….. Peak gr……. PV stenosis: � None � Mild � Moderate � Severe Mean gr…….. Peak gr……. Subvalvular PV stenosis: � None � Mild � Moderate � Severe Mean gr…….. Peak gr……. Supra PV stenosis: � None � Mild � Moderate � Severe Mean gr…….. Peak gr……. Aortic stenosis: � None � Mild � Moderate � Severe Mean gr…….. Peak gr……. 5. Hemodynamics / Shunts: � No � Yes If yes, then level and direction: � atrial � ventricle � great artery � L-R � R-L � Bidir. Peak VSD gradients………………mmHg RV systolic pressure……………...mmHg Assuming RA pressure of ……….mmHg K. LABORATORY RESULTS AT LAST FOLLOW UP Details TSH…………...... . ………… Creatinine… ......................... Calcium (serum vs ionized) Hemoglobin
Platelet

100
100
L. CARDIAC EVENTS AT LAST FOLLOW UP � No: � Yes:
DATE/DETAILS Acute Coronary Syndrome… � Left heart failure…………..... � Right heart failure................. � Pulmonary embolism............. � Aortic dissection .................. � Syncope …………… ..... � Endocarditis…....................... � Cardiac arrest............. � Stroke/TIA ............................ � Ventricular arrhythmias � Supraventricular arrhythmias � Other � M. FAMILY HISTORY �TOF �Other Congenital heart disease: �Other health problems (please specify what):

101
101
7 References
ABU-SULAIMAN, R.M. and SUBAIH, B., 2004. Congenital heart disease in infants of diabetic
mothers: echocardiographic study. Pediatric cardiology, 25(2), 137-140.
ANACLERIO, S., DI CIOMMO, V., MICHIELON, G., DIGILIO, M.C., FORMIGARI, R.,
PICCHIO, F.M., GARGIULO, G., DI DONATO, R., DE IORIS, M.A. and MARINO, B., 2004a.
Conotruncal heart defects: impact of genetic syndromes on immediate operative mortality.
Italian Heart Journal : official journal of the Italian Federation of Cardiology, 5(8), 624-628.
APITZ, C., WEBB, G.D. and REDINGTON, A.N., 2009. Tetralogy of Fallot. Lancet, 374(9699),
1462-1471.
ATALLAH J., JOFFE, A.R., ROBERTSON, C.M.T., LEONARD, M., BLAKELY, P.M.,
NETTEL-AGUIRRE, A., SAUVE, R.S., ROSS, D.B., REBEYKA, I.M., WESTERN
CANADIAN COMPLEX PEDIATRIC THERAPIES PROJECT FOLLOW-UP GROUP., 2007.
Two-year general and neurodevelopmental outcome after neonatal complex cardiac surgery in
patients with deletion 22q11.2: a comparative study. The Journal of Thoracic and
Cardiovascular Surgery,134, 772-779.
ATALLAH-YUNES, N.H., KAVEY, R.E., BOVE, E.L., SMITH, F.C., KVESELIS, D.A.,
BYRUM, C.J. and GAUM, W.E., 1996. Postoperative assessment of a modified surgical
approach to repair of tetralogy of Fallot. Long-term follow-up. Circulation, 94(9 Suppl), II22-6.
BASSETT, A.S., CHOW, E.W., HUSTED, J., WEKSBERG, R., CALUSERIU, O., WEBB,
G.D. and GATZOULIS, M.A., 2005. Clinical features of 78 adults with 22q11 Deletion
Syndrome. American Journal of Medical Genetics, 138(4), 307-313.
BASSETT, A.S., HODGKINSON, K., CHOW, E.W., CORREIA, S., SCUTT, L.E. and
WEKSBERG, R., 1998. 22q11 deletion syndrome in adults with schizophrenia. American
journal of medical genetics, 81(4), 328-337.

102
102
BASSETT, A.S., SCHERER, S.W. and BRZUSTOWICZ, L.M., 2010. Copy Number Variations
in Schizophrenia: Critical Review and New Perspectives on Concepts of Genetics and Disease.
The American Journal of Psychiatry, .
BASSETT, A., CHOW, E., HUSTED, J., HODGKINSON, K., OECHSLIN, E., HARRIS, L. and
SILVERSIDES, C., 2009. Premature death in adults with 22q11.2 deletion syndrome. Journal of
Medical genetics, 46(5), 324-330.
BASSON, C.T., BACHINSKY, D.R. and LIN, R.C., 1997. Mutations in human TBX5 cause
limb and cardiac malformation in Holt-Oram syndrome. Nature genetics, 15, 30-35.
BEARDEN, C.E., VAN ERP, T.G., MONTEROSSO, J.R., SIMON, T.J., GLAHN, D.C.,
SALEH, P.A., HILL, N.M., MCDONALD-MCGINN, D.M., ZACKAI, E., EMANUEL, B.S.
and CANNON, T.D., 2004. Regional brain abnormalities in 22q11.2 deletion syndrome:
association with cognitive abilities and behavioral symptoms. Neurocase, 10(3), 198-206.
BELL, R., RANKIN, J., DONALDSON, L.J. and NORTHERN CONGENITAL
ABNORMALITY SURVEY STEERING GROUP, 2003. Down's syndrome: occurrence and
outcome in the north of England, 1985-99. Paediatric and perinatal epidemiology, 17(1), 33-39.
BILOUS, R.W. and TUNBRIDGE, W.M., 1988. The epidemiology of hypothyroidism--an
update. Bailliere's Clinical Endocrinology and Metabolism, 2(3), 531-540.
BLAKE, K.D. and PRASAD, C., 2006. CHARGE syndrome. Orphanet journal of rare disease1,
34.
BONEVA, R.S., BOTTO, L.D., MOORE, C.A., YANG, Q., CORREA, A. and ERICKSON,
J.D., 2001. Mortality associated with congenital heart defects in the United States: trends and
racial disparities, 1979-1997. Circulation, 103(19), 2376-2381.
BOTTO, L.D., MAY, K., FERNHOFF, P.M., CORREA, A., COLEMAN, K., RASMUSSEN,
S.A., MERRITT, R.K., O'LEARY, L.A., WONG, L.Y., ELIXSON, E.M., MAHLE, W.T. and
CAMPBELL, R.M., 2003. A population-based study of the 22q11.2 deletion: phenotype,
incidence, and contribution to major birth defects in the population. Pediatrics, 112(1 Pt 1), 101-
107.

103
103
BRAUNWALD E, LIBBY P, BONOW RO, MANN DL, ZIPES DP. Braunwald’s heart disease:
a textbook of cardiovascular medicine. Saunders. Philadelphia. 2007.
BRICKER, J.T., 1995. Sudden death and tetralogy of Fallot. Risks, markers, and causes.
Circulation, 92(2), 158-159.
BRUNI, L., ANGELETTI, B., PASCALE, E., TOZZI, M.C., GIAMMARIA, P., VERNA, R.
and D'AMBROSIO, E., 1996. Exclusion of Treacher Collins Franceschetti syndrome in a subject
with tetralogy of Fallot and cryptorchidism. Clinical genetics, 50(2), 89-92.
BURKE, B.A., JOHNSON, D., GILBERT, E.F., DRUT, R.M., LUDWIG, J. and WICK, M.R.,
1987. Thyrocalcitonin-containing cells in the Di George anomaly. Human pathology, 18(4), 355-
360.
BURN, J., BRENNAN, P., LITTLE, J., HOLLOWAY, S., COFFEY, R., SOMERVILLE, J.,
DENNIS, N.R., ALLAN, L., ARNOLD, R., DEANFIELD, J.E., GODMAN, M., HOUSTON,
A., KEETON, B., OAKLEY, C., SCOTT, O., SILOVE, E., WILKINSON, J., PEMBREY, M.
and HUNTER, A.S., 1998. Recurrence risks in offspring of adults with major heart defects:
results from first cohort of British collaborative study. Lancet, 351(9099), 311-316.
CALZOLARI, E., GARANI, G., COCCHI, G., MAGNANI, C., RIVIERI, F., NEVILLE, A.,
ASTOLFI, G., BARONCINI, A., GARAVELLI, L., GUALANDI, F., SCORRANO, M. and
BOSI, G., 2003. Congenital heart defects: 15 years of experience of the Emilia-Romagna
Registry (Italy). European journal of epidemiology, 18(8), 773-780.
CAMPBELL, L.E., DALY, E., TOAL, F., STEVENS, A., AZUMA, R., CATANI, M., NG, V.,
VAN AMELSVOORT, T., CHITNIS, X., CUTTER, W., MURPHY, D.G. and MURPHY, K.C.,
2006. Brain and behaviour in children with 22q11.2 deletion syndrome: a volumetric and voxel-
based morphometry MRI study. 129, 1218-1228.
CARVALHO, J.S., SHINEBOURNE, E.A., BUSST, C., RIGBY, M.L. and REDINGTON,
A.N., 1992. Exercise capacity after complete repair of tetralogy of Fallot: deleterious effects of
residual pulmonary regurgitation. British heart journal, 67(6), 470-473.
CASCOS, A.S., 1971. Genetics of Fallot's tetralogy. British heart journal, 33(6), 899-904.

104
104
CASSIDY, S.C. and ALLEN, H.D., 1991. Tetralogy of Fallot in triplet siblings. American
journal of cardiology, 67(16), 1442-1444.
CHESSA, M., BUTERA, G., BONHOEFFER, P., ISERIN, L., KACHANER, J., LYONNET, S.,
MUNNICH, A., SIDI, D. and BONNET, D., 1998. Relation of genotype 22q11 deletion to
phenotype of pulmonary vessels in tetralogy of Fallot and pulmonary atresia-ventricular septal
defect. Heart, 79(2), 186-190.
CHOW, E.W., ZIPURSKY, R.B., MIKULIS, D.J. and BASSETT, A.S., 2002. Structural brain
abnormalities in patients with schizophrenia and 22q11 deletion syndrome. Biological
psychiatry, 51(3), 208-215.
CLARK, K.L., YUTZEY, K.E. and BENSON, D.W., 2006. Transcription factors and congenital
heart defects. Annual Review of Physiology, 68, 97-121.
COHEN, E., CHOW, E.W., WEKSBERG, R. and BASSETT, A.S., 1999. Phenotype of adults
with the 22q11 deletion syndrome: A review. American journal of medical genetics, 86(4), 359-
365.
COSTAIN, G., SILVERSIDES, C.K., MARSHALL, C.R., SHAGO, M., COSTAIN, N. and
BASSETT, A.S., 2010. 13q13.1-q13.2 deletion in tetralogy of Fallot: Clinical report and a
literature review. International journal of cardiology, .
CUNEO, B.F., 2001. 22q11.2 deletion syndrome: DiGeorge, velocardiofacial, and conotruncal
anomaly face syndromes. Current opinion in pediatrics, 13(5), 465-472.
DABIZZI, R.P., CAPRIOLI, G., AIAZZI, L., CASTELLI, C., BALDRIGHI, G., PARENZAN,
L. and BALDRIGHI, V., 1980. Distribution and anomalies of coronary arteries in tetralogy of
fallot. Circulation, 61(1), 95-102.
DABIZZI, R.P., TEODORI, G., BARLETTA, G.A., CAPRIOLI, G., BALDRIGHI, G. and
BALDRIGHI, V., 1990. Associated coronary and cardiac anomalies in the tetralogy of Fallot. An
angiographic study. European heart journal, 11(8), 692-704.

105
105
DAVLOUROS, P.A., KILNER, P.J., HORNUNG, T.S., LI, W., FRANCIS, J.M., MOON, J.C.,
SMITH, G.C., TAT, T., PENNELL, D.J. and GATZOULIS, M.A., 2002. Right ventricular
function in adults with repaired tetralogy of Fallot assessed with cardiovascular magnetic
resonance imaging: detrimental role of right ventricular outflow aneurysms or akinesia and
adverse right-to-left ventricular interaction. Journal of the American College of Cardiology,
40(11), 2044-2052.
DEMASO, D.R., CAMPIS, L.K., WYPIJ, D., BERTRAM, S., LIPSHITZ, M. and FREED, M.,
1991. The impact of maternal perceptions and medical severity on the adjustment of children
with congenital heart disease. Journal of pediatric psychology, 16(2), 137-149.
DENTICE, M., CORDEDDU, V., ROSICA, A., FERRARA, A.M., SANTARPIA, L.,
SALVATORE, D., CHIOVATO, L., PERRI, A., MOSCHINI, L., FAZZINI, C., OLIVIERI, A.,
COSTA, P., STOPPIONI, V., BASERGA, M., DE FELICE, M., SORCINI, M., FENZI, G., DI
LAURO, R., TARTAGLIA, M. and MACCHIA, P.E., 2006. Missense mutation in the
transcription factor NKX2-5: a novel molecular event in the pathogenesis of thyroid dysgenesis.
The Journal of clinical endocrinology and metabolism, 91(4), 1428-1433.
DEVRIENDT, K., SWILLEN, A., FRYNS, J.P., PROESMANS, W. and GEWILLIG, M., 1996.
Renal and urological tract malformations caused by a 22q11 deletion. Journal of medical
genetics, 33(4), 349.
DIGILIO, M.C., MARINO, B., GIANNOTTI, A., TOSCANO, A. and DALLAPICCOLA, B.,
1997. Recurrence risk figures for isolated tetralogy of Fallot after screening for 22q11
microdeletion. Journal of medical genetics, 34(3), 188-190.
DRISCOLL, D.A., BUDARF, M.L. and EMANUEL, B.S., 1992. A genetic etiology for
DiGeorge syndrome: consistent deletions and microdeletions of 22q11. American journal of
human genetics, 50(5), 924-933.
DRISCOLL, D.A., SALVIN, J., SELLINGER, B., BUDARF, M.L., MCDONALD-MCGINN,
D.M., ZACKAI, E.H. and EMANUEL, B.S., 1993. Prevalence of 22q11 microdeletions in
DiGeorge and velocardiofacial syndromes: implications for genetic counselling and prenatal
diagnosis. Journal of medical genetics, 30(10), 813-817.

106
106
DRISCOLL, D.A., SPINNER, N.B., BUDARF, M.L., MCDONALD-MCGINN, D.M.,
ZACKAI, E.H., GOLDBERG, R.B., SHPRINTZEN, R.J., SAAL, H.M., ZONANA, J. and
JONES, M.C., 1992. Deletions and microdeletions of 22q11.2 in velo-cardio-facial syndrome.
American journal of medical genetics, 44(2), 261-268.
ELDADAH, Z.A., HAMOSH, A., BIERY, N.J., MONTGOMERY, R.A., DUKE, M., ELKINS,
R. and DIETZ, H.C., 2001. Familial Tetralogy of Fallot caused by mutation in the jagged1 gene.
Human molecular genetics, 10(2), 163-169.
ELIEZ, S., ANTONARAKIS, S.E., MORRIS, M.A., DAHOUN, S.P. and REISS, A.L., 2001.
Parental origin of the deletion 22q11.2 and brain development in velocardiofacial syndrome: a
preliminary study. Archives of genetics in psychiatry, 58(1), 64-68.
ELIEZ, S., BARNEA-GORALY, N., SCHMITT, J.E., LIU, Y. and REISS, A.L., 2002.
Increased basal ganglia volumes in velo-cardio-facial syndrome (deletion 22q11.2). Biological
psychiatry, 52(1), 68-70.
ELIEZ, S., SCHMITT, J.E., WHITE, C.D. and REISS, A.L., 2000. Children and adolescents
with velocardiofacial syndrome: a volumetric MRI study. American journal of psychiatry,
157(3), 409-415.
ESKEDAL, L., HAGEMO, P., ESKILD, A., AAMODT, G., SEILER, K.S. and THAULOW, E.,
2004. A population-based study of extra-cardiac anomalies in children with congenital cardiac
malformations. Cardiology in the young, 14(6), 600-607.
EVANS, W.N., 2008. "Tetralogy of Fallot" and Etienne-Louis Arthur Fallot. Pediatric
cardiology, 29(3), 637-640.
FERENCZ, C., NEILL, C.A., BOUGHMAN, J.A., RUBIN, J.D., BRENNER, J.I. and PERRY,
L.W., 1989. Congenital cardiovascular malformations associated with chromosome
abnormalities: an epidemiologic study. Journal of pediatrics, 114(1), 79-86.
FERENCZ, C., RUBIN, J.D., MCCARTER, R.J., BOUGHMAN, J.A., WILSON, P.D.,
BRENNER, J.I., NEILL, C.A., PERRY, L.W., HEPNER, S.I. and DOWNING, J.W., 1987.

107
107
Cardiac and noncardiac malformations: observations in a population-based study. Teratology,
35(3), 367-378.
FINE, S.E., WEISSMAN, A., GERDES, M., PINTO-MARTIN, J., ZACKAI, E.H.,
MCDONALD-MCGINN, D.M. and EMANUEL, B.S., 2005. Autism spectrum disorders and
symptoms in children with molecularly confirmed 22q11.2 deletion syndrome. Journal of autism
and developmental disorders, 35(4), 461-470.
FRANCANNET, C., LANCASTER, P.A., PRADAT, P., COCCHI, G. and STOLL, C., 1993.
The epidemiology of three serious cardiac defects. A joint study between five centres. European
journal of epidemiology, 9(6), 607-616.
FRASER, C.D.,JR., MCKENZIE, E.D. and COOLEY, D.A., 2001. Tetralogy of Fallot: surgical
management individualized to the patient. Annals in thoracic surgery, Annals of thoracic
surgery, 71(5), 1556-61; discussion 1561-3.
FREEDOM RM, SHI-JOON Y, MIKAILIAN H, WILLIAMS WG. The natural and modified
history of congenital heart disease. Blackwell publishing. New York. 2004. pp 186-211.
FRIEDMAN, M.J. and SHARIEFF, G.Q., 2006. Seizures in children. Pediatric clinics of
North.America, 53(2), 257-277.
FUNG, W.L., CHOW, E.W., WEBB, G.D., GATZOULIS, M.A. and BASSETT, A.S., 2008.
Extracardiac features predicting 22q11.2 deletion syndrome in adult congenital heart disease.
International journal of cardiology, 131(1), 51-58.
GATZOULIS, M.A., BALAJI, S., WEBBER, S.A., SIU, S.C., HOKANSON, J.S., POILE, C.,
ROSENTHAL, M., NAKAZAWA, M., MOLLER, J.H., GILLETTE, P.C., WEBB, G.D. and
REDINGTON, A.N., 2000. Risk factors for arrhythmia and sudden cardiac death late after repair
of tetralogy of Fallot: a multicentre study. Lancet, 356(9234), 975-981.
GATZOULIS, M.A., TILL, J.A., SOMERVILLE, J. and REDINGTON, A.N., 1995.
Mechanoelectrical interaction in tetralogy of Fallot. QRS prolongation relates to right ventricular
size and predicts malignant ventricular arrhythmias and sudden death. Circulation, 92(2), 231-
237.

108
108
GATZOULIS MA, WEBB GD, DAUBENEY PEF. Diagnosis and management of adult
congenital heart disease. Churchill Livingstone. 2003. Philadelphia. pp 315-326.
GERDES, M., SOLOT, C., WANG, P.P., MOSS, E., LAROSSA, D., RANDALL, P.,
GOLDMUNTZ, E., CLARK, B.J.,3RD, DRISCOLL, D.A., JAWAD, A., EMANUEL, B.S.,
MCDONALD-MCGINN, D.M., BATSHAW, M.L. and ZACKAI, E.H., 1999. Cognitive and
behavior profile of preschool children with chromosome 22q11.2 deletion. American journal of
medical genetics, 85(2), 127-133.
GERSONY WM, ROSENBAUM MS. Congenital heart disease in the adult. McGraw-Hill
Companies. 2002. pp. 145-166.
GHAI, A., SILVERSIDES, C., HARRIS, L., WEBB, G.D., SIU, S.C. and THERRIEN, J., 2002.
Left ventricular dysfunction is a risk factor for sudden cardiac death in adults late after repair of
tetralogy of Fallot. Journal of the American College of Cardiology, 40(9), 1675-1680.
GIBSON, S. and LEWIS, K.C., 1952. Congenital heart disease following maternal rubella during
pregnancy. Archives of pediatrics and adolescent medicine, 83(3), 317-319.
GIRIRAJAN, S., ROSENFELD, J.A., COOPER, G.M., ANTONACCI, F., SISWARA, P.,
ITSARA, A., VIVES, L., WALSH, T., MCCARTHY, S.E., BAKER, C., MEFFORD, H.C.,
KIDD, J.M., BROWNING, S.R., BROWNING, B.L., DICKEL, D.E., LEVY, D.L., BALLIF,
B.C., PLATKY, K., FARBER, D.M., GOWANS, G.C., WETHERBEE, J.J., ASAMOAH, A.,
WEAVER, D.D., MARK, P.R., DICKERSON, J., GARG, B.P., ELLINGWOOD, S.A., SMITH,
R., BANKS, V.C., SMITH, W., MCDONALD, M.T., HOO, J.J., FRENCH, B.N., HUDSON, C.,
JOHNSON, J.P., OZMORE, J.R., MOESCHLER, J.B., SURTI, U., ESCOBAR, L.F., EL-
KHECHEN, D., GORSKI, J.L., KUSSMANN, J., SALBERT, B., LACASSIE, Y., BISER, A.,
MCDONALD-MCGINN, D.M., ZACKAI, E.H., DEARDORFF, M.A., SHAIKH, T.H., HAAN,
E., FRIEND, K.L., FICHERA, M., ROMANO, C., GECZ, J., DELISI, L.E., SEBAT, J., KING,
M.C., SHAFFER, L.G. and EICHLER, E.E., 2010. A recurrent 16p12.1 microdeletion supports a
two-hit model for severe developmental delay. Nature genetics, 42(3), 203-209.

109
109
GOLDBERG, R., MOTZKIN, B., MARION, R., SCAMBLER, P.J. and SHPRINTZEN, R.J.,
1993. Velo-cardio-facial syndrome: a review of 120 patients. American journal of medical
genetics, 45(3), 313-319.
GOLDMUNTZ, E., CLARK, B.J., MITCHELL, L.E., JAWAD, A.F., CUNEO, B.F., REED, L.,
MCDONALD-MCGINN, D., CHIEN, P., FEUER, J., ZACKAI, E.H., EMANUEL, B.S. and
DRISCOLL, D.A., 1998. Frequency of 22q11 deletions in patients with conotruncal defects.
Journal of the American college of cardiology, 32(2), 492-498.
GOLDMUNTZ, E., GEIGER, E. and BENSON, D.W., 2001. NKX2.5 mutations in patients with
tetralogy of fallot. Circulation, 104(21), 2565-2568.
GOODSHIP, J., CROSS, I., LILING, J. and WREN, C., 1998. A population study of
chromosome 22q11 deletions in infancy. Archives of Disease in Childhood, 79(4), 348-351.
GOODSHIP, J., CROSS, I., SCAMBLER, P. and BURN, J., 1995. Monozygotic twins with
chromosome 22q11 deletion and discordant phenotype. Journal of medical genetics, 32(9), 746-
748.
GOTHELF, D., PRESBURGER, G., ZOHAR, A.H., BURG, M., NAHMANI, A., FRYDMAN,
M., SHOHAT, M., INBAR, D., AVIRAM-GOLDRING, A., YESHAYA, J., STEINBERG, T.,
FINKELSTEIN, Y., FRISCH, A., WEIZMAN, A. and APTER, A., 2004. Obsessive-compulsive
disorder in patients with velocardiofacial (22q11 deletion) syndrome. American journal of
medical genetics, 126B(1), 99-105.
GREENWAY, S.C., PEREIRA, A.C., LIN, J.C., DEPALMA, S.R., ISRAEL, S.J., MESQUITA,
S.M., ERGUL, E., CONTA, J.H., KORN, J.M., MCCARROLL, S.A., GORHAM, J.M.,
GABRIEL, S., ALTSHULER, D.M., QUINTANILLA-DIECK MDE, ARTUNDUAGA, M.A.,
EAVEY, R.D., PLENGE, R.M., SHADICK, N.A., WEINBLATT, M.E., DE JAGER, P.L.,
HAFLER, D.A., BREITBART, R.E., SEIDMAN, J.G. and SEIDMAN, C.E., 2009. De novo
copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nature
genetics, 41(8), 931-935.

110
110
GREENWOOD, R.D., ROSENTHAL, A., PARISI, L., FYLER, D.C. and NADAS, A.S., 1975.
Extracardiac abnormalities in infants with congenital heart disease. Pediatrics, 55(4), 485-492.
GREIG, F., PAUL, E., DIMARTINO-NARDI, J. and SAENGER, P., 1996. Transient congenital
hypoparathyroidism: resolution and recurrence in chromosome 22q11 deletion. Journal of
pediatrics, 128(4), 563-567.
GRIFFIN, H.R., HALL, D.H., TOPF, A., EDEN, J., STUART, A.G., PARSONS, J., PEART, I.,
DEANFIELD, J.E., O'SULLIVAN, J., BABU-NARAYAN, S.V., GATZOULIS, M.A.,
BU'LOCK, F.A., BHATTACHARYA, S., BENTHAM, J., FARRALL, M., RIVERON, J.G.,
BROOK, J.D., BURN, J., CORDELL, H.J., GOODSHIP, J.A. and KEAVNEY, B., 2009.
Genetic variation in VEGF does not contribute significantly to the risk of congenital
cardiovascular malformation. PloS one, 4(3), e4978.
GUCER, S., INCE, T., KALE, G., AKCOREN, Z., OZKUTLU, S., TALIM, B. and CAGLAR,
M., 2005. Noncardiac malformations in congenital heart disease: a retrospective analysis of 305
pediatric autopsies. Turkish journal of pediatrics, 47(2), 159-166.
GUPTA, D., SAXENA, A., KOTHARI, S.S., JUNEJA, R., RAJANI, M., SHARMA, S. and
VENUGOPAL, P., 2001. Detection of coronary artery anomalies in tetralogy of Fallot using a
specific angiographic protocol. American journal of cardiology, 87(2), 241-4, A9.
HARRISON, D.A., SIU, S.C., HUSSAIN, F., MACLOGHLIN, C.J., WEBB, G.D. and
HARRIS, L., 2001. Sustained atrial arrhythmias in adults late after repair of tetralogy of fallot.
American journal of cardiology, 87(5), 584-588.
Harlan BJ, Starr A, Harwin FM. Manual of cardiac surgery. Second edition. Springer-Verlag.
New York. 1995.
HICKEY, E.J., VELDTMAN, G., BRADLEY, T.J., GENGSAKUL, A., MANLHIOT, C.,
WILLIAMS, W.G., WEBB, G.D. and MCCRINDLE, B.W., 2009. Late risk of outcomes for
adults with repaired tetralogy of Fallot from an inception cohort spanning four decades.
European journal of cardiothoracic surgery, 35(1), 156-64; discussion 164.

111
111
HOFFMAN, J.I., 1995. Incidence of congenital heart disease: I. Postnatal incidence. Pediatric
cardiology, 16(3), 103-113.
HOVELS-GURICH, H.H., KONRAD, K., SKORZENSKI, D., NACKEN, C., MINKENBERG,
R., MESSMER, B.J. and SEGHAYE, M.C., 2006. Long-term neurodevelopmental outcome and
exercise capacity after corrective surgery for tetralogy of Fallot or ventricular septal defect in
infancy. Annals of thoracic surgery, 81(3), 958-966.
INTERNATIONAL SCHIZOPHRENIA CONSORTIUM, 2008. Rare chromosomal deletions
and duplications increase risk of schizophrenia. Nature, 455(7210), 237-241.
JAWAD, A.F., MCDONALD-MCGINN, D.M., ZACKAI, E. and SULLIVAN, K.E., 2001.
Immunologic features of chromosome 22q11.2 deletion syndrome (DiGeorge
syndrome/velocardiofacial syndrome). Journal of pediatrics, 139(5), 715-723.
JENKINS, K.J., CORREA, A., FEINSTEIN, J.A., BOTTO, L., BRITT, A.E., DANIELS, S.R.,
ELIXSON, M., WARNES, C.A. and WEBB, C.L., 2007. Noninherited risk factors and
congenital cardiovascular defects: current knowledge: a scientific statement from the American
Heart Association Council on Cardiovascular Disease in the Young: endorsed by the American
Academy of Pediatrics. Circulation, 115(23), 2995-3014.
Jones KL. Smith`s recognizable patterns of human malformation. Sixth edition. Philadelphia.
Elsevier Saunders; 2006.
JUNG, S.C., KIM, S.S., YOON, K.S. and LEE, J.S., 1999. Prevalence of congenital
malformations and genetic diseases in Korea. Journal of human genetics, 44(1), 30-34.
KAO, A., MARIANI, J., MCDONALD-MCGINN, D.M., MAISENBACHER, M.K., BROOKS-
KAYAL, A.R., ZACKAI, E.H. and LYNCH, D.R., 2004. Increased prevalence of unprovoked
seizures in patients with a 22q11.2 deletion. American journal of medical genetics, 129A(1), 29-
34.

112
112
KARAMLOU, T., MCCRINDLE, B.W. and WILLIAMS, W.G., 2006. Surgery insight: late
complications following repair of tetralogy of Fallot and related surgical strategies for
management. Nature clinical practice cardiovascular medicine, 3(11), 611-622.
KARAMLOU, T., SILBER, I., LAO, R., MCCRINDLE, B.W., HARRIS, L., DOWNAR, E.,
WEBB, G.D., COLMAN, J.M., VAN ARSDELL, G.S. and WILLIAMS, W.G., 2006. Outcomes
after late reoperation in patients with repaired tetralogy of Fallot: the impact of arrhythmia and
arrhythmia surgery. Annals of thoracic surgery, 81(5), 1786-93; discussion 1793.
KARKERA, J.D., LEE, J.S., ROESSLER, E., BANERJEE-BASU, S., OUSPENSKAIA, M.V.,
MEZ, J., GOLDMUNTZ, E., BOWERS, P., TOWBIN, J., BELMONT, J.W., BAXEVANIS,
A.D., SCHIER, A.F. and MUENKE, M., 2007. Loss-of-function mutations in growth
differentiation factor-1 (GDF1) are associated with congenital heart defects in humans. American
journal of human genetics, 81(5), 987-994.
KARR, S.S., BRENNER, J.I., LOFFREDO, C., NEILL, C.A. and RUBIN, J.D., 1992. Tetralogy
of Fallot. The spectrum of severity in a regional study, 1981-1985. American journal of diseases
of children, 146(1), 121-124.
KATES, W.R., BURNETTE, C.P., JABS, E.W., RUTBERG, J., MURPHY, A.M., GRADOS,
M., GERAGHTY, M., KAUFMANN, W.E. and PEARLSON, G.D., 2001. Regional cortical
white matter reductions in velocardiofacial syndrome: a volumetric MRI analysis. Biological
psychiatry, 49(8), 677-684.
KATES, W.R., MILLER, A.M., ABDULSABUR, N., ANTSHEL, K.M., CONCHELOS, J.,
FREMONT, W. and ROIZEN, N., 2006. Temporal lobe anatomy and psychiatric symptoms in
velocardiofacial syndrome (22q11.2 deletion syndrome). The journal of American academy of
child and adolescent psychiatry, 45(5), 587-595.
KAWAME, H., ADACHI, M., TACHIBANA, K., KUROSAWA, K., ITO, F., GLEASON,
M.M., WEINZIMER, S., LEVITT-KATZ, L., SULLIVAN, K. and MCDONALD-MCGINN,
D.M., 2001. Graves' disease in patients with 22q11.2 deletion. Journal of pediatrics, 139(6),
892-895.

113
113
KEENAN, G.F., SULLIVAN, K.E., MCDONALD-MCGINN, D.M. and ZACKAI, E.H., 1997.
Arthritis associated with deletion of 22q11.2: more common than previously suspected.
American journal of medical genetics, 71(4), 488.
KHAIRY, P., ABOULHOSN, J., GURVITZ, M.Z., OPOTOWSKY, A.R., MONGEON, F.P.,
KAY, J., VALENTE, A.M., EARING, M.G., LUI, G., GERSONY, D.R., COOK, S., TING,
J.G., NICKOLAUS, M.J., WEBB, G., LANDZBERG, M.J., BROBERG, C.S. and FOR THE
ALLIANCE FOR ADULT RESEARCH IN CONGENITAL CARDIOLOGY (AARCC), 2010.
Arrhythmia Burden in Adults With Surgically Repaired Tetralogy of Fallot. A Multi-Institutional
Study. Circulation, .
Kirklin JW, Barratt-Boyes BG. Cardiac surgery: morphology, diagnostic criteria, natural history,
techniques, results, and indications. Second edition. Churchill Livingstone. New York. 1993.
KIRSHBOM, P.M., JAGGERS, J.J. and UNGERLEIDER, R.M., 1999. Tetralogy of fallot with
absent pulmonary valve: simplified technique for homograft repair. Journal of thoracic and
cardiovascular surgery, 118(6), 1125-1127.
KOBAYASHI, J., KAWASHIMA, Y., MATSUDA, H., NAKANO, S., MIURA, T., TOKUAN,
Y. and ARISAWA, J., 1991. Prevalence and risk factors of tricuspid regurgitation after
correction of tetralogy of Fallot. Journal of thoracic and cardiovascular cardiology, 102(4), 611-
616.
KOVACS, A.H., SAIDI, A.S., KUHL, E.A., SEARS, S.F., SILVERSIDES, C., HARRISON,
J.L., ONG, L., COLMAN, J., OECHSLIN, E. and NOLAN, R.P., 2009. Depression and anxiety
in adult congenital heart disease: predictors and prevalence. International journal of cardiology,
137(2), 158-164.
KUCIENE, R. and DULSKIENE, V., 2008. Selected environmental risk factors and congenital
heart defects. Medicina, 44(11), 827-832.
KYBURZ, A., BAUERSFELD, U., SCHINZEL, A., RIEGEL, M., HUG, M., TOMASKE, M.
and VALSANGIACOMO BUCHEL, E.R., 2008. The fate of children with microdeletion

114
114
22q11.2 syndrome and congenital heart defect: clinical course and cardiac outcome. Pediatric
cardiology, 29(1), 76-83.
LAMBRECHTS, D., DEVRIENDT, K., DRISCOLL, D.A., GOLDMUNTZ, E., GEWILLIG,
M., VLIETINCK, R., COLLEN, D. and CARMELIET, P., 2005. Low expression VEGF
haplotype increases the risk for tetralogy of Fallot: a family based association study. Journal of
medical genetics, 42(6), 519-522.
LAMMER, E.J., CHAK, J.S., IOVANNISCI, D.M., SCHULTZ, K., OSOEGAWA, K., YANG,
W., CARMICHAEL, S.L. and SHAW, G.M., 2009. Chromosomal abnormalities among children
born with conotruncal cardiac defects. Birth defects research A clinical and molecular
teratology, 85(1), 30-35.
LEV, M. and ECKNER, F.A., 1964. The Pathologic Anatomy of Tetralogy of Fallot and Its
Variations. Chest, 45, 251-261.
LICHT, D.J., WANG, J., SILVESTRE, D.W., NICOLSON, S.C., MONTENEGRO, L.M.,
WERNOVSKY, G., TABBUTT, S., DURNING, S.M., SHERA, D.M., GAYNOR, J.W.,
SPRAY, T.L., CLANCY, R.R., ZIMMERMAN, R.A. and DETRE, J.A., 2004. Preoperative
cerebral blood flow is diminished in neonates with severe congenital heart defects. Journal of
thoracic and cardiovascular surgery, 128(6), 841-849.
LILING, J., CROSS, I., BURN, J., DANIEL, C.P., TAWN, E.J. and PARKER, L., 1999.
Frequency and predictive value of 22q11 deletion. Journal of medical genetics, 36(10), 794-795.
LILLEHEI, C.W., LEVY, M.J., ADAMS, P. and ANDERSON, R.C., 1964. Corrective Surgery
for Tetralogy of Fallot. Long-Term Follow-up by Postoperative Recatheterization in 69 Cases
and Certain Surgical Considerations. Journal of thoracic and cardiovascular surgery, 48, 556-
576.
LIN, A.E., BASSON, C.T., GOLDMUNTZ, E., MAGOULAS, P.L., MCDERMOTT, D.A.,
MCDONALD-MCGINN, D.M., MCPHERSON, E., MORRIS, C.A., NOONAN, J., NOWAK,
C., PIERPONT, M.E., PYERITZ, R.E., ROPE, A.F., ZACKAI, E. and POBER, B.R., 2008.

115
115
Adults with genetic syndromes and cardiovascular abnormalities: clinical history and
management. Genetic medicine, 10(7), 469-494.
LINDSAY, E.A., 2001. Chromosomal microdeletions: Dissecting del22q11 syndrome. Nature
reviews genetics, 2(11), 858-868.
LURIE, I.W., KAPPETEIN, A.P., LOFFREDO, C.A. and FERENCZ, C., 1995. Non-cardiac
malformations in individuals with outflow tract defects of the heart: the Baltimore-Washington
Infant Study (1981-1989). American journal of medical genetics, 59(1), 76-84.
MAHLE, W.T., PARKS, W.J., FYFE, D.A. and SALLEE, D., 2003. Tricuspid regurgitation in
patients with repaired Tetralogy of Fallot and its relation to right ventricular dilatation. American
journal of cardiology, 92(5), 643-645.
MARDEN, P.M., SMITH, D.W. and MCDONALD, M.J., 1964. Congenital Anomalies in the
Newborn Infant, Including Minor Variations. A Study of 4,412 Babies by Surface Examination
for Anomalies and Buccal Smear for Sex Chromatin. Journal of pediatrics, 64, 357-371.
MARELLI, A.J., MACKIE, A.S., IONESCU-ITTU, R., RAHME, E. and PILOTE, L., 2007.
Congenital heart disease in the general population: changing prevalence and age distribution.
Circulation, 115(2), 163-172.
MARINO, B., DIGILIO, M.C., GRAZIOLI, S., FORMIGARI, R., MINGARELLI, R.,
GIANNOTTI, A. and DALLAPICCOLA, B., 1996. Associated cardiac anomalies in isolated and
syndromic patients with tetralogy of Fallot. American journal of cardiology, 77(7), 505-508.
MATSUOKA, R., KIMURA, M., SCAMBLER, P.J., MORROW, B.E., IMAMURA, S.,
MINOSHIMA, S., SHIMIZU, N., YAMAGISHI, H., JOH-O, K., WATANABE, S., OYAMA,
K., SAJI, T., ANDO, M., TAKAO, A. and MOMMA, K., 1998. Molecular and clinical study of
183 patients with conotruncal anomaly face syndrome. Human genetics, 103(1), 70-80.
MCDONALD-MCGINN, D.M., GRIPP, K.W., KIRSCHNER, R.E., MAISENBACHER, M.K.,
HUSTEAD, V., SCHAUER, G.M., KEPPLER-NOREUIL, K.M., CIPRERO, K.L.,
PASQUARIELLO, P.,JR., LAROSSA, D., BARTLETT, S.P., WHITAKER, L.A. and ZACKAI,

116
116
E.H., 2005. Craniosynostosis: another feature of the 22q11.2 deletion syndrome. American
journal of medical genetics, 136A(4), 358-362.
MCDONALD-MCGINN, D.M., KIRSCHNER, R., GOLDMUNTZ, E., SULLIVAN, K.,
EICHER, P., GERDES, M., MOSS, E., SOLOT, C., WANG, P., JACOBS, I., HANDLER, S.,
KNIGHTLY, C., HEHER, K., WILSON, M., MING, J.E., GRACE, K., DRISCOLL, D.,
PASQUARIELLO, P., RANDALL, P., LAROSSA, D., EMANUEL, B.S. and ZACKAI, E.H.,
1999. The Philadelphia story: the 22q11.2 deletion: report on 250 patients. Journal of genetic
counseling, 10(1), 11-24.
MCDONALD-MCGINN, D.M., LAROSSA, D., GOLDMUNTZ, E., SULLIVAN, K., EICHER,
P., GERDES, M., MOSS, E., WANG, P., SOLOT, C., SCHULTZ, P., LYNCH, D., BINGHAM,
P., KEENAN, G., WEINZIMER, S., MING, J.E., DRISCOLL, D., CLARK, B.J.,3RD,
MARKOWITZ, R., COHEN, A., MOSHANG, T., PASQUARIELLO, P., RANDALL, P.,
EMANUEL, B.S. and ZACKAI, E.H., 1997. The 22q11.2 deletion: screening, diagnostic
workup, and outcome of results; report on 181 patients. Genetic testing, 1(2), 99-108.
MCDONALD-MCGINN, D.M., TONNESEN, M.K., LAUFER-CAHANA, A., FINUCANE, B.,
DRISCOLL, D.A., EMANUEL, B.S. and ZACKAI, E.H., 2001. Phenotype of the 22q11.2
deletion in individuals identified through an affected relative: cast a wide FISHing net! Genetics
in medicine : official journal of the American College of Medical Genetics, 3(1), 23-29.
MCELHINNEY, D.B., KRANTZ, I.D., BASON, L., PICCOLI, D.A., EMERICK, K.M.,
SPINNER, N.B. and GOLDMUNTZ, E., 2002. Analysis of cardiovascular phenotype and
genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome.
Circulation, 106(20), 2567-2574.
MEBERG, A., HALS, J. and THAULOW, E., 2007. Congenital heart defects--chromosomal
anomalies, syndromes and extracardiac malformations. Acta paediatrica, 96(8), 1142-1145.
MEFFORD, H.C., SHARP, A.J., BAKER, C., ITSARA, A., JIANG, Z., BUYSSE, K., HUANG,
S., MALONEY, V.K., CROLLA, J.A., BARALLE, D., COLLINS, A., MERCER, C., NORGA,
K., DE RAVEL, T., DEVRIENDT, K., BONGERS, E.M., DE LEEUW, N., REARDON, W.,
GIMELLI, S., BENA, F., HENNEKAM, R.C., MALE, A., GAUNT, L., CLAYTON-SMITH, J.,

117
117
SIMONIC, I., PARK, S.M., MEHTA, S.G., NIK-ZAINAL, S., WOODS, C.G., FIRTH, H.V.,
PARKIN, G., FICHERA, M., REITANO, S., LO GIUDICE, M., LI, K.E., CASUGA, I.,
BROOMER, A., CONRAD, B., SCHWERZMANN, M., RABER, L., GALLATI, S., STRIANO,
P., COPPOLA, A., TOLMIE, J.L., TOBIAS, E.S., LILLEY, C., ARMENGOL, L.,
SPYSSCHAERT, Y., VERLOO, P., DE COENE, A., GOOSSENS, L., MORTIER, G.,
SPELEMAN, F., VAN BINSBERGEN, E., NELEN, M.R., HOCHSTENBACH, R., POOT, M.,
GALLAGHER, L., GILL, M., MCCLELLAN, J., KING, M.C., REGAN, R., SKINNER, C.,
STEVENSON, R.E., ANTONARAKIS, S.E., CHEN, C., ESTIVILL, X., MENTEN, B.,
GIMELLI, G., GRIBBLE, S., SCHWARTZ, S., SUTCLIFFE, J.S., WALSH, T., KNIGHT, S.J.,
SEBAT, J., ROMANO, C., SCHWARTZ, C.E., VELTMAN, J.A., DE VRIES, B.B.,
VERMEESCH, J.R., BARBER, J.C., WILLATT, L., TASSABEHJI, M. and EICHLER, E.E.,
2008a. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. The
New England journal of medicine, 359(16), 1685-1699.
MERSCHER, S., FUNKE, B., EPSTEIN, J.A., HEYER, J., PUECH, A., LU, M.M., XAVIER,
R.J., DEMAY, M.B., RUSSELL, R.G., FACTOR, S., TOKOOYA, K., JORE, B.S., LOPEZ, M.,
PANDITA, R.K., LIA, M., CARRION, D., XU, H., SCHORLE, H., KOBLER, J.B.,
SCAMBLER, P., WYNSHAW-BORIS, A., SKOULTCHI, A.I., MORROW, B.E. and
KUCHERLAPATI, R., 2001. TBX1 is responsible for cardiovascular defects in velo-cardio-
facial/DiGeorge syndrome. Cell, 104(4), 619-629.
MICHIELON, G., MARINO, B., FORMIGARI, R., GARGIULO, G., PICCHIO, F., DIGILIO,
M.C., ANACLERIO, S., ORICCHIO, G., SANDERS, S.P. and DI DONATO, R.M., 2006.
Genetic syndromes and outcome after surgical correction of tetralogy of Fallot. Annals of
thoracic surgery, 81(3), 968-975.
MILLER, D.T., ADAM, M.P., ARADHYA, S., BIESECKER, L.G., BROTHMAN, A.R.,
CARTER, N.P., CHURCH, D.M., CROLLA, J.A., EICHLER, E.E., EPSTEIN, C.J., FAUCETT,
W.A., FEUK, L., FRIEDMAN, J.M., HAMOSH, A., JACKSON, L., KAMINSKY, E.B., KOK,
K., KRANTZ, I.D., KUHN, R.M., LEE, C., OSTELL, J.M., ROSENBERG, C., SCHERER,
S.W., SPINNER, N.B., STAVROPOULOS, D.J., TEPPERBERG, J.H., THORLAND, E.C.,
VERMEESCH, J.R., WAGGONER, D.J., WATSON, M.S., MARTIN, C.L. and LEDBETTER,

118
118
D.H., 2010. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test
for individuals with developmental disabilities or congenital anomalies. American Journal of
Human Genetics, 86(5), 749-764.
MING, J.E., MCDONALD-MCGINN, D.M., MEGERIAN, T.E., DRISCOLL, D.A., ELIAS,
E.R., RUSSELL, B.M., IRONS, M., EMANUEL, B.S., MARKOWITZ, R.I. and ZACKAI, E.H.,
1997. Skeletal anomalies and deformities in patients with deletions of 22q11. American journal
of medical genetics, 72(2), 210-215.
MOMMA, K., 2010. Cardiovascular anomalies associated with chromosome 22q11.2 deletion
syndrome. The American Journal of Cardiology, 105(11), 1617-1624.
MOMMA, K., TAKAO, A., MATSUOKA, R., IMAI, Y., MUTO, A., OSAWA, M. and
TAKAYAMA, M., 2001. Tetralogy of Fallot associated with chromosome 22q11.2 deletion in
adolescents and young adults. Genetic medicine, 3(1): 56-60.
MOSS, E.M., BATSHAW, M.L., SOLOT, C.B., GERDES, M., MCDONALD-MCGINN, D.M.,
DRISCOLL, D.A., EMANUEL, B.S., ZACKAI, E.H. and WANG, P.P., 1999.
Psychoeducational profile of the 22q11.2 microdeletion: A complex pattern. Journal of
pediatrics, 134(2), 193-198.
MURPHY, J.G., GERSH, B.J., MAIR, D.D., FUSTER, V., MCGOON, M.D., ILSTRUP, D.M.,
MCGOON, D.C., KIRKLIN, J.W. and DANIELSON, G.K., 1993. Long-term outcome in
patients undergoing surgical repair of tetralogy of Fallot. New England journal of medicine,
329(9), 593-599.
MURPHY, K.C., JONES, L.A. and OWEN, M.J., 1999. High rates of schizophrenia in adults
with velo-cardio-facial syndrome. Archives of general psychiatry, 56(10), 940-945.
NEILL, C.A. and CLARK, E.B., 1994. Tetralogy of Fallot. The first 300 years. Texas heart
institute journal, 21(4), 272-279.
NIWA, K., SIU, S.C., WEBB, G.D. and GATZOULIS, M.A., 2002. Progressive aortic root
dilatation in adults late after repair of tetralogy of Fallot. Circulation, 106(11), 1374-1378.

119
119
NOLLERT, G., FISCHLEIN, T., BOUTERWEK, S., BOHMER, C., DEWALD, O., KREUZER,
E., WELZ, A., NETZ, H., KLINNER, W. and REICHART, B., 1997a. Long-term results of total
repair of tetralogy of Fallot in adulthood: 35 years follow-up in 104 patients corrected at the age
of 18 or older. Journal of thoracic and cardiovascular surgery, 45(4), 178-181.
NOLLERT, G., FISCHLEIN, T., BOUTERWEK, S., BOHMER, C., KLINNER, W. and
REICHART, B., 1997b. Long-term survival in patients with repair of tetralogy of Fallot: 36-year
follow-up of 490 survivors of the first year after surgical repair. Journal of the American college
of cardiology, 30(5), 1374-1383.
NOLLERT, G.D., DABRITZ, S.H., SCHMOECKEL, M., VICOL, C. and REICHART, B.,
2003. Risk factors for sudden death after repair of tetralogy of Fallot. The annals of thoracic
surgery, 76(6), 1901-1905.
NORA, J.J., 1968. Multifactorial inheritance hypothesis for the etiology of congenital heart
diseases. The genetic-environmental interaction. Circulation, 38(3), 604-617.
NORA, J.J. and NORA, A.H., 1978. The evolution of specific genetic and environmental
counseling in congenital heart diseases. Circulation, 57(2), 205-213.
NORGAARD, M.A., LAURIDSEN, P., HELVIND, M. and PETTERSSON, G., 1999. Twenty-
to-thirty-seven-year follow-up after repair for Tetralogy of Fallot. European journal of
cardiothoracic surgery, 16(2), 125-130.
OECHSLIN, E.N., HARRISON, D.A., HARRIS, L., DOWNAR, E., WEBB, G.D., SIU, S.S. and
WILLIAMS, W.G., 1999. Reoperation in adults with repair of tetralogy of fallot: indications and
outcomes. Journal of thoracic and cardiovascular surgery, 118(2), 245-251.
OSKARSDOTTIR, S., PERSSON, C., ERIKSSON, B.O. and FASTH, A., 2005. Presenting
phenotype in 100 children with the 22q11 deletion syndrome. European journal of pediatrics,
164(3), 146-153.
PAPOLOS, D.F., FAEDDA, G.L., VEIT, S., GOLDBERG, R., MORROW, B.,
KUCHERLAPATI, R. and SHPRINTZEN, R.J., 1996. Bipolar spectrum disorders in patients

120
120
diagnosed with velo-cardio-facial syndrome: does a hemizygous deletion of chromosome 22q11
result in bipolar affective disorder? American journal of psychiatry, 153(12), 1541-1547.
PARK, I.S., KO, J.K., KIM, Y.H., YOO, H.W., SEO, E.J., CHOI, J.Y., GIL, H.Y. and KIM, S.J.,
2007. Cardiovascular anomalies in patients with chromosome 22q11.2 deletion: a Korean
multicenter study. International journal of cardiology, 114(2), 230-235.
PERLOFF JK. The Clinical recognition of congenital heart disease. Third edition. Philadelphia.
W.B. Saunders Company. 1987. pp 404-442.
PERLOFF JK, CHILD JS. Congenital heart disease in adults. W.B. Saunders Company.
Philadelphia. 1991. pp 165-187.
PITT, D.B., 1962. A family study of Fallot's tetrad. Australasian annals of medicine, 11, 179-
183.
PIZZUTI, A., SARKOZY, A., NEWTON, A.L., CONTI, E., FLEX, E., DIGILIO, M.C.,
AMATI, F., GIANNI, D., TANDOI, C., MARINO, B., CROSSLEY, M. and DALLAPICCOLA,
B., 2003. Mutations of ZFPM2/FOG2 gene in sporadic cases of tetralogy of Fallot. Human
mutation, 22(5), 372-377.
POLANCZYK, G., DE LIMA, M.S., HORTA, B.L., BIEDERMAN, J. and ROHDE, L.A., 2007.
The worldwide prevalence of ADHD: a systematic review and metaregression analysis. The
American Journal of Psychiatry, 164(6), 942-948.
RAO, B.N., ANDERSON, R.C. and EDWARDS, J.E., 1971. Anatomic variations in the
tetralogy of Fallot. American heart journal, 81(3), 361-371.
RAUCH, R., HOFBECK, M., ZWEIER, C., KOCH, A., ZINK, S., TRAUTMANN, U., HOYER,
J., KAULITZ, R., SINGER, H. and RAUCH, A., 2010. Comprehensive genotype-phenotype
analysis in 230 patients with tetralogy of Fallot. Journal of medical genetics, 47(5), 321-331.
RITTLER, M., PAZ, J.E. and CASTILLA, E.E., 1996. VACTERL association, epidemiologic
definition and delineation. American journal of medical genetics, 63(4), 529-536.

121
121
ROCCHINI, A.P., ROSENTHAL, A., FREED, M., CASTANEDA, A.R. and NADAS, A.S.,
1977. Chronic congestive heart failure after repair of tetralogy of Fallot. Circulation, 56(2), 305-
310.
ROME, J.J., MAYER, J.E., CASTANEDA, A.R. and LOCK, J.E., 1993. Tetralogy of Fallot with
pulmonary atresia. Rehabilitation of diminutive pulmonary arteries. Circulation, 88(4), 1691-
1698.
ROOS-HESSELINK, J., PERLROTH, M.G., MCGHIE, J. and SPITAELS, S., 1995. Atrial
arrhythmias in adults after repair of tetralogy of Fallot. Correlations with clinical, exercise, and
echocardiographic findings. Circulation, 91(8), 2214-2219.
ROWE, S.A., ZAHKA, K.G., MANOLIO, T.A., HORNEFFER, P.J. and KIDD, L., 1991. Lung
function and pulmonary regurgitation limit exercise capacity in postoperative tetralogy of Fallot.
Journal of the American college of cardiology, 17(2), 461-466.
RYAN, A.K., GOODSHIP, J.A., WILSON, D.I., PHILIP, N., LEVY, A., SEIDEL, H.,
SCHUFFENHAUER, S., OECHSLER, H., BELOHRADSKY, B., PRIEUR, M., AURIAS, A.,
RAYMOND, F.L., CLAYTON-SMITH, J., HATCHWELL, E., MCKEOWN, C., BEEMER,
F.A., DALLAPICCOLA, B., NOVELLI, G., HURST, J.A., IGNATIUS, J., GREEN, A.J.,
WINTER, R.M., BRUETON, L., BRONDUM-NIELSEN, K. and SCAMBLER, P.J., 1997.
Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a
European collaborative study. Journal of medical genetics, 34(10), 798-804.
SARUBBI, B., PACILEO, G., PISACANE, C., DUCCESCHI, V., IACONO, C., RUSSO, M.G.,
IACONO, A. and CALABRO, R., 2000. Exercise capacity in young patients after total repair of
Tetralogy of Fallot. Pediatric cardiology, 21(3), 211-215.
SCAMBLER, P.J., 2010. 22q11 deletion syndrome: a role for TBX1 in pharyngeal and
cardiovascular development. Pediatric cardiology, 31(3), 378-390.
SCUTT, L.E., CHOW, E.W., WEKSBERG, R., HONER, W.G. and BASSETT, A.S., 2001.
Patterns of dysmorphic features in schizophrenia. American Journal of Medical Genetics, 105(8),
713-723.

122
122
SHPRINTZEN, R.J., GOLDBERG, R., GOLDING-KUSHNER, K.J. and MARION, R.W.,
1992. Late-onset psychosis in the velo-cardio-facial syndrome. American journal of medical
genetics, 42(1), 141-142.
SHPRINTZEN, R.J., GOLDBERG, R.B., LEWIN, M.L., SIDOTI, E.J., BERKMAN, M.D.,
ARGAMASO, R.V. and YOUNG, D., 1978. A new syndrome involving cleft palate, cardiac
anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome. Cleft palate
journal, 15(1), 56-62.
SIMON, T.J., DING, L., BISH, J.P., MCDONALD-MCGINN, D.M., ZACKAI, E.H. and GEE,
J., 2005. Volumetric, connective, and morphologic changes in the brains of children with
chromosome 22q11.2 deletion syndrome: an integrative study. Neuroimage, 25(1), 169-180.
SMITH, C.A., DRISCOLL, D.A., EMANUEL, B.S., MCDONALD-MCGINN, D.M., ZACKAI,
E.H. and SULLIVAN, K.E., 1998. Increased prevalence of immunoglobulin A deficiency in
patients with the chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial
syndrome). Clinical and diagnostic laboratory immunology, 5(3), 415-417.
SONG, M.S., HU, A., DYHAMENAHALI, U., CHITAYAT, D., WINSOR, E.J., RYAN, G.,
SMALLHORN, J., BARRETT, J., YOO, S.J. and HORNBERGER, L.K., 2009. Extracardiac
lesions and chromosomal abnormalities associated with major fetal heart defects: comparison of
intrauterine, postnatal and postmortem diagnoses. Ultrasound in obstetrics and gynecology,
33(5), 552-559.
STARR, J.P., 2010. Tetralogy of fallot: yesterday and today. World journal of surgery, 34(4),
658-668.
SWILLEN, A., DEVRIENDT, K., LEGIUS, E., EYSKENS, B., DUMOULIN, M., GEWILLIG,
M. and FRYNS, J.P., 1997. Intelligence and psychosocial adjustment in velocardiofacial
syndrome: a study of 37 children and adolescents with VCFS. Journal of medical genetics,
34(6), 453-458.

123
123
SWILLEN, A., DEVRIENDT, K., LEGIUS, E., PRINZIE, P., VOGELS, A., GHESQUIERE, P.
and FRYNS, J.P., 1999. The behavioural phenotype in velo-cardio-facial syndrome (VCFS):
from infancy to adolescence. Journal of genetic counseling, 10(1), 79-88.
SWILLEN, A., VOGELS, A., DEVRIENDT, K. and FRYNS, J.P., 2000. Chromosome 22q11
deletion syndrome: update and review of the clinical features, cognitive-behavioral spectrum,
and psychiatric complications. American journal of medical genetics, 97(2), 128-135.
THOM, T., HAASE, N., ROSAMOND, W., HOWARD, V.J., RUMSFELD, J., MANOLIO, T.,
ZHENG, Z.J., FLEGAL, K., O'DONNELL, C., KITTNER, S., LLOYD-JONES, D., GOFF,
D.C.,JR., HONG, Y., ADAMS, R., FRIDAY, G., FURIE, K., GORELICK, P., KISSELA, B.,
MARLER, J., MEIGS, J., ROGER, V., SIDNEY, S., SORLIE, P., STEINBERGER, J.,
WASSERTHIEL-SMOLLER, S., WILSON, M. and WOLF, P., 2006. Heart disease and stroke
statistics--2006 update: a report from the American Heart Association Statistics Committee and
Stroke Statistics Subcommittee. Circulation, 113(6), e85-151.
TOZZI, R., HERNANZ-SCHULMAN, M., KILEY, R., GENIESER, N., AMBROSINO, M.,
PINTO, R. and DOYLE, E., 1989. Congenital pulmonary steal associated with Tetralogy of
Fallot, right aortic arch and an isolated left carotid artery. Pediatric cardiology, 19(6-7), 449-
451.
URETZKY, G., PUGA, F.J., DANIELSON, G.K., HAGLER, D.J. and MCGOON, D.C., 1982.
Reoperation after correction of tetralogy of Fallot. Circulation, 66(2), I202-8.
VAN ARSDELL, G.S., MAHARAJ, G.S., TOM, J., RAO, V.K., COLES, J.G., FREEDOM,
R.M., WILLIAMS, W.G. and MCCRINDLE, B.W., 2000. What is the optimal age for repair of
tetralogy of Fallot? Circulation, 102(19), III123-9.
VAN ENGELEN, K., TOPF, A., KEAVNEY, B.D., GOODSHIP, J.A., VAN DER VELDE, E.
T., BAARS, M.J., SNIJDER, S., MOORMAN, A.F., POSTMA, A.V. and MULDER, B.J., 2010.
22q11.2 Deletion Syndrome is under-recognised in adult patients with tetralogy of Fallot and
pulmonary atresia. Heart, 96(8), 621-624.

124
124
WEETMAN, A.P. and MCGREGOR, A.M., 1994. Autoimmune thyroid disease: further
developments in our understanding. Endocrine reviews, 15(6), 788-830.
WEISMANN, C.G. and GELB, B.D., 2007. The genetics of congenital heart disease: a review of
recent developments. Current opinion in cardiology, 22(3), 200-206.
WELLS, G.L., BARKER, S.E., FINLEY, S.C., COLVIN, E.V. and FINLEY, W.H., 1994.
Congenital heart disease in infants with Down's syndrome. Southern medical association, 87(7),
724-727.
WERNOVSKY, G., 2006. Current insights regarding neurological and developmental
abnormalities in children and young adults with complex congenital cardiac disease. Cardiology
in the young, 16 Suppl 1, 92-104.
WERNOVSKY, G., SHILLINGFORD, A.J. and GAYNOR, J.W., 2005. Central nervous system
outcomes in children with complex congenital heart disease. Current opinion in cardiology,
20(2), 94-99.
WESSEL, H.U., CUNNINGHAM, W.J., PAUL, M.H., BASTANIER, C.K., MUSTER, A.J. and
IDRISS, F.S., 1980. Exercise performance in tetralogy of Fallot after intracardiac repair. Journal
of thoracic and cardiovascular surgery, 80(4), 582-593.
WHITTEMORE, R., WELLS, J.A. and CASTELLSAGUE, X., 1994. A second-generation study
of 427 probands with congenital heart defects and their 837 children. Journal of the American
College of Cardiology, 23(6), 1459-1467.
WILSON, D.I., BURN, J., SCAMBLER, P. and GOODSHIP, J., 1993. DiGeorge syndrome: part
of CATCH 22. British heart journal, 30(10), 852-856.
WU, H.Y., RUSNACK, S.L., BELLAH, R.D., PLACHTER, N., MCDONALD-MCGINN,
D.M., ZACKAI, E.H. and CANNING, D.A., 2002. Genitourinary malformations in chromosome
22q11.2 deletion. Journal of urology, 168(6), 2564-2565.
WYSE, R.K., AL-MAHDAWI, S., BURN, J. and BLAKE, K., 1993. Congenital heart disease in
CHARGE association. Pediatric cardiology, 14(2), 75-81.

125
125
YAMAGISHI, H., ISHII, C., MAEDA, J., KOJIMA, Y., MATSUOKA, R., KIMURA, M.,
TAKAO, A., MOMMA, K. and MATSUO, N., 1998. Phenotypic discordance in monozygotic
twins with 22q11.2 deletion. American Journal of Medical Genetics, 78(4), 319-321.
YAP, S.C. and HARRIS, L., 2009. Sudden cardiac death in adults with congenital heart disease.
Expert reviews of cardiovascular therapy. 7(12), 1605-1620.
YETMAN, A.T., LEE, K.J., HAMILTON, R., MORROW, W.R. and MCCRINDLE, B.W.,
2001. Exercise capacity after repair of Tetralogy of Fallot in infancy. American journal of
cardiology, 87(8), 1021-3; A5.
ZELTSER, I., JARVIK, G.P., BERNBAUM, J., WERNOVSKY, G., NORD, A.S., GERDES,
M., ZACKAI, E., CLANCY, R., NICOLSON, S.C., SPRAY, T.L. and GAYNOR, J.W., 2008b.
Genetic factors are important determinants of neurodevelopmental outcome after repair of
tetralogy of Fallot. The Journal of thoracic and cardiovascular surgery, 135(1), 91-97.
ZHANG, K.Z., SUN, Q.B. and CHENG, T.O., 1986b. Holt-Oram syndrome in China: a
collective review of 18 cases. American Heart Journal, 111(3), 572-577.
ZIDERE, V., TSAPAKIS, E.G., HUGGON, I.C. and ALLAN, L.D., 2006. Right aortic arch in
the fetus. Ultrasound in obstetrics and gynecology, 28(7), 876-881.