the clinical, characterisation cases lethal perinatal type
TRANSCRIPT

12Med Genet 1996;33:132-136
The clinical, molecular, and pathologicalcharacterisation of a family with two cases oflethal perinatal type 2 Gaucher disease
Ellen Sidransky, Nahid Tayebi, Barbara K Stubblefield, William Eliason,Angelina Klineburgess, Gian-Paolo Pizzolato, Jeremiah N Cox, Josiane Porta,Armand Bottani, Celia D DeLozier-Blanchet
AbstractIt has recently been emphasised that asubset of patients with type 2 Gaucherdisease die in the neonatal period. Thisreport describes an Afghani family withtwo conceptuses having severe, prenatallydetected Gaucher disease. Mutationalanalysis showed that the family carrieda known complex allele which includedmutations at amino acids L444P, A456P,and V460V. Although glucocerebrosidaseRNA was present, an affected fetus hadvirtually no glucocerebrosidase cross re-active material on western analyses. Thesevere clinical course and pathology ob-served in these patients resemble that ofthe null allele Gaucher mouse, and suggestthat the absence ofglucocerebrosidase ac-tivity results in early death.(J Med Genet 1996;33:132-136)
Key words: Gaucher disease; hydrops fetalis; gluco-cerebrosidase.
National Institute ofMental Health, NIH,Bethesda, MD, USAE SidranskyN TayebiB K StubblefieldW EliasonA Klineburgess
Departnent ofPathology, GenevaUniversity Hospital,Geneva, SwitzerlandG-P PizzolatoJ N CoxJ Porta
Division of MedicalGenetics, GenevaUniversity Hospital,Geneva, SwitzerlandA BottaniC D DeLozier-Blanchet
Correspondence to:Dr Sidransky, ClinicalNeuroscience Branch,National Institute of MentalHealth, Building 49, RoomBIEE16, 49 Convent DriveMSC 4405, Bethesda,MD 20892-4405, USA.Received 19 April 1995Revised version acceptedfor publication10 October 1995
Until recently type 2 (acute neuronopathic)Gaucher disease was considered stereotypic inpresentation, with neurological deteriorationand death by the age of 2 to 3 years.' However,the generation of a null allele, knock-outGaucher mouse23 led to increased recognitionof type 2 patients with a much more aggressivephenotype, who die as neonates. Although 22neonates with type 2 Gaucher disease have beenreported to our knowledge, including some whopresented with hydrops fetalis or congenitalichthyosis,'24' there has not been com-prehensive analysis of glucocerebrosidase mut-ations, mRNA, and protein in these patients.In this report we describe the molecular char-acterisation of a family in which two con-ceptuses had severe, prenatally detectedGaucher disease.
Case reportsAlthough consanguinity cannot be proven, theproband's parents both come from the samesmall, isolated, and inbred Afghani population.They had one healthy son. The second preg-nancy (case 1) was complicated by hydramniosdeveloping between 27 and 30 weeks of preg-nancy. Prenatal ultrasound detected severehydrops fetalis with bilateral hydrothorax andfetal hypokinesia with multiple joint con-
tractures. Since fetal blood sampling indicatedthrombocytopenia and abnormal liver en-zymes, labour was induced at 33 weeks' gest-ation. A 1970 g male child (fig 1) measuring40 cm in length was delivered by caesareansection. He exhibited joint contractures, hepa-tosplenomegaly, pulmonary hypoplasia, mus-cular atrophy, and dysmorphic features thatincluded a flattened nose with small nares andmalformed ears. The skin appeared tight andshiny, especially over the face, neck, ears,thorax, and extremities. The infant died inthe first hour of life. The diagnosis of type 2Gaucher disease was based on the presence ofGaucher cells in many organs from necropsymaterial. The parents were subsequently con-firmed by enzyme analysis to be heterozygotesfor Gaucher disease (mother had 62 1% andfather had 66 7% of control glucocerebrosidaseactivity). During a third pregnancy (case 2),the prenatal diagnosis of Gaucher disease wasmade by enzyme assay on cultured amniocytesobtained at week 15. Neither hydrops nor jointcontractures was found in the 640 g, 30 cmmale fetus which was aborted at 23 weeks'gestation.
Figure 1 Postmortem photograph of case 1.
132
on February 8, 2022 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.33.2.132 on 1 February 1996. D
ownloaded from

Lethal perinatal type 2 Gaucher disease
Materials and methodsPATIENT SAMPLESPathological specimens were obtained at nec-ropsy from cases 1 and 2. Amniocytes wereobtained from case 2. Fibroblast lines werederived, with informed consent, from ad-ditional patients with type 1, type 2, and type3 Gaucher disease.
MOUSE SAMPLESFresh tissues obtained at necropsy from neo-natal type 2 Gaucher mice were immediatelyfrozen in liquid nitrogen.
DNA PREPARATION AND SOUTHERN BLOTHYBRIDISATIONGenomic DNA was prepared from cultured celllines from patients and controls as previouslydescribed.6 Human genomic DNA was di-gested with BamHI, electrophoresed on a 1%agarose gel, transferred to supported nitro-cellulose membranes (Schleicher & Schuell,Keene NH), ultraviolet light cross linked, andthen hybridised to a glucocerebrosidase cDNAprobe as previously described.7 Genomic DNAwas also digested with SstII and electro-phoresed on a 0-6% I.D.NATM agarose gel(FMC Rockland ME), transferred, and hy-bridised as described above.
RNA PREPARATION AND NORTHERN BLOTHYBRIDISATIONTotal RNA was isolated from frozen cell pelletsfrom case 2 and controls using RNAzol`B(Biotecx Houston, TX). Approximately 17 ptgof total RNA from each sample was electro-phoresed on a 1% agarose formaldehyde gel,transferred to a supported nitrocellulose mem-brane, and ultraviolet light cross linked. Themembrane was prehybridised using 33 ztl/cm2blot ofQuik hybridisation solution (Stratagene,Lajolla, CA) at 680C for 20 minutes, and thenhybridised at 680C for one hour using a 32pdCTP random labelled full length gluco-cerebrosidase cDNA probe.7 The blot waswashed twice in a 2 x SSC/0 1% (W/V) SDSat room temperature for 15 minutes and oncewith 0 1 x SSC/0 1% (W/V) SDS at 60°C for30 minutes. The membrane was exposed toKodak XAR film with an intensifying screen at- 800C overnight.
MUTATION ANALYSISScreening for the N370S, L444P, R463C, and84insG mutations was performed as previouslydescribed.8 A 1290 bp genomic glucocerebro-sidase segment, spanning exons 9 to 11, wasselectively amplified using oligonucleotideprimers that did not amplify the pseudogeneregion (forward primer 5'-AACATGAT-TCCCTATCTTC-3 from intron 8, reverseprimer 5'ACCACCTAGAGGGGAAAGTG-3' from untranslated exon 1 1).8 This amplifiedDNA fragment was isolated with GeneCleanII (Bio 101 LaJolla, CA) and cycle sequencingwas performed according to the Taq di deoxyT'
terminator kit (Applied Biosystems, PorterCity, IA). The PCR product was purified usingan AQUA Select-D-50 column (5' Prime, 3'Prime, Boulder, CO), vacuum dried, re-suspended in loading buffer, denatured at90°C, and loaded on a 4-75% acrylamide,8 3 mol/l urea and 1 x TBE gel. Results wereanalysed using the 373 DNA Sequencing Ana-lysis software package (Applied Biosystem,Porter City, IA).Forward oligonucleotide primers for se-
quencing were 59F 5'-CACAGGGCTGA-CCTACCCA-3' (located in intron 8); 142F5'-GCAGGAGTTATGGGGTGGGTC-3' (lo-cated in intron 9); 878F 5'-GTGGGCTGAA-GACAGCGTTGG-3' (located in intron 10).
PROTEIN STUDIESCell extracts were prepared from frozen humancell pellets and from mouse tissue or fibroblastsas previously described.3 Glucocerebrosidaseactivity was measured using 4-methyl-umbelliferyl-,B-D-glucopyranoside as sub-strate.9 Nine micrograms of protein from eachsample were electrophoresed on denaturing12% polyacrylamide gels, and the separatedproteins were transferred to Immobilon Pmembranes. The membranes were incubated
I..
Figure 2 (Top) Histological appearance of a lymph nodefrom case 1, showing Gaucher cells (H and E, x 65).(Bottom) EM picture of a Gaucher cell from a lymphnode ( x 15 000).
133
on February 8, 2022 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.33.2.132 on 1 February 1996. D
ownloaded from
Sidransky, Tayebi, Stubblefield, Eliason, Klineburgess, Pizzolato, Cox, Porta, Bottani, DeLozier-Blanchet
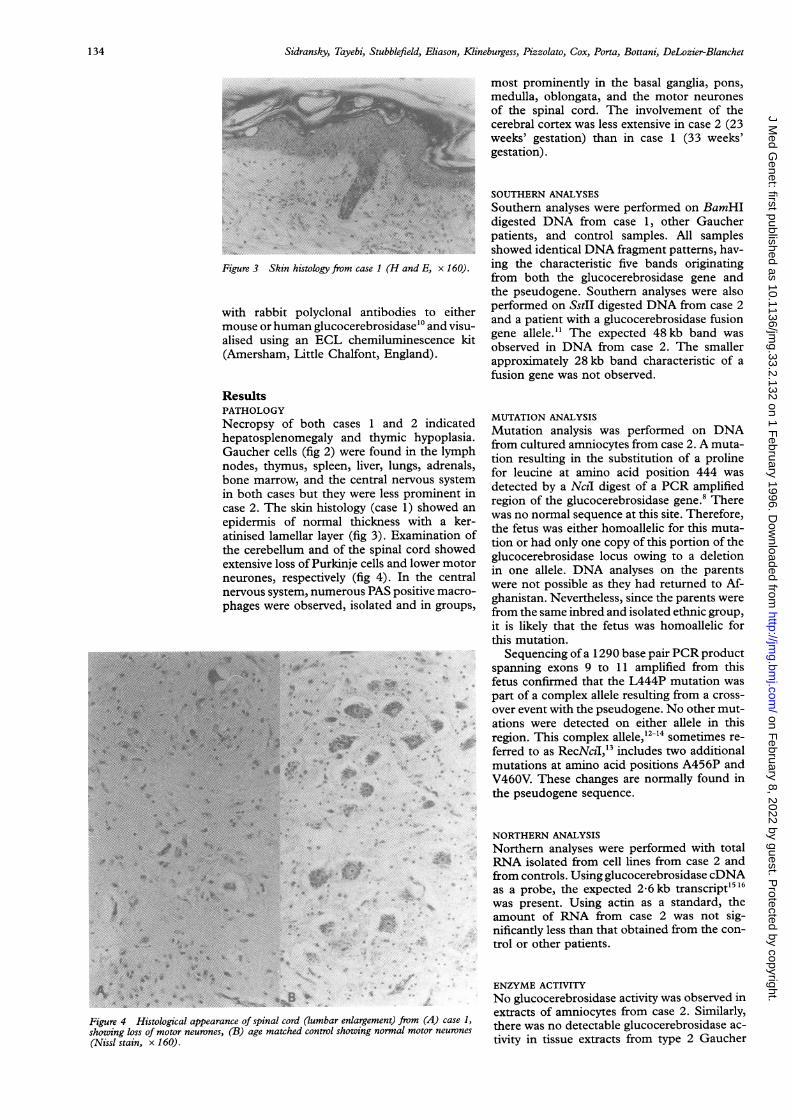
Figure 3 Skin histology from case 1 (H and E, x 160).
with rabbit polyclonal antibodies to eithermouse or human glucocerebrosidaselo and visu-alised using an ECL chemiluminescence kit(Amersham, Little Chalfont, England).
ResultsPATHOLOGYNecropsy of both cases 1 and 2 indicatedhepatosplenomegaly and thymic hypoplasia.Gaucher cells (fig 2) were found in the lymphnodes, thymus, spleen, liver, lungs, adrenals,bone marrow, and the central nervous systemin both cases but they were less prominent incase 2. The skin histology (case 1) showed an
epidermis of normal thickness with a ker-atinised lamellar layer (fig 3). Examination ofthe cerebellum and of the spinal cord showedextensive loss of Purkinje cells and lower motorneurones, respectively (fig 4). In the centralnervous system, numerous PAS positive macro-phages were observed, isolated and in groups,
hi.~~~~~~~~~~~~~~~~~~~~~~-p; h.;;....... I N,
;
*:.Sa:
*1 .*.
.
Figure 4 Histological appearance of spinal cord (lumbar enlargement) from (A) case 1,showing loss of motor neurones, (B) age matched control showing normal motor neurones
(Nissl stain, x 160).
most prominently in the basal ganglia, pons,medulla, oblongata, and the motor neuronesof the spinal cord. The involvement of thecerebral cortex was less extensive in case 2 (23weeks' gestation) than in case 1 (33 weeks'gestation).
SOUTHERN ANALYSESSouthern analyses were performed on BamHIdigested DNA from case 1, other Gaucherpatients, and control samples. All samplesshowed identical DNA fragment patterns, hav-ing the characteristic five bands originatingfrom both the glucocerebrosidase gene andthe pseudogene. Southern analyses were alsoperformed on SstII digested DNA from case 2and a patient with a glucocerebrosidase fusiongene allele." The expected 48 kb band wasobserved in DNA from case 2. The smallerapproximately 28 kb band characteristic of afusion gene was not observed.
MUTATION ANALYSISMutation analysis was performed on DNAfrom cultured amniocytes from case 2. A muta-tion resulting in the substitution of a prolinefor leucine at amino acid position 444 wasdetected by a Nczl digest of a PCR amplifiedregion of the glucocerebrosidase gene.8 Therewas no normal sequence at this site. Therefore,the fetus was either homoallelic for this muta-tion or had only one copy of this portion of theglucocerebrosidase locus owing to a deletionin one allele. DNA analyses on the parentswere not possible as they had returned to Af-ghanistan. Nevertheless, since the parents werefrom the same inbred and isolated ethnic group,it is likely that the fetus was homoallelic forthis mutation.
Sequencing of a 1290 base pair PCR productspanning exons 9 to 11 amplified from thisfetus confirmed that the L444P mutation waspart of a complex allele resulting from a cross-over event with the pseudogene. No other mut-ations were detected on either allele in thisregion. This complex allele,12-14 sometimes re-ferred to as RecNcil,'3 includes two additionalmutations at amino acid positions A456P andV460V. These changes are normally found inthe pseudogene sequence.
NORTHERN ANALYSISNorthern analyses were performed with totalRNA isolated from cell lines from case 2 andfrom controls. Using glucocerebrosidase cDNAas a probe, the expected 2-6 kb transcript15 16was present. Using actin as a standard, theamount of RNA from case 2 was not sig-nificantly less than that obtained from the con-trol or other patients.
ENZYME ACTIVITYNo glucocerebrosidase activity was observed inextracts of amniocytes from case 2. Similarly,there was no detectable glucocerebrosidase ac-tivity in tissue extracts from type 2 Gaucher
134
on February 8, 2022 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.33.2.132 on 1 February 1996. D
ownloaded from

Lethal perinatal type 2 Gaucher disease
kDa
116.3
97.4
66.3 - -
55.4 --
36.5
310 -
215 -
Figure 5 Weste?protein extracts. Jcell line generatec3 blank, lane 4 r
fibroblasts from a
amnwcytes fromwith antibody towere incubated w
mice. In contwell as manypatients show
WESTERN ANAI
Western anal'from cell line
patient, and a
cell lines geneand normalcerebrosidasetected in extiGaucher mic(
Discussion
A surprisingease has beer
type 2 patientdisease. In thDNA mutatio
fetuses havingand biochemi
scribed in typiThe mutant
the L444P, Abeen describe
state. 12-14 Allmay result frc
combination,conversion. 11-1
description of
for this compllethal, with paJewish ancesti
scribed as honwas suggestedmutations in 1
terms "fusion
terchangeablyor other evide:
of a fusion allele. Using the restriction en-donuclease SstII, which cleaves at one site up-stream from the functional gene and at a secondsite downstream from the pseudogene,"1 wehave shown by Southern analysis ofDNA fromthe present family that a fusion gene was notpresent. While the family history makes it un-likely, it is still possible that there may be adeletion of the glucocerebrosidase gene in thesecond allele that is not detectable on ourSouthern blots.The close proximity of the human gluco-
cerebrosidase pseudogene 16 kb downstreamfrom the functional gene is a potential sourcefor the generation of mutant Gaucher alleles.The patients described here appear to havesuffered the result of a crossover event oc-
l l curring in intron 9, an apparently critical region1 2 3 4 5 6 of the glucocerebrosidase gene, resulting inn blot analysis of glucocerebrsidase mutations at amino acid positions L444P,Lane 1 normal murinefibroblasts, lane 2 L456P, and V460V. In the type 2 GaucherI from the null allele Gaucher mouse, lane mouse a severe phenotype was experimentallynormal human fibroblasts, lane 5 generated by replacing intron 9 and portionsl type 1 Gaucher patient, lane 6 g b rcase 2. Lanes 1 and 2 were incubated of exons 9 and 10 with a neomycin cassette.'murine glucocerebrosidase. Lanes 4-6 It is possible that the recombination event in)ith antibody to human glucocerebrosidase. the cases described in this report disrupts a
region with a regulatory, enhancer, or RNAstability element or another contiguous
trast, most type 1 and type 3, as gene.202'longer surviving type 2 Gaucher While homozygosity for this mutant allele inresidual enzyme activity.'718 the two known cases appears to be predictive
of very severe Gaucher disease, none of sevenother neonates with this phenotype studied by
LYSIS our group or others share this genotype.42223yses were performed on extracts We postulate that at least one other unknowns from case 2, a type 1 Gaucher mutation or rearrangement associated withnormal subject, as well as from such severe disease must exist, and thus are~rated from type 2 Gaucher mice continuing the search for mutations in the re-murine fibroblasts. No gluco- gions flanking the glucocerebrosidase gene.cross reactive material was de- Although a glucocerebrosidase transcript was
racts from case 2 or the type 2 present in cells from case 2, no significant(fig 5). glucocerebrosidase cross reactive material was
detected on western blot. This suggests eitherthat the protein was not produced, or that itwas very unstable. Reports regarding stability
recent discovery in Gaucher dis- of the protein from the complex allele RecNcilthe recognition of a subset of are conflicting.'21' One group has suggested
ts with pre- or perinatal onset of the production of a stable protein with verylis report we have identified the low enzymatic activity." Another possibility,ns in a family with 23 and 33 week that of reduced amounts of a very unstable,clinical, pathological, molecular, protein, appears to be more likely.'2cal features resembling those de- Gaucher disease, particularly this severee 2 Gaucher mice. lethal subgroup, should be considered in fe-allele in these fetuses, containing tuses and neonates with hydrops fetalis, ich-L456P, and V460V changes, has thyosis, or joint contractures. Both hydropsd primarily in the heterozygous and arthrogryposis were present in the fetusthree of the observed mutations diagnosed at 33 weeks' gestation, whereas theyom an unequal homologous re- apparently had not yet developed in the fetusa fusion gene, or gene aborted at 23 weeks. The histological ab-
4 We have found only one other normality of the skin observed in case 1 sub-a fetus presumably homoallelic stantiates that the analysis of epidermis can beex of point mutations, a prenatal particularly valuable in the diagnosis of neo-irents of Macedonian/Ashkenazi natal Gaucher disease.24ry.5Although that case was de- In both the fetuses described here and theaozygous for a fusion gene which null allele Gaucher mouse, the absence of en-to account for some or all of the zyme was incompatible with long survival.neonates,' the authors used the Neuropathological studies of both sibs showedgene" and "complex allele" in- findings very similar to those in the null alleleand provided no Southern blot mouse, in that both had an apparent rostal-nce to show the actual presence caudal progression of lesions with lipid ac-
135
on February 8, 2022 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.33.2.132 on 1 February 1996. D
ownloaded from

Sidransky, Tayebi, Stubblefield, Eliason, Klineburgess, Pizzolato, Cox, Porta, Bottani, DeLozier-Blanchet
cumulation varying within different brain re-
gions.25 Further pathological and biochemicalstudies of the severely affected type 2 Gauchermice may offer a unique opportunity to transferinsights from the murine model towards a bet-ter understanding of the pathogenesis of thedisease, perhaps leading to development ofnewtreatments for type 2 Gaucher disease.
We thank Professor N Herschkowitz for the clinical biochemicalanalysis as well as Drs P Extermann, M Bader, C Pag, and ISzalay for patient care. The fibroblast line from the patient witha fusion gene was kindly provided by Dr E Beutler. The murinecell lines were kindly provided by Dr C McKinney. Helpfuldiscussions with Drs E Ginns and B Martin are greatly ap-preciated. The experienced technical assistance of P Williamsis gratefully acknowledged. The authors also thank K Kuhnsand E Alzona for their secretarial assistance.
1 Fredrickson DS, Sloan HR. Gaucher disease. In: StanburyJB, Wyngaarden JB, Frederickson DS, eds. The metabolicbasis of inherited disease. New York: McGraw-Hill, 1978:731-59.
2 Sidransky E, Sherer D, Ginns El. Gaucher disease in theneonate: a distinct Gaucher phenotype is analogous toa mouse model created by targeted disruption of theglucocerebrosidase gene. Pediatr Res 1992;32:494-8.
3 Tybulewicz V, Tremblay ML, LaMarca ME, et al. Targeteddisruption of the mouse glucocerebrosidase gene: de-velopment of an animal model of Gaucher disease. Nature1992;357:407-10.
4 Sherer DM, Metlay L, Sinkin RA, Mongeon C, Lee RE,Woods JR. Congenital ichthyosis with restrictive derm-opathy and Gaucher's disease: a new syndrome with as-sociated prenatal diagnostic and pathology findings. ObstetGynecol 1993;81:843-4.
5 Strasberg PM, Skomorowski MA, Warren IB, Hilson WL,Callahan JW, Clarke JTR. Homozygous presence of thecrossover (fusion gene) mutation identified in a type II
Gaucher disease fetus: is this analogous to the Gaucherknock-out mouse model? Biochem Med Metabol Biol 1994;53:16-21.
6 Maniatis T, Fritsch EF, Sambrook J. Molecular cloning: a
laboratory manual. New York: Cold Spring HarborLaboratory Press, 1982.
7 Tsuji S, Choudary PV, Martin BM, Mayor JA, BarrangerJA, Ginns EI. A mutation in the human glucocerebrosidasegene in neuronopathic Gaucher's disease. N Engl Med1987;316:570-5.
8 Sidransky E, Bottler A, Stubblefield B, Ginns El. DNAmutational analysis of type 1 and type 3 Gaucher patients:how well do mutations predict phenotype? Hum Mutat1994;357:407-10.
9 Beutler E, Kuhl WL. Detection of the defect of Gaucher'sdisease and its carrier state in peripheral blood leukocytes.Lancet 1970;i:612-13.
10 Ginns EI, Brady RO, Pirruccello S, et al. Mutations ofglucocerebrosidase: discrimination ofneurologic and non-neurologic phenotypes of Gaucher disease. Proc NatlAcadSci USA 1982;79:5607-10.
11 Zimran A, Sorge J, Gross E, Kubitz M, West C, Beutler E.A glucocerebrosidase fusion gene in Gaucher disease. 7Clin Invest 1990;85:219-22.
12 Ohashi T, Hong CM, Weiler S, et al. Characterization ofhuman glucocerebrosidase from different mutant alleles._7 Biol Chem 1991;266:3661-7.
13 Horowitz M, Zimran A. Mutations causing Gaucher disease.Hum Mutat 1994;3: 1-11.
14 Latham T, Grabowski GA, Theophilus BDM, Smith FI.Complex alleles of the acid 0-glucocerebrosidase gene inGaucher disease. Am J Hum Genet 1990;47:79-86.
15 Reiner 0, Horowitz M. Differential expression ofthe humanglucocerebrosidase gene. Gene 1988;73:469-78.
16 Graves PN, Grabowski GA, Ludman MD, Palese P, SmithFI. Human acid beta-glucosidase: northem blot and Slnuclease analysis of mRNA from HeLa cells and normaland Gaucher disease fibroblasts. Am J Hum Genet 1986;39:763-74.
17 Wenger DA, Olson GC. Heterogeneity in Gaucher disease.In: Callahan JW, Cowten JA, eds. Lysosomes and lysosomalstorage disorders. New York: Raven Press, 1981:157-71.
18 Fabbro D, Desnick RJ, Grabowski GA. Gaucher disease:genetic heterogeneity within and among the subtypes de-tected by immunoblotting. Am J Hum Genet 1987;40:15-31.
19 Zimran A, Horowitz M. RecTL: a complex allele of theglucocerebrosidase gene associated with a mild clinicalcourse of Gaucher disease. Am J Med Genet 1994;50:74-8.
20 Ginns EI, Winfield S, Sidransky E, Long GC, Bomstein P.Characteristics of a novel gene at the Gaucher diseaselocus spanning the regions between the glucocerebrosidasepseudogene and thrombospondin. AmIHum Genet 1994;53:A325
21 Bornstein P, McKinney CE, LaMarca ME, et al. Targeteddisruption of a novel gene contiguous to both gluco-cerebrosidase and thrombospondin 3 results in an em-bryonic lethal phenotype in the mouse. Proc NatlAcad SciUSA 1995;92:4547-51.
22 Lewis BD, Nelson PV, Robertson EF, Morris CP. Mutationanalysis of 28 Gaucher disease patients: the Australasianexperience. Am _7 Med Genet 1994;49:218-23.
23 Tayebi N, Stubblefield B, Eliason W, Klineburgess A, GinnsEl, Sidransky E. Molecular analysis of severe type 2Gaucher disease. Pediatr Res 1995;37:153A.
24 Holleran WM, Ginns EI, Menon GK, et al. Epidermalconsequences of glucocerebrosidase deficiency. _7 Clin In-vest 1994;93:1756-64.
25 Willemsen R, Tybulewicz V, Sidransky E, et al. A biochemicaland ultrastructural evaluation of the type 2 Gauchermouse. Mol Chem Neuropathol 1995;24:179-92.
136
on February 8, 2022 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.33.2.132 on 1 February 1996. D
ownloaded from