the chemoselective catalytic oxidation of alcohols,ds322sz8050/... · 2011-09-22 · v...
TRANSCRIPT

THE CHEMOSELECTIVE CATALYTIC OXIDATION OF ALCOHOLS,
DIOLS, AND POLYOLS TO KETONES AND HYDROXYKETONES
A DISSERTATION
SUBMITTED TO THE DEPARTMENT OF CHEMISTRY
AND THE COMMITTEE ON GRADUATE STUDIES
OF STANFORD UNIVERSITY
IN PARTIAL FULFILLMENT OF THE REQUIREMENTS
FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY
Ronald Michael Painter
March 2011

http://creativecommons.org/licenses/by-nc/3.0/us/
This dissertation is online at: http://purl.stanford.edu/ds322sz8050
© 2011 by Ronald Michael Painter. All Rights Reserved.
Re-distributed by Stanford University under license with the author.
This work is licensed under a Creative Commons Attribution-Noncommercial 3.0 United States License.
ii

I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Robert Waymouth, Primary Adviser
I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Justin Du Bois
I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Barry Trost
Approved for the Stanford University Committee on Graduate Studies.
Patricia J. Gumport, Vice Provost Graduate Education
This signature page was generated electronically upon submission of this dissertation in electronic format. An original signed hard copy of the signature page is on file inUniversity Archives.
iii

iv
Abstract The chemoselective oxidation of vicinal diols to α-hydroxyketones is a challenge in
organic syntheses because not only does the diol need to be oxidized selectively to a
monocarbonyl compound, but diols are also prone to overoxidation and oxidative
cleavage. Employing a cationic palladium complex, [(neocuproine)Pd(OAc)]2(OTf)2 (1),
we were able to demonstrate the selective oxidation of glycerol to dihydroxyacetone
mediated by either benzoquinone or O2 as the terminal oxidant, an accomplishment that
has little precedent in homogeneous catalysis. Mechanistic studies were undertaken to
uncover the nature of this remarkable chemoselectivity. Kinetic and deuterium-labeling
studies implicate reversible β-hydride elimination from isomeric Pd alkoxides and
turnover-limiting displacement of the dihydroxyacetone product by benzoquinone. We
successfully extended this methodology to other terminal 1,2-diols and symmetric vicinal
1,2-diols and have carried out aerobic oxidation of these substrates catalyzed by 1.
Examination of the reactivity of 1 with conformationally-restricted 1,2-cyclohexanediols
suggests that the diol must chelate to the Pd center for effective oxidation to the
hydroxyketone product.
In a separate project, we have also investigated the electrocatalytic reduction of dioxygen
by several dinuclear copper complexes, an important reaction for fuel cell applications.

v
Acknowledgments
My parents, Mike and Shannan Painter, have long played a crucial role in shaping me for
who I am today. I would never have gotten as far in my education if it hadn't been for
their undying efforts to provide me with the best education possible. Being the parents of
a deaf child has its own challenges, and they have risen to the task by being outstanding
parents and role models. They have provided me with many opportunities that are
available to few other deaf children, and I would not be who I am today without them.
For that, they have earned my unending gratitude.
Perhaps the most important person in my graduate career at Stanford has been my
advisor, Professor Robert M. Waymouth. He has demonstrated time and again his
patience and understanding with me especially when it comes to working with a deaf
graduate student. In addition, he is honestly one of the (if not the) smartest people I
know, and has made himself available as a indispensable resource for me to learn
chemistry from. Moreover, he has been an excellent mentor and advisor for the people
who work in his lab, and I am very proud to have him as my advisor.
My colleagues in the Waymouth lab have been instrumental to making the lab
operational. While I have never had the opportunity to personally collaborate with
anyone in the lab on a project, I am confident that every person that I have worked with
in the lab will go on to be successful scientists.

vi
I could not have been successful in my Stanford graduate career without the opportunity
to have worked with several outstanding ASL interpreters. When I came to Stanford in
my first year, I had already studied chemistry for four years, but none of the interpreters
in the area have had that luxury. They have spent countless hours learning how to
mediate the transfer of chemical information, and that is not an easy task - organic
chemistry is practically a language of its own! I am proud to have gotten to know six
interpreters on a professional and personal basis: Debbie Mancuso and Mary Walsh, who
have been with me since the very beginning (2004-2010), Laura Winick (2004-2005),
Debby Kajiyama (2005-2010), Alicia Davidovich (2004-2010), and Joseph Cartwright
(2008-2010). Stanford's Office of Accessible Education has contracted their services
through an outstanding interpreter agency, Deaf Services Palo Alto, of which Janet Lewis
is the owner. Janet has shown the utmost care for providing the very best interpreters for
her clients, and I am confident that I would not be as successful as I have been without
these six fantastic interpreters.
I'd also like to thank the members of my dissertation committee, Professor Barry M.
Trost and Professor Justin Du Bois. They are both extremely bright and dedicated
chemists, and have taught me much over the last six years. The chemistry department's
student services officer, Roger Kuhn, has been an indispensable resource for me relating
to various aspects of my career at Stanford, and I am grateful for his assistance over the
years. Our lab's adminstrative associate, Dewi Fernandez, has been instrumental to
keeping the lab running smoothly - nothing would get done without her. Patricia Dwyer

vii
has done the herculean task of putting together chemistry events, and despite her always
having a full plate, she always remembers to set a chair aside for my interpreters to use.
Over the past two years, I have been proud to be part of the inaugural cohort of fellows
for the Diversifying Academia, Recruiting Excellence fellowship program. The other
eleven members of the group have been nothing but outstanding colleagues who have
enriched my understanding of the importance of diversity in academia and provided me
with support during the last portion of my career at Stanford. Anika Green and Chris
Golde have put in so much work to making the fellowship program successful, and I am
grateful to them for giving us their time and resources to help us succeed in our future
academic careers.
Finally, I would like to give heartfelt thanks to my partner, William White. When we
met four and a half years ago, I never dreamed that I would be so lucky to have a partner
who has been extremely supportive and encouraging despite the many, many, many
frustrations that I've had in my Stanford graduate career. His being a biology major and
now a medical student has helped me broaden my interests in science, and I know that I
will continue to learn things from him as time goes on. I don't know where I will be
going or what I will be doing after I graduate from Stanford, but I am happy to face the
new challenges before us together.

viii
Preface
"I do not write books; I write pages."
-Dan Fante

ix
Table of Contents
ABSTRACT .................................................................................................................. iv
ACKNOWLEDGMENTS ........................................................................................... v
PREFACE .................................................................................................................... viii
TABLE OF CONTENTS ............................................................................................ ix
LIST OF TABLES ....................................................................................................... xii
LIST OF FIGURES ..................................................................................................... xiii
LIST OF SCHEMES ................................................................................................... xiv
SYMBOLS AND ABBREVIATIONS ........................................................................ xvi
CHAPTER 1. An overview of the chemoselective oxidation of vicinal diols to
hydroxyketones.
1.1 Introduction ........................................................................................................ 1
1.2 Transition-metal catalyzed alcohol oxidation: mechanism ................................ 2
1.3 Intermolecular selectivity in alcohol oxidation .................................................. 5
1.4 Intramolecular selectivity in alcohol oxidation .................................................. 8
1.5 The oxidation of vicinal diols to hydroxyketones .............................................. 9
1.6 References .......................................................................................................... 14

x
CHAPTER 2. The selective catalytic oxidation of glycerol to dihydroxyacetone.
2.0 Preface ................................................................................................................ 18
2.1 Introduction ........................................................................................................ 18
2.2 Results ................................................................................................................ 21
2.3 Discussion .......................................................................................................... 24
2.4 Discussion of reaction mechanism...................................................................... 26
2.5 Conclusion and future directions ....................................................................... 28
2.6 Experimental section .......................................................................................... 28
2.7 References .......................................................................................................... 42
CHAPTER 3 The selective catalytic oxidation of diols and polyols to
hydroxyketones.
3.0 Preface ................................................................................................................ 45
3.1 Introduction ........................................................................................................ 45
3.2 The oxidation of activated diols ......................................................................... 46
3.3 The oxidation of aliphatic, unactivated diols ..................................................... 48
3.4 Stereoelectronic effects on cyclohexane-1,2-diol oxidation .............................. 52
3.5 Conclusions and future directions ...................................................................... 56
3.6 Experimental section .......................................................................................... 57
3.7 References .......................................................................................................... 68

xi
CHAPTER 4 The electrocatalytic reduction of dioxygen using dinuclear copper
complexes.
4.0 Preface ................................................................................................................ 70
4.1 Introduction ........................................................................................................ 70
4.2 Towards a dicopper electrocatalyst .................................................................... 72
4.3 The 3,5-di(2-pyridyl)pyrazole ligand system .................................................... 74
4.4 A 3,6-di(2-pyridylthio)pyrazine dicopper complex ........................................... 81
4.5 Some 3,5-di(2-pyridyl)-1,2,4-triazole ligand systems ....................................... 81
4.6 Conclusions and future directions ...................................................................... 85
4.7 Experimental section: ligand syntheses ............................................................. 86
4.8 Experimental section: electrochemical studies .................................................. 87
4.9 References .......................................................................................................... 88
APPENDIX
A.0 General remarks .................................................................................................. 90
A.1 1H NMR spectrum of the oxidation products for trans,trans-3-methyl-1,2- cyclohexanediol. ................................................................................................. 91 A.2 1H NMR spectrum of the oxidation products for trans,cis-3-methyl-1,2- cyclohexanediol. ................................................................................................. 92

xii
List of Tables
Table 2.1 Catalytic oxidation of glycerol and 1,2-propanediol with complex 1 .......... 22
Table 3.1 The Pd-catalyzed oxidation of 4'-substituted phenylethane-1,2-diols ......... 47
Table 3.2 NMR-scale screening of the oxidation for a variety of alcohols, diols, and polyols with 1 and benzoquinone in CD3CN/D2O at room temperature ...... 49 Table 3.3 The oxidation of six polyols to hydroxyketones on a 2 mmol scale ............ 52

xiii
List of Figures
Figure 2.1 Comparison of conversion vs. time for 1,2-propanediol with air or O2 as the terminal oxidant ........................................................................................... 32 Figure 2.2 First order kinetic plot for oxidation of 1,2-propanediol ............................. 37
Figure 2.3 Second order kinetic plot for oxidation of 1,2-propanediol ......................... 38
Figure 2.4 Plot of kobs vs. [Pd] ....................................................................................... 38
Figure 2.5 Plot of initial rate vs. [BQ] ........................................................................... 39
Figure 2.6 Second order plots for d0, d1, and d2-1,2-propanediols ................................ 40
Figure 2.7 Plot of 1/kobs vs. [HOAc] .............................................................................. 41
Figure 4.1 Cyclic voltammogram of (dppy)Cu2(OAc)3 at pH 4.7 ................................. 76
Figure 4.2 Dependences of the peak current on scan rate for (dppy)Cu2(OAc)3 at (a) its redox potential at -170 mV and (b) its electrocatalytic O2 reduction peak with maximum current at -25 mV ........................................................................ 76 Figure 4.3 (a) Voltammograms of (dppy)Cu2(OAc)3 with varying rotation rates for the rotating disk electrode; (b) Plot of the current at -650 mV as a function of the square root of the rotation rate for the disk electrode ................................... 78 Figure 4.4 Cyclic voltammograms at pH 4.7 for (a) the (4-NO2dppy)Cu2(OAc)3 complex and (b) the (4-NH2dppy)Cu2(OAc)3 complex .............................................. 79 Figure 4.5 Cyclic voltammograms of a copper 3,6-dipyridyl-1,2,5,6-dihydrotetrazine complex using (a) N2 saturated solution; (b) air saturated solution ............. 82 Figure 4.6 Cyclic voltammograms of a copper 3,6-dipyridyl-1,2,5,6-tetrazine complex using (a) N2 saturated solution; (b) air saturated solution ............................ 83 Figure 4.7 Cyclic voltammograms of a copper 4-amino-3,5-dipyridyl-1,2,4-triazole complex using (a) N2 saturated solution; (b) air saturated solution ............. 83 Figure 4.8 Cyclic voltammograms of a copper 3,5-dipyridyl-1,2,4-triazole complex using (a) N2 saturated solution; (b) air saturated solution ..................................... 84

xiv
List of Schemes Scheme 1.1 Multiple oxidation products from glycerol oxidation .............................. 1 Scheme 1.2 Several possible mechanisms for hydride abstraction from alcohols ...... 3 Scheme 1.3 Studies on the β-hydride elimination from metal alkoxides .................... 4 Scheme 1.4 Methods for the preparation of α-hydroxyketones .................................. 10 Scheme 2.1 Catalyst systems ...................................................................................... 20 Scheme 2.2 Selective oxidation of glycerol to dihydroxyacetone .............................. 21 Scheme 2.3 Oxidation of deuterium-labeled 1,2-propanediols ................................... 25 Scheme 2.4 Proposed mechanism for the catalytic oxidation of 1,2-propanediol ...... 26 Scheme 2.5 Stoichiometric oxidation of 1,2-propanediol with 1 ................................ 27 Scheme 3.1 The [(neocuproine)Pd(OAc)]2(OTf)2 complex ........................................ 46 Scheme 3.2 Preparation of substituted phenylethane-1,2-diols and their oxidation to the corresponding α-hydroxyacetophenone product ..................................... 46 Scheme 3.3 Preparation of 4-tert-butylcyclohexane-1,2-diols .................................... 53 Scheme 3.4 Preparation and reactivity of 3-methyl-1,2-cyclohexanediols ................. 54 Scheme 3.5 The oxidation of 4-tert-butylcyclohexane1,2-diols by 1 ......................... 55 Scheme 3.6 The oxidation of 3-methyl-1,2-cyclohexanediols by 1 ............................ 56 Scheme 4.1 Proposed dinuclear copper electrocatalysts ............................................. 72 Scheme 4.2 Proposed catalytic cycle for the reduction of dioxygen ........................... 73 Scheme 4.3 The oxidation of water to dioxygen with a dinuclear Ru complex .......... 74 Scheme 4.4 Synthesis of the 3,5-di(2-pyridyl)pyrazole ligand ................................... 75 Scheme 4.5 Preparation of 4-substituted dppy ligands ............................................... 79 Scheme 4.6 Preparation of 3,6-di(2-pyridylthio)pyrazine dicopper tetrachloride ...... 81

xv
Scheme 4.7 Preparation of 3,5-di(2-pyridyl)-1,2,4-triazole ....................................... 82 Scheme 4.8 Preparation of 3,5-di(2-pyridyl)-1,2,4-triazole derivatives ..................... 85

xvi
Symbols and abbreviations
Anal. Calcd calculated elemental analysis
aq. aqueous
β-H beta-hydride/hydrogen
BQ benzoquinone
DHA dihydroxyacetone
dmso dimethylsulfoxide
eq. equivalents
ESI electron-spray ionization
Et ethyl
h hours
HOAc acetic acid
KIE kinetic isotope effect
M molar
Me methyl
MeO methoxy
mM millimolar
MS mass spectroscopy
m/z mass to charge ratio
neocuproine 2,9-dimethyl-1,10-phenanthroline
NHE normal hydrogen electrode
NMR nuclear magnetic resonance

xvii
OAc acetate
OTf trifluoromethanesulfonate
PD or PG propylene glycol / 1,2-propanediol
Ph phenyl
ppm parts per million
RDE rotating disk electrode
Rf retention factor
RT room temperature
s seconds
TLC thin layer chromatography
V volt

1
Chapter 1
An overview of the chemoselective oxidation of vicinal diols to hydroxyketones
1.1 Introduction
The oxidation of alcohols to carbonyl compounds is a fundamental and important
transformation in organic synthesis; such a broadly useful reaction necessarily carries
with it countless reagents that can serve in this capacity.1-4 One inherent challenge that
comes with further development of this reaction is the ability for a reagent to selectively
oxidize one alcohol group in the presence of other alcohol functional groups in the same
molecule.5 In the case of chemoselective oxidation of polyols, several difficulties present
themselves: lack of chemoselectivity, lack of control over the level of oxidation taking
place (i.e. overoxidation), and, in the case of vicinal diols, oxidative cleavage of the
carbon-carbon bond. Scheme 1.1 gives an illustrative example of these complications as
they relate to glycerol (a problem that is specifically addressed in Chapter 2).
Scheme 1.1. Multiple oxidation products from glycerol oxidation
HO OH
OH
HO OH
O
H
O
HO
OH
OH
O
HO
OH
OH
O
HO
O
HO
O
OH
O

2
This chapter is designed to give an overview of the fundamental problem of the
chemoselective oxidation of polyols. The chapter will first delineate studies that have
been done on intermolecular selectivity for primary vs. secondary alcohols, and then
discuss chemoselective intramolecular examples of diols where the two alcohol groups
are remote from each other. Finally, this chapter will discuss the specific case of the
oxidation of 1,2-diols, where oxidative cleavage of this moiety is especially problematic.
1.2. Transition-metal catalyzed alcohol oxidation: mechanism
A common mechanism for homogeneous transition metal-mediated alcohol oxidations
involves hydride abstraction from the β position of a metal alkoxide intermediate. This
can take place via an electrophilic metal center (such as Pd) to form a metal hydride (vide
infra), or, in the case of some metal oxos, via decomposition of metal esters (i.e. chromic
acid oxidation) to form a metal hydroxo complex.6-10 Another class of homogeneous
transition metal-catalyzed alcohol oxidations where hydride abstraction takes place
invokes a transfer hydrogenation mechanism where there is at least a stoichiometric
amount of sacrificial hydride acceptor present in the reaction solution.11-13 Finally, there
are some examples of alcohol oxidations that go through a radical mechanism for
abstraction of the alcohol’s β hydrogen (i.e. permanganate oxidation).14-17 The latter two
classes of alcohol oxidation reagents are beyond the scope of this chapter, though they
are known to chemoselectively oxidize certain types of alcohol functional groups.5

3
Scheme 1.2. Several possible mechanisms for hydride abstraction from alcohols.
There have been few studies that compare the rates for β-H elimination of transition
metal alkoxides derived from primary and secondary alcohols, owing to the difficulty in
preparing stable metal alkoxide complexes. Milstein has synthesized a number of
(Me3P)3Ir(H)(Cl) alkoxides and studied the rates for Ir-H2 formation by 31P NMR; he
reports that the rates for decomposition of these Ir alkoxide complexes follows the trend:
isopropoxide > ethoxide >> methoxide.18 Bergman has made a series of stable
Cp*Ir(Ph)(PMe3) alkoxides that only undergo β-H elimination when a catalytic amount
of cationic Ir is introduced to the solution as an hydride acceptor.19 In this case, the Ir
methoxide complex is decomposed immediately at ambient temperatures where the
neopentyl analog requires 4 h, likely because of the increased difficulty for the cationic Ir
catalyst to access the Ir alkoxide and subsequently abstract the β-hydride. Hartwig has
studied the thermolysis of trans-(Ph3P)2Ir(CO)(alkoxide) complexes; in contrast to the
preceding two studies, there is an unusual lack of dependence for the nature of the
alkoxide ligand on the rate constant at 95ºC.20 This is attributed to the relatively open
R OH
R O[M]
H
R O[M]
H O
[M]
OH+R H
O
[M]
H+
R H
O
R O[M]
H O
R'
R' O[M] +
R H
O
R O[M]
H
R O[M]
H
[M]
H+
R H
O

4
nature of the coordination around Ir after dissociation of the PPh3 ligand, such that steric
bulk plays a much smaller role in the β-H elimination rates of these alkoxides.
Scheme 1.3. Studies on the β-hydride elimination from metal alkoxides.
In a similar vein, Cámpora has shown that, when (dippe)Ni(CH3)(alkoxide) complexes
are thermolyzed at 60ºC, there is little difference in the initial β-hydride elimination rates
IrMe3PMe3P Cl
PMe3
RCH2OHIr
Me3PMe3P OCH2R
PMe3
H
Cl
IrMe3PMe3P H
PMe3
H
Cl
+R H
O
IrMe3P
PhOCH2R
0.1 eq.
IrMe3P
PhOTf
IrMe3P
PhO
H
R
[Ir+]
IrMe3P
PhH
IrMe3P
PhO
R
H
IrOC
Ph3P OCH2RPPh3
95ºCIr
OCPh3P H
PPh3
+R H
O
PNi
P
iPriPr
iPr iPr
CH3
OCH2Ph
cat. NiIII-HNi
R3P
R3P H
OCH2PhCH3
CH4
PNi
P
iPriPr
iPr iPr
OCH2Ph
Ph H
O
+ PPh3
- PPh3 OC
IrPh3P
OCH2R
N N ArAr
PdO
O
O
H Ph
N N ArAr
PdO
O
H
Ph H
O

5
for the complexes derived from benzyl alcohol and 1-phenylethanol.21 The proposed
mechanism involves a catalytic amount of a NiI/III complex where β-H elimination takes
place from a three-coordinate NiI alkoxide that is presumably not sensitive to the steric
bulk on the alkoxide. Finally, Sigman has conducted elegant theoretical and
experimental studies on the energetics of β-H elimination of alkoxides from a N-
heterocyclic carbene-Pd complex using benzyl alcohol and 1-phenylethanol as
substrates.22 He has found experimentally that there is little difference in ∆G‡ for the two
substrates (21.1 and 21.2 kcal/mol, respectively) at 50ºC.
1.3 Intermolecular selectivity in alcohol oxidation
The oxidation of vicinal diols to hydroxyketones represents an intramolecular selectivity
challenge, as it requires the oxidation of the secondary alcohol in the presence of a
primary alcohol. It is therefore useful to compare this type of selectivity to
intermolecular chemoselective alcohol oxidations that have been reported in the
literature.
A variety of transition metal catalysts have been reported for alcohol oxidation, and, in
contrast to stoichiometric reagents, the large majority of catalysts that show any
selectivity favor primary alcohols over secondary alcohols, generally for steric reasons.
For instance, Naoto has reported a water-soluble dimeric Ru2(OAc)3(CO3) complex that
aerobically oxidizes an equimolar mixture of 1- and 2-decanol to a 14:1 mixture of
aldehyde and ketone products.23 Ishii has demonstrated that this same equimolar mixture
is aerobically oxidized by Ru(PPh3)3Cl2 and hydroquinone as cocatalyst to a 28:1 mixture

6
of the corresponding products;24 Sheldon has demonstrated that this same catalyst with
TEMPO favors the oxidation of benzyl alcohol versus 1-phenylethanol by a factor of 20
while a mixture of 1- and 2-octanol gives the aldehyde product in a 8:1 ratio.25 Katsuki
has shown that (salen)Ru(Cl)(NO) complexes can, once photolyzed, efficiently and
aerobically oxidize primary aliphatic and benzylic alcohols to the corresponding
aldehyde; secondary aliphatic alcohols are practically unreactive (>30 times slower than
primary alcohols), and 1-phenylethanol is 12 times slower than 1-decanol to react under
these conditions.26-28 Finally, a Ru-Co-hydroxyapatite catalyst can aerobically oxidize a
mixture of 1- and 2-octanols to give the carbonyl products in a 3:1 selectivity for the
aldehyde product.29
Copper systems have also shown a preference for primary vs. secondary alcohols. For
instance, Semmelhack's early work on CuCl/TEMPO oxidation of alcohols shows that
secondary alcohols are very slow to oxidize under catalytic conditions: a mixture of
cyclohexanol and 1-octanol gives solely the aldehyde in 60% yield after two hours.30
Further mechanistic studies demonstrate that the copper is responsible for the oxidation
of alcohol to product, and not the TEMPO cocatalyst. For comparison, the reaction of 1-
and 2-octanol with catalytic TEMPO and stoichiometric NaOCl is nine times slower for
2-octanol versus 1-octanol.31 Similarly, Sheldon has reported on the ability of
Cu(bpy)Br2/TEMPO to oxidize benzyl alcohol in the presence of 1-phenylethanol
selectively to benzaldehyde in 63% yield.32 Wieghardt has reported a hydroquinone-
based copper catalyst for the oxidation of primary alcohols at ambient temperatures;
secondary and benzylic alcohols are unreactive under the catalytic conditions.33

7
Palladium-catalyzed alcohol oxidants typically do not have as dramatic of a difference in
chemoselectivity for aerobic alcohol oxidation though most of the studies conducted with
these systems have been limited to benzylic alcohols. For instance, aerobic oxidation
with Sheldon's (neocuproine)Pd(OAc)2 catalyst favors primary alcohols in up to 3:1
selectivity for the aldehyde product. Aerobic oxidation employing Pd(OAc)2 in dmso
qualitatively favors primary benzylic and allylic alcohols, as base additives are needed to
promote oxidation of secondary activated alcohols.34 Shimazu has reported a Pd catalyst
supported on a solid mixture of Ni and Zn hydroxides; aerobic oxidation of benzylic
alcohols under forcing conditions strongly favors the aldehyde product in a 29:1 ratio.35
In contrast, PdCl2-polyoxometallate complexes show a modest selectivity (2:1) selectivity
for primary versus secondary aliphatic alcohols.36 Pd(OAc)2 can also be made
electrocatalytic with benzoquinone as the redox mediator; 1-phenylethanol is five times
slower to react compared to benzyl alcohol though both substrates are eventually
oxidized to the ketone and aldehyde in 60% and 77% yield, respectively.37
There are few examples where transition metal catalysts show a strong preference for
secondary alcohols in the presence of primary ones. For instance, V2O5 can catalyze the
aerobic oxidation of a mixture of cyclohexanol and 1-decanol, furnishing cyclohexanone
as the sole product in 87% after 16 hours with a trace amount of decanal.38 A
heterogeneous Ru-Co-Al-CO3 hydrotalcite catalyst can aerobically oxidize a mixture of
1- and 2-octanol to give the ketone in 82% yield; the primary alcohol is unreactive under
these conditions.39 Finally, Navarro has reported a N-heterocyclic carbene Pd-allyl
chloride complex for the oxidation of secondary alcohols with chlorobenzene as the

8
terminal oxidant; primary aliphatic alcohols are unreactive with this catalyst, and primary
benzylic alcohols are also ineffective substrates for this system.40 Employing Ni instead
of Pd with this carbene catalyst and using a higher catalyst loading shows similar
reactivity.
1.4 Intramolecular selectivity for alcohol oxidation
The majority of stoichiometric alcohol oxidants show little to no selectivity for primary
versus secondary alcohols because of the small magnitude of difference in reaction rates
for oxidation of either functional group. For those that do show any selectivity,
secondary alcohols are typically favored over primary alcohols.5 For instance, potassium
ferrate, or barium or potassium manganate in the presence of copper sulfate and alumina
under phase transfer conditions cleanly oxidizes the secondary alcohol group in terminal
1,3-diols without overoxidation of the molecule.41-42 These observations, and others, is
the subject of an excellent review by Arterburn, who lists numerous examples of
stoichiometric reagents that demonstrate superb intramolecular selectivity.5 Relevant
examples include the oxidation of diols with positive halogen reagents, peroxide-
mediated molybdate catalysts, dioxiranes, or transfer hydrogenation catalysts to obtain
the corresponding hydroxyketone in high yields. One of the most impressive
transformations reported in this review is the ability for dimethyldioxirane to selectively
oxidize 1,2,3-cyclohexanetriol to the 2,3-dihydroxycyclohexanone product in greater than
90% yield.43

9
There are similarly few examples of transition-metal catalyzed oxidations that show
intramolecular selectivity for alcohol oxidation. For instance, Pd(OAc)2 in the presence
of triethylamine under certain conditions can show moderate intramolecular selectivity
for 6-hydroxy-1-heptanol favoring the ketone product in a 6:1 ratio but overoxidation to
the dicarbonyl compound is a significant problem.44 This system can also oxidize
primary and secondary allylic alcohols in the presence of primary aliphatic alcohols by
taking advantage of the superior binding properties that allylic alcohols offer over
saturated aliphatic alcohols.45 More examples of selective transition-metal catalyzed
oxidation of diols are supplied in the next section regarding oxidation of vicinal diols.
1.5 The oxidation of vicinal diols to hydroxyketones
Alpha-hydroxy ketones can be prepared by oxidation of the enol ether of the
corresponding ketone by peracids,46 α-oxidation of ketones,47 acyloin condensation,48
from epoxides,49 or by the direct oxidation of alkenes.50-52 However, there are few
examples of general protocols that can oxidize vicinal diols to the hydroxyketone without
overoxidation to the diketone or oxidative cleavage.
As mentioned in Section 1.2, there are several stoichiometric reagents that can effect the
oxidation of diols to hydroxyketones. An early example is Fétizon's reagent, which is
silver carbonate supported on Celite; this protocol is limited to symmetric diols, however,
and despite the mild conditions for oxidation, an excess of silver carbonate is required.53
Pyridinium chlorochromate has been reported to oxidize vicinal diols to hydroxyketones
for certain substrates, but oxidative cleavage of the product is commonly observed.54

10
Scheme 1.4. Methods for the preparation of α-hydroxyketones.
Positive halogen reagents, such as N-bromosuccinimide, N-bromoacetamide,
iodoxybenzoic acid or Dess-Martin periodinane, sodium bromate mediated by sodium
bisulfite or cerium ammonium nitrate, tetraethylammonium trichloride, or
bromine/aqueous base have all shown selective oxidation of 1,2-diols to the
hydroxyketone, but they suffer from concomitant overoxidation, functional group
incompatibilites, and/or lack of selectivity.5 An effective protocol for the stoichiometric
oxidation of 1,2-diols is reaction of the diol with dibutyltin oxide in hot methanol to form
the stannylene acetal and subsequent oxidation with bromine, but one equivalent of each
toxic reagent is required.55 An electrocatalytic version of this reaction has been reported
where a mixture of dimethyltin chloride (0.1 equiv) and diol substrate in methanol is
electrolyzed at an unspecified (but presumably high) potential.56 Finally,
dimethyldioxirane is a powerful oxidant for a variety of diols and selectively oxidizes
R
O
R
OH
RR
OSiR3
RR
R
O
R
R OR'
O
2
R
R
O
R
OH
R
OH

11
secondary alcohols in the presence of primary alcohols, as is the case for terminal 1,2-
diols.43 Beyond this, the oxidation of internal vicinal diols is neither predictable nor well-
behaved, and the preceding high selectivity is lost in some of these latter substrates.
More recently, Konwar has demonstrated that a combination of dimethyl sulfoxide,
hydrazine, and iodine can selectively oxidize terminal 1,2-diols to hydroxyketones in
moderate yields.57
Few catalytic examples are known that efficiently oxidize vicinal diols to the
hydroxyketone product. Molybdate- or tungstate-based catalysts with either hydrogen
peroxide or tert-butylhydroperoxide as the terminal oxidant are known to effect this
oxidation, but cyclic diols are prone to C-C bond cleavage under catalytic conditions.5
Ikariya reports that Cp*Ir(diamine)(Cl) complexes are capable of aerobically and
selectively oxidizing 1,2-diols, albeit with lower yields.58 Similarly, Martin reports that
Fe(NO3)2 promoted by FeBr3 can aerobically oxidize 1,2-octanediol to the hydroxyketone
product in 74% yield.59 Very recently, Oberhauser and Lee have reported on the ability of
N-heterocyclic carbene Pd complexes to aerobically oxidize 1,2-propanediol and 1,2-
butanediol selectively to the hydroxyketone product in moderate yields.60
The electrocatalytic oxidation of aliphatic diols on Pt-based electrodes can, under
carefully controlled conditions, produce hydroxyketones, but this method is prone to
overoxidation to diketone compounds and oxidative cleavage.61 De Giovani has
demonstrated the ability for (dppe)Ru(polypyridyl) complexes to electrocatalytically
oxidize 1,2-butanediol to 1-hydroxy-2-butanone at 1.18 V vs. NHE in 62% yield.62

12
Romero has shown that trans-Ru(terpy)(O)2(OH2)2+ can oxidize this same substrate to
give the hydroxyketone in 78% yield at 0.8 V; other Ru-polypyridyl complexes were also
selective but gave the product in lower yields.63 Finally, Ishii has shown that Co(acac)3
in the presence of N-hydroxyphthalamide and 3-chlorobenzoic acid can oxidize various
diols to the hydroxyketone product; however, oxidation of 1,2-diol substrates leads to
oxidative cleavage as the major product in most cases.64
Heterogeneous catalysts can selectively oxidize diols to hydroxyketones. Taylor and
Hutchings has reported on the ability for Au-Pd nanoparticles supported on CeO2 to
aerobically oxidize 1,2-butanediol at 160ºC to the hydroxyketone product with a turnover
frequency of 2150 h-1.65 Hache has reported an Au-Pd catalyst supported on TiO2 to
selectively oxidize 1,2-butanediol at 160ºC, with a somewhat lower turnover frequency of
1,520 h-1.66 These two reports are noteworthy because aliphatic alcohols are typically
very poor substrates for heterogeneous Au-Pd catalysts.
1,2-diols are challenging substrates for catalytic oxidation of alcohols due to their
propensity to form stable chelate complexes with various metals. In the case of Pd-
catalyzed alcohol oxidations, similar chelating substrates are ineffective substrates for
Pd(OAc)2/triethylamine and N-heterocyclic carbene Pd complexes,44 and
Pd(OAc)2/sparteine systems.67-68 Indeed, a series of Pd diolate complexes are known that
are quite stable,69 and, upon subjection to elevated temperatures, oxidative cleavage takes
place instead of oxidation to the hydroxyketone product.70

13
1.6 References
(1) Sheldon, R. A.; Arends, I. W. C. E.; Dijksman, A. Catal. Today 2000, 57, 157.
(2) Stahl, S. S. Angew. Chem. Int. Ed. 2004, 43, 3400.
(3) Schultz, M. J.; Sigman, M. S. Tetrahedron 2006, 62, 8227.
(4) Sheldon, R. A.; Arends, I. W. C. E.; Ten Brink, G. J.; Dijksman, A. Acc. Chem.
Res. 2002, 35, 774.
(5) Arterburn, J. B. Tetrahedron 2001, 57, 9765.
(6) Rocek, J.; Aylward, D. E. J. Am. Chem. Soc. 1975, 97, 5452.
(7) Rocek, J.; Radkowsk.Ae J. Am. Chem. Soc. 1973, 95, 7123.
(8) Wiberg, K. B.; Schafer, H. J. Am. Chem. Soc. 1969, 91, 927.
(9) Wiberg, K. B.; Schafer, H. J. Am. Chem. Soc. 1969, 91, 933.
(10) Lee, D. G.; Spitzer, U. A.; Cleland, J.; Olson, M. E. Can. J. Chem. 1976, 54,
2124.
(11) Noyori, R.; Hashiguchi, S. Acc. Chem. Res. 1997, 30, 97.
(12) Graves, C. R.; Campbell, E. J.; Nguyen, S. T. Tetrahedron - Asymmetry 2005, 16,
3460.
(13) Degraauw, C. F.; Peters, J. A.; Vanbekkum, H.; Huskens, J. Synthesis 1994, 1007.
(14) Whitesid.Gm; Sadowski, J. S.; Lilburn, J. J. Am. Chem. Soc. 1974, 96, 2829.
(15) Lee, D. G.; Chen, T. J. Org. Chem. 1991, 56, 5341.
(16) Marko, I. E.; Giles, P. R.; Tsukazaki, M.; Chelle-Regnaut, I.; Gautier, A.; Brown,
S. M.; Urch, C. J. J. Org. Chem. 1999, 64, 2433.
(17) Rocek, J.; Radkowsk.Ae J. Org. Chem. 1973, 38, 89.
(18) Blum, O.; Milstein, D. J. Am. Chem. Soc. 1995, 117, 4582.

14
(19) Ritter, J. C. M.; Bergman, R. G. J. Am. Chem. Soc. 1998, 120, 6826.
(20) Zhao, J.; Hesslink, H.; Hartwig, J. F. J. Am. Chem. Soc. 2001, 123, 7220.
(21) Matas, I.; Campora, J.; Palma, P.; Alvarez, E. Organometallics 2009, 28, 6515.
(22) Mueller, J. A.; Goller, C. P.; Sigman, M. S. J. Am. Chem. Soc. 2004, 126, 9724.
(23) Komiya, N.; Nakae, T.; Sato, H.; Naota, T. Chem. Commun. 2006, 4829.
(24) Hanyu, A.; Takezawa, E.; Sakaguchi, S.; Ishii, Y. Tetrahedron Lett. 1998, 39,
5557.
(25) Dijksman, A.; Marino-Gonzalez, A.; Payeras, A. M. I.; Arends, I. W. C. E.;
Sheldon, R. A. J. Am. Chem. Soc. 2001, 123, 6826.
(26) Miyata, A.; Murakami, M.; Irie, R.; Katsuki, T. Tetrahedron Lett. 2001, 42, 7067.
(27) Egami, H.; Shimizu, H.; Katsuki, T. Tetrahedron Lett. 2005, 46, 783.
(28) Egami, H.; Onitsuka, S.; Katsuki, T. Tetrahedron Lett. 2005, 46, 6049.
(29) Yamaguchi, K.; Mori, K.; Mizugaki, T.; Ebitani, K.; Kaneda, K. J. Am. Chem.
Soc. 2000, 122, 7144.
(30) Semmelhack, M. F.; Schmid, C. R.; Cortes, D. A.; Chou, C. S. J. Am. Chem. Soc.
1984, 106, 3374.
(31) Anelli, P. L.; Biffi, C.; Montanari, F.; Quici, S. J. Org. Chem. 1987, 52, 2559.
(32) Gamez, P.; Arends, I. W. C. E.; Sheldon, R. A.; Reedijk, J. Adv. Synth. Catal.
2004, 346, 805.
(33) Chaudhuri, P.; Hess, M.; Weyhermuller, T.; Wieghardt, K. Angew. Chem. Int. Ed.
1999, 38, 1095.
(34) Peterson, K. P.; Larock, R. C. J. Org. Chem. 1998, 63, 3185.

15
(35) Hara, T.; Ishikawa, M.; Sawada, J.; Ichikuni, N.; Shimazu, S. Green Chem. 2009,
11, 2034.
(36) Barats, D.; Neumann, R. Adv. Synth. Catal. 2010, 352, 293.
(37) Amatore, C.; Cammoun, C.; Jutand, A. Synlett 2007, 2173.
(38) Velusamy, S.; Punniyamurthy, T. Org. Lett. 2004, 6, 217.
(39) Matsushita, T.; Ebitani, K.; Kaneda, K. Chem. Commun. 1999, 265.
(40) Berini, C.; Brayton, D. F.; Mocka, C.; Navarro, O. Org. Lett. 2009, 11, 4244.
(41) Kim, K. S.; Song, Y. H.; Lee, N. H.; Hahn, C. S. Tetrahedron Lett. 1986, 27,
2875.
(42) Kim, K. S.; Chung, S. J.; Cho, I. H.; Hahn, C. S. Tetrahedron Lett. 1989, 30,
2559.
(43) Bovicelli, P.; Truppa, D.; Sanetti, A.; Bernini, R.; Lupattelli, P. Tetrahedron
1998, 54, 14301.
(44) Schultz, M. J.; Hamilton, S. S.; Jensen, D. R.; Sigman, M. S. J. Org. Chem. 2005,
70, 3343.
(45) Batt, F.; Bourcet, E.; Kassab, Y.; Fache, F. Synlett 2007, 1869.
(46) Rubottom, G. M.; Vazquez, M. A.; Pelegrin.Dr Tetrahedron Lett. 1974, 4319.
(47) Moriarty, R. M.; Hu, H.; Gupta, S. C. Tetrahedron Lett. 1981, 22, 1283.
(48) Finley, K. T. Chem. Rev. 1964, 64, 573.
(49) Tsuji, T. Bull. Chem. Soc. Jpn. 1989, 62, 645.
(50) Sakaue, S.; Sakata, Y.; Nishiyama, Y.; Ishii, Y. Chem. Lett. 1992, 289.
(51) Murahashi, S. I.; Saito, T.; Hanaoka, H.; Murakami, Y.; Naota, T.; Kumobayashi,
H.; Akutagawa, S. J. Org. Chem. 1993, 58, 2929.

16
(52) Plietker, B. J. Org. Chem. 2004, 69, 8287.
(53) Fetizon, M.; Golfier, M.; Louis, J. M. J. Chem. Soc. Chem. Commun. 1969, 1102.
(54) Cisneros, A.; Fernandez, S.; Hernandez, J. E. Synthetic Commun. 1982, 12, 833.
(55) Ueno, Y.; Okawara, M. Tetrahedron. Lett. 1976, 4597.
(56) Maki, T.; Iikawa, S.; Mogami, G.; Harasawa, H.; Matsumura, Y.; Onomura, O.
Chem. Eur. J. 2009, 15, 5364.
(57) Gogoi, P.; Sarmah, G. K.; Konwar, D. J Org. Chem. 2004, 69, 5153.
(58) Arita, S.; Koike, T.; Kayaki, Y.; Ikariya, T. Chemistry - an Asian Journal 2008, 3,
1479.
(59) Martin, S. E.; Suarez, D. F. Tetrahedron Lett. 2002, 43, 4475.
(60) Bettucci, L.; Bianchini, C.; Oberhauser, W.; Hsiao, T. H.; Lee, H. M. J. Mol.
Catal. A - Chem. 2010, 322, 63.
(61) Hilmi, A.; Belgsir, E. M.; Leger, J. M.; Lamy, C. J. Electroanal. Chem. 1997,
435, 69.
(62) Sussuchi, E. M.; de Lima, A. A.; De Giovani, W. F. J. Mol. Catal. A - Chem.
2006, 259, 302.
(63) Navarro, M.; De Giovani, W. F.; Romero, J. R. J. Mol. Catal. A - Chem. 1998,
135, 249.
(64) Iwahama, T.; Yoshino, Y.; Keitoku, T.; Sakaguchi, S.; Ishii, Y. J. Org. Chem.
2000, 65, 6502.
(65) Miedziak, P. J.; Tang, Z. R.; Davies, T. E.; Enache, D. I.; Bartley, J. K.; Carley,
A. F.; Herzing, A. A.; Kiely, C. J.; Taylor, S. H.; Hutchings, G. J. J. Mater. Chem.
2009, 19, 8619.

17
(66) Enache, D. I.; Edwards, J. K.; Landon, P.; Solsona-Espriu, B.; Carley, A. F.;
Herzing, A. A.; Watanabe, M.; Kiely, C. J.; Knight, D. W.; Hutchings, G. J.
Science 2006, 311, 362.
(67) Mueller, J. A.; Jensen, D. R.; Sigman, M. S. J. Am. Chem. Soc. 2002, 124, 8202.
(68) Trend, R. M.; Stoltz, B. M. J. Am. Chem. Soc. 2008, 130, 15957.
(69) Fox, S. G.; Gillard, R. D. Polyhedron 1988, 7, 349.
(70) Wang, A.; Jiang, H. F. J. Org. Chem. 2010, 75, 2321.

18
Chapter 2
The selective catalytic oxidation of glycerol to dihydroxyacetone
2.0 Preface
This chapter describes research done by David Pearson and me. D. Pearson made the
initial discovery that glycerol could be oxidized to dihydroxyacetone under the catalytic
conditions reported. I subsequently did most of the studies outlined in this chapter,
including the preparation of all deuterated compounds. D. Pearson did a benzoquinone
dependence study and an acetic acid dependence study on the reaction rate.
2.1 Introduction
The advent of biodiesel as an attractive and renewable alternative to dwindling fossil fuel
supplies has led to an increased market for its consumption on a global scale. A major
consequence from this recent development has been a corresponding acute increase in the
supply of glycerol as the major byproduct of this process.1-2 Glycerol is an attractive and
versatile feedstock as it is nontoxic, edible, and biodegradable, and it can be used as a
building block for value-added chemicals.3-5 The development of novel, selective
chemistry that can provide new applications to glycerol-derived products to meet the
increased supply of glycerol itself remains a key challenge.5-7

19
The chemoselective, catalytic transformation of glycerol to dihydroxyacetone remains an
unsolved problem. Dihydroxyacetone is currently produced on an industrial scale by
microbial oxidation of glycerol with Gluconobacter oxydans, with the major limitations
to this process being long reaction times and difficulty in removing unreacted glycerol
from the desired product.8-9 While there has been considerable effort in investigating the
oxidation of glycerol to glyceric acid, very few systems have shown any selectivity
towards the formation of dihydroxyacetone.10 Kimura has shown that a bismuth-
promoted platinum catalyst supported on carbon at acidic pH leads to an initial selectivity
of 80% for dihydroxyacetone but the catalyst is eventually deactivated by glyceric acid
that is formed as a byproduct.11 Gallezot reports a 37% yield of DHA at 75% conversion
using a similar Bi/Pt catalyst system with significant amounts of overoxidation
byproducts, mainly glyceric acid.12 Very recently, Varma has reported obtaining DHA in
48% yield at 80% conversion with this Bi/Pt catalyst using 30 psig O2 at 80ºC.13 Finally,
Crimmina reports an electrochemical method that produces DHA in 65% yield after 20
hours, but longer reaction times led to a concomitant increase in overoxidation
products.14
To the best of our knowledge, there are no homogeneous catalytic systems that can
promote the selective formation of dihydroxyacetone in satisfactory yields and
conversions. Farnetti has recently demonstrated that a (PNP)Ir(cod)(H) complex can
dehydrogenate glycerol through a transfer hydrogenation mechanism with benzaldehyde
as the hydride acceptor, but yields are low (< 25% yield after 3 h at 100ºC).15-16 In
addition, Wolfson has reported on the use of glycerol as a sacrificial hydrogen donor for

20
Ru-catalyzed transfer hydrogenation of ketones and suggests that dihydroxyacetone is
formed selectively, but there is no evidence that this is actually observed.17
A variety of palladium complexes are known to be robust catalysts for alcohol
oxidation.18-25 We have developed a catalyst inspired from Sheldon's work on aerobic
oxidation of alcohols with (neocuproine)Pd(OAc)2 in dmso. While acetate is a competent
internal base for deprotonation of the alcohol substrate, we envisioned that a
noncoordinating ion would be necessary to provide the requisite open coordination site
for β-hydrogen elimination of the Pd alkoxide complex. Consequently, we have reported
the use of our cationic dimeric palladium complex [(neocuproine)Pd(OAc)]2(OTf)2 (1) in
the aerobic alcohol oxidation of 2-heptanol with high initial rates at ambient
temperature.26 More recently, we have demonstrated that 1 is a competent catalyst for the
dehydrogenation of methanol to methyl formate at 50ºC.27 Key features of our proposed
mechanism for both transformations include a noncoordinating counterion that provides
an open coordination site for the binding of alcohols, and an internal base for the requisite
deprotonation of the bound alcohol.
Scheme 2.1. Catalyst systems.
N NPd
AcO OAc
N NPd
AcO
N NPd
H3CCN NCCH3
(OTf)2
2+
(OTf)2
2+
1 2 3

21
Herein we describe the chemoselective catalytic oxidation of glycerol and 1,2-
propanediol with the Pd complex 1 in the presence of either benzoquinone or air as a
terminal oxidant. We show that these vicinal polyols exhibit both faster rates and higher
chemoselectivities than other primary and secondary alcohols, enabling the rapid
chemoselective oxidation of glycerol to dihydroxyacetone under very mild conditions
(RT, 1 atm air).
Scheme 2.2. Selective oxidation of glycerol to dihydroxyacetone.
2.2. Results
The catalytic oxidation of glycerol with 5 mole % Pd (2.5 mol % 1) and 3 equivalents of
benzoquinone (BQ) in acetonitrile at RT proceeds with 97% conversion in 24 h with
>96% selectivity to dihydroxyacetone (Table 1). The nature of the solvent has a
significant influence on the rate: addition of water to the solvent as a 7/1 CH3CN/H2O
(v/v) mixture results in a significant acceleration in the reaction rate and complete
conversion of glycerol in 3 h. When the reaction is conducted in dmso, the oxidation is
complete within 15 minutes with complete selectivity for dihydroxyacetone. The
selective oxidation of glycerol with 1 can readily be operated on a 10 mmol scale in wet
acetonitrile: oxidation of 0.92 g (10 mmol) of glycerol afforded dihydroxyacetone in 92%
yield after chromatography, or 58% yield after crystallization of the product as its dimer.

22
Despite high reaction rates and conversions with dmso as a solvent, the dihydroxyacetone
product was inseparable from the solvent and thus could not be isolated in pure form.
Table 2.1. Catalytic oxidation of glycerol[a] and 1,2-propanediol with complex 1.
Entry Solvent Diol Oxidant Time [h]
Conversion [%]
Selectivity [%]
Yield [%]
1 CH3CN glycerol BQ 24 97 99 2 CH3CN/H2O[b] glycerol BQ 3 97 96 3 dmso glycerol BQ 0.25 97 99 4 dmso 1,2-PD BQ 0.3 98 96 5 dmso glycerol air 24 47 80 6 CH3CN/H2O [c] glycerol BQ 4 97 92
7 CH3CN/H2O [d] glycerol O2 4 95 69 8 CH3CN/H2O [e] glycerol air 18 73 [a] standard conditions: 0.1 mmol glycerol, 0.3 mmol BQ, 5 mol % Pd, 0.7 mL solvent, 23 ºC. [b] 7:1 CH3CN:H2O. [c] 10:1 CH3CN:H2O, 10 mmol scale. [d] 10 mol % Pd, 1atm O2, 10:1 CH3CN:H2O, 1 mmol scale [e]10 mol % Pd, sparged with air, 10:1 CH3CN:H2O, 10 mmol scale
Significantly, the selective oxidation of glycerol can also be carried out aerobically. The
oxidation of glycerol in wet acetonitrile under a balloon of O2 with 10 mol% Pd (1 mmol
scale) affords dihydroxyacetone in 69% isolated yield; on a larger scale (10 mmol
glycerol) under a continuous stream of air, dihydroxyacetone can be isolated in 73%
isolated yield after chromatography. Monitoring the progress of the reaction by NMR
with aliquots of the reaction solution reveals that there is little difference in reaction rate
for either case. However, it is considerably more convenient to use O2 because, in large-
scale reactions, the rate-limiting mass transfer of air diffusing throughout the solution
becomes a significant problem; if the O2 concentration is not high enough in the solution

23
to promote turnover of the Pd catalyst, the latter decomposes to Pd black and precludes
further conversion of the alcohol substrate to the desired oxidation product.
Attempts to perform the aerobic oxidation of glycerol at lower catalyst concentrations led
to high selectivity for dihydroxyacetone, but low conversions: with 5 mol% Pd under a
balloon of air only 47% conversion was observed after 24 hours in CD3CN/D2O.
Competitive oxidative decomposition of the catalyst is a likely cause of the lower
conversions and yields: the 1H NMR spectrum of the final reaction mixture exhibited
resonances characteristic of the Pd carboxylate 2 that we have previously shown to be
inactive for alcohol oxidation. Thus, high conversions of glycerol to dihydroxyacetone
can be achieved under aerobic conditions, but only at relatively high Pd concentrations.
The oxidation of glycerol and 1,2-propanediol is faster and more selective than that of
1,3-diols or a mixture of primary/secondary alcohols. Under similar conditions (5 mol%
Pd, 3 equiv. BQ, dmso, 23 °C), oxidation of glycerol is complete within 15 minutes and
oxidation of 1,2-propanediol to hydroxyacetone is complete within 20 minutes in dmso
(Table 1). In contrast, oxidation of a 1:1 mixture of 1-heptanol and 2-heptanol was both
slower and non-selective, requiring 10 hours to reach 78% conversion and affording a
45:55 ratio of the ketone/aldehyde. Similarly, oxidation of 1,3-butanediol proceeded to
only 55% conversion after 4 h, yielding a 2:3 mixture of the ketone and aldehyde
products.

24
2.3. Discussion
The high chemoselectivity for the oxidation of the secondary alcohol of glycerol in the
presence of two primary alcohols is noteworthy. While many stoichiometric oxidants
exhibit a preference for secondary over primary alcohols, few chemoselective catalytic
alcohol oxidations are known.
The lower rates and selectivities observed in the inter- and intramolecular competition
experiments suggest that vicinal diols exhibit unusual reactivity with 1. The kinetics of
1,2-propanediol oxidation with benzoquinone were monitored by 1H NMR in dmso-d6.
With 1.5 - 3.0 equivalents of benzoquinone (relative to diol), the disappearance of diol
conforms to a mixed second-order kinetics analysis (eq. 1):
(1)
where [BQ] and [PG] are the concentrations of benzoquinone and 1,2-propanediol
respectively, and t = time in seconds. Plots of kobs vs. [Pd] and the initial rates vs. [BQ]
confirm that the rates are first order in both [Pd] and [BQ] for [BQ] ≤ 0.3 M, yielding a
rate law (eq. 2)
(2)
where kobs = k'[Pd] and k' = 1.9(3) M-2s-1 in dmso-d6 at 23ºC.
In the presence of three equivalents of benzoquinone in either acetonitrile-d3 or dmso-d6,
a trace amount of lactaldehyde (< 5%) is observed to build up during the course of the
reaction, but disappears after approximately 80% conversion. The relative concentration

25
of lactaldehyde appears to inversely correlate with the amount of water present in the
reaction, as the highest amount of lactaldehyde (5% of the mass balance) is observed in
dry acetonitrile.
While NMR studies indicate that lactaldehyde is formed during the course of the
oxidation of 1,2-propanediol, deuterium-labeling studies suggest that generation of a
mixture of hydroxyacetone/lactaldehyde and subsequent isomerization of the aldehyde
does not contribute significantly to the high selectivity for hydroxyacetone. Catalytic
oxidation of 2-d-1,2-propanediol with 1 in dmso-d6 affords unlabeled hydroxyacetone
(<1% d-scrambling) and oxidation of 1-d2-1,2-propanediol yields d2-hydroxyacetone with
96% selectivity at 98% conversion. These experiments suggest that liberation of free
lactaldehyde, followed by Pd- or acid-catalyzed tautomerization is not a major
contributor to the high selectivity for hydroxyacetone. The second-order rate constants
for oxidation of 2-d-1,2-propanediol and that for the undeuteriated diol are within
experimental error (kH/kD = 1.0(2)), implicating that β-H elimination is not rate limiting.
However, an inverse isotope effect of kH/kD = 0.7(2) is evident from the ratio of rate
constants for 1-d2-1,2-propanediol and 1,2-propanediol (Fig. 2).
Scheme 2.3. Oxidation of deuterium-labeled 1,2-propanediols.
HO
OH
D
HO
OH
D D
HO
O
D D
HO
O
HO
O
D
kH/kD = 1.0(2)+ < 1%
kH/kD = 0.7(2)

26
2.4. Discussion of reaction mechanism.
On the basis of previous work, we propose that isomeric Pd alkoxides are formed by
liberation of acetic acid from the cationic Pd acetate derived from dimeric 1. β-H
elimination from the alkoxides would generate a Pd hydride that reacts with
benzoquinone to generate a cationic Pd hydroquinone complex. Reaction of this
hydroquinone complex with the diol regenerates the Pd alkoxides.
Scheme 2.4. Proposed mechanism for the catalytic oxidation of 1,2-propanediol.
To investigate the role of proton-transfer equilibria on the rate, we investigated the
kinetics of 1,2-propanediol oxidation with benzoquinone in the presence of 5, 10, and 20
mol% acetic acid (HOAc, relative to diol) and found that the rates are inverse first order
in [HOAc] (k' = k"/[HOAc]). This is consistent with the reversible generation of the
alkoxide from the reaction of the cationic Pd acetate (Fig. 3). The first-order dependence
on benzoquinone implies that reoxidation of Pd0 or the Pd-H is rate-limiting in dmso.
This is unusual for Pd-mediated alcohol oxidation, but consistent with the absence of a
primary kinetic isotope effect. The origin of the inverse secondary isotope effect is not
clear at present.
N NPd
OH
HO
N NPd
OH
HO
N NPd
O OH
N NPd
HO O
N NPd
O
HO
H
N NPd
O
HHO
H
O
O
H
O
HO
O
HO
N NPd
O
OH
1
HO OHHOAc
N NPd
AcO S
HO OH
HO
OH
(S = solvent)
1/2
H

27
We have also conducted one-turnover experiments with stoichiometric Pd and 1,2-
propanediol in dmso-d6. In this experiment, we observed 40% conversion with a 4:1
selectivity in favor of hydroxyacetone after 18 hours with the appearance of a Pd mirror
(presumably from the accumulation of (neocuproine)Pd0 after deprotonation of the Pd
hydride). Addition of 25 equivalents of carbon tetrachloride in an attempt to trap the Pd-
H species resulted in lower conversion (25%) and the same 4:1 selectivity observed in the
previous experiment. We were able to identify the Pd products as a mixture of
(neocuproine)PdCl2 and dmso-solvated chloropalladium species by comparison to
authentic samples. A key observation is that, when benzoquinone is added to either
reaction mixture, the previously reported high selectivity for hydroxyacetone is restored.
Scheme 2.5. Stoichiometric oxidation of 1,2-propanediol with 1.
The higher selectivities observed for the oxidation of glycerol/1,2-propanediol relative to
1- and 2-heptanol implies that the product-determining steps for the intra- and
OH
OH
0.5 equiv. Pd dimer25 equiv. CCl4
dmso-d6, rtOH
O
CHO
OH+
4 : 1
OH
OH
0.5 equiv. Pd dimer
3 equiv. benzoquinone
dmso-d6, rtOH
O
CHO
OH+
> 25 : 1
25% conversionafter 18 hours
> 97% conversionafter 20 minutes
OH
OH 0.5 equiv. Pd dimer
dmso-d6, rtOH
O
CHO
OH+
4 : 1
40% conversionafter 18 hours
+ Pd mirror

28
intermolecular oxidations are different. One possibility is that β-H elimination is not the
sole product-determining step; but that both the reversible formation of the Pd alkoxides
and β-H elimination contribute to the selectivities. Alternatively, if β-H elimination were
reversible, selective displacement of the bound ketone from the Pd-H intermediate could
explain the high selectivity for hydroxyketone formation. Further kinetic and mechanistic
studies are ongoing to test these hypotheses.
2.5. Conclusion and future directions.
In summary, glycerol is selectively and rapidly oxidized to dihydroxyacetone with the
cationic Pd catalyst 1 using benzoquinone or oxygen as the terminal oxidant. Vicinal
diols appear to be privileged substrates with this catalyst system, and are oxidized with
high rates and selectivities to hydroxyketones. Studies to explore the mechanism and
generality of the oxidation of polyols are currently underway.
2.6. Experimental section.
General considerations The dimeric Pd complex 1 was prepared as previously reported.26 All alcohols were
obtained commercially, stored over 3Å molecular sieves, and used without further
purification. CD3CN and dmso-d6 were obtained from Cambridge Isotope Laboratories,
distilled from CaH2, and stored over 3Å molecular sieves. Acetonitrile was dried by
passage through a pair of alumina columns, and collected and stored under N2.
Benzoquinone was purified by Soxhlet extraction with heptane and subsequent
recrystallization, or sublimed three times under vacuum at ambient temperature.

29
Thin-layer chromatography (TLC) was conducted with Whatman precoated silica gel
plates (0.25 mm, PE SIL E/UV) and visualized with potassium permanganate staining.
Flash column chromatography was performed as described by Still et al.28 using Silicycle
SiliaFlash silica gel 60 (40-63 µm mesh).
1H NMR spectra were recorded on a Varian Mercury-400 (400 MHz) or Varian Inova-
500 (500 MHz) spectrometer and are reported in ppm using residual solvent as an internal
reference (CD3CN: 1.93 ppm, dmso-d6: 2.49 ppm). The data is reported as: s = singlet, d
= doublet, t = triplet, q = quartet, p = quintet, m = multiplet; coupling constant(s) in Hz,
integration. Proton-decoupled 13C NMR spectra were recorded on a Varian Mercury-400
(100 MHz) or Varian Inova-500 (125 MHz) spectrometer, and are reported in ppm using
residual solvent as an internal reference (dmso-d6: 39.5 ppm).
Reaction optimization
Table 1, entry 1: Glycerol (9 mg, 0.1 mmol), benzoquinone (32 mg, 0.3 mmol), and p-
xylene (10.6 mg, 0.1 mmol) were weighed in a tared 1 dram vial and dissolved in 0.7 mL
CD3CN. The yellow solution was transferred to a tared NMR tube containing 2.6 mg 1
(2.5 µmol), the tube shaken, and the reaction monitored by 1H NMR. Dihydroxyacetone
was identified by its characteristic 1H NMR resonance at 4.16 ppm.29
Table 1, entry 2: This was carried out exactly as in entry 1, but 0.1 mL D2O and 0.7 mL
CD3CN was used as the solvent.

30
Table 1, entry 3: This was carried out exactly as in entry 1, but 0.7 mL dmso-d6 was
used as the solvent.
Table 1, entry 4: This was carried out exactly as in entry 1, but 7.6 mg 1,2-propanediol
was used as the substrate, and 0.7 mL dmso-d6 was used as the solvent. Hydroxyacetone
was identified by 1H NMR (dmso-d6): 2.02 ppm (s, 3 H) and 4.02 ppm (s, 2 H).30
Table 1, entry 5: Glycerol (0.92 g, 10 mmol) and benzoquinone (3.24 g, 30 mmol) were
dissolved in a mixture of 60 mL CH3CN and 6 mL H2O. 260 mg 1 (0.25 mmol) was then
added to the solution resulting in a reddish-brown solution. The solution was stirred at
room temperature until complete consumption of glycerol was evident by TLC. The
solution was then poured into 300 mL diethyl ether to precipitate out the catalyst and then
filtered through a plug of silica (60 g), eluting with ether and collecting 30 mL fractions
until the eluent was colorless. The dihydroxyacetone was then eluted with acetone, and
the acetone solution was concentrated. If any dihydroxyacetone coeluted with
benzoquinone, it was purified by column chromatography using acetone as the eluent.
The acetone solution was concentrated to yield 0.83 g of dihydroxyacetone as a colorless
and extremely hygroscopic oil (92%).
Alternatively, the acetonitrile solution was poured into 300 mL diethyl ether, filtered, and
concentrated. The residue was taken up in 30 mL acetonitrile, seeded with 5 mg
dihydroxyacetone dimer, and allowed to stand overnight at 5ºC. The white precipitate

31
was filtered, washed with acetonitrile, and dried to yield 410 mg of the dihydroxyacetone
dimer, characterized by comparison to an authentic sample. A second crop could be
obtained by concentrating the mother liquor and dissolving the residue in a minimal
amount of diethyl ether, seeding with 5 mg dihydroxyacetone dimer, and allowing to
stand at 5ºC overnight. This results in 110 mg of dihydroxyacetone dimer as a white
powder, with an overall net yield of 522 mg (58%). 31
Table 1, entry 6: Glycerol (0.92 g, 10 mmol) was dissolved in a mixture of 60 mL
CH3CN and 6 mL H2O in a 100 mL round-bottom flask fitted with a rubber septum. The
solution was saturated with air by sparging with a stream of air through a needle for 20
minutes, then 520 mg 1 (0.5 mmol) was added to the solution, resulting in an orange
solution. The reaction was monitored via TLC until complete consumption of glycerol
was observed. The solution was directly filtered through a plug of 60 g silica, eluting
with acetonitrile, and the acetonitrile solution was concentrated to obtain 663 mg
dihydroxyacetone as a colorless oil (73%).
This reaction was also carried out on a 1 mmol scale (92 mg glycerol) as described
above, but with a balloon of O2 as the terminal oxidant. Upon consumption of glycerol
by TLC, the solution was filtered through a plug of silica (10 g) eluting with acetonitrile
to remove catalyst, and concentrated to obtain 62 mg of dihydroxyacetone (69%).
Qualitative comparison of reaction rates with air or O2: 60.8 mg 1,2-propanediol (0.8
mmol) was dissolved in 5.6 mL 9:1 (v/v) CD3CN/D2O in a 20 mL vial sealed with a

32
rubber septum, and sparged with dioxygen or air (via a balloon attached to a syringe
affixed with a 18-gauge needle) for 20 minutes. 41.6 mg catalyst (40 µmol) was then
added to the clear solution, and the balloon quickly replaced. 0.5 mL aliquots of the
reaction solution were taken over time and monitored by 1H NMR. Using p-xylene as an
internal standard was ineffective in this experiment as it appears to slowly be lost to
evaporation over time.
Figure 2.1. Comparison of conversion vs. time for 1,2-propanediol oxidation with air or
O2 as the terminal oxidant.
Mechanistic studies
Intermolecular selectivity: oxidation of 1-heptanol and 2-heptanol: 1-heptanol (5.8
mg, 0.05 mmol) and 2-heptanol (5.8 mg, 0.05 mmol), along with benzoquinone (32 mg,
0.3 mmol) and p-xylene (10.6 mg, 0.1 mmol) were dissolved in either dmso-d6 or CD3CN
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0 50 100 150 200 250 300
Conversion
Time (minutes)
air
O2

33
(0.7 mL). This solution was transferred to a tared NMR tube containing 2.6 mg 1 (5
µmol Pd), the tube shaken, and the course of the reaction monitored by 1H NMR.
Integration of the alpha-CH2 protons of both carbonyl compounds (1.49 ppm for
heptaldehyde, 1.43 ppm for 2-heptanone) reveals a 55:45 ratio of aldehyde to ketone in
dmso-d6, and a 1:1 ratio for both products in CD3CN after 10 hours.
Intramolecular selectivity: oxidation of 1,3-butanediol: 1,3-butanediol (9 mg, 0.1
mmol) and benzoquinone (32 mg, 0.3 mmol) were dissolved in 0.7 mL CD3CN and the
solution transferred to a tared NMR tube containing 2.6 mg 1 (5 µmol Pd). The reaction
was monitored by 1H NMR by integration of the CH3 resonances for the diol (1.10 ppm,
d), and the aldehyde (1.16 ppm, d) and ketone products (2.09 ppm, s). After 4 hours,
there was 45% conversion, with the product mixture containing 42% 4-hydroxy-2-
butanone31 and 58% 3-hydroxybutanal.32
Preparation of 1,1-d2-1,2-propanediol: 700 mg LiAlD4 (16.7 mmol) was dissolved in
30 mL THF at 0ºC. 2 g rac-lactide (13.9 mmol) was dissolved in 10 mL THF and added
to the stirring solution over 5 minutes. The colorless solution was stirred at 0ºC for one
hour then at 23ºC for one hour longer. The solution was cooled again to 0ºC and
quenched by adding 0.7 mL water, 2.1 mL 15% aqueous NaOH, and finally 0.7 mL
water, and stirred overnight. The aluminum salts were filtered, washed thoroughly with
THF, and the filtrate concentrated to obtain 2.1 g of a slightly yellow oil that was pure by
1H NMR (97% yield).

34
1H NMR (500 MHz, dmso-d6): 0.98 (d, 3H, J = 6.5 Hz), 3.53 (q, 1 H, J = 5.5 Hz), 4.39 (d,
1 H, J = 4.5 Hz), 4.41 (s, 1H) 13C NMR (125 MHz, dmso-d6): 20.0, 66.5 (p, J = 21 Hz),
67.1
HRMS (m/z): M+ calc’d for C3H6D2NaO2, 101.0548; found, 101.0547
Preparation of 2-d-1,2-propanediol: 813 mg LiAlD4 (19.4 mmol) was dissolved in 20
mL diethyl ether at 0ºC. 2 g acetoxyacetone (17.2 mmol) was added to the stirring
solution dropwise over 5 minutes, and the solution heated to reflux for 4 h. The solution
was cooled to 23ºC, diluted with 20 mL diethyl ether, and quenched with 1 mL water, 3
mL 15% aqueous NaOH, and 1 mL water, and the slurry stirred for one hour. The
aluminum salts were filtered out, washed thoroughly with THF, and the slightly yellow
filtrate concentrated. The residue was purified by column chromatography (60 g silica,
CH3CN eluent, Rf = 0.45) to obtain 740 mg of a colorless oil (56% yield).
1H NMR (500 MHz, dmso-d6): 0.97 (s, 3 H), 3.13 (dd, 1 H, J = 5.6 Hz, 10.5 Hz), 3.23
(dd, 1 H, J = 5.6 Hz, 10.5 Hz), 4.38 (s, 1 H), 4.46 (t, 1 H, J = 6 Hz) 13C NMR (125 MHz,
dmso-d6): 19.9, 66.7 (t, J = 21 Hz), 67.2
HRMS (m/z): M+ calc’d for C3H7DNaO2, 100.0485; found, 100.0487
Representative kinetic run: A stock solution was made consisting of 43.4 mg 1,2-
propanediol (0.57 mmol), 183 mg benzoquinone (1.70 mmol), and 56.1 mg p-xylene (0.5
mmol) dissolved in enough dmso-d6 to make a solution with 4 mL total volume. 0.7 mL

35
of this stock solution was dispensed into a tared NMR tube consisting of 2.6 mg 1 (2.5
µmol) and monitored by NMR.
A 400 MHz Varian Mercury NMR spectrometer was used to record the spectra for every
kinetic run. The longest T1 relaxation time for all reagents in solution is 6.9 seconds for
the benzoquinone protons. Each spectrum during a typical kinetic run was recorded 60
seconds apart via a programmed array, with each spectrum consisting of one pulse with a
d1 delay of 45 seconds to ensure complete relaxation of all protons. Relevant peaks: 1,2-
propanediol (0.93 ppm, d, 3 H, J = 6 Hz); lactaldehyde (1.14 ppm, d, 3 H, J = 7.2 Hz);
hydroxyacetone (2.02 ppm, s, 3 H); p-xylene (2.21 ppm, s, 6 H); hydroquinone (6.58
ppm, s, 4 H); benzoquinone (6.80 ppm, s, 4 H). All peak areas were integrated relative to
the CH3 proton for p-xylene.
The kinetics were analyzed by mixed second-order plots that show the consumption of
alcohol and benzoquinone as a function of time. The relevant equation for this
dependence is described as follows:
where: [BQ]0 and [BQ]t are the concentrations of benzoquinone at time t = 0 and t= t,
respectively;
[PG]0 and [PG]t are the concentration of 1,2-propanediol at time t = 0 and t= t,
respectively; kobs is the observed rate constant; t is time in seconds

36
Error analysis: Due to the long delay times, error in estimates of integration are assumed
to 2%. Standard propagation of error algebra,33 and least-squares analysis was used to
obtain estimates of the variance of each variable:
where
σ = error in a specified variable
y = the y-axis of the mixed second-order plot = ln(BQ/PG) = ln(BQ) - ln(PG)
PG and BQ refer to the integrations from the NMR spectra.
For the linear fits, a least squares analysis7 was used; y = kt + b
for
Kinetics
Shown in Figure S1 is a representative first-order plot for the consumption of 1,2-
propanediol in dry dmso-d6 as a function of time

37
Figure 2.2. First order kinetic plot for oxidation of 1,2-propanediol
Shown in Figure 2.3 are mixed second-order plots for the consumption of 1,2-
propanediol and benzoquinone as a function of time in dry dmso-d6.
There is a linear dependence of kobs on the concentration of Pd in the reaction solution:
where:
[Pd] is the concentration of Pd in the reaction solution (assumed to be a monomeric
species)
k' is the rate constant of the reaction independent of [Pd], [BQ], and [PD].
!"#"$%$$&'("
)*"#"$%+++&&"
$"
$%'"
,"
,%'"
-"
-%'"
&"
&%'"
$" ,$$" -$$" &$$" .$$" '$$" /$$" 0$$" 1$$" +$$"
!"#$%&'()*&'() +,"
-./"%.0$,"
10234"526/2"'784"&9:);+<=>?%=<?/@,"
!
kobs
= k'[Pd]

38
Figure 2.3. Second order kinetic plot for oxidation of 1,2-propanediol.
Shown in Figure 2.4 is a plot of Pd concentration versus kobs (determined through
Equation 1 and the slope of the second order plot in Figure 2.4) is shown below:
Figure 2.4: Plot of kobs vs. [Pd].
!"#"$%&''()"
*+"#",%&-(,$"
,"
,%,,("
,%,,."
,%,,/"
,%,,-"
,%,$"
,%,$("
,%,$."
,%,$/"
,%,$-"
,%,01,," $%,02,3" (%,02,3" 3%,02,3" .%,02,3" 4%,02,3" /%,02,3" '%,02,3" -%,02,3" &%,02,3"
5678"9:
;2$"8;2$<"
=>?@"9:<"
!"#$%$&'"()*"+)&+,**-$.*/0)1**

39
Shown in Figure 2.6 is a plot of initial rate vs. [BQ] concentration, confirming the first
order dependence in BQ for 0.2M < [BQ] < 0.4M
Figure 2.5. Plot of initial rate vs. [BQ].
Oxidation of d-labeled 1,2-propanediols: 7.7 mg of 2-d1-1,2-propanediol or 7.8 mg of
1-d2-1,2-propanediol (0.1 mmol), 32 mg benzoquinone (0.3 mmol), and 8.8 mg p-xylene
(0.08 mmol) was dissolved in dmso-d6 to make a total volume of 0.7 mL. This was then
transferred to a tared NMR tube containing 2.6 mg 1 (2.5 µmol), and monitored by 1H
NMR, as above.
In the d1-alcohol experiments, the principal product was hydroxyacetone with peaks at
2.02 ppm (s, CH3) and 4.02 ppm (s, CH2). No incorporation of deuterium at the alpha
carbon could be observed by either 1H, 13C or 2H NMR. In the d2-alcohol experiments,
the principal product was 1-d2-hydroxyacetone with a peak at 2.02 ppm (s, CH3). A very
small triplet could be observed at the shoulder of the peak at 2.02 ppm, representing the
d1-hydroxyacetone product, but this was < 1% of the mass balance of the reaction.
!"#"$%$$&'("
)*"#"$%+++,-"
$"
$%$$$&"
$%$$$."
$%$$$,"
$%$$$'"
$%$$$/"
$%$$$0"
$%$$$1"
$" $%&" $%." $%," $%'" $%/" $%0" $%1"
!"#$%&'($)&*+,-.&
/012&
!"#$&'($)&*345&67"8.&8-%&/012&

40
The kinetic isotope effect for each of the two deuterated alcohols was determined by
directly calculating kH/kD, both obtained through mixed second-order plots. Shown in
Figure S5 are mixed second-order plots for d0(kobs = 1.1(1) x 10-2 M-1s-1) , d1(kobs = 1.1(1)
x 10-2 M-1s-1), and d2 (kobs = 1.5(1) x 10-2 M-1s-1) 1,2-propanediols:
Figure 2.6. Second order plots for d0, d1, and d2 1,2-propanediols

41
Shown in Figure 2.8 is a plot of the mixed second-order rate constant 1/kobs vs. added
[HOAc]:
Figure 2.7. Plot of 1/kobs vs. [HOAc]

42
2.7 References
(1) Tyson, K. S.; Bozell, J.; Wallace, R.; Petersen, E.; Moens, L.; (National
Renewable Energy Laboratory NREL/TP-510-34796, Boulder , Co, June 2004,
available at www1.eere.energy.gov/biomass/pdfs/34796.pdf): 2004.
(2) Gong, C. S.; Du, J. X.; Cao, N. J.; Tsao, G. T. Applied Biochemistry and
Biotechnology 2000, 84-6, 543.
(3) Werpy, T.; Petersen, G.; (US Department of Energy, Oak Ridge, TN, August
2004, available at www.eere.energy.gov/biomass/pdfs/35523.pdf): 2004.
(4) Zhou, C. H. C.; Beltramini, J. N.; Fan, Y. X.; Lu, G. Q. M. Chem. Soc. Rev. 2008,
37, 527.
(5) Behr, A.; Eilting, J.; Irawadi, K.; Leschinski, J.; Lindner, F. Green Chem. 2008,
10, 13.
(6) Christensen, C. H.; Rass-Hansen, J.; Marsden, C. C.; Taarning, E.; Egeblad, K.
Chemsuschem 2008, 1, 283.
(7) Pagliaro, M.; Ciriminna, R.; Kimura, H.; Rossi, M.; Della Pina, C. Eur. J. Lipid
Sci. Tech. 2009, 111, 788.
(8) Hekmat, D.; Bauer, R.; Neff, V. Process Biochemistry 2007, 42, 71.
(9) Mishra, R.; Jain, S. R.; Kumar, A. Biotechnology Advances 2008, 26, 293.
(10) Pagliaro, M.; Ciriminna, R.; Kimura, H.; Rossi, M.; Della Pina, C. Angew. Chem.
Int. Ed. 2007, 46, 4434.
(11) Kimura, H. Applied Catalysis A - General 1993, 105, 147.
(12) Fordham, P.; Garcia, R.; Besson, M.; Gallezot, P. Studies in Surface Science and
Catalysis 1996, 101, 161.

43
(13) Hu, W. B.; Knight, D.; Lowry, B.; Varma, A. Ind Eng Chem Res 2010, 49, 10876.
(14) Ciriminna, R.; Palmisano, G.; Della Pina, C.; Rossi, M.; Pagliaro, M. Tetrahedron
Lett. 2006, 47, 6993.
(15) Farnetti, E.; Kaspar, J.; Crotti, C. Green Chem. 2009, 11, 704.
(16) Crotti, C.; Kaspar, J.; Farnetti, E. Green Chem. 2010, 12, 1295.
(17) Wolfson, A.; Dlugy, C.; Shotland, Y.; Tavor, D. Tetrahedron Lett. 2009, 50,
5951.
(18) Lloyd, W. G. J. Org. Chem. 1967, 32, 2816.
(19) Nishimura, T.; Kakiuchi, N.; Onoue, T.; Ohe, K.; Uemura, S. J. Chem. Soc.
Perkin Trans. 1 2000, 1915.
(20) Stahl, S. S. Angew. Chem. Int. Ed. 2004, 43, 3400.
(21) Stoltz, B. M. Chem. Lett. 2004, 33, 362.
(22) Sigman, M. S.; Jensen, D. R. Acc. Chem. Res. 2006, 39, 221.
(23) Schultz, M. J.; Sigman, M. S. Tetrahedron 2006, 62, 8227.
(24) ten Brink, G. J.; Arends, I. W. C. E.; Sheldon, R. A. Adv. Synth. Catal. 2002, 344,
355.
(25) Arends, I. W. C. E.; ten Brink, G. J.; Sheldon, R. A. J. Mol. Catal. A - Chem.
2006, 251, 246.
(26) Conley, N. R.; Labios, L. A.; Pearson, D. M.; McCrory, C.; Chidsey, C. E. D.;
Waymouth, R. M. Organometallics 2007, 26, 5447.
(27) Pearson, D. M.; Waymouth, R. M. Organometallics 2009, 28, 3896.
(28) Still, W. C.; Kahn, M.; Mitra, A. J. Org. Chem. 1978, 43, 2923.
(29) Kobayashi, Y.; Takahashi, H. Spectrochimica Acta A 1979, 35, 307.

44
(30) Glushonok, G. K.; Glushonok, T. G.; Maslovskaya, L. A.; Shadyro, O. I. Russ. J.
Gen. Chem. 2003, 73, 1027.
(31) Shei, C. T.; Chien, H. L.; Sung, K. Synlett 2008, 1021.
(32) Hintermann, L.; Kribber, T.; Labonne, A.; Paciok, E. Synlett 2009, 2412.
(33) Bevington, P. R.; Robinson, D. K. Data Reduction and Error Analysis for the
Physical Sciences; 2nd ed.; McGraw-Hill: New York, 1992.

45
Chapter 3
The selective catalytic oxidation of diols and polyols to hydroxyketones
3.0 Preface
The experiments conducted in this chapter were performed by Steven M. Banik and me.
S. M. Banik conducted all of the initial screening of the diols discussed in this chapter,
and I scaled up all of the diol oxidations. I did all of the other studies reported in this
chapter.
3.1 Introduction
The controlled and chemoselective oxidation of polyols to carbonyl compounds remains
a fundamental challenge in organic synthesis.1 The problems associated with selective
oxidation of alcohol functional groups in polyols include oxidative cleavage of a diol
group, overoxidation to polycarbonyl compounds, and/or lack of chemoselectivity for one
alcohol group over another. This problem, and the solutions that have been developed to
address these problems, has been discussed in detail in Chapter 1 of this thesis.
We have recently disclosed the chemoselective catalytic oxidation of glycerol to
dihydroxyacetone in high conversions, selectivities, and yields by a cationic
(neocuproine)Pd(OAc)(OTf) catalyst developed in our laboratory (Chapter 2).2 The
chemoselectivity behind this transformation is remarkable in the face of many efforts by

46
other investigators towards the selective oxidation of glycerol to value-added products.3
The aerobic oxidation with a heterogeneous Pt/Bi catalyst leads to the best selectivity
reported to date for dihydroxyacetone with 48% yield at 80% conversion.4 Herein, we
report the extension of our diol oxidation methodology to other vicinal diols and polyols,
with additional mechanistic insight into the nature of our catalyst’s intrinsic
chemoselectivity for α-hydroxyketone products.
Scheme 3.1. The [(neocuproine)Pd(OAc)]2(OTf)2 complex (1).
3.2 The oxidation of activated diols
We began this study by investigating the oxidation of a series of para-substituted-1,2-
phenylethanediols, which are readily accessible from the corresponding para-substituted
styrene by the Sharpless dihydroxylation protocol. In addition, by observing the
oxidation of these substrates, we anticipated the ability to construct a Hammett plot and
gain further mechanistic insight into the chemoselective oxidation of 1,2-diols to the
corresponding hydroxyketone.
Scheme 3.2. Preparation of substituted phenylethane-1,2-diols and their oxidation to the
corresponding α-hydroxyacetophenone product.
N NPd
AcO
2
2+
(OTf)2
AD-mix alpha
tBuOH/H2O, rt
OH
OH
CD3CN/D2O, rt
O
OH2.5 mol% 1
3 eq. benzoquinone
Y Y Y
Y = CH3O, CH3, Cl, CF3, NO2

47
We discovered that the aryl-substituted 1,2-diols exhibit poor chemoselectivity under the
catalytic conditions (5 mol% Pd, 3 equivalents benzoquinone, 9:1 acetonitrile:water, RT).
In contrast to glycerol and 1,2-propanediol, we observed overoxidation of the initial 2-
hydroxyacetophenone product in all cases to phenylglyoxal (as the hydrate), and, after
longer reaction times, to phenylglyoxalic acid. The product distribution for the various
substituted phenylethanediols after two hours is shown in Figure 3.1 - no significant
amount of phenylglyoxalic acid could be detected at this point.
Table 3.1. The Pd-catalyzed oxidation of para-substituted phenylethane-1,2-diols.a
Substituent Conversion (%) Hydroxyketone (%) Glyoxal (%)
methoxy 55 21 33 methyl 71 10 60 chloro 61 8 53 nitrob 40 30 --
a Conditions: 0.1 mmol substrate, 5 µmol (5 mol%) Pd, 0.3 mmol benzoquinone, 0.63 mL CD3CN/0.07 mL D2O, rt, 2 hours. b after 6 hours of reaction time.
As can be discerned from Table 3.1, the reactivities of the four diols screened are similar,
with the methoxy- and nitro-substituted diols being somewhat more selective than the
other two diols. We attribute this similarity in reactivity of the diols as evidence for our
hypothesis that β-hydride elimination of the Pd alkoxides to the carbonyl product is not
the turnover-limiting step of the catalytic cycle for diol oxidation (see Chapter 2 for the
proposed mechanism for 1,2-diol oxidation by 1). The methoxy and nitro-substituted
diols' somewhat lower reactivities under the catalytic conditions is possibly due to
binding of the substituent to the Pd center, inhibiting the reaction's progress. Due to the
lower conversion for these two diols, it is plausible that the selectivities for the
hydroxyketone product is higher due to a corresponding lower reactivity for the

48
hydroxyketone product for overoxidation. Sheldon has shown that, despite obtaining an
excellent linear free energy relationship with (neocuproine)Pd(OAc)2-catalyzed aerobic
oxidation of a range of substituted benzyl alcohols (ρ = -0.58), p-methoxybenzyl alcohol
is significantly less reactive than predicted from this correlation.5 The dramatic lack of
reactivity for the nitro-substituted diol relative to the other diols is unclear at present.
We also investigated the oxidation of 3-butene-1,2-diol under the prescribed conditions,
and discovered that it gave a complex mixture of products, none of which could be
readily identified. We attribute this mixture to several possible side reactions:
overoxidation of the substrate, Wacker-type oxidation of the olefin, and possible allylic
rearrangement. Thus, we conclude that diols with an activated C-H bond (allylic,
benzylic) are poor substrates for our catalyst and prone to overoxidation.
3.3. The oxidation of aliphatic, unactivated diols.
We wish to report in this section the comparable effectiveness of this catalyst system for
other vicinal diols. Oxidation of the 1,2-diol with 5 mol% Pd (2.5 mol% dimer) and 3
equivalents of benzoquinone in 9:1 (v/v) CD3CN/D2O at room temperature afforded the
hydroxyketone as the major product with little to no overoxidation to the dicarbonyl
compound. Table 3.2 shows the results of the NMR-scale oxidations conducted for a
variety of substrates after 2 hours.

49
Table 3.2. NMR-scale screening of the oxidation for a variety of alcohols, diols, and polyols with 1 and benzoquinone in CD3CN/D2O at room temperaturea. Entry Substrate Conversion (%) NMR yield(%) Major product
1
95 95
2
94 59
3
62 62
4
>99 (in dmso-d6)
72
5
57 38
6
87 76
7
0 0 -
8
55 55
9
94 94
a 0.1 mmol substrate, 2.5 µmol 1 (5 mol% Pd), 0.3 mmol benzoquinone, 0.7 mL 9:1 (v/v)
CD3CN/D2O, RT, 2 hours. Note: all of these substrates were screened by Steven M. Banik. As previously reported,2 1,2-propanediol is oxidized under the catalytic conditions to
hydroxyacetone in two hours with 95% conversion and 95% yield (entry 1). Replacing
the methyl group with a more sterically demanding group (entry 2) does not affect the
conversion, but the selectivity is lower; the NMR spectrum of the final product mixture
OH
OH
OH
O
OH
OH
HOOH
O
HO
OHHO
OH
OH
OH
O
HO
OH
O
OH
O
O
OH
OH
O
OH
O
OH

50
shows two products, the second of which cannot clearly be identified. 1,2,4-butanetriol
and meso-erythritol (1,2,3,4-butanetetraol) were efficiently oxidized to the
hydroxyketone product shown with minimal amounts of overoxidation products (entries 3
and 4, respectively). However, the oxidation of 1,3,5-pentanetriol (entry 5) was
significantly slower and less selective than 1,2,4-butanetriol, suggesting that the
chemoselective oxidation characteristic of this system is unique to 1,2-diols.
We hypothesized that the selectivity for 1,2-diol oxidation that comes with our catalytic
system was partly due to the ability for the diol to chelate efficiently to Pd and effect
conversion to the hydroxyketone. To test this hypothesis, we subjected 1-methoxy-2-
propanol (entry 6) to our catalytic conditions and discovered that reaction rates and
conversions were similar to the 1,2-propanediol case. However, the dimethylamino-
substituted analog (entry 7) appears to bind irreversibly to Pd; a dark red solid
immediately precipitated out of solution when the substrate was added.
We also tested cyclic 1,2-diols and discovered that these diols were equally effective
substrates for oxidation. cis-Cyclopentane-1,2-diol and trans-cyclohexane-1,2-diol
produce the corresponding racemic hydroxyketone product with similar selectivities,
despite the former being somewhat less reactive than the latter. However, the geometry
of substituted cyclohexane-1,2-diols has a significant effect on reactivity and selectivity
(vide infra). We attribute the equal selectivity observed for either geometry in the case of
the unsubstituted diols to rapid interconversion between both possible conformers such
that a favorable geometry for chelation to Pd is readily accessible. In contrast, when 1,2-

51
butanediol was subjected to the catalytic conditions, we observed an approximately 1:1
mixture of 3-hydroxy-2-butanone and 2,3-butanedione products. This suggests that
cyclic internal 1,2-diols are superior to acyclic substrates; the reasons for this are unclear
at present.
To evaluate the synthetic potential of this chemoselective oxidation, the oxidations were
carried out on a preparative scale. We chose six substrates to be subjected to the reaction
conditions (Table 3.3), and in preliminary scale-up attempts (2 mmol scale), we found it
difficult to separate the hydroxyketone product from the large excess of benzoquinone.
We have previously reported that glycerol can be aerobically oxidized to
dihydroxyacetone but at lower conversions and higher catalyst loading due to known
catalyst decomposition.2 Despite this liability, the diols we chose were selectively
oxidized to the hydroxyketone products in three hours, and were readily isolated in pure
form by filtering out the catalyst through a silica plug (Figure 3.3) followed by
chromatography if necessary. The yields for the polyols did not improve when subjected
to longer reaction times. A remarkable observation is that, while the aerobic oxidation of
acyclic vicinal diols typically stops at ~80% conversion, the cyclohexane-1,2-diols
screened were significantly more reactive with nearly quantitative conversion to the
hydroxyketone product in less than 2 hours.

52
Table 3.3. The oxidation of six polyols to hydroxyketones on a 2 mmol scale.a
Entry Substrate Product Isolated yield (%)
1
76
2
75
3
71
4
65
5
95
6
94
a Conditions: 2 mmol diol, 0.1 mmol 1 (10 mol% Pd), 1 atm O2 balloon, 9 mL CH3CN/1 mL H2O, rt, 3 hours
3.4 Stereoelectronic effects on cyclohexane-1,2-diol oxidation
We conducted two additional studies to probe the reactivity of cyclic diols to the
hydroxyketone product. First, we prepared conformationally restricted cyclohexane-1,2-
diols to probe whether there was a significant effect of diol geometry on reactivity under
catalytic conditions. Second, we conducted preliminary screenings of unsymmetrical 3-
methyl-1,2-cyclohexanediol compounds to determine whether the oxidation of these
substrates would be regioselective for one hydroxyketone over another.
OH
OH
O
OH
O
OH O
O
OH
OH
HOOH
O
HO
OHHO
OH
OH
OH
O
HO
OH
OH
OH OH
O
OH
OH OH
O

53
We prepared three of the four possible geometric isomers for 4-tert-butylcyclohexane-
1,2-diol from 4-tert-butylcyclohexene (Scheme 3.2). The conformationally locked trans
diaxial cyclohexanediol was accessible through regioselective ring-opening of the
cyclohexene oxide formed from performic acid followed by alkaline hydrolysis of the
formate ester products. Osmium-catalyzed dihydroxylation of 4-tert-butylcyclohexene
gave both cis diastereomers in a 1:1 ratio that was inseparable by chromatography despite
a previous report outlining their successful separation via their benzoate diesters.6
Scheme 3.3. Preparation of 4-tert-butylcyclohexane-1,2-diols.
Subjection of the diaxial cyclohexanediol to our catalytic conditions with benzoquinone
as the terminal oxidant resulted in very little reaction even after extended reaction times
(< 20% conversion over 2 days). This suggests that chelation of the diol to Pd that takes
place via an accessible diol geometry is necessary for efficient oxidation to take place.
Since the cis-diols could unfortunately not be isolated in pure form, they were carried
forward as a 1:1 mixture. Subjection of the mixture to the same catalytic conditions
resulted in a complex mixture of products that could not be readily identified.
OH
1. MsCl, Et3N
2. collidine, !
H2O2, HCO2H, 45ºC
then KOH/H2O
AD-mix alpha
tBuOH/H2O, rt
tBu
OH
OH
tBu OH
OH
tBu
OH
OH+

54
Two of the four possible unsymmetric 3-methyl-1,2-cyclohexanediol diastereomers were
prepared by sodium borohydride reduction of the corresponding diketone in cold ethanol
(Scheme 3.3).
Scheme 3.4. Preparation and reactivity of 3-methyl-1,2-cyclohexanediols
Each diastereomer was readily separable from the other by chromatography and was
subjected to the catalytic conditions using benzoquinone as the terminal oxidant. The
cis-diol was oxidized with good chemoselectivity, with the 2-hydroxy-6-
methylcyclohexanone product predominating over the other possible regioisomer. The
oxidation of the trans-diol was unfortunately not regioselective, with both ketone
products being present in an ~1:1 mixture. It appears that the regiochemistry of these two
oxidations is also sensitive to the ability of the diols to chelate to the Pd center; in the
latter case, the axial alcohol is preferentially oxidized over the equatorial alcohol,
reminiscent of other alcohol oxidation systems.7-8
The propensity for only specific cyclohexanediols to undergo chemoselective oxidation
to the hydroxyketone deserves comment. The current hypothesis is that an accessible
O
O NaBH4
EtOH/H2O
Me
OHOH
+ MeHO
OH
2.5 mol% 1
3 eq. benzoquinone
CD3CN/D2O, rt
2.5 mol% 1
3 eq. benzoquinone
CD3CN/D2O, rt
Me
OHO
Me
OHO
+Me
HOO

55
chelating geometry for the diols is required for the reaction to proceed with this catalytic
system (Scheme 3.5); for the diaxial isomer, no chelation is possible without forcing the
diol to assume an extremely unfavorable conformer with an axial tert-butyl group. In
contrast, for the cis-diastereomer shown, chelation is possible and the reaction proceeds
normally to afford what is likely the product shown in the Scheme.
Scheme 3.5. The oxidation of 4-tert-butylcyclohexane-1,2-diols by 1.
The oxidation of the 3-methyl-1,2-cyclohexanediols, shown in Scheme 3.6, follows a
similar hypothesis; the trans-diol can chelate to Pd, but the β-hydrogen for either
alkoxide are roughly equally accessible, leading to the observed lack of selectivity for
either hydroxyketone product. In contrast, the β-hydrogen for the axial alcohol in the cis-
diol case is significantly more accessible than that for the equatorial alcohol group,
leading to the observed regiochemistry.
tBu
OH
OH
N NPd
AcO NCCH3
+
- AcOH
+ AcOH
tBu
OH
O
N NPd
NCCH3
//tBu
OH
O
tBu
OH
OH N NPd
AcO NCCH3
- AcOH
+ AcOH
N NPd
+
tBu
O OH
tBuOH
O

56
Scheme 3.6. The oxidation of 3-methyl-1,2-cyclohexanediols by 1.
3.5. Conclusions and future directions.
We have successfully demonstrated the ability of our catalyst system to oxidize a variety
of alcohols, diols, and polyols selectively to the hydroxyketone product. The aerobic
oxidation of polyols is an attractive catalytic transformation, but further studies need to
be conducted to improve conversions and yields with this system. Employing a more
oxidatively-resistant ligand for this catalyst system that affords the same or better
chemoselectivity for the hydroxyketone product would be an advance in this direction.
We have also conducted preliminary mechanistic investigations to determine how
stereoelectronic effects affect the competency of the catalyst system for diol oxidation.
More mechanistic studies need to be conducted to better understand the origin of our
catalyst system's ubiquitous reactivity with vicinal diols.
OH
OH N NPd
AcO NCCH3
- AcOH
+ AcOH
N NPd
+O OH OH
O
H
HMe
MeMe
OH N NPd
AcO NCCH3
- AcOH
+ AcOH
N NPd
+
OH
O
Me
Me
OHO
Me
HO
HH
+
Me
O
OH

57
Being able to oxidize biomass-derived polyols, such as carbohydrates, selectively to a
single ketone product would be an impressive and very desirable achievement with this
catalyst system. Finally, developing a chiral catalyst system for the stereospecific
oxidation of chiral or prochiral diols to optically pure ketones is a desirable goal in our
laboratory. Investigations in both directions are currently underway in our laboratory.
3.6. Experimental section
The preparation of [(neocuproine)Pd(OAc)]2(OTf)2 has previously been described.9 All
alcohol substrates and solvents were obtained commercially and used without further
purification. CD3CN and dmso-d6 were obtained from Cambridge Isotope Laboratories
and used without further purification.
Thin-layer chromatography (TLC) was conducted with Whatman precoated silica gel
plates (0.25 mm, PE SIL E/UV) and visualized with potassium permanganate staining
except in the case of methoxyacetone, where a 2,4-dinitrophenylhydrazine stain was
used. Flash column chromatography was performed as described by Still et al.24 using
Silicycle SiliaFlash silica gel 60 (40-63 µm mesh).
1H NMR spectra were recorded on a Varian Mercury-400 (400 MHz) or Varian Inova-
500 (500 MHz) spectrometer and are reported in ppm using residual solvent as an internal
reference (CD3CN: 1.93 ppm, dmso-d6: 2.49 ppm). The data is reported as: s = singlet, d
= doublet, t = triplet, q = quartet, p = quintet, m = multiplet; coupling constant(s) in Hz,
integration. Proton-decoupled 13C NMR spectra were recorded on a Varian Mercury-400

58
(100 MHz) or Varian Inova-500 (125 MHz) spectrometer, and are reported in ppm using
residual solvent as an internal reference (dmso-d6: 39.5 ppm).
Para-substituted phenylethane-1,2-diols: These diols were prepared on a 1 mmol scale
from the corresponding styrene by the Sharpless dihydroxylation protocol with AD-mix
α, as described in the literature.25 The NMR spectra of each diol prepared was identical
to that previously reported in the literature.10-12
Representative procedure [for 1-(4-methoxyphenyl)ethane-1,2-diol]:
1.4 g AD-mix α (1 mmol) was added to 5 mL tert-butyl alcohol and 5 mL water and
stirred until completely dissolved. 4-methoxystyrene (134 mg, 1 mmol) was then added
to the biphasic yellow solution, and the solution stirred vigorously for 18 hours. Solid
sodium sulfite (1.5 g, 12 mmol) was added to the now heterogeneous solution and the
reaction stirred for 1 hour longer. The product was extracted from the now gray solution
with 4 x 5 mL EtOAc, and the extracts dried over MgSO4, filtered, and evaporated. The
residue was purified by chromatography (5 g SiO2, EtOAc) to yield a white solid (159
mg, 95% yield) that was pure by 1H and 13C NMR; the spectra were consistent with that
reported in the literature.10
1-(4-methylphenyl)ethane-1,2-diol: 118 mg of 4-methylstyrene yielded 149 mg diol as a
white solid (98%). Spectra were consistent with that reported in the literature.11

59
1-(4-chlorophenyl)ethane-1,2-diol: 139 mg of 4-chlorostyrene yielded 145 mg diol as a
white solid (84%). Spectra were consistent with that reported in the literature.11
1-(4-nitrophenyl)ethane-1,2-diol: 149 mg of 4-nitrostyrene yielded 175 mg diol as a
white solid (96%). Spectra were consistent with that reported in the literature.12
Oxidation of 4'-substituted phenylethane-1,2-diols: 0.1 mmol of the diol, 32 mg (0.3
mmol) benzoquinone, and 10.6 mg p-xylene (0.1 mmol, internal standard) were dissolved
in 0.63 mL CD3CN and 0.7 mL D2O. This solution was transferred to a tared NMR tube
containing 2.6 mg (5 µmol Pd) [(neocuproine)Pd(OAc)]2(OTf)2, the NMR tube capped
and shaken, and the reaction monitored by 1H NMR. The 3 ppm to 6 ppm window was
examined for the presence of the following compounds: diol (~3.8 ppm, 2 H),
hydroxyketone (~4.8 ppm, 2 H), and phenylglyoxal hydrate (~5.9 ppm, 1 H); these peaks
were integrated against the methyl peak of p-xylene (2.2 ppm). These chemical shifts are
based on spectra obtained of authentic samples for the corresponding unsubstituted
compounds; the products arising from oxidation of these four substituted diols are not
known in the literature. Over time, the mass balance based on these three peaks
decreases but that of the aromatic region remains roughly constant - this is presumably
because of the formation of the phenylgloyxalic acid product, which only has NMR peaks
in the aromatic region.

60
NMR screening of the oxidation of diols in Figure 3.2
A representative procedure is outlined for the oxidation of 1,2-propanediol. 1,2-
Propanediol (7.6 mg, 0.1 mmol) is carefully weighed into a vial containing p-xylene
(15.2 mg, 0.143 mmol). To this vial is added 0.7 mL of 9:1 v/v CD3CN:D2O. The
mixture is transferred to an NMR tube and an initial reference 1H NMR is taken. Into a
vial is weighed 2.6 mg [(neocuproine)Pd(OAc)]2(OTf)2 (2.5 µmol, 5 mol% Pd) and 32.4
mg benzoquinone (0.3 mmol). The NMR tube mixture is carefully transferred by pipette
to this vial, and the resulting brown mixture is then transferred back to the NMR tube. A
1H NMR is taken after 2 hours to determine product conversion and product yield. Yield
is determined by comparison to the p-xylene internal standard concentration. All
products had spectra consistent with that reported in the literature.
In the case of meso-erythritol (1,2,3,4-butanetetraol), the substrate was not soluble in 9:1
(v/v) CD3CN/D2O, so 0.7 mL dmso-d6 was used in place of CD3CN/D2O.
Representative procedure for the aerobic oxidation of diols and polyols on a 2 mmol
scale.
232 mg (2 mmol) cis-cyclohexanediol was dissolved in 9 mL CH3CN and 1 mL H2O.
The vial was sealed with a rubber septum, and the solution was placed under an O2-filled
balloon (prepared by affixing an O2-filled balloon onto one end of a disposable plastic
syringe, then affixing a 20-gauge needle on the other end, followed by piercing the
septum with the needle). The solution was bubbled with O2 for 20 minutes, then 104 mg
(0.2 mmol) [(neocuproine)Pd(OAc)]2(OTf)2 was added to the reaction solution, and the

61
septum quickly replaced. The reaction was monitored by TLC (EtOAc, KMnO4 stain) for
disappearance of alcohol (Rf = 0.48) and emergence of hydroxyketone (Rf = 0.73).
After the reaction is complete, the solution is evaporated to near-dryness with rotary
evaporation (bath temperature 50ºC), resulting in a viscous opaque brown oil. 10 mL
ethyl acetate is then added to this residue and this residue is triturated for 1 hour with
vigorous stirring. The now yellow supernatant is then filtered through a plug of silica gel
(5 g) and the product eluted with ethyl acetate. The 10 mL fractions containing product
were combined and evaporated to yield 215 mg of an pale yellow oil that gradually
solidified into a white solid (95%). Spectra of the hydroxyketone product were consistent
with that in the literature.17
1-hydroxy-2-butanone
180 mg of 1,2-butanediol, when subjected to the procedure outlined above, yielded a 4:1
mixture of the ketone and diol by 1H NMR. This was further purified by chromatography
(5 g SiO2, 2:1 hexanes:EtOAc to 1:1 hexanes:EtOAc) to obtain 134 mg of the
hydroxyketone product as a colorless oil (76%). Spectra of this compound were
consistent with that in the literature.13
1-methoxy-2-propanone (methoxyacetone)
180 mg 1-methoxy-2-propanol was subjected to the representative procedure outlined
above to obtain 130 mg of the ketone isolated as a colorless oil (74%). Spectra of this
compound were consistent with that in the literature.14

62
1,4-dihydroxy-2-butanone
212 mg 1,2,4-butanetriol was subjected to the representative procedure outlined above to
obtain 146 mg isolated as a colorless oil (71%). Spectra of this compound were consistent
with that in the literature.15
1,3,4-trihydroxy-2-butanone (erythrulose)
244 mg meso-erythritol was subjected to the representative procedure outlined above,
with the following modifications: 9 mL CH3CN and 2 mL H2O were used as the reaction
solvent, and the hydroxyketone product was extracted from the product mixture with 3 x
10 mL EtOAc. 156 mg erythrulose was isolated as a colorless oil (65%). Spectra of this
compound were consistent with that in the literature.15
2-hydroxycyclohexanone
232 mg cis-1,2-cyclohexanediol yielded 215 mg (95%) of the hydroxyketone as a white
solid. Oxidation of 232 mg trans-1,2-cyclohexanediol yields 211 mg (94%) as a white
solid. Spectra of the hydroxyketone product were consistent with that in the literature.17
Preparation of 4-tert-butylcyclohexene: This preparation was based on the procedure
by Sicher and coworkers;18 however, they did not specify reagent quantities or solvent
volumes, so the procedure is outlined below. Sicher's method for preparing the mesylate
intermediate was also much lower-yielding, so it was prepared by a different and more
convenient method.19

63
4-tert-butylcyclohexanol mesylate: 1.56 g 4-tert-butylcyclohexanol (2.3:1 cis/trans
mixture) was dissolved in 50 mL dichloromethane and cooled to 0ºC. Triethylamine (2.1
mL, 15 mmol) was added in one portion followed by dropwise addition of
methanesulfonyl chloride (0.85 mL, 11 mmol) via an additional funnel. The solution was
stirred at 0ºC for one hour; a white precipitate came out of solution. The solution was
quenched by slow addition of 1 mL water, and then 30 mL water was added to the
solution; the biphasic mixture was transferred to a separatory funnel where the aqueous
layer was separated and discarded. The dichloromethane layer was washed with 1 x 30
mL 10% aqueous HCl, 1 x 30 mL water, and 1 x 30 mL saturated aqueous sodium
bicarbonate. The organic layer was dried over magnesium sulfate, filtered, and the
solvent evaporated to obtain 2.31 g of a slightly yellow oil that solidified into a white,
crystalline solid (99%) that was used without further purification. Spectra of this
compound were consistent with that found in the literature.20
4-tert-butylcyclohexene
4-tert-butylcyclohexanol mesylate (3.0 g, 12.8 mmol) was dissolved in 15 mL collidine
and refluxed for 2 hours (150ºC bath temperature, exothermic reaction!). The solution
turned orange-brown and a brown oil separated out. After cooling to room temperature,
the solution was poured into 25 mL 6 M aqueous HCl and stirred until the exothermic
reaction subsided. 20 mL pentane was added to the cool soution and the solution
transferred to a separatory funnel (the flask was washed with an additional 20 mL
pentane and added to the funnel). The pentane layer was separated and the aqueous
solution was extracted with 3 x 40 mL pentane. The pentane extracts were washed with 1

64
x 40 mL 10% aqueous HCl, 1 x 40 mL water, and 1 x 40 mL saturated aqueous sodium
bicarbonate. The pentane extracts were dried with magnesium sulfate, filtered, and the
solvent evaporated. 1.6 g of a clear, colorless oil was obtained (91%), which was
combined with two other runs and distilled under house vacuum (b.p. 80ºC / ~ 40 mm
Hg). Spectra of this compound were consistent with that reported in the literature.20
Preparation of 3-cis-4-trans-1-tert-butylcyclohexanediol (diaxial diol):
30% aqueous hydrogen peroxide (0.42 mL, 3.7 mmol) and 2 mL 95% aqueous formic
acid were combined in a 20 mL scintillation vial, and immersed in an ambient
temperature water bath. Under vigorous stirring, 415 mg 4-tert-butylcyclohexene (3
mmol) was added to the solution in ten 50 µL portions over 30 minutes, a new portion
being added once most of the previous portion had come into solution. After addition
was complete, the solution was heated to 45ºC for 1 hour with stirring, then allowed to
stir at room temperature overnight. 1 mL of concentrated aqueous NaOH (4 g in 7.5 mL
water) was then added to the reaction solution (exothermic). The aqueous solution was
heated to 45ºC, and 2 mL ethyl acetate was added to the warm solution. After stirring
vigorously to mix the layers, the ethyl acetate layer was drawn off with a pipet. This was
repeated six more times, each with 2 mL ethyl acetate. The extracts were dried (sodium
sulfate), filtered, and evaporated to yield a clear oil that was chromatographed on silica
gel (30 g, 10:1 hexanes:EtOAc) to yield 316 mg of a colorless oil that solidified on
standing (61% yield). Rf = 0.3 (10:1 hexanes:EtOAc). Spectra were consistent with that
reported in the literature.21

65
Preparation of a mixture of 3-cis-4-cis-1-tert-butylcyclohexanediol and 3-trans-4-
trans-1-tert-butylcyclohexanediol:
4.2 g AD-mix α (3 mmol) and 285 mg methanesulfonamide (3 mmol) was added to 15
mL tert-butyl alcohol and 15 mL water, and stirred until completely dissolved. 4-tert-
butylcyclohexene (415 mg, 3 mmol) was then added to the biphasic yellow solution, and
the solution stirred vigorously for 18 hours. Solid sodium sulfite (4.5 g, 48 mmol) was
added to the now heterogeneous solution and the reaction stirred for 1 hour longer. The
product was extracted from the now gray solution with 4 x 15 mL EtOAc. The extracts
were washed with 3 x 10 mL 1 M aqueous NaOH to remove the sulfonamide, and the
organic extracts dried over MgSO4, filtered, and evaporated. The cinchona ligand was
removed by chromatography (20 g SiO2, EtOAc) to yield a white solid (400 mg, 77%
yield) containing a mixture of both diols that were inseparable by chromatography.
The diastereomers (400 mg, 2.3 mmol) were converted to their benzoate diesters with
980 mg benzoyl chloride (6.96 mmol) and 10 mg 4-(dimethylamino)pyridine in 8 mL
pyridine. After stirring overnight at room temperature, 10 mL 6 M aqueous HCl was
added, and, after the exotherm subsided, 10 mL dichloromethane was added and the
contents poured into a separatory funnel. The aqueous solution was extracted with 3 x 10
mL dichloromethane, and the extracts washed with 1 x 20 mL 10% aqueous HCl, 1 x 20
mL water, and 1 x 20 mL saturated aqueous bicarbonate. The extracts were dried
(magnesium sulfate), filtered, and the solvent evaporated to yield a white solid that was
chromatographed on silica (30 g, 20:1 hexanes:EtOAc). The diester so obtained (489 mg,

66
56%) also could not be separated by chromatography; 1H NMR shows a roughly 1:1
mixture of both diastereomers.
Preparation of 1-cis-2-cis-3-methylcyclohexanediol and 1-cis-2-trans-3-
methylcyclohexanediol.
This preparation was based on a textbook preparation for sodium borohydride reduction
of alcohols.23
1 g 3-methyl-1,2-cyclohexanedione (7.9 mmol) was dissolved in 10 mL methanol at 0ºC,
and a solution of 400 mg sodium borohydride (10.5 mmol) in 2 mL 2 M aqueous NaOH,
also cooled to 0ºC, was added in 200 µL portions to the solution. After addition was
complete, the solution was warmed to room temperature, and vigorously stirred overnight
at that temperature. After the reaction was complete, 5 mL water was added to the
viscous solution and the product extracted with 3 x 5 mL ethyl acetate. The extracts were
washed with 1 x 5 mL brine, dried over magnesium sulfate, filtered, and the solvent
evaporated. The two diol products were purified by column chromatography (50 g SiO2,
3:1 hexanes:ethyl acetate -> 1:1 hexanes:ethyl acetate) to obtain the diol products as
colorless oils. The cis,cis isomer was the less polar isomer (Rf = 0.4, 1:1 hexanes:ethyl
acetate), while the cis,trans isomer was more polar (Rf = 0.25, 1:1 hexanes:ethyl acetate).
Spectra were consistent with that reported in the literature.22

67
Oxidation of cyclohexanediol substrates: representative procedure
cis, cis-3-methyl-1,2-cyclohexanediol (13 mg, 0.1 mmol) was carefully weighed into a
vial, and 15.2 mg p-xylene (0.143 mmol) and 32.4 mg benzoquinone (0.3 mmol) were
added. These compounds were dissolved in 0.7 mL of 9:1 v/v CD3CN:D2O. This
solution was then transferred to a tared NMR tube containing 2.6 mg
[(neocuproine)Pd(OAc)]2(OTf)2 (2.5 µmol, 5 mol% Pd). The reaction was monitored by
1H NMR after 3 hours to determine product conversion and product yield.
The hydroxyketone products (1-hydroxy-3-methyl-2-cyclohexanone, A, and 2-hydroxy-
3-methyl-1-cyclohexanone, B) are not known in the literature, and no definitive
assignments could be made for either product. When the trans-diol substrate was
subjected to the procedure above, two products emerged in a roughly 1:1 ratio (based on
the peak corresponding to the methyl group for each compound). In contrast, when the
cis-diol substrate was oxidized, one product was strongly favored in a ~10:1 ratio, the
major product being tentatively assigned as A based on the relative chemical shift of the
methyl peaks for the two hydroxyketone products.
An NMR of the two reactions can be found in the Appendix. Page 91 contains the 1H
NMR spectrum from oxidation of the trans-diol to give a mixture of both A and B. Page
92 contains the 1H NMR spectrum from oxidation of the cis-diol to give predominantly
one product, tentatively assigned as A.

68
3.7 References
(1) Arterburn, J. B. Tetrahedron 2001, 57, 9765.
(2) Painter, R. M., Pearson, D. M., Waymouth, R. M. Angew. Chem. Int. Ed. 2010, in
press.
(3) Pagliaro, M.; Ciriminna, R.; Kimura, H.; Rossi, M.; Della Pina, C. Angew. Chem.
Int. Ed. 2007, 46, 4434.
(4) Hu, W. B.; Knight, D.; Lowry, B.; Varma, A. Ind Eng Chem Res 2010, 49, 10876.
(5) ten Brink, G. J.; Arends, I. W. C. E.; Hoogenraad, M.; Verspui, G.; Sheldon, R. A.
Adv. Synth. Catal. 2003, 345, 497.
(6) Maki, T.; Iikawa, S.; Mogami, G.; Harasawa, H.; Matsumura, Y.; Onomura, O.
Chem. Eur. J. 2009, 15, 5364.
(7) Schreiber, J.; Eschenmoser, A. Helv. Chim. Acta. 1955, 38, 1529.
(8) Trost, B. M.; Masuyama, Y. Tetrahedron Lett. 1984, 25, 173.
(9) Conley, N. R.; Labios, L. A.; Pearson, D. M.; McCrory, C.; Chidsey, C. E. D.;
Waymouth, R. M. Organometallics 2007, 26, 5447.
(10) Junttila, M. H.; Hormi, O. E. O. J. Org. Chem. 2007, 72, 2956.
(11) Griffith, J. C.; Jones, K. M.; Picon, S.; Rawling, M. J.; Kariuki, B. M.; Campbell,
M.; Tomkinson, N. C. O. J. Am. Chem. Soc. 2010, 132, 14409.
(12) Kim, J.; De Castro, K. A.; Lim, M.; Rhee, H. Tetrahedron 2010, 66, 3995.
(13) Matsumoto, T.; Ohishi, M.; Inoue, S. J. Org. Chem. 1985, 50, 603.
(14) Wang, X. L.; Liu, R. H.; Jin, Y.; Liang, X. M. Chem. Eur. J. 2008, 14, 2679.
(15) Kaptein, B.; Barf, G.; Kellogg, R. M.; Vanbolhuis, F. J. Org. Chem. 1990, 55,
1890.

69
(16) Simonov, A. N.; Matvienko, L. G.; Pestunova, O. P.; Parmon, V. N.;
Komandrova, N. A.; Denisenko, V. A.; Vas'kovskii, V. E. Kinetics and Catalysis
2007, 48, 550.
(17) Zhang, W.; Shi, M. Chem. Commun. 2006, 1218.
(18) Sicher, J.; Sipos, F.; Tichy, M. Collect. Czech. Chem. Commun. 1961, 26, 847.
(19) Crosslan.Rk; Servis, K. L. J. Org. Chem. 1970, 35, 3195.
(20) Aciro, C.; Claridge, T. D. W.; Davies, S. G.; Roberts, P. M.; Russell, A. J.;
Thomson, J. E. Org. Biomol. Chem. 2008, 6, 3751.
(21) Trainor, R. W.; Deacon, G. B.; Jackson, W. R.; Giunta, N. Aust. J. Chem. 1992,
45, 1265.
(22) Ziffer, H.; Seeman, J. I.; Highet, R. J.; Sokoloski J. Org. Chem. 1974, 39, 3698.
(23) Vogel, A. I.; Furniss, B. S. Vogel's Textbook of Practical Organic Chemistry, 5th
ed. Longman Scientific & Technical, Wiley: London, New York, 1989.
(24) Still, W. C.; Kahn, M.; Mitra, A. J. Org. Chem. 1978, 43, 2923.
(25) Sharpless, K. B., Amberg, W., Bennani, Y. L., Crispino, G. A., Hartung, J., Jeong,
K.-S., Kwong, H.-L., Morikawa, K., Wang, Z.-M., Xu, D., Zhang, X.-L. J. Org.
Chem. 1992, 57, 2768.

70
Chapter 4
The electrocatalytic reduction of dioxygen using dinuclear copper complexes
4.0 Preface
This chapter describes research done by Charles C. L. McCrory and me. I prepared all of
the ligands and the complexes studied in this chapter and did the preliminary
electrochemical screening of all three ligand systems reported here with the assistance of
C. C. L. McCrory. C. C. L. McCrory did more detailed characterization of the
electrochemistry for the 3,5-di(2-pyridyl)-pyrazole ligand system himself, including the
Koutecky-Levich analysis described later in the chapter.
4.1 Introduction
The four-electron reduction of dioxygen to water is a fundamental reaction in biological
systems and low temperature fuel cell devices.1-3 The standard redox potential of
dioxygen at 1.23 V showcases its thermodynamic potency as a terminal oxidant in fuel
cells. However, limitations relating to reaction inefficiency, voltage losses, and poor
mass transfer of O2 to the electrode results in at least a 30% loss in cell efficiency at the
cathode. Because of these difficulties, the development of a system that is able to rapidly
and efficiently reduce dioxygen at the cathode remains a significant and unsolved
problem.

71
State-of-the-art fuel cells routinely employ platinum nanoparticulate electrodes, and
while they are effective catalysts for the reduction of oxygen, they require an
overpotential of at least 350 mV to operate at acceptable current densities.3 Furthermore,
less than 10% of the total platinum metal of a given electrode is catalytically active at the
surface of the electrode. Because of the high cost of this precious metal coupled with the
necessity for operating the cathode at a relatively large overpotential, a more effective
solution to the dioxygen reduction problem needs to be developed.
Heller and coworkers have shown that fungal laccase enzymes, when tethered to the
surface of a graphite electrode, are able to reduce dioxygen to water at an overpotential of
only 70 mV at high current densities.4-6 These enzymes, which employ a trinuclear
copper active site, operate at a turnover frequency of 2.1 dioxygen molecules reduced per
laccase enzyme per second. This closely resembles the turnover frequency for platinum
electrodes (2.5 O2 molecules reduced per surface Pt atom per second), but the former
electrode operates at one-fifth of the overpotential for the latter electrode.3 Synthetic
mononuclear copper phenanthroline complexes adsorbed on the surface of a graphite
surface have demonstrated promising catalytic activity with a high turnover frequency of
10 O2 molecules per Cu catalyst per second, but unfortunately, this reaction also operates
at high overpotentials (1.1 V).7-10 Given the efficiency of laccase enzymes at dioxygen
reduction, synthetic dinuclear dicopper complexes are a viable next step towards solving
the problem of rapid dioxygen reduction at low overpotentials.

72
4.2 Towards a dicopper electrocatalyst
Chidsey and coworkers has shown that mononuclear phenanthroline copper complexes
are competent oxygen reduction electrocatalysts.11 The redox potential at this
electrocatalytic reduction takes place is quite sensitive to the nature of the ligand; adding
electron-withdrawing groups to the ligand or sterically demanding groups ortho to the 2/9
positions of the ligand allows the reaction to be operated at more positive potentials (200-
300 mV vs. NHE, compared to 10 mV for phenanthroline). However, due to very
unfavorable and inefficient binding of dioxygen to these copper complexes, the reaction
rate for dioxygen reduction drops off significantly relative to the unsubstituted
phenanthroline copper complex (0.4-2 s-1 compared to 16 s-1).
Scheme 4.1. Proposed dinuclear copper electrocatalysts.
Thus, we envisioned that the proposed dicopper complexes in Scheme 2.1 would be
efficient electrocatalysts for dioxygen reduction because of the unique binding
environment that dinucleating ligands offer. We propose that using dinuclear copper
complexes would be more effective electrocatalysts to this end for the following reasons:
(1) doubling the number of CuI sites would facilitate dioxygen binding to the complex;
(2) the adjacent, uncoordinated CuI center would help to stabilize the reduced CuI
peroxide radical species by binding to it; and (3) the presence of two CuI centers that both
N N N N
Cu Cu
AcO OAcO O
N N N
RN
N
Cu Cu
AcO OAcO O
N N
S S
N NCu CuOH
Cl Cl
Cl

73
bind to the reduced dioxygen ligand would facilitate electron transfer to effect further
reduction of dioxygen to water.
With these features in mind, the proposed mechanism for dicopper-mediated dioxygen
reduction is shown in Scheme 4.2. The bis-CuII complex is reduced by two electrons to a
bis-CuI intermediate, which then reversibly binds to dioxygen. After oxygen binds to one
of the CuI centers in the catalyst, a single-electron reduction of dioxygen takes place to
generate a CuII superoxide radical. The superoxide radical can then immediately bind to
the adjacent CuI center and be reduced to a bis-CuII peroxide species.
Scheme 4.2. Proposed catalytic cycle for the reduction of dioxygen.
The structure of this peroxide interemediate deserves some comment. Crystal structures
of known 3,5-di(2-pyridyl)pyrazolato dicopper(II) complexes reveal that the two copper
N N N N
CuI CuI
H2O OH2
N N N N
CuI CuI
H2O OH2H2OO2
N N N N
CuII CuI
H2O OH2H2OO
O
OH2
N N N N
CuII CuII
H2O OH2O O
N N N N
CuI CuI
H2O OH2OH
OH
N N N N
CuII CuII
H2O OH2OH2H2O
H2O
2 H+
2 e-
2 e-
H2O
3+

74
centers are approximately 4.05 Å apart.12-13 Known dicopper complexes bearing a "end-
on" peroxo ligand have an average Cu-Cu distance of 4.36 Å, while those that have an
"side-on" peroxo ligand have an average Cu-Cu distance of 3.51 Å.1 Thus, we propose
that the peroxo ligand binds to the dicopper complex in an "end-on" fashion since that is
a more geometrically accessible intermediate.
In any event, the bis-CuII peroxide intermediate can go through one of two pathways: (1)
the peroxo ligand is protolyzed and released from the CuI complex as hydrogen peroxide,
or (2) the peroxo ligand undergoes two-electron reduction and subsequent protolysis to
form the bis-CuII aquo complex. In either pathway, the initial bis-CuII aquo complex is
regenerated, closing the catalytic cycle.
4.3. The 3,5-di(2-pyridyl)pyrazole ligand system
We began this study by investigating the synthesis of dicopper complexes employing the
3,5-di(2-pyridyl)-pyrazole (dppy) ligand. A variety of homobimetallic and
heterobimetallic complexes are known for this ligand system, including dicopper
complexes.14 Particularly relevant to the preceding discussion for the reduction of
dioxygen is Llobet's report of a bis-Ru complex being able to oxidize water, the reverse
reaction (Scheme 4.3).15
Scheme 4.3. The oxidation of water to dioxygen with a dinuclear Ru complex.
N N N NRu Ru
L L
L = terpyridine
3+
OH2 H2O
-4 H+
-4 e-
N N N NRu Ru
L L
3+
O O
1.23 V
2 H2O O2
N N N NRu Ru
L L
3+
OH2 H2O

75
He proposes that both RuII aquo centers are oxidized to RuIV-oxos, and that, when the
final electron for the four-electron process is removed at 1.23 V vs. SCE, a synergistic
phenomenon takes place with expulsion of both oxo ligands on Ru as dioxygen. Key
evidence for this hypothesis is the fact that this final electron-transfer step (with oxygen
evolution) is first-order in catalyst; in addition, independent synthesis of an unlabeled bis-
Ru aquo complex and its electrolysis in 18O-labeled water produces 16O2 as 99.5% of the
isotopic mixture.16-17 A common feature that delineates bimetallic dppy complexes is
that a bridging ligand readily coordinates to both metal centers - this is central to our
hypothesis that the bound dioxygen will bridge both Cu centers as a superoxide, enabling
rapid reduction of this substrate to hydrogen peroxide or water.
Scheme 4.4. Synthesis of the 3,5-di(2-pyridyl)pyrazole ligand.
The dppy ligand is readily synthesized in two steps from the cross-condensation of
methyl 2-picolinate and 2-acetylpyridine in toluene and subsequent cyclization of the 1,3-
diketone with hydrazine hydrate in hot benzene (Scheme 4.4).18 We were able to prepare
the dicopper complex by reaction of the ligand with Cu(OAc)2 in ethanol, as reported by
Pons and coworkers,19 though the paramagnetic nature of the bis-CuII complex defied
ready characterization. Elemental analysis of the complex suggests that the compound
synthesized is (dppy)Cu2(OAc)3*H2O, and based on known dppy copper complexes, we
propose that one acetate ligand bridges both Cu centers.
N N NH N
H2NNH2
benzene
refluxN
O O
N
NaOEt
tolueneN
O
+
N
O
OCH3

76
Figure 4.1. Cyclic voltammogram of (dppy)Cu(OAc)3 at pH 4.7.
Figure 4.2. Dependences of the peak current on scan rate for (dppy)Cu(OAc)3 at (a) its
redox potential at -170 mV and (b) its electrocatalytic O2 reduction peak with maximum
current at -25 mV.

77
The electrochemistry of the dicopper complex adsorbed on an edge-plane graphite
surface is shown in Figure 4.1 (acetate buffer, pH 4.7, using Bu4NClO4 as electrolyte).
When the aqueous solution is saturated with N2, the complex demonstrates a single,
reversible, two- electron peak at -170 mV vs. NHE (Figure 4.2a). This suggests that,
once one CuII center is reduced, the other CuII is then rapidly reduced - hence, the redox
peaks for each of the CuII centers are not well-resolved. The peak current at this potential
is linear with the scan rate, confirming that the species is adsorbed onto the surface.20
Switching the N2-saturated buffer solution to an air-saturated solution effects a new broad
peak with its onset at 170 mV and a maximum current at -25 mV - this peak appears to
represent the electrocatalytic reduction of dioxygen. The peak current at -25 mV varies
with the square root of the scan rate, indicating that a diffusing species is being reduced.20
To better characterize the electrocatalytic reduction taking place, a rotating-disk electrode
was used to more precisely control the mass transport rate of oxygen diffusing to the
surface of an electrode. By varying the rotation rate of the electrode and scanning the
electrode at given rotation rates, a sigmoidal curve can be obtained that represents the
steady-state current at that rotation rate if oxygen binding to the dicopper complex at the
surface of the electrode is rate-limiting. Plotting the steady-state current at a given
potential (-650 mV) as a function of the square root of the rotation rate should then give a
straight line, with the slope being proportional to the number of electrons in the reduction
and the intercept being the current that would arise in the absence of diffusion.20

78
Figure 4.3. (a) Voltammograms of (dppy)Cu(OAc)3 with varying rotation rates for the
rotating disk electrode; (b) Plot of the current at -650 mV as a function of the square root
of the rotation rate for the disk electrode. Dashed lines are theoretical lines representing a
2-electron and 4-electron process (labeled "2 e-" and "4 e-", respectively).
By using the Koutecky-Levich equation,20 the slope of the line so obtained can be
compared to theoretical values. Analyzing such a plot shows that the electrocatalytic
dioxygen reduction reaction more closely matches the theoretical curve for a four-
electron reduction than that for a two-electron reduction. This suggests that the dicopper
complex catalyzes the reduction of oxygen directly to water instead of stopping at the
hydrogen peroxide stage.
We also examined the effect of substituted dppy ligands for dioxygen reduction.
Nitration of the dppy ligand under standard conditions (HNO3/H2SO4, 55ºC) selectively
nitrated the pyrazole ring at the 4-position with no evidence of nitration on either pyridine
ring. Reduction of the nitro group under several conditions (SnCl2/aq. HCl; H2, Pd/C,
EtOAc) only led to decomposition of the ligand, so preparation of the aminopyrazole was
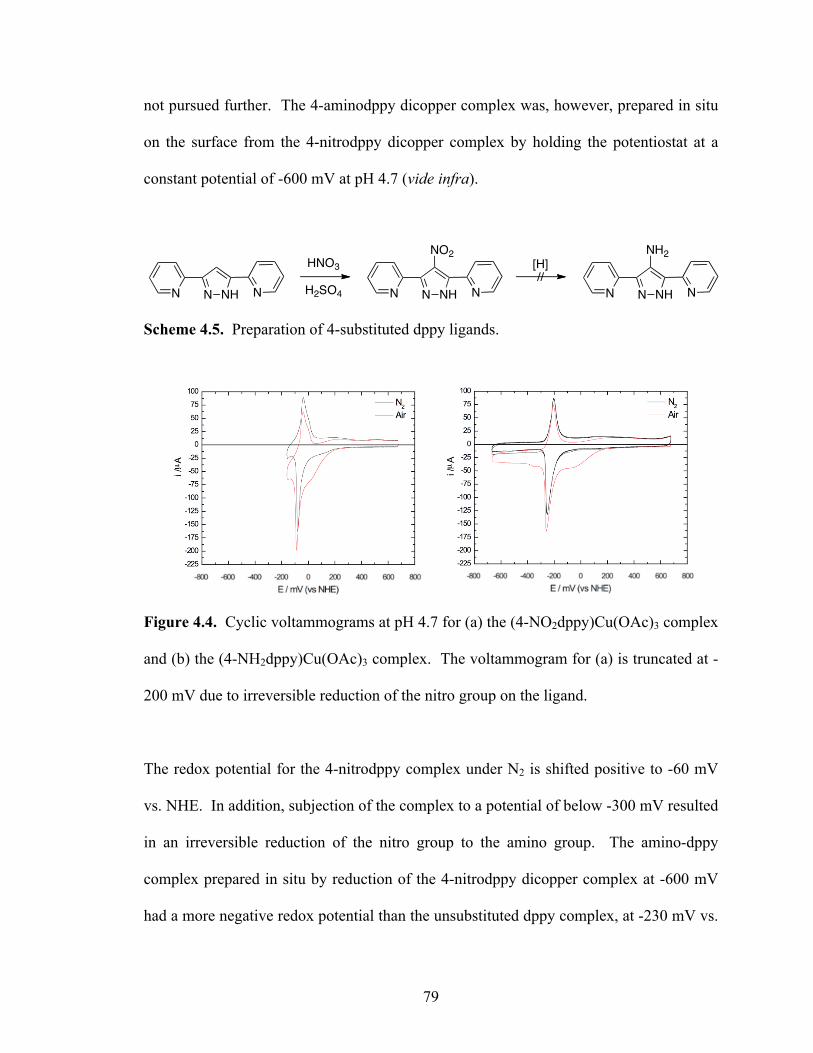
79
not pursued further. The 4-aminodppy dicopper complex was, however, prepared in situ
on the surface from the 4-nitrodppy dicopper complex by holding the potentiostat at a
constant potential of -600 mV at pH 4.7 (vide infra).
Scheme 4.5. Preparation of 4-substituted dppy ligands.
Figure 4.4. Cyclic voltammograms at pH 4.7 for (a) the (4-NO2dppy)Cu(OAc)3 complex
and (b) the (4-NH2dppy)Cu(OAc)3 complex. The voltammogram for (a) is truncated at -
200 mV due to irreversible reduction of the nitro group on the ligand.
The redox potential for the 4-nitrodppy complex under N2 is shifted positive to -60 mV
vs. NHE. In addition, subjection of the complex to a potential of below -300 mV resulted
in an irreversible reduction of the nitro group to the amino group. The amino-dppy
complex prepared in situ by reduction of the 4-nitrodppy dicopper complex at -600 mV
had a more negative redox potential than the unsubstituted dppy complex, at -230 mV vs.
N N NH N
HNO3
H2SO4 N N NH N
NO2
N N NH N
NH2[H]//

80
NHE. What is particularly striking about these three complexes is that, despite the
dependence of the complex's redox potential on the nature of the ligand, there is
practically no difference in the onset potential for dioxygen reduction (~160-170 mV vs.
NHE) or redox potential where there is a maximum current (~ -25 mV vs. NHE). It is not
clear why there is a dependence of the ligand on the redox potential, but not for the
potential of the electrocatalytic reaction - it is possible, however, that the perturbation of
the electronics of the ligand does not significantly affect the rate of binding for oxygen to
the dicopper complex.
There are two directions that can be taken with the dinuclear ligand system. First, it
would be worthwhile to compare the redox potential of the dppy dicopper complex
against dppy ligands that are more sterically demanding. For instance, the 2,9-
dimethylphenanthroline copper complex shows a positive shift of 280 mV for the
electrocatalytic reaction relative to the phenanthroline copper complex but with a
concomitant 10-fold drop in reaction rate.11 However, the six-step synthesis of the
analogous dimethyl dppy complex from 2,6-lutidine (since 6-methyl-2-picolinic acid is
not commercially available)19 was extremely troublesome and ultimately failed to give a
pure sample of the desired ligand for electrocatalytic studies. Second, the negative redox
potentials for the dppy copper complexes could be due to the anionic nature of the
pyrazolate ring, so neutral dinucleating ligands should shift the redox potential to more
positive potentials. To this end, two neutral ligand systems were investigated for the
electrocatalytic reduction of dioxygen.

81
4.4. A 3,6-di(2-pyridylthio)-pyrazine dicopper complex
Thompson has reported neutral pyridazine dicopper complexes with a more favorable
redox potential of 0.6 V vs. NHE.21-22 Little is known about these complexes' reactivity
towards dioxygen though it is a competent and stable catalyst for the aerobic oxidation of
3,5-di-tert-butylcatechol to the orthoquinone product. This ligand system is also
attractive because the ligand can easily be prepared in one step by nucleophilic aromatic
displacement with various thiols on 3,6-dichloropyrazine. The modular nature of this
preparation could then give rise to a library of ligands from a variety of 2-
mercaptopyridine compounds for screening towards the electrocatalytic reduction of
dioxygen. However, in our hands, the preparation of Thompson's reported copper
complex was not reproducible, and the in-situ preparation of the copper complex on the
surface of the electrode failed to give satisfactory results, likely due to the ligand's very
poor solubility in polar solvents. Thus, the investigation of this ligand system was
abandoned.
Scheme 4.6. Preparation of 3,6-di(2-pyridylthio)pyrazine copper tetrachloride
4.5. Some 3,5-di(2-pyridyl)-1,2,4-triazole ligand systems
The second neutral ligand system investigated in this project is a triazole-based ligand
reported by Brooker and coworkers,23-29 but no copper complexes are known employing
this ligand. This ligand is accessible in three steps from 2-cyanopyridine; the
N N
S S
N NCu CuClCl Cl
ClN N
S S
N N
N N
ClCl
N SH
+
EtOH
reflux
CuCl2
EtOH

82
intermediate tetrazine-based ligands were also screened.30 While all of the ligands were
readily prepared and adsorbed well on the graphite surface, none of the copper complexes
showed promising electrochemistry. The CVs of each of the four copper acetate
complexes were complex, indicating poorly-defined coordination chemistry, and had
peaks at negative potentials (-200 mV vs. NHE). The complexes are able to reduce
dioxygen, but consistently did so at very negative potentials (-200 mV to -400 mV vs.
NHE).
Scheme 4.7. Preparation of 3,5-di(2-pyridyl)-1,2,4-triazole.
Figure 4.5. Cyclic voltammograms of a copper 3,6-dipyridyl-1,2,5,6-dihydrotetrazine
complex using (a) N2 saturated solution; (b) air-saturated solution.
N CN
H2NNH2
reflux
N
HN N
NH
NN
NaNO2
HOAc
N
N N
N
NN
aq. HCl!
N N N
N
N
NH2NaNO2
HNO3 N N N
HN
N

83
Figure 4.6. Cyclic voltammograms of a copper 3,6-dipyridyl-1,2,5,6-tetrazine complex
using an (a) N2-saturated solution; (b) air-saturated solution.
Figure 4.7. Cyclic voltammograms of a copper 4-amino-3,5-dipyridyl-1,2,4-triazole
complex using an (a) N2-saturated solution; (b) air-saturated solution.

84
Figure 4.8. Cyclic voltammograms of a copper 3,5-dipyridyl-1,2,4-triazole complex
using an (a) N2-saturated solution; (b) air-saturated solution.
To minimize the issues associated with multiple possible coordination environments for
the dicopper triazole complexes, the nitrogen atom at the 4-position of the triazole ring
needs to be alkylated. However, the triazole ring cannot be regioselectively N-alkylated
at the 4-position based on literature precedent; alkylation at the undesired 2 position of
the triazole ring is strongly favored.31 We took on two approaches to circumvent this
issue. First, reaction of 2,6-dipyridyl-4-amino-1,2,4-triazole with 2,5-
dimethoxytetrahydrofuran would produce 2,6-dipyridyl-4-pyrrolyl-1,2,4-triazole, a
known compound; however, the reported preparation could not be reproduced in our
hands. Second, a longer synthetic route to prepare the 2,6-dipyridyl-4-methyl-1,2,4-
triazole in 4 steps from 2-picoline and methyl 2-picolinate24 proceeded smoothly until the
final condensation step, which produced an intractable reaction mixture. Given the
difficulty in the preparation of these heterocycles, this ligand system was also abandoned.

85
Scheme 4.8. Preparation of 3,5-dipyridyl-1,2,4-triazole derivatives.
4.6 Conclusions and future directions
We have synthesized a number of dinucleating ligands and formed the corresponding
dicopper complexes, and characterized their ability to electrocatalytically reduce
dioxygen at the surface of a graphite electrode. The dipyridylpyrazole copper complex is
the most promising of the dicopper systems investigated since it reduces dioxygen at the
most positive potentials of all of the ligand systems examined. This system needs to be
further studied to better understand the role of both copper centers in the reduction
reaction since it is not clear how this dicopper complex is an improvement over other
mononuclear copper complexes previously studied.
To this end, several useful comparisons can be made with the dipyridylpyrazole system.
First, if a 4-substituted dipyridyltriazole system becomes more synthetically accessible
N N N
N
N
NH2
OMeO OMe
dioxane/AcOH, !
N N N
N
N
N
N
S8, Na2S
reflux N
S
NHMe
NaOEt, EtOH
then EtBr, 50ºC N
S
NHMe
N
O
OMe
H2NNH2
! N
O
NHNH2
n-BuOH
!N
S
NHMeN
O
NHNH2+
N N N
N
N
Me

86
with well-behaved copper coordination chemistry, then a direct comparison of these two
ligand systems can be made, since the dipyridyltriazole copper complex should be
reduced at a more positive potential than the dppy copper complex. Second, the
preparation of more sterically demanding dppy ligands would serve as an useful probe for
examining the binding of dioxygen to the dicopper complex, as Chidsey has done with
his studies of the electrochemistry for phenanthroline copper complexes.11 Third, it
would be useful to directly compare the dipyridylpyrazole ligand system with a
monopyridylpyrazole ligand, the latter of which should form a monocopper complex.
This comparison would be the most direct of the three since it explicitly evaluates
whether a dinuclear copper complex is an improvement in terms of reaction rate and/or
overpotentials over a mononuclear copper complex with a similar ligand environment.
4.7. Experimental considerations: ligand syntheses
All starting materials and solvents were obtained commercially and used without further
purification unless otherwise specified. The ligands were prepared as previously
reported: 3,5-di(2-pyridyl)pyrazole33, 4-nitro-3,5-di(2-pyridyl)pyrazole34, and 3,6-di(2-
pyridylthio)pyrazine.21
3,5-di(2-pyridyl)pyrazolatodicopper(II) triacetate hydrate was prepared identically as
reported.19
Elemental analysis: calc'd for C19H21Cu2N4O7: C, 41.91; H, 3.89; N, 10.29, found: C,
41.86; H, 3.77; N, 10.84. An alternative formula corresponds to Cu(dppy)(OAc)2(OH),
calc'd for C17H17Cu2N4O5: C, 42.15; H, 3.54; N, 11.57.

87
Complexation of 3,6-(2-pyridylthio)pyrazine with two equivalents of CuCl2*2 H2O in
ethanol yielded a green solid that was filtered, and then precipitated from ethanol/ether
and dried in vacuo.
This product had the following elemental analysis data: calc'd for C14H10Cl4Cu2N4S2: C,
29.64; H, 1.78; N, 9.88, found: C, 25.39; H, 1.80; N, 8.31. Note that Thompson's
reported analysis results is also inconsistent with his reported synthesis: (C, 32.30; H,
2.88; N, 9.42).21
4.8. Experimental considerations: electrochemical
measurements
All electrochemical measurements were identical to that reported in Chidsey's studies of
mononuclear copper phenanthroline complexes,11 and their experimental considerations
are reproduced here:
Edge-plane graphite disk electrodes with a 0.195 cm2 macroscopic surface area
purchased from Pine Instrument Company were used. The electrodes were
ground by hand with a 600-grit silicon carbide paper followed by sonication in
pure water before each ligand deposition. Ligands were adsorbed onto the
electrode surface by exposing the surface to 1 mM acetonitrile solutions; the
ligands were complexed with CuII by exposure to 1 M aqueous Cu(NO3)2
solutions and then rinsing the electrode with water. Electrochemical
measurements were recorded with a Pine Instrument Company AFCBP1
bipotentiostat with a MSR rotator, an auxilary Pt-mesh electrode, and a

88
Ag/AgCl/NaCl(saturated) reference electrode. The reference was calibrated
against Cu(NO3)2 in 1 M KBr and all reported values are referenced to NHE.
All electrochemical measurements were conducted in an aqueous acetate buffer (1:1
glacial acetic acid/sodium acetate, each at 0.04 M concentration). In addition, sodium
perchlorate was used as the electrolyte, and was present in the buffered solution at 0.1 M
concentration. Prior to recording electrochemical measurements, the solution was purged
with either air or N2 for at least 20 minutes, and for the latter case, a blanket of N2 was
maintained throughout the measurement. Cyclic voltammograms were recorded with a
scan rate of 100 mV/s.
4.9. References
(1) Mirica, L. M.; Ottenwaelder, X.; Stack, T. D. P. Chem. Rev. 2004, 104, 1013.
(2) Carrette, L.; Friedrich, K. A.; Stimming, U. Chemphyschem 2000, 1, 162.
(3) Ralph, T. R. H., M.P. Platinum Metals Review 2002, 46, 3.
(4) Barton, S. C.; Kim, H. H.; Binyamin, G.; Zhang, Y. C.; Heller, A. J. Am. Chem.
Soc. 2001, 123, 5802.
(5) Barton, S. C.; Kim, H. H.; Binyamin, G.; Zhang, Y. C.; Heller, A. J. Phys. Chem.
B 2001, 105, 11917.
(6) Soukharev, V.; Mano, N.; Heller, A. J. Am. Chem. Soc. 2004, 126, 8368.
(7) Lei, Y. B.; Anson, F. C. Inorg. Chem. 1994, 33, 5003.
(8) Lei, Y. B.; Anson, F. C. Inorg. Chem. 1995, 34, 1083.
(9) Zhang, J. J.; Anson, F. C. J. Electroanal. Chem. 1992, 341, 323.

89
(10) Zhang, J. J.; Anson, F. C. Electrochim. Acta. 1993, 38, 2423.
(11) McCrory, C. C. L.; Ottenwaelder, X.; Stack, T. D. P.; Chidsey, C. E. D. J. Phys.
Chem. A 2007, 111, 12641.
(12) Pons, J.; Lopez, X.; Casabo, J.; Teixidor, F.; Caubet, A.; Rius, J.; Miravitlles, C.
Inorg. Chim. Acta. 1992, 195, 61.
(13) Munakata, M.; Wu, L. P.; Yamamoto, M.; KurodaSowa, T.; Maekawa, M.;
Kawata, S.; Kitagawa, S. J. Chem. Soc. Dalton Trans. 1995, 4099.
(14) Klingele, J.; Dechert, S.; Meyer, F. Coord. Chem. Rev. 2009, 253, 2698.
(15) Sens, C.; Romero, I.; Rodriguez, M.; Llobet, A.; Parella, T.; Benet-Buchholz, J. J.
Am. Chem. Soc. 2004, 126, 7798.
(16) Romain, S.; Bozoglian, F.; Sala, X.; Llobet, A. J. Am. Chem. Soc. 2009, 131,
2768.
(17) Bozoglian, F.; Romain, S.; Ertem, M. Z.; Todorova, T. K.; Sens, C.; Mola, J.;
Rodriguez, M.; Romero, I.; Benet-Buchholz, J.; Fontrodona, X.; Cramer, C. J.;
Gagliardi, L.; Llobet, A. J. Am. Chem. Soc. 2009, 131, 15176.
(18) Pons, J.; Lopez, X.; Benet, E.; Casabo, J.; Teixidor, F.; Sanchez, F. J. Polyhedron
1990, 9, 2839.
(19) Pons, J.; Sanchez, F. J.; Labarta, A.; Casabo, J.; Teixidor, F.; Caubet, A. Inorg.
Chim. Acta. 1993, 208, 167.
(20) Bard, A. J.; Faulkner, L. R. Electrochemical Methods: Fundamentals and
Applications; 2nd ed.; Wiley: New York, 2001.
(21) Woon, T. C.; Mcdonald, R.; Mandal, S. K.; Thompson, L. K.; Connors, S. P.;
Addison, A. W. J. Chem. Soc. Dalton Trans. 1986, 2381.

90
(22) Chen, L. Q.; Thompson, L. K.; Bridson, J. N. Inorg. Chim. Acta 1993, 214, 67.
(23) Klingele, M. H.; Noble, A.; Boyd, P. D. W.; Brooker, S. Polyhedron 2007, 26,
479.
(24) Klingele, M. H.; Brooker, S. Eur. J. Org. Chem. 2004, 3422.
(25) Kitchen, J. A.; White, N. G.; Boyd, M.; Moubaraki, B.; Murray, K. S.; Boyd, P.
D. W.; Brooker, S. Inorg. Chem. 2009, 48, 6670.
(26) Kitchen, J. A.; Noble, A.; Brandt, C. D.; Moubaraki, B.; Murray, K. S.; Brooker,
S. Inorg. Chem. 2008, 47, 9450.
(27) Klingele, M. H.; Boyd, P. D. W.; Moubaraki, B.; Murray, K. S.; Brooker, S. Eur.
J. Inorg. Chem. 2005, 910.
(28) Klingele, M. H.; Brooker, S. Coord. Chem. Rev. 2003, 241, 119.
(29) Klingele, M. H.; Boyd, P. D. W.; Moubaraki, B.; Murray, K. S.; Brooker, S. Eur.
J. Inorg. Chem. 2006, 573.
(30) Geldard, J. F.; Lions, F. J. Org. Chem. 1965, 30, 318.
(31) Atkinson, M. R.; Polya, J. B. J. Chem. Soc. 1954, 141.
(32) Levine, R.; Sneed, J. K. J. Am. Chem. Soc. 1951, 73, 5614.
(33) Teixidor, F.; Garcia, R.; Pons, J.; Casabo, J. Polyhedron 1988, 7, 43.
(34) Zhang, W.; Liu, J. H.; Jin, K.; Sun, L. H. J. Heterocyclic Chem. 2006, 43, 1669.

91
Appendix A.0 General remarks The following two pages are the 1H NMR spectra of the oxidation products
for trans,trans-3-methyl-1,2-cyclohexanediol and trans,cis-3-methyl-1,2-
cyclohexanediol. See section 3.4 for discussion and section 3.6 for
experimental details (p. 68).
Below are the peaks that do not correspond to either the cyclohexanediol
starting material or hydroxycyclohexanone products:
1.93 ppm CHD2CN residual solvent peak (quintet, 1 H)
4.31 ppm HDO solvent peak (s, 1 H)
6.67 ppm hydroquinone (C-H) (s, 4 H)
6.81 ppm benzoquinone (C-H) (s, 4 H)

STA
ND
AR
D
PR
OTO
N
PA
RA
ME
TER
S
Pu
lse
Se
qu
en
ce
: s
2p
ul
So
lve
nt:
c
d3
cn
A
mb
ien
t te
mp
era
ture
IN
OV
A-5
00
"u
i50
O1
'
Pu
lse
30
.5
de
gre
es
A
cq
. ti
me
4
.00
0
se
c
Wid
th 8
00
0.0
H
z
16
re
pe
titi
on
s
OB
SE
RV
E
Hl,
4
99
.75
12
04
4
MH
z D
ATA
P
RO
CE
SS
ING
F
T
Siz
e 6
55
36
T
ota
l ti
me
1 m
in,
4 s
ec
92
A.1. Proton NMR spectrum of the oxidation products for trans,trans-3-methyl-1,2-cyclohexanediol.

STA
ND
AR
D P
RO
TON
P
AR
AM
ETE
RS
Pu
lse
Se
qu
en
ce
: s
2p
ul
So
lve
nt:
c
d3
cn
A
mb
ien
t te
mp
era
ture
IN
OV
A-5
00
"
ui5
00
"
Pu
lse
30
.5
de
gre
es
A
cq
. ti
me
4
.00
0
se
c
Wid
th 8
00
0.0
H
Z
16
re
pe
titi
on
s
OB
SE
RV
E
Hl,
49
9.7
51
20
44
M
Hz
DA
TA
PR
OC
ES
SIN
G
FT
s
ize
65
53
6
To
tal
tim
e 1 m
in,
4 s
ec
93
A.2. Proton NMR spectrum of the oxidation products for trans,cis-3-methyl-1,2-cyclohexanediol.