the cellular source and target of il-21 in k/bxn ... · the cellular source and target of il-21 in...
TRANSCRIPT

of June 8, 2018.This information is current as
K/BxN Autoimmune ArthritisThe Cellular Source and Target of IL-21 in
Katharine E. Block and Haochu Huang
http://www.jimmunol.org/content/191/6/2948doi: 10.4049/jimmunol.1301173August 2013;
2013; 191:2948-2955; Prepublished online 19J Immunol
Referenceshttp://www.jimmunol.org/content/191/6/2948.full#ref-list-1
, 17 of which you can access for free at: cites 45 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2013 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 8, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on June 8, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
The Cellular Source and Target of IL-21 in K/BxNAutoimmune Arthritis
Katharine E. Block* and Haochu Huang†,‡
IL-21 is a pluripotent cytokine that regulates B cell and plasma cell differentiation and is thought be an autocrine factor for
follicular helper T cell (TFH) and Th17 differentiation. Although IL-21 has been implicated in autoimmune diseases, its relevant
cellular source and target cells have not been well characterized. We investigated this issue in the K/BxN mouse model of
autoimmune arthritis. Adoptive transfer of KRN-transgenic CD4+ T cells into appropriate hosts drives germinal center (GC)
formation and autoantibody production against glucose-6-phosphate isomerase, leading to joint inflammation and destruction. By
comparing transfer of T or B cells deficient in IL-21 or IL-21R, we were able to dissect the contribution of each cell type. T cells
deficient in IL-21 did not induce GC formation or autoantibody production, but they went through normal TFH differentiation.
However, T cells lacking IL-21R induced Ab titers, GC B cell frequency, and arthritis development similar to wild-type T cells,
suggesting that IL-21 is not required for TFH differentiation and function. IL-21 acts on B cells, because IL-21R expression on
B cells was required to induce disease. In contrast, Th17 cells, a T cell subset that also produces IL-21 and can provide help to
B cells, are not required for the GC response and arthritis. These data have implications in developing effective therapies for
rheumatoid arthritis and other Ab-mediated autoimmune diseases. The Journal of Immunology, 2013, 191: 2948–2955.
Interleukin-21, a member of the common g-chain–signalingfamily of cytokines, plays an important role in lymphocyteactivation, survival, and differentiation (1). IL-21 produc-
tion is restricted to activated T cells, such as follicular helperT cells (TFH), Th17 cells, and NKT cells. The receptor for IL-21is widely expressed on a variety of cell types, including B cells,activated T cells, NK cells, and dendritic cells. IL-21 promotesB cell proliferation, Ig class switching and production, and plasmacell differentiation (2). IL-21 also enhances the proliferation ofT cells stimulated through their TCRs (3) and was shown tobe an autocrine growth factor for TFH and Th17 cell differen-tiation (4–8).The TFH subset, a canonical producer of IL-21, is controlled by the
transcription factor Bcl6. Changes in chemokine receptor expressionallow TFH to migrate from the T cell zone into B cell follicles.Expression of cell surface molecules promote cell–cell contacts withB cells presenting cognate Ag. It is in these intimate interactions thatIL-21 from TFH is thought to act on B cells to promote germinalcenter (GC) and plasma cell differentiation (reviewed in Refs. 9, 10).In addition to TFH, the Th17 cell subset produces IL-21. Th17 isa dominant proinflammatory T cell subset, controlled by the tran-scription factor RORgt, and is involved in a number of autoimmune
diseases (reviewed in Ref. 11). Th17 cells were shown to directlyinteract with and help B cells (12) and promote spontaneous GCformation in autoimmune BXD2 mice (13).IL-21 is important in a number of animal models of systemic
lupus erythematosus and rheumatoid arthritis (14–18). Accord-
ingly, an association of certain IL-21 and IL-21R alleles with
a risk for systemic lupus erythematosus in humans was reported
(19, 20). In the NOD mouse model, IL-21 is required for the
development of type I diabetes (21, 22). Given the complex bio-
logical functions of IL-21, it is important to understand the rele-
vant cells producing and responding to the cytokine in the context
of B cell–mediated autoimmunity.We investigated this question of the relevant targets of IL-21 using
the K/BxN model of rheumatoid arthritis. K/BxN mice develop
arthritis by 4 wk of age (23). The disease is initiated by KRN TCR-
transgenic CD4+ T cells that recognize a peptide from the ubiqui-
tously expressed self-protein glucose-6-phosphate isomerase (GPI)
presented by the NOD-derived MHC class II molecule I-Ag7 (24).
Activated KRN T cells drive B cells to form GCs and to pro-
duce anti-GPI IgG autoantibodies, which induce joint pathology (25).
K/BxN mice develop spontaneous disease, unlike other models
of arthritis that involve Ag vaccination with adjuvants and can
affect TFH development (26). The advantage of the model for these
studies is that the disease can be induced by transferring naive KRN
T cells into T cell–deficient hosts expressing the MHC class II
molecule I-Ag7 (25, 27).In this study, we used this cell-transfer approach of the K/BxN
model to determine the source and action of IL-21 in arthritis. We
showed that T cells deficient in IL-21 did not induce GC formation
or autoantibody production, but they went through normal TFH
differentiation. However, T cells lacking IL-21R induced similar
Ab titer, GC B cell frequency, and arthritis development as wild-
type (WT) T cells, suggesting that IL-21 is not required for TFH
differentiation and function. IL-21 must act on B cells, because
IL-21R expression on B cells was required to induce disease.
Surprisingly, Th17 cells are not required for arthritis development,
stressing the importance of IL-21 production specifically from the
*Committee on Immunology, University of Chicago, Chicago, IL 60637;†Section of Rheumatology, Department of Medicine, University of Chicago, Chicago,IL 60637; and ‡Knapp Center for Lupus and Immunology Research, University ofChicago, Chicago, IL 60637
Received for publication May 1, 2013. Accepted for publication July 15, 2013.
This work was supported by National Institutes of Health Grant R01 AI087645(to H.H.). K.E.B. was supported in part by National Institutes of Health/National Instituteof Allergy and Infectious Diseases Grant 2T32AI007090.
Address correspondence and reprint requests to Dr. Haochu Huang, Section of Rheu-matology, Department of Medicine, Knapp Center for Lupus and Immunology Re-search, 924 East 57th Street, R412, University of Chicago, Chicago, IL 60637.E-mail address: [email protected]
Abbreviations used in this article: GC, germinal center; GPI, glucose-6-phosphateisomerase; PNA, peanut agglutinin; TFH, follicular helper T cell; WT, wild-type.
Copyright� 2013 by TheAmericanAssociation of Immunologists, Inc. 0022-1767/13/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1301173
by guest on June 8, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

TFH subset. These results have implications for developing ef-fective therapies for rheumatoid arthritis and other Ab-mediatedautoimmune diseases.
Materials and MethodsMice
KRN TCR-transgenic mice (23), IL-21R2/2 mice (28), and RORgtGFP/GFP
mice (29) were maintained on C57/BL6 background. IL-212/2 mice(B6;129S5-Il21tm1Lex, obtained from the Mutant Mouse Regional ResourceCenter, Davis, CA) were maintained on a B6;129S5 mixed background.KRN was crossed to IL-21R2/2 or IL-212/2 mice to generate K/IL-21R2/2
or K/IL-212/2 mice, respectively. TCR Ca2/2 BxN mice used as hosts wereF1 of Ca2/2 B6 and Ca2/2 NOD (25). IL-21R knockout mice crossed toB6.H2g7-congenic mice (30) were used as donors for purified B cells. Allexperiments were approved by the Institutional Animal Care and UseCommittee of the University of Chicago.
Cell-transfer induction of arthritis
CD4+ cells were isolated from splenocytes using AutoMACS positiveselection program. A total of 1 3 106 cells in sterile DMEM was injectedinto the tail vein of Ca2/2 BxN mice. B cells were enriched from spleno-cytes by depleting T cells with anti-CD90.2 Ab (clone 53-2.1; BioLegend),followed by rabbit complement (Cedarlane).
Abs and flow cytometry
Anti-KRN TCRa–specific Ab 3-4G-B7 (J. Perera, B. Stadinski, X. Liu,E. Huseby, and H. Huang, manuscript in preparation) was labeled withAlexa Fluor 647 or biotin. Abs used were against CXCR5, Bcl6, and Fas(BD Biosciences); CD45.1, TCRb, GL-7, CD19, IL-17A, and B220(eBioscience); and CD4 and PD-1 (BioLegend). Intracellular staining forBcl-6 was performed using the Foxp3 intracellular staining kit from eBio-science, according to the manufacturer’s protocol. Intracellular staining forIL-17 was performed using the intracellular staining kit from BD Bio-sciences after cells were stimulated with PMA and ionomycin, as described(12). Multicolor flow cytometric analysis was performed using a FACSCanto(BD Biosciences). Data analysis was conducted using FlowJo software(Tree Star).
Immunohistochemistry staining of splenic sections
Sections of frozen spleen (5 mm) were thawed, rehydrated, and thenstained. Peanut agglutinin (PNA; Alexa Fluor 488), anti-mouse IgD (PE),and anti-mouse Vb6 (Alexa Fluor 647) or anti-KRN TCRa (Alexa Fluor647) were used. Images were taken on an Axiovert200m microscope(Zeiss) and visualized with ImageJ.
ELISA for anti-GPI total IgG
Ninety-six–well plates were coated with 5 mg/ml recombinant GPI in PBSovernight at 4˚C and blocked with 1% BSA 0.05% Tween-20 in PBS atroom temperature. A serial dilution of the samples added to the plate wasdetected with a biotinylated goat anti-mouse IgG (subclasses 1+2a+2b+3)Fcg– or goat anti-mouse IgM Fcm fragment–specific Ab, followed by al-kaline phosphatase–conjugated streptavidin (all from Jackson ImmunoR-esearch). Samples were developed with phosphatase substrate (Sigma) andwere read at 405 nm. A four-parameter variable slope was fitted to the datapoints, and the EC50 (inflection point) for a standard sample was calculatedfrom this nonlinear regression. Serum titers were calculated as the serumdilution (x value) that gave the calculated EC50 (y value) based on thefitted nonlinear regression for each sample. Samples where the curve couldnot be fitted because of low signal (low Ab binding) are indicated as ND(not detectable), and a titer of 1 was assigned for statistical comparisons.All analyses were conducted using Prism 5.0b software (GraphPad).
Statistical analysis
Normally distributed data were analyzed by the unpaired t test using Prism5.0b software (GraphPad).
ResultsTFH differentiation in the cell-transfer model of autoimmunearthritis
To investigate the role of IL-21 in a cell-specific manner, we tookadvantage of the cell-transfer model of the K/BxN mouse. NaiveCD4+ KRN T cells are isolated from healthy KRN/B6 mice (KRN
maintained on C57/BL6 background) and transferred into Ca2/2
BxN hosts (TCR Ca2/2 on B6xNOD F1 background) (25, 27).These hosts lack ab T cells and express the MHC class II alleleI-Ag7, which is required for the KRN TCR to recognize a peptidefrom the self-Ag GPI. Transferred KRN T cells are activated andinduce high titers of anti-GPI IgG Abs, resulting in ankle swellingand joint remodeling. To follow autoreactive TFH differentiationand GC response, TFH and GC B cells were characterized afterT cell transfer. For comparison, naive KRN/B6 T cells were trans-ferred into Ca2/2 B6 hosts. Because Ca2/2 B6 mice do not carrythe MHC class II allele I-Ag7, KRN T cells do not precipitatedisease upon transfer. This allowed us to verify that TFH differ-entiation is dependent on Ag recognition rather than lymphopenia-induced homeostatic proliferation. PD-1+ CXCR5+ or Bcl6+ CXCR5+
TFH were identified in Ca2/2 BxN hosts but not in Ca2/2 B6hosts or KRN/B6 mice (Fig. 1A). In the following experiments,we used CXCR5 and intracellular Bcl6 to mark TFH, because thesemarkers showed a more distinct pattern. Consistent with the TFH
staining, GC B cells (identified as GL-7+ Fas+) were induced inCa2/2 BxN hosts, whereas Ca2/2 B6 hosts had a small GCpopulation, similar to what was observed in naive KRN/B6 mice(Fig. 1B).
IL-21 production by T cells is required to induce arthritis
To determine whether IL-21 production by T cells acts on T cellsin an autocrine manner to induce arthritis, we compared naiveCD4+ KRN T cells purified from WT K/B6 mice (denoted as “K”in the figures) with those purified from KRN mice deficient inIL-21 (referred to as K/IL-212/2) or IL-21R (referred to as K/IL-21R2/2) after transfer into Ca2/2 BxN hosts. In the K/IL-212/2
transfer, KRN T cells could not produce IL-21; in the K/IL-21R2/2
transfer, KRN T cells were able to produce IL-21 but unable toreceive IL-21R signaling. These two groups allowed us to testwhether IL-21 production by autoreactive T cells is required, as
FIGURE 1. KRN T cell transfer leads to TFH and GC B cell differen-
tiation upon Ag recognition. (A) CD4+ cells from KRN/B6 (K) mice were
transferred into Ca2/2 BxN or Ca2/2 B6 hosts, and splenocytes were
analyzed 8 d later. Transferred cells, identified as CD4+ TCRb+, were as-
sessed for TFH differentiation by CXCR5 and PD-1 expression, as well as
CXCR5 and intracellular Bcl6 expression. Naive CD4+ TCRb+ cells in
KRN/B6 splenocytes were used as controls. (B) GC B cells were assessed
by Fas and GL-7 expression in the CD19+ population. Results are repre-
sentative of three independent experiments.
The Journal of Immunology 2949
by guest on June 8, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

well as whether autoreactive T cells require IL-21 as an autocrinefactor for disease. As shown in Fig. 2A, K/IL-212/2 CD4+ T cellstransferred into Ca2/2 BxN hosts did not induce arthritis. Incontrast, K/IL-21R2/2 CD4+ T cells induced severe arthritis withthe same kinetics as the WT KRN T cells. We determined the anti-GPI IgG titers both early (8 d) and late (29–31 d) in disease. WTKRN T cells and K/IL-21R2/2 T cells induced high titers of anti-GPI IgG at both time points. In contrast, anti-GPI IgG titers weretwo to three orders of magnitude lower in the K/IL-212/2 T cell–transfer model (Fig. 2B). These data demonstrate that IL-21 pro-duction by T cells is crucial for IgG Ab response and arthritis butthat IL-21R is not required on T cells.To test the role of IL-21 in extrafollicular response in this model,
we determined anti-GPI IgM and IgG at earlier time points aftertransferring WT KRN or K/IL-212/2 splenocytes. In the WT KRNtransfer, anti-GPI IgM titers were elevated by day 4 and continuedto increase over time; this occurred before IgG titers increased. Incontrast, both anti-GPI IgM and IgG titers remained low in K/IL-212/2 transfers (Fig. 2C). These results suggest that IL-21 is re-quired in even the early extrafollicular response in this model.
IL-21 is not required for TFH differentiation in vivo
We next compared the fate of transferred T cells and TFH differ-entiation in all three transfer settings. Congenic markers ontransferred cells (CD45.2+) and Ca2/2 BxN host cells (CD45.1+/CD45.2+) allowed us to identify the transferred T cells as theCD45.12 CD4+ population (Fig. 3A). Eight days after cell transfer,just after disease onset, there was a small but significant increase inthe percentage and number of K/IL-212/2 T cells compared withWT KRN T cells in the spleen. The percentage and number of K/IL-21R2/2 T cells were comparable to WT KRN T cells. At 29–31 d,when disease was fully established in WT KRN and K/IL-21R2/2
T cell–transfer mice, there was no significant difference in thepercentage and numbers of transferred T cells among the three
transfer groups. These data suggest that the survival of transferredCD4+ KRN T cells was not affected by their ability to produce orrespond to IL-21.To determine how TFH differentiation was affected, CXCR5 and
intracellular Bcl6 staining were used to identify TFH. WT KRN,K/IL-21R2/2, and K/IL-212/2 T cells differentiated into TFH atsimilar frequencies in the spleen 8 d after transfer (Fig. 3B). Theabsolute number of TFH from K/IL-212/2 animals was transientlyhigher than that from WT donors as a result of the higher totalnumber of CD4+ T cells (Fig. 3A). However, by 29–31 d, K/IL-212/2 TFH percentage and numbers decreased to half of those inWT KRN-transfer mice. This presumably reflects the defects inthe maintenance phase of TFH differentiation (31), given that therewere no GCs formed in these mice (see later discussion). How-ever, there were comparable numbers of WT and K/IL-21R2/2
TFH 8 d after transfer, and a small, but not statistically significant,decrease in TFH numbers in K/IL-21R2/2 cells compared with inWT at days 29–31 (Fig. 3B). These data suggest that IL-21 is nota requisite autocrine factor for KRN TFH differentiation.
IL-21 is required for GC formation
Although TFH differentiation was normal in all transfers, anti-GPIIgG Ab production was severely impaired following the transferof K/IL-212/2 T cells (Fig. 2B). Therefore, we investigated GCformation and T cell migration by immunofluorescence on spleensections. As shown in Fig. 4A, there were abundant GCs in bothKRN WT and K/IL-21R2/2 T cell–transfer mice. In contrast, GCsin K/IL-212/2 T cell–transfer mice were rarely observed, and thefew GCs were amorphous and dimly labeled with PNA. The num-ber of GCs was counted from multiple spleen sections from mul-tiple mice (Fig. 4B). KRN WT and K/IL-21R2/2 T cell transferinduced equivalent numbers of GCs/section. GC size was mea-sured; KRN WT mice had slightly larger GCs on average com-pared with K/IL-21R2/2 T cell–transfer mice. We also assessed
FIGURE 2. T cell production of IL-21 is required for arthritis development. (A) A total of 13 106 CD4+ cells purified from KRN/B6 (K), KRN IL-21R2/2
(K/IL-21R2/2), or KRN IL-212/2 (K/IL-212/2) mice were transferred into Ca2/2 BxN hosts; ankle thickening was measured over time. Results shown are
from two experiments (n = 6–8 mice/group). Mean and SD are shown. (B) Anti-GPI IgG titers were measured by ELISA from serum collected 8 and 29–31 d
posttransfer. Mean is shown as the horizontal line within each group; each symbol represents an individual mouse. Day-eight results are from four
experiments (n = 5–10 mice/group). Results for days 29–31 are from the experiment described in (A). Lines between groups indicate the statistical com-
parison. (C) A total of 8 3 106 splenocytes from KRN/B6 (K) or KRN IL-212/2 (K/IL-212/2) mice was transferred into Ca2/2 BxN hosts, and serum was
collected on the days indicated. Anti-GPI IgM and IgG titers were measured by ELISA. Mean is shown as the horizontal line within each group; each symbol
represents an individual mouse (n = 4–5 mice/group). *p, 0.05, **p, 0.01, ***p, 0.001, Student t test. ND, Not detectable; ns, not significant (p. 0.05).
2950 ROLE OF IL-21 IN AUTOIMMUNE ARTHRITIS
by guest on June 8, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
the GC B cells by flow cytometry (Fig. 4C). Consistent with theimmunohistology, there was a significant increase in the GL-7+
Fas+ GC B cell population in KRN WT and K/IL-21R2/2 Tcell–transfer mice compared with Ca2/2 BxN hosts withouttransfers, whereas K/IL-212/2 T cells induced very few GL-7+
Fas+ GC B cells.To determine whether the lack of GCs in the K/IL-212/2 transfer
model was due to defective TFH migration into the B cell follicles ordefective T–B interactions, we examined spleen sections for thepresence of transferred T cells in B cell follicles. As shown in Fig.4D, staining with an anti-KRN Va4–specific Ab 3-4G-B7 (or anti-TCR Vb6, data not shown) revealed KRN T cells throughout theB cell follicle in all transfers. These data suggest that the lack of GCsin K/IL-212/2 is due to impaired T cell help and not T cell migration.
Th17 cells are not essential for GC responses and arthritis
IL-21 is not exclusively a TFH cytokine. The Th17 subset producesIL-21 in addition to IL-17A, IL-17F, and IL-22 (11). Th17 cells
were shown to drive the formation of spontaneous GCs in auto-immune responses of the BXD2 mice (13), and they can provideeffective help to B cells (12, 32). Therefore, we tested the con-tribution of Th17 cells in initiating GC responses and arthritis. K/B6 mice were crossed to RORgtGFP/GFP mice (33) to generate K/RORgtGFP/GFP mice. In these mice, GFP insertion inactivatesRORgt expression, and Th17 differentiation is defective. CD4+
T cells were purified from either K/RORgt+/+ or K/RORgtGFP/GFP
mice and transferred into Ca2/2 BxN hosts. As shown in Fig. 5A,K/RORgt+/+ and K/RORgtGFP/GFP CD4+ T cells induced arthritiswith a similar kinetics and severity. There also was no differencein the serum anti-GPI IgG titers (Fig. 5B). The transferred K/RORgtGFP/GFP cells were indeed deficient in generating Th17 cellsas determined by IL-17 intracellular staining after in vitro stim-ulation (Fig. 5C). However, there was no difference in the dif-ferentiation of TFH (Fig. 5D). These results demonstrate that Th17cells and their production of IL-21 are not essential for GCresponses and arthritis development, supporting the conclusionthat TFH are the major source of IL-21 production.It was shown previously that Th17 cell development is correlated
with disease induction in K/BxNmice (34). We investigated whetherthere were differences in Th17 cell development and distribution inK/BxN mice versus the transfer model. Cells isolated from thespleen, draining lymph nodes (pooled popliteal and inguinal lymphnodes), and mesenteric lymph nodes stimulated in vitro revealeda similar profile of IL-17A production from K/BxN mice andCa2/2 BxN hosts after KRN T cell transfer (Fig. 5E). IL-17Aproduction was detected from KRN T cells identified with theanti-KRN Va4-specific Ab 3-4G-B7. However, non-KRN T cellswere the major IL-17A producers in both K/BxN mice and hostmice that received KRN/B6 cell transfer.
B cells require IL-21R signaling to initiate GCs
Because IL-21 production by T cells is necessary for GC formation,it suggests that the target of IL-21 is the B cell. To directly test theimportance of IL-21R expression on B cells, we performed a Band T cell cotransfer experiment. B cells purified from IL-21R+/2
or IL-21R2/2 mice expressing I-Ag7/b MHC class II alleles weretransferred along with naive K/B6 CD4+ cells into Rag12/2
B6xNOD F1 hosts. Most mice that received IL-21R+/2 B cellsdeveloped arthritis, whereas mice that received IL-21R2/2 B cellsnever showed signs of arthritis (Fig. 6A). The disease states werereflected in the dramatic difference in anti-GPI IgG titers betweenthese two groups of mice (Fig. 6B). There was a moderate decreasein TFH in hosts receiving IL-21R2/2 B cells compared with thosereceiving IL-21R+/2 B cells (Fig. 6C). This result is consistent withwhat was observed in the K/IL-212/2 T cell transfer: TFH numbersdecreased after 29–30 d, likely due to a lack of interaction with GCB cells to promote TFH maintenance. There was a more dramaticdecrease in GC B cells in hosts receiving IL-21R2/2 B cells com-pared with those receiving IL-21R+/2 B cells (Fig. 6D). Immuno-histological analysis of GCs on spleen section confirmed the resultsobtained by flow cytometry, although there were few B cell folliclesand smaller GCs in general compared with T cell transfer into Ca2/2
BxN hosts (data not shown). This is not surprising considering thatRag12/2mice have defective follicular structures and a much smallerpopulation of transferred B cells. These data support the con-clusion that IL-21R signaling in B cells is essential for GC for-mation, Ab production, and arthritis development, and GC B cellsare required for TFH maintenance.
DiscussionIL-21 is a pleiotropic cytokine affecting a diverse array of cell types(2). Without conditional deletion of IL-21 or IL-21R, it has been
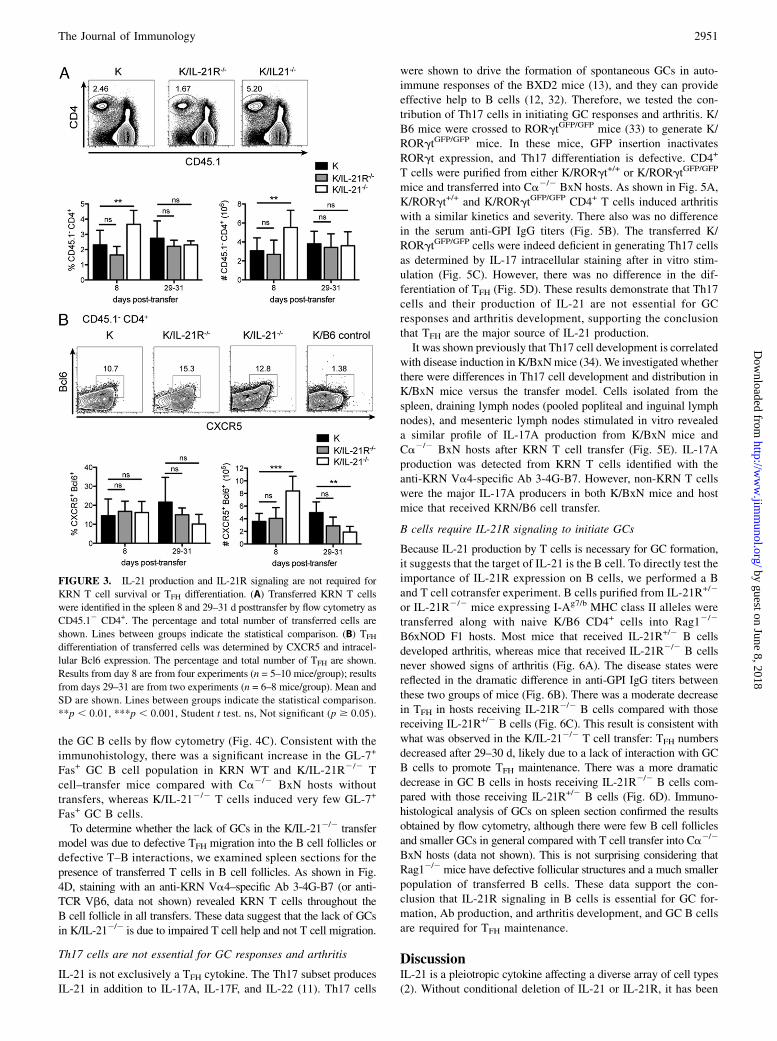
FIGURE 3. IL-21 production and IL-21R signaling are not required for
KRN T cell survival or TFH differentiation. (A) Transferred KRN T cells
were identified in the spleen 8 and 29–31 d posttransfer by flow cytometry as
CD45.12 CD4+. The percentage and total number of transferred cells are
shown. Lines between groups indicate the statistical comparison. (B) TFH
differentiation of transferred cells was determined by CXCR5 and intracel-
lular Bcl6 expression. The percentage and total number of TFH are shown.
Results from day 8 are from four experiments (n = 5–10 mice/group); results
from days 29–31 are from two experiments (n = 6–8 mice/group). Mean and
SD are shown. Lines between groups indicate the statistical comparison.
**p , 0.01, ***p , 0.001, Student t test. ns, Not significant (p $ 0.05).
The Journal of Immunology 2951
by guest on June 8, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
difficult to evaluate the specific roles of IL-21 in autoimmunity.We used a cell-transfer system based on the K/BxN mouse modelof autoimmune arthritis to address these roles in TFH differentia-tion and B cell activation. Our results demonstrate that IL-21production by T cells is important in disease induction. How-ever, IL-21 is not required for TFH differentiation, maintenance, orfunction. There was no defect in T cell survival when KRN T cellswere deficient in either IL-21 or IL-21R; in fact, survival wasincreased 1 wk after transfer in IL-212/2 T cells (Fig. 3A). Asimilar increase was observed in IL-212/2 mice after immuniza-tion with NP-KLH (35), although the mechanism for enhanced
survival is unclear. Both IL-212/2 and IL-21R2/2 KRN T cellsproliferated, differentiated into TFH, and were able to migrate intothe B cell follicles in an Ag-specific manner, because this processdid not take place when KRN T cells were transferred into Ca2/2
B6 hosts that do not express the self-peptide–MHC complex.In an earlier study using IL-21R2/2 K/BxN mice, it was shown
that there were fewer CD4+ T cells in the spleen and joint-draininglymph nodes compared with normal K/BxN mice (36). IL-21R2/2
K/BxN mice did not develop arthritis, and it was attributed toa requirement of IL-21 by KRN T cells for homeostatic prolifer-ation. The different conclusions from our study and the earlier
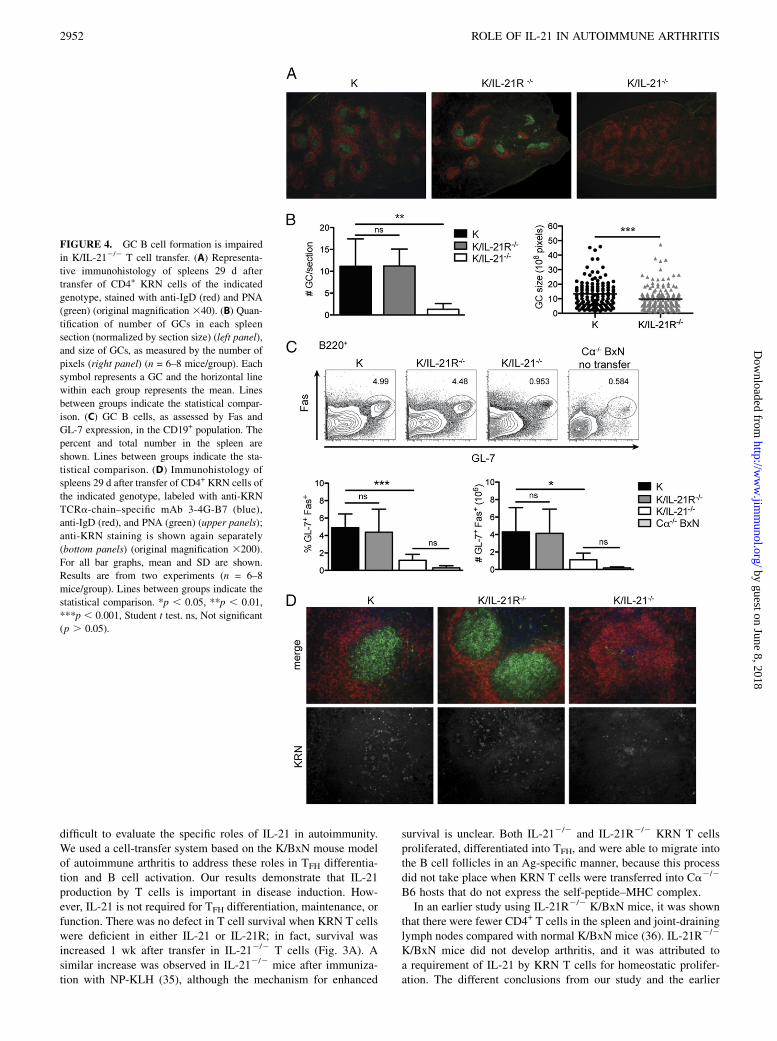
FIGURE 4. GC B cell formation is impaired
in K/IL-212/2 T cell transfer. (A) Representa-
tive immunohistology of spleens 29 d after
transfer of CD4+ KRN cells of the indicated
genotype, stained with anti-IgD (red) and PNA
(green) (original magnification 340). (B) Quan-
tification of number of GCs in each spleen
section (normalized by section size) (left panel),
and size of GCs, as measured by the number of
pixels (right panel) (n = 6–8 mice/group). Each
symbol represents a GC and the horizontal line
within each group represents the mean. Lines
between groups indicate the statistical compar-
ison. (C) GC B cells, as assessed by Fas and
GL-7 expression, in the CD19+ population. The
percent and total number in the spleen are
shown. Lines between groups indicate the sta-
tistical comparison. (D) Immunohistology of
spleens 29 d after transfer of CD4+ KRN cells of
the indicated genotype, labeled with anti-KRN
TCRa-chain–specific mAb 3-4G-B7 (blue),
anti-IgD (red), and PNA (green) (upper panels);
anti-KRN staining is shown again separately
(bottom panels) (original magnification 3200).
For all bar graphs, mean and SD are shown.
Results are from two experiments (n = 6–8
mice/group). Lines between groups indicate the
statistical comparison. *p , 0.05, **p , 0.01,
***p , 0.001, Student t test. ns, Not significant
(p . 0.05).
2952 ROLE OF IL-21 IN AUTOIMMUNE ARTHRITIS
by guest on June 8, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

study highlight the complex biological function of IL-21 and theproblem of understanding the direct versus indirect mechanisms intotal knockout animals.We found no difference in the GC B cell response between KRN
WT and K/IL-21R2/2 T cell–transfer mice by Ab titers or GCB cell formation by flow cytometry or histology, demonstratingthat there is no functional defect in TFH in the absence of IL-21Rsignaling. These results contrast with a study of lupus-like diseaseinduced in a chronic graft-versus-host model, which found that
GC B cells were less frequent and GCs were smaller when IL-21R2/2 T cells were transferred (17). The differential dependenceon IL-21R signaling may be related to the difference in frequencyor affinity of alloreactive T cells, because it was shown that naiveAg-specific Th cells with TCRs of higher affinity preferentiallydifferentiate into the CXCR5hi “resident” TFH compartment (37).Studies on the role of IL-21 in TFH differentiation and GC
formation led to different conclusions in mice immunized withprotein Ags. GC formation was relatively unaffected in IL-212/2
or IL-21R2/2 mice in some studies (35, 38, 39), whereas a moreprofound effect on TFH and GCs was found in other studies (5, 8).It was suggested that the different results obtained in these studiesmight be explained by the different types of Ag or adjuvant, theavidity of TCR involved for peptide–MHC, or the timing ofanalysis (35, 39). Our conclusion that IL-21 is not required forTFH differentiation, but rather acts on GC B cells, is consistentwith studies in mixed bone marrow chimeras (35, 39, 40). GCformation appears to be highly dependent on IL-21 in the K/BxNand other models of spontaneous autoimmune diseases (14, 41),although IL-21 does not play any role in the RoquinSanroque mousemodel of lupus (42). Therapeutic intervention of the IL-21–sig-naling pathway has been explored in animal models (15, 16, 43).However, the efficacy was found to be variable and partial. Forexample, repeated treatment of BXSB-Yaa mice with a soluble IL-21R–Fc fusion protein had minimal effect on lupus symptoms andsurvival, even though IL-21R2/2 BXSB-Yaa mice have no sign ofdisease (14). This variation could be attributed to the partial ef-fectiveness of IL-21R–Fc in blocking IL-21 signaling. The evidencethat TFH development is not dependent on IL-21 in autoimmunityraises the possibility that an efficient inhibition of ongoing GC B cellresponse may be difficult to achieve in practice with partial effec-tiveness of IL-21 blockade. It is tempting to speculate that IL-21blockade is more effective in cases in which TFH is more dependenton the cytokine. IL-21 blockade together with a therapy targetingT cells might be most beneficial for treating certain Ab-mediatedautoimmune diseases.
FIGURE 5. Th17 cells are dispensable for arthritis. (A) KRN spleno-
cytes purified from WT KRN/B6 (K/RORgt+/+) or KRN RORgt-deficient
(K/RORgtGFP/GFP) mice were transferred into Ca2/2 BxN hosts. Ankle
thickening was monitored over time. (B) Anti-GPI IgG titers were detected
by ELISA. Horizontal lines represent the mean. (C) Splenocytes were
cultured in PMA and ionomycin with brefeldin A for 5 h, and IL-17A
production was detected by intracellular staining. The percentage and total
number of IL-17A–producing transferred cells (CD45.12 CD4+) in the
spleen are shown. (D) The percentage and total number of TFH (CXCR5+
Bcl6+) of the CD45.12 CD4+ transferred cell gate in the spleen are shown.
Data (mean + SD) are representative of two independent experiments (n =
4–5 mice/group). (E) Cells from the spleen, draining lymph nodes (dLN;
pooled popliteal and inguinal lymph nodes), and mesenteric lymph nodes
(mLN) from K/BxN mice and Ca2/2 BxN hosts 20 d after KRN/B6 cell
transfer were stimulated and assessed for IL-17A production, as in (C).
Representative of one experiment with two mice/group. *p , 0.05, **p ,0.01, Student t test. ns, Not significant (p . 0.05).
FIGURE 6. B cells require expression of IL-21R for GC formation and
autoantibody production. (A) IL-21R+/2 or IL-21R2/2 I-Ag7/b splenic B
cells and CD4+ KRN/B6 T cells were transferred into Rag12/2 BxN hosts.
Ankle thickening was monitored over time. (B) Anti-GPI IgG titers were
detected by ELISA, analyzed 37–40 d after transfer. Horizontal lines
represent the mean. (C) The percentage of GC B cells (GL-7+ Fas+) of the
B220+ gate and (D) the percentage of TFH (CXCR5+ Bcl6+) of the TCRb+
CD4+ gate were determined by flow cytometry. Mean and SD are shown.
Results shown are from two experiments (n = 8 mice/group). **p ,0.01, Student t test. ns, Not significant (p . 0.05).
The Journal of Immunology 2953
by guest on June 8, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

Arthritis development in K/BxN mice is dependent on gut mi-crobiota, particularly the colonization of segmented filamentousbacteria. Because the KRN-transfer model involves different strainsof mice as the source of donor cells and hosts, the potential dif-ference in their gut microbiota might be a confounding factor in theinterpretation of our experiments. However, we think this is notlikely to be the case. In our experiments, we always used littermatesfor our cell-transfer hosts, dividing the hosts housed in the samecage for different KRN-genotype transfers. We used donors ofdifferent genotypes housed in the same cage when possible. Fur-thermore, the gut microbiota of K/IL-212/2 and K/IL-21R2/2
donor mice should be very similar because they are defective in thesame pathway.Th17 cells are now recognized to interact with Ag-specific
B cells as potential B cell helpers (12, 13). Therefore, it wasimportant to investigate whether IL-21 required for disease in-duction was produced by TFH or from Th17 cells that enter theB cell follicle to initiate GC. By eliminating the Th17 subsetthrough RORgtGFP/GFP, K/RORgtGFP/GFP T cells did not dramati-cally alter disease kinetics or severity, suggesting that IL-21 isderived from TFH but not Th17 cells.At first glance, the result that Th17 cells are not essential for
disease induction seems unexpected, because Th17 cells weresuggested to play an important role in this disease model. Th17 cellinduction correlated with disease onset, and neutralizing Ab againstIL-17A prevented disease in K/BxN mice (34). IL-17R2/2 B cellswere defective in differentiating into GC B cells, suggesting thatthey are the targets of IL-17. However, other lymphocyte andinnate-like cell populations, including gd T cells and some re-cently characterized innate lymphoid cells, are major producers ofIL-17 (44, 45). The intriguing possibility that IL-17 producedconstitutively by innate lymphoid cells plays an important role isbeing investigated.
AcknowledgmentsWe thank Dr. Michael Grusby for IL-21R2/2 mice; Drs. Diane Mathis and
Christophe Benoist for KRN-transgenic mice, B6.H2g7-congenic mice,
Ca2/2 B6 mice, and Ca2/2 NOD mice; and Xiao Liu and Crystal Rayon
for help with mice.
DisclosuresThe authors have no financial conflicts of interest.
References1. Spolski, R., and W. J. Leonard. 2008. Interleukin-21: basic biology and impli-
cations for cancer and autoimmunity. Annu. Rev. Immunol. 26: 57–79.2. Ettinger, R., S. Kuchen, and P. E. Lipsky. 2008. The role of IL-21 in regulating
B-cell function in health and disease. Immunol. Rev. 223: 60–86.3. Parrish-Novak, J., S. R. Dillon, A. Nelson, A. Hammond, C. Sprecher,
J. A. Gross, J. Johnston, K. Madden, W. Xu, J. West, et al. 2000. Interleukin 21and its receptor are involved in NK cell expansion and regulation of lymphocytefunction. Nature 408: 57–63.
4. Nurieva, R., X. O. Yang, G. Martinez, Y. Zhang, A. D. Panopoulos, L. Ma,K. Schluns, Q. Tian, S. S. Watowich, A. M. Jetten, and C. Dong. 2007. Essentialautocrine regulation by IL-21 in the generation of inflammatory T cells. Nature448: 480–483.
5. Vogelzang, A., H. M. McGuire, D. Yu, J. Sprent, C. R. Mackay, and C. King.2008. A fundamental role for interleukin-21 in the generation of T follicularhelper cells. Immunity 29: 127–137.
6. Suto, A., D. Kashiwakuma, S.-I. Kagami, K. Hirose, N. Watanabe, K. Yokote,Y. Saito, T. Nakayama, M. J. Grusby, I. Iwamoto, and H. Nakajima. 2008. De-velopment and characterization of IL-21-producing CD4+ T cells. J. Exp. Med.205: 1369–1379.
7. Korn, T., E. Bettelli, W. Gao, A. Awasthi, A. Jager, T. B. Strom, M. Oukka, andV. K. Kuchroo. 2007. IL-21 initiates an alternative pathway to induce proin-flammatory T(H)17 cells. Nature 448: 484–487.
8. Nurieva, R. I., Y. Chung, D. Hwang, X. O. Yang, H. S. Kang, L. Ma, Y.-H. Wang,S. S. Watowich, A. M. Jetten, Q. Tian, and C. Dong. 2008. Generation of Tfollicular helper cells is mediated by interleukin-21 but independent of T helper1, 2, or 17 cell lineages. Immunity 29: 138–149.
9. Crotty, S. 2011. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29:621–663.
10. King, C., S. G. Tangye, and C. R. Mackay. 2008. T follicular helper (TFH) cellsin normal and dysregulated immune responses. Annu. Rev. Immunol. 26: 741–766.
11. Korn, T., E. Bettelli, M. Oukka, and V. K. Kuchroo. 2009. IL-17 and Th17 Cells.Annu. Rev. Immunol. 27: 485–517.
12. Mitsdoerffer, M., Y. Lee, A. Jager, H.-J. Kim, T. Korn, J. K. Kolls, H. Cantor,E. Bettelli, and V. K. Kuchroo. 2010. Proinflammatory T helper type 17 cells areeffective B-cell helpers. Proc. Natl. Acad. Sci. USA 107: 14292–14297.
13. Hsu, H.-C., P. Yang, J. Wang, Q. Wu, R. Myers, J. Chen, J. Yi, T. Guentert,A. Tousson, A. L. Stanus, et al. 2008. Interleukin 17-producing T helper cellsand interleukin 17 orchestrate autoreactive germinal center development in au-toimmune BXD2 mice. Nat. Immunol. 9: 166–175.
14. Bubier, J. A., T. J. Sproule, O. Foreman, R. Spolski, D. J. Shaffer, H. C. Morse,III, W. J. Leonard, and D. C. Roopenian. 2009. A critical role for IL-21 receptorsignaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaamice. Proc. Natl. Acad. Sci. USA 106: 1518–1523.
15. Young, D. A., M. Hegen, H. L. M. Ma, M. J. Whitters, L. M. Albert, L. Lowe,M. Senices, P. W. Wu, B. Sibley, Y. Leathurby, et al. 2007. Blockade of theinterleukin-21/interleukin-21 receptor pathway ameliorates disease in animalmodels of rheumatoid arthritis. Arthritis Rheum. 56: 1152–1163.
16. Herber, D., T. P. Brown, S. Liang, D. A. Young, M. Collins, and K. Dunussi-Joannopoulos. 2007. IL-21 has a pathogenic role in a lupus-prone mouse modeland its blockade with IL-21R.Fc reduces disease progression. J. Immunol. 178:3822–3830.
17. Nguyen, V., I. Luzina, H. Rus, C. Tegla, C. Chen, and V. Rus. 2012. IL-21promotes lupus-like disease in chronic graft-versus-host disease through bothCD4 T cell- and B cell-intrinsic mechanisms. J. Immunol. 189: 1081–1093.
18. Odegard, J. M., B. R. Marks, L. D. DiPlacido, A. C. Poholek, D. H. Kono,C. Dong, R. A. Flavell, and J. Craft. 2008. ICOS-dependent extrafollicular helperT cells elicit IgG production via IL-21 in systemic autoimmunity. J. Exp. Med.205: 2873–2886.
19. Sawalha, A. H., K. M. Kaufman, J. A. Kelly, A. J. Adler, T. Aberle, J. Kilpatrick,E. K. Wakeland, Q.-Z. Li, A. E. Wandstrat, D. R. Karp, et al. 2008. Geneticassociation of interleukin-21 polymorphisms with systemic lupus erythematosus.Ann. Rheum. Dis. 67: 458–461.
20. Webb, R., J. T. Merrill, J. A. Kelly, A. Sestak, K. M. Kaufman, C. D. Langefeld,J. Ziegler, R. P. Kimberly, J. C. Edberg, R. Ramsey-Goldman, et al. 2009. Apolymorphism within IL21R confers risk for systemic lupus erythematosus.Arthritis Rheum. 60: 2402–2407.
21. Spolski, R., M. Kashyap, C. Robinson, Z. Yu, and W. J. Leonard. 2008. IL-21signaling is critical for the development of type I diabetes in the NOD mouse.Proc. Natl. Acad. Sci. USA 105: 14028–14033.
22. King, C., A. Ilic, K. Koelsch, and N. Sarvetnick. 2004. Homeostatic expansion ofT cells during immune insufficiency generates autoimmunity. Cell 117: 265–277.
23. Kouskoff, V., A. S. Korganow, V. Duchatelle, C. Degott, C. Benoist, andD. Mathis. 1996. Organ-specific disease provoked by systemic autoimmunity.Cell 87: 811–822.
24. Matsumoto, I., A. Staub, C. Benoist, and D. Mathis. 1999. Arthritis provoked bylinked T and B cell recognition of a glycolytic enzyme. Science 286: 1732–1735.
25. Korganow, A. S., H. Ji, S. Mangialaio, V. Duchatelle, R. Pelanda, T. Martin,C. Degott, H. Kikutani, K. Rajewsky, J. L. Pasquali, et al. 1999. From systemicT cell self-reactivity to organ-specific autoimmune disease via immunoglobulins.Immunity 10: 451–461.
26. Malherbe, L., L. Mark, N. Fazilleau, L. J. McHeyzer-Williams, andM. G. McHeyzer-Williams. 2008. Vaccine adjuvants alter TCR-based selec-tion thresholds. Immunity 28: 698–709.
27. LaBranche, T. P., C. L. Hickman-Brecks, D. M. Meyer, C. E. Storer, M. I. Jesson,K. M. Shevlin, F. A. Happa, R. A. Barve, D. J. Weiss, J. C. Minnerly, et al. 2010.Characterization of the KRN cell transfer model of rheumatoid arthritis (KRN-CTM), a chronic yet synchronized version of the K/BxN mouse. Am. J. Pathol.177: 1388–1396.
28. Kasaian, M. T., M. J. Whitters, L. L. Carter, L. D. Lowe, J. M. Jussif, B. Deng,K. A. Johnson, J. S. Witek, M. Senices, R. F. Konz, et al. 2002. IL-21 limits NKcell responses and promotes antigen-specific T cell activation: a mediator of thetransition from innate to adaptive immunity. Immunity 16: 559–569.
29. Eberl, G., S. Marmon, M.-J. Sunshine, P. D. Rennert, Y. Choi, and D. R. Littman.2004. An essential function for the nuclear receptor RORgamma(t) in the gen-eration of fetal lymphoid tissue inducer cells. Nat. Immunol. 5: 64–73.
30. Luhder, F. F., P. P. Hoglund, J. P. J. Allison, C. C. Benoist, and D. D. Mathis.1998. Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) regulates theunfolding of autoimmune diabetes. J. Exp. Med. 187: 427–432.
31. Choi, Y. S., R. Kageyama, D. Eto, T. C. Escobar, R. J. Johnston, L. Monticelli,C. Lao, and S. Crotty. 2011. ICOS receptor instructs T follicular helper cellversus effector cell differentiation via induction of the transcriptional repressorBcl6. Immunity 34: 932–946.
32. Hickman-Brecks, C. L., J. L. Racz, D. M. Meyer, T. P. LaBranche, andP. M. Allen. 2011. Th17 cells can provide B cell help in autoantibody inducedarthritis. J. Autoimmun. 36: 65–75.
33. Ivanov, I. I., B. S. McKenzie, L. Zhou, C. E. Tadokoro, A. Lepelley, J. J. Lafaille,D. J. Cua, and D. R. Littman. 2006. The orphan nuclear receptor RORgammatdirects the differentiation program of proinflammatory IL-17+ T helper cells.Cell 126: 1121–1133.
34. Wu, H.-J., I. I. Ivanov, J. Darce, K. Hattori, T. Shima, Y. Umesaki, D. R. Littman,C. Benoist, and D. Mathis. 2010. Gut-residing segmented filamentous bacteriadrive autoimmune arthritis via T helper 17 cells. Immunity 32: 815–827.
2954 ROLE OF IL-21 IN AUTOIMMUNE ARTHRITIS
by guest on June 8, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

35. Zotos, D. D., J. M. Coquet, Y. Y. Zhang, A. A. Light, K. K. D’Costa,A. A. Kallies, L. M. Corcoran, D. I. Godfrey, K.M. Toellner, M. J. Smyth, et al.2010. IL-21 regulates germinal center B cell differentiation and proliferationthrough a B cell-intrinsic mechanism. J. Exp. Med. 207: 365–378.
36. Jang, E., S.-H. Cho, H. Park, D.-J. Paik, J. M. Kim, and J. Youn. 2009. A positivefeedback loop of IL-21 signaling provoked by homeostatic CD4+CD252 T cellexpansion is essential for the development of arthritis in autoimmune K/BxNmice. J. Immunol. 182: 4649–4656.
37. Fazilleau, N., L. J. McHeyzer-Williams, H. Rosen, and M. G. McHeyzer-Williams. 2009. The function of follicular helper T cells is regulated by thestrength of T cell antigen receptor binding. Nat. Immunol. 10: 375–384.
38. Ozaki, K., R. Spolski, C. G. Feng, C.-F. Qi, J. Cheng, A. Sher, H. C. Morse, III,C. Liu, P. L. Schwartzberg, and W. J. Leonard. 2002. A critical role for IL-21 inregulating immunoglobulin production. Science 298: 1630–1634.
39. Linterman, M. A., L. Beaton, D. Yu, R. R. Ramiscal, M. Srivastava, J. J. Hogan,N. K. Verma, M. J. Smyth, R. J. Rigby, and C. G. Vinuesa. 2010. IL-21 actsdirectly on B cells to regulate Bcl-6 expression and germinal center responses. J.Exp. Med. 207: 353–363.
40. Bessa, J., M. Kopf, and M. F. Bachmann. 2010. Cutting edge: IL-21 and TLRsignaling regulate germinal center responses in a B cell-intrinsic manner. J.Immunol. 184: 4615–4619.
41. Rankin, A. L., H. Guay, D. Herber, S. A. Bertino, T. A. Duzanski, Y. Carrier,S. Keegan, M. Senices, N. Stedman, M. Ryan, et al. 2012. IL-21 receptor isrequired for the systemic accumulation of activated B and T lymphocytes inMRL/MpJ-Fas(lpr/lpr)/J mice. J. Immunol. 188: 1656–1667.
42. Linterman, M. A., R. J. Rigby, R. K. Wong, D. Yu, R. Brink, J. L. Cannons,P. L. Schwartzberg, M. C. Cook, G. D. Walters, and C. G. Vinuesa. 2009. Follicularhelper T cells are required for systemic autoimmunity. J. Exp. Med. 206: 561–576.
43. Bubier, J. A., S. M. Bennett, T. J. Sproule, B. L. Lyons, S. Olland, D. A. Young,and D. C. Roopenian. 2007. Treatment of BXSB-Yaa mice with IL-21R-Fc fu-sion protein minimally attenuates systemic lupus erythematosus. Ann. N. Y.Acad. Sci. 1110: 590–601.
44. Cua, D. J., and C. M. Tato. 2010. Innate IL-17-producing cells: the sentinels ofthe immune system. Nat. Rev. Immunol. 10: 479–489.
45. Sutton, C. E., L. A. Mielke, and K. H. Mills. 2012. IL-17-producing gd T cellsand innate lymphoid cells. Eur. J. Immunol. 42: 2221–2231.
The Journal of Immunology 2955
by guest on June 8, 2018http://w
ww
.jimm
unol.org/D
ownloaded from