the business case for supplier quality and the dqsa enforcement and administrative actions resulting...
TRANSCRIPT

The Business Case for Supplier Quality and the DQSA
Enforcement and Administrative Actions Resulting from Inadequate Oversight and Control of Suppliers
Jacqueline R. Berman, Esq.
Associate
Morgan, Lewis & Bockius LLP

Supplier Quality
Obligations and Why You Should Care

3

Current Legal Landscape
4
• Current Good Manufacturing Practices– FFDCA § 501
• Automatic adulteration if fail to comply with cGMPs• Implementation of oversight and controls over
manufacturing
– Selected Regulations • 21 C.F.R. § 200.10(b) (Contract manufacturers an
extension of manufacturer’s own facility)• 21 C.F.R. § 211.22 (Quality unit ultimate authority for
product quality)• 21 C.F.R. § 211.84 (Incoming inspection and testing)• 21 C.F.R. § 211.184 (Component records)

Potential Regulatory Consequences of Poor Supplier Quality Control
5
• Adverse Inspectional Findings• Warning Letters• Product Recalls• Cessation of Manufacturing• Seizure• Injunction• Criminal Actions• False Claims Act Actions• Government Contracting

Warning Letter Examples
6
• Greer Laboratories Inc. (4/21/2014)– Failure to establish reliability of supplier’s certificate of
testing through validation
• Sanquin Plasma Products (8/29/2013)– Failure to perform incoming testing on supplier
components
• Jabones Pardo S.A. (8/22/2013)– Failure to perform incoming testing, failure to establish
reliability of supplier’s analyses, failure to conduct identity test when relying on supplier analyses

New Focus on Data Integrity• Trifarma S.p.A. (7/7/2014)
– Failure to maintain complete data– Failure to prevent unauthorized access or change to
data/prevent data omission
• Tianjin Zhongan Pharmaceutical Co., Ltd (6/10/2014)– Failure to maintain appropriate documentation to record
manufacturing operations performed on individual pieces of equipment
7

New Consequences of Inadequate Supplier Controls
8
Inadequate supplier controls will require additional product
oversight beginning January 2015

Selected DQSA Requirements• Specifics will depend on role in the supply chain• Some requirements more administrative/procedural
– Record keeping and information transfer– Product identifier– Authorized trading partners
• Other requirements related to supplier quality– Suspect and illegitimate product identification,
investigation, quarantine, and notification– Requests for information– Returned products
9

Some DQSA Practical Implications
• DQSA compliance obligations will substantially affect contractual provisions in sales and related documents– Current commercial terms likely affected– New contracts for outsourced functions requiring supplier
controls
• New SOPs• Training and Auditing• Potential False Claims Act claims arising out of
certifications
10

Selecting and Working with Third Parties
Lori F. Hirsch
Managing Counsel
Merck Sharp & Dohme Corp.
December 9, 2014

Why utilize Third Parties?
• Need for specialized capabilities• Product lifecycle management • Market access • Cost optimization

Selecting a Mate

Things to consider before you enter a Marriage
1. Don't marry the person you think you can live with. Marry the one you can't live without!
2. Don't marry someone who has characteristics that you feel are intolerable!
3. Do not marry impulsively!
4. Make sure that each partner has an iron-willed determination to make it work!

Effective Due Diligence is Essential!
• Qualifications – does the third party have the ability to do what you need them to do?
• Experience – does the third party have the right experience to do what you need them to do?
• Expectations – have you clearly defined what the third party will do, including expectations, standards, etc.
• Compliance – does the third party understand cGMPs and have appropriate systems, processes and people?
• Capacity – does the third party have available capacity?• Culture – does the culture at the third party match your company’s
culture?• Determination – are you both determined to make this relationship
work?

Some Due Diligence Areas
• Manufacturing capabilities - facilities, processes, systems and people
• Management team who are experienced and competent, including quality personnel
• Communication skills• Regulatory agency experience and history• Understanding of Quality System/GMP requirements • Willingness to provide access to the manufacturing site, as needed,
including management visits and audits.

Realities of Working with a Third Party
• A company retains accountability for the acts or omissions of the third party.– Despite the fact that control has been surrendered to the third
party– Only the third party has the direct access to the contracted
activity.
• A company has limited monitoring and influence.– Critical to select the right third party.

Commercial Agreement
• The “what”• Contract must be clear as to roles, responsibilities and
obligations.– Remember: terms will often be interpreted, several
years later, by people not party to the original negotiations.
– Consider cultural and language differences.• Terms should be explicit, where possible. • Dispute resolution/escalation defined.
• Process for changes should be defined.

Quality Agreement
• The “How” • The Quality Agreement should be standalone.
– Allows the quality provisions to be changed from time to time without opening the entire agreement.
• Should include such things as:– Rights to visit during manufacturing of Companies’ products,
audits, etc.– Requirements for prior approval of any changes to the third
party’s facility, equipment, materials or components– Notification of any out-of-specification (OOS) test results and
stability failures– The ability to evaluate any deviations and/or investigations as
well as the review of manufacturing batch records– Recall decisions and regulatory notifications.

And, the marriage begins . . .

Are you ready to manage the relationship?
• Do you have the right people with the right skills to manage these third party relationships in a way that ensures patient safety and security of supply?– Increasingly, these relationships are the
subject of regulatory scrutiny.

On-going oversight
• Meeting Company’s expectations• Oversight of manufacturing processes• Site visits• Performance/Quality Metrics• Periodic Review of Manufacturing and
Quality records, including test data• Escalation Process for managing supplier
deviations

23
Calibrated Quality Oversight Model
• Introduce risk-based management to the Third Party Quality Oversight Process– Define process for determining level of Quality oversight
• Recognize and leverage the development, operational and compliance capabilities of Third Parties
• Objective assessment that drives consistent management of Third Parties
• Assessment consistency across the life cycle of the Third Party relationship (identify, select, manage)
Go
al
Dri
ve
rs
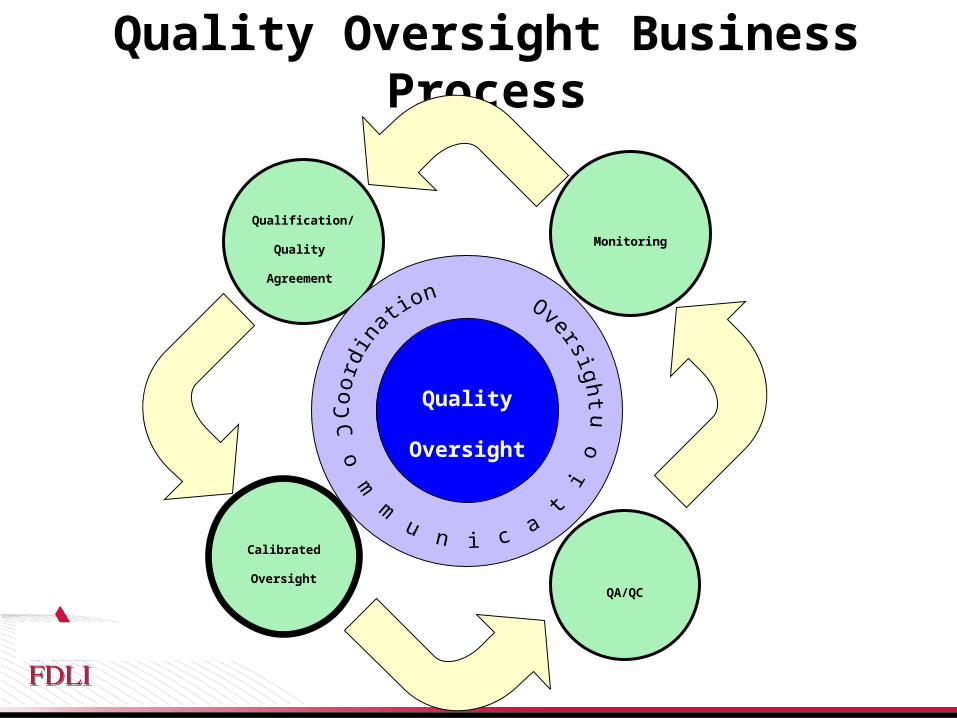
Quality Oversight Business Process
Qualification/
Quality
Agreement
Monitoring
QA/QC
Calibrated
Oversight
Quality
Oversight

25
Calibrated Quality Oversight Model
Process– Determine appropriate level of oversight based on:
• Quality Systems implementation• Contracted activity (e.g. API vs. sterile pharmaceutical)• Technology ownership and complexity• Historical experience / knowledge of third party• Performance (metrics, relationship)• Compliance performance / history
– Process Utilizes Assessment Tool• Defined criteria consistently applied• Information repository for ongoing assessment
– Results in Recommendation Memo– Ongoing Assessment

Assessment Design
Strategic Quality Risk Management Model requires assessment of risk for … – FDA Six Systems (30 Sub-
systems)• Quality Management• Facilities & Equipment • Materials• Production• Packaging & Labeling• Laboratory Control
– Two additional assessment elements• Technology Complexity &
Ownership• Compliance
To provide the users with guidance on . . .
– Degree of presence in the third party’s facility.
– Responsibility for control of starting materials.
– Level of review/oversight on planned and unplanned (deviations) changes.
– Level of review of batch documents.
– Responsibility for product testing.– Responsibility for release of
product for “further processing.”

Quality Oversight LevelsRISK ASSESSMENT LEVEL
PRODUCT TESTING
OPERATIONS RAW MATERIALS
DEVIATION MANAGEMENT
PRODUCT RELEASE
HIGH All testing by Company– third party is not eligible for inclusion in the reduced testing program
Full time presence of Company overseeing third party operations
Materials supplied and cleared by Company
All deviations/ changes reviewed and approved by Company
Release by Company
MEDIUM All testing by receiving sites.Third party is not eligible for inclusion in the reduced testing program
Companies’ personnel only present at critical stages (e.g. validations)
Company specifies sources but third party may clear for use
Only designated changes (listed in Quality Agreement appendix) are approved by Company and deviations are reviewed by Company
Release by Company
LOW Third party is eligible for inclusion in the reduced testing program
No Company presence other than routine audits and visits
Materials sourced and controlled by third party unless otherwise specified in the Quality Agreement
Company reviews deviations and changes as defined in the Quality Agreement
Release by Company


Questions

Current Challenges with Data Integrity and Managing Suppliers
A Changing, Global Environment
©2014 Lachman Consultant Services, Inc. All rights reserved.
Roy Sturgeon
President
Lachman Consultants
Westbury, NY 11590

Legal Notice
The information displayed on these presentation slides is for the sole private use of the attendees of the seminar at which these slides were presented. Lachman Consultant Services, Inc. (“Lachman Consultants”) makes no representations or warranties of any kind, either express or implied, with respect to the contents and information presented. All original contents, as well as the compilation, collection, arrangement and assembly of information provided on these presentation slides, including but not limited to the analysis and examination of information herein, are the exclusive property of Lachman Consultants protected under copyright and other intellectual property laws. These presentation slides and the content and information contained herein may not be displayed, distributed, reproduced, modified, transmitted, used or reused, without the express written permission of Lachman Consultants.
31

Agenda
Supplier Quality Management Process
Data Integrity and 21 CFR Part 211
Potential Regulatory Consequences
Laboratory Audit Methodology
Proactive Action Plan
32

Has Something Changed?
Increased Use of CMOs,
Globally
Requires Proactive
Management of CMOs
33
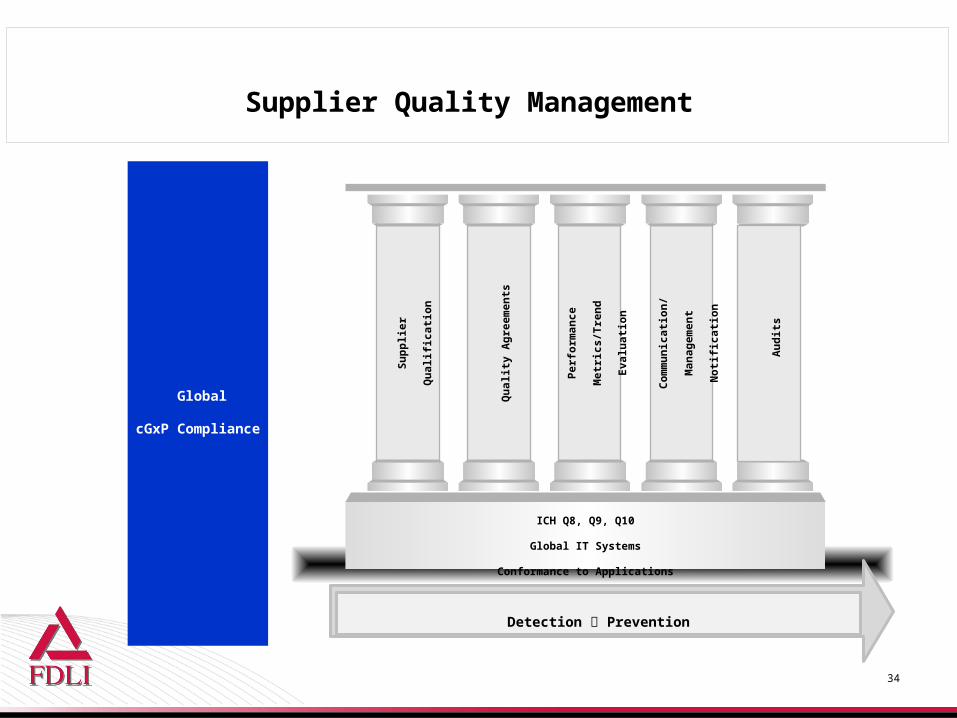
Supplier Quality Management
ICH Q8, Q9, Q10
Global IT Systems
Conformance to Applications
Su
pp
lie
r Q
ua
lifi
ca
tio
n
Qu
ali
ty A
gre
em
en
ts
Pe
rfo
rma
nc
e
Me
tric
s/T
ren
d E
va
lua
tio
n
Co
mm
un
ica
tio
n/
Ma
na
ge
me
nt
No
tifi
ca
tio
n
Au
dit
s
Global
cGxP Compliance
Detection Prevention
34

Critical Success Factor-Supplier Quality
Management-Data Integrity Detection
35

Integrity
A Hierarchy of Drivers. Digital image. Web. 24 Nov. 2014. <http://thebestbrew.files.wordpress.com/2009/06/hierarchyofdrivers_small.jpg>.
36

GMP Regulatory Requirements for Data
Integrity Per 21CFR 211
Instruments must be qualified and fit for purpose [§211.160(b), §211.63] Software must be validated [§211.63] Any calculations used must be verified [§211.68(b)] Data generated in an analysis must be backed up [§211.68(b)] Reagents and reference solutions are prepared correctly with appropriate records
[§211.194(c)] Methods used must be documented and approved [§211.160(a)] Methods must be verified under actual conditions of use [§211.194(a)(2)] Data generated and transformed must meet the criterion of scientific soundness
[§211.160(a)] Test data must be accurate and complete and follow procedures [§211.194(a)] Data and the reportable value must be checked by a second individual to ensure
accuracy, completeness and conformance with procedures [§211.194(a)(8)]
37

Potential Regulatory Consequences of Fraud
Stop Production/Distribution … some or all products based on nature of evidence and results of ongoing District and Company/3rd-party audits
“Voluntary” Recall (Class II) of affected products
Downgrade Therapeutic Equivalence Rating … promptly changed the product’s therapeutic equivalence rating to “not equivalent”, thereby becoming non-interchangeable and without a market
Withdraw ANDA Approval … initiated administrative proceedings to withdraw ANDA approval … unless “voluntarily” withdrawn
Suspension of Future Approvals … OGD continued review of pending ANDAs, supplements and annual reports for products not directly implicated; however, final approval withheld based on general integrity concerns (“Alert List”)
Application Integrity Policy … CDER Director notifies company that all NDA/ANDA reviews have been suspended pending the company’s compliance with the conditions of the AIP
38

What to Look for When Assessing Suppliers?
Fraud - The Big Three: Altered Data
Overwriting of data in chromatography data systemsManipulation of integrations to achieve a passing result
Omitted DataSelective reporting of data for release decisionsUndocumented Sample Trial Injections
Manufactured DataCreation of replacement or dummy weight tapes
39

Audit Methodology
Use the element of surprise:
• Visit laboratory areas as soon as possible and unannounced if possible.• Spend the majority of the audit time in the laboratory before conducting
the more methodical lab systems audit (SOP reviews etc.)• Shift direction of coverage at times when possible rather than following
the sequence of unit operations.• Target for review difficult or complex methods and systems in the lab,
and those associated with deviations, OOS investigations, complaints, FARs, etc.
• As each area in the lab is examined, interview an analyst and/or supervisor. The purpose is to look for evidence of improper data handling, documentation and other practices, and whether the analyst/supervisor have adequate understanding of relevant GMP practices.
40

Audit Methodology (Cont’d)
As soon as possible, visit the laboratory (or other target area) to observe operations and identify targets of interest for immediate evaluation.
Areas that should be targeted include Raw data vs. reports vs. sample/instrument logs vs. other forms of secondary
documentation Identification and control of samples, aliquots, and standards Contemporaneous entries versus delayed or back-dating Condition and control of instrumentation; Suitability of the lab environment; Applicable Part 11 requirements; and Rigor of QC/QA independent data review procedures and practices
41

Audit Methodology:
Computer Systems & Electronic Records
Determine if standalone and networked systems are subject to Part 11 controls. Initially, target standalone instruments to determine nature of testing performed and data being stored, potential for overwriting data, backup practices, audit trails, access privileges, password protection, data review practices, etc.
For standalone systems, check Nature of data stored and, who has access, how the system is
being used If any application programs reside on the system, such as Word,
Access or Excel, and if so, check for unauthorized storage of data, reports, production or test records, SOPs, etc. If these programs exist, ask the user to log onto the system and open document folders
42

Audit Methodology:
Computer Systems & Electronic Records (cont’d)
Check procedures for granting user privileges and whether approvals are documented. Check whether privileges are updated when employees change
position or terminate their employment.
Check on use of individual Passwords, User IDs, Electronic Signatures, and verify they are not being shared.
Check on frequency of system back-ups, whether at appropriate intervals (e.g., immediate, daily).
43

Audit Methodology:
Computer Systems & Electronic Records (cont’d)
Ask users to log onto networked and standalone systems, and check audit trail functions Verify that the audit trail function cannot be overridden or turned off.
Determine if electronic data can be modified, overwritten or deleted from network or standalone systems. Determine if QC or other units have the ability to modify or delete data. Determine the role of the IT coordinator in managing electronic data.
Determine who has the ability to make changes to hardware/software configurations in standalone and networked systems and whether changes are being documented and approved in accordance with change control procedures, and are appropriately validated.
44

Audit Methodology:
Lab Documentation Practices
Review documents, logs and records on lab benches, desktops, shelves, etc., especially those that are in use, to determine if entries appear to be complete, contemporaneous and, where possible, consistent with instrument outputs and settings.
Carefully examine laboratory balance weight tapes. Are all tapes and type on tapes the same or similar color? For a particular set of related weighings, are the elapsed times even possible (Six weight determinations on an analytical balance in 60 seconds.)? Are the weights recorded “too good to be true” (four weight determinations exactly the same in 60 – 120 second time period)?
Open drawers, lockers and physical folders to look for test reports, raw data, obsolete SOPs and test methods, and handwritten entries on docs indicative of not directly entering data into official records (e.g., instrument parameters; weights; results; etc.)
45

Audit Methodology:
Lab Documentation Practices (cont’d)
Examine waste containers to determine whether contents include original data, instrument output, test records, test reports, etc.
Determine where documents intended for shredding are stored; examine documents that are pending shredding. There should be no routine access to paper shredders in the laboratory.
Observe from a distance whether analysts and supervisors appear to be making contemporaneous entries into records.
46

Audit Methodology:
Sample Custody
Look for samples in drawers, lockers, refrigerators and other locations where storage of samples is unexpected.
Look for samples, aliquots and vials that are not adequately labeled.
Open refrigerators and freezers to evaluate conditions, settings, samples, vials, etc., and look for inadequate sample identification and/or storage conditions.
Test and stability sample reconciliation.
47

Audit Methodology:
Training Program Review overall adequacy of the training program (e.g., individual
employee training records, curricula, training schedules, course content, assessments).
Check training records of individual employees who are associated with manufacturing deviations, and with deficiencies identified during the course of the audit.
Check training program and training records for temporary/contract employees.
Review training programs and records of employee training related to computer security (e.g., Part 11 requirements, password use, user access, record retention, change control, use of PCs/standalone computers).
Review the training program and procedures for Good Documentation Practices to determine if the issues of Authenticity and Falsification data are adequately and forcefully addressed.
48

Audit Methodology:
Data Integrity Audit Interviews Examples of Interview Questions:
What is this record, and what is being entered? What is the source of the information that is being entered? When and How was the information determined? How soon after the information is determined are the entries made? What source documentation or instrument outputs support these entries? How do you become aware of missing or mistaken entries after the fact? How do you correct missing or mistaken entries? What are the applicable procedures for preparing this documentation? Are the procedures clear and consistent with actual practice? Have you been trained on the current procedures? Do you and the other analysts use the same practices? Is there anything else you would like to explain?
49

Points to Consider
When Evaluating Data Integrity Findings What systems are deficient or require improvement in the
findings presented in the following section?
Is the finding a case of Data Integrity or bad Laboratory practices/GMP non-compliance? Or both?
What is the potential impact of the finding?
If incidence of GMP compliance, how could it potentially impact the integrity of the data?
For each finding, what action might be taken?
50

Preparation of Proactive Action Plan
Based on the information presented, it should be possible to prepare a proactive Action Plan for Data Integrity
Action Plan starts with an outline of planned activities by system, especially in the laboratory
Identification of lab systems requiring enhancements can be identified by auditing the high risk areas discussed today.
• Most difficult area to audit is the identification of trial injections, if any. Typically, this requires a focused dedicated forensic assessment of chromatography data systems and audit trails.
51

Preparation of Proactive Action Plan (Cont’d)
Plan would take the form of System by System listing of areas (using the laboratory as an example), including but not limited to:• Chromatography Data Systems – Security & Controls• Standalone Instruments• Balance Usage• Sample custody and reconciliation• Chromatographic Data – Trial Injections
For each system the gaps identified by the recommended assessment and the associated action steps to address the gaps should be documented, along with the resources assigned to corrective and preventative actions.
52

Thank You!
Frances M. Zipp
President
Westbury, NY 11590
516-222-6222
Email:[email protected]
www.LachmanConsultants.com
53