telomerase activates transcription of cyclin d1 gene...
TRANSCRIPT

RESEARCH ARTICLE
Telomerase activates transcription of cyclin D1 gene through aninteraction with NOL1Juyeong Hong1, Ji Hoon Lee1,2 and In Kwon Chung1,2,*
ABSTRACTTelomerase is a ribonucleoprotein enzyme that is required for themaintenance of telomere repeats. Although overexpression oftelomerase in normal human somatic cells is sufficient to overcomereplicative senescence, the ability of telomerase to promotetumorigenesis requires additional activities that are independent ofits role in telomere extension. Here, we identify proliferation-associated nucleolar antigen 120 (NOL1, also known as NOP2) asa telomerase RNA component (TERC)-binding protein that is found inassociation with catalytically active telomerase. Although NOL1 ishighly expressed in the majority of human tumor cells, the molecularmechanism by which NOL1 contributes to tumorigenesis remainedunclear. We show that NOL1 binds to the T-cell factor (TCF)-bindingelement of the cyclin D1 promoter and activates its transcription.Interestingly, telomerase is also recruited to the cyclin D1 promoter ina TERC-dependent manner through the interaction with NOL1,further enhancing transcription of the cyclin D1 gene. Depletion ofNOL1 suppresses cyclin D1 promoter activity, thereby leading toinduction of growth arrest and altered cell cycle distributions.Collectively, our findings suggest that NOL1 represents a new routeby which telomerase activates transcription of cyclin D1 gene, thusmaintaining cell proliferation capacity.
KEY WORDS: Telomerase, NOL1, Cyclin D1, Transcriptionalactivation, Tumor cell marker
INTRODUCTIONTelomeres, the specialized nucleoprotein complexes located at theends of eukaryotic chromosomes, are essential for maintenance ofchromosome stability and genome integrity (Blackburn, 2001;Smogorzewska and de Lange, 2004). Telomeric DNA is tightlyassociated with the six-subunit protein complex shelterin, whichprevents chromosomal ends from being recognized as DNAdamage (Palm and de Lange, 2008; Sfeir and de Lange, 2012). Inthe absence of a telomere maintenance pathway, most humansomatic cells show a progressive loss of telomeric DNA with eachround of cell division due to the end replication problem (Lingneret al., 1995; Blasco et al., 1997). The maintenance of telomererepeats in most eukaryotic organisms requires telomerase, whichadds telomere repeats onto the 3′ ends of linear chromosomes byreverse transcription (Autexier and Lue, 2006; Bianchi and Shore,2008). Human telomerase consists of telomerase reversetranscriptase (hTERT), telomerase RNA component (TERC) and
several additional proteins including dyskerin, TCAB1 (alsoknown as WRAP53), pontin and reptin (Egan and Collins,2012; Venteicher et al., 2008, 2009). Telomerase expression isvery low in most human somatic cells but upregulated in manyhuman cancer cells and stem cells, suggesting that activation oftelomerase supports the continued cell proliferation (Kim et al.,1994).
Although overexpression of telomerase is sufficient to overcomereplicative senescence (Bodnar et al., 1998), recent studies havesuggested that besides its reverse transcriptase activity, telomerasehas the noncanonical functions that contribute to cancerdevelopment and progression (Stewart et al., 2002; Li andTergaonkar, 2014). Ectopic expression of telomerase in humanmammary epithelial cells results in enhanced expression ofgrowth-promoting genes (Smith et al., 2003). Transgenicinduction of TERT in mouse skin epithelium has been shown tocause proliferation of quiescent stem cells (Sarin et al., 2005). Thisfunction for TERT is independent of reverse transcriptase activity(Choi et al., 2008). In addition, TERT has been found to directlyinteract with BRG1 (also known as SMARCA4) and activatetranscription of Wnt/β-catenin-dependent genes such as cyclin D1and Myc (Park et al., 2009). However, the proposed noncanonicalrole of TERT in the Wnt/β-catenin signaling cascade has beencontroversial. Several studies have reported a lack of physicalassociation of TERT with BRG1 or β-catenin (Listerman et al.,2014), as well as no apparent effect of TERT deficiency onphenotypes associated with Wnt signaling in TERT-knockout mice(Strong et al., 2011). Although TERT appears to regulate theexpression of growth-promoting genes, this event might not besolely promoted by Wnt signaling. Indeed, TERT has beenreported to bind to the NF-κB p65 subunit (also known as RelA)and activate NF-κB-dependent gene expression (Ghosh et al.,2012).
Given that the large size of human telomerase suggests theexistence of additional components, we performed a large-scaleaffinity purification to identify proteins that interact withtelomerase. Here, we identify proliferation-associated nucleolarantigen p120 (NOL1, also known as NOP2) as a TERC-bindingprotein. NOL1 was originally identified as a RNA-binding andnucleolar-specific protein that is highly expressed in the majorityof human malignant tumor cells but not in normal resting cells(Ochs et al., 1988; Jhiang et al., 1990; Fonagy et al., 1992, 1993).Although NOL1 has been implicated as a tumor cell marker(Gorczyca et al., 1992), the molecular mechanism by which NOL1contributes to tumorigenesis is poorly understood. We show thatNOL1 binds to the T-cell factor (TCF)-binding element (TBE) ofthe cyclin D1 promoter and activates its transcription. Telomeraseis also recruited to the cyclin D1 promoter through the interactionwith NOL1, further enhancing transcription of cyclin D1 gene.These results suggest a new role for telomerase as a modulator ofNOL1-dependent transcriptional activation in human cancer cells.Received 25 September 2015; Accepted 15 February 2016
1Department of Integrated Omics for Biomedical Science, College of Life Scienceand Biotechnology, Yonsei University, Seoul 120-749, Korea. 2Department ofSystems Biology, College of Life Science and Biotechnology, Yonsei University,Seoul 120-749, Korea.
*Author for correspondence ([email protected])
1566
© 2016. Published by The Company of Biologists Ltd | Journal of Cell Science (2016) 129, 1566-1579 doi:10.1242/jcs.181040
Journal
ofCe
llScience

RESULTSIdentification of NOL1 as an hTERT-interacting factorTo identify proteins that interact with hTERT, we expressed Flag-tagged hTERT in HEK293 cells and isolated hTERT complex usinglarge-scale affinity purification. Proteins co-purified with Flag-hTERT were identified by nano-liquid chromatography-tandemmass spectrometry (nano LC-MS/MS). Among the knowntelomerase components, TCAB1 and nucleolin were enriched inthe hTERT complex (Fig. 1A). In addition, analysis of a bandmigrating with an approximate relative molecular mass of 120 kDawas identified as NOL1, a highly conserved, nucleolar-specificRNA-binding protein (Ochs et al., 1988; Jhiang et al., 1990). Giventhat NOL1 has been detected in proliferating tissues but not innormal resting cells, it has been implicated as a tumor cell marker.Thus, we wanted to investigate the role of NOL1 in telomerasefunction.To determine whether hTERT and NOL1 associate in vivo,
HEK293 cells were co-transfected with Flag–hTERT and NOL1–
V5 expression vectors and subjected to immunoprecipitation.NOL1–V5 was specifically bound to Flag–hTERT that wasimmunoprecipitated from HEK293 cells (Fig. 1B). Reciprocalimmunoprecipitation showed that Flag–hTERT was detected inanti-V5 immunoprecipitates, indicating that hTERT associateswith NOL1 in mammalian cells. Interestingly, the interactionbetween Flag–hTERT and NOL1–V5 was disrupted by RNase Atreatment of the extract, which degrades TERC. EndogenousNOL1 was immunoprecipitated by endogenous hTERT, andthis association was also disrupted by RNase A treatment(Fig. 1C), suggesting that NOL1 can associate with hTERTthrough TERC binding in intact cells. These findings werefurther verified by immunoprecipitation experiments with U2OScells, which lack endogenous hTERT and TERC (Jegou et al.,2009). In this cellular background, Flag–hTERT did not interactwith NOL1-V5 owing to a lack of TERC (Fig. 1D). Takentogether, these results suggest that NOL1 is a new TERC-bindingprotein.
Fig. 1. Identification of NOL1 as an hTERT-interacting protein. (A) Lysates fromHEK293 cells expressing Flag–hTERT wereimmunoprecipitated with anti-Flag antibodyand assayed for protein binding byCoomassie staining of a SDS-PAGE gel.Binding proteins were identified by nano-LC-MS/MS. The position of molecular sizemarkers are shown in kDa. Seventeenunique peptides for NOL1 identified from themass spectrometry are shown. (B) HEK293cells expressing Flag–hTERT and NOL1–V5were subjected to immunoprecipitation (IP)with anti-Flag and anti-V5 antibodies,followed by immunoblotting with anti-V5 andanti-Flag antibodies. The indicated extractswere treated with 0.1 mg/ml RNase A duringimmunoprecipitation to degrade TERC.(C) HEK293 cells were subjected toimmunoprecipitation with anti-hTERTantibody, followed by immunoblotting withanti-NOL1 antibody. IgG was used as anegative control. (D) Telomerase-negativeU2OS cells expressing Flag–hTERT andNOL1–V5 were subjected toimmunoprecipitation with anti-Flag antibody,followed by immunoblotting with anti-V5antibody.
1567
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 1566-1579 doi:10.1242/jcs.181040
Journal
ofCe
llScience

NOL1 associates with hTERT in a TERC-dependent mannerTo determine the domain in hTERT that is responsible forNOL1 interaction, we assessed binding of NOL1–V5 by
immunoprecipitating a series of deletion fragments ofhTERT (Fig. 2A). As shown in Fig. 2B, NOL1–V5 wasimmunoprecipitated only by the hTERT fragments containing
Fig. 2. See next page for legend.
1568
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 1566-1579 doi:10.1242/jcs.181040
Journal
ofCe
llScience

amino acid residues 1–589. Because this region contains theTERC-binding domain (TRBD), we examined whether theTRBD is required for NOL1 binding to hTERT. The resultsshowed that removing the TRBD on hTERT abolishedNOL1 binding (Fig. 2C,D), further supporting the idea thatthe association of NOL1 with hTERT is dependent on TERC.To map the region in NOL1 that is required for TERCbinding, we generated deletion constructs lacking a coiled-coildomain or a putative rRNA methyltransferase (MTase) domain(Fig. 2E) (Koonin, 1994; Gustafson et al., 1998). Flag–hTERT
immunoprecipitated NOL1 fragments encompassing aminoacid residues 380–583 (Fig. 2F), indicating that the rRNAmethyltransferase domain is essential for TERC binding.
The finding that NOL1 associates with hTERT through TERCwasfurther verified by immunoprecipitation experiments. HEK293 cellswere transfected with either NOL1–V5 or NOL1-E–V5 (the minimalTERC-binding domain) and subjected to immunoprecipitation,followed by semi-quantitative RT-PCR to detect TERC. The resultsshowed that TERC was specifically immunoprecipitated by NOL1-V5 and NOL1-E-V5 (Fig. 3A). Dyskerin, which has been shown tointeract with TERC, was used as a positive-binding control (Lee et al.,2014). To further demonstrate that NOL1 directly interacts withTERC, we performed a GST pulldown assay with in vitro transcribedTERC. GST–NOL1-E, but not the control GST protein, boundefficiently to in vitro transcribed TERC, as did GST–dyskerin(Fig. 3B).
NOL1 associates with catalytically active telomerase butdoes not affect telomerase enzymatic activityGiven that NOL1 associates with hTERT through TERC binding,we determined whether NOL1 is a telomerase holoenzyme subunit.HEK293 cells were co-transfected with NOL1–V5 and Flag–hTERT, Flag–TCAB1 or Flag–dyskerin, and subjected toimmunoprecipitation. NOL1–V5 was immunoprecipitated byFlag–TCAB1 and Flag–dyskerin, as observed for Flag–hTERT(Fig. 4A). We next examined whether NOL1 associates withcatalytically active telomerase. HEK293 cells expressing Flag–NOL1 or other Flag-tagged telomerase components were subjectedto immunoprecipitation with anti-Flag antibody and analyzed fortelomerase activity by a telomeric repeat amplification protocol(TRAP) assay. Immunoprecipitates of Flag–NOL1 containedtelomerase activity (Fig. 4B), as did those of Flag–hTERT, Flag–
Fig. 3. NOL1 directly interacts with TERC. (A) Lysates from HEK293 cells expressing NOL1–V5 or NOL1-E–V5 or Flag–dyskerin were immunoprecipitated(IP) with anti-V5 or anti-Flag antibodies, followed by immunoblotting (IB) with anti-V5 and anti-Flag antibodies and semi-quantitative RT-PCR to detect TERC. TheV5–NOL1 fragments and Flag–dyskerin are indicated by arrows. The asterisks mark the positions of nonspecific immunoglobulin chains. The position ofmolecular size markers are shown in kDa. (B) GST, GST–NOL1-E or GST–dyskerin were immobilized on glutathione–Sepharose and incubated with in vitrotranscribed TERC. Bound TERC was detected by semi-quantitative RT-PCR. The purified GST fusion proteins were visualized by Coomassie Blue staining.In vitro transcribed TERC was stained with ethidium bromide.
Fig. 2. Identification of the domains in NOL1 and hTERT that are requiredfor their interactions. (A) Schematic representation of the region of hTERTinvolved in NOL1 binding. The approximate positions of the reversetranscriptase motifs analyzed are indicated. TRBD, TERC-binding domain.(B) Lysates from HEK293 cells expressing the various Flag–hTERT fragmentsand NOL1-V5 were immunoprecipitated with anti-Flag antibody, followed byimmunoblotting (IB) with anti-NOL1 antibody. The Flag–hTERT fragments areindicated by arrows. The asterisks mark the positions of nonspecificimmunoglobulin chains. The position of molecular size markers are shown inkDa. (C) Schematic representation of the mutant construct of hTERT in whichthe TRBD was deleted (ΔTRBD). The approximate positions of the reversetranscriptase motifs analyzed are indicated. (D) Removing the TRBD onhTERT abolishes NOL1 association. Lysates from HEK293 cells expressingNOL1–V5 and either Flag–hTERT or Flag–ΔTRBD were immunoprecipitated(IP) with anti-Flag antibody, followed by immunoblotting with anti-V5 antibody.(E) Schematic representation of the region of NOL1 involved in hTERT binding.The approximate positions of the nuclear localization signal (NLS), coiled-coildomain and putative rRNA methyltransferase (MTase) motif are indicated.(F) Lysates from HEK293 cells expressing the various NOL1–V5 domains andFlag–hTERT were immunoprecipitated with anti-Flag antibody, followed byimmunoblotting with the anti-V5 antibody. The NOL1–V5 domainsimmunoprecipitated with Flag–hTERT are indicated by arrows. The asterisksmark the positions of nonspecific immunoglobulin chains. The position ofmolecular size markers are shown in kDa.
1569
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 1566-1579 doi:10.1242/jcs.181040
Journal
ofCe
llScience

TCAB1 and Flag–dyskerin, indicating that NOL1 is a component ofcatalytically active telomerase.To examine an involvement of NOL1 in telomerase function, the
expression of endogenous NOL1 was stably depleted in HeLa S3cells using short hairpin RNA (shRNA) produced from a retroviralvector. NOL1-knockdown cells maintained the reduced levels ofNOL1 throughout the duration of the experiments (see below).Depletion of NOL1 did not affect the levels of telomerasecomponents (Fig. 4C) and shelterin proteins (Fig. 4D). We alsofound that depletion (Fig. 4E) or overexpression of NOL1 (Fig. S1)did not affect telomerase activity. Taken together, these resultsindicate that although NOL1 associates with catalytically activetelomerase, it has no direct regulatory effect on telomeraseenzymatic activity.
NOL1 did not affect the subnuclear localization oftelomeraseTelomerase undergoes a highly elaborate, stepwise processof assembly and trafficking within the nucleus (Lee et al.,2014). If NOL1 is required for assembly and trafficking of activetelomerase, we would expect NOL1 depletion to impair thesubnuclear localization of hTERT. To examine this possibility,we depleted NOL1 in HeLa S3 cells and performed indirectimmunofluorescence staining to determine the subnuclearlocalization of endogenous hTERT. Given that telomerasesynthesizes telomeres specifically during S phase (Lee et al.,2010; Tomlinson et al., 2006), HeLa S3 cells were synchronized atS phase using a double thymidine block (Lee et al., 2010). In theshRNA control cells, the majority of hTERT was found to localize
Fig. 4. NOL1 associates with catalyticallyactive telomerase. (A) Lysates from HEK293cells expressing NOL1–V5, together with Flag–hTERT, Flag–TCAB1 or Flag–dyskerin wereimmunoprecipitated (IP) with anti-Flag antibody,followed by immunoblotting with anti-V5 antibody.(B) Lysates from HEK293 cells expressing Flag–NOL1, Flag–hTERT, Flag–TCAB1 or Flag–dyskerin were immunoprecipitated with anti-Flagantibody and analyzed for telomerase activity bythe TRAP assay. (C) HeLa S3 cells expressingcontrol shRNA (shControl) or NOL1 shRNAs(shNOL1-1 and shNOL1-2) were subjected toimmunoblotting, to measure the protein levels oftelomerase components, and semi-quantitativeRT-PCR, to detect the mRNA levels of telomerasecomponents and TERC. (D) HeLa S3 cellsexpressing shControl or shNOL1 were subjectedto immunoblotting to measure the levels ofshelterin proteins. (E) HeLa S3 cells expressingshControl or shNOL1 were analyzed fortelomerase activity by the TRAP assay. To testRNA-dependent extension, RNaseA (0.25 mg/ml)was added to the extracts before the primerextension reaction when indicated. ITAS, internaltelomerase assay standard.
1570
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 1566-1579 doi:10.1242/jcs.181040
Journal
ofCe
llScience

to nucleoli (Fig. S2A,B). The nucleolar localization of hTERTwas not affected by NOL1 depletion. Telomerase has been shownto accumulate in Cajal bodies prior to telomere elongation(Venteicher et al., 2009; Venteicher and Artandi, 2009). Thus,we determined whether NOL1 depletion affects colocalization ofhTERT with Cajal bodies during S phase. Cajal body localizationof hTERT was not affected by NOL1 depletion, as indicated bydual staining with a coilin-specific antibody (Fig. S2C,D). Theseresults demonstrate that NOL1 does not affect the intranucleartrafficking of hTERT.Dysfunctional telomeres are recognized by the canonical DNA
damage signaling pathway, and the resulting telomere-dysfunction-induced foci (TIFs) represent the foci of DNA damage responsefactors that coincide with telomeres (Takai et al., 2003; D’Adda diFagagna et al., 2003). To determine the role of NOL1 in telomere-damage pathway, the telomeric foci for 53BP1 and thephosphorylated H2AX marker (γH2AX) were examined inNOL1-knockdown cells. Depletion of NOL1 did not induce moretelomere-damage foci in the nucleus compared to the control cells(Fig. S3), indicating that NOL1 is not involved in the control of aDNA damage response at telomeres.
NOL1 activates transcription of the cyclin D1 geneNOL1 is a proliferation-related nucleolar protein that is highlyexpressed in the majority of human malignant tissues (Ochs et al.,1988; Jhiang et al., 1990). It is expressed in the early G1 phase andpeaks in S phase (Fonagy et al., 1992, 1993; Gorczyca et al., 1992).Although the expression of NOL1 has been shown to be inducedrapidly following growth stimulation and produce tumors in thenude mice (Perlaky et al., 1992), the mechanism by which NOL1exerts these effects is poorly understood. To investigate the role ofNOL1 in the control of cell cycle and cell proliferation, weexamined the effect of NOL1 depletion on the expression of cell-cycle-dependent and proliferation-controlling genes such as cyclinD1 and Myc (Musgrove et al., 2011; Sears, 2004). Interestingly,depletion of NOL1 caused a clear reduction in the expression ofcyclin D1 as shown by immunoblot analysis, as well as in the levelsof cyclin D1 mRNA as demonstrated by semi-quantitative RT-PCRexperiments, suggesting that NOL1 regulates the expression ofcyclin D1 gene at the transcription level (Fig. 5A). By contrast, theexpression of c-Myc was not affected by NOL1 depletion,suggesting that the effect of NOL1 is specific to the promoter. Wealso found that overexpression of NOL1 increased the expression ofcyclin D1 but did not influence the expression of c-Myc (Fig. S1A).It has been reported that telomerase modulates Wnt/β-cateninsignaling by acting as a transcriptional cofactor at Wnt target genes(Park et al., 2009). Thus, we examined whether NOL1-mediatedtranscriptional activation of cyclin D1 gene is dependent ontelomerase. The effect of NOL1 depletion on the cyclin D1expression was examined in telomerase-negative U2OS cells.Essentially, similar results to those seen in telomerase-positiveHeLa S3 cells were obtained in this cell line (Fig. 5B), indicatingthat NOL1-dependent activation of cyclin D1 transcription occursregardless of telomerase expression.
Telomerase stimulates transcription of cyclin D1 genethrough the interaction with NOL1The cyclin D1 promoter contains several distinct transcription-factor-binding sites targeted by different signaling pathways (Pestellet al., 1999). To determine the NOL1-responsive element, a series of5′ cyclin D1 promoter deletion constructs were transfected in thepresence of NOL1 expression. As shown in Fig. 5C, cyclin D1
transcription was induced about four-fold by NOL1 overexpressioncompared to the vector control. The proximal 85-base region, whichcontains the TBE, was essential for activation of the cyclin D1promoter, suggesting that the TCF-binding site is a NOL1-responsive element.
We next examined the effect of NOL1 overexpression on TCFtranscriptional activity by transfecting cells with the TCF-sensitiveluciferase reporter vector (TOP-Flash) or TCF-insensitive controlvector (FOP-Flash) in HeLa CCL2 cells. The results showed thatectopic expression of NOL1 increased TOP-Flash activity but notFOP-Flash activity (Fig. 5D). Interestingly, ectopic expression ofhTERT alone also induced TOP-Flash activity, which was furtherincreased by co-expression of NOL1. To verify these findings,telomerase-positive H1299 and MCF7 cells were transfected withthe −964 promoter luciferase reporter vector together with NOL1–V5 or Flag–hTERT or both. Overexpression of either NOL1 orhTERT led to an increase in cyclin D1 promoter activity comparedto the vector control (Fig. 5E,F). When both proteins were co-expressed, we observed an additive effect on cyclin D1 promoteractivity. These results suggest that both NOL1 and hTERT arerequired for efficient cyclin D1 transcription.
Although NOL1 stimulates transcription of the cyclin D1 geneindependently of hTERT, it is unclear whether hTERT alone issufficient to activate cyclin D1 promoter activity without NOL1binding. To test this possibility, dependence of NOL1 wasexamined in telomerase-negative U2OS cells. As shown inFig. 5G, cyclin D1 promoter activity was increased by NOL1overexpression but not by hTERT overexpression. Moreover, theadditive effect was not observed upon overexpression of bothproteins. These results could be due to a lack of TERC in U2OScells. Taken together, these data suggest that telomerase promotestranscription of cyclin D1 gene through the interaction with NOL1.
BothNOL1 and hTERTassociatewith the cyclin D1 promoterat the TBETo determine whether both NOL1 and hTERT are recruited tothe TBE of cyclin D1 promoter, we carried out chromatinimmunoprecipitation (ChIP). HeLa S3 cells expressing Flag–NOL1(or empty vector) and hTERT shRNAs (or control shRNA) werecross-linked with formaldehyde, followed by immunoprecipitationwith anti-Flag antibody. The immunoprecipitated chromatin wasused as a template to amplify the TBE in the cyclin D1 promoter. Theresults showed that anti-Flag antibody immunoprecipitated theTBE-containing fragments when Flag–NOL1 was overexpressed(Fig. 6A). This TBE signal was not altered by depletion ofendogenous hTERT, indicating that NOL1 binds to the TBEindependently of hTERT. No amplification was observed when theimmunoprecipitated chromatin was used to amplify the 3′-untranslated region (3′-UTR) (Fig. 6A). Because transcriptionalactivation byNOL1 is specific to the promoter, we examinedwhetherNOL1 is recruited to the Myc promoter and found that the Flag–NOL1 ChIP signal was not detected at the TBE of theMyc promoter(Fig. 6B). We next examined the effect of hTERT depletion on thecyclin D1 transcription. In HeLa CCL2 cells expressing emptyvector, depletion of hTERT reduced cyclin D1 promoter activity(Fig. 6C). However, in cells expressing Flag–NOL1, cyclin D1promoter activity was not significantly affected by depletion ofhTERT, further supporting the idea that NOL1 activates transcriptionof cyclin D1 regardless of hTERT expression.
To determine whether the occupancy of the cyclin D1 promoterby hTERT is dependent on NOL1, HeLa S3 cells expressing Flag–hTERT (or empty vector) and NOL1 shRNAs (or control shRNA)
1571
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 1566-1579 doi:10.1242/jcs.181040
Journal
ofCe
llScience

Fig. 5. hTERT stimulates transcription of cyclin D1 gene through the interaction with NOL1. (A) HeLa S3 and (B) U2OS cells expressing control shRNA(shControl) or NOL1 shRNAs (shNOL1-1 and shNOL1-2) were subjected to immunoblotting, to measure the protein levels of cyclin D1 and c-Myc, and semi-quantitative RT-PCR to detect the mRNA levels. (C) The structures of the promoter luciferase (luc) constructs containing various lengths of upstream fragmentsfrom the cyclin D1 gene are shown on the left. The binding sites of known transcription factors are indicated. The results of the luciferase assay are shown on theright. HeLa CCL2 cells were co-transfected with the promoter luciferase constructs together with NOL1–V5 or empty vector. The firefly luciferase activity wasnormalized against the Renilla luciferase activity. Results are mean±s.d. of three independent experiments. (D) The effects of NOL1 and hTERT overexpressionon TCF transcriptional activity. HeLa CCL2 cells were transfected with TOP-Flash or FOP-Flash luciferase reporter vectors together with NOL1–V5, Flag–hTERTor both. The firefly luciferase activity was normalized against the Renilla luciferase activity. Results are mean±s.d. of three independent experiments. (E–G) Theluciferase assay in H1299 (E), MCF7 (F) and U2OS cells (G) co-transfected with the −964 promoter luciferase reporter vector together with NOL1–V5, Flag–hTERT or both. Results are mean±s.d. of three independent experiments. Statistical analyses were performed using a two-tailed Student’s t-test.
1572
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 1566-1579 doi:10.1242/jcs.181040
Journal
ofCe
llScience

were subjected to ChIP. Flag–hTERT associated with the TBE-containing fragment of the cyclin D1 promoter in cells expressingcontrol shRNA (Fig. 6D). When endogenous NOL1 was depleted,the ability of Flag–hTERT to bind to the TBE fragment wasabrogated. These results suggest that hTERT can be recruited to thecyclin D1 promoter through the interaction with NOL1.Intriguingly, the faint but consistent ChIP signal of Flag–hTERTwas detected at the TBE fragment of the Myc promoter in a NOL1-independent manner (Fig. 6E). We also examined the effect ofNOL1 depletion on the cyclin D1 transcription. As expected, cyclinD1 promoter activity was reduced by NOL1 depletion in HeLaCCL2 cells (Fig. 6F). Even in the presence of overexpression ofhTERT, cyclin D1 promoter activity was also reduced by NOL1
depletion (Fig. 6F), suggesting that hTERT activates cyclin D1transcription in a NOL1-dependent manner.
To further validate the co-occupancy of NOL1 and hTERT in thecyclin D1 promoter, we performed a ChIP-re-ChIP assay (Qiu et al.,2013). HeLa S3 cells expressing Flag–hTERT and NOL1–V5 weresubjected to ChIP with anti-Flag antibody, followed by re-ChIPusing anti-V5 antibody. Flag–hTERT alone bound to the cyclin D1TBE (Fig. 7A). Interestingly, this binding was increased by co-expression of NOL1–V5. These results are consistent with theprevious findings that co-expression of both proteins led to anadditive effect on cyclin D1 promoter activity (see Fig. 5E,F). Whena sequential ChIP (re-ChIP) assay was performed, we observed thatFlag–hTERT associates with the cyclin D1 TBE only in the
Fig. 6. NOL1 and hTERT associate with the cyclin D1 promoter at the TBE. (A,B) HeLa S3 cells were transfected with Flag–NOL1 (or empty vector) togetherwith hTERT shRNA (shhTERT-1 or shhTERT2) or control shRNA, and ChIP analyses were performed using anti-Flag antibody. The recruitment of NOL1 to thecyclin D1 TBE (A) orMyc TBE (B) was quantified by performing a gel-based semi-quantitative RT-PCR assay. The 3′-untranslated region (3′-UTR) was used as anegative control. (C) A luciferase assay of cyclin D1 transcription in HeLa CCL2 cells transfected with Flag–NOL1 (or empty vector) together with hTERT shRNA(or control shRNA). The firefly luciferase activity was normalized against the Renilla luciferase activity. Results are mean±s.d. of three independent experiments.(D,E) HeLa S3 cells were transfected with Flag–hTERT (or empty vector) together with NOL1 shRNA (or control shRNA), and ChIP analyses were performedusing anti-Flag antibody. The recruitment of NOL1 to the cyclin D1 TBE (D) orMyc TBE (E) was quantified by performing a gel-based semi-quantitative RT-PCRassay. The 3′-UTR was used as a negative control. (F) A luciferase assay of cyclin D1 transcription in HeLa CCL2 cells transfected with Flag–hTERT (or emptyvector) together with NOL1 shRNA (or control shRNA). The firefly luciferase activity was normalized against theRenilla luciferase activity. Results are mean±s.d.of three independent experiments.
1573
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 1566-1579 doi:10.1242/jcs.181040
Journal
ofCe
llScience
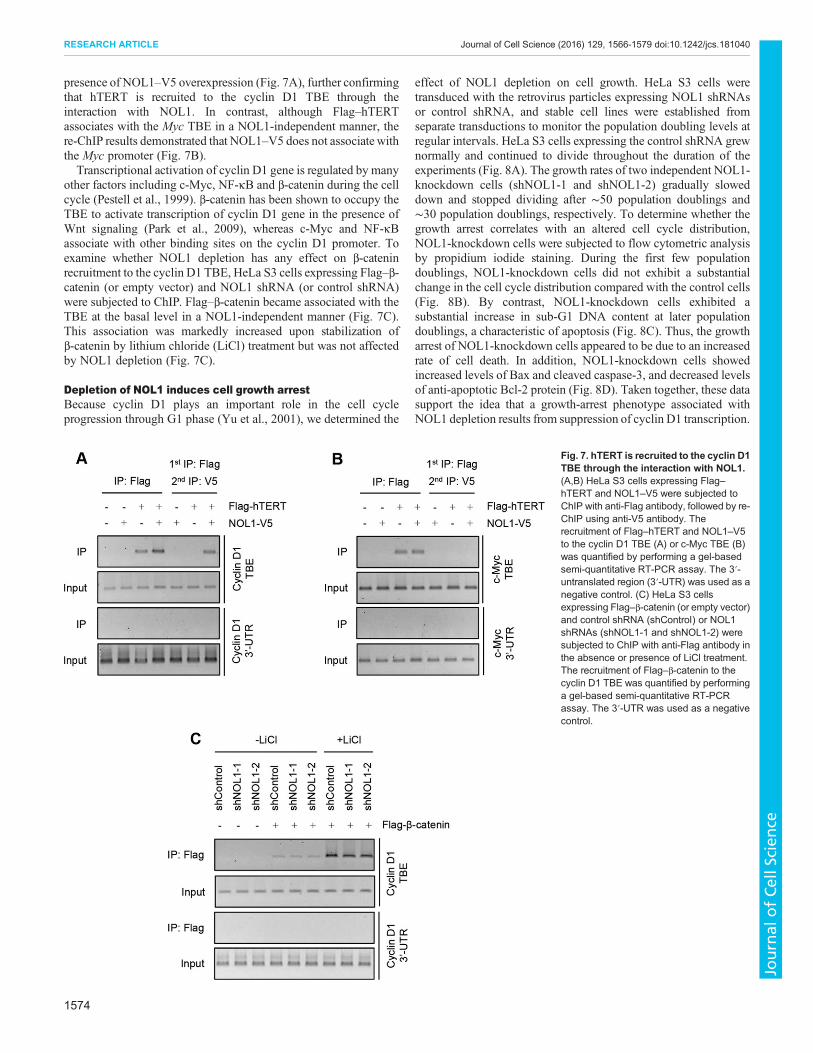
presence of NOL1–V5 overexpression (Fig. 7A), further confirmingthat hTERT is recruited to the cyclin D1 TBE through theinteraction with NOL1. In contrast, although Flag–hTERTassociates with the Myc TBE in a NOL1-independent manner, there-ChIP results demonstrated that NOL1–V5 does not associatewiththe Myc promoter (Fig. 7B).Transcriptional activation of cyclin D1 gene is regulated by many
other factors including c-Myc, NF-κB and β-catenin during the cellcycle (Pestell et al., 1999). β-catenin has been shown to occupy theTBE to activate transcription of cyclin D1 gene in the presence ofWnt signaling (Park et al., 2009), whereas c-Myc and NF-κBassociate with other binding sites on the cyclin D1 promoter. Toexamine whether NOL1 depletion has any effect on β-cateninrecruitment to the cyclin D1 TBE, HeLa S3 cells expressing Flag–β-catenin (or empty vector) and NOL1 shRNA (or control shRNA)were subjected to ChIP. Flag–β-catenin became associated with theTBE at the basal level in a NOL1-independent manner (Fig. 7C).This association was markedly increased upon stabilization ofβ-catenin by lithium chloride (LiCl) treatment but was not affectedby NOL1 depletion (Fig. 7C).
Depletion of NOL1 induces cell growth arrestBecause cyclin D1 plays an important role in the cell cycleprogression through G1 phase (Yu et al., 2001), we determined the
effect of NOL1 depletion on cell growth. HeLa S3 cells weretransduced with the retrovirus particles expressing NOL1 shRNAsor control shRNA, and stable cell lines were established fromseparate transductions to monitor the population doubling levels atregular intervals. HeLa S3 cells expressing the control shRNA grewnormally and continued to divide throughout the duration of theexperiments (Fig. 8A). The growth rates of two independent NOL1-knockdown cells (shNOL1-1 and shNOL1-2) gradually sloweddown and stopped dividing after ∼50 population doublings and∼30 population doublings, respectively. To determine whether thegrowth arrest correlates with an altered cell cycle distribution,NOL1-knockdown cells were subjected to flow cytometric analysisby propidium iodide staining. During the first few populationdoublings, NOL1-knockdown cells did not exhibit a substantialchange in the cell cycle distribution compared with the control cells(Fig. 8B). By contrast, NOL1-knockdown cells exhibited asubstantial increase in sub-G1 DNA content at later populationdoublings, a characteristic of apoptosis (Fig. 8C). Thus, the growtharrest of NOL1-knockdown cells appeared to be due to an increasedrate of cell death. In addition, NOL1-knockdown cells showedincreased levels of Bax and cleaved caspase-3, and decreased levelsof anti-apoptotic Bcl-2 protein (Fig. 8D). Taken together, these datasupport the idea that a growth-arrest phenotype associated withNOL1 depletion results from suppression of cyclin D1 transcription.
Fig. 7. hTERT is recruited to the cyclin D1TBE through the interaction with NOL1.(A,B) HeLa S3 cells expressing Flag–hTERT and NOL1–V5 were subjected toChIP with anti-Flag antibody, followed by re-ChIP using anti-V5 antibody. Therecruitment of Flag–hTERT and NOL1–V5to the cyclin D1 TBE (A) or c-Myc TBE (B)was quantified by performing a gel-basedsemi-quantitative RT-PCR assay. The 3′-untranslated region (3′-UTR) was used as anegative control. (C) HeLa S3 cellsexpressing Flag–β-catenin (or empty vector)and control shRNA (shControl) or NOL1shRNAs (shNOL1-1 and shNOL1-2) weresubjected to ChIP with anti-Flag antibody inthe absence or presence of LiCl treatment.The recruitment of Flag–β-catenin to thecyclin D1 TBE was quantified by performinga gel-based semi-quantitative RT-PCRassay. The 3′-UTR was used as a negativecontrol.
1574
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 1566-1579 doi:10.1242/jcs.181040
Journal
ofCe
llScience

As a control, we also examined the effect of cyclin D1 depletion onthe cell cycle status (Fig. S4A). Depletion of cyclin D1 led to anincrease in the proportion of cells in sub-G1 phase compared to thecontrol cells (Fig. S4B,C).
DISCUSSIONGiven that normal human somatic cells express very low levels oftelomerase, they have a limited proliferative lifespan and ultimatelyenter a non-dividing state of replicative senescence (Bodnar et al.,1998). Although ectopic expression of telomerase is sufficient toextend lifespan, recent studies have suggested that the ability oftelomerase to promote tumorigenesis requires additional activitiesthat are independent of its role in telomere extension (Stewart et al.,
2002; Li and Tergaonkar, 2014). Here, we identify NOL1 as a newTERC-binding protein that is found in association with catalyticallyactive telomerase. Given that NOL1 has no direct regulatory effecton the assembly and trafficking of telomerase or its enzymaticactivity, it is likely that NOL1 is involved in a non-telomericfunction of telomerase. We show that NOL1 activates transcriptionof cyclin D1 gene by binding to the TBE. Telomerase is alsorecruited to the cyclin D1 promoter through the interaction withNOL1, further enhancing transcription of cyclin D1 gene. Thesedata suggest that NOL1 represents a new pathway by whichtelomerase activates transcription of the cyclin D1 gene.
Besides its primary role in telomere extension, telomerase hasbeen demonstrated to have non-canonical functions in signaling
Fig. 8. Depletion of NOL1 induces cell growth arrest. (A) Cell growth curves of HeLa S3 cells stably expressing control shRNA (shControl) or NOL1shRNAs (shNOL1-1 and shNOL1-2). HeLa S3 cells were infected with the retrovirus particles to establish stable cell lines. Stable cells were replated every3–4 days tomaintain log-phase growth and calculate the growth rate, with day 0 representing the first dayafter puromycin selection. (B) Flow cytometric analysis ofHeLa S3 cells stably expressing control shRNA or NOL1 shRNAs. Cells were stained with propidium iodide after a few or and many population doublings (PD),followed by FACS analysis. (C) The percentage of total cells in sub-G1 phase of the cell cycle is shown. Results aremean±s.d. of three independent experiments.(D) HeLa S3 cells stably expressing control shRNA or NOL1 shRNAs were analyzed by immunoblotting to measure the protein levels of Bcl2, Bax and cleavedcaspase-3. (E) Proposed model for the two fates of telomerase during its assembly, telomere extension and transcriptional activation.
1575
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 1566-1579 doi:10.1242/jcs.181040
Journal
ofCe
llScience

pathways that influence human tumorigenesis (Choi et al., 2008;Park et al., 2009; Ghosh et al., 2012). Telomerase has been shown tobind the NF-κB p65 subunit and localize to the promoters of asubset of NF-κB target genes (Ghosh et al., 2012). Inhibition oftelomerase reduces the expression of NF-κB-dependent genes,suggesting that telomerase acts as a transcriptional modulator of theNF-κB signaling cascade in cancer cells. Telomerase has also beenfound to act as a transcriptional modulator of Wnt/β-cateninsignaling pathway and enhance the expression of Wnt target genes(Park et al., 2009). Moreover, overexpression of alternativelyspliced variants that lack telomerase activity stimulate cellproliferation by activating Wnt signaling (Hrdlicková et al.,2012). Given that Wnt signaling target genes are also regulated byother signaling pathways (Guo andWang, 2009), the mechanism bywhich telomerase enhances the expression of growth-promotinggenes cannot be solely dependent on Wnt signaling. Recently, ithas been reported that telomerase regulates Myc-dependentoncogenesis by stabilizing Myc levels on chromatin (Koh et al.,2016). Taken together, these findings suggest that telomerasecontributes to activation of growth-promoting genes throughmultiple signaling pathways in cancer. In this work, we show thattelomerase interacts with NOL1 and promotes transcription of thecyclin D1 gene in the absence of Wnt or NF-κB signaling. WhereasNOL1 alone is sufficient to bind the cyclin D1 promoter andpromote its transcription, hTERT is recruited to the cyclinD1 promoter through its interaction with NOL1, suggesting thattelomerase activates cyclin D1 transcription in a NOL1-dependentmanner. Whereas TERC is not required for telomerase-dependenttranscriptional activation of Wnt/β-catenin signaling, it is essentialfor transcriptional activation by NOL1 and telomerase. Moreover,domain mapping analysis has revealed that the TERC-bindingdomain in hTERT is required for NOL1 binding, further supportingthe idea that the association between NOL1 and hTERT isdependent on TERC. Thus, a functional interplay between NOL1and telomerase modulates the prolonged expression of the cyclinD1 gene that is crucial for the maintenance of cell proliferation.Interestingly, it has been recently reported that non-canonical NF-κB signaling can upregulate mutant hTERT promoter activity alongwith ETS transcription factors (Li et al., 2015). Reactivated hTERTcan induce the binding of NOL1 to the cyclin D1 promoter bysetting a potential feed-forward loop in cell proliferation.NOL1 is a proliferation-related nucleolar protein that is highly
expressed inmostmalignant tumor cells but not in normal resting cells(Ochs et al., 1988; Jhiang et al., 1990). Given that telomerase isinitially assembled in the nucleolus (Lee et al., 2014), the finding thatNOL1 is a new component of catalytically active telomerase suggeststhat the nucleolus could be the site where NOL1 associates with thetelomerase holoenzyme. Based on data presented in this work, wepropose a model for the two fates of telomerase during its initialassembly, telomere extension and transcriptional activation (Fig. 8E).The assembly of the active telomerase holoenzyme occurs in a highlyelaborate, stepwise fashion (Lee et al., 2014). After transcription, aTERC molecule assembles with a preformed dyskerin complex, andthe subsequent assembly of a TERC–dyskerin ribonucleoprotein(RNP) with hTERT occurs specifically during the S phase in thenucleolus. For telomere extension, telomerase associateswithTCAB1and is transported to Cajal bodies (Venteicher et al., 2009; VenteicherandArtandi, 2009).Telomerase-containingCajal bodies are loaded ontelomeric chromatin to elongate telomere repeats. By contrast, whenNOL1 is recruited to the telomeraseRNP in the nucleolus, theNOL1–telomerase complex binds to the cyclin D1 promoter at the TBE.Given that both TCAB1 andNOL1 associatewith telomerase through
the interaction with TERC, these two proteins might compete forbinding to telomerase in the nucleolus. The outcome of thiscompetition likely determines which of the two fates of telomeraseis favored. Although we cannot rule out the possibility that bothproteins exist in the same telomerase RNP, it is yet unclear whatfractions of the telomerase RNP contain NOL1 or TCAB1. Recently,it has been reported that there are several hundred copies of telomeraseRNP in a human cancer cell (Xi and Cech, 2014; Akıncılar et al.,2015). Furthermore, the two telomerase components, hTERT andTERC, appear to be in excess of telomerase RNPs, suggesting theexistence of unassembled telomerase components. Thus, it will beinteresting to investigate howmanymolecules of NOL1 exist in a celland how many of these are associated with TERC.
NOL1 has been shown to be expressed early in the G1 phase andpeaks during the S phase (Fonagy et al., 1992, 1993; Gorczyca et al.,1992). Thus, NOL1-dependent transcriptional activation of cyclinD1 gene might occur in a cell-cycle-dependent manner. Whentelomerase is upregulated in cancer cells, telomerase could interactwith NOL1 and occupy the TBE of the cyclin D1 promoter tofurther enhance gene expression. Thus, NOL1 plays an importantrole in cell cycle progression through G1 phase and is implicated asa tumor cell marker. By contrast, repressing cyclin D1 expression byNOL1 depletion prevents the tumor cells exiting from G1 phase,reversing tumor characteristics. Consistent with this idea, depletionof NOL1 induced a growth arrest in telomerase-positive HeLa S3cells. This growth arrest was accompanied by several featuresconsistent with the induction of apoptosis, including a substantialincrease in sub-G1 DNA content, an increase in the levels of Baxand cleaved caspase-3, and a decrease in the levels of anti-apoptoticBcl2. These findings suggest that NOL1 plays a key role in thecontrol of cell cycle progression through transcriptional activationof cyclin D1 gene. Overall, our results provide an insight into thenew function of NOL1 as an important regulator of cell cycle andcell proliferation, as well as the non-canonical mechanism by whichtelomerase promotes cell proliferation in cancer cells.
MATERIALS AND METHODSCell culture and plasmidsHuman cervical carcinoma HeLa cells and human embryonic kidneyHEK293 cells were grown in Dulbecco’s modified Eagle’s mediumcontaining 10% fetal bovine serum with 100 units/ml penicillin and100 μg/ml streptomycin in 5% CO2 at 37°C. Human osteosarcoma U2OScells were grown in McCoy’s modified medium with 10% fetal bovineserum, 100 units/ml penicillin and 100 μg/ml streptomycin in 5% CO2 at37°C. Theexpressionvectors forNOL1–V5andFlag–NOL1wereconstructedby inserting the full-lengthNOL1 cDNA into pcDNA3.1/V5-His (Invitrogen)and p3xFlag-CMV 7.1 plasmid (Sigma-Aldrich), respectively. The Flag–hTERT expression vector was constructed by cloning the full-length hTERTcDNAinto a pCMV-Tag2 vector (Stratagene). The human cyclinD1promoterluciferase plasmids were constructed by inserting the various promoterfragments into pGL4.20[luc2/Puro] vector (Promega). The expression vectorswere transiently transfected using Lipofectamine-PLUS reagent according tothe manufacturer’s protocol (Invitrogen).
Peptide identification using LC-MS/MSNano LC-MS/MS analysis was performed with a nano HPLC system(Agilent) as previously described (Her and Chung, 2013).
Immunoprecipitation and immunoblottingImmunoprecipitation and immunoblot analyses were performed asdescribed previously (Lee et al., 2004). Briefly, cells were lysed with lysisbuffer (0.5% NP-40, 0.5% Triton X-100, 150 mM NaCl, 2 mM EDTA, pH8.0, 50 mM Tris-HCl, pH 7.5 and 10% glycerol) supplemented with aproteinase inhibitor cocktail (Roche) for 15 min at 4°C, followed by
1576
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 1566-1579 doi:10.1242/jcs.181040
Journal
ofCe
llScience

centrifugation (16,000 g) for 15 min at 4°C. Lysates were pre-cleared withprotein-A–Sepharose beads (GE Healthcare) for 30 min at 4°C. Aftercentrifugation, the supernatants were incubated with primary antibodies at4°C overnight, followed by incubation with protein-A–Sepharose beads for1 h at 4°C. After binding, the beads were washed extensively with lysisbuffer and subjected to immunoblot analysis. Immunoprecipitation andimmunoblotting were performed using anti-Flag (1:10,000, cat. no. F-1804,Sigma-Aldrich), anti-V5 (1:5000, cat. no. 46-0575, Invitrogen), anti-NOL1(1:2000, cat. no. NBP1-92192, Novus Biologicals), anti-cyclin D1 (1:2000,cat. no. ab16663, Abcam), anti-c-Myc (1:1000, cat. no. sc-764, Santa CruzBiotechnology), anti-TCAB1 (1:3000, cat. no. ab99376, Abcam), anti-dyskerin (1:3000, cat. no. sc-48794, Santa Cruz Biotechnology), anti-hTERT (1:1000, cat. no. 600-401-252, Rockland), anti-tubulin (1:1000, cat.no. sc-8035, Santa Cruz Biotechnology), anti-TRF1 (1:1000, cat. no. sc-1977, Santa Cruz Biotechnology), anti-TRF2 (1:1000, cat. no. D1Y5D, CellSignaling), anti-RAP1 (1:3000, cat. no. A300-306A, Bethyl Laboratory),anti-POT1 (1:3000, cat. no. ab21382, Abcam), anti-TPP1 (1:1000, cat. no.ab54685, Abcam), anti-TIN2 (1:1000, cat. no. ab136997, Abcam), anti-Bcl2 (1:1000, cat. no. sc-492, Santa Cruz Biotechnology), anti-Bax(1:1000, cat. no. sc-493, Santa Cruz Biotechnology), anti-cleavedcaspase-3 (1:1000, cat. no. 9661, Cell Signaling) antibodies as specified.All the immunoblots are representatives of at least three experiments, whichdemonstrated similar results.
Semi-quantitative RT-PCRTotal RNA was isolated using Easy BLUE (Intron). The reversetranscription reaction was performed with 1 μg of total RNA using the M-MLV reverse transcriptase (Promega), and cDNA was used for semi-quantitative RT-PCR. The following primers were used: hTERT (forward,5′-CGGAAGAGTGTCTGGAGCAA-3′ and reverse, 5′-GGATGAAGC-GGAGTCTGGA-3′), TERC (forward, 5′-TCTAACCCTAACTGAGAA-GGGCGTAG-3′ and reverse, 5′-GTTTGCTCTAGA ATGAACGGTGG-AAG-3′), dykerin (forward, 5′-ACAGGGTGAAGAGTTCTGGCACAT-3′and reverse, 5′-TGAAGGTGAGGCTTCCCAACTCAA-3′), cyclin D1(forward, 5′-CACACGGACTACAGGGGAGT-3′ and reverse, 5′-CACA-GGAGCTGGTGTTCCAT-3′), c-Myc (forward, 5′-AATGAAAAGGCC-CCCAAGGTAGTTATCC-3′ and reverse, 5′-GTCGTTTCCGCAACAA-GTCCTCTTC-3′), and GAPDH (forward, 5′-CTCAGACACCATGGGG-AAGGTGA-3′ and reverse, 5′-ATGATCTTGAGGCTGTTGTCATA-3′).
GST pulldown assay with in vitro transcribed TERCTERC transcripts were prepared by in vitro transcription using aMAXIscriptT7/T3 Kit and cleared by MEGAclear Kit according to the manufacturer’srecommendation (Ambion). For GST pulldown assays, GST fusion proteins(2 μg) were incubated with in vitro transcribed TERC (200 ng) in reactionbuffer (10 mM HEPES, 3 mM KCl, 150 mM NaCl, 1 mM MgCl2)supplemented with a proteinase inhibitor cocktail (Roche) and RNasin(20 U/ml, Promega). Complexes between GST fusion proteins and TERCwere isolated with glutathione–Sepharose-4B (GE Healthcare) and used forsemi-quantitative RT-PCR to detect TERC.
Telomerase assayThe telomeric repeat amplification protocol (TRAP) was used as previouslydescribed (Kim et al., 2003).
Chromatin immunoprecipitation assayHeLa S3 cells transfected with either Flag–NOL1 or Flag–hTERT werecross-linked with 1% formaldehyde in phosphate-buffered saline (PBS) for30 min at room temperature and neutralized with addition of 125 mMglycine. Cells were lysed in lysis buffer (50 mM Tris-HCl, pH 8.0, 1% SDS,10 mM EDTA) supplemented with a proteinase inhibitor cocktail (Roche).Chromatin was sonicated to obtain chromatin fragments with an average sizeof 600 bp, as assessed by electrophoresis on a 1% agarose gel. Sonicatedsamples were used for ChIP immunoprecipitation with Flag M2 agarosebeads (Sigma-Aldrich) overnight at 4°C and pulled down by centrifugation(16,000 g) for 60 s. Chromatin–Flag-M2-agarose complexes weresequentially washed once with low-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl, pH 8.0), high-salt
buffer (low-salt buffer containing 500 mM NaCl), LiCl buffer (0.25 MLiCl, 0.5% NP-40, 0.5% Na-deoxycholate, 1 mM EDTA, 10 mM Tris-HCl,pH 8.0), and twice with TE buffer. For the re-ChIP assay, immunecomplexes were eluted from the agarose beads using 20 mM dithiothreitoland used for a secondary immunoprecipitation. DNA and proteins wererecovered after incubation in 1% SDS and 0.1 M NaHCO3 at 65°C, for 2 h.Then, NaCl was added to a 10 mM final concentration and protein–DNAcross-links were reversed by incubating samples at 65°C overnight. Finally,samples were digested with proteinase K at 45°C for 1 h. The isolated DNAswere analyzed by semi-quantitative RT-PCR. The primers for amplificationof the TBE and 3′-UTR regions of each gene promoter were as follows:cyclin D1TBE (forward, 5′-CGCTCCCATTCTCTGCCGGG-3′ and reverse,5′-CCGCGCTCCCTCGCGCTCTT-3′), cyclin D1 3′-UTR (forward, 5′-C-AAGAGAAGATTACCGCCCGAG-3′ and reverse, 5′-TCCCCAGCCTTT-TTGACACC-3′), c-Myc TBE (forward, 5′-CGTCTAGCACCTTTGATTT-CTCCC-3′ and reverse, 5′-CTCTGCCAGTCTGTACCCCACCGT-3′), andc-Myc 3′-UTR (forward, 5′-CTAATGTATCACAAAGTCCTTTA-3′ andreverse, 5′-GTGATCTGCTCTGTTAGCTTTTGA-3′).
Dual luciferase reporter assayHeLa cells were transiently co-transfected with cyclin D1 promoterluciferase reporter constructs, together with empty vector or NOL1-V5.At 24 h after transfection, cells were lysed, and firefly and Renilla luciferaseactivities were measured using the dual-luciferase reporter assay system(Promega). The firefly luciferase activity for each sample was normalizedbased on transfection efficiency as measured by Renilla luciferase activity.The results are expressed with the standard deviation from the mean of threeindependent experiments.
Establishment of stable cell linesThe retrovirus vectors were constructed by cloning the shRNA-expressingoligonucleotides targeting NOL1 (5′-GATCCCCGCGTTGCTGCCCAT-TGAAATTTTCAAGAGAAATTTCAATGGGCAGCAACGCTTTTTA-3′for shNOL1-1; 5′-GATCCCCGGACGATGCTGATACGGTATTTTCAA-GAGAAATACCGTATCAGCATCGTCCTTTTTA-3′ for shNOL1-2) intopSUPER.retro.puro vector (OligoEngine). The retrovirus vectors expressingNOL1 shRNAs were co-transfected with pGP (for gag-pol expression) andpE-ampho (for env expression) into HEK293T packaging cells according tothe manufacturer’s instructions (Takara). After 48 h, the culture super-natants were harvested and filtered through a 0.45 μm filter. To generatestable cell lines, HeLa S3 cells were transduced with the viral supernatantscontaining 4 μg/ml polybrene (Sigma-Aldrich). After selection with 1 μg/mlpuromycin (Gibco) for 2 weeks, multiple independent single clones wereisolated and checked for NOL1 expression.
Fluorescence-activated cell sorting analysisHeLa S3 cells were washed with PBS and fixed for 30 min in ice-cold 70%ethanol. The fixed cells were resuspended in PBS containing RNase A(200 μg/ml) and propidium iodide (50 μg/ml), and incubated in the dark for30 min at room temperature. Cell cycle progression was monitored by flowcytometry using a FACScan flow cytometer (BD Biosciences).
Immunofluorescence and telomere fluorescence in situhybridizationCells grown on glass coverslips were fixed, permeabilized and blocked asdescribed previously (Abreu et al., 2011). Cells were incubated with rabbitanti-TERT (500 ng/ml, cat. no. 600-401-252, Rockland), mouse anti-coilin(2 μg/ml, cat. no. ab11822, Abcam), and mouse anti-nucleolin (200 ng/ml,cat. no. sc-17826, Santa Cruz Biotechnology) for 16 h at 4°C. Afterthorough washing with PBS, cells were incubated with Alexa-Fluor-488-conjugated anti-rabbit immunoglobulin (green) (cat. no. A-11034, ThermoFisher) and Alexa-Fluor-568-conjugated anti-mouse immunoglobulin (red)(cat. no. A-11004, Thermo Fisher) for 1 h in the dark. The coverslips weremounted on microscope slides using Vectashield mounting medium withDAPI (Vector Laboratories). Immunofluorescence images were capturedusing a confocal laser-scanning microscope LSM 510 (Carl Zeiss).Telomere FISH staining was performed with Cy3-(CCCTAA)3 PNAprobe (Panagene) as previously described (van Steensel et al., 1998).
1577
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 1566-1579 doi:10.1242/jcs.181040
Journal
ofCe
llScience

AcknowledgementsWe are grateful to Sun Ah Jeong, Prabhat Khadka, Joonyoung Her and Yu YoungJeong for technical assistance and helpful comments on the manuscript.
Competing interestsThe authors declare no competing or financial interests.
Author contributionsJ.H. and J.H.L. designed and performed experiments, analyzed data and assisted inmanuscript preparation. I.K.C. conceived and designed the experiments, analyzeddata and wrote the manuscript.
FundingThis work was supported in part by a grant from the National Research Foundation ofKorea [grant number NRF-2013M3A9B6076413 to I.K.C.]; and by a YonseiUniversity internal grant [grant number 2014-22-0096 to I.K.C.].
Supplementary informationSupplementary information available online athttp://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.181040/-/DC1
ReferencesAbreu, E., Terns, R. M. and Terns, M. P. (2011). Visualization of human telomeraselocalization by fluorescence microscopy techniques. Methods Mol. Biol. 735,125-137.
Akıncılar, S. C., Low, K. C., Liu, C. Y., Yan, T. D., Oji, A., Ikawa, M., Li, S. andTergaonkar, V. (2015). Quantitative assessment of telomerase components incancer cell lines. FEBS Lett. 589, 974-984.
Autexier, C. and Lue, N. F. (2006). The structure and function of telomerase reversetranscriptase. Annu. Rev. Biochem. 75, 493-517.
Bianchi, A. and Shore, D. (2008). How telomerase reaches its end: mechanismof telomerase regulation by the telomeric complex. Mol. Cell 31, 153-165.
Blackburn, E. H. (2001). Switching and signaling at the telomere. Cell 106,661-673.
Blasco, M. A., Lee, H.-W., Hande, M. P., Samper, E., Lansdorp, P. M., DePinho,R. A. and Greider, C. W. (1997). Telomere shortening and tumor formation bymouse cells lacking telomerase RNA. Cell 91, 25-34.
Bodnar, A. G., Ouellette, M., Frolkis, M., Holt, S. E., Chiu, C.-P., Morin, G. B.,Harley, C. B., Shay, J. W., Lichtsteiner, S. andWright,W. E. (1998). Extension oflife-span by introduction of telomerase into normal human cells. Science 279,349-352.
Choi, J., Southworth, L. K., Sarin, K. Y., Venteicher, A. S., Ma, W., Chang, W.,Cheung, P., Jun, S., Artandi, M. K., Shah, N. et al. (2008). TERT promotesepithelial proliferation through transcriptional control of a Myc- and Wnt-relateddevelopmental program. PLoS Genet. 4, e10.
D’Adda di Fagagna, F., Reaper, P. M., Clay-Farrace, L., Fiegler, H., Carr, P., VonZglinicki, T., Saretzki, G., Carter, N. P. and Jackson, S. P. (2003). A DNAdamage checkpoint response in telomere-initiated senescence. Nature 426,194-198.
Egan, E. D. and Collins, K. (2012). Biogenesis of telomerase ribonucleoproteins.RNA 18, 1747-1759.
Fonagy, A., Swiderski, C., Dunn, M. and Freeman, J. W. (1992). Antisense-mediated specific inhibition of p120 protein expression prevents G1- to S-phasetransition. Cancer Res. 52, 5250-5256.
Fonagy, A., Swiderski, C., Wilson, A., Bolton, W., Kenyon, N. and Freeman,J. W. (1993). Cell cycle regulated expression of nucleolar antigen p120 in normaland transformed human fibroblasts. J. Cell Physiol. 154, 16-27.
Ghosh, A., Saginc, G., Leow, S. C., Khattar, E., Shin, E. M., Yan, T. D., Wong, M.,Zhang, Z., Li, G., Sung,W.-K. et al. (2012). Telomerase directly regulates NF-kB-dependent transcription. Nat. Cell Biol. 14, 1270-1281.
Gorczyca, W., Bruno, S., Melamed, M. R. and Darzynkiewicz, Z. (1992). Cellcycle-related expression of p120 nucleolar antigen in normal human lymphocytesand in cells of HL-60 and MOLT-4 leukemic lines: effects of methotrexate,camptothecin, and teniposide. Cancer Res. 52, 3491-3494.
Guo, X. and Wang, X.-F. (2009). Signaling cross-talk between TGF-beta/BMP andother pathways. Cell Res. 19, 71-88.
Gustafson, W. C., Taylor, C. W., Valdez, B. C., Henning, D., Phippard, A.,Ren, Y., Busch, H. and Durban, E. (1998). Nucleolar protein p120 containsan arginine-rich domain that binds to ribosomal RNA. Biochem. J. 331,387-393.
Her, Y. R. and Chung, I. K. (2013). p300-mediated acetylation of TRF2 isrequired for maintaining functional telomeres. Nucleic Acids Res. 41,2267-2283.
Hrdlickova, R., Nehyba, J. and Bose, H. R. (2012). Alternatively splicedtelomerase reverse transcriptase variants lacking telomerase activity stimulatecell proliferation. Mol. Cell. Biol. 32, 4283-4296.
Jegou, T., Chung, I., Heuvelman, G., Wachsmuth, M., Gorisch, S. M., Greulich-Bode, K. M., Boukamp, P., Lichter, P. and Rippe, K. (2009). Dynamics oftelomeres and promyelocytic leukemia nuclear bodies in a telomerase-negativehuman cell line. Mol. Biol. Cell 20, 2070-2082.
Jhiang, S. M., Yaneva, M. and Busch, H. (1990). Expression of humanproliferation-associated nucleolar antigen p120. Cell Growth Differ. 1,319-324.
Kim, N. W., Piatyszek, M. A., Prowse, K. R., Harley, C. B., West, M. D., Ho, P. L.,Coviello, G. M., Wright, W. E., Weinrich, S. L. and Shay, J. W. (1994). Specificassociation of human telomerase activity with immortal cells and cancer. Science266, 2011-2015.
Kim, J. H., Kim, J. H., Lee, G. E., Lee, J. E. and Chung, I. K. (2003). Potentinhibition of human telomerase by nitrostyrene derivatives. Mol. Pharmacol. 63,1117-1124.
Koh, C. M., Khattar, E., Leow, S. C., Liu, C. Y., Muller, J., Ang, W. X., Li, Y.,Franzoso, G., Li, S., Guccione, E. et al. (2016). Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J. Clin.Invest. 125, 2109-2122.
Koonin, E. V. (1994). Prediction of an rRNA methyltransferase domain inhuman tumor-specific nucleolar protein p120. Nucleic Acids Res. 22,2476-2478.
Lee, G. E., Yu, E. Y., Cho, C. H., Lee, J., Muller, M. T. and Chung, I. K. (2004).DNA-protein kinase catalytic subunit-interacting protein KIP binds telomerase byinteracting with human telomerase reverse transcriptase. J. Biol. Chem. 279,34750-34755.
Lee, J. H., Khadka, P., Baek, S. H. and Chung, I. K. (2010). CHIP promoteshuman telomerase reverse transcriptase degradation and negatively regulatestelomerase activity. J. Biol. Chem. 285, 42033-42045.
Lee, J. H., Lee, Y. S., Jeong, S. A., Khadka, P., Roth, J. and Chung, I. K.(2014). Catalytically active telomerase holoenzyme is assembled in the densefibrillar component of the nucleolus during S phase. Histochem. Cell Biol. 141,137-152.
Li, Y. and Tergaonkar, V. (2014). Noncanonical functions of telomerase: implicationsin telomerase-targeted cancer therapies. Cancer Res. 74, 1639-1644.
Li, Y., Zhou, Q.-L., Sun, W., Chandrasekharan, P., Cheng, H. S., Ying, Z.,Lakshmanan, M., Raju, A., Tenen, D. G., Cheng, S. Y. et al. (2015). Non-canonical NF-κB signalling and ETS1/2 cooperatively drive C250T mutant TERTpromoter activation. Nat. Cell Biol. 17, 1327-1338.
Lingner, J., Cooper, J. P. and Cech, T. R. (1995). Telomerase and DNA endreplication: no longer a lagging strand problem? Science 269, 1533-1534.
Listerman, I., Gazzaniga, F. S. andBlackburn, E. H. (2014). An investigation of theeffects of the core protein telomerase reverse transcriptase on Wnt signaling inbreast cancer cells. Mol. Cell. Biol. 34, 280-289.
Musgrove, E. A., Caldon, C. E., Barraclough, J., Stone, A. and Sutherland,R. L. (2011). Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 11,558-572.
Ochs, R. L., Reilly, M. T., Freeman, J. W. and Busch, H. (1988). Intranucleolarlocalization of human proliferating cell nucleolar antigen p120. Cancer Res. 48,6523-6529.
Palm, W. and de Lange, T. (2008). How shelterin protects mammalian telomeres.Annu. Rev. Genet. 42, 301-334.
Park, J.-I., Venteicher, A. S., Hong, J. Y., Choi, J., Jun, S., Shkreli, M., Chang,W.,Meng, Z., Cheung, P., Ji, H. et al. (2009). Telomerase modulates Wnt signallingby association with target gene chromatin. Nature 460, 66-72.
Perlaky, L., Valdez, B. C., Busch, R. K., Larson, R. G., Jhiang, S. M., Zhang,W. W., Brattain, M. and Busch, H. (1992). Increased growth of NIH/3T3 cells bytransfection with human p120 complementary DNA and inhibition by a p120antisense construct. Cancer Res. 52, 428-436.
Pestell, R. G., Albanese, C., Reutens, A. T., Segall, J. E., Lee, R. J. andArnold, A. (1999). The cyclins and cyclin-dependent kinase inhibitors inhormonal regulation of proliferation and differentiation. Endocr. Rev. 20,501-534.
Qiu, R., Yang, Y., Zhao, H., Li, J., Xin, Q., Shan, S., Liu, Y., Dang, J., Yu, X., Gong,Y. et al. (2013). Signal transducer and activator of transcription 6 directly regulateshuman ORMDL3 expression. FEBS J. 280, 2014-2026.
Sarin, K. Y., Cheung, P., Gilison, D., Lee, E., Tennen, R. I., Wang, E., Artandi,M. K., Oro, A. E. and Artandi, S. E. (2005). Conditional telomerase inductioncauses proliferation of hair follicle stem cells. Nature 436, 1048-1052.
Sears, R. C. (2004). The life cycle of C-myc: from synthesis to degradation. CellCycle 3, 1131-1135.
Sfeir, A. and de Lange, T. (2012). Removal of shelterin reveals the telomere end-protection problem. Science 336, 593-597.
Smith, L. L., Coller, H. A. and Roberts, J. M. (2003). Telomerase modulatesexpression of growth-controlling genes and enhances cell proliferation. Nat. CellBiol. 5, 474-479.
Smogorzewska, A. and de Lange, T. (2004). Regulation of telomerase bytelomeric proteins. Annu. Rev. Biochem. 73, 177-208.
Stewart, S. A., Hahn, W. C., O’Connor, B. F., Banner, E. N., Lundberg, A. S.,Modha, P., Mizuno, H., Brooks, M. W., Fleming, M., Zimonjic, D. B. et al.
1578
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 1566-1579 doi:10.1242/jcs.181040
Journal
ofCe
llScience

(2002). Telomerase contributes to tumorigenesis by a telomere length-independent mechanism. Proc. Natl. Acad. Sci. USA 99, 12606-12611.
Strong, M. A., Vidal-Cardenas, S. L., Karim, B., Yu, H., Guo, N. and Greider,C. W. (2011). Phenotypes in mTERT+/− and mTERT−/− mice are due to shorttelomeres, not telomere-independent functions of telomerase reversetranscriptase. Mol. Cell. Biol. 31, 2369-2379.
Takai, H., Smogorzewska, A. and de Lange, T. (2003). DNA damage foci atdysfunctional telomeres. Curr. Biol. 13, 1549-1556.
Tomlinson, R. L., Ziegler, T. D., Supakorndej, T., Terns, R. M. and Terns, M. P.(2006). Cell cycle-regulated trafficking of human telomerase to telomeres. Mol.Biol. Cell 17, 955-965.
van Steensel, B., Smogorzewska, A. and de Lange, T. (1998). TRF2 protectshuman telomeres from end-to-end fusions. Cell 92, 401-413.
Venteicher, A. S. and Artandi, S. E. (2009). TCAB1: driving telomerase to Cajalbodies. Cell Cycle 8, 1329-1331.
Venteicher, A. S., Meng, Z., Mason, P. J., Veenstra, T. D. and Artandi, S. E.(2008). Identification of ATPases pontin and reptin as telomerase componentsessential for holoenzyme assembly. Cell 132, 945-957.
Venteicher, A. S., Abreu, E. B., Meng, Z., McCann, K. E., Terns, R. M., Veenstra,T. D., Terns, M. P. and Artandi, S. E. (2009). A human telomerase holoenzymeprotein required for Cajal body localization and telomere synthesis. Science 323,644-648.
Xi, L. and Cech, T. R. (2014). Inventory of telomerase components in human cellsreveals multiple subpopulations of hTR and hTERT. Nucleic Acids Res. 42,8565-8577.
Yu, Q., Geng, Y. and Sicinski, P. (2001). Specific protection against breast cancersby cyclin D1 ablation. Nature 411, 1017-1021.
1579
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 1566-1579 doi:10.1242/jcs.181040
Journal
ofCe
llScience