synthesis, characterization and surface properties of poly(lactic acid)–perfluoropolyether block...
TRANSCRIPT

50
7
Research ArticleReceived: 22 July 2009 Revised: 14 April 2010 Accepted: 17 June 2010 Published online in Wiley Online Library: 17 December 2010
(wileyonlinelibrary.com) DOI 10.1002/pi.2982
Synthesis, characterization and surfaceproperties of poly(lactic acid)–perfluoropolyether block copolymersAkhilesh Singh,a† Amit K Naskar,b‡ Dahlia Haynes,b Michael J Drewsa
and Dennis W Smith Jra,b∗
Abstract
Laboratory-scale synthesis and morphological and surface energy characterization of triblock A–B–A copolymers based onpoly(lactic acid) (PLA; A segment) containing various block lengths of perfluoropolyether (PFPE; B segment) at 5 wt% PFPEcontent are reported. Incorporation of PFPE segments in PLA lowers significantly both the polar and dispersive components oftotal surface energy. Total surface energy is lowered from ca 35 to ca 17 mN m−1 on copolymerization of PLA with 5 wt% PFPE.Thermal analysis data reveal that lower molecular weight PFPE segments lower significantly the glass transition, crystallizationand melting temperatures of the PLA matrix. Although block length variation of the PFPE segment does not affect surfaceenergies of copolymer films, smaller PFPE segments increase significantly the low-temperature modulus as observed fromdynamic mechanical analysis.c© 2010 Society of Chemical Industry
Keywords: polylactide; perfluoropolyether; triblock copolymer; dynamic mechanical analysis; surface energy
INTRODUCTIONPoly(lactic acid) (PLA), a biodegradable and biocompatiblealiphatic polyester, is well known in the medical and pharmaceu-tical fields and is now rapidly evolving as a commodity polymer.PLA is commercially synthesized from lactide, a renewable re-source monomer derived from corn starch, and is a potentialalternative to petroleum-based commodity polymers as the lat-ter face problems associated with waste disposal and increasedcost.1 – 7 PLA is a semicrystalline and melt-processable polymerwith glass transition and melting temperatures ranging from 50 to60 ◦C and 170 to 180 ◦C, respectively. It exhibits mechanical prop-erties somewhat comparable to those of petroleum-based plasticssuch as polystyrene and polypropylene; however, neat PLA is arelatively brittle material.4,6,8 The future prospects of PLA dependmainly on the cost and availability of raw material, energy con-sumed in producing the monomer, cost of further modification,ease of processing and acceptance of the processed material.9
In order to diversify PLA and deliver tailor-made properties withpreferred crystallinity, surface morphology, hydrolytic degrada-tion and other useful characteristics for commodity applications,its cost-effective modification has been pursued. Such modifi-cation can be achieved by copolymerization, blending and/orplasticization with other polymers.10 – 13
The ring-opening polymerization (ROP) of lactide can be carriedout using complexes of aluminium, iron, tin, yttrium, zinc andvarious other metals.14 – 18 Tin(II) 2-ethylhexanoate (Sn(Oct)2),one of the most commonly used catalysts/initiators for bulkpolymerization of lactones because of its high efficiency, is usedas the catalyst in the present work.2,19,20 Various mechanismsof PLA polymerization have been investigated.2,21 Since ROP isnot initiated by Sn(Oct)2 and octoate has to be converted to an
alkoxide, the polymerization is very sensitive to the relative contentof hydroxyl impurities thus explaining the difficulties reported forcontrolled synthesis.2,22,23 Even when there is no added alcoholor protic source in the system, impurities present in the monomerand/or catalyst may cause increased initiation of the monomerresulting in lower molecular weight polymer.24 Impurities suchas water, lactic acid and lactoyl lactic acid are present in themonomer and catalyst.25,26 Work has been accomplished toimprove the properties of PLA polymerized under controlled levelof impurities.27
Fluorinated polymers have excellent properties such as chem-ical inertness, low coefficient of friction and low surfaceenergy.28 Semifluorinated hybrid materials can be prepared byblending, copolymerization or chemical reaction of fluorinatedreagents with polymers such as polyacrylates, polymethacrylate,polystyrene, poly(ε-caprolactone), polyurethane and others.29 – 33
∗ Correspondence to: Dennis W Smith Jr, School of Material Science andEngineering, Clemson University, Clemson, SC 29634, USA.E-mail: [email protected]
† Present address: Tetramer Technologies LLC, 657 S Mechanic Street, Pendleton,SC 29670, USA.
‡ Present address: Materials Science and Technology Division, Oak Ridge NationalLaboratory, Oak Ridge, TN 37831-6053, USA.
a School of Material Science and Engineering, Clemson University, Clemson, SC29634, USA
b Department of Chemistry and Center for Optical Materials Science andEngineering Technologies (COMSET), Clemson University, Clemson, SC 29634,USA
Polym Int 2011; 60: 507–516 www.soci.org c© 2010 Society of Chemical Industry

50
8
www.soci.org A Singh et al.
Copolymerization requires relatively large amounts of fluorinatedmonomers for a considerable change in surface properties of theresultant copolymer and can lead to an expensive material.28
Surface modification of pre-molded articles with fluorinated ma-terials is also an expensive technique.34 Blending of small amountsof fluorinated materials with nonfluorinated polymeric materialscan enhance the surface properties by segregation of fluorinatedgroups on the surface; however, thermodynamic separation of twophases can lead to poor mechanical properties.28,35,36 PLA-based‘green’ products are now commercially available. However, thoseproducts are facing market challenges due to their high cost. Thus,our overarching objective in the work reported here was to tailorPLA-based material to make it a value-added specialty product.
In this article, we report the modification of the surfaceand bulk properties of PLA using telechelic poly(fluoroalkyleneoxide)s with reactive hydroxyl groups as macrointiator leadingto polylactide-block-poly(fluoroalkylene oxide)-block-polylactidetriblock copolymer (FluoroPLA). The ability of the fluorinatedsegments to migrate towards/on the surface thereby enrichingthe surface with fluorine even at very low concentration has beendemonstrated in previous studies.28,37 – 39 We previously utilized asimilar concept to prepare perfluoropolyether (PFPE)-modifiedPLA triblock copolymers for achieving desired amphiphobicsurface characteristics.40 At 5 wt% PFPE loading, the blockcopolymer exhibited improved mechanical properties. As acontinuation of this work, laboratory-scale synthesis (100–200 gbatch) of FluoroPLAs using macroinitiators of various block lengthsat various monomer/initiator concentrations keeping the PFPEloading at ca 5 wt% was conducted. This article discusses the effectof macroinitiator block length on molecular weight and thermal,dynamic mechanical, crystallographic and surface properties.
EXPERIMENTALMaterialsL-Lactide (LA) was generously donated by Poly-Med Inc.(Pendleton, SC) and was recrystallized from ethyl acetateand vacuum-dried to remove solvent and moisture. α,ω-Diol-poly[(tetrafluoroethylene oxide)-co-(difluoromethylene oxide)](Fomblin Z DOL), a PFPE with number-average molecular weights(Mn) of 1500, 2000 and 4200 g mol−1, was generously donated bySolvay-Solexis (Italy). PFPEs were colorless clear liquids with a den-sity of 1.8 g cm−3, x/y ≈ 1 and hydroxyl functionality of ca 1.8–1.9(determined from NMR analysis; Solvay-Solexis, Fomblin Z deriva-tives, product data sheet). The generalized chemical structure ofPFPE and the polymerization reaction with lactide monomer areshown in Scheme 1. All other chemicals and reagents were pur-chased from Fisher or Sigma Aldrich and were used as receivedunless otherwise stated.
Synthesis of homopolymer and block copolymerDetailed small-scale synthesis and purification methodologiesof FluoroPLAs are described elsewhere.40 A mixture of 10 mLof toluene, 0.42 g of Sn(Oct)2 and 5 g of PFPE was preparedand shaken well. Then LA (100 g) and the above mixture wereadded to the reaction vessel, sealed and blanketed with aninert atmosphere (1.2–1.4 bar (120–140 kPa) of argon). Thetemperature was gradually (ca 2 ◦C min−1) raised to 130 ◦C, andheld for 24 h. The reaction mixture was slowly stirred at 20 rpmthroughout the polymerization process. After polymerization, thereactor was allowed to cool to room temperature and a solidlight yellow mass was removed. The reaction product was then
OO
O OO
F F F F
F F F F F F
OH
x yqp
FluoroPLA
(PLA-PFPE-PLA Block Copolymer)
OH
O
O
O
O
+ HOO
O OOH
F F F F
F F F F F Fx y
PFPEL-Lactide
130 °C24 h
120–140 kPa (argon)in
Parr high pressure reactor
Sn2+
O
-O
2
Tin(II) 2-ethylhexanoate
O
O
(p+q)/2
Scheme 1. Polymerization of L-lactide with Sn(Oct)2 as catalyst/initiatorand PFPE as macroinitiator.
dissolved in chloroform followed by dropwise precipitation inswirling methanol at room temperature. The precipitated materialwas dried under vacuum at 70 ◦C for 1 h. Various batches of thecopolymer of PLA and PFPE were prepared with ca 5 wt% PFPE ofvarious molecular weights (Mn = 1.5, 2.0, 4.2 kg mol−1).
In the following discussion, the abbreviation FluoroPLA(y) isused to designate a copolymer that contains PFPE segmentswith Mn of y kg mol−1. A control PLA homopolymer batch wassynthesized without any PFPE in the reaction mixture.
CharacterizationMn and weight-average molecular weight (Mw) of all of thepolymers were characterized using a Waters Breeze gel permeationchromatography (GPC) system (Waters, Milford, MA) interfacedwith a Polymer Lab (Amherst, MA) PL-ELS 2100 evaporative lightscattering detector. Two Waters columns were used in series(Styragel HR4E and HR5E). The mobile phase was chloroform(Burdick & Jackson, HPLC grade) at 1 cm3 min−1. Ten polystyrenestandards (Polysciences Inc., Warrington, PA) of Mn in the range 106
to 436 g mol−1 (dispersity Mw/Mn ≤ 1.1) were used to calibratethe system.
NMR spectra were obtained using a JEOL (Tokyo, Japan) Eclipse+300 MHz NMR spectrometer. Concentrated solutions (ca 10% w/v)of polymer in CDCl3 were used to obtain 1H NMR, 13C NMR and 19FNMR spectra for FluoroPLAs.
DSC data were collected using a TA Instruments (New Castle, DE)Q1000 DSC instrument. Data were analyzed using TA InstrumentsUniversal Analysis 2000 version 4.1D software. Samples (6–8 mg instandard aluminium pans) were initially heated to 225 ◦C to erasethe thermal history and then cooled to −50 ◦C, and re-heated from−50 to 225 ◦C. A heating rate of 10 ◦C min−1 was used for all scansegments. The glass transition temperatures (Tg) were obtained asthe inflection point of the step transition. Peak temperature valuesof the endothermic and exothermic peak maxima are reported asmelting and crystallization temperatures, and the integral of thepeak areas was used to calculate heats of fusion and crystallization,respectively.
wileyonlinelibrary.com/journal/pi c© 2010 Society of Chemical Industry Polym Int 2011; 60: 507–516
50
9
Poly(lactic acid)–perfluoropolyether block copolymers www.soci.org
TGA was conducted using a TA Instruments 2950 TGA. Datawere analyzed using TA Instruments Universal Analysis 2000version 4.1D software. A heating rate of 10 ◦C min−1 was appliedto samples from room temperature to 400 ◦C under a nitrogenpurge.
XRD patterns of the polymers were collected at room temper-ature using a SCINTAG XDS 2000 (Scintag Inc., Cupertino, CA)diffractometer with Cu Kα radiation at a wavelength of 1.54 Å. Theinstrument was operated at 40 kV and 40 mA with a collimatordiameter of 0.5 mm. Solution-cast polymer films were scanned at2◦ min−1 (2θ value) from 6◦ to 60◦. Data were analyzed usingDMSNT version 1.37 software.
Dynamic mechanical analysis was performed using a DMS 210tension module (Seiko Instruments Inc., Japan) with specimendimensions of 40 mm × 10 mm and an effective gauge lengthof 20 mm. Samples were evaluated over a temperature range of−130 to 125 ◦C at a heating rate of 2 ◦C min−1 at a frequency of1 Hz and a deformation amplitude of 10 µm. Data were analyzedusing EXSTAR6000 software.
Silicon wafers were oxidized by immersion in piranha solution(3 parts 95–98% H2SO4 + 1 part 30% H2O2) at 80 ◦C for45 min. Solutions of 2% (w/v) homopolymer and copolymer inchloroform were prepared and filtered through a Whatman 0.2 µmpolytetrafluoroethylene membrane syringe filter. Dip-coating ofoxidized silicon wafers was carried out using a dip coater (MayerFeintechnik D-3400, Gottingen, Germany). Dip-coated sampleswere air dried and then the films were investigated using aDimension 3100 (Veeco Inc., Woodbury, NY) AFM instrumentequipped with a Nanoscope IIIa controller. All AFM characterizationexperiments were performed using a silicon AFM tip (MicroMashInc.; nominal force constant of 40 N m−1, tip radius <10 nm) innon-contact (tapping) mode.
Static contact angle measurements were performed using thesessile drop method on a Drop Shape Analysis (KRUSS Instruments,Hamburg, Germany) system. Liquid drops with an average volumebetween 5 and 10 µL were placed on dip-coated polymer filmsand the equilibrium contact angles were measured after anequilibration time of 30 s.
RESULTS AND DISCUSSIONSynthesis and characterizationThe synthesis of ABA triblock copolymer of PFPE and PLA wascarried out by ROP of LA using Sn(Oct)2 as initiator (Scheme 1).Sn(Oct)2 is an effective catalyst for ROP of lactones and widelyaccepted as a contaminant in PLA for a variety of commercialapplications.2,41 The effect of PFPE loading on copolymer thermaland surface properties has been reported earlier.40
Controlled ROP of six- and seven-membered cyclic esterscan be carried out using alkoxides formed by the reaction ofcarboxylate group- and hydroxyl group-containing initiators.16
Propagation takes place by nucleophilic attack of the alcoholateactive species at the ester group leading to acyl oxygen bondscission.42 Hydroxyl group-containing species, in addition to theirinitiator behavior, can also act as transfer agents resulting in abroadening of the molecular weight distribution of the resultantpolymer.43,44 Many parameters such as composition of feed,temperature of polymerization, solvent, extent of conversionand relative rate of reactions determine the favored mechanisticpathway.16,44 – 46 There is the possibility of the formation of diblockto multiblock copolymers depending on the relative reactivityof various generated species. A higher hydroxyl content of the
0
0.2
0.4
0.6
0.8
1
14 15 16 17 18
Nor
mal
ized
sig
nal
Elution time, min
PLAFluoroPLA(4.2k)FluoroPLA(2.0k)FluoroPLA(1.5k)
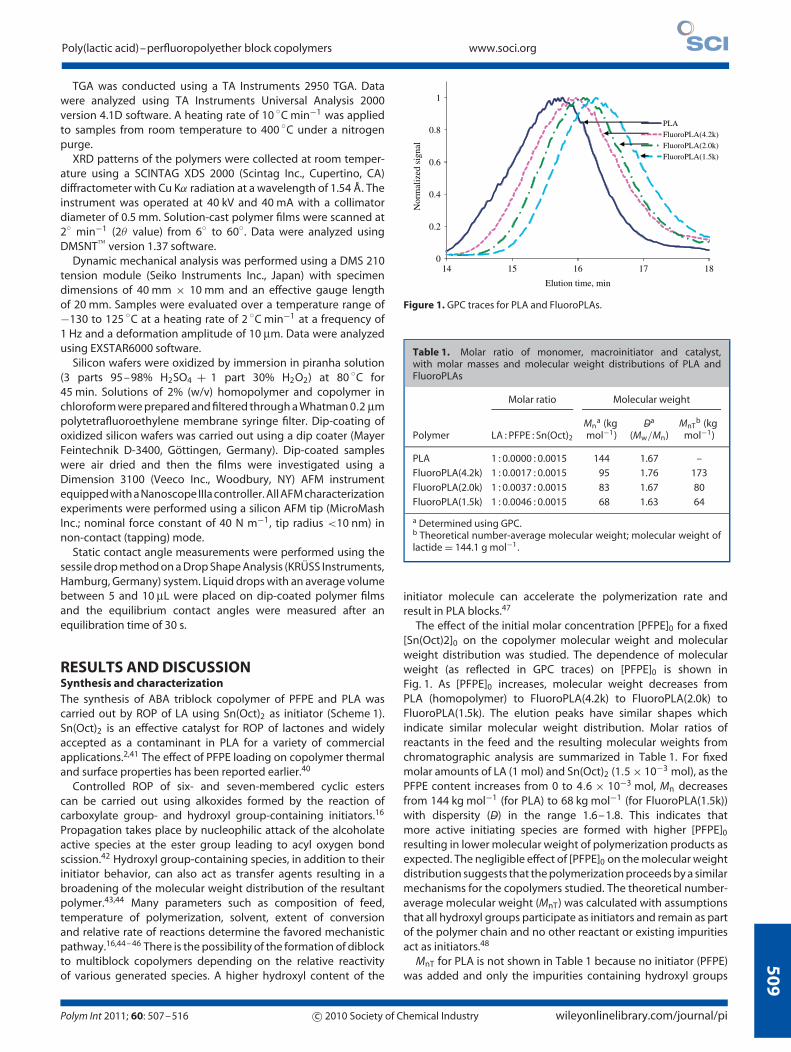
Figure 1. GPC traces for PLA and FluoroPLAs.
Table 1. Molar ratio of monomer, macroinitiator and catalyst,with molar masses and molecular weight distributions of PLA andFluoroPLAs
Molar ratio Molecular weight
Polymer LA : PFPE : Sn(Oct)2
Mna (kg
mol−1)–Da
(Mw/Mn)MnT
b (kgmol−1)
PLA 1 : 0.0000 : 0.0015 144 1.67 –
FluoroPLA(4.2k) 1 : 0.0017 : 0.0015 95 1.76 173
FluoroPLA(2.0k) 1 : 0.0037 : 0.0015 83 1.67 80
FluoroPLA(1.5k) 1 : 0.0046 : 0.0015 68 1.63 64
a Determined using GPC.b Theoretical number-average molecular weight; molecular weight oflactide = 144.1 g mol−1.
initiator molecule can accelerate the polymerization rate andresult in PLA blocks.47
The effect of the initial molar concentration [PFPE]0 for a fixed[Sn(Oct)2]0 on the copolymer molecular weight and molecularweight distribution was studied. The dependence of molecularweight (as reflected in GPC traces) on [PFPE]0 is shown inFig. 1. As [PFPE]0 increases, molecular weight decreases fromPLA (homopolymer) to FluoroPLA(4.2k) to FluoroPLA(2.0k) toFluoroPLA(1.5k). The elution peaks have similar shapes whichindicate similar molecular weight distribution. Molar ratios ofreactants in the feed and the resulting molecular weights fromchromatographic analysis are summarized in Table 1. For fixedmolar amounts of LA (1 mol) and Sn(Oct)2 (1.5 × 10−3 mol), as thePFPE content increases from 0 to 4.6 × 10−3 mol, Mn decreasesfrom 144 kg mol−1 (for PLA) to 68 kg mol−1 (for FluoroPLA(1.5k))with dispersity (–D) in the range 1.6–1.8. This indicates thatmore active initiating species are formed with higher [PFPE]0
resulting in lower molecular weight of polymerization products asexpected. The negligible effect of [PFPE]0 on the molecular weightdistribution suggests that the polymerization proceeds by a similarmechanisms for the copolymers studied. The theoretical number-average molecular weight (MnT) was calculated with assumptionsthat all hydroxyl groups participate as initiators and remain as partof the polymer chain and no other reactant or existing impuritiesact as initiators.48
MnT for PLA is not shown in Table 1 because no initiator (PFPE)was added and only the impurities containing hydroxyl groups
Polym Int 2011; 60: 507–516 c© 2010 Society of Chemical Industry wileyonlinelibrary.com/journal/pi

51
0
www.soci.org A Singh et al.
0
50
100
150
200
250
0 0.001 0.002 0.003 0.004 0.005
Mol
ecul
ar W
eigh
t (M
n)/1
000,
g m
ol–1
Initial [PFPE/LA], (mol mol–1)
Mn, calculated Mn, experimental
Initiating Impurities Dominated Region Controlled Mol Wt Region
Figure 2. Comparison of experimentally obtained Mn with theoreticallycalculated values and their dependence on initial [PFPE/LA].
result in the polymerization of the LA. The concentration ofhydroxyl group-containing impurities was calculated from themolar mass of PLA and molecular weight of the resulting polymerunder the assumption that each hydroxyl group initiates growthof a polymer chain and the impurities are monofunctional. Thecalculated impurity concentration is ca 10−3 mol per mol of LA(or reaction mixture) and it should be noted that the impurityconcentration changes with the functionality of the impurities.
MnT and Mn are shown as a function of the initial ratio of [PFPE]to [LA] in Fig. 2. Discrepancies in the MnT and Mn values are possiblydue to the presence of initiating impurities such as water, lacticacid and lactoyl lactic acid.25,26 The difference in molecular weightsdecreases as the relative concentration of PFPE over impuritiesincreases. For [PFPE]>>[impurities], controlled molecular weightcan be obtained but very high [PFPE] will have detrimental effectson the final molecular weights of triblock copolymers. Hence, theconcentration of PFPE should be carefully determined dependingon the required copolymer molecular weight and desired surfacecharacteristics. It should be noted that Mn are calculated usingpolystyrene standards. The hydrodynamic volume of polystyreneand PFPE-incorporated PLA would be different. However, thatcalibration error should have caused uniform error for all theFluoroPLA samples with the fixed (5 wt%) fraction of PFPE segment.
The 1H NMR, 19F NMR and 13C NMR spectra of FluoroPLA(1.5k)are shown in Fig. 3. The 1H NMR spectrum has intense signalsat 1.55 and 5.15 ppm corresponding, respectively, to CH3 andCH.49 The multiplets in the range 4.3–4.7 ppm are assignedto–CF2 –CH2O–of PFPE in FluoroPLA and these multiplets areshifted from their original position at 3.5 ppm for the PFPEinitiator.40 The presence of the PFPE block in the copolymerwas confirmed by the appearance of peaks around −53,−80 and −89 ppm in the 19F NMR spectrum. In PFPE, theperfluoromethylene oxide units show three signals (−51.7, −53.3,−55.0 ppm) while the perfluoroethylene oxide units give twosignals (−88.7, −90.5 ppm) and two end-group signals appearat −80.7 and −82.7 ppm.50 Upon copolymerization, these end-group signals are shifted to −77.5 and −79.5 ppm, respectively.51
In the 13C NMR spectrum, expected signals from PLA blocks arevery distinct; however, the signals from PFPE block are very weakas the FluoroPLA contains only 5 wt% PFPE segment.
Morphology of PLA and FluoroPLARepresentative DSC thermograms for PLA and FluoroPLAs arecompared in Fig. 4. Characteristic transitions such as the glass
transition step, the exothermic crystallization peak and theendothermic melting peak can be observed. Thermal analysisdata for PLA and FluoroPLAs are given in Table 2. The glasstransition, melting, crystallization and degradation temperaturesfor the FluoroPLAs depend on the PFPE segment length. With adecrease in the PFPE segment molecular weight, the temperaturesfor all transitions are lowered. There is a difference of ca10 ◦C between respective transitions of PLA and FluoroPLA(1.5k).The transition temperatures for the polymers follow the trend:TPLA > TFluoroPLA(4.2k) > TFluoroPLA(2.0k) > TFluoroPLA(1.5k).
The relationship between Tg and copolymer molecular weight iswell explained by Fox et al.52 and Ajroldi et al.53 The dependenceof Tg on the molecular weight is very strong below a criticalmolecular weight (MC) of polymer (due to the large free volumeassociated with the chain ends); however, above MC, Tg ispractically independent of molecular weight. The molecularweights of FluoroPLAs are much higher than the reported MC
(9.6 × 103 g mol−1) for PLA.54 Hence, the decrease in molecularweight from PLA to FluoroPLA(1.5k) is not responsible forlowering of Tg. This lower temperature shift in Tg can beexplained by appreciable mixing between PFPE and PLA segments(Tg,PFPE ≈ −115 to −122 ◦C and Tg,PLA ≈ 60 ◦C)55 of phase-separated block copolymers:56
1
Tg= mPFPE
Tg,PFPE+ mPLA
Tg,PLA(1)
where mPFPE and mPLA are the mass fractions of PFPE and PLAsegments in the block copolymer, respectively. Although apparentmass fractions of PFPE segments in the copolymer compositionsare ca 5%, Tg of the PFPE segments with lower molecular weightis expected to be lower than that of the higher molecular weightPFPE due to increased free volume effect caused by more endgroups in the former.51
The depression in the melting point (Tm) can be theoreticallyexplained by the equation
1
Tm− 1
Tm,0= R
�Hm,0χPFPE (2)
where Tm,0 is the melting point of pure crystalline PLA homopoly-mer, �Hm,0 is the heat of fusion per mole of crystalline PLA, Ris the molar gas constant and χPFPE is the mole fraction of PFPEincorporated into crystalline PLA domains.56,57 From Table 1, it isclear that the mole fraction of PFPE in the copolymer compositionsignificantly increases from FluroPLA(4.2k) to FluoroPLA(1.5k).
The enthalpies of fusion and crystallization are higher forFluoroPLAs than for PLA due to the nucleating action of PFPEleading to a higher level and a higher rate of crystallization ofFluoroPLAs.58 This suggests that the activation energy of diffusionof PLA segments in the crystallization domains decreases as thePFPE segmental molecular weight decreases.59 The depressionin melting points of FluoroPLAs is responsible for the decreasein crystallization temperature as crystallization can occur onlyafter the temperature has fallen below the theoretical meltingtemperature.60
Representative TGA thermograms for PLA, PFPE and FluoroPLAsare compared in Fig. 5. The curve showing the lowest temperatureweight loss is that for PFPE of molecular weight of 4200 g mol−1
and is very similar to the degradation of hydroxyl-terminatedPFPE as reported earlier.61 The thermal decomposition of PFPEresults in low-molecular-weight gaseous products with the main
wileyonlinelibrary.com/journal/pi c© 2010 Society of Chemical Industry Polym Int 2011; 60: 507–516

51
1
Poly(lactic acid)–perfluoropolyether block copolymers www.soci.org
Figure 3. 1H NMR spectrum (top), 19F NMR spectrum (middle) and 13C NMR spectrum (bottom) of FluoroPLA(1.5k) in CDCl3 (300 MHz). Scale in ppm.
Figure 4. Thermal transitions of PLA and FluoroPLAs from DSC measure-ments.
component being hexafluoropropylene (CF2 CF–CF3).61 Theonset of degradation is strongly dependent on the molecularweight of PFPE with the same chemical structure and molecularweight distribution.61 Degradation begins at lower temperaturesfor block copolymer with lower molecular weight PFPE.
Based on the TGA results, neat PLA exhibits higher thermalstability than the FluoroPLAs unless the latter are modifiedwith stabilizing additives. FluoroPLA(1.5k) shows the loweststability among the FluoroPLAs. The decrease in thermal stabilityof FluoroPLAs can be attributed to the incorporation of thecomparatively low thermal stability PFPE segments in the PLAbackbone. In addition, the differences observed in the degradationtemperatures of FluoroPLAs could be due to the molecular weighteffect on the degradation of PFPE as previously discussed. Adecrease of ca 50 ◦C in the degradation temperatures (temperatureat 10% weight loss) of PLA and FluoroPLA(1.5k) is observed.
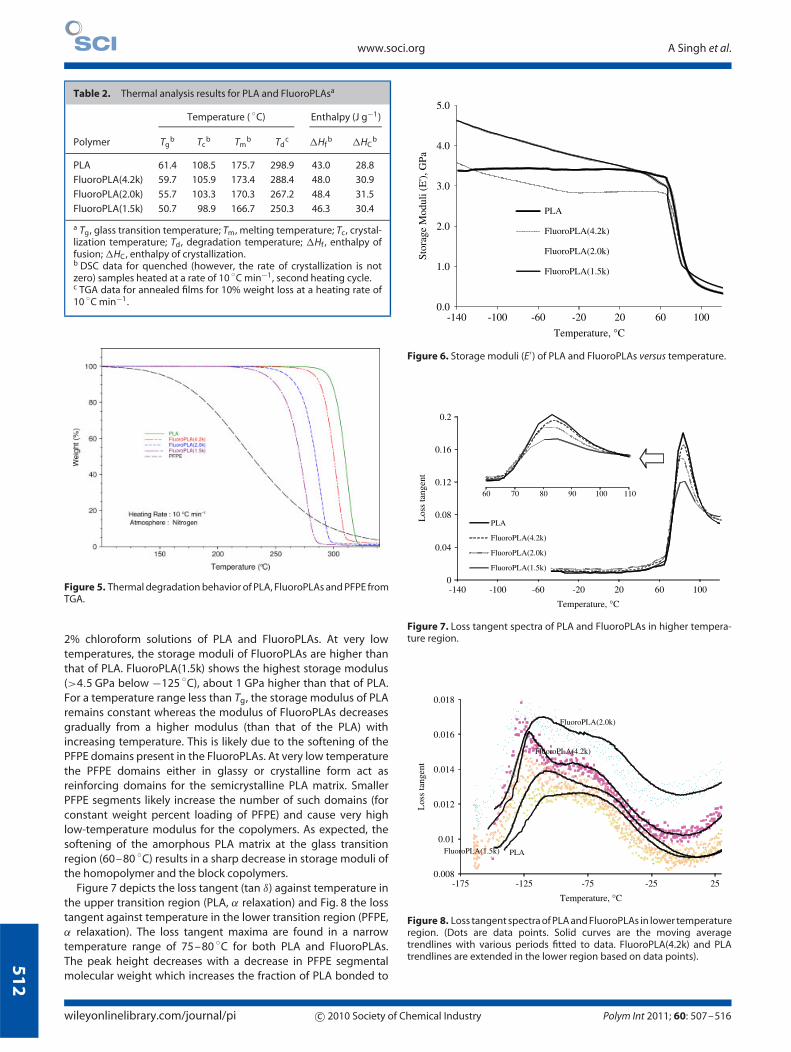
Figure 6 shows the storage moduli of PLA and FluoroPLAs as afunction of temperature. Analysis was performed on annealed(annealing temperature of 90 ◦C) solution-cast films from ca
Polym Int 2011; 60: 507–516 c© 2010 Society of Chemical Industry wileyonlinelibrary.com/journal/pi
51
2
www.soci.org A Singh et al.
Table 2. Thermal analysis results for PLA and FluoroPLAsa
Temperature ( ◦C) Enthalpy (J g−1)
Polymer Tgb Tc
b Tmb Td
c �Hfb �HC
b
PLA 61.4 108.5 175.7 298.9 43.0 28.8
FluoroPLA(4.2k) 59.7 105.9 173.4 288.4 48.0 30.9
FluoroPLA(2.0k) 55.7 103.3 170.3 267.2 48.4 31.5
FluoroPLA(1.5k) 50.7 98.9 166.7 250.3 46.3 30.4
a Tg, glass transition temperature; Tm, melting temperature; Tc, crystal-lization temperature; Td, degradation temperature; �Hf , enthalpy offusion; �HC, enthalpy of crystallization.b DSC data for quenched (however, the rate of crystallization is notzero) samples heated at a rate of 10 ◦C min−1, second heating cycle.c TGA data for annealed films for 10% weight loss at a heating rate of10 ◦C min−1.
Figure 5. Thermal degradation behavior of PLA, FluoroPLAs and PFPE fromTGA.
2% chloroform solutions of PLA and FluoroPLAs. At very lowtemperatures, the storage moduli of FluoroPLAs are higher thanthat of PLA. FluoroPLA(1.5k) shows the highest storage modulus(>4.5 GPa below −125 ◦C), about 1 GPa higher than that of PLA.For a temperature range less than Tg, the storage modulus of PLAremains constant whereas the modulus of FluoroPLAs decreasesgradually from a higher modulus (than that of the PLA) withincreasing temperature. This is likely due to the softening of thePFPE domains present in the FluoroPLAs. At very low temperaturethe PFPE domains either in glassy or crystalline form act asreinforcing domains for the semicrystalline PLA matrix. SmallerPFPE segments likely increase the number of such domains (forconstant weight percent loading of PFPE) and cause very highlow-temperature modulus for the copolymers. As expected, thesoftening of the amorphous PLA matrix at the glass transitionregion (60–80 ◦C) results in a sharp decrease in storage moduli ofthe homopolymer and the block copolymers.
Figure 7 depicts the loss tangent (tan δ) against temperature inthe upper transition region (PLA, α relaxation) and Fig. 8 the losstangent against temperature in the lower transition region (PFPE,α relaxation). The loss tangent maxima are found in a narrowtemperature range of 75–80 ◦C for both PLA and FluoroPLAs.The peak height decreases with a decrease in PFPE segmentalmolecular weight which increases the fraction of PLA bonded to
0.0
1.0
2.0
3.0
4.0
5.0
-140 -100 -60 -20 20 60 100
Stor
age
Mod
uli (
E'),
GPa
Temperature, °C
PLA
FluoroPLA(4.2k)
FluoroPLA(2.0k)
FluoroPLA(1.5k)
Figure 6. Storage moduli (E′) of PLA and FluoroPLAs versus temperature.
0
0.04
0.08
0.12
0.16
0.2
-140 -100 -60 -20 20 60 100
Los
s ta
ngen
t
Temperature, °C
PLA
FluoroPLA(4.2k)
FluoroPLA(2.0k)
FluoroPLA(1.5k)
60 70 80 90 100 110
Figure 7. Loss tangent spectra of PLA and FluoroPLAs in higher tempera-ture region.
0.008
0.01
0.012
0.014
0.016
0.018
-175 -125 -75 -25 25
Los
s ta
ngen
t
Temperature, °C
PLAFluoroPLA(1.5k)
FluoroPLA(4.2k)
FluoroPLA(2.0k)
Figure 8. Loss tangent spectra of PLA and FluoroPLAs in lower temperatureregion. (Dots are data points. Solid curves are the moving averagetrendlines with various periods fitted to data. FluoroPLA(4.2k) and PLAtrendlines are extended in the lower region based on data points).
wileyonlinelibrary.com/journal/pi c© 2010 Society of Chemical Industry Polym Int 2011; 60: 507–516

51
3
Poly(lactic acid)–perfluoropolyether block copolymers www.soci.org
0
0.2
0.4
0.6
0.8
1
13 15.5 18 20.5 23
Nor
mal
ized
sig
nal
2θ (°)
PLA
FluoroPLA(4.2k)
FluoroPLA(2.0k)
FluoroPLA(1.5k)
(010)
(110), (200)
(205)
(203)
Figure 9. XRD plots of annealed PLA and FluoroPLA films.
PFPE (junction segment) and thus such PLA segments have highsegmental mobility. These fractions of modified PLA segmentswill have lower relaxation transition temperature than the neatPLA segments. Therefore, FluoroPLA with low molecular weightPFPE segments show a very broad and less intense loss tangentspectrum. The change in magnitude of the loss tangent is alsoindicative of a possible difference in crystallinities.60 Fig. 7 suggeststhat the FluoroPLAs could be more crystalline than the PLA.Slightly higher enthalpies of fusion (Table 2) and crystallization ofthe copolymers in comparison to that of the neat PLA supportthe above argument. As shown in Fig. 8, the relaxation transitionsfound in the lower temperature region (−150 to −25 ◦C) arepossibly due to the contributions from α relaxation of the PFPE(−120 to −90 ◦C)55,62,63 and the broad β relaxation of PLA (ca−50 ◦C).64
XRD patterns of annealed PLA and FluoroPLA solution-cast filmsare shown in Fig. 9. The PLA and FluoroPLAs exhibit diffraction
peaks at 2θ values of 14.7◦, 16.5–16.7◦, 19.0◦ and 22.2◦, wherethese peaks are assigned to reflections due to the (010), (200) and(110), (203) and (015) crystalline planes, respectively. These peaksare characteristic of the PLA α-crystal orthorhombic cell with thefollowing nominal dimensions: a = 1.060 nm, b = 0.605 nm andc = 2.880 nm.13,65,66 This suggests that PLA and FluoroPLAs havesimilar crystal structures and it appears that there is no significantalteration in the crystal structure of PLA due to the incorporationof PFPE blocks. Minor shift in lattice d spacing could be dueto differential annealing and residual stress present in the filmsprepared by solution-casting method.
Surface characteristicsAFM analysis in non-contact (tapping) mode was performed ondip-coated films. Figure 10 shows the phase images as observedfor the PLA and FluoroPLA films. A clear distinction betweenPLA and FluoroPLA phase images can be observed. However,the differences in the phase images of FluoroPLAs themselvesare not very obvious. The difference in the phase images ofthe PLA and FluoroPLAs can be attributed to contrasts in theviscoelastic, surface modulus and adhesion properties of PLA andPFPE segments. The corresponding topographic images (shownin the graphical abstract) display a very flat topography for neatPLA and a distinct feature with ‘hill and valley’ topography for theFluoroPLA copolymers.
Surface phenomena such as adsorption, wetting and adhesionare governed by the surface energy of materials. Surface energyof polymers depends on the surface microstructures. Organicpolymers, in general, are low-surface-energy materials and directdetermination of surface energy is difficult due to poor mobilityof molecules in the solid state. Contact angle measurements onpolymer surfaces with chosen liquids can be used to determine thesurface energy of polymers.67 The Owens–Wendt–Rabel–Kaelblemethod was used to investigate the surface energies of PLA and
Figure 10. AFM phase images of dip-coated PLA, FluoroPLA(4.2k), FluoroPLA(2.0k) and FluoroPLA(1.5k). Pre-oxidized silicon wafers were dip-coated in2% solutions of PLA and FluoroPLAs in chloroform.
Polym Int 2011; 60: 507–516 c© 2010 Society of Chemical Industry wileyonlinelibrary.com/journal/pi

51
4
www.soci.org A Singh et al.
Table 3. Static contact angle values in degrees (meana ± standard deviation) for PLA and FluoroPLAs
Polymer Water Glycerol Formamide Methylene iodide n-Hexadecane
PLA 74.3 ± 0.6 70.6 ± 1.9 59.4 ± 0.7 39.9 ± 0.3 22.6 ± 2.0
FluoroPLA(4.2k) 104.2 ± 0.5 96.2 ± 2.5 89.6 ± 1.7 73.7 ± 0.3 58.8 ± 1.9
FluoroPLA(2.0k) 104.1 ± 0.4 99.2 ± 1.0 94.1 ± 0.5 77.5 ± 1.0 58.8 ± 2.7
FluoroPLA(1.5k) 104.7 ± 0.5 101.2 ± 0.4 93.3 ± 1.1 75.8 ± 0.8 60.6 ± 4.1
a Mean of at least five measurements.
FluoroPLAs.68,69 Based on the theoretical consideration of Fowkesof attractive forces at interfaces, it is proposed that the surfaceenergy of a solid (γ s) can be broken into its polar (γ p
s ) and dispersive(γ d
s ) components and that these components are independent andadditive in nature.68,69 The surface energy components of a solidsurface can be correlated to its contact angle (θ ) with a liquid ofknown surface energy components (γ p
l , γ dl ) using
1 + cos θ
2
γ l√γ d
l
=√
γ ds +
√γ
ps
√γ
pl√
γ dl
(3)
By measuring the contact angle of a liquid with known dispersiveand polar components of surface energy on a polymer surface, thesurface energy components of the polymer can be evaluated from
Eqn (3). Plots of [(1 + cos θ )/2] × γ l/
√γ d
l versus√
γpl /
√γ d
l would
be linear, with slope and intercept giving the surface energy data.Surface energy components of the polymer surface are measuredby the Kaelble (average of five measurements for five liquids) andRabel (regression) analysis methods.69 – 71
The contact angles of five liquids on dip-coated PLA andFluoroPLA films were measured. Five liquids were selected tocover the whole range of polarity from highly polar to non-polar(water, glycerol, formamide, methylene iodide, n-hexadecane).Table 3 gives the contact angle values for PLA and FluoroPLAs forthe various liquids. A significant difference in the contact anglesof PLA and FluoroPLAs can be seen for all test liquids. FluoroPLAsshow higher contact angles than PLA for all liquids, although thereis not a significant difference between the contact angles amongthe FluoroPLAs as the loading of PFPE segments remains nearlyconstant. We have found previously that by incorporating PFPEsegments in the PLA molecules the surface contact angle valuesfor a specific liquid increases sharply at low loadings (1 wt%) andthen levels off.40
Surface energy components of the PLA and FluoroPLA polymersare summarized in Table 4. For PLA, the dispersive componentsof surface energy obtained using the Kaelble and Rabel methodsare 29.7 and 26.9 mN m−1, respectively, and the respective polarcomponents of surface energy are 10.5 and 8.2 mN m−1. ForFluoroPLAs, the ranges of dispersive component of surface energyobtained using the Kaelble and Rabel methods are 15.2–17.2 and15.0–16.6 mN m−1, respectively, and the respective ranges of polarcomponent of surface energy are 1.7–2.2 and 1.1–1.2 mN m−1.There is no considerable effect of incorporation of various blocklengths of PFPE on the surface energies of FluoroPLAs becausethe overall fluorine content remains nearly the same irrespectiveof block length. The surface energies of FluoroPLAs are closeto those obtained for commercial fluorinated materials such aspolytetrafluoroethylene (Teflon).70,72
Table 4. Surface energy and its components (mN m−1) for PLA andFluoroPLAs
Kaelble (average) Rabel (regression)
Polymer Dispersive Polar Total Dispersive Polar Total
PLA 29.7 10.5 40.2 26.9 8.2 35.1FluoroPLA(4.2k) 17.2 2.0 19.2 16.6 1.1 17.7FluoroPLA(2.0k) 15.2 1.7 16.8 15.0 1.2 16.2FluoroPLA(1.5k) 16.4 2.2 18.5 15.1 1.1 16.2
30
40
50
60
70
80
90
100
PLA
FluoroPLA(4.2k)
FluoroPLA(2.0k)
FluoroPLA(1.5k)
Water Glycerol Formamide MethyleneIodide
n-Hexadecane
Wor
kof
adhe
sion
(m
N m
–1)
Figure 11. Work of adhesion for PLA and FluoroPLAs using five test liquids.
The wettability of a liquid droplet on a polymer surface can bequantified by the work of adhesion (WA) expressed as
WA = 2
(√γ d
s γ dl +
√γ
ps γ
pl
)(4)
As the contact angle increases for a given test liquid, WA
decreases, and the surface becomes repellent in nature. Figure 11shows WA for PLA and FluoroPLAs using water, glycerol,formamide, methylene iodide and n-hexadecane and calculatedusing Eqn (4) based on surface energy data from the Kaelblemethod. The work of adhesion for FluoroPLAs is lower thanthat of PLA in all five liquids. For water, the difference betweenthe work of adhesion of PLA and FluoroPLAs is ca 40 mNm−1. A similar difference is observed for glycerol. The differencedecreases from formamide to methylene iodide to n-hexadecane.The lowest difference of ca 15 mN m−1 is observed in case ofnon-polar n-hexadecane as only the dispersive forces contribute.Hence FluoroPLAs have better hydrophobicity and lipophobicitythan PLA.
wileyonlinelibrary.com/journal/pi c© 2010 Society of Chemical Industry Polym Int 2011; 60: 507–516

51
5
Poly(lactic acid)–perfluoropolyether block copolymers www.soci.org
CONCLUSIONSLaboratory-scale synthesis of PLA and FluoroPLAs was successfullycarried out by ROP of lactide using telechelic hydroxyl-terminatedPFPE as macroinitiator and Sn(Oct)2 as catalyst/initiator. Highmolecular weight (>60 000 g mol−1) copolymers (FluoroPLAs)of PLA and PFPE were obtained with controlled moleculardistribution. Molecular weights of the copolymers were dependenton the molar content of PFPE in the feed.
As expected, the copolymers showed lower glass transition,melting and degradation temperatures than the homopolymer(PLA). The extent of lowering of the transition temperatureswas strongly dependent on the molecular weight of the PFPEsegments. Smaller PFPE segments caused a large internalplasticization effect.
Surprisingly, FluoroPLAs showed higher heats of fusion anddegrees of crystallization indicative of higher crystallinity andprobably higher rates of crystallization than those of PLA. Theincrease in crystallization of FluoroPLAs is possibly due to thenucleating behavior of PFPE segments, which decreases theactivation energy of diffusion of PLA segments at crystallizationsites. Dynamic mechanical analysis showed that the tailoredFluoroPLAs can have higher low-temperature modulus than neatPLA in spite of the fact that the FluoroPLAs have lower molecularweights than the neat PLA. Smaller PFPE segment domains, inglassy or crystalline form, at lower temperatures likely act asreinforcing domains for the PLA matrix.
Incorporation of low-surface-energy segments in the PLA blockmodified markedly its surface morphology and surface properties.Surface energy dropped from 35–40 to 16–20 mN m−1 withvery small amounts of PFPE. FluoroPLAs exhibited significanthydrophobic and lipophobic properties compared to PLA. Thesenew polymers may find application in novel low-temperature-resistant amphiphobic renewable material-based coatings andplastics.
ACKNOWLEDGEMENTThe authors are grateful to the National Science Foundationthrough the Center for Advanced Engineering Fibers and Films(NSF-ERC) and the Department of Commerce through the NationalTextile Center (NTC). We also thank Dr Ivan Wlassics and SolvaySolexis SpA for their generous donation of Fomblins Z Dol
materials and Dr Igor Luzinov and group for the AFM facility. DWSmith Jr is a Cottrell Scholar of Research Corporation.
REFERENCES1 Gross RA and Kalra B, Science 297:803–807 (2002).2 Albertsson A-C and Varma IK, Biomacromolecules 4:1466–1486 (2003).3 Sinclair RG, J Macromol Sci A A33:585–597 (1996).4 Radano CP, Baker GL and Smith MR, J Am Chem Soc 122:1552–1553
(2000).5 Amass W, Amass A and Tighe B, Polym Int 47:89–144 (1998).6 Mecking S, Angew Chem Int Ed 43:1078–1085 (2004).7 Mehta R, Kumar V, Bhunia H and Upadhyay SN, Polym Rev 45:325–349
(2005).8 Huang J, Lisowski MS, Runt J, Hall ES, Kean RT, Buehler N, et al,
Macromolecules 31:2593–2599 (1998).9 Erhard G, Designing with Plastics. Hanser Gardner, Cincinnati, OH,
pp. 22–23 (2006).10 Grijpma DW, Van Hofslot RDA, Super H, Nijenhuis AJ and Pennings AJ,
Polym Eng Sci 34:1674–1684 (1994).11 Anderson KS, Lim SH and Hillmyer MA, J Appl Polym Sci 89:3757–3768
(2003).
12 Piorkowska E, Kulinski Z, Galeski A and Masirek R, Polymer47:7178–7188 (2006).
13 Abayasinghe NK, Glaser S, Perera PKU and Smith DWJ, J Polym Sci A:Polym Chem 43:5257–5266 (2005).
14 Dubois P, Jacobs C, Jerome R and Teyssie P, Macromolecules24:2266–2270 (1991).
15 Kricheldorf HR and Boettcher C, Makromol Chem Macromol Symp73:47–64 (1993).
16 Kowalski A, Duda A and Penczek S, Macromolecules 33:689–695(2000).
17 Chamberlain BM, Jazdzewski BA, Pink M, Hillmyer MA and Tolman WB,Macromolecules 33:3970–3977 (2000).
18 Cheng M, Attygalle AB, Lobkovsky EB and Coates GW, J Am Chem Soc121:11583–11584 (1999).
19 Kricheldorf HR, Kreiser-Saunders I and Stricker A, Macromolecules33:702–709 (2000).
20 Kricheldorf HR, Kreiser-Saunders I and Boettcher C, Polymer36:1253–1259 (1995).
21 Ryner M, Stridsberg K, Albertsson A-C, von Schenck H and Svensson M,Macromolecules 34:3877–3881 (2001).
22 Penczek S, Duda A, Szymanski R and Biela T, Macromol Symp 153:1–15(2000).
23 Dubois P, Ropson N, Jerome R and Teyssie P, Macromolecules29:1965–1975 (1996).
24 Zhang X, MacDonald DA, Goosen MFA and McAuley KB, J Polym Sci A:Polym Chem 32:2965–2970 (1994).
25 Nijenhuis AJ, Grijpma DW and Pennings AJ, Macromolecules25:6419–6424 (1992).
26 Diez FV, Sastre H and Coca J, Ind Eng Chem Res 27:845–847 (1988).27 Kricheldorf HR, Rost S, Wutz C and Domb A, Macromolecules
38:7018–7025 (2005).28 Pilati F, Toselli M, Messori M, Priola A, Bongiovanni R, Malucelli G, et al,
Macromolecules 32:6969–6976 (1999).29 Hillmyer MA and Lodge TP, J Polym Sci A: Polym Chem 40:1–8 (2002).30 Radhakrishnan K, Switek KA and Hillmyer MA, J Polym Sci A: Polym
Chem 42:853–861 (2004).31 Synytska A, Appelhans D, Wang ZG, Simon F, Lehmann F, Stamm M,
et al, Macromolecules 40:297–305 (2007).32 Temtchenko T, Turri S, Novelli S and Delucchi M, Prog Org Coat
43:75–84 (2001).33 Tonelli C, Trombetta T, Scicchitano M and Castiglioni G, J Appl Polym
Sci 57:1031–1042 (1995).34 Hopkins J and Badyal JPS, J Phys Chem 99:4261–4264 (1995).35 Thomas RR, Lloyd KG, Stika KM, Stephans LE, Magallanes GS,
Dimonie VL, et al, Macromolecules 33:8828–8841 (2000).36 Koh K, Sugiyama S, Morinaga T, Ohno K, Tsujii Y, Fukuda T, et al, Mac-
romolecules 38:1264–1270 (2005).37 Bottino FA, Di Pasquale G, Pollicino A, Pilati F, Toselli M and Tonelli C,
Macromolecules 31:7814–7819 (1998).38 Pilati F, Toselli M, Re A, Bottino FA, Pollicino A and Recca A, Mac-
romolecules 23:348–350 (1990).39 Toselli M, Messori M, Bongiovanni R, Malucelli G, Priola A, Pilati F, et al,
Polymer 42:1771–1779 (2001).40 Haynes D, Naskar AK, Singh A, Yang C-C, Burg KJ, Drews M, et al,
Macromolecules 40:9354–9360 (2007).41 Stevels WM, Ankone MJK, Dijkstra PJ and Feijen J, Macromol Chem
Phys 196:3687–3694 (1995).42 Duda A, Biela T, Libiszowski J, Penczek S, Dubois P, Mecerreyes D, et al,
Polym Degrad Stab 59:215–222 (1998).43 Slomkowski S, Sosnowski S and Gadzinowski M, Polym Degrad Stab
59:153–160 (1998).44 Kowalski A, Duda A and Penczek S, Macromolecules 33:7359–7370
(2000).45 Libiszowski J, Kowalski A, Duda A and Penczek S, Macromol Chem Phys
203:1694–1701 (2002).46 Bongiovanni R, Malucelli G, Messori M, Pilati F, Priola A, Tonelli C, et al,
J Polym Sci A: Polym Chem 43:3588–3599 (2005).47 Korhonen H, Helminen A and Seppala JV, Polymer 42:7541–7549
(2001).48 Tsuji H, Miyase T, Tezuka Y and Saha SK, Biomacromolecules 6:244–254
(2005).49 Espartero JL, Rashkov I, Li SM, Manolova N and Vert M, Macromolecules
29:3535–3539 (1996).50 Karis TE, Marchon B, Hopper DA and Siemens RL, J Fluorine Chem
118:81–94 (2002).51 Turri S, Barchiesi E and Levi M, Macromolecules 28:7271–7275 (1995).
Polym Int 2011; 60: 507–516 c© 2010 Society of Chemical Industry wileyonlinelibrary.com/journal/pi

51
6
www.soci.org A Singh et al.
52 Fox J, Thomas G and Flory PJ, J Appl Phys 21:581–591 (1950).53 Ajroldi G, Marchionni G and Pezzin G, Polymer 40:4163–4164 (1999).54 Zhang J-F and Sun X, Poly(lactic acid)-based bioplastics, in Bio-
degradable Polymers for Industrial Applications, ed. by Smith R. CRCPress, Washington, DC, pp. 251–288 (2005).
55 Turri S, Sanguineti A and Levi M, Macromol Chem Phys 198:3215–3228(1997).
56 Sperling LH, Introduction to Physical Polymer Science, 3rd edition. Wiley-Interscience, New York, pp. 295–362 (2001).
57 Keller A, Hikosaka M, Rastogi S, Toda A, Barham PJ and Goldbeck-Wood G, J Mater Sci 29:2579–2604 (1994).
58 Tonelli C, Pilati F, Toselli M, Turturro A and Gattiglia E, Fluoromodifiedpolyesters having improved processability. US Patent5686522 (1997).
59 Huang C-I, Tsai S-H and Chen C-M, J Polym Sci B: Polym Phys44:2438–2448 (2006).
60 Ehrenstein GW, Riedel G and Trawiel P, Thermal Analysis of Plastics:Theory and Practice. Hanser Gardner, Cincinnati, OH, pp. 236–299(2004).
61 Hoshino M, Kimachi Y and Terada A, J Appl Polym Sci 62:207–215(1996).
62 Meroni G, Pogliani C, Zompatori A, Trombetta T and Tonelli C,Perfluoropolyether building blocks: very efficient polymermodifiers, in Proceedings of the 8th Polymers for AdvancedTechnologies International Symposium, Budapest, 13–16 September(2005).
63 Turri S, Sanguineti A and Levi M, Macromol Chem Phys 198:3215–3228(1997).
64 Starkweather HW, Avakian P, Fontanella JJ and Wintersgill MC,Macromolecules 26:5084–5087 (1993).
65 Kobori Y, Iwata T, Doi Y and Abe H, Biomacromolecules 5:530–536(2004).
66 Brizzolara D, Cantow H-J, Diederichs K, Keller E and Domb AJ,Macromolecules 29:191–197 (1996).
67 Ho CC and Khew MC, Langmuir 16:1407–1414 (2000).68 Owens DK and Wendt RC, J Appl Polym Sci 13:1741–1747 (1969).69 Contact Angle. [Online]. Available : http://kruss.de [25 November 2010].70 Kaelble DH and Cirlin EH, J Polym Sci B: Polym Phys 9:363–368 (1971).71 Kaelble DH, Physical Chemistry of Adhesion. Wiley-Interscience, New
York, pp. 149–189 (1971).72 Legeay G, Coudreuse A, Legeais J-M, Werner L, Bulou A, Buzare J-Y,
et al, Eur Polym J 34:1457–1465 (1998).
wileyonlinelibrary.com/journal/pi c© 2010 Society of Chemical Industry Polym Int 2011; 60: 507–516