synthesis and electronic structures and linear optics of solid state compound srb2o4
TRANSCRIPT

CHINESE JOURNAL OF CHEMISTRY 2001, 19, 641-6t.6 641
Synthesis and Electronic Structures and Linear Optics of Solid State Compound SrB204
ZHANG, Hao(%i$) CHENG, Wen-Dan'(%%C) ZHENG, Fa-Kun(#&&l) CHEN , Jiu-Tong( I% A #i ) Fujiun Institllte o f Research on the Structure o f Matter, Chinese Academy o f Sciences, State K ~ Y Laboratory o f Structural Chemistry, Futhou , Fujian 350002, China
The cluster ( Sr&04 )* existing in crystalline states is employed to model the electronic structure and linear optical properties of solid state compound Sr&O,. 'Ihis compound is synthesized by high temperature solution reaction, and it crystallizes in the orthorhombic space group Pbcn with cell dimensions a = 1.1995(3), b=0.4337(1), ~=0 .6575(1 ) ~ 1 , V=0.34202 d, and Z = 4 , p=15.14 mi', D,,=3.36 g / d . The dynamic refractive indices are obtained in terms of INDO/SCI following combination with the SumOver-States method. A width of the calculated gap is 4.424 eV between the valence band and conduction band, and the calculated average refrac- tive index is 1.980 at a wavelength of 1.065 pm. "he charge transfers from d- anion orbitals to S?' cation orbitals make the signEcant contributions to linear polarizability in t e rn of analyses of atomic state density contributing to the valence and conduction bands.
Keywords ic stmctures. refractive index
Strontium metaborate Sr&OJ, crystal and electron-
compounds(3:1, 2 : 1 , 1 ~ 1 , 1 :2 , 1 : 3 ) in this sys- The luminescence of tetraborate SrB44 has been
studied, 91'0 and the polycrystalline phases and the single crystal structure with a space group Pbcn have been de- termined for strontium metaborate s ~ ~ o ~ . I ' * ~ ~ III this paper, we report the synthesis of SrE$04 in high temper- ature solution reaction with the different experimental routes from Ref. 12 and the p w t h condition of single crystal without seed crystals. We also report the calcu- lated electronic structure and linear polarizability of mi- cro-species (SrE$O4)2, and the estimated refractive in- dex of bulk SrE$Od. It is found that the charge transfers from the oxygen atomic orbital to strontium atomic orbital make the significant contributions to the polarizability .
Experimental and computational procedures
Preparation Introduction
In recent years, strontium borate has become a fo- cus of interest due to its posibility being a new optical material. Powder diffraction andyses and single crystal structural determination have been done, and linear op- tical properties have been studied for strontium pyrobo- rate Sr2b0,.1*2 Optical properties of the SIO-~O~ system and the phase equilibrium diagram as determined by the DTA technique have been reported for the five
The mixture containing appropriate amounts of the agents SrC0,(97.0%), LiC0,(98.0%), and H 3 B 4 (99. 0% ) , was heated in open platinum crucible at 1175 K , and cooled to 875 K, then air quenched to room temperature. During these processes, it is possible that the LiC0, was decomposed into Liz0 and CQ, and the Liz0 was a flux in p w i n g single crystals of SrbO,. In the experimental routes of L m et al . , the SK03 and &03 were used as starting materials, and the highest
* E-mail: cwd@ ms. fjirsrn. ac. cn
Received November 21, 2000; revised and accepted March 14, 2001. Project supported by the National Natural Science Foundation of China ( N o . 29973048). the Foundation of State Key Laboratory of Stnirtural Chemisfp (No. 200027).

642 Solid state compound ZHANG et al.
temperature of reaction was at 1428 K . I'
X-Ray a!eterminutwn
A crystal with approximate dimensions 0.20 x 0.15 x 0. 10 mm3 was selected for single crystal X-ray diffraction. The cell parameters and calculated volume: a = 1.1%5(3), b = 0.4337( l ) , c = 0.6575( 1 ) nm, V = 0 . 3 4 3 0 2 nm3, 2 = 4 , p = 15.14 cm-I, l'"= 173.24 and Dcalcd = 3 . 3 6 g/cm3. The final full-matrix least-squares refinement for 33 variable parameters con- verged to R = 4 . 4 0 % , R,=5.73% ( W = 1 / [ ( 2 ( F ) + (0.02F)' + 1 .O]) , S = 1.53, ( M a ) , = O.OOO4, neutral atomic scattering factors were taken from Cromer and Waber.I3 The maximum and minimum peaks on the final different Fourier map are 1.42 and - 0.96 x Id e/nm3 , respectively.
Compuiatwnal details
Some obtained results from electronic structural cal- culations were employed to compute microscopic polariz- ability, and the electronic structural calculations of the cluster (micro-species of the title compound) were based on an all-valence-electron, semi-empirical INDO self- consistent field (SCF) molecular orbital (MO) p m e - dure with configuration interaction ( CI) modified by Zemer and coworkers. I4-I7 'here are the one-center core integral U,, resonance integral pPv, two-electron inte- gral y,, , overlap integral S, and density matrix element P,, in the matrix element of Fock operator under the IN- DO approximation. The INDO model as employed herein included all one-center, two-electron integrals, and two- center, two-electron integrals ypu. 'he one-center two- electron integrals 7, were chosen from the Pariser ap-
twocenter two electron integrals were calculated using the Mataga-Nishimoto formula,'9 7, = 1. 2/[ RM t
2.4/(yw t y,)] in the spectroscopic version of tND0 method. The Slater orbital exponents [, which are taken from reference,B and the other calculation parameters are listed in note." The molecular orbital calculations were performed by the restricted Hartree-Fork method. The ground state, which was obtained from calculated results of the SCF, was taken as the state of reference in the CI. Only single-substituted determinants relative to
proximation, 18 y, = FO(p,u) = IF', - EA,, and the
the p u n d state configuration were considered and only singlet spin-adapted configurations needed to be included in the CI calculations. 'Ihe ground state and all excited states had the multiplicity of one. The electron was pro- moted from the thirteen highest occupied orbitals to the thirteen lowest unoccupied orbitals and the configuration space was constructed by these 26 active orbitals. The wavefunctions and energy eigenvalues of the excited states were determined by solving the secular equation related to configuration coefficients. 'Ihe dipole and transition moment matrix elements were expressed as a sum of one-electron integrals.
'Ihe tensor components of the polarizability a ( w ) with frequency dependence for the micro-species of the title compound was calculated by the sum-over-states (SOS) method as follows :
The unit ( SrE&04)2 consisting in extended oxide crystal S&04 is selected as model of cluster as shown in Fig. 1 . The calculations of electronic structures and po- larizabilities are based on the crystallographic structural data for this cluster.
Results and discussion
Ctystal s t m t m
The structure of Srk0, has been reported by Kim
et al . , '' and herein its more precision of determination is given by using 610 reflections with I > 30( I ) and some other structural details are mentioned. The struc- ture is based on the [ SK&] group linking to each other by sharing O( 2) **-O( 2) edge to form layers parallelling to the bc plane. Interlayer sites are occupied by the B atoms, and the [BQ] p u p is linked through sharing an O(1) atom localized in a comer to form the [ B q ] chains along the b-axis and bridges the neighboring lay- ers to foim the three-dimension framework ( Fig. 1 ) . In this crystal structure, the coordination polyhedron of the Sr atom may be described as a distorted dodecahedron and the S r O distances vary from 0.2507 ( 4 ) to 0.2734 ( 4 ) nni with an averige value of 0.2620 nrn, and it is larger than the expected value 0.259 nm calcu-

Vol. 19 NO. 7 2001 Chinese Journal of Chemistry 643
lated from the crystal radii for seven-coordinate S?' ion.22 It is noted that in the [ BOz] chains the distances between the B atom and the bridging 0 ( 1 ) atom (0.1391(7) or 0.1403(7) nm) are longer than the dis- tance between the B atom and the terminal 0 ( 2 ) atom (0.1319(7) MI). The two 0 atoms have different coor- dinations. The O ( 1 ) is a p3 atom linking two B atoms and one Sr atom and the sum of the B-O( 1)-Sr and B- O( 1)-B angles is 360". The O(2) is a p4 atom bonding one B atom and three Sr atoms with the B-O(2)-Sr and Sr-O(2)-Sr angles ranging from 93.2(3) to 126.1(4)". The atomic coordinates, and the selected bond distances and angles are listed in Table 1 and Table 2 , respective-
1Y -
fig. 1 M i c m t d model (upper) and crystal structure (lower) of SrhO,.
Table 1 Atomic coordinates and equivalent isotropic displacement parameters ( x nm2)
Atom X Y 2 U,O
Sr 0.5 0.2377( I ) 0.75 0.00558(8)
B 0.8058(5) 0.169( I ) 0 . 6 w I ) 0.0072(9)
O( I ) 0.7107(3) 0.3507(9) 0.64%(8) 0.0116(8) O ( 2 ) 0.9088(3) 0.2642(8) 0.5930(6) 0.0081(6)
' U,=(1/3S,S,U,,n,~n,'n,tr,.
Electronic structures
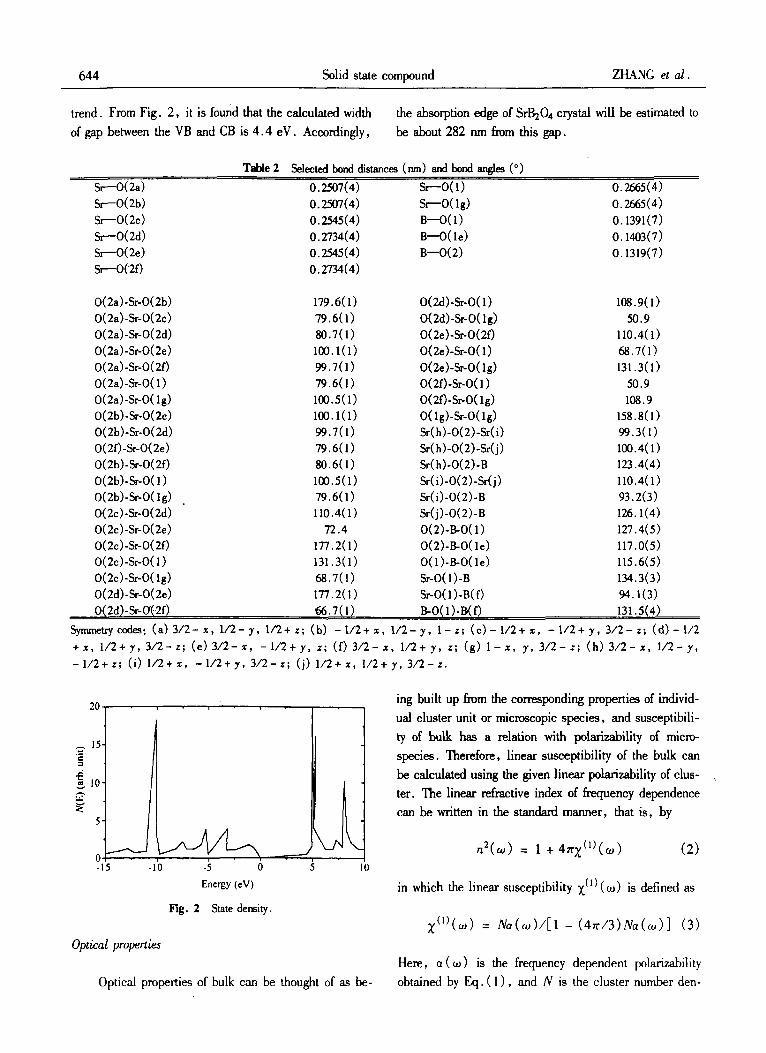
The calculations of electronic structures are based on the INDOIS quantum chemical method for the cluster (Sr&O,), . The calculated energy bands are divided in- to four zones and the topmost level in the valence band is regarded as a reference in order to convenience of de- scriptions. The zone of energy between - 22.1 and - 33.2 eV is a lower valence band one and this zone is attributable to the 2s orbitals of oxygen and boron atoms. There is 74%-85% 0-2s and less than 4 % B-2s or- bital character in this zone. It is approximately assigned as an s valence band. The second zone from - 16.3 to 0.00 eV is a contribution from 52 % -98 % 0-2p orbital character, but mixed with < 32% B-2p orbital character and it is an upper valence band zone. These two band zones are fully occupied by electrons. 'Ihe closer the top edge of valence band ( VB) , the more the contributions are from the 0-2p orbitals. The atomic state density of the upper VB zone, i . e . the calculated second zone, is displayed in Fig. 2. It is found that the band between the - 16.3 and - 9.8 eV is the contributions from 0-
bonding interactions between the 0-2p and B-2p or- bitals. For example, the peak in the energy about - 11 .O eV consists of 68% 0-2p and 25% B-2p or- bitals. ?he 'three peaks localized in the energies - 8.2- - 2.5 eV can be assigned as the x-bonding in- teractions between the 0-2p and B-2p orbitals. For ex- ample, the peak in the energy about - 7.5 eV consists of 61% 0-2p and 14% B-2p orbitals. The band be- tween the energies - 1 . 7 - 0 . 0 eV is assigned as the 2p nonbonding orbitals of the oxygen atoms. For instance, the peak in the energy about - 1 . 7 eV consists of 92% 0-2p orbital character. The third zone is a lower con- duction band (CB) and its atomic state density is shown on the right side of Fig. 2 . The peaks localized in the energy region between 4 . 4 and 10.9 eV are all the con- tributions from the strontium ionic state ( > 88% orbital characters). The fourth zone from 12.1 to 20.5 eV is made up of the antibonding interactions between the boron ( > 49% of 2s and 2p characters) and oxygen ( ( 4 7 % of 2p character) atomic valence orbitals, and this zone is not shown in Fig. 2 . The last two zones are completely empty ones, and the more the energy levels towanls the top edge of CB are, the more the contribu- tions frnm the B valence orbitals are. It is noted Lhat these assignments for atomic states are only a general
644 Solid state compound ZHANC et d.
trend. From Fig. 2, it is found that the calculated width of gap between the VB and CB is 4.4 eV. Accordingly,
the absorption edge of Srb04 crystal will be estimated to be about 282 nm from this gap.
0(2a)-Sr-O(2b) O( 2a)-Sr-O(2c) 0(2a)-Sr-O(2d) O(za)-Sr-O(Ze) O( 2a)-Sr-O(2f) O( 2a)-Sr-O( 1 ) O(2a)-Sr-O( Ig) 0(2b)-Sr-O(2c)
O( 2f)-Sr-O( 2e) 0(2b)-Sr-O( 2f) 0(2b)-Sr-O( 1) 0(2b)-Sr-O(lg) . O( 2c)-Sr-O(2d) O( 2c) -Sr-O( 2e) O(2c)-Sr-O(2f) O( 2c)-Sr-O( 1) O(2c)-Sr-O( Ig) 0(2d)-Sr-O(2e)
O( 2b)-S-O( 2d)
179.6( 1 ) 79.6(1) 80.7(1)
99.7(1) 79.6(1) 100.5(1) 100.1(1) 99.7(1) 79.6(1) 80.6( 1) 100.5(1) 79.6(1) 1 10.4( 1 )
12.4
131.3( 1) 68.7( 1)
100.1(1)
177.2( 1)
177.2( 1)
0(2d)-Sr-O( 1) 0(2d)-Sr-O( lg) O( Ze)-Sr-O( 2f) O(ze)-Sr-O( 1) O(Ze)-Sr-O( Ig) O( 2f)-Sr-O( 1 ) O(X)-Sr-O( Ig) O(lg)-Sr-O(lg) Sr( h)-0(2)-Sr( i ) Sr( h)-0(2)-Sr(j) Sr( h)-0(2)-B Sr( i)-O(2)-Sr(j) Sr( i)-0(2)-B Sr(j)-0(2)-B
0(2)-B-O( le) O( l)-B-O(le) Sr-O( 1)-B Sr-O( 1)-B( f )
0(2)-B0(1)
108.9( 1) 50.9
110.4(1) 68.7(1) 131.3( 1)
50.9 108.9
158.8( 1) 99.3(1) 1OO.4( 1) 123.4(4) 110.4( 1) 93.2(3) 126.1(4) 127.4( 5 ) 117.0(5) 115.6( 5 ) 134.3(3) 94.1(3)
O( 2d)-Sr-U( 2f) 66.7(1) B - 0 1 - ( ) B( f ) 131.5(4) Symmetry codes: (a) 312- z , 112- y, 1/2+ r ; (b) - 1/2+ z , 1/2- y. 1 - z; (c) - 1/2+ z , - 1/2+ y , 3/2- z ; (d) - 1/2 + z , 1 / 2 + y , 3 / 2 - 2 ; ( e ) 3 / 2 - 2 , - 1 / 2 + y , z; ( f ) 3 / 2 - r , 1 / 2 + y , I ; (g) 1 - Z , y , 3 / 2 - z ; ( h ) 3 / 2 - z , 1 /2 -y , -1 /2+z ; (i) I n + % , -1 /2+y , 312-2; (j) 1 /2+z , 1 /2+y , 3 /2- r .
Energy (W
Fig. 2 State density
ing built up from the componding properties of individ- ual cluster unit or microscopic species, and susceptibili- ty of bulk has a relation with polarizability of micro- species. Therefore, linear susceptibility of the bulk can be calculated using the given linear polarizability of clus- ter. The linear r ek t ive index of frequency dependence can be written in the standard manner, that is, by
~
in which the linear susceptibility x ( l ) ( w ) is defined as
Optical properties
Optical properties of bulk can be thought of as be- Here, a ( w ) is the frequency dependent polarizability obtained by Eq. ( 1 ) , and N is the cluster number den-
Vol. 19 No. 7 2001 Chinese Journal of Chemistry 645
a 40:
.- 0 . - G 30- 9 .- 3 '
sity. The practical form of N is obtained by the product of mass density and Avogadro's constant divided by mo- lar mass. For ( SrE$04 1 2 , with a molecular formula weight of 346.48 dmol and a mass density of 3 .36 g / cm3, we obtain N = 5 . 847 x 1021 cmJ. Before at- tempting to compute the variation of the refractive index against wavelengths for the bulk SrkO,, it is necessary to investigate the behavior of the convergence in the summation of excited states of cluster ( Sr&04)2 and check if the results calculated from the INDO/SCI method are reliable. Fig. 3 shows the plots of the calcu- lated average polarizability < a > versus the number of states for the cluster of the title solid compound at the input energies of 0.0 and 1 . 2 eV . Although the conver- gence is slow, the line is stable after summation over 150 states. From analyses combined with Fig. 3 with the states contributing to polarizability, we can find that the polarizabilities are mainly contributions from charge transfers from the 0-2p orbitals to Sr valence orbitals (4d, 5s. 5p) . For instances, the 5th state and the 31th state have the greatest contributions from the 94% con- figuration, +w33, and the 54% configuration, +32-42,
respectively. The configuration +w33 is formed by an electron from the % molecular orbital ( M O ) composed of 92% 0-2p orbital, to the ( ~ 3 ~ MO composed of 89% 5 - 5 s orbital, and the configuration +32-.42 is formed by an electron from the ( ~ 3 ~ MO composed of 84% 0-2p or- bital to the ( ~ 4 2 MO composed of 29% Sr-Ss, 27% Sr-5p and 33% Sr-4d orbital characters. The 151th state has the major contributions from the 55% configurations +21-39, and the 161th state has the major contributions
from the 72% configurations +w41. The configuration +2,-39 is formed by an electron from the MO com-
posed of 74% 0-2p and 12% B-2p orbitals, to the q+g MO composed of 45% Sr-5p and 41 % Sr-4d orbitals, and the configuration is formed by an electron from the % MO composed of 74% 0-2p and 17% B-2p orbitals, to the (p41 MO composed of 18% Sr-Ss, 53% Sr-5p and 18% Sr-4d orbitals.
The average dynamic refractive index of < n (0) > is calculated by Eqs . (2) and ( 3 ) using a given average polarizability < u > = ( urr t a, t a,)/3. The refrac- tive index < n ( W) > of bulk SrbO, from the wave- lengths of 0 . 40 to 2 . 50 pn has been calculated and plotted in Fig. 4 . The refractive index of SrE$04 crystal has not been observed, and the calculated resuhs are
0.0 eV 1.165 eV TI
only compared with that of power. The observed refrac- tive indexes of power, n, , n, , n, are individually 1.632, 1.650, 1.660 and the average refractive index is 1.647 ,= the calculated refractive indexes, n,, n, , n, are individually 1.405, 1.812, 2.157, and the aver- age < n ( w ) > is about 1.980 at a wavelength of 1.065 p for SrE$04 bulk. 'Ihis value may be an overestima- tion due to that the approximations of calculated method and cluster modeling are applied in this studies.
50, . 1
Number of states
Fig. 3 Polarizabilities < a > of cluster model.
4.0
0.5 I .o I .5 2.0 2.5 Wavelength, )i (pm)
Ffg. 4 Calculated dynamic refractive index < n ( w ) > .
References
1 Lin, Q . S . ; Cheng, W . D . ; Chen, J.T.; Huang, J.S. J . sol. stntechem. 1999,144,u).
2 B a d , H . ; Schuckmann, W . Nem Jaharb. Mineral. Monotsh. 1%6, 8 , 253.
3 Hart, P. B . ; Smallwood, S. E. F. J . Inorg. Nucl. Chern. 1962, 2 4 , 1047.
4 5 Weir, C . E.; Schroeder R . A . J . Res. N d b w . Stand.
Krogh-Moe, J . Actn Chem. Sand. 1964, 18, 2055.
.Sect. A . 1964, 68. 465.
1 7 . 314. 6 Block. S . ; PerlotT. A . ; Weir, C . E . Ac~n Cryst. 1964.

646 Solid state compound ZHANC et d.
7
8
9
10
11 12
13
14
15
16
17
Koskentalo, T . ; Leskla, M . ; Niinisttr, L. Ma&. Res. Bull. w15, 20, 265.
Witpnann, H.; Henog, G. Z. Phy5. Chem. 1964, 225, 197. Meijerink, A . ; Nuyten, J . ; Blasse, G. J . Lwnin. 1989, 44, 19. Blase, G . ; Dirksen, G. J . ; Meijerink, A. Chem. Phys. Len. 1990, 167, 41. Demer, P. D. A c t a C y t . 1969, B25, 1001. Kim, J.B.; Lee, K.S.; Suh, L.H.; Lee, J .H.; Park, J.R. ; Shen, Y.H. Acta Cryst. 1%, C52, 498. C m r , D. T . ; Waber, J. T. in "INemoabnol Tabkcfor X-ray CrystauogTaphy", Vol. IV, Table 2. 2A, Kynoch Press, Birmingham, 1974, p. 71. Bacon, A . D.; Zemer, M. C. h r . Chim. A& 1979, 53, 21. Zemer, M. C . ; hvw, G. H. : Kirchner, R. F.; Mueller- Westerhoff, U . T. 1. Am. Chem. SOC. 1980, 102, 589. Anderson, W. P . ; Edwards, E. D . ; Zemer, M . C. In- org. Chem. 1986, 25, 2728. Anderson, W. P.; Cundari, T. R . ; Zemer, M. C. Int .
J. QLLMtwn Chem. 1991, 39 , 31. 18 Pariser, R. 1. Chem. Phys. 1953, 2 1 , 568. 19 Motaga, N . ; Nishimoto, K. Z. Phys. Chem. 1957, 13,
140. 20 Lin, Q.S.; Cheng, W.D.; Chen, J .S . ; Huang, J.S. J .
sol. statecha. 1999,144,30. The input parameten are as follows: Slater exponents Sr: t&,=l.214K1, \;,=2.058A1, B: ~ , , , = 1 . 3 o o A " ,
0: L,2p = 2.275 K' ; vduence state ionization energies Sr : I s s= -5.84eV, Isp= -3.76 eV, I*= -3.66 eV, B: I z , = - 14.05 eV, I % = - 8.70 eV, 0: I h = -32.90 eV , I*, = - 17.28 eV ; Coulomb repulsion integrals Sr: yss, = ~ ~ , , = y ~ ~ ~ = 3 . 7 5 eV, yw=5.31 eV, yMI=yMP= 4.53 eV, B: y=8 .68 eV, 0: y = 13.00 eV; resonance integralsSr: pS*=pS,= -1.88eV, pM= -10 .05eV, B: p = - 17.00 eV, 0: y = -34.20 eV. ShannonR. D. ActaCrysdlog~. 1976, A 3 2 , 751. Donnay, J. D. H.; Ondik, H. M. in"CrystalDarnDeter- minative T&!a " , Polydrystal Book Service, Pittsburgh, 1973, Vol. II, P. 0-63 .
21
22 23
(E2ooO11251 JLWG, X . H . ; LING, J . )