swiss medical forum 10/2016 · a. burri, v. garelli, n. petitpierre ... pr nicolas rodondi, berne...
TRANSCRIPT

SwissMedical Forum
Offizielles Fortbildungsorgan der FMHOrgane officiel de la FMH pour la formation continueBollettino ufficiale per la formazione della FMHOrgan da perfecziunament uffizial da la FMH www.medicalforum.ch
With extended abstracts from “Swiss Medical Weekly”
10
9. 3
. 201
6
241 S. Hofmann, A. Buser, A. TaegtmeyerDéficit en glucose-6- phosphate déshydrogénase
245 A. Burri, V. Garelli, N. PetitpierreEn suivant les rails du tram
249 C. Burkhardt, C. Neuwirth, M. WeberScreening von Kognitions- und Verhaltensänderungen bei der Amyotrophen Lateralsklerose
236 A. Durovic, S. Tschudin-SutterClostridium difficile: une mise à jour
FMS – SMF Forum Médical Suisse – Forum Medico Svizzero – Forum Medical Svizzer – Schweizerisches Medizin-Forum

Et ailleurs…?
A. de Torrenté
235 Emphysème hétérogène grave: un nouveau traitement? Articles de revue
A. Durovic, S. Tschudin-Sutter
236 Clostridium difficile: une mise à jourClostridium difficile est la cause infectieuse la plus fréquente de diarrhée d’origine nosocomiale, et son importance en tant qu’agent pathogène des maladies diarrhéiques contractées en ambulatoire ne cesse d’augmenter. Les infections à C. difficile sont associées à une morbidité et une mortalité élevées, et s’accompagnent d’un large spectre de symptômes cliniques.
S. Hofmann, A. Buser, A. Taegtmeyer
241 Déficit en glucose-6-phosphate déshydrogénaseIl est de plus en plus fréquent de rencontrer au cabinet ou à l’hôpital des patients qui présentent un déficit en glucose-6-phosphate déshydrogénase et pour lesquels il est important de savoir quels médicaments sont potentiellement dangereux. Cet article de revue décrit le déficit en glucose-6-phosphate déshydrogénase, son diagnostic et les médicaments qui sont à éviter.
Quel est votre diagnostic?
A. Burri, V. Garelli, N. Petitpierre
245 En suivant les rails du tramIl s’agit d’une patiente de 73 ans qui présente depuis trois mois une toux incoercible accompagnée d’expectorations abondantes. Elle décrit aussi une dyspnée progressive, actuellement de stade II selon NYHA, et ne signale pas d’état fébrile.La patiente est connue pour un état de dénutrition protéino-calorique, une ostéoporose, et une rectocolite ulcé ro-hémorragique depuis 30 ans.
Recherche
C. Burkhardt, C. Neuwirth, M. Weber
249 Screening von Kognitions- und Verhaltensänderungen bei der Amyotrophen LateralskleroseDie bisher gängigen neuropsycho logischen Tests zur Untersuchung fronto-temporaler Funktionen bei ALS wurden nicht eigens für die ALS-Erkrankten entwickelt und weisen dadurch keine An passung an die besonderen körperlichen Einschränkungen dieser Patienten auf. Die Durchführung und Auswertung wird hierdurch erschwert oder gar verunmöglicht. Mit dem ECAS wurde ein schnell und einfach anwendbares klinisches Screening-Tool entwickelt.
SOMMAIRE 233
Rédaction
Pr Nicolas Rodondi, Berne (Rédacteur en chef); Dr Nadja Pecinska, Bâle (Managing editor); Pr David Conen, Bâle; Pr Martin Krause, Münsterlingen; Pr Klaus Neftel, Berne; Pr Antoine de Torrenté, La Chaux-de-Fonds; Pr Gérard Waeber, Lausanne; PD Dr Maria Monika Wertli, Berne
Rédacteurs conseil
Pr Reto Krapf, Lucerne; Pr Ludwig T. Heuss, Zollikerberg; Dr Pierre Périat, Bâle; Pr Rolf A. Streuli, Langenthal
Membres-adjoints à la rédaction
Dr Sebastian Carballo, Genève; Dr Daniel Franzen, Zurich; Dr Francine Glassey Perrenoud, La Chaux-de-Fonds; Dr Markus Gnädinger, Steinach; Dr Matteo Monti, Lausanne; Dr Sven Streit, Berne; PD Dr Ryan Tandjung, Zurich

Casuistiques
A. Koster-Rusch, J. Capraro, W. Nagel, T. Clerici, S. J. Stoeckli, F. Forrer, M. Brändle, S. Bilz
253 Bilaterale Halsschwellung bei einer jungen FrauParagangliome des Kopf- und Halsbereichs («Glomus tumoren») sind seltene neuroendokrine Tumoren, die von den parasympathischen Paraganglien ausgehen.
T. Rauer, T. De Zulueta, U. Caspar, B. Wagner, M. Zünd
256 Das histiozytäre SarkomDiese äusserst seltene und häufig aggressive Non-Langerhanszell-Histiozytose erfordert sowohl in der Diagnosestellung als auch in der Therapieplanung eine sehr enge interdisziplinäre Zusammenarbeit.
Extended abstracts from SMW
New articles from the online journal “Swiss Medical Weekly” are presented after page 258.
SOMMAIRE 234
ImpressumSwiss Medical Forum – Forum Médical SuisseOrgane officiel de formation continue de la Fédération des médecins suisses FMH et de la Société Suisse de Méde-cine Interne
Adresse de la rédaction: Ruth Schindler, Assistante de la rédaction FMS, EMH Editions Médicales Suisses SA, Farnsburgerstrasse 8, 4132 Muttenz, tél. +41 (0)61 467 85 55, fax +41 (0)61 467 85 56,[email protected], www.medicalforum.ch
Soumission en ligne des manuscrits:http://www.edmgr.com/smf
Editions: EMH Editions Médicales Suisses SA, Farnsburgerstrasse 8, 4132 Muttenz, tél. +41 (0)61 467 85 55, fax +41 (0)61 467 85 56, www.emh.ch
Marketing EMH / annonces: Dr Karin Würz, Responsable communication et marketing, tél. +41 (0)61 467 85 49, fax +41 (0)61 467 85 56, [email protected]
Abonnements membres FMH: FMH Fédération des médecins suisses, Elfenstrasse 18, 3000 Berne 15, tél. +41 (0)31 359 11 11, fax +41 (0)31 359 11 12, [email protected]
Autres abonnements: EMH Editions Médicales Suisses SA, abonnements, Farnsburgerstrasse 8, 4132 Muttenz, tél. +41 (0)61 467 85 75, fax +41 (0)61 467 85 76, [email protected]
Prix d‘abonnement: avec Bulletin des médecins suisses 1 an CHF 395.– / étudiants CHF 198.– plus frais de port; sans Bulletin des médecins suisses 1 an CHF 175.– / étudiants CHF 88.– plus frais de port
(abonnements de courte durée voir www.medicalforum.ch)
ISSN: version imprimée: 1424-3784 / version en ligne: 1424-4020Paraît le mercredi
© EMH Editions Médicales Suisses SA (EMH), 2016. Le Forum Médical Suisse est une publication «open-acess» de EMH. Sur la base de la licence Creative Commons «Attribution – Pas d’Utilisation Commerciale – Pas de Modification 4.0 International», EMH accorde à tous les utilisateurs le droit, illimité dans le temps, de reproduire, distribuer et communiquer cette créa-tion au public, selon les conditions suivantes: (1) Citer le nom de l’auteur; (2) ne pas utiliser cette création à des fins commerciales; (3) ne pas modifier, transformer ou adapter cette création. L’utilisation à des fins commercialespeut être possible uniquement après
obtention explicite de l’autorisation de EMH et sur la base d’un accord écrit.
Note: Toutes les données publiées dans ce journal ont été vérifiées avec le plus grand soin. Les publications signées du nom des auteurs reflètent tout l’opinion de ces derniers, pas forcé-ment celle de la rédaction du FMS. Les doses, indications et formes d’application mentionnées doivent en tous les cas être comparées aux notices des médicaments utilisés, en particulier pour les médicaments récemment autorisés.
Production: Schwabe AG, Muttenz, www.schwabe.ch
Photo de couverture: © CDC / James Archer

Et ailleurs…?Antoine de Torrenté
Emphysème hétérogène grave: un nouveau traitement?
La questionMalgré l’optimisation des traitements médicaux, les patients avec une BPCO sévère restent gravement handicapés. Chez certains patients très sélectionnés, la résection chirurgicale des parties pulmonaires les plus gravement atteintes permet d’améliorer la fonction pulmonaire, l’état de santé général et même la survie. Mais cette chirurgie pratiquée sur des patients très fragiles reste grevée d’une mortalité de 5% environ. Pour éviter la chirurgie, il est possible de placer par bronchoscopie des valves unidirectionnelles (pas d’insufflation possible) réduisant l’hyperinflation du lobe concerné et conduisant parfois jusqu’au collapsus de celuici, un résultat recherché. Idéalement, les scissures pulmonaires doivent être intactes permettant l’affaissement lobaire désiré. Cette technique estelle moins dangereuse que la chirurgie?
La méthodeLes patients concernés avaient un VEMS 1s <50% de la valeur prédite, un volume résiduel
>150% et un périmètre de marche de 6 minutes <450 m. Tous les patients recevaient un traitement médical optimal. Le CT pulmonaire devait montrer un emphysème hétérogène avec un lobe visé presque entièrement détruit et une scissure intacte. L’étude était randomisée, masquée, un groupe recevant une ou plusieurs valves et le groupe placebo subissant une bronchoscoopie sans insertion de valve. L’issue primaire était le % de différence du VEMS 1s entre l’incorporation dans l’étude et à 3 mois. Parmi les issues secondaires, on note la capacité d’endurance mesurée sur bicyclette ergométrique, la distance de marche en 6 mn et le changement de l’état de santé (questionnaire de St George pour les patients avec BPCO).
Les résultats25 patients ont reçu une médiane de 3 valves et 25 ont servi de contrôle. Parmi la foule de données recueillies, il faut noter à 3 mois une augmentation du VEMS de 8,7 vs 2,8% du groupe contrôle, p = 0,032. En faveur du groupe avec valve, on note aussi une augmentation significative de l’endurance et du périmètre de marche. Les complications ont tou
tefois été nombreuses: 2 pneumonies dans chaque groupe. Dans le groupe actif, 2 valves ont dû être retirées et 5 ont été spontanément ex pectorées. Il y a eu deux décès dans le groupe actif et 0 dans le groupe contrôle.
Problèmes et commentaireL’étude à montré que l’exclusion d’un lobe ventilatoirement inutile et délétère par des valves unidirectionnelles améliorait la fonction pulmonaire. On peut relever le courage des patients prêts à subir une bronchoscopie «pour rien». Pourtant, on ne peut pas vraiment tirer de conclusion quant à la supériorité de la méthode comparée à la chirurgie de résection tant dans l’efficacité que dans la sécurité. En consultant le graphique d’augmentation du VEMS, ont voit clairement que seuls 5 patients sur 25 ont vraiment bénéficié de la méthode avec une augmentation du VEMS de >50%. En résumé, pour une affection gravissime, la réduction du volume pulmonaire reste une option. Le choix de la méthode dépend probablement de l’expérience des équipes: chirurgie ou valve? Davey C, et al. Lancet. 2015 Sep 12;386(9998): 1066–73.
Prévention des adénomes colo-rectauxDes études épidémiologiques avaient suggéré que des concentrations sériques élevées de vitamine D et des suppléments de calcium diminuaient les risques de néoplasie colorectaux. ~2300 patients ayant eu des adénomes sans lésions résiduelles après coloscopie ont été randomisés pour recevoir 1000 U de vitamine D/j et 1200 mg de calcium/j ou aucun traitement. Une coloscopie 3 à 5 ans plus tard a découvert des adénomes chez 43 % des patients dans les deux groupes donc sans avantage pour le traitement «préventif». La question semble résolue…Baron JA, et al. N Engl J Med. 2015 Oct 15;373(16): 1519–30.
Médicaments et Internet: le bazar!Une estimation de l’OMS révèle que 50% des médicaments vendus en ligne ne sont pas conformes quant aux standard pharmaceutiques: sous dosage, contaminations diverses. Environ 5 millions d’Américains achèteraient des médicaments en ligne pour 70 à 200 milliards de $ par an. Fournisseurs principaux: l’Inde et la Chine. Les conséquences peuvent être dramatiques sur un plan individuel ou
sur le plan de santé publique, surtout en ce qui concerne les antimalariques. Clark F. Lancet. 2015 Oct 3;386(10001):1327–8.
Spironolactone (Aldactone®) et hypertension résistante300 patients avec une hypertension résistante (TA systolique au cabinet >140 mm Hg) déjà sous triple thérapie ont reçu en plus de leur traitement pendant des périodes de 12 semaines de la spironolactone (25 à 50 mg), de la doxazosine (Cardura®), du bisoprolol (Concor®) ou un placebo. La spironolactone réduit significativement la TA systolique de près de 9 mm Hg. Attention à l’hyperkaliémie! Williams B, et al. Lancet. 2015 Sep 18. pii:S0140–6736(15)00257–3.
Acide urique, goutte et risque cardiovasculaireL’hyperuricémie et la goutte sont associées à un risque cardiovasculaire plus élevé. D’environ 2650 patients goutteux tirés d’une cohorte de 40 000, 55% recevaient un traitement par un médicament hypouricémiant et 45% n’étaient pas traités. Les patients non traités avaient un risque augmenté de décès cardio
vasculaire, HR 2,4. Les patients traités avaient un HR de 0,5 de décès toutes causes confondues. Fautil aussi traiter les patients avec un acide urique élevé mais sans goutte? Probablement…Chen JH, et al. J Rheumatol. 2015 Sep;42(9): 1694–701.
Devenir à long terme des interventions pour anévrisme de l’aorte abdominalePrès de 80 000 patients >65 ans ayant subi soit une intervention par prothèse endovasculaire soit une intervention ouverte ont été appariés. La mortalité opératoire était de 1,6% pour l’intervention endovasculaire contre 5,2%. La conver sion en opération ouverte a baissé de 2,2% en 2001 vs 0,3% en 2008. Mais sur 8 ans de suivi une rupture aortique est survenue chez 5,4% dans le groupe endovasculaire contre 1,4%. L’avantage de survie du groupe endovasculaire disparaît après 3 ans de suivi et rejoint la courbe des interventions ouvertes. Ces chiffres sont importants pour faciliter une décision éclairée! Shermerhorn ML, et al. N Engl J Med. 2015 Jul 23; 373(4):328–38.
ET AILLEURS…? 235
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):235

ARTICLE DE REVUE 236
La cause infectieuse la plus fréquente de diarrhée nosocomiale
Clostridium difficile: une mise à jourAna Durovic, Sarah Tschudin-Sutter
Klinik für Infektiologie und Spitalhygiene, Universitätsspital Basel
Clostridium difficile est la cause infectieuse la plus fréquente de diarrhée d’origine nosocomiale, et son importance en tant qu’agent pathogène des maladies diar-rhéiques contractées en ambulatoire ne cesse d’augmenter. Les infections à C. diffi-cile sont associées à une morbidité et une mortalité élevées, et s’accompagnent d’un large spectre de symptômes cliniques, allant de la diarrhée légère à la colite toxique sévère. Les formes d’évolution clinique sévère et le taux élevé de récidive constituent des défis thérapeutiques particuliers.
Principes de base
Introduction/contexteUne infection à Clostridium difficile (ICD) provoque diarrhée aiguë et colite. Son agent pathogène a été dé-couvert pour la première fois en 1935, faisant partie in-tégrante de la flore intestinale normale chez le nou-veau-né. C’est seulement en 1978 que ce bâtonnet anaérobie Gram-positif a été identifié comme respon-sable de la colite pseudomembraneuse. Sa capacité de sporulation permet à la bactérie de survivre plusieurs mois sur des surfaces inertes, même dans des condi-tions environnementales défavorables. La plupart des désinfectants et des antibiotiques ne sont pas en me-sure de détruire ces formes résistantes. Dès lors, des germes revivifiables sur les mains du personnel soi-gnant ou sur les surfaces de contact représentent une source d’infection potentielle en milieu hospitalier.Après absorption, les spores migrent dans le côlon sous une forme végétative productrice de toxines et peuvent ainsi exercer leur effet pathogène sur leur hôte. Le principal facteur de risque du développement d’une ICD est une précédente exposition antibiotique, en particulier aux céphalosporines, quinolones, pénicil-lines et clindamycine. Ces substances modifient la composition de la flore intestinale normale et offrent ainsi à C. difficile, résistant à une multitude d’antibio-tiques différents, un avantage de survie. D’autres fac-teurs de risque sont la prise d’antiacides (inhibiteurs de la pompe à protons et antagonistes des récepteurs H2), les hospitalisations précédentes, la durée d’hospitalisa-tion, l’âge, les maladies sous-jacentes sévères, les opéra-tions de chirurgie viscérale et l’alimentation par sonde.Dans le côlon, C. difficile produit avant tout deux toxines (les toxines A et B) qui provoquent une réac-
tion inflammatoire neutrophile sévère. Celle-ci en-traîne diarrhée, érosions de la muqueuse intestinale et formation de pseudomembranes.La plupart des souches produisent les deux toxines simul tanément; une seule suffit cependant à déclen-cher une diarrhée ou une colite. Certaines souches sé-crètent en outre la toxine binaire, produite le plus sou-vent par des ribotypes hypervirulents et qui constitue donc un facteur de virulence, bien que sa pertinence reste floue. Les souches qui ne produisent pas de toxines peuvent coloniser l’intestin mais ne sont pas pathogènes.Ana Durovic
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):236–240

ARTICLE DE REVUE 237
EpidémiologieDepuis le début du XXIe siècle, l’épidémiologie de l’ICD a considérablement évolué. C’est principalement aux Etats-Unis et au Canada que des épidémies d’ICD parti-
culièrement sévères ont été observées dans un nombre croissant d’hôpitaux. Ces infections étaient imputables à une souche spécifique appelée PCR-ribotype 027 hy-pervirulent. Le ribotypage PCR est un procédé de ty-page moléculaire utilisé afin d’analyser la propagation de différentes souches de C. difficile. Le PCR-ribotype 027 s’est ensuite largement répandu en Grande-Bre-tagne et dans d’autres pays européens. Au cours des dernières années, une augmentation significative des ICD contractées en ambulatoire a été documentée. Ceci pourrait s’expliquer notamment par des hospitalisa-tions plus courtes et une administration croissante d’antibiotiques en milieu ambulatoire. Les souches hyper virulentes sont potentiellement associées aux ICD chez les patients à faible risque tels que les femmes en postpartum et les enfants.
Manifestations cliniquesLe spectre clinique d’une ICD s’étend de la colonisation asymptomatique aux états septiques les plus sévères avec mégacôlon toxique. Le degré de sévérité de la diar-rhée varie de minime à profus. La fièvre, les douleurs abdominales et les spasmes sont des co-symptômes possibles. La formule sanguine révèle souvent une leu-cocytose. Le tableau 1 regroupe les principales défini-tions ainsi que les manifestations cliniques de l’ICD.
Diagnostic [1]Le pilier du diagnostic de l’ICD est la mise en évidence de toxine A ou B dans les selles. La méthode de réfé-rence est la culture sélective avec mise en évidence de toxine dans la culture cellulaire. En raison de l’énorme quantité de travail et du temps nécessaires, les résul-tats ne sont bien souvent disponibles qu’après 48 heures. Cette méthode a donc été en grande partie abandon-née au profit de procédés plus rapides et moins coû-teux. Entre-temps, les tests immuno-enzymatiques (EIA) se sont établis en tant qu’alternative. Grâce à ces tests standardisés, il est possible de mettre en évidence un antigène de Clostridium (glutamate déshydrogé-nase, GDH) et certaines toxines en l’espace de 1 heure, directement à partir des selles. La mise en évidence de GDH est très sensible (tab. 2); l’enzyme est cependant également sécrétée par des souches C. difficile non pro-ductrices de toxine et ce test ne peut donc pas être réa-lisé comme test unique. La sensibilité de la mise en évi-dence de toxine au moyen des EIA est beaucoup trop faible pour un diagnostic fiable (tab. 2), de sorte que les directives actuelles recommandent pour l’investiga-tion diagnostique un modèle à deux niveaux. A cette fin, différentes combinaisons de tests sont possibles. Une des options réside dans l’analyse préalable de chaque échantillon de selles à la recherche de GDH,
Tableau 2: Sensibilité et spécificité des différents tests diagnostiques pour la mise en évidence de Clostridium difficile [1].
Sensibilité Spécificité
Mise en évidence de C. difficile (souches positives ou non aux toxines)
EIA pour mise en évidence de la GDH 0,71–1,00 0,67–1,00
Mise en évidence de toxines
EIA pour mise en évidence de toxine A et/ou B 0,31–0,99 0,65–1,00
QPCR pour mise en évidence du gène de toxine B 0,86–1,00 0,94–1,00
Abréviations: EIA = test immuno-enzymatique; GDH = glutamate déshydrogénase; QPCR = PCR quantitative, ou réaction en chaîne par polymérase en temps réel.
Tableau 1: Définitions et manifestations cliniques des infections à Clostridium difficile (ICD) [2].
Manifestations cliniques des ICD
– Diarrhée d’intensité variable avec/sans réaction systémique.– Typiquement en rapport avec un traitement antibiotique.– Evolutions sévères avec colite pseudomembraneuse, mégacôlon
toxique, iléus.
Episodes d’ICD – Manifestations cliniques typiques de l’ICD avec mise en évidence de toxines libres et de C. difficile dans les selles, sans signe d’une autre cause de diarrhée.
– Mise en évidence histologique/coloscopique d’une colite pseudo-membraneuse.
Récidive d’ICD – Survenue d’un nouvel épisode d’ICD symptomatique dans les 8 semaines après le début des premiers épisodes (à condition que les symptômes des premiers épisodes disparaissent après le trai-tement).
ICD légère – Diarrhée causée par C. difficile sans symptôme d’infection sévère/compliquée.
Les patients sans signe d’une ICD sévère, âgés de ≥65 ans, présentant une maladie sous-jacente sévère, recevant un traitement de soins intensifs ou un traitement immunosuppresseur ont un risque élevé de souffrir d’une évolution sévère.
ICD sévère – Episode d’ICD accompagné de ≥1 symptôme(s) de colite sévère ou d’une évolution compliquée avec réaction inflammatoire systé-mique sévère:
Observations cliniques:1. Fièvre >38,5 °C, frissons2. Instabilité hémodynamique ou insuffisance respiratoire3. Signes cliniques d’une péritonite ou d’un iléus
Observations de laboratoire:1. Leucocytose >15 × 109/l, déviation à gauche >20% neutrophiles
polynucléaires2. Elévation du taux créatinine sérique de >50% par rapport
à la valeur initiale3. Faible taux d’albumine sérique <30 g/l4. Elévation du taux de lactate sérique ≥5 mmol/l
Coloscopie ou sigmoïdoscopie:Mise en évidence endoscopique d’une colite pseudomembraneuse
Examen d’imagerie: Epaississement de la paroi colique, dilatation des boucles coliques, imbibition des tissus adipeux entourant le côlon, ascite (sans autre cause)
ICD sévère avec complications
– ICD sévère avec réaction systémique sévère et choc entraînant un traitement intensif, une colectomie ou le décès.
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):236–240

ARTICLE DE REVUE 238
puis de procéder dans un second temps à la recherche de toxine A et B des échantillons positifs. La QPCR (PCR quantitative, ou réaction en chaîne par polymérase en temps réel) est de plus en plus utilisée pour le diagnos-tic. Disponible entre-temps en tant que système com-mercial, elle met en évidence les C. difficile productrices de toxine avec une grande fiabilité (tab. 2).La présentation clinique constitue un prérequis essentiel pour l’interprétation des résultats de laboratoire. Malgré un traitement réussi, C. difficile peut persister dans les selles pendant quelque temps; chez certaines personnes, le germe fait même partie de la flore intestinale normale. Les analyses de selles de consistance normale, en particu-lier pour le contrôle thérapeutique, n’apportent ainsi quasiment aucune information et devrai ent donc, pour des raisons financières, être évitées.
Traitement [2] La stratégie thérapeutique d’une ICD dépend du degré de sévérité clinique. En principe, il est nécessaire pour chaque pose de diagnostic de réévaluer de manière cri-tique le traitement antibiotique déclencheur, et de le stopper en l’absence d’indication de maintien. Il est né-cessaire de procéder à une substitution volumique et électrolytique, et d’éviter les médicaments antipéristal-tiques. L’indication d’éventuels antiacides devrait être re-considérée. Le traitement standard des formes d’évolu-tion légère reste l’administration orale de métronidazole ou de vancomycine (tab. 3). En raison de ses propriétés pharmacologiques, la vancomycine est considérée comme supérieure au métronidazole dans le traite-ment de l’ICD sévère. Avec des taux de récidive allant jusqu’à 20%, les traitements courants ne sont toutefois pas encore optimaux. Les deux anti biotiques en particu-lier retardent la reconstitution de la microflore en raison de leur spectre d’action. Un traitement par probiotiques n’est actuellement pas recommandé.
PréventionUne utilisation rationnelle des antibiotiques est proba-blement la principale mesure préventive contre une ICD.En complément, des mesures d’hygiène visant à empê-cher le contact avec la bactérie (en particulier une hy-giène des mains stricte) doivent être prises, avant tout chez les patients stationnaires. En cas d’ICD connue, le port de gants et de tabliers à usage unique permet de réduire davantage la transmission. Les toilettes ne doivent pas être utilisées par d’autres personnes, et ce au moins jusqu’à la normalisation de la consistance des selles. Idéalement, ces patients devraient être hos-pitalisés en chambre individuelle. Actuellement, les traitements préventifs et le dépistage de porteurs asymptomatiques ne sont pas conseillés.
Nouvelles connaissances et développements actuels
Incidence et prévalenceA l’échelle internationale, de grands efforts sont entre-pris afin de saisir davantage l’ampleur de l’ICD. Les chiffres publiés jusqu’à présent affichent une incidence élevée persistante de ces infections à l’échelle mon-diale. Dans une étude réalisée récemment et portant sur la prévalence des infections nosocomiales aux Etats-Unis, C. difficile a été classé comme l’agent patho-gène le plus fréquent [3]. En 2008, une étude épidémio-logique menée dans 34 pays européens a révélé une incidence moyenne de 4,1 pour 10 000 journées d’hos-pitalisation. La Suisse se trouvait légèrement au-dessus de la moyenne européenne (4,8/10 000). Globalement, les taux recueillis variaient largement selon les pays et les hôpitaux participants [4].La grande faiblesse de telles évaluations statistiques est l’épidémiologie rapidement changeante de l’ICD. En outre, afin d’être en mesure d’évaluer la réelle étendue de la maladie, des investigations diagnostiques cohé-rentes et appropriées sont nécessaires. L’étude EUCLID [5], récemment publiée, montre qu’il existe encore un grand potentiel d’amélioration. Selon les estimations, 23% des ICD ne sont pas diagnostiquées à l’échelle eu-ropéenne, simplement parce que aucune indication de procéder à des investigations plus poussées n’est po-sée. Ce problème est en outre accentué par le recours à
des méthodes de laboratoire insuffisantes dans cer-taines cliniques. Le taux d’infection rapporté dans les pays participants pour les années 2011/12 était en moyenne de 7 cas pour 10 000 jours d’hospitalisation. Pourtant, en raison du pourcentage élevé d’ICD non diagnostiquées, le taux d’infection réel pour la même période pourrait être bien plus élevé. Dans ce contexte, il est clair que l’étendue réelle de C. difficile est vraisem-blablement largement sous-estimée.
Changement d’épidémiologieLa proportion d’ICD contractées en ambulatoire est pro-bablement plus élevée qu’envisagée jusqu’à présent. Ces infections contractées en dehors du milieu hospitalier touchent plutôt des patients jeunes moins exposés aux antibiotiques ou aux autres facteurs de risque. Récem-ment, des rapports ont fait état d’ICD chez les femmes enceintes ou les enfants. Les raisons de cette évolution sont encore indéterminées. Peut-être s’explique-t-elle par de nouveaux réservoirs (aliments, animaux) ou en-core par des avantages sélectifs de certaines souches.
L’étendue réelle de Clostridium difficile est vraisemblablement largement sous-estimée.
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):236–240

ARTICLE DE REVUE 239
Corrélation entre ribotype et manifestations cliniquesLe regain d’attention pour le typage des souches C. diffi-cile isolées apporte de nouvelles informations sur leurs propriétés et leur présentation clinique.Des comparaisons génomiques sur plusieurs années montrent une grande diversité et une évolution rapide des ribotypes dominants. Aucune réponse claire ne peut pour l’instant être apportée à la question de savoir dans quelle mesure le ribotype est corrélé à la sévérité de l’infection. Jusqu’à présent, les rapports portant sur les souches hypervirulentes avec une plus grande inci-dence et un moins bon pronostic proviennent en grande partie de données récoltées lors d’épidémies. Des ana-lyses de ces souches sans recrudescence récente n’ont pour l’instant pas permis d’établir un lien certain entre le ribotype, la sévérité de la maladie et la mortalité.Les PCR-ribotypes 027 et 078 sont considérés comme les principaux ribotypes hypervirulents. La souche NAP1/027/B1 (ou PCR-ribotype 027) a été identifiée
comme responsable de certaines épidémies au Canada et aux Etats-Unis au début du millénaire; elle est de-puis associée à un tableau clinique sévère et à des taux élevés de récidive. La souche a été identifiée entre-temps comme cause de l’ICD, et ce à l’échelle mondiale. Le PCR-ribotype 078 semble être plus présent en Eu-rope et se retrouve avant tout dans les ICD chez les pa-tients jeunes, dans les ICD contractées en ambulatoire ainsi que dans les ICD chez l’animal.Des mesures in vitro standardisées infirment de plus en plus l’ancien concept d’hypervirulence par une pro-duction excessive de toxine. En revanche, la quantité significativement plus grande de spores est frappante, et pourrait expliquer la capacité de propagation plus importante et les taux élevés de récidive de telles souches.Une occasion unique de comparaison directe s’est pré-sentée au cours d’une épidémie, avec l’apparition si-multanée de deux ribotypes différents. Une de ces souches (le PCR-ribotype 027) est considérée comme hypervirulente, l’autre (le PCR-ribotype 017) ne com-porte aucun marqueur de virulence. Toutefois, le taux de mortalité à 30 jours était élevé pour les deux souches (027: 26%; 017: 23%). Les infections par d’autres souches non épidémiques affichaient en revanche une mortalité de seulement 3%. Les auteurs avancent l’hy-pothèse que la mortalité accrue ne serait pas due aux ribotypes mais plutôt l’expression d’une maladie sous-jacente sévère avec réceptivité accrue aux infections.En raison de la discordance des données disponibles relatives à l’association entre différents ribotypes et degré de sévérité clinique de la maladie, la European Society of Clinical Microbiology and Infectious Diseases (ESCMID) recommande actuellement d’adapter le trai-tement de l’ICD en fonction des signes cliniques et non en fonction de la souche déclencheuse [2].
Nouvelles options thérapeutiquesLa fidaxomicine est autorisée en Suisse depuis mai 2014. Cette substance appartient à une nouvelle classe d’antibiotiques, les macrocycliques. L’inhibition de l’ARN polymérase produit un effet bactéricide contre C. difficile, avec un spectre d’action réduit et des concen-trations élevées dans les selles. Ce médicament est capa ble in vitro d’inhiber la production de spores et de toxines de C. difficile. Deux études randomisées ont montré que la fidaxomicine est aussi efficace que la vancomycine dans le traitement de l’ICD mais qu’elle présente des taux de récidive significativement plus faibles [2]. Etant donné qu’il n’existe pratiquement au-cune absorption systémique de la fidaxomicine, cette substance se distingue par son profil favorable en ce qui concerne les interactions et les effets indésirables.
Tableau 3: Recommandations pour le traitement d’une infection à Clostridium difficile (ICD) [2].
Degré de sévérité
Traitement recommandé
ICD légère Traitement antibiotiqueMétronidazole 3 × 500 mg par voie orale pdt. 10 j (degré d’évidence A-I) ouVancomycine 4 × 125 mg par voie orale pdt. 10 j (degré d’évidence B-I) ou
Pas de traitement antibiotique (degré d’évidence C-II)Condition: situation non endémique et relation claire avec un traitement antibiotique déclencheur – Arrêt du traitement déclencheur– Surveillance clinique étroite de l’évolution sur 48 heures – Début d’un traitement antibiotique en cas de dégradation clinique
ICD sévère Vancomycine 4 × 125 mg par voie orale pdt. 10 j (degré d’évidence A-I) ouFidaxomicine 2 × 200 mg par voie orale pdt. 10 j (degré d’évidence B-I)
Première récidive ou risque élevé de récidive
Vancomycine 4 × 125 mg par voie orale pdt. 10 j (degré d’évidence B-I) ouFidaxomicine 2 × 200 mg par voie orale pdt. 10 j (degré d’évidence B-I) ouMétronidazole 3 × 500 mg par voie orale pdt. 10 j (degré d’évidence C-I)
Récidives multiples
Vancomycine 4 × 125 mg par voie orale pdt. 10 j (degré d’évidence B-II)Suivi d’une pulsothérapie/tapering* ouFidaxomicine 2 × 200 mg par voie orale pdt. 10 j (degré d’évidence B-II)
En l’absence de réponse après un traitement antibiotique répété, une transplantation fécale combinée à un traitement antibiotique oral est recommandée (degré d’évidence A-I)
Traitement oral impossible
ICD légèreMétronidazole 3 × 500 mg par voie i.v. pdt. 10 j (degré d’évidence A-II)
ICD sévèreMétronidazole 3 × 500 mg par voie i.v. pdt. 10 j (degré d’évidence A-II) combiné à des lavements à la vancomycine 500 mgdans 100 ml de NaCl 4×/j ou combiné à la vancomycine 500 mg dans 100 ml de NaCl 4×/j par sonde gastrique pendant 10 j (degré d’évidence B-III)
* Pulsothérapie: vancomycine 125–500 mg/j tous les 2–3 jours pendant au moins 3 semaines.Tapering: réduction graduelle de la dose à 125 mg/j
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):236–240

ARTICLE DE REVUE 240
Les effets indésirables les plus fréquemment rapportés étaient les vomissements, les nausées et la constipa-tion. L’administration concomitante de ciclosporine, de kétoconazole, d’érythromycine, de clarithromycine, de vérapamil, de dronédarone et d’amiodarone est dé-conseillée (www.compendium.fr). Le coût du traite-ment par fidaxomicine est plus élevé que celui des trai-tements standards, raison pour laquelle cette molécule n’est actuellement pas recommandée en Suisse chez les patients atteints d’une ICD légère; son emploi est ce-pendant à envisager chez les patients présentant un risque de récidive (tab. 3). Dans les directives euro-péennes, la fidaxomicine est, au même titre que la vancomycine, recommandée comme alternative au traitement d’une ICD légère, avec un degré d’évidence toutefois plus faible que le métronidazol [2].L’administration d’anticorps monoclonaux orientés contre les toxines A et B de C. difficile, en complément du traitement standard par métronidazol ou vancomy-cine, a également montré une réduction significative
du taux de récidive. Actuellement, les preuves relatives à son utilisation sont encore insuffisantes [2].La transplantation de selles d’un donneur sain en-traîne une rapide amélioration de la fonction intesti-nale normale. Avec des taux de réussite de plus de 90%, la transplantation fécale s’établit de plus en plus comme alternative thérapeutique pour les épisodes d’ICD récidivants [2]. Si les facteurs et les types de bac-téries responsables étaient mieux identifiés, cela ou-vrirait d’immenses perspectives en vue d’un traite-ment ciblé et de la prophylaxie de l’ICD et d’autres maladies intestinales.
Nouvelles mesures préventivesDes vaccins préventifs contre l’ICD font actuellement l’objet de recherches. La base de ces travaux est un en-semble d’études qui ont montré une association entre la colonisation asymptomatique par C. difficile et des taux élevés d’anticorps IgG contre la toxine A de C. difficile. De plus, les patients qui développent une réponse immuni-taire contre la toxine A au cours d’une ICD présentent un risque de récidive plus faible. Différents groupes de recherche travaillent donc sur des vaccins censés main-tenir une concentration sérique élevée d’anticorps contre les toxines de C. difficile. Une approche alterna-tive consiste en l’administration orale de spores bacté-riennes inactivées qui devraient entraîner une réponse immunitaire mucosale contre C. difficile [6].
Perspectives
C. difficile n’est pas seulement un des germes les plus fréquents des infections contractées en milieu hospi-talier, il s’est aussi imposé comme un agent pathogène majeur à l’origine de diarrhées en milieu ambulatoire. Une des principales mesures préventives est la mise en place d’une utilisation plus restrictive des antibio-tiques dans tous les domaines.A l’avenir, l’épidémiologie dynamique de C. difficile continuera à constituer un défi. La propagation de dif-férents ribotypes et leurs associations avec le degré de sévérité clinique de la maladie doivent continuer à faire l’objet de recherches car la compréhension de telles corrélations constitue le fondement de nouvelles recommandations préventives et thérapeutiques.De nouvelles options thérapeutiques telles que la fi-daxomicine, les anticorps monoclonaux et la trans-plantation fécale représentent des options très pro-metteuses pour réduire les taux de récidive élevés de l’ICD. De futures recommandations thérapeutiques doivent par conséquent déterminer quels sont les pa-tients à même de tirer le plus grand bénéfice de ces nouveaux traitements.
Correspondance: PD Dr Sarah Tschudin- Sutter, MSc FMH Infektiologie und Innere Medizin Klinik für Infektiologie und Spitalhygiene Universitätsspital Basel Petersgraben 4 CH-4031 Basel Sarah.Tschudin[at]usb.ch
L’essentiel pour la pratique• Clostridium difficile est un des principaux agents pathogènes d’infections
nosocomiales, et une des principales causes de diarrhée contractée en
milieu hospitalier.
• Au cours des dernières années, la proportion d’infections à C. difficile
(ICD) contractées en ambulatoire a considérablement augmenté; les
patien ts dont le profil de risque est faible peuvent également en être
touchés sans exposition précédente aux antibiotiques.
• L’état actuel des données relatives à la corrélation entre ribotype et de-
gré de sévérité de la maladie est contradictoire. Le traitement devrait
par conséquent continuer à s’orienter en fonction du degré de sévérité
clinique.
• Le principal facteur de risque du développement d’une ICD est une ex-
position précédente aux antibiotiques. D’autres facteurs de risque sont
la prise d’antiacides, les hospitalisations, l’âge, une maladie sous-
jacente sévère, les opérations de chirurgie viscérale et l’alimentation
par sonde.
• Les examens microbiologiques sont judicieux uniquement chez les
patien ts symptomatiques. Le pilier du diagnostic de l’ICD est la mise
en évidence de toxine A ou B dans les selles. La sensibilité de la mise en
évidence de toxines au moyen des tests immuno-enzymatiques (EIA)
est beaucoup trop faible pour un diagnostic fiable, de sorte que les
direc tives actuelles recommandent pour l’investigation diagnostique
un modèle à deux niveaux.
• Le traitement doit s’adapter au degré de sévérité de la maladie. Les nou-
velles options thérapeutiques s’attachent principalement à la lutte
contre les récidives.
• Une utilisation rationnelle et restrictive des antibiotiques représente la
mesure préventive la plus efficace. Des mesures d’hygiène supplémen-
taires aident à limiter la transmission en milieu hospitalier.
Disclosure statement S. Tschudin-Sutter est membre du Fidaxomicin Advisory Board (Astellas) et a perçu des fonds de recherchede la part d’Astellas, Pfizer International Operations et du Fonds national suisse.
Illustration de couverture CDC / James Archer
Références La liste complète et numérotée des références est disponible en annexe de l’article en ligne sur www.medicalforum.ch.
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):236–240

LITERATUR / RÉFÉRENCES Online-Appendix
Références
1 Crobach MJ, Dekkers OM, Wilcox MH, Kuijper EJ. European Society of Clinical Microbiology and Infectious Diseases (ESCMID): data review and recommendations for diagnosing Clostridium difficile-infection (CDI). Clin Microbiol Infect. 2009;15:1053–66.
2 Debast SB, Bauer MP, Kuijper EJ, Committee. European
Society of Clinical Microbiology and Infectious Diseases: update of the treatment guidance document for Clostridium difficile infection. Clin Microbiol Infect 2014;20 Suppl 2:1–26.
3 Magill SS, Edwards JR, Bamberg W, Beldavs ZG, Dumyati G,
Kainer MA, et al. Multistate point-prevalence survey of health care-associated infections. N Engl J Med. 2014;370:1198–208.
4 Bauer MP, Notermans DW, van Benthem BH, Brazier JS,
Wilcox MH, Rupnik M, et al. Clostridium difficile infection in Europe: a hospital-based survey. Lancet. 2011;377:63–73.
5 Davies KA, Longshaw CM, Davis GL, Bouza E, Barbut F,
Barna Z, et al. Underdiagnosis of Clostridium difficile across Europe: the European, multicentre, prospective, biannual, point-prevalence study of Clostridium difficile infection in hospitalised patients with diarrhoea (EUCLID). Lancet Infect Dis. 2014;14:1208–19.
6 Ivarsson ME, Leroux J, Castagner B. Investigational new
treatments for Clostridium difficile infection. Drug Discov Today 2014.
SWISS MEDICAL FORUM

ARTICLE DE REVUE 241
Un déficit enzymatique avec différentes conséquences
Déficit en glucose-6-phosphate déshydrogénaseSarah Hofmanna, b, Andreas Buserc, Anne Taegtmeyera
a Klinik für Klinische Pharmakologie & Toxikologie, Universitätsspital Baselb Klinik für Innere Medizin, Universitätsspital Baselc Blutspendezentrum beider Basel & Klinik für Hämatologie, Universitätsspital Basel
Il est de plus en plus fréquent de rencontrer au cabinet ou à l’hôpital des patients qui présentent un déficit en glucose-6-phosphate déshydrogénase et pour lesquels il est important de savoir quels médicaments sont potentiellement dangereux. Cet article de revue décrit le déficit en glucose-6-phosphate déshydrogénase, son diag-nostic et les médicaments qui sont à éviter.
Introduction
Le déficit en glucose-6-phosphate déshydrogénase (G6PD) est le déficit enzymatique congénital le plus fréquent à l’échelle mondiale. Env. 400 millions de personnes en sont atteintes, avec des manifestations clini ques variables. En raison de la mondialisation, du com por tement migratoire et des habitudes de voyage, il est aussi de plus en plus fréquent d’observer des cas de déficit en G6PD en Suisse.
Histoire et épidémiologie
C’est en 1926 que Cordes a décrit pour la première fois une anémie hémolytique induite par la primaquine, le seul médicament antipaludique disponible à l’époque [1]. Il aura fallu 20 ans de plus pour que, au cours de la Seconde Guerre mondiale, de la guerre de Corée et de la guerre d’Indochine, des cas d’hémolyse sous prima-quine soient régulièrement rapportés alors que la pri-maquine était massivement utilisée chez les militaires. Cette affection était alors qualifiée de «primaquin-sen-sitive syndrome» [2]. Ce n’est qu’au milieu des années 1950 que le déficit enzymatique et le mécanisme patho-génique ont été découverts par Carson et son équipe [3]. Depuis lors, env. 400 variants de l’enzyme G6PD et plus de 180 mutations du gène ont été identifiés. La classification de l’Organisation mondiale de la Santé (OMS) repose sur l’activité enzymatique. La classe I de l’OMS correspond à un déficit enzymatique sévère (<10% de l’activité enzymatique normale) se manifes-tant par une anémie hémolytique chronique. La classe II correspond également à un déficit enzymatique sé-vère, mais qui se manifeste sur le plan clinique par une
anémie hémolytique intermittente. La classe III corres-pond quant à elle à un déficit enzymatique modéré (10–60% de l’activité enzymatique normale), avec une anémie hémolytique survenant le plus souvent dans le cadre d’une infection ou d’une exposition médicamen-teuse. Les classes IV et V ne sont pas pertinentes sur le plan clinique et sont associées à une activité enzyma-tique quasi normale ou augmentée.Le déficit enzymatique est transmis sur un mode héré-ditaire récessif lié au chromosome X. En Afrique sub-saharienne, le variant G6PD A est le plus fréquent. En Afrique de l’Ouest et en Afrique centrale, 10–15% de la Sarah Hofmann
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):241–244

ARTICLE DE REVUE 242
population en sont atteints; une prévalence similaire est retrouvée chez les Afro-Américains. Les sujets cau-casiens présentent principalement le variant G6PD mé-diterranéen, avec une fréquence accrue dans les ré-gions côtières de Sardaigne et de Grèce (20–35%), ainsi qu’au Moyen-Orient (juifs kurdes 60–70%). En Asie du Sud-Est également, la fréquence est variable (fig. 1) [4]. En raison de la pré valence élevée dans les régions où le paludisme est (ou a été) endémique, il est supposé que le déficit en G6PD confère un avantage sélectif vis-à-vis de l’infection à Plasmodium falciparum. Différents au-teurs sont par venus à démontrer ce lien [5–7]. Le méca-nisme responsable de l’inhibition de la croissance du parasite ou de la phagocytose accrue des érythrocytes infectés en cas de déficit en G6PD reste toujours in-connu.
Mécanisme pathogénique
La G6PD protège les cellules contre les dommages oxy-datifs via la voie des pentoses phosphates (fig. 2). Etant donné que les érythrocytes ne connaissent pas d’autre voie que la voie des pentoses phosphates pour s’appro-visionner en nicotinamide adénine dinucléotide phos-phate réduit (NADPH), un déficit en G6PD est associé à des dommages des constituants cellulaires provoqués par les oxydants et il se produit une hémolyse. La G6PD catalyse la première étape, elle oxyde le glucose-6- phosphate en 6-phosphogluconate et elle réduit le nico tinamide adénine dinucléotide phosphate (NADP) en NADPH. Le NADPH réduit le disulfure de glutathion (GSSG) en glutathion (GSH). Le glutathion réduit est le principal antioxydant et il est capable de réduire le peroxyde d’hydrogène (H2O2) généré par les oxydants en eau (H₂O), le rendant ainsi inoffensif.En cas de déficit en G6PD, il se produit une accumula-tion d’oxydants et une détérioration des érythrocytes. Différents facteurs influencent le degré de sévérité de l’hémolyse:1. Age des érythrocytes: L’enzyme G6PD fonctionnant
normalement a une demi-vie de 62 jours. Les jeunes érythrocytes présentent une activité encore quasi normale de la G6PD. Les patients porteurs d’un va-riant G6PD A ont normalement une hémolyse légère (classe III de l’OMS) qui se limite aux érythrocytes plus âgés car, chez ces patients, la demi-vie de la G6PD est réduite à 13 jours. Le variant G6PD médi-terranéen est plus instable, avec une demi-vie de la G6PD s’élevant à quelques heures et ainsi une hémo lyse plus prononcée (classe II de l’OMS) [8].
2. Dose médicamenteuse: Lorsqu’un médicament est le déclencheur du stress oxydatif, la dose de ce médi-cament est coresponsable de la sévérité de l’hémo-lyse. Par ex., la prise de 45 mg de primaquine peut être à l’origine d’une hémolyse sévère, alors qu’une dose de 15 mg peut donner lieu à une hémolyse qui n’est guère pertinente sur le plan clinique [9].
3. Génétique: Il existe 187 mutations connues du gène de la G6PD et au moins 35 des allèles mutés sont poly morphes. Le mode de transmission héréditaire récessif lié au chromosome X entraîne lui aussi une variabilité du degré de sévérité, selon qu’un déficit enzymatique homozygote ou hétérozygote est pré-sent chez la femme ou chez l’homme [10].
Manifestations cliniques
Différents facteurs déclenchants peuvent être à l’origine d’un stress oxydatif: infections, substances chimiques, médicaments et aliments (par ex. fèves). En règle géné-
Figure 1: Répartition du déficit en G6PD. La prévalence en Suisse est <0,5%.
Source: [21]. Reproduction avec l’aimable autorisation d’Elsevier.
<0–5%0,5–2,9%3–6,9%7–9,9%10–11,9%15–26%
Figure 2: Voie des pentoses phosphates. Elimination des oxydants (H2O2 en H2O)
dans les érythrocytes.
NADP = nicotinamide adénine dinucléotide phosphate, NADPH = nicotinamide adénine
dinucléotide phosphate réduit, GSH = glutathion réduit, GSSG = glutathion oxydé.
Glucose-6-phosphate NADP GSH H2O2
H2OGSSGNADPH6-phosphogluconate
Glucose-6-phosphate déshydrogénase
Oxydant
GSSG-réductase
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):241–244

ARTICLE DE REVUE 243
rale, une hémolyse se produit après 5–72 heures, s’ac-compagnant d’un ictère, d’une fatigue, d’une pâleur et d’une hématurie. En outre, des douleurs abdominales, des maux de dos et/ou des céphalées peuvent égale-ment survenir, de même que des nausées et de la fièvre. Ces symptômes peuvent être d’intensité variable, allant d’épisodes hémolytiques légers à des crises hémoly-tiques nécessitant des transfusions. Une amélioration clinique spontanée devrait se produire en l’espace de 5–10 jours. Toutefois, en fonction du variant enzy ma-tique, une anémie hémolytique chronique peut être présente. Une hémoglobinopathie concomitante, par ex. une thalassémie ou une drépanocytose, peut égale-ment influencer l’évolution clinique.Dans de rares cas, un ictère peut déjà s’observer quelques jours après la naissance. Le tableau clinique se distingue de l’incompatibilité rhésus typique: en cas de déficit en G6PD, l’ictère est rarement visible dès la naissance, mais il se manifeste typiquement entre le 2e et le 3e jour. Par ailleurs, par rapport à la sévérité de l’anémie, l’ictère est nettement plus prononcé en cas de déficit en G6PD qu’en cas d’incompatibilité rhé-sus [11]. Des données provenant des Etats-Unis ont montré que 21% de tous les cas d’ictère nucléaire étaient associés à un déficit en G6PD [12]. Ce constat soulève la question de savoir si un test de détection du déficit en G6PD devrait être intégré au programme de dépistage néonatal. En Suisse, ce test ne fait pas partie du programme de dépistage néonatal, sauf en pré-sence d’un ictère.
Diagnostic
Le déficit en G6PD est en premier lieu un diagnostic cli-nique (fig. 3). Une anamnèse précise permet de déter-miner si un stress oxydatif s’est produit avant la mani-festation de l’hémolyse ou si une autre cause d’anémie hémolytique est probable (par ex. valve aortique méca-nique, séjour dans une zone tropicale). Un hémo-gramme différentiel et un dosage de la bilirubine (di-recte et indirecte), de la lactate déshydrogénase (élevée) et de l’haptoglobine (basse) fournissent des indices et peuvent confirmer les soupçons. Un test de Coombs direc t négatif est utile pour exclure une anémie hémo-lytique immunitaire. Un déficit en G6PD peut d’une part être confirmé par un frottis sanguin révélant une constellation typique (corps de Heinz, hématies mor-dues [«bite cells»]) et d’autre part, l’activité de la G6PD peut être mesurée par analyses biochimiques. Une acti vité normale au moment de l’hémolyse n’exclut toutefois pas un déficit en G6PD, car le recrutement de jeunes érythrocytes à activité enzymatique peut se traduire par une activité enzymatique faussement normale. Dans cette situation, il est recommandé de répéter la mesure après 3 mois car, à ce moment-là, il y a un équilibre entre les érythrocytes jeunes et âgés.
Traitement et prévention
En cas de survenue d’une anémie hémolytique liée à un déficit en G6PD, il convient bien sûr d’arrêter le mé-dicament déclencheur, si cela est possible. En fonction
Figure 3: Proposition d’algorithme diagnostique en cas de suspicion d’hémolyse liée à un déficit en G6PD.
Anamnèse:Valve cardiaque mécanique, agents chimiques/toxiques, transfusions, séjours dans des zones tropicales, infections préalables, anamnèse personnelle et familiale, médicaments
Recherche de l’étiologie
Frottis sanguin(fragmentocytes?
bite cells?microsphérocytes)
ConfirmationDétermination quantitative de l’activité de la glucose-6- phosphate déshydrogénase
–
Confirmation d’une anémie hémolytique
Analyses de base– Hémogramme différentiel, y compris réticulocytes– Bilirubine (directe, indirecte)– LDH , haptoglobine – Test de Coombs direct
+ Anémie hémolytique immunitaire possible
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):241–244

ARTICLE DE REVUE 244
du degré de sévérité, une transfusion sanguine est dans de rares cas nécessaire. Les patients se réta-blissent en l’espace de 5–10 jours et l’hémogramme se normalise avec le temps. Il n’existe pas de traitement spécifique.Il est avant tout essentiel d’expliquer l’affection au patie nt et d’éviter les facteurs déclenchants à l’avenir. Jusqu’à présent, il n’existe pas de consensus quant aux médicaments déclencheurs et compte tenu du poly-morphisme génétique, il n’est guère possible de déter-miner précisément quel patient réagira à quel médica-ment et à quel point. Aucun lien entre la structure chimique du médicament et le mécanisme patho-génique n’a pour l’heure été trouvé et il est donc égale-ment impossible de se prononcer sur la base de ces
Tableau 1: Médicaments qui devraient être évités chez les patients ayant un déficit en G6PD (d’après Youngster et Luzzatto) [10, 13].
Catégorie Hémolyse prévisible («unsafe»)
Hémolyse possible
Antipalu-diques
Primaquine Dapsone
Chloroquine (Nivaquine®) Quinine (Limptar®)
Antalgiques Phénazopyridine Acide acétylsalicylique (Aspirin®) Paracétamol (Dafalgan®, Panadol®)
Antibiotiques Nitrofurantoïne (Uvamin®)
Sulfasalazine (Salazopyrin®)Sulfadiazine (Flammazine®, Ialugen plus®)Cotrimoxazole(triméthoprime/sulfaméthoxazole, Bactrim®, Nopil®)*Quinolones (ciprofloxacine, norfloxcacine, lévofloxacine)*
Autres Rasburicase (Fastutrec®)Bleu de méthylèneBleu de toluidine
IsoniazideAcide ascorbique (vitamine C)Glibenclamide (Daonil®)Vitamine K (Konakion®)Dinitrate d’isosorbide (Isoket®)Succimer (Succicaptal®)
Substances chimiques/ aliments
Colorants d’aniline (colorants dérivés du goudron de houille)Naphtalène (boules de naphtaline)HennéFèves
* Médicaments considérés comme «unsafe» d’après Luzzatto [10].
L’essentiel pour la pratique• En raison de l’augmentation des flux migratoires, il faut s’attendre à ce
que davantage de personnes ayant un déficit en glucose-6-phosphate
déshydrogénase (G6PD) se fassent soigner en Suisse.
• Les patients ayant un déficit en G6PD peuvent se présenter avec une
crise hémolytique, raison pour laquelle il est essentiel de connaître le
diagnostic et le traitement dans cette situation d’urgence.
• Pour la prise en charge de ces patients au cabinet médical, il est néces-
saire de connaître les médicaments à éviter.
Correspondance: Sarah Hofmann Klinik für Innere Medizin Universitätsspital Basel CH-4031 Basel sarah.hofmann[at]usb.ch
connaissances. En 2010, Youngster [13] a publié une evi-dence-based review, dans laquelle il a réalisé une vaste recherche littéraire et l’a analysée. Ce faisant, il est ap-paru que les données relatives à de nombreux médica-ments que l’on soupçonnait de pouvoir déclencher une hémolyse chez les patients ayant un déficit en G6PD étaient très maigres. Par ex., une hémolyse sous para-cétamol a certes été documentée dans quelques études de cas, mais la plupart du temps, le médicament avait été clairement surdosé ou les patients avaient une fièvre élevée, ce qui ne permettait pas de déterminer avec certitude quel était le facteur déclenchant [14–18]. Deux études réalisées chez des enfants ayant un déficit en G6PD n’ont pas été en mesure de mettre en évidence une hémolyse après l’administration de paracétamol [19, 20]. Au vu de l’utilisation très fréquente et large-ment répandue du paracétamol et de ces études de cas peu nombreuses, on peut partir du principe que ce médi cament, s’il est administré à des doses normales, peut sans problème être utilisé chez les patients ayant un déficit en G6PD. En se basant sur les travaux de Luz-zatto et Youngster [10, 13], le tableau 1 présente les médi caments qui ont provoqué de façon répétée des hémo lyses chez les patients ayant un déficit en G6PD et sont dès lors qualifiés de «unsafe». Parmi ces médica-ments, seuls la nitrofurantoïne (Uvamin®, Furadan-tin®) et la rasburicase (Fasturtec®) ainsi que le bleu de méthylène et le bleu de toluidine sont disponibles en Suisse.Une détermination de l’activité de la G6PD avant l’ini-tiation d’un traitement par dapsone ou primaquine est uniquement nécessaire en cas d’anamnèse person-nelle et familiale concordante ou de suspicion clinique. Pour le bleu de méthylène et la rasburicase, qui sont administrés dans des situations d’urgence, il n’y a sou-vent pas suffisamment de temps pour réaliser cette analyse au préalable.Par ailleurs, l’association «Associazione Italiana Fa-vismo – Deficit di G6PD» (G6PD Deficiency favism asso-ciation) tient un site Internet (http://g6pd.org) qui contient des listes des médicaments «sûrs» et «ris-qués» ainsi que des informations destinées aux pa-tients, à leurs proches et aux professionnels de la santé.
Disclosure statementLes auteurs ne déclarent aucun conflit d’intérêts financier ou per-sonnel en rapport avec cet article.
Photo de couvertureWikimedia Commons.
RéférencesLa liste complète et numérotée des références est disponible en annexe de l’article en ligne sur www.medicalforum.ch.
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):241–244

LITERATUR / RÉFÉRENCES Online-Appendix
Literatur / Références
1. Cordes W. Experiences with plasmochin in malaria. Med Depart 15th Annual Report. Boston United Fruit Co; 1926:66-71
2. Dern RJ, Beutler E, Alving AS. The hemolytic effect of primaquine. V. Primaquine sensitivity as a manifestation of a multiple drug sensitivity. J Lab Clin Med. 1955 Jan;45(1):30-9.
3. Alving AS, Carson PE, Flanagan CL, Ickes CE. Enzymatic deficiency in primaquine-sensitive erythrocytes. Science. 1956 Sep 14;124(3220):484-5
4. Nkhoma ET1, Poole C, Vannappagari V, Hall SA, Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood Cells Mol Dis. 2009 May-Jun;42(3):267-78.
5. Ruwende C, Khoo SC, Snow RW, Yates SN, Kwiatkowski D, Gupta S, Warn P, Allsopp CE, Gilbert SC, Peschu N, et al. Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature. 1995 Jul 20;376(6537):246-9.
6. Luzzatto L, Usanga FA, Reddy S. Glucose-6-phosphate dehydrogenase deficient red cells: resistance to infection by malarial parasites. Science. 1969 May 16;164(3881):839-42.
7. Cappadoro M, Giribaldi G, O'Brien E, Turrini F, Mannu F, Ulliers D, Simula G, Luzzatto L, Arese P. Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)-deficient erythrocytes parasitized by Plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood. 1998 Oct 1;92(7):2527-34
8. Piomelli S, Corash LM, Davenport DD, Miraglia J, Amorosi EL. In vivo lability of glucose-6-phosphate dehydrogenase in GdA- and GdMediterranean deficiency. J Clin Invest. 1968 Apr;47(4):940-8.
9. Kellermeyer RW, Tarlov AR, Brewer GJ, Carson PE, Alving AS. Hemolytic effect of therapeutic drugs. Clinical considerations of the primaquine-type hemolysis. JAMA. 1962 May 5;180:388-94.
10. Luzzatto L, Seneca E. G6PD deficiency: a classic example of pharmacogenetics with on-going clinical implications. Br J Haematol. 2014 Feb;164(4):469-80.
11. Kaplan M, Hammerman C, Glucose-6-phosphate dehydrogenase deficiency: a hidden risk for kernicterus.
Semin Perinatol. 2004;28(5):356. 12. L Johnson, V K Bhutani, K Karp, E M Sivieri and S M
Shapiro Clinical report from the pilot USA Kernicterus Registry (1992 to 2004) Journal of Perinatology (2009) 29, S25–S45).
13. Youngster I, Arcavi L, Schechmaster R, Akayzen Y, Popliski H, Shimonov J, Beig S, Berkovitch M. Medications and glucose-6-phosphate dehydrogenase deficiency: an evidence-based review. Drug Saf. 2010 Sep 1;33(9):713-26.
14. Sklar GE, Hemolysis as a potential complication of acetaminophen overdose in a patient with glucose-6-phosphate dehydrogenase deficiency , Pharmacotherapy. 2002 May;22(5):656-8.
15. Wright RO, Perry HE, Woolf AD, Shannon MW. Hemolysis after acetaminophen overdose in a patient with glucose-6-phosphate dehydrogenase deficiency. J Toxicol Clin Toxicol. 1996;34(6):731-4.
16. Phillpotts S, Tash E, Sen S. Glucose-6-phosphate dehydrogenase deficiency: an unusual cause of acute jaundice after paracetamol overdose. Eur J Haematol. 2014 Mar 29
17. Oliver M, Coton T, Badens C, Dehan C, Lena-Russo D, Moalic JL. Homozygous G6PD deficiency and propacetamol induced hemolysis. Haematologica. 2001 Sep;86(9):987-8
18. Ruha AM, Seldem B. Hemolytic anemia after acetaminophen overdose in patient with glucose-6-phosphate dehydrogenase deficiency. Am J Med. 2001 Feb 15;110(3):240-1.
19. Cottafava F, Nieri S, Franzone G, Sanguinetti M, Bertolazzi L, Ravera G. [Double-blind controlled comparison of placebo and paracetamol in patients with G-6-PD deficiency]. Pediatr Med Chir. 1990 Nov-Dec;12(6):631-7. Italian.
20. Najafi N, Van de Velde A, Poelaert J. Potential risks of hemolysis after short-term administration of analgesics in children with glucose-6-phosphate dehydrogenase deficiency. J Pediatr. 2011 Dec;159(6):1023-8.
21. Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008 Jan 5;371(9606):64-74. Review.
SWISS MEDICAL FORUM

QUEL EST VOTRE DIAGNOSTIC? 245
Toux productive et perte pondérale
En suivant les rails du tramAmélie Burria, Valentina Garellib, Nicolas Petitpierreb
a Service de Médecine interne, Centre Hospitalier Universitaire Vaudois CHUV, Lausanneb Service de Pneumologie, Centre Hospitalier Universitaire Vaudois CHUV, Lausanne
Présentation du cas
Il s’agit d’une patiente de 73 ans qui développe depuis trois mois une toux incoercible accompagnée d’expec-torations abondantes, présente jour et nuit mais aug-mentant en position couchée. Elle décrit aussi une dys-pnée progressive, actuellement de stade II selon NYHA (New York Heart Association), et ne signale pas d’état fébrile. La patiente est connue pour une dénutrition protéino-calorique, une ostéoporose et une rectocolite ulcé ro-hémorragique (RCUH) depuis 30 ans, actuellement contrôlée par le traitement. Elle a été victime d’une dis-section carotidienne avec un accident ischémique transitoire en 2011 et d’un accident de la voie publique entraînant un traumatisme crânien sévère et des sé-quelles neurologiques (troubles de la marche, tremor) en 1975. Son traitement habituel est le suivant: clopido-grel, simvastatine, lévétiracétam, mésalazine, thia-mine, alprazolam.A l’examen clinique, la patiente a un indice de masse corporelle à 18 kg/m2. La saturation en oxygène est de 95% à l’air ambiant. L’auscultation pulmonaire révèle quelques râles fins et des ronchi diffus.
1) Lequel de ces examens ne fait pas partie du bilan initial?
a) Radiographie du thorax
b) Cultures d’expectorations
c) Fonctions pulmonaires
d) Bronchoscopie
Cette patiente non tabagique présente une toux chro-nique. La première étape est de rechercher une cause médicamenteuse, en particulier un traitement par un inhibiteur de l’enzyme de conversion (IEC). Nous ne re-levons aucun argument clinique suggérant un écoule-ment postérieur ou un reflux gastro-œsophagien. Un traitement d’épreuve par inhibiteur de la pompe à pro-ton n’a pas amélioré la symptomatologie. A part la toux, nous n’avons pas d’élément pour un asthme. Une radiographie du thorax est effectuée, qui met en évi-dence un infiltrat interstitiel et alvéolaire basal droit avec un bronchogramme aérique (fig. 1). La spirométrie est dans la norme, sans syndrome obs-tructif, mais la patiente tousse beaucoup durant les manœuvres.
En raison de la bronchorrhée, une culture des expecto-rations est demandée, qui retrouve de la flore oro-pha-ryngée. La bronchoscopie ne fait pas partie du bilan initial de la toux chronique.Au vu de la symptomatologie et des infiltrats radio-logiques, le diagnostic de pneumonie communautaire est suspecté et un traitement de cefuroxime (250 mg 2×/j pendant 10 jours) est introduit.Trois semaines plus tard, la patiente décrit une dimi-nution de la toux mais reste gênée par des quintes, surtout nocturnes. La radiographie est répétée. Elle montre des opacités nouvelles et des images en «rails de tram» (fig. 2).
Figure 1: Infiltrat interstitiel et alvéolaire basal droit,
présence d’un bronchogramme aérique (flèche).
Figure 2: Petits infiltrats alvéolaires au niveau basal droit,
avec image en «rails de tram» (flèches).
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):245–248

QUEL EST VOTRE DIAGNOSTIC? 246
Ces images font suspecter des bronchectasies. Un scan-ner thoracique est réalisé, qui confirme la présence de bronchectasies associées à des infiltrats au niveau du lobe moyen et inférieur droit (fig. 3).
2) Lequel de ces examens ne fait pas partie du bilan initial
des bronchectasies?
a) Tb-spot et culture de mycobactéries
b) Dosage des immunoglobulines G (IgG) sériques
c) Dosage sérique des immunoglobulines E (IgE) totales et des
précipitines aspergillaires
d) Recherche de mutation du gène CFTR (Cystic fibrosis trans-
membrane conductance regulator)
La bronchectasie est définie comme une dilatation per-manente d’une bronche. Le diagnostic est retenu, sur un scanner thoracique à haute résolution, en présence d’un diamètre interne de la bronche plus grand que le diamètre du vaisseau qui l’accompagne. Le bilan de-mandé dépend de la présentation clinique [1]. Face à des bronchectasies diffuses, nous évoquons chez cette patiente les diagnostics différentiels suivants: une immunodéficience primaire ou secondaire (hypo-gammaglobulinémie primaire, infection par le virus de l’immunodéficience humaine [HIV]), des séquelles d’une pathologie infectieuse respiratoire, une infec-tion à mycobactéries atypiques, une association avec une maladie de système (polyarthrite rhumatoïde, Sjö-gren, etc…) ou avec une maladie inflammatoire chro-nique de l’intestin (MICI).Une aspergillose broncho-pulmonaire allergique, peu vraisemblable en l’absence d’antécédent d’asthme, sera tout de même recherchée.
Une maladie génétique (mucoviscidose, dyskinésie ci-liaire primitive) semble moins probable compte tenu de l’âge et de l’absence de symptomatologie oto-rhino-laryngologique ou digestive associée. Le bilan effectué comprend un dosage des IgG et leurs sous-classes, un Tb-spot, un test HIV, les IgE totales et spécifiques pour Aspergillus fumigatus, les précipitines aspergillaires, ainsi qu’un bilan auto-immun et micro-biologique.L’ensemble de ces examens est normal, y compris les cultures d’expectorations, qui resteront négatives (bactéries classiques, mycobactéries et champignons).Devant la persistance des symptômes et des infiltrats pulmonaires malgré un traitement antibiotique, une bronchoscopie est effectuée, qui révèle une muqueuse trachéale et bronchique très inflammatoire avec des sécrétions muco-purulentes abondantes (fig. 4).La répartition cellulaire du lavage broncho-alvéolaire (LBA) montre une prédominance neutrophilique (33%). A la culture du LBA, on retrouve une flore oro-pharyn-gée (103 germes/millilitre) et dans l’aspiration bron-chique, un Serratia marcescens, motivant une tentative de traitement par sulfaméthoxazole/triméthoprime pendant 3 semaines. Une culture de mycobactéries est demandée, qui restera négative.Deux mois plus tard, la toux a diminué, mais n’a pas complètement disparu. L’auscultation pulmonaire et les fonctions pulmonaires sont inchangées. Quelques jours plus tard, la patiente présente un pre-mier épisode d’hémoptysies (5–6 cuillères à soupe). Elle signale, de plus, une perte pondérale sans trouble du transit associé.
Figure 3: Scanner thoracique: bronchectasies (flèche) et infiltrats péribronchiques.
A: Coupe axiale. B: Reconstruction dans l’axe d’une bronche.
A B
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):245–248

QUEL EST VOTRE DIAGNOSTIC? 247
Le scanner thoracique, réalisé en urgence en raison des hémoptysies, met en évidence une progression des bronchectasies, associée à de multiples infiltrats alvéo-laires (fig. 5). En résumé, nous sommes face à une progression rapide des bronchectasies, l’aspect macroscopique évoque une inflammation chronique et le bilan étiologique habi tuel est sans particularité.
3) Quel diagnostic suspectez-vous en premier lieu?
a) Une infection pulmonaire à mycobactéries atypiques
b) Une mucoviscidose
c) Des bronchectasies secondaires à la RCUH
d) Une aspergillose broncho-pulmonaire allergique
Une progression aussi rapide de bronchectasies est tout à fait inhabituelle. Après avoir raisonnablement exclu une infection, une immunodéficience ou une connectivite, nous retenons le diagnostic de bronchec-tasies secondaires à la RCUH. Une atteinte bronchique liée à une MICI, alors qu’il n’y a aucun signe d’activité de la maladie au niveau intestinal, est un phénomène connu [2].
4) Quel traitement proposez-vous?
a) Corticothérapie systémique puis corticostéroïdes inhalés
b) Corticoïdes inhalés seuls
c) Mésalazine
d) Infliximab
Nous débutons une corticothérapie par prednisone (30 mg/j) associée à une prophylaxie de l’ostéoporose et de la pneumonie à Pneumocystis jiroveci.Deux mois plus tard, l’évolution est bonne avec amen-dement de la toux et une reprise pondérale, permet-tant le sevrage progressif de la prednisone.En parallèle, un traitement de corticoïdes inhalés est instauré (budésonide 400 μg 2×/j), dans l’espoir qu’il facilite le sevrage de la corticothérapie systémique.
Discussion
Les maladies inflammatoires chroniques de l’intestin (MICI) ont de nombreuses manifestations extra-intes-tinales, principalement articulaires, oculaires et cuta-nées.Bien qu’il semble y avoir une prévalence élevée d’ano-malies fonctionnelles ou radiologiques subcliniques [3, 4], les atteintes respiratoires symptomatiques sont rares. Il y a une grande varié té de lésions et de manifes-tations cliniques. Tout le système respiratoire peut être atteint: les voies aériennes, de la trachée aux bron-chioles, le parenchyme pulmonaire, la plèvre et la vas-
Figure 4: Bronche souche droite: muqueuse très inflamma-
toire avec des sécrétions muco-purulentes revenant du lobe
supérieur droit.
Figure 5: A/C: premier scanner thoracique, B/D: deuxième scanner 3 mois plus tard
(mêmes ni veaux de coupe). Apparition de nouvelles bronchectasies (flèches), progres-
sion des bronchectasies (tête de flèche). On note par ailleurs des épaississements des
parois bronchiques et des infiltrats alvéolaires.
A
C
B
D
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):245–248

QUEL EST VOTRE DIAGNOSTIC? 248
cularisation pulmonaire. L’inflammation des voies aé-riennes est l’atteinte la plus fréquente, et peut évoluer vers des bronchectasies, des sténoses sévères ou une bronchiolite.Les atteintes bronchiques sont plus fréquemment asso ciées à la RCUH qu’à la maladie de Crohn.Outre les atteintes directement attribuables à la mala-die inflammatoire, on peut aussi rencontrer des effets secondaires pulmonaires du traitement comme une infection opportuniste ou une pneumopathie intersti-tielle. 80 à 85% des patients développent des symptômes res-piratoires après le diagnostic de la MICI. Les manifesta-tions respiratoires peuvent être la présentation initiale de la maladie, mais elles peuvent aussi survenir des an-nées après le diagnostic, voire même chez des patients en rémission. L’apparition d’une atteinte respiratoire après une colectomie a souvent été décrite. La physiopathologie n’est pas complètement élucidée, mais est probablement en lien avec l’origine embryon-naire commune de l’épithélium respiratoire et colique (cellules à mucus et des glandes sous-muqueuses).
Concernant la prise en charge, les corticostéroïdes re-présentent le traitement de premier choix, contraire-ment aux autres formes de bronchectasies. Ils sont en général administrés par voie systémique avec un relais de corticoïdes inhalés à hautes doses. Dans les atteintes plus légères, les corticoïdes inhalés peuvent suffire.En plus du traitement anti-inflammatoire, la prise en charge habituelle des bronchectasies est nécessaire: physiothérapie de drainage bronchique, éventuelle-ment à l’aide d’aérosols de NaCl, immunisations contre la grippe saisonnière et le pneumocoque, traitement précoce et prolongé des infections bronchiques. L’azi-thromycine, souvent utilisée dans les bronchectasies d’autres origines, n’a pas été spécifiquement étudiée dans cette population. Elle est parfois prescrite empiri-quement en cas d’exacerbations répétées.Le pronostic est en général bon mais il existe des attein tes pulmonaires cortico-résistantes. Les options thérapeutiques sont alors limitées. Des lavages bron-chiques avec de la methylprednisolone ont été décrits. Il semble que l’effet des traitements immunosupres-seurs classiques des MICI (azathioprine, mercapto pu-rine et méthotrexate) soit mitigé.
Disclosure statementLes auteurs n’ont déclaré aucun lien financier ou personnel en rapport avec cet article.
Références1 Espinosa, V, Rochat T, Bronchiectasies chez l’adulte: recherche
d’une étiologie. Rev Med Suisse. 2013;9(407):2155–9.2 Camus Ph, Colby TV, Bronchiectasis associated with inflammatory
bowel disease., in ERS monograph. Bronchiectasis, Floto RA et Haworth CS Editors. 2011, European Respiratory Society: Plymouth. p. 163–77.
3 Herrlinger KR, Noftz MK, Dalhoff K, Ludwig D, Stange EF, Fellermann K, Alterations in pulmonary function in inflamma-tory bowel disease are frequent and persist during remission. Am J Gastroenterol. 2002;97(2):377–81.
4 Desai D, Patil S, Udwadia Z, Maheshwari S, Abraham P, Joshi Al, Pulmonary manifestations in inflammatory bowel disease: a prospective study. Indian J Gastroenterol. 2011;30(5):225–8.
L’essentiel pour la pratique
Chez les patients atteints de MICI, en plus des manifestions extra-intesti-
nales habituelles, il faut penser aux complications pulmonaires, en parti-
culier aux atteintes trachéo-bronchiques. Ces complications pulmonaires
peuvent survenir alors que la maladie digestive est quiescente, ou après
une colectomie. Cela rend le rapprochement entre les deux maladies par-
fois difficile. Il est important de repérer les bronchectasies secondaires
aux MICI, car une corticothérapie peut entraîner une excellente réponse.
Réponses aux questions:
Question 1: d. Question 2: d. Question 3: c. Question 4: a.
Correspondance: Dr Amélie Burri Service de Médecine Interne CHUV CH-1011 Lausanne amelie.burri[at]chuv.ch
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):245–248

RECHERCHE 249
Validierungsstudie einer Version des ECAS im Schweizer Hochdeutsch
Screening von Kognitions- undVerhaltensänderungen beider Amyotrophen LateralskleroseChristian Burkhardt, Christoph Neuwirth, Markus Weber
Muskelzentrum/ALS Clinic, Kantonsspital St. Gallen
Hintergrund
Die Amyotrophe Lateralsklerose (ALS) ist die häufigsteMotoneuronerkrankung beim Erwachsenen. Sie führt durch Degeneration des ersten und zweiten motori-schen Neurons zu fortschreitenden Lähmungen. In denletzten beiden Dekaden änderte sich die Wahrnehmungder Erkrankung radikal. Neben Paresen und Spastiksind kognitive Defizite und Verhaltensänderungen als häufige Symptome dieser Multisystem-Erkrankung akzeptiert, wenn auch gegenwärtig noch nicht in den Diagnosekriterien verankert. Auf Basis von geneti-schen (z.B. c9orf72-Expansion) und histomorphologi-schen (z.B. mit TDP-43-positiven Einschlüssen im ZNS) Gemeinsamkeiten konnte die Brücke zwischen der ALSund der fronto-temporalen Demenz (FTD) geschlossen werden [1]. Beide Erkrankungen bilden somit ein Kon-tinuum mit variabler phänotypischer Präsentation.Frontotemporale Störungen der ALS decken ein breites klinisches Spektrum von Symptomen ab. Einige Pa-tienten weisen lediglich in kognitiven TeilbereichenDefizite (kognitive Beeinträchtigungen, ALS-ci) auf. Ebenso werden isolierte Verhaltensänderungen (Ver-haltensstörungen, ALS-bi) beobachtet, und einige Pa-tienten präsentieren ein Mischbild aus beiden Berei-chen. Nur eine Minderheit der ALS-Patienten (5–15%)erfüllt die Kriterien einer «ausgewachsenen» ALS-FTD. Für ALS-ci schwanken die Prozentangaben von 15–60%[2]. Die Defizite bei der ALS-ci liegen meist in der Wort-flüssigkeit (fluency), Beeinträchtigung von exekutivenFunktionen, eingeschränkten Fähigkeiten zur Planung und Bearbeitung von Problemstellungen, Aufmerk-samkeits- und Konzentrationsstörungen sowie dem Multitasking. Bei der ALS-bi reicht das Spektrum vongeringfügigen, kaum wahrnehmbaren Verhaltens- änderungen mit wenig Einfluss auf das tägliche Leben bis hin zu Verhaltensauffälligkeiten einer frontalenDemenz. Hier sind sowohl Minussymptome wie Apa-thie, Interessenverlust, Verlust an Empathie und sozial-
emotionalen Beziehungen als auch Plussymptome wie Reizbarkeit und eine unzureichende Impulskontrolle zu nennen. Faktoren, welche die grosse Heterogenitätder Symptompräsentation dieses Kontinuums der ALS-FTD in der Literatur erklären, liegen zum einen in denunterschiedlichen Erkrankungsstadien der Patienten,der hohen genetischen Variabilität sowie der sehr unterschiedlichen diagnostischen Testbatterien, die zur Detektion der fronto-temporalen Defizite ein-gesetzt werden. Die bisher gängigen neuropsycho-logischen Tests zur Untersuchung fronto-temporalerFunktionen bei ALS wurden nicht eigens für dieALS-Erkrankten entwickelt und weisen dadurch keineAnpassung an die besonderen, körperlichen Ein-schränkungen dieser Patienten, vor allem in spätenKrankheitsstadien, auf. Die Durchführung und Aus-wertung wird hierdurch erschwert oder gar verun-möglicht. Oftmals werden mit diesen Tests nur einzelne kognitive Teilbereiche geprüft, was bei der Heterogeni-tät der Patienten zu einer unzureichenden Empfindlich-keit des Screeningtools führen kann. Fronto-tempo-rale Dysfunktionen können in nahezu allen Bereichendes täglichen Lebens des Patienten bei Interaktion mit Angehörigen und medizinischem Personal sowie beiEntscheidungsprozessen Relevanz zeigen und die Lebensqualität von Patienten und Angehörigen beein-trächtigen. Zudem stellt eine FTD einen Risikofaktorfür ein verkürztes Überleben dar. So ist zum Beispieldie Adaptation an die fortschreitenden körperlichen Einschränkungen sowie die Fähigkeit zu Problem-lösungen vermindert. Patienten stossen rasch an die Grenzen ihrer Ressourcen bei der Anwendung von Kom-munikations-, Ernährungs- und Beatmungshilfen. Be-reits das Wissen und Verständnis der Angehörigen und Pflegenden um die kognitiven Defizite als Teil der Er-krankung entlastet und hilft, den Alltag besser zu be-wältigen. Mit dem ECAS (Edinburgh Cognitive andBehavioural ALS Screen) wurde kürzlich von Abrahamset al. [3] ein schnell und einfach anwendbares klinisches Screening-Tool zur Aufdeckung von fronto-tempora-Christian Burkhardt
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):249–252

RECHERCHE 250
len Auffälligkeiten bei ALS-Patienten entwickelt, dasauch in fortgeschrittenen Krankheitsstadien mit motorischen Einschränkungen anwendbar ist. Seine Sensitivität und Spezifität ist vergleichbar mit einerdetaillierten neuropsychologischen Testung.
Zielsetzung und Hypothese
Das Ziel der vorliegenden Studie war es, eine Versiondes ECAS im Schweizer Hochdeutsch für den klinischen Gebrauch zu validieren. Es handelt sich hierbei um eine Übersetzung ins schweizerische Deutsch. Es liegtauch eine hochdeutsche Version des ECAS vor, die sichetwas von dieser unterscheidet. Die Studienergebnissesind Teil einer bereits publizierten Untersuchung einer multinationalen Kooperation zur Harmonisierungund Standardisierung von Biomarkern bei ALS [4].
Methodik
Die Studie wurde bei Einverständnis der Teilnehmernach Prüfung der Ethikkommision des Kantons St.Gallenin Einklang mit den ethischen Standards der Helsinki- Deklaration von 1964 durchgeführt. Die englische Versiondes ECAS wurde durch ein zertifiziertes Übersetzungs-büro ins Schweizer Hochdeutsch übersetzt. Zur Prüfung der Stimmigkeit der Übersetzung erfolgte eine Rück-übersetzung durch eine zweisprachige Linguistin insEnglische und ein Vergleich zur Ursprungsversion. Der ECAS (Abb. 1) beinhaltet neben einem ALS-spezi-fischen Abschnitt (exekutive Funktionen, Wortflüssig-keit, Sprache und soziale Kognition) einen nicht-ALS-spezifischen Abschnitt (Gedächtnis, visuell-räumliches Verständnis) zur Abgrenzung von anderen Demenz-formen. Zusätzlich beinhaltet der ECAS die Befragung von engen Bezugspersonen bezüglich Verhaltensände-rungen in fünf charakteristischen Kompetenzberei-chen (Enthemmung, Apathie, Empathie, Stereotypien,Hyperoralität) und für psychotische Symptome [3]. Der ECAS kann entweder in einer schriftlichen oder münd-lichen Version erfolgen, um den Einfluss von motori-schen Handicaps auf das Testergebnis zu minimieren. Bei 40 Patienten mit Diagnose einer ALS gemäss den revidierten El-Escorial-Kriterien wurde der ECAS ange-wandt. Diese Ergebnisse wurden mit jenen von 49 ge-sunden Kontrollpersonen zur Festlegung der Grenz-werte (zwei Standardabweichungen unterhalb desMittelwerts der Kontrollen) verglichen. Wir definier-ten vier Untergruppen für die Cut-off-Werte in Bezug auf Alter und Ausbildung, da beide Parameter einen grossen Einfluss auf Leistungen in kognitiven Tests haben (Tab. 1). Vierunddreissig enge Bezugspersonen wurden hinsichtlich Verhaltensänderungen der Pa-tienten befragt. Die Untersuchung erfolgte durch einenNeurologen oder eine nicht-neuropsychologisch aus-gebildete Study Nurse. Die Testdauer betrug im Durch-schnitt 25 Minuten.
Ergebnisse
Die Basischarakteristika der Patienten und Kontrollensind in Tabelle 2 aufgeführt. ALS-Patienten wiesen vorallem in den Bereichen der exekutiven Funktionenund der Sprache Einschränkungen gegenüber gesun-den Kontrollpersonen auf. Auffälligkeiten der Wort-flüssigkeit waren seltener nachweisbar. Jedoch zeigten einige ALS-Patienten auch in nicht-ALS-spezifischen Bereichen wie Gedächtnis Veränderungen im Ver-gleich zu den Kontrollprobanden. Insgesamt konntenbei 7/40 Patienten in einem oder mehreren Bereichenkognitive Defizite nachgewiesen werden. Weitaus häu-figer, bei 17/34 Patienten, wurden durch die Angehöri-
Abbildung 1: Beispielseite des Edinburgher kognitiven ALS-Testverfahrens (ECAS).
Nachdruck mit freundlicher Genehmigung von Professor Sharon Abrahams,
Chair of Neuropsychology & Consultant Clinical Neuropsychologist, Edinburgh.
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):249–252
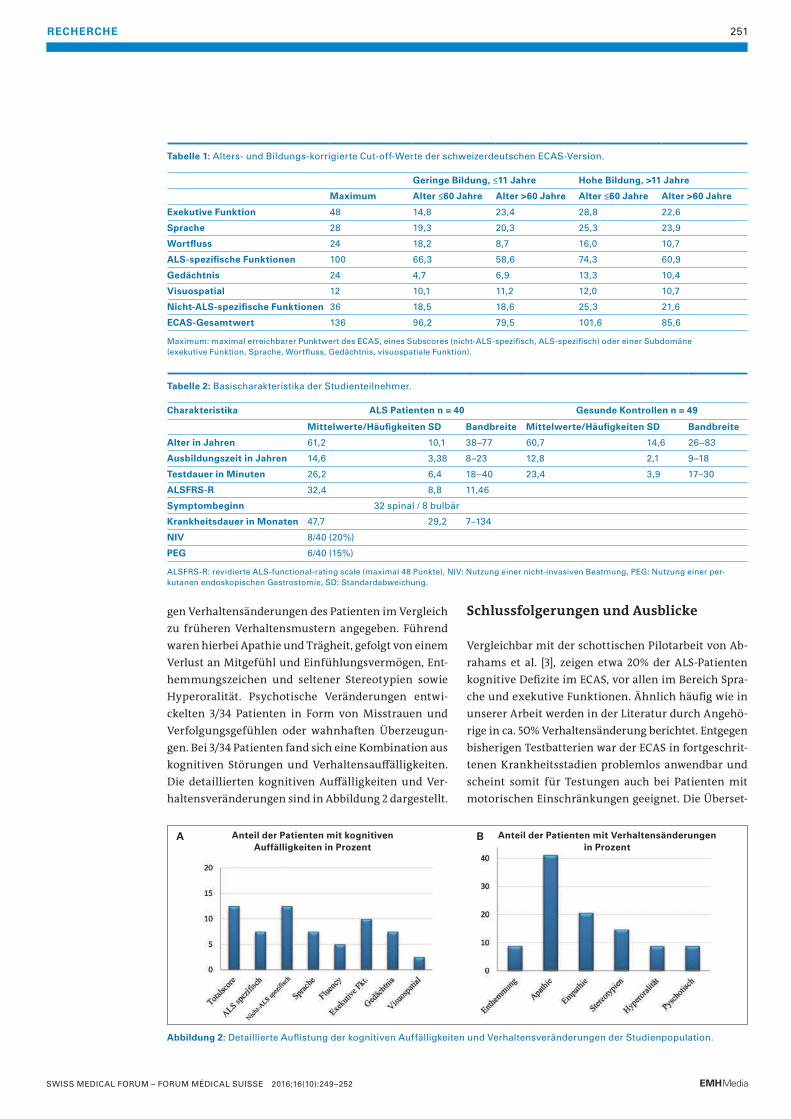
RECHERCHE 251
gen Verhaltensänderungen des Patienten im Vergleichzu früheren Verhaltensmustern angegeben. Führend waren hierbei Apathie und Trägheit, gefolgt von einem Verlust an Mitgefühl und Einfühlungsvermögen, Ent-hemmungszeichen und seltener Stereotypien sowie Hyperoralität. Psychotische Veränderungen entwi-ckelten 3/34 Patienten in Form von Misstrauen und Verfolgungsgefühlen oder wahnhaften Überzeugun-gen. Bei 3/34 Patienten fand sich eine Kombination auskognitiven Störungen und Verhaltensauffälligkeiten. Die detaillierten kognitiven Auffälligkeiten und Ver-haltensveränderungen sind in Abbildung 2 dargestellt.
Schlussfolgerungen und Ausblicke
Vergleichbar mit der schottischen Pilotarbeit von Ab-rahams et al. [3], zeigen etwa 20% der ALS-Patienten kognitive Defizite im ECAS, vor allen im Bereich Spra-che und exekutive Funktionen. Ähnlich häufig wie inunserer Arbeit werden in der Literatur durch Angehö-rige in ca. 50% Verhaltensänderung berichtet. Entgegenbisherigen Testbatterien war der ECAS in fortgeschrit-tenen Krankheitsstadien problemlos anwendbar undscheint somit für Testungen auch bei Patienten mit motorischen Einschränkungen geeignet. Die Überset-
Tabelle 1: Alters- und Bildungs-korrigierte Cut-off-Werte der schweizerdeutschen ECAS-Version.
Geringe Bildung, ≤11 Jahre Hohe Bildung, >11 Jahre
Maximum Alter ≤60 Jahre Alter >60 Jahre Alter ≤60 Jahre Alter >60 Jahre
Exekutive Funktion 48 14,8 23,4 28,8 22,6
Sprache 28 19,3 20,3 25,3 23,9
Wortfluss 24 18,2 8,7 16,0 10,7
ALS-spezifische Funktionen 100 66,3 58,6 74,3 60,9
Gedächtnis 24 4,7 6,9 13,3 10,4
Visuospatial 12 10,1 11,2 12,0 10,7
Nicht-ALS-spezifische Funktionen 36 18,5 18,6 25,3 21,6
ECAS-Gesamtwert 136 96,2 79,5 101,6 85,6
Maximum: maximal erreichbarer Punktwert des ECAS, eines Subscores (nicht-ALS-spezifisch, ALS-spezifisch) oder einer Subdomäne (exekutive Funktion, Sprache, Wortfluss, Gedächtnis, visuospatiale Funktion).
Tabelle 2: Basischarakteristika der Studienteilnehmer.
Charakteristika ALS Patienten n = 40 Gesunde Kontrollen n = 49
Mittelwerte/Häufigkeiten SD Bandbreite Mittelwerte/Häufigkeiten SD Bandbreite
Alter in Jahren 61,2 10,1 38–77 60,7 14,6 26–83
Ausbildungszeit in Jahren 14,6 3,38 8–23 12,8 2,1 9–18
Testdauer in Minuten 26,2 6,4 18–40 23,4 3,9 17–30
ALSFRS-R 32,4 8,8 11,46
Symptombeginn 32 spinal / 8 bulbär
Krankheitsdauer in Monaten 47,7 29,2 7–134
NIV 8/40 (20%)
PEG 6/40 (15%)
ALSFRS-R: revidierte ALS-functional-rating scale (maximal 48 Punkte), NIV: Nutzung einer nicht-invasiven Beatmung, PEG: Nutzung einer per-kutanen endoskopischen Gastrostomie, SD: Standardabweichung.
Abbildung 2: Detaillierte Auflistung der kognitiven Auffälligkeiten und Verhaltensveränderungen der Studienpopulation.
A BAnteil der Patienten mit kognitivenAuffälligkeiten in Prozent
Anteil der Patienten mit Verhaltensänderungenin Prozent
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):249–252

RECHERCHE 252
zung des ECAS ins Schweizer Hochdeutsch stellt damit ein schnelles und einfaches Werkzeug zur Aufdeckung von fronto-temporalen Dysfunktionen bei ALS dar. BeiNachweis fronto-temporaler Defizite sollten entspre-chende patientenorientierte, massgeschneiderte Pro-gramme in einem multidisziplinären Setting entwi-ckelt werden, um den Patienten und ihren AngehörigenHilfen anbieten zu können. Insbesondere benötigendiese eine Unterstützung bei therapeutischen Ent-scheidungsprozessen, da die daraus resultierenden Konsequenzen vom Patienten nicht immer eingeord-net werden können (z.B. Auswahl aus einer limitierten Anzahl von Alternativen, Meiden impulsiver Entschei-dungen und ausführliche Information über Konse-quenzen). Eine revidierte ECAS-Version mit entspre-chenden Cut-off-Werten ist unter http://ecas.network verfügbar.
VerdankungenWir bedanken uns bei den beteiligten Study Nurses: Bea Goldman undUrsula Schneider. Wir möchten uns bei unseren ALS-Patienten undihren Bezugspersonen sowie den gesunden Kontrollprobanden fürdie Teilnahme an der Studie bedanken. Zudem Bedanken wir uns für
die Unterstützung von Sharon Abrahams, Thomas Bak und Dorothee Lulé bei der Ausarbeitung der schweizerdeutschen Übersetzung undbei Maren Carbon für die Hilfe bei der Rückübersetzung. Diese Studie ist Teil eines EU-geförderten Joint Programme – NeurodegenerativeDisease Research (JPND SNF-Nr. SNF 31ND30_141622 (SOPHIA)) undwird unterstützt durch den Schweizerischen Nationalfonds zur Förde-rung der wissenschaftlichen Forschung.
Disclosure statementThis work was supported by the Swiss ALS Foundation and the EU Joint Programme – Neurodegenerative Disease Research (JPND) projects (grant number SNF 31ND30_141622 (SOPHIA)). The authors report no disclosures with the submitted manuscript.
Literatur1 Ng AS, Rademakers R, Miller BL. Frontotemporal dementia: a
bridge between dementia and neuromuscular disease. Ann N YAcad Sci; 1338;71–93.
2 Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology; 65;586–90.
3 Abrahams S, Newton J, Niven E, Foley J, Bak TH. Screening for cog-nition and behaviour changes in ALS. Amyotroph Lateral SclerFrontotemporal Degener; 15;9–14.
4 Lule D, Burkhardt C, Abdulla S, Bohm S, Kollewe K, Uttner I, et al.The Edinburgh Cognitive and Behavioural Amyotrophic LateralSclerosis Screen: A cross-sectional comparison of establishedscreening tools in a German-Swiss population. Amyotroph LateralScler Frontotemporal Degener; 16;16–23.
Korrespondenz: Christian BurkhardtMuskelzentrum/ALS Clinic Kantonsspital St.GallenGreithstrasse 20 CH-9007 St.Gallenchristian.burkhardt[at] kssg.ch
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):249–252

CASUISTIQUES 253
Was drückt da auf die Hirnnerven?
Bilaterale Halsschwellung bei einer jungen FrauAnita Koster-Ruscha, Joël Capraroa, Wolfgang Nagelb, Thomas Clericib, Sandro J. Stoecklic, Flavio Forrerd, Michael Brändlea, Stefan Bilza
a Klinik für Endokrinologie/Diabetologie, Kantonsspital St. Gallenb Klinik für Chirurgie, Kantonsspital St. Gallenc HNO-Klinik, Kantonsspital St. Gallend Klinik für Radiologie und Nuklearmedizin, Kantonsspital St. Gallen
Fallbericht
AnamneseDie 32-jährige Patientin berichtete über rezidivierende Infekte im Bereich des oberen Respirationstrakts in den letzten eineinhalb Jahren mit intermittierender Heiserkeit. Zudem beklagte sie ein progredientes Druck-gefühl und eine zunehmende sichtbare Schwellung am Hals rechtsbetont. Tachykardien oder erhöhte Blut-druckwerte lagen keine vor. Die Familienanamnese war bezüglich Tumorerkrankungen bland.
StatusDie Patientin präsentierte sich in gutem Allgemeinzu-stand mit normalen Vitalparametern. Zervikal beidseits kaudal des Kieferwinkels fanden sich pulsierende, wenig verschiebliche, indolente Raumforderungen. Hirnner-venausfälle bestanden keine. Der Endolarynx zeigte sich mit symmetrisch beweglichen Stimmlippen ebenso unauffällig wie der weitere internistische Status.
BefundeSonographisch wurden angulär beidseits hypoecho-gene, inhomogene, unregelmässig begrenzte Raum-forderungen dargestellt. Im MRI (Abb. 1) bestätigte sich der Verdacht auf bilaterale Glomus-caroticum-Tumo-ren. Das folgende 18F-FDOPA-PET (Abb. 2) zeigte eine inten sive Radionuklidspeicherung im Bereich der Läsi-onen, fand aber keine Hinweise auf weitere Paragang-liome, Metastasen oder ein Phäochromozytom. Die freien Plasmametanephrine einschliesslich des Me-tho xytyramins waren normwertig. Auch bei ana-mnestisch fehlenden Hinweisen für eine hereditäre Erkran kung wurde eine Mutationsanalyse des SDHB-, SDHC- und SDHD-Gens durchgeführt, die eine Keim-bahnmutation des SDHD-Gens zeigte.
DiagnoseBilaterale Glomus-caroticum-Tumoren aufgrund ei-ner Keimbahnmutation im Succinat-Dehydrogenase-(SDHD)-Gen.
Therapie und VerlaufAufgrund der Beschwerden der Patientin, der fortge-schrittenen Ausdehnung und des jungen Alters wurde die Indikation zur Operation gestellt. Im Wissen um die mögliche Morbidität der Operation wegen der Tu-morausdehnung erfolgte primär die einseitige Resek-tion der dominanten Seite. Im Situs fand sich ein die Karotisgabel umgebender und bis an die Schädelbasis reichender Tumor mit vollständiger Ummauerung des N. vagus, N. glossopharyngeus, N. hypo glossus und N. accessorius. Dieser konnte von den Ge fäs sen, die nicht infiltriert waren, problemlos abpräpa riert wer-den, die entsprechenden Nerven wurden intraoperativ erhalten – dennoch manifestierten sich postoperativ partielle oder vollständige Ausfälle derselben.Die histologische Untersuchung zeigte ein Para-gangliom mit einem Proliferationsindex (Mib-1) von 5% sowie einer Positivität für Somatostatinrezeptoren (Subtyp 2, SSTR2). Insbesondere aufgrund der Vaguspa-rese mit der entsprechenden Schluckstörung im Sinne von Aspirationen und der Heiserkeit aufgrund der
Anita Koster-Rusch
Abbildung 1: T1-gewichtete MR-Sequenz nach Gadolinium-
Gabe: Nachweis zweier irregulär begrenzter, intensiv
Kontrastmittel-aufnehmender zervikaler Raumforderungen.
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):253–255
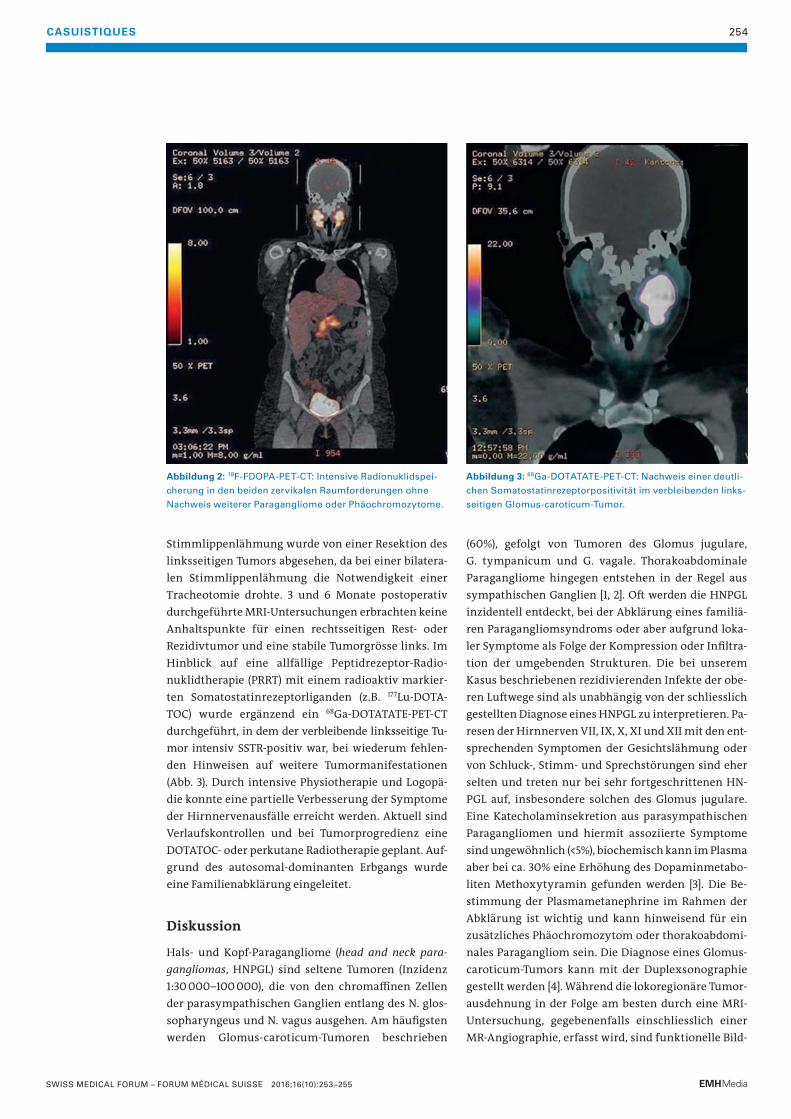
CASUISTIQUES 254
Stimmlippenlähmung wurde von einer Resektion des linksseitigen Tumors abgesehen, da bei einer bilatera-len Stimmlippenlähmung die Notwendigkeit einer Tra cheo tomie drohte. 3 und 6 Monate postoperativ durchgeführte MRI-Untersuchungen erbrachten keine Anhaltspunkte für einen rechtsseitigen Rest- oder Rezi divtumor und eine stabile Tumorgrösse links. Im Hinblick auf eine allfällige Peptidrezeptor-Radio-nuklidtherapie (PRRT) mit einem radioaktiv markier-ten Somatostatinrezeptorliganden (z.B. 177Lu-DOTA-TOC) wurde ergänzend ein 68Ga-DOTATATE-PET-CT durchgeführt, in dem der verbleibende linksseitige Tu-mor intensiv SSTR-positiv war, bei wiederum fehlen-den Hinweisen auf weitere Tumormanifestationen (Abb. 3). Durch intensive Physiotherapie und Logopä-die konnte eine partielle Verbesserung der Symptome der Hirnnervenausfälle erreicht werden. Aktuell sind Verlaufskontrollen und bei Tumorprogredienz eine DOTATOC- oder perkutane Radiotherapie geplant. Auf-grund des autosomal-dominanten Erbgangs wurde eine Fami lien abklärung eingeleitet.
Diskussion
Hals- und Kopf-Paragangliome (head and neck para-gangliomas, HNPGL) sind seltene Tumoren (Inzidenz 1:30 000–100 000), die von den chromaffinen Zellen der parasympathischen Ganglien entlang des N. glos-so pharyngeus und N. vagus ausgehen. Am häufigsten werden Glomus-caroticum-Tumoren beschrieben
(60%), gefolgt von Tumoren des Glomus jugulare, G. tympanicum und G. vagale. Thorakoabdominale Paragang liome hingegen entstehen in der Regel aus sympa thischen Ganglien [1, 2]. Oft werden die HNPGL inzidentell entdeckt, bei der Abklärung eines familiä-ren Paragangliomsyndroms oder aber aufgrund loka-ler Sym ptome als Folge der Kompression oder Infiltra-tion der umgebenden Strukturen. Die bei unserem Kasus beschriebenen rezidivierenden Infekte der obe-ren Luftwege sind als unabhängig von der schliesslich gestellten Diagnose eines HNPGL zu interpretieren. Pa-resen der Hirnnerven VII, IX, X, XI und XII mit den ent-sprechenden Symptomen der Gesichtslähmung oder von Schluck-, Stimm- und Sprechstörungen sind eher selten und treten nur bei sehr fortgeschrittenen HN-PGL auf, insbesondere solchen des Glomus jugulare. Eine Katecholaminsekretion aus parasympathischen Paragangliomen und hiermit assoziierte Symptome sind ungewöhnlich (<5%), biochemisch kann im Plasma aber bei ca. 30% eine Erhöhung des Dopaminmetabo-liten Methoxytyramin gefunden werden [3]. Die Be-stimmung der Plasmametanephrine im Rahmen der Abklärung ist wichtig und kann hinweisend für ein zusätzliches Phäochromozytom oder thorakoabdomi-nales Paragangliom sein. Die Diagnose eines Glomus-caroticum-Tumors kann mit der Duplexsonographie gestellt werden [4]. Während die lokoregionäre Tumor-ausdehnung in der Folge am besten durch eine MRI-Untersuchung, gegebenenfalls einschliesslich einer MR-Angiographie, erfasst wird, sind funktionelle Bild-
Abbildung 2: 18F-FDOPA-PET-CT: Intensive Radionuklidspei-
cherung in den beiden zervikalen Raumforderungen ohne
Nachweis weiterer Paragangliome oder Phäochromozytome.
Abbildung 3: 68Ga-DOTATATE-PET-CT: Nachweis einer deutli-
chen Somatostatinrezeptorpositivität im verbleibenden links-
seitigen Glomus-caroticum-Tumor.
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):253–255

CASUISTIQUES 255
gebungsverfahren (18F-FDOPA-PET-CT oder 68Ga-DOTA-TATE-PET-CT, Sensitivität >95%) sensitiver, um weitere Tumormanifestationen zu erfassen, und im nächsten Schritt empfohlen [5]. Ca. 35% der HNPGL werden im Rahmen eines hereditären Paragangliomsyndroms als Folge einer Keimbahnmutation in einem der für den Succinat-Dehydrogenase-(SDHx-)Komplex der mito-chondrialen Atmungskette kodierenden Gene beob-achtet. Am häufigsten sind Mutationen des SDHD-Gens (>50%) gefolgt von SDHB- (20–35%) und SDHC- (15%) Mutationen. Die Penetranz der Erkrankung ist hoch, Menschen mit einer SDHD-Mutation haben ein 75%- Risiko, im Verlauf des Lebens ein HNPGL zu entwickeln. Die Vererbung ist autosomal dominant, wobei bei der SDHD-Mutation die Krankheit nur auftritt, wenn die Mutation vom Vater vererbt wird (sog. maternal imprin-ting). Eine genetische (SDHB-, -C- und -D-Mutationen, evtl. SDHA und -AF2) und gegebenenfalls Familien-abklärung ist somit wünschenswert, de-novo-Muta-tionen gelten als Rarität. SDHx-Mutationen führen zu einer Akkumulation von Succinat, der Bildung von reaktiven Sauerstoffspezies und letztlich einer Akti-vierung des HIFα(hypoxia-inducible-factor α)-Signal-weges, der die Tumorentstehung auf mehreren Ebenen begünstigt [1]. Die immunhistochemische Untersu-chung der Expression der verschiedenen SDH-Proteine in chirurgisch entfernten Paragangliomen erlaubt ebenso Rückschlüsse auf eine zugrundeliegende SDHx-Mutation und kann, so diese Abklärung noch nicht erfolgt ist, für die genetische Untersuchung weg-weisend sein [2]. Maligne Paragangliome – diese Dia-gnose kann nur durch das Auftreten von Metastasen gestellt werden – sind bei einer SDHD-Mutation selten (<5%), werden hingegen bei SDHB-assoziierten Tumo-ren in bis zu 30% beobachtet [1]. Mit SDHx-Mutationen können Nierenzellkarzinome (SDHB ca. 14%, SDHD ca.
8%), Gastrointestinale Stromatumoren (GIST) und sel-ten Hypophysenadenome assoziiert sein [3].Prinzipiell können HNPGL bei Beschwerdefreiheit mit-tels MRI beobachtet werden. Dies gilt aufgrund der Operationsmorbidität vor allem für fortgeschrittene temporale Paragangliome, Glomus-vagale-Tumoren und fortgeschrittene Glomus-caroticum-Tumoren. Hier bei spielen auch das Alter und allfällige Komorbi-ditäten des Patienten eine wichtige Rolle. Therapeu-tisch kommen eine Operation, Radiotherapie und Pep-tidrezeptor-Radionuklidtherapie (PRRT) in Betracht. Kleinere HNPGL bei jüngeren Patienten sollen primär operiert werden, wobei bei bilateralen Tumoren ein zweizeitiges Vorgehen angezeigt ist. Bei Malignitäts-verdacht, vor allem bei einer nachgewiesenen SDHB-Mutation, soll ebenso eine Operation angestrebt wer-den. Insbesondere bei älteren Patienten mit ausgedehnten Tumoren hat sich die primäre Bestrah-lung etabliert (konventionelle Radiotherapie oder Ra-diochirurgie). Die PRRT ist besonders für multiple oder maligne Paragang liome ein vielversprechendes Ver-fahren. Für jeden Patienten muss in Abhängigkeit vom Lokalbefund, allfälliger weiterer Paragangliome, des Malignitätsverdachtes sowie der genetischen Konstel-lation ein individuelles Therapiekonzept erstellt wer-den. Auch nach der initialen Therapie sind regelmä-ssige Kontrollen nötig, da noch über Jahre Rezidive, metachrome Paragangliome oder Metastasen auftre-ten können. Eine genetische Beratung ist obligat. Bei im Rahmen eines genetischen Screenings entdeckten asymptomatischen Trägern einer SDHx-Mutation soll jährlich eine klinische und biochemische Untersu-chung sowie alle zwei bis drei Jahre eine MRI-Bildge-bung (Schädelbasis bis Becken) erfolgen [1].
Disclosure statementDie Autoren haben keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag deklariert.
Literatur1 Taïeb D, Kaliski A, Boedeker CC, Martucci V, Fojo T, Adler JR, et al.
Current approaches and recent developments in the management of head and neck paragangliomas. Endocr Rev. 2014 Oct;35(5):795–819.
2 Favier J, Amar L, Gimenez-Roqueplo A-P. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015 Feb;11(2):101–11.
3 Benn DE, Robinson BG, Clifton-Bligh RJ. 15 YEARS OF PARAGANGLI-OMA: Clinical manifestations of paraganglioma syndromes types 1–5. Endocr Relat Cancer. 2015 Aug;22(4):T91–103.
4 Stoeckli SJ, Schuknecht B, Alkadhi H, Fisch U. Evaluation of para-gangliomas presenting as a cervical mass on color-coded Doppler sonography. The Laryngoscope. 2002 Jan;112(1):143–6.
5 Castinetti F, Kroiss A, Kumar R, Pacak K, Taieb D. 15 YEARS OF PARAGANGLIOMA: Imaging and imaging-based treatment of pheochromocytoma and paraganglioma. Endocr Relat Cancer. 2015 Aug;22(4):T135–45.
Schlussfolgerung für die PraxisHNPGL sind seltene neuroendokrine Tumoren. Die Diagnose wird durch
anatomische (MRI) und funktionelle (18F-FDOPA- oder 68Ga-DOTATATE-
PET-CT) bildgebende Verfahren bestätigt. Das Behandlungskonzept wird
von einem interdisziplinären Team unter Berücksichtigung der Klinik, Be-
funde aus der Bildgebung und des Resultats der genetischen Abklärung –
in 35% liegt ein familiäres Paragangliomsyndrom als Folge einer SDHx-
Mutation vor – festgelegt. Hierbei kommen eine «wait-and-scan»-Strategie,
eine Operation, eine Radiotherapie oder eine Radionuklidtherapie in Frage.
Bei Nachweis einer SDHx-Mutation ist ein Familienscreening anzustreben.
Korrespondenz: Anita Koster-Rusch Klinik für Allgemeine Innere Medizin und Hausarztmedizin Departement Innere Medizin Kantonsspital St. Gallen Rorschacherstrasse 95 CH-9007 St. Gallen Anita.Koster-Rusch[at] kssg.ch
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):253–255

CASUISTIQUES 256
Eine Herausforderung für alle Beteiligten
Das histiozytäre SarkomThomas Rauera, Teresa De Zuluetab, Uwe Casparc, Béatrice Wagnerd, Michael Zünda
a Chirurgische Klinik, Zuger Kantonsspital; b Onko-Zentrum, Medizinische Klinik, Zuger Kantonsspital; c Radiologie, Zuger Kantonsspital;d Pathologisches Institut, Luzerner Kantonsspital
Fallbericht
Ein 56-jähriger, HIV-1-positiver Patient (CDC-Stadium A3) stellte sich mit einer dreiwöchigen Anamnese ei-ner indolenten, grössenprogredienten Schwellung imBereich der linken Brust vor. Klinisch wurde die Verdachtsdiagnose eines Lipoms gestellt. Die bildgebende Diagnostik mittels Sonogra-phie und thorakoabdominaler Computertomographie(Abb. 1) zeigte einen solitären, 3,5 × 2 × 4 cm grossen, gutvaskularisierten Tumor im Bereich der linken Pektora-lismuskulatur. Die Stanzbiopsie ergab den dringenden Verdacht auf das Vorliegen eines histiozytären Sar-koms. Im Rahmen einer interdisziplinären Fallbesprechungwurde wegen des unifokalen Tumorbefalls die Indika-tion zur primären radikalen Resektion gestellt. Esfolgte die En-bloc-Resektion des Tumors mit Subkutis und dem grössten Teil des Musculus pectoralis majorbis auf die Thoraxwand unter Mitnahme des Musculus pectoralis minor unterhalb des Tumors. Hierdurch wurde eine R0-Resektion erreicht. Die histologischeAufarbeitung des Resektats (Abb. 2) bestätigte die Diag-nose eines histiozytären Sarkoms mit Expression der histiozytären Marker Faktor XIIIa und CD 68; typi-scherweise fehlte eine Expression der follikulär-dend-ritischen, Langerhanszell- und myeloischen Marker CD 21, CD 1a und MPO sowie spezifischer B- und T-Zellmar-ker, Epithelmarker und Marker der muskulären Diffe-renzierung. Bei komplikationslosem Verlauf konnte der Patient amdritten postoperativen Tag nach Hause entlassen wer-den. Eine adjuvante Strahlentherapie, die im Rahmeneiner interdisziplinären Tumorkonferenz empfohlen wurde, lehnte der Patient ab.In der Nachsorgekontrolle nach 4 Monaten zeigte sichsowohl in der thorakalen Computertomographie alsauch im anschliessendem F-18 FDG-PET/CT (Abb. 3) einsolitärer axillärer Lymphknotenbefall links (3×3×4 cm) ohne Hinweis auf ein Lokalrezidiv oder weitere nodale/extranodale Manifestationen. In der daraufhin durch-geführten therapeutisch/diagnostischen Axilladissek-tion wurden neben dem befallenen Lymphknoten wei-tere 12 tumorfreie Lymphknoten entfernt.
Postoperativ schloss sich eine Bestrahlung der gesamten linken Axilla Level 1 mit einer Gesamtdosis von 40 Gy (20×2 Gy) analog einer Lymphombehandlung an. In denNachsorgekontrollen nach 4 und 8 Monaten zeigte sich in der zerviko-thorako-abdominalen Computertomo-graphie (Abb. 4) erfreulicherweise kein Hinweis auf einLokalrezidiv oder neue nodale/extranodale Manifesta-tionen.
Diskussion
Das histiozytäre Sarkom ist kein Sarkom, sondern einemaligne Erkrankung hämatopoetischen Ursprungs aus-gehend von den reifen Gewebehistiozyten [1, 2]. Es han-delt sich um eine sehr seltene und häufig aggressive Non-Langerhanszell-Histiozytose noch nicht vollstän-dig geklärter Pathogenese, die weniger als 1% aller Non-Hodgkin-Lymphome darstellt [1]. Bisher sind knapp über 40 Fälle in der Literatur beschrieben worden [2]. Das histiozytäre Sarkom kann ohne eindeutige Ge-schlechterbevorzugung (männlich/weiblich 1,5 : 1) in je-
Thomas Rauer
Abbildung 1: Thorakoabdominales CT, sagittaler Strahlen-
gang: Tumor in der linken Pektoralismuskulatur (Pfeil).
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):256–258

CASUISTIQUES 257
dem Lebensalter auftreten, von der Kindheit bis ins Greisenalter, wobei die Mehrheit der Fälle in einem Al-ter von 40–50 Jahren diagnostiziert wird [2, 3].Theoretisch kann jedes Organ befallen sein. In der Pub-likation mit der bisher grössten Fallzahl [2] traten die meisten histiozytären Sarkome im Bereich der Extre-mitäten auf; ein Befall von Gastrointestinaltrakt, Haut und Weichteilen sowie Lymphknoten wurde aber eben-falls beschrieben. Das klinische Beschwerdebild ist dementsprechend sehr variabel, systemische Sym-ptome wie Fieber oder Gewichtsverlust treten relativhäufig auf [4]. Aufgrund der Seltenheit und der nicht ganz geklärtenPathogenese kann die Diagnosestellung sowohl für denKliniker als auch für den erfahrenen Pathologen her-ausfordernd sein [2, 4]. Zur Diagnosebestätigung ist einhistologischer und insbesondere immunhistochemi-scher Nachweis einer histiozytären Differenzierung mit CD45-Expression unter Ausschluss von lymphatischen, epithelialen, melanozytären oder dendritischen Phäno-typen wie im vorliegenden Fall notwendig [2, 5]. In der Serie von Gounder et al. [2] waren sämtliche histiozytä-ren Sarkome positiv für CD 68, S100 und CD 163. Bisherkonnte – anders als beim Burkitt-Lymphom – nochkeine direkte Assoziation zwischen dem histiozytären Sarkom und einer HIV-Infektion nachgewiesen werden [6]. Interessanterweise sind histiozytäre Sarkome mit
vermehrtem Auftreten von Lymphomen (diffusesgrosszelliges B-Zell-Lymphom, chronisch lymphatische Leukämie, follikuläres Lymphom) assoziiert [2]. Eine standardisierte Therapie gibt es angesichts derSeltenheit der Erkrankung nicht. Bei einer unifokalenErkrankung wird die radikale Tumorexzision mit ggf. adjuvanter Strahlentherapie und/oder Chemotherapie empfohlen. Optimale Strahlendosis und Bestrahlungs-volumen sind unklar [3]. Bei einer multifokalen Er-krankung wird in der Literatur eine kombinierte Che-motherapie analog einer Lymphombehandlung nachCHOP, ABVD oder ICE-Schema vorgeschlagen [2]. Da beidieser hämatopoetischen Erkrankung ein PTEN-Ver-lust wahrscheinlich pathogenetisch mitverantwort-lich ist, erstaunt nicht, dass histiozytäre Sarkome auf«multitargeted» Tyrosinkinase-Inhibitoren sowie aufmTOR-Inhibitoren ansprechen [2].Generalisierte histiozytäre Sarkome zeigen häufig ei-nen aggressiven Verlauf mit hoher Mortalität undsprechen nur insuffizient auf eine Systemtherapie an[2, 3, 5]. Eine Tumorgrösse über 3,5 cm sowie eine mul-tifokale Erkrankung haben die ungünstigste Prog-nose mit einem medianen Überleben von knapp 12Monaten [3] bis zu 2,5 Jahren [2]. Bei einigen Patienten mit lokalisierten, radikal operierten histiozytärenSarkomen können allerdings längere Überlebensra-ten erreicht werden [2].
Abbildung 2: Tumorgewebe, 400-fache Vergrösserung: atypische Mitosen (Pfeil).
Korrespondenz: Dr. med. Thomas RauerZuger Kantonsspital Landhausstrasse 11 CH-6340 Baar Thomas.Rauer[at]zgks.ch
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):256–258

CASUISTIQUES 258
Im vorliegenden Fall wurde auf eine primäre Resektion der lokoregionären, klinisch nicht befallenen Lymph-knoten verzichtet und in Anlehnung an die wenigen bis-her publizierten Fälle eine adjuvante Strahlentherapie zur Reduktion der Lokalrezidivrate empfohlen. Wie be-reits erwähnt, wurde diese vom Patienten abgelehnt.Der vier Monate später nachgewiesene solitäre Lymph-knotenbefall wurde mittels regionaler Lymphadenekto-mie in therapeutisch/diagnostischer Absicht behandeltund anschliesssend bestrahlt. Acht Monate später ist der Patient erfreulicherweise weiterhin tumorfrei.
Disclosure statementDie Autoren haben keine finanziellen oder persönlichen Verbindungen im Zusammenhang mit diesem Beitrag deklariert.
Literatur1 Jaffe ES, Harris NL, Stein H, Vardiman JW (eds). Pathology and
Genetics of Tumors of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2001.
2 Gounder M, Desai V, Kuk D, Agaram N, Arcila M, Durham B, et al.Impact of surgery, radiation and systemic therapy on outcomes of patients with dendritic cell and histiocytic sarcomas. Eur J Cancer. 2015;51(16):2413–22.
3 Hornick JL, Jaffe ES, Fletcher CD. Extranodal histiocytic sarcoma: clinicopathologic analysis of 14 cases of a rare epithelioidmalignancy. Am J Surg Pathol. 2004;28:1133.
4 Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al.World Health Organization Classification of Tumors of Haemato-poietic and Lymphoid Tissues. Lyon: IARC Press; 2008.
5 Vos JA, Abbondanzo SL, Barekman CL, Andriko JA, Miettinen M, Aguilera NA. Histiocytic sarcoma: a study of five cases includingthe histiocyte marker CD163. Modern Pathology. 2005;18:693–704.
6 Narita K, Noro R, Seike M, et al. Succesful treatment of histiocyticsarcoma and concurrent HIV infection using a combination of CHOP and antiretroviral therapy. Intern Med. 2013;52(24):2805–9.
Abbildung 3: PET-CT mit 330 MBq F-18 FDG: solitärer axillärer Lymphknotenbefall
links (Pfeil).
Abbildung 4: Thorakales CT, axialer Strahlengang, 8 Monate postoperativ.
Das Wichtigste für die PraxisDas histiozytäre Sarkom ist eine äusserst seltene und häufig aggressive Non-Langerhanszell-Histiozytose,
die sowohl in der Diagnosestellung als auch in der Therapieplanung eine Herausforderung darstellt und
deshalb eine sehr enge interdisziplinäre Zusammenarbeit erfordert.
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(10):256–258