sulfur dioxide exposure: a way to improve the oxidation catalyst performance
TRANSCRIPT

Sulfur Dioxide Exposure: A Way To Improve the Oxidation CatalystPerformanceXavier P. Auvray and Louise Olsson*
Competence Center for Catalysis Chemical Engineering, Chalmers University of Technology, SE-412 96 Goteborg, Sweden
ABSTRACT: The oxidation of nitric oxide and propylene was studied over a model alumina-supported platinum catalyst. Twotreatments (22 h at 250 °C and 2 h at 800 °C) involving sulfur dioxide were performed in order to understand the effect of SO2aging. The role and stability of sulfur species stored during aging were investigated by the reduction of sulfated samples at 500 °Cand temperature-programmed reduction (TPR) up to 800 °C. The low activity obtained after aging without reduction revealedthe poisoning effect of surface sulfur species. The reduction at 500 °C released half of the surface species, which increased thecatalytic activity for both NO and C3H6 oxidation. The TPR removed the most stable sulfur species and improved the activityduring cooling. The catalyst aged in SO2 at high temperature showed the greatest activity for both reactions because of SO2exposure and low sulfur storage.
■ INTRODUCTIONThe catalytic system aiming to reduce the emission of harmfulcompounds from combustion engine exhausts is more andmore complex. As a matter of fact, diesel and gasoline lean-burnengines require a combination of several catalytic units in orderto treat their wide variety of pollutants in their characteristicoxidative exhausts. The first catalyst needed is commonly calleda diesel oxidation catalyst (DOC), the functions of which areoxidation of CO and residual hydrocarbons as well asconversion of NO to NO2. The latter reaction is motivatedby the great power of NO2 to oxidize soot that is trapped onthe next catalytic unit: the diesel particulate filter and the highactivity reached by currently developed NOx removal systemswhen they are fed with an equimolar mixture of NO and NO2.As a part of an exhaust after-treatment system, DOCs
undergo harsh thermal treatment and are exposed to catalystpoisons through their lifetime. SO2 is one of these poisonspresent in small amounts in vehicle exhausts with major impacton the catalyst activity. It originates from the lubricating oil andcombustion of sulfur-containing hydrocarbon present in thefuel. After-treatment catalysts are sensitive to SO2 because itcan store on the surface. Thus, SO2 is a well-known source ofdeactivation for NOx traps1−3 C3H6 oxidation,4,5 and COoxidation.5,6 The main reason is the competition of SO2 for theactive sites and the formation of stable sulfate species on thecatalyst surface, which decreases the number of accessible sites.This sulfate storage is important on metal oxides such asalumina, which is a very common support used in the DOCformulation, and the presence of platinum, also a majorcomponent of DOC, promotes sulfate formation.7
However, SO2 promotion has been noted for variousreactions such as propane oxidation,8−10 NH3-SCR,
11 HC-SCR,12 and NO oxidation.13,14 Several reasons are suggested forthat: promotion of sintering, reduction of small oxidized metalparticles, and modification of the reaction mechanism.15 Corroet al.16 suggested that adsorbed SO2 inhibited propaneoxidation but promoted the same reaction after the formationof aluminum sulfates at temperatures above 300 °C. Theformation of crystalline aluminum sulfate was observed in the
promotion of alkane combustion over sulfated Pt/Al2O3.17
Deshmukh et al.6 observed a promoting effect of SO2 exposurefor C3H6 oxidation but suppression of CO oxidation overcerium oxide. Auvray et al.13 observed that catalysts aged in thepresence of SO2 showed enhanced NO oxidation activity. Oneeffect of SO2, impacting the activity, was to promote platinumsintering. Low-temperature sintering is made possible by theaddition of SO2 to the gas feed and results in particles with abroad size distribution, while SO2 aging at high temperatureforms large particles of comparable size. Low-temperature agingyields high activity for NO oxidation while maintaining arelatively high dispersion.13 However, the role of SO2 storedduring 22 h of aging at 250 °C on the subsequent performanceis unknown. In addition, it is not previously investigated whateffect this long SO2 treatment at low temperature has on otherreactions occurring in the DOC, like, for example, propeneoxidation.In this work, we examine the properties of SO2 that are
responsible of the previously noted amelioration of Pt/Al2O3
activity for the NO oxidation reaction. In addition, we examinealso the effect of these treatments on propene oxidation.Various SO2 treatments and reduction procedures wereperformed on a model catalyst in a flow reactor. The activityof the catalyst for NO oxidation and propylene oxidation wasthen evaluated by temperature-programmed reaction duringboth a controlled heating and a controlled cooling. The effectof the different kinds of sulfur species on hysteresis was thusstudied. A catalyst was stepwise aged at high temperature in aninert atmosphere in order to evaluate the role played by metaldispersion.
Received: July 8, 2013Revised: August 28, 2013Accepted: September 18, 2013Published: September 18, 2013
Article
pubs.acs.org/IECR
© 2013 American Chemical Society 14556 dx.doi.org/10.1021/ie402153u | Ind. Eng. Chem. Res. 2013, 52, 14556−14566

■ EXPERIMENTAL SECTION
All experiments were carried out in a flow reactor composed ofa quartz tube (Ø = 22 mm) surrounded by a heating coilconnected to a power supplier in order to heat the gas flow.The inlet flow was supplied and metered by a set of mass flowcontrollers (Bronckhorst). Two thermocouples (K-type) wereinserted in the reactor: one, upstream of the catalyst, served asthe reference for temperature control, and the other one,inserted in the center channel of the monolithic sample,recorded the actual catalyst temperature. The outflow gaseswere analyzed by mass spectrometry (MS; Hiden Analytical)and Fourier transform infrared spectroscopy (MKS Instru-ments, MultiGas 2030). Figure 1 gives an overview of theexperimental sequence.
Preparation of Pt/Al2O3. The Pt/Al2O3 catalyst powderwas prepared by wet impregnation of a solution of alumina(Puralox SBa-200, Sasol) previously calcined for 2.5 h at 750°C in air in order to prevent morphological changes after metaldeposition. The pH of the alumina slurry was maintained at 4by adding a diluted solution of HNO3. The slurry was keptunder constant stirring. The platinum-containing solution wasprepared by dilution of Pt(NO3)2 (15.46 wt %; HeraeusGmbH) in ultrapure water (Advantage A10, Millipore). Theaqueous solution of platinum was added dropwise to thealumina slurry and stirred for 1 h. The pH was then checkedand adjusted to pH 2. After 3 h under stirring, the solution wasquickly frozen and dried under vacuum (Scanvac Coolsafe,Labogene). The resulting Pt/Al2O3 powder was calcined for 2 hat 500 °C in air. The amount of precursors was calculated to geta powder with an eventual Pt loading of 1 wt %.The samples were prepared by deposition of the catalyst
powder described above onto monoliths. The monoliths (Ø =20 mm; length = 22 mm) were extracted from a commercialhoneycomb cordierite structure with a channel density of 400cpsi. A blend of powder containing 80 wt % of the catalyst and20 wt % of a binder (Disperal P2, Sasol) was diluted in anethanol solution (water-to-ethanol ratio of 1:1). The monolithswere dipped into the obtained slurry under vigorous stirring.After that, a homogeneous covering of the monolith walls by
the solution was obtained, the excess of solution was removed,and the deposited layer was dried and calcined with an air gunat 550 °C for 1 min. The dipping and drying operation wasrepeated until the correct amount of washcoat (≈500 mg) wasdeposited onto the internal walls of the monolith. Themonoliths obtained were calcined in air at 500 °C for 2 h.Before any experiments, the fresh catalysts were degreened for2 h at 500 °C with a gas flow containing 500 ppm of NO and8% O2 in argon.
Elemental Analysis. One monolithic sample, labeled M-ICP, was prepared as described above and aged in the presenceof SO2, NO, and O2 in order to deposit sulfur species accordingto the SO2 aging procedure at low temperature (see below).The sample was then divided into three parts: one was keptunchanged; one was reduced in a flow reactor at 500 °C for 1 hwith 2% H2 in argon; the last one was desulfated by performingTPR from room temperature to 800 °C in 2% H2, with aheating rate of 5 °C/min. The three samples obtained (sulfated,reduced at 500 °C, and reduced at 800 °C) were crushed andanalyzed by inductively coupled plasma atomic emissionspectroscopy (ICP-AES) in order to measure the platinumand sulfur contents after aging and the different reduction steps(Table 1).
Dispersion Measurement: CO Chemisorption. COadsorption on platinum was performed at 25 °C in the flowreactor. The catalyst was first heated to 450 °C, where it wasexposed for 30 min to a flow of 1 L/min containing 2% H2,with argon as the inert balance. After rapid cooling in the samereducing stream, pure argon flowed through the catalyst for 10min in order to remove traces of H2. The CO adsorption wascarried out in two pulses composed of 100 ppm of CO (1 L/min) for 20 and 10 min, respectively. Under the first pulse, COadsorbed on platinum and was also weakly physisorbed. Thecatalyst was then flushed for 500 s with an argon flow toremove weakly bonded CO molecules. Under the secondexposure, platinum atoms were already occupied by chem-isorbed CO, and incoming CO was only physisorbed. Theoutlet concentration was monitored, and the CO uptake ofeach pulse could be found by integration. The first adsorptionrepresents the total uptake of CO, and the adsorptionquantified in the second pulse corresponds to the physisorbedCO and response of the empty reactor. Thus, subtraction of theweakly bonded CO from the first uptake yields to theadsorption of chemisorbed CO. The number of platinumatoms on the surface (Ptsurf) is correlated to the amount of COchemisorbed. On platinum, CO can bind both linearly andbridged, and we have therefore used a factor of 0.8 CO/Ptaccording to Foger et al.18 and Olsson et al.19 The dispersionvalues of fresh and aged catalysts are reported in Table 3. Onaged catalysts, the dispersion was performed after reduction ofthe sample at 500 °C (2 h with 2% H2).
Activity Tests. After the CO chemisorption experiment, theNO and propylene oxidation activities were evaluated in wetconditions. The temperature was raised to 100 °C in argonbefore a mixture of 8% O2, 500 ppm of NO, and 5% H2O inargon (total flow 3 L/min) was flown through the catalyst. Thetemperature was kept constant for 30 min in order to reachsaturation. Then the temperature was linearly increased at arate of 5 °C/min up to 500 °C. This temperature was held for ashort time (10 min) in order to get a stable final temperatureand decreased at a rate of −5 °C/min to 100 °C.The propylene oxidation test was carried out following a
similar procedure. First, the temperature was increased from
Figure 1. Series of experiments carried out on M-Pt and M-Pt2 (1 wt% Pt/Al2O3).
Industrial & Engineering Chemistry Research Article
dx.doi.org/10.1021/ie402153u | Ind. Eng. Chem. Res. 2013, 52, 14556−1456614557

100 to 150 °C (130 °C for very active samples) with a flowcontaining 5% H2O and argon and a constant total flow (3 L/min). A total of 500 ppm of C3H6 and 8% O2 were thenintroduced to the gas mixture, and the temperature was slowlyincreased (1 °C/min) up to 230 °C. Because propyleneoxidation is an exothermic reaction, a slow heating rate waschosen to avoid a rapid increase of the intracatalyst temperatureand, as a consequence, fast variation of the conversion. Thetemperature was decreased with a rate of −1 °C/min.Aging in SO2 at Low Temperature. Two Pt/Al2O3
samples (M-Pt and M-Pt2) were aged at 250 °C according tothe same procedure. The reactor was heated to 100 °C in argonin order to start the flow of 5% H2O, 500 ppm of NO, and 8%O2. The total flow was 3 L/min and was kept constantthroughout aging. The temperature was then raised to 250 °Cat a rate of 10 °C/min. When the aging temperature wasreached, 30 ppm of SO2 was added and these conditions weremaintained for 22 h. Although the two samples M-Pt and M-Pt2tend to be identical, it should be mentioned that they wereprepared from different batches and might therefore presentslightly different behavior. Two different sulfur removalprocedures were used. The first method was denoted reductionand contained 1 h with 2% H2 in argon at 500 °C. The secondprocedure, called desulfation, was TPR from room temperatureto 800 °C in 2% H2, with a heating rate of 5 °C/min. Forclarity, extensions to the catalysts’ names were added toindicate their aging status: -D for degreened, -A for aged, -ARfor aged and reduced at 500 °C, and -ATPR for aged andreduced according to the TPR procedure. The series ofexperiments are shown in Figure 1.Aging in SO2 at High Temperature. One Pt/Al2O3
sample (M-SO2, same batch as M-Pt2) was exposed to adifferent aging gas flow containing only 30 ppm of SO2,balanced with argon. This aging step was carried out for 2 h at800 °C. The total flow was 1 L/min; the heating and coolingphases were conducted under an argon flow. This aging at hightemperature in the absence of O2 and H2O was performed toevaluate the intrinsic effect of SO2 aging with minimal storage.High-Temperature Aging in Pure Argon. One Pt/Al2O3
sample (M-Ar, same batch as M-Pt2) was consecutively aged inargon at multiple temperatures: 2 h at 600 °C followed by 2 hat 700 °C and 2 h at 800 °C. The dispersion and catalyticactivity were measured after each of these three steps. Thisprocedure is used to study the effect of the temperature on themetallic dispersion as well as the relationship between thedispersion and activity.
■ RESULTS AND DISCUSSIONFresh Catalyst Characteristics. The results of the activity
tests, after degreening (-D), of the sample denoted as M-Pt areshown in Figure 2. The conversion is plotted against thecatalyst temperature during the heating phase as well as thecooling phase of the experiment in order to notice a possiblehysteresis phenomenon. The NO conversion reached amaximum of 48% (see Table 4) when the temperature wasramped up, whereas it was only 39% during the controlledcooling phase (Figure 2). This typical profile for NO oxidation,observed by Krocher et al.,20 was studied by Hauptmann et al.21
and named inverse hysteresis. It was attributed to the reversibleformation of platinum oxide. During the temperature ramp, ahigh concentration of NO2 is produced and is very oxidizing. Inthese conditions, platinum is oxidized to form platinum oxides,which are less active for NO oxidation. Therefore, the activity
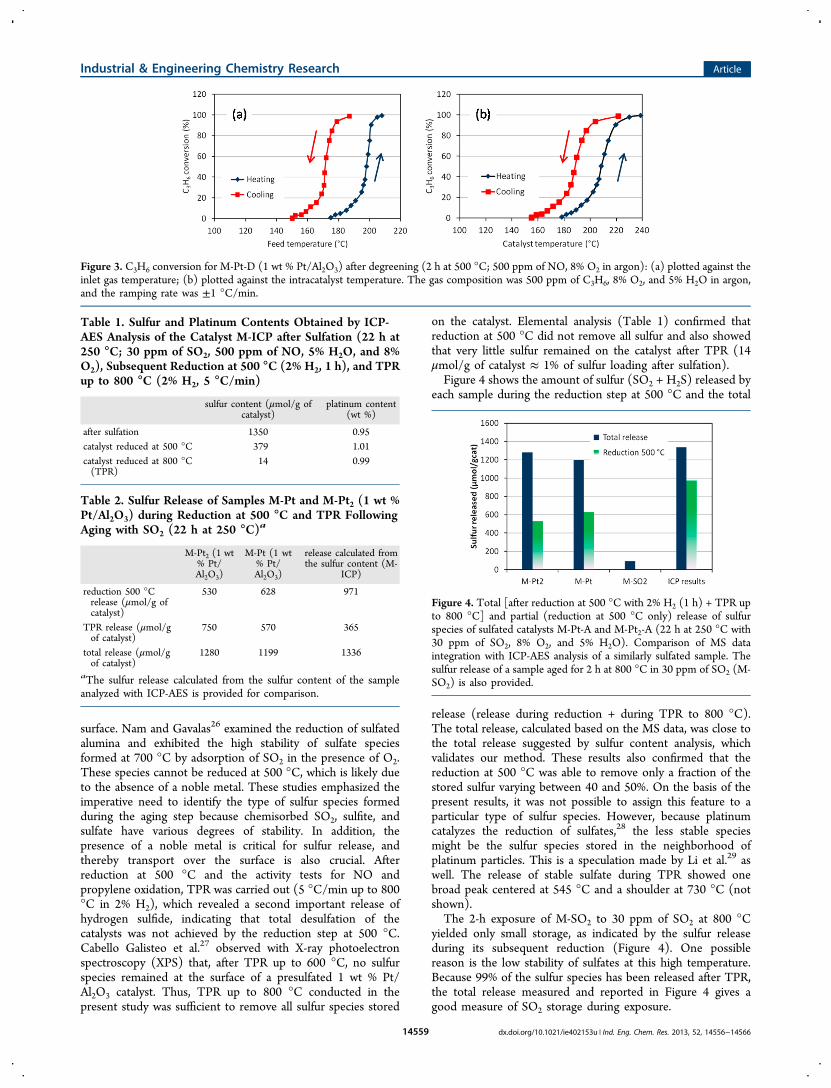
during the ramp down is lower. Inverse hysteresis wasreproduced and was also observed by Hauff et al.,22 whoconfirmed the reversible formation of platinum oxide as thecause of the hysteresis. The propylene conversion displayed inFigure 3 also presents a hysteresis, but unlike for NO oxidation,the catalyst is more active when the temperature is decreasing.The difference of 50% conversion temperature (T50) is 20 °C(Table 4). Propylene combustion creates an intracatalysttemperature gradient,4 and also the exact axial placement ofthe thermocouple inside the monolith channel can varybetween different samples. Therefore, the conversion wasplotted versus the feed temperature, as in Figure 3 a, in order tofacilitate the comparison between samples. Nevertheless, Figure3 b shows the same propene conversion plotted against thecatalyst temperature. The trend is similar, and the curves areshifted to higher temperature because of exothermiccombustion. Thus, after propene combustion light-off, thecatalyst temperature is higher than the inlet gas temperature.
Sulfur Uptake and Release. During 22 h of aging, thecatalysts are exposed to 30 ppm of SO2, 500 ppm of NO, 8%O2, and 5% H2O at a temperature of 250 °C. These conditionsare favorable for SO2 to store on the catalysts.20,23 Thesimultaneous adsorption of NO promotes the formation ofsurface sulfates, as evidenced by Xie et al.24 The sulfur contentdetermined by ICP-AES after aging is reported in Table 1. Withthe help of a mass spectrometer, quantification of outlet H2Sand SO2 during the reduction steps was also performed. Thereduction after aging was carried out at 500 °C for 1 h with 2%H2 balanced by argon. The intracatalyst temperature measuredwas ca. 470 °C during this reduction. The sulfur speciesdesorption was completed within a short time after H2 wasturned on, and zero desorption was noted at the end of thestep. H2S and SO2 released in comparable amounts. Theamounts released by M-Pt-A and M-Pt2-A at 500 °C werecalculated as 628 and 530 μmol/g of catalyst, respectively, whilethe ICP analysis suggests a release of 971 μmol/g of catalyst(Table 2). This difference might be due to the difference in thepretreatment between the analyzed sample and the samplesdevoted to activity measurement. Indeed, the analyzed samplewas sulfated without degreening and reduction, while M-Pt andM-Pt2 have been degreened for 2 h at 500 °C, reduced fordispersion measurement, and tested for NO and C3H6oxidation before sulfation. The results of Luo et al.25 regardingthe regeneration of sulfated catalysts showed that most sulfurreleases from Pt/Al2O3 below 500 °C with only 1% H2 and thatplatinum catalyzed the reduction of sulfate on the alumina
Figure 2. NO conversion for M-Pt-D (1 wt % Pt/Al2O3) afterdegreening (2 h at 500 °C; 500 ppm of NO, 8% O2 in argon). The gascomposition was 500 ppm of NO, 8% O2, and 5% H2O in argon, andthe ramping rate was ±5 °C/min.
Industrial & Engineering Chemistry Research Article
dx.doi.org/10.1021/ie402153u | Ind. Eng. Chem. Res. 2013, 52, 14556−1456614558
surface. Nam and Gavalas26 examined the reduction of sulfatedalumina and exhibited the high stability of sulfate speciesformed at 700 °C by adsorption of SO2 in the presence of O2.These species cannot be reduced at 500 °C, which is likely dueto the absence of a noble metal. These studies emphasized theimperative need to identify the type of sulfur species formedduring the aging step because chemisorbed SO2, sulfite, andsulfate have various degrees of stability. In addition, thepresence of a noble metal is critical for sulfur release, andthereby transport over the surface is also crucial. Afterreduction at 500 °C and the activity tests for NO andpropylene oxidation, TPR was carried out (5 °C/min up to 800°C in 2% H2), which revealed a second important release ofhydrogen sulfide, indicating that total desulfation of thecatalysts was not achieved by the reduction step at 500 °C.Cabello Galisteo et al.27 observed with X-ray photoelectronspectroscopy (XPS) that, after TPR up to 600 °C, no sulfurspecies remained at the surface of a presulfated 1 wt % Pt/Al2O3 catalyst. Thus, TPR up to 800 °C conducted in thepresent study was sufficient to remove all sulfur species stored
on the catalyst. Elemental analysis (Table 1) confirmed thatreduction at 500 °C did not remove all sulfur and also showedthat very little sulfur remained on the catalyst after TPR (14μmol/g of catalyst ≈ 1% of sulfur loading after sulfation).Figure 4 shows the amount of sulfur (SO2 + H2S) released by
each sample during the reduction step at 500 °C and the total
release (release during reduction + during TPR to 800 °C).The total release, calculated based on the MS data, was close tothe total release suggested by sulfur content analysis, whichvalidates our method. These results also confirmed that thereduction at 500 °C was able to remove only a fraction of thestored sulfur varying between 40 and 50%. On the basis of thepresent results, it was not possible to assign this feature to aparticular type of sulfur species. However, because platinumcatalyzes the reduction of sulfates,28 the less stable speciesmight be the sulfur species stored in the neighborhood ofplatinum particles. This is a speculation made by Li et al.29 aswell. The release of stable sulfate during TPR showed onebroad peak centered at 545 °C and a shoulder at 730 °C (notshown).The 2-h exposure of M-SO2 to 30 ppm of SO2 at 800 °C
yielded only small storage, as indicated by the sulfur releaseduring its subsequent reduction (Figure 4). One possiblereason is the low stability of sulfates at this high temperature.Because 99% of the sulfur species has been released after TPR,the total release measured and reported in Figure 4 gives agood measure of SO2 storage during exposure.
Figure 3. C3H6 conversion for M-Pt-D (1 wt % Pt/Al2O3) after degreening (2 h at 500 °C; 500 ppm of NO, 8% O2 in argon): (a) plotted against theinlet gas temperature; (b) plotted against the intracatalyst temperature. The gas composition was 500 ppm of C3H6, 8% O2, and 5% H2O in argon,and the ramping rate was ±1 °C/min.
Table 1. Sulfur and Platinum Contents Obtained by ICP-AES Analysis of the Catalyst M-ICP after Sulfation (22 h at250 °C; 30 ppm of SO2, 500 ppm of NO, 5% H2O, and 8%O2), Subsequent Reduction at 500 °C (2% H2, 1 h), and TPRup to 800 °C (2% H2, 5 °C/min)
sulfur content (μmol/g ofcatalyst)
platinum content(wt %)
after sulfation 1350 0.95catalyst reduced at 500 °C 379 1.01catalyst reduced at 800 °C(TPR)
14 0.99
Table 2. Sulfur Release of Samples M-Pt and M-Pt2 (1 wt %Pt/Al2O3) during Reduction at 500 °C and TPR FollowingAging with SO2 (22 h at 250 °C)a
M-Pt2 (1 wt% Pt/Al2O3)
M-Pt (1 wt% Pt/Al2O3)
release calculated fromthe sulfur content (M-
ICP)
reduction 500 °Crelease (μmol/g ofcatalyst)
530 628 971
TPR release (μmol/gof catalyst)
750 570 365
total release (μmol/gof catalyst)
1280 1199 1336
aThe sulfur release calculated from the sulfur content of the sampleanalyzed with ICP-AES is provided for comparison.
Figure 4. Total [after reduction at 500 °C with 2% H2 (1 h) + TPR upto 800 °C] and partial (reduction at 500 °C only) release of sulfurspecies of sulfated catalysts M-Pt-A and M-Pt2-A (22 h at 250 °C with30 ppm of SO2, 8% O2, and 5% H2O). Comparison of MS dataintegration with ICP-AES analysis of a similarly sulfated sample. Thesulfur release of a sample aged for 2 h at 800 °C in 30 ppm of SO2 (M-SO2) is also provided.
Industrial & Engineering Chemistry Research Article
dx.doi.org/10.1021/ie402153u | Ind. Eng. Chem. Res. 2013, 52, 14556−1456614559
Aged Catalysts. The activity and characteristics of thesamples M-Pt-AR and M-Pt2-AR after aging with 30 ppm ofSO2 (22 h at 250 °C) and subsequent reduction at 500 °C (1 hwith 2% H2) are discussed in this section. Although aging wasperformed at low temperature, dispersion of the two catalystsM-Pt-AR and M-Pt2-AR dropped to a very low value (Table 3).
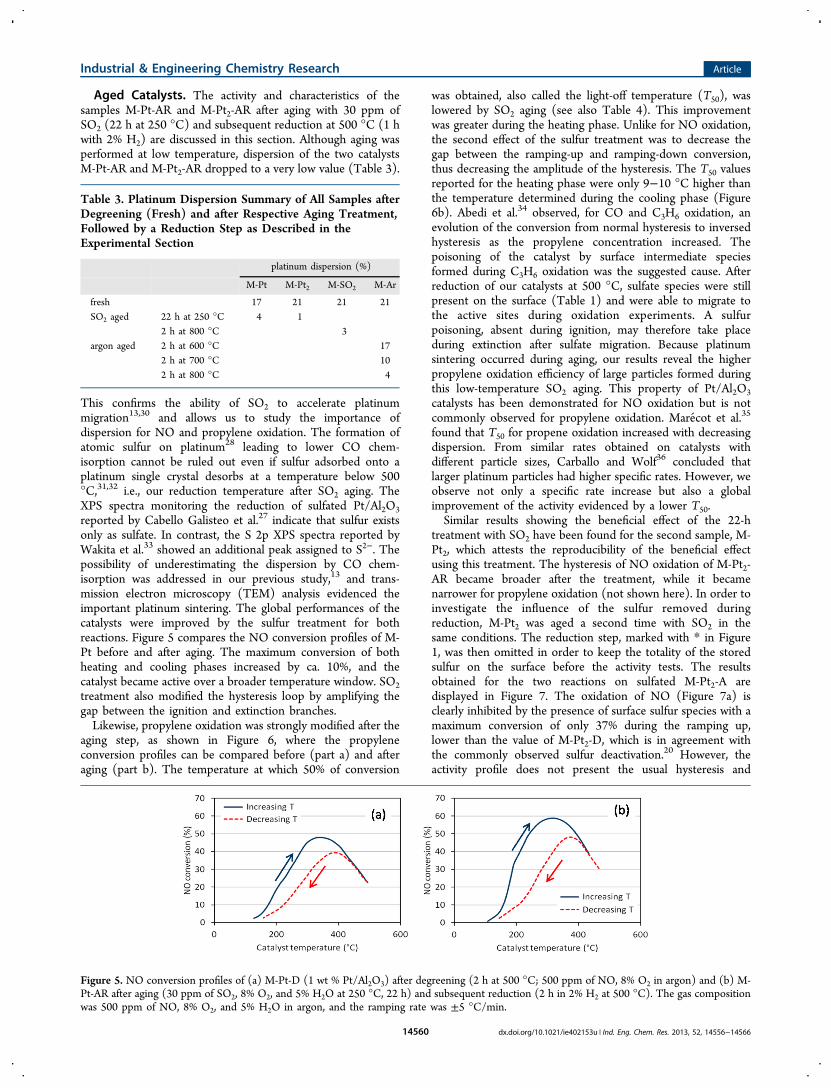
This confirms the ability of SO2 to accelerate platinummigration13,30 and allows us to study the importance ofdispersion for NO and propylene oxidation. The formation ofatomic sulfur on platinum28 leading to lower CO chem-isorption cannot be ruled out even if sulfur adsorbed onto aplatinum single crystal desorbs at a temperature below 500°C,31,32 i.e., our reduction temperature after SO2 aging. TheXPS spectra monitoring the reduction of sulfated Pt/Al2O3reported by Cabello Galisteo et al.27 indicate that sulfur existsonly as sulfate. In contrast, the S 2p XPS spectra reported byWakita et al.33 showed an additional peak assigned to S2−. Thepossibility of underestimating the dispersion by CO chem-isorption was addressed in our previous study,13 and trans-mission electron microscopy (TEM) analysis evidenced theimportant platinum sintering. The global performances of thecatalysts were improved by the sulfur treatment for bothreactions. Figure 5 compares the NO conversion profiles of M-Pt before and after aging. The maximum conversion of bothheating and cooling phases increased by ca. 10%, and thecatalyst became active over a broader temperature window. SO2treatment also modified the hysteresis loop by amplifying thegap between the ignition and extinction branches.Likewise, propylene oxidation was strongly modified after the
aging step, as shown in Figure 6, where the propyleneconversion profiles can be compared before (part a) and afteraging (part b). The temperature at which 50% of conversion
was obtained, also called the light-off temperature (T50), waslowered by SO2 aging (see also Table 4). This improvementwas greater during the heating phase. Unlike for NO oxidation,the second effect of the sulfur treatment was to decrease thegap between the ramping-up and ramping-down conversion,thus decreasing the amplitude of the hysteresis. The T50 valuesreported for the heating phase were only 9−10 °C higher thanthe temperature determined during the cooling phase (Figure6b). Abedi et al.34 observed, for CO and C3H6 oxidation, anevolution of the conversion from normal hysteresis to inversedhysteresis as the propylene concentration increased. Thepoisoning of the catalyst by surface intermediate speciesformed during C3H6 oxidation was the suggested cause. Afterreduction of our catalysts at 500 °C, sulfate species were stillpresent on the surface (Table 1) and were able to migrate tothe active sites during oxidation experiments. A sulfurpoisoning, absent during ignition, may therefore take placeduring extinction after sulfate migration. Because platinumsintering occurred during aging, our results reveal the higherpropylene oxidation efficiency of large particles formed duringthis low-temperature SO2 aging. This property of Pt/Al2O3catalysts has been demonstrated for NO oxidation but is notcommonly observed for propylene oxidation. Marecot et al.35
found that T50 for propene oxidation increased with decreasingdispersion. From similar rates obtained on catalysts withdifferent particle sizes, Carballo and Wolf36 concluded thatlarger platinum particles had higher specific rates. However, weobserve not only a specific rate increase but also a globalimprovement of the activity evidenced by a lower T50.Similar results showing the beneficial effect of the 22-h
treatment with SO2 have been found for the second sample, M-Pt2, which attests the reproducibility of the beneficial effectusing this treatment. The hysteresis of NO oxidation of M-Pt2-AR became broader after the treatment, while it becamenarrower for propylene oxidation (not shown here). In order toinvestigate the influence of the sulfur removed duringreduction, M-Pt2 was aged a second time with SO2 in thesame conditions. The reduction step, marked with * in Figure1, was then omitted in order to keep the totality of the storedsulfur on the surface before the activity tests. The resultsobtained for the two reactions on sulfated M-Pt2-A aredisplayed in Figure 7. The oxidation of NO (Figure 7a) isclearly inhibited by the presence of surface sulfur species with amaximum conversion of only 37% during the ramping up,lower than the value of M-Pt2-D, which is in agreement withthe commonly observed sulfur deactivation.20 However, theactivity profile does not present the usual hysteresis and
Table 3. Platinum Dispersion Summary of All Samples afterDegreening (Fresh) and after Respective Aging Treatment,Followed by a Reduction Step as Described in theExperimental Section
platinum dispersion (%)
M-Pt M-Pt2 M-SO2 M-Ar
fresh 17 21 21 21SO2 aged 22 h at 250 °C 4 1
2 h at 800 °C 3argon aged 2 h at 600 °C 17
2 h at 700 °C 102 h at 800 °C 4
Figure 5. NO conversion profiles of (a) M-Pt-D (1 wt % Pt/Al2O3) after degreening (2 h at 500 °C; 500 ppm of NO, 8% O2 in argon) and (b) M-Pt-AR after aging (30 ppm of SO2, 8% O2, and 5% H2O at 250 °C, 22 h) and subsequent reduction (2 h in 2% H2 at 500 °C). The gas compositionwas 500 ppm of NO, 8% O2, and 5% H2O in argon, and the ramping rate was ±5 °C/min.
Industrial & Engineering Chemistry Research Article
dx.doi.org/10.1021/ie402153u | Ind. Eng. Chem. Res. 2013, 52, 14556−1456614560
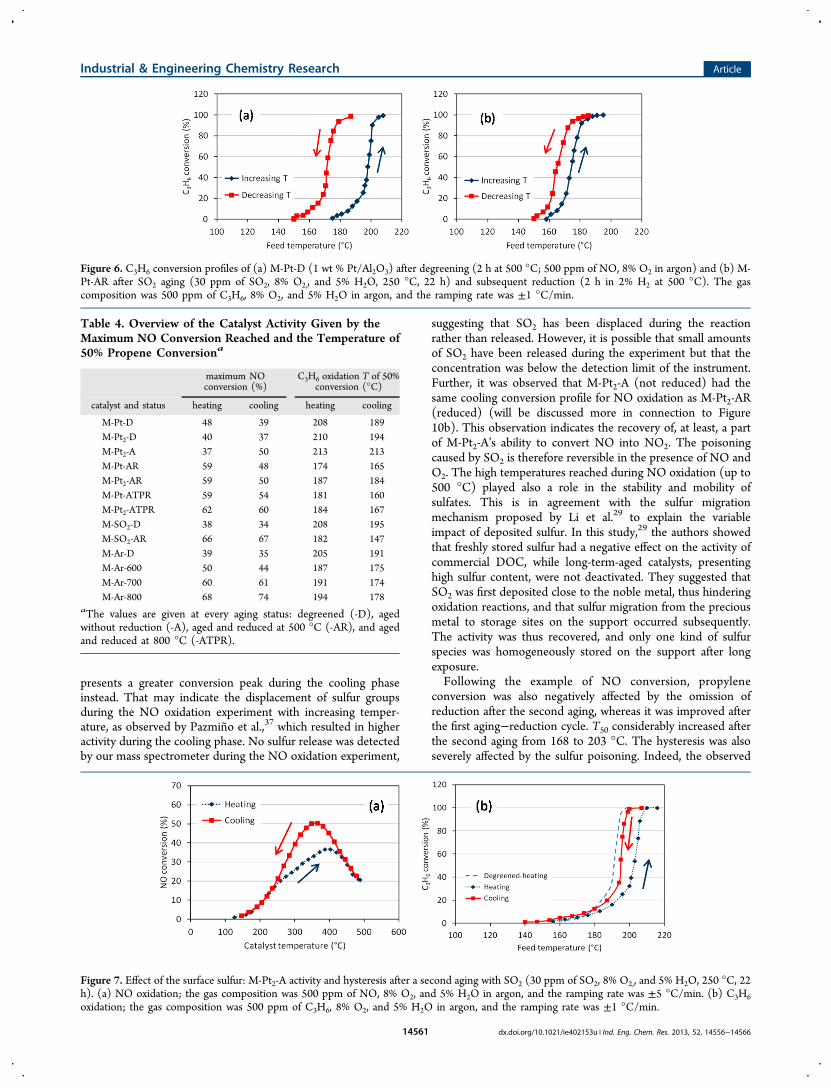
presents a greater conversion peak during the cooling phaseinstead. That may indicate the displacement of sulfur groupsduring the NO oxidation experiment with increasing temper-ature, as observed by Pazmino et al.,37 which resulted in higheractivity during the cooling phase. No sulfur release was detectedby our mass spectrometer during the NO oxidation experiment,
suggesting that SO2 has been displaced during the reactionrather than released. However, it is possible that small amountsof SO2 have been released during the experiment but that theconcentration was below the detection limit of the instrument.Further, it was observed that M-Pt2-A (not reduced) had thesame cooling conversion profile for NO oxidation as M-Pt2-AR(reduced) (will be discussed more in connection to Figure10b). This observation indicates the recovery of, at least, a partof M-Pt2-A’s ability to convert NO into NO2. The poisoningcaused by SO2 is therefore reversible in the presence of NO andO2. The high temperatures reached during NO oxidation (up to500 °C) played also a role in the stability and mobility ofsulfates. This is in agreement with the sulfur migrationmechanism proposed by Li et al.29 to explain the variableimpact of deposited sulfur. In this study,29 the authors showedthat freshly stored sulfur had a negative effect on the activity ofcommercial DOC, while long-term-aged catalysts, presentinghigh sulfur content, were not deactivated. They suggested thatSO2 was first deposited close to the noble metal, thus hinderingoxidation reactions, and that sulfur migration from the preciousmetal to storage sites on the support occurred subsequently.The activity was thus recovered, and only one kind of sulfurspecies was homogeneously stored on the support after longexposure.Following the example of NO conversion, propylene
conversion was also negatively affected by the omission ofreduction after the second aging, whereas it was improved afterthe first aging−reduction cycle. T50 considerably increased afterthe second aging from 168 to 203 °C. The hysteresis was alsoseverely affected by the sulfur poisoning. Indeed, the observed
Figure 6. C3H6 conversion profiles of (a) M-Pt-D (1 wt % Pt/Al2O3) after degreening (2 h at 500 °C; 500 ppm of NO, 8% O2 in argon) and (b) M-Pt-AR after SO2 aging (30 ppm of SO2, 8% O2,, and 5% H2O, 250 °C, 22 h) and subsequent reduction (2 h in 2% H2 at 500 °C). The gascomposition was 500 ppm of C3H6, 8% O2, and 5% H2O in argon, and the ramping rate was ±1 °C/min.
Table 4. Overview of the Catalyst Activity Given by theMaximum NO Conversion Reached and the Temperature of50% Propene Conversiona
maximum NOconversion (%)
C3H6 oxidation T of 50%conversion (°C)
catalyst and status heating cooling heating cooling
M-Pt-D 48 39 208 189M-Pt2-D 40 37 210 194M-Pt2-A 37 50 213 213M-Pt-AR 59 48 174 165M-Pt2-AR 59 50 187 184M-Pt-ATPR 59 54 181 160M-Pt2-ATPR 62 60 184 167M-SO2-D 38 34 208 195M-SO2-AR 66 67 182 147M-Ar-D 39 35 205 191M-Ar-600 50 44 187 175M-Ar-700 60 61 191 174M-Ar-800 68 74 194 178
aThe values are given at every aging status: degreened (-D), agedwithout reduction (-A), aged and reduced at 500 °C (-AR), and agedand reduced at 800 °C (-ATPR).
Figure 7. Effect of the surface sulfur: M-Pt2-A activity and hysteresis after a second aging with SO2 (30 ppm of SO2, 8% O2,, and 5% H2O, 250 °C, 22h). (a) NO oxidation; the gas composition was 500 ppm of NO, 8% O2, and 5% H2O in argon, and the ramping rate was ±5 °C/min. (b) C3H6oxidation; the gas composition was 500 ppm of C3H6, 8% O2, and 5% H2O in argon, and the ramping rate was ±1 °C/min.
Industrial & Engineering Chemistry Research Article
dx.doi.org/10.1021/ie402153u | Ind. Eng. Chem. Res. 2013, 52, 14556−1456614561

hysteresis for M-Pt2-A was smaller than that for M-Pt2-AR andM-Pt2-D. When conversion was plotted against the catalysttemperature (not shown), the extinction curve followed strictlythe ignition curve. Contrary to NO oxidation, the aged catalystdid not retrieve its previous activity during the ramping-downphase and was still far below its degreened and reducedperformance. These results show that SO2 poisoning isreversible for NO oxidation but irreversible for C3H6combustion. It should, however, be stressed that propyleneoxidation was only ramped up to 230 °C and for NO oxidationto 500 °C. The higher temperature during the ramp-up for NOoxidation could be the reason for the removal of poisoningspecies from platinum. The results also demonstrate the dualeffect of sulfur, which is detrimental when the catalyst issaturated with sulfur and beneficial after reduction of thecatalyst involving the elimination of a sulfur fraction,supposedly located on platinum and in its vicinity.Desulfated Catalysts. Because the partial removal of sulfur
improved the catalytic performance of M-Pt2-A, a secondreduction aiming at complete desulfation of the catalysts wasconducted. TPR carried out up to 800 °C has almostcompletely removed stable sulfate species remaining on thecatalyst (see Tables 1 and 2) and produced H2S. The activitytest results obtained after this treatment (M-Pt-ATPR) arediscussed below and compared with the ones obtained on M-Pt-D and M-Pt-AR. Desulfation did not improve the activity ofeither catalyst for NO oxidation during the ramping-up phase(Figures 8a and 9a). It is likely that the stable sulfur speciesremoved by TPR to 800 °C were stored on alumina and didnot influence NO oxidation, as observed by Li et al.,29 but, once
again, the nature of the sulfur species cannot be determined.Krocher et al.20 also noted the negligible influence of theremaining stable sulfates on NO oxidation. Reduction at 500°C was sufficient to remove sulfur adsorbed on platinumbecause the catalysts present the same activity after partial andcomplete desulfation. However, NO conversion measuredduring extinction was better after TPR (Figures 8b and 9b).The activity improvement is more significant on M-Pt2. As aconsequence, M-Pt2-ATPR showed a narrower NO conversionhysteresis loop after desulfation. While aging in the presence ofSO2 decreased dispersion and improved the activity, desulfationdid not improve the maximum conversion reached (whichoccurred in the heating ramp). However, the latter treatmentled to narrowing of the hysteresis loop, which ensures a morestable performance independent of temperature variation andin addition higher conversion during cooling.Total reduction of the sample (TPR to 800 °C) also affected
the catalyst ability to oxidize propylene. As seen in Figure 9a,the light-off temperature of M-Pt-ATPR was similar to aged andreduced M-Pt-AR. However, the 50% conversion during thecooling phase was attained at a much lower temperature on M-Pt-TPR (Figure 9b). TPR removed the sulfur speciesresponsible for deactivation during extinction. The gap of T50
between the heating and cooling phases grew from 8 to 24.5 °Cfor M-Pt-AR and M-Pt-ATPR, respectively (Table 4). As forNO oxidation, complete desulfation had a limited impact onpropylene oxidation during ignition. However, it resulted in amajor improvement of the activity during the cooling phase,indicated by a lower T50.
Figure 8. NO conversion of M-Pt at three stages: degreened, aged with SO2 + NO + O2, and reduced and desulfated after TPR during (a) rampingup (5 °C/min) and (b) ramping down (5 °C/min). The feed composition was 500 ppm of NO + 8% O2 + 5% H2O balance with argon for a totalflow of 3 L/min.
Figure 9. Propylene conversion of M-Pt at three stages: degreened, aged with SO2 + NO + O2, and reduced and desulfated after TPR during (a)ramping up (1 °C/min) and (b) ramping down (1 °C/min). The feed composition was 500 ppm of C3H6 + 8% O2 + 5% H2O balance with argonfor a total flow of 3 L/min.
Industrial & Engineering Chemistry Research Article
dx.doi.org/10.1021/ie402153u | Ind. Eng. Chem. Res. 2013, 52, 14556−1456614562
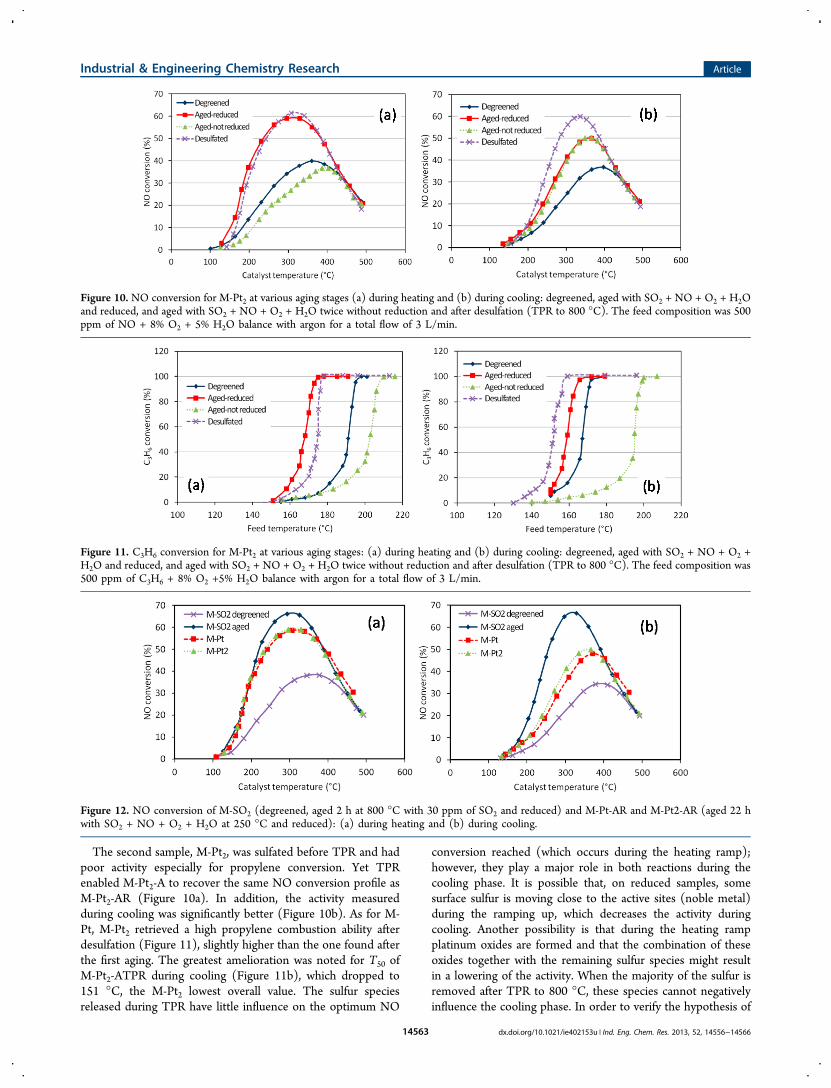
The second sample, M-Pt2, was sulfated before TPR and hadpoor activity especially for propylene conversion. Yet TPRenabled M-Pt2-A to recover the same NO conversion profile asM-Pt2-AR (Figure 10a). In addition, the activity measuredduring cooling was significantly better (Figure 10b). As for M-Pt, M-Pt2 retrieved a high propylene combustion ability afterdesulfation (Figure 11), slightly higher than the one found afterthe first aging. The greatest amelioration was noted for T50 ofM-Pt2-ATPR during cooling (Figure 11b), which dropped to151 °C, the M-Pt2 lowest overall value. The sulfur speciesreleased during TPR have little influence on the optimum NO
conversion reached (which occurs during the heating ramp);however, they play a major role in both reactions during thecooling phase. It is possible that, on reduced samples, somesurface sulfur is moving close to the active sites (noble metal)during the ramping up, which decreases the activity duringcooling. Another possibility is that during the heating rampplatinum oxides are formed and that the combination of theseoxides together with the remaining sulfur species might resultin a lowering of the activity. When the majority of the sulfur isremoved after TPR to 800 °C, these species cannot negativelyinfluence the cooling phase. In order to verify the hypothesis of
Figure 10. NO conversion for M-Pt2 at various aging stages (a) during heating and (b) during cooling: degreened, aged with SO2 + NO + O2 + H2Oand reduced, and aged with SO2 + NO + O2 + H2O twice without reduction and after desulfation (TPR to 800 °C). The feed composition was 500ppm of NO + 8% O2 + 5% H2O balance with argon for a total flow of 3 L/min.
Figure 11. C3H6 conversion for M-Pt2 at various aging stages: (a) during heating and (b) during cooling: degreened, aged with SO2 + NO + O2 +H2O and reduced, and aged with SO2 + NO + O2 + H2O twice without reduction and after desulfation (TPR to 800 °C). The feed composition was500 ppm of C3H6 + 8% O2 +5% H2O balance with argon for a total flow of 3 L/min.
Figure 12. NO conversion of M-SO2 (degreened, aged 2 h at 800 °C with 30 ppm of SO2 and reduced) and M-Pt-AR and M-Pt2-AR (aged 22 hwith SO2 + NO + O2 + H2O at 250 °C and reduced): (a) during heating and (b) during cooling.
Industrial & Engineering Chemistry Research Article
dx.doi.org/10.1021/ie402153u | Ind. Eng. Chem. Res. 2013, 52, 14556−1456614563
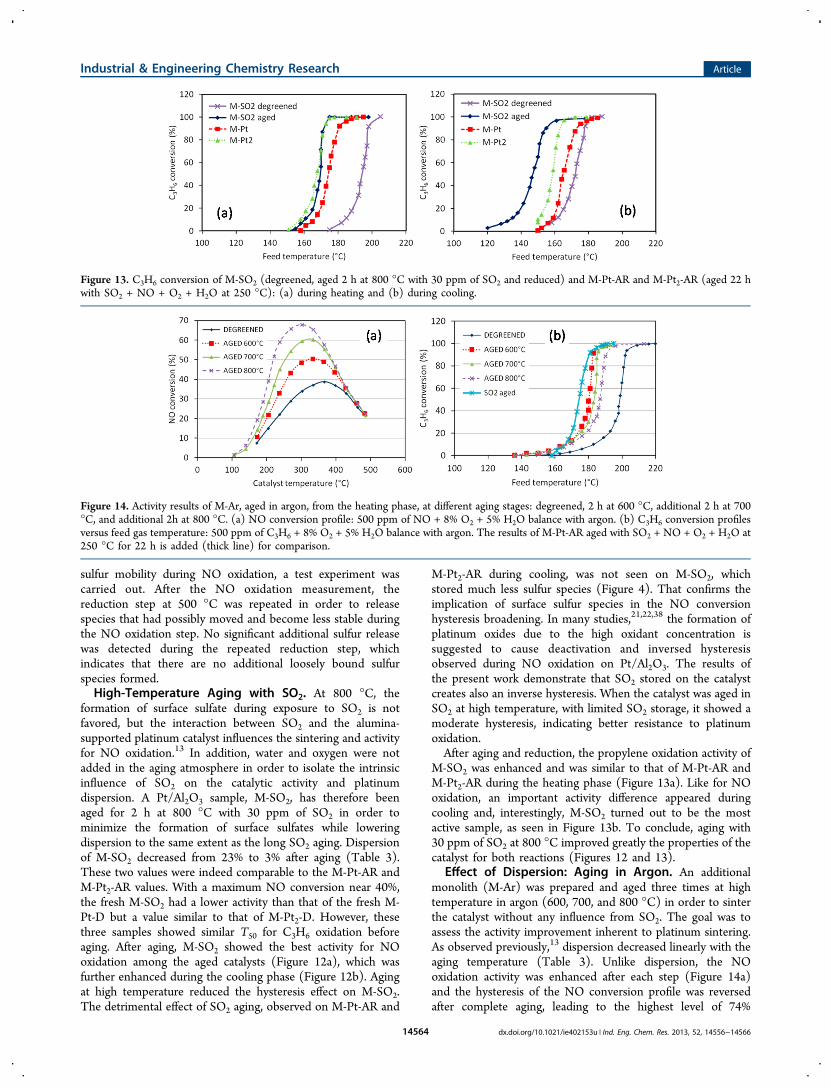
sulfur mobility during NO oxidation, a test experiment wascarried out. After the NO oxidation measurement, thereduction step at 500 °C was repeated in order to releasespecies that had possibly moved and become less stable duringthe NO oxidation step. No significant additional sulfur releasewas detected during the repeated reduction step, whichindicates that there are no additional loosely bound sulfurspecies formed.High-Temperature Aging with SO2. At 800 °C, the
formation of surface sulfate during exposure to SO2 is notfavored, but the interaction between SO2 and the alumina-supported platinum catalyst influences the sintering and activityfor NO oxidation.13 In addition, water and oxygen were notadded in the aging atmosphere in order to isolate the intrinsicinfluence of SO2 on the catalytic activity and platinumdispersion. A Pt/Al2O3 sample, M-SO2, has therefore beenaged for 2 h at 800 °C with 30 ppm of SO2 in order tominimize the formation of surface sulfates while loweringdispersion to the same extent as the long SO2 aging. Dispersionof M-SO2 decreased from 23% to 3% after aging (Table 3).These two values were indeed comparable to the M-Pt-AR andM-Pt2-AR values. With a maximum NO conversion near 40%,the fresh M-SO2 had a lower activity than that of the fresh M-Pt-D but a value similar to that of M-Pt2-D. However, thesethree samples showed similar T50 for C3H6 oxidation beforeaging. After aging, M-SO2 showed the best activity for NOoxidation among the aged catalysts (Figure 12a), which wasfurther enhanced during the cooling phase (Figure 12b). Agingat high temperature reduced the hysteresis effect on M-SO2.The detrimental effect of SO2 aging, observed on M-Pt-AR and
M-Pt2-AR during cooling, was not seen on M-SO2, whichstored much less sulfur species (Figure 4). That confirms theimplication of surface sulfur species in the NO conversionhysteresis broadening. In many studies,21,22,38 the formation ofplatinum oxides due to the high oxidant concentration issuggested to cause deactivation and inversed hysteresisobserved during NO oxidation on Pt/Al2O3. The results ofthe present work demonstrate that SO2 stored on the catalystcreates also an inverse hysteresis. When the catalyst was aged inSO2 at high temperature, with limited SO2 storage, it showed amoderate hysteresis, indicating better resistance to platinumoxidation.After aging and reduction, the propylene oxidation activity of
M-SO2 was enhanced and was similar to that of M-Pt-AR andM-Pt2-AR during the heating phase (Figure 13a). Like for NOoxidation, an important activity difference appeared duringcooling and, interestingly, M-SO2 turned out to be the mostactive sample, as seen in Figure 13b. To conclude, aging with30 ppm of SO2 at 800 °C improved greatly the properties of thecatalyst for both reactions (Figures 12 and 13).
Effect of Dispersion: Aging in Argon. An additionalmonolith (M-Ar) was prepared and aged three times at hightemperature in argon (600, 700, and 800 °C) in order to sinterthe catalyst without any influence from SO2. The goal was toassess the activity improvement inherent to platinum sintering.As observed previously,13 dispersion decreased linearly with theaging temperature (Table 3). Unlike dispersion, the NOoxidation activity was enhanced after each step (Figure 14a)and the hysteresis of the NO conversion profile was reversedafter complete aging, leading to the highest level of 74%
Figure 13. C3H6 conversion of M-SO2 (degreened, aged 2 h at 800 °C with 30 ppm of SO2 and reduced) and M-Pt-AR and M-Pt2-AR (aged 22 hwith SO2 + NO + O2 + H2O at 250 °C): (a) during heating and (b) during cooling.
Figure 14. Activity results of M-Ar, aged in argon, from the heating phase, at different aging stages: degreened, 2 h at 600 °C, additional 2 h at 700°C, and additional 2h at 800 °C. (a) NO conversion profile: 500 ppm of NO + 8% O2 + 5% H2O balance with argon. (b) C3H6 conversion profilesversus feed gas temperature: 500 ppm of C3H6 + 8% O2 + 5% H2O balance with argon. The results of M-Pt-AR aged with SO2 + NO + O2 + H2O at250 °C for 22 h is added (thick line) for comparison.
Industrial & Engineering Chemistry Research Article
dx.doi.org/10.1021/ie402153u | Ind. Eng. Chem. Res. 2013, 52, 14556−1456614564

attained during the cooling phase (Table 4). This activity valuefor M-Ar-800 was higher than that for the catalysts treated withSO2 at 250 °C at comparable dispersion (0−5%). This resultconfirms the need for large particles to optimize the conversionof NO.39,40 Figure 14b shows the propylene conversion profilesof M-Ar as well as M-Pt-AR. The first aging at 600 °C provokeda moderate dispersion loss, from 21% to 17% and increased theactivity. As for the catalysts aged in SO2, the activity of M-Arwas better for propylene combustion after sintering at 600 °C,but the two subsequent aging steps reversed this trend becausethe activity decreased after both of them. However, the globaleffect of the full procedure remained positive because T50 wasstill lower after complete aging than the one measured on thedegreened catalyst. This result shows that the propyleneoxidation efficiency is altered by severe platinum sinteringcaused by thermal aging. For propylene oxidation, M-Pt-AR hasa higher activity than aged M-Ar when considering the heatingphase (Figure 14b). The improvement observed on propeneconversion during heating for samples aged in SO2 cantherefore only partly be attributed to decreased dispersion.The reason is likely due to the chemical effect that steersplatinum migration and particle formation during aging,resulting in particle shape, morphology, and size distributionthat are more active. In a previous study,13 we examined theparticles after low-temperature SO2 + NO + O2 aging withTEM and observed a broad particle-size distribution, which wasnot the case for platinum aged at high temperature in argon.However, the results are different during the cooling phase,
where M-Ar had higher activity than M-Pt-AR aged at 250 °Cwith SO2 + NO + O2 + H2O. The reason for this is likely thenegative effect of stored sulfate that migrates during the heatingphase on M-Pt-AR. This hypothesis was confirmed by the factthat after desulfation both M-Pt-TPR and M-Pt2-ATPRexhibited much better properties than M-Ar for C3H6 oxidationduring both cooling and heating. This means that platinumparticles sintered in a SO2-containing environment have abetter intrinsic activity than those aged in argon, at similardispersion.Role of SO2. In this section, the impact of surface sulfur
during oxidation reactions is discussed. SO2 is often seen as apoison because it adsorbs easily on oxide surfaces and metals,which causes deactivation of catalysts. On the other hand,several studies dealing with the beneficial effect of sulfur andsulfur-assisted catalytic reactions have been published.9,12,41 Leeet al.8 have described the beneficial effect of SO2 for propaneoxidation. In our experiments, the catalysts are exposed to SO2+ NO + O2 + H2O during the aging process at 250 °C only andare subsequently reduced in order to remove the stored sulfurin two steps. The first reduction at 500 °C released the lessstable sulfates, supposedly close to the platinum particles, whichrepresented only 40−50% of the total amount stored. ThenTPR was performed to eliminate the most stable species, storedon the alumina support, and evaluate their impact on theactivity. As demonstrated with the sample M-Pt2, theperformance of the catalyst for both NO and C3H6 oxidationis greatly lowered when the catalyst is not reduced aftersulfation. This result, combined with the amelioration seen afteraging and reduction, indicates the strong inhibiting effect of theless stable surface species. These adsorbed SOx hinder thecombustion of propylene, probably by blocking the active sites.The low temperature applied during the propylene oxidationexperiment makes the adsorbed SOx stable and does not permitthe propylene to compete for the blocked sites, whereas the
high temperature reached (500 °C) and the presence of NO,O2, and H2O in the feed during the NO oxidation test weresufficient conditions to displace these sulfur species. Despitetheir large amount, the stable sulfur species had a minorinfluence on the activity during the ignition phase. The coolingphase, however, revealed that their presence on the agedcatalysts was detrimental for both reactions. The presence ofstable sulfur broadened the inversed hysteresis loop displayedon NO conversion curves and narrowed the hysteresis observedfor propylene oxidation. The complete desulfation led to amajor activity improvement during the cooling phase for bothreactions. Even though catalysts show a better activity whensulfur has been removed, the presence of SO2 in the agingatmosphere is necessary to obtain such activity. Indeed, M-Araged at 600 °C showed a lower performance toward C3H6
oxidation than M-Pt-AR and M-Pt2-AR aged in SO2 + NO + O2
+ H2O at 250 °C. Further aging at higher temperature evendecreased the M-Ar activity. Interestingly, superior activity forpropene oxidation was obtained after aging M-SO2 with 30ppm of SO2 at 800 °C for 2 h (Figure 14b), which confirms theimportant role of SO2 during aging. T50 for propene oxidationdecreased from 194 °C for the degreened catalyst to 169 °Cduring heating.
■ CONCLUSIONS
The activity enhancement of alumina-supported platinumcatalysts by SO2 aging has been shown for NO oxidation andpropylene combustion. The reduction treatments carried outenabled us to distinguish two types of surface sulfur specieswith different properties. The less stable species, locatedprobably on and around platinum, were removed by reductionat 500 °C, and these species greatly lowered the activity of thecatalysts. The second type of species was more stable anddecomposed during TPR up to 800 °C. The removal of thesespecies, supposedly sulfates stored on alumina support, did notaffect the performance during ignition but improved the activityduring extinction. This effect was more pronounced forpropylene oxidation. As a consequence, the observed hysteresisfor both oxidation reactions was modified by SO2 exposure andreduction treatments. Because surface sulfates decreased theactivity during cooling, the NO conversion inverse hysteresisgot larger after aging, while the propylene conversion hysteresisgot smaller. After desulfation treatment, the NO oxidationhysteresis loop was reduced and C3H6 conversion hysteresisbecame large again.The high-temperature aging in argon showed that the
catalyst with large particles was more active for NO oxidationthan when it was fresh and well-dispersed. Although the firstaging at 600 °C decreased the light-off temperature ofpropylene combustion, further platinum sintering in argondecreased the catalyst activity. At comparable low dispersionthe catalysts aged in the presence of SO2 + NO + O2 + H2O at250 °C presented a better propylene oxidation activity than thecatalyst aged in argon. In addition, the best activity was foundfor the sample aged with SO2 only at 800 °C. One can concludethat the remarkable properties of catalysts aged with SO2 comefrom the SO2-assisted sintering that creates platinum particleswith appropriate size and probably other features likemorphology and structure that improve the catalytic activity.
Industrial & Engineering Chemistry Research Article
dx.doi.org/10.1021/ie402153u | Ind. Eng. Chem. Res. 2013, 52, 14556−1456614565

■ AUTHOR INFORMATIONCorresponding Author*Tel: +46(0)31-772 4390. Fax: +46(0)31-772 3035. E-mail:[email protected] authors declare no competing financial interest.
■ ACKNOWLEDGMENTSFunding from the Swedish Foundation for Strategic Research(F06-0006) and the Swedish Energy agency is acknowledged.This work was carried out at the Competence Centre forCatalysis, Chalmers University of Technology. KCK isfinancially supported by Chalmers University of Technology,the Swedish Energy Agency, and the member companies: ABVolvo, Volvo Car Corp., Scania CV AB, Haldor Topsoe A/S,and ECAPS AB.
■ REFERENCES(1) Choi, J. S.; Partridge, W. P.; Daw, C. S. Appl. Catal., B 2007, 77(1−2), 145−156.(2) Limousy, L.; Mahzoul, H.; Brilhac, J. F.; Gilot, P.; Garin, F.;Maire, G. Appl. Catal., B 2003, 42 (3), 237−249.(3) Engstrom, P.; Amberntsson, A.; Skoglundh, M.; Fridell, E.;Smedler, G. Appl. Catal., B 1999, 22 (4), L241−L248.(4) Russell, A.; Henry, C.; Currier, N. W.; Yezerets, A.; Epling, W. S.Appl. Catal. A 2011, 397 (1−2), 272−284.(5) Kolli, T.; Kanerva, T.; Huuhtanen, M.; Vippola, M.; Kallinen, K.;Kinnunen, T.; Lepisto, T.; Lahtinen, J.; Keiski, R. L. Catal. Today2010, 154 (3−4), 303−307.(6) Deshmukh, S. S.; Zhang, M.; Kovalchuk, V. I.; d’Itri, J. L. Appl.Catal., B 2003, 45 (2), 135−145.(7) Summers, J. C. Environ. Sci. Technol. 1979, 13 (3), 321−325.(8) Lee, A. F.; Wilson, K.; Lambert, R. M.; Hubbard, C. P.; Hurley, R.G.; McCabe, R. W.; Gandhi, H. S. J. Catal. 1999, 184 (2), 491−498.(9) Skoglundh, M.; Ljungqvist, A.; Petersson, M.; Fridell, E.; Cruise,N.; Augustsson, O.; Jobson, E. Appl. Catal., B 2001, 30 (3−4), 315−328.(10) Yao, H. C.; Stepien, H. K.; Gandhi, H. S. J. Catal. 1981, 67 (1),231−236.(11) Amiridis, M. D.; Wachs, I. E.; Deo, G.; Jehng, J. M.; Kim, D. S. J.Catal. 1996, 161 (1), 247−253.(12) Angelidis, T. N.; Kruse, N. Appl. Catal., B 2001, 34 (3), 201−212.(13) Auvray, X.; Pingel, T.; Olsson, E.; Olsson, L. Appl. Catal., B2013, 129 (0), 517−527.(14) Olsson, L.; Karlsson, H. Catal. Today 2009, 147, S290−S294.(15) Hinz, A.; Skoglundh, M.; Fridell, E.; Andersson, A. J. Catal.2001, 201 (2), 247−257.(16) Corro, G.; Montiel, R.; Vazquez, L. C. Catal. Commun. 2002, 3(11), 533−539.(17) Gawthrope, D. E.; Lee, A. F.; Wilson, K. Catal. Lett. 2004, 94(1−2), 25−29.(18) Foger, K.; Anderson, J. R. Appl. Surf. Sci. 1979, 2 (3), 335−351.(19) Olsson, L.; Abul-Milh, M.; Karlsson, H.; Jobson, E.;Thormahlen, P.; Hinz, A. Top. Catal. 2004, 30−31 (1), 85−90.(20) Krocher, O.; Widmer, M.; Elsener, M.; Rothe, D. Ind. Eng.Chem. Res. 2009, 48 (22), 9847−9857.(21) Hauptmann, W.; Votsmeier, M.; Gieshoff, J.; Drochner, A.;Vogel, H. Appl. Catal., B 2009, 93 (1−2), 22−29.(22) Hauff, K.; Tuttlies, U.; Eigenberger, G.; Nieken, U. Appl. Catal.,B 2012, 123−124, 107−116.(23) Ottinger, N. A.; Toops, T. J.; Pihl, J. A.; Roop, J. T.; Choi, J.-S.;Partridge, W. P. Appl. Catal., B 2012, 117−118 (0), 167−176.(24) Xie, Y.; Chen, Y.; Ma, Y.; Jin, Z. J. Hazard. Mater. 2011, 195 (0),223−229.(25) Luo, J.-Y.; Kisinger, D.; Abedi, A.; Epling, W. S. Appl. Catal., A2010, 383 (1−2), 182−191.
(26) Nam, S. W.; Gavalas, G. R. Appl. Catal. 1991, 74 (1), 53−64.(27) Cabello Galisteo, F.; Mariscal, R.; Lopez Granados, M.; ZafraPoves, M. D.; Fierro, J. L. G.; Kroger, V.; Keiski, R. L. Appl. Catal., B2007, 72 (3−4), 272−281.(28) Apesteguia, C. R.; Garetto, T. F.; Borgna, A. J. Catal. 1987, 106(1), 73−84.(29) Li, J.; Kumar, A.; Chen, X.; Currier, N.; Yezerets, A. SAE (Tech.Pap.) 2013, 2.(30) Jones, J. M.; Dupont, V. A.; Brydson, R.; Fullerton, D. J.; Nasri,N. S.; Ross, A. B.; Westwood, A. V. K. Catal. Today 2003, 81 (4),589−601.(31) Streber, R.; Papp, C.; Lorenz, M. P. A.; Hofert, O.; Darlatt, E.;Bayer, A.; Denecke, R.; Steinruck, H. P. Chem. Phys. Lett. 2010, 494(4−6), 188−192.(32) Zebisch, P.; Stichler, M.; Trischberger, P.; Weinelt, M.;Steinruck, H. P. Surf. Sci. 1997, 371 (2−3), 235−244.(33) Wakita, H.; Kani, Y.; Ukai, K.; Tomizawa, T.; Takeguchi, T.;Ueda, W. Appl. Catal., A 2005, 283 (1−2), 53−61.(34) Abedi, A.; Hayes, R.; Votsmeier, M.; Epling, W. S. Catal. Lett.2012, 142 (8), 930−935.(35) Marecot, P.; Fakche, A.; Kellali, B.; Mabilon, G.; Prigent, P.;Barbier, J. Appl. Catal., B 1994, 3 (4), 283−294.(36) Carballo, L. M.; Wolf, E. E. J. Catal. 1978, 53 (3), 366−373.(37) Pazmino, J. H.; Miller, J. T.; Mulla, S. S.; Nicholas Delgass, W.;Ribeiro, F. H. J. Catal. 2011, 282 (1), 13−24.(38) Olsson, L.; Fridell, E. J. Catal. 2002, 210 (2), 340−353.(39) Benard, S.; Retailleau, L.; Gaillard, F.; Vernoux, P.; Giroir-Fendler, A. Appl. Catal., B 2005, 55 (1), 11−21.(40) Xue, E.; Seshan, K.; Ross, J. R. H. Appl. Catal., B 1996, 11 (1),65−79.(41) Olsson, L.; Karlsson, H. Catal. Today 2009, 147 (Suppl), S290−S294.
Industrial & Engineering Chemistry Research Article
dx.doi.org/10.1021/ie402153u | Ind. Eng. Chem. Res. 2013, 52, 14556−1456614566