suementary inrmatin - media.nature.com · département de chimie, laboratoire de chimie...
TRANSCRIPT

NATURE CHEMISTRY | www.nature.com/naturechemistry 1
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S1
Supplementary Information
Synthesis of giant globular multivalent glycofullerenes as potent inhibitors in a model of Ebola virus infection
Antonio Muñoz,§ David Sigwalt,¥,£ Beatriz M. Illescas,§ Joanna Luczkowiak,‡ Laura
Rodríguez,§ Iwona Nierengarten,¥ Michel Holler,¥ Jean‐Serge Remy,£ Kevin Buffet,¶
Stéphane P. Vincent,¶ Javier Rojo,†,* Rafael Delgado,‡,* Jean‐François Nierengarten,¥,*
Nazario Martín§,,*
§Departamento de Química Orgánica, Facultad de Química, Universidad Complutense, 28040
Madrid, Spain, e‐mail: [email protected]; † Glycosystems Laboratory, Instituto de
Investigaciones Químicas (IIQ), CSIC – Universidad de Sevilla, Av. Américo Vespucio 49, Seville
41092 Spain. Tel: + 34 954489568; FAX +34 954460165; e‐mail: [email protected]; £ Laboratory V‐SAT (CAMB UMR 7199, CNRS), Labex Medalis, Université de Strasbourg, 74
Route du Rhin, 67401 Illkirch‐Graffenstaden, France; ¶ University of Namur (FUNDP),
Département de Chimie, Laboratoire de Chimie Bio‐Organique, rue de Bruxelles 61, B‐5000
Namur, Belgium; ‡Laboratorio de Microbiología Molecular, Instituto de Investigación Hospital
12 de Octubre (imas12) 28041 Madrid, Spain, e‐mail: [email protected]; ¥ Laboratoire de Chimie des Matériaux Moléculaires, Université de Strasbourg et CNRS (UMR
7509), Ecole Européenne de Chimie, Polymères et Matériaux, 25 rue Becquerel, 67087
Strasbourg, France, e‐mail: [email protected]; IMDEA‐Nanoscience, Campus
Cantoblanco, 28049 Madrid, Spain.
Table of Contents ........................................................................................................................ S1 Synthesis and Characterization .................................................................................................. S2 DLS Analysis .............................................................................................................................. S44 TEM .......................................................................................................................................... S47 XPS ............................................................................................................................................ S48 Biological assays ....................................................................................................................... S50 Cytotoxicity studies .................................................................................................................. S51
Synthesis of giant globular multivalent glycofullerenes as potent inhibitors in a model of Ebola virus infection
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 2
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S2
Synthesis and Characterization General Reagents and solvents were purchased as reagent grade and used without further purification. Compounds 4,1 6,2 8,3 10,4 11,5 13a,6 13b,7 and 168 were prepared according to previously reported procedures. All reactions were performed in standard glassware under an inert Ar atmosphere. Evaporation and concentration were done at water aspirator pressure and drying in vacuum at 10‐2 Torr. Column chromatography: silica gel 60 (230‐400 mesh, 0.040‐0.063 mm) was purchased from E. Merck. Thin Layer Chromatography (TLC) was performed on glass sheets coated with silica gel 60 F254 purchased from E. Merck, visualization by UV light. IR spectra (cm‐1) were measured on a Perkin Elmer Spectrum One. NMR spectra were recorded on a Bruker AC 300, AC 400, AMX‐500 or AMX‐700 equipped with cryoprobe, with solvent peaks as reference. MALDI‐TOF‐mass spectra were carried out on a Bruker BIFLEX
TM or a
Bruker ULTRAFLEX III matrix‐assisted laser desorption time‐of‐flight mass spectrometer. Copper analysis was carried out using a Varian ICP‐MS.
1 J. Iehl, R. Pereira de Freitas, B. Delavaux‐Nicot, J.‐F. Nierengarten, Chem. Commun. 2008, 2450‐2452. 2 J.‐F. Nierengarten, J. Iehl, V. Oerthel, M. Holler, B. M. Illescas, A. Munoz, N. Martin, J. Rojo, M. Sanchez‐Navarro, S. Cecioni, S. Vidal, K. Buffet, M. Durka, S. P. Vincent, Chem. Commun. 2010, 46, 3860‐3862. 3 H. Sekiguchi, K. Muranaka, A. Osada, S. Ichikawa, A. Matsuda, Bioorganic & Medicinal Chemistry 2010, 18, 5732‐5737. 4 F. Wessendorf, J.‐F. Gnichwitz, G. H. Sarova, K. Hager, U. Hartnagel, D. M. Guldi, A. Hirsch J. Am. Chem. Soc. 2007, 129, 16057‐16071. 5 J. Iehl, R. Pereira de Freitas, J.‐F. Nierengarten Tetrahedron Lett. 2008, 49, 4063. 6 a) E. Arce, P. M. Nieto, V. Díaz, R. García‐Castro, A. Bernad, J. Rojo, Bioconjug. Chem. 2003, 14, 817; b) Lindhorst, T. K.; Kötter, S.; Krallmann‐Wenzel U.; Ehlers, S. J. Chem. Soc., Perkin Trans. 1 2001, 823. 7 Davis, B.G.; Maughan, M.A.T.; Ullman, A.; Jones, J.B. Tetrahedron: Asymm. 2000, 11, 245. 8 M. Sánchez‐Navarro, A. Muñoz, B. M. Illescas, J. Rojo, N. Martín Chem. Eur. J. 2011, 17, 766‐769.
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 3
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S3
Compound 2
Ethylmalonyl chloride (1.70 mL, 13 mmol) was added dropwise to a solution of 1 (1.7 mL, 20 mmol) and pyridine (1.3 mL, 20 mmol) in dry CH2Cl2 (100 mL) at 0° C under Argon. After 1 h, the mixture was allowed to slowly warm to room temperature (within 1 h), then stirred for 18 h and evaporated. Column chromatography (SiO2, CH2Cl2/cyclohexane, 4:1) gave 2 (3.69 g, 99%). Colorless oil. IR (neat): 2175 (C≡C), 1733 (C=O). 1H NMR (CDCl3, 300 MHz), δ: 4.23 (q, J = 7 Hz, 2H), 4.20 (t, J = 7 Hz, 2H), 3.37 (s, 2H), 2.32 (t, J = 7 Hz, 2H), 1.87 (m, 2H), 1.29 (t, J = 7 Hz, 2H), 0.14 (s, 9H). 13C NMR (CDCl3, 75 MHz), δ: 165.7, 165.6, 105.4, 84.6, 63.2, 60.7, 40.7, 26.7, 16.4, 15.6, ‐0.8.
1H NMR spectrum of compound 2 (CDCl3, 300 MHz, 298 K)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 4
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S4
13C NMR spectrum of compound 2 (CDCl3, 75 MHz, 298 K)
DEPT 135 spectrum of compound 2 (CDCl3, 75 MHz, 298 K)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 5
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S5
Compound 3
DBU (0.6 ml, 3.5 mmol) was added to a stirred solution of C60 (998 mg, 1.4 mmol), 2 (375 mg, 1.4 mmol) and I2 (528 mg, 2.0 mmol) in dry toluene (1 L) under argon. The resulting solution was stirred for 16 h, then filtered through a short plug of SiO2 (CH2Cl2) and evaporated. Column chromatography (SiO2, CH2Cl2/cyclohexane, 3:2) gave 3 (675 mg, 49%). Brown solid. IR (neat): 2176 (C≡C), 1747 (C=O). UV‐Vis (CH2Cl2): 326 (38000), 426 (2800), 486 (1540), 688 (180). 1H NMR (CDCl3, 300 MHz), δ: 4.56 (m, 4H), 2.46 (t, J = 7 Hz, 2H), 2.07 (m, 2H), 1.51 (t, J = 7 Hz, 3H), 0.15 (s, 9H). 13C NMR (CDCl3, 75 MHz), δ: 162.65, 162.6, 145.4, 144.3, 144.25, 144.2, 143.9, 143.65, 143.6, 143.55, 143.0, 142.95, 142.2, 142.1, 142.0, 141.0, 140.9, 140.05, 140.0, 138.2, 137.9, 104.2, 85.1, 70.6, 64.8, 62.1, 51.1, 26.7, 15.7, 13.4, ‐0.7. MALDI‐MS: 988.5 ([M]+, calcd for C73H20O4Si: 988.11).
1H NMR spectrum of compound 3 (CDCl3, 300 MHz, 298 K)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 6
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S6
13C NMR spectrum of compound 3 (CDCl3, 75 MHz, 298 K)
DEPT 135 spectrum of compound 3 (CDCl3, 75 MHz, 298 K)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 7
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S7
MS (MALDI‐TOF) of compound 3
IR spectrum of compound 3
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 8
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S8
Compound 5
OO
OO
OO
OO
O
O O
OO
O OO
OO
O
OO
OO O
SiMe3N3N3
N3
N3
N3
N3
N3 N3
N3
N3
5 DBU (0.5 ml, 3 mmol) was added to a stirred solution of 3 (140 mg, 0.14 mmol), CBr4 (4.7 g, 14
mmol) and 4 (459 mg, 1.7 mmol) in ODCB (30 mL) at room temperature. The resulting solution
was stirred for 72 h, then filtered through a short plug of SiO2 (CH2Cl2) and evaporated. Column
chromatography (SiO2, cyclohexane/AcOEt, 2:1) gave 5 (220 mg, 67%). Caution: owing to its
high number of azide residues, this compound must be handled with special care. Upon
evaporation, compound 5 has never been dried under high vacuum and the use of metallic
spatula avoided. Furthermore, this compound has been always prepared on a small scale.
Orange glassy product. IR (neat): 2176 (C≡C), 2092 (N3), 1739 (C=O). 1H NMR (CDCl3, 300 MHz),
δ: 4.35 (m, 24H), 3.41 (m, 20H), 2.34 (t, J = 7 Hz, 2H), 1.95 (m, 22H), 1.34 (t, J = 7.0 Hz, 3H), 0.15
(s, 9H). 13C NMR (CDCl3, 75 MHz), δ: 163.4, 145.7, 141.2, 105.185.7, 69.0, 65.5, 63.8, 63.1, 47.8,
45.3, 27.9, 27.4, 16.4, 14.0, 0.0.
1H NMR spectrum of compound 5 (CDCl3, 300 MHz, 298 K)
© 2015 Macmillan Publishers Limited. All rights reserved
NATURE CHEMISTRY | www.nature.com/naturechemistry 9
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S9
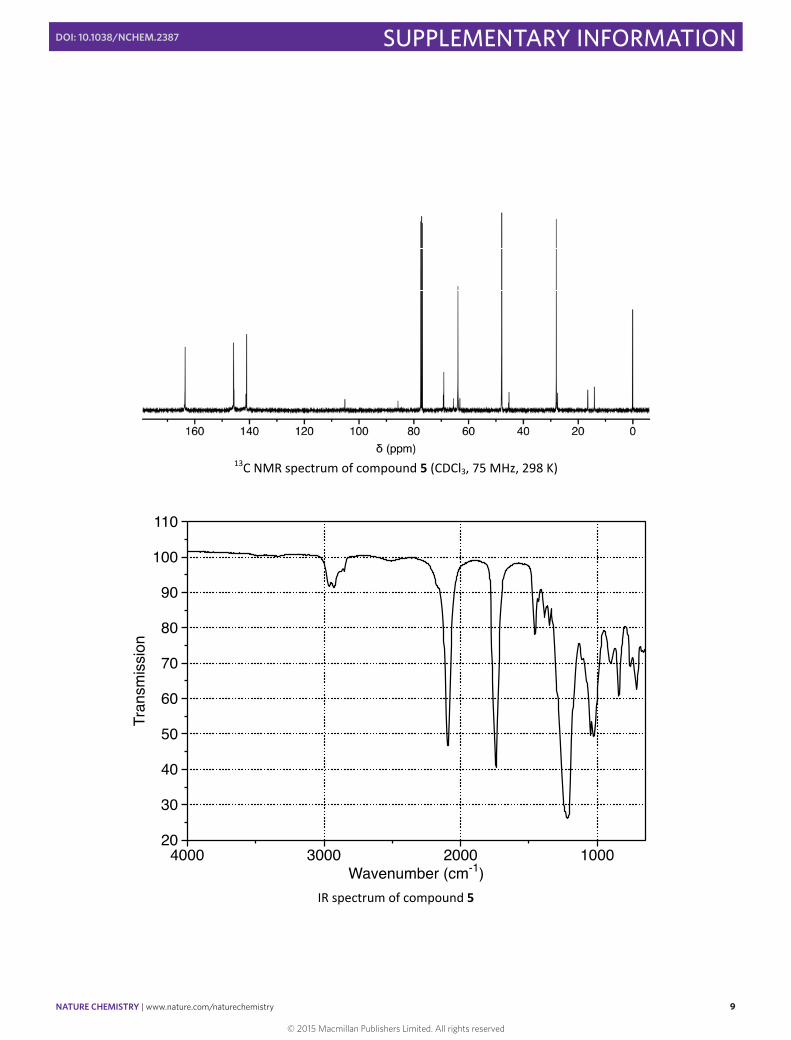
13C NMR spectrum of compound 5 (CDCl3, 75 MHz, 298 K)
IR spectrum of compound 5
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 10
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S10
Compound 7
A mixture of CuSO4.5H2O (15 mg, 94 mol), sodium ascorbate (30 mg, 152 mol), 5 (367 mg, 157 mol) and 6 (412 mg, 1.89 mmol) in THF/H2O (7 mL, 1:1) was heated 2 h under microwave irradiation (100°C). The product was precipitated by addition of MeOH (20 mL). The precipitate was washed with MeOH and dried under reduced pressure. Gel permeation chromatography (Sephadex G‐50, H2O/MeOH; 90:10) gave 7 (647 mg, 91%). Orange powder. IR (neat): 3308 (OH), 1738 (C=O). 1H NMR (400 MHz, D2O), δ: 8.05 (m, 10H), 4.80 (m), 4.43 (m), 3.75 (m), 2.25 (m), 1.16 (m), 0.15 (s, 9H, TMS). 13C NMR (100 MHz, D2O), δ: 163.8 (broad), 145.6 (broad), 141.2 (broad), 125.1 (broad), 99.6, 73.0, 70.7, 70.1, 66.7, 64.6, 60.9, 58.3, 47.6, 45.6, 38.9, 31.8, 28.7, 13.7, 0.1. MALDI‐MS: 4465.6 (8%, M‐TMS+Na+, calcd for C205H216O84N30Na: 4465.3), 4120.5 (8%, M‐1/2 malonate+Na+, calcd for C192H197O76N27Na: 4119.2), 3759.4 (34%, M‐1 malonate+Na+, calcd for C178H177O68N24Na: 3761.1), 3414.2 (26%, M‐1 malonate‐1/2malonate+Na+, calcd for C165H157O60N21Na: 3415.0), 3054.2 (36%, M‐2 malonates+Na+, calcd for C151H136O52N18Na: 3056.8), 2708.9 (21%, M‐2 malonates‐1/2malonate+Na+, calcd for C138H117O44N15Na: 2710.7), 2349.7 (17%, M‐TMS‐3 malonates+Na+, calcd for C124H97O36N12Na: 2352.6), 2003.6 (8%, M‐3 malonates‐1/2malonate+Na+, calcd for C111H77O28N9Na: 2006.5), 1644.4 (6%, M‐4 malonates+Na+, calcd for C97H57O20N6Na: 1648.3).
© 2015 Macmillan Publishers Limited. All rights reserved
NATURE CHEMISTRY | www.nature.com/naturechemistry 11
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S11
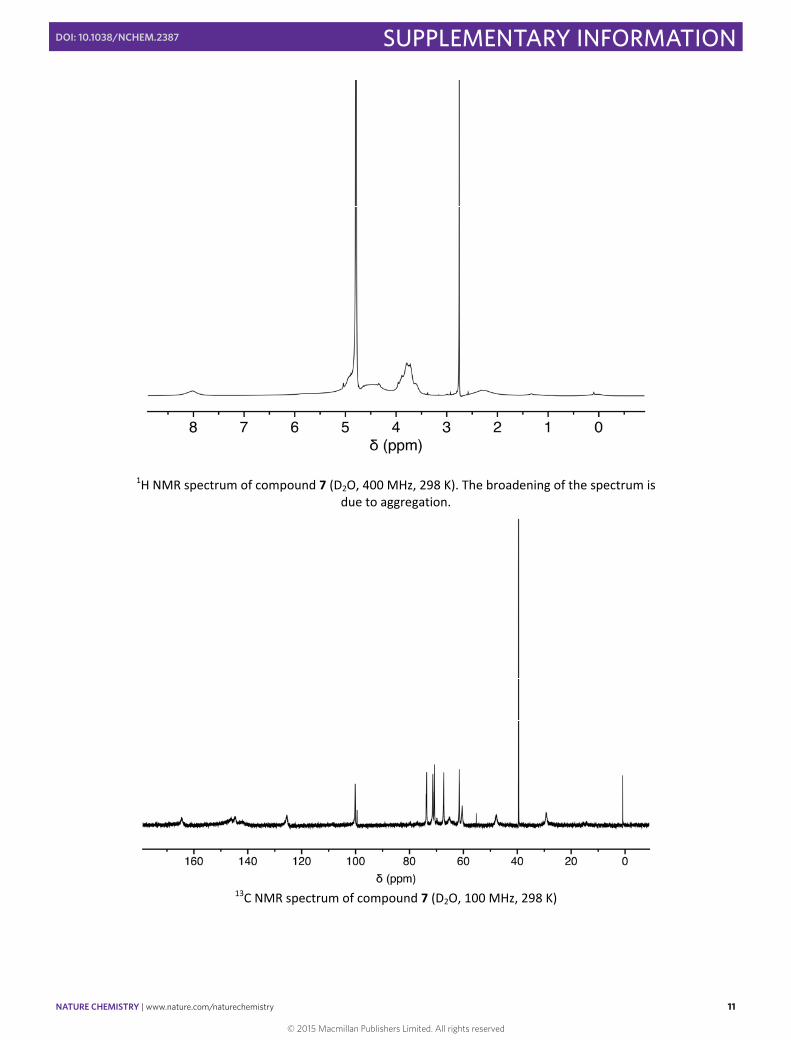
1H NMR spectrum of compound 7 (D2O, 400 MHz, 298 K). The broadening of the spectrum is due to aggregation.
13C NMR spectrum of compound 7 (D2O, 100 MHz, 298 K)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 12
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S12
DEPT 135 spectrum of compound 7 (D2O, 100 MHz, 298 K)
MS (MALDI‐TOF) of compound 7. A series of typical fragments resulting from successive retro‐Bingel reactions are observed. Further fragments resulting from the cleavage of a malonic ester unit followed by a decarboxylation are also observed.
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 13
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S13
IR spectrum of compound 7
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 14
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S14
Compound 9
A 1 M solution of TBAF in THF (0.1 mL, 1 mmol) was added to a mixture of CuSO4.5H2O (3 mg, 19 mol), sodium ascorbate (12 mg, 60 mol), 7 (273 mg, 60 mol) and 8 (481 mg, 1.14 mmol) in THF/H2O (3.5 mL, 0.5:3). The resulting mixture was heated for 1.5 h at 80°C under microwave irradiation. THF and the major part of water were evaporated. Then the crude product was precipitated by addition of a mixture of acetone/CH2Cl2 and extensively washed with CH2Cl2 and acetone. This procedure was repeated three times. A gel permeation chromatography (Sephadex G‐50, H2O/MeOH; 90:10) gave 9 as a brown‐orange glassy solid (254 mg, 87 %). IR (neat) 3324 (OH), 2107 (N3), 1738 (C=O) cm‐1; 1H NMR (400 MHz, D2O), : 8.05 (m, 11H; triazole), 4.80(m), 4.43 (m), 3.66 (m), 2.25 (m), 1.24 (m). 13C NMR (100 MHz, D2O), δ: 163.6 (broad), 146.6 (broad), 143.8 (broad), 125.0 (broad), 99.5, 97.8 73.5, 73.0, 71.5, 70.7, 70.2, 70.1, 69.7, 69.3 66.7, 65.6, 64.2, 60.9, 59.8, 50.3, 49.0, 47.2, 39.2, 28.8, 31.8, 28.7, 21.2, 13.6.
1H NMR spectrum of compound 9 (D2O, 400 MHz, 298 K)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 15
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S15
13C NMR spectrum of compound 9 (D2O, 100 MHz, 298 K)
DEPT 135 spectrum of compound 9 (D2O, 100 MHz, 298 K)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 16
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S16
IR spectrum of compound 9
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 17
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S17
Compound 12
OO
OO
OO
O
O
O
O O
OO
O OO
OO
O
O
O
OO O
12
Br
DBU (1.02 g, 8 mmol) was added to a solution of 10 (400 mg, 0.4 mmol), 11 (0.94 g, 4 mmol) and CBr4 (7 g, 21.3 mmol) in dry toluene (400 mL) under argon atmosphere. The mixture was kept with stirring and under argon atmosphere for 72 h, after which a Na2S2O3 solution (150 mL) was added. The organic solution was washed with HCl 1M (2 x 150 mL), water (2 x 150 mL) and brine (150 mL), dried (Na2SO4) and evaporated. Column chromatography (SiO2, CH2Cl2/ AcOEt 100:5) yielded 12 (428 mg, 49 %) as a red solid. FTIR (KBr): 3292 (≡CH), 2117 (C≡C) 1742 (C=O) cm‐1; 1H NMR (300 MHz, CDCl3), δ: 4.55 – 4.10 (m, 24H), 3.44 – 3.23 (m, 2H), 2.39 – 2.14 (m, 19H), 2.05 – 1.69 (m, 30H), 1.48 – 1.15 (m, 12H). 13C NMR (75 MHz, CDCl3), δ: 163.8, 146.1, 140.8, 82.2, 70.1, 69.1, 65.6, 63.0, 33.7, 32.50, 28.4, 27.2, 24.8, 17.1, 16.5, 15.1, 14.2. MS (MALDI‐TOF) calculated for [M]+ C136H87O24Br= 2185.04; found: 2184.5.
1H NMR spectrum of 12 (300 MHz, CDCl3, 298 K)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 18
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S18
13C NMR spectrum of 12 (75 MHz, CDCl3, 298 K)
MS (MALDI‐TOF) of compound 12
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 19
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S19
IR spectrum of 12
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 20
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S20
Compound 14a
O
HO
HO O
HO
HO
OO
OO
OO
OO
O
O O
OO
O OO
OO
O
OO
OO O
O
HO
HO
OHOHO
OHO
OH
O
HO OH OHO
OH
O
HOOH
O
OH
HO
O
HO
HO
O
OHHO
O
OH
HO
OOH
HO
O
OHHO
O OH
HO
O
OHHO
O
OH
OH
O OH
HO
NNN
NNN
NNN
NNN
NNN
NNN
NN N
NN N
N NN
N NN
Br
O
OH
OHO
OH
OH
14a
A mixture of 12 (35 mg, 0.016 mmol), 13a (73 mg, 0.29 mmol), CuBr∙S(CH3)2 (16 mg, 0.08 mmol), sodium ascorbate (24 mg, 0.12 mmol) and a piece of copper metal wire in DMSO (2 mL) was deoxygenated and kept under argon with vigorous stirring for 48 h. The crude reaction was filtered through a QuadraSilTM Mercaptopropyl column. Et2O was added to the mixture and the precipitate was centrifuged and extensively washed with AcOEt, then dried under high vacuum to give compound 14a (64 mg, 86 %) as a dark red solid. FTIR (KBr): 3386 (OH), 1741 (C=O) cm‐1; 1H NMR (700 MHz, DMSO‐d6), δ: 7.84 (s, 10H), 4.61 (m, 12H), , 4.52 – 4.26 (m, 44H), 3.93 (s, 8H), 3.77 (s, 8H), 3.58 (m, 8H), 3.53 – 3.12 (m, 12H), 3.03 – 2.98 (m, 96H), 2.06 (s, 24H), 1.41 – 1.05 (m, 32H); 13C NMR (175 MHz, DMSO‐d6), δ: 163.3, 146.1, 145.5, 141.2, 122.8, 100.2, 74.5, 71.3, 70.4, 69.2, 67.2, 65.3, 61.6, 55.1, 49.6, 28.2, 21.8, 14.3. MS (MALDI‐TOF) calculated for [M]+ C216H237BrN30O84= 4673,44; found: 4697 [M+Na]+
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 21
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S21
1H NMR spectrum of 14a (700 MHz, DMSO‐d6, 298 K)
13C NMR spectrum of 14a (175 MHz, DMSO‐d6, 298 K)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 22
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S22
DEPT 135 spectrum of 14a (175 MHz, DMSO‐d6, 298 K)
MS (MALDI‐TOF) of compound 14a
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 23
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S23
IR spectrum of 14a
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 24
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S24
Compound 15a
A mixture of compound 14a (140 mg, 0.03 mmol) and sodium azide (8 mg, 0.120 mmol) in DMSO (2 mL) was deoxygenated with argon for 5 min and heated 3 h under microwave irradiation (70°C). AcOEt was added and the precipitate was centrifuged, washed with MeOH and MeOH/AcOEt 3:1, then dried under high vacuum to give compound 15a (117 mg, 84%) as a red solid. FTIR (KBr): 3385 (OH), 2099 (N3), 1740 (C=O) cm‐1; 1H NMR (700 MHz, DMSO‐d6), δ: 7.85 (s, 10H), 4.68 (m, 12H), , 4.59 – 4.09 (m, 44H), 3.91 (s, 8H), 3.76 (s, 8H), 3.61 (m, 8H), 3.53 (s, 8H), 3.46 – 3.25 (m, 96H), 3.12 (m, 4H), 2.04 (m, 24H), 1.36 – 0.98 (m, 32H); 13C NMR (175 MHz, , DMSO‐d6), δ: 163.3, 146.1, 145.8, 141.4, 122.7, 100.2, 78.6, 74.5, 71.2, 70.5, 67.2, 65.3, 61.6, 52.5, 52.0, 49.6, 28.2, 21.8, 14.4; MS (MALDI‐TOF) calculated for [M]+ C216H237N33O84= 4636,53; found: 4659 [M+Na]+.
© 2015 Macmillan Publishers Limited. All rights reserved
NATURE CHEMISTRY | www.nature.com/naturechemistry 25
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S25
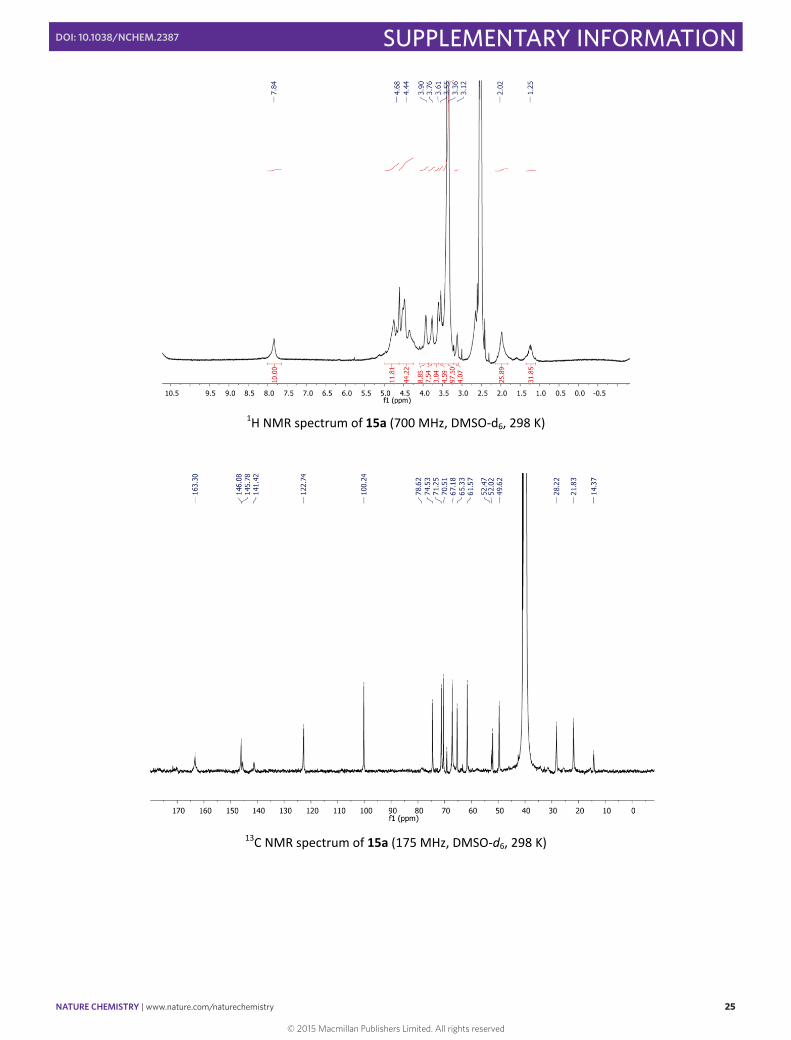
1H NMR spectrum of 15a (700 MHz, DMSO‐d6, 298 K)
13C NMR spectrum of 15a (175 MHz, DMSO‐d6, 298 K)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 26
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S26
DEPT 135 spectrum of 15a (175 MHz, DMSO‐d6, 298 K)
MS (MALDI‐TOF) of compound 15a
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 27
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S27
IR spectrum of 15a
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 28
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S28
Compound 14b
A mixture of 12 (35 mg, 0.016 mmol), 13b (72 mg, 0.29 mmol), CuBr∙S(CH3)2 (16 mg, 0.08 mmol), sodium ascorbate (25 mg, 0.12 mmol) and a piece of copper metal wire in DMSO (2 mL) was deoxygenated and kept under argon with vigorous stirring for 48 h. The crude reaction was filtered through a QuadraSilTM Mercaptopropyl column. Et2O was added to the mixture and the precipitate was centrifuged and extensively washed with AcOEt, then dried under high vacuum to give compound 14b (64 mg, 86 %) as a dark red solid. FTIR (KBr): 3422 (OH), 1740 (C=O) cm‐1; 1H NMR (700 MHz, DMSO‐d6), δ: 7.93 (s, 10H), 5.01–4.72 (m, 12H), , 4.64–4.18 (m, 44H), 3.86 (s, 8H), 3.78 (s, 8H), 3.61–3.53 (m, 16H), 3.41–3.18 (m, 96H), 3.09 (m, 4H), 2.10 (s, 24H), 1.40–0.95 (m, 32H); 13C NMR (175 MHz, DMSO‐d6), δ: 177.1, 167.8, 163.1, 147, 146.1, 145, 140.8, 124.7, 123.2, 103.9, 97.8, 82.7, 75.7, 73.9, 70.8, 69.1, 68.8, 68.5, 67.3, 63.5, 61, 60.2, 49.9, 28.1, 21.9, 15.5; MS (MALDI‐TOF) calculated for [M]+ C216H237BrN30O84= 4673.44; found: 4674.
O
HO
HOO
HO OH
OO
OO
OO
OO
O
O O
OO
O OO
OO
O
OO
OO O
O
HO
OHOHO
OH
OHO
OH
O
HO
HO
OHO
OH
O
OH
HO
O
OH
HO
O
HO
OH
O
OHHO
O
HO
OH
OOH
HO
O
HO
OH O OH
HO
O
OH
OH
O
OH
OHO OH
HO
NNN
NNN
NNN
NNN
NNN
NNN
NN N
NN N
N NN
N NN
Br
O
OH
OHO
OHHO
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 29
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S29
1H NMR spectrum of 14b (700 MHz, DMSO‐d6, 298 K)
13C NMR spectrum of 14b (175 MHz, DMSO‐d6, 298 K)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 30
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S30
MS (MALDI‐TOF) of compound 14b
IR spectrum of 14b
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 31
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S31
Compound 15b
A mixture of compound 14b (30 mg, 0.007 mmol) and sodium azide (5 mg, 0.077 mmol) in DMSO (1.5 mL) was deoxygenated with argon for 5 min and heated 3 h under microwave irradiation (70°C). AcOEt was added and the precipitate was centrifuged, washed with MeOH and MeOH/AcOEt 3:1, then dried under high vacuum to give compound 15a (26 mg, 81%) as a red solid. FTIR (KBr): 3123 (OH), 2097 (N3), 1740 (C=O) cm‐1; 1H NMR (700 MHz, DMSO‐d6), δ: 7.83 (s, 10H), 4.84– 4.72(m, 12H), , 4.64 – 4.16 (m, 44H), 3.94 (s, 8H), 3.74 (s, 8H), 3.63–3.52 (m, 16H), 3.41–3.18 (m, 96H), 2.10 (s, 24H), 1.40–0.95 (m, 32H); 13C NMR (175 MHz, DMSO‐d6), δ: 163.4, 146.1, 145.5, 141.2, 123.1, 100.2, 78.2, 77.9, 74.5, 71.3, 70.5, 67.2, 65.1, 61.4, 52.4, 49.4, 28.3, 25.4, 21.9, 14.2; MS (MALDI‐TOF) calculated for [M]+ C216H237N33O84= 4636,53; found: 4638.
15b
O
HO
HOO
HO OH
OO
OO
OO
OO
O
O O
OO
O OO
OO
O
OO
OO O
O
HO
OHOHO
OH
OHO
OH
O
HO
HO
OHO
OH
O
OH
HO
O
OH
HO
O
HO
OH
O
OHHO
O
HO
OH
OOH
HO
O
HO
OH O OH
HO
O
OH
OH
O
OH
OHO OH
HO
NNN
NNN
NNN
NNN
NNN
NNN
NN N
NN N
N NN
N NN
N3
O
OH
OHO
OHHO
© 2015 Macmillan Publishers Limited. All rights reserved
NATURE CHEMISTRY | www.nature.com/naturechemistry 32
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S32
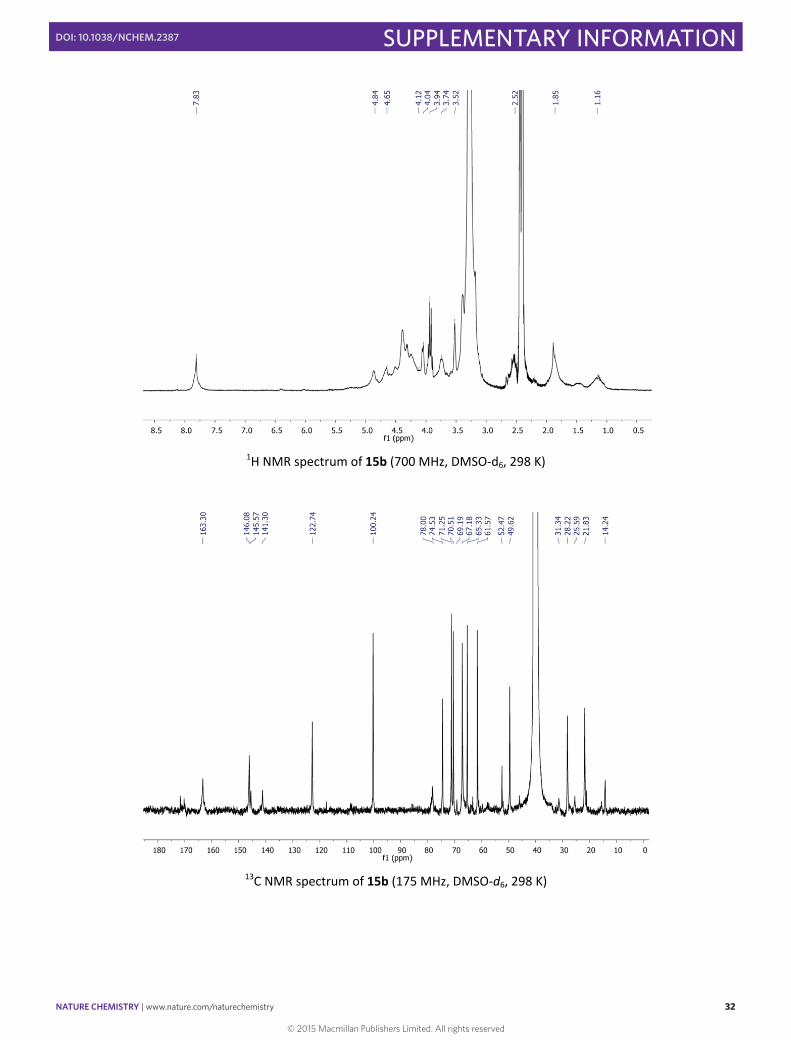
1H NMR spectrum of 15b (700 MHz, DMSO‐d6, 298 K)
13C NMR spectrum of 15b (175 MHz, DMSO‐d6, 298 K)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 33
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S33
MS (MALDI‐TOF) of compound 15b
IR spectrum of 15b
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 34
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S34
Compound 17a
A mixture of hexakis‐adduct 16 (1.5 mg, 6∙10‐4 mmol), compound 15a (68 mg, 0.014 mmol), CuBr∙S(CH3)2 (20 mg, 0.08 mmol), sodium ascorbate (25 mg, 0.12 mmol) and a piece of copper metal wire in DMSO (2 mL) was deoxygenated and kept under argon with vigorous stirring for 48 h. The crude reaction was filtered through a QuadraSilTM Mercaptopropyl column. Et2O was added to the mixture and the precipitate was centrifuged and extensively washed with AcOEt, then dried under high vacuum to give compound 17a (25 mg, 73%) as a red solid. FTIR (KBr): 3419 (OH), 1740 (C=O) cm‐1; 1H NMR (700 MHz, DMSO‐d6), δ: 7.82 (s), 4.76 (m), 4.61 (m), 4.50 (m), 4.35 (m), 3.93 (s), 3.73 (m), 3.60 (m), 3.57 – 3.50 (m), 3.12 (m), 2.80 – 2.59 (m), 2.03 (m), 1.30 (m); 13C NMR (175 MHz, DMSO‐d6), δ: 163.8, 146.9, 146.4, 145.4, 141.6, 123.3, 122.9, 100.2, 78.7, 74.7, 73.4, 73, 70.2, 69.3, 68.6, 67.6, 65.6, 62.3, 61.9, 60.5, 49.8, 45.2, 42.2, 34.2, 28.2, 26.1, 22.1, 14.2. XPS: % atomic: C (284.6 eV) = 73.84, O (531.6 eV) = 20.84, N (399.63 eV) = 5.33.
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 35
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S35
1H NMR spectrum of 17a (700 MHz, DMSO‐d6, 298 K)
13C NMR spectrum of 17a (175 MHz, DMSO‐d6, 298 K)
© 2015 Macmillan Publishers Limited. All rights reserved
NATURE CHEMISTRY | www.nature.com/naturechemistry 36
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S36
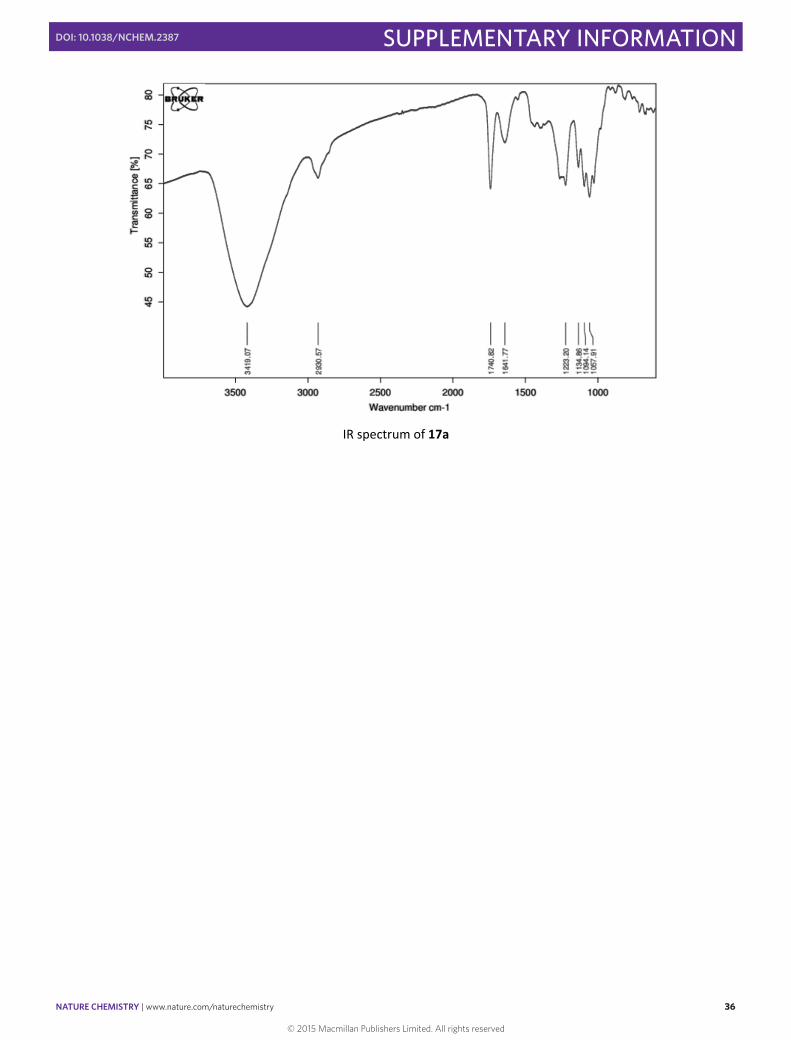
IR spectrum of 17a
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 37
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S37
Compound 17b
A mixture of hexakis‐adduct 16 (1.3 mg, 6∙10‐4 mmol), compound 15b (70 mg, 0.015 mmol), CuBr∙S(CH3)2 (18 mg, 0.08 mmol), sodium ascorbate (25 mg, 0.12 mmol) and a piece of copper metal wire in DMSO (2 mL) was deoxygenated and kept under argon with vigorous stirring for 48 h. The crude reaction was filtered through a QuadraSilTM Mercaptopropyl column. Et2O was added to the mixture and the precipitate was centrifuged and extensively washed with AcOEt, then dried under high vacuum to give compound 17b (27 mg, 79%) as a red solid. FTIR (KBr): 3423 (OH), 1738 (C=O) cm‐1; 1H NMR (700 MHz, DMSO‐d6), δ: 7.93 (s), 4.98 – 4.79 (m), 4.60 – 4.50 (m), 4.35 (m), 3.93 – 3.87 (m), 3.73 – 3.69 (m), 3.63–3.54 (m), 3.12–2.94 (m), 2.79–2.63 (m), 2.12 – 1.98 (m), 1.45 – 1.21 (m); 13C NMR (175 MHz, DMSO‐d6), δ: 163.8, 147.7, 146.4, 146.0, 141.5, 124.4, 123.0, 100.5, 74.8, 73.7, 71.7, 70.7, 69.6, 67.6, 65.6, 61.8, 61.1, 49.8, 46.3, 42.9, 33.3, 28.5, 25.7, 22.3, 14.5.
OO
OO
OO
OO
O
O O
OO
O OO
OO
O
OO
OO O
O O
RORO
RO
RO
OR
OR
ORO
O
OO
O
OOR
O
ORO
OO
O
ORO
X
NNN
OO
OROR
OR
OR
RO
RO
ROO
O
OO
O
ORO
O
ROO
OO
O
RO O
X
N NN
O
O
OR ORRO
RO
OROR
RO
O
O
O O
O
O OR
OOR
OOO
O
RO
OX
NNN
OO
OR
OR
OROR
RORO
RO
O
O
OO
O
O
ORO OR
O
OO
O
ROO
X
NNN
OO
OR
OR
OROR
RORO
RO
O
O
OO
O
O
ORO OR
O
OO
O
ROO
X
NNN
O
O
OR ORRO
RO
OROR
RO
O
O
O O
O
O OR
OOR
OOO
O
RO
O
XN
NN
OO
OROR
OR
OR
RO
RO
ROO
O
OO
O
ORO
O
ROO
OO
O
RO O
X
N NN
O O
RORO
RO
RO
OR
OR
ORO
O
OO
O
OOR
O
ORO
OO
O
ORO
X
NNN
O
O
ROROOR
OR
RORO
OR
O
O
OO
O
ORO
ORO
OO O
O
OR
O
XNNN
OO
RO
RO
RO RO
OR OR
OR
O
O
OO
O
O
ROORO
O
OO
O
ORO
X
NN N
OO
RO
RO
RO RO
OR OR
OR
O
O
OO
O
O
ROORO
O
OO
O
ORO
X
NN N
O
O
ROROOR
OR
RORO
OR
O
O
OO
O
ORO
ORO
OO O
O
OR
OX
NNN
17b -X- = -(CH2)6-, R = OHO
HO
OH
HO
O NN N
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 38
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S38
1H NMR spectrum of 17b (700 MHz, DMSO‐d6, 298 K)
13C NMR spectrum of 17b (175 MHz, DMSO‐d6, 298 K)
© 2015 Macmillan Publishers Limited. All rights reserved
NATURE CHEMISTRY | www.nature.com/naturechemistry 39
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S39
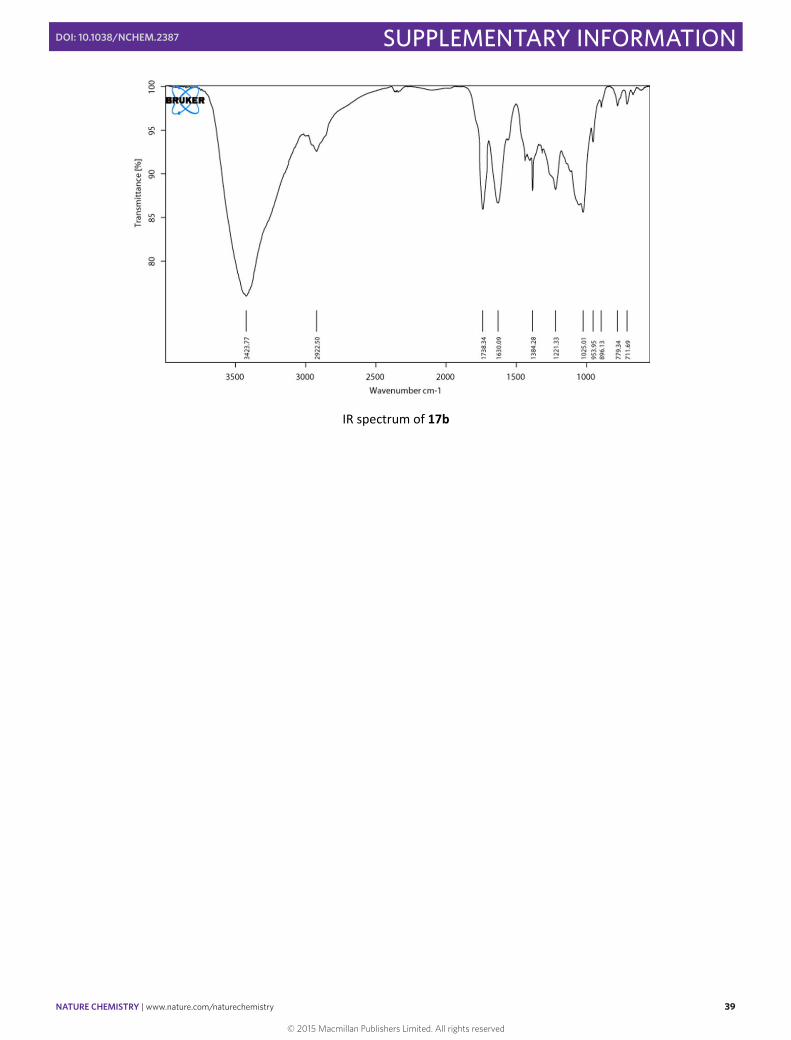
IR spectrum of 17b
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 40
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S40
Compound 17c
A mixture of 9 (254 mg, 52 mol), 16 (10.1 mg, 3 μmol), CuSO4.5H2O (1 mg, 6 mol) and sodium ascorbate (3 mg, 15 mol) in THF/H2O (3.5 mL, 0.5:3) was heated at 80°C under microwave irradiation in a sealed tube. After 2 hours, the mixture was evaporated. Exclusion
column chromatography (Econo‐Pac® 10 DG, BIO‐RAD, H2O) followed by a dialysis against milli‐Q water (Molecular Weigth CutOff: 25000) gave 17c (155 mg, 76 %). Brown‐orange glassy solid. IR (neat) 3251 (OH), 1732 (C=O) cm‐1; 1H NMR (400 MHz, DMSO‐d6), δ: 8.10 (s, 120H), 7.83 (s, 24H), 4.90‐3.18 (m, 1272H), 3.85‐3.28 (m, 744H), 2.74‐2.63 (m, 48H), 2.38‐2.11 (m, 240H), 2.08‐1.91 (m, 48H), 1.25‐1.11 (m, 36H). 13C NMR (100 MHz, DMSO‐D6), δ: 162.9, 145.5, 143.7, 140.6, 124.0, 122.3, 99.0, 74.1, 70.9, 70.2, 69.8, 69.7 (two peaks), 69.6, 69.2, 68.7, 67.0, 64.2, 61.3, 59.1, 50.0, 49.2, 46.1, 44.2, 28.6, 27.7, 21.3, 13.7.
OO
OO
OO
OO
O
O O
OO
O OO
OO
O
OO
OO O
O O
RORO
RO
RO
OR
OR
ORO
O
OO
O
OOR
O
ORO
OO
O
ORO
X
NNN
OO
OROR
OR
OR
RO
RO
ROO
O
OO
O
ORO
O
ROO
OO
O
RO O
X
N NN
O
O
OR ORRO
RO
OROR
RO
O
O
O O
O
O OR
OOR
OOO
O
RO
OX
NNN
OO
OR
OR
OROR
RORO
RO
O
O
OO
O
O
ORO OR
O
OO
O
ROO
X
NNN
OO
OR
OR
OROR
RORO
RO
O
O
OO
O
O
ORO OR
O
OO
O
ROO
X
NNN
O
O
OR ORRO
RO
OROR
RO
O
O
O O
O
O OR
OOR
OOO
O
RO
O
XN
NN
OO
OROR
OR
OR
RO
RO
ROO
O
OO
O
ORO
O
ROO
OO
O
RO O
X
N NN
O O
RORO
RO
RO
OR
OR
ORO
O
OO
O
OOR
O
ORO
OO
O
ORO
X
NNN
O
O
ROROOR
OR
RORO
OR
O
O
OO
O
ORO
ORO
OO O
O
OR
O
XNNN
OO
RO
RO
RO RO
OR OR
OR
O
O
OO
O
O
ROORO
O
OO
O
ORO
X
NN N
OO
RO
RO
RO RO
OR OR
OR
O
O
OO
O
O
ROORO
O
OO
O
ORO
X
NN N
O
O
ROROOR
OR
RORO
OR
O
O
OO
O
ORO
ORO
OO O
O
OR
OX
NNN
17c -X- = R =NNN
O O6 N
N NOHO
HO
HOHO
O
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 41
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S41
1H NMR spectrum of compound 17c (DMSO‐d6, 400 MHz, 298 K)
13C NMR spectrum of compound 17c (DMSO‐d6, 100 MHz, 298 K)
© 2015 Macmillan Publishers Limited. All rights reserved
NATURE CHEMISTRY | www.nature.com/naturechemistry 42
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S42
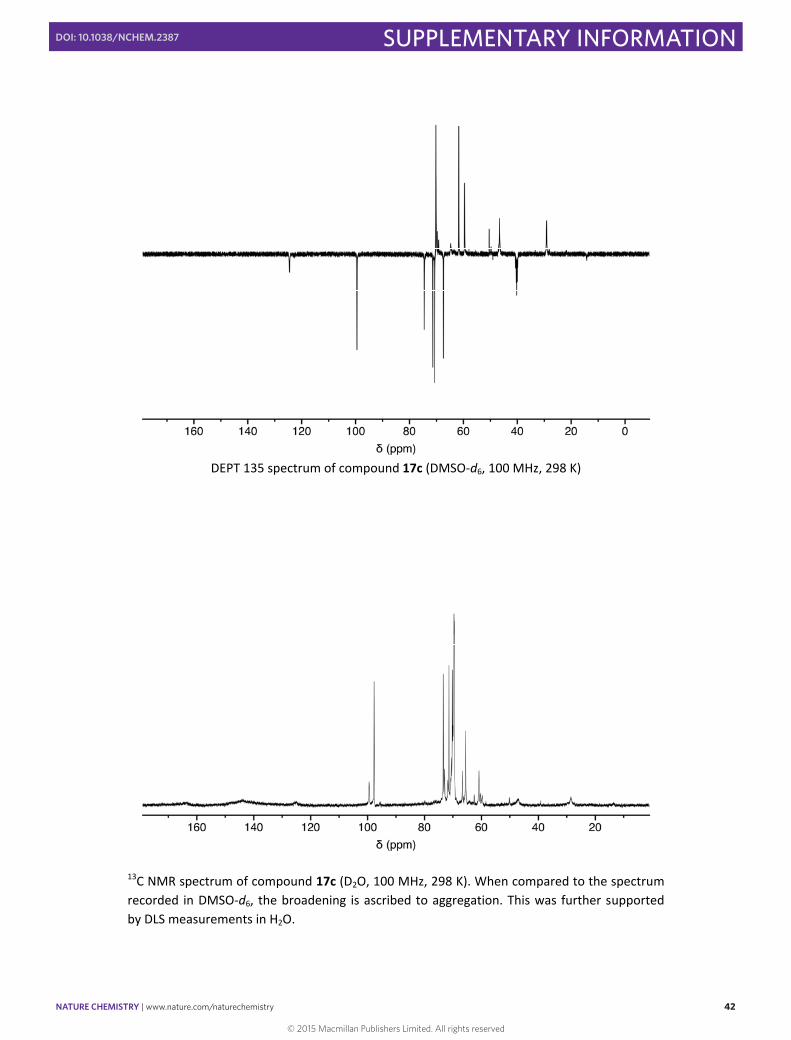
DEPT 135 spectrum of compound 17c (DMSO‐d6, 100 MHz, 298 K)
13C NMR spectrum of compound 17c (D2O, 100 MHz, 298 K). When compared to the spectrum recorded in DMSO‐d6, the broadening is ascribed to aggregation. This was further supported by DLS measurements in H2O.
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 43
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S43
IR spectrum of compound 17c
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 44
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S44
DLS analysis
For compounds 17a‐c, Dynamic Light Scattering measurements were carried out on an ALV GSC08 correlator working in a cross correlation mode with an Ar + laser operating at λ = 514.5 nm. The output signals were obtained with backscatter detection at an angle of 90o and processed with a digital correlator that computed intensity‐intensity autocorrelation of the scattered light. Measurements were made in a 1‐cm path‐length round quartz cell maintained at 298 K. Solution samples of 0.1 mg/mL were filtered through nylon Acrodisc syringe filters (Pall Life Sciences) with 0.2‐μm pore size.
17a, 0.01M 17a, 0.1M
© 2015 Macmillan Publishers Limited. All rights reserved
NATURE CHEMISTRY | www.nature.com/naturechemistry 45
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S45
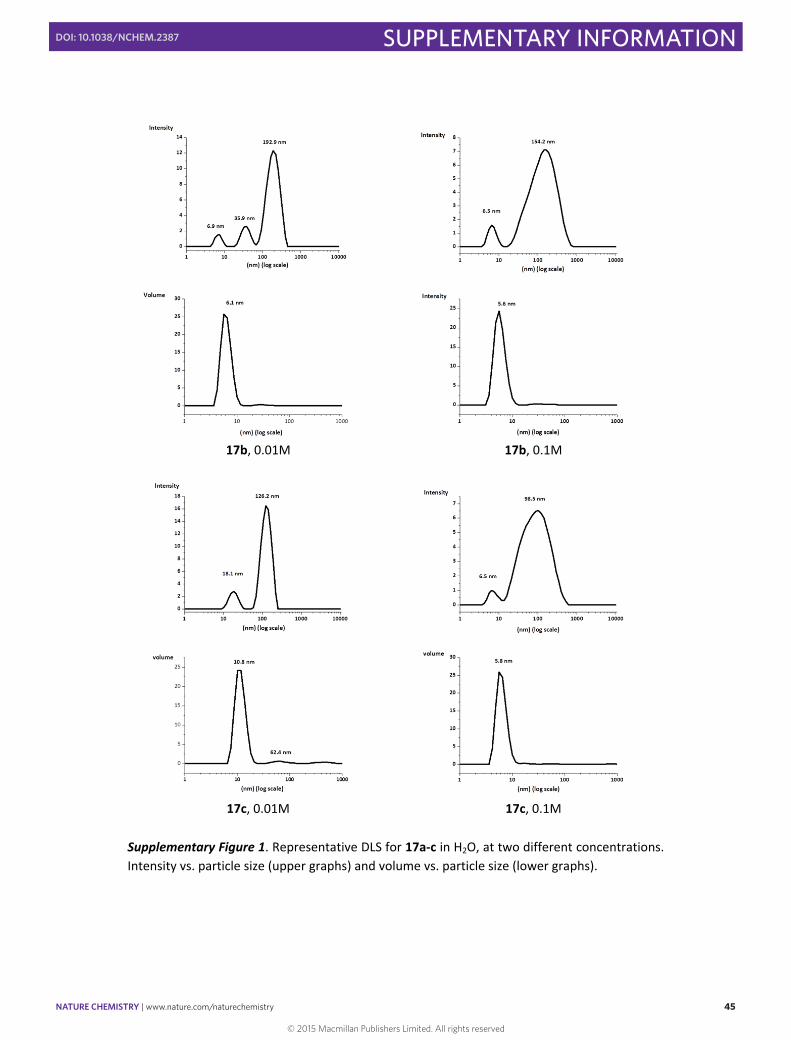
17b, 0.01M 17b, 0.1M
17c, 0.01M 17c, 0.1M
Supplementary Figure 1. Representative DLS for 17a‐c in H2O, at two different concentrations. Intensity vs. particle size (upper graphs) and volume vs. particle size (lower graphs).
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 46
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S46
17a
17b
17c
Supplementary Figure 2. Representative DLS for 17a‐c in DMSO, 0.1 mg/mL. Intensity vs. particle size (left graphs) and volume vs. particle size (right graphs).
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 47
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S47
TEM
For 17a‐c, TEM images were obtained using a JEOL 2100 microscope operating at 200 kV. A solution of 17a‐c in water (0.01 mg/mL or 0.1 mg/mL) was dropped onto a holey carbon copper grid (200 mesh), and the solvent was allowed to evaporate before analysis.
Supplementary Figure 3. TEM images for compounds 17a‐c upon deposition of 0.01 and 0.1 mg/mL solutions in H2O. In all cases, spherical aggregates are observed, ranging from one to several molecules.
nm
160
180
200
220
240
260
280
300
0 2 4 6 8nm
nm
40
60
80
100
120
140
0 1 2 3 4 5 6nm
20 nm20 nm nm
950
1000
1050
1100
1150
1200
1250
0 1 2 3 4nm
nm
600
650
700
750
800
0 1 2 3 4 5 6 7 8nm
20 nm20 nm nm
1000
1100
1200
1300
1400
1500
0 2 4 6 8nm
20 nm20 nm
nm
6200
6400
6600
6800
7000
7200
7400
0 2 4 6 8 10 12nm
17 a ‐ 0.1 g/L
17 b ‐ 0.1 g/L
17 b ‐ 0.01 g/L
17 c ‐ 0.1 g/L
17 c ‐ 0.01 g/L
17 a ‐ 0.01 g/L
© 2015 Macmillan Publishers Limited. All rights reserved
NATURE CHEMISTRY | www.nature.com/naturechemistry 48
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S48
XPS
XPS analyses were obtained on a SPECS GmbH (PHOIBOS 150 9 MCD) spectrometer working in the constant analyzer energy mode and a non‐monochromatic aluminium X‐ray source (1486.61 eV) powered at 200 W and a voltage of 12 eV. For recording both survey and high resolution spectra pass energies of 75 and 25 eV were applied. Survey data were acquired from kinetic energies of 1487 – 400 eV with 0.1 eV of energy step and 100 ms dwell time per point. The high resolution scans were registered around the significant emission lines with 0.1 eV steps and 100 ms dwell time per point. The spectrometer control and data handling were monitored using SpecsLab Version 2.48 software. Binding energies were calibrated relatively to the C 1s peak at 284.6 eV and the atomic ratios were computed from experimental intensity rations. The samples were introduced in the XPS apparatus as powder coated over a polycarbonate membrane.
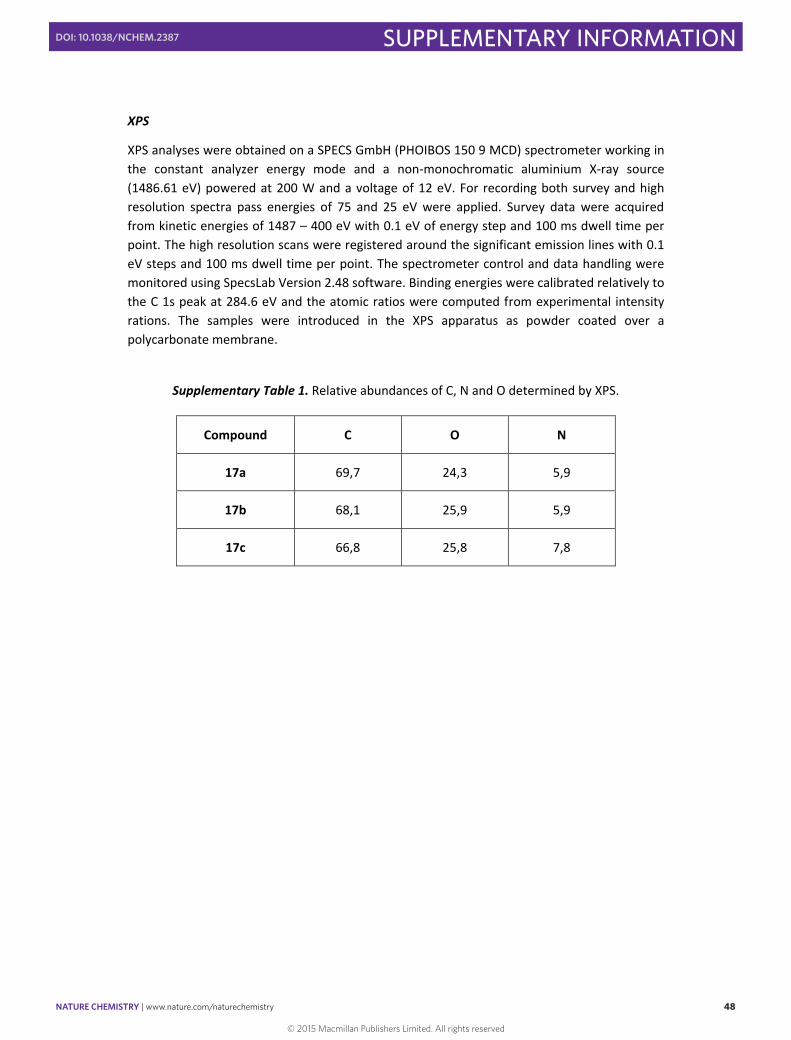
Supplementary Table 1. Relative abundances of C, N and O determined by XPS.
Compound C O N
17a 69,7 24,3 5,9
17b 68,1 25,9 5,9
17c 66,8 25,8 7,8
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 49
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S49
020040060080010000
100000
200000
300000
400000
500000
600000
700000
C 1s
O 1s
Binding Energy (eV)
N 1s400
Binding Energy (eV)
C-NN-N-N
Supplementary Figure 4. XPS survey spectrum of compound 17a (up), 17b (medium) and 17c (down) with the N 1s deconvoluted components (inset right for each compound).
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 50
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S50
Biological assays
C60 (12Man) C60 (36 Man)
OO
O
O
O
O
O
O
O
O O
O
O
OO
O
O
O
O
O
O
OO O
NNN
NNN
N NN
NN
N
NN NN N
N
NNN
NN N
NNN
NNN
NNN NN
N
O O
O
O
O
O
OO
O
O
O
O
OOO
O
O
OHOH
OHOH
N NN
N NNO
N NN
O
O
O
O
OHOH
OHOH
O
OHOH
OHOH
O
O
O
O
O
OHOH
OH
OH
N NN
NNN
O
NN
N
O
O
O
O
OHOH
OH
OH
O
HOOHOH
OH
O
O
OO
O
HOHO OH
OH
NN
NNN
N
O
NNN O
OO
O
HOHO OH
OHO
HOHO OH
OH
O
O
OO
O
HOHO
OH
OH
NNN
NNN
ON
NN
OO
O
O
HOHO
OH
OH
O
HOHO
OHOH
O
O
OO
OHOHO
OHOH
NNN
NNN
ONN
N
O
O
O
OHOHO
OHOH
OHOHO
OH OH
OO
O
O
OHOHO
HO HO
NN
N
NNN
ONNN
O
O
O
O
HOHO
HO HO
O
HOHO
HO HO
OO
O
O
O
HOHO
HOHO NN
N
NNN O
NNN
O
O
O
O
HOHO
HOHO
O
HOHO
HO
HO O
O
O
OO
OHHO
HO
HO
NNN
NNN
O
NNN
O
O
OO
OHHOHO
HO
O
OHHO
HO
HO
O
O
OO
O
OHOHHO
HO
NNN
NNN
O
NN N
O
OO
O
OHOHHO
HO
O
OHOHHO
HO
O
O
O O
O
OHOH
HO
HO
NN N
NN N
ONN N
O O
O
O
OHOH
HO
HO
O
OHOH
HOHO
O
O
OO
O OHOH
HOHO
NN N
NN N
O NNN
O
O
O
O OHOH
HOHO
O OHOH
OHHO
OO
O
O
OOH
OH
OHOH
NNN
N NN
O N NN
O
O
O
O
OHOH
OHOH
O
OHOH
OHOH
OO
O
O
O
O
O
O
O
O
O
O
C60LL (36 Man)
Supplementary Figure 5. Chemical structures of multivalent glycofullerenes studied in inhibition experiments with pseudotyped Ebola virus particles.
© 2015 Macmillan Publishers Limited. All rights reserved
NATURE CHEMISTRY | www.nature.com/naturechemistry 51
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2387
S51
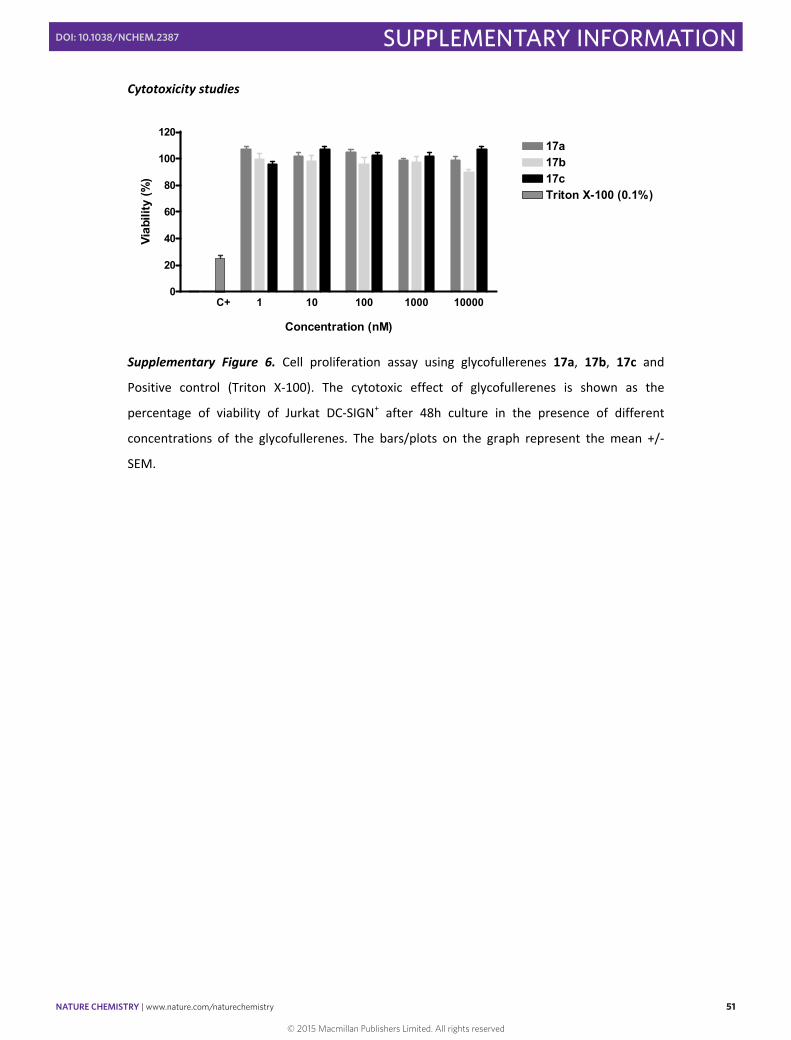
Cytotoxicity studies
1 10 100 1000 100000
20
40
60
80
100
12017a17b17cTriton X-100 (0.1%)
C+
Concentration (nM)
Viab
ility
(%)
Supplementary Figure 6. Cell proliferation assay using glycofullerenes 17a, 17b, 17c and
Positive control (Triton X‐100). The cytotoxic effect of glycofullerenes is shown as the
percentage of viability of Jurkat DC‐SIGN+ after 48h culture in the presence of different
concentrations of the glycofullerenes. The bars/plots on the graph represent the mean +/‐
SEM.
© 2015 Macmillan Publishers Limited. All rights reserved