su.diva-portal.orgsu.diva-portal.org/smash/get/diva2:1153263/fulltext01.pdf · antibiotics that we...
TRANSCRIPT



ANTIBIOTIC UPTAKE INGRAM-NEGATIVE BACTERIA
Claudio Muheim


Antibiotic uptake inGram-negative bacteria
Claudio Muheim

©Claudio Muheim, Stockholm University 2017 ISBN print 978-91-7797-041-5ISBN PDF 978-91-7797-042-2 Printed in Sweden by Universitetsservice US-AB, Stockholm 2017Distributor: Department of Biochemistry and Biophysics, Stockholm University

To the decentralized future.

List of publications
I. YfgM is an Ancillary Subunit of the SecYEG Translocon in Escherichia coli Götzke H, Palombo I, Muheim C, Perrody E, Genevaux P, Kudva R, Müller M, Daley DO. J Biol Chem (2014) 289(27), 19089-19097
II. Identification of Putative Substrates for the Periplasmic
Chaperone YfgM in Escherichia coli Using Quantitative Proteomics Götzke H, Muheim C, Maarten AF, Heck AJ, Maddalo G, Daley DO. Mol Cell Proteomics (2015) 14(1), 216–226
III. Increasing the Permeability of Escherichia coli using
MAC-13243 Muheim C, Götzke H, Eriksson AU, Lindberg S, Lauritsen I, Nørholm MHH, Daley DO. Submitted
IV. Identification of a Fragment-Based Scaffold that Inhibits the
Glycosyltransferase WaaG from Escherichia coli. Muheim C, Bakali A, Engström O, Wieslander Å, Daley DO, Widmalm G.
Antibiotics (2016) 5(1), 10

Author’s contribution to the publications
I. I performed the antibiotic disc diffusion assays and was involved in the interpretation of the results.
II. I cloned the plasmid constructs and performed the acid-stress
assays.
III. I was involved in the design of all experiments. I performed all of the experiments apart from the high-throughput screen. I contributed significantly in writing the final manuscript.
IV. I designed and performed all experiments apart from the in vitro lipid binding assay and the O-Deacylation of LPS. I contributed significantly in writing the final manuscript.

Abstract
The increasing emergence and spread of antibiotic-resistant bacteria is a
serious threat to public health. Of particular concern are Gram-negative
bacteria such as Escherichia coli, Acinetobacter baumannii, Klebsiella
pneumoniae or Pseudomonas aeruginosa. Some of these strains are resistant
to a large number of antibiotics and thus our treatment options are rapidly
declining. In addition to the increasing number of antibiotic-resistant
bacteria, a major problem is that many of the antibiotics at our disposal are
ineffective against Gram-negative bacteria. This is partly due to the
properties of the outer membrane (OM) which prevents efficient uptake. The
overarching goal of this thesis was to investigate how the OM of the Gram-
negative bacterium E. coli could be weakened to improve the activity of
antibiotics.
In the first two papers of my thesis (paper I + II), I investigated the
periplasmic chaperone network which consists of the two parallel pathways
SurA and Skp/DegP. This network is essential for the integrity of the OM
and strains lacking either SurA or Skp are defective in the assembly of the
OM, which results in an increased sensitivity towards vancomycin and other
antimicrobials. We identified a novel component of the periplasmic
chaperone network, namely YfgM, and showed that it operates in the same
network as Skp and SurA/DegP. In particular, we demonstrated that deletion
of YfgM in strains with either a surA or skp background further
compromised the integrity of the OM, as evidenced by an increased
sensitivity towards vancomycin.
In the remaining two papers of my thesis (paper III + IV), the goal was to
characterize small molecules that permeabilize the OM and thus could be

used to improve the activity of antibiotics. Towards this goal, we performed
a high-throughput screen and identified an inhibitor of the periplasmic
chaperone LolA, namely MAC-13243, and showed that it can be used to
permeabilize the OM of E. coli (paper III). We further demonstrated that
MAC-13243 can be used to potentiate the activity of antibiotics which are
normally ineffective against E. coli. In the last paper of my thesis (paper IV),
we undertook a more specific approach and wanted to identify an inhibitor
against the glycosyltransferase WaaG. This enzyme is involved in the
synthesis of LPS and genetic inactivation of WaaG results in a defect in the
OM, which leads to an increased sensitivity to various antibiotics. In this
paper, we identified a small molecular fragment (compound L1) and showed
that it can be used to inhibit the activity of WaaG in vitro.
To summarize, this thesis provides novel insights into how the OM of the
Gram-negative bacterium E. coli can be weakened by using small molecules.
We believe that the two identified small molecules represent important first
steps towards the design of more potent inhibitors that could be used in
clinics to enhance the activity of antibiotics.

Contents
List of publications .................................................................................... viii
Author’s contribution to the publications ................................................. ix
Abstract .......................................................................................................... x
Contents ...................................................................................................... xii
Abbreviations ............................................................................................. xiv
Introduction ................................................................................................. 16
1.1 Antibiotics .......................................................................................... 16
1.2 Antibiotic resistance ........................................................................... 18
1.3 The antibiotic crisis ............................................................................ 20
1.3.1 The lack of novel antimicrobials ................................................ 21
1.3.2 The excessive use and misuse of antibiotics ............................... 22
1.3.3 The lack of infection control and surveillance programs ............ 23
1.4 The emerging threat of Gram-negative bacteria ................................. 24
1.5 Structural overview of the Gram-negative cell envelope ................... 25
1.5.1 The outer membrane ................................................................... 26
1.5.2 The periplasmic space ................................................................. 28
1.5.3 The inner membrane ................................................................... 28
1.6 Antibiotic transport across the Gram-negative cell envelope ............. 29
1.6.1 Porin-mediated antibiotic uptake ................................................ 29
1.6.2 Diffusion across the LPS ............................................................ 30
1.6.3 Diffusion across the periplasmic space and IM .......................... 31
1.6.4 Efflux pumps .............................................................................. 33

1.7 Antibiotic adjuvants ........................................................................... 34
1.8 Additional targets for antibiotic adjuvants ......................................... 37
Summary of the papers .............................................................................. 44
Conclusions and future perspectives ......................................................... 57
Populärvetenskaplig sammanfattning....................................................... 61
Acknowledgements ..................................................................................... 62
References .................................................................................................... 64

Abbreviations
A. baumannii Acinetobacter baumannii
ABC ATP-binding cassette
BAM -barrel assembly machinery
CL Cardiolipin
E. coli Escherichia coli
EPIs Efflux pump inhibitors
HEP L-glycero-D-manno-heptose
IM Inner membrane
KDO 3-deoxy-D-manno-oct-2-ulosonic acid
K. pneumoniae Klebsiella pneumoniae
LPS Lipopolysaccharide
MATE Multidrug and toxic compound extrusion
MIC Minimal Inhibitory Concentration
MDR Multidrug resistance
MFS Major facilitator superfamily
NPN N-phenyl-1-naphthylamine
OM Outer membrane
OMP Outer membrane protein
OS Oligosaccharide
PA N Phenylalanine-arginine -naphthylamide
PE Phosphatidylethanolamine
PG Phosphatidylglycerol
P. aeruginosa Pseudomonas aeruginosa
PMBN Polymyxin B nonapeptide
RND Resistance-nodulation-division

SMR Small multidrug resistance
TPR Tetratricopeptide repeat
WT Wild type

16
Introduction
1.1 Antibiotics
Microbes, such as bacteria and fungi synthesize antibiotics and secrete them
into the environment to inhibit growth of competing microbes. Millions of
years of chemical warfare between rivalling species formed an arsenal of
antibiotics that we use today to treat bacterial infections (1, 2). Based on
their antimicrobial activity, antibiotics can be broadly classified into two
groups: bacteriostatic antibiotics arrest the growth of bacteria but do not kill
them whereas bactericidal antibiotics induce cell death (Figure 1). However,
it is often difficult to classify an antibiotic into one of these categories since
antibiotics can have both bacteriostatic and bactericidal activity. Various
factors such as growth conditions, bacterial density, antibiotic concentration
or antibiotic exposure time influence the antimicrobial activity (3, 4). Thus,
antibiotics are also categorized into different classes, based on their
structure, mechanism or spectrum of activity. One of the most prominent
classifications is based on the chemical structure, since antibiotics with
structural similarities typically show similar effects regarding their activity
and toxicity.

17
Figure 1. Antimicrobial effect of bacteriostatic and bactericidal antibiotics.
Based on their antimicrobial effect, antibiotics can either inhibit bacterial growth
(green curve) or induce cell death (red curve). Various factors such as growth
conditions, bacterial density, antibiotic concentration or antibiotic exposure time
influence the antimicrobial activity. Figure adapted from (5). Reprinted with
permission.
Despite approximately 3,000 known antibiotics, most of their cellular targets
have not been characterized yet (6). Interestingly, those antibiotic targets
which have been identified can be assigned to a few cellular processes (7).
These include cellular processes which are essential for the survival of
bacteria such as DNA replication, RNA transcription, protein translation,
cell wall synthesis, the cell membrane and a few other metabolic pathways
(Figure 2).
One of the most successful antibiotic classes are the -lactams. -lactams are
a class of broad-spectrum antibiotics which inhibit cell wall synthesis (8). In
particular, -lactams prevent crosslinking of the peptidoglycan units by
inhibiting peptide bond formation which is catalysed by penicillin-binding
proteins. Many different classes of -lactam antibiotics have been introduced
into the clinics which include the penicillins, the cephalosporins, the
cephamycins, the carbapenems and the monobactams. However, despite
their unarguable success, -lactams and other antibiotics have been

18
challenged by the problem of increasing bacterial resistance. As a
consequence, these antibiotics need to be constantly evolved in order to
evade bacterial resistance.
Figure 2. Antibiotic classes inhibit various cellular processes. Most antibiotic classes
inhibit only a few cellular processes. These are often conserved among bacteria and
include DNA replication, transcription, translation, cell wall synthesis, the cell membrane
and a few other metabolic pathways (not shown).
1.2 Antibiotic resistance
In the traditional sense, antibiotic resistance occurs when a bacterial species
acquires the ability to resist the action of an antibiotic. This process is also
known as acquired resistance and can either happen by mutation of the
bacterial chromosome or by horizontal gene transfer of foreign DNA coding
for antibiotic resistance elements. Horizontal gene transfer is particularly
problematic since it largely contributes to the spread of antibiotic resistance
elements within and even across bacterial species (9). On the other hand,
bacterial species can be naturally resistant to certain antibiotics. These

19
species have the ability to withstand the action of an antibiotic due to its
inherent structural or functional properties. These properties are encoded in
the bacterial genome and ensure that bacteria are intrinsically resistant to
certain antibiotics.
The general mechanisms which confer resistance to antibiotics are depicted
in Figure 3. These include (1) utilizing an alternative metabolic pathway
which bypasses the inhibited one, (2) modification of the antibiotic target
that prevents or reduces binding of the antibiotic, (3) overproduction of the
antibiotic target, (4) enzymatic inactivation or modification of the antibiotic,
(5) active efflux of the antibiotic by efflux pumps and (6) reduced antibiotic
uptake by a decreased permeability of the outer membrane (OM) (10).
Figure 3. Antibiotic resistance mechanisms. Bacterial cells have general mechanisms to
resist the action of an antibiotic. These include (1) bypassing the inhibited pathway, (2)
alteration of the target, (3) amplification of the target, (4) enzymatic inactivation or
modification of the antibiotic, (5) increased antibiotic efflux and (6) reduced antibiotic
uptake. Figure adapted from (10). Reprinted with permission.

20
1.3 The antibiotic crisis
The discovery of the -lactam antibiotic penicillin by the Scottish scientist
Alexander Fleming in 1928 is considered as a key event in modern medicine
(11). After being introduced into the clinics in the 1940s, penicillin saved
thousands of lives. For example, it was used to treat pneumococcal wound
infections that were caused by battlefield injuries, and as a consequence, the
survival rate of injured soldiers significantly improved (12). The great
success of penicillin initiated a golden rush in antibiotic drug discovery.
More than 20 new antibiotic classes were discovered from 1940 to 1962
(Figure 4) (13).
Figure 4. Timeline of antibiotic discovery. Figure adapted from (14).
Reprinted with permission.
Notably, most antibiotics which are currently in clinical use were discovered
during this period. However, despite ongoing efforts, this productive phase
abruptly ended in the 1960s and since then only two new antibiotic classes

21
were clinically approved for human use, namely the oxazolidinones in 2000
and the lipopeptides in 2003 (15). These two classes were discovered
decades ago (Figure 4) and illustrate how long it can take until a novel
antibiotic class enters into the market. It is estimated that we need a further
20 novel antibiotic classes to support modern medicine during the next 50
years (13).
There are several reasons which are believed to be the driving forces of the
antibiotic crisis. As previously described, the number of novel antibiotic
classes entering into the market has been steadily declining over the last few
decades. In addition, the number of multidrug resistant bacteria has been
increasing due to an excessive use of antibiotics. Finally, the global spread
of multidrug resistant bacteria has been increasing due to an enhanced global
connectivity. In the next few sections, I will discuss each of these points and
explain some of the major problems.
1.3.1 The lack of novel antimicrobials
The antibiotic pipeline is drying out and the situation is deteriorating (16).
After the antibiotic gold rush ended in the 1960s, pharmaceutical companies
shifted their focus on analogue development because of a gradually
declining number of novel antibiotic classes. The toxicity risks of analogues
were considered to be more predictable compared to new antibiotic classes
(13). Despite the risk of cross-resistance, analogue development yielded a
number of new antibiotics that kept up with the emergence of bacterial
resistance until the last two decades (13). Since then, however, many
pharmaceutical companies have completely abandoned antibiotic drug
discovery and development programs. It is no longer considered to be an
attractive investment because of regulatory obstacles, increasingly restricted

22
antibiotic use and uncertainties about emerging resistance (17–19).
Additional efforts to develop new platforms for antibiotic drug discovery,
such as high-throughput screening against defined targets or rational drug
design were largely unsuccessful (6, 20). Although there is increasing
evidence that pharmaceutical companies are restarting antibiotic drug
discovery programs, it remains unclear if the antibiotic pipeline can be filled
to its current need (21).
1.3.2 The excessive use and misuse of antibiotics
Antibiotics are among the most commonly purchased drugs (22). There is
clear evidence that antibiotic resistance is a direct consequence of excessive
antibiotic use (23–25). It is estimated that about half of all antibiotics
prescribed to patients are not required or inadequately prescribed (26).
Antibiotic misuse is especially problematic in countries which have no
regulations regarding antibiotic dispensation. In these countries, antibiotics
are sold without prescription and dispensed by people lacking medical
knowledge (27). Another major problem is the use of antibiotics as growth
supplements in livestock (28, 29). In this common practice, a sub-therapeutic
concentration of antibiotic is mixed to the feed to improve the feed
efficiency. Some of these growth promotors belong to antibiotic classes
which are used in human medicine and it has been reported that this can lead
to cross-resistance (30). Another and often neglected problem is the
environmental contamination with antibiotics by pharmaceutical production
facilities in developing countries. Antibiotic waste products end up
unfiltered in natural waters, which causes a selection pressure on bacteria
that eventually leads to the emergence of antibiotic-resistant bacteria (31).

23
1.3.3 The lack of infection control and surveillance
programs
Antibiotic resistance is an international problem and the increase in global
connectivity is partly responsible for the transmission of multidrug resistant
bacteria across borders. Multidrug-resistant bacteria often end up in
healthcare facilities where they spread among patients and healthcare
professionals (32). In addition, there is often a lack of infection control and
surveillance programs in these facilities which further promotes the
transmission of drug-resistant bacteria (33). As a consequence, thousands of
people die every year from hospital-acquired infections (34). Hence, various
antimicrobial stewardship programs have been initiated to address this issue
(35–37). These programs are coordinated at a national and international level
and address points to limit transmission rates within healthcare facilities and
community settings as well as monitoring antibiotic resistance and antibiotic
use.

24
1.4 The emerging threat of Gram-negative bacteria
It is evident that we are entering into a period with a lack of novel antibiotics
to treat bacterial infections. The discovery of novel antibiotics is not keeping
up with the emergence of antibiotic resistant bacteria. Although resistance to
antibiotics is a general problem, the increasing number of infections caused
by multidrug resistant Gram-negative bacteria is alarming (38, 39). Highly
resistant pathogens such as Escherichia coli, Acinetobacter baumannii,
Klebsiella pneumoniae or Pseudomonas aeruginosa are among the most
critical threats (26, 40). These pathogens are of particular concern in both
healthcare facilities and community settings (41). It is no longer uncommon
that some of these strains are resistant to nearly all available antibiotics.
Thus, the number of treatment options rapidly decline and clinicians are
forced to use older drugs, such as the last-resort antibiotic colistin, which has
been associated with neuro- and nephrotoxicity (42). The situation is
alarming and we need new strategies to tackle the problem of Gram-negative
resistance. It is worth mentioning that the unique architecture of the Gram-
negative cell envelope is partly responsible for the problem. A combination
of limited antibiotic uptake across the OM and efflux of antibiotics by
multidrug efflux pumps limit the activity of many antimicrobials. Since the
work covered in this doctoral thesis has aimed to focus on the antibiotic
uptake problem, the following sections will introduce a structural overview
of the Gram-negative cell envelope whilst describing the properties that
make the cell envelope largely impermeable to most antibiotics.

25
1.5 Structural overview of the Gram-negative cell
envelope
The cell envelope is a complex structure which is involved in various
cellular processes, such as protection of the cell and maintenance of cellular
shape (43). It is composed of three essential layers, namely an OM that faces
towards the exterior, a periplasmic space that includes a thin layer of
peptidoglycan and an inner membrane (IM) that separates the periplasm
from the cytosol (Figure 5).
Figure 5. The cell envelope of Gram-negative bacteria. The Gram-negative cell
envelope consists of an OM that provides a barrier to the extracellular environment, a
periplasmic space which includes a thin layer of peptidoglycan and an IM that separates
the periplasmic space from the cytosol. The OM contains lipopolysaccharides (LPS) and
various outer membrane proteins (OMPs), which have a characteristic -barrel structure.
The IM contains integral membrane proteins as well as peripherally attached membrane
proteins. In addition, both membranes contain various lipoproteins which face towards the
periplasmic side. Adapted from (44). Reprinted with permission.
26
1.5.1 The outer membrane
The OM is the outermost barrier, which separates the extracellular
environment from the inner compartment of the bacterial cell. It is an
asymmetric lipid bilayer that consists of two leaflets: the inner leaflet
predominantly contains phospholipids, in particular
phosphatidylethanolamine (PE) and phosphatidylglycerol (PG), as well as a
small fraction of cardiolipin (CL) (45). The outer leaflet faces towards the
exterior and is mainly composed of the glycolipid lipopolysaccharide (LPS)
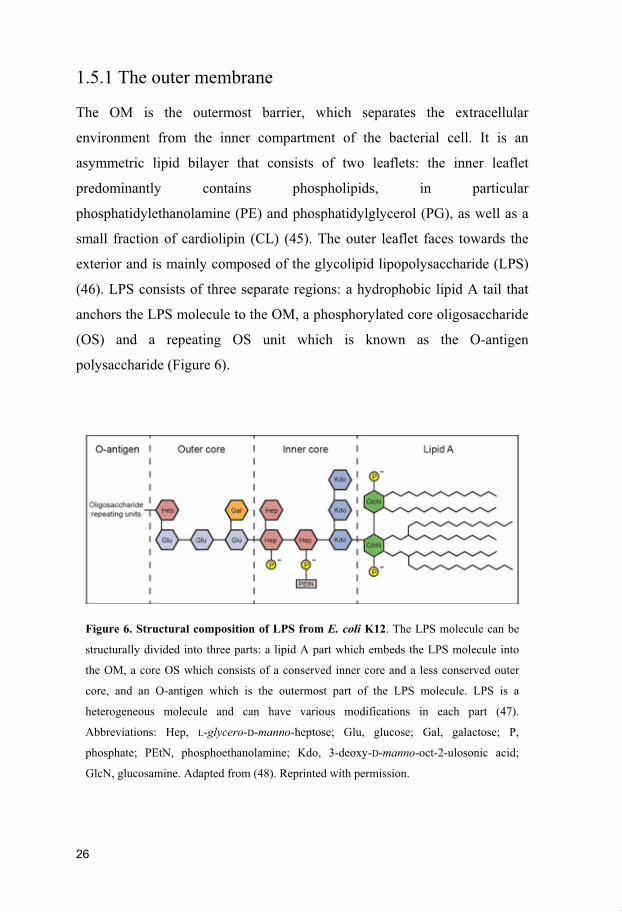
(46). LPS consists of three separate regions: a hydrophobic lipid A tail that
anchors the LPS molecule to the OM, a phosphorylated core oligosaccharide
(OS) and a repeating OS unit which is known as the O-antigen
polysaccharide (Figure 6).
Figure 6. Structural composition of LPS from E. coli K12. The LPS molecule can be
structurally divided into three parts: a lipid A part which embeds the LPS molecule into
the OM, a core OS which consists of a conserved inner core and a less conserved outer
core, and an O-antigen which is the outermost part of the LPS molecule. LPS is a
heterogeneous molecule and can have various modifications in each part (47).
Abbreviations: Hep, L-glycero-D-manno-heptose; Glu, glucose; Gal, galactose; P,
phosphate; PEtN, phosphoethanolamine; Kdo, 3-deoxy-D-manno-oct-2-ulosonic acid;
GlcN, glucosamine. Adapted from (48). Reprinted with permission.

27
The lipid A part (also known as endotoxin) is highly hydrophobic and
activates the adaptive immune response (49). It consists of a phosphorylated
diglucosamine backbone with covalently linked acyl chains that anchor the
LPS molecule into the OM. The structure of lipid A varies among bacterial
species and modification of the lipid A component can affect the endotoxic
activity (47). For example, lipid A modifying enzymes affect the number,
position and length of acyl chains as well as the phosphorylation pattern or
the sugar composition of the disaccharide backbone (50). The core OS is
attached to the lipid A and divided into two parts: the inner core is largely
conserved in Gram-negative bacteria and composed of the sugars 3-deoxy-D-
manno-oct-2-ulosonic acid (Kdo) and L-glycero-D-manno-heptose (Hep).
The outer core is less conserved and composed of different hexose sugars.
Both the inner and outer core can be modified by the addition of phosphates,
pyrophosphates, various phospholipids and amino acids (47). The outermost
part of the LPS molecule is the O-antigen which is linked to the core OS.
The O-antigen is the major antigen targeted by the host immune response
and consists of repeating units of oligosaccharides with high variability (51).
Each of these repeating units contains one to eight glycosyl residues and
varies between sequence, chemical linkage and sugar composition. The OM
is also spanned by outer membrane proteins (OMPs). OMPs have a
characteristic -barrel structure and mediate transport, signalling and other
vital functions (52). Some of these OMPs form water-filled channels which
are also known as porins. Porins regulate the uptake of nutrients, ions and
other molecules such as antibiotics (53, 54). The OM also contains
lipoproteins, which are anchored through an N-terminal lipid moiety to the
inner leaflet of the OM. In E. coli, there are ~ 100 OM lipoproteins which
play important roles in the assembly of the OM (55). The most abundant
lipoprotein in E. coli is Lpp with more than 500,000 copies per cell (43). Lpp
provides structural stability to the cell envelope by covalently linking the
OM with the underlying peptidoglycan layer (Figure 5) (56, 57).

28
1.5.2 The periplasmic space
The periplasmic space is an aqueous, protein-filled environment that lies
between the IM and OM (Figure 5). This compartment contains various
proteins and polysaccharides that regulate cellular processes such as nutrient
uptake, detoxification of harmful compounds or protein transport and quality
control (58). It also includes a thin layer of peptidoglycan that stabilizes the
cell membranes against internal osmotic pressures (43). The peptidoglycan
layer is composed of linear glycan strands that consist of alternating units of
N-acetylglucosamine and N-acetylmuramic acid, which are connected by -
(1,4)-glycosidic bonds (59). The glycan strands are crosslinked by short
peptide linkages which create a net-like polymeric structure.
1.5.3 The inner membrane
The IM separates the periplasm from the cytosol and is composed of two
leaflets that form a symmetric phospholipid bilayer. The phospholipid
composition of the IM is comparable to the inner leaflet of the OM. In E.
coli, the IM consists of 70 – 80 % PE, 15 – 20 % PG and a small fraction of
CL (60, 61). The IM is composed of various proteins that mediate essential
cellular functions such as selective transport, energy metabolism, cell
division, motility, and signalling (62). IM proteins are generally divided into
two classes: integral membrane proteins contain one or more -helical
transmembrane segments that span the phospholipid bilayer. Peripherally
attached membrane proteins do not span the IM but instead adhere to one of
the leaflets via electrostatic, hydrophobic or non-covalent interactions.
Another group of proteins which interacts with the IM are lipoproteins.
Lipoproteins are anchored to the outer leaflet of the IM through their N-
terminal lipid moieties and involved in various processes of the bacterial cell
envelope (63).

29
1.6 Antibiotic transport across the Gram-negative
cell envelope
All antibiotics need to cross one or both membranes of the cell envelope in
order to reach their intracellular target. In addition to the physical barriers
provided by the bacterial membranes, multiple efflux pumps reduce the
intracellular antibiotic concentration. The first and most relevant barrier that
antibiotics encounter is provided by the OM (53, 64). Antibiotics have two
choices to cross this OM: they can either diffuse through water-filled porin
channels or permeate through the LPS-containing leaflet of the OM. In this
section, I will address how the various barriers of the cell envelope limit the
efficient uptake of certain antibiotics in Gram-negative bacteria.
1.6.1 Porin-mediated antibiotic uptake
The uptake of hydrophilic antibiotics < 600 Daltons (herein termed as small-
scaffold antibiotics) is largely regulated by water-filled channels known as
porins (Figure 7) (53, 54, 65). Porins can be classified into different groups
according to their structure, regulation, expression and activity (66). General
diffusion porins form trimers of 16-stranded -barrels and allow the passage
of hydrophilic molecules with limited substrate selectivity (54, 64). In
contrast, substrate-specific porins form trimers of 18-stranded -barrels and
are selective for specific substrate classes such as higher oligosaccharides
(53). Most porins which are involved in antibiotic transport belong either to
the OmpC or OmpF subfamilies (54). These porins represent the main entry
pathway for small, hydrophilic antibiotics such as -lactams or
fluoroquinolones (54, 67). E. coli has three major diffusion porins, namely
OmpC, OmpF and PhoE. OmpC and OmpF porins have a slight preference

30
for cations whereas PhoE prefers inorganic phosphate and anions (53, 65).
Other factors such as molecular shape, number of rotatable bonds and the
presence of an ionisable nitrogen group also affect diffusion across porin
channels (68).
Bacterial cells have different strategies to acquire resistance to antibiotics
which diffuse through porin channels. This can be caused by exchanges in
the porin type, changes in the expression profile of porins or by mutations or
modifications which affect the channel properties of porins (54). For
example, a study showed that clinical isolates of K. pneumonia had a
reduced OM permeability after antibiotic treatment due to a change in the
expression of porins from large channel size OmpF to the smaller channel
size OmpC (69). In addition, some Gram-negative bacteria such as P.
aeruginosa possess a high intrinsic resistance towards many antibiotics (e.g.
-lactams) which is partly caused by the low number of general diffusion
porins (53).
1.6.2 Diffusion across the LPS
Antibiotics larger than 600 Daltons (herein called large-scaffold antibiotics)
do not efficiently cross porin channels (Figure 7). They can permeate
through the LPS-containing OM but their diffusion is often limited. It has
been shown that lipophilic molecules permeate much less efficiently across
the OM bilayer than through a standard phospholipid bilayer (70, 71). In
addition, the LPS-containing bilayer largely restricts the diffusion of
hydrophilic molecules. Hence, the OM is an effective barrier against both
hydrophobic as well as hydrophilic molecules (53, 72).
The impermeability of the OM can be mainly attributed to the LPS layer.
This layer is stabilized by divalent cations, such as Mg2+ and Ca2+, which

31
cross-bridge negatively charged LPS molecules and thus stabilize the lateral
interaction between neighbouring LPS molecules (53). Displacement of the
divalent cations by the chelator EDTA destabilizes the LPS layer (73).
Subsequently, LPS is released into the medium and it is assumed that
phospholipids from the inner leaflet compensate for the loss. As a
consequence, Gram-negative bacteria become sensitized towards
hydrophobic antibiotics including erythromycin, rifampicin and novobiocin
(74). The core region of LPS plays an important role and contributes to the
barrier function (64). Strains expressing full-length LPS (termed smooth
LPS) are intrinsically resistant to hydrophobic and large-scaffold antibiotics.
However, strains expressing truncated LPS (termed rough or deep rough
LPS) are more susceptible (64, 75). Deep rough mutants which have the
most truncated core are also highly susceptible to lipophilic agents such as
detergents, bile salts and some antibiotics (64).
1.6.3 Diffusion across the periplasmic space and IM
Antibiotics that have crossed the OM arrive in the periplasmic space. This
compartment is the site of action for antibiotics such as the -lactams or the
glycopeptide antibiotic vancomycin. Little is known about the diffusion of
antibiotics through this compartment but it is believed that the periplasm
represents no major barrier for antibiotics (76). Those antibiotics destined for
the cytosol need to penetrate the IM. The phospholipid bilayer of the IM is
largely permeable to lipophilic antibiotics (67, 77). Thus, relatively
hydrophobic antibiotics such as the macrolides, lincosamides,
oxazolidinones, pleuromutilins streptogramins A and B, the elfamycins, and
rifamycins appear to cross the IM by simple diffusion (78). In contrast, the
IM is largely impermeable to large, uncharged polar molecules and all

32
charged molecules including ions (79). Those antibiotics which belong to
these categories need to cross the IM by specific uptake systems. For
example, D-cycloserine is transported across the IM via the D-alanine
transport system and coupled to the proton motive force (80). Another
example is fosfomycin which is transported across the IM by using the
glycerol-3-phosphate or the hexose phosphate transporters (81). Transport of
aminoglycosides requires both the electrochemical gradient across the IM
and the electron flow through the respiratory chain (82). Despite the
characterization of a few transport systems, antibiotic transport across the IM
remains poorly characterized. Notably, it has also been suggested that all
hydrophilic antibiotics traverse the IM by solute transport systems (83).
Figure 7. Antibiotic uptake in Gram-negative bacteria. Small-scaffold antibiotics (<
600 Da) generally permeate the OM by passively diffusing through non-specific OMPs.
In contrast, large-scaffold antibiotics (> 600 Da) diffuse through the LPS-containing OM
to gain access to the cell interior. However, their diffusion is limited and thus their uptake
inefficient. Both large and small scaffold antibiotics can either diffuse across the IM or
they can be inadvertently taken up by membrane-embedded transporters.

33
1.6.4 Efflux pumps
Efflux pumps are conserved in nearly all bacterial species and the genes
encoding for efflux pumps are either located on the bacterial chromosome or
on transmissible elements such as plasmids (84). Bacterial efflux pumps can
be classified into several families based on the number of components, the
number of transmembrane-spanning regions and the energy source they use
to transport their substrate (85). Efflux pumps can be specific for a substrate
or they can expel a broad range of structurally diverse compounds (86).
There are five families of efflux pumps which are associated with multidrug
resistance (MDR): these include the ATP-binding cassette (ABC)
superfamiliy, the major facilitator superfamily (MFS), the multidrug and
toxic compound extrusion (MATE) family, the small multidrug resistance
(SMR) family and the resistance-nodulation-division (RND) family (85).
The ABC family transporters use the energy of ATP-hydrolysis for drug
export whereas the others are antiporters and dependent on the H+ (or Na+)
proton gradient (87). The major relevant efflux pumps in Gram-negative
bacteria belong to the RND-type family which is often associated with
multidrug resistance in clinical isolates (67). Efflux pumps of the RND-type
family are organised as tripartite systems which are composed of an IM
transporter, a periplasmic adapter protein and an OM protein. RND-type
efflux systems are polyspecific and expel a broad range of substrates (88).
Two of the best studied RND-type efflux pumps in Gram-negative bacteria
are the AcrAB-TolC system from E. coli and the MexAB-OprM system
from P. aeruginosa. The AcrAB-TolC efflux pump from E. coli is
constitutively expressed and has a broad substrate profile which includes
antimicrobials such as fluoroquinolones, lipophilic -lactams,
chloramphenicol, rifampicin, novobiocin, tetracycline and fusidic acid (85,
89, 90). Several Acr efflux pumps have been chraracterized in E. coli (91–
93). However, AcrAB-TolC has been found to be the most relevant one in
clinical isolates (94–96). In P. aeruginosa, several RND-type efflux pumps

34
have been characterized among which MexAB-OprM is the major multidrug
efflux system (97). MexAB-OprM is consitutively expressed in wild type
(WT) P. aeruginosa and involved in the transport of various antimicrobials
such as fluoroquinolones, -lactams, macrolides, tetracyclines, trimethoprim,
sulfamides and chloramphenicol (98). MexAB-OprM contributes
significantly to the high intrinsic resistance and overexpression of it has been
associated with MDR in clinical isolates of P. aeruginosa (99).
1.7 Antibiotic adjuvants
As previously stated, the development of novel antibiotics is not sufficient to
keep up with the increasing number of antibiotic resistant bacteria. Thus,
there is a pressing need to explore alternative strategies to tackle the issue.
One alternative strategy which has received increased attention over the past
few years is the development of adjuvants to potentiate the activity of
existing antibiotics (100–102). Antibiotic adjuvants are compounds which
normally have little or no intrinsic antimicrobial activity. However, when
used in combination with an antibiotic, adjuvants enhance the activity of the
antibiotic. Antibiotic adjuvants can have different cellular targets but they
act by either reversing acquired resistance or sensitizing strains that are
intrinsically resistant (100). Most adjuvants that have been identified
potentiate antibiotics by either inhibiting antibiotic resistance elements (e.g.
inactivation of -lactamases), blocking bacterial efflux pumps, increasing the
permeability of the bacterial cell envelope, or by interfering with signal
systems that are involved in antibiotic resistance (102). In this section, I
would like to focus on a few antibiotic adjuvants which belong to one of
these categories and have been shown to potentiate antibiotics in Gram-
negative bacteria.

35
-lactamase inhibitors are clinically used to improve the activity of -lactam
antibiotics. One of the most successful drugs is Augmentin© which is a
combination of clavulanic acid (a -lactamase inhibitor) and amoxicillin (a
-lactam antibiotic). Augmentin© is used to restore or extend the
antimicrobial activity of amoxicillin in -lactamase-producing strains such
as E. coli and K. pneumoniae (103). Despite the clinical use of Augmentin©
for more than 30 years, the emergence of resistance in clinical isolates has
been very low (104). Potentiation of antibiotics by inactivation of -
lactamases has been of ongoing interest and a few additional drugs are
currently in development (105).
Efflux Pumps are another target to potentiate the activity of antibiotics.
Since most antibiotics are susceptible to active efflux, the use of efflux pump
inhibitors (EPIs) could make antibiotics more effective by increasing their
intracellular concentration. A number of EPIs have been identified in Gram-
negative bacteria (106, 107). One prominent example is the peptidomimetic
compound phenylalanine-arginine -naphthylamide (PA N), which inhibits
the Mex efflux pumps in P. aeruginosa and the homolog AcrAB-TolC in E.
coli (108). PA N potentiated the activity of the fluoroquinolone antibiotic
levofloxacin in P. aeruginosa (108). In particular, PA N reduced the MIC
(Minimal Inhibitory Concentration) of levofloxacin about 8-fold in WT
strains and up to 64-fold in strains overexpressing efflux pumps. PA N also
decreased the MICs of various antibiotics including chloramphenicol,
nalidixic acid, ofloxaxin, cloxaxillin and erythromycin in K. pneumonia
strains that are either naturally sensitive or inherently resistant against these
antibiotics (109). However, despite its good in vitro activity against
clinically relevant multidrug efflux pumps, PA N and its derivatives did not
enter into clinics due to acute toxicity in pre-clinical trials (110).
Another class of antibiotic adjuvants target the bacterial cell envelope. For
example, the OM permeabilizer polymyxin B nonapeptide (PMBN) has been
shown to potentiate various antibiotics in Gram-negative bacteria (74).

36
PMBN improved the activity of erythromycin and novobiocin in mice
infected with either K. pneumonia or P. aeruginosa (111). A combination of
PMBN with either avibactam, cefazidime or cefazidime-avibactam increased
the antimicrobial activity against clinical isolates of E. coli, K. pneumoniae
and E. aerogenes (112). However, despite good in vitro activity against
Gram-negative bacteria, PMBN has been shown to cause a similar toxicity
profile as polymyxin B (113).
Of recent interest are the findings of Wright and colleagues (114): they
screened a collection of previously approved drugs for potentiators of the
tetracycline antibiotic minocycline against E. coli, P. aeruginosa and S.
aureus. The authors identified several non-antibiotic compounds which had
antibiotic adjuvant properties. For example, the anti-diarrheal medication
loperamide improved the activity of minocycline and other tetracycline
antibiotics in either E. coli or P. aeruginosa. Loperamide dissipates the
electrical component of the proton motive force across the bacterial
membrane of Gram-negative bacteria. As a consequence, loperamide
increases antibiotic influx of tetracycline antibiotics. The combination of
loperamide and minocycline was shown to be highly synergistic in a
Salmonella in vivo model. The same group performed an additional screen to
identify antibiotic adjuvants of the aminocoumarin antibiotic novobiocin,
which is normally ineffective against Gram-negative bacteria (115). They
identified four compounds that were shown to be synergistic with
novobiocin in either E. coli or P. aeruginosa. Two of these compounds,
namely A22 and pivmecillinam alter bacterial cell shape by blocking the
cytoskeleton protein MreB and inhibiting peptidoglycan synthesis,
respectively.

37
1.8 Additional targets for antibiotic adjuvants
The number of antibiotic adjuvants is most likely not sufficient to solve the
antibiotic crisis. However, recent genetic studies have provided novel targets
for antibiotic adjuvants by identifying a large number of genes that
contribute to the intrinsic resistance of Gram-negative bacteria. For example,
a transposon mutant library has been used to identify genes that are
responsible for intrinsic resistance to various antibiotics in Acinetobacter
baylyi (116). Other studies have used transposon mutant libraries of P.
aeruginosa to screen for enhanced sensitivity against tobramycin or
ciprofloxacin (117, 118). In addition, Fajardo et al. screened two different
transposon-tagged insertion libraries of P. aeruginosa for increased
susceptibility to six antimicrobials belonging to different structural families
(119). Similar studies have also been done in E. coli. For example, Tamae et
al. and Liu et al. screened an E. coli knockout collection to look for mutants
which are more susceptible to various antibiotics (75, 120). Taken together,
these genetic studies have identified a large number of additional targets that
could be inhibited by adjuvants to improve the activity of various antibiotics.
In this section, I will focus on the work of Liu et al. who screened an E. coli
knockout collection of close to 4,000 strains, each lacking a different non-
essential gene (75). Their goal was to identify strains that were more
susceptible to a panel of 22 antimicrobials. In doing so, the authors
determined an antibiotic susceptibility profile for each mutant. Out of these
4,000 strains, they identified 283 that showed enhanced sensitivity to at least
one or more of the 22 tested antibiotics. These strains could be classified into
7 different categories based on the cellular function of the deleted gene
(Figure 8).

38
Figure 8. Functional categorisation of the E. coli K12 deletion mutants that are
hypersensitive to antibiotics. 283 E. coli strains showed increased sensitivity to at least
one or more of the 22 antibiotics. The strains were grouped into 7 different categories
based on the cellular function of the deleted gene. These include category 1: DNA
replication, recombination and repair, category 2: transport, efflux, cell wall and cell
membrane synthesis, category 3: protein synthesis, category 4: central metabolic
reactions, category 5: regulation, category 6: prophage-carried genes and cell adhesion,
and category 7: unassigned gene products. Data taken from (75). Reprinted with
permission.
Interestingly, 96 of the 283 strains (34 %) could be classified into the
category which is involved in transport, efflux, cell wall and cell membrane
synthesis (Figure 8), indicating that components of the cell envelope restrict
the action of many antibiotics. Table 1 shows the sensitivity profiles of each
of the 96 strains that belong to this category. Most of these mutants showed
enhanced sensitivity to several antibiotics. Strikingly, some of these mutants
such as the deletion strains acrA or tolC were more susceptible to nearly
all of the tested antibiotics (Table 1). The products of the two genes are
components of the multidrug efflux pump AcrAB-TolC. Namely, AcrA is
the periplasmic adapter protein of AcrAB-TolC whereas TolC represents the
OM efflux channel.

39

40
Table 1. Antibiotic sensitivity profiles of the 96 E. coli K12 knockout strains that are
involved in transport, efflux, cell wall and cell membrane synthesis. Sensitivity
profiles were categorized into three different groups with strong susceptibilities in dark
shades, medium susceptibilities in lighter shades and weak susceptibilities in lightest
shade. CIP, Ciprofloxacin; ENX, Enoxacin; NIT, Nitrofurantoin; MTR, Metronidazole;
SFX, Sulfamethoxazole; RIF, Rifampicin; GEN, Gentamicin; TOB, Tobramycin; NEO,
Neomycin; STR, Streptomycin; SPT, Spectinomycin; TET, Tetracycline; VAN,
Vancomycin; AMP, Ampicillin; RAD, Cephradine; FOX, Cefoxitin; ATM, Azetreonam,
CST; Colistin; CHL, Chloramphenicol; ERY, Erythromycin; FUS, Fusidic acid; TRI,
Triclosan. Large-scaffold antibiotics rifampicin, vancomycin and erythromycin are boxed
in red. Data taken from Liu et al (75). Reprinted with permission.

41
Since one of the goals of my doctoral thesis was to improve the activity of
those antibiotics which do not efficiently cross the OM, I was particularly
interested in the mutants that showed increased susceptibility to the large-
scaffold antibiotics rifampicin, vancomycin and erythromycin (Table 1).
Initially I focused on the glycopeptide antibiotic vancomycin. Vancomycin
is largely ineffective against Gram-negative bacteria because it does not
efficiently penetrate the OM. However, as shown in Table 2, there are 60
non-essential protein targets in E. coli whose inactivation improves the
activity of vancomycin. Notably, some of these mutants have previously
been shown to have increased sensitivity towards vancomycin. For example,
the hypersensitive mutant surA was 125-fold more sensitive to vancomycin
than the corresponding WT strain (Table 2). In addition, the surA mutant
became also sensitized to other large-scaffold antibiotics including
rifampicin and erythromycin (Table 1). SurA is a periplasmic chaperone that
is involved in the folding and transport of periplasmic proteins. Strains
lacking SurA are defective in the assembly of the OM, which results in an
increased sensitivity towards vancomycin and other antimicrobials (121,
122). Other mutants which have been previously described to be more
sensitive towards vancomycin include bamB and smpA ( bamE) (Table
2). They encode for the lipoproteins BamB and BamE which are part of the
BAM ( -barrel assembly machinery) complex. This complex catalyses the
folding and insertion of OMPs and is essential for the integrity of the OM
(123). Mutants lacking either BamB or BamE have a severe defect in the
OM which results in an increased sensitivity to various antibiotics including
vancomycin (124, 125).

42
Gene MIC
( g/ml)
Functional
category
Protein name
BW25113 wt 500 - -
surA 4 2 Peptidyl-prolyl cis-trans isomerase SurA
smpA 70 2 OM protein assembly factor BamE
bamB 100 2 OM protein assembly factor BamB
envC 100 2 Murein hydrolase activator EnvC
lpxL 100 2 Lipid A biosynthesis lauroyl acetyltransferase
tatC 100 2 Twin arginine protein translocation system
tolR 100 2 Colicin transport; Tol-Pal system component
ydcS 100 2 Putative ABC transporter periplasmic-binding protein YdcS
yciM 100 7 Lipopolysaccharide assembly protein B
recA 150 1 DNA strand exchange and recombination protein
envZ 150 2 Osmolarity sensor protein EnvZ
lpxM 150 2 Lipid A biosynthesis myristoyltransferase
nlpC 150 2 Probable endopeptidase NlpC
pal 150 2 Peptidoglycan-associated lipoprotein Pal
proW 150 2 Glycine betaine/L-proline transport system permease protein ProW
rfaC 150 2 Lipopolysaccharide heptosyltransferase I
ybgF 150 2 Periplasmic TolA-binding protein
yhdP 150 2 Uncharacterized protein YhdP
dnaK 150 2 Chaperone protein DnaK
hlpA 150 2 Periplasmic chaperone Skp
hscA 150 2 Iron-sulfur cluster biosynthesis chaperone HscA
hscB 150 2 Co-chaperone for iron-sulfur cluster biosynthesis
rimK 150 3 Ribosomal protein S6 modification protein
rlpA 150 3 Endolytic peptidoglycan transglycosylase RlpA
tufA 150 3 Translation elongation factor Tu 1
yfgC 150 3 -barrel assembly-enhancing protease
aceE 150 4 Pyruvate dehydrogenase E1 component
pgaC 150 4 Poly- -1,6-N-acetyl-D-glucosamine synthase
ycjU 150 4 -phosphoglucomutase
ygcO 150 4 Ferredoxin-like protein YgcO
dksA 150 5 RNA polymerase-binding transcription factor DksA
fur 150 5 Ferric uptake regulation protein
rsmF 150 5 Ribosomal RNA small subunit methyltransferase F
xapR 150 5 HTH-type transcriptional regulator XapR
yciT 150 5 Uncharacterized HTH-type transcriptional regulator YciT
ylcG 150 6 Uncharacterized protein YlcG
ydhT 150 7 Uncharacterized protein YdhT
recB > 150 1 RecBCD enzyme subunit RecB
ftsP > 150 1 Cell division protein FtsP
fepC > 150 2 Ferric enterobactin transport ATP-binding protein FepC
qmcA > 150 2 Protein QmcA
tatB > 150 2 Sec-independent protein translocase protein TatB
tonB > 150 2 Ton complex subunit TonB
yheL > 150 2 Sulfurtransferase complex subunit TusB
elaD > 150 3 Protease ElaD
rpmJ > 150 3 50S ribosomal protein L36
rpsO > 150 3 30S ribosomal protein S15
rrmJ > 150 3 Ribosomal RNA large subunit methyltransferase E

43
Table 2. List of E. coli K12 deletion strains which showed increased sensitivity to
vancomycin. Category 1: DNA replication, recombination and repair, category 2:
transport, efflux, cell wall and cell membrane synthesis, category 3: protein synthesis,
category 4: central metabolic reactions, category 5: regulation, category 6: prophage-
carried genes and cell adhesion, and category 7: unassigned gene products. Data taken
from (75). Reprinted with permission.
Interestingly, most of the remaining mutants in Table 2 had not been
previously described to show increased sensitivity towards vancomycin.
Notably, some of these mutants such as lpxL, lpxM, rfaC or yciM are
involved in LPS biosynthesis. Both LpxL and LpxM are acyltransferases
which transfer either a laurate or myristate chain onto the Kdo2-lipid A
precursor (126). RfaC (also known as WaaC) is a heptosyltransferase which
transfers the first heptose sugar onto the Kdo2 moiety of the LPS inner core
(127). YciM has been recently characterized as a modulator of LPS levels by
negatively regulating the biosynthesis of lipid A (128).
Based on the findings of this study, we reasoned that a small molecule which
inhibits any of the targets listed in Table 2 could be used as an antibiotic
adjuvant to enhance the activity of vancomycin. Since many vancomycin-
sensitive strains are also more susceptible to other antibiotics (Table 1), we
believe that such a small molecule inhibitor could potentially be used to
enhance the activity of several antibiotics. Hence, we carried out a high-
throughput screen in paper III to identify such small molecules.
yheM > 150 3 Protein TusC
yheN > 150 3 Sulfurtransferase TusD
rppH > 150 3 RNA pyrophosphohydrolase
cls > 150 4 Cardiolipin synthase A
cydB > 150 4 Cytochrome bd-I ubiquinol oxidase subunit 2
fdx > 150 4 2Fe-2S ferredoxin
ytjC > 150 4 Probable phosphoglycerate mutase GpmB
gpmM > 150 4 2,3-bisphosphoglycerate-independent phosphoglycerate mutase
iscS > 150 4 Cysteine desulfurase IscS
hns > 150 5 DNA-binding protein H-NS
ybgT > 150 7 Cytochrome bd-I ubiquinol oxidase subunit X
yjjY > 150 7 Uncharacterized protein YjjY

44
Summary of the papers
The overarching goal of my thesis was to understand how the OM barrier of
E. coli could be weakened. One approach that I took was to investigate the
feasibility of using small molecules to increase the permeability of the OM.
We reasoned that small molecules which permeabilize the OM could be used
as adjuvants or lead molecules for adjuvants to improve the activity of large-
scaffold antibiotics.
As a prelude to this, I initially investigated the periplasmic chaperone
network which is required for the biogenesis of OMPs. (Figure 9, left panel).
OMPs are synthesized on cytosolic ribosomes as precursors with an N-
terminal signal sequence and then transported across the IM by the SecYEG
translocase (129). After translocation and cleavage of the signal sequence,
newly exported OMPs are transported from the IM to the OM by the
periplasmic chaperone network SurA, Skp and DegP (130). The periplasmic
chaperone network, in particular SurA and Skp, is essential for the integrity
of the OM and strains lacking either of them are more sensitive to various
large-scaffold antibiotics including vancomycin (121, 122, 131).

45
Figure 9. Three major pathways required for the biogenesis of the OM in Gram-
negative bacteria. Left panel: OMPs are synthesized on cytosolic ribosomes and
subsequently translocated across the IM by the SecYEG translocase. Unfolded OMPs are
trafficked across the periplasmic space by the periplasmic chaperone network SurA, Skp
and DegP, and delivered to the -barrel assembly machinery (BAM) complex, which
folds and integrates OMPs into the OM. Middle panel: Lipoproteins are trafficked across
the IM via the SecYEG translocase and subsequently processed at the outer leaflet of the
IM. Processed lipoproteins destined to the OM are extracted from the IM by the ABC
transporter LolCDE, and then transferred to the periplasmic chaperone LolA. LolA
traffics lipoproteins across the periplasmic space to the OM lipoprotein LolB which
incorporates lipoproteins into the OM. Right panel: Lipopolysaccharide synthesis occurs
at the inner leaflet of the IM. Nascent LPS molecules are translocated across the IM by
the ABC transporter MsbA. Subsequently, LPS molecules are extracted from the IM by
the LptBCFG complex and then translocated across the periplasmic space via a
transenvelope bridge. Once arrived at the OM, LPS molecules are integrated into the OM
by the LptD-LptE complex. Adapted from (132). Reprinted with permission.

46
Before I joined the Daley lab, my former coworker Jörg Götzke identified a
protein with no annotated function, namely YfgM, which we thought might
be a novel member of this periplasmic chaperone network. He was able to
show that YfgM forms a complex with the periplasmic chaperone PpiD at
the SecYEG translocon (Figure 9, left panel). At this point, the function of
YfgM remained unknown. PpiD has been characterized as a member of the
periplasmic chaperone network, which assists in the release of newly
translocated OMPs proteins by preventing their premature aggregation (133,
134). Since YfgM interacts with PpiD at the SecYEG translocon, we
reasoned that YfgM might also be a part of this chaperone network.
In paper I, we had a closer look at the bacterial chromosome and found that
yfgM is located upstream of bamB. As previously mentioned, BamB is a
subunit of the BAM complex which is required for the integrity of the OM
(Figure 9, left panel). Strains lacking BamB have a defect in their OM and
thus are susceptible towards vancomycin. Hence, I was interested to
investigate if strains lacking YfgM also have a defect in their OM. To
address this question, we engineered strains that were lacking YfgM, PpiD
or other periplasmic chaperones such as SurA, Skp or DegP. We then
analyzed the integrity of the OM in these strains by performing an antibiotic
disc diffusion assay. In this assay, a filter disk containing a sub-inhibitory
concentration of vancomycin was spotted onto a lawn of bacteria (Figure
10A). Strains which have an intact OM, such as WT E. coli, are intrinsically
resistant towards vancomycin. Hence, no growth inhibition zone is observed
around the filter disk (Figure 10A, top panel). In contrast, strains with a
defect in their OM, such as surA or skp, have a compromised OM. In
these strains, vancomycin can access its target in the periplasmic space,
which results in growth inhibition around the filter disk (Figure 10A, lower
panel).

47
Figure 10. Disc diffusion assay to investigate the integrity of the OM. (A) WT E. coli
strains are intrinsically resistant towards vancomycin. Hence, no zone of growth inhibition is
observed around the filter disk (top panel). Strains with a defect in their OM, such as the
surA strain, became sensitized towards vancomycin which results in a growth inhibition
zone around the filter disk (bottom panel). (B) Disc diffusion assays were performed in WT E.
coli and various deletion strains to investigate the integrity of the OM. Figure 10B taken from
(135). Reprinted with permission.
Our data showed that deletion of the gene encoding for YfgM did not cause
any obvious defects in the OM (Figure 10B). In addition, we observed the
same phenotype for strains that were either lacking PpiD or DegP (Figure
10B). However, as previously reported, deletion of either SurA or Skp
destabilized the OM, which increased the sensitivity towards vancomycin
(Figure 10B).
Since periplasmic chaperones often have overlapping functions, their role in
the periplasmic chaperone network can only be elucidated by deleting them
in combination. For example, genetic deletion of ppiD and degP results in a
temperature-sensitive phenotype (134). Thus, we investigated if YfgM had
overlapping functions with other periplasmic chaperones. To address this
question, we engineered double knockout strains that were lacking YfgM in

48
combination with either PpiD, DegP, Skp or SurA. By using the vancomycin
susceptibility assay, we investigated the integrity of the OM in these strains
and found that deletion of YfgM in strains with either a ppiD or degP
background did not cause any obvious defects in the OM (Figure 10B).
However, deletion of YfgM in strains with either a skp or surA
background further compromised the integrity of the OM, as evidenced by
an increased growth inhibition zone around the filter disk containing
vancomycin (Figure 10B). Hence, these experiments suggest that YfgM is a
novel periplasmic chaperone which operates in the same network as Skp and
SurA.
In a follow-up study (paper II), we wanted to further understand the
function of YfgM by identifying its putative substrates. We hypothesized
that substrates of YfgM would be incorrectly folded or trafficked when
YfgM was absent from the cell, and therefore more prone to proteolytic
degradation. In this study, we used a comparative proteomic approach to
quantify the steady-state levels of proteins in strains lacking yfgM. Since our
previous study indicated that YfgM has overlapping functions with SurA and
Skp, we also included strains into the proteomic analysis that were lacking
yfgm together with either skp or surA, to exclude compensatory effects
caused by the major periplasmic chaperones.
Surprisingly, the proteomic analysis did not identify any changes in the
levels of OMPs or lipoproteins. However, we identified 9 IM proteins and 7
periplasmic proteins whose abundance was significantly changed. One key
finding was that a few proteins involved in the adaptation to gastrointestinal
stress (e.g. acid-resistance) were lower in abundance in strains lacking
YfgM. For example, the periplasmic chaperone HdeB which is involved in
the acid-stress response was lower in abundance in all three strains lacking
YfgM. To investigate if HdeB was misfolded / mistargeted in strains lacking
yfgM and thus turned over faster, we performed pulse-chase experiments. In

49
these pulse-chase experiments, we could show that HdeB was turned-over
faster in strains lacking yfgM. The identification of HdeB and other cell
envelope proteins as potential substrates of YfgM will be a valuable resource
for follow-up experiments that aim to decipher the function of YfgM.
Paper I + II provided novel insights into the periplasmic chaperone network
and how it contributes to the permeability of the OM. However, the findings
of these two studies do not provide any direct application that could be used
to improve the activity of antibiotics. In paper III our goal was to identify a
small molecule which causes the OM to be more permeable and thus might
be used as an antibiotic adjuvant. Initially, we wanted to identify an inhibitor
against the periplasmic chaperone SurA. Hence, we performed a high-
throughput screen in which we monitored growth of WT E. coli in the
presence of a sub-inhibitory concentration of vancomycin (150 μg ml-1) and
28,000 small molecules. We reasoned that a small molecule that inhibits
SurA would increase the activity of vancomycin, which would result in a
growth arrest. I established the assay conditions whereas the high-throughput
screen was performed by our collaboration partner from the Department of
Chemistry at Umeå University. The high-throughput screen identified one
promising molecule, namely MAC-13243. This small molecule has been
previously identified as an inhibitor of the periplasmic chaperone LolA
(136). LolA is an essential protein that functions as a periplasmic shuttle that
transports lipoproteins from the IM to the OM (Figure 9, middle panel).
All lipoproteins destined for the OM are synthesized in the cytoplasm as
precursors with an N-terminal sequence. After translocation through the
SecYEG translocon, lipoproteins are processed at the periplasmic side of the
IM, which involves sequential modification of a cysteine residue and
cleavage of the signal peptide by the lipoprotein-specific signal peptidase
LspA (63). Processed lipoproteins remain either attached to the IM or they
are extracted from the IM by the ABC transporter LolCDE (Figure 9, middle

50
panel). Subsequently, the periplasmic chaperone LolA captures lipoproteins
from the LolCDE complex and shuttles them across the periplasmic space to
the OM. At the OM, lipoproteins are transferred from LolA to LolB, which
localizes them to the OM.
Since inhibition of LolA by MAC-13243 leads to decreased levels of OM
lipoproteins (136), we hypothesized that this might affect the integrity of the
OM. To explore this possibility, I established an NPN (N-phenyl-1-
naphthylamine) uptake assay to investigate if the small molecule MAC-
13243 could be used to increase the permeability of the OM. The NPN
uptake assay is a commonly used method to analyze the integrity of the OM
(137). For example, WT E. coli strains have an intact OM and thus the
hydrophobic dye NPN cannot efficiently cross the OM. However, when the
OM is damaged, NPN can access phospholipids in the IM and the inner
leaflet of the OM, which results in prominent fluorescence. Using this assay,
I demonstrated that MAC-13243, when used at sub-inhibitory
concentrations, permeabilizes the OM of WT E. coli in a concentration-
dependent manner (Figure 11).
Figure 11. NPN uptake in WT E. coli cells. Left panel: WT E. coli cells were exposed to
different concentrations of MAC-13243 and NPN uptake was monitored. Right panel: The
increase in fluorescence was considered to be due to increased permeability of the OM
since the amount of MAC-13243 did not reduce cell viability. Abbreviations: MIC =
Minimal Inhibitory Concentration. Figure taken from paper III.

51
Since MAC-13243 permeabilized the OM of WT E. coli cells, I was curious
to investigate if MAC-13243 could also be used as an adjuvant for
antibiotics which do not efficiently cross the OM. To address this question, I
performed a series of checkerboard assays. The checkerboard assay is a
commonly used method to evaluate interactions between two drugs. In brief,
I found that MAC-13243 worked synergistically with either erythromycin or
novobiocin, meaning that there was added benefit when those two drugs
were used in combination (Figure 12).
Figure 12. MAC-13243 works synergistically with various large-scaffold antibiotics.
Heat plots illustrate inhibition of growth of WT E. coli in the presence of MAC-13243
and vancomycin, rifampicin, erythromycin or novobiocin. Growth percentage of E. coli is
shown with different colours where black represents 100% growth and red 0% growth.
Figure taken from paper III.
At this point, it remained unclear if the leaky phenotype, which we observed
after treating WT E. coli cells with MAC-13243, was caused by inhibition of
LolA. Since MAC-13243 is not stable in aqueous solution and a structural
analogue of its degradation product S-(4-chlorobenzyl)isothiourea, namely
A22, has been reported to inhibit the eukaryotic actin-homolog MreB
(Figure 17), it remained unclear what caused the cells to become more
permeable (138). To address this question, I used the CRISPRi system to
reduce the intracellular levels of LolA and showed that partial depletion of

52
LolA was sufficient to induce the permeable phenotype, as evidenced by an
increased permeability of the OM.
To summarize this study, we showed that inhibition of lipoprotein trafficking
could be an attractive target for small molecules. By performing a high-
throughput screen, we identified an inhibitor of the periplasmic chaperone
LolA, namely MAC-13243, and showed that this small molecule can be used
as an adjuvant to improve the activity of certain large-scaffold antibiotics.
In a parallel approach, we have collaborated with the Widmalm group from
the organic chemistry section at Stockholm University. Our common goal
was to break the OM permeability barrier by using small molecules which
target the synthesis of LPS (paper IV). Previous to this study, the Widmalm
group identified 3 small molecules in a fragment-based screen for inhibitors
against the glycosyltransferase WaaG (Figure 13A) (139). These molecular
scaffolds, namely L1-L3, have low affinity for WaaG and compete with its
natural substrate UDP-Glc for binding. This was of particular interest for us
since WaaG is a key enzyme in the synthesis of LPS. The peripherally
attached IM protein WaaG adds the first glucose residue to the outer core of
the growing LPS molecule (Figure 6).
Figure 13. Small molecular fragments L1-L3 and schematic representation of the
glucose transfer by WaaG. (A) L1, 4-(2-amino-1,3-thiazol-4-yl)phenol; L2, 4-(1H-
pyrrol-1-yl)benzoic acid; L3, 2-(1H-pyrrol-1-ylmethyl)pyridine; (B) The
glycosyltransferase WaaG transfers a 14C-glucose residue from UDP-Glc* to the waaG
LPS acceptor molecule. Figure 13A taken from (142). Reprinted with permission.

53
WaaG is essential for the stability of the OM and deletion of the gene
encoding for WaaG results in an inability to synthesize the outer core and the
O-antigen (Figure 14) (140). As a consequence, strains lacking WaaG
become more sensitive to several antibiotic classes (Table 1, formerly known
as rfaG) (75). We hypothesized that a small molecule that inhibits WaaG
could be used as an antibiotic adjuvant. Thus, in paper IV I explored if any
of these small molecular scaffolds could be used to inhibit WaaG in vitro. I
developed an activity assay for WaaG using 14C-labeled UDP-glucose and
LPS that had been purified from a waaG strain (Figure 13B).
Figure 14. Molecular dynamics simulations of OmpF trimer intercalated in OMs of E.
coli. OmpF trimer was intercalated in either E. coli rough LPS (left panel), E. coli K12 core
LPS (middle panel) or E. coli R1 core LPS with five repeating units of O6-antigen (right
panel). Lipid A is illustrated as pink spheres. Core sugars (gray) and O-antigen
polysaccharides (orange) are illustrated as stick models. The inner leaflet consists of PPPE
(blue spheres), PVPG (orange spheres), and PVCL2 (magenta spheres). Ca2+ ions are depicted
as cyan small spheres, K+ ions as green small spheres and Cl- ions as magenta small spheres.
Abbreviations: PPPE, 1-palmitoyl(16:0)-2-palmitoleoyl(16:1cis-9)-
phosphatidylethanolamine; PVPG, 1-palmitoyl(16:0)-2-vacenoyl(18:1 cis-11)
phosphatidylglycerol; PVCL2, 1,10-palmitoyl-2,20-vacenoyl cardiolipin. Figure taken from
(141). Reprinted with permission.
54
When I started to develop the assay for WaaG, I initially had problems with
its low activity. Since WaaG is a peripherally attached IM protein, I explored
if its activity could be improved by the addition of various lipids. After
several rounds of optimization, I discovered that I could improve the activity
of WaaG by including two E. coli membrane lipids, namely PG and CL, and
the non-ionic detergent CHAPS to the reaction mixture. Using these
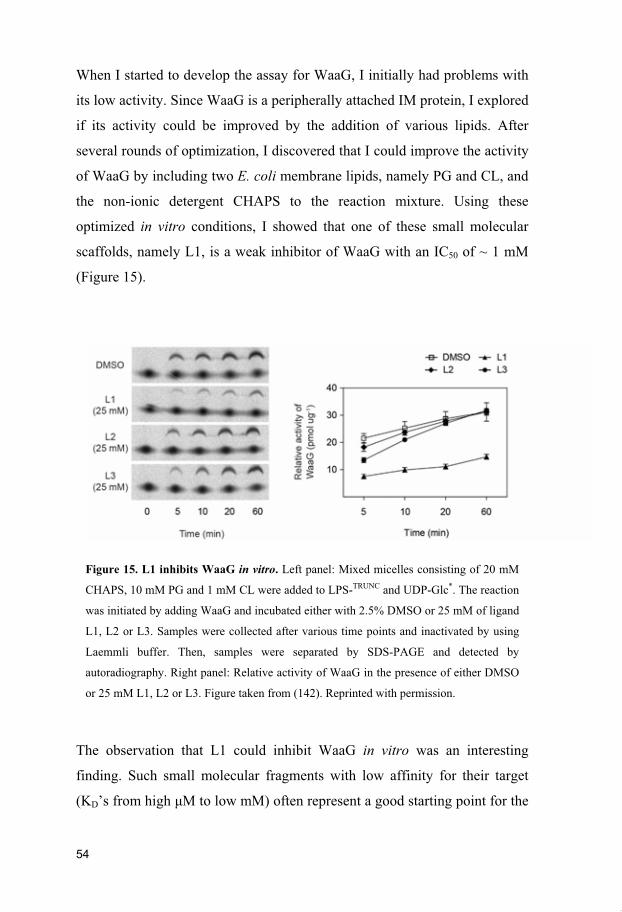
optimized in vitro conditions, I showed that one of these small molecular
scaffolds, namely L1, is a weak inhibitor of WaaG with an IC50 of ~ 1 mM
(Figure 15).
Figure 15. L1 inhibits WaaG in vitro. Left panel: Mixed micelles consisting of 20 mM
CHAPS, 10 mM PG and 1 mM CL were added to LPS-TRUNC and UDP-Glc*. The reaction
was initiated by adding WaaG and incubated either with 2.5% DMSO or 25 mM of ligand
L1, L2 or L3. Samples were collected after various time points and inactivated by using
Laemmli buffer. Then, samples were separated by SDS-PAGE and detected by
autoradiography. Right panel: Relative activity of WaaG in the presence of either DMSO
or 25 mM L1, L2 or L3. Figure taken from (142). Reprinted with permission.
The observation that L1 could inhibit WaaG in vitro was an interesting
finding. Such small molecular fragments with low affinity for their target
(KD’s from high M to low mM) often represent a good starting point for the

55
design of a high-affinity inhibitor (143, 144). In this process, also known as
fragment-based drug design, small molecular fragments with low affinity for
their target are chemically elaborated or linked to produce a high affinity
inhibitor. Since L1 could be used to inhibit WaaG in vitro, we expanded
chemical space around L1 (and also L2 - L3), and created a fragment-based
library that included an additional 17 small molecular fragments (Figure 16,
unpublished data). At this point, the aim was to identify additional small
molecular fragments that either compete with the natural substrate UDP-Glc
for binding or inhibit WaaG in the in vitro activity assay.
Figure 16. Small molecular scaffold library L1-20.

56
Hence, I tested all small molecular fragments in the in vitro activity assay to
evaluate their inhibitory activity. Surprisingly, the results indicated that none
of the additional fragments had inhibitory activity (data not shown). These
findings were surprising since some of these fragments are structurally
closely related to L1 (Figure 16). The Widmalm group is currently
investigating by NMR spectroscopy if any of the additional small molecular
fragments compete with the natural substrate UDP-Glc for binding. Our
preliminary results indicate that one of these fragments, namely L8, also
binds to WaaG (data not shown). Despite the fact that the expanded library
did not contain a more potent inhibitor than L1, we could identify at least
one additional small molecular fragment that binds to WaaG. This provides
further insight for the design of a potent inhibitor against WaaG.

57
Conclusions and future perspectives
Gram-negative bacteria have developed sophisticated mechanisms to protect
themselves against noxious molecules such as antibiotics. Two of the major
mechanisms that limit the activity of many antibiotics include active efflux
by efflux pumps and reduced uptake across the OM barrier. The presented
doctoral thesis had the objective to investigate how the reduced uptake
across the OM barrier could be improved by destabilizing the OM. To
address this problem, I initially started investigating the periplasmic
chaperone network and how it contributes to the permeability of the OM
(paper I). In this paper, we identified a novel component of the SecYEG
translocon, namely YfgM, and showed that it operates in the same pathway
as the periplasmic chaperone network SurA/Skp. However, YfgM plays only
a minor role in this network since strains lacking YfgM did not have any
obvious defects in the OM. The molecular function of YfgM remains to be
determined but we speculate that it might act as a docking platform for the
periplasmic chaperones SurA/Skp. Interestingly, the periplasmic domain of
YfgM contains tetratricopeptide repeat (TPR) domains whose function has
not been determined yet. These domains are often involved in protein-
protein interactions and thus it might be interesting to further investigate the
function of the TPR domains in YfgM (145). There is also evidence that Skp
interacts with OMPs during their early translocation through the SecYEG
translocon (146). Hence, it is possible that Skp might dock to the SecYEG
translocon or a protein in close vicinity to it.
To better understand the function of YfgM, we performed a comparative
proteomic approach to identify potential substrates of YfgM (paper II). We
hypothesized that strains lacking YfgM might have a changed OMP profile

58
since YfgM operates in the same network as SurA/Skp. Although we did not
observe any significant changes in the levels of OMPs and lipoproteins in
strains lacking yfgM, the proteomic data revealed an unexpected insight into
the physiological role of YfgM. We identified a number of proteins that are
involved in acid-stress response, which were lower in abundance in strains
lacking YfgM. These findings were unexpected since YfgM had not been
shown to be induced during acid-stress previously (147–150). In an
additional experiment, we could confirm that strains devoid of YfgM had a
decreased survival rate at low pH. At this stage, it remains unclear if the
decreased survival rate at low pH in strains lacking YfgM is caused by a
defect in the cell envelope or by another secondary effect.
To address the reduced uptake of antibiotics across the OM, we performed a
high-throughput screen to look for small molecules that destabilize the OM
(paper III). In this study, we identified an inhibitor of the periplasmic
chaperone LolA, named MAC-13243. This small molecule had been
previously identified as a novel antimicrobial lead (136). However, I could
show that MAC-13243 can also be repurposed to an antibiotic adjuvant. I
observed that MAC-13243 synergized with the large-scaffold antibiotics
novobiocin and erythromycin, but not with vancomycin and rifampicin. At
this point, it remains unclear to us why MAC-13243 works synergistically
with some large-scaffold antibiotics but not with others. It is worth noting
that Krishnamoorthy et al. reported that the activity of these 4 antibiotics is
significantly limited by the OM and/or by active efflux (72). Surprisingly,
they found that the OM presented no major obstacle for novobiocin whereas
active efflux drastically reduced its activity. In the case of rifampicin and
vancomycin, they found that the OM barrier significantly limited their
activity whereas inactivation of active efflux only minor improved their
activity. In the case of erythromycin, a combination of both reduced uptake
and active efflux limited its activity.

59
Despite the preliminary findings, I believe that MAC-13243 has certain
limitations in its current form that prevent it from being used as an antibiotic
adjuvant. First of all, the synergistic interactions between MAC-13243 and
either novobiocin or erythromycin were moderate and only observed under
specific conditions. One of the major limitations of MAC-13243 might be its
limited stability in aqueous solution (151). MAC-13243 degrades in aqueous
solution into one molecule of 3,4-dimethoxyphenethylamine, two molecules
of formaldehyde and one molecule of S-(4-chlorobenzyl)isothiourea (Figure
17A). Thus, an attempt to improve the chemical stability of MAC-13243
could improve its activity as an antibiotic adjuvant. This could be achieved
by stabilizing the central triazine ring of MAC-13243, which is prone to
hydrolysis (151).
Figure 17. Degradation of MAC-13243 in aqueous solution. (A) MAC-13243 is
degraded into one molecule 3,4-dimethoxyphenethylamine, two molecules of
formaldehyde and one molecule S-(4-chlorobenzyl)isothiourea (151). Both MAC-13243
and the degradation product S-(4-chlorobenzyl)isothiourea bind to LolA (151) (B) An
analogue of the degradation product, named A22 or S-(4-dichlorobenzyl)isothiourea, has
been shown to bind to LolA. Interestingly this compound is an inhibitor of the
cytoskeletal protein MreB (138). Figure adjusted from (151). Reprinted with permission.

60
Interestingly, one of the degradation products of MAC-13243, namely S-(4-
chlorobenzyl)isothiourea, is a structural analogue of compound A22, which
has been identified as an inhibitor of the cytoskeletal protein MreB (Figure
17B) (138). Taylor et al. showed that A22 works synergistically with both
novobiocin and rifampicin in E. coli (115). Hence, the design of a molecule
which inhibits both LolA and MreB could be an interesting concept to
further enhance the synergistic effects of MAC-13243.
I personally think that it would be most interesting to expand on the findings
of paper IV. In this paper, I showed that the small molecular fragment,
namely compound L1, can be used to inhibit the glycosyltransferase WaaG
in vitro. However, due to its weak inhibitory activity, L1 is not a molecule
that can be used in its current form as an antibiotic adjuvant. However, we
believe that L1 represents a good lead fragment for the design of a more
potent inhibitor against WaaG. By expanding chemical space around L1, we
could identify an additional molecule, namely L8 (Figure 16), that binds to
WaaG in vitro. In a next optimization round, we plan to systematically
evaluate the chemical space around these two molecules to gain further
insight into the properties that are required for binding to and inhibiting
WaaG.

61
Populärvetenskaplig sammanfattning
Antibiotika anses vara en av de viktigaste upptäckterna inom
humanmedicinen. Sedan de infördes in i klinikerna på 1940-talet har de
framgångsrikt använts för att bota bakteriella infektioner. På grund av sin
omfattande användning och missbruk under de senaste decennierna har dock
många bakteriearter som inte längre är känsliga mot de flesta antibiotika
uppstått. Dessa så kallade multiresistenta bakterier har utvecklat olika
tillvägagångssätt för att inaktivera antibiotika. Följaktligen har vi ont om
behandlingsalternativ och är ofta tvungna att använda äldre antibiotika som
är mindre säkra.
Resistens mot antibiotika är ett allmänt problem. Gramnegativa bakterier är
dock arter som är särskilt bekymmersamma. Dessa bakterier dödar tusentals
människor årligen och anses därför vara ett stort hot mot folkhälsan. Ett av
de största problemen är att många antibiotika är ineffektiva mot
gramnegativa bakterier. Detta beror dels på en fysisk barriär, nämligen det
yttre membranet, som hämmar effektivt upptag av många antibiotika.
I den presenterade doktorsavhandlingen undersökte jag hur dessa fysiska
barriärer kunde försvagas. Det ultimata målet med min avhandling var att
hitta små molekyler som skulle kunna användas för att bryta denna fysiska
barriär. Vi resonerade att dessa små molekyler skulle kunna användas för att
förbättra upptagningen av de antibiotika som normalt inte effektivt passerar
yttermembranet.

62
Acknowledgements
I would like to take the opportunity to thank my supervisor Daniel Daley for his excellent supervision during the past 5 years. I really enjoyed it to be part of your group. Thanks mate! I also would like to thank our current group members Kiavash, Rageia, Zoe and Patrick, and the former group members Jörg, Stephen, Bill and Isolde. It was very pleasant to work with all of you and I think we have a really good ambience in the group. Special thanks to my co-supervisor Göran Widmalm and his current group members Jonas and Alessandro. It was fun to work together with you guys! Also a special thanks to our former collaborators Klaas, Spyridon, Amin, Olof and Åke. I am also grateful to my former students Lars Nilsson, Johanna Hultgren, Muna Amanuel, Andreas Dunge and Anja Blümler. Thanks to our neighbors from the Jan-Willem de Gier group: Thomas, Zhe, Henry and Alex. Special thanks to my table soccer friend Thomas for never giving up and Alex the Greek for all the insightful philosophical discussions. Thanks to the Rob Daniels group: Henrik, Hao, Dan, Rebecca and the former group members Johan and Diogo. It was always nice to see other people in the lab on the weekends. A big thanks to the Gunnar von Heijne and Ingmarie Nilsson group: Renuka, Grant, Felix, Ioanna, Luigi, Nir, José, Nina and Åsa. Special thanks to my bongo friend Mr. Felix – it was a pleasure to explore Africa with you! Thanks to the Tara Hessa group: Tara, Theresa, Genia and Anastasia. Cool group, good people!

63
Thanks to the Elzbieta Glaser group: Pedro and Beata for always being in a good mood and Elzbieta for never-ending but interesting discussions at the lunch table. Many thanks to Brzezinski and Ädelroth group members Jacob, Johan, Tobias, Camilla, Ingrid, Max, Nathalie and Pia. Great thanks to my climbing and sailing friend Max (aka sailor man) – I knew it was a good decision to have a friend with a sailing boat! We truly had some good laughs and I will never forget the large number of fish we caught - zero! Thanks to the David Drew group members Emmanuel, Mathieu, Aziz, Povilas, Pascal and Magnus. Bsunderä Dank äm bärtigä Päsci und sim Schätzi dr Regula - heb sorg zunnärä! Du bisch wahrhaftig en Glückspilz! Sogar dr Andi us Rümlang wär nidisch! Thanks to the Martin Ott group members Hannah, Mama, Magdalena, Tamara and Braulio for the discussions about the book exam and other stuff. Special thanks to Alex Tuuling, Maria Sallander, Anita Hjelm, Lotta Sturell, Karin Häggbom Sandberg, Malin Kamb, Håkan Thorén, Matthew Bennett, Ann-Britt Rönnell and Peter Nyberg for running DBB in the background. Last but not least, a very special thanks to my family and friends back home for their great support and visits to Stockholm. Looking forward to see you again!

64
References
1. Hall, B. G., Barlow, M. Evolution of the serine -lactamases: past, present and future. Drug Resist. Updat. 7, 111–123 (2004).
2. Baltz, R. A. Antibiotic discovery from Actinomycetes: Will a renaissance follow the decline and fall? SIM News 55, 188–96 (2005).
3. Udekwu, K. I., Parrish, N., Ankomah, P., Baquero, F., Levin, B. R. Functional relationship between bacterial cell density and the efficacy of antibiotics. J. Antimicrob. Chemother. 63, 745–757 (2009).
4. Li, J. et al. Antimicrobial Activity and Resistance: Influencing Factors. Front. Pharmacol. 8, (2017).
5. Scholar, E. M., Pratt, W. B. The Antimicrobial Drugs. 2nd edition. Oxford University Press, New York (2000).
6. Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 12, 371–387 (2013). 7. Kohanski, M. A., Dwyer, D. J., Collins, J. J. How antibiotics kill bacteria: from targets to
networks. Nat. Rev. Microbiol. 8, 423–435 (2010). 8. Kong, K.F., Schneper, L., Mathee, K. -lactam Antibiotics: From Antibiosis to Resistance and
Bacteriology. APMIS 118, 1–36 (2010). 9. Barlow, M. What antimicrobial resistance has taught us about horizontal gene transfer. Methods
Mol. Biol. 532, 397–411 (2009). 10. Coates, A., Hu, Y., Bax, R., Page, C. The future challenges facing the development of new
antimicrobial drugs. Nat. Rev. Drug Discov. 1, 895–910 (2002). 11. Fleming, A. On the Antibacterial Action of Cultures of a Penicillium, with Special Reference to
their Use in the Isolation of B. influenzae. Br. J. Exp. Pathol. 10, 226–236 (1929). 12. Lobanovska, M., Pilla, G. Penicillin’s Discovery and Antibiotic Resistance: Lessons for the
Future? Yale J. Biol. Med. 90, 135–145 (2017). 13. Coates, A. R., Halls, G., Hu, Y. Novel classes of antibiotics or more of the same? Br. J.
Pharmacol. 163, 184–194 (2011). 14. Silver, L. L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 24, 71–109 (2011). 15. Wenzel, R. P. The Antibiotic Pipeline – Challenges, Costs, and Values. N. Engl. J. Med. 351,
523–526 (2004). 16. Spellberg, B. The future of antibiotics. Crit. Care 18, 228 (2014). 17. Bakken, J. S. Antibiotic partners promote discovery. Nature 537, 167–167 (2016). 18. Piddock, L. J. V. The crisis of no new antibiotics – What is the way forward? Lancet Infect. Dis.
12, 249–253 (2012). 19. Michael, C. A., Dominey-Howes, D., Labbate, M. The Antimicrobial Resistance Crisis: Causes,
Consequences, and Management. Front. Public Health 2, (2014). 20. Payne, D. J., Gwynn, M. N., Holmes, D. J., Pompliano, D. L. Drugs for bad bugs: confronting the
challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6, 29–40 (2007). 21. An antibiotic comeback ? Nat. Rev. Drug Discov. 13, 165–165 (2014). 22. Col, N. F., O’Connor, R. W. Estimating Worldwide Current Antibiotic Usage: Report of Task
Force 1. Rev. Infect. Dis. 9, S232–S243 (1987). 23. Barbosa, T. M., Levy, S. B. The impact of antibiotic use on resistance development and
persistence. Drug Resist. Updat. 3, 303–311 (2000).

65
24. Goossens, H., Ferech, M., Vander Stichele, R., Elseviers, M., ESAC Project Group. Outpatient antibiotic use in Europe and association with resistance: a cross-national database study. Lancet Lond. Engl. 365, 579–587 (2005).
25. Riedel, S. et al. Antimicrobial use in Europe and antimicrobial resistance in Streptococcus pneumoniae. Eur. J. Clin. Microbiol. 26, 485–490 (2007).
26. Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States, 2013. Available at: http://www.cdc.gov/drugresistance/threat-report-2013/index.html, Accessed 20.09.2017
27. Ayukekbong, J. A., Ntemgwa, M., Atabe, A. N. The threat of antimicrobial resistance in developing countries: causes and control strategies. Antimicrob. Resist. Infect. Control 6, 47 (2017).
28. Witte, W. Selective pressure by antibiotic use in livestock. Int. J. Antimicrob. Agents 16, 19–24 (2000).
29. Bengtsson, B., Wierup, M. Antimicrobial Resistance in Scandinavia after a Ban of Antimicrobial Growth Promoters. Anim. Biotechnol. 17, 147–156 (2006).
30. Butaye, P., Devriese, L. A., Haesebrouck, F. Antimicrobial Growth Promoters Used in Animal Feed: Effects of Less Well Known Antibiotics on Gram-Positive Bacteria. Clin. Microbiol. Rev. 16, 175–188 (2003).
31. Larsson, D. G. J. Pollution from drug manufacturing: review and perspectives. Phil. Trans. R. Soc. B. 369, (2014).
32. Chemaly, R. F. et al. The role of the healthcare environment in the spread of multidrug-resistant organisms: update on current best practices for containment. Ther. Adv. Infect. Dis. 2, 79–90 (2014).
33. Sydnor, E. R. M., Perl, T. M. Hospital Epidemiology and Infection Control in Acute-Care Settings. Clin. Microbiol. Rev. 24, 141–173 (2011).
34. The evolving threat of antimicrobial resistance: options for action. World Health Organization: Geneva, Switzerland, 2012. Available at: http://apps.who.int/iris/bitstream/10665/44812/1/9789241503181_eng.pdf, Accessed: 23.09.2017
35. Allerberger, F., Gareis, R., Jindrák, V., Struelens, M. J. Antibiotic stewardship implementation in the EU: the way forward. Expert Rev. Anti Infect. Ther. 7, 1175–1183 (2009).
36. Allerberger, F., Lechner, A., Wechsler-Fördös, A., Gareis, R. Optimization of Antibiotic Use in Hospitals – Antimicrobial Stewardship and the EU Project ABS International. Chemotherapy 54, 260–267 (2008).
37. Barlam, T. F. et al. Implementing an Antibiotic Stewardship Program: Guidelines by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America. Clin. Infect. Dis. 62, 51-77 (2016).
38. Waterer, G. W., Wunderink, R. G. Increasing threat of Gram-negative bacteria. Crit. Care Med. 29, 75-81 (2001).
39. Zowawi, H. M. et al. The emerging threat of multidrug-resistant Gram-negative bacteria in urology. Nat. Rev. Urol. 12, 570–584 (2015).
40. Boucher, H. W. et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48, 1–12 (2009).
41. Peleg, A. Y., Hooper, D. C. Hospital-acquired infections due to gram-negative bacteria. N. Engl. J. Med. 362, 1804–1813 (2010).
42. Falagas, M. E., Kasiakou, S. K. Toxicity of polymyxins: a systematic review of the evidence from old and recent studies. Crit. Care 10, R27 (2006).
43. Silhavy, T. J., Kahne, D., Walker, S. The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Biol. 2, a000414 (2010).
44. Brown, L., Wolf, J. M., Prados-Rosales, R., Casadevall, A. Through the wall: extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat. Rev. Microbiol. 13, 620–630 (2015).
45. Morein, S., Andersson, A., Rilfors, L., Lindblom, G. Wild-type Escherichia coli cells regulate the membrane lipid composition in a ‘window’ between gel and non-lamellar structures. J. Biol. Chem. 271, 6801–6809 (1996).

66
46. Whitfield, C., Trent, M. S. Biosynthesis and export of bacterial lipopolysaccharides. Annu. Rev. Biochem. 83, 99–128 (2014).
47. Raetz, C. R. H., Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700 (2002).
48. Clifton, L. A. et al. Asymmetric phospholipid: lipopolysaccharide bilayers; a Gram-negative bacterial outer membrane mimic. J. R. Soc. Interface 10, (2013).
49. Medzhitov, R., Janeway, C. J. Innate Immunity. N. Engl. J. Med. 343, 338–344 (2000). 50. Raetz, C. R., Reynolds, C. M., Trent, M. S., Bishop, R. E. Lipid A modification systems in gram-
negative bacteria. Ann. Rev. Biochem. 76, 295–329 (2007). 51. Wang, L., Wang, Q., Reeves, P. R. The variation of O-antigens in gram-negative bacteria.
Subcell. Biochem. 53, 123–152 (2010). 52. Koebnik, R., Locher, K. P., Van Gelder, P. Structure and function of bacterial outer membrane
proteins: barrels in a nutshell. Mol. Microbiol. 37, 239–253 (2000). 53. Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol.
Biol. Rev. 67, 593–656 (2003). 54. Pagès, J. M., James, C. E., Winterhalter, M. The porin and the permeating antibiotic: a selective
diffusion barrier in Gram-negative bacteria. Nat. Rev. Microbiol. 6, 893–903 (2008). 55. Grabowicz, M., Silhavy, T. J. Redefining the essential trafficking pathway for outer membrane
lipoproteins. Proc. Natl. Acad. Sci. 114, 4769–4774 (2017). 56. Hirota, Y., Suzuki, H., Nishimura, Y., Yasuda, S. On the process of cellular division in
Escherichia coli: a mutant of E. coli lacking a murein-lipoprotein. Proc. Natl. Acad. Sci. 74, 1417–1420 (1977).
57. Suzuki, H. et al. Murein-lipoprotein of Escherichia coli: a protein involved in the stabilization of bacterial cell envelope. Mol. Gen. Genet. 167, 1–9 (1978).
58. Merdanovic, M., Clausen, T., Kaiser, M., Huber, R., Ehrmann, M. Protein quality control in the bacterial periplasm. Annu. Rev. Microbiol. 65, 149–168 (2011).
59. Schleifer, K. H., Kandler, O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 36, 407–477 (1972).
60. Yamagami, A., Yoshioka, T., Kanemasa, Y. Differences in Phospholipid Composition between Wild Strains and Streptomycin Resistant Mutants of Certain Enteric Bacteria. Jpn. J. Microbiol. 14, 174–176 (1970).
61. Yasuhiro, K., Yuzuru, A., Shoshichi, N. Composition and turnover of the phospholipids in Escherichia coli. Biochim. Biophys. Acta 144, 382–390 (1967).
62. Facey, S. J., Kuhn, A. Biogenesis of bacterial inner-membrane proteins. Cell. Mol. Life Sci. 67, 2343–2362 (2010).
63. Okuda, S., Tokuda, H. Lipoprotein Sorting in Bacteria. Annu. Rev. Microbiol. 65, 239–259 (2011).
64. Delcour, A. H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta 1794, 808–816 (2009).
65. Delcour, A. H. Solute uptake through general porins. Front. Biosci. 8, 1055–1071 (2003). 66. Masi, M., Pagès, J. M. Structure, Function and Regulation of Outer Membrane Proteins Involved
in Drug Transport in Enterobactericeae: the OmpF/C – TolC Case. Open Microbiol. J. 7, 22–33 (2013).
67. Li, X. Z., Plésiat, P., Nikaido, H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 28, 337–418 (2015).
68. Richter, M. F. et al. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 545, 299–304 (2017).
69. Hasdemir, U. O., Chevalier, J., Nordmann, P., Pagès, J. M. Detection and Prevalence of Active Drug Efflux Mechanism in Various Multidrug-Resistant Klebsiella pneumoniae Strains from Turkey. J. Clin. Microbiol. 42, 2701–2706 (2004).
70. Vaara, M., Plachy, W. Z., Nikaido, H. Partitioning of hydrophobic probes into lipopolysaccharide bilayers. Biochim. Biophys. Acta 1024, 152–158 (1990).
71. Plésiat, P., Nikaido, H. Outer membranes of gram-negative bacteria are permeable to steroid probes. Mol. Microbiol. 6, 1323–1333 (1992).

67
72. Krishnamoorthy, G. et al. Breaking the Permeability Barrier of Escherichia coli by Controlled Hyperporination of the Outer Membrane. Antimicrob. Agents Chemother. 60, 7372–7381 (2016).
73. Hancock, R. E. W. Alterations in Outer Membrane Permeability. Annu. Rev. Microbiol. 38, 237–264 (1984).
74. Vaara, M. Agents that increase the permeability of the outer membrane. Microbiol. Rev. 56, 395–411 (1992).
75. Liu, A. et al. Antibiotic sensitivity profiles determined with an Escherichia coli gene knockout collection: generating an antibiotic bar code. Antimicrob. Agents Chemother. 54, 1393–1403 (2010).
76. Hancock, R. E., Bell, A. Antibiotic uptake into gram-negative bacteria. Eur. J. Clin. Microbiol. 7, 713–720 (1988).
77. Zgurskaya, H. I., Löpez, C. A., Gnanakaran, S. Permeability Barrier of Gram-Negative Cell Envelopes and Approaches To Bypass It. ACS Infect. Dis. 1, 512–522 (2015).
78. Silver, L. L. A Gestalt approach to Gram-negative entry. Bioorg. Med. Chem. 24, 6379–6389 (2016).
79. Yang, N. J., Hinner, M. J. Getting Across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins. Methods Mol. Biol. 1266, 29–53 (2015).
80. Wargel, R. J., Hadur, C. A., Neuhaus, F. C. Mechanism of D-cycloserine action: transport mutants for D-alanine, D-cycloserine, and glycine. J. Bacteriol. 105, 1028–1035 (1971).
81. Chopra, I., Ball, P. Transport of antibiotics into bacteria. Adv. Microb. Physiol. 23, 183–240 (1982).
82. Taber, H. W., Mueller, J. P., Miller, P. F., Arrow, A. S. Bacterial uptake of aminoglycoside antibiotics. Microbiol. Rev. 51, 439–457 (1987).
83. Chopra, I. Molecular mechanisms involved in the transport of antibiotics into bacteria. Parasitology 96 Suppl, 25-44 (1988).
84. Piddock, L. J. V. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin. Microbiol. Rev. 19, 382–402 (2006).
85. Piddock, L. J. V. Multidrug-resistance efflux pumps not just for resistance. Nat. Rev. Microbiol. 4, 629–636 (2006).
86. Sun, J., Deng, Z., Yan, A. Bacterial multidrug efflux pumps: Mechanisms, physiology and pharmacological exploitations. Biochem. Biophys. Res. Commun. 453, 254–267 (2014).
87. Du, D., van Veen, H. W., Murakami, S., Pos, K. M., Luisi, B. F. Structure, mechanism and cooperation of bacterial multidrug transporters. Curr. Opin. Struct. Biol. 33, 76–91 (2015).
88. Nikaido, H., Pagès, J. M. Broad Specificity Efflux pumps and Their Role in Multidrug Resistance of Gram Negative Bacteria. FEMS Microbiol. Rev. 36, 340–363 (2012).
89. Okusu, H., Ma, D., Nikaido, H. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J. Bacteriol. 178, 306–308 (1996).
90. Nikaido, H. Multiple antibiotic resistance and efflux. Curr. Opin. Microbiol. 1, 516–523 (1998). 91. Rosenberg, E. Y., Ma, D., Nikaido, H. AcrD of Escherichia coli is an aminoglycoside efflux
pump. J. Bacteriol. 182, 1754–1756 (2000). 92. Zgurskaya, H. I., Nikaido, H. Multidrug resistance mechanisms: drug efflux across two
membranes. Mol. Microbiol. 37, 219–225 (2000). 93. Nishino, K., Yamaguchi, A. Analysis of a complete library of putative drug transporter genes in
Escherichia coli. J. Bacteriol. 183, 5803–5812 (2001). 94. Mazzariol, A., Tokue, Y., Kanegawa, T. M., Cornaglia, G., Nikaido, H. High-level
fluoroquinolone-resistant clinical isolates of Escherichia coli overproduce multidrug efflux protein AcrA. Antimicrob. Agents Chemother. 44, 3441–3443 (2000).
95. Oethinger, M., Kern, W. V., Jellen-Ritter, A. S., McMurry, L. M., Levy, S. B. Ineffectiveness of topoisomerase mutations in mediating clinically significant fluoroquinolone resistance in Escherichia coli in the absence of the AcrAB efflux pump. Antimicrob. Agents Chemother. 44, 10–13 (2000).

68
96. Webber, M. A., Piddock, L. J. V. Absence of Mutations in marRAB or soxRS in acrB-Overexpressing Fluoroquinolone-Resistant Clinical and Veterinary Isolates of Escherichia coli. Antimicrob. Agents Chemother. 45, 1550–1552 (2001).
97. Stoitsova, S. O., Braun, Y., Ullrich, M. S., Weingart, H. Characterization of the RND-Type Multidrug Efflux Pump MexAB-OprM of the Plant Pathogen Pseudomonas syringae. Appl. Environ. Microbiol. 74, 3387–3393 (2008).
98. Dreier, J., Ruggerone, P. Interaction of antibacterial compounds with RND e ux pumps in Pseudomonas aeruginosa. Front. Microbiol. 6, (2015).
99. Ziha-Zarifi, I., Llanes, C., Köhler, T., Pechere, J. C., Plesiat, P. In Vivo Emergence of Multidrug-Resistant Mutants of Pseudomonas aeruginosa Overexpressing the Active Efflux System MexA-MexB-OprM. Antimicrob. Agents Chemother. 43, 287–291 (1999).
100. Bernal, P., Molina-Santiago, C., Daddaoua, A., Llamas, M. A. Antibiotic adjuvants: identification and clinical use. Microb. Biotechnol. 6, 445–449 (2013).
101. Wright, G. D. Antibiotic Adjuvants: Rescuing Antibiotics from Resistance. Trends Microbiol. 24, 862–871 (2016).
102. Melander, R. J., Melander, C. Antibiotic adjuvants. In: Topics in Medicinal Chemistry. Springer, Berlin, Heidelberg. (2017).
103. White, A. R. et al. Augmentin (amoxicillin/clavulanate) in the treatment of community-acquired respiratory tract infection: a review of the continuing development of an innovative antimicrobial agent. J. Antimicrob. Chemother. 53, i3–20 (2004).
104. Leflon-Guibout, V., Speldooren, V., Heym, B., Nicolas-Chanoine, M. H. Epidemiological Survey of Amoxicillin-Clavulanate Resistance and Corresponding Molecular Mechanisms in Escherichia coli Isolates in France: New Genetic Features of blaTEM Genes. Antimicrob. Agents Chemother. 44, 2709–2714 (2000).
105. Butler, M. S., Blaskovich, M. A., Cooper, M. A. Antibiotics in the clinical pipeline at the end of 2015. J. Antibiot. 70, 3–24 (2017).
106. Venter, H., Mowla, R., Ohene-Agyei, T., Ma, S. RND-type drug e ux pumps from Gram-negative bacteria: molecular mechanism and inhibition. Front. Microbiol. 6, (2015).
107. Pagès, J. M., Masi, M., Barbe, J. Inhibitors of efflux pumps in Gram-negative bacteria. Trends Mol. Med. 11, 382–389 (2005).
108. Lomovskaya, O. et al. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrob. Agents Chemother. 45, 105–116 (2001).
109. Pagès, J. M. et al. Efflux Pump, the Masked Side of -Lactam Resistance in Klebsiella pneumoniae Clinical Isolates. PLOS ONE 4, e4817 (2009).
110. Lomovskaya, O. et al. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrob. Agents Chemother. 45, 105–116 (2001).
111. Ofek, I. et al. Antibacterial synergism of polymyxin B nonapeptide and hydrophobic antibiotics in experimental gram-negative infections in mice. Antimicrob. Agents Chemother. 38, 374–377 (1994).
112. Pagès, J. M., Peslier, S., Keating, T. A., Lavigne, J. P., Nichols, W. W. Role of the Outer Membrane and Porins in Susceptibility of -Lactamase-Producing Enterobacteriaceae to Ceftazidime-Avibactam. Antimicrob. Agents Chemother. 60, 1349–1359 (2015).
113. Zabawa, T. P., Pucci, M. J., Parr, T. R., Lister, T. Treatment of Gram-negative bacterial infections by potentiation of antibiotics. Curr. Opin. Microbiol. 33, 7–12 (2016).
114. Ejim, L. et al. Combinations of antibiotics and nonantibiotic drugs enhance antimicrobial efficacy. Nat. Chem. Biol. 7, 348–350 (2011).
115. Taylor, P. L., Rossi, L., De Pascale, G., Wright, G. D. A forward chemical screen identifies antibiotic adjuvants in Escherichia coli. ACS Chem. Biol. 7, 1547–1555 (2012).
116. Gomez, M. J., Neyfakh, A. A. Genes Involved in Intrinsic Antibiotic Resistance of Acinetobacter baylyi. Antimicrob. Agents Chemother. 50, 3562–3567 (2006).
117. Lee, S. et al. Targeting a bacterial stress response to enhance antibiotic action. Proc. Natl. Acad. Sci. 106, 14570–14575 (2009).

69
118. Breidenstein, E. B. M., Khaira, B. K., Wiegand, I., Overhage, J., Hancock, R. E. W. Complex Ciprofloxacin Resistome Revealed by Screening a Pseudomonas aeruginosa Mutant Library for Altered Susceptibility. Antimicrob. Agents Chemother. 52, 4486–4491 (2008).
119. Fajardo, A. et al. The Neglected Intrinsic Resistome of Bacterial Pathogens. PLOS ONE 3, e1619 (2008).
120. Tamae, C. et al. Determination of antibiotic hypersensitivity among 4,000 single-gene-knockout mutants of Escherichia coli. J. Bacteriol. 190, 5981–5988 (2008).
121. Lazar, S. W., Kolter, R. SurA assists the folding of Escherichia coli outer membrane proteins. J. Bacteriol. 178, 1770–1773 (1996).
122. Rouvière, P. E., Gross, C. A. SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev. 10, 3170–3182 (1996).
123. Hagan, C. L., Silhavy, T. J., Kahne, D. -Barrel membrane protein assembly by the Bam complex. Annu. Rev. Biochem. 80, 189–210 (2011).
124. Ruiz, N., Falcone, B., Kahne, D., Silhavy, T. J. Chemical conditionality: a genetic strategy to probe organelle assembly. Cell 121, 307–317 (2005).
125. Sklar, J. G. et al. Lipoprotein SmpA is a component of the YaeT complex that assembles outer membrane proteins in Escherichia coli. Proc. Natl. Acad. Sci. 104, 6400–6405 (2007).
126. Vorachek-Warren, M. K., Ramirez, S., Cotter, R. J., Raetz, C. R. A triple mutant of Escherichia coli lacking secondary acyl chains on lipid A. J. Biol. Chem. 277, 14194–14205 (2002).
127. Kadrmas, J. L., Raetz, C. R. Enzymatic synthesis of lipopolysaccharide in Escherichia coli. Purification and properties of heptosyltransferase i. J. Biol. Chem. 273, 2799–2807 (1998).
128. Mahalakshmi, S., Sunayana, M. R., SaiSree, L., Reddy, M. yciM is an essential gene required for regulation of lipopolysaccharide synthesis in Escherichia coli. Mol. Microbiol. 91, 145–157 (2014).
129. Tsirigotaki, A., De Geyter, J., Šoštaric, N., Economou, A., Karamanou, S. Protein export through the bacterial Sec pathway. Nat. Rev. Microbiol. 15, 21–36 (2017).
130. Sklar, J. G., Wu, T., Kahne, D., Silhavy, T. J. Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev. 21, 2473–2484 (2007).
131. Schäfer, U., Beck, K., Müller, M. Skp, a Molecular Chaperone of Gram-negative Bacteria, Is Required for the Formation of Soluble Periplasmic Intermediates of Outer Membrane Proteins. J. Biol. Chem. 274, 24567–24574 (1999).
132. Grabowicz, M., Silhavy, T. J. Envelope Stress Responses: An Interconnected Safety Net. Trends Biochem. Sci. 42, 232–242 (2017).
133. Antonoaea, R., Fürst, M., Nishiyama, K. & Müller, M. The Periplasmic Chaperone PpiD Interacts with Secretory Proteins Exiting from the SecYEG Translocon. Biochemistry 47, 5649–5656 (2008).
134. Matern, Y., Barion, B., Behrens-Kneip, S. PpiD is a player in the network of periplasmic chaperones in Escherichia coli. BMC Microbiol. 10, 251 (2010).
135. Götzke, H. et al. YfgM is an ancillary subunit of the SecYEG translocon in Escherichia coli. J. Biol. Chem. 289, 19089–19097 (2014).
136. Pathania, R. et al. Chemical genomics in Escherichia coli identifies an inhibitor of bacterial lipoprotein targeting. Nat. Chem. Biol. 5, 849–856 (2009).
137. Loh, B., Grant, C., Hancock, R. E. Use of the fluorescent probe 1-N-phenylnaphthylamine to study the interactions of aminoglycoside antibiotics with the outer membrane of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 26, 546–551 (1984).
138. Bean, G. J. et al. A22 disrupts the bacterial actin cytoskeleton by directly binding and inducing a low-affinity state in MreB. Biochemistry 48, 4852–4857 (2009).
139. Landström, J. et al. Small molecules containing hetero-bicyclic ring systems compete with UDP-Glc for binding to WaaG glycosyltransferase. Glycoconj. J. 29, 491–502 (2012).
140. Yethon, J. A., Vinogradov, E., Perry, M. B., Whitfield, C. Mutation of the lipopolysaccharide core glycosyltransferase encoded by waaG destabilizes the outer membrane of Escherichia coli by interfering with core phosphorylation. J. Bacteriol. 182, 5620–5623 (2000).
141. Patel, D. S. et al. Dynamics and Interactions of OmpF and LPS: Influence on Pore Accessibility and Ion Permeability. Biophys. J. 110, 930–938 (2016).

70
142. Muheim, C. et al. Identification of a Fragment-Based Scaffold that Inhibits the Glycosyltransferase WaaG from Escherichia coli. Antibiot. 5, (2016).
143. Hajduk, P. J., Greer, J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug Discov. 6, 211–219 (2007).
144. de Kloe, G. E., Bailey, D., Leurs, R., de Esch, I. J. P. Transforming fragments into candidates: small becomes big in medicinal chemistry. Drug Discov. Today 14, 630–646 (2009).
145. Cerveny, L. et al. Tetratricopeptide Repeat Motifs in the World of Bacterial Pathogens: Role in Virulence Mechanisms. Infect. Immun. 81, 629–635 (2013).
146. Harms, N. et al. The early interaction of the outer membrane protein PhoE with the periplasmic chaperone Skp occurs at the cytoplasmic membrane. J. Biol. Chem. 276, 18804–18811 (2001).
147. Maurer, L. M., Yohannes, E., Bondurant, S. S., Radmacher, M., Slonczewski, J. L. pH Regulates Genes for Flagellar Motility, Catabolism, and Oxidative Stress in Escherichia coli K12. J. Bacteriol. 187, 304–319 (2005).
148. House, B. et al. Acid-stress-induced changes in enterohaemorrhagic Escherichia coli O157:H7 virulence. Microbiol. Read. Engl. 155, 2907–2918 (2009).
149. Stincone, A. et al. A systems biology approach sheds new light on Escherichia coli acid resistance. Nucleic Acids Res. 39, 7512–7528 (2011).
150. Kannan, G. et al. Rapid acid treatment of Escherichia coli: transcriptomic response and recovery. BMC Microbiol. 8, 37 (2008).
151. Barker, C. A. et al. Degradation of MAC13243 and studies of the interaction of resulting thiourea compounds with the lipoprotein targeting chaperone LolA. Bioorg. Med. Chem. Lett. 23, 2426–2431 (2013).