studies on the role of cyclic amp in platelet function
TRANSCRIPT

STUDIES ON THE ROLE OF CYCLIC AMP IN PLATELET FUNCTION *
Brian Cole, G. Alan Robison,+ and Robert C. Hartmann
Departments of Physiology, Pharmacology, and Medicine Vanderbilt University School of Medicine
Nashville, Tenn. 37203
Introduction
Evidence that cyclic AMP plays an important physiological role as an inhibitor of platelet aggregation has been reviewed recently by Salzman and Levine,' and will be briefly summarized here.
The prostaglandins, which inhibit platelet aggregation in response to ADP and other were shown by Scott and colleagues5vG to be potent stimulants of platelet adenyl cyclase, and this has been confirmed by others.'. i-' In line with this, the prostaglandins also increase the intracellular level of cyclic AMP in intact platelets.lo The order of potency of the prostaglandins in stimulating platelet adenyl cyclase and increasing the level of cyclic AMP has been found to be the same as their order of potency as inhibitors of platelet aggregation.
by a process that appears to be mediated by an interaction with adrenergic a-receptors.l2 Sutherland l5
had previously suggested that a-adrenergic effects might be associated with a fall in the level of cyclic AMP, and indeed such an effect has now been demonstrated in several systems,' 1 including, at least under some conditions, human blood platelets.lO. 15. lfi This effect may be secondary to inhibition of adenyl cyclase activity.', have reported that ADP also produces a fall in the level of cyclic AMP in these cells.
Still another piece of evidence is that exogenous cyclic AMP itself inhibits platelet aggregation, with the dibutyryl derivative being more effective than the parent nuc1eotide.l. ' This is relatively weak evidence when viewed in isolation, because, with the exception of ADP, other adenine nucelotides also inhibit platelet aggregation. It is thought that these other nucleotides may become effective only after conversion to adenosine, which is the most potent inhibitor of aggregation of the naturally occurring adenine compounds.'
An important missing link in this chain of evidence has been the lack of any demonstration that the prostaglandins and agents that inhibit phosphodi- esterase act synergistically to inhibit platelet aggregation. The methylxanthines have been reported to oppose the aggregating effect of epinephrine," and caffeine and prostaglandin E, (PGE,) do act synergistically to increase the accumulation of cyclic AMP in broken cell preparations.R The latter type of synergism should be reflected at the functional level, and one of the aims of the present research was to obtain evidence on this point.
Epinephrine stimulates platelet aggregation
In addition, Salzman and Neri
These investigations were supported, in part, by grants AM-14240 and HE-03509 from the National Institutes of Health.
i Investigator, Howard Hughes Medical Institute.
477

478 Annals New York Academy of Sciences
A second aim of this research was to investigate in more detail the cyclic AMP-lowering effects of epinephrine and ADP, with a view toward determining whether the effects of these agents on aggregation could be under- stood in terms of their effects on cyclic AMP. Some preliminary results of this research were reported at the 54th Annual Meeting of the Federation of American Societies for Experimental Biology.17
Methods and Materials
Blood was collected through an 18-gauge needle attached to a 20-inch venotube from normal human donors and mixed with %O volume of 3.2% sodium citrate. Platelet-rich plasma (PRP) was collected by centrifugation of the whole blood at 120 x g for eight minutes at room temperature. The PRP was pooled and recentrifuged as before to eliminate white and red blood cell contamination. Platelet-free plasma (PFP) was obtained by spinning the red cell suspension at 10,000 x g for five minutes at room temperature and plasma collected. Platelets were counted on a Coulter counter, and the platelet con- centration then adjusted with PFP to 200,000 to 300,000 plateletslcmm.
Platelet aggregation was measured using a 4-ml aliquot of PRP. Epineph- rine, ADP, or other agents were added in volumes of 200 X or less to achieve the final concentrations as reported. Distilled water was generally used as a control, and the total volume of additions never exceeded 600 A per sample. The samples were preincubated for five minutes with PGE,, theophylline, or water before the aggregating agents were added. Aggregation was measured using a Chronolog platelet aggregometer, and the data analyzed according to the method of Baumgartner and Born.lR
For experiments in which cyclic AMP was measured, the samples were poured into 0.1 N HCI in an operating Waring blender, and cyclic AMP assayed in purified extracts essentially as described by Butcher and coworkers.'" Most of the data reported in this communication were corrected by subtracting the amount of cyclic AMP initially present in plasma as determined by assaying an aliquot of the appropriate PFP.
The PGE, used in this study was a gift from Dr. J. E. Pike of the Upjohn Co. Stock solutions were prepared by dissolving 10 mglml PGE, in 95% ethanol and then diluting 1:100 with a solution containing 0.1 mg/ml K,CO, and 63 pglml bovine serum albumin. Further dilutions were made with dis- tilled water. /-Epinephrine HCI was purchased from K and K Labs. A stock solution of 5 x lo-' M was prepared with 40 pglml bovine serum albumin added. Phentolamine mesylate was a gift from Dr. A. J. Plummer of the Ciba Pharmaceutical Company. It was dissolved in distilled water immediately before use. ADP was purchased from the Sigma Chemical Company. It was dissolved in 0.01 M HEPES at pH 6.8 and stored at -70" C until the day of use.
Results
The tracings from a typical aggregation experiment are shown in FIGURE 1. Epinephrine was added to each sample, as indicated by the arrows. The left- hand tracing, obtained in the absence of any added inhibitor, is typically

Cole et al.: Platelet Function 479
S M l N ,
CON [EP~]
The0 PGEl PGE I The0
FIGURE 1 . Effect of theophylline and PGE, on the platelet aggregation response to epinephrine. Theophylline (1 mM), or PGE, (0.13 pM), or both were added five minutes before the addition of 2.3 x 10.' M epinephrine (epi), and aggregation mea- sured as described in text. Arrows indicate time of addition of epinephrine. Control curve (CON) represents effect of epi in absence of theophylline or PGEI.
biphasic. At this concentration of epinephrine (2.3 x M ) , the maximal response has always been reached within ten minutes. Theophylline added five minutes earlier reduced the response to epinephrine slightly in this experi- ment, whereas PGE, caused an obvious reduction in the response. The addition of theophylline and PGE, together completely prevented the response to epinephrine.
The results of 17 similar experiments, analyzed by the method of Baum- gartner and Born, are summarized in TABLE 1. The mean height of the first phase of the response to epinephrine was 30.2 mm. The apparent slight reduc- tion in the presence of theophylline was not significant at the 5 % level. The
TABLE 1
ANALYSIS OF FIRST PHASE AGGREGATION RESPONSE
Agent Response (mm*)
Epi (2.3 x M) 30.2 2 1.9 Epi (2.3 x lO-'M), Theo (1.0 mM) 26.8 2 5.1
O & O Epi (2.3 x M ) , PGEl (0.13 1M) 6.2 & 1.0 Epi (2.3 x M ) , PGE, (0.13 pM), The0 ( 1.0 mM)
* Av & SEM.
480 Annals New York Academy of Sciences
addition of PGE, did decrease the response significantly, while the addition of PGE, and theophylline together, as in FIGURE 1, completely prevented the response.
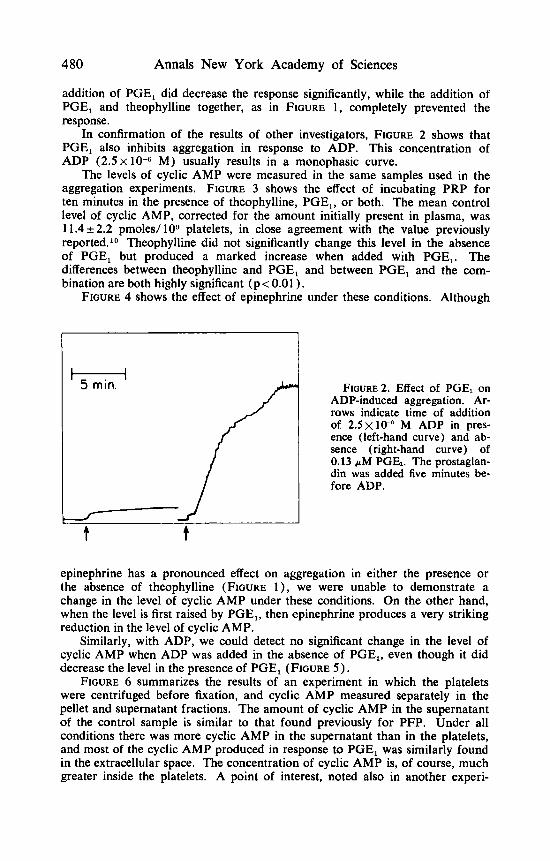
In confirmation of the results of other investigators, FIGURE 2 shows that PGE, also inhibits aggregation in response to ADP. This concentration of ADP (2.5 x
The levels of cyclic AMP were measured in the same samples used in the aggregation experiments. FIGURE 3 shows the effect of incubating PRP for ten minutes in the presence of theophylline, PGE,, or both. The mean control level of cyclic AMP, corrected for the amount initially present in plasma, was 1 1.4 & 2.2 pmoles/ loD platelets, in close agreement with the value previously reported.1° Theophylline did not significantly change this level in the absence of PGE, but produced a marked increase when added with PGE,. The differences between theophylline and PGE, and between PGE, and the com- bination are both highly significant (p < 0.01 ).
FIGURE 4 shows the effect of epinephrine under these conditions. Although
M) usually results in a monophasic curve.
H
/ f 5 min. FIGURE^. Effect of PGEl on ADP-induced aggregation. Ar- rows indicate time of addition of 2 . 5 ~ 1 0 - " M ADP in pres- ence (left-hand curve) and ab- sence (right-hand curve) of 0.13 p M PGE,. The prostaglan- din was added five minutes be- fore ADP.
epinephrine has a pronounced effect on aggregation in either the presence or the absence of theophylline (FIGURE l ) , we were unable to demonstrate a change in the level of cyclic AMP under these conditions. On the other hand, when the level is first raised by PGE,, then epinephrine produces a very striking reduction in the level of cyclic AMP.
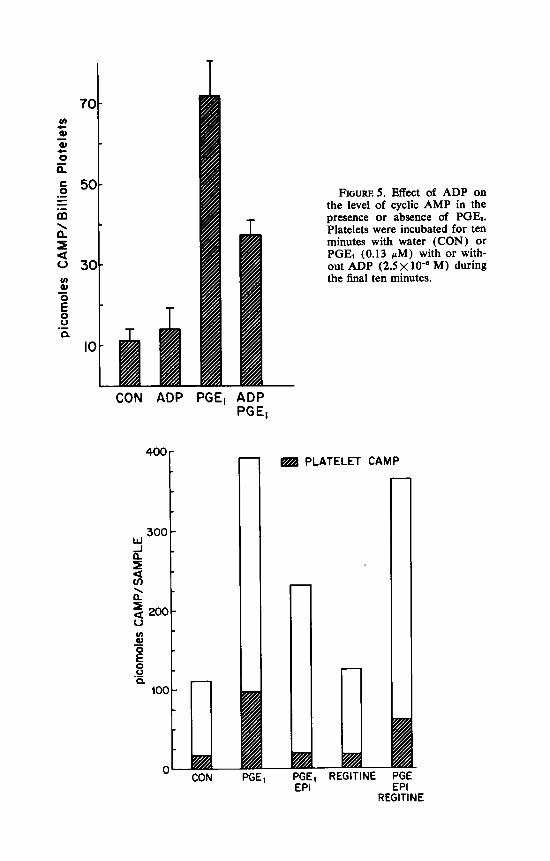
Similarly, with ADP, we could detect no significant change in the level of cyclic AMP when ADP was added in the absence of PGE,, even though it did decrease the level in the presence of PGE, (FIGURE 5 ) .
FIGURE 6 summarizes the results of an experiment in which the platelets were centrifuged before fixation, and cyclic AMP measured separately in the pellet and supernatant fractions. The amount of cyclic AMP in the supernatant of the control sample is similar to that found previously for PFP. Under all conditions there was more cyclic AMP in the supernatant than in the platelets, and most of the cyclic AMP produced in response to PGE, was similarly found in the extracellular space. The concentration of cyclic AMP is, of course, much greater inside the platelets. A point of interest, noted also in another experi-

T 140-
5 120- - al 0 c
z i 100- c 0 .- - *= 80- m a 5 60- 2 40- E
n
\
u u)
0
0 .g 20-
CON THEO PGE, THEO PGE I
FIGURE 3. Effect of PGE, on platelet cyclic AMP levels in the presence and absence of theophylline. Platelet-rich plasma was incubated for ten minutes with the addition of water (CON), 1 mM theophylline (THEO), 0.13 pM prostaglandin El (PGE,), or both THEO and PGE, together. Samples were immediately homogenized in the presence of 0.1 M HCI, and cyclic AMP measured in purified extracts. Values plotted represent cyclic AMP in the samples less the amount initially present in platelet-free plasma, and are the means of 14 separate experiments.
CON EPI THEO THEO PGEl PGEI PGEl PGEI EPI EPI THEO THEO
EPI FIGURE 4. Effect of 2.3 x lo-% M epinephrine (EPI) on cyclic AMP levels in pres-
ence or absence of theophylline and PGE,. This Figure includes data shown in FIGURE 3, but also shows the effect of epinephrine added five minutes after the other agents and incubated five minutes with epinephrine before fixation.
T
300 W A 0,
cn 5 a' = 200- 4
T
-
FIGURES. Effect of ADP on the level of cyclic AMP in the presence or absence of PGE,. Platelets were incubated for ten minutes with water (CON) or PGE, (0.13 pM) with or with- out ADP ( 2 . 5 ~ lo-' M) during the final ten minutes.
CON ADP PGEl
!,ool n
ADP PG El
" CON PG
PLATELET CAMP
EF
1 REG11 ii PGE-
EPI REGlTlNE

Cole et al.: Platelet Funct ion 483
ment, is that some of the cyclic A M P that disappears in response to epineph- rine appears to come from the extracellular component. However, more experi- ments will be required to establish this point. Lastly, this Figure shows that phentolamine inhibits the cyclic AMP-lowering effect of epinephrine, as reported previously. lo
PGE 0.13uM 0 + 0 + + + + + EPI 2.2 to-) M o o + - I - - + + + + SECONDS 7 15 30 60 120
FIGURE 7. Time-course of the cyclic AMP-lowering effect of epinephrine. The samples represented by the first three bars were incubated for seven minutes with the indicated concentrations of PGE, or epinephrine. The samples represented by the last five bars were incubated for five minutes with PGE, and then for the indicated number of seconds with epinephrine. Samples were fixed and assayed as described in legend for FIGURE 3.
The results of a time-course experiment are shown in FIGURE 7, illustrating the striking rapidity with which the level of platelet cyclic AMP falls in response to epinephrine. It can be seen that within seven seconds of the addition of epinephrine the concentration of cyclic A M P fell almost to base-line level, after having been first elevated by PGE,. The results of a similar experiment in
FIGURE 6. Distribution of cyclic AMP between platelets and plasma under several conditions. Platelet-rich plasma was incubated for ten minutes in the presence or absence of PGE, with or without epinephrine (EPI) or phentolamine (REGITINE) present during the last five minutes. Samples were then centrifuged at 4°C for five minutes, and the supernatant and pellets frozen separately. Each sample contained 1.96 x 10" platelets in a total volume of 4 ml. The open portion of each bar represents cyclic AMP in the supernatant and the cross-hatched portion represents that present in the pellet. The concentration of PGE, (1 .3 pM) in these experiments was supra- maximal. The concentration of epinephrine was 2 .3 x and of phentolamine 5x 10-5 M.

484 Annals New York Academy of Sciences
u 10 n
-
, . . 1 . . 1 . . 1
which ADP was included are shown in FIGURE 8. From this it can be seen that ADP appears to act somewhat less rapidly than epinephrine under these same conditions.
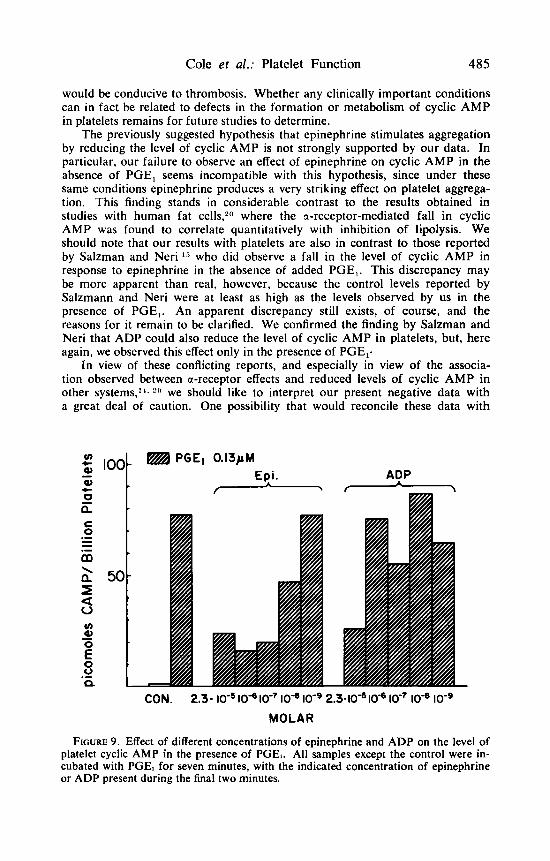
Finally, FIGURE 9 depicts the results of an experiment in which platelets were incubated with PGE, and different concentrations of either epinephrine or ADP. It would appear that under these conditions epinephrine is more potent than ADP insofar as its ability to lower the level of cyclic AMP is concerned.
Discussion
The demonstration that PGE, and theophylline act synergistically to increase the level of platelet cyclic AMP and to inhibit aggregation can be added to the evidence previously summarized that the prostaglandins inhibit platelet aggregation by increasing the level of cyclic AMP. All of the available evidence supports this hypothesis and there is no evidence that is incompatible with it. This in turn lends additional support to the suggestion made by many investi- gators that the level of cyclic AMP in platelets is probably an important de- terminant of the tendency of these cells to aggregate. Abnormally high levels would interfere with normal clot formation, whereas abnormally low levels
Cole et al.: Platelet Function 485
would be conducive to thrombosis. Whether any clinically important conditions can in fact be related to defects in the formation or metabolism of cyclic AMP in platelets remains for future studies to determine.
The previously suggested hypothesis that epinephrine stimulates aggregation by reducing the level of cyclic AMP is not strongly supported by our data. In particular, our failure to observe an effect of epinephrine on cyclic AMP in the absence of PGE, seems incompatible with this hypothesis, since under these same conditions epinephrine produces a very striking effect on platelet aggrega- tion. This finding stands in considerable contrast to the results obtained in studies with human fat cells,2n where the a-receptor-mediated fall in cyclic AMP was found to correlate quantitatively with inhibition of lipolysis. We should note that our results with platelets are also in contrast to those reported by Salzman and Neri l 5 who did observe a fall in the level of cyclic AMP in response to epinephrine in the absence of added PGE,. This discrepancy may be more apparent than real, however, because the control levels reported by Salzmann and Neri were at least as high as the levels observed by us in the presence of PGE,. An apparent discrepancy still exists, of course, and the reasons for it remain to be clarified. We confirmed the finding by Salzman and Neri that ADP could also reduce the level of cyclic AMP in platelets, but, here again, we observed this effect only in the presence of PGE,.
In view of these conflicting reports, and especially in view of the associa- tion observed between a-receptor effects and reduced levels of cyclic AMP in other systems,". we should like to interpret our present negative data with a great deal of caution. One possibility that would reconcile these data with
u) Q, z 0 V .- - 1 P
PGEl 0.13pM Epi. - ADP
CON. 2.3- lo-' lod lo-' loqe lo-' 2.3-10" lo-' lo-' lo-'
MOLAR
FIGURE 9. Effect of different concentrations of epinephrine and ADP on the level of platelet cyclic AMP in the presence of PGEI. All samples except the control were in- cubated with PGE, for seven minutes, with the indicated concentration of epinephrine or ADP present during the final two minutes.

486 Annals New York Academy of Sciences
the fact that a-receptors have been shown to mediate a fall in the level of cyclic AMP in every system studied (at least under some conditions) would be that these receptors are part of some system that not only reduces the level of cyclic AMP but also does something else. According to this hypothesis, adrenergic a-receptors would be analogous to the receptors for insulin in rat liver and fat cells.21. 22 If this were the case, it is clear that some a-receptor effects could result from a lack of cyclic AMP, whereas other could be caused by “something else.” The observation that epinephrine can interact with a-receptors to inhibit platelet adenyl cyclase in homogenates under some con- ditions would not necessarily be incompatible with this hypothesis.
The other possibility, of course, is that epinephrine did reduce the level of cyclic AMP in our experiments in the absence of PGE,, but, for any of several reasons, we simply failed to see it. It seems possible, for example, that a substantial fraction of the cyclic AMP that we measured under control con- ditions was bound in a metabolically inactive form, creating a background sufficiently high that small but functionally important changes could not be detected against it. We think, in any event, that more experiments will be needed before we can draw any stronger conclusions.
Although the ability of ADP to reduce the level of cyclic AMP under any conditions is of interest, it seems unlikely, for several reasons, that ADP and epinephrine ‘stimulate platelet aggregation by precisely the same mechanism. First, the synergism that has been observed between these agents I t would seem to imply more than one mechanism, and the results of preliminary experi- ments in our laboratory indicate that they do not act synergistically to reduce the level of cyclic AMP. A second reason is that the responses to these agents are not identical, in that aggregation in response to epinephrine is associated with a reduction in cell volume, whereas that seen in response to ADP is associated with platelet swelling.I~ 4
Finally, we should like to mention our preliminary finding that adenosine itself is capable of increasing the level of cyclic AMP in platelets, at least under some conditions. This effect appears to depend very critically on the concentration of adenosine used. Sattin and Rall 23 had previously found that adenosine could increase the level of cyclic AMP in guinea pig brain slices, an effect that could, interestingly enough, be prevented by theophylline. Whether theophylline will prevent the effect on platelet cyclic AMP and whether this effect can be related in any way to the ability of adenosine to inhibit platelet aggregation are questions currently being investigated.
Summary
Prostaglandin E, and theophylline, which stimulate platelet adenyl cyclase and inhibit platelet phosphodiesterase, respectively, act synergistically to raise the level of platelet cyclic AMP and to inhibit platelet aggregation. These findings support the hypothesis that these agents inhibit aggregation by way of cyclic AMP and further support the view that cyclic AMP plays an important role in regulating platelet function. Epinephrine and ADP were found to reduce the level of platelet cyclic AMP if the level was first raised by PGE,, but had no apparent effect on the level of cyclic AMP in the absence of PGE,. Since epinephrine causes platelets to aggregate under these conditions, the data do not support the hypothesis that this effect occurs as the result of a fall in

Cole et al.: Platelet Function 487
the level of cyclic AMP. However, the possibility that epinephrine did reduce the level of cyclic AMP in these experiments, but that this was obscured by a technical artifact. has not been ruled out.
Acknowledgments
We should like to thank Bonnie Freeman and Julie Totty for their excellent technical assistance.
References
1. SALZMAN, E. W. & L. LEVINE. 1971. J. Clin. Invest. 5 0 131. 2. KLOEZE, J. 1967. In Prostaglandins. (Nobel Symposium 2). S. Bergstrom &
3. WEEKS, J. R., N. C. SEKHAR & D. W. DUCHARME. 1969. J. Pharm. Pharmacol.
4. MUSTARD, J. F. & M. A. PACKHAM. 1970. Pharmacol. Rev. 22: 97. 5. BUTCHER, R. W., R. E. SCOTT & E. W. SUTHERLAND. 1967. The Pharmacologist
6. SCOTT, R. E. 1970. Blood 35: 519. 7. WOLFE, S. M. & N. R. SCHULMAN. 1969. Biochem. Biophys. Res. Comm. 35: 265. 8. MARQUIS, N. R., R. L. VIGDAHL & P. A. TAVORMINA. 1969. Biochem. Biophys.
9. MOSKOWITZ, J., J. P. HARWOOD, W. D. REID & G. KRISHNA. 1970. Fed. Proc.
10. ROBISON, G. A., A. ARNOLD & R. C. HARTMANN. 1969. Pharmacol. Res. Comm.
11. ARDLIE, N. G., G. GLEW & C. J. SCHWARTZ. 1966. Nature 212: 415. 12. O’BRIEN, J. R. 1963. Nature 200: 763. 13. SUTHERLAND, E. W. 1965. In Pharmacology of Cholinergic and Adrenergic
Transmission. G. B. Koelle, W. W. Douglas & A. Carlsson, Eds. : 317. Mac- millan (Pergamon). New York, N.Y.
14. ROBISON, G. A. & E. W. SUTHERLAND. 1970. Circ. Res. 26 (Suppl. I): 1-147. 15. SALZMAN, E. W. & L. L. NERI. 1969. Nature 224: 601. 16. MARQUIS, N. R., J. A. BECKER & R. L. VIGDAHL. 1970. Biochem. Biophys. Res.
17. COLE, B., G. A. ROBISON & R. C. HARTMANN. 1970. Fed. Proc. 29: 316 Abs. 18. BAUMGARTNER, H. R. & G. V. R. BORN. 1968. Nature 218: 137. 19. BUTCHER, R. W., R. J. Ho, H. C. MENG & E. W. SUTHERLAND. 1965. J. Biol.
20. ROBISON, G. A,, P. E. LANCLEY & T. W. BURNS. 1971. Biochem. Pharmacol. In
21. PARK, C. R., 0. B. CROFFORD & T. KONO. 1968. J. Gen. Physiol. 52: 296s. 22. VILLAR-PALASI, C., N. D. GOLDBERG, J. S. BISHOP, F. Q. NUTTALL, K. K.
SCHLENDER & J. LAMER. 1970. In Biogenic Amines as Physiological Regula- tors. J. J. Blum, Ed. : 161-180. Prentice-Hall. Englewmd Cliffs, N.J.
B. Samuelson, Eds. : 241-252. Interscience. New York, N.Y.
21: 103.
9: 172.
Res. Comm. 3 6 965.
29: 602 (abstr.)
1: 325.
Comm. 39: 783.
Chem. 240 4515.
press.
23. SATTIN, A. & T. W. RALL. 1970. Mol. Pharmacol. 6 13.

DISCUSSION
W. F. H. M. Mommaerts, Moderator
University of California Medical School Los Angeles, Calif.
QUESTION: Are there really such things as a- and p-adrenergic receptors? DR. MAYER: There are at least two types of receptors in muscle. In
skeletal muscle blood vessels there are both a- and preceptors on the smooth muscle, the former mediating constriction, the latter dilation. These can be demonstrated in the classical manner using blocking agents. Cholinergic sympathetic nerve fibers have also been demonstrated. In the experiments described in our paper we are presumably dealing with receptors that exist on the surface of the skeletal muscle membrane. It is conceivable that there are a-receptors here also, but this seems unlikely in view of the fact that the only response that we and others have observed is the stimulation of glycogenolysis, which follows the pattern by which 8-adrenergic stimulation is classically demonstrable. Both in vivo and with adenyl cyclase preparations in vitro, isoproterenol is the most potent and norepinephrine the least potent. Also, 8-blocking agents are effective while a-blocking agents are not. How- ever, since a-blocking agents prevent the vasoconstrictor effect of norepineph- rine and epinephrine, they can potentiate the effects of these two agents by allowing more of them to get to the receptors. This was demonstrated by Hornbrook and Brody several years ago.
DR. NAHAS: In your slide of the gracilis muscle preparation there was a transducer present. I wonder if you have any record of the functional events following administration of isoproterenol, especially in relation to the biochemical alterations you discussed.
DR. MAYER: The experiments I presented involve no functional changes in the muscle in terms of changes in contraction, since these muscles are at rest and isoproterenol does not stimulate contraction. On the other hand, evidence from the literature indicates that fairly large doses of catecholamines increase twitch tension and the duration of the active state when this type of muscle (i.e., fast-contracing muscle) is simultaneously stimulated elec- trically. The purpose of the transducer is to provide a measurement of developed tension during electrical stimulation of the gracilis muscle. So far we have found that tetanic electrical stimulation does not alter the biochemical responses to isoproterenol. We have not yet investigated the effect of cate- cholamines on twitch tension in relation to stimulation of the glycogenolytic system.
DR. NAHAS: It is still fair to say, isn't it, that whatever adrenergic effects upon contraction exist, they are very small compared to the cardiac effects?
DR. MAYER: Yes. Catecholamines affects calcium exchange in resting skeletal muscle, but this is not necessarily coupled with contraction.
DR. I. KLEIN (New York University, New York, N.Y.): Dr. Mayer's very nice formulation brings to mind the paper by Danforth, Helmreich, and Cori of a number of years ago, in which they showed that electrical stimu- lation per se, at least in their preparation, stimulated phosphorylase b kinase
488

Mommaerts : Discussion 489
and that this stimulation took place even in the presence of propranolol. How does this fit in with the formulation that you presented here?
DR. MAYER: It fits in rather nicely. Time didn’t permit me to discuss these observations by Danforth and his colleagues. However, they didn’t demonstrate phosphorylase kinase activation, but measured phosphorylase b to a transformation, i.e., phosphorylase kinase activity. In the presence of a /3-blocking agent, electrical stimulation led to an increase in the latter, but, as Posner, Stein, and Krebs later discovered, without cyclic AMP formation. More recently, Drummond, Harwood, and Powell confirmed this and also showed a lack of phosphorylase kinase activation. Danforth and his colleagues had postulated, as early as 1962, that the key factor was calcium. Thus, evidence has been accumulating that the activation of phosphorylase kinase that takes place in response to electrical stimulation is not due to transformation from the nonactivated to the activated state, i.e., transformation from a less phosphorylated to a more phosphorylated enzyme, but rather that calcium stimulates the catalytic activity of phos- phorylase kinase.
DR. KLEIN: Would that be referred to as phosphorylase a then, or would it still be phosphorylase b?
DR. MAYER: Presumably, phosphorylase kinase does not have catalytic activity in resting muscle because there isn’t enough free calcium available. But during the process of excitation-contraction coupling, enough calcium is made available to activate the enzyme, but without the formation of new covalent bonds, i.e., without transformation to the phosphorylated form.’
DR. J. LARNER: Is Dr. Villar-Palasi here? He has recently demonstrated that the inactive or nonactive form of the kinase is under very definite control by energy charge and he has shown that the enzyme is essentially “turned on” at decreasing energy charge.’ This could be at least in part the mechanism of control of phosphorylase interconversion during muscular contraction.
DR. MAYER: I wonder if Dr. Levey would comment on what the relation- ship is between the activity of adenyl cyclase and how much cyclic AMP is produced. The reason I ask this question is that in the dog heart in situ and also in the isolated rat heart, large doses of epinephrine cause the formation of concentrations of cyclic AMP in excess of what is required either for maximal activation of phosphorylase or maximal contraction of cardiac muscle. Therefore, is there any functional significance to a reduction in adenyl cyclase? How much does adenyl cyclase have to be reduced before it becomes a limiting factor?
DR. G. S. LEVEY: The only good evidence on this subject that I know of comes from the data reported by Grahame-Smith and his colleague^.^ They demonstrated that approximately 1% of the actual cyclic AMP produced by ACTH was necessary for maximal steroidogenesis. Drs. Skelton and Epstein and myself encountered this problem when we examined adenyl cyclase activity in hearts from hypothyroid cats. We found a 30% reduction in the total amount of adenyl cyclase present in these hearts without any alteration in the physiologic responses of the papillary muscles from the same hearts. I think we can say from our studies that certainly a 30% decrease means nothing. If the increase in intracellular cyclic AMP necessary for a physiologic response is small, it would be impossible to predict the significance of a decrease in the amount of adenyl cyclase. However, the

490 Annals New York Academy of Sciences
crucial point here is that the physiologic parameter as measured by the papillary muscle response would also be altered if the cyclase were significantly decreased. In the absence of a physiologic alteration, we probably could say nothing about the decrease in adenyl cyclase activity.
QUESTION: Have there been any guesses as to how phosphatidylserine abolishes adenyl cyclase activity? And what do you add to dextran to make it blue?
DR. LEVEY: I didn’t add anything to it. Blue dextran is commercially available. In answer to the first question, I don’t understand the mechanism of the phosphatidylserine inhibition of the fluoride stimulation.
DR. W. A. PETTINGER (Hoflmann-La Roche, Nutley, N . J . ) : We’ve had experience reversing congestive heart failure of different types in the dog using a potent phosphodiesterase inhibitor, RO 7-2956. This compound was dramatically effective.4 I’m curious as to whether your system may at least in the hernodynamic sense be reversed by a drug like theophylline?
DR. LEVEY: We haven’t studied it. DR. R. WALTER (Mount Sinai School of Medicine, New York City, N . Y . ) :
I have a comment on the earlier question of how much cyclic AMP is required to attain a maximal physiological response. We studied this problem by comparing the action of neurohypophyseal hormones and synthetic analogs with two types of toad urinary bladder preparations: (1) at the level of the intact tissue and (2) at the level of a broken-cell adenyl cyclase preparation of the bladder. When 8-arginine-vasotocin (the natural water balance principle of the toad, and therefore a full agonist in the intact bladder) was compared with a partial agonist (a peptide which elicits a lower maximal hydroosmotic response in the intact bladder) in the broken-cell preparation, the hormone was found capable of stimulating cyclic AMP production approximately ten times as much as the partial agonist. Thus, it can be con- cluded that only partial receptor occupation-or synchronously, partial en- hancement of adenyl cyclase activity-is required to elicit a full physiologic response.
DR. T. W. RALL: I would like to offer a few comments regarding smooth muscle. Of course I would like to agree that the relaxing effects of the catecholamines on smooth muscle are mediated through the aegis of cyclic AMP. However, I feel the available evidence for this, including the evidence presented today, is full of pitfalls. I don’t argue with the evidence itself, but I do argue against the idea that it is conclusive, and I think we have to be cautious in trying to interpret it. First of all, when one measures the absolute level of cyclic AMP in smooth muscle, in this case rabbit thoracic aorta, one sees that the changes during contraction and relaxation are very small. Secondly, and perhaps more importantly, we’re dealing with a com- posite tissue, a tissue which is not uniform in its composition. It may be made up of a number of different cell types, each with differing amounts of cyclic AMP under control conditions and with different responses to the catecholamines or methylxanthines under experimental conditions. This is especially important if one is trying to compare the ratio of phosphodiesterase to adenyl cyclase as one progresses down the arterial tree. I submit that this does not allow you to derive any conclusions about what the physiological properties of the tissue would be.
Potentiation by theophylline may also be hard to interpret, unless you can show some reverse effects. For example, what agents which will con-

Mommaerts: Discussion 49 1
tract the aorta are not reversed by theophylline? Dr. Triner showed a slide where phenykphrine in the presence of theophylline produced accentuated relaxation. I’ve done dose-response curves with histamine and serotonin as well as the catecholamines, and found that in the presence of theophylline the dose-response curves for contraction are shifted to the right, meaning that in the presence of theophylline you have to add more of the agent in order to produce the same amount of contraction. Now either all of these agents are producing cyclic AMP, and therefore the opposing relaxing effect is being potentiated by theophylline, or else theophylline has a non- specific effect which would be expressed in the presence of any agent which causes contraction. So, I think we need more data before we can consider this point nailed down. I would like to think it’s nailed down but I don’t believe it is.
DR. R. F. FURCHGOTT (Downstate Medical Center, Brooklyn, N . Y . ) : I think Dr. Triner has presented us with some very provocative evidence. His results to this point are not inconsistent with the hypothesis that cyclic AMP is the mediator of relaxation produced by catecholamines and a number of other agents in smooth muscle. One would naturally feel more sanguine about the hypothesis if there were data showing a positive temporal relationship between the degree of relaxation and the tissue concentration of cyclic AMP during the time-course of action of a relaxing agent, but such data are not presently available.
Dr. Triner has used certain selected relaxing agents, but there are others to try. For example, on the rabbit aortic strip, 5’-AMP and adenosine are about as potent as theophylline as relaxants, and inorganic nitrite and glyceryl- trinitrate are much more potent. The question arises whether we are going to invoke cyclic AMP as a mediator in the relaxation produced by all these agents.
On the basis of pharmacological characteristics, there appear to be different “subtypes” of 8-receptors in different tissues. On the rabbit aortic strip, the potency ratio of epinephrine to norepinephrine for relaxation is about 50: 1 whereas, on small coronary arteries of the dog, according to David Bohr, this potency ratio is about 1 : 10. In both kinds of blood vessels the catecholamines are acting on 8-receptors. If the hypothesis about cyclic AMP as a second messenger for relaxation is correct, then one would expect to find a high potency ratio of epinephrine to norepinephrine for activation of cyclase in the rabbit artery, and a low potency ratio for activation in dog coronary arteries.
My final comment is that one should be wary of other actions besides the inhibition of phosphodiesterase that may be exerted by the agents selected as inhibitors of this enzyme. For example, when aortic strips are exposed to papaverine at concentrations about M, the stimulating effects of even strong agonists like phenylephrine are unsurmountably inhibited, whereas with the other relaxing agents I mentioned this is not the case.
DR. PETTINGER: I think I have some evidence that relates to several of these compounds. Dr. Hoffer at Hoffmann-La Roche has synthesized a series of phosphodiesterase inhibitors with a spectra of potency up to 3,000 times that of theophylline. The vasodilatory and antihypertensive activity of this series of compounds correlates (p < 0.01 ) with the phosphodiesterase inhibiting activity very closely. This correlation within a series of very similar com- pounds suggests that the cyclic AMP system is a mediator of some drug-induced vasodilatory activities.5
DR. MAYER: But how much phosphodiesterase inhibition is required

492 Annals New York Academy of Sciences
before the enzyme becomes a rate-limiting step? In heart and in skeletal muscle, with cyclic AMP concentrations in the micromolar range, that is, in physiological concentrations, there is an excess of phosphodiesterase over hormone-stimulatable adenyl cyclase such that the enzyme has to be inhibited at least 90% before it is likely to be rate-limiting in the metabolism of cyclic AMP.
DR. L. VOLICER (Boston University School of Medicine, Boston, Mass.) : I would like to present additional information in support of the role of cyclic AMP in vascular smooth muscle. At the 1970 Fall ASPET Meeting we reported that it is possible after gentle homogenization to stimulate adenyl cyclase activity in the rat aorta by isoproterenol.x Norepinephrine caused only minor stimulation of adenyl cyclase activity, which was not statistically signifi- cant. In addition to the effects of catecholamines, we observed inhibition of adenyl cyclase activity in broken cell preparations from the rat aorta by angiotensin. Angiotensin also inhibited incorporation of labeled adenine into cyclic AMP in the rat tail artery in vitro. Theophylline increased the amount of labeled CAMP but did not influence the decrease of cyclic AMP level caused by angiotensin. Isoproterenol caused increased incorporation of labeled adenine into the cyclic AMP and this effect was blocked by addition of propranolol in the same way as Dr. Triner already reported. In our experiments, however, the increase in the cyclic AMP level due to norepinephrine was not only blocked by propranolol but was also reversed to a significant decrease. We expressed the total cyclic AMP activity as percent of ATP-ADP activity present in the same sample rather than as an absolute amount. I wonder if this difference in calculation can explain why Dr. Triner did not see a decrease of cyclic AMP level after treatment with norepinephrine and propranolol.
DR. MOMMAERTS: Are there any questions or comments applicable to Dr. Cole's paper?
DR. M. B. ZUCKER ( N e w York University, New York City, N . Y . ) : There is a third difference I could mention between epinephrine and ADP. which is that epinephrine has very little, really no, aggregating effects in animal platelets, it has this effect only in humans. I wanted to ask Dr. Cole if he had any idea of where the high level of cyclic AMP in plasma comes from, what its stability in plasma is, and finally whether he used heparin or citrate as an anticoagulant? Could he comment on their relative merits?
DR. COLE: We used citrate as an anticoagulant for no better reason than that is what we started using. Since then some people at the Mead Johnson Research Center have shown that platelet adenyl cyclase activity is affected by changing the calcium levels. I don't know whether the citrate concentration we're using modifies the calcium level enough to cause major changes in cyclic AMP or not. I see no reason why heparin could not be used. Indeed it might be better, since its action is not thought to involve calcium. Regarding your other question, the level of cyclic AMP is stable for up to 20 minutes, the longest we have investigated it. The high level in response to prostaglandin E, in these experiments definitely came from the platelets. We don't know whether it just leaks out or is actively extruded.
DR. G. DUBOFF (University of Michigan, Ann Arbor, Mich.) : Dr. Penner and I have also been interested in the role of prostaglandins in platelet function. But I must make a note of caution here regarding your comments about the possible involvement of cyclic AMP in blood coagulation. So far as I know, nothing has been done regarding cyclic AMP or adenyl cyclase and the plasma

Mommaerts: Discussion 493
factors, which are the major features in the coagulation phenomena. Of course the platelet does play a role, but it’s a very minor role. In the case of the platelets in hemophilia A and B, we have not been able to show any significant difference between the effect of PGE, in inhibiting aggregation and PGE, in enhancing aggregation in patients with hemophilia of this type. I notice that no one here has referred to PGE,. which enhances aggregation. If PGE, is really doing what it does by way of cyclic AMP, why is it that no one is using PGE, to show that it counteracts the effect of PGE, via the cyclic AMP system?
showed that PGE, increases cyclic AMP in human platelets, although it was much less effective than PGE,. This was in line with the work of Sekhar and his colleagues at Upjohn, showing that PGE, inhibited the aggregation of human platelets. I believe the work that most people remember, showing the opposite effect, was reported by Kloeze, but this was not done with human platelets. As for the relation of adenyl cyclase to plasma factors, I am not aware of any direct evidence that cyclic AMP is involved in the production of these factors. HOW- ever, I think the evidence that epinephrine increases factor VIII levels by way of adrenergic 8-receptors makes this very much worth investigating. It’s just that we chose to look at platelets. They are a very desirable system to investigate from our point of view.
I would like to correct the impression possibly created by the previous commentator, regarding the importance of this work. The really critical thrombus that kills a lot of us is a white thrombus, not a blood clot, and therefore is primarily based on the kind of study that’s going on here. It’s a frightfully important problem in the question of keeping people alive a bit longer.
DR. COLE: Work reported from our lab last year
DR. H. WEISENBERGER (Beth Israel Hospital, Boston, Mass.) :
References
1. BROSTROM, C. 0. & E. G. KREBS. 1971. J. Biol. Chem. 246 1961. 2. VILLAR-PALASI, C. & S. H. WEI. 1970. Proc. Nat. Acad. Sci. 67: 345. 3. GRAHAM€-SMITH, D. G., R. W. BUTCHER, R. L. NEY & E. W. SUTHERLAND. 1967.
4. OSBORNE, M. W., J. J. WENCER & R. A. MOE. 1971. J. Pharmacol. Exp. Therap.,
5. PETTINGER, W. A., G. T. BAUTZ, G. A. WIGGAN & H. SHEPPARD. 1970. The
6. VIGDAHL, R. L., N. R. MARQUIS & P. A. TAVORMINA. 1969. Biochem. Biophys.
7. ROBISON, G. A., A. ARNOLD & R. C. HARTMANN. 1969. Pharmacol. Res. Com-
8. VOLICER, L. & S. HYNIE. 1970. Pharmacologist 12: 262.
J. Biol. Chem. 242: 5535.
176: 174.
Pharmacologist 12: 291.
Res. Commun. 37: 409.
mun., 1: 325.