structural/electronic properties and reaction energies of a series of mono- and bis-uranyl dihalides...
TRANSCRIPT

ORIGINAL PAPER
Structural/electronic properties and reaction energiesof a series of mono- and bis-uranyl dihalides equatoriallycoordinated by N/O ligands
Jun Yao & Yong-Ming Wang & Qing-Jiang Pan &
Yuan-Ru Guo & Hong-Xing Zhang
Received: 25 March 2014 /Accepted: 12 May 2014# Springer-Verlag Berlin Heidelberg 2014
Abstract Monometallic (UO2)(X)2(L)3 (L=pyridine (py),X=F (1), Cl (2), Br (3) and I (4); L=tetrahydrofuran (thf),X=Cl (5); L=pyrrole (pl), X=Cl (6)) as well as bimetallic[(UO2)(μ2-X)(X)(L)2]2 (L=py, X=F (7), Cl (8), Br (9) and I(10); L=thf, X=Cl (11); L=pl, X=Cl (12); μ2=doublybridged) were examined using relativistic density functionaltheory. With changing from F, Cl, Br to I irregardless of inmono- or bis-uranyl complexes, bond lengths of U=O werecalculated to be decreasing, resulting from strengthening ofaxial U=O bonds while weakening equatorial X→U coordi-nation. This is further evidenced by calculated bond orders ofU=O and stretching vibrational frequencies. A similar situa-tion was is found in 2, 5 and 6 as well as in 8, 11 and 12, whereN/O ligands are varied but the chlorine atoms are retained.The present study reveals that all these complexes have U(f)-character low-lying unoccupied orbitals, and their π*(U=O)antibonds are located on higher-energy orbitals. Complex 1was calculated to show σ(U=O) bonding character for HO-MO, and pyridine-character for other occupied orbitals; the
fluorine ligand occurs in a relatively low-energy region. Incontrast, the π(p) characters of heavier halogen atoms signif-icantly contribute to most frontier molecular orbitals of 2, 3and 4. Unlike this electronic feature of 2, complexes 5 and 6exhibit mainly thf and pyrrole characters, respectively, fortheir high-lying occupied orbitals. Electronic structures ofbisuranyl complexes 7–12, albeit a little more complicated,are revealed to be similar to those of the correspondingmonouranyl complexes. Finally, energies of formation reac-tions of the above complexes were calculated and comparedwith available experimental results.
Keywords N/O coordinating uranyl halide . Electronicstructure . Environmental effects . Relativistic DFT
Introduction
Apart from its important role in the manufacture of nuclearweapons and the generation of nuclear power, uranium ex-hibits a rich chemistry and forms various complexes withother elements because of the accessibility of its of s, p, dand f orbitals to chemical bonding [1–3]. Among diverseuranium species, the hexavalent linear uranyl ion is the mostprevalent and most thermodynamically stable. Uranyl and itsderivatives are major components of nuclear waste [1]. Uranylderivatives are highly soluble and mobile as well as easilybiologically available, and thus are key players in long-termenvironmental risk [1, 4]. Therefore, it is necessary to under-stand the structures of uranyl complexes, as well as theirphysicochemical properties and relationships.
The uranyl ion displays much more active coordinationchemistry in its equatorial plane [5–14] due to the extraordi-narily chemically robust U=O bond [3]. In most processingand environmental conditions, the uranyl species exists as alinear cation saturated equatorially by four to six ligands
Electronic supplementary material The online version of this article(doi:10.1007/s00894-014-2305-6) contains supplementary material,which is available to authorized users.
J. Yao :Y.<M. Wang :Q.<J. Pan (*)Key Laboratory of Functional Inorganic Material Chemistry ofEducation Ministry, School of Chemistry and Materials Science,Heilongjiang University, Harbin 150080, Chinae-mail: [email protected]
Y.<R. Guo (*)Key Laboratory of Bio-based Material Science and Technology ofEducation Ministry, College of Material Science and Engineering,Northeast Forestry University, Harbin 150040, Chinae-mail: [email protected]
H.<X. ZhangState Key Laboratory of Theoretical and Computational Chemistry,Institute of Theoretical Chemistry, Jilin University,Changchun 130023, China
J Mol Model (2014) 20:2305DOI 10.1007/s00894-014-2305-6

[7–19]. A large number of ligands [7–40], such as halide,pyridine, THF, porphyrin, expanded porphyrin andcalixpyrroles, have been used for uranyl complexation. Somestructurally simple and stable as well as easily synthesizeduranyl complexes are increasingly attractive, because of theirpotential as starting materials to develop and enrich uranylchemistry [8, 9, 12, 17, 18, 41]. For example, (UO2)(X)2(L)3and [(UO2)(μ2–X)(X)(L)2]2 (X=F, Cl, Br, I et al.; L=pyridine(py), tetrahydrofuran (thf), H2O, et al.; μ2=doubly bridged)have been prepared with various synthetic approaches, andmany of these complexes have been characterized structurallyand spectroscopically [9, 12, 15–17, 42]
Such findings present the opportunity to deeply understandthe electronic structures and relevant properties of uranylcomplexes. Although experimental spectroscopic techniquesachieve success in many aspects, it would be much better tounite theoretical methodology to further complement andperfect them, especially considering the constraints imposedon experimental uranyl chemistry by its chemical toxicity,radioactivity and scarcity [2, 3, 43–46]. Current quantumchemical methods have been developed to correctly describethe electron correlation effects and relativistic effects of theheavy metal and environmental effects. For example, Li et al.[47] calculated the electronic properties of UO2X3
− (X=F, Cl,Br and I) using density functional theory (DFT) and ab initiomethods; the charge transfer in UO2X4
− was addressed byRuipérez and co-workers [48]; Pierloot et al. [49] rationalizedthe electronic spectra of UO2Cl2(acetone)3 based on acomplete-active-space type wavefunction. Differing from theabove gas-phase calculations, we examine here the electronicstructures of (UO2)(X)2(L)3 and [(UO2)(μ2–X)(X)(L)2]2 [X=F, Cl, Br, and I; L=py, thf and pyrrole (pl)] in their surround-ing media, i.e., considering the environmental effects aroundthe uranyl complex. Further, the equatorial ligand type andnumber of uranyl ions will be varied.
Computational methods and details
In this work, we investigated monometallic (UO2)(X)2(L)3,where L is fixed as py and X varies from F (1), Cl (2), Br (3) toI (4). Then, we used Cl atoms, and L adopts thf (5) and pl (6),which can be compared with 2. The case is similar for bime-tallic [(UO2)(μ2–X)(X)(L)2]2 (L=py, X=F (7), Cl (8), Br (9)and I (10); X=Cl, L=thf (11); X=Cl, L=pl (12). In thesecomplexes, two halogen atoms are trans to each other. Ofthese complexes, 2, [15] 4, [9] 5 [12] and 11 [16] have beencharacterized experimentally by X-ray crystal diffraction.
The structures of complexes 1–12 have been fully opti-mized in the gas phase without any symmetry constraintsusing Priroda code (Version 6) [50–54]. These calculationswere carried out with the generalized gradient approximation(GGA) PBE functional [55], and all-electron correlation-
consistent Gaussian basis sets of double-ς polarized qualityfor the large component and corresponding kinetically bal-anced basis sets for the small components [51]. Relativisticeffects were implemented using a scalar relativistic four-component all-electron (AE) approach [53, 56], which isbased on the full Dirac equation but with spin-orbit projectedout [57] and neglected. Previous work by us [58–61] andothers [47–49] indicated that inclusion of scalar relativity isreasonable for calculations of structural properties and reac-tion energies. Analytical frequency calculations were per-formed on the basis of these optimized geometries. No imag-inary frequency is found, indicative of their local minimanatures of the stationary points on the potential energy surface.Besides obtaining thermodynamic data, frequency resultswere also used to simulate the vibrational spectra viaLorentzian broadening. Population-based (Mayer) [62] bondorders were calculated.
As experimental uranyl complexes were synthesized andcharacterized either in the solid state or in the solution, we willconsider environmental effects on their electronic properties inthe calculations. So, we have used the self-consistent reactionfield (the COSMO [63] model as implemented in the ADF2010.02 code [64–66]) to simulate medium environment ofreal complex. The dielectric constant (ε) of 78.39 was appliedwith an integration parameter of 6.0 for COSMO. Klamt radiiwere used for the main group atoms (H=1.30 Å, C=2.00 Å,N=1.83Å, O=1.72Å, F=1.76Å, Cl=2.05 Å, Br=2.22Å andI=2.38 Å) [67] and for the uranium atom (1.70 Å) [58–61,68]. The scalar ZORA relativistic approach of van Lentheet al. [69–72] was employed, associated with the all-electronSlater-type TZP basis sets. In the work, the single-point cal-culationswere carried out building on the gas-phase optimizedgeometries. This approximation is feasible because of a slightchange from geometries optimized in the gas phase to thoseoptomized in the various media considered here [61].
Results and discussion
Structures and vibrational spectra
Optimized geometry parameters and bond orders are listed inTable 1. The structures of 1–12 are depicted in Figs. 1 and 2.The monometallic complexes 1–6 are seen to adopt apseudopentagonal bipyramidal fashion, where N/O atoms of3 L and two nonadjacent halogen atoms form the equatorialplane. The analogous bimetallic complexes 7–12 exhibit adimeric geometry formed by edge-sharing pentagonal bipyr-amids. Regarding pyridine-coordinating bisuranyl complexes,the calculations reveal U1–X2–X2′–U2 dihedral angles of 7and 8 to be 179°–180°, suggesting that the U2X2 core liesapproximately in the equatorial plane. Moreover, changesfrom pyridine (7) to tetrahydrofuran (11) and pyrrole (12)
2305, Page 2 of 9 J Mol Model (2014) 20:2305
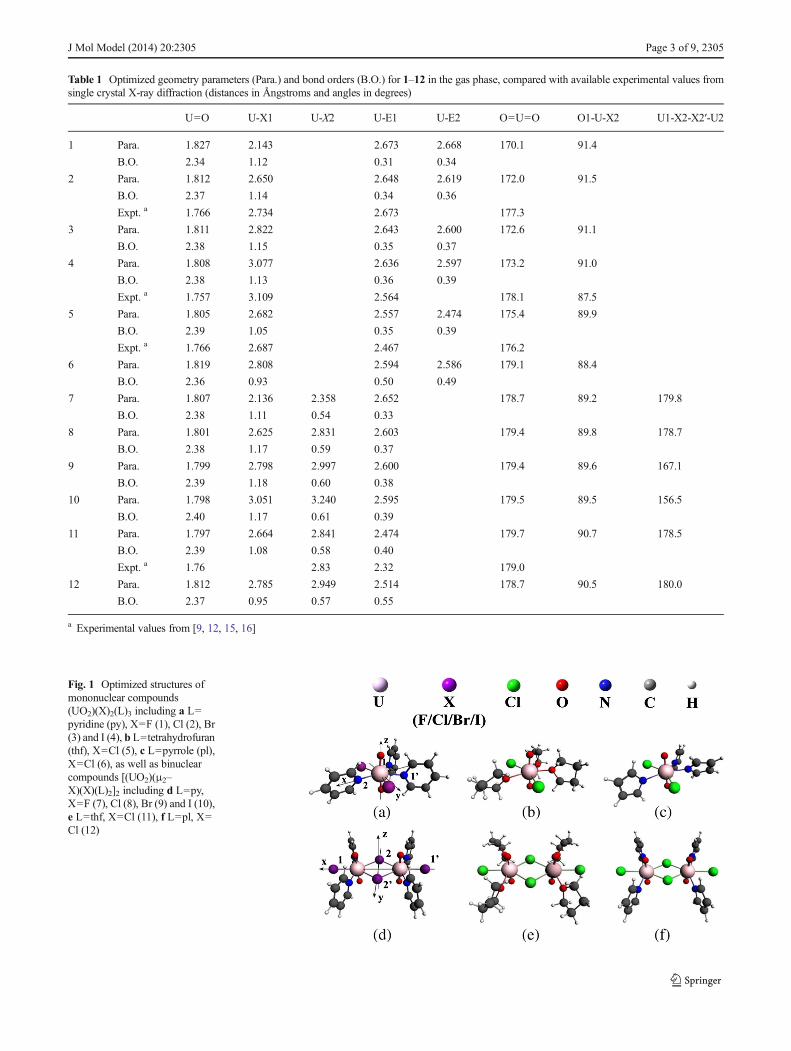
Table 1 Optimized geometry parameters (Para.) and bond orders (B.O.) for 1–12 in the gas phase, compared with available experimental values fromsingle crystal X-ray diffraction (distances in Ångstroms and angles in degrees)
U=O U-X1 U-X2 U-E1 U-E2 O=U=O O1-U-X2 U1-X2-X2′-U2
1 Para. 1.827 2.143 2.673 2.668 170.1 91.4
B.O. 2.34 1.12 0.31 0.34
2 Para. 1.812 2.650 2.648 2.619 172.0 91.5
B.O. 2.37 1.14 0.34 0.36
Expt. a 1.766 2.734 2.673 177.3
3 Para. 1.811 2.822 2.643 2.600 172.6 91.1
B.O. 2.38 1.15 0.35 0.37
4 Para. 1.808 3.077 2.636 2.597 173.2 91.0
B.O. 2.38 1.13 0.36 0.39
Expt. a 1.757 3.109 2.564 178.1 87.5
5 Para. 1.805 2.682 2.557 2.474 175.4 89.9
B.O. 2.39 1.05 0.35 0.39
Expt. a 1.766 2.687 2.467 176.2
6 Para. 1.819 2.808 2.594 2.586 179.1 88.4
B.O. 2.36 0.93 0.50 0.49
7 Para. 1.807 2.136 2.358 2.652 178.7 89.2 179.8
B.O. 2.38 1.11 0.54 0.33
8 Para. 1.801 2.625 2.831 2.603 179.4 89.8 178.7
B.O. 2.38 1.17 0.59 0.37
9 Para. 1.799 2.798 2.997 2.600 179.4 89.6 167.1
B.O. 2.39 1.18 0.60 0.38
10 Para. 1.798 3.051 3.240 2.595 179.5 89.5 156.5
B.O. 2.40 1.17 0.61 0.39
11 Para. 1.797 2.664 2.841 2.474 179.7 90.7 178.5
B.O. 2.39 1.08 0.58 0.40
Expt. a 1.76 2.83 2.32 179.0
12 Para. 1.812 2.785 2.949 2.514 178.7 90.5 180.0
B.O. 2.37 0.95 0.57 0.55
a Experimental values from [9, 12, 15, 16]
Fig. 1 Optimized structures ofmononuclear compounds(UO2)(X)2(L)3 including a L=pyridine (py), X=F (1), Cl (2), Br(3) and I (4), b L=tetrahydrofuran(thf), X=Cl (5), c L=pyrrole (pl),X=Cl (6), as well as binuclearcompounds [(UO2)(μ2–X)(X)(L)2]2 including d L=py,X=F (7), Cl (8), Br (9) and I (10),e L=thf, X=Cl (11), f L=pl, X=Cl (12)
J Mol Model (2014) 20:2305 Page 3 of 9, 2305

have no effect on this dihedral angle. However, the introduc-tion of heavier halogen atoms such as Br (8) and I (9) cause theU2X2 core to deviate from planarity. Almost linear O=U=Oangles were calculated for 1–12, falling within the range of170°–180°.
Close comparison of structures of 1–12 reveals that fluo-rine complexes (1 and 7) have different structural featuresfrom other complexes in which the plane of the L ligand isapproximately parallel to the linear trans-UO2. We presentstable geometries of 1 and 7 in Fig. 2, in contrast to those ofother complexes in Fig. 1. With respect to 1, the wholepyridine ligand between two fluorine atoms is located rightin the equatorial plane; two hydrogen atoms adjacent to the Natom of the pyridine form hydrogen bonds with two fluorineatoms at 2.13 Å; the very strong F→U bonding weakens theN→U bonds, as reflected by the U–N distance of 1 being0.03–0.07 Å longer than those of the analogous bonds in 2–4.Similar to 1, the bimetallic 7 has the longest and weakest U–Nbonds among 7–10. Owing to insufficient space, four planarpyridine ligands tilt toward the equatorial plane with an angleof 24.5° (mean value). All the H⋯F hydrogen bonds werecalculated within 2.31–2.33 Å. Apparently, the present ofthese hydrogen bonds stabilizes complexes 1 and 7. This isnot the case in other complexes with relatively big halogenatoms, which do not favor H⋯X hydrogen bonds. This isbecause the spatial arrangement of the L ligands is the result ofcompetition between U–L bonds and hydrogen bonds. Simul-taneously, the size of the halogen atom is also important.
The U=O distances were calculated to decrease from 1.83to 1.81 Å for 1–4, and from 1.81 to 1.80 Å for 7–10, inTable 1. This conforms to enhancement of the U=O bonds,which results from the weakening of equatorial X→U coor-dination along F, Cl, Br and I. The calculated bond orderssupport the above trend. The U=O bond orders of 2.34–2.40indicate the character of the partial triple bond. Chlorinecomplexes 2, 5 and 6 as well as 8, 11 and 12 differ in donatingability of L ligands. For example, the strong N−(pyrrole)→Ucoordination in 6 lengthens both U–Cl and U=O distances,compared with N(pyridine)→U of 2.
The calculated vibrational spectra in Fig. 3 agree with theabove geometry results. Frequencies of 1–6 ranging from 865to 904 cm−1 are attributed to U=O stretching vibrations [9, 12,
15]. They are comparable to experimentally reported infraredspectra at 925 cm−1 for 2 [9, 15] and 927 cm−1 for 4 [9], andclose to 846 cm−1 for [UO2I2(H2O)2] [17–19] and 870 cm−1
for [UO2(H2O)5]2+ [73]. A pronounced shift to high frequency
is found for the U=O stretches upon changing the halogenatom from F, Cl, Br to I. This results from enhancement of U=O bonds. Two series U=O stretches at 799–827 and 887–914 cm−1 were calculated for bisuranyl complexes 7–12,agreeing well with experimental values of 841 and 875 cm−1
for 11 [16].
Electronic structures
The electronic properties of mono- and bisuranyl 1–12 werecalculated using the PBE functional, TZP basis sets and scalarrelativistic ZORA approach, where the medium environmentsurrounding the complex was considered by the COSMOmodel. In Figs. 4, 5, S1 and S2, we illustrate the energy-correlation diagrams of their characteristic orbitals, comparedwith detailed composition information in Tables 2, 3, and S1–S5.
Similar characters were revealed for unoccupied orbitals ofall the complexes. For example, monometallic 1–6 were cal-culated to show four low-lying unoccupied orbitals mainlywith U(f) characters. This is generally typical of hexavalenturanium complexes. Their π*(U=O) anti-bonding orbitals arefound at about 1.29–1.60 eV further in the virtual band thanthe U(f) orbitals. However, a large difference was calculatedfor characters of occupied orbitals of 1–12.
The bis(fluoro)-tris(pyridyl) dioxouranium complex (1)exhibits high-lying occupied orbitals with the U=O bondsand pyridine characters. Its HOMO is a σ(U=O) bondingorbital, composed by 45.1 % U(fz
3) and 36.7 % O(pz) inTable 2. A very small π[pz(F)], 14.4 %, is involved in theorbital. The combination of pyridine, fluorine and uranyl oxoforms an energetically lower HOMO-1 (H-1). Seven filledorbitals below H-1 are all of pyridine-type. The π(U=O)bonding orbitals occur in H-9, H-10 and H-12–H-14, whileH-11 is featured with σ(U-N) bonds. In contrast, the fluorine-dominant orbitals are present in the very low-energy region.This further confirms strong U–F bonding, as indicated in theabove structural discussion.
The change from fluorine in 1 to heavier chlorine leads to 2having more halogen participation in its occupied orbitals.The first five high-lying filled orbitals (HOMO–H-4) covermore than 50 % π[p(Cl)] composition. Moreover, the σ(U-Cl)bond contributes to H-10, albeit with more σ(U-N) character.And pyridine ligands form H-5–H-7 and some lower-energyorbitals. Linking Table 3 with Fig. 4, complex 2 presents twoσ(U=O) bonding orbitals i.e., H-1 and H-12. The former hasless than 30 % σ(U=O) compositions, but the latter are σ(U=O)-dominant (about 63 %).
Fig. 2 Optimized structures of monometallic (UO2)(F)2(py)3 (1) andbimetallic [(UO2)(μ2–F)(F)(py)2]2 (7)
2305, Page 4 of 9 J Mol Model (2014) 20:2305

Comparison of calculated compositions (Tables 2, 3, S1and S2) reveals that bromine complex (3) and iodine complex(4) have electronic structures similar to 2, except that the Brand I atoms play more significant roles in occupied orbitals oftheir respective complexes than does Cl. This phenomenoncan be observed in high-lying occupied orbitals, say HOMOto H-4. Complex 3 has a π[p(Br)] contribution of more than78 %, and complex 4 has an even higher π[p(I)], above 85 %.Similar to those of 2, two σ(U=O) bonding orbitals were alsocalculated for 3 and 4. Close analyses reveal regular changefor σ(U=O) orbitals of 1–4 along F, Cl, Br and I. Regardingthe higher-energy orbital, about 82 % σ(U=O) compositionand 14 % π[pz(F)] were calculated for HOMO of 1. And lessσ(U=O), about 28 %, and more 51 % π[pz(Cl)] were found inH-1 of 2. In complexes 3 and 4, π[pz(X)] is increased to 84 %and 91 %, respectively, while σ(U=O) is reduced to 14 % and7 %. The opposite change was found in lower-energy orbitalswith σ(U=O)+π[pz(X)] upon increasing halogen size, i.e.,σ(U=O) is becoming predominant, accounting for 63 %,
88 % and 96 % for 2, 3 and 4, respectively. This change hasalso been reflected intuitively by electron density diagrams inFig. 4.
After understanding the regular change of electronic prop-erties of 1–4 featured with the same pyridine ligands anddifferent halogen atoms, we turned to series complexes 2, 5and 6. In these, the chlorine atom is chosen, and then theligand effects of pyridine, thf and pyrrole on electronic struc-tures are taken into account; 2 and 5 differ in N and Ocoordination. The N− donor of pyrrole in 6 is thought to bestronger than the N atom of pyridine in 2. The calculatedresults in Tables 3, S3 and S4 indicate that thf and pyrrolecontribute much more to the HOMOs of 5 and 6, respectively,than the pyridine of 2. HOMO–H-2 of 5 is mainly of thf-character, and HOMO–H-5 of 6 is pyrrole-type-dominant.However, two σ(U=O) bonding orbitals were still calculatedfor them. In Fig. 5, we have correlated these characteristicorbitals.
(a) (b)
500 600 700 800 900 1000 1100 1200
(Cl-pl)
(Cl-thf)
(I-py)
(Br-py)
(Cl-py)
(F-py)
500 550 600 650 700 750 800 850 900 950
(I-py)
(Br-py)
(F-py)
(Cl-pl)
(Cl-thf)
(Cl-py)
Fig. 3 Simulated vibrational spectra of a 1–6 and b 7–12
Fig. 4 Energy-correlation diagrams of mononuclear (UO2)(X)2(py)3where X=F (1), Cl (2), Br (3) and I (4) from PBE/TZP/ZORA/COSMOcalculations
Fig. 5 Energy-correlation diagrams of (UO2)Cl2(L)3 while changing Lfrom py (2) to thf (5) and pl (6) from the PBE/TZP/ZORA/COSMOcalculations, compared with those of bimetallic [(UO2)(μ2-Cl)(Cl)(py)2]2
J Mol Model (2014) 20:2305 Page 5 of 9, 2305

Bimetallic complexes 7–12 were calculated to show morecomplicated electronic structures than corresponding mono-metallic ones. Detailed information of 8 is presented inTable S5 and Fig. 5; 8 has eight U(f)-type unoccupied orbitals,twice as those of monometallic 2. Five σ(U=O)+π[pz(Cl)]occupied orbitals of 8 were observed. Three of them areπ[pz(Cl)]-dominant and the two lower ones are mainlyof σ(U=O)-type, which is similar to those of monome-tallic 2. Additionally, some electron density diagrams ofbimetallic 7–12 are depicted in Figs. S1 and S2.
Reaction energies
The bis(fluoro)-tris(tetrahydrofuran) dioxouranium 5was syn-thesized by Burns and co-workers [12] by the reaction ofexcess ClSiMe3 with UO2Cl2(H2O)3 in the thf solution. Viaelemental analysis of crystals, they found that one of thfligands in 5 is labile, and easily forms bimetallic 11. Thus,we used the following Reactions 1 and 2 to produce uranylcomplexes 1–12. Environmental effects have been shown tobe important in obtaining accurate reaction energies involvingactinide species [58–61]. So free energy of medium
environmental effects (labeled as Gsol) for each complex wascalculated by the COSMO model.
Reaction 1: UO2X2(H2O)3+6XSiMe3+3 L UO2X2(L)3+3O(SiMe3)2+6HX L=py, X=F(1), Cl (2), Br (3) and I(4); X=Cl, L=thf (5) and pl (6)Reaction 2: 2UO2X2(L)3 [(UO2)(μ2-X)(X)(L)2]2+4 LL=py, F(7), Cl (8), Br (9) and I (10); X=Cl, L=thf (11)and pl (12)
In the case of X=F, Cl, Br and I, and L=py, thf and pl, atotal of 12 possible complexes are covered with theUO2X2(L)3 formula; we calculated reaction energies for allof them. The values obtained are given in Table 4, where freeenergies [ΔrG(sol), kcal mol−1] including medium environ-mental effects are plotted in Fig. 6. These show the same trendregardless of L being py, thf or pl. The pyridine and thfcomplexes have quite similar reaction energies, while thenegatively charged pyrrole complexes display much lowervalues. It is worth pointing out that we did not obtain ener-getically stable [UO2I2(pl)3]
3−. One iodine atom is squeezedout of the first coordination sphere of uranyl due to the strongN(pl)→U interaction. This agrees well with experimentally
Table 2 Partial molecular orbital contributions (%) of 1 calculated at the PBE/TZP/ZORA/COSMO level
Orbital Energy (eV) Composition (%) Assignment a
U O F py
L+5 −2.135 56.4(fz2y,fz
3,fz(x2-y2)) 13.9(py) 13.5 π*(f)
L+4 −2.136 72.3(fz2y,fxyz,px) 17.9(px) π*(f)
L+3 −3.430 89.2(fy(3x2-y2),fz(x
2-y2),dxy) 5.5(px)
L+2 −3.586 88.5(fx(x2-3y
2),dx
2-y2,fxyz) 2.4(py)
L+1 −3.730 93.7(fz(x2-y2),fz
2y,fy(3x
2-y2)) 2.4(pz)
LUMO −3.752 95.9(fxyz,fz2y,fx(x
2-3y
2))
HOMO −6.663 45.1(fz3,pz,fz
2y) 36.7(pz) 14.4(pz) σ(f)+π(F)
H-1 −6.750 2.6(py) 11.6(py) 15.5(py) 46.4 py+π(F)
H-2 −6.834 93.3 py
H-3 −6.869 92.4 py
H-4 −6.883 6.6(px) 62.6 py
H-5 −6.943 93.9 py
H-6 −7.428 2.1(py) 85.5 py
H-7 −7.485 3.2(s,dx2-y2) 2.2(pz) 3.7(px) 69.8 py
H-8 −7.580 2.2(px) 89.9 py
H-9 −7.600 4.2(fxyz,dyz) 50.6(py) 41.3(pz) π(f)+π(F)
H-10 −7.632 16.8(fz2y,fy(3x
2-y2),py,dxy,fz
3) 35(py) 23.5(py,px) 11.1 π(f)+σ(U-N)
H-11 −7.680 1.3(dx2-y2) 2.1(pz) 3.9(px) 72.1 σ(U-N)
H-12 −8.045 17.9(fz2x,fx(x
2-3y
2)) 59.4(px) 10.7(px,py) 4.0 π(f)+σ(U-N)
H-13 −8.327 12.6(dxz,fz3) 58.4(px) 18.5(pz) π(d)+π(F)
H-14 −8.463 4.1(fz2y,dz
2,fy(3x2-y2)) 23.4(pz,py) 41.3(px) 9.1 π(f)
a The σ(f), π(f) and π(d) are short for various U=O bonds, where f and d is the major orbital character of uranium involved in the bonds
2305, Page 6 of 9 J Mol Model (2014) 20:2305

obtained uranyl complexes with four equatorial ligands suchas not only UO2I2(H2O)2 [17, 18] and [UO2I4]
2− [19] iodidesbut also strong carbene coordinated UO2Cl2(IMes)2 (IMes=1,3-dimesitylimidazole-2-ylidene) [74]. According to Reac-tion 1, the fluoride complex is the hardest to form among thedifferent halides while keeping L the same. It seems chlorides
Table 3 Partial molecular orbital contributions (%) of 2 calculated at the PBE/TZP/ZORA/COSMO level
Orbitals Energy (eV) Composition (%) Assignment a
U O Cl py
L+5 −2.369 72.4(fz2y) 20.3(py) π*(f)
L+4 −2.381 76.4(fz2x) 19.4(px) π*(f)
L+3 −3.921 87.4(fy(3x2-y2),dxy) 6.0(px)
L+2 −4.007 88.1(fx(x2-3y
2),dx
2-y2) 4.7(px,py)
L+1 −4.086 94.4(fz(x2-y2)) 2.4(pz)
LUMO −4.095 95.9(fxyz,fx(x2-3y
2))
HOMO −6.578 2.6(fx(x2-3y
2)) 50.7(px) 28.0 π(Cl)+py
H-1 −6.600 16.2(fz3,pz,fz(x
2-y2)) 12.1(pz) 65.5(pz,px,py) σ(f)+π(Cl)
H-2 −6.651 3.2(fy(3x2-y2)) 64.3(px,py) 14.2 π(Cl)+py
H-3 −6.672 1.8(fxyz) 8.5(py) 72.5(pz) 3.5 π(Cl)
H-4 −6.851 3.2(py,fy(3x2-y2)) 4.2(py) 61.4(px,py) 17.2 π(Cl)
H-5 −6.859 5.3(pz) 85.2 py
H-6 −6.991 7.6(pz) 73.4 py
H-7 −7.008 1.0(fz3) 91.4 py
H-8 −7.179 1.3(px) 6.4(px) 37.2(px,py) 29.5 π(Cl)+py
H-9 −7.377 8.4(fx(x2-3y
2),dxy) 26.5(py,px) 47.0 σ(U-N)
H-10 −7.572 7.6(fx(x2-3y
2),dx
2-y2) 12.9(py) 60.1 σ(U-N)+σ(U-Cl)
H-11 −7.575 2.8(fz3) 2.5(pz) 2.7(pz) 74.3 py
H-12 −7.674 39.2(fz3,pz) 23.4(pz) 21.4(pz) 2.7 σ(f)+π(Cl)
H-13 −7.873 1.5(dx2-y2) 3.4(pz) 4.5(py) 66.8 py
H-14 −7.908 4.8(dxy) 19.1(px,py) 64.1 σ(U-N)
a The σ(f), π(f) and π(d) are short for various U=O bonds, where f and d is the major orbital character of uranium involved in the bonds
Table 4 Energies (kcal mol−1) of formation reactions ofmonometallic 1–6 in the gas phase and solution, together with those of bimetallic 7–12 thatare transformed from monometallic complexes
ΔrE(gas) a ΔE0(gas)a ΔrG(gas)
a ΔrG(sol)b
UO2X2(H2O)3+6XSiMe3+3 L ⇋ UO2X2(L)3+3O(SiMe3)2+6HX
1 60.15 53.71 65.60 58.66
2 10.61 −2.67 6.03 5.85
3 17.39 0.82 8.76 15.64
4 −2.06 −18.15 −10.01 −0.745 14.94 1.37 12.08 15.17
6 109.46 96.09 103.04 −28.842UO2X2(L)3 ⇋ [(UO2)(μ2-X)(X)(L)2]2+4 L
7 0.29 −0.81 −10.88 −7.848 9.01 7.61 −3.58 1.53
9 11.92 10.50 0.21 5.27
10 15.77 14.15 2.67 7.90
11 6.91 5.46 −9.02 −8.6412 −22.38 −23.68 −32.96 0.95
a ΔrE(gas),ΔrE0(gas) andΔrG(gas) denote the total energy, total energyincluding zero-point vibration energy and free energy of the reaction inthe gas phase, respectivelybΔrG(sol)=ΔrG(gas)+ΔGsol, ΔGsol=∑νBGsol(B), where Gsol(B) wascalculated in the media for each complex (B) in the formation reaction
-30
-20
-10
0
10
20
30
40
50
60
70 67.24
28.63
IBrCl
-21.83
6.53
24.85
-28.84
15.17
-0.74
15.64
5.85
pl
thf
py58.66
F
Fig. 6 Free energies of formation reactions for monometallic(UO2)(X)2(L)3 (L=py, thf and pl; X=F, Cl, Br and I)
J Mol Model (2014) 20:2305 Page 7 of 9, 2305

and iodides are more energetically favorable. So far, chlorineand iodine complexes 2 [15], 4 [9] and 5 [12] were isolatedsuccessfully and their crystalline structures characterized byX-ray crystal diffraction.
Building on Reaction 2, we can evaluate the relative sta-bility of mono- and bimetallic complexes. The implication ofthis is that if the reaction is calculated to be exothermic, thenthe bisuranyl complex is more energetically stable than itscorresponding monouranyl one. Our calculations indicate aregular energetic rising along F, Cl, Br to I whatever L ispyridine, thf or pl, in Fig. 7. So it is suggested that lighthalogen is capable of stabilizing bisuranyl complexes, where-as heavy halogen favors monouranyl complex. For example,all the negative values were calculated for reactions whiletransforming monouranyl to bisuranyl fluorides. It is worthnoting that the transformation energy from monometallic 5 tobimetallic 11 was calculated to be −8.64 kcal mol−1. Thisexothermic process confirms previous experimental results[16] mentioned at the beginning of this section.
Conclusions
In this work, a series of monometallic 1–6 and bimetallic 7–12was investigated for their structural and electronic propertiesas well as reaction energies using relativistic DFT. Based onthese and experimentally reported results of 2, 4, 5 and 11, wecome to the following conclusions:
Among bimetallic 7–12, the fluorides and chlorides wereoptimized to show an extended planarity formed by twouranium and equatorial coordination atoms. Moreover, thisarchitecture is not affected by the variation of py, thf to plligands. Unlike those, heavier Br and I atoms greatly distortthe planar structure. It is also found that the spatial arrange-ment of L depends on cooperative effects of the L→U dative
bond, the H⋯X hydrogen band and the size of the halogenatom. With small size fluorine atoms, for instance, 1 and 7readily form a co-planar feature between the pyridine ligandand the uranyl equatorial plane (Fig. 2). The calculated resultsof bond lengths, bond orders and stretching vibrational fre-quencies indicate that the axial U=O bond can be tuned, tosome degree, by varying equatorial X/L→U coordination.
It is revealed that 1–12 have U(f)-character low-lying un-occupied orbitals and energetically higher π*(U=O) orbitals.Fluorine complex such as in 1 is featured with σ(U=O)bonding HOMO and pyridine-character for other occupiedorbitals; the fluorine ligand makes only a small contributiontomost frontier orbitals. However, heavier halogen atoms playa predominant role in HOMOs, leading to electronic featuresof halogen atoms, (U=O) bonds and L (py, thf and pl).Regular changes were calculated for orbitals with σ(U=O)+π[pz(X)] of 1–4 differing in halogen atoms. For their higher-energy orbital, σ(U=O) composition decreases from 82 % to7 %, while π[pz(X)] increases from 14 % to 91 % along F, Cl,Br to I. The reverse trend is found in lower-energy orbital withσ(U=O)+π[pz(X)] character. It was also found that bimetallicuranyl complexes show electronic structures similar to, but alittle more complicated than, their respective monometallicone.
In addition, energies of formation reactions of 1–12 as wellas structurally relevant complexes (24 complexes in total)suggest that monouranyl chlorides and iodides are energeti-cally favorable. Regarding the relative stability of mono- andbimetallic complexes, light halogen is capable of stabilizingbinuclear complexes, while heavy halogen favors mononucle-ar ones. The present study has theoretically evidenced exper-imental transformation from monometallic 5 to bimetallic 11.
Acknowledgments Q.J.P. is grateful to Dr. Dimitri Laikov for provid-ing us with the Priroda code. This work is supported by the NationalNatural Science Foundation of China (21273063) and the Program forNew Century Excellent Talents in University (NCET-11-0958). TheNatural Science Foundation of Heilongjiang Province (B201318 andLC2011C22), the Program for Innovative Research Team in University(IRT-1237) and the Key Project of Chinese Ministry of Education(211048) are gratefully acknowledged.
References
1. Hashke JM, Stakebake JL (2006) Handling, storage, and dispositionof plutonium and uranium, in: Morss LR, Edelstein NM, Fuger J(eds) The chemistry of the actinide and transactinide elements,Springer, pp 3199
2. Schreckenbach G, Shamov GA (2010) Acc Chem Res 43:193. Denning RG (2007) J Phys Chem A 111:41254. Choppin GR (2007) J Radioanal Nucl Chem 273:6955. Alexander V (1995) Chem Rev 95:2736. Bart SC, Meyer K (2008) Highlights in uranium coordination chem-
istry. In: Albrecht-Schmitt TE (ed) Organometallic and coordinationchemistry of the actinides. Springer, Berlin, p 119
-18
-15
-12
-9
-6
-3
0
3
6
9
7.59
-17.49
-8.64
5.27
-7.84
-6.01
0.95
1.53
7.90
-3.45
1.11
pl
thf
py
IBrClF
Fig. 7 Free energies of formation reactions for bimetallic [(UO2)(μ2–X)(X)(L)2]2 (L=py, thf and pl; X=F, Cl, Br and I)
2305, Page 8 of 9 J Mol Model (2014) 20:2305

7. Berthet JC, Nierlich M, Ephritikhine M (2003) Chem Commun2003:1660
8. Berthet JC, Lance M, Nierlich M, Ephritikhine M (2000) Eur J InorgChem 2000:1969
9. Berthet JC, Nierlich M, Ephritikhine M (2004) Chem Commun2004:870
10. Berthet JC, Nierlich M, Ephritikhine M (2003) Angew Chem-Int Ed42:1952
11. Oldham WJ, Oldham SM, Scott BL, Abney KD, Smith WH, CostaDA (2001) Chem Commun 2001:1348
12. Wilkerson MP, Burns CJ, Paine RT, Scott BL (1999) Inorg Chem 38:4156
13. Vaughn AE, Barnes CL, Duval PB (2007) J ChemCrystallogr 37:77914. Alcock NW, Flanders DJ, Brown D (1985) J Chem Soc-Dalton Trans
1985:100115. Berthet JC, Siffredi G, Thuéry P, EphritikhineM (2009) Dalton Trans
2009:347816. Charpin P, Lance M, Nierlich M, Vigner D, Baudin C (1987) Acta
Crystallogr Sect C: Cryst Struct Commun 43:183217. Crawford M-J, Ellern A, Karaghiosoff K, Mayer P, Noth H, Suter M
(2004) Inorg Chem 43:712018. Crawford M-J, Ellern A, Noth H, Suter M (2003) J Am Chem Soc
125:1177819. Crawford M-J, Mayer P (2005) Inorg Chem 44:554720. Burns CJ, Clark DL, Donohoe RJ, Duval PB, Scott BL, Tait CD
(2000) Inorg Chem 39:546421. Berthet JC, Nierlich M, Ephritikhine M (2004) Dalton Trans 2004:
281422. Villiers C, Thuery P, Ephritikhine M (2008) Angew Chem-Int Ed 47:
589223. Alcock NW, Flanders DJ, Pennington M, Brown D (1988) Acta
Crystallogr Sect C-Cryst Struct Commun 44:24724. Sessler JL, Hemmi G, Mody TD, Murai T, Burrell A, Young SW
(1994) Acc Chem Res 27:4325. Sessler JL, Vivian AE, Seidel D, Burrell AK, Hoehner M, Mody TD,
Gebauer A, Weghorn SJ, Lynch V (2001) Coord Chem Rev 222:27526. Sessler JL, Melfi PJ, Pantos GD (2006) Coord Chem Rev 250:81627. Sessler JL, Seidel D, Vivian AE, Lynch V, Scott BL, Keogh DW
(2001) Angew Chem-Int Ed 40:59128. Sessler JL, Mody TD, DulayMT, Espinoza R, Lynch V (1996) Inorg
Chim Acta 246:2329. Sessler JL, Mody TD, Lynch V (1992) Inorg Chem 31:52930. Arnold PL, Hollis E, Nichol GS, Love JB, Griveau J-C, Caciuffo R,
Magnani N, Maron L, Castro L, Yahia A, Odoh SO, SchreckenbachG (2013) J Am Chem Soc 135:3841
31. Arnold PL, Jones GM, Odoh SO, Schreckenbach G, Magnani N,Love JB (2012) Nat Chem 4:221
32. Jones GM, Arnold PL, Love JB (2012) Angew Chem Int Ed 51:12584
33. Arnold PL, Jones GM, Pan Q-J, Schreckenbach G, Love JB (2012)Dalton Trans 41:6595
34. Arnold PL, Pecharman A-F, Love JB (2011) AngewChem Int Ed 50:9456
35. Arnold PL, Hollis E, White FJ, Magnani N, Caciuffo R, Love JB(2011) Angew Chem Int Ed 50:887
36. Arnold PL, Pecharman A-F, Hollis E, Yahia A, Maron L, Parsons S,Love JB (2010) Nat Chem 2:1056
37. Love JB (2009) Chem Commun 2009:315438. Arnold PL, Patel D, Wilson C, Love JB (2008) Nature 451:31539. Arnold PL, Patel D, BlakeAJ,Wilson C, Love JB (2006) J AmChem
Soc 128:961040. Natrajan L, Burdet F, Pecaut J, Mazzanti M (2006) J Am Chem Soc
128:7152
41. Andersen RA (1979) Inorg Chem 18:20942. Rebizant J, Vandenbossche G, Spirlet MR, Goffart J (1987) Acta
Crystallogr Sect C-Cryst Struct Commun 43:129843. Kaltsoyannis N (2003) Chem Soc Rev 32:944. Kaltsoyannis N (2000) Inorg Chem 39:600945. Kaltsoyannis N, Scott P (1999) The f elements. Oxford University
Press, Oxford46. Gagliardi L, Roos BO (2005) Nature 433:84847. Su J, Phuong Diem D, Qiu Y-H, Liu H-T, Xu C-F, Huang D-L,Wang
L-S, Li J (2013) Inorg Chem 52:661748. Ruiperez F, Wahlgren U (2010) J Phys Chem A 114:361549. van Besien E, Pierloot K, Gorller-Walrand C (2006) Phys chem chem
phys: PCCP 8:431150. Laikov DN (1997) Chem Phys Lett 281:15151. Laikov DN (2000) PhD Thesis, Moscow State University, Moscow52. Laikov DN (2005) Chem Phys Lett 416:11653. Laikov DN, Ustynyuk YA (2005) Russ Chem Bull 54:82054. Laikov DN (2007) J Comput Chem 28:69855. Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:386556. Laikov DN (2000) An Implementation of the Scalar Relativistic
Density Functional Theory for Molecular Calculatios with GaussianBasis Sets, DFT2000 Conference, Menton, France
57. Dyall KG (1994) J Chem Phys 100:211858. Pan Q-J, Odoh SO, Schreckenbach G, Arnold PL, Love JB (2012)
Dalton Trans 41:887859. Pan Q-J, Schreckenbach G, Arnold PL, Love JB (2011) Chem
Commun 47:572060. Pan QJ, Shamov GA, Schreckenbach G (2010) Chem Eur J 16:228261. Pan QJ, Schreckenbach G (2010) Inorg Chem 49:650962. Mayer I (2003) Simple theorems, proof and derivations in quantum
chemistry. Kluwer/Plenum, New York63. Pye CC, Ziegler T (1999) Theor Chem Acc 101:39664. te Velde G, Bickelhaupt FM, Baerends EJ, Fonseca Guerra C, Van
Gisbergen SJA, Snijders JG, Ziegler T (2001) J Comput Chem 22:931
65. Fonseca Guerra C, Snijders JG, te Velde G, Baerends EJ (1998)Theor Chem Acc 99:391
66. Baerends EJ, Ziegler T, Autschbach J, Bashford D, Bérces A,Bickelhaupt FM, Bo C, Boerrigter PM, Cavallo L, Chong DP,Deng L, Dickson RM, Ellis DE, van Faassen M, Fan L, FischerTH, Fonseca Guerra C, Ghysels A, Giammona A, van GisbergenSJA, Götz AW, Groeneveld JA, Gritsenko OV, Grüning M, GusarovS, Harris FE, van den Hoek P, Jacob CR, Jacobsen H, Jensen L,Kaminski JW, van Kessel G, Kootstra F, Kovalenko A, KrykunovMV, van Lenthe E, McCormack DA, Michalak A, Mitoraj M,Neugebauer J, Nicu VP, Noodleman L, Osinga VP, Patchkovskii S,Philipsen PHT, Post D, Pye CC, Ravenek W, Rodríguez JI, Ros P,Schipper PRT, Schreckenbach G, Seldenthuis JS, Seth M, SnijdersJG, Solà M, Swart M, Swerhone D, te Velde G, Vernooijs P, VersluisL, Visscher L, Visser O, Wang F, Wesolowski TA, van WezenbeekEM,Wiesenekker G,Wolff SK,Woo TK, Yakovlev AL (2010) ADF,in, SCM Theoretical Chemistry. Vrije Universiteit, Amsterdam
67. Klamt A, Jonas V, Burger T, Lohrenz JCW (1998) J Phys Chem A102:5074
68. Shamov GA, Schreckenbach G (2006) J Phys Chem A 110:948669. van Lenthe E, Ehlers A, Baerends EJ (1999) J Chem Phys 110:894370. van Lenthe E, Baerends EJ, Snijders JG (1994) J Chem Phys 101:
978371. van Lenthe E, Baerends EJ, Snijders JG (1993) J Chem Phys 99:459772. van Lenthe E, Snijders J, Baerends E (1996) J Chem Phys 105:650573. Nguyentrung C, Begun GM, Palmer DA (1992) Inorg Chem 31:528074. Oldham JrWJ, Oldham SM, Scott BL, Abney KD, SmithWH, Costa
DA (2001) Chem Commun 2001:1348
J Mol Model (2014) 20:2305 Page 9 of 9, 2305