stereoselective block of cardiac sodium channels by...
TRANSCRIPT

1306
Stereoselective Block of Cardiac SodiumChannels by RAC109 in Single Guinea Pig
Ventricular MyocytesCraig W. Clarkson
The effects of the optical stereoisomers of the local anesthetic RAC109 (RAC109-I andRAC109-II) on sodium current in isolated guinea pig ventricular myocytes were investigated byuse of the whole-cell variation of the patch-clamp technique. RAC109-I and RAC109-1Iproduced similar levels of tonic block, but RAC109-I produced a significantly larger use-dependent block on repetitive pulsing to potentials positive to -60 mV. Definition of the timecourses of block development at -20 mV and recovery at -140 and —160 mV indicated thatRAC109-I had a higher affinity for activated and inactivated channels and dissociated moreslowly at hyperpolarized potentials compared with RAC109-II. Removal of fast inactivation bya>-chymotrypsln intensified tonic block but did not reduce use-dependent block by RAC109-I;this finding suggests that channel inactivation is not necessary for use-dependent block. Theguarded-receptor model was used to calculate apparent rate constants of drug binding andunbinding. According to the model, RAC109-I and RAC109-II have significantly differentunbinding rate constants for channels when they exist predominantly in rested, activated, orinactivated states, as well as significantly different binding rate constants when channels areactivated. However, the apparent rates of drug binding to closed (rested and inactivated)channels are not significantly different for the two isomers; this finding indicates that drugbinding to dosed channels is not markedly stereospecific, in contrast to unbinding. The effectsof RAC109 stereoisomers on cardiac sodium channels were also qualitatively similar to thosepreviously reported in nerve; these findings suggest that the binding sites for local anestheticsin both tissue types have a similar structural topography. (Circulation Research 1989;65:1306-1323)
Several recent electrophysiological studies haveshown evidence for competitive interactionsbetween different tertiary amine drugs in
both nerve1-2 and cardiac tissue.3-4 These studieshave provided strong support for the hypothesisthat tertiary aminc local anesthetic agents blocksodium current (INa) in nerve and cardiac tissue bybinding to a specific receptor site associated withthe sodium channel. Additional support for a spe-cific receptor for local anesthetics in nerve tissuehas been obtained from studies of the stereoisomersof the tertiary amine local anesthetic RAC109(RAC109-I and RAC109-II). RAC109-I has beenshown to be two to three times more potent than itsenantiomer RAC109-II in blocking conduction,5
Supported in part by Grant HL-36096 from the NationalInstitutes of Health and a grant from the American HeartAssociation, Louisiana Affiliate.
Address for correspondence: Graig W. Clarkson, Departmentof Pharmacology, School of Medicine, Tulane University, 1430Tulane Avenue, New Orleans, LA 70112.
Received September t5, 1988; accepted June 2, 1989.
blocking INBJ6'7 a nd in inhibiting batrachotoxin bind-
ing to nerve tissue.8 Such differences in potencybetween RAC109-I and RAC109-II are consideredto reflect stereoselective drug binding to a sodiumchannel receptor since both enantiomers have beenfound to have identical membrane-buffer partitioncoefficients,9 as well as identical levels of uptakeinto various biological membranes.9-10
In contrast to the evidence that has been accu-mulated in nerve tissue, there is little electrophysi-ological evidence for stereoselective interaction oftertiary amine drugs with sodium channels in car-diac tissue. Neither the optical isomers of diso-pyramide,11 propranolol,12 or K659213 nor the geo-metrical isomers of quinidine11-14 appear to blockcardiac sodium channels stereoselectively. Further-more, there are several lines of evidence that indi-cate that cardiac sodium channels in nerve andcardiac tissue are not pharmacologically identical.For example, hyperpolarization has been shown toaccelerate the rate of recovery from use-dependentsodium channel block by QX-314, QX-222,15 and
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

Clarkson Block of Na Channels by RAC109 Stereoisomers 1307
lidocaine16-17 in cardiac tissue. In contrast, in thesquid giant axon, hyperpolarization has been shownto slow the rate of recovery from block by QX-314and QX-222 and to have little effect on the rate ofrecovery from block by lidocaine.18 In addition, theeffects of tetrodotoxin on sodium channels in nerveand cardiac tissue have been shown to differ mark-edly in both potency of interaction and effects onchannel gating.19-22 Thus, it is difficult to extrapo-late drug effects observed in one tissue to the othersince there appear to be qualitative differences inresponse.
The primary goal of the present study was todetermine whether the receptor for tertiary aminelocal anesthetics in cardiac tissue displays stereo-selectivity similar to that previously shown in nervepreparations.6-7 To achieve this goal, the blockingeffects of RAC109 stereoisomers on cardiac sodiumchannels were compared in terms of their potencyfor producing tonic and use-dependent block, aswell as their kinetics and voltage dependence ofproducing channel block. Further insight into themolecular mechanisms underlying channel blockand slow recovery from block was also sought inexperiments performed after removal of fast inacti-vation with a-chymotrypsin and through modelingof voltage- and time-dependent drug-channel inter-actions by use of the guarded-receptor model.23-26
Materials and MethodsIsolation of Cardiac Myocytes
The method for dissociation and isolation ofventricular myocytes used in this study is similar tothat described by Mitra and Morad.27 Hearts fromyoung guinea pigs (200-300 g) were removed underpentobarbital anesthesia and quickly mounted bythe aorta on the cannula of a Langendorff perfusionapparatus. Hearts were initially perfused for 5 min-utes at 10-15 ml/min with a calcium-free bicarbonate-buffered solution at 37° C, followed by an 8-minuteperfusion with the same solution supplemented with2 mg/ml collagenase (284 units/mg; type II, Worth-ington Biochemicals, Freehold, New Jersey) and0.28 mg/ml protease (5.6 units/mg; type XIV, SigmaChemical, St. Louis, Missouri). The ventricles werethen cut away from the atria, placed in enzyme-freesolution supplemented with 0.2 mM CaCl2, mincedwith scissors, and gently shaken at 37° C for 5minutes. Cells were harvested by filtering the sus-pension through 200 ^m nylon mesh and by spin-ning the filtrate at 200-300 rpm in a table-topcentrifuge to form a pellet. The supernatant wasdrawn off, and the pellet was resuspended in enzyme-free solution supplemented with 1 mM CaCl2 and 1mg/ml bovine serum albumin (Fraction V, SigmaChemical). This suspension was also centrifuged toform a pellet, and the pellet was resuspended inMinimal Essential Medium supplemented with Ear-le's salts, L-glutamine (292 mg/ml), 3% horse serum,0.1 mg/ml streptomycin, and 100 units/ml penicillin
(GIBCO Laboratories, Grand Island, New York).This final cell suspension was poured into culturedishes and stored in a CO2 incubator at 37° C untilthe cells were used in experiments (within 30 hoursafter isolation). The cells used in experiments wererod-shaped with crisp cross-striations and lackedvisible spots or blebs on their surface. The solutionused for perfusing the heart and cell isolation was aJoklik modified minimum essential medium (GIBCO)equilibrated with 95% O2-5% CO2, titrated to pH 7.2with hydrogen chloride, and warmed to 37° C.
Solutions and DrugsWhen electrical recordings were made on isolated
cells, the tissue chamber was perfused with an"external" solution containing (mM) tetramethyl-ammonium chloride 115, NaCl 25, CsCl 5, CoCl2 2,CaCl2 1.8, MgCl2 1.2, HEPES 20, and glucose 11adjusted to a pH of 7.3 with tetramethylammoniumhydroxide. The glass pipettes (electrodes) werefilled with an Internal solution containing (mM) CsF125, CsCl 20, NaF 5.6, HEPES 5, and EGTA 5adjusted to a pH of 7.2 with CsOH. The stereoiso-mers of RAC109 (RAC109-I and RAC109-II) weresupplied by Ms. Jeanne Johnson (Astra Pharmaceuti-cal Products, Westborough, Massachusetts). RAC109stereoisomers have a pK,, of 9A28 The estimateddistribution coefficient of the base form of RAC109 incod liver oil versus base plus cation in aqueous bufferat pH 7.4 is 2.6.28 a-Chymotrypsin (type II) wasobtained from Sigma Chemical.
Voltage-Clamp RecordingAt the beginning of each experiment, an aliquot
of cells was transferred from a culture dish into ashallow chamber (~1.5-ml volume), which wasmounted on the stage of an inverted microscope.The whole-cell variant of the patch-clamp method29
was used to measure INo from isolated ventricularmyocytes. Glass pipettes were pulled in two stepsand fire-polished until tip openings were 3-4 /xm indiameter. When filled with the standard internalsolution and placed in the external solution, tipresistances were 0.3-0.5 MO. Pipette tips wereplaced centrally along the length of the cell, and agigaohm seal was formed between the pipette tipand the cell membrane. The membrane patch under-neath the pipette tip was then ruptured by applyinga negative pressure transient, which created a con-tinuity between the pipette (internal) solution and thecell cytoplasm. A thermistor was placed near the cellunder study, and the temperature of the externalsolution was cooled to 16° C (±0.5° C) with a ther-moelectric device (model 806-7243-01, Cambion/Midland Ross, Cambridge, Massachusetts).
An Axopatch amplifier (Axon Instruments, Bur-lingame, California) was used for whole-cell voltageclamping. Creation of voltage-clamp pulses anddata acquisition was controlled by an IBM PC/ATcomputer running pClamp software (Axon Instru-ments) interfaced to the amplifier by an 80-kHz
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

1308 Circulation Research Vol 65, No 5, November 1989
Labmaster board (Tecmar, Solon, Ohio). Sodiumcurrents were digitized at sample intervals of 20-200^isec. Between pulse protocols, cells were main-tained at a holding potential of —140 mV to removefully resting inactivation.
Evaluation of MethodTo study accurately drug effects on sodium
current, contaminating ionic currents must be insig-nificant, and voltage-clamp errors related to uncom-pensated series resistance and voltage inhomo-geneity must be reduced to within acceptable levels.In these experiments, the isolation of INa fromcalcium and potassium currents was accomplishedby replacing all K+ with Cs+ and by blockingcalcium channels with external Co2+ and internal F"ions. Under these conditions, steady-state (leak)current at the test potential (—20 mV) was less than0.1 nA, or less than 1-2% of peak INa. No correctionfor leak current was made.
Capacitative transients evoked by 10-mV hyper-polarizing pulses were well-defined by single expo-nential functions with a mean time constant (TC) of85±8 /isec (rc = 13). Values of cell capacitance (Cm)were estimated from capacitative transients usingthe equation: CIO=Q/A V, where A V is the ampli-tude of a voltage step, and Q is the total chargedisplaced during the voltage step. Q was estimatedby integration of the capacitative transient. Themean value of cell capacitance for guinea pig myo-cytes from young animals was 66±4 pF (« = 13). Themean total series resistance (R,) for the pathwaybetween the pipette and cell membrane after rup-ture of the membrane seal was calculated from theequation rc=RJxCm to be approximately 1.3±0.1Mfl (mean±SD, n=13)30 under the experimentalconditions used in this study. Compensation for thisseries resistance was done empirically by applyingelectronic series resistance compensation to a max-imum level of 60-80% before producing currentoscillation. The mean amplitude of the peak IN, nearthe peak of the current-voltage relation in this studywas determined to be 9.2±4.2 nA (n=13) (holdingpotential, -140 mV; test potential, -20 mV). Undersuch conditions, the expected voltage drop acrossthe uncompensated series resistance (A V=INaxR,)is 2-5 mV. An attempt was made to confirm the sizeof the series resistance voltage error based on shiftsin the sodium conductance-voltage relation pro-duced by reductions in current amplitude. Reduc-tion of peak IN, by 44% after changes in the holdingpotential produced only a 1.6±0.5 mV shift of theconductance-voltage relation.30 These results indi-cate that measurement errors due to uncompen-sated series resistance were within acceptable limitsunder these experimental conditions. The estimatedmembrane length constant at the time of peaksodium conductance under these experimental con-ditions has also been calculated to be approximately317 fjbm.30 This is more than five times the distancefrom the pipette tip to the end of a typical myocyte
when the tip is placed centrally along the celllength; thus, problems due to nonuniformity arealso within acceptable limits.
The inclusion of divalent cations commonly usedto block calcium channels (including Co2+) in theexternal solution has recently been reported to reduceINl amplitude.31 The hypothesis that cobalt block ofsodium channels alters the blocking characteristicsof RAC109 stereoisomers was considered. However,in a series of pilot experiments, the observed block-ing effects of RAC109-I were not noticeably differentwhen using an external solution without externalCo2+. In four cells exposed to 50 jtM RAC109-I inthe absence of external Co2+, the mean amplitude oftonic block after a long rest at -140 mV was0.27 ±0.10 of control INa, and the steady-state ampli-tude of use-dependent block after a train of 15 pulsesof 20-msec duration to —20 mV applied at 2 Hz was0.71 ±0.06 of IN, available after a long rest. Thesevalues are not significantly different than thoseobserved in the presence of Co2+ (Table 4).
Data Analysis and Mathematical ModelingCurve fitting and mathematical modeling were
performed on an IBM-PC/AT with programs writ-ten in the ASYST language (Asyst Software Technol-ogies, Rochester, New York). A nonlinear least-squares algorithm using the Gauss-Newton methodwas used to fit exponential functions to experimen-tal data. When the time courses of block develop-ment and recovery were defined, an F test based onthe residual error was used to compare the good-ness of fit for single and double exponential func-tions. A double exponential function was acceptedif it provided a significantly better fit (/><0.05).
A "two-state" version of the guarded-receptorhypothesis23-26 was used to describe the effects ofRAC109 on cardiac sodium channels and to calcu-late apparent association and dissociation rateconstants for drug interaction with channels at theholding (-140 mV) and test (-20 mV) potentials.The theoretical basis for the parameter estimationmethod has been extensively described by Starmerand coworkers.24-26 According to the model, tonicand use-dependent block of sodium channels thatis observed during a train of short depolarizingpulses results from drug interaction with a recep-tor site having diffusion pathways that are guardedby the position of channel activation and inactiva-tion gates. Drug cannot bind to a channel havingall its activation gates closed since activation gatesguard access to the receptor site via both hydro-philic and hydrophobic pathways. Thus, at stronglynegative holding potentials, where the fraction ofchannels having open activation gates (m3 in aHodgkin-Huxley model) is small, only a relativelysmall "tonic" block is observed at low drug con-centrations. When a channel is open, a drug can bindand unbind from the channel receptor via both hydro-philic and hydrophobic pathways. In addition, drugmay continue to bind and unbind from the channel
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

Clarkson Block of Na Channels by RAC109 Stereoisomers 1309
receptor via the hydropjiobic (membrane) pathwayonce the hydrophilic pathway becomes obstructed bya closed inactivation gate. When the channel activa-tion or inactivation gates close in a channel occludedby a charged drug, drug dissociation cannot occuruntil the channel reopens^ or, as for a_tejdiary ajmine,the drug is converted to an uncharged form. Unchargeddrugs are not trapped in rested or inactivated channelssince they can diffuse into the lipid bilayer at any time.One consequence of receptor guarding and drug trap-ping by channel gates is that a pulse-by-pulse accu-mulation of channel block (use-dependent block) ispredicted to occur when channels are repetitivelydepolarized by a train of pulses from a hyperpolarizedpotential.
When drug binding during depolarizing pulsesoccurs primarily to a single channel (e.g., open)state, drug interaction may be considered as twosequential processes described by24
Activation interval: [DJ+U
Recovery interval: [D]+U
where U and B represent the fraction of unblockedand blocked channels, [D] is the drug concentra-tion, kK and kT are binding rate constants, and /, and/r are unbinding rate constants in the activated (a)and recovery (r) intervals, respectively. Duringeach activation or recovery interval, changes inthe proportion of blocked channels is an exponen-tial process denned by the rate constants:
Activation interval: Aa=(fc,[D]H (1)
Recovery interval: A,=(A:r[D]-l-/r) (2)
where A, and Aj are the blocking and unblockingrates associated with the activation and recoveryintervals during the pulse protocol.
According to the model, the different binding andunbinding rate constants observed at the test andholding potentials reflect differences in the positionsof channel gates at the two potentials, as well as apossible Boltzmann term. For charged hydrophilicdrugs that can bind and unbind from the receptoronly when all channel gates are open, Starmer andGrant24 denned kt or Jt,=/nn3h, and lT or /,=/m3h,where k and / represent state-independent rateconstants, and m3 and h indicate the fraction ofchannels with open activation and inactivation gates,respectively (assuming a Hodgkin-Huxley modelfor channel gating). The binding and unbinding rateconstants for charged drugs may also need to bemultiplied by an additional Boltzmann term of theform e"*vmF/RT (where 8 indicates the electricaldistance of the binding site across the transmem-brane field, z is the drug molecule valence, F isFaraday's constant, R is the gas constant, and T is
absolute temperature) to account for the interactionbetween the dipole moment of the drug and themembrane electric field. For uncharged hydropho-bic drugs, Starmer and Grant also defined k, orka=km3, and /r or /„=/ since such agents can onlybind to channels once the activatiorLgates open butcan escape into the lipid bilayer at any time.
Tertiary amines such as RAC109 may not behavesimply as charged or uncharged drugs since theycan interconvert between charged and unchargeddrug forms by protonation and deprotonation at anytime. It has been proposed that the drug-channelinteraction in the presence of a tertiary amine may,therefore, be more completely characterized by thefollowing scheme24-25:
Unchargedinteraction
H
3hexp(-5zVmF/RT) c
where kp and lp represent the protonation and depro-tonation rates of drug within the blocked channel,Dc and Du indicate the concentration of charged (c)and uncharged (u) drug near the receptor site, Bcand Bu are the fraction of channels blocked bycharged and uncharged drug forms, and ka ka, lc,and /u are the rates of binding and unbinding forcharged and uncharged drug forms, which may notbe equal.25 A mathematical description for channelblock by drug mixtures based on the guarded recep-tor model has been derived25 and predicts a biexpo-nential time course for use-dependent accumulationof block and recovery from block when both thecharged and uncharged drug forms contribute sig-nificantly to both channel block and unblock. How-ever, as proposed by Hille,28 block of open oractivated channels may be predominantly or exclu-sively due to binding of the charged form over anaqueous pathway, whereas the lipid soluble neutralform may be the predominant drug form responsiblefor interacting with closed (rested or inactivated)channels. Based on these assumptions, the timecourse of use-dependent block during a pulse trainmay be modeled as a sequence of bimolecularbinding reactions similar to that predicted for a drugof fixed charge; the major caveat is that the appar-ent rate constants derived for drug interaction withopen channels may reflect the binding properties ofa different drug form (i.e., charged) than that respon-sible for drug interaction with closed channels (i.e.,uncharged). In addition, since RAC109 stereoiso-
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

1310 Circulation Research Vol 65, No 5, November 1989
mers are highly charged drugs with protonatedlifetimes of several seconds, it is quite possible thatthe deprotonation process may be rate limiting indetermining the rate of drug egress from closedchannels. In this case, the apparent rate constantsdefining unbinding from rested and inactivated chan-nels (/, and /j) would primarily reflect the rate of drugdeprotonation rather than the actual rate of drugdissociation from the receptor. Further experimen-tal information on the rate of deprotonation ofbound RAC109 molecules is required to test thishypothesis. In this study, only the composite"voltage-dependent" or "state-dependent" rate con-stants are defined since neither the proper gatingmodel for cardiac sodium channels nor the conver-sion rates from charged to uncharged RAC109 withinthe channel were known.
When assuming a two-state model to simulatedrug interaction during a train of pulses, the guarded-receptor model predicts that the time course ofuse-dependent block onset during a train of repeti-tive pulses can be described in terms of a recur-rence relation24:
during the recovery and activation intervals werecalculated by use of the following equations25-26:
bn=bSJ+(b0-bss)e- (3)
where bn is the fraction of channels blocked duringthe n1* pulse, b0 is the fraction of channels initiallyblocked (e.g., after a 3-minute rest at the holdingpotential), bM is the "steady-state" fraction of chan-nels blocked after many pulses, n is the pulsenumber, and A is the blocking rate constant. Theblocking rate constant A can be further denned by
(4)
where A, and A, are the blocking rates associatedwith the activation and recovery intervals duringthe pulse protocol, tr is the interpulse (recovery)interval, and t, is the mean receptor access time.24-26
For analysis, t, was assumed to be equal to 1 msec(i.e., similar to reported values of the mean opentime for single cardiac sodium channels).32'33
Least-squares fits of peak sodium currents duringpulse trains to Equation 3 were used to obtainestimates of the parameters A, bo, and bj,. Estimatesof A, and A, were then computed according toequation 4 by linear regression of A versus t,. by useof data from pulse trains elicited at four to fivedifferent interpulse intervals. The equilibrium levelsof block at the holding (R.) and test potential (A.)were then computed by linear regression of b^(obtained from Equation 3) versus a according tothe equations25:
bM=A.+a(R«-A.)
where a is the blocking parameter and
(5)
From calculated values of Ar, A,, R,, and A., theforward (kr and kB) and reverse (/r and /a) rateconstants defining drug interaction with channels
*r=ArR./[D]fc,=A,A4D]
/ r=Ail-R.)
(7)
(8)
(9)
A.) (10)
To estimate the apparent rate constants definingdrug interaction with channels once they becomeinactivated during a long pulse at -20 mV, it wasassumed that the slow component of time-dependent block characterized by use of the two-pulse protocol (Figure 4) results from drug bindingto a continuously accessible receptor (i.e., via thehydrophobic membrane pathway) as described by
[D]+U
B1.=ft,[D]/(*i[D]+/i) or B,.=A,[D]Ti
*,=B,./(7i[D])
hwhere k\ and I, are apparent rates of drug bindingand unbinding, respectively. During a long depolar-izing pulse, changes in the proportion of blockedchannels is an exponential process defined by therate equations:
(U)
(12)
(13)
(14)
Statistical AnalysisComparisons between two groups of data were
evaluated for statistical significance by paired orunpaired two-tailed Student's t test. Comparisonsbetween more than two groups were evaluated byone-way ANOVA and Scheffe's test for criticaldifference. Data analyzed by linear regression wereevaluated by calculation of the Pearson product-moment correlation coefficient (r) and a t test forsignificance of the linear trend. Differences wereconsidered significant if values of /?<0.05 wereobtained. Results are expressed as mean±SD.
ResultsStereospecificity of Tonic andUse-Dependent Block
The effects of RAC109 isomers RAC109-I andRAC109-II on cardiac IN, were similar to most otherlocal anesthetic agents in that block could be empir-ically divided into two primary components, a tonicblock and a use-dependent block. As illustrated inFigures IB and 1C, on application of a train ofdepolarizing pulses at 1 Hz after a long rest, therewas no change in sodium current in the absence ofdrug. In contrast, after a 15-minute exposure to 50fiM of either RAC109 stereoisomer, there was areduction of INt amplitude during the first pulse after
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

Clarkson Block of Na Channels by RAC109 Stereoisomers 1311
a long rest (tonic block) and an additional use-dependent block of current that became progres-sively larger during each successive pulse until asteady state was approached after 10-15 pulses.
The mean levels of tonic and use-dependent blockat different stimulation rates are shown in Figure1C. In the absence of drug, there was no significantchange in IN> amplitude as the stimulation rate wasincreased up to 5 Hz. In the presence of 50 /AMRAC109-I or RAC109-II, only a stable tonic blockwas observed when preparations were stimulatedinfrequently (e.g., once per 3 minutes), and thelevels of tonic block were not significantly differentfor the two drugs (RAC109-I, 0.30+0.08, n = 10;RAC109-II, 0.24±0.10, n=12) (Figure 1C). How-ever, at stimulation rates greater than 0.1 Hz, therewas a marked use-dependent block that becameprogressively larger at higher stimulation rates withRAC109-I producing a significantly greater blockcompared with RAC109-II at all stimulation ratesbetween 0.1 and 5 Hz (p<0.05).
Voltage-Dependence of Use-Dependent BlockBlock of sodium channels in nerve by local anes-
thetic drugs,34-35 including RAC109,7 has been shownto be a strongly voltage-dependent process. Thevoltage dependence of sodium channel block byRAC109 in cardiac myocytes was characterized byuse of the pulse protocol shown in Figure 2A.Thirty pulses of 2-msec duration were applied tovarious conditioning voltages at 3 Hz to produce asteady-state level of block. The level of blockproduced by the conditioning train was then deter-mined by a test pulse after a 300-msec recoveryinterval to allow unblocked channels to recoverfrom inactivation. In the absence of drug, the con-ditioning train had little effect on the test INa (Figure2B). However, after addition of either stereoiso-mer, channel block was a steep function of mem-brane voltage between -60 and +40 mV. As indi-cated by the smooth curves in Figure 2B, thevoltage dependence of block could be well describedby an equation that has been previously used todescribe block of open channels by local anesthet-ics in nerve35-36:
20 ms
-140mV
I N . lN. (cont»i)=(l - B ) / ( l + e x p [ V c -
(15)where B is the steady-state level of sodium currentremaining at positive potentials, Vc is the membranepotential during the conditioning train, V ^ is themembrane potential at which drug block is halfmaximal, and S is the slope of the voltage-dependent block. Least-squares fits of Equation 15to data obtained in the presence of 50 /iM drugindicated that RAC109-I reduced IN, to a signifi-cantly smaller steady-state level at positive poten-tials (B=0.32±0.06; n=6) compared with RAC-109-11 (B=0.72±0.05 mV; n=5) and that the slopefactor for block by RAC109-I (S=13.7±1.9) was
B
3m»
1.0
0.8as
f o ejo 0.4CD
= 0.2
0.0 L .
>Control
0 1 2 3 4 5Stimulation Rate (Hz)
FIGURE 1. Tonic and use-dependent inhibition of thepeak sodium current (INJ by RAC109-1 and RAC109-1I inguinea pig ventricular myocytes. Panel A: Diagram ofpulse protocol. A train of depolarizing pulses to -20 mVwas applied at different stimulation rates after a 3-minuterest at the holding potential (—140 mV). Panel B: Super-imposed recordings of INa obtained during a train ofpulses applied at 1 Hz before and during exposure to 50iJA RAC109-I or RAC109-II. For the controls, currentsfor only the first and fifteenth pulses are shown. Therewas no noticeable decrease in peak current during repet-itive pulsing under control conditions, whereas in thepresence of drug there was both an initial (tonic) blockobserved on the first pulse, as well as a use-dependentblock that approached a steady state within 10-15pulses.The arrows at the beginning of each family of tracingsindicate the zero current level Panel C: Graph showingthe mean level of peak sodium current after 15 pulses atselected stimulation rates under control conditions (o)and in the presence of 50 /JM RAC109-I (•) and 50 yMRAC109-H (•). Peak lNa amplitudes have been normal-ized to the amplitude of control IN,, obtained after a restof 10 seconds or more. RAC109-Iproduced a significantlygreater block compared with RAC109-II at all stimulationrates between 0.1-5 Hz (p<0.05). Data values representmean±SD (n=4-10).
significantly larger than for RAC109-II(S = 17.6±3.3mV;/?<0.05). The midpoints for voltage-dependentblock (Vmid) were not significantly different(-37.9±3.9 for RAC109-I vs. -34.2±6.7 mV forRAC109-H).
The parameters denning the voltage dependenceof channel block were also similar, but not identi-cal, to those of channel opening or conductance(GN.) [where GN,=IN,/(Vm-VN,), Vm is membranepotential, and VN, is sodium equilibrium potential].GNa was calculated from either peak IN, or integrals
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

1312 Circulation Research Vol 65, No 5, November 1989
B
50/iMRACI 09-11
0.0Lt
-140 -100 -60
Vc(mV)
FIGURE 2. Voltage dependence of use-dependent blockproduced by RAC109 stereoisomers. Panel A: Diagramshowing use-dependent block produced by a train of 30conditioning pulses of2-msec duration at 3 Hz to selectedmembrane potentials (VJ. The amplitude of the peaksodium current (INa) available after the conditioning trainwas determined with a test pulse after a recovery intervalof 300 msec. Panel B: Graph showing the relation betweenpeak INa and conditioning potential. Peak current wasnormalized to its value after a long rest at —140 mV. Datavalues represent mean±SD. O, control (n = ll); m,RAC109-I (n=6); • , RAC109-II fn=5). The amplitude ofblock produced by RAC109-I was significantly greaterthan RAC109-II at all voltages between -60 and +40 mV(p<0.05). The smooth curves represent least-squares fitsto a Boltzmann relation (equation 15). The best-fit valueswereB=0.32, S=13.6mV, andVmkl=-38mVforRAC109-I and B=0.73, S=16.3 mV, and VmU=-34.2 mV forRAC109-II, where B is the steady-state level of INQ
remaining at positive potentials, S is the slope of thevoltage-dependent block, and Vmii is the membrane poten-tial at which drug block is half-maximal.
of IN. • dt during the initial 2 msec or 25 msec ofdepolarizing pulses to potentials from —70 to +20mV. The 2-msec and 25-msec integrals were selectedon the basis that they closely resembled either theconditioning pulse duration (Figure 2A) or an infi-nite integral of INa-dt since INB was almost fullyinactivated within 25 msec at most potentials. Fitsof peak GNl with a Boltzmann equation of the formGNa=GIto/{l+exp[(Vlllld-Vin)]/S} (where GM« is themaximum GN,) had a mean slope factor (S) of6.0 ±1.2 mV and a half-maximal value (V,^) of-34.4±7.9 mV («=9). Thus, although both channelblock and peak GN, were half-maximal at similarpotentials, the slope factor for peak GN, was signif-icantly-smaller ij? <0;05) than that for channels lock;thus, the channel opening -had a steeper voltagedependence than channel block. Similar differences
in the slope factors were also obtained when com-paring channel block with time integrals of GNa (fora 2-msec integral: S=7.3±0.8, Vmid=-21.3±7.5;and for a 25-msec integral: S = 4 . 3 ± l . l ,Vmid=-42.1±7.4;«=9).
Time Course of Recovery FromUse-Dependent Block
The time course of recovery from use-dependentblock was characterized using the pulse protocolshown in Figure 3A. A steady-state level of blockwas first produced by a train of pulses of 50-msecduration to +20 mV at 3 Hz, and the time course ofrecovery from block was defined by use of a singletest pulse evoked after a variable recovery time. Inabsence of drug, recovery of INa from fast inactiva-tion at -140 mV was well described by a singleexponential function having a time constant of7.2±3.1 msec (n = 16). In contrast, in the presenceof 50 /xM of either RAC109-I or RAC109-II, recov-ery of INa after the conditioning train exhibited twodistinct phases: a fast phase and a slow phase(Figures 3B and 3C). In the presence of eitherstereoisomer, the fast phase had a time constantsimilar, although significantly larger, than control(Table 1). The time constant of the slow phase ofrecovery from block at both -140 and -160 mVwas significantly larger in the presence of RAC109-Icompared with RAC109-II (p<0.001) (Table 1).The slow phase of recovery in the presence of drugwas assumed to reflect the time course with whichdrug unbinds or escapes from rested channels. Thefast phase of recovery in the presence of drug mayreflect a combination of recovery from inactivationof drug-free channels as well as some unblockingfrom open channels at -20 mV after block inducedby pulses at +20 mV. Hyperpolarization of themembrane by —20 mV during the recovery period(from —140 to —160 mV) did not significantly alterthe time constant for recovery from block (Table 1).
Time Course of Block DevelopmentTo determine how RAC109 stereoisomers differ
in their binding to sodium channels during a depo-larizing pulse, the time course of block develop-ment was defined by use of a two-pulse protocol.As illustrated in Figure 4A, the membrane wasfirst conditioned by a clamp of variable duration to-20 mV. The level of block produced by thisconditioning pulse was then determined with a testpulse to —20 mV after a fixed recovery interval toallow drug-free channels to recover from inactiva-tion. Figure 4B shows the mean time course ofblock development in the presence of 50 tiMRAC109-I and RAC109-II. In the absence of drug,there was little change in test current amplitudeafter conditioning pulses of up to 15-second dura-tion. However, after addition of either RAG109isomer, there was a progressive decrease in INaamplitude as the conditioning pulse duration wasincreased. Two definable phases of time-dependent
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

Clarkson Block of Na Channels by RAC109 Stereoisomers 1313
Condition Test
140mV
0.1
0.01
0.001
0.001L
60 120 180(ma)
12 24 36 48 60
Recovery Time (s)
FIGURE 3. Time course of recovery from use-dependent block. Panel A: Diagram of steady-state level of use-dependent block first producedby a train of 30 conditioning pulses of 50-msecduration to +20 mVapplied at 3 Hz. The kineticsof recovery from block were then defined bymeasuring the fraction of peak sodium current(lust) available with a test pulse elicited after avariable recovery time (At). Panel B: Graph show-ing the time course of recovery of peak sodiumcurrent under control conditions (o) and in thepresence ofRAC109-I (•) and RAC109-II (•). Forillustration, the test currents have been normal-ized to their steady-state rested value (Im) andplotted semilogarithmicalty as a function of recov-ery time. Panel C: Graph showing the dataobtained at early recovery times on an expandedtime scale. Under control conditions, recoveryfrom inactivation was well described by a singleexponent. In the presence ofRAC109-I or RAC109-II, recovery of current was clearly biphasic andwas well fit by the sum of two exponential func-tions. Full recovery from block required morethan 60 seconds. The recovery time constant (t)for the slow phase (inverse of the slope) wassmaller for RAC109-II compared with RAC109-I.Data indicate mean±SD for control (n=I6),RAC109-I (n=10), and RAC109-II (n=U). Theequations of fit for mean data were l.Oe'1174 msec
for control, 0.31e"1194 na*c+0.65e-"24-7 "c forRAC109-I, and 0.63e-"14-3 l~cc+0.33e-""u ^ forRAC109-II (where t=time).
block were observed, which could be well approx-imated by the sum of two exponentials and aconstant (Afi'"r'+Ase~tJu+A.). The amplitudes (A,,fast phase; As, slow phase; A., steady state) andtime constants (rf, fast phase; T3, slow phase) fortime-dependent block by RAC109-I were Af=0.11±0.03, Tf=2.1±1.5 msec, As=0.74±0.10, TS=14.1±4.6 sec, and A.=0.16±0.09 (/i=8), and those forRAC109-II were A,=0.10±0.05, rf=2.7±1.8 msec,A,=0.54±0.11, T , = 1 5 . 1 ± 3 . 4 sec, and A.=0.36±0.09 (n=7). The time constants for both phasesand the amplitude of the fast phase were not
significantly different for RAC109-I versusRAC109-II, in contrast to the amplitude of theslow phase and the steady-state level of block,which were significantly different for the twoisomers (/?<0.05).
The very large time constant (14-15 seconds)observed for the slow phase of block developmentfor both stereoisomers indicates that this compo-nent of channel block cannot contribute signifi-cantly to the cumulative use-dependent block pre-viously denned by use of trains of brief pulses(Figures 1 and 2). In addition, since use-dependent
TABLE 1. Parameters Defining the Time Course of Recovery From Block
50 ftM RAC109-I 50 RAC109-IIParameter
A,Tf (msec)A,T, (sec)n
-140 mV
0.30±0.1519.5±6.6*0.65+0.1026.8±4.3
9
-160 mV
0.66+0.0920.5±3.8
6
-140 mV
0.65±0.1413.0±6.2*0.32±0.05t15.1±4.6t
11
-160 mV
0.31±0.04t11.9+1.Of
4
Values are mean±SD. Af, amplitude of fast phase; T(, time constant for fast phase; A,, amplitude of slow phase;T,, time constant for slow phase. A( and A, represent the fraction of total available current recovering at the recoverypotential.
'Significantly different from control time constant: 7.2±3.1 msec (n = 16) (p<0.05).tSignificantly different from RAC109-I at the same recovery potential (p<0.05).
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

1314 Circulation Research Vol 65, No 5, November 1989
Condition
0 8 12 16 20
Condition Pulse Duration (s)
FIGURE 4. Biphasic time course of block development at—20 mV. Panel A: Diagram showing block developmentdetermined by a single test pulse applied 300 msec after aconditioning pulse of variable duration (At). The intervalbetween each trial of the pulse protocol was 3 minutes.Panel B: Graph showing the time course of block devel-opment at —20 mV. Under control conditions (o) therewas little change in peak current amplitude after condi-tioning pulses of up to 15-second duration. In the pres-ence of RAC109-I (•) or RAC109-II (•), peak sodiumcurrent (Ilca) was inhibited in two distinct phases havingmean time constants of a few milliseconds and 12 sec-onds. The inset shows the time course of block develop-ment at earlier times on an expanded scale (time inseconds). Data indicate mean±SD for control (n=9),RAC109-I (n=8), and RAC109-II (n=7). The equations offit for mean data were O.lOe-"4-9 m^+0.67e-"'Z0 ^+0.23for RAC109-I and O.lOe'"2-1 •™c+0.45e"1'2-0 ™+0.46 forRAC109-11 (where t=time). Test currents have been nor-malized to their value (Io) in the absence of conditionpulse.
block by the RAC109 stereoisomers requires multi-ple depolarizations to approach steady state (Figure1), the amplitude of the fast phase of block observedduring the two-pulse protocol (Figure 4B) must notreflect a true steady-state level of drug binding to agiven channel state but rather reflects incompletedrug binding to a high-affinity channel state that isonly transiently occupied during a depolarizing pulse.
Effect ofRAC109 on INa Tune CourseAlthough 50 fiM of either RAC109 isomer could
produce a large use-dependent block of IN> duringrepetitive pulsing to -20 mV, neither isomer pro-duced a significant effect on the time course of INt atthis potential (Table 2). In the absence of drug, at-20 mV the decay of INl after its inward peak canbe well described as the sum of a fast and slowexponential function plus a small constant.30 After a15-minute exposure to 50 /iM of either RAC109stereoisomer, neither the time constants nor therelative fractions of available current that decayedwith fast or slow time constants were significantlyaffected by exposure to drug (Table 2). Consistentwith the lack of effect on sodium current kinetics,neither isomer produced a significant change in thetime to peak current after the onset of pulses to - 20mV when compared with control (RAC109-I:-0.06±0.26 msec, n = U; RAC109-II: -0.01±0.14msec, n = 12). These results suggest that, if RAC109stereoisomers bind to open channels during a depo-larizing pulse, they must do so with a rate that isslower than that for channel inactivation.
Effects of Reducing Channel InactivationSince sodium channels are both activating and
inactivating within the first few milliseconds of apulse to —20 mV, the possibility was consideredthat the fast phase of RAC109 block could resultfrom drug binding to a rapidly occupied inactivatedstate. To test this hypothesis, cardiac myocyteswere first pretreated with a-chymotrypsin (0.7 mg/ml applied intracellularly by addition to the pipettesolution), which has been reported to markedlyreduce or eliminate fast inactivation in neuroblast-oma cells.37 After exposure of the internal membranesurface of cardiac myocytes to a-chymotrypsin for30-60 minutes, there was only a very slow andincomplete decay of IN. detectable during pulses of20-100 -msec duration at potentials ranging from-60 mV to +60 mV (Figures 5C and 6). In addition,as illustrated in Figure 5C, RAC109-I was still fullycapable of producing use-dependent block in cellsin which fast inactivation was virtually abolished byenzyme pretreatment. At a 1-Hz stimulation rate,the amplitude of use-dependent block produced by50 .̂M RAC109-I was similar in both normal(0.50±0.10; n = 10) and inactivation-modified cells
TABLE 2. Effect
Condition
ControlRAC109-IRAC109-II
of RAC109 on
A,(iN.,/W0.75±0.110.69±0.070.73+0.18
Kinetics of Sodium
(msec)1.7±0.31.5±0.31.6±0.5
Current Decay at
A,(IN.. / IN..)
0.22±0.100.28±0.O60.25±0.14
- 2 0 mV
(msec)7.7±1.96.9±1.88.9±4.1
A.(IN. . / IN. O )
0.03+0.020.03±0.020.03+.0.04
n
201010
Values are mean+SD. Time constant (T) and amplitude (A) for the fast (f), slow (s), and steady-state («) level ofsodium current (IN.) are estimated from fits of the time course of current decay under control conditions and after15 minutes in 50 MM RACIOS-Lor RAG109-H. Pulses to -20 mV were elicited after a 3-minute rest at -140 mV.Amplitudes are normalized to the extrapolated current amplitude at zero time PN.J - IN.,, current amplitude at fastlevel; 1 ^ , current amplitude at slow level. Values in drug are not significantly different from- control.
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

Clarkson Block of Na Channels by RAC109 Stereoisomers 1315
(0.59±0.09; n=5) (/>«0.05) (compare Figures 5Band 5C). However, the rate of onset of use-dependent block during a train of pulses was signif-icantly accelerated in inactivation-modified cells(Figures 5B and 5C): the pulse constant for theonset of use-dependent Wock for RAC109-I at 1 Hzwas 5.48 ±1.10 pulses in normal cells («=10) and1.88±0.37 pulses in inactivation-modified cells (n =5)(p<0.001). These results suggest that inactivation isnot essential for use-dependent block and that thepresence of an intact inactivation gate may actuallyretard its development. In contrast to use-dependent block, the amplitude of tonic block pro-duced by RAC109-I was significantly increased ininactivation-modified cells (0.54±0.11; n=5) com-pared with normal cells (0.30±0.08; n = lO) (/><0.01).
In addition to producing a use-dependent decreasein peak current amplitude, exposure to 50 /iMRAC109-I also induced a noticeable time-dependentdecay of INa in inactivation-modified cells (whichwas not definable in cells with intact inactivation)(Figures 5C and 6). The kinetics of this time-dependent block of channels were defined by fittingthe ratio of current in drug over the control currentobtained during 50-100-msec pulses to -20 mV(Figure 6C). (Although fitting raw current traces ordifference currents provided similar results, fittingof the current ratio was considered to more accu-rately reflect the drug-induced time-dependent blocksince most cells had a small amount of slowlyinactivating current before drug exposure [see Fig-ure 6B] and there was some overlap in the kineticsof channel activation and channel block.) By use ofthis approach, the time course of drug-inducedblock could be well fit by a single exponentialfunction and a constant (e.g., IN, ra t io=Ae~+B,where t=time) (Figure 6C). The mean amplitude oftime-dependent block (A) was 0.37±0.05, the meantime constant (T) of channel block was 34.0±10.0msec, and the fraction of unblocked current at steadystate (B) was 0.14±0.03 (n=6). The instantaneouslevel of current (A+B) provides an additional esti-mate of channel block before channel activation andhad a mean amplitude of O.5O±O.O5 (n=6).
Mathematical Modeling of RAC109 BlockTo gain further insight into the molecular mech-
anisms responsible for the stereoselectivity of use-dependent and tonic block produced by RAC109isomers, data from pulse train protocols (Figure 7A)were analyzed by a parameter estimation procedurebased on the guarded-receptor model (see "Materi-als and Methods").2*-26 A nonlinear least-squaresalgorithm was used to obtain fits of equation 3 tovalues of peak INo recorded during trains of 20-msecpulses to -20 mV (Figure 7B). From these fits,values of blocking rate (A), tonic block (b0), and thesteady-state level of use-dependent block (b^) wereestimated for four to five different interpulse inter-vals (tr). A linear regression of blocking rate (A)against recovery interval (tr) was then performed by
20 ms
3ms
FIGURE 5. Effect of inactivation modification on sodiumchannel block by RAC109-I. Panel A: Diagram showing atrain of depolarizing pulses to -20 mV applied at 1 Hzafter a 3-minute rest at the holding potential (—140 mV).Panel B: Tracings showing tonic and use-dependent blockproduced by 50 /JM RAC109-1 in a typical cell with intactinactivation. Panel C: Tracings showing tonic and use-dependent block produced in a cell pretreated with 0. 7mglml a-chymotrypsin. Although inactivation was reducedto a negligible level, RAC109-I still produced a tonic anduse-dependent block qualitatively similar to that observedin cells with intact inactivation. The horizontal arrowsindicate the level of zero current.
use of Equation 4 to calculate values of A.t, (theintercept) and Ar (the slope) (Figure 7C). A 1-msecvalue for the activation interval (t,), consistent withestimates of sodium channel mean open time,32-33
was assumed to calculate A,. Values of the blockingparameter (a) were then computed by use of Equa-tion 6, and a linear regression of steady-state block(bM) against a was performed by use of Equation 5to estimate the equilibrium levels of channel blockat the holding potential (R.) and at -20 mV (A.)(Figure 7D). Both types of relations (A vs. t, and b,,vs. a) were found to be well described by a simplelinear function (r>0.9, /><0.05) (Figures 7C and7D), consistent with the theoretical model. Theseestimated model parameters were then used tocalculate the apparent binding and unbinding ratesaccording to Equations 7-10. Mean values of theapparent binding and unbinding rate constants kT, lr,kB, and /, obtained from 10 experiments in RAC109-I and nine experiments in RAC109-H are given inTable 3. Since the estimates of k, and /„ were basedon an assumed value for t., assumption of a differentaccess interval for calculation of A, will result inreciprocal changes in estimated values of km and /,from Equations 8 and 10 (e.g., assuming tg=2 msecresults in values of kt and /, half those shown inTable 3).
The apparent rate constants defining drug bindingand unbinding from inactivated channels (kt and /;)were calculated from Equations 11-14 by use ofestimates of the steady-state level of block(tonic+time-dependent) at -20 mV (B;,) and thetime constant for the slow phase of channel blockobtained by use of a two-pulse protocol (Figure 4).Mean values for Iq and lt are shown in Table 3.
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from
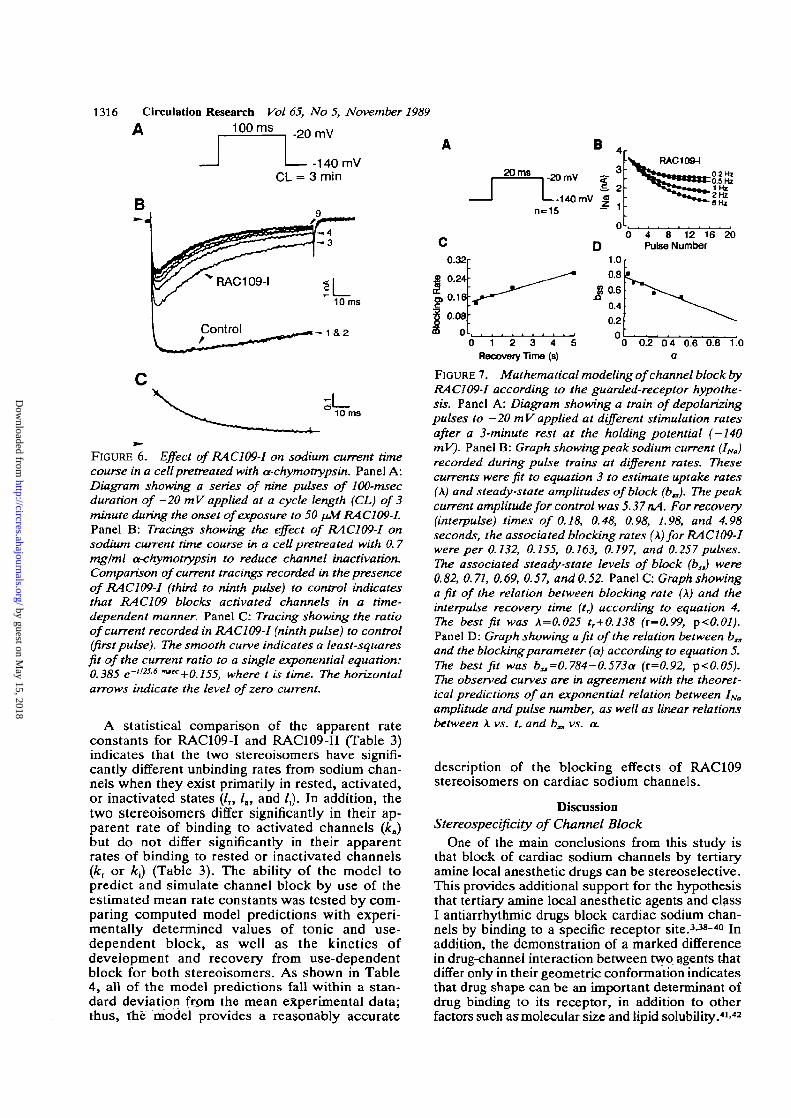
1316 Circulation Research Vol 65, No 5, November 19891OO ms _2Q
140mVCL = 3 min
10 ma
FIGURE 6. Effect of RAC109-1 on sodium current timecourse in a cellpretreated with a-chymotrypsin. Panel A:Diagram showing a series of nine pulses of 100-msecduration of —20 mV applied at a cycle length (CL) of 3minute during the onset of exposure to 50 /uM RAC 109-1.Panel B: Tracings showing the effect of RAC109-1 onsodium current time course in a cell pretreated with 0.7mg/ml a-chymotrypsin to reduce channel inactivation.Comparison of current tracings recorded in the presenceof RAC 109-1 (third to ninth pulse) to control indicatesthat RAC109 blocks activated channels in a time-dependent manner. Panel C: Tracing showing the ratioof current recorded in RAC109-I (ninth pulse) to control(firstpulse). The smooth curve indicates a least-squaresfit of the current ratio to a single exponential equation:0.385 e-"256 n"c+0.155, where t is time. The horizontalarrows indicate the level of zero current.
A statistical comparison of the apparent rateconstants for RAC109-I and RAC109-II (Table 3)indicates that the two stereoisomers have signifi-cantly different unbinding rates from sodium chan-nels when they exist primarily in rested, activated,or inactivated states (/„ /„ and /,). In addition, thetwo stereoisomers differ significantly in their ap-parent rate of binding to activated channels (fcB)but do not differ significantly in their apparentrates of binding to rested or inactivated channels(fcr or k) (Table 3). The ability of the model topredict and simulate channel block by use of theestimated mean rate constants was tested by com-paring computed model predictions with experi-mentally determined values of tonic and use-dependent block, as well as the kinetics ofdevelopment and recovery from use-dependentblock for both stereoisomers. As shown in Table4, all of the model predictions fall within a stan-dard deviation from the mean experimental data;thus, the model provides a reasonably accurate
B
20 ms 20 ms
1L-140mVn-=15
0.32
0.24
, 0.16
0.08
00 1 2 3 4
Recovery Time (a)
4
3
I2I 1
0
1.0
0.8
0.6
0.4
0.2
0
^ ^ RAC109
^*«^"V'iii3CL2 H i~0& Hz
1 Hi2 Z H z•-9HI
4 8 12 16 20Putee Number
02 0 4 0.6 0.8 1.0a
FIGURE 7. Mathematical modeling of channel block byRAC109-I according to the guarded-receptor hypothe-sis. Panel A: Diagram showing a train of depolarizingpulses to -20 mV applied at different stimulation ratesafter a 3-minute rest at the holding potential (—140mV). Panel B: Graph showing peak sodium current (INa)recorded during pulse trains at different rates. Thesecurrents were fit to equation 3 to estimate uptake rates(X) and steady-state amplitudes of block (ba). The peakcurrent amplitude for control was 5.37 nA. For recovery(interpulse) times of 0.18, 0.48, 0.98, 1.98, and 4.98seconds, the associated blocking rates (X) for RAC 109-1were per 0.132, 0.155, 0.163, 0.197, and 0.257 pulses.The associated steady-state levels of block (bj) were0,82, 0.71, 0.69, 0.57, and 0.52, Panel C: Graph showinga fit of the relation between blocking rate (\) and theinterpulse recovery time (tr) according to equation 4.The best fit was X=0.025 tr+0.138 (T-0.99, p<0.01).Panel D: Graph showing a fit of the relation between bn
and the blocking parameter (a) according to equation 5.The best fit was bIJ=0.784-0.573a (r=0.92, p<0.05).The observed curves are in agreement with the theoret-ical predictions of an exponential relation between INa
amplitude and pulse number, as well as linear relationsbetween A vs. tr and ba vs. a.
description of the blocking effects of RAC109stereoisomers on cardiac sodium channels.
DiscussionStereospecificity of Channel Block
One of the main conclusions from this study isthat block of cardiac sodium channels by tertiaryamine local anesthetic drugs can be stereoselective.This provides additional support for the hypothesisthat tertiary amine local anesthetic agents and classI antiarrhythmic drugs block cardiac sodium chan-nels by binding to a specific receptor site.3-38-40 Inaddition, the demonstration of a marked differencein drug-channel interaction between two agents4hatdiffer only in their geometric conformation indicatesthat drug shape can be an important determinant ofdrug binding to its receptor, in addition to otherfactors such as molecular size and lipid solubility.41-42
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

Clarkson Block of Na Channels by RAC109 Stereoisomers 1317
TABLE 3. State-Dependent Rate Constants for RAC109 Stereoisomers
RAC109-I
Mean
SD
RAC109-II
Mean
SD
P
K(/M/msec)
2.09 • 10"1
1.02 • 10"'
4.62 • 10"1
5.63 -10-'
NS
(/msec)
2.24 • 10-=
0.55 • 10"!
6.08 • 10"3
3.14 10"3
<0.0O2
(/iM)
143
104
610
987
NS
k.(/M/msec)
2.52-103
0.68 • 103
1.52-103
0.82-103
<0.01
/.(/msec)
2.86 • 10 2
1.33-10"2
8.00 • 10"2
3.50 • 10-2
<0.001
0*M)
12.3
6.0
60.1
28.1
<0.0005
kx
(/M/msec)
1.32
0.36
1.05
0.21
NS
h(/msec)
7.95 • lO"6
4.88 • 10-*
1.80 lO"5
9.84 • 10"6
<0.03
(MM)
6.36
3.86
17.47
8.51
<0.01
Rate constants indicate apparent binding (k) and unbinding (/) rates to channels rested at -140 mV (r), activated at -20 mV (a), andinactivated at -20 mV (i). No attempt was made to factor out receptor guard and trap functions (see "Materials and Methods").State-dependent dissociation constants (IQ) are mean values of the ratio l/k for all experiments (n=8-10).
This conclusion is also supported by the results ofseveral recent structure-activity studies, which haveshown that physiochemical factors (e.g., pK,, lipidsolubility, and molecular weight) alone can onlypartly account for the ability of different drugs toblock sodium channels.43-45 Interestingly, althoughuse-dependent block by RAC109 was markedlystereoselective, the level of tonic block producedby the two stereoisomers was not significantly dif-ferent (Figure 1 and Table 4). As discussed below,this similarity in tonic block can be attributed to alack of stereospecificity in the binding (association)rates (kr) for RAC109 enantiomers to channels at theholding potential (Table 3).
Two Components of Block DevelopmentRecent voltage-clamp studies on cardiac sodium
channels have provided strong evidence that use-dependent block produced by tertiary amine localanesthetics can result from drug binding to multiplechannel states (e.g., activated and inactivated).30-46-47
Therefore, experiments were performed to clarify
further the molecular basis underlying the stereospec-ificity of RAC109 block.
Characterization of the time course of RAC109block development at —20 mV by use of a two-pulseprotocol (Figure 4) revealed that block associatedwith depolarization consists of two definable com-ponents: a fast component (T{=2-3 msec) and avery slow component (TS= 14-15 sec). This suggeststhat RAC109 stereoisomers have a relatively highaffinity for more than one channel state occupiedduring a depolarizing pulse. Since virtually all sodiumchannels are completely inactivated within less thana hundred milliseconds at -20 mV, the simplestexplanation for the slow component of block is thatit reflects very slow drug binding to inactivatedchannels. Furthermore, the slow component devel-ops so slowly that it cannot significantly contributeto the use-dependent block measured by use of brieftrains of short (2-20-msec) pulses (Figures 1 and 2).Therefore, the use-dependent block defined by useof trains of short pulses must reflect a pulse-by-pulse accumulation of channels blocked via the
TABLE 4. Comparison of Model Predictions With Experimental Data
Tonic block
Steady-state block
5Hz
1.43 Hz
1 Hz
0.5 Hz
0.2 Hz
A (/pulses)
5 H z
1.43 Hz
1 Hz
0.5 Hz
0.2 Hz
T, (sec)
50 /xM RAC109-I
Experiment
0.30±0.08
0.75±0.08
0.68±0.10
0.66±0.09
0.62±0.09
0.54±0.10
0.16±0.04
0.15±0.03
0.19+0.04
0.22±0.04
0.31±0.05
26.8+4.3
Model
0.32
0.74
0.71
0.69
0.64
0.54
0.16
0.18
0.19
0.22
0.32
30.4
5OAIN
Experiment
O.24±0.10
0.43+0.10
0.44±0.09
0.38±0.11
0.35 + 0.11
0.30+0.08
0.17+0.05
0.20+0.01
0.23 + 0.06
0.33 ±0.10
0.61 ±0.45
15.1+4.6
1 RAC109-II
Model
0.28
0.45
0.42
0.40
0.37
0.32
0.17
0.21
0.24
0.32
0.57
11.9
Experimental data are mean±SD (TI=4-10). Values are model predictions computed from the rate constantsshown in Table 3. Steady-state block was produced by a train of 15 pulses of 20-msec duration to -20 mV (holdingpotential=-140mV). A, rate of use-dependent block onset; rr, time constant for recovery from use-dependent blockat -140 mV.
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

1318 Circulation Research Vol 65, No 5, November 1989
fast component. This conclusion is supported bythe observation that the amplitude of channel blockproduced by a series of brief pulses (Figures 1 and2) is much greater than the amplitude of the fastcomponent produced by a single pulse of variableduration (Figure 4, inset). The observation of acomponent of drug binding that develops rapidlybut does not reach steady state during a singledepolarizing pulse indicates that RAC109 isomersmust bind to a channel state that is only transientlyoccupied on membrane depolarization and that thetime constant for drug binding is slower than themean dwell time of the channel in this high-affinitystate.
Since sodium channels fluctuate through a num-ber of conformational states during the first fewmilliseconds of a suprathreshold depolarization,the identity of the high-affinity activated state isnot readily obvious. However, current theoriessuggest that activation-related block may be pri-marily due to the charged (cationic) form of localanesthetics selectively binding to open sodiumchannels.23-34-38'39 Because both RAC109 enanti-omers (pK,=9.4) exist almost exclusively in thecharged form at the pH used (7.2-7.3) and the timecourse of the observed fast component overlapsthat of channel opening, the possibility was con-sidered that this component results from block ofopen channels (which have a mean dwell time ofonly 1-2 msec at 10°-20° C).32-33-48
Drugs believed to block open channels in nerve,including RAC109,7 have been shown to blocksodium channels in a voltage-dependent manner.34-35
Characterization of the voltage dependence ofRAC109 block in cardiac myocytes (Figure 2)revealed that the fast component of block had avoltage dependence that was similar to channelopening in that both phenomena developed andsaturated over a similar voltage range and hadsimilar voltage midpoints (-34 to -38 mV). Incontrast, the voltage dependence of block differedfrom that of channel inactivation, which developsand saturates over a voltage range of -120 to —60mV under these conditions.30-49 Furthermore, theamplitude of use-dependent block was not reducedeven after enzymatic modification of channels, whichprevented inactivation development during shortpulses (Figure 5C). In fact, the rate of use-dependent block onset was accelerated ininactivation-modified cells; this occurrence sug-gests that the closure of the inactivation gate actu-ally retards the development of the fast componentof block. These results indicate that the fast com-ponent of RAC109 block cannot be attributed todrug binding to inactivated channels and must resultfrom binding to either the open state or a preopenclosed state that is closely associated with the openstate (i.e., is also transiently occupied at depolar-ized potentials and is not removed by proteolyticenzymes).
Although the fast component of channel block byRAC109 isomers developed and saturated over asimilar voltage range as that of channel opening(e.g., peak GN.), the voltage dependence for the twophenomena were not identical in that channel blockwas consistently less steep than that of peak GNa.The explanation for this difference is not clear butmight possibly be due to a competitive interactionbetween RAC109 and monovalent cations (e.g.,Na+) for a common binding site. Previous studies innerve have shown that the presence of externalsodium ions can markedly reduce the slope forvoltage-dependent block for QX-314,36 as well assome analogues of disopyramide.44 However, addi-tional studies are needed to confirm this for RAC109.
Although the results seem consistent with blockof open channels, the voltage dependence of blockis much steeper than predicted by previouslyproposed models that account for voltage-dependent open-channel block in terms of aneffect of the transmembrane field on the passivediffusion of cationic drug to a blocking site withinthe sodium channel.34-35 According to this type ofmodel, the slope parameter defining the steepnessof voltage-dependent block (e.g., S in Equation15) can be defined as S=RT/z6F,33 where F, R,and T have their usual meaning, z is the drugmolecule valence, and S is the equivalent electricaldistance of the binding site across the membranefield (0-1) from the inside. In this study, theexperimental conditions were such that RT/F=25mV and z = l . Therefore, the minimal value of Spredicted by this theory (i.e., when 5=1) is 25. Incontrast, RAC109-I and RAC109-II had meanslope factors of 13.6±1.7 and 17.6±3.3 mV, respec-tively. Similar slope factors have also been recentlyreported for block of sodium channels by lidocainein cardiac myocytes30 and for block of sodiumchannels in the squid giant axon by analogues ofdisopyramide.44 These results indicate that thedependence of fast channel block on membranevoltage (Figure 2) may be due primarily to avoltage dependence of the availability of a high-affinity channel state, rather than a voltage depen-dence of the binding and/or unbinding rate con-stants (kt or /,).
Mathematical ModelingPrevious studies using upstroke velocity measure-
ments in cardiac tissue have suggested that theguarded-receptor model can adequately describethe blocking effects of local anesthetic agents oncardiac sodium channels.24-25 However, the abilityof this model to describe and predict the blockingbehavior of drugs based on direct measurements ofcardiac IN, has not been rigorously tested. In thisstudy, the use-dependent effect of RAC109 stereo-isomers on cardiac sodium channels was found tobe adequately simulated by equations based on theguarded-receptor model. As predicted by the model,the onset time course of use-dependent block waswell described by a single exponential function
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

Clarkson Block of Na Channels by RAC109 Stereoisomers 1319
(Figure 7B), and linear relations were observedbetween both the uptake rate (A) and interpulserecovery time (t,), as well as the stimulus parametera and b^ (r>0.9,p<0.05) (Figures 7C and 7D). Inaddition, state-dependent rate constants calculatedfrom the guarded-receptor parameter estimationprocedure provided an accurate description of boththe kinetics and amplitude of block by RAC109stereoisomers (Table 4).
When comparing the model rate constants esti-mated for drug interaction with rested, activated,and inactivated channels (Table 3), one pattern thatbecomes apparent is that the rates of both drugbinding and unbinding from activated channels (ktand /,) are three to four orders of magnitude largerthan those defining drug interaction with channelsthat exist predominantly in rested (k^ and lr) orinactivated (kt and l{) states. Such differences in thestate-dependent rate constants are at least qualita-tively consistent with the guarded-receptor model,which predicts that closure of either the activationor inactivation gates can greatly impede access andegress of drug from its receptor site.23-2* However,when one attempts to account for the differences inthe state-dependent rate constants more quantita-tively by factoring out estimated guard and trapfunctions (m3 and h) to derive the state-independentrate constants, some inconsistencies emerge fromthe analysis. Since the guarded-receptor modelassumes that tonic block at strongly hyperpolarizedpotentials results from drug binding to infrequentlyopening channels23-26 and that kt reflects drug bind-ing to open channels, then estimates of k, forRAC109 stereoisomers should be approximatelyequal to Jt,m3h.. This prediction was tested byestimating the voltage dependence of m3 and h,from least-squares fits of the peak GNl-Vm relation(where peak GNo=m3) and sodium current availabil-ity (h.) curves to Boltzmann equations.30 The meanmidpoints and slope factors for these relations wereVnud=-34±8 mV and S=6.0±1.2 mV (n=9) for thepeak GNs-Vm relation and ho^=-93±3 mV andS=5.0±0.5 mV (n = 16) for the h.-Vm relation. Fromthese values, the estimated levels of m3 and h. at-140 mV are calculated to be m3=2xl0~8 (orm3=lxl0~1() when estimated from an infinite timeintegral of GNl) and h. = 1.0. Multiplying these val-ues for m3 and h« by the values for km shown in Table3 yields predicted values of kT for RAC109-I andRAC109-II of 5xl0"5 and 3xl(r5/M/msec, whichare four orders of magnitude smaller than the valuesof k, calculated from the experimental data (0.2 and0.5/M/msec). This appreciable discrepancy suggeststhat there is an error in either the formulation of themodel or in the estimation of the gating parameters.Although a Boltzmann equation can accuratelydescribe the voltage dependence of GN, (m3) atpotentials where the macroscopic current can beaccurately measured (i.e., positive to —60 mV), it isnot clear whether the same equation can accuratelypredict the frequency of channel opening at much
more hyperpolarized potentials. More precise mea-surements of the frequency of channel openingsbased on single channel recordings may provide amore accurate estimation of the guard parameter(m3) and would help to clarify whether the differencebetween the predicted and calculated values of k, aredue to inaccuracies in model assumptions or inaccu-rate estimates of the guard parameter m3. Values ofkj larger than that predicted by use of an m3 guardparameter could also result if drug access to channelsvia the lipid bilayer pathway was possible after onlypartial channel activation, for example, after openingof a single (m) or pair (m2) of activation gates. Arecent structural model of the excitable sodium chan-nel has postulated that the channel activation gatesare located on four homologous transmembrane seg-ments that are concentrically arranged around thechannel lumen.50 Therefore, it seems reasonable topostulate that rearrangement of only one or twoactivation gates neighboring a drug positioned withinthe membrane may be sufficient to permit lateraldiffusion of drug into an obstructive position withinthe channel lumen, whereas entrance of chargeddrug into the channel lumen via the aqueous pathwaymight still require all activation gates to be open.This variant of the guarded-receptor model couldaccount for the presence of significant tonic block(large kr) produced by lipid soluble drugs at hyper-polarized potentials where the frequency of channelopening may be very low. Comparison of kr valuesfor other lipid soluble and lipid insoluble drugs wouldprovide one means of testing this hypothesis. Finally,it is difficult to rule out the possibility that largevalues for kT could reflect the existence of a separatereceptor for tonic block34-43 or a receptor having astate-dependent conformation as postulated by themodulated-receptor models of Hille38 and Hon-deghem and Katzung.39 Indeed, without firm evi-dence for a quantitative relation between the appar-ent rate constants and channel-gating variables for avariety of drugs, a modulated-receptor explanationfor the state-dependent rate constants seems equallydefensible. Even though there is more than onepossible molecular explanation for the state-dependent rate constants, the close agreementbetween the experimental data and model predic-tions (Table 4) suggests that the procedure for calcu-lating rate constants (Equations 1-14) provides asufficiently accurate description of use-dependentblock and as such may provide useful insight intohow local anesthetic agents differ in their state-dependent interaction with cardiac sodium channels.
Stereoselectivity of Model Rate ConstantsComparison of the estimated rate constants for
RAC109-I and RAC109-II interaction with cardiacsodium channels indicates that the stereoselectivityof use-dependent block observed by use of trains ofbrief pulses results from two factors: significantlydifferent affinities and rate constants (kt and /,)defining drug interaction with activated channels
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

1320 Circulation Research Vol 65, No 5, November 1989
during the test pulse and significantly different ratesof unbinding (/r) during the interpulse interval (Table3). Both of these factors contribute to the differencein observed potency of use-dependent block inducedby brief pulse trains (Figures 1 and 2).
Interestingly, although the two stereoisomers havedifferent binding rates to activated channels (k,),there is no significant stereoselectivity in the appar-ent rates of drug binding to channels when they areheld in predominantly rested or inactivated states(kr and k{) (Table 3). This observation may beaccounted for by Hille's hypothesis38 that localanesthetics interact with closed versus open chan-nels via different pathways: charged local anesthet-ics selectively block open channels by accessing thereceptor via a hydrophilic pathway, whereas un-charged drug forms can interact with closed chan-nels by diffusing to the receptor via a hydrophobicpathway through the lipid bilayer. Although RAC109stereoisomers exist primarily in their charged formin aqueous solution (>99% at pH 7.2-7.3), thepartition coefficient (P) of the uncharged form isfairly high (P=260),28 so that, from the equation:Q=?/(1 + 1(FK'-'H) at pH 7.2-7.3, the amount ofuncharged RAC109 dissolved in the membrane isexpected to be 1.6-2 times higher than the totalamount of drug present in the aqueous phase.28
Therefore, it seems reasonable to expect thatRAC109 stereoisomers could interact with theirreceptor in both uncharged and charged forms.Based on this assumption, one possible explanationfor the observed difference in stereoselectivity of kTand /fcj versus kt is that both kr and k{ reflect bindingof uncharged drug to closed-channel states (pre-open or inactivated), whereas kB reflects binding ofcharged drug over the aqueous pathway. Chargedand uncharged drugs might be expected to havedifferent conformations or orientations when pre-sented to the drug receptor; thus, different stereo-selectivity of binding would result.
Comparison of the ratio of unbinding rates for thetwo stereoisomers (RAC109-I and RAC109-II) forall three channel states (0.37, 0.36, and 0.44 forrested, activated, and inactivated states) indicatesthat there is relatively little state-dependent changein stereoselectivity of the unbinding rates. This isconsistent with the guarded-receptor model, whichassumes that the receptor conformation is static23
and that differences in unbinding rates for differentchannel states result primarily from changes in thepositions of channel gates (drug trapping), whichshould affect both stereoisomers equally.
Role of Channel Activation and InactivationGates in Recovery From Block
For use-dependent block to occur on applicationof repetitive depolarizing pulses, there must beincreased drug binding during each depolarizingpulse as well as slow and incomplete unbindingbetween each pulse at the holding potential. Twopossible molecular mechanisms for slow unbind-
ing at the holding potential include 1) trapping ofcharged drug within the channel by closed activa-tion gates, as previously postulated by numerousinvestigators18'26-34-38-51 or 2) trapping of drug in aninactivated-blocked state due to a drug-inducedhyperpolarizing shift in the voltage dependence ofthe inactivation gating mechanism. 15,38,39,51.52,53
Several pieces of evidence indicate that the slowrate of RAC109 unbinding at the holding potentialsstudied (-140 to -160 mV) cannot be accounted forby drug trapping in an inactivated-blocked state.First, estimation of the voltage shift of inactivationfor RAC109 stereoisomers by use of the equation ofBean ct al16 (A ybl=\dn[KJK(il], where k is theslope factor for the inactivation curve and K& andK& are dissociation constants in activated and inresting states, respectively) predicts a drug-inducedvoltage shift of only -19 to —21 mV (assuming K&and Ktf values shown in Table 3 and k=6 mV30).Under the experimental conditions, the midpoint ofthe inactivation (h.) curve was estimated to be—93±3 mV (n=16). Therefore, clamping the mem-brane to potentials between —140 to —160 mVwould be predicted to completely surmount a drug-induced voltage shift of this magnitude. Hyperpo-larization to potentials that surmount a voltage shiftshould result in rapid recovery of sodium current atthe holding potential and abolishment of use-dependent block when drug trapping in aninactivated-blocked state is the rate-limiting stepregulating channel unblocking.53 Such effects werenot observed experimentally (Figure 1 and Table 1).In addition, use-dependent block remained markedeven after removal of fast inactivation by a-chymotrypsin (Figure 5C). These results indicatethat trapping of channels in an inactivated-blockedstate is not responsible for use-dependent accumu-lation of block when pulsing from strongly hyper-polarized potentials.
In contrast, the observed increase in block onapplication of depolarizing pulses, slow recoveryfrom block at strongly hyperpolarized potentials,and persistence of use-dependent block after removalof fast inactivation can be explained in terms ofreceptor guarding and trapping by the channel acti-vation gates. At strongly hyperpolarized potentials,most channels will have their activation gates closed;this condition will permit only a relatively smalltonic block to develop even after several minutesrest at —140 mV. Closed activation gates will alsoact to trap charged drug within the channellumen.18-24-26 Nevertheless, since RAC109 stereo-isomers are tertiary amines, the lifetime of drugwithin the channel at hyperpolarized holding poten-tials may not be regulated by the low frequency ofchannel openings since these drugs may deproto-nate to an uncharged lipid soluble form while trappedwithin the channel. Assuming a protonation rate (kp)of 5 x 108/M/sec,52 the lifetime of cationic RAC109(l//p) is predicted (from y/p=10p K-)«x to be ~5seconds. However, the lifetime of cationic RAC109
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

Clarkson Block of Na Channels by RAC109 Stereoisomers 1321
may actually be longer than this if the chargedmicroenvironment of the channel increases the pKaof bound drug.54 The protonation-deprotonation reac-tions could be significantly affected by such factorsas local potential fields or proton access paths54 orlowering of local pH due to attraction of charged H+
ions into the channel from the extracellular solutionon hyperpolarization.55 Thus, the slow rate ofRAC109 unbinding from rested channels seemsconsistent with trapping of drug within the channelby closed activation gates although it is necessaryto assume the presence of a higher pKa for bothstereoisomers when they are bound within thechannel.
The conclusion that RAC109 stereoisomers canbecome trapped behind closed activation gates atstrongly hyperpolarized potentials is consistent withthe conclusions of several previous studies thatinvestigated the blocking behavior of other highlycharged drugs on V ^ , in cardiac tissue15-56 andsodium current in nerve.18-34-38 Thus, although stronghyperpolarization may result in relief of block forsome tertiary amine drugs (e.g., lidocaine andquinidine),53 it may not do so for others (e.g.,RAC109 stereoisomers and penticainide56).
Effects of a-Chymotrypsin on Channel BlockIn addition to a lack of removal of use-dependent
block (Figure 5), several other effects of RAC109stereoisomers observed after enzyme treatment alsoseem consistent with predictions made by theguarded-receptor model. For example, recent stud-ies have shown that removal of channel inactivationby agents such as ar-chymotrypsin, trypsin, andAf-bromoacetamide produce both a several-foldincrease in the single channel mean open time,37-38
as well as a —20 mV hyperpolarizing shift in theGN.-VO, relation.37 If it is assumed that tonic blockby RAC109 stereoisomers results, at least in part,from occasional channel openings at the holdingpotential (i.e., m3.>0), such an enzyme-inducedshift of channel activation to more negative poten-tials would be expected to increase significantly thelevel of tonic block. An enzyme-induced increase inmean open time (ta) would also be expected toincrease the onset of use-dependent block (Equa-tion 4). Both of these effects were observed (Figure5). Hypothetically, if the mean open time were tobecome infinitely long (which seems unlikely), thenthe time constants for drug-induced decay (Figure6Q should approach the predicted time constant foractivated-state block (I/A:,=6 msec). The observa-tion of a significantly longer time constant is expectedif there is significant channel flickering betweenopen (activated) and closed states, as has beenobserved after treatments that reduce channelinactivation.57-58
The conclusions drawn from experiments carriedout in cells pretreated with a-chymotrypsin toremove channel inactivation are based largely onthe assumption that a-chymotrypsin has a selective
effect on removing fast inactivation in cardiac cells,similar to its effect in neuroblastoma cells.37 Althoughother effects cannot be ruled out, the observationthat cardiac sodium currents still have a qualita-tively similar sigmoidal onset on depolarization andbrisk deactivation kinetics on repolarization (Fig-ures 5 and 6) suggests that the function of thechannel activation gates in cardiac tissue may berelatively unaffected after a digestion sufficient tovirtually abolish fast channel inactivation in cardiaccells. However, it is difficult to rule out the possi-bility that proteolytic digestion may not also directlymodify the drug receptor or induce structuralchanges to the sodium channel that alter drugaccess and egress from the receptor, as has beensuggested to occur after pronase treatment in squidgiant axons.59 For example, in earlier experimentsusing concentrations of trypsin that reduced sodiumconductance twofold to threefold, little if any time-dependent block by RAC109 could be defined dur-ing 20-msec pulses to -20 mV (although use-dependent block was still observable).50 Thiscontrasts with the results observed in the presentstudy where a milder digestion using a-chymotrypsinwas performed, and both use-dependent and time-dependent block were clearly observable during20-msec pulses (Figure 5C). Thus, strong diges-tion, or different agents, may produce slightlydifferent effects on the drug-receptor reaction. Asimilar conclusion was reached by Wang et al,59
who found that removal of channel inactivation bychloramine T and pronase can affect local anes-thetic block differently in squid giant axons. Nev-ertheless, the observation of use-dependent blockafter abolition of fast inactivation indicates thatchannel inactivation is not obligatory for block ofcardiac sodium channels by local anesthetics andthat it does not regulate channel unblocking atstrongly hyperpolarized potentials.
RAC109 Has Similar Blocking Characteristicsin Nerve and Heart
There are many close similarities between theeffects of RAC109 stereoisomers in cardiac myo-cytes and in nerve. In both tissues RAC109-I hasbeen found to produce two to three times moreuse-dependent block compared with RAC109-II atequimolar concentrations (Figure I)6-7 although bothstereoisomers produce similar levels of tonic block(Figures 1).6>7 In addition, use-dependent block byboth stereoisomers is strongly voltage-dependent inboth tissues; RAC109-I has the steeper voltagedependence (Figure 2).7 Finally, recovery fromuse-dependent block by RAC109-I occurs with atime constant several times slower than for RAC109-II in both tissues (Figure 3).7 These findings suggestthat the local anesthetic binding site in sodiumchannels of nerve and heart may have a similarstructural topography.
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

1322 Circulation Research Vol 65, No 5, November 1989
AcknowledgmentsI wish to thank Jianyi Wang for developing the
software used for curve fitting and graphics, as wellas Dr. Frank Starmer for assistance in incorporatingthe guarded-receptor equations into a computermodel and for constructive criticism of a prelimi-nary version of the manuscript.
References1. Schmidtmayer J, Ulbricht W: Interaction of lidocaine and
benzocaine in blocking sodium channels. Pflugers Arch1980;387:47-54
2. Pichon Y, Schmidtmayer J, Ulbricht W: Mutually exclusiveblockage of sodium channels of myelinated frog nerve fibersby benzocaine and the indole alkaloid ervatamine. NeurosciLett 1981;22:325-330
3. Qarkson CW, Hondeghem LM: Evidence for a specificreceptor site for lidocaine, quinidine, and bupivacaine asso-ciated with cardiac sodium channels in guinea pig ventricularmyocardium. Ore Res 1985;56:496-506
4. Sanchez-Chapula J: Interaction of lidocaine and benzocainein depressing V,,^ of ventricular action potentials. / MolCell Cardiol 1985;17:495-503
5. Akerman SBA, Camougis G, Sandberg RA: Stereoisomer-ism and differential activity in excitation block by localanesthetics. Eur J Pharmacol 1969;8:337-347
6. Hille B, Courtney KR, Durn R: Rate and site of action oflocal anesthetics in myelinated nerve fibers, in Fink BR (ed):Progress in Anesthesiology: Molecular Mechanisms of Anes-thesia. New York, Raven Press, Publishers, 1975, vol 1, pp13-20
7. Yeh JZ: Blockage of sodium channels by stereoisomers oflocal anesthetics, in Fink BR (ed): Progress in Anesthesiol-ogy: Molecular Mechanisms of Anesthesia. New York,Raven Press, Publishers, 1980, vol 2, pp 35-44
8. Postma SW, Catterall WA: Inhibition of binding offH]batrachotoxin A 20a-benzoate to sodium channels bylocal anesthetics. Mol Pharmacol 1984;25:219-227
9. Roth S, Seeman P, Akennan SBA, Chau-Wong M: Theaction and adsorption of local anesthetic enantiomers onerythrocyte and synaptosorae membranes. Biochim BiophysActa 1972;255:199-206
10. Akerman B: Uptake and retention of the enantiomers of alocal anesthetic in isolated nerve in relation to differentdegrees of blocking of nervous conduction. Acta PharmacolToodcol 1973;32:225-236
11. Mirro MJ, Watanabe AM, Bailey JC: Electrophysiologicaleffects of the optical isomcrs of disopyramide and quinidinein the dog: Dependence on stereochemistry. Ore Res 1981;48:867-874
12. Coltart DJ, Meldrum SJ: The effect of racemic propranolol,dextropropranolol and racemic practolol on the human andcanine cardiac transmembrane action potential. Arch IntPharmacodyn Ther 1971;192:188-197
13. Sada H, Ban T: Effects of acebutolol and other structurallyrelated beta adrenergic blockers on transmembrane actionpotential in guinea-pig papillary muscles. / Pharmacol ExpTher 1980;215:507-514
14. Bird LB, Colatsky TJ: Use-dependent block of V ^ , byquinidine in dog Purkinje fibers is not stereospecific (abstract).Circulation 1985;72(suppl IlI):III-38
15. Ointant GA, Hoffman BF: Use-dependent block of cardiacsodium channels by quaternary derivatives of lidocaine.Pflugers Arch 1984;4O0:121-129
16. Bean BP, Cohen CJ, Tsien RW: Lidocaine block of cardiacsodium channels. / Gen Physiol 1983;81:613-642
17. Sanchez-Chapula J, Tsuda Y, Josephson IR: Voltage- anduse-dependent effects of lidocaine on sodium current in ratsingle ventricular Gells. Ore Res 1983^2:557-565
18. Yeh JZ, Tanguy J: Na channel activation gate modulatesslow recovery from use-dependent block by local anesthet-ics in squid giant axens. Biophys J 1985;47r685-694
19. Narahashi T: Chemicals as tools in the study of excitablemembranes. Physiol Rev 1974;54:813-889
20. Baer M, Best PM, Reuter H: Voltage-dependent action oftetrodotoxin in mammalian cardiac muscle. Nature 1976;263:344-345
21. Cohen CJ, Bean BP, Colatsky TJ, Tsien RW: Tetrodotoxinblock of sodium channels in rabbit Purkinje fibers. Interac-tions between toxin binding and channel gating. J GenPhysiol 1981;78:383-341
22. darkson CW, Matsubara T, Hondeghem LM: Evidence forvoltage-dependent block of cardiac sodium channels bytetrodotoxin. J Mol Cell Cardiol 1988;20:1119-1131
23. Starmer CF, Grant AO, Strauss HC: Mechanisms of use-dependent block of sodium channels in excitable membranesby local anesthetics. Biophys J 1984;46:15-27
24. Starmer CF, Grant AO: Phasic ion channel blockade: Akinetic model and parameter estimation procedure. MolPharmacol 1985;28:348-356
25. Starmer CF: Theoretical characterization of ion channelblockade. Competitive binding to periodically accessiblereceptors. Biophys J 1987;52:405-412
26. Starmer CF, Yeh JZ, Tanguy J: A quantitative description ofQX222 blockade of sodium channels in squid axons. BiophysJ 1986;49:913-920
27. Mitra R, Morad M: A uniform enzymatic method for disso-ciation of myocytes from hearts and stomachs of verte-brates. Am J Physiol 1985;249:H1056-H1060
28. Hillc B: The pH-depcndcnt rate of action of local anestheticson the node of Ranvier. J Gen Physiol 1977;69:475-496
29. Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ:Improved patch-clamp techniques for high-resolution cur-rent recording from cells and cell-free membrane patches.Pflugers Arch 1981^91:85-100
30. Clarkson CW, Follmer CH, Ten Eick RE, Hondeghem LM,Yeh JZ: Evidence for two components of sodium channelblock by lidocaine in isolated cardiac myocytes. Ore Res1988;63:869-878
31. Sheets MF, Hanck DA, Fozzard HA: Divalent block ofsodium current in single canine cardiac Purkinje cells(abstract). Biophys J 1988;53:535a
32. Fozzard HA, Hanck DA, Makielski JC, Scanley BE, SheetsMF: Sodium channels in cardiac Purkinje cells. Expcrientia1987;43:1162-1168
33. Kunze DL, Lacerda AE, Wilson DL, Brown AM: CardiacNa currents and the inactivating, reopening, and waitingproperties of single cardiac Na channels. / Gen Physiol1985;86:691-719
34. Strichartz GR: The inhibition of sodium current in myeli-nated nerve by quaternary derivatives of lidocaine. J GenPhysiol 1973;62;37-57
35. Cahalan MD: Local anesthetic block of sodium channels innormal and pronase-treated squid giant axons. Biophys J1978;23:285-310
36. Cahalan MD, Aimers W: Interactions between quaternarylidocaine, the channel gates, and tetrodotoxin. Biophys J1979;27:39-56
37. Gonoi T, Hille B: Gating of sodium channels. Inactivationmodifiers discriminate between models. J Gen Physiol 1987;89:253-274
38. Hille B: Local anesthetics: Hydrophilic and hydrophobicpathways for the drug-receptor reaction. J Gen Physiol1977;69:497-515
39. Hondeghem LM, Katzung BD: Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiacsodium channels. Biochim Biophys Acta 1977;472:373-398
40. Sheldon RS, Cannon NJ, Duff HJ: A receptor for type Iantiarrhythmic drugs associated with rat cardiac sodiumchannels. Ore Res 1987;61:492-497
41. Courtney KR: interval-dependent effects of small antiar-rhythmic drugs on excitability of guinea-pig myocardium. JMol Cell Cardiol 1980;12:1273-1286
42. Courtney KR: Size-dependent kinetics associated with drugblock of sodium current. Biophys J 1984;45:42-44
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

Clarkson Block of Na Channels by RAC109 Stereoisomers 1323
43. Bokesch PM, Post C, Strichartz G: Structure-activity rela-tionship of lidocaine horoologs producing tonic and frequency-dependent impulse blockade in nerve. / Pharmacol ExpTher 1986;237:773-781
44. Yeh JZ, Ten Eick RE: Molecular and structural basis ofresting and use-dependent block of sodium current definedusing disopyramide analogues. Biophys J 1987;51:123-135
45. Ehring GR, Moyer JW, Hondeghem LM: Quantitative struc-ture activity studies of antiarrhythmic properties in a seriesof lidocaine and procainamide derivatives. J Pharmacol ElxpTher 1988;244:479-492
46. Matsubara T, Clarkson C, Hondeghem L: Lidocaine blocksopen and inactivated cardiac sodium channels. NaunynSchmiedebergs Arch Pharmacol 1987;336:224-231
47. Hondeghem LM, Matsubara T: Quinidine blocks cardiacsodium channels during opening and slow inactivation inguinea pig papillary muscle. BrJPharmacol 1988^3:311-318
48. Cachelin AB, De Peyer JE, Kokubun S, Reuter H: Sodiumchannels in cultured cardiac cells. J Physiol (Lond) 1983;340:389-402
49. Follmer CF, Ten Eick RE, Yeh JZ: Sodium current kineticsin cat atrial myocytes. / Physiol (Lond) 1987;384:169-197
50. Guy HR: A model relating the structure of the sodiumchannel to its function. Curr Top Membr Transport 1988;13:289-308
51. Courtney KR: Mechanism of frequency-dependent inhibi-tion of sodium currents in frog myelinated nerve by thelidocaine derivative GEA 968. J Pharmacol Exp Ther 1975;195:225-236
52. Schwarz W, Palade PT, Hille B: Local anesthetics: Effect ofpH on use-dependent block of sodium channels in frogmuscle. Biophys J 1977;20:343-368
53. Hondeghem LM, Katzung BG: Test of a model of antiar-rhythmic drug action. Effect of quinidine and lidocaine onmyocardial conduction. Circulation 1980;61:1217-1224
54. Starmer CF, Courtney KR: Modeling ion channel blockadeat guarded binding sites: Application to tertiary drugs. Am JPhysiol 19S6;251:H848-H856
55. Woodhull AM: Ionic blockage of sodium channels in nerve./ Gen Physiol 1973;61:687-708
56. Carmeliet E: Activation block and trapping of penticainide,a disopyramide analogue, in the Na+ channel of rabbitcardiac Purkinje fibers. Ore Res 1988;63:50-60
57. Horn R, Vandenberg CA, Lange K: Statistical analysis ofsingle sodium channels. Effects of/V-bromoacetamide. Bio-phys J 1984;45:323-335
58. Vandenberg CA, Horn R: Inactivation viewed through singlesodium channels. / Gen Physiol 1984;84:535-564
59. Wang GK, Brodwick MS, Eaton DC, Strichartz GR: Inhibi-tion of sodium currents by local anesthetics in chloromine-T-treated squid axons. J Gen Physiol 1987;89:645-667
60. Clarkson CW: Effects of proteorytic enzymes on cardiacsodium channel inactivation and block by local anesthetics(abstract). Biophys J 1988;53:534a
KEY WORDS • sodium channel • RAC109 • stereoisomers• guarded-receptor hypothesis • antiarrhythmic agents •local anesthetics
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from

C W Clarksonmyocytes.
Stereoselective block of cardiac sodium channels by RAC109 in single guinea pig ventricular
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 1989 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/01.RES.65.5.13061989;65:1306-1323Circ Res.
http://circres.ahajournals.org/content/65/5/1306World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Research Requests for permissions to reproduce figures, tables, or portions of articles originally published inPermissions:
by guest on May 15, 2018
http://circres.ahajournals.org/D
ownloaded from