standard operating procedures for data review by dldcc drc
TRANSCRIPT

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 1 of 32
© Baylor College of Medicine 2005
1. Purpose 1.1. To describe administration issues and required components of data and safety
monitoring by the DLDCC DRC.
2. Scope 2.1. This procedure applies to all DLDCC clinical research protocols under the purview of the
DLDCC DRC.
3. Definitions and Abbreviations 3.1. BCM Baylor College of Medicine 3.2. CRLC Clinical Research Leadership Committee 3.3. DLDCC Dan L. Duncan Cancer Center 3.4. DM Data Manager 3.5. DR Data Review 3.6. DRC Data Review Committee 3.7. IND Investigational New Drug Application 3.8. IRB Institutional Review Board 3.9. PI Principal Investigator 3.10. PRMC Protocol Review and Monitoring Committee 3.11. SAE Serious Adverse Event 3.12. Coordinator DRC Coordinator (or designee, as needed)
4. Materials and Equipment: 4.1. DRC Meeting Workflow Scheme (Appendix I) 4.2. DLDCC Data and Safety Monitoring Plan
5. Description
5.1. Data review of clinical research protocols follows the DLDCC Data and Safety Monitoring Plan (DSMP). 5.1.1. The DSMP describes the type of studies which will be assigned to DRC review.
5.2. DRC Membership
5.2.1 The Coordinator maintains the DRC membership list. 5.2.2 The DRC membership will include the CTSU Director, representative faculty
members, at least one statistician, and the DLDCC Patient Safety Officer. 5.2.3 The DLDCC Director will appoint members for 2 year terms, with an unlimited
number of terms. 5.2.4 Adding and Removing Members
5.2.4.1 The addition/removal of the member should be made to the member list by the Coordinator.
5.2.4.2 The modified list should then be reviewed and approved by the CRLC. 5.2.4.3 Once the modified list has been approved by the CRLC and chair(s),
the following should be sent out as appropriate. A “welcome to the committee” packet should be sent to the new member. The welcome packet should include a welcome letter (Appendix II) and the DRC SOP.

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 2 of 32
© Baylor College of Medicine 2005
A “thank you for serving” letter (Appendix III) should be sent to the removed member.
5.2.4.4 Once the appropriate letter/packet has been sent to the member, email distribution lists should be updated to reflect the change.
5.2.4.5 The new member list should then be uploaded to the appropriate website.
5.3. DRC Administration 5.3.1. All correspondence related to the DRC will be maintained in the DRC binders,
respectively. The Coordinator maintains the DRC binders. 5.3.2. DRC activity is tracked and maintained by the Coordinator.
5.3.2.1. The following information is to be updated immediately following DRC review: • Meeting Date • Determination
5.3.2.2. Updated when applicable: • Response Date • Safe to Continue Date
5.3.3. Scheduling of DRC Meetings 5.3.3.1 The DRC meetings are routinely scheduled once a month. The DRC
chairs or CRLC may schedule special sessions as needed. 5.3.3.2 A monthly report of protocols to be scheduled for review is created by
the Coordinator from DRC records.
5.4. Data Review (DR) Parameters 5.4.1. The frequency of DR for a protocol (e.g., annually, bi-annually) is decided by the
PRMC and stated in the minutes of the PRMC. 5.4.1.1. If a non-interventional study is referred for DRC review, the PRMC
minutes should clearly indicate the rationale. 5.4.1.2. Following initial review by the DRC, the DRC may determine that
continuing review is no longer necessary. 5.4.1.3. The assigned review month will be three months prior to IRB annual
renewal for a non-IND study, or one month prior to IND renewal month for an IND study.
5.4.1.4. If at any time the DRC determines that further review is not necessary, the DRC records should be updated to remove review requirements. The determination will be clearly documented in the minutes. The PI will be notified in writing that DRC review is no longer necessary.
5.4.2. For internally initiated, IND studies: DR of open protocols should continue until the IND is closed, or until the DRC determines that further review is not needed. 5.4.2.1. An IND study is defined as a study that is operating under an internally
initiated IND that is held by a BCM Investigator 5.4.3. For internally initiated, non-IND studies: DR should continue until all protocol
interventions are completed, or the DRC determines that further review is not needed.
5.5. Data Review Procedures
BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 3 of 32
© Baylor College of Medicine 2005
5.5.1. On the first of the month prior to a protocol’s scheduled DR date, the Coordinator creates a notification letter (Appendix IV) for each scheduled protocol. (Note: This should allow the PI approximately 7-8 weeks to prepare his/her DRC report.) 5.5.1.1. The letter will include the submission deadline, which is 10 calendar
days prior to the assigned DRC meeting. 5.5.1.2. The notification letter is sent via email to PI, and copied to the protocol’s
administrative or regulatory contact. A copy of notification letter is maintained in the DRC records.
5.5.1.3. For non-IND studies, a copy of the DRC Report template will be included with the notification letter.
5.5.2. The PI provides the DRC report to the Coordinator 10 calendar days prior to the scheduled DRC meeting date. 5.5.2.1. An acceptable report submission must be signed by the PI, submitted
from the PI’s email, or submitted via email by a designee if the PI is copied on the submission.
5.5.2.2. See Section 5.9 for guidelines for the DRC reports. 5.5.2.3. Delayed reports are handled as follows:
5.5.2.3.1. If the PI fails to provide the report for the meeting the study is scheduled for, the Coordinator will inform the DRC chair(s). A memo should be sent to the study PI from the DRC chair(s) requesting that the study be submitted to the next scheduled DR meeting.
5.5.2.3.2. If the PI fails to provide the report for a second meeting, CRLC will be notified.
5.5.3. Data Review Packets 5.5.3.1. One week prior to DRC meeting, the Coordinator prepares the data
review packets. 5.5.3.2. DR packets include a cover letter (Appendix V) to the DRC members,
and the DRC reports for each study scheduled for review. 5.5.3.3. DR packets are sent via email to DRC members no later than one week
prior to the DRC meeting. 5.5.3.4. A copy of the DR packet is filed in the DRC binder.
5.5.4. Meeting Conduct 5.5.4.1. The DRC meeting will be conducted in two parts:
5.5.4.1.1. An open session in which members of the study team may be present to answer questions from the DRC. The PI will be invited to the open session.
5.5.4.1.2. A closed executive session (without the study PI or study team) in which the DRC discusses interim outcome results, determines what actions are to be taken, and votes.
5.5.4.2. The Principal Investigator cannot act as chair for his/her protocol. 5.5.5. Minimum Review Requirements
5.5.5.1. Each study must be reviewed by a biostatistician, a medical reviewer, and a Chair (or designee).
5.5.5.2. Reviews can be submitted to the DRC for consideration as written comments, in lieu of attendance.
5.5.5.3. At the discretion of the Chair, a review may be deferred to allow for additional reviews or comments, as needed.
BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 4 of 32
© Baylor College of Medicine 2005
5.5.6. DRC Meeting Minutes 5.5.6.1. The Coordinator writes the DRC Meeting Minutes (see template in
Appendix VI). 5.5.6.2. The minutes are sent via email to the DRC meeting chair(s) for
approval. 5.5.6.3. Once approved, the minutes are sent via email to DRC members. 5.5.6.4. The minutes are maintained in the DRC records.
5.5.7. DRC Determinations 5.5.7.1. The DRC will recommend continuation, modification, or halting of the
trial. 5.5.7.2. The DRC may also determine that further DRC review is not needed.
The PI will be notified in writing of this determination. 5.5.7.3. If the DRC determines that the protocol is safe to continue without
modification or comments, the PI will be notified as per Section 5.5.8. 5.5.7.4. If the DRC determines that comments or revisions should be sent to the
PI, it should also determine if the PI’s reply can be administratively reviewed by the Chair, or should be reviewed by the full committee.
5.5.7.5. The Coordinator forwards via email the requests for additional information or clarification from the DRC to the PI and the administrative contact.
5.5.7.6. If study changes are recommended by the DRC, it is the responsibility of the PI to implement these changes (through the IRB and other appropriate regulatory agencies) and notify the DRC after the changes have been made.
5.5.7.7. When a resubmission of the DRC report is requested, a deadline of 30 days is included in the request. The PI is responsible for providing additional information and clarification to the Coordinator. 5.5.7.7.1. Any query that impacts subject safety may require a more
immediate response, at the discretion of the DRC chair(s). 5.5.7.7.2. In all cases, the PI will be notified of the response deadline.
5.5.7.8. Delayed submissions are handled as follows: 5.5.7.8.1. If the PI fails to provide the requested data by the response
deadline, the Coordinator will notify the DRC chair(s). A memo should be sent to the study PI from the DRC chair(s) requesting that the data be provided within 30 days, or sooner at the discretion of the chair(s).
5.5.7.8.2. If the PI fails to submit the requested data after this second notice, the Coordinator will notify the CRLC.
5.5.7.9. If the DRC determines that a study should be halted, this should be implemented immediately unless the determination is to be appealed. Until the matter is fully resolved, the PI must carry out the actions as required by the DRC. Any appeal related to halting a study should be submitted within 30 days of the DRC determination.
5.5.7.10. All queries sent to (and from) the PI are filed in the DRC binder. 5.5.8. Appeals
5.5.8.1. If the PI does not concur with a DRC requirement for modifying or halting a study, the PI may formally appeal the DRC’s decision.

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 5 of 32
© Baylor College of Medicine 2005
5.5.8.1.1. During the appeal process, the original DRC determination (e.g., study suspension) remains in effect.
5.5.8.2. Any appeal to a DRC determination must be received, in writing, within 30 days of the DRC determination.
5.5.8.3. The appeal must include the reason(s) for disagreement and any alternate proposal from the PI.
5.5.8.4. The DRC will meet to review the appeal. The DRC may vote to accept or reject the appeal.
5.5.8.5. If the DRC rejects the appeal, the PI may make an additional appeal as per the DLDCC Data and Safety Monitoring Plan.
5.5.9. Safe to Continue Notification 5.5.9.1. If the DRC has no questions or comments to the PI after reviewing the
DRC report, the Coordinator creates a “safe to continue” memorandum (Appendix VII).
5.5.9.2. If additional information or clarification is requested by the DRC, upon receipt, review and approval of the PI’s response, the Coordinator creates a “safe to continue” memorandum (Appendix VII).
5.5.9.3. The “safe to continue” memorandum is sent by email to the PI and the study’s administrative contact.
5.5.9.4. The “safe to continue” memorandum is filed in the DRC binder. 5.5.9.5. The study PI is responsible for forwarding the DRC determination to the
appropriate regulatory agencies, as required (i.e., IRB, etc.). 5.5.9.6. The Coordinator will update the DRC records to reflect the approval
date.
5.6. Serious Adverse Events (SAEs) 5.6.1. For studies under DRC review: SAEs that are reportable to the IRB (as per BCM
IRB requirements) should be submitted to the DRC at the time of submission to the IRB.
5.6.2. The SAE report should be sent via email to the Coordinator, who will forward the report to the DRC chair(s) and the DLDCC Patient Safety Officer.
5.6.3. The DRC chair(s) will review the SAE to decide if the event should be reviewed by the DRC. 5.6.3.1. If no interim DRC review is needed, there is no further action and the
PI will be notified. 5.6.3.2. If the chair determines that interim DRC review is needed, the SAE
report will be distributed to the DRC members. The membership will determine if any interim actions should occur (i.e., an interim review of all study data, study suspension, audit of the study, etc.).
5.6.3.3. The reported event will also be reviewed at the next DRC meeting. 5.7. At time of study closure, a final report and notice of the closure should be sent to the
DRC. Review of closure requests should focus on whether study procedures and follow up were complete in all subjects as specified in the protocol, not on study objectives and outcomes.
5.8. The CRLC reviews a summary report of DRC reviews at each CRLC meeting. This report is created and updated by the Coordinator.

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 6 of 32
© Baylor College of Medicine 2005
5.9. Required components of data review / DRC Report
5.9.1. Protocols conducted under internal IND: 5.9.1.1. For protocols being conducted under an internal IND, the annual IND
report will be used as the DRC report. See Appendix VIII for a template of IND annual report (including IND Report Appendices I and II).
5.9.1.2. In addition to the IND report, the PI must submit a Protocol Conduct Summary (Appendix IX).
5.9.1.3. Confidential subject identifiers should be used throughout the report. 5.9.1.4. The adverse event table should be cumulative. All adverse events
regardless of type and severity should be listed. 5.9.2. Protocols not conducted under internal IND:
5.9.2.1. See Appendix X for a template of DRC report for protocols not conducted under internal IND. This template will sent with the notification letter for non-IND studies.
5.9.2.2. At a minimum, the DRC report should include the following: Title PI Brief study overview, including study objectives Study Status Expected number of subjects to be treated Number of subjects currently enrolled Toxicity data to date Safety and efficacy data, if available Summary findings to date, including results and dates of any interim analyses Any major amendment(s) planned for the study Recommendations and future plans Statement of protocol conduct
5.10. The following data should be included in any submission sent to a study statistician for
analysis: 5.10.1. Subject ID 5.10.2. Dose Level 5.10.3. Experienced DLT (Y/N) as defined in the protocol and within the time frame of
observation. 6. References:
6.1. DLDCC Data and Safety Monitoring Plan 7. Review and Revisions:
7.1. Initial Version Information Written by: Sarah McNees Reviewed by: Helen Heslop, Martha Mims, Bambi Grilley, Sarah McNees Ratified by CRLC: 05/06/2014 Initial Release Date: 05/06/2014 Changed and this version archived:

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 7 of 32
© Baylor College of Medicine 2005
7.2. Revision Information:
Revised by: Reviewed by: Ratified by CRLC: Changed and this version archived:
7.3. Renewal Information: Year: Reviewed by: Ratified by CRLC:

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 8 of 32
© Baylor College of Medicine 2005
Appendix I DRC Meeting Workflow Schema
Comments requiring a response are emailed to the PI and administrative contact.
PI provides requested report to the Coordinator by the submission deadline, which is 10 days prior to the DRC meeting.
The Coordinator prepares DR packet 1 week prior to meeting date, and distributes the packet to the DRC members. (Previously tabled protocols should be indicated as such.) Two emails are sent out 1 week prior to DRC meeting date: invite email to DRC members, PI & study staff; another email to DRC members with the link to the packet (both emails should contain the agenda). Copy of entire packet is filed in the DRC binder.
For each study, the Coordinator will bring the full protocol and the previous DRC submission and correspondence to the meeting. The Coordinator writes minutes after meeting and sends to the DRC chair for approval. The DRC records are updated for each study. Once the minutes are approved, the Coordinator sends the minutes to the DRC members and files a copy in the DRC binder.
The Coordinator creates safe to continue memo and emails it to the PI and administrative contact. The approval date is updated in the DRC records for each study.
If there is a resubmission required, the PI is given 1 month to get the info to the Coordinator. (If delayed, CRLC will send a memo to the PI.)
PI gives requested info to the Coordinator who forwards it to the DRC chair for approval and updates the DRC records. All correspondence & requests will be filed in the DRC records.
The memo is filed in the DRC records.
The PI is responsible for submitting the DRC findings to regulatory agencies as needed.
DRC records are updated with the approval date listed on the safe to continue memo.
The DRC Coordinator or designee (“Coordinator”) determines the studies due for review, and sends notification letters. A copy of the letter is filed in the DRC binder.

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 9 of 32
© Baylor College of Medicine 2005
Appendix II DRC Welcome Letter
To: (Committee Member) From: (Committee Chair) Date: (Date) We would like to welcome you to the DLDCC Data Review Committee. You have a unique knowledge base that will be beneficial in the evaluation of studies. In addition to the members of the standing committee, the committee membership is somewhat fluid which allows the PI and any other relevant individuals to participate in the discussions. We have added your name to the list of active members on this committee. Please note, your participating in the DRC is covered by BCM’s General Policy on Conflicts of Interest, which may be found on the BCM Policies and Procedures website. The DRC will meet on the fourth Friday of each month from approximately 1:00 pm – 2:00 pm in the conference room located in the Feigin Center, 16th Floor – Room (xx). You will receive meeting invitations via Microsoft Outlook. If there is nothing to review, the meeting will be canceled. The invitations and cancellations are sent from the DRC Coordinator. Please feel free to contact myself or the DRC Coordinator with any questions. Thank you. (DRC Chair’s Name) (DRC Coordinator’s Name) DRC Chair DRC Coordinator (phone) (phone) (email) (email)

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 10 of 32
© Baylor College of Medicine 2005
Appendix III
DRC Thank You Letter
To: (Committee Member) From: (Committee Chair) Date: (Date) Dear: The Dan L. Duncan Cancer Center would like to thank you for your service in the Data Review Committee. We appreciate your time and dedication and wish you the best in your new endeavors.

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 11 of 32
© Baylor College of Medicine 2005
Appendix IV
DRC Notification Letter to PI To: (Principal Investigator) From: (DRC Coordinator) Date: (Date) Re: H#: Title of Protocol The DLDCC Data and Safety Monitoring Plan states that the above study should receive at least annual review by a Data Review Committee (DRC). This protocol will be due for annual renewal with the IRB and/or FDA within the next few months, and therefore is due for DRC review. The purpose of this meeting is to review the study’s data prior to submission to the IRB and/or FDA. The following is a list of information that you will need to provide to the DRC by [date that is 10 calendar days prior to DRC meeting].
♦ Brief study overview, including study objectives ♦ Study Status (pending activation; open to accrual; open but closed to accrual; fully
closed) ♦ Expected number of patients to be enrolled ♦ Subject enrollment/accrual (for protocols including multiple sites please specify which
subjects were enrolled from BCM as compared to other sites) ♦ Toxicity data to date ♦ Safety and efficacy data, if available ♦ Any major amendment planned for the study (if applicable) ♦ Summary of findings to date, interim analysis ♦ Recommendations and future plans
There is no required format for this report, as long as the required information is submitted. If this study is conducted under an internally initiated IND, submission of the proposed FDA annual IND renewal will be sufficient. Additionally, submit a Protocol Conduct Summary; a template is attached to this memorandum. (Appendix IX to be included.) If this study is not conducted under an internally initiated IND, a template DRC report is provided as guidance (attached to this memorandum). (Appendix X to be included.) The review will be conducted on (Date of Meeting) in (Location of Meeting). You are invited to attend the open session of this DRC meeting to present and discuss your study; your attendance is

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 12 of 32
© Baylor College of Medicine 2005
not required. Please let me know if you plan to attend so that we may schedule this review appropriately. If you have any questions, please contact me at (DRC coordinator phone number) or (DRC coordinator email address). Thank you for your cooperation. Cc: (Study Administrative Contact)

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 13 of 32
© Baylor College of Medicine 2005
Appendix V
Cover letter to DRC packet
To: DRC Members
From: (DRC Coordinator) Date: Re: Data Review of: H#: Title of Protocol PI: Enter the PI Name (repeat H# and title as necessary, for multiple studies to be reviewed) Attached is a copy of the DRC Report(s) for the above referenced protocol(s). We will be meeting to review the protocol(s) on Friday, (DATE) at 1:00pm in the Feigin Center 16th Floor Conference Room. If you will be unable to attend, please send your comments to (DRC Coordinator) at (DRC Coordinator email address). Thank you.

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 14 of 32
© Baylor College of Medicine 2005
Appendix VI
DRC Minutes
Minutes of the DLDCC Data Review Committee
Date: [ date ]
Location: Feigin Center, 15th Floor Conference Room A 1530.30
In Attendance: ........ (with Chair(s) or Acting Chair identified)
Guests: ….......
Comments: ……………
Data review of:
1) H# - Study title - PI
2) H# - Study title - PI
3) H# - Study title - PI

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 15 of 32
© Baylor College of Medicine 2005
Appendix VII
Safe to Continue Memorandum
To: PI From: DRC meeting chair Date: Date RE: H#: Study Title The DRC reviewed the data you submitted and determined that the study is safe to continue. You will be notified by the DRC Coordinator when this study is next due for DRC review. Please note, when this study is closed, a final report and notice of closure should be sent to the DRC.

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 16 of 32
© Baylor College of Medicine 2005
Appendix VIII
Template of IND annual report DRC report for protocols being conducted under internal IND
IND ANNUAL REPORT IND NUMBER: REPORT DATE: This date should correspond with the “end date” of the data, not the date of the submission A: INDIVIDUAL STUDY INFORMATION
1.0 Study Title:
2.0 Purpose of Study: (study objectives)
3.0 Study Population: (eligibility criteria) Subjects who meet the following criteria will be eligible to participate: Subjects who meet the following criteria will NOT be eligible to participate:
4.0 Study Status: This study is (pending, active, CNPE, closed)
5.0 Total Number of Evaluable Subjects Initially Planned: (include the number to be treated, not the procurement number)
5.1 The number of subjects entered to date: (This section should include number
of patients enrolled, number of patients procured, and number of patients treated, as applicable. Any discrepancy between the number procured and cell lines manufactured, as specified in Section G, should be clarified. If no patients have been enrolled that can be stated and the table should be deleted.)
Patient ID NOTE: This should be the ID used consistently throughout this report. For any studies where some of the patients have 2 numbers, please include both numbers here.
Age Gender Race Date of Enrollment
Dose Level
Number of Doses
Dates of Treatment (For CAGT INDs Only)

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 17 of 32
© Baylor College of Medicine 2005
5.2 The number of subjects who completed treatment and initial follow-up: Include previous information with a note to the PI to update as needed.
5.3 The number of subjects who did not complete treatment and initial follow-up (and reasons): Include previous information with a note to the PI to update as needed.
5.4 The number of subjects currently on study:
5.5 The number of subjects who received additional doses not specified in the protocol: This section should be included as applicable and should include a brief description such as:
Patient (include CAGT ID), dose, number of doses, and extenuating circumstances.
6.0 Brief description of any available study results including information obtained
pertinent to an understanding of the drug’s action: Include for example, information about dose response, information from controlled trial, and information about bioavailability. This data should not be presented as a listing of individual patient outcomes. Rather, it should be a summary of study outcomes (in aggregate). Tables are recommended. Include a comment on whether the stopping rules were triggered and show the data that supports the conclusion. (The request for this information should be sent to the study statistician and the PI should be copied on the email.)
B: SUMMARY INFORMATION:
1.0 Most frequent and most serious adverse experiences by body system / IND safety reports: (cumulative data) Please see table – Appendix I
2.0 Subjects who died during participation in the study / Cause of death: (Cumulative data. If none, can state none and delete the table.)
Patient ID Date of Death Cause of Death Relationship to Study
3.0 Subjects who dropped out during the course of the investigation in association
with any adverse experience (whether or not the event is determined to be study related): (Cumulative data)
4.0 Summary of all IND safety reports submitted during the past year:

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 18 of 32
© Baylor College of Medicine 2005
1571 Serial # + date sent - SAE
5.0 List of any preclinical studies completed or in progress, with a summary of major preclinical findings during the year: The response here may be: N/A; there may actual data (if it is our product); or you may refer them to the cross-referenced IND, NDA or master file. If there is no new data, the following statement should be included: “All animal safety information available to this file has been provided to the agency as described in 21 CFR 312.32-33. There is no animal model applicable to this product so we have not undertaken any animal studies. Additionally, there is no new in-vitro or in-vivo pre-clinical data pertinent to this product that has not already been submitted to the FDA.”
6.0 Summary of any significant manufacturing / microbiological changes made during
the past year: The response here may be: N/A; a summary of actual data (if it is our product); or you may refer them to the cross-referenced IND, NDA or master file.
C: DESCRIPTION OF GENERAL INVESTIGATIONAL PLAN FOR THE COMING YEAR: D: SUMMARY OF SIGNIFICANT PROTOCOL MODIFICATIONS MADE SINCE LAST ANNUAL
REPORT: All amendments should be listed here (check IND binder and IRB binder to ensure that nothing
is missed). E: CROSS REFERENCE INFORMATION SINCE LAST ANNUAL REPORT: F: OUTSTANDING BUSINESS WITH THE FDA WITH RESPECT TO THIS IND: We have no outstanding business with the FDA at this time. (or state any outstanding business,
if such exists) For studies using investigational products that are manufactured externally, the report ends here with attached appendices as required. For studies using investigational cell or gene therapy products that are manufactured at BCM, please continue as follows:
Clinical trial oversight and monitoring have been conducted per the Monitoring plan previously
submitted to the FDA. Modifications to the plan are attached in Appendix II. For Gene Therapy INDs: G. Annual update to gene therapy product information requested March 6, 2000

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 19 of 32
© Baylor College of Medicine 2005
1. Please provide a list of all lots of all gene therapy products, cell banks (CB), and viral banks (VB) ever produced or generated in your facility for potential use in non-clinical or clinical studies of human gene therapy. Please include the date of manufacture for each, their use (e.g. non-clinical or clinical), and indicate their interrelationships, i.e., which CBs and/or VBs were used to prepare each CB, VB, or product lot. This information is provided in BB MF 11236 2. Please provide a list of all IND files that cross-reference your IND(s) or master file(s). In addition, please confirm all IND(s) or master files that you have obtained authorization to cross reference for support of your IND. This section should include two lists. One that includes the IND files we allow to cross reference, and one that includes the INDs we have permission to cross reference. – Regulatory Coordinator to complete 3. Please submit all lot release data and characterization testing for each lot of product used in clinical trials, and testing information for all master CB, working CB, master VB and/or working VB used during manufacture of your lots. When possible, please submit this information in tabular form including the lot number or identifier, date of manufacture, test, test method, the sensitivity and specificity of test methods when appropriate, specification, and test result. If you have already submitted this information to your file in the past, you are now requested to send it again as part of a manufacturing summary document to your file. This information is provided in BB MF 11236. 4. If any lots of product were produced for, but not used in, clinical studies please describe the reason they were not used. This information is provided in BB MF 11236 5. Please provide a summary of your product manufacturing quality assurance (QA) and quality control (QC) programs. This should consist of a brief approximately three pages) description of your system for preventing, detecting, and correcting deficiencies that may compromise product integrity or function, or may lead to the possible transmission of adventitious infectious agents. Also, identify each individual who has authority over the QA and QC programs and list their duties. Please provide the date of your last QA and QC audits of your manufacturing operations and those of contract manufacturers, vendors, or other partners. Product QA/QC are described in BB MF 11236 previously submitted to the FDA. Modified SOPs related specifically to the manufacture of product used for this IND are attached in Appendix 3. (Obtain modified SOPs from the study PI or appropriate post-doc assigned to the project.) 6. For each clinical trial contained in your IND, please submit a 2-3 page summary of the procedures you have in place to ensure:

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 20 of 32
© Baylor College of Medicine 2005
a. there is adequate monitoring of the clinical investigations to demonstrate the trials(s) are conducted in accordance with the protocol, regulatory requirements and Good Clinical Practices (GCPs); that the rights and well-being of human subjects are protected; and that data reporting, including safety reporting to you (the sponsor), the IRB, and NIH is accurate and complete; and b. you as the sponsor, have adequate oversight over the clinical investigation, as outlined in 21 CFR 312, Subpart D. Please include with your summary an organizational chart identifying each individual responsible for oversight of clinical studies and his or her duties. If you have transferred some or all of these obligations to a Contract Research Organization (CRO), please so indicate, verify that these obligations are being appropriately met, and provide a summary of the CRO’s oversight procedures. Clinical trial oversight and monitoring have been conducted per the Cell/Gene Therapy Monitoring plan previously submitted to the FDA. Modifications to the plan are attached in Appendix 2. Confirm that all modifications to the monitoring plan are attached in Appendix 2. Work with BG as necessary. For Gene Therapy INDs utilizing a Retroviral Vector: 7. Per request of the FDA made in January 2010, we provide the following additional information related to retroviral products:
a. RCR Testing Results (data to be obtained from QA/QC Laboratory Personnel) The table below shows RCR testing results on products administered to patients and follow up blood samples obtained from patients post infusion. Patient CAGT #
Date Infused
Testing on Products Follow up RCR Testing
# XXX XX/XX/XX Products w/QA review date Submitted Results In Vitro Assay -
finding XX/XX/XX XX/XX/XX finding
# XXX XX/XX/XX In Vitro Assay - finding
XX/XX/XX XX/XX/XX finding
b. Detection of Transgene (data to be obtained from the study specific laboratory PI)
The table below shows transgene detection with X = transgene detected 0= no transgene detected
CAGT Number
Product/Cell Type (as specified in protocol)
Week (as specified in protocol)
Month (as specified in protocol)

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 21 of 32
© Baylor College of Medicine 2005
Integration information: Assays with positivity levels <0.5 do not require evaluation of integration sites and such a statement should be added here. Assays with positivity levels ≥0.5 do require evaluation of integration sites and integration data should be included here.
For Cell Therapy INDs: H. Annual Update to Cell Therapy Product Information
1. Following is the product lot number or identifier, date of product manufacture, date(s) of patient administration (if known) or shipping date. If the product is patient-specific (e.g. autologous cells), the tissue or cell source is also indicated. Protocol specific – request from PI and get C of A from GMP QA/QC. 2. Following is a listing of all attempts to prepare cell therapy products for clinical studies that were not, or could not be, used clinically. Included are the product lot number or identifier, and date of product manufacture. If the product is patient-specific (e.g., autologous cells) the tissue or cell source is indicated. The reason the product was not used is also provided. Also, included are the results from any investigation of this outcome, where applicable. Protocol specific – request from PI

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 22 of 32
© Baylor College of Medicine 2005
IND REPORT APPENDIX I
Most frequent and most serious adverse experiences by body system / IND Safety Reports: For Phase I CBER INDs:
Summary Table: Data Current as of: XX/XX/XXXX
Dose Level
Number of Patients
Treated at that Dose
Number of Doses Given
Number of Patients with Grade 3-5 AE
within Toxicity Evaluation Period as
specified in the protocol
Number of Patients with Grade 3-5 AE
within Toxicity Evaluation Period as
specified in the protocol at Least Possibly Related
to the Study Agent
Number of Patients with
Grade 3-4 GvHD (for
protocols in which GvHD
is a risk)
Number of Patients with
a DLT as specified in the protocol
Any Grade 3-5 AEs at least possibly related to the study agent within the toxicity evaluation period as specified in the protocol should be listed:
Body System Patient ID
Event Grade Study Relationship
Onset Date Dose Date
INFUSION RELATED AEs: (AEs that occurred within 24 hours of the infusion that are at least possibly related)
Body System Patient ID
Event Grade Study Relationship
Onset Date Dose Date

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 23 of 32
© Baylor College of Medicine 2005
For studies with patients that received doses where the toxicity information was not to be included in the overall toxicity evaluation, a separate summary table should be done for each subject as follows: Patient (include CAGT ID) Toxicity Data for Additional Dose:
Dose Level
Total Number of
Doses Given
Number of Grade 3-5 AEs within toxicity period
Number of Grade 3-5 AEs within toxicity period
possibly related to study agent
DLT
Y/N
For submission to the DRC only (not submitted to the FDA), provide a line listing of all adverse events, separated by Group and Dose Level. Below is a sample utilizing CTCAE v3; however, events should be organized in a manner that is compatible with the toxicity schema specified in the protocol (e.g., CTVAE v4). For each report, unneeded rows should be deleted and/or rows should be added as necessary.
As of: XX/XX/XXXX
Body System Patient ID Event Grade Study
Relationship Onset Date Dose Date
Allergy/Immunology Auditory/Hearing Blood/Bone Marrow Cardiovascular Coagulation Constitutional Symptoms Dermatology/Skin Endocrine Gastrointestinal Hemorrhage Hepatic Infection/Febrile Neutropenia
Lymphatics Metabolic/Laboratory
Musculoskeletal

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 24 of 32
© Baylor College of Medicine 2005
Neurology Ocular/Visual Pain Pulmonary Renal/Genitourinary Secondary Malignancy Sexual/Reproductive Function
Syndromes For all other INDs: Delete rows not applicable or if there are no AEs; add rows as necessary for the events under each body system.
Data current as of: XX/XX/XXXX
Body System Patient ID Event Grade Study
Relationship Onset Date Dose Date (For CAGT INDs only)
Allergy/Immunology Auditory/Hearing Blood/Bone Marrow Cardiovascular Coagulation Constitutional Symptoms Dermatology/Skin Endocrine Gastrointestinal Hemorrhage Hepatic Infection/Febrile Neutropenia
Lymphatics Metabolic/Laboratory
Musculoskeletal

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 25 of 32
© Baylor College of Medicine 2005
Neurology Ocular/Visual Pain Pulmonary Renal/Genitourinary Secondary Malignancy Sexual/Reproductive Function
Syndromes

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 26 of 32
© Baylor College of Medicine 2005
IND REPORT APPENDIX II
Cell/Gene Therapy Monitoring Plan Update
(Delete for studies that do not have such a plan.)

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 27 of 32
© Baylor College of Medicine 2005
APPENDIX IX
Protocol Conduct Summary Example Required for internal IND studies, in addition to the IND Report
To: DLDCC DRC From: (PI) Date: (Date) Re: H#: Study Title Accrual Information Initial IRB Approval Date: Study Activation Date: Date of First Enrollment: Multi-site (Y/N): Stated Accrual Rate (from the time of PRMC review): Protocol Conduct Statement Describe any problems with protocol conduct, such as any issues with subject eligibility, compliance issues, and deviations/variations from the protocol. Variance reports (if any) should be included.

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 28 of 32
© Baylor College of Medicine 2005
Appendix X DRC Report Template
For protocols not conducted under internal IND NOTE TO PI / DESIGNEE: Instructions and examples within this template are provided in blue font. As you complete this report, please delete the instructions/examples, and replace with your data and information.
1.0 Report Date: This is the “end date” of the data in the report; not the date of submission.
2.0 Study H-number:
3.0 Study Title: 4.0 Principal Investigator: 5.0 Brief Study Overview: Include study objectives and treatment schema, if applicable.
6.0 Inclusion / Exclusion Criteria: Current eligibility criteria.
7.0 Study Summary:
7.1 Current Status: Such as: pending; open to accrual; open but closed to accrual; fully closed
7.2 Activation Date: Date the study was fully open to enrollment (may differ from IRB approval date).
7.3 Date of First Enrollment: 7.4 Multi-Site: Yes / No 7.5 Total Target Accrual: the total number of evaluable subjects planned for the study 7.6 Anticipated Accrual Rate (as stated at the time of PRMC review): state as “ X accruals
over Y months”
8.0 Principal Investigator Summary Statement 8.1 Study Findings: Summarize study findings to date, including any interim analyses (e.g.,
toxicities, efficacy, etc.). 8.2 Future Plans: Describe the future plans for the study. 8.3 Correlative Studies: If applicable, provide preliminary results of correlative studies.
9.0 Study Modifications
Summarize any significant protocol amendments or modifications since the last DRC review, and provide the rationale. Use free text OR complete table below (insert rows as needed).
Amendment Approval Date Amendment Summary and Rationale Version 1.1 Mo/Day/Year
10.0 Statement of Protocol Conduct: Describe any problems with protocol conduct, such as any
issues with subject eligibility, compliance issues, and deviations/variations from the protocol.
BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 29 of 32
© Baylor College of Medicine 2005
11.0 Statistician Summary Statement
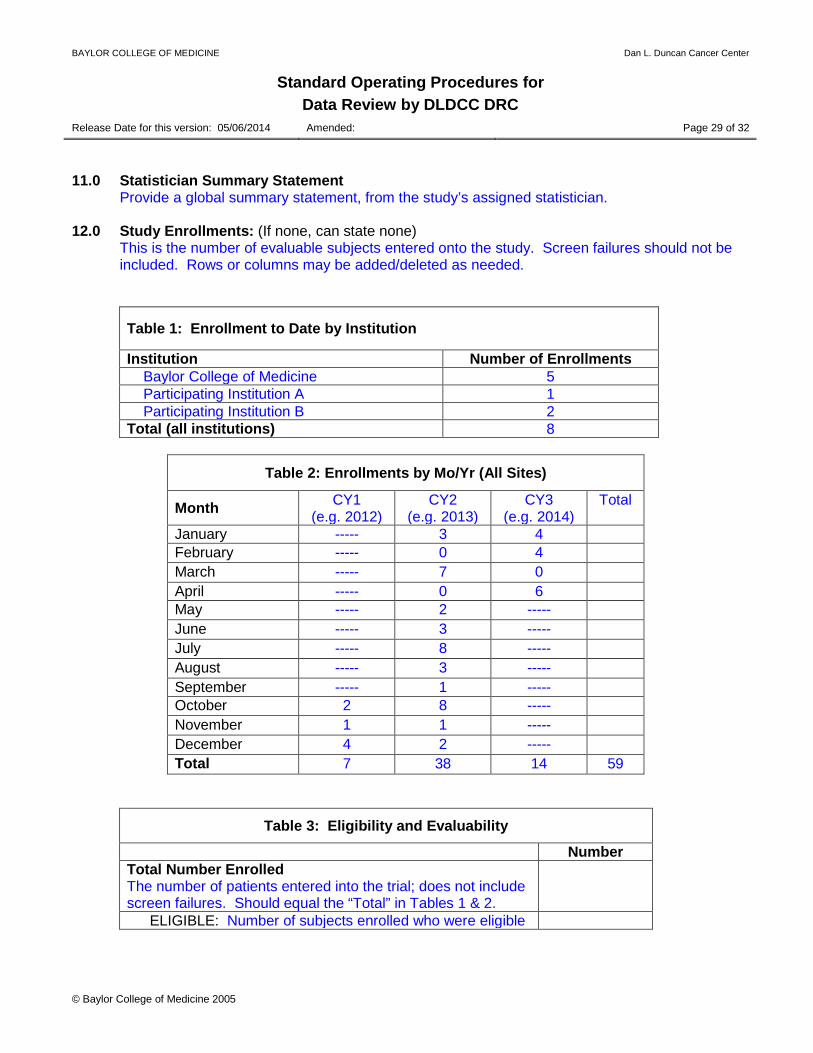
Provide a global summary statement, from the study’s assigned statistician. 12.0 Study Enrollments: (If none, can state none)
This is the number of evaluable subjects entered onto the study. Screen failures should not be included. Rows or columns may be added/deleted as needed.
Table 1: Enrollment to Date by Institution
Institution Number of Enrollments Baylor College of Medicine 5 Participating Institution A 1 Participating Institution B 2 Total (all institutions) 8
Table 2: Enrollments by Mo/Yr (All Sites)
Month CY1 (e.g. 2012)
CY2 (e.g. 2013)
CY3 (e.g. 2014)
Total
January ----- 3 4 February ----- 0 4 March ----- 7 0 April ----- 0 6 May ----- 2 ----- June ----- 3 ----- July ----- 8 ----- August ----- 3 ----- September ----- 1 ----- October 2 8 ----- November 1 1 ----- December 4 2 ----- Total 7 38 14 59
Table 3: Eligibility and Evaluability
Number Total Number Enrolled The number of patients entered into the trial; does not include screen failures. Should equal the “Total” in Tables 1 & 2.
ELIGIBLE: Number of subjects enrolled who were eligible

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 30 of 32
© Baylor College of Medicine 2005
INELIGIBLE: Number of subjects who, after enrollment, were found to be ineligible. For each one, list immediately below and why they were ineligible.
Ineligible Subject 1: describe reason ineligible Ineligible Subject 2: describe reason ineligible
Number Evaluable for Response
EVALUABLE: Number of subjects who have completed the study and are evaluable for response.
INEVALUABLE: Number of subjects who did not complete enough of the study to be evaluable for response.
Inevaluable Subject 1: describe reason inevaluable Inevaluable Subject 2: describe reason inevaluable
TOO EARLY
Number Evaluable for Toxicity EVALUABLE: Number of subjects who are evaluable for toxicity. (Should include all subjects who received study drug.)
INEVALUABLE: Number of subjects who are inevaluable for toxicity.
Inevaluable Subject 1: describe reason inevaluable Inevaluable Subject 1: describe reason inevaluable
TOO EARLY
Table 4: Current Patient Status Current as of [Date]
This table should account for all subjects who are considered enrolled. Arm
(delete columns that are not needed)
Total
A B C Currently receiving therapy / intervention Completed protocol therapy / intervention Removed due to disease progression / relapse Removed due to adverse event / complication Removed due to patient withdrawal of consent Total
Table 5: Subject Characteristics Add any subject characteristics that are important to the evaluation of the data,

BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 31 of 32
© Baylor College of Medicine 2005
such a protocol strata. Irrelevant characteristics can be deleted. Arm
(delete columns that are not needed)
A B C Total Gender
Female Male
Diagnosis (if applicable) Diagnosis A Diagnosis B
Stage (if applicable) I II III
Performance Status (if applicable)
0 1 2 3
Total
13.0 Statistical Analysis
13.1 Does the study have stopping rules? If so, please state the stopping rules, and whether or not the study has met these rules.
13.2 Is an interim analysis planned, or has one been done?
13.2.1 Date of Interim Analysis:
13.2.2 Brief description of Interim Analysis results:
14.0 Efficacy Data (if available): Preferably presented in a table form summarizing the study endpoints.
15.0 Toxicity Data: Complete the table below. Provide a line listing of all adverse events, organized by disease category or system organ class. Below is an example utilizing CTCAE v4; however, events should be organized in a
BAYLOR COLLEGE OF MEDICINE Dan L. Duncan Cancer Center
Standard Operating Procedures for Data Review by DLDCC DRC
Release Date for this version: 05/06/2014 Amended: Page 32 of 32
© Baylor College of Medicine 2005
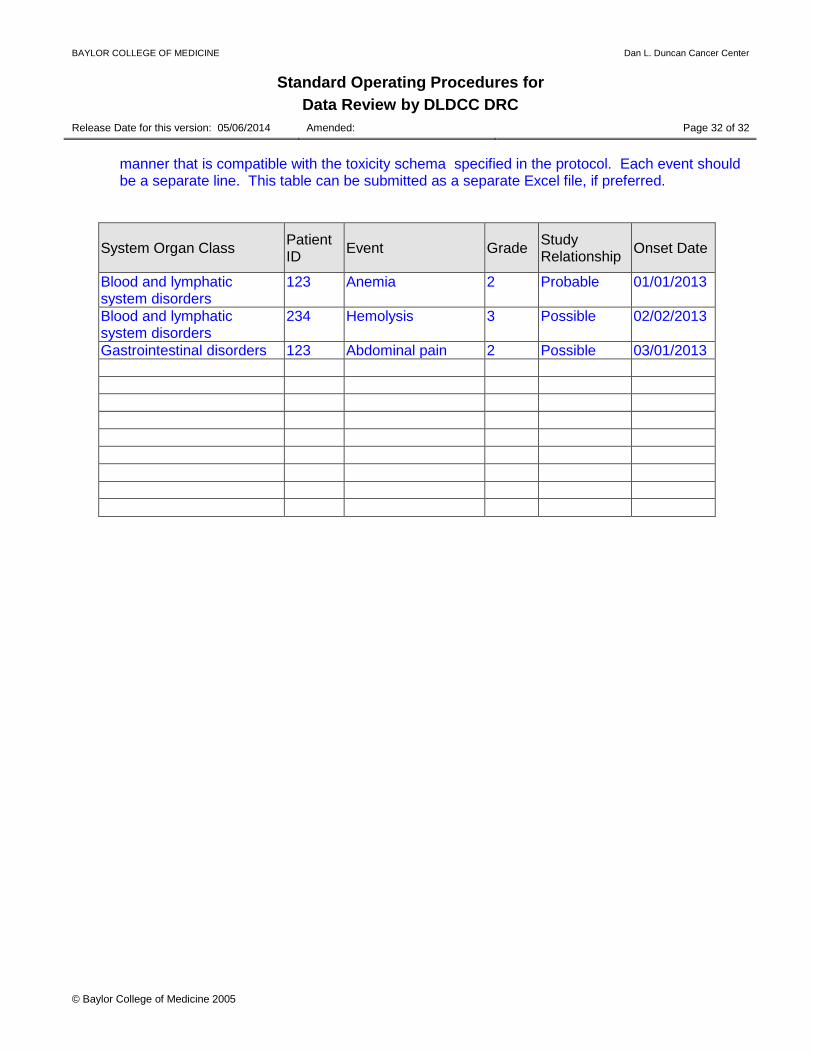
manner that is compatible with the toxicity schema specified in the protocol. Each event should be a separate line. This table can be submitted as a separate Excel file, if preferred.
System Organ Class Patient ID Event Grade Study
Relationship Onset Date
Blood and lymphatic system disorders
123 Anemia 2 Probable 01/01/2013
Blood and lymphatic system disorders
234 Hemolysis 3 Possible 02/02/2013
Gastrointestinal disorders 123 Abdominal pain 2 Possible 03/01/2013