some characteristics ofstreptococcal …jb.asm.org/content/88/2/497.full.pdf · vol. 88, no. 2, p....
TRANSCRIPT

JOURNAL OF BACTERIOLOGYVol. 88, No. 2, p. 497-508 August, 1964Copyright © 1964 American Society for Microbiology
Printed in U.S.A.
SOME CHARACTERISTICS OF STREPTOCOCCAL NICOTINAMIDEADENINE DINUCLEOTIDASE AND STREPTOLYSIN 0'
E. IRENE PENTZ,2 EVA KOT, AND J. J. FERRETTI3Department of Biochemistry, Northwestern University School of Medicine, and Departments of Research
and Therapeutic Radiology, Chicago Wesley Memorial Hospital, Chicago, Illinois
Received for publication 30 March 1964
ABSTRACT
PENTZ, E. IRENE (Northwestern UniversitySchool of Medicine, Chicago, 11.), EVA KOT, ANDJ. J. FERRETTI. Some characteristics of streptococ-cal nicotinamide adenine dinucleotidase andstreptolysin 0. J. Bacteriol. 88:497-508. 1964.-Nicotinamide adenine dinucleotidase produced byStreptococcus pyogenes strain C203U, grown inTodd-Hewitt broth (Difco), was recovered fromdiethylaminoethyl-cellulose columns free fromstreptolysin 0. It contained a trace of deoxyribo-nuclease. The elution peak of nicotinamide ade-nine dinucleotidase was fairly symmetrical; whenit was treated with dinitrofluorobenzene (DNFB)and chromatogrammed on paper in three differentsolvent systems, only one yellow spot was ob-tained. Examination of the enzyme in an ultra-centrifuge presented no moving boundary. Thepore size of two different widths of Visking casingwas estimated by dialyzing proteins or peptidesof known molecular weight and testing the dialy-sate for their appearance. The casing which al-lowed a molecule of 2,500 molecular weight to pass,but not one of 3,000 molecular weight, retained thenicotinamide adenine dinucleotidase. A fragmenthaving no nicotinamide adenine dinucleotidaseactivity did pass through this casing, however, butwas retained by the second casing which retainedmolecules weighing 2,500. Amino acid analyses ofthree different nicotinamide adenine dinucleo-tidase preparations were remarkably similar, andwere characterized by a high content of prolineand glycine and a very low content of sulfur-con-
I Preliminary reports of portions of the datareported here were presented at the meetings ofthe Federation of American Societies for Experi-mental Biology, Atlantic City, N.J., April, 1962,and at the Annual Meeting of the American Soci-ety for Microbiology, Cleveland, Ohio, June, 1963.
2 Present address: Frank H. Neely Nuclear Re-search Center, Georgia Institute of Technology,Atlanta.
3 Present address: Departments of Pediatricsand Biochemistry, College of Medical Sciences,University of Minnesota, Minneapolis.
taining amino acids. Examination by treatmentwith DNFB followed by hydrolysis for the pres-ence of N-terminal amino acids ruled out allamino acids, with the possible exception of argi-nine. The combined evidence suggests that itsmolecular weight is between 2,500 and 20,000.Some data are presented to illustrate the irrevers-ible instability of streptolysin 0; amino acidanalyses for three separate preparations of highactivity indicate a very low content of sulfur-containing amino acids.
Efforts to concentrate and to purify both strep-tolysin 0 and streptococcal nicotinamide adeninedinucleotidase led to further definition of some oftheir chemical and physicochemical properties.It is common knowledge among workers in thefield that both of these substances are highly un-stable and, therefore, exceedingly difficult to con-centrate in adequate supply when classical meth-ods of protein chemistry are applied. Perhaps it isbecause of the high incidence of failure in thisendeavor that their respective behaviors are verypoorly documented in the literature.
Carlson et al. (1957) were able to prepare onlysmall amounts of each substance by means of con-tinuous-flow paper electrophoresis. Pace andPappenheimer (1959), using the same method,successfully prepared nicotinamide adeninedinucleotidase of high activity as long as a highconcentration of ammonium sulfate was left in thefinal product. Because of their instability, it isstandard practice to add bovine serum albuminto dilute solutions of each substance as a protec-tive agent where such procedure does not interferewith the objectives of the experiment. That strep-tolysin 0 and nicotinamide adenine dinucleoti-dase, in their active forms, are separate sub-stances was well substantiated immunologically(Carlson et al., 1957; Pace and Pappenheimer,1959; Halbert, 1958; Halbert and Auerbach,1956; Bernard and Stollerman, 1959). However,they do have several properties in common,
497
on May 14, 2018 by guest
http://jb.asm.org/
Dow
nloaded from

PENTZ, KOT, AND FERRETTI
namely, their instability; their ability to be in-fluenced by reducing compounds, such as cysteineand glutathione (Slade and Knox, 1950; Ayouband Wannamaker, 1963); and, as will be shownhere, some striking differences as well as some
similarities in their amino acid content.It had been previously observed by Carlson
et al. (1957) and Pentz (unputblished data) that allstreptolysin 0 preparations examined also con-
tained nicotinamide adenine dinucleotidase. Asexperiments continued in efforts to plrepare one or
the other of these activities in good yield, withoutthe presence of salt in the final product, a strain(C2031U) of Streptococcus pyogenes, which hadbeen lyophilized for some y-ears, was found not tobe extruding hemolytic activity into the liquidmedium (Todd-Hewitt broth, Difco). This fortui-tous circumstance allowed the successful use ofcellulose columns to prepare the enzyme; thefollowing experiments report a reasonable degreeof success with the method, as well as some of thecharacteristics of this enzyme.
MATERIALS AND MIETHODS
The culture of S. pyogenes used was strainC203U (originally obtained from Alan lBern-heimer). This strain was originally a nonhemo-lytic variant of C203. On continued subculture inTodd-Hewitt broth, it was found to be a goodproducer of streptolysin 0, and the experimentson this lysin reported here were performed with it.A specimen of this culture was lyophilized by themethod of Fry and Greaves (1951) and stored forsome -ears. At the beginning of the attempt topurify nicotinamide adenine dinucleotidase by theuse of diethylaminoethyl (DEAE)-cellulosecolumns, a vial of this stored culture wNas openedand regrown in the Todd-Hewitt broth. Althoughthis sample of the strain is hemolytic whengrown on blood-agar plates, it has not so far pro-
duced a significant titer of any hemolytic activityin the liquid medium.
Nicotinamide adenine dinucleotidase activitywas assayed routinely by modifying the cyanidemethod of Kaplan, Colowick, and Nason (1951).All assays were run at room temperature (25 C)rather than at 37 C, except in the experimentswhere the two assays were compared. For eachassay, 2 mg of material were dissolved in an ap-
propriate amount of phosphate buffer. This was
distributed equally among five or seven cuvettes,each containing a standard amount of diphospho-
pyridine nucleotide. At 1-min intervals, sodiumcyanide was delivered to individual cuvettes; theoptical density of the cuvettes was read at 325mn, and the results were plotted against time. Ifthe first two points fell on the slope of a curve, oneof which was above 60 % of the blank, a curve wasdrawn, a perpendicular was dropped from the60% position, and an estimate was made of theunits present. If no curve could be drawn, furthermaterial had to be used in higher dilution. If moreaccurate assays were ma(de, frequently all of thematerial was used before further manipulationscould be carried out. It was intended to use thissimplified method only during the course of thepreparative steps and then to accurately assayfinal lproducts. Because of repeated low y-ieldsthis was not possible. Comparison of the twomethods led to the establishment of a factor ofapproximately 7 between them. Therefore, all ofthe nicotinamide adenine dinucleotidase unitsreported here should be multiplied by this factorfor comparison between this and other publishedunits. In earlier studies, the alcohol dehydrogen-ase method of Racker (1950) was used also. Thefindings of the two assay l)rocedures were alwayssimilar.The Folin-Ciocalteau method, as modified by
Lowry et al. (1951), was used to determine totalprotein. Deoxyribonuclease was assaved by themethod of Kunitz (1950).
1'roduction of the enzyme was achieved by in-oculating 2-liter amounts of sterile Todd-Hewittbroth with 1O ml of an 8-hr culture and incubatingit for 18 to 24 hr at 37 C. The bacterial growth wAasremoved by centrifugation at room temperature.The supernatant fluid was decanted through apacked, wet layer of shredded, sterilizing filtersheets (Republic Seitz Filter Corp., Newark,N.J.) supported on the plate of a sintered-g,lassfunnel of coarse porosity. To this clear filtratewere added l)hosphate buffer and calcium acetate,as in the method for the preparation of strepto-lysin 0 (Pentz and Shigemura, 1955). The crudeproduct obtained from the first calcium acetateprecip)itation, after elution, dialysis, and lyophi-lization, was used as the starting material for thecolumn separations.
IUltracentrifuge analyses were carried out in aSpinco model E apparatus, with the standardAn-I) analytical head at 59,780 rev/min at 21 C.A 1 cl solution of nicotinamide adenine dinueleo-
498 J. BACTERIOL.
on May 14, 2018 by guest
http://jb.asm.org/
Dow
nloaded from

CHARACTERISTICS OF STREPTOCOCCAL PRODUCTS
tidase, made in phosphate buffer (pH 7.0) of 0.1ionic strength, was examined.
Visking casing was sized by introducing crystal-line ribonuclease (C. E. Boehringer and Soehne),protamine sulfate (Nutritional BiochemicalCorp., Cleveland, Ohio), and hydrolyzed (by themethod of Sanger, 1949) insulin (E. R. Squibb &Sons, New York, N.Y.; USP unmodified) insidevarious casings and dialyzing for 24 hr in a coldroom against deionized water contained in 100-mlgraduate cylinders. At the end of this time, thedialysate was tested for the presence of Folin-Ciocalteau positive material.Amino acid analyses were carried out in a
Beckman-Spinco apparatus with the methodsrecommended in the manual.
Reactions with dinitrofluorobenzene (DNFB;Eastman Chemical Products, Inc., Kingsport,Tenn.), were carried out in 8% sodium bicar-bonate on a shaking machine at room tempera-ture for 24 hr. After extraction with ether andacidification with HCl, the dinitrophenyl (DNP)-tagged materials separate as oils or crystals.
Proteins, including the DNP-labeled proteins,were hydrolyzed by adding 1 ml of 6 N HCl intotest tubes which had been previously drawn out toprovide a narrow neck. The liquid contents werethen frozen, and the tube was evacuated, sealed,and heated at 110 C for 22 hr.
One-dimensional paper chromatography wascarried out in cylindrical glass jars at room tem-perature with the solvent system descending.Two-dimensional paper chromatography wascarried out in double-walled chromatocabs, thepaper being previously washed with the first sol-vent; again, the solvents were descending.The precise method of measuring streptolysin
O was a modification of the method of Herbert(1941) and was described previously (Pentz andShigemura, 1955). Units are referred to asHerbert Units (HU). Estimation against stand-ardized antistreptolysin 0 (supplied by the SerumInstitute, Carshalton, Surrey, England) was alsoused, and these units are referred to as ToddUnits (TU). It was found that 1 TU was approxi-mately equal to 20 HU. In the original Herbert(1941) method the equivalence was 2.5 to 1. Thedifference is due to a difference in the method ofstandardizing the rabbit red blood cells, alsopreviously described (Pentz and Shigemura,1955).The streptolysin 0 preparations used were pre-
pared by the method described by Pentz andShigemura (1955) unless otherwise indicated inthe text, and they had frequently not been carriedthrough all three steps of the procedure.
All electrophoretic analyses were carried out ina conventional apparatus equipped with a Phil-pot-Svensson (Philpot, 1938; Svensson, 1940)optical system. Unless otherwise stated, thebuffer was phosphate of 0.1 ionic strength. Solu-tions were made from lyophilized powder exceptwhere indicated, and all dialyses were carried outat 4 C.
For calculating mobilities, the negatives wereplaced in an enlarger and projected at a standarddistance from the projected image. The image wasthen traced, and distances between the startingboundary and the centers of the peaks were care-fully measured.To prepare nicotinamide adenine dinucleoti-
dase, 50- to 75-mg amounts of crude lyophilizedmaterial having no streptolysin 0 activity weredissolved in 5 ml of 0.0025 M phosphate buffer andadded to the top of a 10-cm column of DEAE-cellulose. [After beginning this work, it was neces-sary to reorder the DEAE-cellulose (Cellex-D;Bio-Rad Laboratories, Richmond, Calif.). Onelot of the replacement material sent produced norecovery of nicotinamide adenine dinucleotidaseactivity from the columns when crude materialthat had been successfully recovered previouslywas applied. A second lot of the cellulose pro-vided about one-quarter of the recovery originallyobtained. The manufacturer was then able toprovide an adequate supply of the original cellu-lose which had been successful (control no.B-1899) to complete these studies. The reason forthis discrepancy in the product is obscure.] Thecellulose had been sized previously in water toremove the fines and then washed with two vol-umes of the first concentration of phosphatebuffer.
RESULTSNicotinamide adenine dinucleotidase. Elution of
the DEAE columns with a step-wise concentra-tion (0.0025, 0.025, 0.05, and 0.25 M) of phosphatebuffer (pH 6.95) at room temperature providedfour separate protein peaks (Fig. 1). Only thefirst peak contained any nicotinamide adeninedinucleotidase activity, usually about 7,000 unitsper mg, but this varied from 3,000 to 12,000 unitsper mg after lyophilization. The yield from this
499VrOL. 88, 1964
on May 14, 2018 by guest
http://jb.asm.org/
Dow
nloaded from
PENTZ, KOT, AND FERRETTI
step was highly variable with different lots of thecrude nicotinamide adenine dinucleotidase prepa-ration. Starting with 50 mg, and dialyzing afterrecovery from the column, anywhere from 2 to 20mg may be recovered.A collection of this material was made, dialyzed
against deionized water, and lyophilized. Thematerial was then examined in an ultracentrifugeon two separate occasions, at 4.5 and 2.1 hr. Inneither case did it present a moving boundary.In each, nicotinamide adenine dinucleotidaseactivity was recovered from the run. This mate-rial was examined for desoxyribonucleic aciddepolymerizing activity and was found to containapproximately 0.2 to 0.4 deoxyribonuclease unitsper mg. The relation of the protein content as wellas the two types of activity are illustrated in Fig.2. The scale describing the deoxyribonuclease wasexaggerated to illustrate the relative position ofthis activity. In fact, only a very small percentageof this material can be present. As will be shownlater, it is insufficient to appear as a separate
2432
12.700
12.000
3,800
3.000
2,500
1632
2,500`7
Ribonuclease
Insulin
Phenylalonyl
Glycyl
Profam ine
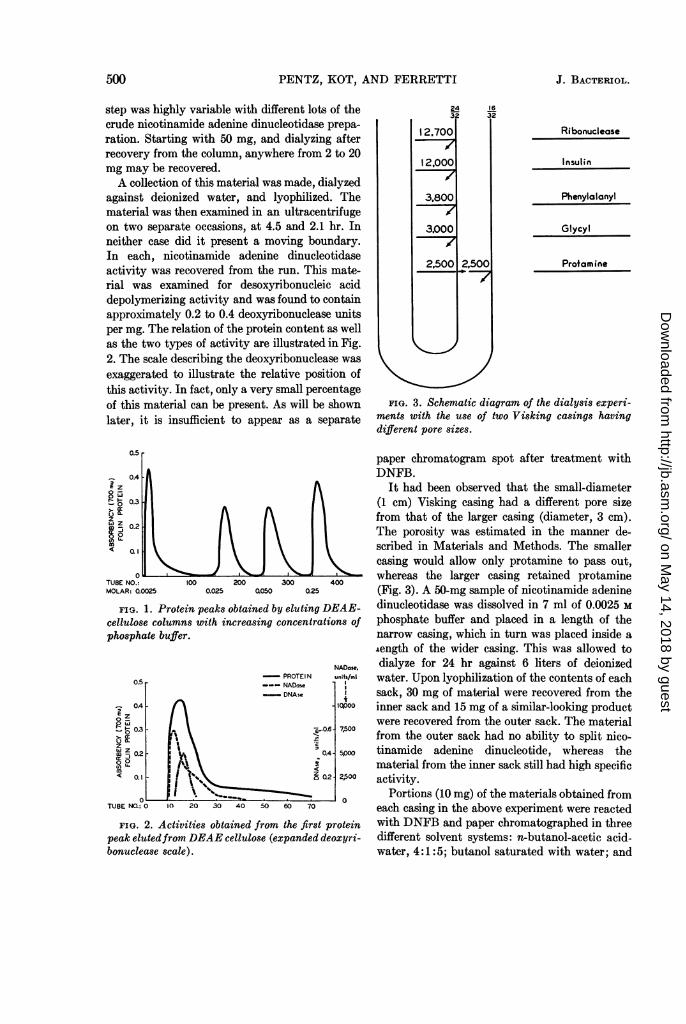
FIG. 3. Schematic diagram of the dialysis experi-ments with the use of two Visking casings havingdifferent pore sizes.
Q.5 -
paper chromatogram spot after treatment with.OA DNFB.
8-Z It had been observed that the small-diametert 8 03l (1 cm) Visking casing had a different pore size-z 0.2 l A A l from that of the larger casing (diameter, 3 cm).tizs 0.2 The porosity was estimated in the manner de-< Ql scribed in Materials and Methods. The smaller
TUBE} \ § \casing would allow only protamine to pass out,
o 0 2 3 400 whereas the larger casing retained protamineTUBE NO. 100 200 300 400MOLAR: 0.0025 0.025 0050 0.25 (Fig. 3). A 50-mg sample of nicotinamide adenine
FIG. 1. Protein peaks obtained by eluting DEAE- dinucleotidase was dissolved in 7 ml of 0.0025 Mcellulose columns with increasing concentrations of phosphate buffer and placed in a length of thephosphate buffer. narrow casing, which in turn was placed inside a
length of the wider casing. This was allowed toNADose. dialyze for 24 hr against 6 liters of deionized
0.5 _ PROTEIN units/mi water. Upon lyophilization of the contents of each- DNAse sack, 30 mg of material were recovered from the
04A . \, inner sack and 15 mg of a similar-looking product
p - \ were recovered from the outer sack. The materialj-0.3 -.0.6 7,500_ k Q3 . \ \ t 5 from the outer sack had no ability to split nico-
M Z 0.2 0A\o0 tinamide adenine dinucleotide, whereas the00.. material from the inner sack still had high specific~~~~~~~~0.1 - 0.2Q 2,500 activity.
N040 0_20 0 Portions (10 mg) of the materials obtained fromTUBE NO.: O In 20 30 40 50 60 70 each casing in the above experiment were reacted
FIG. 2. Activities obtained from the first protein with DNFB and paper chromatographed in threepeak elutedfrom DEAE cellulose (expanded deoxyri- different solvent systems: n-butanol-acetic acid-bonuclease scale). water, 4:1:5; butanol saturated with water; and
500 J. BACTERIOL.
on May 14, 2018 by guest
http://jb.asm.org/
Dow
nloaded from

CHARACTERISTICS OF STREPTOCOCCAL PRODUCTS
.97 .60 .61
FIG. 4. Chromatogram with water-saturated bu-tanol, illustrating spots obtained on examination ofthe dinitrofluorobenzene-treated materials obtainedfrom the inner and outer Visking casings afterdialysis.
TABLE 1. Antino acid analyses of three differentpreparations of nicotinamide adenine
dinucleotidase*
Activity of sample
Amino acid 12,800 7 25 unt 7 u
units per725unt726uismng per mg per mg
1smoles/ ,umolles/ ,umoles/mgt mg mg
Aspartic acid ......... 0.310 0.290 0.329Threonine ............ 0.224 0.204 0.230Serine ................ 0.227 0.212 0.233Proline ............... 0.893 0.950 1.041Glutamic acid ........ 0.624 0.565 0.627Glycine ............... 1.208 1.025 1.341Alanine ............... 0.434 0.431 0.564Valine. 0.318 0.300 0.341Methionine ........... 0.073 0.083 0.072Isoleucine ............ 0.153 0.180 0.170Leucine .............. 0.226 0.117 0.218Tyrosine............. 0.032 0.160 0.035Phenylalanine ........ 0.104 0.434 0.090Lysine ................ 0.265 J 0.310Histidine ............. 0.117 0.129 0.087Arginine .............. 0.186 0.216 0.210Hydroxyproline ....... 0.255 0.301 0.392Hydroxylysine ......0...037 0.034 0.043
* All samples were hydrolyzed for 22 hr.t Average of three analyses.
TABLE 2. Amino acid analyses of Todd-Hewittbroth, three samples of nicotinamide adenine
dinucleotidase, and two control samples
Nicotin- ControlTodd- amide from
Amino acid Hewitt adenine DEAEbroth dinucleo- column
tidase-clm
jAmolesl jimolesl p.inoles/rng rng mng
Glutamic acid.. 1.441 0.624 0.3531.15 0.565 0.609
0.627Proline ............... 0.371 0.893 0.496
0.343 0.950 0.9201.041
Glycine ............... 0.386 1.208 0.9730.314 1.025 0.940
1.341
benzene-1% acetic acid, 1:1 (all one-dimen-sional).
In every case only one yellow spot, other thanthe DNFB, was obtained from each material(Fig. 4).With amino acid analyses as an ultimate objec-
tive, it was thought advisable to test the Todd-Hewitt broth itself for ability to provide a peak ofprotein under similar conditions. Accordingly, 2liters of broth in which no organisms were grown
were subjected to calcium phosphate precipita-tion, elution, dialysis, and lyophilization. Thematerial obtained was applied to DEAE-cellulosecolumns, and a peak of protein was obtained inapproximately the same tube positions of theeffluent fractions as in the active material. Thismaterial had neither nicotinamide adenine dinu-cleotidase nor deoxyribonuclease activity. Thematerial, considered to be a suitable control, was
hydrolyzed as were three different nicotinamideadenine dinucleotidase preparations, and all were
analyzed for their amino acid content. Two differ-ent lots of otherwise untreated Todd-Hewittbroth were also hydrolyzed and examined fortheir amino acid content (Tables 1 and 2).The unhydrolyzed control material was also
reacted with DNFB and examined by one-dimen-sional chromatography. With the use of thebutanol saturated with water system, two spotswere obtained apart from the DNFB itself; one
did not move from the origin and the other movedapproximately 0.5 in., but only after allowing thesolvent to run off the paper. In short, its behavior
VOL. 88, 1964 501
0 0
(Iw N QOUT0DN FB
on May 14, 2018 by guest
http://jb.asm.org/
Dow
nloaded from

PENTZ, KOT, AND FERRETTI
was completely different from that of nicotinam-ide adenine dinucleotidase. The material wasexamined in two more solvent systems, butanol-1% acetic acid (1:1) and butanol-butyl acetate-1%ammonium hydroxide (1:2:3); in each case thebehavior was completely different from that of theactive samples.
Samples of the materials which had been re-tained in the smaller and the larger sacks andtreated with DNFB were hydrolyzed and in turnexamined on the long and short columns of aBeckman-Spinco amino acid analyzer. Under theroutine conditions used, all of the DNP-labeledamino acids come off at the end of the run as asingle broad peak. By comparing these analysesto those obtained from the DNFB-nontreated,hydrolyzed materials, a good approximation ofwhich amino acids had been labeled could bemade. In short, those amino acids which weredecreased in amount in the DNP-labeled analysesmust be appearing as the trailing yellow peak.
Comparative examination of these analysesagainst that of the active material which had beenretained in the inner sack showed it to have lostabout one-half of its lysine, about two-thirds ofits histidine, and about one-fifth of its arginine,and no other amino acids. Comparison of theanalyses of the material in the outer sack showedit to have lost almost all of its lysine, two-thirdsof its histidine, one-quarter of its arginine, andtwo-thirds of its isoleucine. Because of the diffi-culty of having to apply a sample containing anunknown number of dinitrophenol groups to thecolumns, exact comparisons were not possible,and the above information could be consideredonly an approximation.Having no further interest in the inactive ma-
terial obtained from the outer sack, we did nofurther work on it.The DNFB-treated, hydrolyzed nicotinamide
adenine dinucleotidase was then subjected toexhaustive examination by paper chromatogra-phy. The hydrolysate was extracted with etheruntil no further yellow color was transferred intothe ether layer. The aqueous layer was then chro-matographed in n-butanol saturated with water;only two yellow spots were obtained, one ofwhich remained at the origin. DNP-mono-imidazole-L-histidine, mono-O-DNP-tyrosine,and &DNP-L-lysine (Mann Research Laboratory,New York, N.Y.) were purchased and the mono-DNP derivatives of the L isomers (obtained from
the California Foundation for BiochemicalResearch) of hydroxyproline, serine, arginine,cysteic acid, cystine, leucine, isoleucine, phenyl-alanine, glutamic acid, valine, alanine, glycine,methionine, aspartic acid, proline, and threoninewere synthesized by the method of Rao and Sober(1954). The di-DNP derivatives of tyrosine,histidine, and lysine were similarly prepared. Pre-liminary paper chromatographic runs eliminatedall other possibilities for the identity of the non-mobile spot except mono-imidazole histidine. Theidentity of this spot was subsequently shown tobe DNP-mono-imidazole histidine; the possibilityof its being anything else was eliminated by useof the following six different solvent systems:phenol-isoamyl alcohol-water, 1:1:1; pyridine-isoamyl alcohol-1.6 N ammonium hydroxide,6:14:20; benzene-1% acetic acid, 1:1; n-butanol-butyl acetate-1% ammonium hydroxide, 1:2:3;n-butanol saturated with water; and n-butanol-acetic acid-water, 4:1:5. The fact that it was ahistidine derivative was further confirmed byapplying the specific color test for histidine, sulfa-nilamide-sodium nitrite-sodium carbonate (Block,Darrum, and Zweig, 1958) to the paper chroma-togram, which gave a positive result.
One-dimensional paper chromatography inn-butanol-butyl acetate-ammonium hydroxide,1:2:3, indicated that the second spot corre-sponded to e-DNP-lysine. This was confirmed bytwo-dimensional chromatography (Table 3).The ether extract of the DNP-labeled, hydro-
lyzed nicotinamide adenine dinucleotidase wasapplied to paper along with the ether-extractableDNP-labeled amino acids, and was shown to giveonly one spot when run as a one-dimensionalchromatogram in benzene-acetic acid (1:1). Theunknown spot corresponded to none of the ether-extractable amino acids, and was subsequentlyshown to be DNFB itself. Alkaline hydrolysis ofthis material produced no ninhydrin-positivematerial, and the zirconium alizarinate test(Feigl, 1960) specific for fluoride was positivewhen applied to this fraction.
Streptolysin 0. Preliminary electrophoreticstudies indicated that large losses of hemolyticactivity occurred during the manipulationsattendant upon such analyses. Therefore, to con-serve the more concentrated preparations, thefollowing electrophoretic experiments were con-ducted with material having relatively lowhemolytic activity. The material had been pre-
502 J. BACTERIOL.
on May 14, 2018 by guest
http://jb.asm.org/
Dow
nloaded from
CHARACTERISTICS OF STREPTOCOCCAL PRODUCTS
pared by adsorption onto and elution from cal-cium phosphate (Pentz and Shigemura, 1955).The eluates from one precipitation were pooledand dialyzed against 6 liters of 0.067 M phosphatebuffer (pH 7.1) for 16 hr, then against runningdeionized water for 24 hr, and finally against 6liters of distilled water for 16 hr. The product waslyophilized, yielding fluffy yellow powder andhaving a hemolytic activity of 108 HU/mg. Bygrowing lots of 3 and 4 liters it was possible toobtain 5.4 g of this product from 20 to 25 liters ofculture supernatant fluid. It is now known thatthis material probably had nicotinamide adeninedinucleotidase activity, but these experimentswere concerned only with the hemolytic activity.
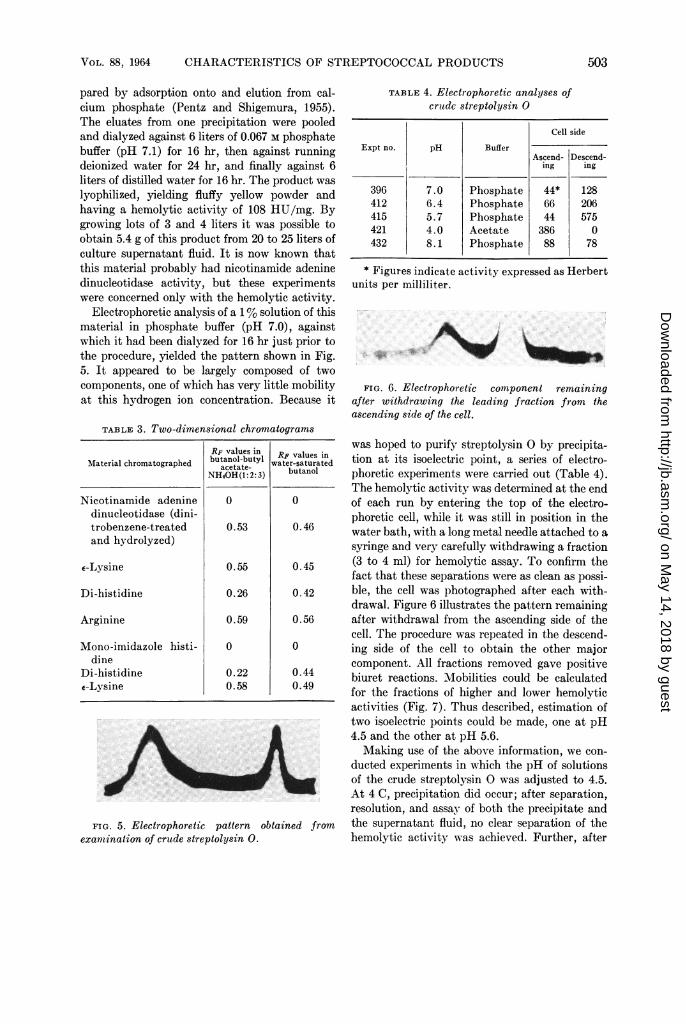
Electrophoretic analysis of a 1% solution of thismaterial in phosphate buffer (pH 7.0), againstwhich it had been dialyzed for 16 hr just prior tothe procedure, yielded the pattern shown in Fig.5. It appeared to be largely composed of twocomponents, one of which has very little mobilityat this hydrogen ion concentration. Because it
TABLE 3. Two-dimensional chromatograms
Material chromatographed
Nicotinamide adeninedinucleotidase (dini-trobenzene-treatedand hydrolyzed)
e-Lysine
Di-histidine
Arginine
Mono-imidazoledine
Di-histidinee-Lysine
histi-
RF values inbutanol-butyl
acetate-NH40H(1 :2:3)
0
0.53
0.55
0.26
0.59
0
0.22
0.58
FIG. 5. Electrophoretic pattern obexamination of crude streptolysin 0.
TABLE 4. Electrophoretic analyses ofcrutdc streptolysin 0
Cell side
Expt no. pH BufferAscend- Descend-
ing ing
396 7.0 Phosphate 44* 128412 6.4 Phosphate 66 206415 5.7 Phosphate 44 575421 4.0 Acetate 386 0432 8.1 Phosphate 88 78
* Figures indicate activity expressed as Herbertunits per milliliter.
W.
FIG. 6. Electrophoretic component remainingafter withdrawing the leading fraction from theascending side of the cell.
was hoped to purify streptolysin 0 by precipita-RiF values in tion at its isoelectric point, a series of electro-wvater-saturated tinpn,
butanol phoretic experiments were carried out (Table 4).The hemolytic activity was determined at the end
0 of each run by entering the top of the electro-phoretic cell, while it was still in position in the
0.46 water bath, with a long metal needle attached to asyringe and very carefully withdrawing a fraction
0.45 (3 to 4 ml) for hemolytic assay. To confirm thefact that these separations were as clean as possi-
0.42 ble, the cell was photographed after each with-drawal. Figure 6 illustrates the pattern remaining
0.56 after withdrawal from the ascending side of thecell. The procedure was repeated in the descend-
0 ing side of the cell to obtain the other majorcomponent. All fractions removed gave positive
0.44 biuret reactions. Mobilities could be calculated0.49 for the fractions of higher and lower hemolytic
activities (Fig. 7). Thus described, estimation oftwo isoelectric points could be made, one at pH4.5 and the other at pH 5.6.Making use of the above information, we con-
ducted experiments in which the pH of solutionsof the crude streptolysin 0 was adjusted to 4.5.At 4 C, precipitation did occur; after separation,resolution, and assay of both the precipitate and
tained from the supernatant fluid, no clear separation of thehemolytic activity was achieved. Further, after
VOL. 88, 1964 503
on May 14, 2018 by guest
http://jb.asm.org/
Dow
nloaded from
PENTZ, KOT, AND FERRETTI
dialysis and lyophilization of each fraction, largelosses in both hemolytic activity as well as totalmaterial were found to occur. Two possibilitiespresented themselves: (i) that protein was leakingthrough the walls of the dialysis sack, or (ii) thatprotein was breaking ul) into small fragmentsduring the course of dialysis and the fragmentswere being lost through the walls of the sack. Thefirst of these possibilities seemed unlikely, becausethe original material had been exhaustivelydialyzed, during its l)reparation, prior to lyophi-lization.To investigate the second possibility, the fol-
owing experiment was carried out. Portions
8
6
z 4
0
-Q
-0.
D2
2
0
2
6
8
o-O Lower hemolytic activity0----O Higher hemolytic activity
2 4pH
6 8
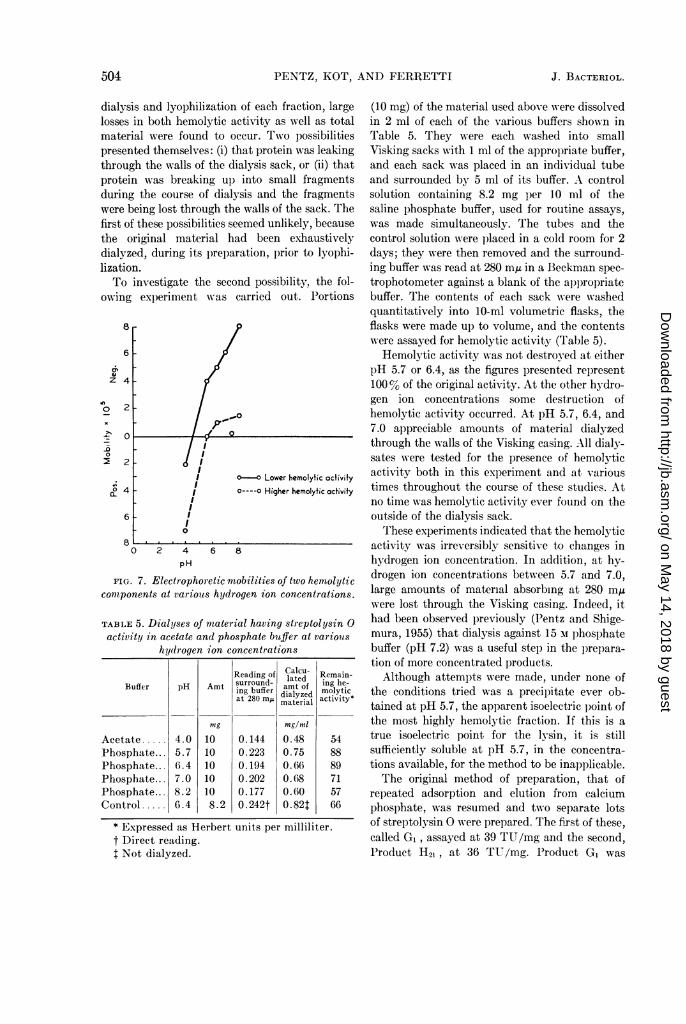
FIG. 7. Electrophoretic mobilities of two hemolyticcomponents at variouts hydrogen ion concentrations.
TABLE 5. Dialyses of material having streptolysin 0activity in acetate and phosphate buffer at variouis
hydrogen ion concentrations
Reading of Calcu- Remain-surround - lated ing eBuffer pH Amt amt of g be-ing bferdialyzed molyticat 280 mA material activity*
mg mng/mlAcetate ..... 4.0 10 0.144 0.48 54Phosphate... 5.7 10 0.223 0.75 88Phosphate... 6.4 10 0.194 0.66 89Phosphate... 7.0 10 0.202 0.68 71Phosphate... 8.2 10 0.177 0.60 57Control . 6.4 8.2 0. 242t 0. 82t 66
* Expressed as Herbert units per milliliter.t Direct reading.I Not dialyzed.
(10 mg) of the material used above wi-ere dissolvedin 2 ml of each of the various buffers shown inTable 5. They were each washed into smallVisking sacks with 1 ml of the appropriate buffer,and each sack was placed in an individual tubeand surrounded by 5 ml of its buffer. A controlsolution containing 8.2 mg per 10 ml of thesaline phosphate buffer, used for routine assays,was made simultaneously. The tubes and thecontrol solution were placed in a cold room for 2days; they were then removed and the surround-ing buffer was read at 280 mA in a Beckman spec-trophotometer against a blank of the appropriatebuffer. The contents of each sack were washedquantitatively into 10-ml volumetric flasks, theflasks were made up to volume, and the contentswere assayed for hemolytic activity (Table 5).
Hemolytic activity was not destroy-ed at eitherpH 5.7 or 6.4, as the figures presented represent100% of the original activity. At the other hYdro-gen ion concentrations some destruction ofhemolytic activity occurred. At pH 5.7, 6.4, and7.0 appreciable amounts of material dialyzedthrough the walls of the Visking casing. All dialy-sates were tested for the presence of hemolyticactivity both in this experiment and at varioustimes throughout the course of these studies. Atno time was hemolytic activity ever found on theoutside of the dialysis sack.
These experiments indicated that the hemolyticactivity was irreversibly sensitive to changes inhydrogen ion concentration. In addition, at hy-drogen ion concentrations between 5.7 and 7.0,large amounts of material absorbing at 280 m,uwere lost through the Visking casing. Indeed, ithad been observed previously (Pentz and Shige-mura, 1955) that dialysis against 15 M lphosphatebuffer (pH 7.2) was a useful step in the prepara-tion of more concentrated products.Although attempts were made, under none of
the conditions tried was a precip)itate ever ob-tained at pH 5.7, the apparent isoelectric point ofthe most highly hemolytic fraction. If this is atrue isoelectric point for the ly3sin, it is stillsufficiently soluble at pH 5.7, in the concentra-tions available, for the method to be inapplicable.The original method of preparation, that of
repeated adsorption and elution from calciumphosphate, was resumed and two separate lotsof streptolysin 0 were prepared. The first of these,called G,, assayed at 39 TU/mg and the second,Product H21, at 36 TU/mg. Product G, was
504 J. BACTERIOL.
on May 14, 2018 by guest
http://jb.asm.org/
Dow
nloaded from
CHARACTERISTICS OF STREPTOCOCCAL PRODUCTS
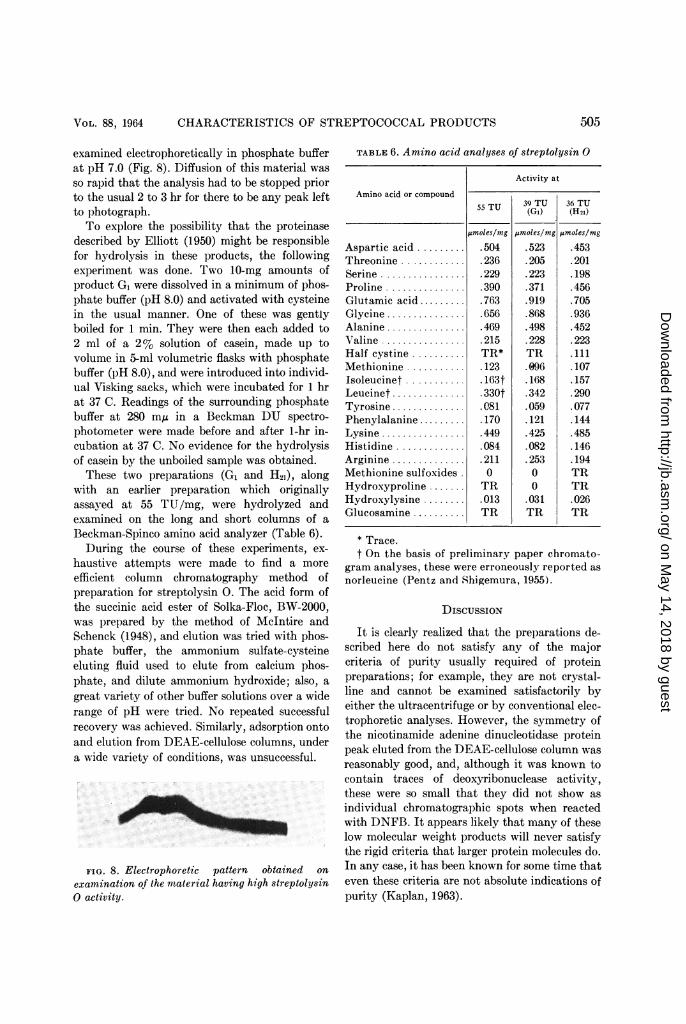
examined electrophoretically in phosphate bufferat pH 7.0 (Fig. 8). Diffusion of this material was
so rapid that the analysis had to be stopped priorto the usual 2 to 3 hr for there to be any peak leftto photograph.To explore the possibility that the proteinase
described by Elliott (1950) might be responsiblefor hydrolysis in these products, the followingexperiment was done. Two 10-mg amounts ofproduct GI were dissolved in a minimum of phos-phate buffer (pH 8.0) and activated with cysteinein the usual manner. One of these was gentlyboiled for 1 min. They were then each added to2 ml of a 2% solution of casein, made up tovolume in 5-ml volumetric flasks with phosphatebuffer (pH 8.0), and were introduced into individ-ual Visking sacks, which were incubated for 1 hrat 37 C. Readings of the surrounding phosphatebuffer at 280 m,u in a Beckman DU spectro-photometer were made before and after 1-hr in-cubation at 37 C. No evidence for the hydrolysisof casein by the unboiled sample was obtained.
These two preparations (G1 and H21), alongwith an earlier preparation which originallyassayed at 55 TU/mg, were hydrolyzed andexamined on the long and short columns of a
Beckman-Spinco amino acid analyzer (Table 6).During the course of these experiments, ex-
haustive attempts were made to find a more
efficient column chromatography method ofpreparation for streptolysin 0. The acid form ofthe succinic acid ester of Solka-Floc, BW-2000,was prepared by the method of McIntire andSchenck (1948), and elution was tried with phos-phate buffer, the ammonium sulfate-cysteineeluting fluid used to elute from calcium phos-phate, and dilute ammonium hydroxide; also, a
great variety of other buffer solutions over a widerange of pH were tried. No repeated successfulrecovery was achieved. Similarly, adsorption ontoand elution from DEAE-cellulose columns, undera wide variety of conditions, was unsuccessful.
FIG. 8. Electrophoretic pattern obtained on
examination of the material having high streptolysin0 activity.
TABLE 6. Amino acid analyses of streptolysin 0
Activity at
Amino acid or compound5s TU 39 TU 36TU5TU (G) (H21)
Amoles/mg ,umoles/mg 4moles/mgAspartic acid ......... .504 .523 .453Threonine ............ .236 .205 .201Serine ................. .229 .223 .198Proline .390 .371 .456Glutamic acid ......... .763 .919 .705Glycine ............... .656 .868 .936Alanine ............... .469 .498 .452Valine ..2.1.215.215 .228 .223Half cystine .......... TR* TR .111Methionine .......... .123 .096 .107Isoleucinet ..... .163t .168 .157Leucinet .............. .330t .342 .290Tyrosine .081 .059 .077Phenylalanine ......... .170 .121 .144Lysine ................ .449 .425 .485Histidine ............. .084 .082 .146Arginine ..... .211 .253 .194Methionine sulfoxides 0 0 TRHydroxyproline ....... TR 0 TRHydroxylysine ........ .013 .031 .026Glucosamine .......... TR TR TR
* Trace.t On the basis of preliminary paper chromato-
gram analyses, these were erroneously reported asnorleucine (Pentz and Shigemura, 1955).
DISCUSSION
It is clearly realized that the preparations de-scribed here do not satisfy any of the majorcriteria of purity usually required of proteinpreparations; for example, they are not crystal-line and cannot be examined satisfactorily byeither the ultracentrifuge or by conventional elec-trophoretic analyses. However, the symmetry ofthe nicotinamide adenine dinucleotidase proteinpeak eluted from the DEAE-cellulose column wasreasonably good, and, although it was known tocontain traces of deoxyribonuclease activity,these were so small that they did not show asindividual chromatographic spots when reactedwith DNFB. It appears likely that many of theselow molecular weight products will never satisfythe rigid criteria that larger protein molecules do.In any case, it has been known for some time thateven these criteria are not absolute indications ofpurity (Kaplan, 1963).
VOL. 88, 1964 505
on May 14, 2018 by guest
http://jb.asm.org/
Dow
nloaded from

PENTZ, KOT, AND FERRETTI
The amino acid analyses for three separatepreparations of nicotinamide adenine dinucleoti-dase (Table 1) were in reasonable agreement, thesamples assaying at 12,800 and 7,260 units per mgbeing very close. One of these preparations con-tained a trace of cystine, but the other twoshowed neither cystine nor cysteic acid, and theircontent of methionine was relatively low. It wouldappear that the very low content of sulfur-con-taining amino acids offers few possibilities forsulfhydryl groups and, therefore, the opportuni-ties for cross-linking and H-bonding by thischemical group are almost negligible. Anotherstriking finding of the amino acid analyses wasthe high content of proline and glycine. These,combined with the presence of a trace of hydroxy-proline and hydroxylysine, suggest that this pep-tide, nicotinamide adenine dinucleotidase, may berelated to collagen. Further, proline, except whenpresent at the end of a peptide chain, provides noNH group to assist in either inter- or intrachainH-bonding. Therefore, some further explanationis provided for the instability of this material.Although all of the amino acids present were in
both the Todd-Hewitt broth in which the organ-ism was grown and in the controls which wereeluted from the DEAE-cellulose column, Table 2illustrates the most striking differences present.The control material, obtained by treating un-inoculated Todd-Hewitt broth with calciumphosphate and then eluting the product obtainedfrom a DEAE-cellulose column, was apparentlynot a uniform product, since the values for glu-tamic acid and proline were widely different. Sucha result is not surprising, because the Todd-Hewitt broth is made from beef heart infusionand, therefore, may vary greatly in its peptideand protein content from batch to batch. Neitheris it too surprising, in view of the plethora ofpossibility, that fractions are found which elutefrom the cellulose columns in a similar position tothe nicotinamide adenine dinucleotidase. Thedata in Table 2, considered by themselves, wouldnot be too convincing, but paper chromatographicexamination of this material after treatment withDNFB made it clear that these products are quitedissimilar from nicotinamide adenine dinucleoti-dase. Further examination of Table 2 shows that,whereas proline and glycine must be concentratedby the bacteria, glutamic acid must be reduced inrelative concentration to produce the nicotinam-ide adenine dinucleotidase.
On the basis of the analyses in Table 1, if oneassumes one residue of tyrosine and one of hy-droxylysine and calculates the relative numbersof other amino acids present, one arrives at anestimated molecular weight of approximately22,600. Because it is known that there are tracesof other material present, this figure is probablysomewhat too high; however, in view of the evi-dence presented in the sizing experiments, it isnot unreasonable. It would appear on the basisof this and the evidence from the dialysis experi-ment that the molecular weight is between 2,500and 20,000.The two DNFB-labeled amino acids, mono-
imidazole histidine and E-lysine, can only be ter-minal amino acids if their carboxyl groups arefree. The possibility that N-DNP-arginine mayoccur has not been exhaustively ruled out. Thepreliminary evidence, obtained by amino acidanalyses of the hydrolyzed, DNP-labeled product,indicated that it might be present. These prepara-tions were routinely kept in a refrigerator, becauseit is well known that continued exposure to lightpromotes their decomposition (Pollara and VonKorff, 1960). Not more than 70% should bedestroyed during the hydrolyses. However, duringthe course of the paper chromatographic examina-tion itself, light was not completely excluded fromthe chromatocab. The cabinet used had a doublewindow at one end only, which received a verysmall amount of indirect light. It is possible thatthe combination of losses from hydrolysis andlight decomposition was sufficient to obscureN-DNP-arginine. The terminal carboxyl groupsare currently being investigated. Should it turnout that no carboxyl groups are found either, thena possible relationship of this material to collagenwould be increased (Society for ExperimentalBiology Symposia, 1955).The electrophoretic experiments with the crude
streptolysin preparation are of some interest.Since a good precipitate was obtained at pH 4.5,it seems probable that this represents a true iso-electric point of some material in the mixture.However, since appreciable hemolytic activitywas obtained in the precipitate, it is not clearwhether the activity was carried out of solutionmechanically with the precipitation of extraneousmaterial or whether it represents the presence ofmore than one molecular species of this hemolyticsubstance.The component having the higher hemolytic
506 J. BACTERIOL.
on May 14, 2018 by guest
http://jb.asm.org/
Dow
nloaded from

CHARACTERISTICS OF STREPTOCOCCAL PRODUCTS
activity in these experiments was also the onewhich, under most of the conditions tested, hadvery little mobility; therefore, these measure-ments have considerably less accuracy. Theisoelectric point for this component is probablysomewhere between 5.5 and 7.0. Thus, failure toobtain a precipitate at pH 5.7 is not too surpris-ing. The combined evidence from Tables 5 and 6makes it clear that the molecule responsible forthe hemolytic activity is quite sensitive tochanges in hydrogen ion concentration. One isled to believe, also, that the substance responsiblefor the hemolytic activity here is different fromthe products of other workers, because their suc-cessful paper curtain electrophoretic preparationswere carried out a pH 7.8 (Carlson et al., 1957;Pace and Pappenheimer, 1959). In the experi-ments reported here, large amounts of hemolyticactivity were destroyed at pH 8.0, and electrc-phoretic examination was the least satisfactory.The amino acid analyses for three separate
preparations of streptolysin 0 were extremelyuniform. These analyses were characterized, also,by an extremely low content of sulfur-containingamino acids, although, when compared with thenicotinamide adenine dinulceotidase analyses, theamount of methionine in relation to glutamicacid and glycine was somewhat higher. The moststriking differences, however, between theseanalyses and those of the nicotinamide adeninedinucleotidase were the greatly reduced contentsof proline and hydroxyproline. With the wisdomof hindsight, it is evident that it would have beenvery helpful to have been able to estimate theamount of nicotinamide adenine dinucleotidasepresent in this material. However, the threepreparations were different in appearance fromthe nicotinamide adenine dinucleotidase in thatthey were light tan in color and did not have thesame fluffy character. In addition, they coagu-lated immediately in the presence of 6 N HC1,prior to hydrolyses, whereas the nicotinamideadenine dinucleotidase samples were immediatelysoluble.
ACKNOWLEDGMENTS
We wish to express our thanks to Arthur Veisfor performing the ultracentrifuge analyses andfor reading the manuscript and making helpfulsuggestions on it. Kenneth Holley merits ourthanks for many of the amino acid analyses. It is
a pleasure to thank E. Albert Zeller for helpfuladvice on the pore sizing of the Visking casing.
This investigation was supported in part byPublic Health Service grants H-3706-C5 and1-SD1-FR-05070-01, and Chicago Wesley Memo-rial Hospital Women's Service Research Fund.
LITERATURE CITED
AyouB, E. M., AND L. W. WANNAMAKER. 1963. Afactor other than streptococcal nicotinamideadenine dinucleotidase which combines withantibody to this enzyme. J. Immunol. 90:793-803.
BERNHARD, G. C., AND G. H. STOLLERMAN. 1959.Serum inhibition of streptococcal diphos-phopyridine nucleotidase in uncomplicatedstreptococcal pharyngitis and in rheumaticfever. J. Clin. Invest. 38:1942-1949.
BLOCK, R. J., E. L. DARRUM, AND G. ZWEIG. 1958.A manual of paper chromatography and paperelectrophoresis, p. 132. Academic Press Inc.,New York.
CARLSON, A. S., A. KELLNER, A. W. BERNHEIMER,AND E. B. FREEMAN. 1957. A streptococcalenzyme that acts specifically upon diphos-phopyridine nucleotide; characterization ofthe enzyme and its separation from strep-tolysin 0. J. Exptl. Med. 106:15-26.
ELLIOTT, S. D. 1950. Streptococcal proteinase andprecursor. J. Exptl. Med. 92:201-218.
FEIGL, F. 1960. Spot tests in organic analysis, p.91-92. Elsevier Publishing. Co., New York.
FRY, R. M., AND R. I. N. GREAVES. 1951. Thesurvival of bacteria during and after drying.J. Hyg. 49:220-246.
HALBERT, S. P. 1958. The use of precipitin analysisfor the study of human streptococcal infec-tions. J. Exptl. Med. 108:385-410.
HALBERT, S. P., AND T. AUERBACH. 1961. The useof precipitin analysis in agar for the study ofhuman streptococcal infections. J. Exptl.Med. 113:131-158.
HERBERT, D. 1941. A simple colorimetric methodfor the estimation of hemolysis and its applica-tion to the study of streptolysin. Biochem. J.(London) 35 (part 2):1116-1123.
KAPLAN, N. 0. 1963. Symposium on multiple formsof enzymes and control mechanisms. I. Mul-tiple forms of enzymes. Bacteriol. Rev. 27:155-169.
KAPLAN, N. O., S. P. COLOWICK, AND A. NASON.1951. Neurospora diphosphopyridine nucleo-tidase. J. Biol. Chem. 191:473-483.
KUNITZ, M. 1950. Crystalline desoxyribonuclease.J. Gen. Physiol. 33:349-362.
LOWRY, 0. H., N. J. ROSEBROUGH, A. L. FARR,
VOL. 88, 1964 507
on May 14, 2018 by guest
http://jb.asm.org/
Dow
nloaded from

508 PENTZ, KOT, AND FERRETTI
AND R. J. RANDALL. 1951. Protein measure-ment with Folin phenol reagent. J. Biol.Chem. 193:265-275.
MCINTIRE, F. C., AND J. R. SCHENCK. 1948. Poly-saccharide acid esters as cation exchangemedia. J. Am. Chem. Soc. 70:1193-1194.
PACE, M. G., ANI) A. M. PAPPENHEIMER, JR. 1959.An immunochemical study of streptococcaldiphosphopyridine nucleotidase. J. Immunol.83:83-86.
PENTZ, E. I., AND Y. SHIGEMURA. 1955. The pro-duction, concentration, and partial character-ization of streptolysin 0. J. Bacteriol. 69:210-214.
PHILPOT, J. S. L. 1938. Direct photography ofultracentrifuge sedimentation curves. Nature141:283-284.
POLLARA, B., AND R. W. VON KORFF. 1960. Thephotodecomposition of dinitrophenyl-aminoacids. Biochim. Biophys. Acta 39:364-367.
J. BACTERIOL.
RACKER, E. 1950. Crystalline alcohol dehydro-genase from bakers yeast. J. Biol. Chem. 184:313-319.
RAO, K. R., AND H. A. SOBER. 1954. Preparationand properties of 2,4-dinitrophenyl-L-aminoacids. J. Am. Chem. Soc. 76:1328-1331.
SANGER, F. 1949. Fractionation of oxidized insulin.Biochem. J. 44:12(6-128.
SLADE, H. 1., AND G. A. KNOX. 1950. Nutritionand the role of reducing agents in the forma-tion of streptolysin 0 by a group A hemoly-ticstreptococcus. J. Bacteriol. 60:301-310.
SOCIETY FOR EXPERIMENTAL BIOLOGY SYMPOSIA.1955. Fibrous proteins and their biologicalsignificance, IX, p. 14 and 18. Academic PressInc., New York.
SVENSSON, H. 1940. Theorie der Beobachtungs-methode der gekreuzten Spalte. Kolloid-Z.90 :141-156.
on May 14, 2018 by guest
http://jb.asm.org/
Dow
nloaded from