soft tissue sarcoma at the turn of the millennium nijhuis, paulus
TRANSCRIPT

University of Groningen
Soft tissue sarcoma at the turn of the millenniumNijhuis, Paulus Henricus Antonius
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2001
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Nijhuis, P. H. A. (2001). Soft tissue sarcoma at the turn of the millennium: aspects of epidemiology,cytogenetics, diagnosis and treatment s.n.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 04-04-2018

Soft tissue sarcoma at the turnof the millennium
Aspects of epidemiology, cytogenetics,
diagnosis and treatment
Paul H.A. Nijhuis

Nijhuis P.H.A.Soft tissue sarcoma at the turn of the millenniumAspects of epidemiology, cytogenetics, diagnosis and treatmentThesis University of GroningenISBN 90-367-1494-x
Cover and lay-out: Ben Mobach/AriëS Grafische vormgevingPrinting: PrintPartners Ipskamp B.V., Enschede

Rijksuniversiteit Groningen
Soft tissue sarcoma at the turnof the millennium
Aspects of epidemiology, cytogenetics,
diagnosis and treatment
Proefschrift
ter verkrijging van het doctoraat in deMedische Wetenschappen
aan de Rijksuniversiteit Groningenop gezag van de
Rector Magnificus, dr. D.F.J. Bosscher,in het openbaar te verdedigen op
maandag 29 oktober 2001om 16.00 uur
door
Paulus Henricus Antonius Nijhuis
geboren op 9 oktober 1961te Heerlen

Promotores: Prof. Dr. H.J. HoekstraProf. Dr. W.M. MolenaarProf. Dr. H. Schraffordt Koops
Beoordelingscommissie: Prof. Dr. D.Th. SleijferProf. Dr. B.G. SzabóProf. Dr. Th. Wobbes (University Medical Center Nijmegen)
ISBN 90-367-1494-x

Paranimfen: Ir. D.W.P. NijhuisDrs. S.E.M. Konijnenberg
All studies of this thesis were designed and carried out at the department of SurgicalOncology at the Groningen University Hospital, in cooperation with the departments ofPathology, Radiotherapy, Medical Oncology, Medical Genetics, and the ComprehensiveCancer Center North-Netherlands, Groningen, The Netherlands.Financial support for the publication of this thesis was kindly provided by Schering-Plough BV and Johnson & Johnson Medical BV.


Ter herinnering aan mijn moederVoor mijn vader
Voor Rian, Amanda en Paul jr.

8
Contentspage
Chapter 1 Introduction and aim of the thesis 9
Chapter 2 Epidemiological aspects of soft tissue sarcomas (STS)-Consequences for the design of clinical STS trials 19
Chapter 3 Long-term results of preoperative intraarterial doxorubicincombined with neoadjuvant radiotherapy, followed by extensivesurgical resection for locally advanced soft tissue sarcomas of theextremities 29
Chapter 4 Clinico-pathological data and prognostic factors in completelyresected AJCC stage I-III liposarcomas 37
Chapter 5 Soft tissue sarcoma- Compliance with guidelines 53
Chapter 6 Prognostic relevance of cytogenetic changes in soft tissue sarcomas 67
Chapter 7 Soft tissue sarcoma: where to go? 81
Chapter 8 Summary 97
Chapter 9 Samenvatting 105
Chapter 10 Dankwoord 113

9
Chapter 1
Introduction and aim of the thesis

10 Introduction and aim of the thesis Chapter 1
Introduction
Soft tissue sarcomas (STS) can be defined as malignant tumors arising from the non-epithelial extra-skeletal tissue of the body, exclusive of the reticulo-endothelial systemand glia. Embryologically, these tumors are derived principally from mesoderm, exceptsome, which derive from the neuroectoderm [1]. The pathogenesis of most STS remainsunknown. Genetic factors (neurofibromatosis I and II, retinoblastoma, Li-Fraumeni syn-drome, Gardner�s syndrome), environmental factors (exposure to environmental car-cinogens as dioxin and some herbicides, and trauma, injury or ionizing radiation in thepast), and immunological factors (immunosuppression after transplant surgery) havebeen identified as etiological factors in STS development [1-11]. As in most other malig-nancies, it is very unlikely that only one of these various factors is causing the disease;a multifactorial etiology seems obvious.
STS are rare tumors, accounting for approximately 1% of all malignant tumors diag-nosed annually (8100 patients in the United States and 422 in the Netherlands) [12,13].There is a slight male preponderance and the incidence is increasing with age [14,15,16].As prognosis varies between the different histological types and even between subtypes,an adequate histopathological classification is crucial. The most recent classification ofsoft tissue tumors is the World Health Organization histological classification (1994),dividing these tumors into 15 categories [17].
Figure 1 presents the distribution of STS according to anatomical site in patients olderthan 16 years who were treated at the Memorial Sloan-Kettering Cancer Center (MSKCC)from 1982-1990. It should be mentioned that patients with visceral and genitourinarySTS were included in that series. Figure 2 shows the distribution according to histo-pathology in the MSKCC series. In accordance with other reports, the most commonhistopathogical types were liposarcoma (LPS), malignant fibrous histiocytoma (MFH),and leiomyosarcoma [15,18,19,20]. Approximately half of the STS occurred in the ex-tremities (Figure 1), where liposarcoma and MFH were most common [19]. In theretroperitoneum and visceral tissues, however, leiomyosarcoma predominated [19].
Figure 1.Distribution of soft tissue sarcomas according to histopathology (Brennan, Ann Surg 1991; 214: 328-336).

11
Unfortunately, there are only relatively few studies on epidemiological aspects of STS,and most of them are center-based. In the northeastern part of Netherlands, all malig-nancies, sarcomas included, are registered by the cancer registry of the ComprehensiveCancer Center North-Netherlands (CCCN), which is population-based, thus having a majoradvantage of avoiding selection bias caused by referral pattern. Collecting and studyingdata from such a cancer registry may provide more insight into STS epidemiology.
During the last decennia, many prognostic clinical factors have been identified in STS[21-39]. One of the most potent factors determining outcome seems to be the histo-pathological (sub)type of the tumor, which may become indiscernible if prognosticallyfavorable tumor (sub)types are reviewed together with less favorable (sub)types, as inmost reports on STS. Liposarcoma, as a group, has a better prognosis than epithelioidsarcoma [21-28], which in turn has a better prognosis than synovial sarcoma [29]. Buteven in the group of liposarcomas prognosis largely differs between the varioushistopathological subtypes [21-24].Besides histological (sub)type, other prognostic characteristics, as age at presentation,gender, tumor size, anatomical site, tumor depth, surgical margin, tumor grade, tumornecrosis, and vascular invasion have been reported [30-39], and the most important oneshave been embedded in the new staging system of the American Joint Committee onCancer (AJCC) (Table 1) [40]. As liposarcoma is one of the most common STS in whichseveral histopathological subtypes can be distinguished, this tumor is particularly suit-able to study prognostic characteristics.In recent years, the interest in the genetic etiology of malignancies has been increasingrapidly, and significant progress has been made in identifying chromosomal abnormali-ties in solid tumors. In several STS, characteristic cytogenetic alterations have been found,which often have diagnostic relevance [1,41-45]. As in other solid neoplasms, as well as insome hematological malignancies, it seems likely that some of these alterations haveprognostic importance in STS, although, at present, such a relation between (specific)cytogenetic alterations and prognosis and survival in STS could not be demonstratedunequivocally [46,47].
Figure 2.Distribution of soft tissue sarcomas according to anatomical site(Brennan, Ann Surg 1991; 214: 328-336).
Chapter 1 Introduction and aim of the thesis

12
Advances in technology have resulted in an important progress in diagnostic tools inSTS. The introduction of the computer tomography scan (CT-scan) caused a major break-through in radiodiagnostics because, for the first time, it was possible to visualize thesoft tissues directly using X-rays [48]. Notwithstanding its possibilities, this techniquealso had its shortcomings (the use of X-rays, direct images limited to only two plains,only information on anatomy and not on metabolism). The development of the mag-netic resonance imaging (MRI) and the currently available spiral CT-scan overcame thefirst two shortcomings of the CT-scan [49,50], but still could not cope with the third one.The positron emission tomography scan (PET-scan) and the single photon emission to-mography (SPECT) made it possible to study the metabolism of tumors [51,52].As with all new technology-driven instruments, these challenging techniques can easilybe used inappropriately, resulting in significant health-care costs [53]. Therefore, thedevelopment of specific diagnostic guidelines is important, especially in rare tumors, asclinical and histopathological presentation widely varies, the experience in individualhospitals is limited, and treatment often is very complex, including many modalities.
In February 1983, a cooperative group for rare tumors, consisting of specialists in surgi-cal oncology, medical oncology, radiotherapeutic oncology and pathology from varioushospitals in the CCCN-region, recognized this problem and developed regional guide-
Introduction and aim of the thesis Chapter 1
Table 1. The American Joint Committee on Cancer (AJCC) soft tissue sarcoma stagingsystem.*
Primary Tumor (T)
TX Primary tumor cannot be assessedT0 No evidence of primary tumorT1 Tumor 5 cm or less in greatest dimension
T1a superficial tumorT1b deep tumor
T2 Tumor more than 5 cm in greatestdimension
T2a superficial tumorT2b deep tumor
Regional Lymph Nodes (N)
NX Regional lymph nodes cannot beassessed
N0 No regional lymph node metastasisN1 Regional lymph node metastasis
Distant metastasis (M)
MX Distant metastasis cannot be assessedM0 No distant metastasis
Histopathologic grade
GX Grade cannot be assessedG1 Well differentiatedG2 Moderately differentiatedG3 Poorly differentiatedG4 Undifferentiated
Stage grouping
Stage IA G1-2, T1a-1b, N0, M0Stage IB G1-2, T2a, N0, M0Stage IIA G1-2, T2b, N0, M0Stage IIB G3-4, T1a-1b, N0, M0Stage IIC G3-4, T2a, N0, M0Stage III G3-4, T2b, N0, M0Stage IV Any G, any T, N1, M0
Any G, any T, N0, M1
* Fleming ID, Cooper JS, Henson DE, et al, Eds.Chapter 22. Soft Tissue Sarcoma. In: AJCC Can-cer Staging Manual. 5th Ed. Philadelphia,Lippincott-Raven Publishers. 1997:149-156

13
lines for the diagnosis and treatment of soft tissue tumors [54]. A few years later, theDutch Sarcoma Group initiated national guidelines for STS diagnosis and treatment[55]. Although compliance with such guidelines is important for various reasons (e.g.appropriateness of practice, health-care savings, and better outcome and survival [53,56-58]), and despite an increase in medical practice guideline development and dissemina-tion, compliance with such guidelines has often been surprisingly low. In malignantdisorders, reports on the adherence to such guidelines are very limited, whereas in softtissue tumors, nothing has been published on this item. Nevertheless, such informationseems very valuable for future guideline development and introduction.
Treatment of STS has changed dramatically during the second half of the last century.Prior to the 1950s and 1960s, most surgeons dealing with STS performed local resec-tions or shell-out procedures, which were associated with an unacceptable high localrecurrence rate of 60-95% [59-63]. In the same period, Bowden and Booher reportedlimb-saving techniques with a low local recurrence risk [64]. In the same year, 1958, thegroup of Stener published the same surgical principles and results [65,66]. Both groupsmight be considered true pioneers in modern STS treatment, because both recognizedthe infiltrative manner of sarcoma growth, explaining the high local recurrence rate aftershell-out procedures. Later, this high local recurrence rate was further explained byEnneking�s theory of sarcoma tumor growth pattern [67]. STS grow in a centrifugal fash-ion, resulting in the formation of an edematous pseudocapsule of compressed, normaltissue and a reactive zone of proliferating mesenchymal cells and neovascularization.Further tumor growth causes a continuous extent of microscopic tumor pseudopodsinto this pseudocapsule, where they form microscopic and macroscopic nodules (satel-lites). Especially in high-grade lesions, such satellites can also be found in surrounding�normal� tissue, far beyond the pseudocapsule (skip metastases). In extremities, wheremost of the STS are located, the tumor extends longitudinally, within the compartment,bounded by fibrous barriers (muscle fascia and aponeurosis, deep fascia and intermus-cular septae). Crossing these barriers is a late phenomenon, and is associated with high-grade lesions [67,68].
Based on these new insights, en-block resections of the entire compartment containingthe tumor or amputations were recommended, resulting not only in a drop of local re-currence rate to 5-30%, but also in a high amputation rate of 40-50% [60,61,68-70].Although these so-called �compartment resections� were widely adopted, the division ofsurgical oncology of the Groningen University Hospital performed only wide local resec-tions followed by external beam radiotherapy (EBRT) or amputation of the affected limb.
Modern STS management started at the end of the seventies and the beginning of theeighties. Suit and Lindberg were the first to demonstrate the importance of adjuvantradiotherapy in STS therapy [71,72]. Their results formed the basis of the famous Na-tional Cancer Institute (NCI) trial by Steven Rosenberg et al., a study that became one ofthe cornerstones in today�s state of the art STS treatment [70]. For the first time, a pro-spective randomized study showed that adequately performed local resection followedby high dose radiotherapy formed a reliable limb-saving treatment, comparable to am-putation with regard to local recurrence, disease-free and overall survival rates.
Chapter 1 Introduction and aim of the thesis

14
Unfortunately, the optimal treatment for locally advanced extremity sarcomas remainedan unsolved problem. One of the most promising approaches at that time was a multi-modality therapy, consisting of preoperative (intraarterial) chemotherapy, immediatelyfollowed by EBRT, and surgical resection. This technique was initiated by Morton andEilber [73], and later further developed by Eilber and co-workers at the UCLA School ofMedicine [74,75]. The sequence of the various therapies was based on the premise thatpreoperative treatment of micrometastases at the periphery of the tumor with intact bloodsupply would enable the surgeon to perform a local surgical procedure. In three sequentialtrials, Eilber et al. showed a high limb salvage rate of 95%, with a low local recurrencerate of approximately 10% [75]. In the early eighties, this treatment strategy was adoptedby the Groningen Sarcoma Working Party, which added postoperative EBRT to thetreatment protocol in case of a marginal resection or involvement of the surgical margin[76]. The multimodality treatment, as initiated by Morton and Eilber, has been associatedwith a substantial short-term morbidity, especially wound complications [75]. Althoughthere has been a growing awareness of potential long-term side-effects of intensified(multimodality) cancer treatment protocols, only very few reports have dealt with thelong-term complications and functional outcome after this intensive STS treatment [75,77-79].
Another way to decrease the number of amputations in locally advanced extremity STShas been hyperthermic isolated limb perfusion (HILP) with cytostatics agents. Severaldrugs have been used (melphalan, the standard drug for HILP in melanoma, doxorubicin,cisplatinum, and other agents), without improvement of local control or disease-freesurvival when compared to the other therapies, as intravenous or intraarterial adriamycinin combination with neoadjuvant radiotherapy followed by local resection [75,80-83].It was not until the early nineties, that significant progress was made by the addition oftumor necrosis factor-alpha (TNF-α) to melphalan in HILP for locally advanced extrem-ity STS [84]. This resulted in a high response rate and high limb salvage rates with anacceptable toxicity level [85]. In the early nineties, the Groningen Sarcoma Working Partychanged the treatment of locally advanced extremity STS in HILP with TNF-α, melphalan,with or without interferon-gamma (IFN-γ), with good results [85]. In 1998, this sarcomagroup published the results of a study on adjuvant EBRT (60-70 Gy) after HILP withmelphalan, TNF-α, and IFN-γ and delayed tumor resection of locally advanced extremitySTS with histopathological viable tumor after resection [86]. It was demonstrated thatthis was feasible and that the addition of EBRT increased local tumor control withoutincreasing treatment morbidity. Recently, Ham et al. highlighted several aspects of mod-ern surgical sarcoma treatment at the Groningen University Hospital [87].
Since the mid eighties, the cancer registry of the CCCN has registered all STS in theCCCN-region, and the Groningen Sarcoma Working Party discusses all sarcomas re-ferred to the Groningen University Hospital. In recent years, many aspects of these tumorshave been studied and reported by this sarcoma group, resulting in several theses [88-90]. Still a variety of questions remain unanswered. The goal of the present thesis is toget more insight into several aspects of this uncommon malignancy.
Introduction and aim of the thesis Chapter 1

15
References1. Enzinger FM, Weiss SW. General considerations. In: Enzinger FM, Weiss SW (Eds). Soft tissue tumors.
Third edition. St. Louis, Mosby, 1995: 1-16.2. Wiklund TA, Blomqvist CP, Raty J, et al. Postirradiation sarcoma: analysis of nationwide cancer registry
material. Cancer 1991; 68: 524-531.3. Mark RJ, Poen J,Tran LM, Fu YS, Selch MT, Parker RG. Postirradiation sarcoma: a single institution study and
a review of the literature. Cancer 1994; 73: 2653-2662.4. Zahm SH, Fraumeni JF, Jr. The epidemiology of soft tissue sarcoma. Semin Oncol 1997; 24: 504-514.5. Hardell L, Eriksson M. The association between soft tissue sarcomas and exposure to phenoxyacetic acids: A new
case-referent study. Cancer 1988; 62: 652-656.6. Kang H, Enzinger FM, Breslin P, et al. Soft tissue sarcoma and military service in Vietnam: a case-control
study. J Natl Cancer Inst 1987; 79: 693-699.7. Fingerhut MA, Halperin WE, Marlow DA, et al. Cancer mortality in workers exposed to 2,3,7,
8-tetrachlorodibenzo-p-dioxin. N Eng J Med 1991; 324: 212-218.8. Saracci R, Kogevinas M, Bertazzi PA, et al. Cancer mortality in workers exposed to chlorophenoxy herbicides
and chlorophenols. Lancet 1991; 338: 1027-1032.9. Eriksson M, Hardell L, Adami HO. Exposure to dioxins as a risk factor for soft tissue sarcoma: a population-
based case-control study. J Natl Cancer Inst 1990; 82: 486-490.10. McClay EF. Epidemiology of bone and soft tissue sarcomas. Semin Oncol 1989; 16: 264-272.11. Malkin D, Li F, Strong L, et al. Germ line P53 mutations in a familial syndrome of breast cancer, sarcomas, and
other neoplasms. Science 1990; 250: 1233-1238.12. Greenlee RT, Murray T, Bolden S, Wingo PA. Cancer statistics, 2000. CaCancer J Clin 2000; 50: 7-33.13. Incidence of cancer in the Netherlands 1996. Visser O, Coebergh JWW, Schouten LJ, Dijck JAAM (Eds). Utrecht,
Vereniging van Integrale Kankercentra 2000.14. Gustafson P. Soft tissue sarcoma. Epidemiology and prognosis in 508 patients. Acta Orthop Scand 1994,
65 (suppl 259), 1- 31.15. Harris M, Hartley AL, Blair V, et al. Sarcomas in North West England. I. Histopathological peer review.
Br J Cancer 1991, 64, 315-320.16. Jane MJ, Hughes PJ. Disease incidence and results of extremity lesion treatment: Mersey Region soft tissue
sarcomas (1975-1985). Sarcoma 1998, 2, 89-96.17. Weiss SW. Histopathological typing of soft-tissue tumors, second edition. (WHO international histopathological
classification of tumors). Berlin, Springer Verlag, 1994.18. Torosian MH, Friedrich C, Godbold J, Hajdu SI, Brennan F. Soft-tissue sarcoma: Initial characteristics and
prognostic factors in patients with and without metastatic disease. Sem Surg Oncol 1988, 4, 13-19.
Chapter 1 Introduction and aim of the thesis
Aim of the thesis
1. To provide more insight into epidemiological aspects of STS, based on a population-basedsurvey.
2. To study the impact of the histopathological heterogeneity on prognosis in one of the mostcommon STS, liposarcoma.
3. To evaluate the necessity of long-term follow-up, especially in case of intensive, multimodalitytreatment protocols, in order to determine long-term effects, which might interfere with theprimary goal of such therapies.
4. To investigate the adherence to (diagnostic) STS guidelines, and to evaluate the role ofcentralization in the diagnostic management of these rare tumors.
5. To look into the (near) future of STS treatment, and to evaluate the prognostic importanceof cytogenetic changes in these tumors.

16
19. Brennan MF, Casper ES, Harrison LB, et al. The role of multimodality therapy in soft-tissue sarcoma. Ann Surg1991; 214: 328-336.
20. Pollock RE, Karnell LH, Menck HR, Winchester DP. The National Cancer Data Base Report on soft tissuesarcoma. Cancer 1996, 78, 2247-2257.
21. Chang HR, Hajdu SI, Collin C, Brennan MF. The prognostic value of histologic subtypes in primary extremityliposarcoma. Cancer 1989; 64: 1514-1520.
22. Chang HR, Gaynor J, Tan C, Hajdu SI, Brennan MF. Multifactorial analysis of survival in primary extremityliposarcoma. World J Surg. 1990; 14: 610-618.
23. Zagars GK, Goswitz MS, Pollack A. Liposarcoma: outcome and prognostic factors following conservationsurgery and radiation therapy. Int J Rad Oncol Biol Phys 1996; 36: 311-319.
24. Pearlstone DB, Pisters PWT, Bold RJ, et al. Patterns of recurrence in extremity liposarcoma. Implications forstaging and follow-up. Cancer 1999; 85: 85-92.
25. Linehan DC, Lewis JJ, Leung D, Brennan MF. Influence of biologic factors and anatomic site in completelyresected liposarcoma. J Clin Oncol 2000; 18:1637-1643.
26. Enzinger FM, Weiss SW. Synovial sarcoma. In: Enzinger FM, Weiss SW (Eds). Soft tissue tumors. Thirdedition. St. Louis, Mosby, 1995: 757-785.
27. Heide HJL van der, Veth RPH, Pruszczynski M, Wobbes T, Hoesel QGCM van, Lemmens JAM. Synovialsarcoma: oncological and functional results. Eur J Surg Oncol 1998; 24: 114-119.
28. Lewis JJ, Antonescu CR, Leung DHY, Blumberg D, Healey JH, Woodruff JM, Brennan MF. Synovial sarcoma:A multivariate analysis of prognostic factors in 112 patients with primary localized tumors of the extremity.J Clin Oncol 2000; 18: 2087-2094.
29. Spillane AJ, Thomas JM, Fisher C. Epithelioid sarcoma: the clinicopathological complexities of this rare softtissue sarcoma. Ann Surg Oncol 2000; 7: 218-225.
30. Markhede G, Angervall L, Stener B. A multivariate analysis of the prognosis after treatment of soft-tissue tumors.Cancer 1982; 49: 1721-1733.
31. Reitan JB, Kaalhus O, Brennhovd IO, Sager EM, Stenwig AE, Talle K. Prognostic factors in liposarcoma.Cancer 1985; 55:2482-2490.
32. Potter DA, Kinsella T, Glatstein E, et al. High-grade soft tissue sarcomas of the extremities. Cancer 1986;58: 190-205.
33. Lawrence W Jr., Donegan WL, Natarajan N, Mettlin C, Beart R, Winchester D. Adult soft tissue sarcomas. AnnSurg 1987; 205: 349-359.
34. Singer S, Corson JM, Gonin R, Labow B, Eberlein TJ. Prognostic factors predictive of survival and localrecurrence for extremity soft tissue sarcoma. Ann Surg 1994; 219: 165-173.
35. Coindre JM, Terrier P, Bui N, et al. Prognostic factors in adult patients with locally controlled soft tissuesarcoma: A study of 546 patients from the French Federation of Cancer Centers Sarcoma Group. J Clin Oncol1996; 14: 869-877.
36. Heslin MJ, Woodruff J, Brennan MF. Prognostic significance of a positive microscopic margin in high-riskextremity soft tissue sarcoma: Implications for management. J Clin Oncol 1996; 14: 473-478.
37. Pisters PW, Leung DH, Woodruff J, Shi W, Brennan MF. Analysis of prognostic factors in 1.041 patients withlocalized soft tissue sarcomas of the extremities. J Clin Oncol 1996; 14: 1679-1689.
38. Rydholm A. Prognostic factors in soft tissue sarcoma. Acta Orthop Scand 1997; 68 [suppl 273]: 148-155.39. Brooks AD, Heslin MJ, Leung DH, Lewis JJ, Brennan MF. Superficial extremity soft tissue sarcoma: an
analysis of prognostic factors. Ann Surg Oncol 1998; 5: 41-47.40. Fleming ID, Cooper JS, Henson DE, et al, Eds. Chapter 22. Soft Tissue Sarcoma. In: AJCC Cancer Staging
Manual. 5th Ed. Philadelphia, Lippincott-Raven Publishers. 1997:149-156.41. Molenaar WM, De Jong B, Buist J, Idenburg VJS, Seruca R, Vos AM, Hoekstra HJ. Chromosomal analysis and
the classification of soft tissue sarcomas. Lab. Invest 1989; 60: 266-274.42. Fletcher JA, Kozakewich HP, Hoffer FA, Lage JM, Weidner N, Tepper R, Pinkus GS, Morton CC, Corson JM.
Diagnostic relevance of clonal cytogenetic aberrations in malignant soft-tissue tumors. N Engl J Med 1991;324: 436-442.
43. Sreekantaiah C, Ladanyi M, Rodriguez E, Chaganti RSK. Chromosomal aberrations in soft tissue tumors:Relevance to diagnosis, classification, and molecular mechanisms. Am J Pathol 1994; 144: 1121-1134.
44. Fletcher CDM, Akerman M, Dal Cin P, et al. Correlation between clinicopathological features and karyotype inlipomatous tumors. A report of 178 cases from the Chromosomes and Morphology (CHAMP) CollaborativeStudy Group. Am J Pathol 1996; 148: 623-630.
Introduction and aim of the thesis Chapter 1

17
45. Plaat BEC, Hollema H, Molenaar WM, et al. Soft tissue leiomyosarcomas and malignant gastrointestinalstromal tumors: differences in clinical outcome and expression of multidrug resistance proteins. J Clin Oncol2000; 18: 3211-3220.
46. Choong PF, Mandahl N, Mertens F, et al. 19p+ marker chromosome correlates with relapse in malignant fibroushistiocytoma. Genes Chromosom Cancer 1996; 16: 88-93.
47. Plaat BEC, Muntinghe FLH, Molenaar WM, et al. Clinical outcome of patients with previously untreated softtissue sarcomas in relation to tumor grade, DNA ploidy and karyotype. Int J Cancer 1997; 74: 396-402.
48. Hounsfield GN. Computerized transverse axial scanning (tomography): Part I. Description of system. Br JRadiol 1973; 46: 1016-1022.
49. Budinger TF, Lauterbur PC. Nuclear Magnetic Resonance technology for medical studies. Science 1984;226: 288-298.
50. Hogeboom WR, Hoekstra HJ, Mooyaart EL, Freling NJ, Schraffordt Koops H. MRI and CT in the preoperativeevaluation of soft tissue tumors. Arch Orthop Trauma Surg 1991; 110: 162-164.
51. Kole AC, Nieweg OE, Ginkel RJ van, et al. PET with L-[1-Carbon-11]-tyrosine for the visualization of tumors andmeasurement of the protein synthesis rate. J Nucl Med 1997; 38: 191-195.
52. Kole AC, Nieweg OE, Ginkel RJ van, et al. Detection of local recurrence of soft tissue sarcoma with positronemission tomography using 18F-fluorodeoxy-glucose. Ann Surg Oncol 1997; 4: 57-63.
53. Walsh GL, Winn RJ. Baseline institutional compliance with NCCN guidelines: non-small-cell lung cancer. Oncology (Huntingt) 1997; 11: 161-170.
54. Richtlijnen voor diagnostiek en behandeling van premaligne en maligne aandoeningen in de IKN-regio 1992. R.Otter (Ed). Medische Advies Raad IKN. ISBN 90-74114-03-2. 394-399.
55. Geel AN van, Unnik JAM van, Keus RB. Diagnosis and treatment of soft tissue tumours: the Dutch nationwide-accepted consensus. Sarcoma 1998; 2: 183-191.
56. Grimshaw JM, Russell IT. Effect of clinical guidelines on medical practice: a systematic review of rigorousevaluations. Lancet 1993; 342: 1317-1322.
57. Wolfe CDA, Tilling K, Bourne HM, Raju KS. Variations in the screening history and appropriateness ofmanagement of cervical cancer in south east England. Eur J Cancer 1996; 32A: 1198-1204.
58. Tilling K, Wolfe CDA, Raju KS. Variations in the management and survival of women with endometrial cancerin south east England. Eur J Gynaec Oncol 1998; 19: 64-68.
59. Cantin J, McNeer GP, Chu FC, Booker RJ. The problem of local recurrence after treatment of soft tissuesarcoma. Ann Surg 1968; 168: 47-53.
60. Shiu MH, Castro EB, Hajdu SI, Fortner JC. Surgical treatment of 297 soft tissue sarcomas of the lowerextremity. Ann Surg 1975; 182: 597-602.
61. Simon MA, Enneking WF. The management of soft-tissue sarcomas of the extremities. J Bone Joint Surg1976; 58A: 317-327.
62. Abbas JS, Holyoko ED, Moore R, Karakousis CP. The surgical treatment and outcome of soft tissuesarcoma. Arch Surg 1981; 116: 765-769.
63. Markhede G, Angervall L, Stener B. A multivariate analysis of the prognosis after surgical treatment ofmalignant soft tissue tumors. Cancer 1982; 49: 1721-1733.
64. Bowden L, Booher RJ. The principles and technique of resection of soft parts for sarcoma. Surgery 1958; 44:963-977.
65. Stener B, Stener I. Malignant tumors of the soft tissues of the thigh. Acta Chir Scand 1958; 115: 457-475.66. Berlin Ö, Stener B, Angervall L, Kindblom LG, Markhede G, Oden A. Surgery for soft tissue sarcoma in the
extremities. A multivariate analysis of the 6-26-years prognosis in 137 patients. Acta Orthop Scand 1990;61: 475-486.
67. Enneking WF, Spanier SS, Malawer MM. The effect of the anatomic setting on the results of surgical proceduresfor soft parts sarcoma of the thigh. Cancer 1981; 47: 1005-1022.
68. Rydholm A. Surgical margins for soft tissue sarcomas. Acta Orthop Scand (suppl 273) 1997; 68: 81-85.69. Eilber FR, Mirra JJ, Grant TT, Weisenburger T, Morton DL. Is amputation necessary for sarcomas?
A seven-years experience with limb-salvage. Ann Surg 1980; 192: 431-437.70. Rosenberg SA, Tepper J, Glatstein E, et al. The treatment of soft tissue sarcomas of the extremities.
Prospective randomized evaluation of (1) limb-sparing surgery plus radiation therapy compared withamputation, and (2) the role of adjuvant chemotherapy. Ann Surg 1982; 96: 305-315.
71. Suit HD, Russell WO, Martin RG. Sarcoma of soft tissue: clinical and histopathologic parameters and responseto treatment. Cancer 1975; 35: 1478-1483.
Chapter 1 Introduction and aim of the thesis

18
72. Lindberg RD, Martin RG, Romsdahl MM. Surgery and postoperative radiotherapy in the treatment of soft tissuesarcomas in adults. Ther Nucl Med 1975; 123: 123-129.
73. Morton DL, Eilber FR, Townsend CM, Grant TT, Mirra J, Weisenburger TH. Limb salvage from amultidisciplinary treatment approach for skeletal and soft tissue sarcomas of the extremity. Ann Surg 1976;184: 268-278.
74. Eilber FR, Giuliano AE, Huth JF, et al. A randomized prospective trial using postoperative adjuvantchemotherapy (Adriamycin) in high grade extremity soft tissue sarcoma. Ann J Clin Oncol 1988; 11: 39-45.
75. Eilber FR, Eckhardt JJ, Rosen G, Fu YS, Seeger LL, Selch MT. Neoadjuvant chemotherapy and radiotherapy inthe multidisciplinary management of soft tissue sarcomas of the extremity. Surg Oncol Clin N Am 1993; 2:611-620.
76. Hoekstra HJ, Schraffordt Koops H, Molenaar WM, Mehta DM, Sleijfer DTh, Dijkhuis G, Oldhoff J.A combination of intraarterial chemotherapy, preoperative and postoperative radiotherapy, and surgery aslimb-salving treatment of primarily unresectable high-grade soft tissue sarcomas of the extremities. Cancer1989; 63: 59-62.
77. Jentzsch K, Binder H, Cramer H, et al. Leg function after radiotherapy for Ewing�s sarcoma. Cancer 1981; 47:1267-1278.
78. Brown AP, Fixen JA, Plowman PN. Local control of Ewing�s sarcoma: An analysis of 67 patients. Br J Radiol1987; 60: 261-268.
79. Stinson SF, DeLaney TF, Greenberg J, et al. Acute and long-term effects on limb function of combinedmodality limb-sparing therapy for extremity soft tissue sarcoma. Int J Radiat Oncol Biol Phys 1991; 21:1492-1499.
80. Krementz ET, Carter RD, Sutherland CM, Hutton I. Chemotherapy of sarcomas of the limbs by regional perfusion.Ann Surg 1977; 185: 555-564.
81. Hoekstra HJ, Schraffordt Koops H, Molenaar WM, Oldhoff J. Results of isolated regional perfusion in thetreatment of malignant soft tissue tumours of the extremities. Cancer 1987; 60: 1703-1707.
82. Klaase JM, Kroon BBR, Benckhuijsen C, Van Geel AN, Albus-Lutter ChE, Wieberdink J. Results of regionalisolated perfusion with cytostatics in patients with soft tissue tumors of the extremities. Cancer 1989; 64: 616-621.
83. Ginkel RJ van, Schraffordt Koops H, Vries EGE de, Molenaar WM, Uges DR, Hoekstra HJ. Hyperthermicisolated limb perfusion with cisplatin in four patients with sarcomas of soft tissue and bone. Eur J Surg 1996; 22:528-531.
84. Lienard D, Delmotte JJ, Renard N, Ewalenko P, Lejeune FJ. High-dose recombinant tumour necrosis factor-alpha in combination with interferon gamma and melphalan in isolation perfusion of the limbs for melanomaand sarcoma. J Clin Oncol 1992; 10: 52-60.
85. Eggermont AMM, Schraffordt Koops H, Lienard D, Kroon BBR, Van Geel AN, Hoekstra HJ, Lejeune FJ.Isolated limb perfusion with high dose tumor necrosis factor-a in combination with interferon-? and melphalanfor nonresectable extremity soft tissue sarcomas: a multicenter trial. J Clin Oncol 1996; 14: 2653-2665.
86. Olieman AFT, Pras E, Ginkel RJ van, Molenaar WM, Schraffordt Koops H, Hoekstra HJ. Feasibility andefficacy of external beam radiotherapy after hyperthermic isolated limb perfusion with TNF-α and melphalanfor limb-saving treatment in locally advanced extremity soft-tissue sarcoma. Int J Rad Oncol Biol Phys1998; 40: 807-814.
87. Ham SJ, Graaf WTA van der, Pras E, Molenaar WM, Berg E van den, Hoekstra HJ. Soft tissue sarcomasof the extremities. A multi-modality diagnostic and therapeutic approach. Cancer Treatment Reviews 1998,24, 373-391.
88. Olieman AFT. Hyperthermic isolated limb perfusion: aspects of morbidity and efficacy. Thesis UniversityGroningen, The Netherlands, Enschede, ISBN 90-367-0986-5.
89. Ham SJ. Current concepts and surgical aspects of extremity bone and soft tissue sarcoma. Thesis UniversityGroningen, The Netherlands, Wageningen, ISBN 90-367-1069-3.
90. Plaat BEC. Soft tissue sarcomas: histopathology and cytogenetics in relation to diagnosis, treatment and clinicaloutcome. Thesis University Groningen, The Netherlands, Enschede, ISBN 90-367-1165-7; 1: 41-54.
Introduction and aim of the thesis Chapter 1

19
Chapter 2
Epidemiological aspects of soft tissue sarcomas (sts)-Consequences for the design of clinical sts trials
Nijhuis P.H.A.1, Schaapveld M.2, Otter R.2, Molenaar W.M.3, van der Graaf W.T.A.4, Hoekstra H.J.1
1. Department of Surgical Oncology, Groningen University Hospital, Groningen, The Netherlands 2. Comprehen-
sive Cancer Center North-Netherlands (CCCN), Groningen, The Netherlands 3. Department of Pathology,
Groningen University Hospital, Groningen, The Netherlands 4. Department of Medical Oncology, Groningen
University Hospital, Groningen, The Netherlands
European Journal of Cancer 1999; 35: 1705-1710

20
Introduction
Soft tissue sarcomas (STS) are relatively rare tumors of different mesenchymal deriva-tion, which account for less than 1% of all cancers in adults, and for approximately 7% ofall childhood malignancies [1-3]. In adults the most common histological types are li-posarcomas (21%), malignant fibrous histiocytomas (MFH) (20%), leiomyosarcomas(20%), fibrosarcomas (11%), and tendosynovial sarcomas (10%) [4]. In children, 70% ofall STS are rhabdomyosarcomas, and in 20% of the remaining non-rhabdomyosarcomasthe histological subtype remains unclassified [2,3].Tumor size, histology, primary site, grade, and the presence of metastatic disease appearto be the most important prognostic factors in the treatment of STS [4]. Only a fewdescriptive epidemiological studies on STS have been published in recent years [4-10].
Patients and methods
For a 6-years period (1989-1995), data on the incidence of primary STS were derivedfrom the population-based cancer registry of the Comprehensive Cancer Center North-Netherlands (CCCN), which covers an area of 2.1 million inhabitants. Cancer registrationat the CCCN started in 1986, but a full coverage of the whole area encompassed by theCCCN was only achieved from 1 January 1989. The main sources for the CCCN cancerregistry are the computerised national pathology databank (PALGA) and the hospitaldischarge databank to which all Dutch hospitals provide information annually on dischargediagnoses of admitted patients. Specially trained CCCN employees prospectively registerthe data from the patients� clinical records. Completeness of records, data consistencyand the possibility of duplicate records are continuously and extensively checked. Basedon the outcomes of studies on the completeness of the cancer registry databanks in otherregional cancer registries in The Netherlands, which operate on the same basis, the overallcompleteness of the cancer registry databank is estimated around 95% [11,12].In this study, information on primary STS was used, with the exception of Kaposi sarco-mas, and urogenital and gastrointestinal STS, thus encompassing ICD-O codes 171.0-171.9, 173.0-173.9, 174.0-174.9, and 158.0 for topography and ICD-O codes 8800-8933,8963, 8990-8991, 9020-9044, 9120-9134, 9141-9340 and 9540-9581 for morphology.For calculation of crude and age-specific incidence rates, the population structure of theCCCN area on 1 January 1992 was used. Age-adjusted rates were calculated using theEuropean Standard Population [13]. Differences in histology, tumor size, number of pa-tients presenting with regional or distant metastasis at time of diagnosis and primarytreatment received were analysed according to anatomical subsite and/or gender usingthe Chi-square test.Tumors were staged according to UICC Tumour Node Metastasis Classification [14]. Fora pathological TNM classification, histologic confirmation was required. For clinical TNMclassification, a set of minimal requirements was used to determine a clinical tumor(cT), clinical node (cN) and clinical metastases (cM). For cT and cN, physical examina-tion, ultrasonography, computed tomography (CT)-scan or magnetic resonance imaging(MRI) was required. To determine cM, physical examination, plain chest X-ray and deter-mination of liver function tests were minimal requirements.Where no TNM classification was available (STS of skin and breast) or TNM classifica-
Epidemiological aspects of soft tissue sarcomas Chapter 2
21
tion was incomplete, the extent of disease (EoD), from clinical and/or pathological infor-mation from the patients� medical records, was used.Surgery, as described below, included different types of primary tumor resection, butexcluded incisional biopsy.
Results
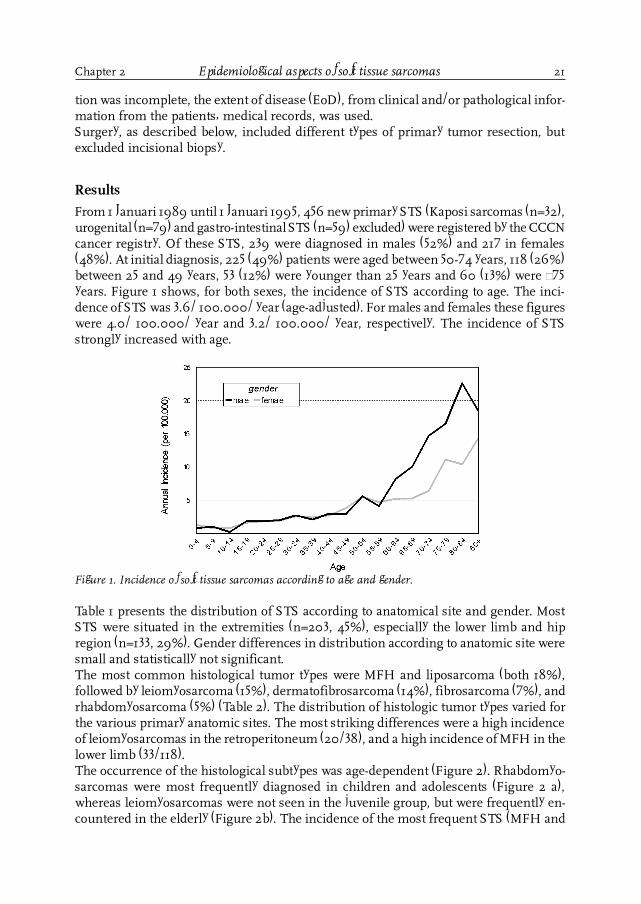
From 1 Januari 1989 until 1 Januari 1995, 456 new primary STS (Kaposi sarcomas (n=32),urogenital (n=79) and gastro-intestinal STS (n=59) excluded) were registered by the CCCNcancer registry. Of these STS, 239 were diagnosed in males (52%) and 217 in females(48%). At initial diagnosis, 225 (49%) patients were aged between 50-74 years, 118 (26%)between 25 and 49 years, 53 (12%) were younger than 25 years and 60 (13%) were ³75years. Figure 1 shows, for both sexes, the incidence of STS according to age. The inci-dence of STS was 3.6/ 100.000/ year (age-adjusted). For males and females these figureswere 4.0/ 100.000/ year and 3.2/ 100.000/ year, respectively. The incidence of STSstrongly increased with age.
Chapter 2 Epidemiological aspects of soft tissue sarcomas
Figure 1. Incidence of soft tissue sarcomas according to age and gender.
Table 1 presents the distribution of STS according to anatomical site and gender. MostSTS were situated in the extremities (n=203, 45%), especially the lower limb and hipregion (n=133, 29%). Gender differences in distribution according to anatomic site weresmall and statistically not significant.The most common histological tumor types were MFH and liposarcoma (both 18%),followed by leiomyosarcoma (15%), dermatofibrosarcoma (14%), fibrosarcoma (7%), andrhabdomyosarcoma (5%) (Table 2). The distribution of histologic tumor types varied forthe various primary anatomic sites. The most striking differences were a high incidenceof leiomyosarcomas in the retroperitoneum (20/38), and a high incidence of MFH in thelower limb (33/118).The occurrence of the histological subtypes was age-dependent (Figure 2). Rhabdomyo-sarcomas were most frequently diagnosed in children and adolescents (Figure 2 a),whereas leiomyosarcomas were not seen in the juvenile group, but were frequently en-countered in the elderly (Figure 2b). The incidence of the most frequent STS (MFH and

22
Table 1. Distribution of STS according to localisation and gender.Localisation Total Male Female
n (%) n (%) n (%)Head and neck 60 13 36 15 24 11Upper limb and shoulder 70 16 45 19 25 12Thorax 48 11 22 9 26 12Abdomen 56 12 24 10 32 15Trunk 10 2 4 1 6 2(Retro)peritoneum 38 8 19 8 19 9Pelvis 33 7 19 8 14 6Lower limb and hip 133 29 64 27 69 32Overlapping sites and NOS 8 2 6 3 2 1
Total 456 100 239 100 217 100
NOS, No other specification.
Table 2. Distribution of STS according to histology and gender.Morphology Total Male Female
n (%) n (%) n (%)MFH (ICD-O 8830) 83 18 48 20 35 16Liposarcoma (ICD-O 885) 82 18 44 18 38 17Leiomyosarcoma (ICD-O 889) 70 15 34 14 36 17Rhabdomyosarcoma (ICD-O 890) 23 5 11 5 12 5Dermatofibrosarcoma (ICD-O 8832) 65 14 32 13 33 15Fibrosarcoma (ICD-O 881) 30 7 18 8 12 6Hemangiosarcoma (ICD-O 912,913,915) 9 2 5 2 4 2Other sarcoma 94 21 47 20 47 22Sarcoma NOS (ICD-O 880) 34 7 18 8 16 7Phyllodes tumor(ICD-O 9020) 11 2 - - 11 5Synovial sarcoma (ICD-O 904) 17 4 11 5 6 3Clear cell sarcoma (ICD-O 9044) 2 0.5 2 1 - -Mesenchymal chondrosarcoma (ICD-O 9240) 3 1 3 1 - -Malignant giant cell tumor of soft parts(ICD-O 9251) 5 1 3 1 2 1Ewing�s sarcoma (ICD-O 9260) 3 1 2 1 1 1Malignant peripheral nerve sheath tumor(ICD-O 9540+9560) 17 4 8 3 9 1Alveolar soft part sarcoma (ICD-O 9581) 2 0.5 - - 2 1
Total 456 100 239 100 217 100
MFH, malignant fibrous histiocytoma. NOS, no other specification
Epidemiological aspects of soft tissue sarcomas Chapter 2

23
liposarcoma) increased with age (Figure 2 c, d).The distribution of T, N, and M stage is presented in Table 3. Skin and breast STS (n=88)were excluded, since no TNM classification applies to these tumors. Overall, 33% of thepatients presented with a T1-tumor, 48% with a T2-tumor, whereas in the remaining19% T-stage was unknown. Notwithstanding the existence of staging guidelines in theCCCN region [15], presence or absence of lymph node involvement was unknown in 210patients (57%). Twelve of 158 patients with a documented N-stage (8%) had lymph nodemetastases at initial presentation. However, using a �best-case-scenario�, in which allunknown N-stages are to be considered as node-negative, the overall incidence of nodalinvolvement would be 3%.
Table 3. TNM-distribution (STS of skin and breast excluded) n=368.
T-stage N %T1 121 33T2 177 48Unknown 70 19
N-stage N %N- 146 40N+ 12 3Unknown 210 57
M-stage N %M- 205 56M+ 34 9Unknown 129 35
5 patients had both lymph node involvement and distant metastases at presentation
Chapter 2 Epidemiological aspects of soft tissue sarcomas
Figure 2 a,b,c,d. Histological distribution of soft tissue sarcomas according to age.
a. b.
c. d.

24
The M-stage was not recorded in 129 patients (35%). At presentation, 34 of 239 patients(14%) with documented M-stage had distant metastases. Using the �best-case-scenario�,the overall incidence of distant metastatic disease would be 9%. Five patients (1%) hadboth lymph node and distant metastases.Lymph node involvement was not related to tumor size (T1: 3.3%, T2: 3.4%, unknown T-stage: 1.4%). Distant metastases, however, were significantly related to T-stage (P<0.01;Table 4). Five per cent (4/83) of patients with a T1-tumor and a documented M-stage haddistant metastases at presentation, in contrast to 17% (23/136) of patients with a T2-tumor and a documented M-stage.
A site-specific description of TNM classification is presented in Table 5. 27 (75%) ofdocumented head/neck STS were T1-tumors, whereas all retroperitoneal STS (n=31) wereT2-tumors. There was no site-specific difference in lymph node involvement, in contrastto distant metastases, which were encountered in 21% of retroperitoneal STS, 14% oflower limb and hip STS, but were absent in head/neck STS. Tumor grade was docu-mented in only 104 patients (23%).
Table 5. TNM-stage and localisation (STS of skin and breast excluded).T1 T2 N+ * M+ � T-unknown N-unknown M-unknown
Localisation n % n % n % n % n % n % n %
Head/neck (n=60) 27 45 9 15 2 10 - - 24 40 39 65 35 58
Upper limb/shoulder (n=70) 26 37 14 20 2 9 1 3 30 43 48 69 40 57
Trunk (n=10) 4 40 5 50 - - 2 22 1 10 7 70 1 10
(Retro)peritoneum (n=38) - - 31 82 - - 5 21 7 18 28 74 14 37
Lower limb/hip (n=133) 40 30 66 50 5 7 13 14 27 20 65 49 40 30
* Rate of lymph node metastases in patients with a documented N-stage. � Rate of distant metastases in patients with a docu-
mented M-stage.
Table 4. Tumor size and distant metastases (STS of skin and breast excluded).T-stage M- M+ M-unknown
n % n % n %T1 (n=121) 79 65 4 3 38 31T2 (n=177) 113 64 23 13 41 23Unknown (n=70) 13 19 7 10 50 71
Epidemiological aspects of soft tissue sarcomas Chapter 2
Table 6 and 7 present the initial treatment according to the extent of disease, for all 456patients, and for patients under 20 years of age, respectively. Overall, 371 patients (81%)received surgical treatment, 42 patients (9%) were not surgically treated, and 43 patients(9%) received no treatment at all. The proportion of patients that received no treatmentwas 3% in children and adolescents (≤ 20 years), 7% in those aged 21-69 years, and 16%in patients above 70 years.

25
Table 7. Primary treatment of STS according to extent of disease (EoD) in children andadolescents.
Total Localised Regional Distant EoD unknown disease metastases metastases
Treatment n % n % n % n % n %
Surgery 14 41 11 48 - - - - 3 75Surgery + radiotherapy 3 9 3 13 - - - - - -Surgery + chemotherapy 3 9 2 9 - - 1 25 - -Surgery + radiotherapy + chemotherapy - - - - - - - - - -Radiotherapy - - - - - - - - - -Chemotherapy 7 21 2 9 3 100 1 25 1 25Radiotherapy + chemotherapy 6 18 4 17 - - 2 50 - -No treatment 1 3 1 4 - - - - - -Total 34 100 23 100 3 100 4 100 4 100
Table 6. Primary treatment of STS according to extent of disease (EoD).Localised Regional Distant EoD unknown disease metastases metastases
Total 0-69 yrs 70 + yrs 0-69 yrs 70 + yrs 0-69 yrs 70 + yrs 0-69 yrs 70 + yrs
Treatment n % n % n % n % n % n % n % n % n %
Surgery 262 57 155 62 78 64 2 33 1 50 6 22 - - 14 54 6 38
Surgery + radiotherapy 82 18 53 21 25 21 - - - - - - - - 4 15 - -
Surgery + chemotherapy 18 4 8 3 4 3 1 17 - - 5 19 - - - - - -
Surgery + radio+chemotherapy 9 2 9 4 - - - - - - - - - - - - - -
Radiotherapy 12 3 2 1 5 4 - - 1 50 2 7 1 13 1 4 - -
Chemotherapy 23 5 8 3 1 1 3 50 - - 7 26 2 25 2 8 - -
Radiotherapy + chemotherapy 7 2 4 2 - - - - - - 3 11 - - - - - -
No treatment 43 9 10 4 9 7 - - - - 4 15 5 63 5 19 10 63
Total 456 100249 100 122 100 6 100 2 100 27 100 8 100 26 100 16 100
Discussion
There are only few epidemiological reports on STS, which are not centered-based [5-10].The CCCN registry used in this study is population-based, that is it contains informationabout all patients diagnosed and treated in a defined area. One of the major advantagesof a population-based registry is the avoidance of selection bias, caused by referral tospecialised centers.The annual incidence of STS (Kaposi�s sarcomas and urogenital and gastrointestinalSTS excluded) was 3.6 per 100.000 inhabitants, demonstrating the rarity of these tumors.In accordance with other reports, there was a slight male predominance [6,7,16]. Nearlyhalf of our patients was older than 65 years (n=187), with the highest age-specific ratesseen in patients over the age of 70 years, indicating that most sarcomas are tumors of theelderly (Fig. 1). Most STS (45%) were located at the extremities, predominantly the lowerextremity and hip region (29%). Pollock and colleagues have published similar findings[10]. A higher rate of limb STS (59.5%) was reported by Lawrence and colleagues [17], but
Chapter 2 Epidemiological aspects of soft tissue sarcomas

26
in this nationwide, multicenter study, anatomical sites such as bone, lymphoid organsand lymph nodes, viscera, and the central nervous system were excluded. Moreover, thisstudy was center-based, which might have led to selection bias.As reported by others [4,6,9,10,18], the most common histological types in our studywere MFH and liposarcoma (both 18%). Other histological types conformed more or lessto modern reports [4,7,9]. However, in the Swedish population-based Cancer Registry,Gustafson [6] encountered more MFH (41%) and less liposarcomas (10%). In the latterregistry, only STS of limb and trunk wall were included, whereas dermatofibrosarcomaswere excluded.
The most obvious age differences in histogenetics were the higher incidence of rhab-domyosarcomas in children and adolescents, and the absence of leiomyosarcomas inthis age group. During childhood and adolescence, the frequency of rhabdomyosarco-mas is equal or greater than that of the other types of STS combined [19]. In the currentseries, 14 out of 34 juvenile STS were rhabdomyosarcomas. As rhabdomyosarcomas arevery rare tumors beyond adolescence, the registration of rhabdomyosarcomas at middleand late age might have been caused by misdiagnosis. The other histological types showedan increasing incidence with increasing age, as demonstrated by others [9].
The relationship between tumor site and tumor size can be explained by the fact thatpalpation of relatively small tumors is easier in the head/neck region than in theretroperitoneum and lower extremity. Most reports on STS of the retroperitoneum and(especially the upper part of the) lower extremity confirm the high incidence of largetumors at presentation [6,20,21].At initial presentation, the rate of lymph node involvement in STS appears to be between3 and 8%. With regard to lymph node metastasis, the �best-case-scenario� seems validbecause many pathological reports tend not to mention normal findings, and becausethe STS types that were encountered most frequently, are rarely associated with nodalinvolvement at initial presentation [4,6,18,22,23], whereas relatively rare STS, such asepithelioid sarcomas, angiosarcomas, synovial sarcomas and rhabdomyosarcomas, havea higher incidence of early lymph node involvement [4,17,23].Fourteen per cent of patients with a documented M-stage had distant metastases at ini-tial presentation. With regard to distant metastatic disease, the �best-case-scenario� seemsless reliable, as reported data show a relatively high incidence of distant metastases atpresentation (7-25%) [4,6,18,20,22]. Nevertheless, it is very difficult to compare thesedata. The best comparable data come from the population-based study of Gustafson,who reported 13% distant metastases at initial presentation [6]. In their nationwidemulticenter survey, Lawrence and colleagues encountered 23% distant metastatic dis-ease at initial presentation [18]. However, they also used a �best-case-scenario�, as in 49%of the patients the M-stage was unknown. Other, center-based series, reported 25% distantmetastases at initial presentation, but these series mainly comprised STS of the lowerextremity [20], or included visceral STS [4], both of which are associated with a muchlarger tumor size, and a higher frequency of metastatic disease. The lowest reportedincidence was from a center-based study by Gaakeer and coworkers, who encountereddistant metastases in 12 of 183 patients (7%) [22]. However, as 80% of their patients werereferred after initial surgery in another hospital, this figure seems to be selection-biased.
Epidemiological aspects of soft tissue sarcomas Chapter 2

27
The most obvious treatment differences were found between children and adolescents(≤20 years) and patients aged above 70 years. In contrast to localised disease, where notreatment difference was demonstrated, treatment in (regional and/or distant) meta-static disease was different between both age groups. In case of metastatic disease, atleast 50% of the older patients were not treated at all and only 20% received some formof chemotherapy, in contrast to the young patients, who were all treated with, at least,chemotherapy.One of the major contributing factors is the age difference in histogenetics, especiallythe higher incidence of rhabdomyosarcomas in children and adolescents, tumors thatare relatively high radio- and chemosensitive. Also, in the elderly, their general healthstate is often weakened due to increasing co-morbidity, thus increasing the risk of mor-bidity and mortality following treatment. Only 5% of patients aged over 70 years receivedchemotherapy, in contrast to 12% in patients aged 21-69 years and 47% in children andadolescents. Many chemotherapeutic (doxorubicin- and/or ifosfamide-based) STS treat-ment protocols exclude patients over the age of 65-70 years. In this study, no less than33% of all patients was older than 70 years, 41% of which was even older than 80 years.Another contributing factor is the anatomical site of the tumor at initial presentation. Byfar the highest age-specific incidence of STS of trunk, lower extremities, and retro-peritoneum is encountered in patients above 70 years (9, 6, and 3/100.000, respec-tively). Tumors at these anatomic sites are known to be large at presentation, and areassociated with a higher incidence of metastatic disease [4,20,21]. Moreover, their sizeand specific location at presentation often makes radical tumor excision difficult or evenimpossible [21]. Especially in retroperitoneal STS, radical surgical excision often necessi-tates removal of adjacent organs such as kidneys (25-70%), colon (20-25%), adrenals (12-25%) or pancreas (8%), and in rare cases even segmental resection of major vessels,exenterations or hemipelvectomy [21]. Most of the older patients with a retroperitonealsarcoma are not candidates for this kind of extended surgical resection, although currentimprovement in anaesthetic techniques and intensive care treatment have decreasedtreatment-related morbidity and mortality. In the present study, the influence of primarytumor site on treatment regimen was obvious in patients over the age of 70 years. Whenthe STS was situated in the retroperitoneum, no less than 44% of these patients receivedno treatment at all, in contrast to 18% in trunk STS, 14% in lower extremity STS, 8% inupper extremity STS, and 3% in head/neck STS.
Data on STS, derived from this population-based cancer registry revealed interestingaspects that may be important for the design of future clinical trials on STS. Sarcomasare rare tumors, with an annual incidence of 3.6/100.000, increasing with age. At initialpresentation, lymph node involvement is rare (3-8%), whereas distant metastases areencountered more frequently (9-14%). As it is extremely difficult to attain a final diagno-sis, even by experienced pathologists [8], and as STS staging generally is not well per-formed, notwithstanding staging guidelines, centralization of STS diagnosis and stag-ing seems advisable. As most of the progress in STS treatment is to be expected frommultimodality treatment protocols, it is important to realize that nearly half of the pa-tients with a newly diagnosed STS are over the age of 65 years. These patients shouldreceive an extensive work-up and a tailored treatment, according to tumor site, extent ofdisease, and co-morbidity, in order to improve overall survival and quality of life [24].
Chapter 2 Epidemiological aspects of soft tissue sarcomas

28
References1. Landis SH, Murray T, Bolden S, Wingo PA. Cancer Statistics 1998. CA Cancer J Clin 1998; 48: 6- 29.2. Marina NM, Krance R, Ribeiro RC, Crist WM. Diagnosis and treatment of the most common solid tumors in
childhood. Prim Care 1992; 19: 871- 889.3. Flamant F, Habrand J-L, Lacombe MJ, Revillon Y. Malignant mesenchymal tumours in childhood. In Peckham
M, Pinedo HM, Veronesi U, eds. Oxford Textbook of Oncology. Oxford, Oxford University Press, 1995,1939-1953.
4. Torosian MH, Friedrich C, Godbold J, Hajdu SI, Brennan F. Soft tissue sarcoma: Initial characteristics andprognostic factors in patients with and without metastatic disease. Sem Surg Oncol 1988; 4: 13-19.
5. Clemente C, Orazi A, Rilke F. The Italian registry of soft tissue tumors. Appl Pathol 1988; 6: 221-240.6. Gustafson P. Soft tissue sarcoma. Epidemiology and prognosis in 508 patients. Acta Orthop Scand 1994; 65
(suppl 259): 1- 31.7. Coebergh JWW, Heijden van der LH, Janssen-Heijnen MLG. Cancer of the soft tissues. In Coebergh JWW,
Heijden van der LH, Janssen-Heijnen MLG eds. Cancer incidence and survival in the southeast of the netherlands1955-1994. A report from the Eindhoven Cancer Registry. ISBN 90-5001-007-5. Eindhoven, The Netherlands,1995, 46-47.
8. Harris M, Hartley AL, Blair V, et al. Sarcomas in North West England. I. Histopathological peer review. Br JCancer 1991; 64: 315-320.
9. Hartley AL, Blair V, Harris M, et al. Sarcomas in North West England. II. Incidence. Br J Cancer 1991;64: 1145-1150.
10. Pollock RE, Karnell LH, Menck HR, Winchester DP. The National Cancer Data Base Report on soft tissuesarcoma. Cancer 1996; 78: 2247-2257.
11. Berkel J. General practitioners and completeness of cancer registry. J Epidemiol Comm Health 1990; 44: 121- 124.12. Schouten LJ, Höppener P, Van den Brandt PA, Knottnerus JA, Jager JJ. Completeness of cancer registration in
Limburg, The Netherlands. Int J Epidemiology 1993; 22: 369-376.13. Waterhouse JAH, Muir C, Correa P, Tomatis L. Cancer incidence in five continents, Vol. III. IARC scientific
publications No. 15. Lyon: International Agency for Research on Cancer, 1976, 453-459.14. Hermanek P, Sobin LH, eds. TNM classification of malignant tumours. UICC/ International Union Against
Cancer. Fourth edition, 2nd revision. Berlin, Springer-Verlag,1992, 90-92.15. Van Geel AN, Van Unnik JAM, Keus RB. Diagnosis and treatment of soft tissue tumours: the dutch nationwide-
accepted consensus. Sarcoma 1998; 2: 183-191.16. Jane MJ, Hughes PJ. Disease incidence and results of extremity lesion treatment: Mersey Region soft tissue
sarcomas (1975-1985). Sarcoma 1998; 2: 89-96.17. Lawrence W Jr, Hays DM, Heyn R, et al. Lymphatic metastases with childhood rhabdomyosarcoma.
Cancer 1987; 60: 910-915.18. Lawrence W Jr., Donegan WL, Natarajan N, Mettlin C, Beart R, Winchester D. Adult soft tissue sarcomas.
Ann Surg 1987; 205: 349-359.19. Hays DM. Rhabdomyosarcoma. Clin Ortop 1993; 289: 36-49.20. Shiu MH, Castro ELB, Hajdu I, Fortner JG. Surgical treatment of 297 soft tissue sarcomas of the lower
extremity. Ann Surg 1975; 182: 597-602.21. Van Dam PA, Lowe DG, McKenzie-Gray B, Shepherd JH. Retroperitoneal soft tissue sarcomas: a review of the
literature. Obst Gyn Surv 1990; 45: 670-682.22. Gaakeer HA, Albus-Lutter ChE, Gortzak E, Zoetmulder FAN. Regional lymph node metastases in patients with
soft tissue sarcomas of the extremities, what are the therapeutic consequences? Eur J Surg Oncol 1988; 14:151-156.
23. Weingrad DW, Rosenberg SA. Early lymphatic spread of osteogenic and soft tissue sarcomas. Surgery 1978;84: 231-240.
24. Ham SJ, Graaf WTA van der, Pras E, Molenaar WM, Berg E van den, Hoekstra HJ. Soft tissue sarcomasof the extremities. A multi-modality diagnostic and therapeutic approach. Cancer Treatment Reviews 1998;
24:373-391.
Epidemiological aspects of soft tissue sarcomas Chapter 2

29
Chapter 3
Long-term results of preoperative intraarterialdoxorubicin combined with neoadjuvant radiotherapy,
followed by extensive surgical resection for locallyadvanced soft tissue sarcomas of the extremities
Nijhuis P.H.A.1, Pras E. 1, Sleijfer D.Th. 3, Molenaar W.M.4, Schraffordt Koops H.1, Hoekstra H.J.1
1. Department of Surgical Oncology, Groningen University Hospital, Groningen, The Netherlands 2. Department
of Radiotherapy, Groningen University Hospital, Groningen, The Netherlands 3. Department of Medical
Oncology, Groningen University Hospital, Groningen, The Netherlands 4. Department of Pathology, Groningen
University Hospital, Groningen, The Netherlands
Radiotherapy and Oncology 1999; 51: 15-19
Chapter 3

30
Introduction
Soft tissue sarcomas (STS) are rare, generally high-grade tumors. Half of the patients dieof metastatic disease [11]. Sixty percent of STS are situated in the extremities and areoften large at the time of initial diagnosis. In the past these locally advanced tumors weretreated with ablative surgery [11]. Rosenberg demonstrated in a prospective randomizedtrial that adequate surgical resection, so-called compartment resection, followed byadjuvant high dose external beam radiotherapy (EBRT), was equivalent to amputation ofthe affected limb with regard to local recurrence, disease free and overall survival [18].In the seventies, Morton and Eilber introduced a combination of intraarterial doxorubicin,preoperative radiotherapy and surgery as a limb-saving treatment for locally advancedsoft-tissue sarcomas of the extremities [7]. This approach was adopted at the GroningenUniversity Hospital in the early eighties, and the preliminary results were promising asreported [10]. The present study focused especially on the long-term morbidity of thisintensive treatment modality in patients with locally advanced STS of the extremities,which were considered primarily irresectable due to their relationship to bone, vascularand/or nerve structures, making a radical local resection impossible.
Patients and Methods
Between 1983 and 1987, eleven patients, nine males and two females, median age 52(range 24-70) years with locally advanced, primarily irresectable high-grade STS of theextremities were treated with continuous intraarterial doxorubicin, followed by preopera-tive radiotherapy and tumor resection, with or without postoperative external beam ra-diotherapy (EBRT). The ultimate goal was to save the affected extremity with good limbfunction and to achieve local tumor control. Tumor histology revealed: four liposarcomas,two synovial sarcomas, three malignant fibrous histiocytomas (MFH), one epithelioidmalignant schwannoma, and one leiomyosarcoma. According to the revised AJCC sta-ging system two patients were classified as stage IIB and nine patients as stage III.Preoperative continuous intraarterial infusion was performed via an intraarterial cath-eter introduced through the contralateral axilla or groin using the Seldinger technique.Doxorubicin was given for three consecutive days at a daily dose of 20 mg/m2 by continu-ous infusion using an IVAC pump. Patients received 5000 IU of heparin subcutane-ously twice a day to prevent thrombosis.Preoperative EBRT started within 24 h after the completion of intraarterial doxorubicinadministration. A total dose of 35 Gy was applied in ten fractions of 350 cGy per day in 12-14 days. Based on preoperative CT scanning, the entire tumor region was irradiated.Within three days after completion of chemo-radiation treatment, the tumor was surgi-cally resected. Microscopical non-radical resections were treated with an additional doseof 20-30 Gy EBRT (200 cGy per fraction). Patients did not receive any further adjuvantsystemic chemotherapy. All the patients were referred to the Department of Rehabilita-tion and received intensive physiotherapy treatment to preserve optimal function of theaffected limb. Regular follow-up was performed according to the EORTC STS guidelinesfor local and distant failures. Short and long-term locoregional side effects of this inten-sive multimodality treatment were recorded. Special attention was paid to late normaltissue damage.
Long-term results of multimodality treatment Chapter 3

31
Results
Inserting and positioning the intraarterial catheter was accomplished without complica-tions in all of the patients. Three patients suffered a severe local skin reaction to thedoxorubicin (27%). This complication appeared to be the consequence of decreased out-flow of doxorubicin towards the tumor region, which in three patients resulted in localskin necrosis in the left groin, right elbow, and the lower abdominal wall, respectively.Preoperative radiotherapy with 10 x 3.5 Gy was well tolerated in nine patients. In two ofthe three patients with skin reactions to doxorubicin, the skin condition deterioratedduring radiotherapy.In ten patients (91%) limb-saving surgery could be performed. In one patient anexarticulation of the hip was necessary due to extensive local growth of an epithelioidmalignant schwannoma of the sciatic nerve. Eight resections had to be classified asmarginal resections, because of a very close relationship with bone, vascular and/or nervestructures, making wide resection at those sites impossible. One resection appeared tobe macroscopically non-radical. Marginal and macroscopical non-radical resections re-ceived additionally 20-30 Gy EBRT (200 cGy per fraction).Wound healing disturbances occurred in two patients (18%). Both patients had alreadyhad skin reactions to the intraarterial doxorubicin, which had been aggravated bypreoperative radiotherapy. In one patient with skin necrosis near the elbow, surgicalreconstruction with an abdominal pedicle flap was performed six weeks after tumorresection. In the other patient, who already had a doxorubicin-induced partial skin necrosisof the lower abdominal wall, the combination of pre- and postoperative radiotherapy andextensive surgery resulted in a wound dehiscence in the cranial part of the thigh, whichdid not show any healing tendency. The patient died 3 months after surgical resectionfrom lung metastases, not demonstrable at the time of diagnosis.During a median follow-up of 84 (range 3-136) months, six patients died (55%). Fivepatients died from metastases after a median follow-up of 42 (range 3-48) months: allhad lung metastases and one patient also had extensive vertebral metastases. The sixthpatient died 84 months after surgical resection from a non-disease-related cerebral haem-orrhage. None of the patients developed a local recurrence.At present, five patients are still alive with a median follow-up of 120 (range 110-136)months. The limb could be saved in all five of them. So far, none of them have shownany signs of local or systemic recurrence of the disease. Actuarial disease-free and overallsurvival is presented in Fig. 1.
Chapter 3 Long-term results of multimodality treatment
Figure 1. Actuarial overall and disease-free survival after combined modalitytreatment for primarily unresectablehigh grade STS.

32
Three of five long-term survivors (60%) developed severe functional limitation of theaffected extremity. In two of these patients, severe late complications occurred. One patientwith a very large myxoid liposarcoma that covered the whole dorsal side of the upper legreceived intra-arterial doxorubicin and 10 x 3.5 Gy external radiotherapy, followed by(marginal) resection. Postoperatively the operation area was irradiated with 14 x 2 Gyusing a moving juncture technique where the abutments were shifted daily. Five yearsafter surgical resection, sensory and motor neuropathy of the sciatic nerve developedand proved to be progressive. After a follow-up of 110 months, the patient has completesensory and motor paralysis over the total sciatic nerve region. A similar case of sensoryand motor neuropathy of the sciatic nerve had also occurred in one of the non-survivors.In the latter patient, neuropathy developed 8 months after surgical resection. Because itwas not clear whether this neuropathy was related to the surgical resection, or whether itwas the result of the treatment combination, the sciatic nerve was explored. A great dealof fibrosis was found in the direct vicinity, but the nerve itself was macroscopically normal.This neuropathy gradually recovered postoperatively, but the patient died shortlyafterwards from distant metastases.The second patient with severe late complications had a liposarcoma covering almost thewhole ventral side of the left upper leg. After the usual preoperative chemo-radiotherapy,marginal surgical resection was performed, followed by 13 x 2 Gy postoperativeradiotherapy. A spontaneous fracture of the affected femur occurred 91 months afterresection. The fracture was stabilized using an intramedullary osteosynthesis. Ten monthsafter fixation, the intramedullary nail broke and re-fracture occurred. The broken nailwas removed and replaced by a new, reamed nail. Recently, 135 months after primaryresection and 44 months after the initial osteosynthesis of the femoral fracture, there areclinical signs of re-fracture caused by malunion.
Discussion
In the past, most soft tissue sarcomas of the extremities were treated by local resection oramputation. However, the local recurrence rate was high (+ 30%) and about 40% of thepatients died from distant metastases [11]. Suit and Lindberg were the first to emphasizethe importance of postoperative radiotherapy for STS [21]. Their results and the resultsof specific compartment resections [13] formed the basis of the National Cancer Institute(NCI) trial, in which amputation was compared to resection followed by high dosepostoperative radiotherapy [18]. For the first time, a prospective randomized study showedthat adequately executed compartment resection followed by high dose radiotherapyformed a reliable limb-saving treatment, with comparable local recurrence, disease-freeand overall survival rates. Nevertheless, treatment of locally advanced sarcomas of theextremities remained an unsolved problem.Doxorubicin was, and still is, one of the few effective cytotoxic drugs for the treatment ofsarcomas. A disadvantage of this drug is its cardiac toxicity [8], but it also has the favour-able property of being a radiosensitizer. In the seventies, Morton and Eilber developedthe concept of intraarterial chemotherapy with doxorubicin, combined with preoperativeradiotherapy followed by tumor resection [7]. Initially, only resectable STS were treatedwith this multimodality treatment, but it soon appeared that locally advanced STS couldalso be treated in this way [10].
Long-term results of multimodality treatment Chapter 3

33
Although the multimodality treatment proved to be very effective for preventing localrecurrence, it also appeared to be associated with considerable morbidity. The additionof radiotherapy to surgical treatment leads to about 10% more or less serious complica-tions, especially wound healing problems, loss of function, pathological fractures, neu-ropathy and angiopathy [14]. Animal experiments and clinical research have shown thatradiotherapy particularly inhibits the early phase of wound healing [5]. In addition, thereseems to be a time-dependent effect. Radiotherapy applied directly preoperatively or post-operatively inhibits wound healing, whereas if it is applied one week after surgery, itdoes not have an unfavourable influence on wound healing [5].The influence of doxorubicin on wound healing is controversial. In animal experiments,doxorubicin inhibited wound healing [4]. However, the dosages used were often higherthan those administered in clinical practice. Other studies demonstrated a potential in-fluence of doxorubicin on radiotherapy [2]. It seems obvious that a combination ofpreoperative doxorubicin, radiotherapy and surgery involves a high risk of wound heal-ing problems.Compared to the literature, the short-term complication rate of 18% is relatively low [7],especially in view of the fact that 82% of our patients additionally received postoperativeradiotherapy with 20-30 Gy.
At the time we adopted this multimodality treatment, nothing was known about latefuture effects, and even nowadays, relatively little is known about the specific long-termresults of this treatment regimen. At the end of the eighties, it became clear that, besidesthe total dose of radiotherapy (as demonstrated by Eilber and coworkers), hypofractionationis associated with increased late normal tissue damage [7]. Interaction between doxorubicinand radiotherapy, known to be responsible for many acute wound healing disturbances,also influences long-term morbidity. Chang et al. drew particular attention to the influenceof the combination of chemotherapy and radiotherapy on the development of contracturesof the joints [3].In the present study, patients received a hypofractionated neoadjuvant radiotherapyscheme in ten fractions, as originally advocated by the Morton and Eilber [7]. It was notuntill the second part of the eighties that the latter group changed this ten-fractions-scheme into an eight-fractions-scheme in order to diminish the high early complicationrate. Fractions, however, still were hypofractionated (3.5 Gy).
Besides the neoadjuvant chemo-radiotherapy regimen, 82% of our patients received ad-ditional postoperative external beam radiotherapy of 20-30 Gy (2 Gy fractions). Althoughthis very intensive treatment resulted in an excellent local control, even in this unfavour-able group of patients whose standard treatment used to be an amputation, 60% of thelong-term survivors has severe functional limitations of the affected limb. Radiobiologi-cal calculations were performed to evaluate whether there was a relation between thebiologically effective dose and late tissue damage. The patient with the sciatic nerve neu-ropathy and the patient with the femoral fracture received a biologically effective dose of122.5 and 119.1 Gy
3, respectively, which forms an explanation for the high incidence of
late effects (Table 1) [1,9,12,17,19,20].In all the long-term survivors, a certain degree of fibrosis developed in the affected ex-tremity. In two cases, it was so slight that they hardly developed any functional com-
Chapter 3 Long-term results of multimodality treatment

34
plaints. In one patient, very severe fibrosis developed, which led to strongly decreasedlimb function. This patient received a biologically effective dose of 109.1 Gy
3.
The occurrence of severe invalidating fibrosis as a late complication has also been de-scribed by others [15].
Partly due to the high morbidity rate, this intensive multimodality treatment was re-placed by hyperthermic isolated regional limb perfusion (HILP) with TNF-α andmelphalan as a limb salvage treatment for these locally advanced STS of the extremities.Presently, this treatment appears to be an attractive treatment option with low morbidityand low local recurrence rates [6]. However, it is not yet possible to draw any conclusionsabout the long-term effects, although, untill now, additional EBRT has not been associ-ated with increased morbidity [16].
Conclusion
In the present series, it appeared that even the very unfavourable group of locally ad-vanced, primarily irresectable sarcomas of the extremities can be treated successfullywith a multimodality limb-saving treatment comprising neoadjuvant chemo-radiotherapy,followed by surgical resection and, in most cases, additional postoperative EBRT. Short-
Long-term results of multimodality treatment Chapter 3
Table 1. Radiation induced late toxicity.Author n Tumor type Dose of EBRT BED a/ß=3 ToxicityJentzsch [12] 29 Ewing 50 Gy (2 Gy/fraction) 83.3 Gy 54%pain, edema,
sarcoma + chemotherapy weakness,
23% contracture
Brown [1] 60 Ewing 60-65 Gy (1.8 Gy/ fraction) 96-104 Gy 5% pathological
sarcoma fracture
Stinson [19] 145 Sarcoma 63 Gy (1.8 Gy/ fraction) 100.8 Gy pain, edema,
muscle weakness
Stoll [20] 33 Breast 63 Gy (5.25 Gy/ fraction) 173.25 Gy 73% peripheral
neuropathy
57.25 Gy (5.25 Gy/ fraction) 157.43 Gy 15% peripheral
neuropathy
Powell [17] 449 Breast 45 Gy (3 Gy/ fraction) 90 Gy 5% peripheral
neuropathy
54 Gy (1.8 Gy/ fraction) 86.4 Gy 1% peripheral
neuropathy
Nijhuis 11 Extremity 35 Gy (3.5 Gy/ fraction) 75.8 Gy 1, 60% fibrosis,
[current series] sarcoma preoperatively (n=11) + 109-122.2 Gy 2 20% pathological
chemotherapy fracture, and
+ 20-30 Gy (2 Gy/ fraction) 20% peripheral
postoperatively (n=9) neuropathy 3
1. Only preoperative radiotherapy 2. Combination of pre- and postoperaative radiotherapy 3. Long-term survivors.

35
term complications, particularly wound healing problems, were substantial. Moreover,in the long-term, 60% of the survivors developed serious functional problems varyingfrom fibrosis to pathological stress fractures and invalidating neuropathy. This high rateof serious long-term complication may indicate, that in comparable intensive treatmentprotocols, it is important to pay special attention to late side effects, which may intervenewith the primary treatment goal, i.e. limb-salvage.
Chapter 3 Long-term results of multimodality treatment
References1. Brown AP, Fixen JA, Plowman PN. Local control of Ewing�s sarcoma: An analysis of 67 patients. Br J Radiol
1987; 60: 261-268.2. Cassaday JR, Richter MP, Piro AJ, Jaffe N. Radiation-adriamycin interactions: preliminary clinical
observations. Cancer 1975; 36: 946-949.3. Chang AE, Steinberg SM, Culnane M, et al. Functional and psychosocial effects of multimodality limb-sparing
therapy in patients with soft tissue sarcomas. J Clin Oncol 1989; 7:1217-1228.4. Cohen SC, Gabelnick HL, Johnson RK, Goldin A. Effects of cyclophosphamide and adriamycin on the healing of
surgical wounds in mice. Cancer 1975; 36: 1277-1281.5. Devereux D, Thibault L, Boretos J, Brennan MF. The quantitative and qualitative impairment of wound
healing of surgical wounds in rats. Cancer 1979; 43: 932-938.6. Eggermont AMM, Schraffordt Koops H, Klausner JM, et al. Isolated limb perfusion with tumor necrosis factor
and melphalan for limb salvage in 186 patients with locally advanced soft tissue extremity sarcomas. Ann Surg1996; 224: 756-765.
7. Eilber FR, Eckhardt JJ, Rosen G, Fu YS, Seeger LL, Selch MT. Neoadjuvant chemotherapy and radiotherapy inthe multidisciplinary management of soft tissue sarcomas of the extremity. Surg Oncol Clin N Am 1993; 2:611-620.
8. Ettinghausen SE, Bonow RO, Palmeri ST, et al. Prospective study of cardiomyopathy induced by adjuvantdoxorubicin therapy in patients with soft tissue sarcomas. Arch Surg 1986; 121: 1445-1451.
9. Gilette EL, Mahler PA, Powers BE, Gilette SM, Vujaskovic Z. Late radiation injury to muscle and peripheralnerves. Int J Radiat Oncol Biol Phys 1995; 31: 1309-1318.
10. Hoekstra HJ, Schraffordt Koops H, Molenaar WM, Mehta DM, Sleijfer DTh, Dijkhuis G, Oldhoff J. Acombination of intraarterial chemotherapy, preoperative and postoperative radiotherapy, and surgery aslimb-salving treatment of primarily unresectable high-grade soft tissue sarcomas of the extremities. Cancer 1989;63: 59-62.
11. Hoekstra HJ, Schraffordt Koops H, Oldhoff J. Soft tissue sarcoma of the extremity. Eur J Surg 1994; 20: 3-6.12. Jentzsch K, Binder H, Cramer H, et al. Leg function after radiotherapy for Ewing�s sarcoma. Cancer 1981; 47:
1267-1278.13. Lawrence WJr, Baker LL, Balch CM. (NIH Concensus Development Panel): Limbsparing treatment of adult
soft tissue sarcomas and osteosarcomas. JAMA 1985; 254: 1791-1794.14. Lindberg RD, Martin RG, Romsdahl MM, Barkley Jr HT. Conservative surgery and postoperative radiotherapy
in 300 adults with soft tissue sarcoma. Cancer 1981; 47: 2391-2397.15. Mason M, Robinson M, Harmer C, Westbury G. Intraarterial Adriamycin, conventionally fractionated
radiotherapy and conservative surgery for soft tissue sarcomas. Clin Oncol R-Coll-Radiol 1992; 4: 32-35.16. Olieman AFT, Pras E, Van Ginkel RJ, Molenaar WM, Schraffordt Koops H, Hoekstra HJ. Feasibility and
efficacy of external beam radiotherapy after hyperthermic isolated limb perfusion with TNF-α and Melphalanfor limb-saving treatment in locally advanced extremity soft tissue sarcoma. Int J Radiat Oncol Biol Phys 1998;40:807-814.
17. Powell S, Cooke J, Parsons C. Radiation-induced brachial plexus injury: Follow-up of two different fractionationschedules. Radiother Oncol 1990; 18: 213-220.
18. Rosenberg SA, Tepper J, Glatstein E, et al. The treatment of soft tissue sarcomas of the extremities. Prospectiverandomized evaluation of (1) limb-sparing surgery plus radiation therapy compared with amputation, and (2)the role of adjuvant chemotherapy. Ann Surg 1982; 96: 305-315.
19. Stinson SF, DeLaney TF, Greenberg J, et al. Acute and long-term effects on limb function of combined modalitylimb-sparing therapy for extremity soft tissue sarcoma. Int J Radiat Oncol Biol Phys 1991; 21: 1492-1499.

36
20. Stoll BA, Andrews JT. Radiation-induced peripheral neuropathy. Br Med J 1966; 1: 834-837.21. Suit HD, Russell WO, Martin RG. Sarcoma of soft tissue: Clinical and histopathologic parameters and response
to treatment. Cancer 1975; 35: 1478-1483.
Long-term results of multimodality treatment Chapter 3

37
Chapter 4
Clinico-pathological data and prognostic factors incompletely resected AJCC stage I-III liposarcomas
Nijhuis P.H.A.1, Sars P.R.A.1, Plaat B.E.C.2, Molenaar W. M.2, Sluiter W.J.3, Hoekstra H. J.1
1.Department of Surgical Oncology, Groningen University Hospital, Groningen, The Netherlands 2. Department of
Pathology, Groningen University Hospital, Groningen, The Netherlands 3. Department of Internal Medicine,
Groningen University Hospital, Groningen, The Netherlands
Annals of Surgical Oncology 2000; 7: 535-543
Chapter 4

38
Introduction
The majority of soft tissue mass is composed of muscle and fatty tissue. As adiposetissue makes up about twenty percent of the body weight, it seems obvious that one ofthe most common soft tissue sarcomas (STS) is liposarcoma (LPS) [1,2,3,4]. However,LPS originate from primitive mesenchymal cells rather than mature fat cells. In fact,these tumors are rare in the subcutaneous fat, a common location of lipomas, and theyare most frequently located in deeper structures [3]. Although LPS are one of the mostcommon STS, accounting for 10-20% of all STS [1,2,3,4], there are only limited recentdata that address epidemiological and treatment related aspects of this entity. Usually,these data are embedded in reports on sarcoma in general, which seems not advisablebecause the biological behavior and prognosis of LPS seems to be different from mostother STS [3,5]. Furthermore, there are strong indications that biological subtype deter-mines the outcome in LPS [6,7]. This diversity in clinical behavior may not becomeapparent if LPS are reviewed together with other STS. The purpose of the present studywas to gain an insight into epidemiological aspects of LPS, to evaluate treatment resultsand to determine prognostic factors for local recurrence, metastasis, disease-free anddisease-specific survival.
Patients and methods
All consecutive liposarcomas that were diagnosed at the Groningen University Hospital,from October 1977 to January 2000, were reviewed regarding the clinicopathologicaldata, treatment, and follow-up. Patients were followed clinically at the Groningen Uni-versity Hospital for a maximum period of 10 years. Data were retrospectively collected bychart review. For patients who were no longer being followed, data had to be collected bycorrespondence with the referring physician. Histopathologically, all tumors were re-viewed, and if necessary revised, by one pathologist with special interest and experiencein STS (WMM). Patients with perioperative signs of regional and/or distant metastaticdisease (AJCC stage IV) and those, in whom the LPS could not be resected completely(R2-resection), were excluded from the study.The extent of diagnostic preoperative work up has changed during the last decades. Cur-rently, preoperative work up of a soft tissue tumor includes magnetic resonance imaging(MRI) or magnetic resonance angiography (MRA) and/or computed tomography (CT)and/or ultrasonography (US) of the tumor site, followed by fine-needle aspiration (FNA),core biopsy, and/or incisional biopsy [8]. In superficially located soft tissue tumors smallerthan 3 cm, and without clinical suspicion of malignancy, an excisional biopsy is per-formed [9]. In case of a histological diagnosis of liposarcoma, a chest CT-scan and a bonescan are performed to rule out metastatic disease [8]. We used the classification describedby Enzinger [3], and recognized four different subtypes: well-differentiated LPS (WDLPS),myxoid LPS (MXLPS), pleiomorphic LPS (PMLPS), and dedifferentiated LPS (DDLPS).Tumors were graded according to Coindre et al [10], and grade was assigned based on thehighest grade presented. Patients were clinically staged according to the latest AJCCstaging guidelines for sarcoma [11].After preoperative work up, the tumor was resected with the intention to perform a widelocal resection. Resection margins were classified as microscopically involved if, on his-
Prognostic factors in liposarcoma Chapter 4

39
tologic examination, tumor cells were detected at the marked surface of the resectionspecimen. In recent years, in cases of microscopic involvement of the margins (R1-resec-tion), especially in high-grade tumors, high-dose adjuvant radiation therapy (50-70 Gy)has been recommended [8]. Chemotherapy was only delivered to eligible patients whoparticipated in different chemotherapy protocols during this time period.In primary LPS, the follow-up period was calculated from the time of histological diag-nosis. In LPS, presenting with a recurrence, the follow-up period was measured fromthe time of histological diagnosis of the recurrence. The local and distant recurrencerates were calculated using the Kaplan-Meier method.Epidemiologic data were analyzed using the Mann-Whitney U-test and Chi-square test.Clinicopathological and treatment related factors were analyzed using a log-rank test forlocal recurrence, disease-free and overall survival. A P value <0.05 was considered statis-tically significant. The relative risk ratio and 95% confidence interval (95% CI) are re-ported.
Results
Epidemiological Characteristics
The study group was composed of 35 men (51%) and 34 women (49%). Fifty-two LPSwere primary tumors (75%). Seventeen patients presented with a local recurrence afterearlier attempts at definitive treatment at outside institutions (25%). In nine of them(53%) it was the first or second recurrence, in four patients (24%), it was the third recur-rence, in two patients (11%), it was the fourth recurrence, in one patient (6%), the sixthrecurrence, and one patient (6%) had a seventh recurrence. Overall, the median age atpresentation was 51 (range 11-80) years. The median age at presentation was not signifi-cantly different in primary or recurrent LPS, 49 (range 11-80) years and 56 (range 35-80)years, respectively.
Clinical characteristics
By far, most patients (n=57) presented with a palpable mass (83%), which in most ofthem (n=46) was painless. At presentation, the median duration of symptoms was 6months for primary LPS, and 3 months for recurrent LPS. There was no gender-relateddifference.The distribution of LPS according to anatomical site is shown in Fig. 1. The majority ofLPS was located in the extremities (n=43; 62%), especially the thigh (n=29; 42%). Al-though 57 tumors (83%) were palpable on clinical examination, there were site-specificdifferences. In the head/neck region, trunk and extremities, most LPS were palpable(80%, 100%, 95%, respectively), whereas in retroperitoneum and buttock respectively64% and 50% of the tumors, were undetectable on clinical examination. Overall, 61 LPSwere situated beneath the fascia (88%). The relation between tumor site and tumor depthwas highly statistically significant (P=0.002), with the highest proportion of superfi-cially located tumors in the head/neck region and in the upper extremity, 60% and 40%,respectively. In retroperitoneum, buttock and leg (nearly) all LPS were deeply seated(Fig. 1).
Chapter 4 Prognostic factors in liposarcoma
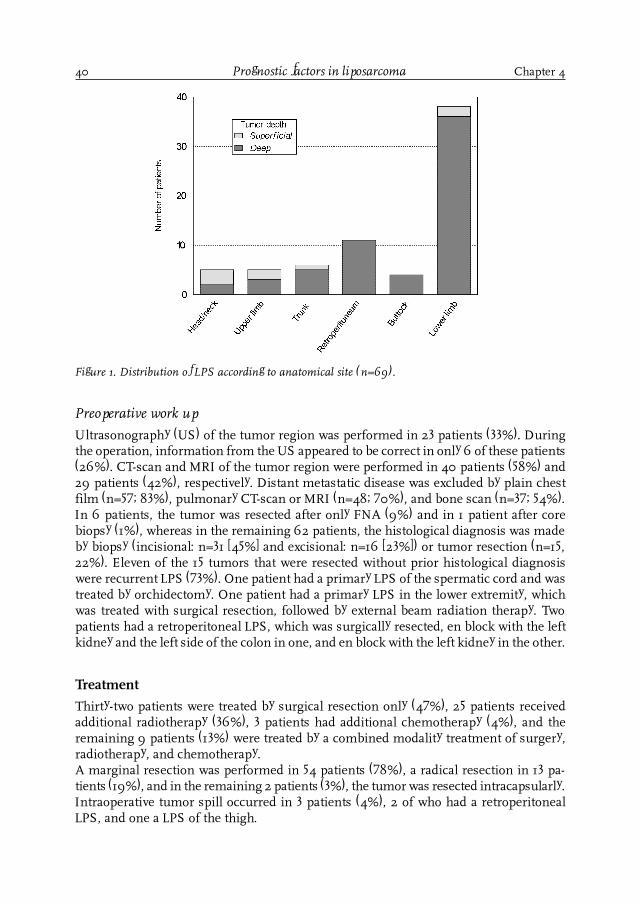
40
Preoperative work up
Ultrasonography (US) of the tumor region was performed in 23 patients (33%). Duringthe operation, information from the US appeared to be correct in only 6 of these patients(26%). CT-scan and MRI of the tumor region were performed in 40 patients (58%) and29 patients (42%), respectively. Distant metastatic disease was excluded by plain chestfilm (n=57; 83%), pulmonary CT-scan or MRI (n=48; 70%), and bone scan (n=37; 54%).In 6 patients, the tumor was resected after only FNA (9%) and in 1 patient after corebiopsy (1%), whereas in the remaining 62 patients, the histological diagnosis was madeby biopsy (incisional: n=31 [45%] and excisional: n=16 [23%]) or tumor resection (n=15,22%). Eleven of the 15 tumors that were resected without prior histological diagnosiswere recurrent LPS (73%). One patient had a primary LPS of the spermatic cord and wastreated by orchidectomy. One patient had a primary LPS in the lower extremity, whichwas treated with surgical resection, followed by external beam radiation therapy. Twopatients had a retroperitoneal LPS, which was surgically resected, en block with the leftkidney and the left side of the colon in one, and en block with the left kidney in the other.
Treatment
Thirty-two patients were treated by surgical resection only (47%), 25 patients receivedadditional radiotherapy (36%), 3 patients had additional chemotherapy (4%), and theremaining 9 patients (13%) were treated by a combined modality treatment of surgery,radiotherapy, and chemotherapy.A marginal resection was performed in 54 patients (78%), a radical resection in 13 pa-tients (19%), and in the remaining 2 patients (3%), the tumor was resected intracapsularly.Intraoperative tumor spill occurred in 3 patients (4%), 2 of who had a retroperitonealLPS, and one a LPS of the thigh.
Prognostic factors in liposarcoma Chapter 4
Figure 1. Distribution of LPS according to anatomical site (n=69).

41
Radiotherapy was applied in 34 patients (49%), most often postoperatively (n=30; 88%).Two patients with a LPS of the thigh received both pre- and postoperative external beamradiation therapy (EBRT), one patient with a primary gluteal LPS received neoadjuvantEBRT, followed by intraoperative radiation therapy (IORT) [12]. One patient with a sec-ond recurrence of a MXLPS of the popliteal fossa, previously treated with surgery and 64Gy EBRT, was treated with surgical resection and 25 Gy IORT [13]. Overall, the mediantotal radiation dose was 60 Gy, ranging from 25 (IORT)-70 Gy.Only a small number of patients (n=12; 17%), who participated in different protocols,received chemotherapy. Radiotherapy and/or chemotherapy related complications wereencountered in 17 of 37 patients (46%). By far, most were minor complications (ery-thema, dermatitis, epidermolysis, mucositis, and wound complications). However, fourpatients developed a neuropathy (11%), which was transient in two, but persisted in theother two patients.Limb salvage was achieved in 45 of 47 limbs (96%) involved (gluteal LPS included). Inone patient presenting with a recurrent LPS of the thigh, an exarticulation of the hip hadto be performed. One patient with a primary gluteal LPS had to be treated by hemipelvec-tomy and intraoperative radiation therapy [12]. During follow-up, an additional patientwho developed a local recurrence could only be salvaged by a high exarticulation of thelower limb, decreasing the cumulative limb salvage rate to 94%.
Histopathology
The revised histopathological diagnoses are presented in Fig. 2, which shows that nearlyhalf of the tumors were MXLPS, and that more than one third were WDLPS. For thevarious anatomical sites, tumor size, as measured by the pathologist, is presented in Fig.3. The largest tumors were encountered in the retroperitoneum (median diameter 25[range 12-46] cm) and lower limb (median diameter 12 [range 2-40] cm). Twenty-four of26 WDLPS (92%) were classified as grade I LPS, the other 2 as grade II (8%), 29 MXLPSwere classified as grade I (85%), and 5 MXLPS as grade II (15%). Three DDLPS weregrade III LPS (50%), whereas the other three were classified as grade II. Two of threePMLPS were classified as grade II, the third as grade III. The relation between histologi-cal subtype and tumor grade seemed to be highly significant (P<0.0001).
Chapter 4 Prognostic factors in liposarcoma
Figure 2. Histological distribution of LPS (n=69).

42
Overall, there was no statistically significant relation between anatomical site and tumorgrade (P=0.19), although retroperitoneal LPS had a significantly higher tumor gradecompared to LPS at other sites (P=0.02). According to the new AJCC staging guidelines[10], 10 LPS were staged as stage Ia (14%), 3 LPS as stage Ib (4%), 52 LPS as stage IIa(76%), and 4 LPS as stage III (6%). LPS of the retroperitoneum, buttock, and thigh hadsignificantly more often stage II and III (P=0.01). Microscopically, free margins (R0-resection) were achieved in 53 patients (77%); in the other 16 patients (23%) marginswere microscopically involved (R1-resection). No patient had a macroscopic tumor leftbehind (R2-resection).
Recurrence and Survival
The duration of follow-up for this cohort of patients ranged from 5 to 251 months withmedian and mean follow-up of 79 and 87 months, respectively.
Local recurrence
During follow-up, 18 of 69 patients (26%) developed a local recurrence after 2-101 months.A total of 28% of local recurrences was evident by 1 year, 44% by 2 years, 72% by 3 years,78% by 4 years, and 89% by 5 years. There were two very late local recurrences after 81and 101 months. Multiple recurrences were common and occurred in 67% of patientswho developed a local recurrence after initial presentation with a primary LPS, and in83% of patients who presented with a recurrent LPS.On univariate analysis, retroperitoneal localization was associated with a significantlyshorter local recurrence-free interval (Table 1, Fig. 4). Retroperitoneal LPS recurred aftera median recurrence-free interval of 48 months. At the end of the study period, five ofthe patients who had had a retroperitoneal LPS had no signs of recurrent disease. Fourpatients died from unresectable local recurrences in absence of distant metastases, and
Prognostic factors in liposarcoma Chapter 4
Figure 3. Tumor size according to anatomical site.

43
one patient died from an unresectable local recurrence and metastases to the lung, verte-brae, and soft tissues. One patient is still alive but has an unresectable local recurrencewithout distant metastases and is likely to suffocate in the near future.
Chapter 4 Prognostic factors in liposarcoma
Table 1. Local recurrence-free interval according to potential prognostic factors.Clinicopathological factor RR 95% CI
Gender Male vs. female 0.69 0.27-1.74Histological subtype WDLPS vs. all other subtypes 2.39 0.79-7.3
All other subtypes vs. DDLPS 1.04 0.14-7.8Primary vs. recurrent presentation 1.36 0.51-3.63Anatomical site All other sites vs. retroperitoneum 3.22* 1.21-8.6Depth Superficial vs. deep 2.69 0.36-20.2Type of resection R1 vs. marginal 1.01 0.32-3.18
R1 vs. radical 1.25 0.28-5.56Tumor diameter ≤10 cm vs. 10-20 cm 1.64 0.53-5.07
≤10 cm vs. >20 cm 2.92 0.94-9.0Grade I vs. II 1.11 0.32-3.85
I vs. III 2.05 0.27-15.6Stage I vs. II 5.32 0.71-40.7
I vs. III 8.55 0.53-137Treatment Surgery vs. surgery + radiotherapy 0.54 0.19-1.54
Surgery vs. surgery + chemotherapy 0.85 0.11-6.55Surgery vs. surgery + radiotherapy +chemotherapy 0a
* Significance, P< .05.a Numbers too small to draw conclusions.
Figure 4. Local recurrence-free interval according to anatomical site.
Distant metastases
Within a range of 4-103 months, 11 patients (16%) developed distant metastases. Of these,27% were evident by 1 year, 55% by 2 years, 82% by 3 years, and 91% by 5 years. One

44
patient developed distant metastases after 103 months. Four patients (36%) with distantmetastases also experienced a local recurrence. In three of them, the local relapse pre-ceded the distant failure by a median interval of 47 (range 4-91) months. In one patient,local and distant relapse occurred simultaneously.The lung was the most common site for metastases (72%), followed by vertebrae (36%),soft-tissues (27%), liver (18%), and brain (9%). None of the 26 WDLPS developed distantmetastases, whereas 3 out of 6 metastasizing MXLPS (50%) did so to the soft tissues.Three patients (all MXLPS) developed extrapulmonary metastases only (soft tissue, spine,and brain).A univariate analysis of prognostic factors with regard to metastasis-free interval is pre-sented in Table 2. Deep tumor location, DDLPS (Relative Risk [RR] 13.6), and tumorgrade II and III (RR 7.8 and 14.6, respectively) were associated with a significantly shortermetastasis-free period (Fig. 5 a,b). After 5 years, DDLPS had a metastasis rate of 78%,with a median metastasis-free interval of 20 months. After 5 years, grade I, II, and IIILPS had a metastasis rate of 8%, 49%, and 50%, respectively. The median metastasis-free interval in grade II an III LPS was 36 and 20 months, respectively. The relative riskof DDLPS was not influenced by tumor depth (RR 11.9, 95% CI: 3.49- 40.7), nor by grade(RR 10.3, 95% CI: 3.01- 35.2). Tumor grade II and III lost significance, when adjusted fordedifferentiated subtype (RR 2.82, 95%CI: 0.76- 10.5 and RR 1.45, 95%CI: 0.27- 7.9,respectively). None of the superficially located tumors metastasized to distant sites.
Survival
A univariate analysis with regard to disease-free interval is presented in Table 3, demon-
Prognostic factors in liposarcoma Chapter 4
Table 2. (Distant) metastasis-free interval according to potential prognostic factors.Clinicopathological factor RR 95% CI
Gender Male vs. female 0.36 0.10-1.37Histological subtype All other subtypes vs. DDLPS 13.6* 4.0-47Primary vs. recurrent presentation 0.28 0.04-2.15Anatomical site All other sites vs. retroperitoneum 0.53 0.07-4.2Depth Superficial vs. deep ∞*Type of resection R1 vs. marginal 3.83 0.49-30.3
R1 vs. radical 1.57 0.10-25.1Tumor diameter ≤ 10 cm vs. 10-20 cm 1.57 0.39-6.26
≤ 10 cm vs. > 20 cm 1.93 0.43-8.6Grade I vs. II 7.81* 2.10-29.1
I vs. III 14.6* 2.67-80I vs. II + III 9.01* 2.64-31
Stage I vs. II + III 0.66 0.17-2.49Treatment Surgery vs. surgery + radiotherapy 0.34 0.07-1.66
Surgery vs. surgery + chemotherapy 1.23 0.15-10.0Surgery vs. surgery + radiotherapy + 0.59 0.15-2.80chemotherapy
* Significance, P< .05.

45
strating a significantly longer disease-free interval in WDLPS (Fig 6), grade I LPS (Fig 7),stage I LPS, and in tumors ≤10 cm. When corrected for tumor size and stage, WDLPSstill had a better disease-free interval (RR 4.4 and 3.6, respectively). When adjusted forgrade, WDLPS lost its significance (RR 2.7). Tumor grade I remained a significant factorwhen adjusted for tumor size (RR 2.6), but lost independency when adjusted for histo-logical subtype (RR 1.3) or stage (RR 1.7). Tumor stage I, corrected for histological subtype,tumor grade or tumor size, lost significance (RR 6.9, 5.2, and 4.2, respectively). Tumorsize lost significance, when adjusted for grade (RR 1.96) or stage (RR1.74), but was notinfluenced by histological subtype WDLPS (RR 4.2).In univariate analysis, dedifferentiation, grade II and III, non-radical resection, and stageII and III were associated with a worse disease-specific survival (Table 4). None of thestage I patients died of the disease, but the numbers are too limited to reach statisticalsignificance. After correction for radicalness of resection, DDLPS (Fig 8a) and grade II-III LPS (Fig 8b) continued to have a significantly worse disease-specific survival (RR 10.2and 7.9, respectively), but when corrected for each other, both factors lost significance.After a radical resection, no patient died of the disease.
Chapter 4 Prognostic factors in liposarcoma
Figure 5. (A) Metastasis-free interval according to tumor grade. (B) Metastasis-free interval according tohistological subtype.
A
B
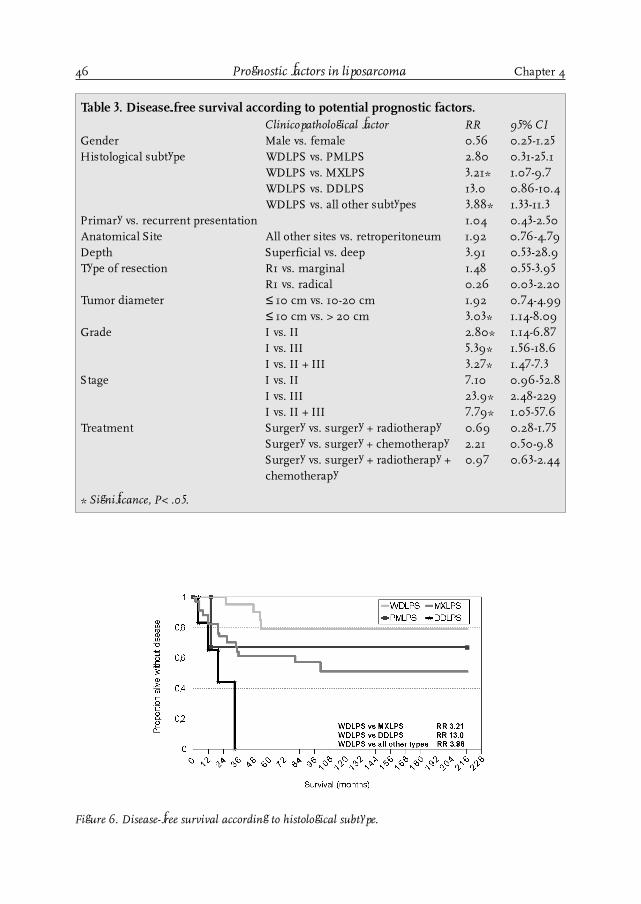
46 Prognostic factors in liposarcoma Chapter 4
Table 3. Disease-free survival according to potential prognostic factors.Clinicopathological factor RR 95% CI
Gender Male vs. female 0.56 0.25-1.25Histological subtype WDLPS vs. PMLPS 2.80 0.31-25.1
WDLPS vs. MXLPS 3.21* 1.07-9.7WDLPS vs. DDLPS 13.0 0.86-10.4WDLPS vs. all other subtypes 3.88* 1.33-11.3
Primary vs. recurrent presentation 1.04 0.43-2.50Anatomical Site All other sites vs. retroperitoneum 1.92 0.76-4.79Depth Superficial vs. deep 3.91 0.53-28.9Type of resection R1 vs. marginal 1.48 0.55-3.95
R1 vs. radical 0.26 0.03-2.20Tumor diameter ≤ 10 cm vs. 10-20 cm 1.92 0.74-4.99
≤ 10 cm vs. > 20 cm 3.03* 1.14-8.09Grade I vs. II 2.80* 1.14-6.87
I vs. III 5.39* 1.56-18.6I vs. II + III 3.27* 1.47-7.3
Stage I vs. II 7.10 0.96-52.8I vs. III 23.9* 2.48-229I vs. II + III 7.79* 1.05-57.6
Treatment Surgery vs. surgery + radiotherapy 0.69 0.28-1.75Surgery vs. surgery + chemotherapy 2.21 0.50-9.8Surgery vs. surgery + radiotherapy + 0.97 0.63-2.44chemotherapy
* Significance, P< .05.
Figure 6. Disease-free survival according to histological subtype.

47Chapter 4 Prognostic factors in liposarcoma
Table 4. Overall (disease-specific) survival according to potential prognostic factors.Clinicopathological factor RR 95% CI
Gender Male vs. female 0.41 0.13-1.33Histological subtype all other subtypes vs. DDLPS 14.4* 4.72-44.1Primary vs. recurrent presentation 0.50 0.11-2.28Anatomical Site All other sites vs. retroperitoneum 2.76 0.85-8.9Depth Superficial vs. deep ∞Type of resection R1 vs. marginal 2.34 0.52-10.6
R1 vs. radical 0*Tumor diameter ≤ 10 cm vs. 10-20 cm 1.91 0.51-7.1
≤ 10 cm vs. > 20 cm 2.60 0.65-10.4Grade I vs. II 8.01* 2.26-28.4
I vs. III 31.7* 7.1-142I vs. II + III 10.7* 3.29-34.6
Stage I vs. II + III ∞Treatment Surgery vs. surgery + radiotherapy 1.43 0.41-5.0
Surgery vs. surgery + chemotherapy 1.95 0.23-16.7Surgery vs. surgery + radiotherapy + 2.34 0.38-7.4chemotherapy
* Significance, P< .05.
Figure 7, Disease-free survival according to tumor grade.

48
Discussion
Liposarcoma (LPS) is the second or third most common soft tissue sarcoma (STS) ofadult life, and the incidence is estimated at 10-20% of all STS [1,2,3,4]. This tumor isprimarily a tumor of adult life, and generally shows a slight preference for the male sex[7,14,15]. In this series, the median age at presentation with a primary or recurrent LPSwas 49 and 56 years, respectively, and the male/female ratio was 1.03. Liposarcoma mostoften occur in the lower extremity (13-68%), and the retroperitoneum is the second mostcommon site (10-36%) [7,16,17]. This was confirmed in our series in which 38 of 69 LPS(55%) were situated in the lower extremity, primarily in the thigh (n=29). Theretroperitoneum was the second most frequent anatomical site (16%). Characteristically,most LPS are deeply seated, and the majority seems to take origin from large intermus-cular connective tissue spaces. Localization in the subcutaneous tissue is rare. However,noteworthy exceptions are the shoulder and head/neck area where tumors of smallersize, and shorter duration of symptoms, may extend into subcutaneous fat [18,19]. Inthis series, 8 of 69 LPS (12%) were subcutaneously seated, with the highest relative
Figure 8. (A) Disease-specific survival according to histological subtype. (B) Disease-specific survivalaccording to tumor grade.
A
B
Prognostic factors in liposarcoma Chapter 4

49
frequency in the head/neck region (60%), and the lowest in lower extremity, buttock,and retroperitoneum (5%, 0%, 0%) (Fig. 1).It is difficult to compare the distribution of histological subtypes in the literature be-cause different classifications have been used and anatomical distributions often vary.Undoubtedly, the myxoid type is by far the most common LPS. It is described mostfrequently in the literature [7,14,19], and was present in almost half of our patients (49%).The distribution of the other histological subtypes was in accordance with the seriesfrom the University of Texas M.D. Anderson Cancer Center (MDACC), published byEvans [7]. The relative frequency of PMLPS in our and the MDACC series was relativelylow (4-5%), compared to some reports in the literature [2,14]. However, the latter series,which report 23%-26% PMLPS, have a different distribution of anatomical sites and/ordid not recognize the dedifferentiated subtype.
After 5 years of follow-up, the overall local recurrence rate in the present series was 27%,which seems to be in accordance with other reports, although the figures should beinterpreted with caution because length of follow-up and distribution of histologicalsubtypes and anatomical sites vary widely [2,15,17]. One of the highest local recurrencerates (85%) is reported in a series by Evans, comparable with this present series, but thatstudy had a much longer follow-up with a minimum of 10 years [7]. This very long fol-low-up may be responsible for the high local recurrence rate because in LPS, which isdifferent from other STS, (very) late recurrences are common [2,17,19]. In this series,only 44% of all local recurrences were evident by 2 years, whereas 11% occured after 5years, the latest of which even occurered 8.5 years after treatment. From the literature,histological subtype (DDLPS and PMLPS) [19,20], retroperitoneal localization [15,21],recurrent presentation [19,21], and involved surgical margins [17,19] have been reportedas independent negative prognostic factors with regard to local recurrence. In the currentseries, retroperitoneal localization was the most important significant factor impairingthe local recurrence-free interval (Table 1). No local recurrences were encountered aftermultimodality treatment (surgery, radiotherapy, and chemotherapy), but numbers weretoo small to draw conclusions. Histological subtype, tumor grade and size, recurrentpresentation, margin status, and stage were not significant prognostic factors for localcontrol.
Once LPS recurred, multiple recurrences were common (overall 72%). LPS tend to recuruncontrollably, especially in the retroperitoneum, and may be fatal through local effects,as reported by others [22,23]. Dedifferentiation in recurrence is reported in well-differen-tiated retroperitoneal LPS and is associated with a poor outcome [6,7,15,22]. This featurewas encountered in 2 of 18 local recurrences (11%), both of which were retroperitonealWDLPS. Both patients died from an unresectable local recurrence, in the absence ofdistant metastases.
After 5 years of follow-up, the overall metastasis rate was 16%. Most metastases becameevident within the first three postoperative years (72%), as reported by others [2,19]. Latedistant failures appeared to be relatively rare because nearly all metastases were evidentby 5 years. As expected, the lung was the most common site for metastases (72%). Thisrelative frequency is comparable with the results of Kindblom [6], but is high compared
Chapter 4 Prognostic factors in liposarcoma

50
to others (38%-57%) [2,7]. The high tendency of MXLPS to metastasize to extrapulmonarysoft-tissue sites, reported in the literature (38-88%) [2,7,19,20,24], was confirmed in ourseries, in which 50% of metastasizing MXLPS did so to soft tissues. WDLPS did notmetastasize to distant sites.
From the literature, high tumor grade [2], pleomorphic subtype [19,20], round cell subtype(a poorly differentiated form of MXLPS) [24,25], and tumor necrosis [2] have been re-ported as independent factors associated with impaired metastasis-free interval. As shownin table 2, and Fig. 5 a,b, DDLPS, tumor grade II and III, and deep tumor location weresignificant determinants of metastatic outcome in the present series. After 5 years, gradeII and III LPS had the highest metastatic rate (49 and 50%, respectively), with the short-est median metastasis-free interval (≤ 3 years). Superficially located tumors did notmetastasize.
Because several different classification systems for LPS have been used, it is very diffi-cult to compare survival data from the literature. Reported independent negative prog-nostic factors are high tumor grade [21,26], tumor necrosis [2], tumor size ≥ 5 cm [19,26],tumor size ≥ 10 cm [21], histological subtype (PMLPS, DDLPS, round cell MXLPS)[14,18,19,20,24,25,27], recurrent presentation [21], and retroperitoneal localization[14,15,17,21]. In the current series, four factors (histological subtype, tumor size, tumorgrade, and tumor stage) were significantly associated with disease-free survival (Table 3,Fig. 6, 7). Patients with WDLPS, tumors ≤ 10 cm, grade I LPS, and AJCC stage I LPS hada significantly longer disease-free survival, but these factors appeared to be associatedwith each other.
Three factors that significantly determined disease-specific survival were histologicalsubtype, tumor grade, and type of resection (Table 4, Fig. 8 a,b). DDLPS, grade II-III, andnon-radical resections had a significantly worse disease-specific survival. After radicalresection, no patient died of the disease. Even when adjusted for type of resection, DDLPSand grade II-III LPS continued to have a significantly worse disease-specific survival,although both factors were associated with each other.
In the current series, we did not study the influence of the round cell subtype, a poorlydifferentiated variation of MXLPS, because round cell LPS was not recognized as a sepa-rate entity. Although strict criteria defining the prognostic significance of the round cellcomponent in MXLPS have not been established [27], there are reports showing that around cell component varying from >25% to even >5% is associated with a poor progno-sis [25,28]. Tumor necrosis was also not analyzed as a separate prognostic factor. How-ever, this factor, together with tumor differentiation and mitosis count, is one of thecornerstones of tumor grade classification, as described by Coindre et al [10]. The prog-nostic importance of retroperitoneal localization could not be demonstrated in this se-ries, but this may be a time-dependent issue, because at the end of the study period, 4 of11 patients (45%) with retroperitoneal LPS had died from their disease; 1 patient had anunresectable local recurrence at the end of the study period, which will be fatal in thenear future (9%). Moreover, in those patients with retroperitoneal STS without evidenceof disease, the duration of follow-up was relatively short.
Prognostic factors in liposarcoma Chapter 4

51
Conclusion
This series of 69 consecutive patients with completely resected, AJCC stage I-III LPSconfirms that liposarcoma is a quite heterogeneous disease, and that its outcome is de-termined to a significant degree by histological subtype, grade, size, stage, depth, andtype of resection. Compared with other soft tissue sarcomas, LPS has a relatively mildbiological behavior, with the exception of large, deeply located, dedifferentiated and/orgrade II-III LPS. With regard to local failure, retroperitoneal localization was an addi-tional negative prognostic factor. However, in contrast to some reports in the literature,we were not (yet) able to demonstrate a significant influence of retroperitoneal localiza-tion on survival.
Chapter 4 Prognostic factors in liposarcoma
References1. Torosian MH, Friedrich C, Godbold J, Hajdu SI, Brennan F. Soft tissue sarcoma: Initial characteristics and
prognostic factors in patients with and without metastatic disease. Sem Surg Oncol 1988; 4:13-19.2. Gustafson P, Rydholm A, Willén H, Baldetorp B, Fernö M, Åkerman M. Liposarcoma: a population-based
epidemiologic and prognostic study of features of 43 patients, including tumor DNA content. Int J Cancer 1993;55: 541-546.
3. Enzinger FM, Weiss SW. Liposarcoma. In: Soft Tissue Tumors. 3d Ed. St.Louis, MO: Mosby; 1995: 431-466.4. Nijhuis PHA, Schaapveld M, Otter R, Molenaar WM, Graaf van der WTA, Hoekstra HJ. Epidemiologic aspects
of soft tissue sarcomas (STS)-Consequences for the design of clinical STS trials. Eur J Cancer 1999; 35:1705-1710.
5. Shiu MH, Castro EB, Hajdu SI, Fortner JG. Surgical treatment of 297 soft tissue sarcomas of the lowerextremity. Ann Surg 1975; 82: 597-602.
6. Kindblom LG, Angervall L, and Svendsen P. Liposarcoma. A clinicopathologic, radiographic and prognosticstudy. Acta Pathol Microbiol Scand (A) 1975; Suppl. 253: 1-71.
7. Evans HL. Liposarcomas and atypical lipomatous tumors: a study of 66 cases followed for a minimum of 10years. Surg Pathol 1988; 1: 41-54.
8. Ham SJ, Graaf van der WTA, Pras E, Molenaar WM, Berg van den E, Hoekstra HJ. Soft tissue sarcoma of theextremities. A multimodality diagnostic and therapeutic approach. Cancer Treatment Reviews 1998; 24:373-391.
9. Geel van AN, Unnik van JAM, Keus RB. Diagnosis and treatment of soft tissue tumours: the Dutch nationwide-accepted consensus. Sarcoma 1998; 2: 183-191.
10. Coindre JM, Trojani M, Contesso G, et al. Reproducibility of a histopathologic grading system for adult soft tissuesarcoma. Cancer 1986; 58: 306-309.
11. Fleming ID, Cooper JS, Henson DE, et al, eds. Soft Tissue Sarcoma. In: AJCC Cancer Staging Manual. 5th Ed.Philadelphia, Lippincott-Raven Publishers. 1997:149-156.
12. Hoekstra HJ, Sindelar WF, Szabo BG, Kinsella TJ. Hemipelvectomy and intraoperative radiotherapy for boneand soft tissue sarcomas of the pelvic girdle. Radiotherapy and Oncology 1995; 37: 160-163.
13. Wijffels RTM, Mehta DM, Spauwen PHM, Hoekstra HJ. Limb-sparing treatment with surgery and IORT for asecond local recurrence of myxoid liposarcoma in the popliteal region, after previous surgery and high-doseradiation. J Surg Oncol 1993; 53: 64-67.
14. Enzinger FM and Weiss SW. Liposarcoma. A study of 103 cases. Virchows Arch Path Anat 1962; 335: 367-388.15. Henricks WH, Chu YC, Goldblum JR, Weiss SW. Dedifferentiated Liposarcoma. A clinicopathological analysis
of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol. 1997; 21: 271-281.16. O�Connor M and Snover DC. Liposarcoma. A review of factors influencing prognosis. Am Surg 1983; 49:
379-384.17. Lucas DR, Nascimento AG, Sanjay KSS, Rock MG. Well-differentiated liposarcoma: the Mayo Clinic
experience with 58 cases. Am J Clin Pathol 1994; 102: 677-683.18. Golledge J, Fisher C, Rhys-Evans PH. Head and neck liposarcoma. Cancer 1995; 76: 1051-1058.

52
19. Zagars GK, Goswitz MS, Pollack A. Liposarcoma: outcome and prognostic factors following conservationsurgery and radiation therapy. Int J Rad Oncol Biol Phys 1996; 36: 311-319.
20. Pearlstone DB, Pisters PWT, Bold RJ, et al. Patterns of recurrence in extremity liposarcoma. Implications forstaging and follow-up. Cancer 1999; 85: 85-92.
21. Linehan DC, Lewis JJ, Leung D, Brennan MF. Influence of biologic factors and anatomic site in completelyresected liposarcoma. J Clin Oncol 2000; 18:1637-1643.
22. Weiss SW, Rao VK. Well-differentiated liposarcoma (atypical lipoma) of deep soft tissue of the extremities,retroperitoneum, and miscellaneous sites: a follow-up study of 92 cases with analysis of the incidence of�dedifferentiation�. Am J Surg Pathol. 1992; 16: 1051-1058.
23. Reitan JB, Kaalhus O, Brennhovd IO, Sager EM, Stenwig AE, Talle K. Prognostic factors in liposarcoma.Cancer 1985; 55: 2482-2490.
24. Spillane AJ, Fisher C, Thomas JM. Myxoid liposarcoma- The frequency and the natural history of nonpulmonarysoft tissue metastases. Ann Surg Oncol 1999; 6: 389-394.
25. Smith TA, Easley KA, Goldblum JR. Myxoid/round cell liposarcoma of the extremities. A clinicopathologicstudy of 29 cases with particular attention to extent of round cell liposarcoma. Am J Surg Pathol 1996; 20:171-180.
26. Chang HR, Gaynor J, Tan C, Hajdu SI, Brennan MF. Multifactorial analysis of survival in primary extremityliposarcoma. World J Surg 1990; 14: 610-618.
27. Mentzel T, Fletcher CDM. Lipomatous tumours of soft tissues: an update. Virchows Arch 1995; 427: 353-363.28. Kilpatrick SE, Doyon J, Choong PFM, Sim FH, Nascimento AG. The clinicopathologic spectrum of myxoid and
round cell liposarcoma. A study of 95 cases. Cancer 1996; 77: 1450-1458.
Prognostic factors in liposarcoma Chapter 4

53
Chapter 5
Soft tissue sarcoma- compliance with guidelines
Nijhuis P.H.A.1, Schaapveld M.2, Otter R.2, Hoekstra H.J.1
1. Department of Surgical Oncology, Groningen University Hospital, The Netherlands.
2. Comprehensive Cancer Center North-Netherlands, Groningen, The Netherlands.
Cancer 2001; 91: 2186-2195
Chapter 5

54
Introduction
Soft tissue sarcomas (STS) are very uncommon tumors, with approximately 8100 newcases annually in the United States and 422 new cases annually in the Netherlands [1,2].Because of the rarity of these tumors in individual institutions, the great variability inclinical and histopathologic presentation, and the complex multimodality (limb-sparing)treatment, standardization of diagnosis and treatment seems necessary. This importantissue was recognized by the national Dutch Cooperative Group for Soft Tissue Tumors,founded in 1991, that organized a nationwide consensus meeting in 1993, resulting innationwide accepted guidelines with regard to the diagnosis and treatment of patientswith STS [3]. Compliance with guidelines is important for various reasons: 1) appropriatepreoperative investigations are vital parts in the process of care, accurate staging is essentialfor planning of appropriate treatment, and explicit guidelines improve clinical practice[4]; 2) frequently, factors other than scientific factors guide physicians in their decisionmaking [5]; 3) adherence to guidelines can result in significant institutional and nationalhealth care savings [6]; and 4) there are indications that the appropriateness of treatment(according to the guidelines) is related to patient outcome and survival [4,7,8]. Despitean increase in the development and dissemination of medical practice guidelines,compliance with the guidelines has often been low. No data have been reported thatdefinitively prove that adherence to guidelines improves care for patients with STS.However, in other medical situations, guidelines have been shown to improve patientcare if development, dissemination, and implementation are appropriate [4].
In the region of the Comprehensive Cancer Center North-Netherlands (CCCN), the firstguidelines for the diagnosis and treatment of patients with STS were developed in Feb-ruary 1983 by a cooperative group for rare tumors, consisting of specialists in surgicaloncology, medical oncology, radiotherapeutic oncology, and pathology from various hos-pitals in the CCCN region [9]. After realization of the first Dutch nationwide acceptedSTS guidelines in 1993, the CCCN guidelines were revised for the first time in 1994 [10].Later revisions took place in 1996 and 1998. All physicians who treated STS patientswithin the CCCN region received a written copy of these (revised) guidelines. It is impor-tant to realize that these Dutch guidelines have not been proven to improve patientsoutcome but have been agreed on by experts in the field and are expected to improveoutcome. This study is an analysis of how well these guidelines are being followed in theCCCN region.
Materials and methods
Between January 1989 and January 1996, 833 patients were diagnosed with primary STSin the CCCN region, an area of 2.1 million inhabitants. From these, 393 STS were excludedbecause of urogenital or gastrointestinal origin, and 40 were excluded because they werediagnosed as Kaposi sarcoma. From the remaining 400 patients with head and neck,trunk (retro)peritoneal, or extremity STS, another 49 patients were excluded from analysisfor various reasons, as listed in Table 1.Cancer registration at the CCCN started in 1986, although full coverage of the wholearea encompassed by the CCCN was achieved only from January 1, 1989. The main
Soft tissue sarcoma- compliance with guidelines Chapter 5

55
sources for the Cancer Registry are the national computerized pathology databank(PALGA) and the hospital discharge databank to which all Dutch hospitals provide infor-mation annually on discharge diagnoses of admitted patients. Specially trained CCCNemployees prospectively register data from the patients� clinical records. For this study,data on diagnosis, treatment, and follow-up were reviewed by CCCN employees and, ifnecessary, revised and completed, based on information recorded in the patients� chart.Within the CCCN district, the Groningen University Hospital is regarded as the referralcenter for patients with sarcomas and is indicated in the text as the center. Four of theother 18 hospitals in the CCCN region are teaching general hospitals, and 14 arenonteaching district general hospitals, all of which will be referred to as district hospi-tals. All district hospitals had access to well trained pathologists. Before 1995, tumorslides were reviewed by one of the STS experts at the Groningen University Hospital ifhistological typing was difficult or if patients were referred to the center. Since 1995, allSTS diagnoses in the CCCN region have been revised and discussed monthly by a panelof pathologists from the various hospitals within the CCCN region under the chairman-ship of experts from the center.
According to the 1983 guidelines, diagnostic work up of patients with STS should startwith an adequate physical examination, especially with regard to the tumor region. Lac-tate dehydrogenase and liver and renal function should be assessed (the latter two ofwhich had been dropped from the guidelines since the 1994 revision). Radiographicexamination should include a conventional X-ray of the tumor, followed by computedtomography (CT) and/or ultrasonography (US) of the tumor region. In 1994, the con-ventional X-ray became an optional examination, whereas magnetic resonance imaging(MRI) of the tumor became the imaging technique of choice. The presence of distantmetastatic disease was examined by pulmonary X-rays, and, if necessary, followed bypulmonary CT scan. A bone scintigraphy was obligatory until 1994, after which it had tobe performed only on indication [11].
Fine-needle aspiration and core biopsy were advised only to differentiate between mes-enchymal, epithelioid or lymphoid origin of the tumor. For definitive histologic diagno-sis, a surgical biopsy procedure was advocated. A (radical) excisional biopsy should beperformed only in patients with small, unsuspicious, superficial tumors. Although nospecific size was mentioned in the 1983 and 1994 guidelines, a small tumor size wasinterpreted as < 3 cm. In patients with larger (≥ 3 cm), suspicious, and/or deeply seatedtumors, an incisional biopsy had to be performed prior to definitive resection. Further-
Chapter 5 Soft tissue sarcoma- compliance with guidelines
Table 1. Reasons for exclusion.Reason nPediatric STS 15Medical chart lost 17Medical chart inaccessible 3Diagnosis postmortem 9Morphology not STS (revised) 3Other 2Total 49

56
more, it was stated in the guidelines that biopsy should be performed preferentially bythe same surgeon who would be responsible for the definitive treatment. In patientswith large tumors and/or tumors located at �difficult� locations, referral to the center wasrecommended.Actions were classified as appropriate, if the investigations performed and the interven-tions undertaken were in agreement with the guidelines, whereas deviations from theguidelines were classified as inappropriate. Comparison between groups was performedusing the chi-square test.
Results
Between January 1989 and January 1996, 351 patients with primary STS were diagnosedin the CCCN region. More than half of the patients were age > 60 years (n=201 patients;57%). Ninety-eight patients were even age > 70 years (28%). Figure 1 presents the agedistribution in patients who underwent biopsy and treatment in the center comparedwith patients who underwent biopsy in a district hospital but who were treated in theuniversity hospital and patients who were diagnosed and treated in district hospitals.There was an obvious age difference between patients who were treated in the center andthose who were treated in the district hospitals. More of the younger patients were treatedin the center, whereas more of the older patients were treated in community hospitals(P<0.001). Furthermore, there was a significant, nearly linear decline in the referral ratewith increasing patient age (P=0.002) (Fig. 2).Malignant fibrous histiocytoma was the most frequent histologic type (24%), followed by
Soft tissue sarcoma- compliance with guidelines Chapter 5
Figure 1. This chart shows the age distribution among patients who were treated at a specialized treat-ment center (Groningen University Hospital; center), referred for treatment to a specialized center (re-ferred), or treated at district general hospitals (district hospitals). The inset at right indicates patient agesin years.

57
liposarcoma (23%), leiomyosarcoma (21%), fibrosarcoma (7%), synovial sarcoma (5%),malignant peripheral nerve sheath tumors (5%), and rhabdomyosarcoma (4%). Six per-cent of the STS tumors were classified as otherwise specified, whereas 5% were classi-fied as not otherwise specified. The distribution of histologic types differed between thethree patient groups, with a higher incidence of synovial sarcomas in the specializedcenter (center: 8 of 59 patients; 14%; referred: 6 of 62 patients; 10%; district hospital: 3of 230 patients; 1%), and a higher incidence of leiomyosarcomas in district hospitals(center: 6 of 59 patients; 10%; referred: 6 of 62 patients; 10%; district hospital: 61 of 230patients; 27%). Most synovial sarcomas (n=12 tumors) were located in the lower limb(71%), especially around the knee (n=6 tumors; 36%). Overall, 17 of 73 leiomyosarcomaswere encountered in the retroperitoneum (23%).
Table 2 shows the distribution of STS according to anatomic site. Overall, most STS werelocated in the extremities (n=159 tumors; 45%). The distribution according to anatomicsite differed significantly between hospital groups (P <0.001). Most obvious were thehigher incidence of lower extremity STS in the center and the higher incidence of STS ofretroperitoneal, abdomen, pelvis, and head and neck in the community hospitals. In thespecialized center, nearly 50% of the STS were located in the lower extremity and hipcompared with only 21% of patients diagnosed and treated in community hospitals. Therelatively highest referral rates were in STS of the lower extremity and hip (45%) andupper limb and shoulder (19%). Fifteen of 40 referred patients with lower limb STS(38%) were referred to the center for hyperthermic isolated limb perfusion with tumornecrosis factor α and melphalan, followed by resection. Thirteen patients were referredfor reexcision of an STS that was diagnosed after an unplanned excision (�whoops-operation�) in a district hospital (33%). Two patients (5%) were referred for primary re-section because of radiographic suspicion of infiltration into bone or vascular structures.In the remaining 10 patients, the reason for referral could not be derived from the medical
Chapter 5 Soft tissue sarcoma- compliance with guidelines
Figure 2. This chart shows the referral pattern according to patient age (in years) among patients whowere or were not referred for treatment to a specialized center (Groningen University Hospital).
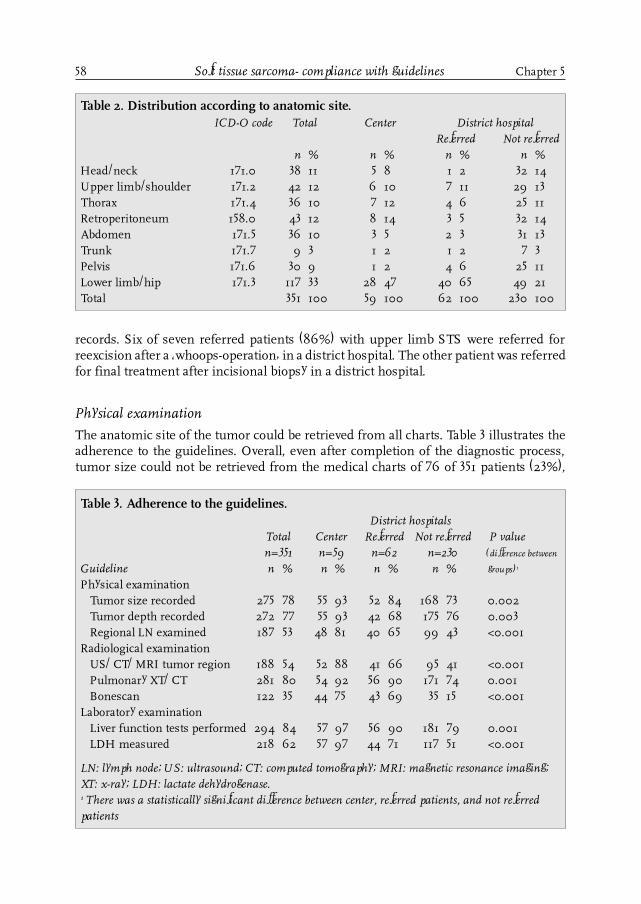
58
records. Six of seven referred patients (86%) with upper limb STS were referred forreexcision after a �whoops-operation� in a district hospital. The other patient was referredfor final treatment after incisional biopsy in a district hospital.
Physical examination
The anatomic site of the tumor could be retrieved from all charts. Table 3 illustrates theadherence to the guidelines. Overall, even after completion of the diagnostic process,tumor size could not be retrieved from the medical charts of 76 of 351 patients (23%),
Soft tissue sarcoma- compliance with guidelines Chapter 5
Table 2. Distribution according to anatomic site.ICD-O code Total Center District hospital
Referred Not referredn % n % n % n %
Head/neck 171.0 38 11 5 8 1 2 32 14Upper limb/shoulder 171.2 42 12 6 10 7 11 29 13Thorax 171.4 36 10 7 12 4 6 25 11Retroperitoneum 158.0 43 12 8 14 3 5 32 14Abdomen 171.5 36 10 3 5 2 3 31 13Trunk 171.7 9 3 1 2 1 2 7 3Pelvis 171.6 30 9 1 2 4 6 25 11Lower limb/hip 171.3 117 33 28 47 40 65 49 21Total 351 100 59 100 62 100 230 100
Table 3. Adherence to the guidelines.District hospitals
Total Center Referred Not referred P valuen=351 n=59 n=62 n=230 (difference between
Guideline n % n % n % n % groups)1
Physical examinationTumor size recorded 275 78 55 93 52 84 168 73 0.002Tumor depth recorded 272 77 55 93 42 68 175 76 0.003Regional LN examined 187 53 48 81 40 65 99 43 <0.001
Radiological examinationUS/ CT/ MRI tumor region 188 54 52 88 41 66 95 41 <0.001Pulmonary XT/ CT 281 80 54 92 56 90 171 74 0.001Bonescan 122 35 44 75 43 69 35 15 <0.001
Laboratory examinationLiver function tests performed 294 84 57 97 56 90 181 79 0.001LDH measured 218 62 57 97 44 71 117 51 <0.001
LN: lymph node; US: ultrasound; CT: computed tomography; MRI: magnetic resonance imaging;XT: x-ray; LDH: lactate dehydrogenase.1 There was a statistically significant difference between center, referred patients, and not referredpatients

59
with a highly significant difference between patients who were diagnosed and treated inthe center, referred patients, and patients who were diagnosed and treated in districthospitals (P =0.002). This difference was age related but was not influenced by differ-ences in anatomic distribution. In patients age < 65 years, tumor size was recordedsignificantly better in the center (P =0.002), whereas, for patients age ≥ 65 years, suchdifference could not be demonstrated (P =0.29). Figure 3 presents the distribution of therecorded tumor size, which was not significantly different between the three groups (P=0.227). Recording of tumor depth differed significantly between the three groups of
Chapter 5 Soft tissue sarcoma- compliance with guidelines
patients: it was worst in patients who were diagnosed and treated in district hospitals(Table 3). In the center, 38 of 59 tumors were deeply seated (64%), and 16 tumors werelocated superficially (28%), whereas, in the remaining 5 patients (8%), the exact tumordepth was described insufficiently or not at all. For patients who initially were diagnosedin a district hospital, these rates were 38 of 292 tumors (28%), 90 of 292 tumors (31%),and 119 of 292 tumors (41%), respectively. In the center, in 31 of 40 STS of the extremi-ties, head and neck, and pelvis, the regional lymph node status was recorded (78%). Forreferred and not referred patients, initially diagnosed in district hospitals, these rateswere 36 of 52 tumors (69%), and 49 of 135 tumors (36%), respectively.
Radiologic examination
Overall, radiologic examination of the tumor region by ultrasonography, CT, or MRI wasperformed in 188 of 351 patients (54%) (Table 3). Because CT/MRI became obligatoryonly after 1994, we examined how the change in guidelines had influenced clinical prac-tice. Before 1994, at least US or CT of the tumor region was performed in 83% of the
Figure 3. This chart shows the recorded tumor size among patients who were treated at a specializedtreatment center (Groningen University Hospital; center), referred for treatment to a specialized center(referred), or treated at district general hospitals (district hospitals).

60
patients who were diagnosed and treated in the center, in 56% of the referred patients,and in 41% of the patients who were diagnosed and treated in the community hospitals.For the period 1994- 1996, these rates were 100%, 83%, and 43%, respectively.Patients who were diagnosed and treated in the specialized center more often under-went a plain pulmonary X-ray and/or pulmonary CT scan (Table 3). Furthermore, in thecenter, most of the patients had both radiologic examinations (42 of 54 patients; 78%),whereas, in the district hospitals, in only 41 of 171 patients were both radiologic examina-tions performed (24%). In the center, 76% of the patients with normal chest X-raysunderwent an additional pulmonary CT scan compared with only 24% of comparablepatients in the district hospitals. In case the chest X-ray was suspect for metastatic dis-ease, then these rates were 83% and 44%, respectively.
Overall, a bone scan was made in only 122 of 351 patients (35%) (Table 3). In patients whowere treated finally in the specialized center, a bone scan was performed significantlymore often. This difference could be demonstrated for all different anatomic sites.According to the guidelines from 1983- 1993, this examination was obligatory in all STSpatients. However, since 1994, bone scans were performed on indication. During theperiod before 1994, 73% of patients who were diagnosed and treated in the center had abone scan compared with 62% of referred patients and only 16% of patients who werediagnosed and treated in district hospitals. During the period 1994-1995, these rateswere 79%, 82%, and 14%, respectively, resulting in an overall increase from 33% in1989-1993 to 39% after 1994.
Laboratory examination
Overall, liver function tests were performed in 294 of 351 patients (84%). Again, thedifference between the three patient groups was significant (Table 3). Although liverfunction tests were abandoned in the 1994 revision of the guidelines, no change in prac-tice was observed. From 1989 to 1993, at least one liver function test was performed in199 of 239 patients (83%), whereas, in the period 1994- 1995, this number increased to95 of 112 patients (85%). The median and mean numbers of liver function tests per-formed were equal in both periods (1989-1993, 4 and 3.4, respectively; 1994- 1996, 4and 3.3, respectively). Serum lactate dehydrogenase also was determined most frequentlyin the specialized center (Table 3).
Diagnostic procedure
Overall, 100 of 233 tumors ≥3 cm (43%) were diagnosed after an unplanned excision(�whoops-operation�). The management of patients with these larger tumors differedsignificantly between the specialized center and community hospitals (P <0.001). Ac-cording to the guidelines, an incisional biopsy had to be performed in patients withthese larger tumors. In the center, this guideline was followed in 32 of 50 patients withtumors ≥ 3 cm (64%) compared with 19 of 47 referred patients with a tumor of that size(40%), and only 34 of 136 patients who ware diagnosed and treated in community hospi-tals (25%). Tables 4 and 5 present the management of patients with tumors ≥ 3 cm accord-ing to anatomic site. The diagnostic management of patients with larger retroperitoneal
Soft tissue sarcoma- compliance with guidelines Chapter 5

61
and abdominal STS did not differ between the specialized center and the communityhospitals (P =0.93). However, the management of patients did differ for those with largerSTS at other sites (P <0.001).
In the center, in four patients with tumors ≥ 3 cm who underwent an unplanned exci-sion, the medical chart explicitly stated the surgeon�s conviction of the benign nature ofthe tumor (8%). Such statements were encountered in the charts of 17 of 183 patientswith a tumor ≥ 3 cm who were diagnosed initially in community hospitals (9%). Five ofthe latter patients were referred to the specialized center for definitive treatment.
To determine the influence of experience on adherence to the guidelines, we divideddistrict hospitals into two groups according to the number of patients diagnosed in thestudy period (≥ 15 or < 15). In six district hospitals, which included the four teachinghospitals, 15 or more patients were diagnosed. Table 6 summarizes the adherence to theindividual guidelines and compares the specialized center with district hospitals, whichare divided into two groups according to patient volume. For all individual guidelines,compliance was significantly better in the specialized center. In district hospitals, pa-tient volume had no significant impact on compliance, except for one important item,
Chapter 5 Soft tissue sarcoma- compliance with guidelines
Table 4. Biopsy procedure in tumors ≥ 3 cm: Retroperitoneal and abdominal STS.Procedure Total Center District hospitals1
n % n % n %Fine-needle aspiration 5 8 1 9 4 7Core biopsy 11 17 1 9 10 19Incisional biopsy 13 20 3 27 10 19Incision-frozen section-excision 2 3 - - 2 4No biopsy prior to excision 26 41 5 46 21 40Other 6 11 1 9 6 11Total 64 100 11 100 53 100
1 Referred and not referred patients were included.
Table 5. Biopsy procedure in tumors ≥ 3 cm: Retroperitoneal and abdominal STSexcluded.Procedure Total Center District hospitals1
n % n % n %Fine-needle aspiration 12 7 - - 12 9Core biopsy 9 5 1 3 8 6Incisional biopsy 69 41 28 71 41 32Incision-frozen section-excision 1 1 1 3 - -No biopsy prior to excision 74 44 9 23 65 50Other 4 2 - - 4 3Total 169 100 39 100 130 100
1 Referred and not referred patients included.

62
i.e., the management of patients with larger tumors. In district hospitals, where fewerthan 15 patients were diagnosed with STS in the 7-years period, significantly more often,an inadequate biopsy or even no biopsy was performed prior to resection of the largerand, thus, suspicious soft tissue tumors (P =0.02). Furthermore, there was a trend to-ward better preoperative imaging in district hospitals with larger patient volumes com-pared with hospitals that had less experience (P =0.056). The difference in the diagnos-tic approach for patients with larger tumors was not influenced by tumor depth, as shownin Table 7. For patients with both superficially located and deeply seated STS measuring≥ 3 cm, the guideline for performing an incisional biopsy was best followed in the spe-cialized center followed by �higher� volume�district hospitals. In �lower volume� districthospitals the compliance rate was less than 20%.
Discussion
Because of the rarity of STS, many surgeons are unfamiliar with these tumors, oftenleading to unforeseen findings during or after an operation for a soft tissue mass. Inad-equate treatment, especially inadequate skin incision, intraoperative tumor spillage,postoperative bleeding, or hematoma, can seriously hamper definitive surgery, that, of-ten, is already very challenging. Furthermore, it can increase the morbidity of the defini-tive resection, increase the necessity for adjuvant radiation therapy, and possibly eveninterfere with the sparing of a limb. Although it seems likely that such inadequate pri-mary resections negatively influence locoregional control or even prognosis, this has not
Soft tissue sarcoma- compliance with guidelines Chapter 5
Table 6. Adherence to individual guidelines according to patient volume.District hospitals
≥15 patients <15 patients P valueTotal Center /7 yrs (n=173) /7 yrs (n=119) (difference
n=351 n=59 n=62 n=230 between
Guideline n % n % n % n % groups)1
Physical examinationTumor size recorded 275 78 55 93 135 78 85 71 0.004Tumor depth recorded 272 77 55 93 131 76 86 72 0.005Regional LN examined 187 53 48 81 84 49 55 46 <0.001
Radiological examinationUS/ CT/ MRI tumor region 188 54 52 88 89 51 47 40 <0.001Pulmonary XT/ CT 281 80 54 92 138 80 89 75 0.03Bonescan 122 35 44 75 48 28 30 25 <0.001
Laboratory examinationLiver function tests performed 294 84 57 97 140 81 97 82 0.01LDH measured 218 62 57 97 101 58 60 50 <0.001
Incisional biopsy in tumors ≥3 cm 85/233 36 32/50 64 40/113 35 13/70 19 <0.001
LN: lymph node; US: ultrasound; CT: computed tomography; MRI: magnetic resonance imaging; XT:x-ray; LDH: lactate dehydrogenase.1 There was a statistically significant difference between center and lower and higher volume districthospitals.

63Chapter 5 Soft tissue sarcoma- compliance with guidelines
been demonstrated in patients with STS, provided that negative surgical margins can beachieved after definitive resection.There are only limited studies with respect to adherence to guidelines. Progress in can-cer treatment can be made only if guidelines are followed. This may explain why cancerpatients who are treated in trial protocols have a better prognosis [12,13]. In patients withcervical and endometrial carcinoma, observational studies in seven districts of the SouthEast Thames Regional Health Authority demonstrated that survival was influenced notonly by biologic and demographic factors but also, independently, by appropriateness ofcare according to guidelines [7,8]. This seems to be a particularly important finding,because, unlike biologic and demographic factors, treatment patterns can be changed.We therefore analyzed the diagnostic process in patients with primary STS in the north-eastern part of the Netherlands, with special attention to differences in adherence to theguidelines according to specialization and experience.
The majority of patients who present with a primary STS, as demonstrated in otherstudies [14,15,16,17], were age > 60 years (57%). Younger patients were treated moreoften in the center, a phenomenon also seen in the referral pattern of patients accordingto age. In contrast to patients ages 15-29 years, of whom 43% were sent to the specializedcenter, only 14% of patients age > 60 years, by far the largest group, were referred. Oneof the reasons might be a more aggressive approach of younger patients compared witha more conservative and often fatalistic attitude toward older patients. Cancer in theelderly is treated surprisingly poorly, and the behavior of the disease in older patientsoften is understood poorly [18]. Several other studies have shown clearly that cancer therapyvaries significantly with age and that the percentage of patients receiving definitive therapydeclines with increasing age [19,20,21]. One of the contributing factors may be that, inolder patients, physicians are less likely to recommend specialist consultation [20].Unfortunately, the current study did not examine this issue. Other factors may beconsidered, although, in the Netherlands, financial issues and difficulties of getting tothe medical center seem to be of minor importance because of excellent public healthinsurance and relatively short traveling distances combined with good public transportfacilities.The distribution of histologic types was in concordance with the literature [14-17]. Most
Table 7. Incisional biopsy procedure in tumors ≥ 3 cm according to tumor depthand patient volume.
District hospitals P value≥15 patients/ <15 patients/ (difference
Total Center 7 yrs 7 yrs between
Tumor depth n % n % n % n % groups)
Superficial 19/55 35 9/15 60 7/19 37 3/21 14 <0.0011
Deep 40/92 43 21/31 68 15/40 38 4/21 19 0.0011
Uncertain or unknown 26/86 30 2/4 50 18/54 33 6/28 21 0.36Total 85/233 36 32/50 64 40/113 35 13/70 19 <0.0011
1 There was a statistically significant difference between center and lower and higher volumedistrict hospitals.

64 Soft tissue sarcoma- compliance with guidelines Chapter 5
obvious differences were the higher incidence of synovial sarcomas in the center and thehigher incidence of leiomyosarcoma in the community hospitals. Synovial sarcomas occurpredominantly in the lower extremity, where they tend to arise in the vicinity of largejoints, especially the knee region. Furthermore, they are most prevalent in younger patients[22], which was shown in the current series. Therefore, the higher incidence of synovialsarcomas in the center can be explained by the higher incidence of younger patients andlower extremity STS. Because leiomyosarcomas have the highest incidence in patientsage > 65 years [17] and significantly more patients age > 65 years were diagnosed initiallyin district hospitals, the observed higher frequency of leiomyosarcomas diagnosed indistrict hospitals was not unexpected.
One of the decisive factors for definitive (surgical) treatment is a correct diagnostic pro-cedure. During the study period, guidelines advised the performance of an incisionalbiopsy for patients with larger tumors. Therefore, the adequacy of the biopsy was evalu-ated in that perspective, although core biopsy has been promoted as a very elegant diag-nostic tool and although some surgeons have suggested that definitive resection withoutbiopsy is the best treatment for patients with STS, especially if the tumors are containedwithin a muscle, provided that a �wide� margin can be obtained. Notwithstanding theguideline, 43% of tumors ≥ 3 cm was diagnosed after excisional biopsy. This seems verydisappointing but is in accordance with the results of a nationwide survey of the man-agement of patients with STS in the United States. In that study, which was carried outunder the auspices of the Commission on Cancer of the American College of Surgeons,49% of patients with STS were diagnosed by excisional biopsy [23].The current study demonstrated a significant difference in the diagnostic managementof patients with larger STS between the specialized center, �higher volume� district hos-pitals and �lower volume� district hospitals, in both superficially located STS and deeplylocated STS. Because we could not demonstrate a difference between the specializedcenter and the other hospitals in the diagnostic approach of larger retroperitoneal andabdominal STS (Table 4), the difference in biopsy procedure in patients with deeplylocated tumors must be explained by different approaches in patients with larger STSlocated at other anatomic sites. Because the appropriateness of the biopsy procedure isvery important for definitive treatment, a concentration of patients with these rare tumorsin a limited number of hospitals seems advisable. Progress in the treatment of patientswith STS can be made only by combined modality treatment, which is another argumentfor treating patients with these tumors in defined centers. In addition, to get more in-sight into the disease itself, modern sophisticated basic research is needed, which can beperformed only in such centers.
A remarkable finding was that, in 9% of patients with STS ≥ 3 cm, the patient�s medicalchart contained an explicit statement of the surgeon�s conviction of the benign nature ofthe tumor, although its greater size should have raised some suspicion. It appears thatthe optimal diagnostic approach for patients with soft tissue masses should be an impor-tant topic for educational programs for practicing surgeons.In their review of baseline institutional compliance with the National ComprehensiveCancer Network guidelines regarding patients with non-small-cell lung carcinoma, Walshand Winn demonstrated excessive, inappropriate, and expensive radiologic testing in

65Chapter 5 Soft tissue sarcoma- compliance with guidelines
50% of patients [6]. Their conclusion was that significant institutional and national healthcare savings would result if clinicians would to use and implement the recommenda-tions put forth in the guidelines. This also can be demonstrated in the current study.Although liver function tests and bone scans were no longer obligatory because of therevisions to the guidelines in 1994, we could not demonstrate a change in clinical prac-tice. Instead of a decline, the number of liver function tests slightly increased by 2%,whereas the number of bone scans increased by 18%. It seems obvious that adherence tothe guidelines would have resulted in considerable savings.In a systematic review of rigorous evaluations of clinical guidelines, Grimshaw and Russellconcluded that explicit guidelines do improve clinical practice and patient outcome ifdevelopment, dissemination, implementation, and evaluation all are appropriate [4]. Intheir classification of clinical guidelines, guidelines with the highest probability of beingeffective are those that are developed internally, with specific educational interventionsand patient-specific reminders at the time of consultation. Conversely, guidelines withthe lowest probability of being effective are those that are developed nationally and aredisseminated by publication in a journal without patient specific feedback but with onlygeneral reminders [24]. According to this classification system, the probability that theCCCN STS guidelines would have been effective seems to be rather low. These guide-lines were developed locoregionally by a cooperative group of oncologic specialists fromvarious hospitals in the CCCN region. Thereafter, written guidelines were sent to alltreating physicians in the region. However, adequate implementation and quality-con-trol programs were lacking. With regard to the dissemination strategy, which was de-signed to ensure that target clinicians had a knowledge of the guidelines, the review byRussell and Grimshaw suggested that the greater the educational component, the greaterthe likelihood that the guidelines would be adopted into clinicians� practices. Specificlocoregional educational initiatives seem to be more effective than mailing to targetedclinicians or publication in professional journals. Concerning implementation strategythat is designed to encourage clinicians to adopt the guidelines, Russell and Grimshawsuggested that strategies specific to individual patients (specific feedback) more likelywould encourage clinician compliance.For the introduction of future guidelines for the diagnosis and treatment of patients withthese rare tumors, knowledge of this complex process and its crucial steps is vital.Guidelines should be developed by those who are to use them, should be disseminatedby enthusiastic educational programs, and should be implemented by a quality-controlprogram with patient specific feedback.
Conclusions
In many aspects of the diagnostic process of STS, existing guidelines are not followed,especially in community hospitals. Adherence to individual guidelines is significantlybetter in specialized centers. Although, in district hospitals, patient volume had no sig-nificant impact on adherence to most guidelines, the biopsy procedure in larger tumorswas significantly better in higher volume hospitals. Because the correct diagnosis, stag-ing, and treatment of patients has been shown to be crucial in the management of manytumors, concentration of patients with STS in a limited number of hospitals, and inten-sified collaboration with specialized centers seem advisable.

66
Because STS is a disease of the elderly, special attention should be paid to the olderpatients, because significantly more often, they are not referred to a specialized centerfor treatment.
References1. Greenlee RT, Murray T, Bolden S, Wingo PA. Cancer statistics, 2000. CaCancer J Clin 2000; 50: 7-33.2. Visser O, Coebergh JWW, Schouten LJ, Dijck JAAM, editors. Incidence of cancer in the Netherlands 1996.
Utrecht: Vereniging van Integrale Kankercentra, 2000.3. Geel van AN, Unnik van JAM, Keus RB. Diagnosis and treatment of soft tissue tumours: the Dutch nationwide-
accepted consensus. Sarcoma 1998; 2:183-191.4. Grimshaw JM, Russell IT. Effect of clinical guidelines on medical practice: a systematic review of rigorous
evaluations. Lancet 1993; 342: 1317-1322.5. Liberati A, Apolone G, Nicolucci A, Confalonieri C, Fossati R, Grilli R, et al. The role of attitudes, beliefs, and
personal characteristics of Italian physicians in the surgical treatment of early breast cancer. Am J Public Health1991; 81: 38-42.
6. Walsh GL, Winn RJ. Baseline institutional compliance with NCCN guidelines: non-small-cell lung cancer.Oncology-Huntingt 1997; 11: 161-170.
7. Wolfe CDA, Tilling K, Bourne HM, Raju KS. Variations in the screening history and appropriateness ofmanagement of cervical cancer in southeast England. Eur J Cancer 1996; 32A: 1198-1204.
8. Tilling K, Wolfe CDA, Raju KS. Variations in the management and survival of women with endometrial cancerin southeast England. Eur J Gynaec Oncol 1998; 19: 64-68.
9. R. Otter, editor. Richtlijnen voor diagnostiek en behandeling van premaligne en maligne aandoeningen in deIKN-regio 1992. Groningen; IKN, 1992. ISBN 90-74114-03-2: 394-399.
10. R. Otter, editor. Richtlijnen voor diagnostiek en behandeling van premaligne en maligne aandoeningen in deIKN-regio 1994. Groningen; IKN, 1992. ISBN 90-74114-06-7: 452-458.
11. Jager PL, Hoekstra HJ, Leeuw AJ, van der Graaf WT, de Vries EG de, Piers DA. Routine bone scintigraphy inprimary staging of soft tissue sarcoma. Cancer 2000; 89: 1726-1731.
12. Stiller CA, Draper GJ. Treatment centre size, entry to trials, and survival in acute lymphoblastic leukaemia.Arch Dis Child 1989; 64: 657-661.
13. Stiller CA. Centralised treatment entry to trials and survival. Br J Cancer 1994; 70: 352-362.14. Hartley AL, Blair V, Harris M, Birch JM, Banerjee SS, Freemont AJ, et al. Sarcomas in North West England. II.
Incidence. Br J Cancer 1991; 64: 1145-1150.15. Gustafson P. Soft tissue sarcoma. Epidemiology and prognosis in 508 patients. Acta Orthop Scand 1994;
65 (suppl 259): 1-31.16. Pollock RE, Karnell LH, Menck HR, Winchester DP. The National Cancer Data Base report on soft tissue
sarcoma. Cancer 1996; 78: 2247-2257.17. Nijhuis PHA, Schaapveld M, Otter R, van der Graaf WTA, Hoekstra HJ. Epidemiologic aspects of soft tissue
sarcomas- consequences for the design of clinical STS trials. Eur J Cancer 1999; 35:1705-1710.18. Fentiman IS, Tirelli U, Monfardini S, Schneider M, Festen J, Cognetti F, et al. Cancer in the elderly: why so
badly treated? Lancet 1990; 335: 1020-1022.19. Samet JM, Hunt WC, Key Ch, Humble ChG, Goodwin JS. Choice of cancer therapy varies with age of patient.
JAMA 1986; 255: 3385-3390.20. Newcomb PA, Carbone PP. Cancer treatment and age: patient perspectives. J Nat Cancer Inst 1993; 85:
1580-1584.21. Goodwin JS, Hunt WC, Samet JM. Determinants of cancer therapy in elderly patients. Cancer 1993; 72:
594-601.22. Enzinger FM, Weiss SW. General considerations. In: Enzinger FM, Weiss SW. Soft tissue tumors. 3rd ed.
St. Louis, Mosby, 1995: 757-786.23. Lawrence W Jr., Donegan WL, Natarajan N, Mettlin C, Beart R, Winchester D. Adult soft tissue sarcomas.
Ann Surg 1987; 205: 349-359.24. Russell IT, Grimshaw JM. The effectiveness of referral guidelines: a review of the methods and findings of
published evaluations. In: Roland M, Coulter A. Hospital Referrals. Oxford: Oxford University Press, 1992:179-211.
Soft tissue sarcoma- compliance with guidelines Chapter 5

67Chapter 6
Chapter 6
Prognostic relevance of cytogenetic changes insoft tissue sarcomas
Nijhuis P.H.A.1, Hoekstra H. J.1, Plaat B.E.C.2, van der Graaf W.T.A.3, Sluiter W.J.4,Molenaar W.M.2, van den Berg E.5
1.Department of Surgical Oncology, Groningen University Hospital, Groningen, The Netherlands 2. Department of
Pathology, Groningen University Hospital, Groningen, The Netherlands 3. Department of Medical Oncology,
Groningen University Hospital, Groningen, The Netherlands 4. Department of Internal Medicin, Groningen
University Hospital, Groningen, The Netherlands 5. Department of Medical Genetics, University of Groningen,
Groningen, The Netherlands
submitted

68
Introduction
The prognosis of soft tissue sarcomas (STS) largely depends on tumor-specific characteris-tics, as histological (sub) type, tumor grade, size and site [1-3]. In recent years, significantprogress has been made in identifying chromosomal abnormalities in solid tumors.Characteristic cytogenetic alterations have been demonstrated in several STS, and oftenhave diagnostic relevance [4-7]. Well-known rearrangements include the translocation t(11;22)(q24; q11.2-12) in Ewing�s sarcoma and primitive neuroectodermal tumors, t(12;16) (q13;p11) in myxoid liposarcomas (MXLPS), t(X;18)(p11.2; q11.2) in synovial sarcomas, andt(2;13)(q35- 37;q14) in alveolar rhabdomyosarcomas [7-9]. Most studies deal with the specificchromosomal rearrangements within histological tumor types and refer to their diagnosticrelevance, whereas only a few report on prognosis of such aberrations [10-14].
The present study analyzes the prognostic significance of cytogenetic changes observedin soft-tissue sarcomas, using a computer-assisted cytogenetic analysis [13]. A databasewas constructed, which permits the detection of statistically significant, non-randomchromosomal aberrations and allows direct comparison of different karyotypes. Specialattention was paid to cytogenetic differences between metastatic and non-metastatic STSand between patients who died of the disease and who did not.
Materials and methods
For this study, material for cytogenetic analysis was obtained from consecutive STS speci-mens submitted for pathologic examination at the Department of Pathology of theGroningen University Hospital from 1984- 1993. All STS were reviewed by a pathologistwith special interest and experience in STS (WMM), and histopathologically classifiedaccording to the criteria described by Enzinger and Weiss [6]. Cytogenetic analysis wasperformed at the department of Medical Genetics at the University of Groningen. Thecriteria for inclusion in the current study were 1) a (reviewed) histological diagnosis of aprimary or locally recurrent malignant mesenchymal tumor, located in the soft tissues,which had not been previously treated with radiotherapy and/or chemotherapy, 2) a suc-cessful karyotype, and 3) the availability of complete clinical follow-up. Mesenchymalproliferations of parenchymal organs were excluded.For genetic analysis, part of the tissue specimen was minced with scalpels, incubated in acollagenase-DNAse solution and cultured in RPMI 1640 supplemented with 16% FCS,glutamine and antibiotics. After short-term culture, cells were harvested, chromosomeswere G-banded using trypsin/pancreatin, and karyotypes were described according to theISCN 1995 Guidelines for Cancer [15]. If more than one tumor per patient was described, itwas decided to use the karyotype of one tumor, preferably the primary tumor, to avoidoverrepresentation.
The database consisted of four main parts related to the described karyotype: 1) patient�scharacteristics, 2) histopathological data, 3) gain and loss of chromosomal material, and 4)structural rearrangements. After interpretation of the karyotype, the gains and losses ofchromosomal material were entered, as described by Plaat et al [13]. Each chromosome wasdivided according to the ideogram at 400 bands level as described by the ISCN, in such a
Prognostic relevance of cytogenetic changes in soft tissue sarcomas Chapter 6

69
way that the net gains and losses in 1p11-13 were summarized in 1p1, changes in 1p21-22were summarized in 1p2, etc. In case of loss in a particular region a �1 was entered. If thesame region was lost in both chromosomes, a �2 was entered. Similarly, if gain occurred inone of the chromosomal regions a +1 was entered, etc. Only changes as compared to theconstitutional karyotype were evaluated.Data were analyzed for the number of tumors with gains or losses in a particular chromo-somal region, and the mean change in chromosomal material per chromosomal region.Mean change in chromosomal material in each of the 86 chromosomal regions wasexpressed as a chromosomal change ratio (CCR), which was defined as the change in aspecific chromosomal region as compared to a normal diploid karyotype. Only full abnormalkaryotypes were used for the analysis of the over- or under representation of chromosomesor chromosomal regions. In the analysis of over- or under representation, marker-chromosomes were not included, because of the fact that these are structurally rearrangedchromosomes, in which no part can be identified. Chromosomal changes were comparedbetween patients with metastasizing STS and those with STS that had not metastasized,and between patients with no evidence of disease and patients who had died from theirdisease. Graphs were constructed to visualize the change in chromosomal material, andthe differences between selected groups.To identify chromosomal regions with significant gains or losses, 95% confidence inter-vals (CI) were determined. Survival curves were calculated by the method of Kaplan andMeier. Cox�s proportional hazards regression model was used to assess the importance ofspecific chromosomal alterations in overall and metastasis-free survival. A P-value <0.05was considered statistically significant.
Results
Patients
Forty-three patients met the inclusion criteria, 23 males and 20 females (53% and 47%,respectively), with a mean age of 45 (range 2-81) years. Most STS (n=38) were primarytumors (88%), the others (n=5) were local recurrences (12%). Liposarcoma (LPS) was themost common histological type (n=20, 47%), followed by synovial sarcoma (n=5, 12%),malignant fibrous histiocytoma (MFH) (n=4, 9%), and rhabdomyosarcoma (n=2, 5%) [Table1].Table 2 presents the patients� status of disease after a median and mean follow-up of 55 and
Chapter 6 Prognostic relevance of cytogenetic changes in soft tissue sarcomas
Table 1. Distribution according to histopathology (n=43).Histopathology nLiposarcoma 20Synovial sarcoma 5Sarcoma nos 5Malignant fibrous histiocytoma 4Rhabdomyosarcoma 2Others 7
Sarcoma nos: sarcoma with no other specification
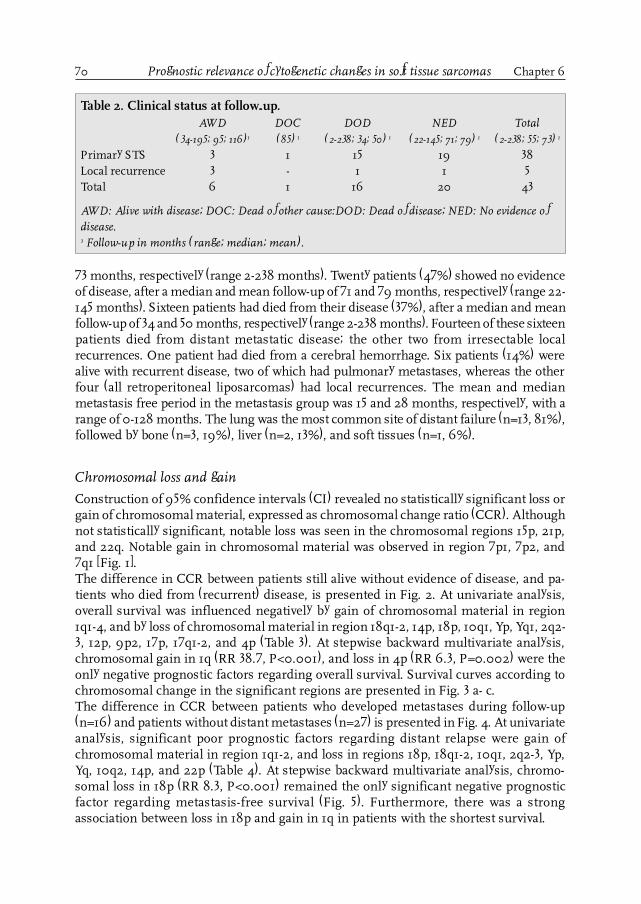
70
73 months, respectively (range 2-238 months). Twenty patients (47%) showed no evidenceof disease, after a median and mean follow-up of 71 and 79 months, respectively (range 22-145 months). Sixteen patients had died from their disease (37%), after a median and meanfollow-up of 34 and 50 months, respectively (range 2-238 months). Fourteen of these sixteenpatients died from distant metastatic disease; the other two from irresectable localrecurrences. One patient had died from a cerebral hemorrhage. Six patients (14%) werealive with recurrent disease, two of which had pulmonary metastases, whereas the otherfour (all retroperitoneal liposarcomas) had local recurrences. The mean and medianmetastasis free period in the metastasis group was 15 and 28 months, respectively, with arange of 0-128 months. The lung was the most common site of distant failure (n=13, 81%),followed by bone (n=3, 19%), liver (n=2, 13%), and soft tissues (n=1, 6%).
Chromosomal loss and gain
Construction of 95% confidence intervals (CI) revealed no statistically significant loss orgain of chromosomal material, expressed as chromosomal change ratio (CCR). Althoughnot statistically significant, notable loss was seen in the chromosomal regions 15p, 21p,and 22q. Notable gain in chromosomal material was observed in region 7p1, 7p2, and7q1 [Fig. 1].The difference in CCR between patients still alive without evidence of disease, and pa-tients who died from (recurrent) disease, is presented in Fig. 2. At univariate analysis,overall survival was influenced negatively by gain of chromosomal material in region1q1-4, and by loss of chromosomal material in region 18q1-2, 14p, 18p, 10q1, Yp, Yq1, 2q2-3, 12p, 9p2, 17p, 17q1-2, and 4p (Table 3). At stepwise backward multivariate analysis,chromosomal gain in 1q (RR 38.7, P<0.001), and loss in 4p (RR 6.3, P=0.002) were theonly negative prognostic factors regarding overall survival. Survival curves according tochromosomal change in the significant regions are presented in Fig. 3 a- c.The difference in CCR between patients who developed metastases during follow-up(n=16) and patients without distant metastases (n=27) is presented in Fig. 4. At univariateanalysis, significant poor prognostic factors regarding distant relapse were gain ofchromosomal material in region 1q1-2, and loss in regions 18p, 18q1-2, 10q1, 2q2-3, Yp,Yq, 10q2, 14p, and 22p (Table 4). At stepwise backward multivariate analysis, chromo-somal loss in 18p (RR 8.3, P<0.001) remained the only significant negative prognosticfactor regarding metastasis-free survival (Fig. 5). Furthermore, there was a strongassociation between loss in 18p and gain in 1q in patients with the shortest survival.
Prognostic relevance of cytogenetic changes in soft tissue sarcomas Chapter 6
Table 2. Clinical status at follow-up.AWD DOC DOD NED Total
(34-195; 95; 116)1 (85) 1 (2-238; 34; 50) 1 (22-145; 71; 79) 1 (2-238; 55; 73) 1
Primary STS 3 1 15 19 38Local recurrence 3 - 1 1 5Total 6 1 16 20 43
AWD: Alive with disease; DOC: Dead of other cause:DOD: Dead of disease; NED: No evidence ofdisease.1 Follow-up in months (range; median; mean).

71Chapter 6 Prognostic relevance of cytogenetic changes in soft tissue sarcomas
Figure 1. Mean CCR with 95% Confidence Interval of all cases
Figure 2. Mean CCR in patients with no evidence of disease (NED) versus patients who died of thedisease (DOD)

72 Prognostic relevance of cytogenetic changes in soft tissue sarcomas Chapter 6
Table 3. Overall Survival: statistically significant chromosomal changes atunivariate analysis.Chromosomal Change Relative Risk (RR) P-value
Gain of 1q1 7.7 <0.001Loss of 14p 6.3 <0.001Loss of 18p 6.3 <0.001Gain of 1q3 6.3 0.001Gain of 1q2 5 0.006Loss of 10q1 5.6 0.009Gain of 1q4 4.4 0.01Loss of Yp 5.6 0.01Loss of Yq 5.6 0.012Loss of 2q2 5 0.013
Chromosomal Change Relative Risk (RR) P-value
Loss of 2q3 5 0.013Loss of 12p 3 0.016Loss of 18q2 4.8 0.029Loss of 9p2 2.2 0.037Loss of 17p 2.2 0.037Loss of 17q1 2.2 0.037Loss of 18q1 7.7 0.044Loss of 4p 3 0.045Loss of 17q2 2.1 0.045
Figure 3a. Overall survival and gain in chromosome 1q
Figure 3b. Overall survival and loss in chromosome 4p
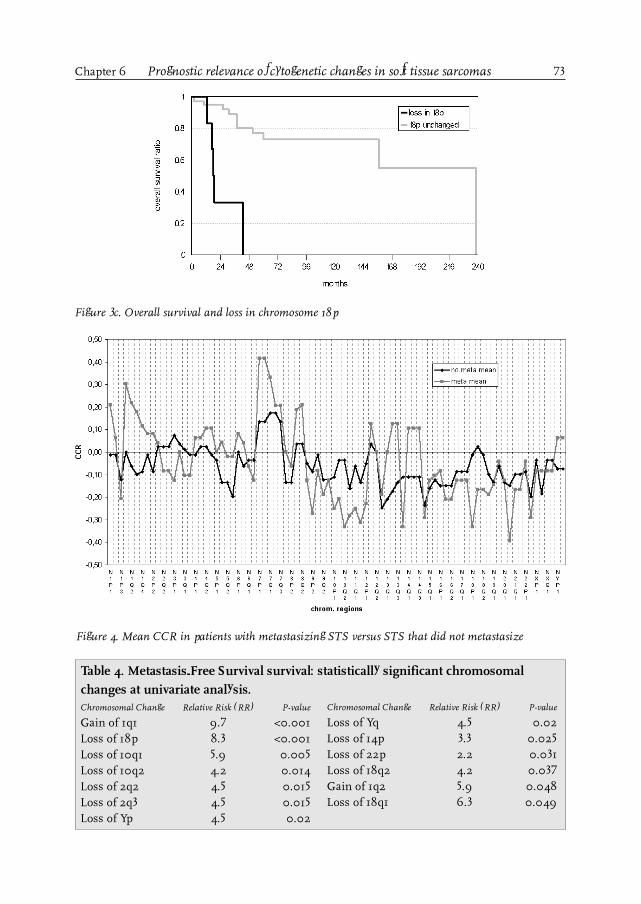
73Chapter 6 Prognostic relevance of cytogenetic changes in soft tissue sarcomas
Figure 3c. Overall survival and loss in chromosome 18p
Figure 4. Mean CCR in patients with metastasizing STS versus STS that did not metastasize
Table 4. Metastasis-Free Survival survival: statistically significant chromosomalchanges at univariate analysis.Chromosomal Change Relative Risk (RR) P-value
Gain of 1q1 9.7 <0.001Loss of 18p 8.3 <0.001Loss of 10q1 5.9 0.005Loss of 10q2 4.2 0.014Loss of 2q2 4.5 0.015Loss of 2q3 4.5 0.015Loss of Yp 4.5 0.02
Chromosomal Change Relative Risk (RR) P-value
Loss of Yq 4.5 0.02Loss of 14p 3.3 0.025Loss of 22p 2.2 0.031Loss of 18q2 4.2 0.037Gain of 1q2 5.9 0.048Loss of 18q1 6.3 0.049

74 Prognostic relevance of cytogenetic changes in soft tissue sarcomas Chapter 6
Discussion
Cytogenetic analysis has demonstrated that both benign and malignant tumors often havecharacteristic chromosomal aberrations and that the karyotype may be important indiagnosis and treatment [5,7,16,17]. STS often reveal a very complex karyotype due to alarge number of chromosomal abnormalities [13]. As all these chromosomal changes, eitheralone or in combination, might be responsible for certain steps in oncogenesis, it is verydifficult to relate these changes to the oncogenetic process. Another problem is the difficultyto interpret and compare different studies, as most of them have presented karyotypes insuch a way that comparison and statistical analysis is hardly possible. To overcome thisproblem, we started a database in which all karyotypes are interpreted and entered in auniform fashion, which makes a computer-assisted analysis of large groups of karyotypespossible, as described by Plaat et al. [13]. Data from the database were linked to clinicaloutcome to determine the prognostic importance of shared cytogenetic abnormalities.
In the present series, overrepresentation of STS with balanced translocations (especiallythe myxoid and round-cell liposarcoma subtype, characterized by the t(12;16) (q13;p11)
Figure 5b. Metastasis-free survival and gain in chromosome 1q
Figure 5a. Metastasis-free survival and loss in chromosome 18p

75
translocation) seems to be of minor importance, as such translocations do not result innet gain or loss of chromosomal material.Correlation between cytogenetic alterations and prognosis has been documented in otherhuman malignancies [18-22]. For STS, however, hardly any data exist on the prognosticrelevance of specific chromosomal aberrations [23-25]. In the present study, univariateanalysis of involved chromosomal changes revealed many regions with chromosomalalterations as negative prognostic factors. Several studies have identified cytogenetic bandsin these regions involved in human tumor development and progression. Specific infor-mation on STS, on the other hand, is very scanty.
In the present study, chromosomal gain in 1q1 was the most important negative prog-nostic factor regarding survival. A relation between alterations at the long arm of chro-mosome 1 and human malignancies has been demonstrated in various tumor types,suggesting the existence of oncogenes (breast cancer [26], prostate cancer [27], and en-dometrium cancer [28]), as well as tumor suppressor genes (breast cancer [26]) andmedulloblastoma [29]). In liposarcomas, there are indications that amplification of geneslocated at 1q21-24, often with concomitant gain in 12q14-21, plays a significant role indevelopment and progression [30]. Furthermore, the long arm of chromosome 1 is aregion of particular interest with regard to metastasis, as it harbors the KiSS-1 metasta-sis-suppressor gene at 1q32, which has been identified as a metastasis-suppressor genein malignant melanoma and breast cancer [31]. However, as gain of chromosomal mate-rial at the long arm of chromosome 1 was a negative prognosticator for metastasis-freesurvival, it seems very unlikely that this metastasis-suppressor gene is involved in themetastatic process of STS.
The other negative prognostic factor regarding survival, loss in 4p, was surprising, asthere is only very limited information on its role in human malignancies [32,33]. In theirsearch for the human homologue of the SH3BP2 protein in bladder cancer, Bell et al.identified an interesting gene at 4p16.3 that is a potential negative regulator of the Ablgene [34], a proto-oncogene that has been related to differentiation and apoptosis inhibi-tion in chondrosarcoma [35]. The association between high malignancy grade, a well-known negative prognostic factor regarding disease-specific survival, and low amountsof apoptosis in STS [36], might further support a potential role of SH3BP2 and Abl genesin STS prognosis.
At univariate analysis, aberrations in many other chromosomal regions were related tosurvival. Because of the magnitude of genes, located at these regions, and the fact that wedid not investigate specific gene products, it is impossible to predict which chromo-somal bands and which genes will be involved, although some regions (9p, chromosome17, and 18p) are very intriguing. The negative prognostic importance of loss in region9p2 was not unexpected, as this region contains the p16 (CDKN2A/ IKN4A) and p15(CDKN2B/ IKN4B) genes, which act as negative regulators of proliferation of normalcells. Deletions or mutations of these CDK (cyclin-dependent kinase)- 4 and 6 inhibitorslead to unchecked cell growth, which has been demonstrated in many human cancertypes [18,21,37]. In Ewing�s sarcoma, Wei et al. demonstrated that p16 deletion was a verystrong negative prognostic factor (P<0.001) [37]. In Wilms� tumors, loss of p16 also seems
Chapter 6 Prognostic relevance of cytogenetic changes in soft tissue sarcomas

76 Prognostic relevance of cytogenetic changes in soft tissue sarcomas Chapter 6
to be of prognostic importance, as it correlates with advanced tumor stage [39]. In STS,however, the prognostic impact of both p16 and p15 alterations remains contradictory.Orlow et al. reported a significant relation between p16 deletions or alterations and poorsurvival in 46 STS (P=0.036 and 0.005, respectively), with alteration of the IKN4A/Bgene being the only statistically significant predictor for poor survival when controllingfor tumor grade and size (P=0.03) [40]. Yao and Meye investigated the role of the p16gene status and expression in STS, and reported a low frequency of deletions andmutations of this gene in STS, in contrast to Simons et al., who demonstrated a loss of9p21 in 55% of MFH [42-44]. Moreover, Yao et al. suggested that CDK4 might act as anoncogene in STS, which is in contrast to findings in other human malignancies whereCDK4 acts as a tumor suppressor gene [41]. In the present study, however, loss of region9p2 is a statistically significant negative prognostic factor (P=0.037), suggesting thelocation of a STS suppressor gene in this region.
The prognostic importance of chromosomal loss at the short arm of chromosome 17 isvery interesting, as some important tumor suppressor genes are located there. TP53,located at 17p13.1, is the most common tumor suppressor gene, altered in many malig-nancies. A study from the Memorial Sloan Kettering Cancer Center demonstrated that17p deletions and p53 mutations were common events in adult STS [44]. Dei Tos et al.demonstrated a 30% incidence of p53 aberrations in myxoid and round cell liposarcoma[45]. However, these studies did not provide any information on the prognostic impor-tance of the p53 alteration. In a series of 113 bone and soft tissue sarcomas, Moussesreported p53 alterations at different frequencies in various sarcomas; furthermore, thesealterations were not associated with prognosis [46]. As p53 expression was not examinedin the present study, we can not be sure whether this gene product is responsible for thepoor prognosis in patients with loss of the short arm of chromosome 17, or whetherother suppressor genes at 17p are involved, as has been reported in sporadic breast can-cer [47].Besides chromosomal loss at the short arm of chromosome 17, also loss at its long armhad prognostic importance. One of the genes that might contribute to this is the proto-oncogene c-erbB-2, at band 17q21.1. Although a correlation between loss of 17q and ahigh degree of amplification of c-erbB-2 has been demonstrated in human breast cancer[48], no reports on its prognostic importance in STS are available.Loss of the short arm of chromosome 18 (18p1) was another important negative prognos-tic factor, both in metastasis-free and overall survival, suggesting the location of (a) puta-tive suppressor gene(s). In the literature, there is some evidence for the presence oftumor suppressor genes on the short arm of chromosome 18, involved in breast carci-noma, NSCLC, and brain tumors [49]. Apart from that, information on the role of 18p inhuman malignancy is extremely scarce.
Except for retroperitoneal STS, survival in STS has been related directly to metastasis. Inhuman malignancies in general, and in STS in particular, the cytogenetic base for me-tastasis remains obscure. As reports revealing possible clues are awaited, the findingthat loss of 18p was the only statistically significant negative prognostic factor regardingmetastasis-free survival is very interesting, as it suggests the location of a putative (sar-coma-) metastasis suppressor gene.

77Chapter 6 Prognostic relevance of cytogenetic changes in soft tissue sarcomas
The computer-assisted approach, used in the present study, is valuable in the analysis oflarge groups of complex karyotypes to detect common chromosomal alterations. Thestrong association between gain in the long arm of chromosome 1 and loss in the shortarm of chromosome 18 in patients with the shortest survival has not been reported be-fore, and its importance in STS prognosis remains unclear.In conclusion, the present study provides indications for correlations between cytoge-netic changes and metastasis and prognosis in STS. Some of these findings confirmearlier reports, whereas many are novel in STS, and need to be confirmed in additionalstudies. The strong association between alterations in the long arm of chromosome 1and the short arm of chromosome 18 in non-survivors is very challenging and may haveprognostic value in STS.
References
1. Pisters PW, Leung DH, Woodruff J, Shi W, Brennan MF. Analysis of prognostic factors in 1.041patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996; 14: 1679-1689.
2. Pisters PWT and Pollock RE. Staging and prognostic factors in soft tissue sarcoma. Sem RadiatOncol 1999; 9: 307-314.
3. Coindre JM, Terrier P, Bui N, et al. Prognostic factors in adult patients with locally controlled softtissue sarcoma: A study of 546 patients from the French Federation of Cancer Centers SarcomaGroup. J Clin Oncol 1996; 14: 869-877.
4. Molenaar WM, Jong de B, Buist J, Idenburg VJS, Seruca R, Vos AM, Hoekstra HJ. Chromosomalanalysis and the classification of soft tissue sarcomas. Lab. Invest 1989; 60: 266-274.
5. Fletcher JA, Kozakewich HP, Hoffer FA, Lage JM, Weidner N, Tepper R, Pinkus GS, Morton CC,Corson JM. Diagnostic relevance of clonal cytogenetic aberrations in malignant soft tissue tumors.N Engl J Med 1991; 324: 436-442.
6. Enzinger FM, and Weiss SW. (eds.) Soft Tissue Tumors, 3rd ed. St. Louis, C.V. Mosby, 1995.7. Dei Toss AP, Dal Cin P. The role of cytogenetics in the classification of soft tissue tumors. Virchows
Arch 1997; 431: 83-94.8. Sreekantaiah Ch, Ladanyi M, Rodriguez E, Chaganti RSK. Chromosomal abberations in soft tissue
tumors. Relevance to diagnosis, classification, and molecular mechanisms. Am J Pathol 1994; 144:1121-1134.
9. Rydholm A. Chromosomal abberations in musculoskeletal tumours: clinical importance. J BoneJoint Surg 1996; 78-B: 501-506.
10. Fletcher CDM, Akerman M, Dal Cin P, et al. Correlation between clinicopathologic features andkaryotype in lipomatous tumors. Am J Pathol 1996; 148: 623-630.
11. Szymanska J, Virolainen M, Tarkkanen M, et al. Overrepresentation of 1q21-23 and 12q13-21 inlipoma-like liposarcomas but not in benign lipomas: a comparative genomic hybridization study.Cancer Genet Cytogenet 1997; 99: 14-18.
12. Mertens F, Fletcher CDM, Dal Cin P, et al. Cytogenetic analysis of 46 pleomorphic soft tissuesarcomas and correlations with morphologic and clinical features: a report of the CHAMP studygroup. Genes, chromosomes Cancer 1998; 22: 16-25.
13. Plaat BEC, Molenaar WM, Mastik MF, Hoekstra HJ, Meerman te GJ, Berg van de E. Computer-assisted cytogenetic analysis of 51 malignant peripheral nerve sheath tumors: sporadic vs.neurofibromatosis type 1-associated malignant schwannomas. Int J Cancer 1999; 83: 171-178.
14. Plaat BEC, Muntinghe FLH, Molenaar WM, et al. Clinical outcome of patients with previously

78
untreated soft tissue sarcomas in relation to tumor grade, DNA ploidy and karyotype. Int J Cancer1997; 74: 396-402.
15. ISCN (1995): An International System for Human Cytogenetic Nomenclature. F. Mitelman (ed);S. Karger, Basel, 1995.
16. Sandberg AA, Turc Carel C. The cytogenetics of solid tumors. Relation to diagnosis, classificationand pathology. Cancer 1987; 59: 387-395.
17. Heim S and Mitelman F. (eds.) Cancer Cytogenetics, 2nd ed. New York, Wiley-Liss, 1995.18. Parris CN, Harris JD, Griffin DK, Cuthbert DK, Silcer AJ, Newbold RF. Functional evidence of
novel tumor suppressor genes for cutaneous malignant melanoma. Cancer Res 1999; 59: 516-520.19. Hermsen MA, Baak JP, Meijer GA, et al. Genetic analysis of 53 lymph node-negative breast
carcinomas by CGH and relation to clinical, pathological, morphometric, and DNA cytometricprognostic factors. J Pathol 1998; 186: 356-362.
20. Tomizawa Y, Adachi J, Kohno T, et al. Prognostic significance of allelic imbalances on chromosome9p in stage I non-small cell lung carcinoma. Clin Cancer Res 1999; 5: 1139-1146.
21. Heyman M, Rasool O, Borgonovo-Brandter L, et al. Prognostic importance of p15INK4B andp16INK4A gene inactivation in childhood acute lymphocytic leukemia. J Clin Oncol 1996; 14:1512-1520.
22. Moch H, Presti Jr JC, Sauter G, Buchholz N, Jordan P, Mihatsch MJ, Waldman FM. Geneticaberrations detected by comparative genomic hybridization are associated with clinical outcome inrenal cell carcinoma. Cancer Res 1996; 56: 27-30.
23. Choong PFM, Mandahl N, Mertens F, et al. 19p+ marker chromosome correlates with relapse inmalignant fibrous histiocytoma. Genes Chromosom Cancer 1996; 16: 88-93.
24. Tarkkanen M, Elomaa I, Blomqvist C, et al. DNA sequence copy number increase at 8q: a potentialnew prognostic marker in high-grade osteosarcoma. Int J Cancer 1996; 84: 114-121.
25. Sanjuan X, Sobel ME, Yang J, Merino MJ. Alveolar soft part sarcoma: the role of prognosticmarkers. Ann Diagn Pathol 2000; 4: 135-142.
26. Bieche I, Champeme MH, Lidereau R. Loss and gain of distict regions of chromosome 1q in primarybreast cancer. Clin Cancer Res 1995; 1: 123-127.
27. Valeri A, Drelon A, Paiss T, et al. Genetic analysis of familial prostatic cancer: localization of a genepredisposing to prostatic cancer (PcaP) on chromosome 1q42.2-43. Prog Urol 1999; 9: 680-688.
28. Suzuki A, Fukushige S, Nagase S, Ohuchi N, Satomi S, Horii A. Frequent gains on chromosomalarms 1q and/or 8q in endometrial cancer. Hum Genet 1997; 100: 629-636.
29. Pietsch T, Koch A, Wiestler OD. Molecular genetic studies in medulloblastomas: evidence for tumorsuppressor genes at the chromosomal regions 1q31-32 and 17p13. Klin Pediatr 1997; 209: 150-155.
30. Szymanska J, Tarkkanen M, Wiklund T, et al. Gains and losses of DNA sequences in liposarcomasevaluated by comparative genomic hybridization. Genes Chromosom Cancer 1996; 15: 89-94.
31. Lee JH, Welch DR. Suppression of metastasis in human breast carcinoma MDA-435 cells aftertransfection with the metastasis suppressor gene, KiSS-1. Cancer Res 1997; 57: 2384-2387.
32. Gryfe R, Swallow C, Bapat B, Redston M, Gallinger S, Couture J. Molecular biology of colorectalcancer. Curr Probl Cancer 1997; 21: 233-300.
33. Kitamura Y, Shimizu K, Tanaka S, Ito K, Emi M. Association of allelic loss on 1q, 4p, 7q, 9p, 9q,and 16q with postoperative death in papillary thyroid carcinoma. Clin Cancer Res 2000; 6:1819-1825.
34. Bell SM, Shaw M, Jou YS, Myers RM, Knowles MA. Identification and characterization of thehuman homologue of SH3BP2, an SH3 binding domain protein within a common region of deletionat 4p16.3 involved in bladder cancer. Genomics 1997; 44: 163-170.
Prognostic relevance of cytogenetic changes in soft tissue sarcomas Chapter 6

79
35. O�Donovan M, Russell JM, O�Leary JJ, Gillan JA, Lawler MP, Gaffney EF. Abl expression, tumourgrade, and apoptosis in chondrosarcoma. Mol Pathol 1999; 52: 341-344.
36. Nakanishi H, Ohsawa M, Naka N, et al. Immunohistochemical detection of bcl-2 and p53 proteinsand apoptosis in soft tissue sarcoma: their correlations with prognosis. Oncology 1997; 54: 238-244.
37. Nakanishi H, Wang XL, Imai FL, et al. Localization of novel tumor suppressor gene loci onchromosome 9p21-22 in oral cancer. Anticancer Res 1999; 19: 29-34.
38. Wei G, Antonescu CR, de Alava E, et al. Prognostic impact of INK4A deletion in Ewing sarcoma.Cancer 2000; 89: 793-799.
39. Arcellana-Panlilio MY, Egeler RM, Ujack E, Pinto A, Demetrick DJ, Robbins SM, Coppes MJ.Decreased expression of the INK4 family of cyclin-dependent kinase inhibitors in Wilms tumor.Genes Chromosom Cancer 2000; 29: 63-69.
40. Orlow I, Drobnjak M, Zhang ZF, Lewis J, Woodruff JM, Brennan MF, Cordon-Cardo C.Alterations of INK4A and INK4B genes in adult soft tissue sarcomas: effect on survival. J NatlCancer Inst 1999; 91: 73-79.
41. Yao J, Pollock RE, Lang A, et al. Infrequent mutation of the p16/MTS1 gene and overexpression ofcyclin-dependent kinase 4 in human primary soft tissue sarcoma. Clin Cancer Res 1998; 4:1065-1070.
42. Meye A, Wurl P, Hinze R, et al. No p16INK4A/CDKN2/MTS1 mutations independent of p53 statusin soft tissue sarcomas. J Pathol 1998; 184: 14-17.
43. Simons A, Schepens M, Jeuken J, et al. Frequent loss of 9p21 (p16(INK4A)) and other genomicimbalances in human malignant fibrous histiocytoma. Cancer Genet Cytogenet 2000; 118: 89-98.
44. Latres E, Drobnjak M, Pollack D, et al. Chromosome 17 abnormalities and TP53 mutations in adultsoft tissue sarcomas. Am J Pathol 1994; 145: 345-355.
45. Dei Tos AP, Piccinin S, Doglioni C, Vukosavljevic T, Mentzel T, Boiocchi M, Fletcher CD.Molecular aberrations of the G1-S checkpoint in myxoid and round cell liposarcoma. Am J Pathol1997; 151: 1531-1539.
46. Mousses S, McAuley L, Bell RS, Kandel R, Andrulis IL. Molecular and immunohistochemicalidentification of p53 alterations in bone and soft tissue sarcomas. Mod Pathol 1996; 9: 1-6.
47. Liscia DS, Morizio R, Venesio T, Palenzona C, Donadio M, Callahan R. Prognostic significance ofloss of heterozygosity at loci on chromosome 17p13.3-ter in sporadic breast cancer is evidence for aputative tumour suppressor gene. Br J Cancer 1999; 80: 821-826.
48. Borresen AL, Ottestad L, Gaustad A, et al. Amplification and protein over-expression of the neu/HER-2/c-erbB-2 proto-oncogene in human breast carcinomas: relationship to loss of gene sequenceson chromosome 17, family history and prognosis. Br J Cancer 1990; 62: 585-590.
49. Tran Y, Benbatoul K, Gorse K, Rempel S, Futreal A, Green M, Newsham I. Novel regions of allelicdeletions on chromosome 18p in tumors of the lung, brain and breast. Oncogene 1998; 17:3499-3505.
Chapter 6 Prognostic relevance of cytogenetic changes in soft tissue sarcomas

80 Prognostic relevance of cytogenetic changes in soft tissue sarcomas Chapter 6

81
Chapter 7
Soft tissue sarcoma: where to go?
Chapter 7

82
Introduction
Although surgery is still the keystone in sarcoma treatment, the role for other specialtiesas radiation oncology, medical oncology, pathology, medical genetics, molecular biology,radiology, nuclear medicine, and epidemiology will continue to increase. Specialists fromthe various disciplines should cooperate in sarcoma working parties in order to improvetreatment results and to stimulate clinical and basic research for future progress in theunderstanding of this rare tumor. In this chapter, some aspects of future sarcomatreatment are highlighted.
Specialization and centralization
Many studies on morbidity and mortality in various surgical procedures have clearlydemonstrated a relationship between outcome and case load. Outcome disparity has notonly been identified among various institutions, but also among surgeons within oneinstitution [1-3]. Ten years ago, McArdle reported on the impact of variability amongsurgeons on postoperative morbidity, mortality, and ultimate survival in surgery forcolorectal cancer [3]. He concluded that only surgeons with a special interest in colorectalsurgery or surgical oncology should undertake such kind of surgery. Later studies con-firmed the prognostic importance of surgical subspeciality training in colorectal sur-gery, but also demonstrated the prognostic importance of case load per surgeon and perinstitution [4,5]. The independent prognostic importance of the treatment center, theindividual surgeon, and patient-volume has also been confirmed in other malignancies[6-9]. At present, considerable effort and resources are spent on large multicenter trialson adjuvant chemo- and/or radiation therapy in the hope to improve cancer survival. Inthese trials, the type of surgical resection is more and more defined, since there is evi-dence that outcome is related to the surgery performed, which may be improved by bettersurgical training and by centralization of difficult surgical procedures. This especiallyholds true for rare tumors as sarcomas, where the experience of the individual instituteis very limited.
The Scandinavian Sarcoma Group has reported on the impact of specialization in STS.In the early eighties, they demonstrated that an adequate resection with associated lowlocal recurrence risk was more often obtained in patients treated in a center compared tothose treated at local hospitals [10]. Later, one of their population-based studies clearlyshowed the benefit for STS patients who were referred to a center before any type ofsurgery had taken place. Patients referred after surgery or not at all, underwent substan-tially more operations, whereas their local recurrence rate was higher [11]. However, in arecent study from the Memorial Sloan-Kettering Cancer Center, reresection seemed tohave no detrimental effect on survival in extremity STS [12]. Moreover, none of the cur-rently available prospective randomized trials supports the hypothesis that better localcontrol enhances survival in sarcoma patients [13-15]. The problem is that the power todetect survival difference in relation to local control is small in the reported randomizedtrials. A large number of patients will be required to determine whether prevention of localrecurrence improves survival [16]. Nevertheless, local control of the primary tumor remainsvery important for the quality of life of cancer patients.
Soft tissue sarcoma: where to go? Chapter 7

83
During the past twenty years, the establishment of a sarcoma working party that has beenresponsible for the multimodality treatment of sarcomas at the Groningen UniversityHospital has led to an improvement in local control at various tumor localisations [17].Nevertheless, specialization should not only focus on sarcoma treatment, but also ondiagnosis, especially the histopathological diagnosis, which is often extremely difficult.Histopathological peer review studies have demonstrated that 6- 24% of registered soft-tissue and bone sarcomas were considered on review not to be sarcomas, whereas agreementon subtype could be achieved in only 53- 75% [18-20]. Experienced pathologists shouldtherefore preferably review histopathological specimens of these rare tumors. Recentprogress in immunohistology and molecular biology has improved sarcoma diagnosis.Knowledge of these techniques will become increasingly important because they may serveas aids in the diagnosis and classification of bone and soft tissue tumors, especially in thedifferential diagnosis of those of confusing nature [21-24]. In order to optimize the use oftumor tissue, it is advisable to submit fresh specimens to the pathologist.
Another argument for specialization in STS is given by the Groningen Sarcoma Group,which reported on the adherence to STS staging guidelines [25]. In a specialized center,compared to non-specialized community hospitals, the case load was higher and thecompliance with the guidelines was significantly better. Special attention should be paidto future guideline development, dissemination, and implementation. Centralization ofSTS treatment in a limited number of hospitals with dedicated multidisciplinary sar-coma working parties appears advisable. In this way, adequate patient volumes can beachieved to ensure a certain threshold of clinical experience, and to enable special fel-lows, not only in surgical oncology, but also in medical oncology, radiotherapy, and pa-thology, to get more familiar with various aspects of the multimodality treatment of theserare tumors.Another important aspect of specialization and centralization of STS treatment and theestablishment of a sarcoma working party is the possibility to perform clinical, basic andexperimental STS research.
Prognostic factors: old and new
During the last decades, many prognostic factors have been identified, and the mostrelevant ones have been incorporated into various staging systems. Of these, the revisedAmerican Joint Committee on Cancer (AJCC) staging system is the most widely used(Table 1). The AJCC STS staging system relies upon histological grade, tumor size, depth,as well as the presence of nodal or distant metastasis [26], all classical parameters withproven value as prognosticators [27-30]. One of the major limitations of the present AJCCstaging system is that it does not take into account the anatomical site, which is animportant determinant of outcome. Patients with retroperitoneal and visceral sarcomashave a worse overall prognosis compared to patients with extremity STS [27,31-33].In addition to the tumor characteristics embedded in the AJCC system, several otherparameters have been identified as prognostic factors [27-29]. Among these, the specifichistological (sub)type of the STS is considered secondary in importance to the histologi-cal grade. STS as fibrosarcoma, leiomyosarcomas and malignant peripheral nerve sheathtumors have a poorer outcome, whereas liposarcomas tend to do better [27,31]. Other negative
Chapter 7 Soft tissue sarcoma: where to go?

84
prognostic factors are local recurrence [11,27], microscopically positive margins [27,29],tumor necrosis [11,34,35], and vascular invasion [11,36,37]. Age over 50 years [27] and malesex [28] have been identified as unfavourable prognostic factors in some series, whereasother series could not confirm this [27,29,38]. Although the AJCC staging system allowsfor the identification of high-risk patients at presentation, i.e. patients with large (> 5 cm),high-grade, deep lesions, one should realize that, with the current clinicopathological factors,clinicians can at best identify (non-metastasized) patients with up to 50% risk of distantfailure, i.e. the patient with a G3T2b lesion [39]. Therefore, future studies will have to focuson the identification of new prognostic indicators that might facilitate the identification ofthe highest-risk subset of patients. Recent research into angiogenesis, molecular biology,and cytogenetics has revealed some new prognostic factors. High intratumoral microvesseldensity [40], high S-phase fraction [41-43], and a high expression of proliferation markers(proliferating cell nucleolar antigen, Ki-67, nucleolar organizer regions) [44-48] have beenassociated with a poor survival. However, as results have not been unequivocal, the use ofthese parameters as clinical indicators of prognosis remains controversial, and prospectivelong-term observational studies in a large number of patients will be required to establishthe clinical impact of these factors. Although the value of DNA ploidy as a prognosticindicator is well established in many other cancers, its prognostic relevance in STS remainsunclear and controversial [37,49-51]. In recent years, more specific chromosome and gene
Table 1. The American Joint Committee on Cancer (AJCC) soft tissue sarcoma stagingsystem.*
Primary Tumor (T)
TX Primary tumor cannot be assessedT0 No evidence of primary tumorT1 Tumor 5 cm or less in greatest dimension
T1a superficial tumorT1b deep tumor
T2 Tumor more than 5 cm in greatestdimension
T2a superficial tumorT2b deep tumor
Regional Lymph Nodes (N)
NX Regional lymph nodes cannot beassessed
N0 No regional lymph node metastasisN1 Regional lymph node metastasis
Distant metastasis (M)
MX Distant metastasis cannot be assessedM0 No distant metastasis
Histopathologic grade
GX Grade cannot be assessedG1 Well differentiatedG2 Moderately differentiatedG3 Poorly differentiatedG4 Undifferentiated
Stage grouping
Stage IA G1-2, T1a-1b, N0, M0Stage IB G1-2, T2a, N0, M0Stage IIA G1-2, T2b, N0, M0Stage IIB G3-4, T1a-1b, N0, M0Stage IIC G3-4, T2a, N0, M0Stage III G3-4, T2b, N0, M0Stage IV Any G, any T, N1, M0
Any G, any T, N0, M1
* Fleming ID, Cooper JS, Henson DE, et al, Eds.Chapter 22. Soft Tissue Sarcoma. In: AJCC Can-cer Staging Manual. 5th Ed. Philadelphia,Lippincott-Raven Publishers. 1997:149-156
Soft tissue sarcoma: where to go? Chapter 7

85
abnormalities have been associated with prognosis. Mutation or functional inactivationof the p53 tumor suppressor gene has been reported in several STS types [30,52-54].Functional inactivation of both the wild type and mutant p53 protein can be achieved byamplification of the murine double minute 2 (MDM2) gene. The presence of p53 genemutations and/or MDM2 amplification seems to be associated with adverse outcome[52,55,56]. Nevertheless, more thorough evaluation is needed. Another tumor suppressorgene often altered in STS is the retinoblastoma gene (RB1). Absence of expression of thisgene has been associated with poor outcome [57], but results have been contradictory [58].The role of oncogenes in initiation or progression of STS seems rather limited, with apossible exception of the myc oncogene. A correlation between expression/amplificationof this gene with higher tumor grade and poorer survival has been reported in STS [59-61].Although new cellular and molecular prognostic factors have been identified, there ispresently no consensus how these factors should be incorporated into clinical practice.Once more, the need for centralization, further consensus development and multicentercollaboration seems obvious because of the rarity of these tumors.
Treatment of STS in the new millennium
As in most solid tumors, surgery will remain the most important modality to cure pa-tients with a primary STS. Minimal invasive techniques and attention for functionaloutcome dominate modern surgery, not only in surgical oncology (for example breastconservative surgery and sentinel lymph node biopsy in breast cancer), but also in gastro-intestinal surgery (various laparoscopic approaches), vascular surgery (endovascularaneurysm repair), and orthopedic surgery (minimal invasive percutaneous plate osteo-synthesis and less invasive stabilization system). In STS, the prospective trial from theNational Cancer Institute, comparing amputation to wide excision and postoperativeradiotherapy (both with adjuvant chemotherapy), was very important for the develop-ment of multimodality, limb-saving treatment [13]. The conclusions of this trial weresupported by other randomized trials of less or more aggressive local treatment of STS[62-65]. Nowadays, limb-sparing surgery is possible in roughly 90% of patients withextremity STS [27,66,67].
Treatment of locally advanced extremity STS remains a challenging problem. As ampu-tation does not influence survival [13], limb-saving techniques have been developed. Nev-ertheless, amputation of the limb still might be a good treatment, especially in sarcomasinvading joints, bones, or neurovascular structures, although involvement of major nervesis not an indication per se for ablative surgery [68].In the seventies, Morton and Eilber developed the multimodality therapy of preoperativeintraarterial chemotherapy (doxorubicin), followed by external beam radiotherapy (EBRT)and surgical resection [69-71]. This treatment has been applied to patients with interme-diate and high-grade extremity STS. In the early days, when patients were treated withpreoperative intraarterial doxorubicin, immediately followed by ten fractions of 3.5 GyEBRT and surgical resection, treatment related morbidity was very high (35%). However,local recurrence and amputation rates were low, 9% and 5% respectively. To decreasemorbidity, the total radiation dose was lowered to 17.5 (5 x 3.5) Gy, which indeed reducedtreatment-related complications, but also significantly increased local failure rate to 15%.
Chapter 7 Soft tissue sarcoma: where to go?

86
An intermediate radiation dose of 28 (8 x 3.5) Gy appeared to achieve an excellent localcontrol rate of 91%, and an acceptable complication rate, with only 6% requiringreoperation. In addition, there was no difference between intraarterial and intravenousadministration routes of doxorubicin [71]. Notwithstanding these good results, whicheven seem to increase by the addition of ifosfamide to the regimen, long-term treatment-related complications can result in disabling limb function [72]. Although the intraarterialchemotherapy, with or without stop flow occlusion has some theoretical advantages (�firstpass� effect and/or decreased drug extraction), to date there is no indication for intra-arterial chemotherapy in the treatment of STS of the extremities [73].
Another technique, used in locally advanced STS of the extremities, is the hyperthermicisolated limb perfusion (HILP). In 1958, Creech and co-workers pioneered the use ofregional isolated perfusion for melanoma of the limb, using an extracorporal circulationsystem [74]. The addition of hyperthermia, based on studies of Cavaliere and Stehlin,yielded the technique of hyperthermic isolated limb perfusion as a new and importanttherapy in the management of (advanced) extremity tumors [75,76]. Krementz was oneof the first to report on the results of this limb-saving treatment modality in locally ad-vanced extremity sarcomas [77]. In STS, melphalan, the standard drug for HILP inmelanoma, has been studied most extensively, although other agents (doxorubicin,cisplatin, etc) have also been applied [78-80]. At the end of the eighties, it looked likeHILP would not achieve a major breakthrough in the treatment of locally advanced ex-tremity sarcomas as results were disappointing and not better than other limb-savingprotocols [71,81,82]. In the early nineties, however, much progress was made by studiesfrom Lejeune and co-workers, who added tumor necrosis factor- alpha (TNF-α) and in-terferon- gamma (IFN-γ) to the HILP regimen with melphalan, resulting in higher localresponse rates, with high limb-salvage rates and acceptable toxicity [83]. Recently,Eggermont published the results of 55 patients with primarily irresectable STS of theextremities, who were perfused in four European centers [84]. Notwithstanding the ratherunfavorable group of patients in the latter study, with 24% multifocal primary or multi-ple recurrent STS, and a median tumor size of 18 cm, treatment response was very good.The complete and partial response rates were 36% and 51%, respectively. After a medianfollow-up of 27 months, limb salvage with good function was achieved in 84% of thepatients [84]. Data from eight European centers showed more or less the same results[85]. Clinical and pathological tumor response and limb salvage rates in patients whoreceived additional IFN-γ was virtually identical to those observed in patients treatedwith HILP with TNF-α and melphalan only. However, systemic toxicity was higher inthe IFN group [85]. In 1998, Olieman demonstrated that in locally advanced extremitySTS, adjuvant EBRT was feasible after HILP and surgical resection. This treatmentresulted in a better local tumor control without increased treatment morbidity [86].However, long-term results have to be awaited. The future HILP-TNF-α research has tobe directed towards dose-reduction studies. Results from these studies are pendant, andmay have further impact on the applicability of TNF-α for other malignancies.Unfortunately, there will still be STS, especially in the extremities, which remainirresectable, because patients are no candidates for HILP. Such tumors might be treatedby high dose EBRT only. In a series of Tepper, EBRT alone could achieve local control in44% of patients for radiation doses greater than 64 Gy, in contrast to only 15% for lower
Soft tissue sarcoma: where to go? Chapter 7

87
doses [87]. Local control rate was better for tumors of 5 cm or less (88%), than for tumorsof 5- 10 cm (53%), or greater than 10 cm (30%). As most (unresectable) STS of the ex-tremities are large at presentation [86,88], very low local control rates may be expectedfrom EBRT only.
Other radiation techniques, as brachytherapy and intraoperative radiotherapy (IORT),have been applied as a radiation boost technique in sarcoma treatment. These modalitiesare attractive, because they involve a much smaller treatment volume than conventionalmethods and because they can be combined with the surgical procedure, whereas con-ventional EBRT can only be started after woung healing. In 1996, Pisters and Brennanfrom MSKCC published the long-term results of a randomized trial on adjuvant radia-tion therapy in STS [14]. Brachytherapy significantly improved the local control rate inhigh-grade lesions. In low-grade STS, however, adjuvant brachytherapy did not improvelocal control, which was in contrast to the findings of a randomized prospective trial onadjuvant EBRT from the National Cancer Institute, demonstrating also a significant de-crease in local recurrence rate in low-grade STS treated with adjuvant EBRT [15]. Im-provement in local control in both the brachytherapy and adjuvant EBRT trial was notassociated with a decrease in the rate of distant metastases or an improved disease-spe-cific survival [14,15]. The experience in IORT for STS is still very limited. This appealingtechnique has been applied especially in retroperitoneal STS [89-92], with only a fewreports on extremity STS [93-95]. All of these studies demonstrated the feasibility of thismethod in combination with surgical resection and adjuvant low-dose EBRT (≤ 40 Gy).In extremity STS, good local control rates have been reported with IORT and low-dosepostoperative EBRT [95]. The only prospective, randomized trial, comparing 20 Gy IORTin combination with low-dose postoperative EBRT (35- 40 Gy) with postoperative high-dose EBRT (50- 55 Gy) in retroperitoneal STS, demonstrated a significantly lower localrecurrence rate in the IORT group, with fewer complications of disabling radiation-re-lated enteritis, but with more radiation-related peripheral neuropathy. However, again,enhanced local control did not translate into prolonged survival [91].In extremity STS, presently available data suggest similar local control rates for pre-operative chemoradiation, pre-operative radiotherapy only, and postoperative radiotherapy[96-101]. In large, high-grade lesions, brachytherapy (and perhaps IORT) may be superiorto conventional EBRT, whereas the opposite seems to be true for large low-grade tumors.For the future, not much progress in local control of STS is expected. However, relativelynew radiotherapy modalities, as three-dimensional conformal radiotherapy, inverseradiotherapy planning, and intensity modulated radiation therapy, seem to provide severaladvantages over conventional radiotherapy with regard to treatment-related morbidity[102-104].
In STS treatment, the role of adjuvant systemic chemotherapy remains quite controver-sial. The most effective single agents in STS are doxorubicin (adriamycin), dacarbazine(DTIC) and ifosfamide [105-109]. Although dose intensification and combination withgrowth factor support can further increase response rates [110-112], the role of such in-tensive high-dose combination chemotherapy remains controversial and side effects arenot negligible [113,114], whereas randomized trials have not shown conclusively whetheradjuvant chemotherapy benefits adult patients with localized STS. At present, the meta-
Chapter 7 Soft tissue sarcoma: where to go?

88
analysis from the Sarcoma Meta-Analysis Collaboration (SMAC) is the most reliable, up-to-date analysis of the effect of doxorubicin-based chemotherapy in localized resectableSTS [115]. Overall, this review suggested that adjuvant doxorubicin-based chemotherapycould lengthen the time alive without recurrence, with a trend towards improved sur-vival. There was a 27% risk reduction in the risk of local failure, with an absolute benefitof 6% at 10 years (81% vs 75%; P=0.016). With regard to distant relapse-free survival, arisk reduction of 30% was reported with an absolute benefit of 10% at 10 years (70% vs60%; P=0.0003). The overall recurrence-free survival at 10 years improved with 10%(55% vs 45%; P=0.0001). However, with regard to the overall survival, the absolute ben-efit at 10 years was only 4% (54% vs 50%; P=0,19). Subgroup analysis demonstrated theclearest evidence of a treatment effect on survival in patients with extremity STS, wherean absolute benefit of 7% at 10 years was reported (P=0.029).
From this and other reviews on adjuvant chemotherapy in STS, it is clear that futurerandomized trials should be larger than those from the past, because modest improve-ments are probably the best that can be expected. This will not be possible without entryof STS patients into sarcoma trials, and large-scale collaboration between research groups.New active systemic agents with limited toxicity are needed. However, most new drugsthat have recently been tested in phase II trials have shown discouraging response rates[116-121].
Other potential ways to improve the efficacy of systemic therapy may be combinationmodalities involving chemotherapy, immunotherapy, biologic response modifiers, andmodulators of multidrug resistance [122-124].An intriguing new approach is the differentiation therapy, which has been proposed asan alternative to conventional anti-tumor therapy because many tumor cells retain someability to differentiate through induction by chemical agents [125]. As differentiation statusis predictive of clinical outcome in many STS, modulation of differentiation may favorclinical behavior. In rhabdomyosarcoma cells, exposure to low concentrations ofactinomycin D and exposure to GR- 891, a novel 5- fluorouracil acyclonucleoside prodrug,led to a terminal process of myogenetic differentiation [126-129]. Tontonoz demonstratedthat human liposarcoma cells could be induced to undergo terminal differentiation bytreatment with troglitazone, a ligand for the peroxisome proliferator-activated receptor-gamma (PPAR-γ) nuclear receptor [130]. Demetri demonstrated in three patients withintermediate to high-grade liposarcomas in vivo troglitazone-induced histological andbiochemical differentiation, with a marked reduction in Ki-67 expression [131]. Clinicaltrials investigating this new technique are currently underway in the MDACC and theDana Farber Cancer Institute [130,131]. The differentiation treatment has the potential ofchanging the outcome of intermediate and high-grade liposarcomas, which may be im-portant especially in the retroperitoneum.Another example of a new alternative treatment strategy is the treatment of unresectableor metastatic gastrointestinal stromal tumors with specific inhibitors of the constitu-tively active mutant c-kit tyrosine kinase that is expressed in most gastrointestinal stro-mal tumors in which it may play a central part in the pathogenesis [132-134].
Soft tissue sarcoma: where to go? Chapter 7

89
Conclusion
The Sarcoma Working Party of the Groningen University Hospital and Faculty of Medi-cine, as well as the Comprehensive Cancer Center North-Netherlands facilitate clinicaltreatment of STS patients and basic STS research. During the last decade, the majorbreakthrough in STS was the introduction of TNF-α in HILP for primarily unresectableSTS of the extremities, increasing the limb-salvage rate to a maximum, without increas-ing treatment-related morbidity or reducing (disease-free) survival.New techniques have been developed, and some are very appealing. Although results fromexperimental and clinical pilot studies have been encouraging, the way to routine clinicaluse is still very long. To stimulate research and to improve treatment results, centralizationand specialization seems important, especially in rare tumors as STS. To answer urgentquestions, e.g. the survival benefit of adjuvant chemotherapy, STS should be treated onlyin trials and sarcoma working parties should be encouraged to collaborate. Only then, wewill be able to dissolve the fog around many aspects of this intriguing tumor, so that we canenter this new millennium with the conviction that the future for STS treatment looksbright.
References
1. Begg CB, Cramer LD, Hoskins WJ, Brennan MF. Impact of hospital volume on operative mortalityfor major cancer surgery. JAMA 1998; 280: 1747-1751.
2. Pellegrini CA, Heck CF, Raper S, Way LW. An analysis of reduced morbidity and mortality ratesafter pancreaticoduodenectomy. Arch Surg 1989; 124: 778-781.
3. McArdle CS, Hole D. Impact of variability among surgeons on postoperative morbidity andmortality and ultimate survival. BMJ 1991; 302: 1501-1505.
4. Hermanek P, Wiebelt H, Staimmer D, Riedl S. Prognostic factors of rectum carcinoma- Experienceof the German Multicentre Study SGCRC. Tumori 1995; 81 Supplement: 60-64.
5. Porter GA, Soskolne CL, Yakimets WW, Newman SC. Surgeon-related factors and outcome inrectal cancer. Ann Surg 1998; 227: 157-167.
6. Matthews HR, Powell DJ, McConkey CC. Effect of surgical experience on the results of resection foroesophageal carcinoma. Br J Surg 1986; 73: 621-623.
7. Sainsbury R, Haward B, Rider L, Johnston C, Round C. Influence of clinician workload andpattern of treatment on survival from breast cancer. Lancet 1995; 345: 1265-1270.
8. Sainsbury R, Rider L, Smith A, MacAdam A on behalf of the Yorkshire Breast Cancer Group. Doesit matter where you live? Treatment variation for breast cancer in Yorkshire. Br J Cancer 1995; 71:1275-1278.
9. Lieberman MD, Kilburn H, Lindsey M, Brennan MF. Relation of perioperative deaths to hospitalvolume among patients undergoing pancreatic resection for malignancy. Ann Surg 1995; 222:638-645.
10. Rydholm A, Berg NO, Persson BM, Åkerman M. Treatment of soft tissue sarcoma should becentralised. Acta Orthop Scand 1983; 54: 333-339.
11. Gustafson P. Soft tissue sarcoma. Epidemiology and prognosis in 508 patients. Acta Orthop Scand1994; Suppl 259: 1-31.
12. Lewis JJ, Leung D, Espat J, Woodruff JM, Brennan MF. Effect of reresection in extremity soft tissuesarcoma. Ann Surg 2000; 231: 655-63.
Chapter 7 Soft tissue sarcoma: where to go?

90
13. Rosenberg SA, Tepper J, Glatstein E, et al. The treatment of soft tissue sarcomas of the extremities.Prospective randomized evaluation of (1) limb-sparing surgery plus radiation therapy comparedwith amputation, and (2) the role of adjuvant chemotherapy. Ann Surg 1982; 96: 305-315.
14. Pisters PWT, Harrison LB, Leung DHY, et al. Long-term results of a prospective randomized trial ofadjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 1996; 14: 859-868.
15. Yang JC, Chang AE, Baker AR, et al. A randomized prospective study of the benefit of adjuvantradiation therapy in the treatment of soft tissue sarcoma of the extremity. J Clin Oncol 1998; 16:197-203.
16. Barr LC, Stotter AT, A�Hern RP. Influence of local recurrence on survival: A controversy reviewedfrom the perspective of soft tissue sarcoma. Br J Surg 1991; 78: 648-650.
17. Ham SJ. Current concepts and surgical aspects of extremity bone and soft tissue sarcoma. ThesisUniversity Groningen, The Netherlands, Wageningen, ISBN 90-367-1069-3.
18. Presant CA, Russell WO, Alexander RW, Fu YS. Soft-tissue and bone sarcoma histopathology peerreview: the frequency of disagreement in diagnosis and the need for second pathology opinions. TheSoutheastern Cancer Study Group experience. J Clin Oncol 1986; 4: 1658-1661.
19. Alvegård Th A and Berg NO, for the Scandinavian Sarcoma Group. Histopathology peer review ofhigh-grade soft tissue sarcoma: the Scandinavian Sarcoma Group experience. J Clin Oncol 1989; 7:1845-1851.
20. Harris M, Hartley AL, Blair V, et al. Sarcomas in North West England: I. Histopathological peerreview. Br J Cancer 1991; 64: 315-320.
21. Singer. New diagnostic modalities in soft tissue sarcoma. Semin Sug Oncol 1999; 17: 111-222.22. Graadt van Roggen JF, Bovee JV, Morreau J, Hogendoorn PC. Diagnostic and prognostic
implications of the unfolding molecular biology of bone and soft tissue tumours. J Clin Oncol 1999;52: 481-489.
23. Bridge JA, Sandberg AA. Cytogenetics and molecular genetic techniques as adjunctive approachesin the diagnosis of bone and soft tissue tumors. Skeletal Radiol 2000; 29: 249-258.
24. Pfeiffer JD, Hill DA, O�Sullivan MJ, Dehner LP. Diagnostic gold standard for soft tissue tumours:morphology or molecular genetics. Histopathology 2000; 37: 485-500.
25. Nijhuis PHA, Schaapveld M, Otter R, Hoekstra HJ. Soft tissue sarcoma- Compliance withguidelines. Cancer 2001; 91: 2186-2195.
26. Fleming ID, Cooper JS, Henson DE, et al, Eds. Chapter 22. Soft Tissue Sarcoma. In: AJCC CancerStaging Manual. 5th Ed. Philadelphia, Lippincott-Raven Publishers. 1997:149-156.
27. Pisters PWT, Leung DHI, Woodruff J, Shi W, Brennan MF. Analysis of prognostic factors in 1041patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996; 14: 1679-1689.
28. Coindre J-M, Terrier P, Bui NB, et al. Prognostic factors in adult patients with locally controlled softtissue sarcoma: a study of 546 patients from the French Federation of Cancer Centers SarcomaGroup. J Clin Oncol 1996; 14: 869-877.
29. Brooks AD, Heslin MJ, Leung DHY, Lewis JJ, Brennan MF. Superficial extremity soft tissuesarcoma: An analysis of prognostic factors. Ann Surg Oncol 1998; 5: 41-47.
30. Levine EA. Prognostic factors in soft tissue sarcoma. Semin Surg Oncol 1999; 17: 23-32.31. Torosian MH, Friedrich C, Godbold J, Hajdu SI, Brennan F. Soft tissue sarcoma: Initial
characteristics and prognostic factors in patients with and without metastatic disease. Sem SurgOncol 1988; 4: 13-19.
32. Dam PA van, Lowe DG, McKenzie-Gray B, Shepherd JH. Retroperitoneal soft tissue sarcomas:A review of the literature. Obstet Gynaecol Surv 1990; 45: 670-682.
33. Hartley AL, Blair V, Harris M, et al. Sarcomas in North West England: III Survival. Br J Cancer
Soft tissue sarcoma: where to go? Chapter 7

91
1992; 66: 685-691.34. Costa J, Wesley RA, Glatstein E, Rosenberg SA. The grading of soft tissue sarcomas. Results of a
clinico-pathologic correlation in a series of 163 cases. Cancer 1984; 53: 530-541.35. Unnik JAM van, Coindre JM, Contesso C, et al. Grading of soft tissue sarcomas: experience of the
EORTC soft tissue and bone sarcoma group. Eur J Cancer 1993; 29A: 2089-2093.36. Trojani M, Contesso G, Coindre JM, et al. Soft tissue sarcomas of adults: study of pathological
prognostic variables and definition of a histopathological grading system. Int J Cancer 1984; 33:37-42.
37. Alvegård TA, Berg NO, Baldetorp B, et al. Cellular DNA content and prognosis of high-grade softtissue sarcoma: the Scandinavian sarcoma group experience. J Clin Oncol 1990; 8: 538-547.
38. Gaynor JJ, Tan CC, Casper ES, et al. Refinement of clinicopathologic staging for localized softtissue sarcoma of the extremity: A study of 423 adults. J Clin Oncol 1992; 10: 1317-1329.
39. Pisters PW, Pollock RE. Staging and prognostic factors in soft tissue sarcoma. Semin Radiat Oncol1999; 9: 307-314.
40. Dirix LY, Vermeulen P, Wever I de, Oosterom AT van. Soft tissue sarcoma in adults. Curr OpinOncol 1997; 9: 348-359.
41. Alho A, Skjeldal S, Melvik JE, Pettersen EO, Eeg Larsen T. The clinical importance of DNAsynthesis and aneuploidy in bone and soft tissue tumors. Anticancer Research 1993; 13: 2383-2388.
42. Huuhtanen RL, Blomqvist CP, Wiklund TA, Virolainen MJ, Elomaa AI, Pan Y, Tribukait B.S-phase fraction of 155 soft tissue sarcomas. Correlation with outcome. Cancer 1996; 77: 1815-1822.
43. Gustafson P, Fernö M, Åkerman M, Baldetorp B, Willén H, Killander D, Rydholm A. FlowcytometricS-phase fraction in soft tissue sarcoma: prognostic importance analysed in 160 patients. Br JCancer 1997; 75: 94-100.
44. Levine EA, Holzmayer T, Bacus S, et al. Evaluation of newer prognostic markers for adult soft tissuesarcomas. J Clin Oncol 1997; 15: 3249-3257.
45. Rudolph P, Kellner U, Chassevent A, Collin F, Bonichon F, Parwaresch R, Coindre JM. Prognosticrelevance of a novel proliferation marker, Ki-S11, for soft tissue sarcoma. Am J Pathol 1997; 150:1997-2007.
46. Hesslin MJ, Cordon-Cardo C, Lewis JJ, Woodruff JM, Brennan MF. Ki-67 detected by MIB-1predicts distant metastasis and tumor mortality in primary, high grade extremity soft tissuesarcoma. Cancer 1998; 83: 490-497.
47. Huuhtanen RL, Blomqvist CP, Wiklund TA, Böhling TO, Virolainen MJ, Tukiainen EJ, TribukaitB. Comparison of the Ki-67 score and S-phase as prognostic variables in soft-tissue sarcoma. Br JCancer 1999; 79: 945-951.
48. Kuratso S, Tomita Y, Myoui A, Uchida A, Ono K, Aozasa K. DNA ploidy pattern and cell cyclestage of tumor cells in soft-tissue sarcomas: clinical implications. Oncology 1995; 52: 363-370.
49. Niggli FK, Powell JE, Parkes SE, et al. DNA ploidy and proliferative activity (S-phase) in childhoodsoft tissue sarcomas: their value as prognostic indicators. Br J Cancer 1994; 69: 1106-1110.
50. Li XQ, Parkekh SG, Rosenberg AE, Mankin HJ. Assessing prognosis for high-grade soft tissuesarcomas: search for a marker. Ann Surg Oncol 1996; 3: 550-557.
51. Ludemann M, Fischer A, Finke M, Wolff-Vorbeck G, Bohm J, Imdahl A. Molecular biologicalobservations in some rare neoplasms: DNA ploidy and cell cycle data in liposarcoma and malignantfibrous histiocytoma. Langenbecks Arch Surg 1999; 384: 209-215.
52. Oliner JD, Kinzler KW, Meltzer PS, et al. Amplification of a gene encoding a p53-associated proteinin human sarcomas. Nature 1992; 358: 80-83.
53. Leach FS, Tokino T, Meltzer PS, et al. p53 Mutation and MDM2 amplification in human soft
Chapter 7 Soft tissue sarcoma: where to go?

92
tissue sarcomas. Cancer Res 1993; 53 (10 Suppl): 2231-2234.54. Hung J, Anderson R. p53: function, mutations and sarcomas. Acta Orthop Scand 1997; 68 (Suppl
273): 68-73.55. Hieken TJ, Das Gupta TK. Mutant p53 expression: A marker of diminished survival in well-
differentiated soft tissue sarcoma. Clin Cancer Res 1996; 2: 1391-1395.56. Nakayama T, Toguchida J, Wadayama B, Kanoe H, Kotoura Y, Sasaki MS. MDM2 gene
amplification in bone and soft tissue tumors: association with tumor progression in differentiatedadipose-tissue tumors. Int J Cancer 1995; 64: 342-346.
57. Cance WG, Brennan MF, Dudas ME, Huang CM, Cordon-Cardo C. altered expression of theretinoblastoma gene product in human sarcomas. N Engl J Med 1990; 323: 1457-1462.
58. Karpeh MS, Brennan MF, Cance WG, et al. Altered patterns of retinoblastoma gene productexpression in adult soft tissue sarcomas. Br J Cancer 1995; 72: 986-991.
59. Barrios C, Castresana JS, Kreicbergs A. Clinicopathologic correlations and short-term prognosis inmusculoskeletal sarcoma with c-myc oncogene amplification. Am J Clin Oncol 1994; 17: 273-276.
60. Sollazzo MR, Benassi MS, Magagnoli G, et al. Increased c-myc oncogene expression in Ewing�ssarcoma: correlation with Ki67 proliferation index. Tumori 1999; 85: 167-173.
61. Shen J, Scotlandi K, Baldini N, et al. Prognostic significance of nuclear accumulation of c-myc andmdm2 proteins in synovial sarcoma of the extremities. Oncology 2000; 58: 253-260.
62. Eilber FR, Giuliano AE, Huth JF, et al. A randomized prospective trial using postoperative adjuvantchemotherapy (Adriamycin) in high grade extremity soft tissue sarcoma. Ann J Clin Oncol 1988;11: 39-45.
63. Harrison LB, Franzese F, Gaynor JJ, Brennan MF. Long-term results of a prospective randomizedtrial of adjuvant brachytherapy in the management of completely resected soft tissue sarcomas of theextremity and superficial trunk. Int J Radiat Oncol Biol Phys 1993; 75: 94-100.
64. Pisters PWT, Harrison LB, Woodruff JM, Gaynor JJ, Brennan MF. A prospective randomized trialof adjuvant brachytherapy in the management of low-grade soft tissue sarcomas of the extremityand superficial trunk. J Clin Oncol 1994; 12: 1150-1155.
65. Yang JC, Chang AE, Baker AR, et al. Randomized prospective study of the benefit of adjuvantradiation therapy in the treatment of soft tissue sarcomas of the extremities. J Clin Oncol 1998; 16:197-203.
66. Eilber FR, Eckhardt JJ, Rosen G, Fu YS, Seeger LL, Selch MT. Neoadjuvant chemotherapy andradiotherapy in the multidisciplinary management of soft tissue sarcomas of the extremity. SurgOncol Clin N Am 1993; 2: 611-620.
67. Mann GR, Lewis JJ, Brennan MF. Adult soft tissue sarcoma. Aust N Z J Surg 1999; 69: 336-343.68. Hoekstra HJ, Geertzen JH. Resectie van de nervus ischiadicus. Zien patiënt en dokter er nog een
been in? Ned Tijdschr Geneesk 2001; in press.69. Morton DL, Eilber FR, Townsend CM, Grant TT, Mirra J, Weisenburger TH. Limb salvage from a
multidisciplinary treatment approach for skeletal and soft tissue sarcomas of the extremity. AnnSurg 1976; 184: 268-278.
70. Eilber FR, Giuliano AE, Huth JF, et al. A randomized prospective trial using postoperative adjuvantchemotherapy (Adriamycin) in high grade extremity soft tissue sarcoma. Ann J Clin Oncol 1988;11: 39-45.
71. Eilber FR, Eckhardt JJ, Rosen G, Fu YS, Seeger LL, Selch MT. Neoadjuvant chemotherapy andradiotherapy in the multidisciplinary management of soft tissue sarcomas of the extremity. SurgOncol Clin N Am 1993; 2: 611-620.
72. Nijhuis PHA, Pras E, Sleijfer D Th, Molenaar WM, Schraffordt Koops H, Hoekstra HJ. Long-term
Soft tissue sarcoma: where to go? Chapter 7

93
results of preoperative intraarterial doxorubicin combined with neoadjuvant radiotherapy, followedby extensive surgical resection for locally advanced soft tissue sarcomas of the extremities. RadiotherOncol 1999; 51: 15-19.
73. Hoekstra HJ. In: Oxford Textbook of Oncology, 2nd ed. Souhami RL, Tannock I, Hohenberger P,Horiot JC, eds. London, Oxford University Press 2001; in press.
74. Creech OJ, Ryan R, Krementz A. Treatment of malignant melanoma by isolation perfusiontechnique. JAMA 1959; 169: 339-343.
75. Cavaliere R, Ciocatto EC, Giovanella BC, et al. Selective heat sensitivity of cancer cells.Biochemical and clinical studies. Cancer 1967; 20: 1351-1381.
76. Stehlin Jr JS, Giovanella BC, De Ipolyi PD, Muenz LR, Anderson RF. Results of hyperthermicperfusion for melanoma of the extremities. Surg Gynecol Obstet 1975; 140: 339-348.
77. Krementz ET, Carter RD, Sutherland CM, Hutton I. Chemotherapy of sarcomas of the limbs byregional perfusion. Ann Surg 1977; 185: 555-564.
78. Hoekstra HJ, Schraffordt Koops H, Molenaar WM, Oldhoff J. Results of isolated regional perfusionin the treatment of malignant soft tissue tumours of the extremities. Cancer 1987; 60: 1703-1707.
79. Rossi CR, Vecchiato A, Foletto M, et al. Phase II study on neoadjuvant hyperthermic antiblasticperfusion with doxorubicin in patients with intermediate or high grade limb sarcomas. Cancer1994; 73: 2140-2146.
80. Ginkel RJ van, Schraffordt Koops H, Vries EGE de, Molenaar WM, Uges DR, Hoekstra HJ.Hyperthermic isolated limb perfusion with cisplatin in four patients with sarcomas of soft tissue andbone. Eur J Surg 1996; 22: 528-531.
81. Suit HD, Mankin HJ, Wood WC, et al. Preoperative, intraoperative, and postoperative radiationin the treatment of primary soft tissue sarcomas. Cancer 1985; 55: 2659-2667.
82. Hoekstra HJ, Schraffordt Koops H, Molenaar WM, Mehta DM, Sleijfer DTh, Dijkhuis G,Oldhoff J. A combination of intraarterial chemotherapy, preoperative and postoperativeradiotherapy, and surgery as limb-salving treatment of primarily unresectable high-grade soft tissuesarcomas of the extremities. Cancer 1989; 63: 59-62.
83. Lienard D, Delmotte JJ, Renard N, Ewalenko P, Lejeune FJ. High-dose recombinant tumournecrosis factor-alpha in combination with interferon gamma and melphalan in isolation perfusionof the limbs for melanoma and sarcoma. J Clin Oncol 1992; 10: 52-60.
84. Eggermont AMM, Schraffordt Koops H, Lienard D, Kroon BBR, Van Geel AN, Hoekstra HJ,Lejeune FJ. Isolated limb perfusion with high dose tumor necrosis factor-α in combination withinterferon-γ and melphalan for nonresectable extremity soft tissue sarcomas: a multicenter trial. JClin Oncol 1996; 14: 2653-2665.
85. Eggermont AMM, Schraffordt Koops H, Klausner JM, et al. Isolated limb perfusion with tumornecrosis factor and melphalan for limb salvage in 186 patients with locally advanced soft tissueextremity sarcomas. The cumulative Multicentre European Experience. Ann Surg 1996; 224:756-765.
86. Olieman AFT, Pras E, Ginkel RJ van, Molenaar WM, Schraffordt Koops H, Hoekstra HJ.Feasibility and efficacy of external beam radiotherapy after hyperthermic isolated limb perfusionwith TNF-α and melphalan for limb-saving treatment in locally advanced extremity soft tissuesarcoma. Int J Rad Oncol Biol Phys 1998; 40: 807-814.
87. Tepper JE, Suit HD. Radiation therapy alone for sarcoma of soft tissue. Cancer 1985; 56: 475-479.88. Nijhuis PHA, Schaapveld M, Otter R, Molenaar WM, Graaf van der WTA, Hoekstra HJ.
Epidemiological aspects of soft tissue sarcomas (STS)- Consequences for the design of clinical STStrials. Eur J Cancer 1999; 35: 1705-1710.
Chapter 7 Soft tissue sarcoma: where to go?

94
89. Kinsella Tj, Sindelar WF, Lack E, Glatstein E, Rosenberg SA. Preliminary results of a randomizedstudy of adjuvant radiation therapy in resectable adult retroperitoneal soft tissue sarcomas. J ClinOncol 1988; 6: 18-25.
90. Willett CG, Tepper JE, Mankin HJ, et al. Intraoperative electron beam therapy for retroperitonealsoft tissue sarcoma. Cancer 1991; 68: 278-283.
91. Sindelar WF, Kinsella TJ, Chen PW, DeLaney ThF, Tepper JE, Rosenberg SA, Glatstein E.Intraoperative radiotherapy in retroperitoneal sarcomas. Final results of a prospective, randomized,clinical trial. Arch Surg 1993; 128: 402-410.
92. Alektiar KM, Hu K, Brennan MF, Harrison LB. High-dose-rate intraoperative radiation therapy(HDR-IORT) for retroperitoneal sarcomas. Int J Radiat Oncol Biol Phys 2000; 47: 157-163.
93. Hoekstra HJ, Mehta DM, Wijffels RTM, Vermey J, Oldhoff J. Local tumor control by intraoperativeradiotherapy (IORT): a pilot experience. Eur J Surg Oncol 1991; 17: 364-369.
94. Hoekstra HJ, Sindelar WF, Szabo BG, Kinsella TJ. Hemipelvectomy and intraoperativeradiotherapy for bone and ssoft tissue sarcomas of the pelvic girdle. Radiather Oncol 1995; 37:160-163.
95. Schwarzbach, Willeke F, Eble M, Wannenmacher M, Lehnert T, Herfarth C. Morbidity and tumorcontrol in limb-saving resection with intraoperative radiotherapy in a multimodality therapyconcept in soft tissue sarcoma. Langenbecks Arch Chir Suppl Kongressbd 1998; 115: 1312-1315.
96. Suit HD, Markin HJ, Wood WC, Proppe KH. Preoperative, intraoperative, and postoperativeradiation in the treatment of primary soft tissue sarcoma. Cancer 1985; 55: 2659-2667.
97. Barkley T, Martin R, Romsdahl M, Pindberg R, Zagars GK. Treatment of soft tissue sarcomas bypreoperative irradiation and conservative surgical resection. Int J Radiat Oncol Biol Phys 1988; 14:693-699.
98. Suit HD. The George Edelstyn memorial lecture: Radiation in the management in the manage-ment of malignant soft tissue tumours. Clin Oncol 1989; 1: 5-10.
99. Robinson MH, Ball AB, Schofield J, Fisher C, Harmer CL, Thomas JM. Preoperative radiotherapyfor initially inoperable extremity soft tissue sarcomas. Clin Oncol 1992; 4: 36-43.
100. Mundt AJ, Awan A, Sibley GS, et al. Conservative surgery and adjuvant radiation therapy in themanagement of adult soft tissue of the extremities: clinical and radiobiological results. Int J RadiatOncol Biol Phys 1995; 32: 977-985.
101. Ngan SY. Radiotherapy in soft tissue sarcoma of the extremities. Acta Orthop Scand 1997; 68(Suppl 273): 112-116.
102. Leibel SA, Ling CC, Kutcher GJ, et al. The biological basis of conformal three-dimensionalradiation therapy. Int J Radiat Oncol Biol Phys 1991; 21: 805-811.
103. Leibel SA, Zelefsky MJ, Kutcher GJ, et al. The biological basis and clinical application ofthree-dimensional conformal external beam radiation therapy in carcinoma of the prostate. SeminOncol 1994; 21: 580-597.
104. Leibel SA, Kutcher GJ, Mohan R, et al. Three-dimensional conformal radiation therapy at theMemorial Sloan-Kettering Cancer Center. Semin Radiat Oncol 1992; 2: 274-289.
105. Bramwell V, Mouridsen H, Santoro G, et al. Cyclophosphamide vs ifosfamide: final report of arandomized phase II trial in adult soft tissue sarcoma. Eur J Cancer 1987; 23: 311-321.
106. Elias AD. Salvage therapy for soft tissue sarcomas. Semin Oncol 1994; 21: 76-81.107. Edmondson JH, Ryan LM, Blum RH, et al. Randomized comparison of doxorubicin alone versus
ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissuesarcomas. J Clin Oncol 1993; 11: 1269-1275.
108. Antman KH. Adjuvant therapy of sarcomas of soft tissue. Semin Oncol 1997; 24: 556-560.
Soft tissue sarcoma: where to go? Chapter 7

95
109. Pápai Zs, Bodoky Gy, Szánto, et al. The efficacy of a combination of etoposide, ifosfamide, andcisplatin in the treatment of patients with soft tissue sarcoma. Cancer 2000; 89: 177-180.
110. Steward WP, Verweij J, Somers R, et al. Granulocyte macrophage colony-stimulating factor allowssafe escalation of dose intensity of chemotherapy in metastatic adult soft tissue sarcoma: a study ofthe European Organizaation for Research and Treatment of Cancer Soft Tissue and Bone SarcomaGroup. J Clin Oncol 1993; 11: 15-21.
111. Frustaci S, Buenadonna A, Galligioni E, et al. Increasing 41-epidoxorubicin and fixed ifosfamidedoses plus granulocyte-macrophage colony-stimulating factors in advanced soft tissue sarcomas: apilot study. J Clin Oncol 1997; 15: 1418-1426.
112. Patel SR, Vadhan-Raj S, Burgess MA, et al. Results of two consecutive trials of dose-intensivechemotherapy with doxorubicin and ifosfamide in patients with sarcomas. Am J Clin Oncol 1998;21: 317-321.
113. Elias AD, Eder JP, Shea T, et al. High-dose ifosfamide with mesna uroprotection: A phase I study.J Clin Oncol 1990; 8: 95-103.
114. Santoro A, Turez T, Mouridsen H, et al. Doxorubicin versus CYVADIC versus doxorubicin plusifosfamide in first-line treatment of advanced soft tissue sarcoma: a randomized study of theEuropean Organizaation for Research and Treatment of Cancer Soft Tissue and Bone SarcomaGroup. J Clin Oncol 1995; 13: 1537-1545.
115. Sarcoma meta-analysis collaboration. Adjuvant chemotherapy for localised resectable soft-tissuesarcomas of adults: meta-analysis of individual data. Lancet 1997; 350: 1647-1654.
116. Bramwell VH, Eisenhauer EA, Blackstein M, et al. Phase II study of topotecan (NSC 609699) inpatients with recurrent or metastatic soft tissue sarcoma. Ann Oncol 1995; 6: 847-849.
117. Edmonson JH, Ebbert LP, Nascimento AG, et al. Phase II study of docetaxel in advanced soft tissuesarcomas. Am J Clin Oncol 1996; 19: 574-576.
118. Fidias P, Demetri G, Harmon DC. Navelbine shows activity in previously treated sarcomapatients: phase II results from MGH/ Dana Farber Partners Cancer Care Study. Proc Am SocClin Oncol 1998; 17: 513a (abstract 1977).
119. Rankin C, Budd GT, Hutchins L, Antman K. SWOG 9518 phase II trial of continuous infusiontopotecan in patients with advanced soft tissue sarcomas. Proc Am Soc Clin Oncol 1998; 17: 523a(abstract 2011).
120. Blay JY, Judson I, Rodenhuis S, et al. Phase II study of raltitrexed (�Tomudex�) for patients withadvanced soft tissue sarcomas refractory to doxorubicin-containing regimens. Anticancer Drugs1999; 10: 873-877.
121. Verweij J, Lee SM, Ruka W, et al. Randomized phase II study of docetaxel versus doxorubicin infirst- and second-line chemotherapy for locally advanced or metastatic soft tissue sarcomas in adults:a study of the European Organization for Research and Treatment of Cancer Soft Tissue and BoneSarcoma Group. J Clin Oncol 2000; 18: 2081-2086.
122. Plager C, Howard J, Papadopoulos NE, et al. A phase II study of 5-FU infusion and interferonalpha in metastatic sarcoma. Proc Am Assoc Cancer Res 1992; 33: 229 (Abstract 1369).
123. Elias AD. High-dose therapy for adult soft tissue sarcoma: dose response and survival. Semin Oncol1998; 25 (suppl 4): 19-23.
124. Lineham DC, Bowne WB, Lewis JJ. Immunotherapeutic approaches to sarcoma. Semin Surg Oncol1999; 17: 72-77.
125. Waxman S, Borden EC, Lotan R, Levens D, Young CW. Differentiation therapy of cancer:laboratory and clinical investigations. Cancer Res 1993; 53: 4109-4115.
126. Molenaar WM, Oosterhuis JW, Kamps WA. Cytological �differentiation� in childhood rhabdomy-
Chapter 7 Soft tissue sarcoma: where to go?

96
osarcomas following polychemotherapy. Hum Pathol 1984; 15: 973-979.127. Molenaar WM, Oosterhuis JW, Oosterhuis AM, Ramaekers FCS. Mesenchymal and muscle
specific intermediate filaments, vimentin and desmin, as markers for differentiation in childhoodrhabdomyosarcomas. Hum Pathol 1985; 16: 838-843.
128. Melguizo C, Prados J, Marchal JA, Aránega AE, Alvarez L, Aránega A. Low concentrations ofactinomycin D cause potentially therapeutic differentiation in human rhabdomyosarcoma cell lineRD. Pathol Res Pract 1996; 192: 188-194.
129. Marchal JA, Prados J, Melguizo C, et al. GR-891: a novel fluorouracil acyclonucleoside prodrug fordifferentiation therapy in rhabdomyosarcoma cells. Br J Cancer 1999; 79: 807-813.
130. Tontonoz P, Singer S, Forman BM, et al. Terminal differentiation of human liposarcoma cellsinduced by ligands for peroxisome proliferator-activated receptor gamma and the retinoid X receptor.Proc Natl Acad Sci USA 1997; 94: 237-241.
131. Demetri GD, Fletcher CD, Mueller E, et al. Induction of solid tumor differentiation by theperoxisome proliferator- activated receptor-gamma ligand troglitazone in patients with liposarcoma.Proc Natl Acad Sci USA 1999; 96: 3951-3956.
132. Hirota S, Isozaki K, Moriyama Y, et al. Gain -of- function mutations of c-kit in humangastrointestinal stromal tumors. Science 1998; 279: 577-580.
133. Lux Ml, ubin BP, Biase TL, et al. KIT extracellular and kinase domain mutations in gastrointestinalstromal tumors. Am J Pathol 2000; 156: 791-795.
134. Joenslu H, Roberts PJ, Saslomo- Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in apatient with a metastatic gastrointestinal stromal tumor. N Eng J Med 2001; 344: 1052-1056.
Soft tissue sarcoma: where to go? Chapter 7

97Chapter 8
Chapter 8
Summary

98
Soft tissue sarcomas (STS) comprise a relatively heterogenic group of malignant tumorsarising in mesenchymal tissues. In 1997, 523 new primary STS were diagnosed in theNetherlands, whereas 219 patients died from this disease in that year. In the UnitedStates, approximately 8100 new STS are diagnosed annually, whereas 4600 patients dieannually from their disease. These figures are relatively disappointing when comparedto testicular cancer, a malignancy with a similar incidence, but a much lower cancerdeath rate. In 1997, 415 new primary testicular cancers were diagnosed in the Nether-lands, whereas �only� 31 patients died from that disease. In the U.S. these numbers are6900 patients and 300 patients, respectively. The unfavourable prognosis of STS is causedmainly by the propensity for metastasis. Moreover, local recurrence rates are relativelyhigh (up to 20%), causing a substantial morbidity. Although STS have been studiedquite extensively, several aspects remain unclear. The goal of the present thesis was toget more insight into some of the unsolved aspects of this uncommon malignancy.
The first part of the introduction in Chapter 1 contains general considerations regardingepidemiology, pathogenesis, and prognostic factors in STS. There is a need for popula-tion-based studies, as most studies on sarcoma are center-based and may therefore bebiased. In recent years, the understanding of the STS biology has improved by advancesin cytogenetics and molecular techniques. The increased insight into the interactionbetween oncogenes, tumor suppressor genes and their regulators may aid in prognosti-cation.One of the most important prognostic factors is the histopathological tumor (sub)type,which may become indiscernible if prognostic favorable tumor (sub)types are reviewedtogether with less favorable (sub)types. Therefore, studies that review the various histo-logical (sub)types seperately are awaited.The second part of the introduction deals with guidelines. Guidelines are very meaning-ful in rare diseases as STS, because the experience in individual hospitals is often lim-ited, whereas treatment often is very complex. In the Netherlands, as in most westerncountries, STS guidelines have been developed and distributed. Nevertheless, nothing isknown about the compliance with these guidelines, although such information seemsessential for the development of future guidelines and quality assurance programs.The last part of the introduction reviews the treatment of STS, which changed dramati-cally during the second half of the last century. Before the mid-fifties, most STS wereexcised by local resections or shell-out procedures, resulting in an unacceptable highlocal failure rate. At the end of the fifties, Bowden, Booher and Stener developed the firstlimb-saving techniques, which were not widely adopted. After Enneking�s theory of lon-gitudinal STS spread within the compartment, wide en-block resections gained popular-ity, resulting in low local recurrence rates, but high amputation rates. The studies of Suitand Lindberg and the famous NCI trial formed the base of true multimodality STS treat-ment.
Unfortunately, the approach of locally advanced STS remained a problem. Morton andEilber developed a successful multimodality protocol, which was associated with a rela-tively high acute morbidity rate. Another very successful treatment for these unfavour-able tumors has been the hyperthermic isolated limb perfusion (HILP), a technique thatwas first described in STS in 1977. The major breakthrough came in the early nineties,
Summary Chapter 8

99
when Lejeune added tumor necrosis factor-alpha (TNF-α) and interferon-gamma (IFN-γ) to the HILP regimen, which resulted in a better local response rate and a high limb-salvage rate with an acceptable toxicity.In recent years, there has been a growing awareness of the potential long-term side effectsof cancer treatment protocols. Studies on intensive multimodality therapies used in locallyadvanced STS should therefore not only report on recurrence rate and short-termmorbidity, but also focus on complications and functional outcome in the long-term, asthese might interfere with the initial goal of the treatment.
The research questions that formed the basis of this thesis are formulated at the end ofthe introduction and are dealt with in the following chapters.In Chapter 2, we describe epidemiological aspects of STS, based on the cancer registry ofthe Comprehensive Cancer Center North-Netherlands (CCCN). The aim of the study wasto gain insight into true epidemiological aspects of STS, and to provide data for thedevelopment of future STS clinical trials. Four hundred and fifty-six primary STS (Kaposi,urogenital and gastrointestinal STS excluded) that were registered from 1989- 1995 bythe cancer registry of the CCCN were analyzed. The age-adjusted incidence of STS was3.6/100.000/year. At initial diagnosis, most patients (n=225) were aged between 50- 74years (49%), whereas 118 patients (26%) were 25- 49 years, 53 patients (12%) were youngerthan 25 years, and 60 patients (13%) were 75 years or older. Most STS were located in theextremities (45%), especially the lower limb and hip region (29%). The most commonhistological tumor types were malignant fibrous histiocytoma (MFH) and liposarcoma(both 18%), followed by leiomyosarcoma (15%), dermatofibrosarcoma (14%), fibrosar-coma (7%), and rhabdomyosarcoma (5%). The incidence of the histological types wasage-dependent. Most of the head/neck STS were T1-tumors, whereas all retroperitonealSTS were T2-tumors. Notwithstanding the existence of staging guidelines in the CCCNregion, presence or absence of lymph node involvement was not recorded in 210 pa-tients (57%). Twelve of 158 patients with a documented N-stage (8%) had lymph nodemetastases at initial presentation. However, using a �best-case-scenario�, in which allunknown N-stages are to be considered as node-negative, the overall incidence of nodalinvolvement would be 3%. The M-stage was not recorded in 129 patients (35%). At pres-entation, 34 of 239 patients (14%) with documented M-stage had distant metastases.Using the �best-case-scenario�, the overall incidence of distant metastatic disease wouldbe 9%. Five patients (1%) had both lymph node and distant metastases at presentation.Although lymph node involvement was not related to tumor size, distant metastaseswere significantly related to T-stage. Distant metastases were encountered in 21% ofretroperitoneal STS, in 14% of lower limb and hip STS, but were absent in head/neckSTS.
Most obvious differences in treatment were found between children and adolescents(≤20 years) and patients above 70 years. In contrast to localized disease, where we couldnot demonstrate any treatment difference, treatment in (regional and/or distant) meta-static disease was different between both groups. Less than 50% of the older patientswith metastatic disease were treated and only 20% received some form of chemotherapy,in contrast to the younger patients, who were all treated with, at least, chemotherapy. Asnearly half of the patients are older than 65 years at initial presentation, and roughly 10%
Chapter 8 Summary

100
are children and adolescents, only 40% of all adult patients presenting with a STS maybe eligible for clinical trials investigating the value of (neo) adjuvant chemotherapy inadult STS. Therefore, either new STS trials should be developed especially for the eld-erly, or future trials should not contain upper age limits. Instead, inclusion of olderpeople should be dependent on other criteria, as WHO performance scale or absence ofcertain co-morbidity.
In Chapter 3, we analyze the long-term results of a very intensive multimodality treat-ment protocol used in locally advanced STS of the extremities. The treatment protocolimplied continuous intraarterial doxorubicin for three consecutive days (20 mg/m2),followed by preoperative radiotherapy (10 x 3.5 Gy) and tumor resection, with or withoutpostoperative external beam radiotherapy (EBRT). This treatment scheme was adoptedat the Groningen University Hospital in the early eighties. Although the acute morbidityof this intensive protocol was well known, hardly anything was known about late compli-cations. In an era, where there is a growing awareness of potential long-term side effectsin cancer survivors, it is necessary to gain more insight into this aspect of cancer treat-ment. We therefore analyzed 11 patients with locally advanced STS who were treatedbetween 1983 and 1987 according to the above mentioned multimodality scheme.Three patients suffered a severe acute local skin necrosis after intraarterial doxorubicin(27%), which deteriorated in two of them after preoperative radiotherapy. Limb-salvagewas possible in ten patients (91%). After a median follow-up of 84 (range 3- 136) months,six patients died (55%), five from metastatic disease and one from a non-disease-relatedcerebral hemorrhage. Five patients were still alive after a median follow-up of 120 (range110- 136) months. Three of them developed disabling functional limitations of the af-fected extremity. In two patients, limitations were caused by severe long-term complica-tions: a complete neuropathy of the sciatic nerve five years after treatment and a sponta-neous fracture of the affected femur 91 months after therapy with no healing tendencysince. The third patient developed a very severe fibrosis of the affected limb, resulting inan invalidating limb function. Potential contributing factors to these problems in thelong-term were discussed: 1) interaction between doxorubicin and radiotherapy, 2)hypofractionated radiation schemes, and 3) addition of postoperative EBRT, leading to ahigh biological effective radiation dose.Although the Groningen Sarcoma Working Party abandoned this treatment regimenafter the introduction of hyperthermic isolated limb perfusion (HILP) with TNF-α andmelphalan, this study demonstrates once more the need for long-term follow-up surveysin order to discover potential side effects that might eventually interfere with the primarytreatment goal.
Although, in general, biological behaviour and prognosis of liposarcomas (LPS) are morefavourable compared to most other STS, prognosis can widely vary depending on tumorcharacteristics, especially histological (sub)type and tumor grade. The study, presentedin Chapter 4, aimed to get more insight into epidemiological aspects of LPS, to evaluatetreatment results and to determine prognostic factors for local recurrence, metastasis,disease-free and disease-specific survival.All consecutive, completely resected AJCC stage I-III LPS, treated at the GroningenUniversity Hospital from 1977- 2000, were analyzed (n=69). The legs and retroperitoneum
Summary Chapter 8

101
were the most common tumor sites (n=38 and n=11, respectively), followed by trunk (n=6),arms (n=5), head/neck (n=5), and buttock (n=4). Twenty-five patients received adjuvantradiotherapy (36%), three patients were treated by resection and chemotherapy (4%), andnine patients were treated by all three modalities (13%). In 53 patients (77%), surgical marginswere microscopically free (R0-resection), whereas in the other 16 patients margins weremicroscopically involved (R1-resection). The most common histological subtypes weremyxoid LPS (n=34; 49%) and well-differentiated LPS (n=26; 38%), followed bydedifferentiated LPS (n=6; 9%), and pleomorphic LPS (n=3; 4%). After a median follow-up of 71 (range 5-231) months, the overall local recurrence and metastasis rate at five yearsafter diagnosis were 27% and 16%, respectively. It was concluded that liposarcoma is aquite heterogeneous disease, and that its outcome is determined to a significant degree byhistological subtype, grade, size, stage, depth, and type of resection. Compared to otherSTS, LPS had a relatively mild biological behavior, with the exception of very large, deeplylocated, dedifferentiated and/or grade II-III LPS. Retroperitoneal localization was anadditional negative prognostic factor for local recurrence. However, in contrast to somereports in the literature, we were not (yet) able to demonstrate a significant influence ofretroperitoneal localization on survival.
As the diagnostic management of STS is essential for definitive treatment, adherence todiagnostic guidelines seems important. Chapter 5 reports on the appropriateness of thediagnostic management of STS in the CCCN-region, according to these guidelines.All primary STS (n=351), registered by the CCCN from January 1989-January 1996, wereanalyzed with regard to adherence to the diagnostic guidelines. Urogenital, gastrointestinalSTS, and Kaposi sarcomas were excluded. In the specialized center (Groningen UniversityHospital), 69% of patients were younger than 60 years, whereas in the eighteen districthospitals, 63% of patients were 60 years or older. With increase of age, referral to thecenter declined in a linear fashion. For all guidelines, adherence was significantly betterin the center. In district hospitals, case load had no significant influence on compliancewith the guidelines, except for the management of STS ≥3 cm. In district hospitals, whereless than 15 patients were treated in the 7-years period, significantly more often aninadequate or even no biopsy procedure was performed prior to resection. As correctdiagnosis, staging and treatment has been shown to be important in the management ofmany tumors, it was concluded that STS should be concentrated in a limited number ofhospitals that collaborate with (the) specialized center(s).Ways to improve development, dissemination, implementation, and evaluation of guide-lines are discussed. Ideally, future guidelines should be developed by those who are touse them, should be disseminated by enthusiastic educational programs, and should beimplemented by a quality control program with patient-specific feedback. Furthermore,special attention should be paid to the older patients, as they were significantly moreoften not referred to a specialized center for treatment.
The aim of the study presented in Chapter 6 was to determine the prognostic signifi-cance of cytogenetic changes in soft tissue sarcomas, using a computer-assisted cytoge-netic analysis. From 1984- 1993, 38 primary STS (88%) and 5 local recurrent STS (12%)were karyotyped successfully. None of these STS was previously treated by chemo- and/or radiotherapy. Liposarcoma was the most frequent STS (47%), followed by synovial
Chapter 8 Summary

102
sarcoma (12%), and MFH (9%). After a median follow-up of 55 months, 20 patients hadno evidence of disease (47%), 6 were alive with recurrent disease (14%), and 17 patientshad died, 16 of which from their disease. At multivariate analysis, chromosomal gain at thelong arm of chromosome 1 (1q) and chromosomal loss at the short arm of chromosome 4(4p) were statistically significant negative prognostic factors regarding survival (RelativeRisk (RR) 39 and 6, respectively). Regarding metastasis-free survival, the only significantnegative prognostic factor was chromosomal loss at the short arm of chromosome 18 (18p),with a relative risk of 8. Furthermore, there was a strong association between loss in 18pand gain in 1q in patients with the shortest survival.This study provides indications for a correlation between cytogenetic changes andmetastasis and prognosis in STS. Some of the findings confirm earlier reports, whereasothers are novel in STS and need to be confirmed in additional studies. The computer-assisted approach used in this study, is valuable in the analysis of large groups of complexkaryotypes in order to detect common chromosomal alterations. The strong associationbetween alterations in the long arm of chromosome 1 and the short arm of chromosome18 in non-survivors is very challenging and may have prognostic value in STS.
In Chapter 7, we look into the near future of STS treatment and discuss some importantissues as centralization and specialization, new prognostic factors, and developmentsthat might be interesting for future STS treatment.In many malignancies, the independent prognostic importance of the treatment institu-tion, the individual surgeon, and case load has been confirmed. At present, when consid-erable effort and resources are spent on large multicenter trials on adjuvant chemo- and/or radiation therapy in the hope to improve cancer survival marginally, there is growingevidence that outcome could be increased much more by improvements in surgery. Suchimprovements may be accomplished by better surgical training and by centralization ofdifficult surgical procedures and multidisciplinary treatment protocols. This especiallyholds true for rare tumors as sarcomas where the experience of the individual institutionsand surgeons is very limited.
With the current clinicopathological prognostic factors, clinicians can identify at bestpatients with approximately a 50% risk of distant failure (G3T2b). Therefore, future studiesshould focus on the identification of new prognostic indicators that might facilitate theidentification of a higher-risk subset of patients. Recent research into angiogenesis,molecular biology, and cytogenetics has revealed some new prognostic factors as highintratumoral microvessel density, high S-phase fraction, overexpression of proliferationmarkers as Ki-67, DNA aneuploidy, mutation or functional inactivation of the p53 tumorsuppressor gene, MDM2 gene amplification, loss of expression of the RB1 gene, andamplification of myc oncogene. At present, there is no consensus how these factors shouldbe incorporated into clinical practice. Thorough future studies are necessary before rou-tine clinical use of these cellular and molecular markers.
The last part of this chapter deals with STS treatment in the new millennium, and startswith a historical overview of the development of modern multimodality treatment.In radiotherapy, relatively new techniques, as brachytherapy, intraoperative radiotherapy,three-dimensional conformal radiotherapy, inverse radiotherapy planning, and intensity
Summary Chapter 8

103
modulated radiation therapy, might be valuable to shorten treatment time, to savesurrounding normal tissues, and/or to enable further dose-escalation.The role of adjuvant systemic chemotherapy is very limited in STS. Combinationmodalities involving chemotherapy, immunotherapy, biological response modifiers, andmodulators of multidrug resistance might improve the efficacy.
At last, differentiation therapy is described as a new and very appealing approach thatmay become an alternative or addition to conventional anti-tumor therapy. This treat-ment that is based on the theory of reprogramming malignant cells to stabilize or evenconvert them into normal tissue, has been studied especially in liposarcoma. The firstpositive results have recently been published.
Many of these new techniques are very appealing, and, although in some of them resultsfrom experimental and clinical pilot studies are very encouraging, the way to routineclinical use is still very long. To improve treatment results and to stimulate research,centralization and specialization seems important, especially in rare tumors as STS. Inorder to develop new treatment strategies, STS should preferably be treated in trials onlyand sarcoma working parties should be encouraged to collaborate more.
Chapter 8 Summary

104 Summary Chapter 8

105
Chapter 8
Samenvatting
Chapter 9 Samenvatting

106
Wekedelensarcomen (WDS) omvatten een relatief heterogene groep kwaadaardigegezwellen die ontstaan uit mesenchymale weefsels. In 1997 werden in Nederland 523nieuwe WDS gediagnosticeerd, terwijl 219 patiënten overleden aan deze ziekte. In deVerenigde Staten worden jaarlijks ongeveer 8100 nieuwe patiënten gediagnosticeerd enoverlijden naar schatting 4600 patiënten aan deze ziekte. Deze cijfers zijn teleurstellendals ze vergeleken worden met bijvoorbeeld zaadbalkanker, een maligniteit met eenvergelijkbare incidentie, maar met een veel lagere sterfte. In 1997 werden in Nederland415 nieuwe testistumoren gediagnosticeerd, terwijl in dat jaar �slechts� 31 patiëntenoverleden aan deze aandoening. In de Verenigde Staten worden jaarlijks ongeveer 6900nieuwe testistumoren geregistreerd en 300 gerelateerde sterfgevallen. De matige prognosevan WDS wordt vooral veroorzaakt door de neiging tot snelle hematogene metastasering.Bovendien recidiveert de tumor vaak lokaal (tot 20%), hetgeen meestal gepaard gaat meteen aanzienlijke morbiditeit. Ofschoon er reeds veel onderzoek is verricht bij WDS, blijfteen aantal aspecten nog onduidelijk. Het doel van dit proefschrift is meer inzicht tekrijgen in een aantal onopgeloste vraagstukken.
Het eerste deel van de inleiding in Hoofdstuk 1 bevat een aantal algemene gegevens overepidemiologie, pathogenese en prognostische factoren bij WDS. Aangezien de meestestudies bij WDS zijn gebaseerd op gegevens uit gespecialiseerde centra waardoor selectiemogelijk is, is er behoefte aan populatiegebaseerd onderzoek.Een van de belangrijkste bekende prognostische factoren is het histopathologische(sub)type van het WDS, hetgeen minder zal opvallen indien prognostisch gunstige(sub)typen tezamen met minder gunstige (sub)typen worden onderzocht. Vandaar dat erbehoefte is aan studies waarbij de verschillende histologische (sub)typen apart wordenonderzocht.Dankzij de vooruitgang in cytogenetische en moleculaire technieken is het inzicht in debiologie van WDS de laatste jaren sterk toegenomen. Dit toegenomen inzicht in de rolen interactie van oncogenen, tumorsuppressorgenen en hun regulatoren kan in de nabijetoekomst een belangrijke rol gaan spelen bij het bepalen van de prognose.
Het tweede deel van de inleiding gaat over richtlijnen. Met name bij zeldzameaandoeningen als WDS lijken richtlijnen erg zinvol aangezien de ervaring met dergelijkeziektebeelden in de meeste ziekenhuizen beperkt is, terwijl de behandeling vaak zeercomplex is. Net als in de meeste westerse landen, zijn in Nederland richtlijnen voor dediagnostiek en behandeling van WDS ontwikkeld en verspreid. Er is echter bijna nietsbekend over de mate waarin die richtlijnen worden gevolgd, ofschoon dergelijkeinformatie van belang lijkt voor het opstellen van toekomstige richtlijnen enkwaliteitsbewakingsprogramma�s.Het laatste deel van de inleiding gaat over de behandeling van WDS die in de tweede helftvan de afgelopen eeuw sterk is veranderd. Tot het midden van de jaren 50 werden demeeste WDS verwijderd via een krappe lokale excisie hetgeen resulteerde in eenonacceptabel hoog lokaal recidief percentage. In de tweede helft van de jaren 50ontwikkelden Bowden, Booher en Stener de eerste extremiteitsparende technieken, dieechter niet veel navolging kregen. Onder invloed van de theorieën van Enneking werdenruime compartimentresecties in toenemende mate uitgevoerd. Dit leidde weliswaar toteen vermindering van het lokaal recidief percentage, maar ook tot een toename van het
Samenvatting Chapter 9

107
aantal amputaties. Een echte multidisciplinaire benadering van WDS kwam echter pas ineen stroomversnelling na publicaties van Suit, Lindberg, en met name Rosenberg.De behandeling van lokaal uitgebreide WDS bleef echter een groot probleem. Morton enEilber ontwikkelden een multidisciplinaire therapie voor deze uitgebreide tumoren, dieweliswaar succesvol was, maar tevens leidde tot een relatief hoge (vroeg-) postoperatievemorbiditeit. Een andere succesvolle behandeling van deze ongunstige tumoren was dehypertherme geïsoleerde extremiteitperfusie (hyperthermic isolated limb perfusion,HILP), die voor WDS het eerst beschreven werd in 1977. De echte doorbraak kwamechter pas in het begin van de jaren 90, toen Lejeune tumor necrose factor-alpha (TNF-α) en interferon-gamma (IFN-γ) aan het perfusie protocol (met Melfalan) toevoegde. Dezetoevoeging leidde tot een betere lokale tumorrespons met een acceptabele toxiciteit, terwijlhet percentage extremiteiten dat gespaard kon worden toenam.
De laatste jaren is er een toenemend bewustzijn dat intensieve kankerbehandelingen opde lange termijn bijwerkingen kunnen hebben. In het licht hiervan zullen intensieve,vaak multidisciplinaire, therapieën zoals bij het lokaal voortgeschreden WDS niet alleenbeoordeeld moeten worden op recidief percentage en vroegtijdige morbiditeit, maar ookop de complicaties en het uiteindelijke functionele resultaat op de lange termijn.De onderzoeksvragen die de basis vormden voor dit proefschrift staan geformuleerd aanhet einde van de inleiding en zullen in de daaropvolgende hoofdstukken aan de ordekomen.
In Hoofdstuk 2 beschrijven we epidemiologische aspecten van WDS, gebaseerd op dekankerregistratie van het Integraal Kanker Centrum Noord-Nederland (IKN). Het doelvan de in dit hoofdstuk beschreven studie was het verkrijgen van meer inzicht in deepidemiologie van WDS en het verzamelen van gegevens voor toekomstige klinischeWDS studies. In dit onderzoek werden 456 nieuwe primaire WDS geanalyseerd, die van1989- 1995 door het IKN waren geregistreerd. Kaposi�s sarcomen en urogenitale engastrointestinale WDS werden hierbij uitgesloten. De leeftijdsgecorrigeerde incidentievan WDS bedroeg 3.6/100.000/jaar. De meeste patiënten (n=225) waren 50- 74 jaar oud(49%), 118 patiënten (26%) waren 25- 49 jaar, 53 patiënten jonger dan 25 jaar (12%) en60 patiënten (13%) 75 jaar of ouder. De meeste WDS (45%) waren gelokaliseerd in deextremiteiten, vooral been en heupregio (29%). Het maligne fibreus histiocytoom (MFH)en het liposarcoom (LPS) waren de meest voorkomende WDS (beide 18%), gevolgd doorleiomyosarcoom (15%), dermatofibrosarcoom (14%), fibrosarcoom (7%), en rhabdomyo-sarcoom (5%). De incidentie van de verschillende histologische typen was gerelateerdaan leeftijdsgroepen. De tumorgrootte bleek gerelateerd aan de lokalisatie van het WDS:de meeste WDS in het hoofd/hals gebied waren T1-tumoren, terwijl alle WDS in hetretroperitoneum T2-tumoren waren. Ondanks het bestaan van IKN-richtlijnen voor destadiëring en therapie van WDS, kon bij 210 patiënten (57%) de regionale klierstatusniet achterhaald worden. Twaalf van de 158 patiënten met een gedocumenteerde regionalelymfklierstatus (8%) hadden bij presentatie een metastase. Uitgaande van het zogenaamde�best case scenario� waarin alle onbekende N-stadia beschouwd worden als kliernegatief,daalt het percentage kliermetastasen bij presentatie naar 3%. Het M-stadium wasonbekend bij 129 patiënten (35%). Bij presentatie hadden 34 van de 239 patiënten (14%)met een gedocumenteerd M-stadium metastasen op afstand.
Chapter 9 Samenvatting

108
Bij toepassing van het �best case scenario� zou het percentage afstandsmetastasen dalennaar 9%. Vijf patiënten (1%) hadden bij presentatie zowel lymfogene als hematogenemetastasen. Het optreden van lymfkliermetastasen bleek niet gerelateerd te zijn aantumorgrootte, in tegenstelling tot het optreden van metastasen op afstand die optraden bij21% van de retroperitoneale WDS en bij 14% van WDS in been of heup, maar afwezigwaren bij hoofd/hals WDS.
De meest opvallende verschillen in behandeling traden op tussen de groep van kinderenen adolescenten (≤ 20 jaar) en de groep patiënten van 75 jaar en ouder. Bij gelokaliseerdeziekte was de behandeling in beide groepen gelijk. In geval van metastasering (lymfogeenen/of hematogeen) was er echter een groot verschil: minder dan 50% van de oudere patiëntenmet gemetastaseerde ziekte werd behandeld, waarbij slechts 20% chemo-therapie kreeg.Dit in tegenstelling tot de jongere patiënten met gemetastaseerde ziekte, die allen behandeldwerden met, ten minste, chemotherapie.Aangezien bij presentatie ongeveer de helft van de patiënten 65 jaar of ouder is en erongeveer 10% kinderen en adolescenten zijn, zal slechts 40% van alle patiënten met eenprimair WDS in aanmerking komen voor inclusie in klinische onderzoeken naarbijvoorbeeld de waarde van adjuvante chemotherapie. In verband hiermee zullen er nieuweWDS onderzoeken moeten worden ontwikkeld die met name gericht zijn op ouderepatiënten, oftewel moeten toekomstige WDS onderzoeken geen leeftijdslimieten meerbevatten. Inclusie van oudere patiënten zou gebaseerd moeten zijn op andere criteria, zoalsde WHO performance scale of de aan- of afwezigheid van bepaalde co-morbiditeit.
In Hoofdstuk 3 worden de lange termijn resultaten geanalyseerd van een zeer intensiefmultidisciplinair protocol voor de behandeling van lokaal uitgebreide WDS van deextremiteiten. Dit protocol bestond uit continue intraarteriële infusie met doxorubicinegedurende 3 opeenvolgende dagen (20 mg/m2), gevolgd door preoperatieve radiotherapie(10 x 3,5 Gy) en tumor resectie, met of zonder postoperatieve adjuvante radiotherapie. Desarcoomwerkgroep van het Academisch Ziekenhuis Groningen (AZG) startte met ditprotocol in het begin van de jaren 80. Ofschoon deze behandeling later geassocieerdbleek met een aanzienlijke morbiditeit op de korte termijn, is er weinig bekend over delange termijn gevolgen. Zeker in de huidige tijd waarin er een groeiend besef is vaneventueel nadelige gevolgen van kankertherapieën op de lange termijn, is het van belangmeer inzicht te krijgen in dit aspect van intensieve behandelprotocollen. Daarom werden11 patiënten met een lokaal uitgebreid WDS van de ledematen geanalyseerd, die tussen1983 en 1987 volgens het eerder beschreven schema waren behandeld in het AZG.Drie patiënten ontwikkelden een acute lokale huidnecrose na intraarteriële infusie metdoxorubicine (27%). Bij twee van hen verergerde dit na preoperatieve bestraling. Bij 10patiënten (91%) kon een extremiteitsparende ingreep worden uitgevoerd. Zes patiënten(55%) overleden na een mediane follow-up van 84 (spreiding 3-136) maanden. Vijf vanhen overleden ten gevolge van afstandsmetastasen en een patiënt overleed ten gevolgevan een hersenbloeding. Na een mediane follow-up van 120 (spreiding 110- 136) maandenwaren 5 patiënten (45%) nog in leven. Bij drie van hen was er een ernstige functionelebeperking van de aangedane extremiteit opgetreden. Bij 2 patiënten waren de beperkingenontstaan ten gevolge van een ernstige late complicatie: een spontane en volledige uitvalvan de nervus ischiadicus die 5 jaar na behandeling optrad en een pathologische fractuur
Samenvatting Chapter 9

109
van het femur dat in het bestralingsveld had gelegen. De fractuur was 91 maanden nabehandeling ontstaan en had geen genezingstendens. De derde patiënt ontwikkelde eenzeer ernstige fibrosering van het aangedane been, resulterend in een invaliderendefunctiebeperking. De mogelijke oorzaken voor deze late complicaties worden besproken:1) interactie tussen doxorubicine en bestraling, 2) de hypogefractioneerde bestralings-schema�s en 3) toevoeging van postoperatieve bestraling hetgeen kan leiden tot een hogebiologisch effectieve dosis.Ofschoon de Groningse sarcoomwerkgroep dit behandelprotocol afschafte na de intro-ductie van HILP met TNF-α en Melfalan, toont deze studie nog eens aan dat er behoefteis aan langdurige follow-up die mogelijke late complicaties aan het licht kan brengen.Dergelijke complicaties kunnen interveniëren met het oorspronkelijke doel van debehandeling, namelijk het behoud van een functionerende extremiteit.
Ofschoon het biologisch gedrag en de prognose van liposarcomen (LPS) in het algemeengunstiger is dan van de meeste andere WDS, kunnen beide sterk variëren afhankelijkvan tumorkarakteristieken als histopathologische (sub)typering en gradering. De studiedie in Hoofdstuk 4 wordt beschreven heeft tot doel meer inzicht te krijgen in deepidemiologie van LPS, de behandeling ervan in het Academisch Ziekenhuis Groningen(AZG) te evalueren, en het vaststellen van prognostische factoren voor lokaal recidief,metastasen, ziekte-vrije en ziekte-specifieke overleving.
Alle opeenvolgende, niet gemetastaseerde LPS (n=69), die van 1977 tot 2000 volledigwaren gereseceerd in het AZG, werden geanalyseerd. De meeste LPS waren gelokaliseerdin benen en retroperitoneum (respectievelijk n=38 en n=11), gevolgd door romp (n=6),armen (n=5), hoofd/hals (n=5), en bil (n=4). Vijfentwintig patiënten (36%) kregenadjuvante radiotherapie, 3 patiënten (4%) werden behandeld met resectie enchemotherapie, en 9 patiënten (13%) werden behandeld met alle drie de modaliteiten.Bij 53 patiënten (77%) waren de chirurgische sneevlakken microscopisch vrij van tumor(R0-resectie), terwijl in de overige 16 patiënten de sneevlakken microscopisch niet vrijvan tumor waren (R1-resectie). Het meest voorkomende histologisch subtype was hetmyxoide LPS (n=34; 49%), gevolgd door het goedgedifferentieerde LPS (n=26; 38%), hetgededifferentieerde LPS (n=6; 9%), en het pleiomorfe LPS (n=3; 4%). Met een medianefollow-up van 71 (spreiding 5- 231) maanden was het 5-jaars lokaal recidief en metastasepercentage respectievelijk 27% en 16%.Deze studie toont aan dat LPS een zeer heterogene tumorsoort is en dat de prognose inbelangrijke mate wordt bepaald door het histologisch subtype, de gradering, detumorgrootte, het AJCC stadium, de lokalisatie en de aard van resectie. Vergeleken metandere WDS hebben LPS een relatief mild biologisch gedrag, met uitzondering vanvolumineuze, subfasciaal gelegen, gededifferentieerde en/of graad II-III LPS.Retroperitoneale LPS hebben een hoger lokaal recidief percentage. Echter, in tegenstellingtot een aantal andere studies, konden wij (nog) niet aantonen dat patiënten met eenretroperitoneaal LPS een slechtere overleving hadden.
Aangezien een correcte diagnostiek bij WDS essentieel is voor het definitieve behandelplan,lijkt het belangrijk bestaande richtlijnen op te volgen.Hoofdstuk 5 handelt over de mate waarin diagnostische richtlijnen binnen de IKN-regio
Chapter 9 Samenvatting

110
worden opgevolgd. Daartoe werden alle primaire WDS die van januari 1989- januari 1996binnen het IKN waren geregistreerd geanalyseerd, waarbij met name gekeken werd naarde mate waarin bestaande richtlijnen werden gevolgd. Kaposi sarcomen en urogenitale engastrointestinale WDS werden uitgesloten. In het gespecialiseerde oncologische centrum(AZG) was 69% van de patiënten jonger dan 60 jaar, in tegenstelling tot de 18 algemeneziekenhuizen binnen de IKN-regio waar 63% van alle patiënten ouder was dan 60 jaar. Bijtoename van de leeftijd van de patiënt nam het verwijspercentage naar het gespecialiseerdecentrum bijna rechtlijnig af. In het oncologisch centrum werden alle richtlijnen significantbeter opgevolgd. Het patiëntenaantal bleek binnen de groep van de algemene ziekenhuizengeen significante invloed te hebben op de mate waarin de richtlijnen werden opgevolgd.Het patiëntenaantal bleek echter wel van invloed op het beleid bij grotere, en dus suspecte,tumoren. In algemene ziekenhuizen waar minder dan 15 patiënten waren behandeld in destudieperiode werd significant vaker een inadequate of zelfs geen biopsie uitgevoerdvoorafgaande aan de �definitieve� resectie.Er werd geconcludeerd dat WDS geconcentreerd zouden moeten worden in een beperktaantal ziekenhuizen die nauw samenwerken met gespecialiseerde centra.Tot slot worden in dit hoofdstuk eventuele mogelijkheden ter verbetering van deontwikkeling, verspreiding, implementatie en evaluatie van richtlijnen besproken.Idealiter zouden richtlijnen moeten worden opgesteld door diegenen die ze ook gaangebruiken, zouden ze verspreid moeten worden via enthousiaste (bij-) scholings-programma�s en zouden ze geïmplementeerd moeten worden in een kwaliteits-bewakingprogramma met patiëntspecifieke feedback.
De studie die in Hoofdstuk 6 wordt beschreven heeft tot doel de prognostische waarde tebepalen van cytogenetisch afwijkingen in WDS, gebruik makend van een computer-geassisteerde analyse. Van 1984- 1993 konden 38 primaire WDS (88%) en 5 lokaalgerecidiveerde WDS (12%) met succes gekaryotypeerd worden. Patiënten mochten niettevoren behandeld zijn met chemo- en/of radiotherapie. In deze studie was LPS hetmeest voorkomende WDS (47%), gevolgd door synoviasarcoom (12%) en MFH (9%). Naeen mediane follow-up van 55 maanden, hadden 20 patiënten (47%) geen tekenen vangerecidiveerde ziekte, waren 6 patiënten (14%) nog in leven met een lokaal recidief en/of afstandsmetastasen en waren 17 patiënten overleden, van wie 16 ten gevolge van deziekte. Bij multivariate analyse bleek dat chromosomale winst op de lange arm vanchromosoom 1 (1q) en chromosomaal verlies op de korte arm van chromosoom 4 (4p)statistisch significante negatief prognostische factoren waren. Voor de metastasevrijeoverleving bleek chromosomaal verlies op de korte arm van chromosoom 18 (18p) eensignificante negatief prognostische factor te zijn. Er bleek een sterke associatie te bestaantussen verlies in 18p en winst in 1q bij patiënten met de kortste overleving.Geconcludeerd werd dat deze studie aanwijzingen gaf voor het bestaan van een relatietussen cytogenetische veranderingen en metastase en prognose bij WDS. Een aantalresultaten ondersteunt eerder gerapporteerde bevindingen, terwijl andere niet eerderbeschreven zijn en bevestigd zullen moeten worden in toekomstige studies. De sterkeassociatie tussen veranderingen op de lange arm van chromosoom 1 en op de korte armvan chromosoom 18 bij patiënten met de kortste overleving is erg intrigerend en zouprognostische waarde kunnen hebben bij WDS. De computergeassisteerde cytogenetischeanalyse die gebruikt werd bleek waardevol voor de analyse van een grotere groep tumoren
Samenvatting Chapter 9

111
met een complex karyotype.
In Hoofdstuk 7 wordt ingegaan op mogelijke ontwikkelingen in de nabije toekomst.Belangrijke aspecten, zoals specialisatie en centralisatie, nieuwe prognostische factorenen nieuwe ontwikkelingen op therapeutisch gebied worden besproken.Bij veel maligniteiten is aangetoond dat het instituut waar de behandeling plaatsvindt,de individuele chirurg en het patiëntenaantal onafhankelijke prognostische factoren zijn.Heden ten dage wordt er veel geld en moeite gestoken in groots opgezette multicentrischeonderzoeken naar de waarde van adjuvante chemo- en/of radiotherapie, die, naarverwachting, de overleving slechts marginaal zullen beïnvloeden. Ondertussen komener steeds meer aanwijzingen, dat verbeteringen op het gebied van de chirurgie wel eensvan grotere invloed zouden kunnen zijn. Dergelijke verbeteringen zouden bereikt kunnenworden door een verbeterde chirurgische opleiding (vervolgopleidingen) en doorcentralisatie van ingewikkelde chirurgische procedures en gecompliceerde multidiscipli-naire behandelprotocollen. Dit geldt met name voor zeldzame tumoren zoals WDS waarde ervaring in de afzonderlijke ziekenhuizen veelal zeer beperkt is.Met de huidige klinische en pathologische prognostische factoren zijn clinici slechts instaat patiënten te identificeren met ongeveer 50% kans op afstandsmetastasering (G3T2b).Om een subgroep te kunnen identificeren met een hoger risico, zullen er nieuwe,specifiekere, prognostische factoren moeten worden vastgesteld. Recente onderzoekenop het gebied van angiogenese, moleculaire biologie en cytogenetica hebben enkele nieuwepotentiële prognostische factoren opgeleverd. Er is echter momenteel geen consensushoe deze factoren in de dagelijkse praktijk zouden moeten worden ingevoerd.Het laatste deel van dit hoofdstuk gaat over de behandeling van WDS in het nieuwemillennium nadat eerst een overzicht gegeven is van de historische ontwikkeling van demoderne multidisciplinaire behandeling van WDS.
In de nabije toekomst zou er in de behandeling van WDS meer gebruik kunnen wordengemaakt van een aantal radiotherapeutische technieken als brachytherapie, intraoperatieveradiotherapie, �three-dimensional radiotherapy�, �inverse radiotherapy planning� en�intensity modulated radiotherapy�. Met behulp van deze technieken zou de totalebehandeltijd verkort kunnen worden, zouden de omgevende weefsels meer gespaardkunnen worden en zou dosisescalatie mogelijk zijn.De rol van adjuvante chemotherapie is bij WDS vrij beperkt. Combinatietherapieën metchemotherapie, immunotherapie, biologische respons modulatoren en medicamentendie de chemotherapieresistentie moduleren lijken effectiever te zijn.Tot slot wordt differentiatie therapie besproken, een intrigerend nieuw behandelings-concept dat gebaseerd is op de herprogrammering van maligne cellen, waardoor dezegestabiliseerd worden of zelfs weer veranderen in normale cellen. De eerste positieveresultaten van experimenteel onderzoek bij LPS zijn recentelijk gepubliceerd.
Veel van deze nieuwe technieken en behandelingen zijn erg intrigerend. De weg naarroutinematige klinische toepassing lijkt echter nog vrij lang, ofschoon de resultaten vaneen aantal experimentele en klinische onderzoeken veelbelovend zijn. Centralisatie enspecialisatie lijken van belang te zijn voor stimulatie van onderzoek op het gebied vanWDS en voor verbetering van de resultaten. WDS zouden bij voorkeur in trials moeten
Chapter 9 Samenvatting

112 Samenvatting Chapter 9
worden behandeld en sarcoomwerkgroepen zouden meer moeten samenwerken, opdatin de nabije toekomst nieuwe behandelingsstrategieën kunnen worden ontwikkeld.

113Dankwoord
Dankwoord

114
De in dit proefschrift beschreven onderzoeken werden uitgevoerd bij de afdelingChirurgische Oncologie van het Academisch Ziekenhuis Groningen en het IntegraalKankercentrum Noord-Nederland (I.K.N.), in nauwe samenwerking met de afdelingenPathologie, Radiotherapie, Medische Oncologie en Klinische Genetica.
In het bijzonder dank ik mijn promotores prof. dr. H.J. Hoekstra, prof. dr. W.M. Molenaaren prof. dr. H. Schraffordt Koops voor het in mij gestelde vertrouwen, hun steun enbegeleiding.
Prof. Dr. H.J. Hoekstra, beste Harald, jouw interesse in de wetenschap is bijna spreek-woordelijk. Het enthousiasme, wetenschappelijk inzicht en niet te vergeten de snelheidwaarmee jij manuscripten weet te reviseren zijn van groot belang geweest bij detotstandkoming van dit proefschrift. Ik beschouw het als een eer jouw eerste promovenduste zijn en hoop dat wij in de toekomst nog vaak mogen samenwerken.
Prof. Dr. W.M. Molenaar, beste Ineke, naast je grote deskundigheid op het gebied vansarcomen zal vooral de prettige samenwerking mij bijblijven. Ondanks je drukke werk-zaamheden kon ik altijd op je rekenen als er weer eens een aantal sarcomen moest wordengereviseerd.
Prof. Dr. H. Schraffordt Koops, beste Heimen, jij was degene die mij naar Groningenhaalde voor de vervolgopleiding Chirurgische Oncologie. Ik ben je dankbaar voor het inmij gestelde vertrouwen en de steun die ik van je mocht ontvangen.
De leden van de beoordelingscommissie, bestaande uit prof. dr. D.Th. Sleijfer, prof. dr.B.G. Szabó en prof. dr.Th. Wobbes (Universitair Medisch Centrum St. Radboud) ben ikdank verschuldigd voor de beoordeling van dit proefschrift.
Een aantal personen wil ik apart noemen voor hun bijdrage aan dit proefschrift.
Drs. M. Schaapveld, beste Michael, jij hebt veel werk verzet bij de bewerking van degrote hoeveelheid data uit de I.K.N. regio. Nu dit proefschrift achter de rug is zullen weeindelijk eens een duik moeten plannen.
Dr. W.T.A. van der Graaf, beste Winette, ik wil je bedanken voor je bijdrage aan eenaantal manuscripten waarbij jouw grote deskundigheid op het gebied van sarcomen meerdan eens bleek. Daarnaast is de prettige omgang met patiënten en collega�s kenmerkendvoor jou.
Dr. W.J. Sluiter, beste Wim, dank voor je hulp bij de statistische analyse van een aantalin het kader van dit proefschrift uitgevoerde studies.
Drs. E. Pras, beste Betty, met veel genoegen kijk ik terug op onze samenwerking.Door gezamenlijk radiotherapeutische planningen uit te werken toonde jij mij het belangvan een goede communicatie tussen radiotherapeut en chirurg en vooral van een adequaatoperatieverslag. Het zou een verplichte stage moeten zijn voor elke chirurg die onco-
Dankwoord Chapter 10

115
logische patiënten behandelt.
Dr. E. van den Berg, beste Eva, het is geen eenvoudig opgave een chirurg iets bij tebrengen over cytogenetica. Jij bent daar evenwel prima in geslaagd. Het meelopen op deafdeling Medische Genetica was zeer zinvol voor een goede begripsvorming. Jouw hulpen enthousiasme waren van doorslaggevende waarde bij de totstandkoming vanHoofdstuk 6.
Dr. R. Otter, beste Renée, ik dank je voor de mogelijkheid om via het I.K.N. data overwekedelensarcomen in Noord-Nederland te kunnen verzamelen. Verder zou ik allemedewerkers van het I.K.N. die hierbij betrokken zijn geweest nogmaals willen bedankenvoor hun inzet.
Dr. B.E.C. Plaat, beste Boudewijn, dank voor je hulp bij het bewerken van gegevensuit de sarcoomdatabank. Ik wens je veel sterkte toe met je opleiding tot kno-arts.
Dr. J.Th.M. Plukker, Prof. Dr. A. Vermey, Dr. R.C.J. Verschueren, Dr. J. de Vries enDrs. R.T.M. Wijffels, beste John, Bert, René, Jaap en Robert, dank voor jullie bijdrage aanmijn opleiding tot chirurg-oncoloog. Bert, jouw suggesties waren van harte welkom.
Prof. Dr. T. Wiggers, beste Theo, in de (te) korte tijd dat ik met jou heb mogen werkenheb jij, niet alleen als professional, maar ook als mens, een grote indruk achtergelaten.
Drs. H. de Vries, beste Harry, met veel plezier kijk ik terug op onze samenwerking.Never forget Harry�s nerve! Ik wens Margreet en jou het allerbeste toe voor de toekomst.Nu jij!
Leden van de werkgroep Wekedelentumoren Noord-Nederland, tijdens onze bijeenkomstenheb ik veel van jullie kunnen leren over de histologie van deze zeldzame tumoren.
Dr. J.G. Prins, als �mijn� opleider leerde u mij de beginselen van het vak. De wijzewaarop u patiënten benaderde en de, in mijn ogen, ongeëvenaarde technischevaardigheden hebben een grote invloed gehad op mijn huidige functioneren.
Cees Coppe en Sylvia Stalman, dank voor jullie hulp bij alles wat voorafging aan ditboekje.
Ben Mobach, beste Ben, ondanks jouw drukke werkzaamheden bood jij mij aan tehelpen met het ontwerpen van de lay-out en de omslag van dit proefschrift. Het resultaattoont in welke bevoorrechte positie ik verkeerde door van jouw kennis en ervaring gebruikte kunnen maken.
Ir. D.W.P. Nijhuis en drs. S.E.M. Konijnenberg, beste Danny en Stephan, het is mij eengroot genoegen dat jullie mijn paranimfen willen zijn. Jullie gevoel voor humor zal eengoede basis vormen voor een geslaagde dag.
Chapter 10 Dankwoord

116
Grote dank ben ik verschuldigd aan mijn ouders die altijd alle vertrouwen hebbengehad in mijn ambities. Zij hebben mij gestimuleerd en in de gelegenheid gesteld om testuderen. Mijn moeder heeft het einde van mijn specialisatie helaas niet mee mogenmaken. In liefdevolle herinnering draag ik dit boekje aan haar op.
Lieve Rian, alleen jij weet hoeveel je voor mij betekent. Dank voor je begrip en steunbij de verwezenlijking van mijn ambities in Groningen. De ruimte die jij me bood, jouwliefde, opgewektheid, en relativerend vermogen zijn onmisbaar geweest bij het bereikenvan de geplande doelen.
Lieve Amanda en Paul, jullie betekenen oneindig veel voor mij. Nu pappa�s boekjeklaar is, kan ik weer meer tijd vrijmaken voor jullie.
Dankwoord Chapter 10