sickle cell anemia
TRANSCRIPT
Dr shamsheer ali p t
moDerateD by:Dr.sheena
siCKle Cell anaemia

I am the red of the blood responsible for respiration;
Deliver oxygen to tissues and return co2 to lungs;
influenced by factors ph, BPG, Cl in my functions;
disturbed in duties by structural abnormalities.
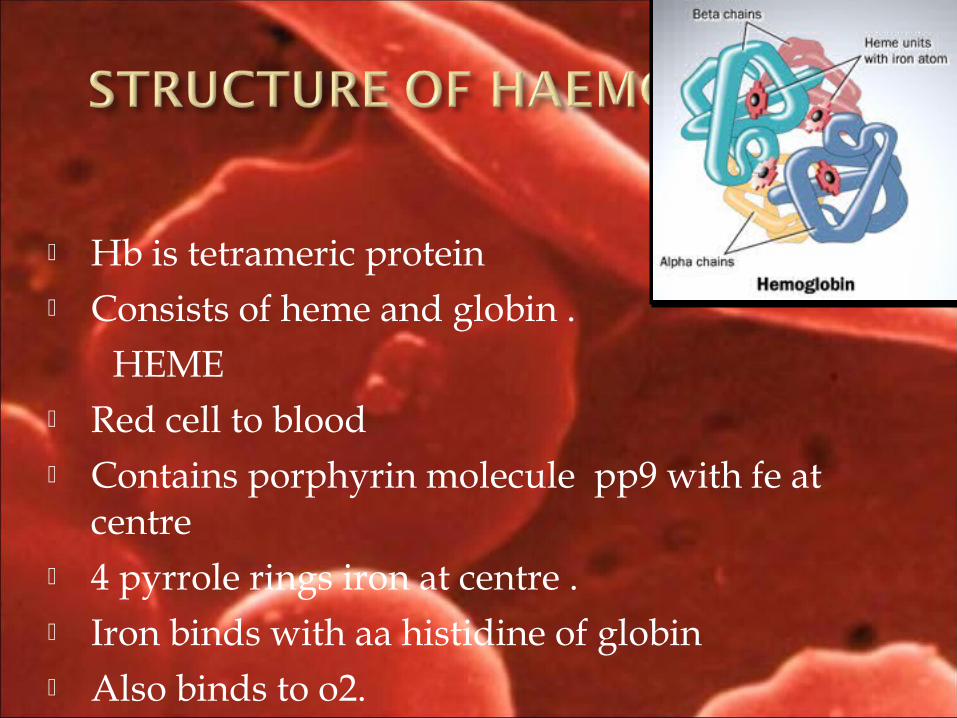
Hb is tetrameric protein Consists of heme and globin . HEME Red cell to blood Contains porphyrin molecule pp9 with fe at
centre 4 pyrrole rings iron at centre . Iron binds with aa histidine of globin Also binds to o2.

globin :consist of two pairs of polypeptide chain. (alpha & beta)
Abnormalities in these proteins leads to hemoglobinopathies
Genes for hemoglobin production lies on chromosome no 16 and 11 .
Chromosome 16 3 genes(zeta,alpha 1 n 2 ) ,chrom 11 5 genes(epsilon,2 gamma,delta genes,beta genes )
Molecular weight of Hb is 64000
Hb A1- 97% of adult haemoglobin, consists of 2 alpha & 2 beta chains.
Hb A2- Consists of 2 alpha & 2 delta chains. Hb F - 70 – 90% at birth, 25% by 1 month and
5% by 6months of age, consists of 2 alpha & 2 gamma chains.

In the year 1904 cardiologist and professor of medicine James B Herrick found peculiar elongated cells in blood of a 20 years old dental student who was suffering from anemia.
Veron Mason in 1922 named SICKLE CELL ANAEMIA.
Linus Pauling in 1940 demonstrated that sickling occurs as a result of abnormality in haemoglobin molecule.
Sickle cell disease is the first genetic disorder whose molecular basis was known.
June 19th is celebrated as World Sickle Cell Day
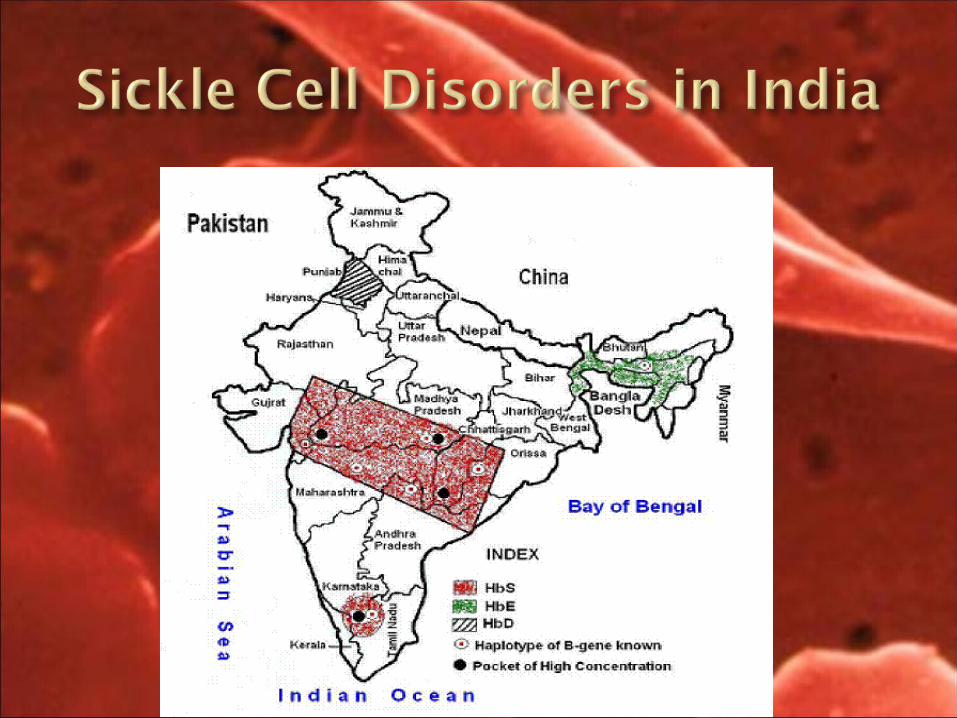

Sickle cell disease in Wayanad, Kerala: gene frequencies and disease characteristics.
Department of Pathology, Medical College, Kerala, India.
A large number of patients with sickle cell anaemia are seen at the Medical College, Calicut from among the tribals and Chetti communities of the adjacent Wayanad district. We carried out a population-based study of gene frequencies and disease characteristics to plan an appropriate intervention.

methoDs:CliniCal examination anD haemoglobin eleCtrophoresis were Done in 1016 subjeCts belonging to the tribal anD Chetti Communities in wayanaD DistriCt, by visiting hamlets anD sChools anD evaluating everyone present at the time of the visit.
results:
the gene frequenCy of haemoglobin s rangeD from 0.019 in KattunayaKan to 0.196 in wayanaDan Chettis. wayanaDan Chettis, Kurumas anD aDiyas showeD a high number of homozygotes with the olDest being 48 years. the survival of homozygotes is longer than what is generally reCorDeD in other states. the Disease was milD in 52.2% of Cases. painful Crises were founD in 43.5% anD splenomegaly anD leg ulCers in 4.3% eaCh. the mean haemoglobin f rate in homozygotes was 25.9%. it was higher in CliniCally milD Cases anD in those showing an absenCe of irreversible siCKle Cells in the peripheral smear.

ConClusions: the survival of patients with siCKle Cell anaemia seems to be higher in Kerala as CompareD to other states. it appears that even small improvements in primary health Care available to the population (as in Kerala) are suffiCient to aChieve this effeCt. integration of Disease Diagnosis anD management into the alreaDy existing health Care Delivery system may leaD to even better survival anD quality of life

1. HOMOZYGOUS AND HETEROZYGOUS SICKLE CELL DISEASES.
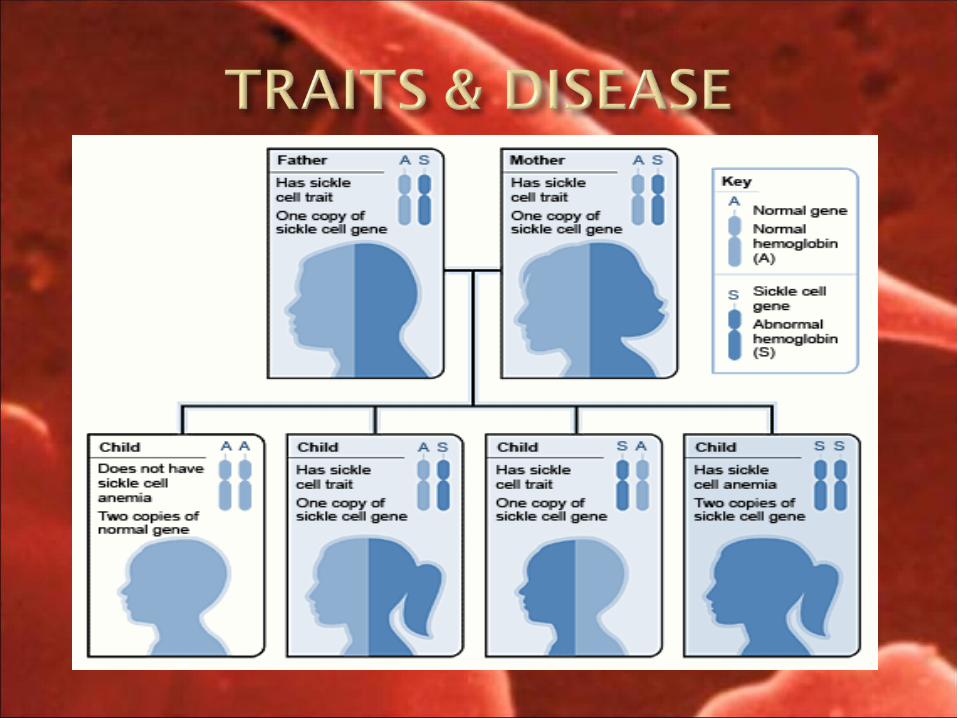
2. HOMOZYGOUS means mutant gene inherrited from both parents (HbSS) i.e sickle cell anemia
3. HETEROZYGOUS means (HbAS) only one gene is affected other one is normal i.e sickle cell trait.
4. Sickle cell disease----sickle cell anemia +sickle cell traits5. Haemoglobin C (HbSC) 6. Haemoglobin E (HbSE)7. Haemoglobin S beta Thalasseamia-This is a mild form
of sickle disorder.

Sickle cell anemia is a autosomal recessive genetic disease that results from the substitution of Valine from Glutamic acid in position 6 of beta globin gene leading to production of defective form of haemoglobin.(Hb S)
Hb S is a structurally defective haemoglobin.
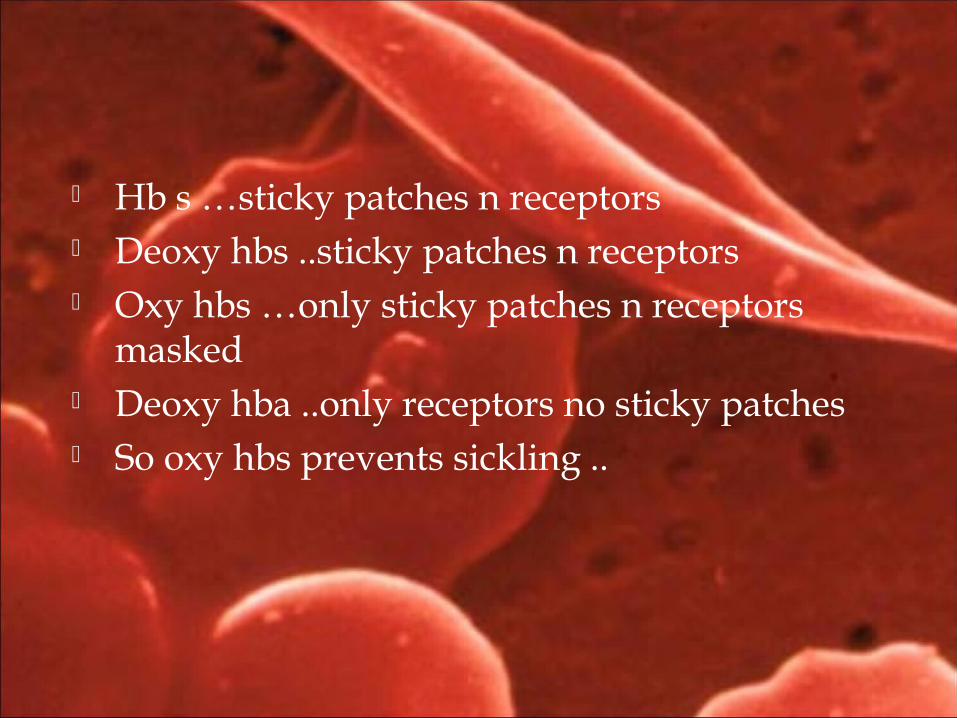
Hb s …sticky patches n receptors Deoxy hbs ..sticky patches n receptors Oxy hbs …only sticky patches n receptors
masked Deoxy hba ..only receptors no sticky patches So oxy hbs prevents sickling ..

Deoxygenation leads to hydrophobic interaction between adjacent Hb S molecules.
Distortion of RBC into sickle form cells Rapid haemolysis Decreased elasticity of cell wall of RBC Decreased life span 10 – 20 days. Clogging of RBC in microcirculation.
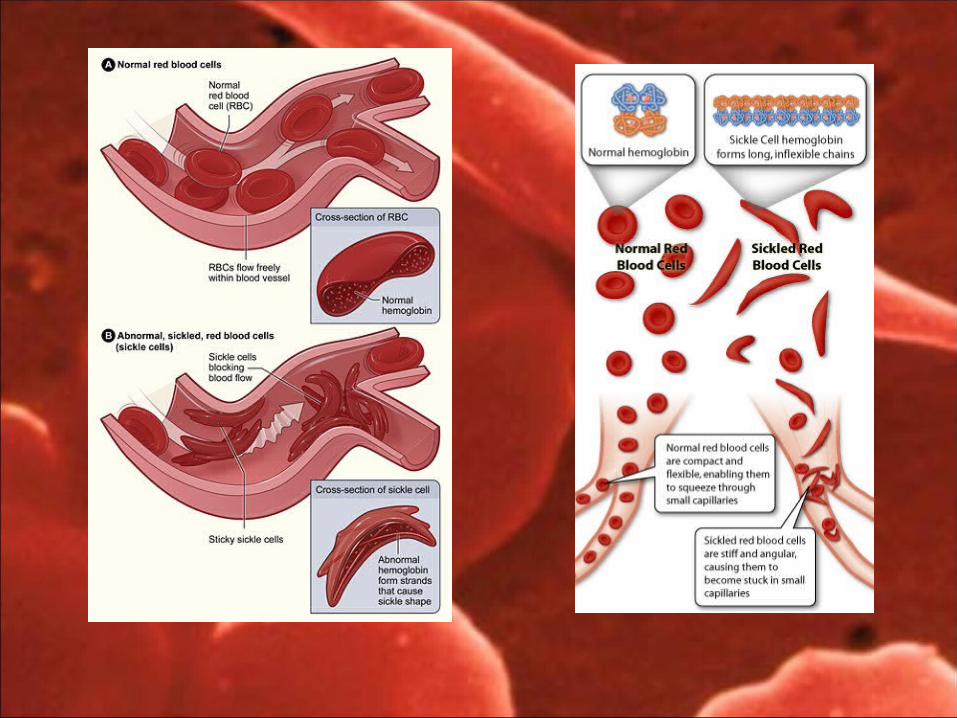
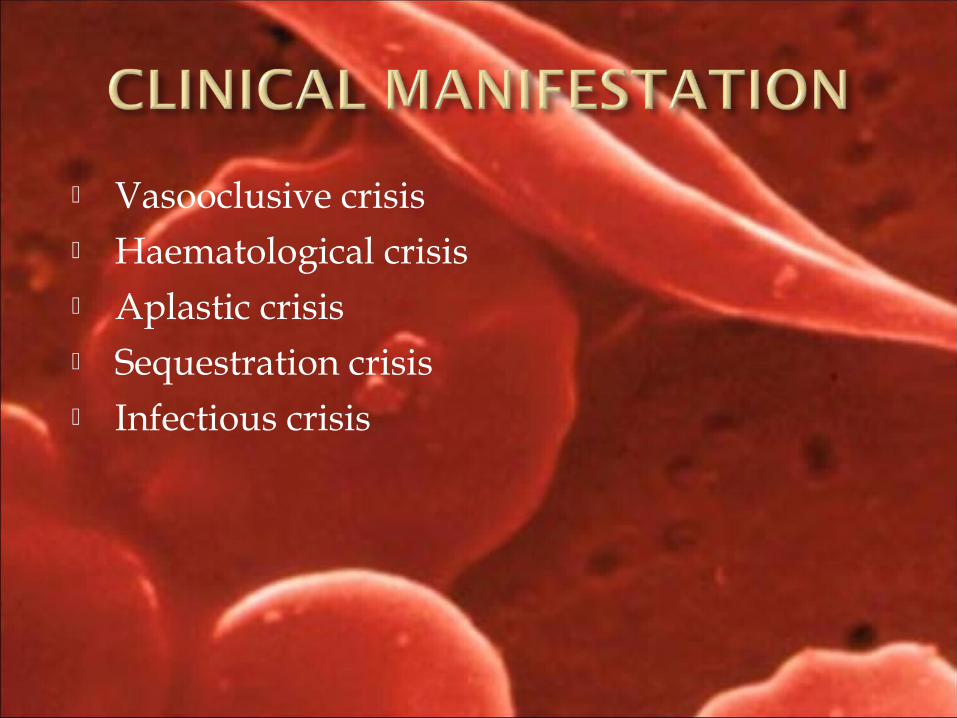
Vasooclusive crisis Haematological crisis Aplastic crisis Sequestration crisis Infectious crisis

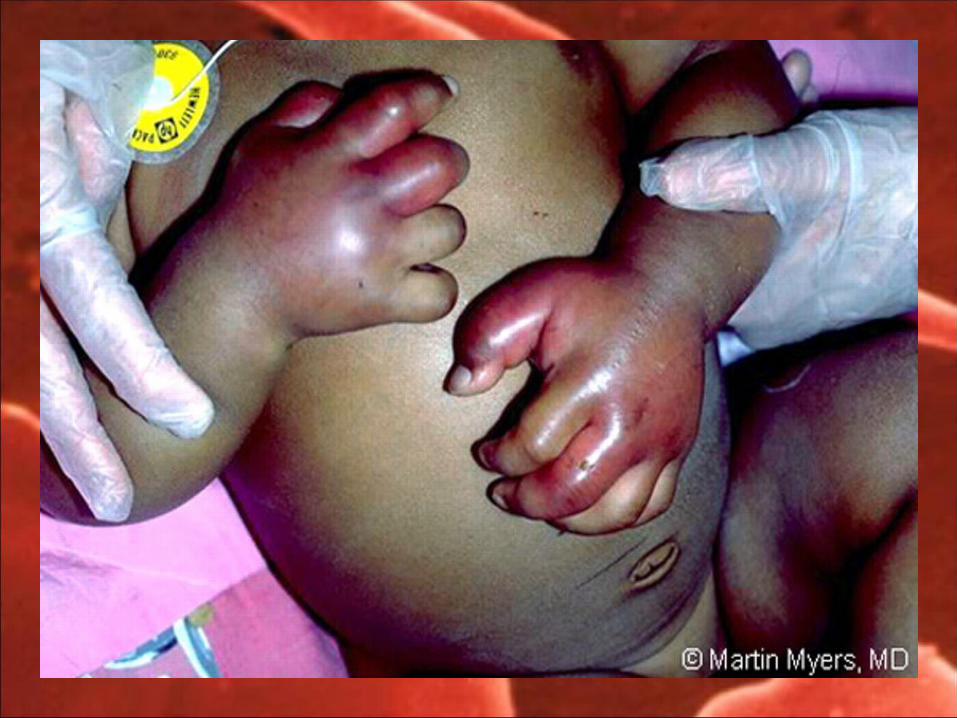
Often first manifestation of sca 50% of children by 2 yrs of age Symmetrical or unilateral swelling of the hands Not to be confused with osteomyelitis as mgt
varies….

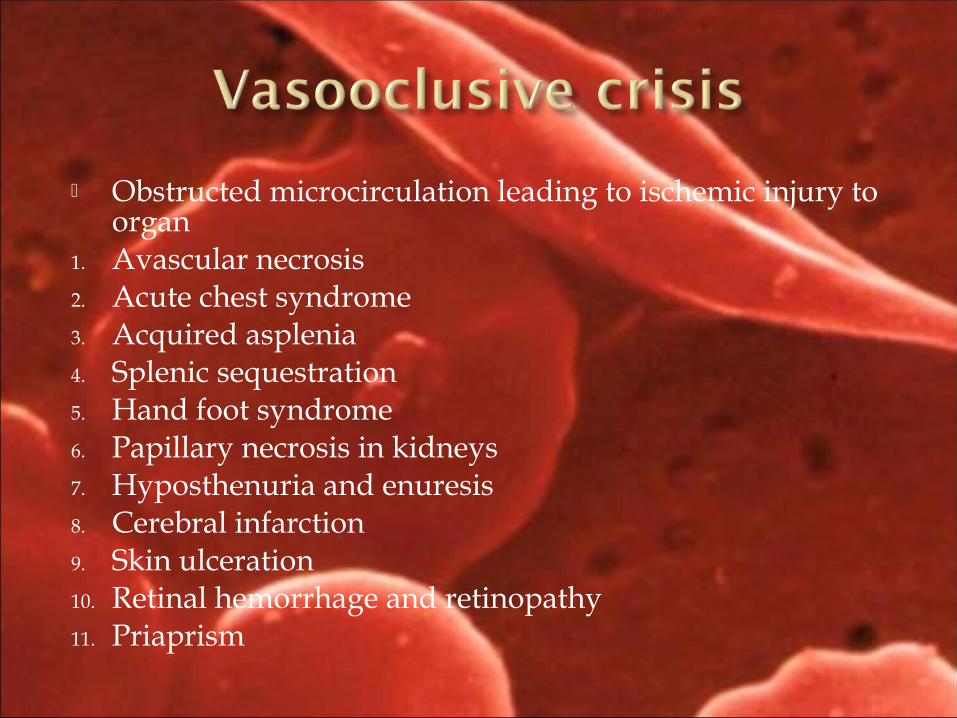
Obstructed microcirculation leading to ischemic injury to organ
1. Avascular necrosis 2. Acute chest syndrome3. Acquired asplenia4. Splenic sequestration5. Hand foot syndrome6. Papillary necrosis in kidneys7. Hyposthenuria and enuresis8. Cerebral infarction9. Skin ulceration10. Retinal hemorrhage and retinopathy11. Priaprism
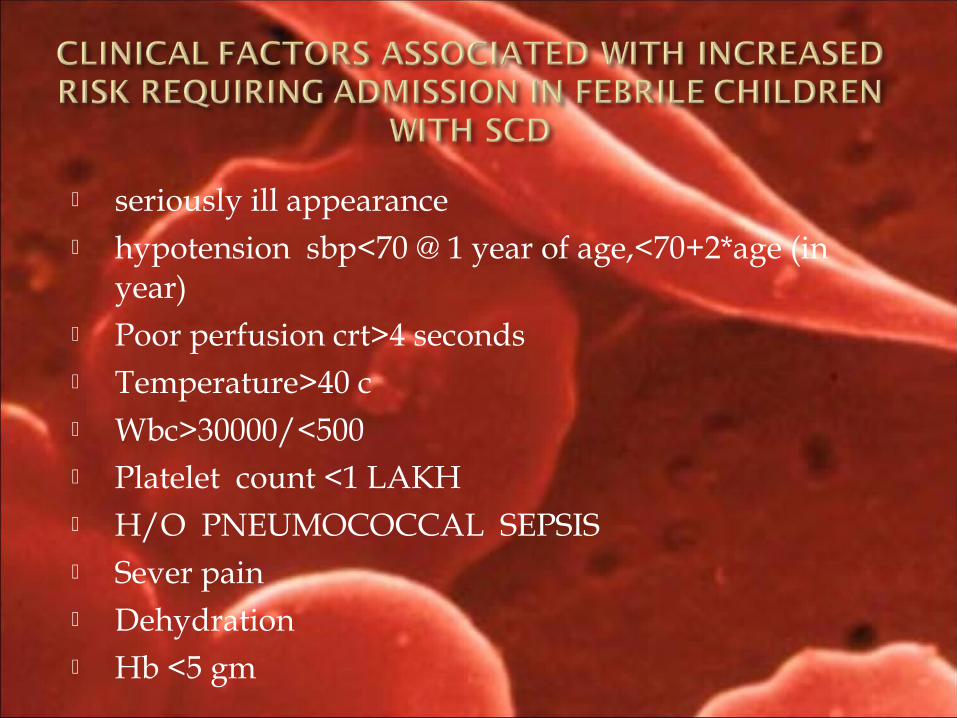
seriously ill appearance hypotension sbp<70 @ 1 year of age,<70+2*age (in
year) Poor perfusion crt>4 seconds Temperature>40 c Wbc>30000/<500 Platelet count <1 LAKH H/O PNEUMOCOCCAL SEPSIS Sever pain Dehydration Hb <5 gm

Acute splenic sequestration, pooling of blood in the engorged spleen
Aplastic crisis – Seen in patients with parovrus B-19 infection or folic acid deficiency leading to decreased marrow erythropoiesis..reticlopenia..dictum
Anemia Pigmented gallstone Jaundice Delayed growth

Infectious crisis is due to functional asplenia and deficiency of opsonin of alternate compliment pathway.. decreased level of serum immunoglobulin M (IGM) increasing susceptibility to infections.
Haemophilius influenzae, streptococcus pneumoniae, mycoplasma pneumoniae, salmonella typhimurium, staphylococcus aureus, and escherichia coli are the common causative microbes.
Common infections include pneumonia, bronchitis, pyelonephritis, cystitis, osteomyelitis, meningitis, and sepsis

Life threatening compli.. Spleen becomes enlarged and engorged with
decline in hb,hypovolemia Etiology not known Parents to be taught to palpate spleen and seek
intervention at the earliest.
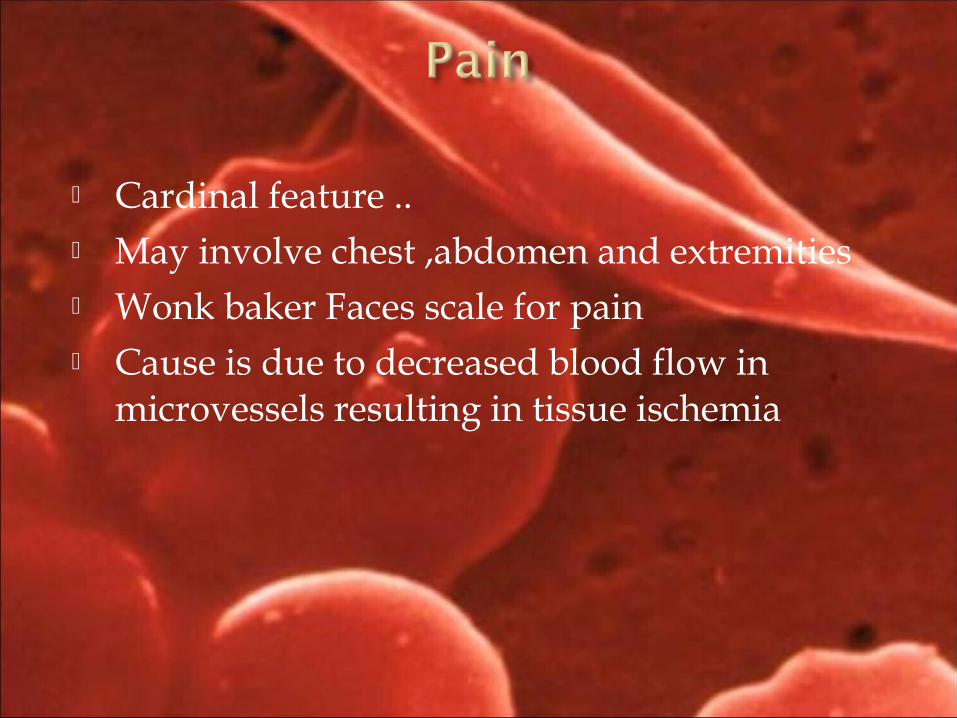
Cardinal feature .. May involve chest ,abdomen and extremities Wonk baker Faces scale for pain Cause is due to decreased blood flow in
microvessels resulting in tissue ischemia
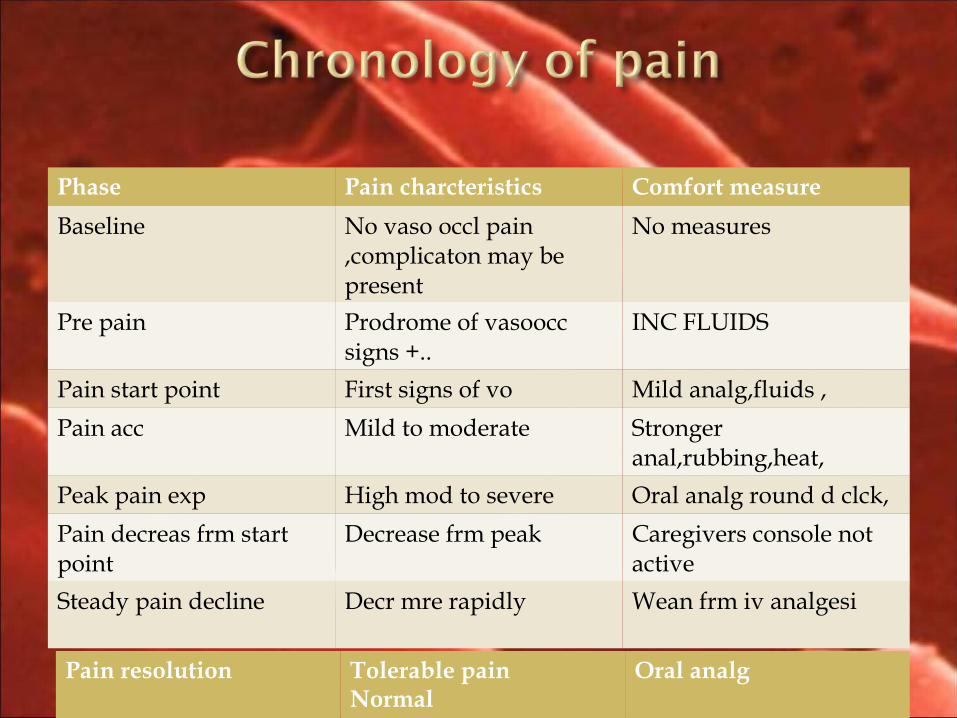
Phase Pain charcteristics Comfort measure
Baseline No vaso occl pain ,complicaton may be present
No measures
Pre pain Prodrome of vasoocc signs +..
INC FLUIDS
Pain start point First signs of vo Mild analg,fluids ,
Pain acc Mild to moderate Stronger anal,rubbing,heat,
Peak pain exp High mod to severe Oral analg round d clck,
Pain decreas frm start point
Decrease frm peak Caregivers console not active
Steady pain decline Decr mre rapidly Wean frm iv analgesi
Pain resolution Tolerable pain Normal
Oral analg

Involuntary penile erection lasting for more than 30 min ..for an hour suggests priapism
Ventral portion of glans of the penis is not involved Types Stuttering and refractory types Former self limiting,intermittent bouts Latter continuous for hour Rx is sitz bath for acute mgt Lasting >4 hrs then aspiration of blood from corpora
cavernosa followed by irrigation with epinephrine Etilefrine is safe drug for priapism prevention.

CVS-Anemia and vasooclusive phenomena causing myocardial ischemia and myocardial infarction, repeated blood transfusion leading to restrictive cardiomyopathy.
Pulmonary-Acute chest syndrome CNS-25% patient have TIA, strokes, cerebral hemorrhage,rpls reversible
post leuco encephalopathy syndrme. Excessive iron stores ..occurs in children receiving regular
transfusion..mri r2* liver , r2 heart . deferasirox Lung disease 2 nd most common Acs…radiodensity acute+fever+respi distress+pain in chest or back
abdomen Hepato biliary system-Gall stone recurrent abdominal pain,
autosplenectomy Urinary system- Haematuria, hyposthenurea and renal failure Ocular complication- Proliferative retinopathy, vitreous hemorrhage and
retinal detachment Orthopedic – Hand foot syndrome, avascular necrosis of hip,
osteomyelitis



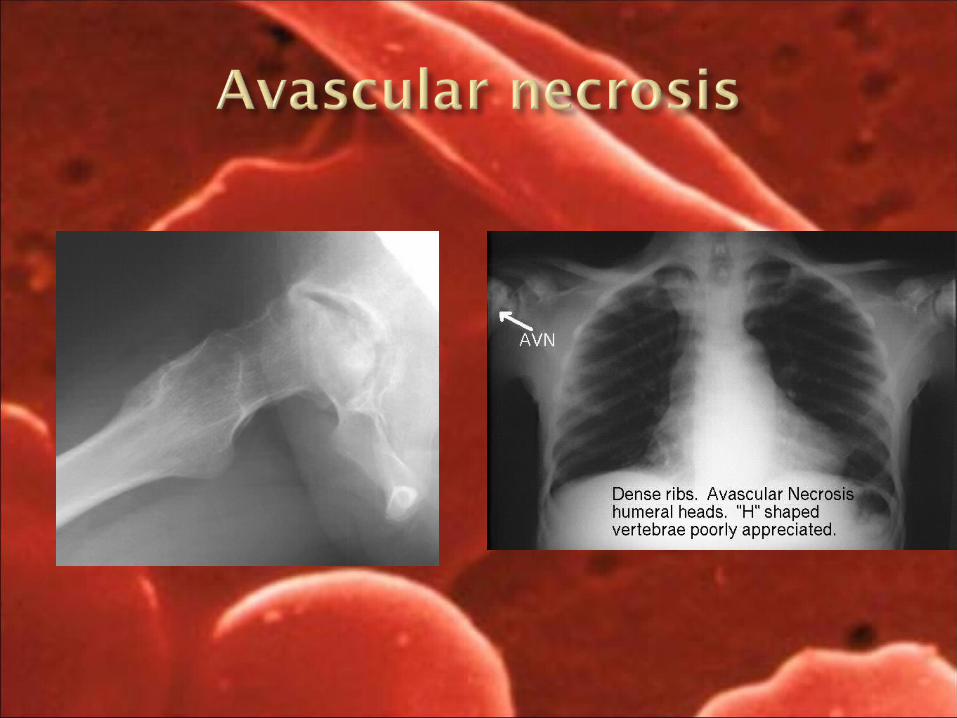

1. Young infants have recurrent edema of the dorsum of hands and feet.
2. Infarction of cortex of long bones lead to prominent signs of local inflammation.
3. Repeated infarction in the joints of large and small bones lead to abnormal angled digits, malformed and frozen joints, particularly at the knee and ankle.
4. Chronic leg ulcer is common in adolescent patients.5. Abdominal examination may reveal splenomegaly if
sequestration is occurring otherwise the spleen is small in size due to autoinfarction
6. Evidence of cholilethiasis is seen in patients as young as 3 years old.
7. By mid childhood most patients are underweight as compared to children of their same age and height.
1. There is a higher rate of spontaneous abortion. A miscarriage may happen up to 25% of the time.
2. There is ahigher rate of babies not surviving to birth or being stillborn. 8-10% .
3. Birth weight is lower than average.4. Infection is more common in women with sickle cell
disease during pregnancy, especially bladder infection.
5. Increased chance of PIH and preeclampsia6. Increased incidence of PPH.

Hb-6-8gm% Reticulocytes high Peripheral smear may show sickle cells Features of hyposplenism :Target cells and
Howell- Jolley bodies seen Sickling test with reducing agent Sodium
metabisulphide-na dithionite Hb electrophoresis High performance liquid chromatography(HPCL) WBC may be elevated Bilirubin may be elevated Urinary cast may be seen or trace of RBC in urine

When subjected to electrophoresis in alkaline medium sickle cell hb move towards anode slow than does adult hb.
Slow mobility of hbs is due to absence of –ve charge caused by loss of glutamate.
Incase sickle cell trait fast moving hba and slow moving hba are seen.

1. Oxygenation2. Pain Management (NSAID, OPIODS)3. Hydroxyurea15-20mg/kg daily upto 35
mg/kg/dose, Folic acid4. Hydration5. Blood Transfusion6. Antibiotics (Penicillin group)7. Steroids8. Bone marrow transplantation9. Gene therapy
Blood transfusion is currently the most effective and proven treatment for severe anemia of SCD, it significantly reduces crisis.
Blood transfusion reduces pain by increasing the number of functioning RBC and by increasing the oxygen caring capacity of blood
Bone marrow transplant is the closest thing possible to the cure of SCA.
Helps in production of healthy RBC from transplanted bone marrow
The success rate is 90 – 95%

Gene therapy is a relatively new idea of inserting genes into the cells of an individual in order to treat hereditary disease such as SCA, in which a defective mutants alleles is replaced with a functional one.
Gene therapy would be the best cure for SCA in future, as of now it is on it’s experimental stage.
Dialysis or kidney transplant for renal failure. Cholecystectomy for pigmented cholelitheasis. Hip replacement for avascular necrosis. Surgery for eye problem. Irrigation surgery for Priapism. Wound care for leg ulcer.

1. Genetic counseling2. Pre implantation genetic diagnosis3. High perfomance liquid chromatography to
detect hb phenotype. In new born ..4. Perenatal testing -amniocentesis (16 – 18 wks)
and chorius villus sampling ( 9 -10 wks)

1. Regular health check up & good hydration.2. Vaccination for pneumonia, meningitis, influenza,
and hepatitis3. Preventing infection – daily dose of penicillin4. Preventing strokes – Transcranial doppler
ultrasound every 3 months.5. Preventing eye damage – Fundus examination
every 3 months.6. Avoid alcohol, smoking, cold climate, high altitude exposure.THERE ARE 4 MILLION SICKLE CELL DISEASED
PATIENTS WORLDWIDE

Malaria caused by plasmodium falciparum . The parasite spends part of its cycle in
erythrocytes.so lysis interupts cycle Mp increases acidity of erythrocytes which in
turn cause increased sickling to 40 % frm nrml 2%.therefore entry of mp cause incresased sickling and lysis
Concn of pottasium is low in sickle cells unfavourable for mp to survey


Hb Gower 1-Confined to embryonic stage, consists of 2 alpha & 2 epsilon chains.
Hb Gower 2-Consists of 2 alpha & 2 zeta chains.
Hb Portland- Found in trace in neonates and intrauterine life.
Hb –Brat’s –Found in small amount in cord blood of neonates with thalasseamia.

New concepts in the field of priapism research suggest that priapism often results from altered vascular homeostatic actions in the penis and is associated with deficient erection control mechanisms on a molecular level.
A leading proposal in this regard is the notion of aberrant signaling of the endothelium-derived nitric oxide and PDE5 signal transduction pathway in the penis.

Additionally, dysfunctional regulatory control of signal transduction systems which interact with this pathway such as adenosine and RhoA/Rho-kinase may contribute to the development of priapism.Recent investigations of opiorphins also demonstrate a role in regulating corporal smooth muscle tone and thereby dysregulation of erection physiology in priapism. These advances have paved the way for understanding this disorder as having a molecular pathogenesis.

The Human Genome Project has provided valuable insight and extensive research advances in the understanding of the human genome and sickle cell disease.

A 3.5-year-old girl from race of Arab reffered to Shafa Hospital with severe anemia, thrombocytopenia, leucocytosis and elevated ESR and LDH. Her parents assigned fever, cough, pallor, weakness and tachypnea from six day ago (Figure 1).
Fig. 1 Chest x ray. Past medical history and familial history of the patient was negative.
On physical examination, she had fever with temperature of 39.5°C, severe pallor, pulse rate of 110 per minute, respiratory rate of 32 per minute and blood pressure of 90/60 mmHg. On abdominal examination, she had hepatosplenomegaly. Rales and rhonchi were in both lung fields on respiratory examination while the other systemic examination was essentially normal. Initial laboratory investigations demonstrated hemoglobin of 4.5 gm/dL, white cell count of 19,000/mm(3) (55% neutrophils, 45% lymphocyte), platelet count of 70,000/mm(3) and an erythrocyte sedimentation rate of 45 mm/hour. Renal function tests and urinalysis were normal. A chest radiograph revealed bilateral haziness. For decline of malignancy, bone marrow aspiration was done and discussed reactive bone marrow due to infection.
ootnotes

After one day a dactylitis was present in hands of the patient (Figure 2).
Fig. 2 Dactylitis. In follow up of Hb electrophoresis, Hb s was 80%, Hb F: 18%, and
Hb A2: 2%. Finally in peripheral blood smear, sickling of RBC was detected and the patient was diagnosed with sickle cell anemia and acute splenic sequestration crisis which was associated with acute chest syndrome treated with wide spectrum antibiotic (cefotaxim and erythromycin) and transfusion exchange (Figure 3). The patient was discharged with stable clinical state after 8 days.
F
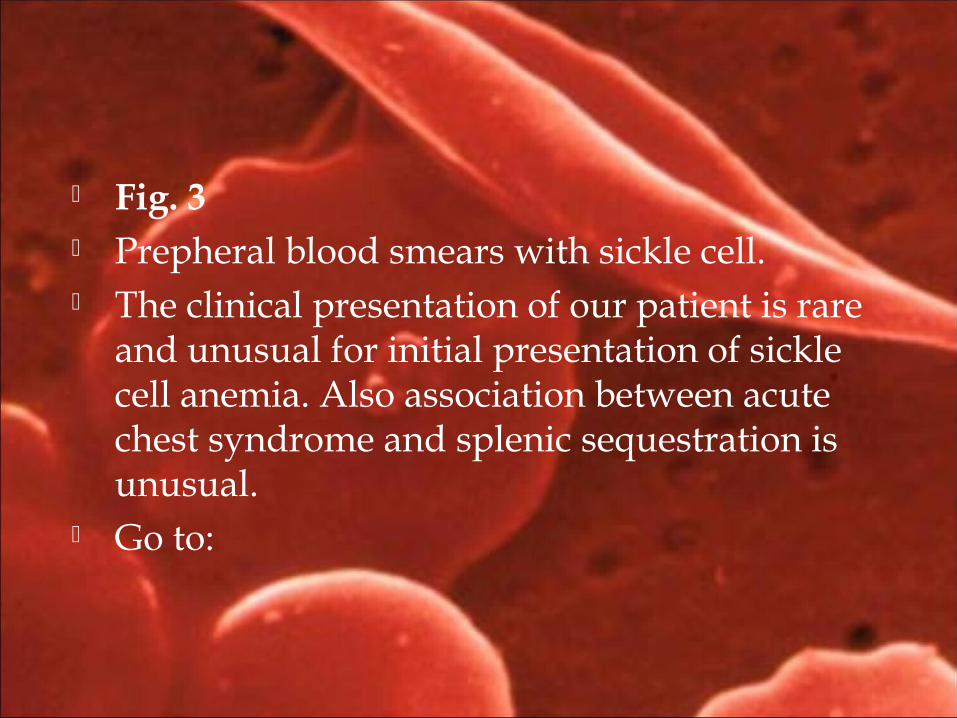
Fig. 3 Prepheral blood smears with sickle cell. The clinical presentation of our patient is rare
and unusual for initial presentation of sickle cell anemia. Also association between acute chest syndrome and splenic sequestration is unusual.
Go to:

Nelson 19th edition Biochemistry U satyanarayanan William’s hematology www.pubmed.com.
THANKs…