shianna weilcornell lecture march26 2013 - cornell...
TRANSCRIPT

Clinical Sequencing
Kevin Shianna, PhD Senior Vice President, Sequencing Opera;ons New York Genome Center

Background
• Next Genera;on Sequencing – $1000 exome/$4000 genome
• Sequence a genome in a day (maybe hours?) • Can we iden;fy, annotate and interpret unique (rare) variants?

NGS to iden;fy muta;ons in:
• Mendelian disease genes • Undiagnosed gene;c condi;ons • Complex diseases • Non-‐invasive prenatal tes;ng (NIPT)
-‐Freeman-‐Sheldon syndrome -‐Autosomal dominant – MYH3
-‐June 2009
Nonsense mutations and splice-site disruptions are often assumedto be deleterious, but have a broad range of potential fitnesseffects25–27. Our non-redundant cSNP catalogue included 225 non-sense mutations (112 novel) and 102 splice-site disruptions (49novel). Excluding 86 nonsense alleles that are common in this dataset (two or more observations) or in a recent study25 (.5% allelefrequency), our genome-wide estimate (projected to 30 Mb) for theaverage number of relatively rare mutations introducing prematurenonsense codons in an individual genome was 10 for non-Africans(n 5 8) and 20 for Yoruba (n 5 4). However, these are probablyoverestimates, given that our catalogue of common nonsense muta-tions remains incomplete.
Short insertions and deletions (indels) in coding sequence arelikely to be functionally important when they cause frameshifts,but are difficult to detect with short reads. We developed and appliedan approach for identifying indels from our unpaired 76 bp reads. Intotal, 664 coding indels were called in one or more individuals. Onaverage, 166 coding indels were called per individual, of which 63%were previously annotated in dbSNP (Supplementary Table 3). Toassess our sensitivity, we compared our data for NA18507 to datapublished previously12. The majority (73%) of their coding indelswere also observed in our data (136 of 187). To assess specificity,we attempted PCR and Sanger sequencing of 28 novel coding indelschosen at random. Of 21 successful assays, 20 coding indels wereverified and 1 was a false positive. We anticipate that future use ofpaired-end reads will improve detection of coding indels.
The shape of the distribution of coding indel lengths was consist-ent with other studies10,20 as well as across the 12 exomes (Fig. 1d),demonstrating a preference for multiples of 3 (‘3n’). Of the 664coding indels observed here, 65% were 3n in length. The allele fre-quency distribution for novel indels relative to annotated indels wasmarkedly shifted towards rarer variants (Supplementary Fig. 4).However, the length histograms for novel versus annotated codingindels were similar (Supplementary Fig. 5), reinforcing the notionthat our set of novel coding indels is not excessively contaminatedwith false positives (as these would not be expected to have theobserved 3n bias). Excluding indels that were common in this data
set (two or more observations), the average number of relatively rareframeshifting indels identified per individual was 8 for non-Africans(n 5 8) and 17 for Yoruba (n 5 4).
The number of synonymous, missense, nonsense, splice site, fra-meshifting indel and non-frameshifting indel variants observed ineach individual (as well as the size of the subsets that are novel andsingleton observations) is presented in Supplementary Table 4. Alsoshown are the average numbers of variants of each class for non-Africans and Yoruba.
Phenotypes inherited in an apparently Mendelian pattern oftenlack sufficiently sized pedigrees to pinpoint the causal locus. Weevaluated whether exome sequencing could be applied to identifydirectly the causative gene underlying a monogenic human disease(FSS), that is, with neither linkage data nor candidate gene analysis.Even in this simple scenario for ‘whole exome/genome genetics’, thekey challenge that arises immediately is that the large number ofapparently private mutations present by chance in any single humangenome makes it difficult to identify which variant is causal, evenwhen only considering non-synonymous variants. This hurdle wasovercome recently in the context of hereditary pancreatic cancer byrestricting focus only to nonsense mutations and also resequencingtumour DNA from the same individual, but this approach greatlylimits sensitivity and is only relevant to a subset of mechanismswithin one disease class28.
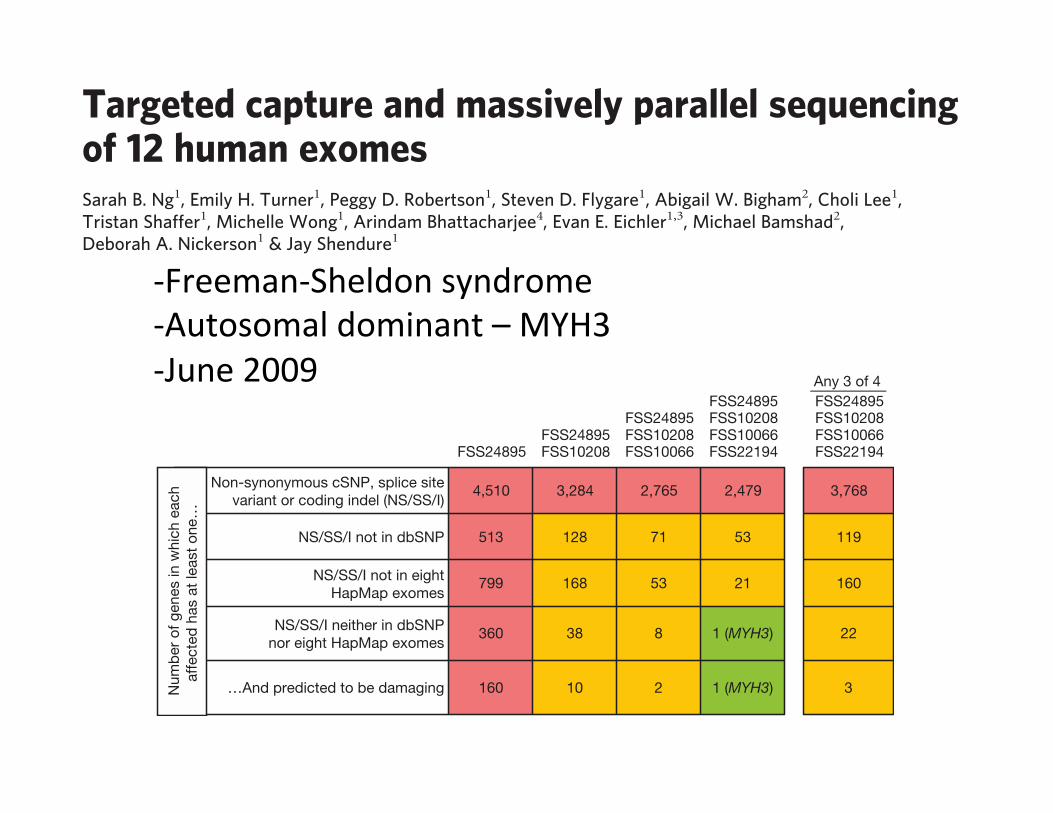
To quantify this background of non-causal variants in our exomedata, we first investigated how many genes had one or more non-synonymous cSNPs, splice site disruptions or coding indels in one orseveral FSS exomes (Fig. 2, row 1). Simply requiring that a genecontain variants in multiple affected individuals was clearly insuf-ficient, as over 2,000 candidate genes remained even after intersectingfour FSS exomes. We then applied filters to remove presumablycommon variants, as these are unlikely to be causative. RemovingdbSNP-catalogued variants from consideration reduced the numberof candidates considerably (Fig. 2, row 2). Remarkably, the eightHapMap exomes provided a filter nearly equivalent to dbSNP(Fig. 2, row 3). Combining the two catalogues had a synergistic effect(Fig. 2, row 4), such that the candidate list could be narrowed to asingle gene (MYH3, identified previously by a candidate geneapproach as causative for FSS5). Specifically, MYH3 is the onlygene where: (1) at least one (but not necessarily the same) non-synonymous cSNP, splice-site disruption or coding indel is observedin all four individuals with FSS; (2) the mutations are not in dbSNP,nor in the eight HapMap exomes. Taking the predicted deleterious-ness of individual mutations into account served as an effective filteras well (Fig. 2, row 5), but was not required to identify MYH3. Ranges
0
2,000
4,000
6,000
8,000
10,000
12,000
1 2 3 4 5 6 7 8 9 10 11 12
0
10
20
30
40
50
60
70
0102030405060708090
100
ba
dc
Num
ber o
f var
iant
s
0
2,000
4,000
6,000
8,000
10,000
12,000
Num
ber o
f var
iant
s
Number of observations of minor allele
1 1 2 3 4 5 6 7 8 9 1011121314152 3 4 5 6 7 8 9 10 11 12Number of observations of minor allele
1 2 3 4 5 6 7 8 9 10 11 12Number of observations of minor allele
Frac
tion
of e
ach
varia
nt c
lass
(%)
SynonymousBenign non-synonymousPossibly damaging non-synonymousProbably damaging non-synonymous
AnnotatedNovel
SynonymousNon-synonymous
Ave
rage
num
ber o
f ind
els
Length of coding indels (bases)
3n indelsNon-3n indels
Figure 1 | Minor allele frequency and coding indel length distributions.a, The distribution of minor allele frequencies is shown for previouslyannotated versus novel cSNPs. b, The distribution of minor allelefrequencies is shown for synonymous versus non-synonymous cSNPs. c, Thedistribution of minor allele frequencies (by proportion, rather than count) isshown for synonymous cSNPs (n 5 21,201) versus non-synonymous cSNPspredicted to be benign (n 5 13,295), possibly damaging (n 5 3,368), orprobably damaging (n 5 2,227) by PolyPhen24. d, The distribution of lengthsof coding indel variants is shown (average numbers per exome). Error barsindicate s.d.
FSS24895FSS24895 FSS10208
FSS24895 FSS10208 FSS10066
FSS24895 FSS10208 FSS10066 FSS22194
Any 3 of 4FSS24895 FSS10208 FSS10066 FSS22194
Non-synonymous cSNP, splice sitevariant or coding indel (NS/SS/I)
4,510 3,284 2,765 2,479 3,768
NS/SS/I not in dbSNP 513 128 71 53 119
NS/SS/I not in eightHapMap exomes
799 168 53 21 160
NS/SS/I neither in dbSNPnor eight HapMap exomes
360 38 8 1 (MYH3) 22
…And predicted to be damaging 160 10 2 1 (MYH3) 3Num
ber o
f gen
es in
whi
ch e
ach
affe
cted
has
at l
east
one
…
Figure 2 | Direct identification of the causal gene for a monogenic disorderby exome sequencing. Boxes list the number of genes with one or more non-synonymous cSNP, splice-site SNP, or coding indel (NS/SS/I) meetingspecified filters. Columns show the effect of requiring that one or more NS/SS/I variants be observed in each of one to four affected individuals. Rowsshow the effect of excluding from consideration variants found in dbSNP,the eight HapMap exomes, or both. Column five models limited geneticheterogeneity or data incompleteness by relaxing criteria such that variantsneed only be observed in any three of four exomes for a gene to qualify.
LETTERS NATURE | Vol 461 | 10 September 2009
274 Macmillan Publishers Limited. All rights reserved©2009
LETTERS
Targeted capture and massively parallel sequencingof 12 human exomesSarah B. Ng1, Emily H. Turner1, Peggy D. Robertson1, Steven D. Flygare1, Abigail W. Bigham2, Choli Lee1,Tristan Shaffer1, Michelle Wong1, Arindam Bhattacharjee4, Evan E. Eichler1,3, Michael Bamshad2,Deborah A. Nickerson1 & Jay Shendure1
Genome-wide association studies suggest that common geneticvariants explain only a modest fraction of heritable risk for com-mon diseases, raising the question of whether rare variants accountfor a significant fraction of unexplained heritability1,2. AlthoughDNA sequencing costs have fallen markedly3, they remain far fromwhat is necessary for rare and novel variants to be routinely iden-tified at a genome-wide scale in large cohorts. We have thereforesought to develop second-generation methods for targeted sequen-cing of all protein-coding regions (‘exomes’), to reduce costs whileenriching for discovery of highly penetrant variants. Here we reporton the targeted capture and massively parallel sequencing of theexomes of 12 humans. These include eight HapMap individualsrepresenting three populations4, and four unrelated individualswith a rare dominantly inherited disorder, Freeman–Sheldon syn-drome (FSS)5. We demonstrate the sensitive and specific identifica-tion of rare and common variants in over 300 megabases of codingsequence. Using FSS as a proof-of-concept, we show that candidategenes for Mendelian disorders can be identified by exome sequen-cing of a small number of unrelated, affected individuals. Thisstrategy may be extendable to diseases with more complex geneticsthrough larger sample sizes and appropriate weighting of non-synonymous variants by predicted functional impact.
Protein-coding regions constitute ,1% of the human genome or,30 megabases (Mb), split across ,180,000 exons. A brute-forceapproach to exome sequencing with conventional technology6 isexpensive relative to what may be possible with second-generationplatforms3. However, the efficient isolation of this fragmentary geno-mic subset is technically challenging7. The enrichment of an exomeby hybridization of shotgun libraries constructed from 140 mg ofgenomic DNA to seven microarrays was described previously8. Toimprove the practicality of hybridization capture, we developed aprotocol to enrich for coding sequences at a genome-wide scale start-ing with 10 mg of DNA and using two microarrays. Our initial targetwas 27.9 Mb of coding sequence defined by CCDS (the NCBIConsensus Coding Sequence database)9. This curated set avoids theinclusion of spurious hypothetical genes that contaminate broaderexome definitions10. The target is reduced to 26.6 Mb on exclusion ofregions that are poorly mapped with our anticipated read lengthowing to paralogous sequences elsewhere in the genome(Supplementary Data 1).
We captured and sequenced the exomes of eight individuals previ-ously characterized by the HapMap4 and Human Genome StructuralVariation11 projects. We also analysed four unrelated individualsaffected with Freeman–Sheldon syndrome (FSS; Online MendelianInheritance in Man (OMIM) #193700), also called distal arthro-gryposis type 2A, a rare autosomal dominant disorder caused by
mutations in MYH3 (ref. 5). Unpaired, 76 base-pair (bp) reads12
from post-enrichment shotgun libraries were aligned to the referencegenome13. On average, 6.4 gigabases (Gb) of mappable sequence wasgenerated per individual (20-fold less than whole genome sequencingwith the same platform12), and 49% of reads mapped to targets(Supplementary Table 1). After removing duplicate reads thatrepresent potential polymerase chain reaction artefacts14, the averagefold-coverage of each exome was 513 (Supplementary Fig. 1). Onaverage per exome, 99.7% of targeted bases were covered at leastonce, and 96.3% (25.6 Mb) were covered sufficiently for variant call-ing ($83 coverage and Phred-like15 consensus quality $30). Thiscorresponded to 78% of genes having .95% of their coding basescalled (Supplementary Fig. 2 and Supplementary Data 2). The aver-age pairwise correlation coefficient between individuals for gene-by-gene coverage was 0.87, consistent with systematic bias in coveragebetween individual exomes.
False positives and false negatives are critical issues in genomicresequencing. We assessed the quality of our exome data in four ways.First, comparing sequence-based calls for the eight HapMap exomesto array-based genotyping, we observed a high concordance withboth homozygous (99.94%; n 5 219,077) and heterozygous(99.57%; n 5 43,070) genotypes (Table 1). Second, we comparedour coding single-nucleotide polymorphism (cSNP) catalogue to,1 Mb of coding sequence determined in each of the eightHapMap individuals by molecular inversion probe (MIP) captureand direct resequencing16. At coordinates called in both data sets,99.9% of all cSNPs (n 5 4,620) and 100% of novel cSNPs(n 5 334) identified here were concordant, consistent with a low falsediscovery rate. Third, we compared the NA18507 cSNPs identifiedhere to those called by recent whole genome sequencing of thisindividual12, and found substantial overlap (Supplementary Fig. 3).The relative numbers of cSNPs called by only one approach, and theproportions of these represented in dbSNP, indicate that exomesequencing has equivalent sensitivity for cSNP detection comparedto whole genome sequencing. Fourth, we compared our data tocSNPs in high-quality Sanger sequence of single haplotype regionsfrom fosmid clones of the same HapMap individuals17. Most fosmid-defined cSNPs (38 of 40) were at coordinates with sufficient coveragein our data for variant calling. Of these, 38 of 38 were correctlyidentified as variant.
A comparison of our data to past reports on exonic18 or exomic8
array-based capture revealed roughly equivalent capture specificity,but greater completeness in terms of coverage and variant calling(Supplementary Table 2). These improvements probably arise froma combination of greater sequencing depth and differences in arraydesigns and in experimental conditions for capture. Within the set of
1Department of Genome Sciences, 2Department of Pediatrics, University of Washington, 3Howard Hughes Medical Institute, Seattle, Washington 98195, USA. 4Agilent Technologies,Santa Clara, California 95051, USA.
Vol 461 | 10 September 2009 | doi:10.1038/nature08250
272 Macmillan Publishers Limited. All rights reserved©2009

-‐Kabuki Syndrome -‐10 probands -‐Autosomal Dominant? -‐1/32,000 -‐April 2010
790 VOLUME 42 | NUMBER 9 | SEPTEMBER 2010 NATURE GENETICS
We demonstrate the successful application of exome sequencing1–3 to discover a gene for an autosomal dominant disorder, Kabuki syndrome (OMIM%147920). We subjected the exomes of ten unrelated probands to massively parallel sequencing. After filtering against existing SNP databases, there was no compelling candidate gene containing previously unknown variants in all affected individuals. Less stringent filtering criteria allowed for the presence of modest genetic heterogeneity or missing data but also identified multiple candidate genes. However, genotypic and phenotypic stratification highlighted MLL2, which encodes a Trithorax-group histone methyltransferase4: seven probands had newly identified nonsense or frameshift mutations in this gene. Follow-up Sanger sequencing detected MLL2 mutations in two of the three remaining individuals with Kabuki syndrome (cases) and in 26 of 43 additional cases. In families where parental DNA was available, the mutation was confirmed to be de novo (n = 12) or transmitted (n = 2) in concordance with phenotype. Our results strongly suggest that mutations in MLL2 are a major cause of Kabuki syndrome.
Kabuki syndrome is a rare, multiple malformation disorder characterized by a distinctive facial appearance (Supplementary Fig. 1), cardiac anomalies, skeletal abnormalities, immunological defects and mild to moderate mental retardation. Originally described in 1981 (refs. 5,6), Kabuki syndrome has an estimated incidence of 1 in 32,000 (ref. 7), and approximately 400 cases have been reported worldwide. The vast majority of reported cases have been sporadic, but parent-to-child transmission in more than a half dozen instances8 suggests that Kabuki syndrome is an autosomal dominant disorder. The relatively low number of cases, the lack of multiplex families and the pheno-typic variability of Kabuki syndrome have made the identification of the gene(s) underlying this disorder intractable to conventional approaches of gene discovery, despite aggressive efforts.
We sequenced the exomes of ten unrelated individuals with Kabuki syndrome: seven of European ancestry, two of Hispanic ancestry and one of mixed European and Haitian ancestry (Supplementary Fig. 1 and Supplementary Table 1). Enrichment was performed by hybridi-zation of shotgun fragment libraries to custom microarrays followed by massively parallel sequencing1–3. On average, 6.3 gigabases of sequence were generated per sample to achieve 40! coverage of the mappable, targeted exome (31 Mb). As with our previous studies, we focused our analyses here primarily on nonsynonymous variants, splice acceptor and donor site mutations and coding indels, anticipating that synonymous variants were far less likely to be pathogenic. We also predicted that variants underlying Kabuki syndrome are rare, and therefore likely to be previously unidentified. We defined variants as previously unidentified if they were absent from all datasets used for comparison, including dbSNP129, the 1000 Genomes Project, exome data from 16 individuals previously reported by us2,3 and 10 exomes sequenced as part of the Environmental Genome Project (EGP).
Under a dominant model in which each case was required to have at least one previously unidentified nonsynonymous vari-ant, splice acceptor and donor site mutation or coding indel vari-ant in the same gene, only a single candidate gene (MUC16) was shared across all ten exomes (Table 1 and Supplementary Table 2). However, we considered MUC16 as a likely false positive due to its extremely large size (14,507 amino acids). Potential explanations for our failure to find a compelling candidate gene in which newly identified variants were seen in all affected individuals included: (i) Kabuki syndrome is genetically heterogeneous and therefore not all affected individuals will have mutations in the same gene; (ii) we failed to identify all mutations in the targeted exome; and (iii) some or all causative mutations were outside of the targeted exome, for example, in noncoding regions or unannotated genes. To allow for a modest degree of genetic heterogeneity and/or missing data, we conducted a less stringent analysis by looking for candidate genes shared among subsets of affected individuals. Specifically, we searched
Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndromeSarah B Ng1,7, Abigail W Bigham2,7, Kati J Buckingham2, Mark C Hannibal2,3, Margaret J McMillin2, Heidi I Gildersleeve2, Anita E Beck2,3, Holly K Tabor2,3, Gregory M Cooper1, Heather C Mefford2, Choli Lee1, Emily H Turner1, Joshua D Smith1, Mark J Rieder1, Koh-ichiro Yoshiura4, Naomichi Matsumoto5, Tohru Ohta6, Norio Niikawa6, Deborah A Nickerson1, Michael J Bamshad1–3 & Jay Shendure1
1Department of Genome Sciences, University of Washington, Seattle, Washington, USA. 2Department of Pediatrics, University of Washington, Seattle, Washington, USA. 3Seattle Children’s Hospital, Seattle, Washington, USA. 4Department of Human Genetics, Nagasaki University Graduate School of Biomedical Sciences, Nagasaki, Japan. 5Department of Human Genetics, Yokohama City University Graduate School of Medicine, Yokohama, Japan. 6Research Institute of Personalized Health Sciences, Health Sciences University of Hokkaido, Hokkaido, Japan. 7These authors contributed equally to this work. Correspondence should be addressed to J.S. ([email protected]) or M.J.B. ([email protected]).
Received 28 April; accepted 21 July; published online 15 August 2010; addendum published after print 7 January 2011; doi:10.1038/ng.646
L E T T E R S

NATURE GENETICS VOLUME 42 | NUMBER 9 | SEPTEMBER 2010 791
for subsets of x out of 10 exomes having 1 previously unidentified variant in the same gene, with x = 1 to x = 10. For x = 9, x = 8 and x = 7, previously unidentified variants were shared in 3 genes, 6 genes and 16 genes, respectively (Table 1). However, there was no obvious way to rank these candidate genes.
We speculated that genotypic and/or phenotypic stratification would facilitate the prioritization of candidate genes identified by subset analysis. Specifically, we assigned a categorical rank to each individual with Kabuki syndrome based on a subjective assessment of the presence of, or similarity to, the canonical facial characteris-tics of Kabuki syndrome (Supplementary Fig. 1) and the presence of developmental delay and/or major birth defects (Supplementary Table 1). The highest-ranked individual was one of a pair of mono-zygotic twins with Kabuki syndrome. We then categorized the func-tional impact (that is, nonsense versus nonsynonymous substitution, splice-site disruption and frameshift compared to in-frame indel) of each newly identified variant in candidate genes shared by each subset of two or more ranked cases. Manual review of these data high-lighted distinct, previously unidentified nonsense variants in MLL2 in each of the four highest-ranked cases. After sequential analysis of phenotype-ranked cases with a loss-of-function filter, MLL2 was the only candidate gene remaining after addition of the second individual (Table 2). We found no such variant in MLL2 in the individual with Kabuki syndrome ranked fifth; hence, the number of candidate genes dropped to zero after the individual ranked fourth in the set (Table 2). However, we found a 4-bp deletion in the individual ranked sixth, and we found nonsense variants in the individuals ranked seventh and ninth. Thus, exome sequencing identified a nonsense substitution or frameshift indel in MLL2 in seven of the ten individuals with Kabuki syndrome analyzed here.
Retrospectively, we applied a loss-of-function filter to the subset analysis of exome data (Table 1), and at x = 7, found MLL2 to be the only candidate gene. We also developed a post hoc ranking of candidate genes based on the functional impact of the variants present (variant score) and the rank of the cases in which each variant was observed (case score). When this was applied to the exome data as a combined metric, MLL2 emerged as the top candidate gene (Supplementary Fig. 2).
In parallel with these analyses, we applied genomic evolutionary rate profiling (GERP)9 to the exome data. GERP uses mammalian genome alignments to define a rejected sub-stitution score for each variant regardless of functional class. We have previously shown that
the quantitative ranking of candidate genes by the rejected substitution scores of their vari-ants can facilitate the exome-based analysis of Mendelian disorders10. Following subset analy-sis with GERP-based ranking, MLL2 remained on the candidate list up to x = 8, ranking third in a list of 11 candidate genes at this threshold (Table 3 and Supplementary Fig. 3). Notably, the additional MLL2 variant contributing to this analysis (such that MLL2 was still consid-ered at x = 8) was a synonymous substitution with a rejected substitution score of 0.368 in the individual ranked fifth.
We sought to confirm all newly identified variants in MLL2, particularly because loss-
of-function variants identified through massively parallel sequencing have a high prior probability of being false positives. All seven loss-of-function variants in MLL2 were validated by Sanger sequencing. We further analyzed the three cases in which we did not initially find a loss-of-function variant in MLL2, first by array comparative genomic hybridization (aCGH) to determine any gross structural changes and then by Sanger sequencing of all exons of MLL2 in case of false nega-tives by exome sequencing. Because an average of 96% of the coding bases in MLL2 were called at sufficient quality and coverage for single nucleotide variant detection, we anticipated that any missed variants were more likely to be indels because of the higher coverage required for confident indel detection in short-read sequence data. Indeed, although aCGH did not find any structural variants in the region, Sanger sequencing did identify frameshift indels in two of these three cases (specifically, the cases ranked eighth and tenth).
Ultimately, loss-of-function mutations in MLL2 were identified in nine out of ten cases in the discovery cohort (Fig. 1), making this gene a compelling candidate for Kabuki syndrome. For validation, we screened all 54 exons of MLL2 in 43 additional cases by Sanger sequenc-ing. Previously unidentified nonsynonymous, nonsense or frameshift mutations in MLL2 were found in 26 of these 43 cases (Fig. 1 and Supplementary Table 3). In total, through either exome sequencing or targeted sequencing of MLL2, 33 distinct MLL2 mutations were iden-tified in 35 of 53 families (66%) with Kabuki syndrome (Fig. 1 and Supplementary Table 3). In each of 12 cases for which DNA from both parents was available, the MLL2 variant was found to have occurred de novo. Three mutations were found in two individuals each. One of these three mutations was confirmed to have arisen de novo in one of the cases, indicating that some mutations in individuals with Kabuki syndrome are recurrent. In addition, MLL2 mutations (resulting in p.4527K>X and p.5464T>M) were also identified in each of two fami-lies in which Kabuki syndrome was transmitted from parent to child.
Table 1 Number of genes common to any subset of x affected individuals.Subset analysis (any x of 10) 1 2 3 4 5 6 7 8 9 10
NS/SS/I 12,042 8,722 7,084 6,049 5,289 4,581 3,940 3,244 2,486 1,459Not in dbSNP129 or 1000 Genomes
7,419 2,697 1,057 488 288 192 128 88 60 34
Not in control exomes 7,827 2,865 1,025 399 184 90 50 22 7 2Not in either 6,935 2,227 701 242 104 44 16 6 3 1Is loss-of-function (non-sense or frameshift indel)
753 49 7 3 2 2 1 0 0 0
The number of genes with at least one nonsynonymous variant (NS), splice-site acceptor or donor variants (SS) or coding indel (I) are listed under various filters. Variants were filtered by presence in dbSNP or 1000 Genomes (not in dbSNP129 or 1000 genomes) and control exomes (not in control exomes) or both (not in either); control exomes refer to those from 8 Hapmap3, 4 FSS3, 4 Miller2 and 10 EGP samples. The number of genes found using the union of the intersection of x individuals is given.
Table 2 Number of genes common in sequential analysis of phenotypically ranked individualsSequential analysis 1 +2 +3 +4 +5 +6 +7 +8 +9 +10
NS/SS/I 5,282 3,850 3,250 2,354 2,028 1,899 1,772 1,686 1,600 1,459Not in dbSNP129 or 1000 Genomes
687 214 145 84 63 54 42 40 39 34
Not in control exomes 675 134 50 26 13 13 8 5 4 2Not in either 467 89 34 18 9 8 4 4 3 1Is loss-of-function (non-sense/frameshift indel)
25 1 1 1 0 0 0 0 0 0
Variants were filtered as in Table 1. Exomes were added sequentially to the analysis by ranked phenotype; for example, column “+3” shows the number of genes at the intersection of the three top ranked cases (Supplementary Fig. 1). The gene with at least one NS/SS/I in all individuals is MUC16, which is very likely to be a false positive due to its extreme length (14,507 amino acids).
L E T T E R S
NATURE GENETICS VOLUME 42 | NUMBER 9 | SEPTEMBER 2010 791
for subsets of x out of 10 exomes having 1 previously unidentified variant in the same gene, with x = 1 to x = 10. For x = 9, x = 8 and x = 7, previously unidentified variants were shared in 3 genes, 6 genes and 16 genes, respectively (Table 1). However, there was no obvious way to rank these candidate genes.
We speculated that genotypic and/or phenotypic stratification would facilitate the prioritization of candidate genes identified by subset analysis. Specifically, we assigned a categorical rank to each individual with Kabuki syndrome based on a subjective assessment of the presence of, or similarity to, the canonical facial characteris-tics of Kabuki syndrome (Supplementary Fig. 1) and the presence of developmental delay and/or major birth defects (Supplementary Table 1). The highest-ranked individual was one of a pair of mono-zygotic twins with Kabuki syndrome. We then categorized the func-tional impact (that is, nonsense versus nonsynonymous substitution, splice-site disruption and frameshift compared to in-frame indel) of each newly identified variant in candidate genes shared by each subset of two or more ranked cases. Manual review of these data high-lighted distinct, previously unidentified nonsense variants in MLL2 in each of the four highest-ranked cases. After sequential analysis of phenotype-ranked cases with a loss-of-function filter, MLL2 was the only candidate gene remaining after addition of the second individual (Table 2). We found no such variant in MLL2 in the individual with Kabuki syndrome ranked fifth; hence, the number of candidate genes dropped to zero after the individual ranked fourth in the set (Table 2). However, we found a 4-bp deletion in the individual ranked sixth, and we found nonsense variants in the individuals ranked seventh and ninth. Thus, exome sequencing identified a nonsense substitution or frameshift indel in MLL2 in seven of the ten individuals with Kabuki syndrome analyzed here.
Retrospectively, we applied a loss-of-function filter to the subset analysis of exome data (Table 1), and at x = 7, found MLL2 to be the only candidate gene. We also developed a post hoc ranking of candidate genes based on the functional impact of the variants present (variant score) and the rank of the cases in which each variant was observed (case score). When this was applied to the exome data as a combined metric, MLL2 emerged as the top candidate gene (Supplementary Fig. 2).
In parallel with these analyses, we applied genomic evolutionary rate profiling (GERP)9 to the exome data. GERP uses mammalian genome alignments to define a rejected sub-stitution score for each variant regardless of functional class. We have previously shown that
the quantitative ranking of candidate genes by the rejected substitution scores of their vari-ants can facilitate the exome-based analysis of Mendelian disorders10. Following subset analy-sis with GERP-based ranking, MLL2 remained on the candidate list up to x = 8, ranking third in a list of 11 candidate genes at this threshold (Table 3 and Supplementary Fig. 3). Notably, the additional MLL2 variant contributing to this analysis (such that MLL2 was still consid-ered at x = 8) was a synonymous substitution with a rejected substitution score of 0.368 in the individual ranked fifth.
We sought to confirm all newly identified variants in MLL2, particularly because loss-
of-function variants identified through massively parallel sequencing have a high prior probability of being false positives. All seven loss-of-function variants in MLL2 were validated by Sanger sequencing. We further analyzed the three cases in which we did not initially find a loss-of-function variant in MLL2, first by array comparative genomic hybridization (aCGH) to determine any gross structural changes and then by Sanger sequencing of all exons of MLL2 in case of false nega-tives by exome sequencing. Because an average of 96% of the coding bases in MLL2 were called at sufficient quality and coverage for single nucleotide variant detection, we anticipated that any missed variants were more likely to be indels because of the higher coverage required for confident indel detection in short-read sequence data. Indeed, although aCGH did not find any structural variants in the region, Sanger sequencing did identify frameshift indels in two of these three cases (specifically, the cases ranked eighth and tenth).
Ultimately, loss-of-function mutations in MLL2 were identified in nine out of ten cases in the discovery cohort (Fig. 1), making this gene a compelling candidate for Kabuki syndrome. For validation, we screened all 54 exons of MLL2 in 43 additional cases by Sanger sequenc-ing. Previously unidentified nonsynonymous, nonsense or frameshift mutations in MLL2 were found in 26 of these 43 cases (Fig. 1 and Supplementary Table 3). In total, through either exome sequencing or targeted sequencing of MLL2, 33 distinct MLL2 mutations were iden-tified in 35 of 53 families (66%) with Kabuki syndrome (Fig. 1 and Supplementary Table 3). In each of 12 cases for which DNA from both parents was available, the MLL2 variant was found to have occurred de novo. Three mutations were found in two individuals each. One of these three mutations was confirmed to have arisen de novo in one of the cases, indicating that some mutations in individuals with Kabuki syndrome are recurrent. In addition, MLL2 mutations (resulting in p.4527K>X and p.5464T>M) were also identified in each of two fami-lies in which Kabuki syndrome was transmitted from parent to child.
Table 1 Number of genes common to any subset of x affected individuals.Subset analysis (any x of 10) 1 2 3 4 5 6 7 8 9 10
NS/SS/I 12,042 8,722 7,084 6,049 5,289 4,581 3,940 3,244 2,486 1,459Not in dbSNP129 or 1000 Genomes
7,419 2,697 1,057 488 288 192 128 88 60 34
Not in control exomes 7,827 2,865 1,025 399 184 90 50 22 7 2Not in either 6,935 2,227 701 242 104 44 16 6 3 1Is loss-of-function (non-sense or frameshift indel)
753 49 7 3 2 2 1 0 0 0
The number of genes with at least one nonsynonymous variant (NS), splice-site acceptor or donor variants (SS) or coding indel (I) are listed under various filters. Variants were filtered by presence in dbSNP or 1000 Genomes (not in dbSNP129 or 1000 genomes) and control exomes (not in control exomes) or both (not in either); control exomes refer to those from 8 Hapmap3, 4 FSS3, 4 Miller2 and 10 EGP samples. The number of genes found using the union of the intersection of x individuals is given.
Table 2 Number of genes common in sequential analysis of phenotypically ranked individualsSequential analysis 1 +2 +3 +4 +5 +6 +7 +8 +9 +10
NS/SS/I 5,282 3,850 3,250 2,354 2,028 1,899 1,772 1,686 1,600 1,459Not in dbSNP129 or 1000 Genomes
687 214 145 84 63 54 42 40 39 34
Not in control exomes 675 134 50 26 13 13 8 5 4 2Not in either 467 89 34 18 9 8 4 4 3 1Is loss-of-function (non-sense/frameshift indel)
25 1 1 1 0 0 0 0 0 0
Variants were filtered as in Table 1. Exomes were added sequentially to the analysis by ranked phenotype; for example, column “+3” shows the number of genes at the intersection of the three top ranked cases (Supplementary Fig. 1). The gene with at least one NS/SS/I in all individuals is MUC16, which is very likely to be a false positive due to its extreme length (14,507 amino acids).
L E T T E R S

Clinical sequencing: best case scenario Aug 2010
Making a definitive diagnosis: Successful clinicalapplication of whole exome sequencing in a child with
intractable inflammatory bowel diseaseElizabeth A. Worthey, PhD1,2, Alan N. Mayer, MD, PhD2,3, Grant D. Syverson, MD2,
Daniel Helbling, BSc1, Benedetta B. Bonacci, MSc2, Brennan Decker, BSc1, Jaime M. Serpe, BSc2,Trivikram Dasu, PhD2, Michael R. Tschannen, BSc1, Regan L. Veith, MSc2, Monica J. Basehore, PhD4,
Ulrich Broeckel, MD, PhD1,2,3, Aoy Tomita-Mitchell, PhD1,2,3, Marjorie J. Arca, MD3,5,James T. Casper, MD2,3, David A. Margolis, MD2,3, David P. Bick, MD1,2,3, Martin J. Hessner, PhD1,2,
John M. Routes, MD2,3, James W. Verbsky, MD, PhD2,3, Howard J. Jacob, PhD1,2,3,6,and David P. Dimmock, MD1,2,3
Purpose: We report a male child who presented at 15 months withperianal abscesses and proctitis, progressing to transmural pancolitiswith colocutaneous fistulae, consistent with a Crohn disease-like illness.The age and severity of the presentation suggested an underlyingimmune defect; however, despite comprehensive clinical evaluation, wewere unable to arrive at a definitive diagnosis, thereby restrictingclinical management. Methods: We sought to identify the causativemutation(s) through exome sequencing to provide the necessary addi-tional information required for clinical management. Results: Aftersequencing, we identified 16,124 variants. Subsequent analysis identi-fied a novel, hemizygous missense mutation in the X-linked inhibitor ofapoptosis gene, substituting a tyrosine for a highly conserved andfunctionally important cysteine. X-linked inhibitor of apoptosis was notpreviously associated with Crohn disease but has a central role in theproinflammatory response and bacterial sensing through the NOD sig-naling pathway. The mutation was confirmed by Sanger sequencing ina licensed clinical laboratory. Functional assays demonstrated an in-creased susceptibility to activation-induced cell death and defectiveresponsiveness to NOD2 ligands, consistent with loss of normalX-linked inhibitor of apoptosis protein function in apoptosis and NOD2signaling. Conclusions: Based on this medical history, genetic andfunctional data, the child was diagnosed as having an X-linked inhibitorof apoptosis deficiency. Based on this finding, an allogeneic hemato-poietic progenitor cell transplant was performed to prevent the devel-opment of life-threatening hemophagocytic lymphohistiocytosis, inconcordance with the recommended treatment for X-linked inhibitor of
apoptosis deficiency. At !42 days posttransplant, the child was able toeat and drink, and there has been no recurrence of gastrointestinaldisease, suggesting this mutation also drove the gastrointestinal disease.This report describes the identification of a novel cause of inflammatorybowel disease. Equally importantly, it demonstrates the power of exomesequencing to render a molecular diagnosis in an individual patient inthe setting of a novel disease, after all standard diagnoses were ex-hausted, and illustrates how this technology can be used in a clinicalsetting. Genet Med 2011:13(3):255–262.
Key Words: genomic, personalized, medicine, clinical, immunodeficiency
Over the last year, a number of publications have reportedthe use of exome or genome sequencing in patients.1–6
Most of these studies made use of disease cohorts or familiesand do not report functional assays or a change in treatment. Wereport the use of whole exome sequencing to reach a clinicaldiagnosis and alter treatment in a single child with a life-threatening but previously undefined form of inflammatorybowel disease (IBD) (AHC [OMIM# 266600]).7
The patient is a male who initially presented at 15 monthswith poor weight gain and a perianal abscess. The abscessenlarged, drained spontaneously, but failed to close despiteseveral rounds of oral, and then parenteral, antibiotics. Hesubsequently developed diarrhea and weight loss, despite sup-plemental enteral feedings, and his condition continued to de-teriorate over a period of 6 months, with referral to our hospitalat 30 months. He had a weight of 8.1 kg, length 81.2 cm, andbody mass index of 12.7 (all "3 percentile), indicating severestunting and malnutrition. Examination under anesthesia showedperineal fistulae and deep fissures. Initial endoscopy showed arectal stricture and linear ulcers of the rectum; the sigmoidcolon and proximal bowel were healthy. Biopsy showed focalactive proctitis with ulceration. The child was treated withnasoenteric feeds and infiximab for a presumptive diagnosis ofCrohn disease.
Despite treatment, the perineal fistulae persisted, and newones developed threatening the scrotum. A diverting sigmoidcolostomy was performed to divert fecal material and facilitatefistulae closure. The colostomy and mucus fistula failed toincorporate, and new fistulae developed. Although the perianalfistula and the mucosa of the defunctionalized distal limb re-covered, the afferent limb became inflamed, eventually involv-ing the entire colon, but not the terminal ileum or upper gas-trointestinal (GI) tract. The patient was started on long-termtotal parenteral nutrition using a peripherally inserted central
From the 1Human and Molecular Genetics Center; 2The Department ofPediatrics, The Medical College of Wisconsin, Milwaukee; 3The Children’sHospital of Wisconsin, Wauwatosa, Wisconsin; 4Molecular Diagnostic Lab-oratory, Greenwood Genetic Clinic, Greenwood, South Carolina; 5The De-partment of Surgery; and 6The Department of Physiology, The MedicalCollege of Wisconsin, Milwaukee, Wisconsin.
Elizabeth A. Worthey, PhD, Human and Molecular Genetics Center and theDepartment of Pediatrics, The Medical College of Wisconsin, 8701 WatertownPlank Road, Milwaukee, WI 53226. E-mail: [email protected].
Disclosure: The authors declare no conflict of interest.
Supplemental digital content is available for this article. Direct URL citationsappear in the printed text and are provided in the HTML and PDF versions ofthis article on the journal’s Web site (www.geneticsinmedicine.org).
The first two authors contributed equally to this work.
Submitted for publication August 12, 2010.
Accepted for publication November 23, 2010.
Published online ahead of print December 17, 2010.
DOI: 10.1097/GIM.0b013e3182088158
BRIEF REPORT
Genetics IN Medicine • Volume 13, Number 3, March 2011 255
Nicholas Volker

Clinical sequencing: best case scenario -‐15 month old presented with Crohn disease like illness -‐Age/severity suggested immune defect -‐At 4 years of age
-‐Hundreds of surgeries -‐Over 700 days in the hospital -‐Tested mul;ple candidate genes = normal -‐Treatments working, but not long term solu;on
-‐Immune recons;tu;on?
-‐Aggressive approach -‐Unknown mechanism
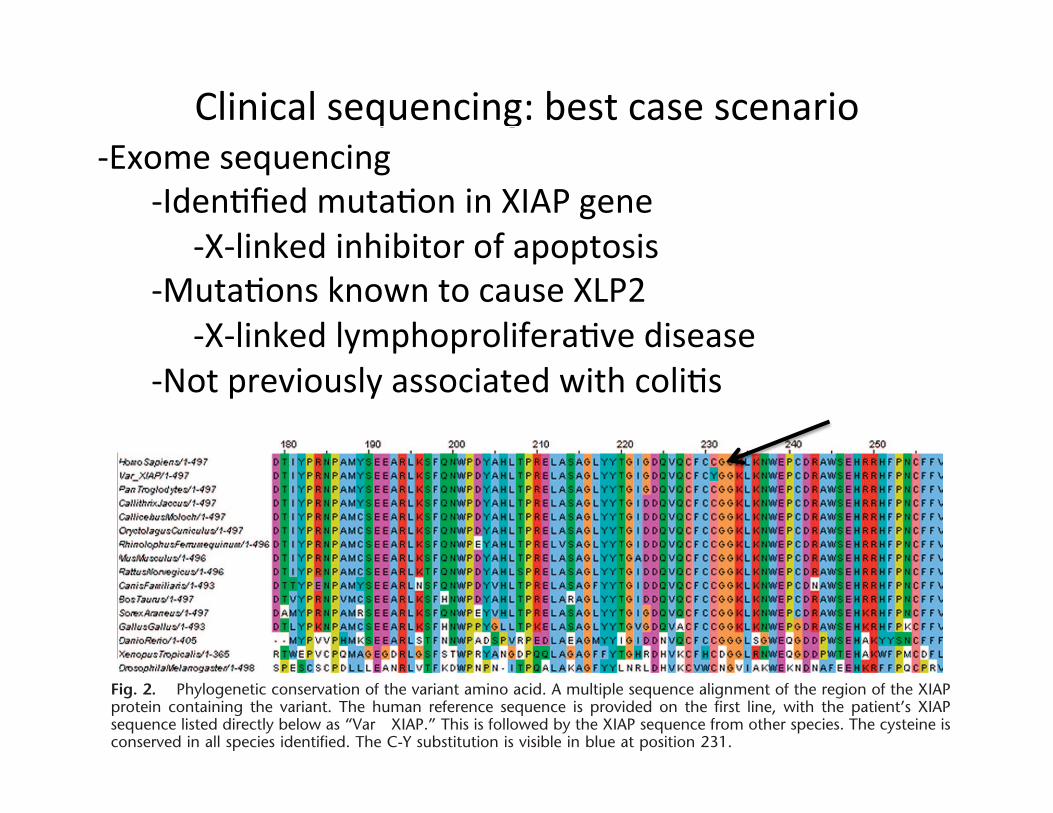
Clinical sequencing: best case scenario -‐Exome sequencing
-‐Iden;fied muta;on in XIAP gene -‐X-‐linked inhibitor of apoptosis -‐Muta;ons known to cause XLP2 -‐X-‐linked lymphoprolifera;ve disease -‐Not previously associated with coli;s
Manual inspection of a subset of !2000 variants confirmedapproximately 0.65% as likely false positives; the majority ofthese were polynucleotide tract errors, and the remainder weremissassemblies of reads to regions sharing high-sequence iden-tity. Unsurprisingly, the majority of these misassemblies existedeither in low-complexity regions or in regions highly conservedamong members of protein families. Variants found in a smallnumber of genes selected for their clinical significance wereevaluated by Sanger sequencing. In all instances, the base callswere concordant across both technologies.
Table 1, category A, provides a summary of the total num-bers and numbers of novel variations broken down by bothlocation and variation class (insertions, deletions, and substitu-tions). As expected, the majority of variants identified weresubstitutions; insertions were the least common across all cat-egories. A larger percentage of the novel variants were inser-tions or deletions rather than substitutions when compared withthe previously identified variants. A small number of thesenonsynonymous substitutions resulted in the production of stop
codons; all homozygous examples were either previously iden-tified or resulted in a stop in a protein commonly found in thepopulation to harbor stop codons (Table 1, category A).
To reduce the search space, we hypothesized, based on theseverity and unique clinical presentation, that this case waslikely a recessive disorder caused by a hemizygous or homozy-gous mutation, or compound heterozygote. Sixty-six genes con-taining potential compound heterozygous mutations (wherevariants were nonsynonymous and predicted to be damaging byPolyphen, a tool that predicts the impact of an amino acidsubstitution on the structure and function of a human protein)were identified (Table 1, category B) and investigated; none ofthese candidates remained after exclusion based on novelty andsequence conservation. Seventy homozygous/hemizygous non-synonymous variants were identified (Table 1C); eight werenovel (when compared against all publicly available data sets)and predicted to be damaging by PolyPhen. Only two of thesewere highly conserved. One variant, in GSTM1, was excludedbecause this gene has a high null genotype frequency in the
Fig. 2. Phylogenetic conservation of the variant amino acid. A multiple sequence alignment of the region of the XIAPprotein containing the variant. The human reference sequence is provided on the first line, with the patient’s XIAPsequence listed directly below as “Var XIAP.” This is followed by the XIAP sequence from other species. The cysteine isconserved in all species identified. The C-Y substitution is visible in blue at position 231.
Fig. 3. Clinical confirmation in the child and mother. The region of the XIAP gene surrounding the mutation in both thechild and the mother was sequenced using the BigDye Terminator Cycle Sequencing kit and analyzed on an ABI3730XLautomated DNA sequencer. The Sanger sequence trace from a normal human control is shown at the top. Hemizygosityat the candidate locus is confirmed in the child (middle panel). The mother is heterozygous at this locus (bottom panel).
Genetics IN Medicine • Volume 13, Number 3, March 2011 Clinical exome sequencing in IBD
Genetics IN Medicine • Volume 13, Number 3, March 2011 259

Clinical sequencing: best case scenario -‐Func;onality
-‐Central role in proinflammatory response -‐Ac;va;on of NFkB followed by ac;va;on of proinflammatory cytokines -‐NOD signaling/mediated programmed cell death
-‐Treatment
-‐Hemopoie;c progenitor cell transplant -‐42 days poseransplant – eat and drank normally -‐Complete resolu;on of coli;s -‐No recurrence of gastrointes;nal disease

Filtering process -‐Quality scores/coverage -‐dbSNP -‐1000 Genomes -‐Exome variant server (EVS)
-‐Over 5000 exomes (by frequency) -‐Database of genomic variants (DGV) -‐Affected/unaffected/trios -‐Addi;onal probands/families -‐Online Medelian Inheritance in Man (OMIM) -‐Human Gene Muta;on Database (HGMD) -‐PolyPhen-‐2/SIFT

HUMAN GENET I C S
Whole-Genome Sequencing for OptimizedPatient ManagementMatthew N. Bainbridge,1,2 Wojciech Wiszniewski,3 David R. Murdock,1 Jennifer Friedman,4,5
Claudia Gonzaga-Jauregui,3 Irene Newsham,1 Jeffrey G. Reid,1 John K. Fink,6,7
Margaret B. Morgan,1 Marie-Claude Gingras,1 Donna M. Muzny,1 Linh D. Hoang,8
Shahed Yousaf,8 James R. Lupski,1,3,9,10 Richard A. Gibbs1,3*
Whole-genome sequencing of patient DNA can facilitate diagnosis of a disease, but its potential for guidingtreatment has been under-realized. We interrogated the complete genome sequences of a 14-year-old fraternaltwin pair diagnosed with dopa (3,4-dihydroxyphenylalanine)–responsive dystonia (DRD; Mendelian Inheritance inMan #128230). DRD is a genetically heterogeneous and clinically complex movement disorder that is usuallytreated with L-dopa, a precursor of the neurotransmitter dopamine. Whole-genome sequencing identifiedcompound heterozygous mutations in the SPR gene encoding sepiapterin reductase. Disruption of SPR causesa decrease in tetrahydrobiopterin, a cofactor required for the hydroxylase enzymes that synthesize the neuro-transmitters dopamine and serotonin. Supplementation of L-dopa therapy with 5-hydroxytryptophan, a serotoninprecursor, resulted in clinical improvements in both twins.
INTRODUCTIONSubclassification of phenotypically similar but genetically hetero-geneous conditions by identifying underlying causative allelescan be pivotal for precise disease diagnosis and treatment. High-throughput sequencing of patient genomes could potentially fa-cilitate the diagnosis of rare diseases. Identified variants can becross-checked with databases for previous associations with disease,and benign variants with high allele frequency can be eliminatedfrom consideration using population-variation databases (1). The re-maining variants can be assessed for their effects on genes and thosegenes can be assessed for their association with disease. These ap-proaches require integrated and accurate databases as well as bestpractices guidelines (2).
Dopa (3,4-dihydroxyphenylalanine)–responsive dystonia (DRD)[Mendelian Inheritance in Man (MIM) #128230], formally knownas “hereditary dystonia with marked diurnal variation” (Segawa dys-tonia), is a genetically and clinically heterogeneous movement disorder(3). DRD typically begins in childhood after a period of normal devel-opment and frequently manifests variable severity during the day (re-duced dystonia upon awakening, increased dystonia by midday). Thedifferential diagnosis for DRD includes early-onset parkinsonism,cerebral palsy, and early-onset primary dystonia (4–6). The clinicaldiagnosis of DRD is based on neurological presentation, age of onsetand progression of the disease, mode of inheritance, concentrations ofneurotransmitter metabolites and pterins (cofactors for neurotransmitter-
producing enzymes) in the cerebrospinal fluid (CSF), and the degreeof responsiveness to L-dopa treatment. Sustained clinical benefit fromvery low dose L-dopa administration is a clinical hallmark of DRD.However, a range of clinical responses to L-dopa therapy have beendocumented (7) and L-dopa therapy alone may not be sufficient forcomplete alleviation of clinical symptoms.
DRD can be inherited as either an autosomal dominant or recessivetrait and is associated with mutations in genes encoding guanosine5!-triphosphate (GTP) cyclohydrolase (GCH1), tyrosine hydroxylase(TH), and sepiapterin reductase (SPR) (Fig. 1). GCH1 and SPR areenzymes of the tetrahydrobiopterin (BH4) biosynthesis pathway. BH4serves as a cofactor for tyrosine and tryptophan hydroxylases in theinitial biosynthesis of the neurotransmitters dopamine, noradrenaline,and serotonin. TH converts tyrosine to L-dopa, a precursor of dopamineand noradrenaline (8) (Fig. 1). In a study of 64 patients diagnosed withDRD, ~83% of cases were caused by autosomal dominant or de novopointmutations and deletions inGTP cyclohydrolase, whereas autosomalrecessive cases were caused by mutations in tyrosine hydroxylase (~5%),sepiapterin reductase (~3%), or parkin (encoded by the PARK2 gene, agene implicated in juvenile-onset Parkinson disease) (~3%). Five percentof DRD cases had unknown genetic causes (9). Molecular genetic testinghas proved a valuable tool for diagnosing DRD; however, until recently,clinical molecular genetic assays were limited to the identification ofmutations in the TH and GCH1 genes (10). Heterozygous deletion ofthe entire TH gene, which potentially results in decreased endogenousdopamine production, has also been reported in a patient with adult-onset Parkinson disease (11), a common movement disorder caused byloss of dopamine-producingneurons in the brain’s nigrostriatal pathway.
Here, we studied a fraternal twin pair diagnosed with DRD, who hadno identified deleterious variants in the TH or GCH1 genes. Sequencingof the SPR gene was not available through a clinical laboratory at thetime this study was initiated and was not performed (see Materialsand Methods). Because the primary candidate genes for DRD wereeliminated, we used high-throughput sequencing (12, 13) to inter-rogate the whole genomes of the male and female twin to identify po-tential causative genetic variants.
1Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030,USA. 2Department of Structural and Computational Biology and Molecular Biophysics,Baylor College of Medicine, Houston, TX 77030, USA. 3Department of Molecular andHuman Genetics, Baylor College of Medicine, Houston, TX 77030, USA. 4Departmentsof Neurosciences and Pediatrics, University of California, San Diego, CA 92093, USA.5Rady Children’s Hospital, San Diego, CA 92123, USA. 6Department of Neurology,University of Michigan, Ann Arbor, MI 48109, USA. 7Geriatric Research Education andClinical Center, Ann Arbor Veterans Affairs Medical Center, Ann Arbor, MI 48105, USA.8Life Technologies, Carlsbad, CA 92008, USA. 9Department of Pediatrics, BaylorCollege of Medicine, Houston, TX 77030, USA. 10Texas Children’s Hospital, Houston, TX77030, USA.*To whom correspondence should be addressed. E-mail: [email protected]
R EPORT
www.ScienceTranslationalMedicine.org 15 June 2011 Vol 3 Issue 87 87re3 1
on
Mar
ch 6
, 201
3st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m
-‐Fraternal twins (male/female) – 14 years old -‐Dopa responsive dystonia
-‐gene;cally heterogeneous -‐clinically complex movement disorder -‐L-‐Dopa treatment
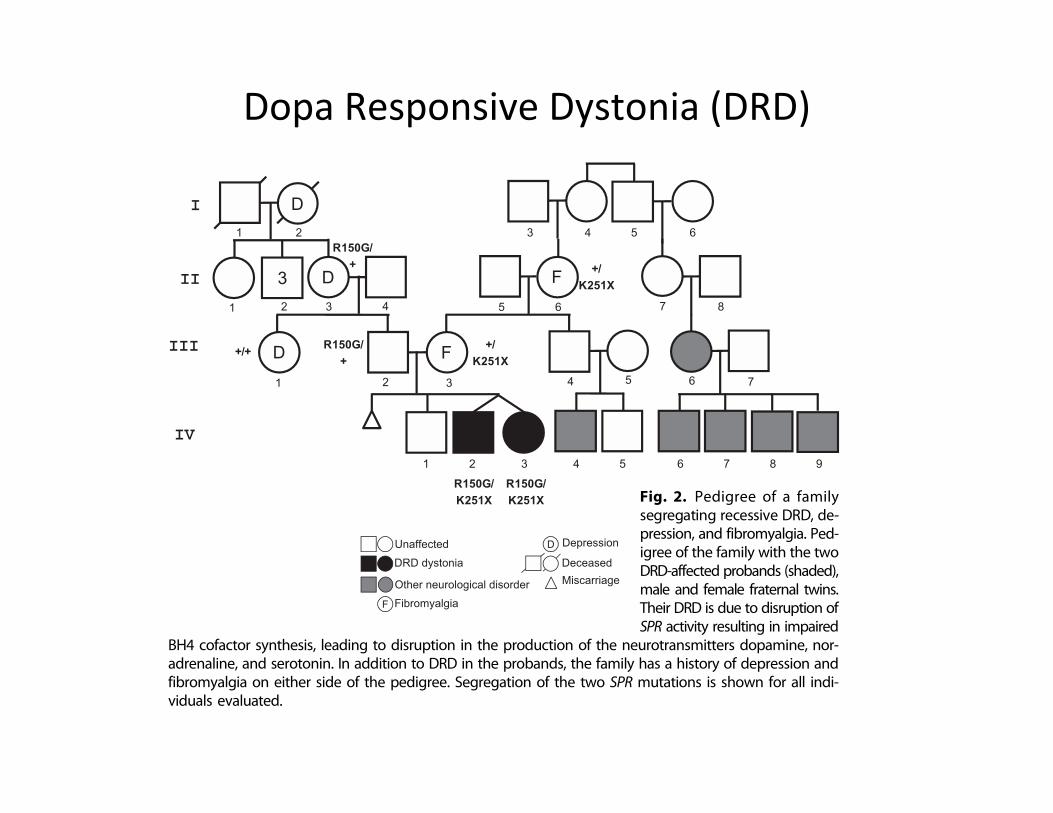
Dopa Responsive Dystonia (DRD)
The identified variants in SPR were NM_003124:c.448A>G (chro-mosome 2: 72,969,094, p.Arg150Gly) and NM_003124:c.751A>T(chromosome 2:72,972,139, p.Lys251X). The former mutation oc-curs in a b strand secondary structure element in close proximity toa substrate binding region, and the latter results in a truncation ofthe last 10 amino acid residues of SPR destroying one entire bstrand (17). Both mutations have been previously identified in two
Caucasian families (18, 19), but in bothcases, the mutation was homozygousrather than a compound heterozygote.Functional studies for each of the pu-tative pathogenic variants were reportedand found to be deleterious to SPR ac-tivity (18, 19). In these studies, SPR ac-tivity was measured with a biochemicalassay either using skin fibroblasts takenfrom the patient or by cloning the mu-tated gene in a bacterial vector and sub-sequent purification. Disruption of SPRprevents the regeneration of BH4, whichis an important cofactor for the produc-tion of both dopamine and serotonin(Fig. 1). Thus, the recommended treat-ment of DRD caused by SPR mutationsis with both the dopamine precursorL-dopa, which the twins were already pre-scribed, and the serotonin precursor5-hydroxytryptophan (5-HTP), whichthe twins were not receiving. Both com-pounds can readily cross the blood-brainbarrier. The serotonin pathway may befurther enhanced by the addition of selec-tive serotonin reuptake inhibitors (SSRIs)used to treat depression.
Validation and segregationTo test for a pattern of segregation of these alleles that is consistentwith their causative role in DRD, we designed oligonucleotide primersto correspond to sequences that flank both mutations, and we usedthem to PCR (polymerase chain reaction) amplify and capillarysequence all members of the immediate family. Both mutations wereconfirmed as compound heterozygous mutations in the affected twins,and the c.751A>T (p.Lys251X) nonsense mutation and the c.448A>G(p.Arg150Gly) missense mutation were found in the heterozygousstate in the mother and father, respectively. Neither mutation wasfound in the unaffected sibling, although the individual alleles wereidentified in members of previous generations (Fig. 3).
Efficacy of 5-HTP treatmentAs a consequence of the molecular diagnosis, the treatments for the maleand female twin were modified to include 5-HTP (0.8 and 1.2 mg/kg,respectively). They have been on this therapy for ~4 months at the timeof writing. Both patients underwent periodic follow-up visits at the sametime of day with one physician (J.F.) who assessed the impact of themedications. According to the physician report, both patients showedthe first signs of improvement after 1 to 2 weeks, and their conditionreached a plateau after 2 months of therapy. The male DRD patientreported improved focus in school, as well as improved coordinationin athletics. Further, the male showed reduced drooling and hand tremor,and objective evidence for the latter was provided by serial handwritingsamples (fig. S1). The female twin reported reduced frequency of laryn-geal spasms, improved sleep and focus, and improved tolerance for exer-cise and was able to resume participation in sports after a 14-monthabsence. In the female DRD patient, there were also reduced choreiform
3
D
D
D
F
F
Unaffected
DRD dystonia
Other neurological disorder
Fibromyalgia
Depression
Miscarriage
F
D
Deceased
I
II
III
IV
R150G/
K251X
R150G/
K251X
+/
K251X
+/
K251X
R150G/
+
+/+R150G/
+
1
1
2 3 4 5
2 3 75 6 8
6
1 2 3 4 5 6 7
1 2 3 4 5 6 7 8 9
4
Fig. 2. Pedigree of a familysegregating recessive DRD, de-pression, and fibromyalgia. Ped-igree of the family with the twoDRD-affected probands (shaded),male and female fraternal twins.Their DRD is due to disruption ofSPR activity resulting in impaired
BH4 cofactor synthesis, leading to disruption in the production of the neurotransmitters dopamine, nor-adrenaline, and serotonin. In addition to DRD in the probands, the family has a history of depression andfibromyalgia on either side of the pedigree. Segregation of the two SPR mutations is shown for all indi-viduals evaluated.
Table 1. Single-nucleotide variants in the sequence of the DRD twins.All high-quality variants first observed from primary alignment ofsequence reads to the current reference haploid human genome werethen filtered for coding regions and annotated if they cause proteincoding mutations. These variants were filtered further against data-bases of known and common variation to enrich for rare variants,which are more likely to be disease-causing. Finally, candidate geneswere identified, under a recessive inheritance model, by homozygosityor by identifying genes that harbor two or more variants.
Nucleotide variants IV-2(male subject)
IV-3(female subject) Shared
All variants 2,427,038 2,504,162 1,631,770
% dbSNP129 88.7 88.1
Variant density (bp!1) 1/1112 1/1078
Coding 13,352 14,961 9531
Nonsynonymous 6432 7141 4605
Rare nonsynonymous 174 175 77
Candidate genes 6 9 3
R E PORT
www.ScienceTranslationalMedicine.org 15 June 2011 Vol 3 Issue 87 87re3 3
on
Mar
ch 6
, 201
3st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m

Dopa Responsive Dystonia (DRD)
The identified variants in SPR were NM_003124:c.448A>G (chro-mosome 2: 72,969,094, p.Arg150Gly) and NM_003124:c.751A>T(chromosome 2:72,972,139, p.Lys251X). The former mutation oc-curs in a b strand secondary structure element in close proximity toa substrate binding region, and the latter results in a truncation ofthe last 10 amino acid residues of SPR destroying one entire bstrand (17). Both mutations have been previously identified in two
Caucasian families (18, 19), but in bothcases, the mutation was homozygousrather than a compound heterozygote.Functional studies for each of the pu-tative pathogenic variants were reportedand found to be deleterious to SPR ac-tivity (18, 19). In these studies, SPR ac-tivity was measured with a biochemicalassay either using skin fibroblasts takenfrom the patient or by cloning the mu-tated gene in a bacterial vector and sub-sequent purification. Disruption of SPRprevents the regeneration of BH4, whichis an important cofactor for the produc-tion of both dopamine and serotonin(Fig. 1). Thus, the recommended treat-ment of DRD caused by SPR mutationsis with both the dopamine precursorL-dopa, which the twins were already pre-scribed, and the serotonin precursor5-hydroxytryptophan (5-HTP), whichthe twins were not receiving. Both com-pounds can readily cross the blood-brainbarrier. The serotonin pathway may befurther enhanced by the addition of selec-tive serotonin reuptake inhibitors (SSRIs)used to treat depression.
Validation and segregationTo test for a pattern of segregation of these alleles that is consistentwith their causative role in DRD, we designed oligonucleotide primersto correspond to sequences that flank both mutations, and we usedthem to PCR (polymerase chain reaction) amplify and capillarysequence all members of the immediate family. Both mutations wereconfirmed as compound heterozygous mutations in the affected twins,and the c.751A>T (p.Lys251X) nonsense mutation and the c.448A>G(p.Arg150Gly) missense mutation were found in the heterozygousstate in the mother and father, respectively. Neither mutation wasfound in the unaffected sibling, although the individual alleles wereidentified in members of previous generations (Fig. 3).
Efficacy of 5-HTP treatmentAs a consequence of the molecular diagnosis, the treatments for the maleand female twin were modified to include 5-HTP (0.8 and 1.2 mg/kg,respectively). They have been on this therapy for ~4 months at the timeof writing. Both patients underwent periodic follow-up visits at the sametime of day with one physician (J.F.) who assessed the impact of themedications. According to the physician report, both patients showedthe first signs of improvement after 1 to 2 weeks, and their conditionreached a plateau after 2 months of therapy. The male DRD patientreported improved focus in school, as well as improved coordinationin athletics. Further, the male showed reduced drooling and hand tremor,and objective evidence for the latter was provided by serial handwritingsamples (fig. S1). The female twin reported reduced frequency of laryn-geal spasms, improved sleep and focus, and improved tolerance for exer-cise and was able to resume participation in sports after a 14-monthabsence. In the female DRD patient, there were also reduced choreiform
3
D
D
D
F
F
Unaffected
DRD dystonia
Other neurological disorder
Fibromyalgia
Depression
Miscarriage
F
D
Deceased
I
II
III
IV
R150G/
K251X
R150G/
K251X
+/
K251X
+/
K251X
R150G/
+
+/+R150G/
+
1
1
2 3 4 5
2 3 75 6 8
6
1 2 3 4 5 6 7
1 2 3 4 5 6 7 8 9
4
Fig. 2. Pedigree of a familysegregating recessive DRD, de-pression, and fibromyalgia. Ped-igree of the family with the twoDRD-affected probands (shaded),male and female fraternal twins.Their DRD is due to disruption ofSPR activity resulting in impaired
BH4 cofactor synthesis, leading to disruption in the production of the neurotransmitters dopamine, nor-adrenaline, and serotonin. In addition to DRD in the probands, the family has a history of depression andfibromyalgia on either side of the pedigree. Segregation of the two SPR mutations is shown for all indi-viduals evaluated.
Table 1. Single-nucleotide variants in the sequence of the DRD twins.All high-quality variants first observed from primary alignment ofsequence reads to the current reference haploid human genome werethen filtered for coding regions and annotated if they cause proteincoding mutations. These variants were filtered further against data-bases of known and common variation to enrich for rare variants,which are more likely to be disease-causing. Finally, candidate geneswere identified, under a recessive inheritance model, by homozygosityor by identifying genes that harbor two or more variants.
Nucleotide variants IV-2(male subject)
IV-3(female subject) Shared
All variants 2,427,038 2,504,162 1,631,770
% dbSNP129 88.7 88.1
Variant density (bp!1) 1/1112 1/1078
Coding 13,352 14,961 9531
Nonsynonymous 6432 7141 4605
Rare nonsynonymous 174 175 77
Candidate genes 6 9 3
R E PORT
www.ScienceTranslationalMedicine.org 15 June 2011 Vol 3 Issue 87 87re3 3
on
Mar
ch 6
, 201
3st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m

Dopa Responsive Dystonia (DRD)
-‐Compound het in SPR -‐Supplement with 5-‐HTP (L-‐dopa)
RESULTS
Clinical presentationThe patients were two affected 14-year-old fraternal twins, who werediagnosed with DRD at age 5 after L-dopa was found to alleviate theclinical symptoms of dystonia in one twin. The subjects were born at36 weeks of gestation after a pregnancy complicated by a hyperco-agulable state in their mother that required heparin treatment. Theperinatal history was uneventful. Well-child evaluations in the firstyear of life revealed generalized hypotonia and global developmentaldelay. These clinical observations prompted an initial evaluation in-cluding imaging studies of the brain (magnetic resonance imaging)that revealed periventricular leukomalacia in the male patient andbasic metabolic tests that were normal in both twins. CSF was notobtained before treatment with L-dopa. The female twin was moreseverely affected and subsequently developed dystonic movements,hypokinesia, rigidity, tremor, oculogyric crises (ocular dystonic move-ments), and seizures. Her brother had milder disease symptoms andwas originally diagnosed with static encephalopathy (cerebral palsy).However, later serial examinations showed the appearance of progres-sive subtle dystonia at age 5 years. Whereas the female patient hada diurnal fluctuation of neurological symptoms with less severe symp-
toms in the morning and more severe symptoms in the afternoon, themale patient did not.
At age 5 years, a trial of L-dopa/carbidopa at a ratio of 10:100, one-quarter tablet a day increasing to one-quarter a tablet three times perday over several days, reduced clinical symptoms by day 3 but was ac-companied by mild dyskinesia. The dosage, therefore, was reducedinitially but then was reinstated. Both patients are in middle schoolfollowing a regular curriculum and have excellent academic perform-ance despite reportedly a reduced attention span. At age 14 years andon L-dopa/carbidopa 25:100 three times per day, the affected male wasfound to have mild tremor and dystonic posturing of the hands uponneurological examination. His sister demonstrated slightly unsteadygait; mild choreiform movements in the tongue; mild dysphonia; milddystonia in the neck, shoulder, and hands; and mild bradykinesia. Herhistory is also significant for respiratory difficulties thought to be sec-ondary to intermittent laryngospasm. The immediate family historywas negative for movement disorders or other neurological diseasesapart from fibromyalgia and depression. The diagnosis of recessivedystonia in the probands was complicated by the presence of a firstcousin with reported juvenile seizures and a third cousin and her fourchildren diagnosed with an unspecified neurological disorder (Fig. 2).
Genome variationDNA was extracted from peripheral blood cells obtained from bothaffected twins, an unaffected sibling, and their parents. DNA fromthe twins was subjected to whole-genome sequencing on the SOLiDplatform. In total, 178.4 giga–base pairs (Gbp) of sequence data wasproduced and aligned to the human reference genome, resulting in anaverage sequence coverage of 29.4 and 30.0 for the male and femaletwin, respectively (59-fold for sites shared by both twins).
A set of putative, high-quality sequence differences between eachtwin and the reference genome (hg18) was identified, and variantsshared by both twins were subsequently analyzed. About 90% of thevariants discovered were also identified in the dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/), with one variant discoveredper 1100 bp on average across the genome, which is similar to re-ported values (14). The degree of variant overlap confirmed that thetwins were dizygotic, consistent with the model of recessive dystoniain this family. The variant data were next filtered to allow removalof likely benign variants with the dbSNP v.129 (15) and ThousandGenomes (1) databases of common and likely non–disease-causingmutations, as well as the Baylor Human Genome Sequencing Center’smaintained database of common variants from other sequencing proj-ects. Finally, mutations that caused nonsynonymous changes to pro-tein products were classified (Table 1). There were no remaining rarehomozygous mutations shared between both twins, and no large ge-nomic regions with stretches of homozygous mutations, which is con-sistent with the absence of consanguinity.
After overlapping shared mutations, filtering, and genetic annotation,only three genes were identified that contained two or more predictedamino acid–altering heterozygous mutations (table S1). One of these(ZNF544) encodes a computationally predicted zinc finger protein withno known function or targets, another predicts an open reading frame(C2orf16), and the third is the SPR gene encoding sepiapterin reduc-tase. Subsequent automated annotation of these genes by comparisonto the MIM disease database (16) indicated a known association of SPRwith DRD and no associations of either of the other two genes with anydisease.
Guanosine triphosphate (GTP)
Dihydroneopterin triphosphate
6-Pyruvoyl-tetrahydropterin
Tetrahydrobiopterin (BH4)
Quinoid-dihydrobiopterin
Pterin-4a-carbinolamine
L-dopa
Tyrosine
TH
GCH1
PTPS
SPR DHPR PCD
Tryptophan
5-Hydroxytryptophan (5-HTP)
Phenylalanine
Tyrosine
TPHPAH
Serotonin
5-Hydroxyindoleacetic acid
Dopamine
Homovanillic acid Norepinephrine
Fig. 1. Metabolic pathways of neurotransmitter production. DRD has beenassociatedwithmutations in the genes encoding GTP cyclohydrolase (GCH1),tyrosine hydroxylase (TH), and sepiapterin reductase (SPR) (boxed), which areenzymes associated with production of the neurotransmitters dopamine andserotonin. The catalytic action of GCH1 is the rate-limiting step in productionof tetrahydrobiopterin (BH4), a cofactor for the tyrosine and tryptophan hy-droxylases. Disruption of the GCH1 gene can cause autosomal dominantDRD. Autosomal recessive DRD is caused by mutations in TH and SPR. Both5-hydroxytryptophan (5-HTP) and dopamine production are disrupted bymutations in SPR, whereas only dopamine production is disrupted by muta-tions in TH.
R E PORT
www.ScienceTranslationalMedicine.org 15 June 2011 Vol 3 Issue 87 87re3 2
on
Mar
ch 6
, 201
3st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m

Trio sequencing
• 40 or so de novo coding variants per genome – Aier close inspec;on/valida;on only a frac;on are real (most only have 0-‐3)
• Rare diseases and Complex diseases

NATURE GENETICS VOLUME 44 | NUMBER 9 | SEPTEMBER 2012 1033
L E T T E R S
This study was funded in part by grants from the AHCF (to K.J.S., S.P.R. and T.M.N.); the ENRAH for SMEs Consortium under the European Commission Sixth Framework Programme; the Institut National de la Santé et de la Recherche Médicale (to S.N. and B.F.); the Centre National de la Recherche Scientifique (to S.N. and B.F.); the University Pierre and Marie Curie (to S.N. and B.F.); the Association Française Contre les Myopathies (to S.N. and B.F.); the Association Française de l’Hémiplégie Alternante (to S.N., A.M.J.M.v.d.M. and B.d.V.); AISEA Onlus (to F.G. and G.N.); the Center for Human Genome Variation; the Wellcome Trust (084730 to S.M.S.); the National Center for Research Resources (UL1RR025764 to the University of Utah Center for Clinical and Translational Sciences; K.J.S.); the NIH (1T32HL105321-01 to C.H.); the University of Luxembourg Institute for Systems Biology Program (to C.H.) and the Center for Medical Systems Biology established in The Netherlands Genomics Initiative and The Netherlands Organisation for Scientific Research (project 050-060-409 to A.M.J.M.v.d.M. and M.D.F.). S.N. is a recipient of a Contrat d’Interface from Assistance Publique-Hôpitaux de Paris.
AUTHOR CONTRIBUTIONSE.L.H., Y.H., S.M.S., M.A.M. and D.B.G. conceived and designed the study. Genetic data were generated and analyzed by E.L.H., K.J.S., Y.H., F.G., S.N., B.d.V., F.D.T., S.F., E.A., L.D.P., C.H., L.B.J., K.V.S., C.E.G., L.L., G.N., A.A. A.M.J.M.v.d.M and D.B.G. DNA samples and phenotypic information for AHC patients were collected, compiled and analyzed by K.J.S., F.G., S.N., N.M.W., B.d.V., F.D.T., B.F., S.H., E.P., M.T.S., T.M.N., L.V., S.P.R., K.J.M., K.S., L.J.P., J.H., M.D.F., A.M.B., G.K.H., C.M.W., D.W., B.J.L., P.U., M.D.K., I.E.S., G.N., A.A., S.M.S., M.A.M., the European AHC Genetics Consortium, the I.B.AHC Consortium and the ENRAH for SMEs Consortium. E.L.H., A.M.J.M.v.d.M., S.M.S., M.A.M. and D.B.G. wrote the paper. All authors reviewed the compiled manuscript.
COMPETING FINANCIAL INTERESTSThe authors declare competing financial interests: details are available in the online version of the paper.
Published online at http://www.nature.com/doifinder/10.1038/ng.2358. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
1. Verret, S. & Steele, J.C. Alternating hemiplegia in childhood: a report of eight patients with complicated migraine beginning in infancy. Pediatrics 47, 675–680 (1971).
2. Bourgeois, M., Aicardi, J. & Goutieres, F. Alternating hemiplegia of childhood. J. Pediatr. 122, 673–679 (1993).
3. Mikati, M.A., Kramer, U., Zupanc, M.L. & Shanahan, R.J. Alternating hemiplegia of childhood: clinical manifestations and long-term outcome. Pediatr. Neurol. 23, 134–141 (2000).
4. Sweney, M.T. et al. Alternating hemiplegia of childhood: early characteristics and evolution of a neurodevelopmental syndrome. Pediatrics 123, e534–e541 (2009).
5. Panagiotakaki, E. et al. Evidence of a non-progressive course of alternating hemiplegia of childhood: study of a large cohort of children and adults. Brain 133, 3598–3610 (2010).
6. Rho, J.M. & Chugani, H.T. Alternating hemiplegia of childhood: insights into its pathophysiology. J. Child Neurol. 13, 39–45 (1998).
7. Neville, B.G. & Ninan, M. The treatment and management of alternating hemiplegia of childhood. Dev. Med. Child Neurol. 49, 777–780 (2007).
8. Mikati, M.A. et al. A syndrome of autosomal dominant alternating hemiplegia: clinical presentation mimicking intractable epilepsy; chromosomal studies; and physiologic investigations. Neurology 42, 2251–2257 (1992).
9. Swoboda, K.J. et al. Alternating hemiplegia of childhood or familial hemiplegic migraine? A novel ATP1A2 mutation. Ann. Neurol. 55, 884–887 (2004).
10. Bassi, M.T. et al. A novel mutation in the ATP1A2 gene causes alternating hemiplegia of childhood. J. Med. Genet. 41, 621–628 (2004).
11. Vanmolkot, K.R. et al. Novel mutations in the Na+/K+ ATPase pump gene ATP1A2 associated with familial hemiplegic migraine and benign familial infantile convulsions. Ann. Neurol. 54, 360–366 (2003).
12. De Fusco, M. et al. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump 2 subunit associated with familial hemiplegic migraine type 2. Nat. Genet. 33, 192–196 (2003).
13. Rebhan, M., Chalifa-Caspi, V., Prilusky, J. & Lancet, D. GeneCards: integrating information about genes, proteins and diseases. Trends Genet. 13, 163 (1997).
14. de Carvalho Aguiar, P. et al. Mutations in the Na+/K+ ATPase 3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron 43, 169–175 (2004).
15. Anselm, I.A., Sweadner, K.J., Gollamudi, S., Ozelius, L.J. & Darras, B.T. Rapid-onset dystonia-parkinsonism in a child with a novel ATP1A3 gene mutation. Neurology 73, 400–401 (2009).
16. Svetel, M. et al. Rapid-onset dystonia-parkinsonism: case report. J. Neurol. 257, 472–474 (2010).
17. Kamm, C. et al. Novel ATP1A3 mutation in a sporadic RDP patient with minimal benefit from deep brain stimulation. Neurology 70, 1501–1503 (2008).
18. Blanco-Arias, P. et al. A C-terminal mutation of ATP1A3 underscores the crucial role of sodium affinity in the pathophysiology of rapid-onset dystonia-parkinsonism. Hum. Mol. Genet. 18, 2370–2377 (2009).
19. Ogawa, H., Shinoda, T., Cornelius, F. & Toyoshima, C. Crystal structure of the sodium-potassium pump Na+/K+ ATPase with bound potassium and ouabain. Proc. Natl. Acad. Sci. USA 106, 13742–13747 (2009).
20. Bellus, G.A. et al. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am. J. Hum. Genet. 56, 368–373 (1995).
21. Cooper, D.N. & Youssoufian, H. The CpG dinucleotide and human genetic disease. Hum. Genet. 78, 151–155 (1988).
22. Clapcote, S.J. et al. Mutation I810N in the 3 isoform of Na+/K+ ATPase causes impairments in the sodium pump and hyperexcitability in the CNS. Proc. Natl. Acad. Sci. USA 106, 14085–14090 (2009).
23. Stenson, P.D. et al. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 21, 577–581 (2003).
24. Jain, E. et al. Infrastructure for the life sciences: design and implementation of the UniProt website. BMC Bioinformatics 10, 136 (2009).
25. Pruitt, K.D. et al. The consensus coding sequence (CCDS) project: identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 19, 1316–1323 (2009).
Erin L Heinzen1,2,35, Kathryn J Swoboda3,4,35, Yuki Hitomi1,35, Fiorella Gurrieri5, Sophie Nicole6–8, Boukje de Vries9, F Danilo Tiziano5, Bertrand Fontaine6–8,10, Nicole M Walley1, Sinéad Heavin11, Eleni Panagiotakaki12, the European Alternating Hemiplegia of Childhood (AHC) Genetics Consortium13, the Biobanca e Registro Clinico per l’Emiplegia Alternante (I.B.AHC) Consortium13, the European Network for Research on Alternating Hemiplegia (ENRAH) for Small and Medium-sized Enterpriese (SMEs) Consortium13, Stefania Fiori5, Emanuela Abiusi5, Lorena Di Pietro5, Matthew T Sweney3, Tara M Newcomb3, Louis Viollet4, Chad Huff14, Lynn B Jorde14, Sandra P Reyna4, Kelley J Murphy4, Kevin V Shianna1,2, Curtis E Gumbs1, Latasha Little1, Kenneth Silver15,16, Louis J Ptác̆ek17,18, Joost Haan19,20, Michel D Ferrari20, Ann M Bye21, Geoffrey K Herkes22, Charlotte M Whitelaw23, David Webb24, Bryan J Lynch25, Peter Uldall26, Mary D King25, Ingrid E Scheffer11,27,28, Giovanni Neri5, Alexis Arzimanoglou12,29,30, Arn M J M van den Maagdenberg9,20, Sanjay M Sisodiya31,36, Mohamad A Mikati32,33,36 & David B Goldstein1,34,36
1Center for Human Genome Variation, Duke University School of Medicine, Durham, North Carolina, USA. 2Department of Medicine, Duke University School of Medicine, Durham, North Carolina, USA. 3Department of Pediatrics, University of Utah, Salt Lake City, Utah, USA. 4Department of Neurology, University of Utah, Salt Lake City, Utah, USA. 5Instituto di Genetica Medica, Università Cattolica del Sacro Cuore, Policlinico A. Gemelli, Rome, Italy. 6Université Pierre et Marie Curie, Centre de Recherche de l’Institut du Cerveau et de la Moelle épinière UMR S975, Paris, France. 7Institut National de la Santé et de la Recherche Médicale, U975, Paris, France. 8Centre National de la Recherche Scientifique, UMR7225, Paris, France. 9Department of Human Genetics, Leiden University Medical Centre, Leiden, The Netherlands. 10Assistance Publique-Hôpitaux de Paris, Département de Neurologie & Centre de Référence Canalopathies Musculaires, Groupe Hospitalier de la Pitié-Salpêtrière, Paris, France. 11Department of Medicine, University of Melbourne, Austin Health, Melbourne, Australia. 12Epilepsy, Sleep and Pediatric Neurophysiology Department, Woman-Mother-Child Hospital, University Hospitals of Lyon (HCL), Lyon, France. 13A full list of members and affiliations appears at the end of this paper. 14Department of Human Genetics, Eccles Institute of Human Genetics, University of Utah, Salt Lake City, Utah, USA. 15Department of Neurology, Comer Children’s Hospital, University of Chicago, Chicago, Illinois, USA. 16Department of Pediatrics, Comer Children’s Hospital, University of Chicago, Chicago,
1030 VOLUME 44 | NUMBER 9 | SEPTEMBER 2012 NATURE GENETICS
L E T T E R S
Alternating hemiplegia of childhood (AHC) is a rare, severe neurodevelopmental syndrome characterized by recurrent hemiplegic episodes and distinct neurological manifestations. AHC is usually a sporadic disorder and has unknown etiology. We used exome sequencing of seven patients with AHC and their unaffected parents to identify de novo nonsynonymous mutations in ATP1A3 in all seven individuals. In a subsequent sequence analysis of ATP1A3 in 98 other patients with AHC, we found that ATP1A3 mutations were likely to be responsible for at least 74% of the cases; we also identified one inherited mutation in a case of familial AHC. Notably, most AHC cases are caused by one of seven recurrent ATP1A3 mutations, one of which was observed in 36 patients. Unlike ATP1A3 mutations that cause rapid-onset dystonia-parkinsonism, AHC-causing mutations in this gene caused consistent reductions in ATPase activity without affecting the level of protein expression. This work identifies de novo ATP1A3 mutations as the primary cause of AHC and offers insight into disease pathophysiology by expanding the spectrum of phenotypes associated with mutations in ATP1A3.
AHC was first characterized as a distinct syndrome in 1971, with a report that described eight patients with episodes of intermittent hemiplegia on alternating sides of the body, developmental delay, dystonia and choreoathetosis beginning in infancy1. Since then, spe-cific diagnostic criteria have more clearly defined the classic parox-ysmal and interictal neurological manifestations associated with this disease2–6. AHC affects approximately 1 in 1 million individuals7, with most cases occurring sporadically5,8–10. Although the etiology of AHC is usually unknown, a missense mutation in ATP1A2 was reported in one case of atypical familial alternating hemiplegia9,10; however, the clinical presentation of some of the family members with the ATP1A2 mutation was more consistent with familial hemiplegic migraine9, which is caused by mutations in ATP1A2 (refs. 11,12). To our knowledge, no cases of sporadic AHC have yet been attributed to ATP1A2 mutations.
In this study, we used next-generation sequencing (NGS) of the exomes or whole genomes of ten individuals with AHC and, where possible, their unaffected parents. We identified and confirmed rare (minor allele frequency (MAF) <0.01%) mutations in ATP1A3 (encod-ing the sodium-potassium (Na+/K+) ATPase 3 subunit, also known as ATP1A3) in eight of ten probands, and we showed that the mutations
had occurred de novo in the seven patients for whom parental DNA was available. The ATP1A3 mutations included five distinct non-synonymous mutations, one of which was found in four patients with AHC (Supplementary Table 1). To further investigate ATP1A3 in the two unexplained AHC probands, we looked for structural variants in the whole-genome sequence data, and we Sanger sequenced the protein- encoding exons to find single-nucleotide and insertion-deletion vari-ants missed by whole-genome sequencing; neither analysis identified candidate causal ATP1A3 mutations in these individuals. Given the rarity of functional de novo mutations, the occurrence of seven de novo mutations in the same gene in seven patients with AHC provides strong genetic evidence that mutations in ATP1A3 cause sporadic AHC.
We next Sanger sequenced the protein-encoding exons of ATP1A3 in an additional cohort of 95 individuals with AHC. In these 95 patients, we identified rare ATP1A3 mutations in 74 patients (Table 1); these mutations were found to have arisen de novo in the 59 patients with sporadic AHC for whom parental DNA was available. Including samples sequenced with NGS, we identified a total of 19 different ATP1A3 mutations in 82 of 105 (78%) patients studied. The majority of these mutations were located in or near regions encoding trans-membrane domains (Fig. 1). Seven of the mutations were identified in multiple cases of AHC; in particular, those giving rise to amino acid substitutions D801N and E815K were identified in 36 (34%) patients and 19 (18%) patients, respectively (Table 1). One of the 95 patients evaluated had a familial form of alternating hemiplegia, first described in 1992 (ref. 8). In this individual, we identified a rare ATP1A3 mutation (giving rise to I274N) affecting the encoded cytoplasmic domain that cosegregates with the AHC phenotype (Fig. 2; see Supplementary Note for phenotypic details of affected family members).
Thirteen of the 18 ATP1A3 mutations seen in sporadic AHC cases were confirmed to be de novo (Supplementary Table 1). We also observed rare ATP1A3 variants in 15 patients with sporadic AHC for whom parental DNA was not available, and it is possible that some of these mutations are inherited benign polymorphisms. This is unlikely given the rarity of functional variants in ATP1A3, but we can con-servatively estimate the number of patients with pathogenic ATP1A3 mutations by considering as pathogenic only those mutations observed as de novo in at least one patient. Under this criterion, 11 of the 15 patients have a pathogenic ATP1A3 mutation. We can, therefore, con-clude that at least 74% of patients with sporadic, typically presenting AHC studied here harbor disease-causing mutations in ATP1A3.
De novo mutations in ATP1A3 cause alternating hemiplegia of childhood
A full list of authors and affiliations appears at the end of the paper.
Received 16 April; accepted 28 June; published online 29 July 2012; doi:10.1038/ng.2358
1030 VOLUME 44 | NUMBER 9 | SEPTEMBER 2012 NATURE GENETICS
L E T T E R S
Alternating hemiplegia of childhood (AHC) is a rare, severe neurodevelopmental syndrome characterized by recurrent hemiplegic episodes and distinct neurological manifestations. AHC is usually a sporadic disorder and has unknown etiology. We used exome sequencing of seven patients with AHC and their unaffected parents to identify de novo nonsynonymous mutations in ATP1A3 in all seven individuals. In a subsequent sequence analysis of ATP1A3 in 98 other patients with AHC, we found that ATP1A3 mutations were likely to be responsible for at least 74% of the cases; we also identified one inherited mutation in a case of familial AHC. Notably, most AHC cases are caused by one of seven recurrent ATP1A3 mutations, one of which was observed in 36 patients. Unlike ATP1A3 mutations that cause rapid-onset dystonia-parkinsonism, AHC-causing mutations in this gene caused consistent reductions in ATPase activity without affecting the level of protein expression. This work identifies de novo ATP1A3 mutations as the primary cause of AHC and offers insight into disease pathophysiology by expanding the spectrum of phenotypes associated with mutations in ATP1A3.
AHC was first characterized as a distinct syndrome in 1971, with a report that described eight patients with episodes of intermittent hemiplegia on alternating sides of the body, developmental delay, dystonia and choreoathetosis beginning in infancy1. Since then, spe-cific diagnostic criteria have more clearly defined the classic parox-ysmal and interictal neurological manifestations associated with this disease2–6. AHC affects approximately 1 in 1 million individuals7, with most cases occurring sporadically5,8–10. Although the etiology of AHC is usually unknown, a missense mutation in ATP1A2 was reported in one case of atypical familial alternating hemiplegia9,10; however, the clinical presentation of some of the family members with the ATP1A2 mutation was more consistent with familial hemiplegic migraine9, which is caused by mutations in ATP1A2 (refs. 11,12). To our knowledge, no cases of sporadic AHC have yet been attributed to ATP1A2 mutations.
In this study, we used next-generation sequencing (NGS) of the exomes or whole genomes of ten individuals with AHC and, where possible, their unaffected parents. We identified and confirmed rare (minor allele frequency (MAF) <0.01%) mutations in ATP1A3 (encod-ing the sodium-potassium (Na+/K+) ATPase 3 subunit, also known as ATP1A3) in eight of ten probands, and we showed that the mutations
had occurred de novo in the seven patients for whom parental DNA was available. The ATP1A3 mutations included five distinct non-synonymous mutations, one of which was found in four patients with AHC (Supplementary Table 1). To further investigate ATP1A3 in the two unexplained AHC probands, we looked for structural variants in the whole-genome sequence data, and we Sanger sequenced the protein- encoding exons to find single-nucleotide and insertion-deletion vari-ants missed by whole-genome sequencing; neither analysis identified candidate causal ATP1A3 mutations in these individuals. Given the rarity of functional de novo mutations, the occurrence of seven de novo mutations in the same gene in seven patients with AHC provides strong genetic evidence that mutations in ATP1A3 cause sporadic AHC.
We next Sanger sequenced the protein-encoding exons of ATP1A3 in an additional cohort of 95 individuals with AHC. In these 95 patients, we identified rare ATP1A3 mutations in 74 patients (Table 1); these mutations were found to have arisen de novo in the 59 patients with sporadic AHC for whom parental DNA was available. Including samples sequenced with NGS, we identified a total of 19 different ATP1A3 mutations in 82 of 105 (78%) patients studied. The majority of these mutations were located in or near regions encoding trans-membrane domains (Fig. 1). Seven of the mutations were identified in multiple cases of AHC; in particular, those giving rise to amino acid substitutions D801N and E815K were identified in 36 (34%) patients and 19 (18%) patients, respectively (Table 1). One of the 95 patients evaluated had a familial form of alternating hemiplegia, first described in 1992 (ref. 8). In this individual, we identified a rare ATP1A3 mutation (giving rise to I274N) affecting the encoded cytoplasmic domain that cosegregates with the AHC phenotype (Fig. 2; see Supplementary Note for phenotypic details of affected family members).
Thirteen of the 18 ATP1A3 mutations seen in sporadic AHC cases were confirmed to be de novo (Supplementary Table 1). We also observed rare ATP1A3 variants in 15 patients with sporadic AHC for whom parental DNA was not available, and it is possible that some of these mutations are inherited benign polymorphisms. This is unlikely given the rarity of functional variants in ATP1A3, but we can con-servatively estimate the number of patients with pathogenic ATP1A3 mutations by considering as pathogenic only those mutations observed as de novo in at least one patient. Under this criterion, 11 of the 15 patients have a pathogenic ATP1A3 mutation. We can, therefore, con-clude that at least 74% of patients with sporadic, typically presenting AHC studied here harbor disease-causing mutations in ATP1A3.
De novo mutations in ATP1A3 cause alternating hemiplegia of childhood
A full list of authors and affiliations appears at the end of the paper.
Received 16 April; accepted 28 June; published online 29 July 2012; doi:10.1038/ng.2358

Alterna;ng Hemiplegia of Childhood (AHC)
• Intermieent episodes of hemiplegia on alterna;ng sides of the body, abnormal eye movement
• Symptoms before 18 months of age.
• Developmental delay
• 1 in 1,000,000

Exome sequencing of seven AHC trios
• De novo muta;ons in ATP1A3
– Sodium potassium ATPase subunit
• All nonsynonymous muta;ons
• Muta;ons in ATP1A3 previously known to cause rapid-‐onset dystonia-‐parkinsonism (DYT12)
– Loss/decreased protein levels • AHC-‐causing muta;ons
– Consistent reduc;on of ATPase ac;vity – Normal protein expression level

Two AHC causing muta;ons affect amino acids also affected by DYT12-‐causing muta;ons

ORIGINAL ARTICLE
Clinical application of exome sequencing inundiagnosed genetic conditionsAnna C Need,1 Vandana Shashi,2 Yuki Hitomi,1 Kelly Schoch,2
Kevin V Shianna,1 Marie T McDonald,2 Miriam H Meisler,3 David B Goldstein1,4
ABSTRACTBackground There is considerable interest in the use ofnext-generation sequencing to help diagnose unidentifiedgenetic conditions, but it is difficult to predict thesuccess rate in a clinical setting that includes patientswith a broad range of phenotypic presentations.Methods The authors present a pilot programme ofwhole-exome sequencing on 12 patients withunexplained and apparent genetic conditions, along withtheir unaffected parents. Unlike many previous studies,the authors did not seek patients with similarphenotypes, but rather enrolled any undiagnosedproband with an apparent genetic condition whenpredetermined criteria were met.Results This undertaking resulted in a likely geneticdiagnosis in 6 of the 12 probands, including theidentification of apparently causal mutations in fourgenes known to cause Mendelian disease(TCF4, EFTUD2, SCN2A and SMAD4) and one generelated to known Mendelian disease genes (NGLY1).Of particular interest is that at the time of this study,EFTUD2 was not yet known as a Mendelian diseasegene but was nominated as a likely cause based on theobservation of de novo mutations in two unrelatedprobands. In a seventh case with multiple disparateclinical features, the authors were able to identifyhomozygous mutations in EFEMP1 as a likely cause formacular degeneration (though likely not for otherfeatures).Conclusions This study provides evidence thatnext-generation sequencing can have high success ratesin a clinical setting, but also highlights key challenges.It further suggests that the presentation of knownMendelian conditions may be considerably broader thancurrently recognised.
INTRODUCTIONWhole-genome and whole-exome sequencing haveproven remarkably successful in identifying thecauses of Mendelian diseases. These analyses havegenerally depended on the availability of more thanone unrelated affected individual and/or linkageevidence in at least one family. However, next-generation sequencing (NGS) has also succeeded inidentifying causes of genetic conditions even whenthey are seen in only a single patient.1e3
Consequently, there is growing interest in theintroduction of NGS into the clinic to aid in thediagnosis of conditions for which no genetic causecan be found with targeted testing or chromosomalarrays. However, in a clinical setting, patients with
undiagnosed genetic conditions tend to presentwith a wide range of clinical features, and it is oftennecessary to consider each patient’s genome indi-vidually, rather than looking for common disruptedgenes in multiple cases with a similar phenotype. Itis not clear what success rate NGS approaches willachieve in providing genetic diagnoses in this morechallenging setting. In this study, we have evalu-ated the use of NGS to provide genetic diagnosesusing 12 parent-child trios in which the child hadcongenital anomalies and/or intellectual disabilitiesdue to unexplained conditions presumed to begenetic. Importantly, the patients were chosen tobe representative of a clinical sample of undiag-nosed genetic conditions, in that they were notselected for genetic tractability or phenotypichomogeneity.
METHODSExome sequencing was performed on each patientand both parents using the Illumina HiSeq2000platform and the Agilent SureSelect Human AllExon 50Mb Kit. Detailed methods for laboratorywork can be found in the online supplementarymethods.
Study populationThe research protocol was approved by the DukeInstitutional Review Board, and all human partici-pants or their guardians gave written informedconsent. Twelve families (child, mother and father)were recruited through the genetics clinic at DukeUniversity Medical Center based on whether theirchild met two or more of the following criteria:(1) unexplained intellectual disability and/or devel-opmental delay; (2) one major congenital anomaly;(3) 2e3 minor congenital anomalies; and (4) facialdysmorphisms. In addition, the families wererequired to meet the following eligibility require-ments: (1) both biological parents available fortesting; (2) previous clinically indicated genetictesting, including a chromosomal microarray(Affymetrix 6.0, http://www.affymetrix.com),had been normal; and (3) no evidence of effects ofteratogens, birth asphyxia or non-accidental trauma.Subjects were not eligible if the mother was preg-nant at the time of enrolment. Finally, results wereonly returned to patients and/or patient familiesfollowing confirmation of detected variants ina CLIA certified laboratory. Controls were subjectsenrolled in Center for Human Genome Variationstudies through Duke Institutional Review Boardapproved protocols (n!830).
< Additional materials arepublished online only. To viewthese files please visit thejournal online (http://jmg.bmj.com/content/49/6.toc).1Center for Human GenomeVariation and Department ofMedicine, Duke UniversitySchool of Medicine, Durham,North Carolina, USA2Department of Pediatrics,Section of Medical Genetics,Duke University, Durham, NorthCarolina, USA3Department of HumanGenetics, University ofMichigan, Ann Arbor, Michigan,USA4Department of MolecularGenetics and Microbiology,Duke University School ofMedicine, Durham, NorthCarolina, USA
Correspondence toDr David Goldstein, Center forHuman Genome Variation, DukeUniversity School of Medicine,Box 91009, Durham, NC 27708,USA; [email protected]
AN and VS contributed equallyto this work.
Received 10 February 2012Revised 14 March 2012Accepted 2 April 2012Published Online First11 May 2012
This paper is freely availableonline under the BMJ Journalsunlocked scheme, see http://jmg.bmj.com/site/about/unlocked.xhtml
J Med Genet 2012;49:353e361. doi:10.1136/jmedgenet-2012-100819 353
New disease loci
group.bmj.com on March 22, 2013 - Published by jmg.bmj.comDownloaded from

Documentation of a functional effect on splicing will berequired to confirm pathogenicity of this variant. The EFTUD2variant in trio 7 is a frameshift INDEL causing the prematuretermination of the protein at the end of exon 9 (residue 222/962). This study thus identified EFTUD2 as a leading candidatefor explaining the conditions in these children. Subsequent tothis work, Lines and colleagues13 very recently reported ananalysis of 12 patients with Mandibulofacial Dysostosis withmicrocephaly, and found that all have de novo mutations inEFTUD2. On examination, both these patients show similaritiesto the children in this report, and the patient from trio sevenfits the condition very closely.
Trio 2: NGLY1Screening for compound heterozygous variants revealed thatpatient 2 had inherited a frameshift variant in the last exon ofNGLY1 from his mother, and a nonsense mutation in exon 8from his father. NGLY1 encodes N-glycanase 1, which is involvedin the degradation of misfolded glycoproteins. N-glycanase 1 hasnot been associated with a specific disorder, but the phenotypeof this child is consistent with a congenital disorder of glyco-
sylation (table 1), and transferring isoelectric focusing andN-glycan analyses have been normal on repeated testing. Tofurther explore the effect of these variants, we compared NGLY1protein expression in leucocytes extracted from blood from thepatient, his parents and three controls. Both parents showedreduced expression compared with controls, and the patient hadbarely discernible levels of NGLY1 (figure 1). Dysfunction ofNGLY1 would be expected to result in abnormal accumulationof misfolded glycoproteins due to impaired degradation. In ourpatient, liver biopsy showed an amorphous unidentified substancethroughout the cytoplasm, suggestive of stored material in theliver cells. It is to be noted that extensive testing for lysosomalstorage had also been pursued in this child, and all the results hadbeen normal. Further cellular assays are underway to bettercharacterise this mutation.
Trio 3: SMAD4A de novo non-synonymous mutation was identified in SMAD4in trio 3, resulting in an isoleucine to valine substitution atamino acid position 500 (I500V). This variant has recently beenreported to be the causal variant in approximately half of all
Table 1 Demographic and clinical features of sequenced patients
Trio Sex Age Race SymptomsGenetic tests performed clinicallybefore enrolment in study
1 M 8 Indian Developmental delay, possible autism, microcephaly,dysmorphic features, spine abnormalities, sensorineuralhearing loss
Chromosome microarray (paternally inherited 15q13.3dup), Fragile X
2 M 3 European-American Developmental delay, multifocal epilepsy, involuntarymovements, abnormal liver function, absent tears
Chromosomes, chromosome microarray, Niemann-Picktype C, hepatocerebral mDNA depletion panel (POLG1,DGUOK, MPV17), ataxia with oculomotor apraxia type 2(SETX), Allgrove Syndrome, ataxia telangectasia (ATM),Rett (MECP2), alphad1 antitrypsin (AAT), congenitaldisorder of glycosylation (transferrin isoelectric focusingand N-glycan analysis), metabolic tests (Tay Sachs,Sandhoff, mannosidosis, mucolipidosis II, Krabbe,metachromatic leukodystrophy, adrenoleukodystrophy,GAMT, plasma amino acids, plasma acylcarnitine, urineorganic acids).
3 M 3 European-American Developmental delay, autism, coarctation of the aorta,tethered cord, congenital nystagmus and strabismus
Chromosome microarray (maternally inherited 15q26.3deletion), Smith-Lemli-Opitz, Aarskog
4 F adult European-American multiple congenital abnormalities and macular degeneration Chromosome microarray (2 stretches of loss ofheterozygosity on chromosome 2), Fragile X(premutation carrier)
5 F 12 European-American Severe intellectual disability, autism, bilateral hyperpronatedfeet, facial dysmorphisms
Chromosomes, chromosome microarray, Rett, Angelmanmethlyation, Fragile X, Cohen Syndrome
6 M 18 European-American Intellectual disability, epilepsy, panhypopituitarism,hypertension, bifid great toe, vertebral segmentationanomalies and sagittal cleft of the vertebra, hypoplastic 13thrib, and delayed bone age
Chromosomes, chromosome microarray, Borgeson-Forssman-Lehman syndrome
7 M 2 European-American Microcephaly, facial asymmetry, acyanotic Tetralogy ofFallot; history of small muscular ventricular septal defect;right aortic arch with mirror image branching; malformedright ear with hearing loss, bifid uvula, cleft soft palate
Chromosome microarray, CHARGE (CHD7)
8 M 16 European-American Severe intellectual disability, dysmorphic features evident,bicuspid aortic valve, bilateral coronal craniosynostoses,quadriplegic cerebral palsy, bilateral inguinal hernias, G-tubeplacement and obstructive sleep apnoea
Chromosome microarray, craniosynostosis syndromes(FGFR2), non-syndromic craniosynostosis (FGFR3)Saethre-Chotzen syndrome (TWIST)
9 F 4 Algerian Developmental delay, bilateral congenital cataracts andstrabismus, ventricular and atrial septal defects, a unilateralclubfoot, and unilateral choanal atresia
Chromosome microarray (Long stretch of loss ofheterozygosity on chromosome 17), CHARGE (CHD7),PAX6, 7-dehydrocholesterol and cholesterol levels
10 M 11 European-American Attention deficit hyperactivity disorder, language delays,coarse facial features, bilateral mandibular cysts, low muscletone
Chromosome microarray, Costello (H-RAS), Gorlin(PTCH), Comprehensive Noonan sequencing array(BRAF, HRAS, KRAS MAPT2K1, MAPTK2, PTPN11,RAF1, SHOC2 and SOS1), MPS panel
11 M 9 European-American Severe intellectual disability, developmental delay, seizures/infantile spasms, hypotonia and minor dysmorphisms
Chromosomes, chromosome microarray (familial Xp11.4duplication), acylcarnitine profile, plasma amino acids,urine organic acids, creatine/guanidinoacetate analysis inurine and blood
12 F 4 European-American Speech delay, borderline microcephaly, failure to thrive,dysplastic nails, ventricular septal defect and hip dysplasia
Chromosomes, chromosome microarray
J Med Genet 2012;49:353e361. doi:10.1136/jmedgenet-2012-100819 355
New disease loci
group.bmj.com on March 22, 2013 - Published by jmg.bmj.comDownloaded from
Trio/Pa;ent 5 • De novo muta;on in TCF4 – Transcrip;on Factor 4 – Pie Hopkins syndrome (PHS) – Haploinsufficiency of TCF4 – Atypical PHS?
Nav1.2, within the sequence WNIFDF that is highly conservedin mammalian and invertebrate voltage-gated sodium channels(figure 3). In the bacterial sodium channel, the correspondingsequence is WSLFDF, and the recently determined crystalstructure indicates that this aspartate residue (D80) can form ahydrogen bond with a positive (arginine) gating charge intransmembrane segment S4.32 Conversion of this aspartate tothe non-polar glycine residue would prevent this interaction,potentially impairing regulation of channel opening. Theseconsiderations strongly indicate the pathogenicity of thismutation.
Further support for the role of this mutation comes from theclosely related sodium channel SCN1A. SCN1A and SCN2Aarose by gene duplication during vertebrate evolution, and retain87% amino acid sequence identity (1747/2005) with mostdivergence in non-transmembrane domains. A de novo mutationin the corresponding residue of SCN1A, D1608Y, was found ina patient with severe myoclonic epilepsy of infancy, whichlike our patient is characterised by infantile seizures andintellectual disability.33 Three additional missense mutationsin transmembrane segment D4S3 of SCN1A have been identi-fied in patients with epilepsy (http://www.molgen.ua.ac.be/SCN1AMutations/), further demonstrating the pathogenicpotential of this transmembrane segment of the protein.
SCN2A is not routinely included in DNA testing for epilepsybecause mutations of SCN1A are much more common.
Interesting findingsIn the remaining six cases, no variants judged as likely to becausal for most or all features were identified, although in twocases one or more interesting candidate variants were found.
Trio 4Exome sequencing revealed several regions of homozygosityincluding several homozygous variants in EFEMP1 (two intronicSNVs and a 39UTR INDEL), a gene in which heterozygousmutations are known to cause early onset maculopathies.34e36
Subsequent to this finding, it was judged that the patient’sretinal phenotype of bilateral and symmetric distribution ofdrusenoid deposits most likely reflects dysregulation of thefunction of EFEMP1 (E Heon, personal communication). A real-time reverse transcriptase PCR assay indicated that the levelof EFEMP1 expression in blood is too low to assess any effectsof the variant on controls. This patient also carries a de novonon-synonymous coding SNV with a PolyPhen score of 0.999 inthe gene ATP6AP2. This gene encodes the (pro)renin receptorand has multiple functions in the eye, heart, kidney, centralnervous system and other tissues.37e39 This patient highlightsthe fact that some subjects who would undergo NGS may verywell have more than one underlying diagnosis, and that allcausative variants may not be detected.
Trio 6A de novo variant was observed in the 59 consensus splice siteof exon 9 of the HNRNPU gene, which encodes HnRNP U.This gene is in the critical target region for the seizure pheno-type of patients with microdeletion of 1q43e44,40 41 a highlyvariable syndrome characterised by speech delay, intellectualdisability and seizures. In mice, HnRNP U has been shown tobe linked to preaxial polydactyly caused by abnormal expressionof SHH during limb development,42 and normal HnRNPU expression is essential for embryonic development.43 We havebeen unable to demonstrate a functional effect of the de novovariant in blood, but it remains possible that it affects expressionof a particular isoform, perhaps in a tissue-specific mannerduring development. In addition, this patient has a de novomutation in SMAD1, a gene that partners with SMAD4 in bonemorphogenetic protein signal transduction.44 Given the associ-ation of de novo SMAD4 mutations with a spontaneous clini-cally heterogeneous developmental disorder (see above), it is
Figure 2 Expression of TCF4 variant and wild-type (WT) protein in COS-7 cells. The variant protein (V) is only seen in the presence of proteasomeinhibitors. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Figure 3 The SCN2A mutation, D1598G, is located in transmembranesegment 3 of the sodium channel protein domain 4. This residue isconserved in vertebrate, invertebrate DM (Drosophila) and bacterial(NaChBac) sodium channels. The D to Y mutation at the correspondingposition of SCN1A was identified in a patient with severe myoclonicepilepsy (SME) of childhood, an early onset epileptic encephalopathywith features similar to the affected individual in trio 11 h, human; f, fish.
Figure 1 Expression of endogenous NGLY1 protein in peripheral bloodmononuclear cells from patient, parents and three unrelated healthycontrols. The protein expression level in the patient is less than both parentsand healthy controls. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
J Med Genet 2012;49:353e361. doi:10.1136/jmedgenet-2012-100819 357
New disease loci
group.bmj.com on March 22, 2013 - Published by jmg.bmj.comDownloaded from

Important lessons
• Variability in clinical presenta;on of gene;c disorders
• Ability to interpret unique variants is a work in progress
• Func;onal work is necessary
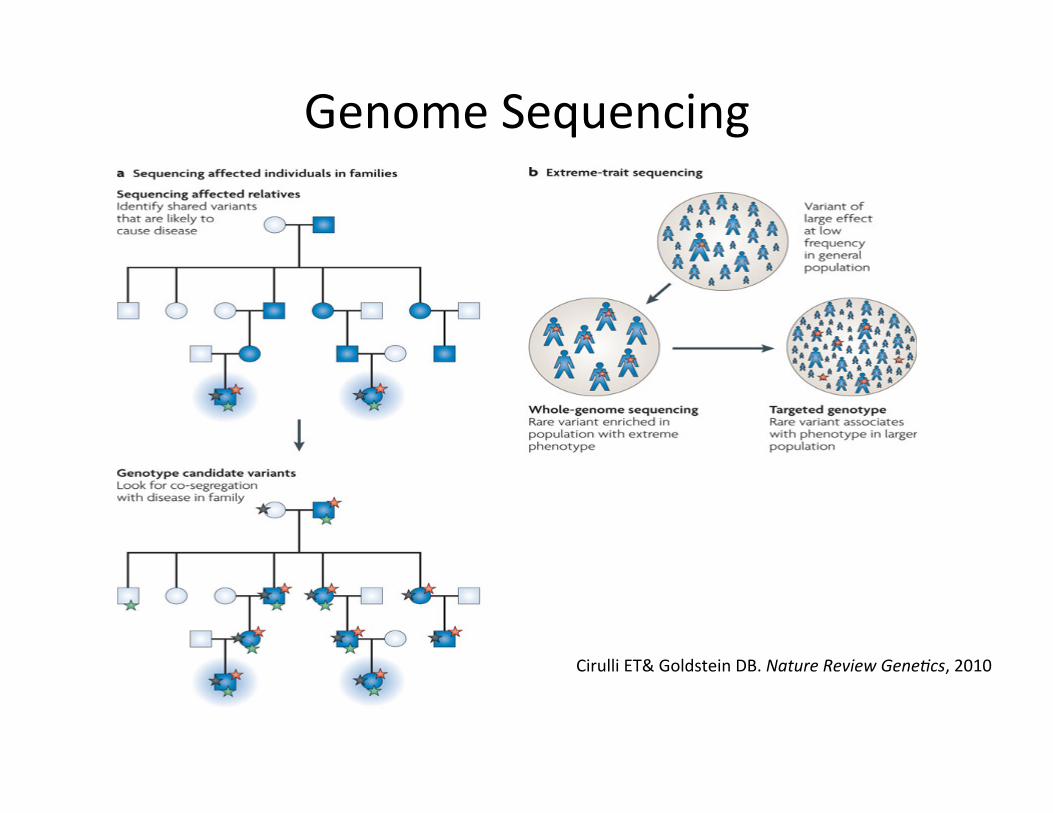
Genome Sequencing
Cirulli ET& Goldstein DB. Nature Review Gene.cs, 2010

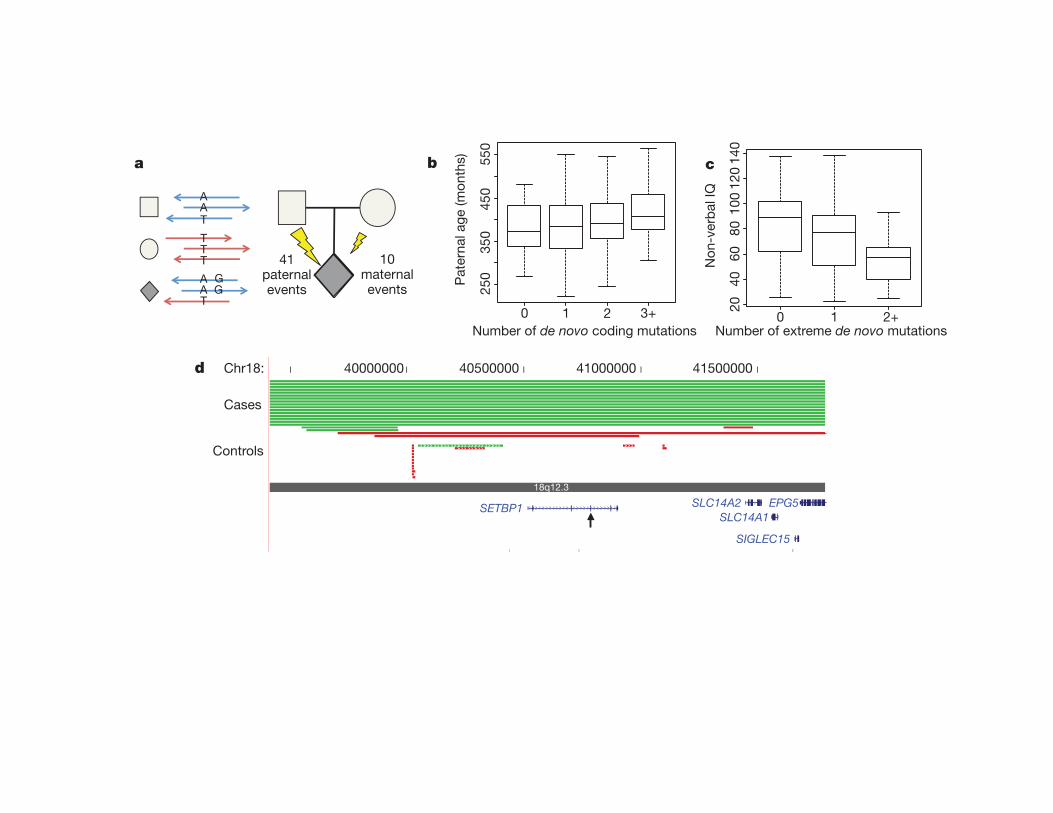
Mendelian or ASD loci (n 5 57), or de novo CNVs that intersect genes(n 5 5) (Fig. 1c and Supplementary Discussion). NVIQ, but not VIQ,decreased significantly (P , 0.01) with increased number of events.Covariant analysis of the samples with CNV data showed that thisfinding was strengthened, but not exclusively driven, by the presenceof either de novo or rare CNVs (Supplementary Fig. 5).
Among the de novo events, we identified 62 top ASD risk con-tributing mutations based on the deleteriousness of the mutations,functional evidence, or previous studies (Table 1). Probands with thesemutations spanned the range of IQ scores, with only a modest non-significant trend towards individual’s co-morbid with intellectualdisability (Supplementary Figs 1 and 6). We observed recurrent,protein-disruptive mutations in two genes: NTNG1 (netrin G1) andCHD8 (chromodomain helicase DNA binding protein 8). Given theirlocus-specific mutation rates, the probability of identifying two inde-pendent mutations in our sample set is low (uncorrected, NTNG1:P , 1.2 3 1026; CHD8: P , 6.9 3 1025) (Supplementary Fig. 7,Supplementary Table 8 and Methods). NTNG1 is a strong biologicalcandidate given its role in laminar organization of dendrites and axonalguidance12 and was also reported as being disrupted by a de novo trans-location in a child with Rett’s syndrome, without MECP2 mutation13.Both de novo mutations identified here are missense (p.Tyr23Cys andp.Thr135Ile) at highly conserved positions predicted to disrupt proteinfunction, although there is evidence of mosaicism for the former muta-tion (Supplementary Table 3).
CHD8 has not previously been associated with ASD and codes foran ATP-dependent chromatin-remodelling factor that has a signifi-cant role in the regulation of both b-catenin and p53 signalling14,15. Wealso identified de novo missense variants in CHD3 as well as CHD7(CHARGE syndrome, OMIM 214800), a known binding partner ofCHD8 (ref. 16). ASD has been found in as many as two-thirds ofchildren with CHARGE, indicating that CHD7 may contribute to anASD syndromic subtype17.
We identified 30 protein-altering de novo events intersecting withMendelian disease loci (Supplementary Table 3) as well as inheritedhemizygous mutations of clinical significance (Supplementary Table 9).
The de novo mutations included truncating events in syndromicintellectual disability genes (MBD5 (mental retardation, autosomaldominant 1, OMIM 156200), RPS6KA3 (Coffin–Lowry syndrome,OMIM 303600) and DYRK1A (the Down’s syndrome candidategene, OMIM 600855)), and missense variants in loci associated withsyndromic ASD, including CHD7, PTEN (macrocephaly/autismsyndrome, OMIM 605309) and TSC2 (tuberous sclerosis complex,OMIM 613254). Notably, DYRK1A is a highly conserved genemapping to the Down’s syndrome critical region (SupplementaryFig. 8). The proband here (13890) is severely cognitively impairedand microcephalic, consistent with previous studies of DYRK1Ahaploinsufficiency in both patients and mouse models18.
Twenty-one of the non-synonymous de novo mutations map toCNV regions recurrently identified in children with developmentaldelay and ASD (Supplementary Table 10), such as MBD5 (2q23.1 dele-tion syndrome), SYNRG (17q12 deletion syndrome) and POLRMT(19p13.3 deletion)19. There is also considerable overlap with genes dis-rupted by single de novo CNVs in children with ASD (for example,NLGN1 and ARID1B; Supplementary Table 11). Given the priorprobability that these loci underlie genomic disorders, the disruptivede novo SNVs and small indels may be pinpointing the possible majoreffect locus for ASD-related features. For example, we identified a com-plex de novo mutation resulting in truncation of SETBP1 (SET bindingprotein 1), one of five genes in the critical region for del(18)(q12.2q21.1)syndrome (Fig. 1d), which is characterized by hypotonia, expressivelanguage delay, short stature and behavioural problems20. Recurrentde novo missense mutations at SETBP1 were recently reported to becausative for a distinct phenotype, Schinzel–Giedion syndrome,probably through a gain-of-function mechanism21, indicating diversephenotypic outcomes at this locus depending on mutation mechanism.
Several of the mutated genes encode proteins that directly interact,suggesting a common biological pathway. From our full list of genescarrying truncating or severe missense mutations (126 events from all209 families), we generated a protein–protein interaction (PPI) net-work based on a database of physical interactions (SupplementaryTable 12)22. We found 39% (49 of 126) of the genes mapped to a highly
0 1 2+
2040
6080
100
120
140
ba
d Chr18: 40000000 40500000 41000000 41500000
18q12.3
SETBP1 SLC14A2SLC14A1
SIGLEC15
EPG5
Cases
Controls
c
Pat
erna
l age
(mon
ths)
Number of de novo coding mutations 0 1 2 3+
250
350
450
550
Number of extreme de novo mutations
Non
-ver
bal I
Q
41paternalevents
10maternalevents
AAT
TTT
T
AA
GG
Figure 1 | De novo mutation events in autism spectrum disorder.a, Haplotype phasing using informative markers shows a strong parent-of-origin bias with 41 of 51 de novo events occurring on the paternally inheritedhaplotype. Arrows represent sequence reads from paternal (blue) or maternal(red) haplotypes. b, c, Box and whisker plots for 189 SSC probands. b, Thepaternal estimated age at conception versus the number of observed de novopoint mutations (0, n 5 53; 1, n 5 65; 2, n 5 44; 31, n 5 27). c, Decreased non-verbal IQ is significantly associated with an increasing number of extreme
mutation events (0, n 5 138; 1, n 5 41; 21, n 5 10), both with and withoutCNVs (Supplementary Discussion). d, Browser images showing CNVsidentified in the del(18)(q12.2q21.1) syndrome region. The truncating pointmutation in SETBP1 occurs within the critical region, identifying the likelycausative locus. Each red (deletion) and green (duplication) line represents anidentified CNV in cases (solid lines) versus controls (dashed lines), witharrowheads showing point mutation.
LETTER RESEARCH
1 0 M A Y 2 0 1 2 | V O L 4 8 5 | N A T U R E | 2 4 7
Macmillan Publishers Limited. All rights reserved©2012
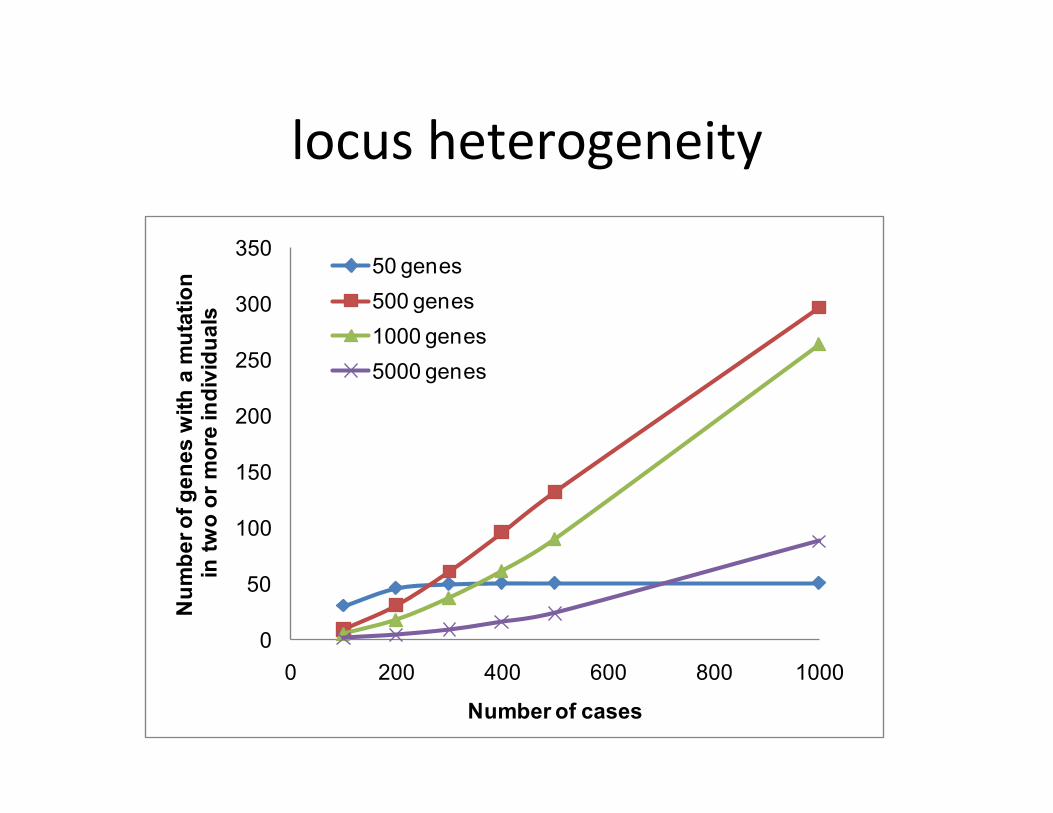
locus heterogeneity
0
50
100
150
200
250
300
350
0 200 400 600 800 1000
Num
ber o
f gen
es w
ith a
mut
atio
nin
two
or m
ore
indi
vidu
als
Number of cases
50 genes500 genes1000 genes5000 genes

Clinical Sequencing in the NICU

-‐Hard to diagnose -‐Time sensi;ve! -‐Symptom and sign assisted genome analysis
(SSAGA)
D IAGNOST I CS
Rapid Whole-Genome Sequencing for Genetic DiseaseDiagnosis in Neonatal Intensive Care UnitsCarol Jean Saunders,1,2,3,4,5* Neil Andrew Miller,1,2,4* Sarah Elizabeth Soden,1,2,4*Darrell Lee Dinwiddie,1,2,3,4,5* Aaron Noll,1 Noor Abu Alnadi,4 Nevene Andraws,3
Melanie LeAnn Patterson,1,3 Lisa Ann Krivohlavek,1,3 Joel Fellis,6 Sean Humphray,6 Peter Saffrey,6
Zoya Kingsbury,6 Jacqueline Claire Weir,6 Jason Betley,6 Russell James Grocock,6
Elliott Harrison Margulies,6 Emily Gwendolyn Farrow,1 Michael Artman,2,4 Nicole Pauline Safina,1,4
Joshua Erin Petrikin,2,3 Kevin Peter Hall,6 Stephen Francis Kingsmore1,2,3,4,5†
Monogenic diseases are frequent causes of neonatal morbidity and mortality, and disease presentations are oftenundifferentiated at birth. More than 3500 monogenic diseases have been characterized, but clinical testing is avail-able for only some of them and many feature clinical and genetic heterogeneity. Hence, an immense unmet needexists for improved molecular diagnosis in infants. Because disease progression is extremely rapid, albeit hetero-geneous, in newborns, molecular diagnoses must occur quickly to be relevant for clinical decision-making. We de-scribe 50-hour differential diagnosis of genetic disorders by whole-genome sequencing (WGS) that featuresautomated bioinformatic analysis and is intended to be a prototype for use in neonatal intensive care units. Ret-rospective 50-hour WGS identified known molecular diagnoses in two children. Prospective WGS disclosed potentialmolecular diagnosis of a severe GJB2-related skin disease in one neonate; BRAT1-related lethal neonatal rigidity andmultifocal seizure syndrome in another infant; identified BCL9L as a novel, recessive visceral heterotaxy gene (HTX6) in apedigree; and ruled out known candidate genes in one infant. Sequencing of parents or affected siblings expedited theidentification of disease genes in prospective cases. Thus, rapid WGS can potentially broaden and foreshorten differ-ential diagnosis, resulting in fewer empirical treatments and faster progression to genetic and prognostic counseling.
INTRODUCTIONGenomic medicine is a new, structured approach to disease diagnosisand management that prominently features genome sequence infor-mation (1). Whole-genome sequencing (WGS) by next-generationsequencing (NGS) technologies has the potential for simultaneous,comprehensive, differential diagnostic testing of likely monogenic ill-nesses, which accelerates molecular diagnoses and minimizes the du-ration of empirical treatment and time to genetic counseling (2–7).Indeed, in some cases, WGS or exome sequencing provides moleculardiagnoses that could not have been ascertained by conventional single-gene sequencing approaches because of pleiotropic clinical presenta-tion or the lack of an appropriate molecular test (7–9).
Neonatal intensive care units (NICUs) are especially suitable forearly adoption of diagnostic WGS because many of the 3528 mono-genic diseases of known cause are present during the first 28 days oflife (10). In the United States, more than 20% of infant deaths arecaused by congenital malformations, deformations, and chromosomalabnormalities that cause genetic diseases (11–13). Although this pro-portion has remained unchanged for the past 20 years, the preciseprevalence of monogenic diseases in NICUs is poorly understood be-cause ascertainment rates are low. Serial gene sequencing is too slowto be clinically useful for NICU diagnosis. Newborn screens, while
rapid, identify only a few genetic disorders for which inexpensive testsand cost-effective treatments exist (14, 15). Further complicating diag-nosis is the fact that the full clinical phenotype may not be manifestin newborn infants (neonates), and genetic heterogeneity can be im-mense. Thus, acutely ill neonates with genetic diseases are often dis-charged or deceased before a diagnosis is made. As a result, NICUtreatment of genetic diseases is usually empirical, may lack efficacy,may be inappropriate, or may cause adverse effects.
NICUs are also suitable for early adoption of genomic medicinebecause extraordinary interventional efforts are customary and inno-vation is encouraged. Indeed, NICU treatment is among the mostcost-effective of high-cost health care, and the long-term outcomes ofmost NICU subpopulations are excellent (16–18). In genetic diseasesfor which treatments exist, rapid diagnosis is critical for timely deliveryof interventions that lessen morbidity and mortality (14–17, 19, 20).For neonatal genetic diseases without effective therapeutic interven-tions, of which there aremany (21), timely diagnosis avoids futile inten-sive care and is critical for research to develop management guidelinesthat optimize outcomes (22). In addition to influencing treatment, neo-natal diagnosis of genetic disorders and genetic counseling can spareparents diagnostic odysseys that instill inappropriate hope or perpetuateneedless guilt.
Two recent studies exemplify the diagnostic and therapeutic usesof NGS in the context of childhood genetic diseases. WGS of fraternaltwins concordant for 3,4-dihydroxyphenylalanine (dopa)–responsivedystonia revealed known mutations in the sepiapterin reductase(SPR) gene (3). In contrast to other forms of dystonia, treatment with5-hydroxytryptamine and serotonin reuptake inhibitors is beneficial inpatients with SPR defects. Application of this therapy in appropriatecases resulted in clinical improvement. Likewise, extensive testing
1Center for Pediatric Genomic Medicine, Children’s Mercy Hospital, Kansas City, MO 64108,USA. 2Department of Pediatrics, Children’s Mercy Hospital, Kansas City, MO 64108, USA.3Department of Pathology, Children’s Mercy Hospital, Kansas City, MO 64108, USA. 4Schoolof Medicine, University of Missouri-Kansas City, Kansas City, MO 64108, USA. 5University ofKansas Medical Center, Kansas City, KS 66160, USA. 6Illumina Inc., Chesterford Research Park,Little Chesterford, CB10 1XL Essex, UK.*These authors contributed equally to this work.†To whom correspondence should be addressed. E-mail: [email protected]
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 3 October 2012 Vol 4 Issue 154 154ra135 1
on
Febr
uary
25,
201
3st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m

50 hrs from pa;ent consent to diagnosis Nevertheless, there was good concordance between the genotypes ofvariants detected by rapid WGS (using the HiSeq 2500 and CASAVA)and targeted sequencing (using exome enrichment, the HiSeq 2000, andGATK/GSNAP)—99.5% (UDT002), 99.9% (UDT173), and 99.7%(CMH064) (Table 2)—further indicating that rapid WGS is highly con-cordant with an established genotyping method (33). In subsequentstudies, the rapid WGS technique used CASAVA for alignment andvariant detection.
Genomic variants were characterized with respect to functionalconsequence and zygosity with a new software pipeline [RapidUnderstanding of Nucleotide variant Effect Software (RUNES), fig.S2] that analyzed each sample in 2.5 hours. Samples contained a meanof 4.00 ± 0.20 million (SD) genomic variants, of which a mean of 1.87 ±0.09 million (SD) were associated with protein-encoding genes (Table 1).Less than 1% of these variants (mean, 10,848 ± 523 SD) were also of afunctional class that could potentially be disease causative (Table 1)(25–27). Of these, ~14% (mean, 1530 ± 518 SD) had an allele frequen-cy that was sufficiently low to be a candidate for being causative in anuncommon disease (<1% allele frequency in 836 individuals sequencedat CMH) (42). Last, of these, ~71% (mean, 1083 ± 240 SD) were also ofa functional class that was likely to be disease causative [AmericanCollege of Medical Genetics (ACMG) categories 1 to 3] (Table 1). Thisset of variants was evaluated for disease causality in each patient, withpriority given to variants within the candidate genes that had beennominated by an individual patient presentation.
Retrospective analysesPatient UDT002 was a male who presented at 13 months of age withhypotonia, developmental regression. Brain magnetic resonance imag-ing (MRI) showed diffuse white matter changes suggesting leukodys-trophy. Three hundred fifty-two disease genes were nominated byone of the three clinical terms hypotonia, developmental regression, orleukodystrophy; 150 disease genes were nominated by two terms; and9 disease genes were nominated by all three terms (table S3). Only16 known pathogenic variants had allele frequencies in dbSNP and theCMH cumulative database that were consistent with uncommon dis-ease mutations. Of these, only two variants mapped to the nine can-didate genes; the variants were both compound heterozygous (verifiedby parental testing) substitution mutations in the gene that encodes thea subunit of the lysosomal enzyme hexosaminidase A [HEXA Chr15:72,641,417T>C (gene symbol, chromosome number, chromosomecoordinate, reference nucleotide > variant nucleotide), c.986+3A>G(transcript coordinate, reference nucleotide, variant nucleotide), andChr15:72,640,388C>T, c.1073+1G>A]. The c.986+3A>G alters a 5!exon–flanking nucleotide and is a knownmutation that causes Tay-Sachsdisease (TSD), a debilitating lysosomal storage disorder [OnlineMendelian Inheritance in Man (OMIM) number 272800]. The varianthad not previously been observed in our database of 651 individuals ordbSNP, which is relevant because mutation databases are contaminatedwith some common polymorphisms, and these can be distinguished fromtrue mutations on the basis of allele frequency (33). The c.1073+1G>Avariant is a known TSD mutation that affects an exonic splice donor site(dbSNP rs76173977). The variant has been observed only once beforein our database of 414 samples, which is consistent with an allele frequen-cy of a causative mutation in an orphan genetic disease. Thus, the knowndiagnosis of TSD was confirmed in patient UDT002 by rapid WGS.
Patient UDT173 was a male who presented at 5 months of age withdevelopmental regression, hypotonia, and seizures. Brain MRI showed
Fig. 1. STAT-Seq. Summary of the steps and timing of STAT-Seq, result-ing in an interval of 50 hours between consent and delivery of a pre-liminary, verbal diagnosis. t, hours.
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 3 October 2012 Vol 4 Issue 154 154ra135 3
on
Febr
uary
25,
201
3st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m

Non-‐Invasive Prenatal Tes;ng (NIPT)

Noninvasive prenatal diagnosis of fetal chromosomalaneuploidy by massively parallel genomic sequencingof DNA in maternal plasmaRossa W. K. Chiua,b, K. C. Allen Chana,b, Yuan Gaoc,d, Virginia Y. M. Laua,b, Wenli Zhenga,b, Tak Y. Leunge,Chris H. F. Foof, Bin Xiec, Nancy B. Y. Tsuia,b, Fiona M. F. Luna,b, Benny C. Y. Zeef, Tze K. Laue, Charles R. Cantorg,1,and Y. M. Dennis Loa,b,1
aCentre for Research into Circulating Fetal Nucleic Acids, Li Ka Shing Institute of Health Sciences, Departments of bChemical Pathology and eObstetrics andGynaecology, and fCentre for Clinical Trials, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong SAR, China; cCenter for the Study ofBiological Complexity and dDepartment of Computer Science, Virginia Commonwealth University, Richmond, VA 23284; and gSequenom, Inc., San Diego, CA92121
Contributed by Charles R. Cantor, October 22, 2008 (sent for review September 29, 2008)
Chromosomal aneuploidy is the major reason why couples opt forprenatal diagnosis. Current methods for definitive diagnosis rely oninvasive procedures, such as chorionic villus sampling and amniocen-tesis, and are associated with a risk of fetal miscarriage. Fetal DNA hasbeen found in maternal plasma but exists as a minor fraction amonga high background of maternal DNA. Hence, quantitative perturba-tions caused by an aneuploid chromosome in the fetal genome to theoverall representation of sequences from that chromosome in ma-ternal plasma would be small. Even with highly precise single mole-cule counting methods such as digital PCR, a large number of DNAmolecules and hence maternal plasma volume would need to beanalyzed to achieve the necessary analytical precision. Here wereasoned that instead of using approaches that target specific geneloci, the use of a locus-independent method would greatly increasethe number of target molecules from the aneuploid chromosome thatcould be analyzed within the same fixed volume of plasma. Hence, weused massively parallel genomic sequencing to quantify maternalplasma DNA sequences for the noninvasive prenatal detection of fetaltrisomy 21. Twenty-eight first and second trimester maternal plasmasamples were tested. All 14 trisomy 21 fetuses and 14 euploid fetuseswere correctly identified. Massively parallel plasma DNA sequencingrepresents a new approach that is potentially applicable to all preg-nancies for the noninvasive prenatal diagnosis of fetal chromosomalaneuploidies.
Down syndrome ! Solexa sequencing ! trisomy 21
The testing of fetal chromosomal aneuploidies is the predomi-nant reason why many pregnant women opt for prenatal
diagnosis. Conventional methods for definitive prenatal diagnosisof these disorders involve the invasive sampling of fetal materialsthrough amniocentesis and chorionic villus sampling, with a risk forthe fetus (1). Many workers tried to develop noninvasive ap-proaches. Methods based on ultrasound scanning and maternalserum biochemical markers (2) have proved to be useful screeningtests. However, they detect epiphenomena instead of the corepathology of chromosomal abnormalities. They have limitationssuch as a narrow gestational window of applicability and the needto combine multiple markers, even over different time points, toarrive at a clinically useful sensitivity and specificity profile.
For the direct detection of fetal chromosomal and geneticabnormalities from maternal blood, early work focused on therelatively difficult isolation of the rare fetal nucleated cells frommaternal blood (3–5). The discovery of cell-free fetal nucleic acidsin maternal plasma in 1997 opened up new possibilities (6, 7).However, the fact that fetal DNA represents only a minor fractionof total DNA in maternal plasma (8), with the majority beingcontributed by the pregnant woman herself, has offered consider-able challenge. Recently, a number of approaches have beendeveloped. One strategy targets a fetal-specific subset of nucleic
acids in maternal plasma, e.g., placental mRNA (9–11) and DNAmolecules bearing a placental-specific DNA methylation signature(12–14). The fetal chromosomal dosage is then assessed by allelicratio analysis of SNPs within the targeted molecules. These strat-egies are called the RNA–SNP allelic ratio approach (11) and theepigenetic allelic ratio approach (14). These allelic ratio-basedmethods can be used only for fetuses heterozygous for the analyzedSNPs. Thus, multiple markers are needed to enhance the popula-tion coverage of the methods.
To develop a polymorphism-independent method for the detec-tion of fetal chromosomal aneuploidies from maternal plasma, ourgroup has recently outlined the principles for the measurement ofrelative chromosome dosage (RCD) using digital PCR (15). DigitalRCD aims to measure the total (maternal plus fetal) amount of aspecific locus on a potentially aneuploid chromosome in maternalplasma, e.g., chromosome 21 (chr21) in trisomy 21 (T21), andcompares it to that on a reference chromosome. Hence, fetal T21is diagnosed by detecting the small increment in the total amountof the chr21 gene locus contributed by the trisomic chr21 in the fetusas compared with a gene locus on a reference chromosome. Theproportional increment in chr21 sequences is expectedly smallbecause fetal DNA contributes only a minor fraction of DNA inmaternal plasma (8). To reliably detect the small increase, a largeabsolute number of chr21 and reference chromosome sequences ofthe loci targeted by the digital PCR assays need to be analyzed andquantified with high precision. The number of molecules requiredfor RCD increases by four times, for every twofold reduction in thefractional concentration of circulating fetal DNA. Thus, for cases inwhich the fractional concentration for circulating fetal DNA is low,e.g., during early gestation, relatively large volumes of maternalplasma may be needed. One way is to perform multiplex analysis ofmultiple genetic loci. However, the optimization of highly multi-plexed digital PCR might be challenging. If fluorescence reportersare used, one would also quickly run out of reporters for distin-guishing the products from the various loci.
Author contributions: R.W.K.C., K.C.A.C., and Y.M.D.L. designed research; R.W.K.C.,K.C.A.C., Y.G., V.Y.M.L., W.Z., B.X., N.B.Y.T., and F.M.F.L. performed research; T.Y.L. andT.K.L. collected clinical samples; R.W.K.C., K.C.A.C., V.Y.M.L., C.H.F.F., B.C.Y.Z., C.R.C., andY.M.D.L. analyzed data; and R.W.K.C. and Y.M.D.L. wrote the paper.
Conflict of interest statement: R.W.K.C., K.C.A.C., N.B.Y.T., F.M.F.L., B.C.Y.Z., C.R.C., andY.M.D.L. have filed patent applications on the detection of fetal nucleic acids in maternalplasma for noninvasive prenatal diagnosis. Part of this patent portfolio has been licensedto Sequenom. C.R.C. is Chief Scientific Officer of and holds equities in Sequenom. Y.M.D.Lis a consultant to and holds equities in Sequenom.
Freely available online through the PNAS open access option.1To whom correspondence may be addressed. E-mail: [email protected] or [email protected].
This article contains supporting information online at www.pnas.org/cgi/content/full/0810641105/DCSupplemental.
© 2008 by The National Academy of Sciences of the USA
20458–20463 ! PNAS ! December 23, 2008 ! vol. 105 ! no. 51 www.pnas.org"cgi"doi"10.1073"pnas.0810641105

To overcome the above limitations, we propose to use a methodindependent of any particular gene locus to quantify the amount ofchr21 sequences in maternal plasma. When a locus-independentmethod is used, potentially every DNA fragment originating fromthe aneuploid chromosome could contribute to the measurement ofthe amount of that chromosome. Therefore, for any fixed volumeof maternal plasma, the number of quantifiable sequences would bemuch greater than the number of DNA molecules that could serveas templates for detection by gene locus-specific assays. Hence,precise detection of the over- or underrepresentation of sequencesfrom an aneuploid chromosome could be more readily achieved.We previously (15) proposed that the recently available massivelyparallel genomic sequencing (MPGS) platforms (16, 17) might beadaptable as an approach to quantify DNA sequences for thenoninvasive prenatal diagnosis of fetal chromosomal aneuploidy. Inthis study, we demonstrate the use of the ‘‘Solexa’’ sequencingtechnique (Illumina) (18) for this purpose.
ResultsProcedural Framework. The procedural framework of using MPGSfor noninvasive fetal chromosomal aneuploidy detection in mater-nal plasma is schematically illustrated in Fig. 1. In this study, we usedthe sequencing-by-synthesis Solexa method (18). As the maternalplasma DNA (maternal and fetal) molecules were already frag-mented in nature (19), no further fragmentation was required. Oneend of the clonally expanded copies of each plasma DNA fragmentwas sequenced and processed by standard postsequencing bioin-formatics alignment analysis for the Illumina Genome Analyzer,which uses the Efficient Large-Scale Alignment of NucleotideDatabases (ELAND) software. The purpose of the alignment wasto simply determine the chromosomal origin of the sequencedplasma DNA fragments and details about their gene-specific loca-tion were not required. The number of sequence reads originatingfrom any particular chromosome was then counted and tabulatedfor each human chromosome. In this study, we counted onlysequences that could be mapped to just one location in therepeat-masked reference human genome with no mismatch, i.e.,deemed as a ‘‘unique’’ sequence in the human genome. We termedthese sequences as U0–1–0–0 on the basis of values in a number offields in the data output files of the ELAND sequence alignmentsoftware (Illumina) (see Materials and Methods).
We then determined the percentage contribution of uniquesequences mapped to each chromosome by dividing the U0–1–0–0count of a specific chromosome by the total number of U0–1–0–0sequence reads generated in the sequencing run for the testedsample to generate a value termed % chrN, when the chromosomeof interest is chrN. To determine if a tested maternal plasma samplebelonged to a T21 pregnancy, we calculated the z-score of % chr21of the tested sample. The z-score refers to the number of standarddeviations from the mean of a reference data set. Hence, for a T21fetus, a high z-score for % chr21 was expected when compared withthe mean and standard deviation of % chr21 values obtained frommaternal plasma of euploid pregnancies.
For this procedure to be effective for noninvasive prenatal fetalchromosomal aneuploidy detection, a number of assumptions needto be met. First, MPGS needs to be sensitive enough to capture andgenerate sequence reads for the small fraction of fetal DNA inmaternal plasma alongside the background maternal DNA. Sec-ond, the pool of plasma DNA fragments captured for sequencingneeds to be a representative sample of the total DNA pool withsimilar interchromosomal distribution to that in the original ma-ternal plasma. Third, there should be no major bias in the ability tosequence DNA fragments originating from each chromosome.When these assumptions hold, then the % chrN values should bereflective of the genomic representation of the maternal and fetalDNA fragments in maternal plasma. Furthermore, if both thematernal and the fetal genomes are evenly represented in maternalplasma, the proportional contribution of plasma DNA sequences
per chromosome should in turn bear correlation with the relativesize of each chromosome in the human genome. If the % chrNvalues could be determined precisely enough by sequencing andcounting a large enough pool of plasma DNA sequences, wehypothesize that we would be able to discriminate perturbations inthe quantitative representation of sequences mapped to the aneu-ploid chromosomes in a maternal plasma sample from a pregnancyinvolving a fetus with the said aneuploidy. We set out to test eachof these assumptions.
Detection of Fetal DNA in Maternal Plasma. If MPGS could sequencefetal DNA in maternal plasma, one should be able to detect chrYDNA from plasma of women carrying male fetuses. Plasma samplesobtained from four pregnant women carrying euploid fetuses (threemales and one female) were processed using the beta ChIP-Seqprotocol from Illumina, which included amplification of the adap-tor-ligated DNA fragments both before and after (i.e., two roundsof amplification) a gel electrophoresis-based size fractionation stepas described in supporting information (SI) Text.
Fig. 1. Schematic illustration of the procedural framework for using mas-sively parallel genomic sequencing for the noninvasive prenatal detection offetal chromosomal aneuploidy. Fetal DNA (thick red fragments) circulates inmaternal plasma as a minor population among a high background of mater-nal DNA (black fragments). A sample containing a representative profile ofDNA molecules in maternal plasma is obtained. In this study, one end of eachplasma DNA molecule was sequenced for 36 bp using the Solexa sequencing-by-synthesis approach. The chromosomal origin of each 36-bp sequence wasidentified through mapping to the human reference genome by bioinformat-ics analysis. The number of unique (U0–1–0–0, see text) sequences mapped toeach chromosome was counted and then expressed as a percentage of allunique sequences generated for the sample, termed % chrN for chromosomeN. Z-scores for each chromosome and each test sample were calculated usingthe formula shown. The z-score of a potentially aneuploid chromosome isexpected to be higher for pregnancies with an aneuploid fetus (cases E–Hshown in green) than for those with a euploid fetus (cases A–D shown in blue).
Chiu et al. PNAS ! December 23, 2008 ! vol. 105 ! no. 51 ! 20459
MED
ICA
LSC
IEN
CES

GENOMICS
Noninvasive Whole-Genome Sequencing of aHuman FetusJacob O. Kitzman,1* Matthew W. Snyder,1 Mario Ventura,1,2 Alexandra P. Lewis,1 Ruolan Qiu,1
LaVone E. Simmons,3 Hilary S. Gammill,3,4 Craig E. Rubens,5,6 Donna A. Santillan,7
Jeffrey C. Murray,8 Holly K. Tabor,5,9 Michael J. Bamshad,1,5 Evan E. Eichler,1,10 Jay Shendure1*
Analysis of cell-free fetal DNA in maternal plasma holds promise for the development of noninvasive prenatalgenetic diagnostics. Previous studies have been restricted to detection of fetal trisomies, to specific paternallyinherited mutations, or to genotyping common polymorphisms using material obtained invasively, for example,through chorionic villus sampling. Here, we combine genome sequencing of two parents, genome-wide maternalhaplotyping, and deep sequencing of maternal plasma DNA to noninvasively determine the genome sequenceof a human fetus at 18.5 weeks of gestation. Inheritance was predicted at 2.8 ! 106 parental heterozygous siteswith 98.1% accuracy. Furthermore, 39 of 44 de novo point mutations in the fetal genome were detected, albeitwith limited specificity. Subsampling these data and analyzing a second family trio by the same approach in-dicate that parental haplotype blocks of ~300 kilo–base pairs combined with shallow sequencing of maternalplasma DNA is sufficient to substantially determine the inherited complement of a fetal genome. However,ultradeep sequencing of maternal plasma DNA is necessary for the practical detection of fetal de novo mutationsgenome-wide. Although technical and analytical challenges remain, we anticipate that noninvasive analysis ofinherited variation and de novo mutations in fetal genomes will facilitate prenatal diagnosis of both recessiveand dominant Mendelian disorders.
INTRODUCTIONOn average, ~13% of cell-free DNA isolated frommaternal plasma dur-ing pregnancy is fetal in origin (1). The concentration of cell-free fetalDNA in the maternal circulation varies between individuals, increasesduring gestation, and is rapidly cleared postpartum (2, 3). Despite thisvariability, cell-free fetal DNA has been successfully targeted for non-invasive prenatal diagnosis including for development of targeted assaysfor single-gene disorders (4). More recently, several groups have dem-onstrated that shotgun,massively parallel sequencing of cell-freeDNAfrom maternal plasma is a robust approach for noninvasively diag-nosing fetal aneuploidies such as trisomy 21 (5, 6).
Ideally, it should be possible to noninvasively predict the whole-genome sequence of a fetus to high accuracy and completeness, poten-tially enabling the comprehensive prenatal diagnosis of Mendeliandisorders and obviating the need for invasive prenatal diagnostic proce-dures such as chorionic villus samplingwith their attendant risks. How-ever, several key technical obstacles must be overcome for this goal tobe achieved using cell-freeDNA frommaternal plasma. First, the sparserepresentationof fetal-derived sequences poses the challenge of detectinglow-frequency alleles inherited from the paternal genomeaswell as those
arising from de novomutations in the fetal genome. Second,maternalDNApredominates in themother’s plasma,making it difficult to assessmaternally inherited variation at individual sites in the fetal genome.
Recently, Lo et al. showed that fetal-derived DNA is distributedsufficiently evenly in maternal plasma to support the inference of fetalgenotypes, and furthermore, they demonstrated how knowledge ofparental haplotypes could be leveraged to this end (7). However, theirstudy was limited in several ways. First, the proposedmethod dependedon the availability of parental haplotypes, but at the time of their work,no technologies existed to measure these experimentally on a genome-wide scale. Therefore, an invasive procedure, chorionic villus sampling,was used to obtain placental material for fetal genotyping. Second, pa-rental genotypes and fetal genotypes obtained invasively were used toinfer parental haplotypes. These haplotypes were then used in combi-nation with the sequencing of DNA from maternal plasma to predictthe fetal genotypes. Although necessitated by the lack of genome-widehaplotyping methods, the circularity of these inferences makes it diffi-cult to assess how well the method would perform in practice. Third,their analysis was restricted to several hundred thousand parentally het-erozygous sites of common single-nucleotide polymorphisms (SNPs)represented on a commercial genotyping array. These common SNPsare only a small fraction of the severalmillion heterozygous sites presentin each parental genome and include few of the rare variants that pre-dominantly underlie Mendelian disorders (8). Fourth, Lo et al. did notascertain de novo mutations in the fetal genome. Because de novo mu-tations underlie a substantial fraction of dominant genetic disorders,their detection is critical for comprehensive prenatal genetic diagnostics.Therefore, although the Lo et al. study demonstrated the first successfulconstruction of a genetic map of a fetus, it required an invasive proce-dure and did not attempt to determine the whole-genome sequenceof the fetus. We and others recently demonstrated methods for exper-imentally determining haplotypes for both rare and common variation
1Department of Genome Sciences, University of Washington, Seattle, WA 98195, USA.2Department of Biology, University of Bari, Bari 70126, Italy. 3Department of Obstetricsand Gynecology, University of Washington, Seattle, WA 98195, USA. 4Division of ClinicalResearch, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA. 5De-partment of Pediatrics, University of Washington School of Medicine, Seattle, WA 98195,USA. 6Global Alliance to Prevent Prematurity and Stillbirth, an initiative of SeattleChildren’s, Seattle, WA 98101, USA. 7Department of Obstetrics and Gynecology, Univer-sity of Iowa Hospitals and Clinics, Iowa City, IA 52242, USA. 8Department of Pediatrics,University of Iowa, Iowa City, IA 52242, USA. 9Treuman Katz Center for PediatricBioethics, Seattle Children’s Research Institute, Seattle, WA 98101, USA. 10HowardHughes Medical Institute, Seattle, WA 98195, USA.*To whom correspondence should be addressed. E-mail: [email protected] (J.S.);[email protected] (J.O.K.)
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 6 June 2012 Vol 4 Issue 137 137ra76 1
on
Mar
ch 6
, 201
3st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m

assuming 13% fetal content). To assess these, we performed a site-by-site log-odds test; this amounted to taking the observationof one ormorereads matching the paternal-specific allele at a given site as evidence ofits transmission and, conversely, the lack of suchobservations as evidenceof nontransmission (Fig. 1C). In contrast tomaternal-only heterozygoussites, this simple site-by-site model was sufficient to correctly predictinheritance at 1.1 ! 106 paternal-only heterozygous sites with 96.8%accuracy (Table 2).We anticipate that accuracy could likely be improvedby deeper sequence coverage of the maternal plasma DNA (fig. S2) or,alternatively, by taking a haplotype-based approach if high–molecularweight genomic DNA from the father is available.
We next considered transmission at sites heterozygous in both par-ents. We predicted maternal transmission at such shared sites phasedusing neighboring maternal-only heterozygous sites in the same hap-lotype block. This yielded predictions at 576,242 of 631,721 (91.2%) ofshared heterozygous sites with an estimated accuracy of 98.7% (Table 2).Although we did not predict paternal transmission at these sites, we an-ticipate that analogous to the case ofmaternal transmission, this could bedone with high accuracy given paternal haplotypes.We note that sharedheterozygous sites primarily correspond to common alleles (fig. S3),which are less likely to contribute toMendelian disorders in nonconsan-guineous populations.
0 5 10 15
A
D
WGS
WGS(validation)
WGS +haplotypes
B
Plasma
13%87%
Dilution pool whole-genome phasing
A
G
C
A
G
C
Father
Father
Using individual sites Using maternal haplotypes
Mother
Mother
GG
CC
AA
CC
Haplotype A transmitted
Haplotype B transmitted
Plasma read fraction of maternal-specific alleles
Plasma paternal-specific reads/site
ACMSD
90/93(96.8%)
3/93(3.2%)
T
CMaternal plasma reads
chr2:135,596,281
C
Father
T C T A CT C T A C
Mother
T C A CYp.Leu10Pro
Offspring
T
C
paternal-specificShared
Allele transmitted
0.3 0.4 0.5 0.6 0.3 0.4 0.5 0.6
Fig. 1. Experimental approach. (A) Sequenced individuals in a family trio.Maternal plasma DNA sequences were ~13% fetal-derived on the basis ofread depth at chromosomeY and alleles specific to each parent.WGS,whole-genome shotgun. (B) Inheritance ofmaternally heterozygous alleles inferredusing long haplotype blocks. Among plasma DNA sequences, maternal-specific alleles are more abundant when transmitted (expected, 50% versus43.5%), but there is substantial overlap between the distributions of allelefrequencies when considering sites in isolation (left histogram: yellow,shared allele transmitted; green, maternal-specific allele transmitted).Taking average allele balances across haplotype blocks (right histogram)provides much greater separation, permitting more accurate inference ofmaternally transmitted alleles. (C) Histogram of fractional read depth
among plasma data at paternal-specific heterozygous sites. In the over-whelming majority of cases when the allele specific to the father was notdetected, the opposite allele had been transmitted (96.8%, n = 561,552).(D) De novo missensemutation in the gene ACMSD detected in 3 of 93 ma-ternal plasma reads and later validated by PCR and resequencing. The mu-tation, which is not observed in dbSNP nor among coding exons sequencedfrom >4000 individuals as part of the National Heart, Lung, and Blood In-stitute Exome Sequencing Project (http://evs.gs.washington.edu), createsa leucine-to-proline substitution at a site conserved across all alignedmammalian genomes (University of California, Santa Cruz, GenomeBrowser)in a gene implicated in Parkinson’s disease by genome-wide associationstudies (25).
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 6 June 2012 Vol 4 Issue 137 137ra76 3
on
Mar
ch 6
, 201
3st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m
on a genome-wide scale (9–12).Here,we set out to integrate the haplotype-resolved genome sequence of a mother, the shotgun genome sequenceof a father, and the deep sequencing of cell-free DNA inmaternal plasmato noninvasively predict the whole-genome sequence of a fetus.
RESULTS
We set out to predict the whole-genome sequence of a fetus in each oftwo mother-father-child trios (I1, a first trio at 18.5 weeks of gestation;G1, a second trio at 8.2 weeks of gestation). We focus here primarily onthe trio for which considerably more sequence data were generated(I1) (Table 1).
In brief, the haplotype-resolved genome sequence of the mother(I1-M) was determined by first performing shotgun sequencing of ma-ternal genomic DNA from blood to 32-fold coverage (coverage =median-fold coverage ofmapping reads to the reference genome after discardingduplicates). Next, by sequencing complex haploid subsets of maternalgenomic DNA while preserving long-range contiguity (9), we directlyphased 91.4% of 1.9 ! 106 heterozygous SNPs into long haplotypeblocks [N50 of 326 kilo–base pairs (kbp)]. The shotgun genome se-quence of the father (I1-P) was determined by sequencing of paternalgenomic DNA to 39-fold coverage, yielding 1.8 ! 106 heterozygousSNPs. However, paternal haplotypes could not be assessed because onlyrelatively low–molecular weight DNA obtained from saliva was availa-ble. ShotgunDNA sequencing libraries were also constructed from 5mlof maternal plasma (obtained at 18.5 weeks of gestation), and thiscomposite of maternal and fetal genomes was sequenced to 78-foldnonduplicate coverage. The fetus was male, and fetal content in theselibraries was estimated at 13% (Fig. 1A). To properly assess the accuracyof our methods for determining the fetal genome solely from samplesobtained noninvasively at 18.5 weeks of gestation, we also performedshotgun genome sequencing of the child (I1-C) to 40-fold coveragevia cord blood DNA obtained after birth.
Our analysis comprised four parts: (i) predicting the subset of“maternal-only” heterozygous variants (homozygous in the father)transmitted to the fetus; (ii) predicting the subset of “paternal-only”heterozygous variants (homozygous in the mother) transmitted tothe fetus; (iii) predicting transmission at sites heterozygous in both par-ents; (iv) predicting sites of de novomutation—that is, variants occurringonly in the genome of the fetus. Allelic imbalance in maternal plasma,manifesting across experimentally determined maternal haplotypeblocks, was used to predict their maternal transmission (Fig. 1B). Theobservation (or lack thereof) of paternal alleles in shotgun librariesderived frommaternal plasmawas used to predict paternal transmission
(Fig. 1C). Finally, a strict analysis of alleles rarely observed in maternalplasma, but never in maternal or paternal genomic DNA, enabled thegenome-wide identification of candidate de novo mutations (Fig. 1D).Fetal genotypes are trivially predicted at sites where the parents are bothhomozygous (for the same or different allele).
We first sought to predict transmission at maternal-only hetero-zygous sites. Given the fetal-derived proportion of ~13% in cell-freeDNA, the maternal-specific allele is expected in 50% of reads alignedto such a site if it is transmitted versus 43.5% if the allele shared withthe father is transmitted. However, even with 78-fold coverage of thematernal plasma “genome,” the variability of sampling is such that site-by-site prediction results in only 64.4% accuracy (Fig. 2). We thereforeexamined allelic imbalance across blocks of maternally heterozygoussites defined by haplotype-resolved genome sequencing of the mother(Fig. 1B). As anticipated given the haplotype assembly N50 of 326 kbp,the overwhelming majority of experimentally defined maternal hap-lotype blocks were wholly transmitted, with partial inheritance in asmall minority of blocks (0.6%, n = 72) corresponding to switch errorsfromhaplotype assembly and to sites of recombination.Wedeveloped ahidden Markov model (HMM) to identify likely switch sites and thusmore accurately infer the inherited alleles at maternally heterozygoussites (Figs. 3 and 4 and Supplementary Materials). With the use of thismodel, accuracy of the inferred inherited alleles at 1.1 ! 106 phased,maternal-only heterozygous sites increased from98.6 to 99.3% (Table 2).Remaining errors were concentrated among the shortest maternalhaplotype blocks (fig. S1), which provide less power to detect allelicimbalance in plasma DNA data compared with long blocks. Amongthe top 95% of sites ranked by haplotype block length, prediction accu-racy rose to 99.7%, suggesting that remaining inaccuracies can be miti-gated by improvements in haplotyping.
We performed simulations to characterize how the accuracy ofhaplotype-based fetal genotype inference depended on haplotypeblock length, maternal plasma sequencing depth, and the fraction offetal-derived DNA. To mimic the effect of less successful phasing, wesplit the maternal haplotype blocks into smaller fragments to create aseries of assemblies with decreasing contiguity.We then subsampled arange of sequencing depths from the pool of observed alleles in ma-ternal plasma and predicted the maternally contributed allele at eachsite as above (Fig. 5A). The results suggest that inference of the inheritedallele is robust either to decreasing sequencingdepth ofmaternal plasmaor to shorter haplotype blocks, but not both. For example, using only10% of the plasma sequence data (median depth = 8x) in conjunctionwith full-length haplotype blocks, we successfully predicted inheritanceat 94.9% of maternal-only heterozygous sites.We achieved nearly iden-tical accuracy (94.8%) at these sites when highly fragmented haplotypeblocks (N50 = 50 kbp) were used with the full set of plasma sequences.We next simulated decreased proportions of fetal DNA in the maternalplasma by spiking in additional depth of both maternal alleles at eachsite and subsampling from these pools, effectively diluting away the sig-nal of allelic imbalance used as a signature of inheritance (Fig. 5B).Again, we found the accuracy of the model to be robust to either lowerfetal DNA concentrations or shorter haplotype blocks, but not both.
We next sought to predict transmission at paternal-only hetero-zygous sites. At these sites, when the father transmits the shared allele,the paternal-specific allele should be entirely absent among the fetal-derived sequences. If instead the paternal-specific allele is transmitted,it will on average constitute half the fetal-derived reads within thematernal plasma genome (about five reads given 78-fold coverage,
Table 1. Summary of sequencing. Individuals sequenced, type ofstarting material, and final fold coverage of the reference genome afterdiscarding PCR or optical duplicate reads.
Individual Sample Depth of coverage
Mother (I1-M) Plasma (5 ml, gestationalage 18.5 weeks)
78
Whole blood (<1 ml) 32
Father (I1-P) Saliva 39
Offspring (I1-C) Cord blood at delivery 40
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 6 June 2012 Vol 4 Issue 137 137ra76 2
on
Mar
ch 6
, 201
3st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m
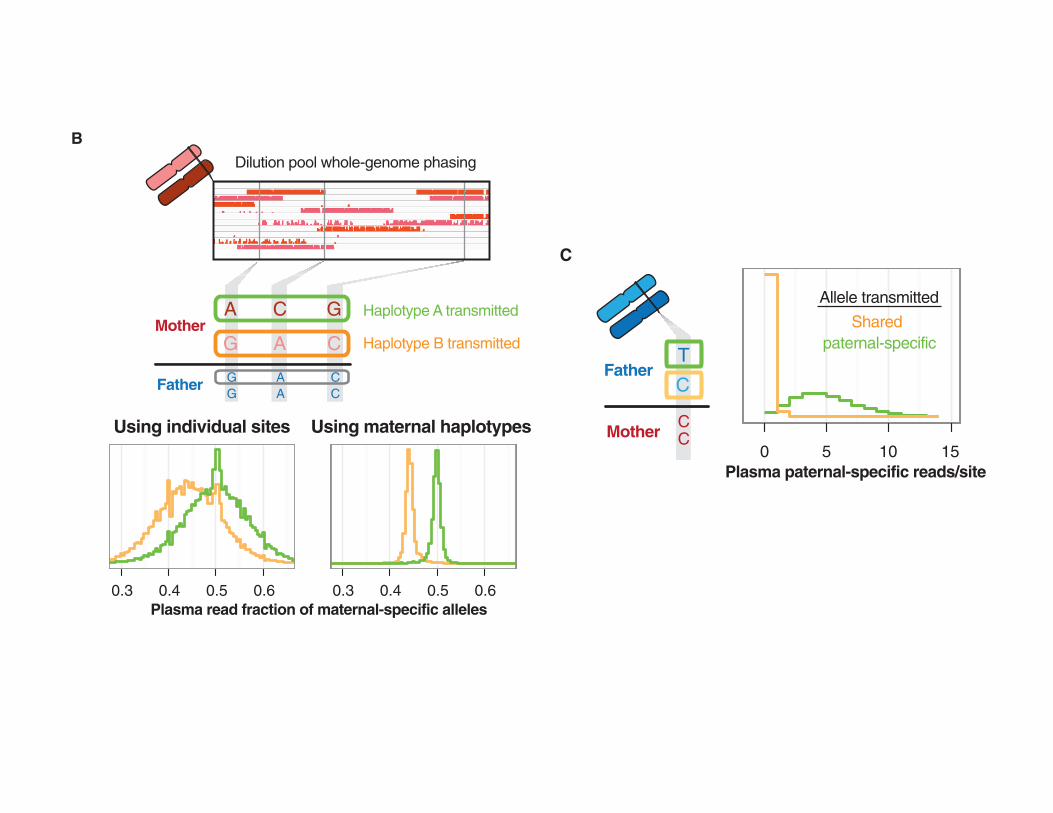
assuming 13% fetal content). To assess these, we performed a site-by-site log-odds test; this amounted to taking the observationof one ormorereads matching the paternal-specific allele at a given site as evidence ofits transmission and, conversely, the lack of suchobservations as evidenceof nontransmission (Fig. 1C). In contrast tomaternal-only heterozygoussites, this simple site-by-site model was sufficient to correctly predictinheritance at 1.1 ! 106 paternal-only heterozygous sites with 96.8%accuracy (Table 2).We anticipate that accuracy could likely be improvedby deeper sequence coverage of the maternal plasma DNA (fig. S2) or,alternatively, by taking a haplotype-based approach if high–molecularweight genomic DNA from the father is available.
We next considered transmission at sites heterozygous in both par-ents. We predicted maternal transmission at such shared sites phasedusing neighboring maternal-only heterozygous sites in the same hap-lotype block. This yielded predictions at 576,242 of 631,721 (91.2%) ofshared heterozygous sites with an estimated accuracy of 98.7% (Table 2).Although we did not predict paternal transmission at these sites, we an-ticipate that analogous to the case ofmaternal transmission, this could bedone with high accuracy given paternal haplotypes.We note that sharedheterozygous sites primarily correspond to common alleles (fig. S3),which are less likely to contribute toMendelian disorders in nonconsan-guineous populations.
0 5 10 15
A
D
WGS
WGS(validation)
WGS +haplotypes
B
Plasma
13%87%
Dilution pool whole-genome phasing
A
G
C
A
G
C
Father
Father
Using individual sites Using maternal haplotypes
Mother
Mother
GG
CC
AA
CC
Haplotype A transmitted
Haplotype B transmitted
Plasma read fraction of maternal-specific alleles
Plasma paternal-specific reads/site
ACMSD
90/93(96.8%)
3/93(3.2%)
T
CMaternal plasma reads
chr2:135,596,281
C
Father
T C T A CT C T A C
Mother
T C A CYp.Leu10Pro
Offspring
T
C
paternal-specificShared
Allele transmitted
0.3 0.4 0.5 0.6 0.3 0.4 0.5 0.6
Fig. 1. Experimental approach. (A) Sequenced individuals in a family trio.Maternal plasma DNA sequences were ~13% fetal-derived on the basis ofread depth at chromosomeY and alleles specific to each parent.WGS,whole-genome shotgun. (B) Inheritance ofmaternally heterozygous alleles inferredusing long haplotype blocks. Among plasma DNA sequences, maternal-specific alleles are more abundant when transmitted (expected, 50% versus43.5%), but there is substantial overlap between the distributions of allelefrequencies when considering sites in isolation (left histogram: yellow,shared allele transmitted; green, maternal-specific allele transmitted).Taking average allele balances across haplotype blocks (right histogram)provides much greater separation, permitting more accurate inference ofmaternally transmitted alleles. (C) Histogram of fractional read depth
among plasma data at paternal-specific heterozygous sites. In the over-whelming majority of cases when the allele specific to the father was notdetected, the opposite allele had been transmitted (96.8%, n = 561,552).(D) De novo missensemutation in the gene ACMSD detected in 3 of 93 ma-ternal plasma reads and later validated by PCR and resequencing. The mu-tation, which is not observed in dbSNP nor among coding exons sequencedfrom >4000 individuals as part of the National Heart, Lung, and Blood In-stitute Exome Sequencing Project (http://evs.gs.washington.edu), createsa leucine-to-proline substitution at a site conserved across all alignedmammalian genomes (University of California, Santa Cruz, GenomeBrowser)in a gene implicated in Parkinson’s disease by genome-wide associationstudies (25).
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 6 June 2012 Vol 4 Issue 137 137ra76 3
on
Mar
ch 6
, 201
3st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m
assuming 13% fetal content). To assess these, we performed a site-by-site log-odds test; this amounted to taking the observationof one ormorereads matching the paternal-specific allele at a given site as evidence ofits transmission and, conversely, the lack of suchobservations as evidenceof nontransmission (Fig. 1C). In contrast tomaternal-only heterozygoussites, this simple site-by-site model was sufficient to correctly predictinheritance at 1.1 ! 106 paternal-only heterozygous sites with 96.8%accuracy (Table 2).We anticipate that accuracy could likely be improvedby deeper sequence coverage of the maternal plasma DNA (fig. S2) or,alternatively, by taking a haplotype-based approach if high–molecularweight genomic DNA from the father is available.
We next considered transmission at sites heterozygous in both par-ents. We predicted maternal transmission at such shared sites phasedusing neighboring maternal-only heterozygous sites in the same hap-lotype block. This yielded predictions at 576,242 of 631,721 (91.2%) ofshared heterozygous sites with an estimated accuracy of 98.7% (Table 2).Although we did not predict paternal transmission at these sites, we an-ticipate that analogous to the case ofmaternal transmission, this could bedone with high accuracy given paternal haplotypes.We note that sharedheterozygous sites primarily correspond to common alleles (fig. S3),which are less likely to contribute toMendelian disorders in nonconsan-guineous populations.
0 5 10 15
A
D
WGS
WGS(validation)
WGS +haplotypes
B
Plasma
13%87%
Dilution pool whole-genome phasing
A
G
C
A
G
C
Father
Father
Using individual sites Using maternal haplotypes
Mother
Mother
GG
CC
AA
CC
Haplotype A transmitted
Haplotype B transmitted
Plasma read fraction of maternal-specific alleles
Plasma paternal-specific reads/site
ACMSD
90/93(96.8%)
3/93(3.2%)
T
CMaternal plasma reads
chr2:135,596,281
C
Father
T C T A CT C T A C
Mother
T C A CYp.Leu10Pro
Offspring
T
C
paternal-specificShared
Allele transmitted
0.3 0.4 0.5 0.6 0.3 0.4 0.5 0.6
Fig. 1. Experimental approach. (A) Sequenced individuals in a family trio.Maternal plasma DNA sequences were ~13% fetal-derived on the basis ofread depth at chromosomeY and alleles specific to each parent.WGS,whole-genome shotgun. (B) Inheritance ofmaternally heterozygous alleles inferredusing long haplotype blocks. Among plasma DNA sequences, maternal-specific alleles are more abundant when transmitted (expected, 50% versus43.5%), but there is substantial overlap between the distributions of allelefrequencies when considering sites in isolation (left histogram: yellow,shared allele transmitted; green, maternal-specific allele transmitted).Taking average allele balances across haplotype blocks (right histogram)provides much greater separation, permitting more accurate inference ofmaternally transmitted alleles. (C) Histogram of fractional read depth
among plasma data at paternal-specific heterozygous sites. In the over-whelming majority of cases when the allele specific to the father was notdetected, the opposite allele had been transmitted (96.8%, n = 561,552).(D) De novo missensemutation in the gene ACMSD detected in 3 of 93 ma-ternal plasma reads and later validated by PCR and resequencing. The mu-tation, which is not observed in dbSNP nor among coding exons sequencedfrom >4000 individuals as part of the National Heart, Lung, and Blood In-stitute Exome Sequencing Project (http://evs.gs.washington.edu), createsa leucine-to-proline substitution at a site conserved across all alignedmammalian genomes (University of California, Santa Cruz, GenomeBrowser)in a gene implicated in Parkinson’s disease by genome-wide associationstudies (25).
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 6 June 2012 Vol 4 Issue 137 137ra76 3
on
Mar
ch 6
, 201
3st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m

assuming 13% fetal content). To assess these, we performed a site-by-site log-odds test; this amounted to taking the observationof one ormorereads matching the paternal-specific allele at a given site as evidence ofits transmission and, conversely, the lack of suchobservations as evidenceof nontransmission (Fig. 1C). In contrast tomaternal-only heterozygoussites, this simple site-by-site model was sufficient to correctly predictinheritance at 1.1 ! 106 paternal-only heterozygous sites with 96.8%accuracy (Table 2).We anticipate that accuracy could likely be improvedby deeper sequence coverage of the maternal plasma DNA (fig. S2) or,alternatively, by taking a haplotype-based approach if high–molecularweight genomic DNA from the father is available.
We next considered transmission at sites heterozygous in both par-ents. We predicted maternal transmission at such shared sites phasedusing neighboring maternal-only heterozygous sites in the same hap-lotype block. This yielded predictions at 576,242 of 631,721 (91.2%) ofshared heterozygous sites with an estimated accuracy of 98.7% (Table 2).Although we did not predict paternal transmission at these sites, we an-ticipate that analogous to the case ofmaternal transmission, this could bedone with high accuracy given paternal haplotypes.We note that sharedheterozygous sites primarily correspond to common alleles (fig. S3),which are less likely to contribute toMendelian disorders in nonconsan-guineous populations.
0 5 10 15
A
D
WGS
WGS(validation)
WGS +haplotypes
B
Plasma
13%87%
Dilution pool whole-genome phasing
A
G
C
A
G
C
Father
Father
Using individual sites Using maternal haplotypes
Mother
Mother
GG
CC
AA
CC
Haplotype A transmitted
Haplotype B transmitted
Plasma read fraction of maternal-specific alleles
Plasma paternal-specific reads/site
ACMSD
90/93(96.8%)
3/93(3.2%)
T
CMaternal plasma reads
chr2:135,596,281
C
Father
T C T A CT C T A C
Mother
T C A CYp.Leu10Pro
Offspring
T
C
paternal-specificShared
Allele transmitted
0.3 0.4 0.5 0.6 0.3 0.4 0.5 0.6
Fig. 1. Experimental approach. (A) Sequenced individuals in a family trio.Maternal plasma DNA sequences were ~13% fetal-derived on the basis ofread depth at chromosomeY and alleles specific to each parent.WGS,whole-genome shotgun. (B) Inheritance ofmaternally heterozygous alleles inferredusing long haplotype blocks. Among plasma DNA sequences, maternal-specific alleles are more abundant when transmitted (expected, 50% versus43.5%), but there is substantial overlap between the distributions of allelefrequencies when considering sites in isolation (left histogram: yellow,shared allele transmitted; green, maternal-specific allele transmitted).Taking average allele balances across haplotype blocks (right histogram)provides much greater separation, permitting more accurate inference ofmaternally transmitted alleles. (C) Histogram of fractional read depth
among plasma data at paternal-specific heterozygous sites. In the over-whelming majority of cases when the allele specific to the father was notdetected, the opposite allele had been transmitted (96.8%, n = 561,552).(D) De novo missensemutation in the gene ACMSD detected in 3 of 93 ma-ternal plasma reads and later validated by PCR and resequencing. The mu-tation, which is not observed in dbSNP nor among coding exons sequencedfrom >4000 individuals as part of the National Heart, Lung, and Blood In-stitute Exome Sequencing Project (http://evs.gs.washington.edu), createsa leucine-to-proline substitution at a site conserved across all alignedmammalian genomes (University of California, Santa Cruz, GenomeBrowser)in a gene implicated in Parkinson’s disease by genome-wide associationstudies (25).
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 6 June 2012 Vol 4 Issue 137 137ra76 3
on
Mar
ch 6
, 201
3st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m

NGS caveats for clinical sequencing
• Short read technology/aligning to reference genome
• SNV vs indels, CNV…… • Small repeats/repe;;ve sequence • Phasing
• Interpreta;on of unique variants!